 Jing Lu
Jing Lu Taotao Liang
Taotao Liang Ping Li2
Ping Li2 Qingsong Yin
Qingsong Yin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 06 February 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1086803
This article is part of the Research TopicThe Immunosuppressive Tumor Microenvironment and Strategies to Revert its Immune Regulatory Milieu for Cancer ImmunotherapyView all 15 articles
The tumor microenvironment (TME) is implicated in tumorigenesis, chemoresistance, immunotherapy failure and tumor recurrence. Multiple immunosuppressive cells and soluble secreted cytokines together drive and accelerate TME disorders, T cell immunodeficiency and tumor growth. Thus, it is essential to comprehensively understand the TME status, immune cells involved and key transcriptional factors, and extend this knowledge to therapies that target dysfunctional T cells in the TME. Interferon regulatory factor 4 (IRF4) is a unique IRF family member that is not regulated by interferons, instead, is mainly induced upon T-cell receptor signaling, Toll-like receptors and tumor necrosis factor receptors. IRF4 is largely restricted to immune cells and plays critical roles in the differentiation and function of effector cells and immunosuppressive cells, particularly during clonal expansion and the effector function of T cells. However, in a specific biological context, it is also involved in the transcriptional process of T cell exhaustion with its binding partners. Given the multiple effects of IRF4 on immune cells, especially T cells, manipulating IRF4 may be an important therapeutic target for reversing T cell exhaustion and TME disorders, thus promoting anti-tumor immunity. This study reviews the regulatory effects of IRF4 on various immune cells in the TME, and reveals its potential mechanisms, providing a novel direction for clinical immune intervention.
The occurrence and development of tumors highly depend on the surrounding matrix environment, called the tumor microenvironment (TME). The oncogene proteins expressed by tumor cells stimulate and induce the abnormal activation of effector T cells (1, 2). Multiple soluble tumor-derived products, such as the chemokines CCL2, CCL5 and the cytokines IL10 and TGFβ, etc., recruit tumor-associated macrophages (TAMs) (3–6) and myeloid-derived suppressor cells (MDSCs) (7) into the TME, and lead to the impairment of differentiation, maturation and function of dendritic cells (DCs) (8, 9). These factors in turn jointly aggravates TME disorders, inhibits the anti-tumor immunity of effector T cells, and induces T cell exhaustion and the development of regulatory T (Treg) cells (2). As a result, apart from genetic deficiencies, the immunosuppressive TME is considered to be involved in tumorigenesis (10), chemoresistance, immunotherapy failure and even tumor recurrence (2, 6).
Given this reliance on the TME, there is an opportunity for anti-tumor immunotherapies that work by targeting TME components and their signaling pathways (11, 12). Although tremendous progress has been made in the past few years, including immune checkpoint inhibitors (13), bispecific antibodies (14) and chimeric antigen receptor (CAR) T cells (15), many studies focusing on elements of the TME have failed to show promising efficacy in patients, particularly with sustainable efficacy (16–18). Therefore, the development of new immunotherapies may also require consideration of the key transcription regulatory factors involved in multiple components and processes in the TME.
Interferon regulatory factor 4 (IRF4) is a member of the interferon regulatory factor (IRF) family, and its unique characteristics and the importance in multiple biological processes have been highlighted by oncology and immunology. It first serves as an oncogene or a tumor suppressor in multiple types of lymphoid neoplasms (19–21). In addition, intriguingly, accumulating studies have demonstrated that IRF4 is a central determinant of differentiation, activation and effector function for various immune cells (22, 23). IRF4 is essential for the sustained differentiation and proliferation of CD8+ cytotoxic T cells (CTLs) and T helper 1 (Th1) cells, promoting anti-tumor immunity. In parallel, IRF4 is also involved in T cell exhaustion in specific biological contexts (24, 25). In contrast, it plays an important role in the differentiation and function of various immunosuppressive cells, such as Th2 cells, Treg cells, TAMs and MDSCs, establishing an immunosuppressive TME to inhibit anti-tumor immunity and favor the immune escape and survival of tumor cells (3–5, 7) (Figure 1). Thus, an in-depth understanding of the effects and potential mechanisms of IRF4 in a variety of immune cells and a disordered TME may provide new directions for clinical immune intervention.
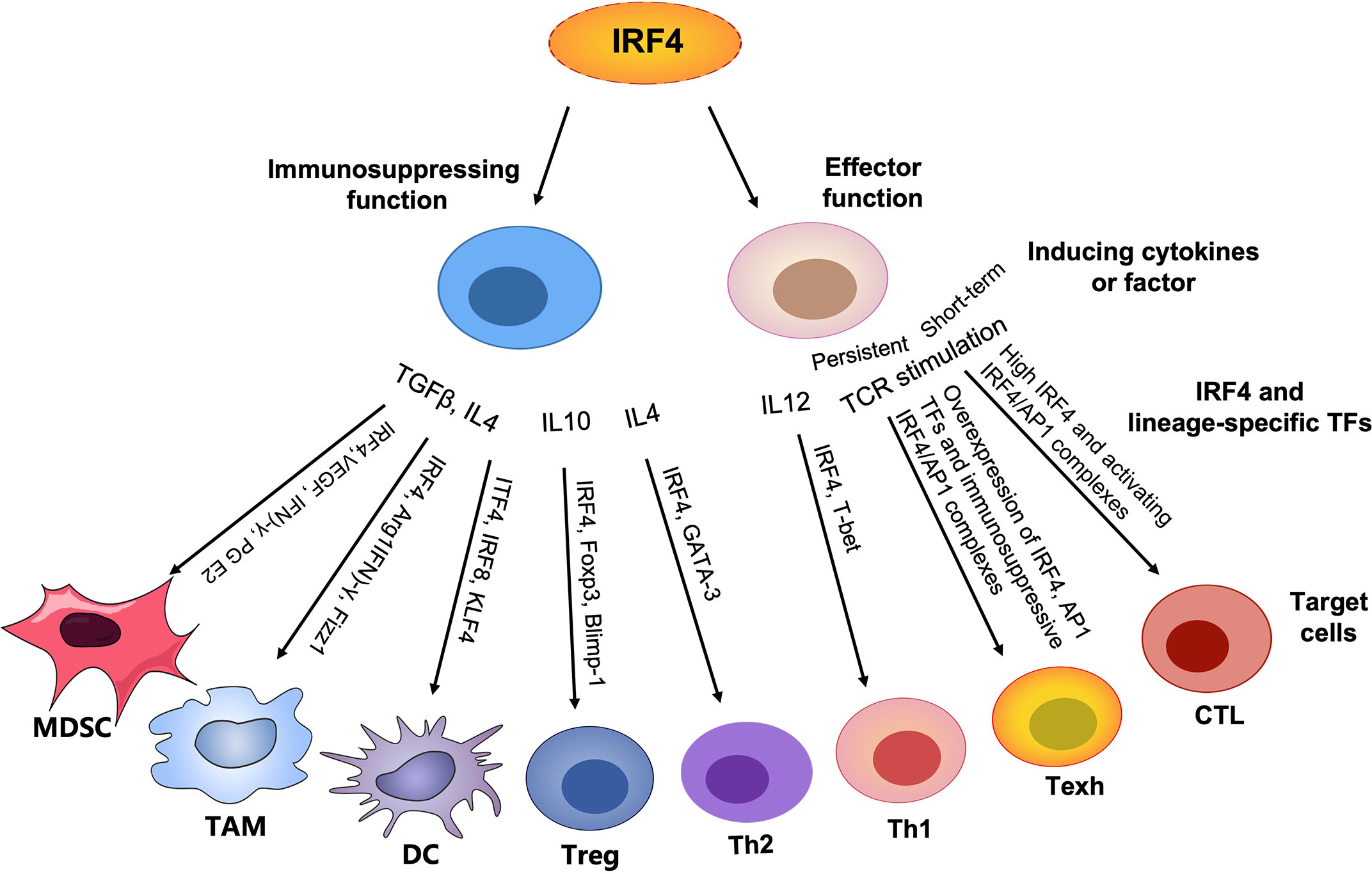
Figure 1 Graphical abstract. In contrast to lineage-specific TFs, IRF4 plays an important role in T cell differentiation and function by regulating the expression of corresponding transcription factors (TFs) to control the generation of other lineages, particularly the differentiation and proliferation of effector T cells, promoting anti-tumor immunity. However, persistently high expression of IRF4 and AP1 family members leads to overabundance of IRF4/AP1 complexes to drive T cell exhaustion. In addition, IRF4 plays an important role in the development and function of a series of immunosuppressive cells, such as MDSCs, TAMs, immature DCs, Treg cells and Th2 cells, maintaining immune homeostasis and in parallel establishing an immunosuppressive TME and inhibiting anti-tumor immunity.
The IRF family consists of nine members (IRF1-IRF9) in mammals that play important roles in regulating innate and adaptive immune responses. Unlike other IRFs, IRF4 is a unique family member that is not regulated by interferons (IFNs) (22), instead, is mainly induced upon T-cell receptor (TCR) signaling, Toll-like receptors (TLRs; such as TLR4 and TLR9) and tumor necrosis factor receptors. The expression of IRF4 is restricted to immune cells, including T and B cells, macrophages and DCs (19, 22). In naïve T cells, IRF4 is expressed at low levels (23); however, following TCR signaling it is immediately induced and mediates critical immune responses by interacting with upstream signaling pathways, such as the TCR signaling, and its diverse binding partners (26).
IRF4 is composed of three structural domains: a variable C-terminal functional regulatory domain, a highly conserved N-terminal DNA-binding domain and an intermediate compact linker domain (22, 27, 28) (Figure 2). IRF4 interacts with numerous DNA-binding domains to play corresponding functions as a homodimer or heterodimer (29). IRF4 binds to interferon-stimulated response elements (ISREs) to regulate the activation of interferon-stimulated genes (ISGs) as a homodimer. However, the formation of heterodimeric complexes containing IRF4 depends largely on the target cell type. For instance, IRF4 engages activator protein 1 (AP1)-IRF composite elements (AICE) as a heterodimer mainly in T cells, germinal center B cells and plasma cells (23, 28). Whereas the binding of IRF4 with erythroblast transformation (ET)-specific transcription factors (TFs) is largely restricted to B cells and DCs. Of note, the binding of IRF4 to AICE requires AP1 family TFs, including basic leucine zipper transcription factor ATF-like (BATF), BATF3 and Jun family members, such as JunB, c-Jun, for high-affinity interaction (23, 30–33). These TFs form ternary complexes through physical interaction to coordinately regulate the differentiation and function of T cells, as well as T cell exhaustion, in a special microenvironment (24, 33–35).

Figure 2 Schematic diagram of IRF4 structure. IRF4 consists of three structural domains: a highly conserved N-terminal DNA-binding domain (DBD), a variable C-terminal IRF association domain (IAD) and an intermediate linker domain (ILD). The DBD is characterized by five conserved tryptophans enabling it to form a helix–loop–helix motif that facilitates DNA binding. IAD is a protein–protein interaction domain that mediates the interaction of IRF4 with itself or multiple distinct transcription factors. IAD also contains a C-terminal auto-inhibitory region (AR) which physically interacts with DBD and results in low DNA binding affinity.
Collectively, IRF4 can signal to regulate diverse transcriptional programs through complexes containing ET or AP1 TF motifs in different cell types depending on the corresponding cellular context, particularly T cell exhaustion in the TME, thus suggesting new directions for improving anti-tumor immunity by modulating IRF4-dependent transcription.
According to different functions, CD4+ T cells can be divided into CD4+ effector T cells, including Th1, Th2 and Th17 cells, which predominantly promote the immune response, T follicular helper cells (Tfh), which orchestrate antibody responses (26), and Treg cells, which are characterized by their inhibition of the immune response and maintenance of immune tolerance (26, 36, 37). In contrast to lineage-specific TFs (e.g., T-bet for Th1, GATA3 for Th2, RORγt for Th17, B-cell lymphoma 6 (Bcl6) for Tfh and Foxp3 for Treg), TCR signaling-induced IRF4 plays an important role in Th cell differentiation and function by regulating the expression of corresponding TFs to control the generation of other lineages, thus determining the fate of Th cells (23, 26, 29, 38).
Th cell differentiation is regulated by the coordinated functions of distinct cytokines and transcription factors. A recent study has demonstrated that increased IRF4 promotes the differentiation of CD4+ CD25low Teff cells, including Th1, Th2 and Th17 cells, at the expense of Tfh cells (26). In fact, the development and differentiation of Tfh cells only needs an appropriate amount IRF4 in addition to specific TFs, including Bcl-6 and signal transducer and activator of transcription 3 (STAT3) (26, 39). B-lymphocyte-induced maturation protein 1 (Blimp1) is a critical antagonist for Tfh cell differentiation, but it is an important TF for other Th cells, including Th1, Th2, Th17 and Treg cells (40). It has been found that IRF4 cooperates with STAT3 to activate Blimp1 (41), and lack of IRF4 in CD4+ T cells reduces binding to STAT3, resulting in Tfh deficiency (41, 42).
Increasing studies have shown that IRF4 regulates Th17 cell development (43–45). IRF4 knockout decreases the expression of RORγt, a specific TF in Th17 cells (45, 46), which leads to a decrease in Th17 counts, in line with a reduction in serum IL17 and IL21 (47). Likewise, IRF4 deficiency also results in the impairment of Th2 cell differentiation and function by reducing GATA3 and IL4, as well as growth factor independence 1 (Gfi1), a transcriptional repressor required by Th2 cells (48, 49), instead, can promote the T-bet expression and skew toward Th1 cells (48), suggesting that IRF4 plays a pivotal role in the development of Th2 cells rather than Th1 cells. Additionally, IRF4 deficiency inevitably impairs the development of Th2 cells (49). Collectively, IRF4 regulates the differentiation and function of diverse Th subsets that mainly depend on its expression level as well as lineage-specific TFs (26).
Treg cells are indispensable for maintaining immune tolerance (37, 50); nevertheless, they also impair anti-tumor capability and promote tumor growth, particularly tumor-infiltrating Treg cells (51). Foxp3 is a lineage-defining TF for Tregs and the key regulator of its development and function (52, 53). IRF4, which acts downstream of Foxp3, can physically and functionally interact with Foxp3 and cooperate with BATF3 to regulate Foxp3 expression (54, 55), which instructs effector Treg cell differentiation and immune suppression (56). Moreover, Blimp1 is a target of Foxp3 in Treg cells, and it is directly induced by IRF4 (57, 58). Accordingly, lack of IRF4 in Treg cells suppresses Blimp1 expression, and more intriguingly, leads to decreases in multiple Treg-related molecules, such as inducible T cell costimulatory (ICOS), IL10 and IL1 receptor 11 (IL1RL1), confirming that IRF4 cooperates with Blimp1 to regulate the differentiation and function of Treg cells (56, 58).
Additionally, compared with IRF4-deficient Treg cells, IRF4+ Treg cells overexpress BATF, IKAROS family zinc finger 2 (IKZF2), Ki67, ICOS and inhibitory molecules, such as programmed cell death protein 1 (PD1) and T cell immunoreceptor with immunoglobulin and ITIM domain (TIGIT) (38), exhibiting a highly activated phenotype and strong inhibitory effects in several tumors (59–61). In particular, an increase in intratumoral IRF4+ Treg cells with superior suppressive activity was significantly correlated with early tumor recurrence and poor disease-free survival (DFS) and overall survival (OS) (38). Accordingly, inhibition of IRF4 severely impaired the development and function of Treg cells at the tumor-infiltrating sites and significantly repressed tumor growth in a mouse model (38, 51). Collectively, growing evidence implicates IRF4 plays a central role in the differentiation and immunosuppressive activity of Treg cells in the TME, and IRF4+ Treg cells definitely inhibit anti-tumor immunity. Therefore, specifically targeting IRF4 in Treg cells may reverse the tumor microenvironment from immunosuppression to immune activation against tumor cells, which may become an effective anti-tumor therapeutic strategy.
CD8+ T cells play critical roles in adaptive immunity. Antigen stimulation drives naïve CD8+ T cells to rapidly undergo a step-by-step process of early activation, clonal expansion and differentiation (62–65). In addition to early activation, IRF4 participates in the entire process of differentiation and function of effector CD8+ T cells (66, 67). Intriguingly, the amount and duration of IRF4 expression determine the fate of CD8+ T cells, which are differentiated into CD8+ effector T cells or exhausted T cells (24, 67–69).
Following antigen stimulation, naïve CD8+ T cells are differentiated into a large number of antigen-specific short-lived effector cells (SLECs) (62, 63), exerting cytotoxic activity (Figure 3A). Mechanically, antigen stimulation drives the expression of TCR responsive factor IRF4 (68). Next, IRF4 combined with AP1 family TFs form an activating IRF4/AP1 complex, which integrates TCR and costimulatory signals to induce the production of a series of effector cytokines. After antigen clearance, the expression of IRF4 decreased, followed by an increase in expression of stemness-like gene T cell factor 7 (Tcf7; encoding TCF1) (Figure 3B), and further producing memory precursor cells (MPECs) and TCF1+ memory-like T cells to rapidly function in the secondary response (64, 65) (Figure 3A).
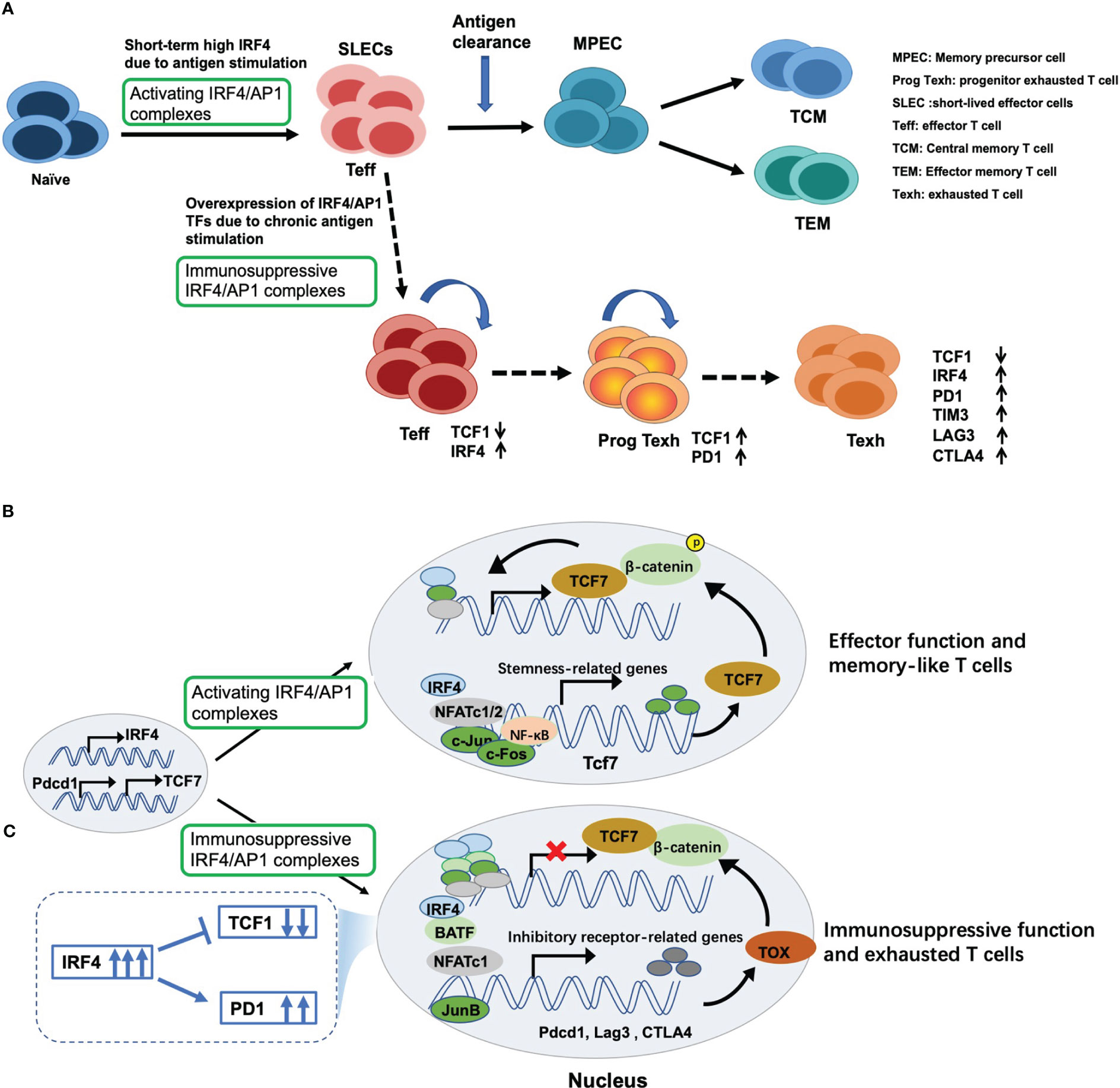
Figure 3 Dual regulatory effects of IRF4 on T cell immunity and underlying mechanisms. (A) Antigen stimulation drives and induces the expression of IRF4, which contributes to maintaining the expansion and sustained differentiation of effector CD8+ T cells. However, sustained overexpression of IRF4 due to chronic antigen stimulation drives CD8+ T cell exhaustion. Mechanically, (B) once antigen stimulation, IRF4 is induced and combined with its binding partners to form activating IRF4/AP1 complexes, thus inducing the production of effector cytokines and exerting cytotoxic activity. Once antigen clearance, the expression of IRF4 decreases, followed by an increase in expression of stemness-like gene TCF7 expression, thus producing TCF1+ memory-like T cells; (C) however, persistent overexpression of IRF4 and AP1 family members, such as BATF, BATF3 and JunB, leads to the formation of immunosuppressive IRF4/AP1 complexes, which opens multiple exhaustion-related chromatin regions, promoting the expression of inhibitory receptors and NR4A and TOX family members, which inhibits TCF7 expression and eventually drives CD8+ T cell exhaustion.
The intensity of TCR signaling regulates the expression of IRF4 (66, 70). High levels of IRF4 in CD8+ T cells contribute to the clonal expansion of SLECs, which are critical for maintaining effective anti-tumor immunity (71) and acute pathogen control (64). Interestingly, ectopic expression of IRF4 remarkably enhances the clonal expansion and effector cytokine production of T cells induced by low-intensity TCR signaling (69). Conversely, selective knockout of IRF4 in peripheral CD8+ T cells leads to progressive loss of the effector function of CD8+ T cells (72–74). The RNA-binding protein Roquin1, a key target upstream of IRF4, can effectively inhibit the expansion of CD8+ T cells (75). Accordingly, lack of Roquin1 can significantly promote the proliferation of CD8+ T cells by upregulating IRF4 (71). However, if IRF4 is also deficient, the expansion-promoting effects caused by Roquin1 deficiency is completely abolished (71). Therefore, the Roquin-IRF4 axis may also serve as a potential target for enhancing anti-tumor immunity.
IRF4 also converts TCR affinity into appropriate transcriptional programs, linking metabolic function to T cell clone expansion and effector differentiation (76) by regulating the expression of key molecules required for aerobic glycolysis on effector T cells, including hypoxia inducible factor1 α (HIF1α) and forkhead box protein o1 (Foxo1) (77). Compared with weak or low-affinity TCR stimulation, strong or high-affinity TCR stimulation contributes to increased glucose uptake in an IRF4-dependent manner (78). Taken together, IRF4 regulates the expansion and differentiation of effector CD8+ T cells by translating the TCR signal and converting it to metabolic function.
Not surprisingly, similar to initial antigen stimulation, IRF4 overexpression significantly induces an increase in the cytotoxicity of memory CD8+ T cells (32, 68, 79). By contrast, IRF4 deficiency may cause memory CD8+ T cells to produce but not proliferate (68), which results in impairment of the effector function (32, 72, 79). So far, at least three types of memory CD8+ T cells have been defined: central memory T (TCM) cells, effector memory T (TEM) cells and tissue-resident memory T (TRM) cells (80). Compared with TEM cells, TRM cells express higher levels of IRF4, and their formation and maintenance are IRF4 dependent (32). IRF4 deletion leads to an increase in TEM cells and a decrease in TRM cells, but it does not affect the total number of memory T cells (32). Thus, targeting IRF4 may strongly reduce the number of TRM cells, thus substantially weakening transplant rejection (81).
In addition, recent studies have shown that TCF1 is essential for maintaining CD8+ TCM cells and serves as a positive biomarker for prolonged survival and effective responses to PD1 inhibitors in various solid tumors and hematological malignancies (82–85). Undoubtedly, high-level IRF4 is beneficial to the initial effector function, but sustained overexpression of IRF4 inhibits the expression of TCF1, which further damages the production of antigen-specific TCM cells and is not conductive to the rapid effect function in recall responses (24). Collectively, accumulating studies have demonstrated that IRF4 is indispensable for robust proliferation and the effector function of memory T cells in recall responses.
High IRF4 is essential for maintaining the differentiation and expansion of effector CD8+ T cells (68, 72). However, too much is as bad as too little. Persistent antigen stimulation due to tumor or chronic viral infection can cause constitutively high expression of IRF4, which in turn induces CD8+ T cell exhaustion (24). There are several characteristics of exhausted CD8+ T cells (Figures 3A, 4): (1) up-regulation of multiple inhibitory receptors (86), (2) progressive loss of effector function and impaired differentiation of potential memory T cells (85, 87), (3) decreased production of cytokines involved in chemotaxis, adhesion and migration, and (4) metabolic deficiency (88). Thus, functional exhaustion is probably due to both active suppression and passive defects in signaling and metabolism.
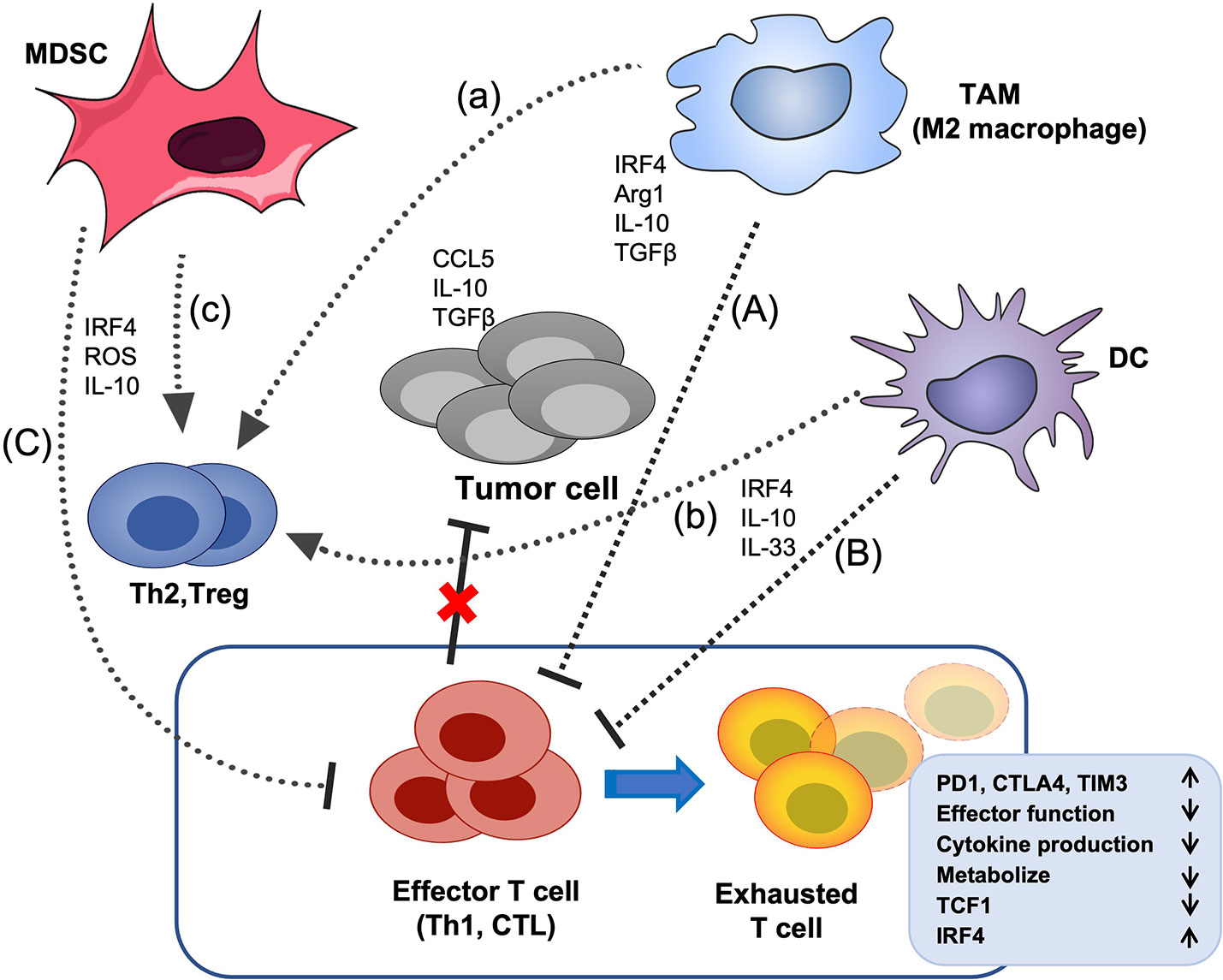
Figure 4 The effects of IRF4 on the crosstalk between immunosuppressive cells and T cells in the TME. Tumor cells and multiple soluble chemokines and cytokines recruit and induced various immunosuppressive cells, such as MDSCs, TAMs and DCs to the TME, which further aggravate the TME disorder and promote tumor growth. These myeloid derived immunosuppressive cells can suppress the effector function of CTL and Th1 cells and promote the differentiation of Treg cells and Th2 cells. In addition, tumor-related antigens stimulate the abnormal activation of effector T cells, ultimately, lead to the CD8+ T cell exhaustion, characterized by up-regulation of multiple inhibitory receptors, progressive loss of effector function and impaired differentiation of memory T cells, etc. IRF4 plays critical roles in the generation of various immunosuppressive cells, and the above crosstalk between myeloid derived immunosuppressive cells and effector T cells (A–C) and Treg cells (a-c) in the TME. The black arrow indicates promotion, the black horizontal line indicates inhibition, and the red cross indicates that the anti-tumor activity of effector T cells is impaired.
Studies have demonstrated that the epigenetic and transcriptional programs driving CD8+ T cell exhaustion are triggered by sustained antigen-dependent activation of TCR signaling, leading to two events: (1) the sustained overexpression of TCR-responsive IRF4 and its binding partners, mainly AP1 family members, including BATF, BATF3, JunB and JunD (24, 35, 89–92), as well as nuclear factor of activated T cells (NFAT), a key regulator of T cell activation (93), followed by (2) sustained expression of multiple exhaustion-related molecules (24). Specifically, overexpressed IRF4 binding with AP1 family members or NFAT leads to an overabundance of IRF4/AP1 complexes or NFAT homodimers that are recruited to specific DNA sites to open multiple exhaustion-related chromatin regions, including inhibitory receptors, such as PD1, T-cell immunoglobulin and mucin domain 3 (TIM3) and cytotoxic T lymphocyte antigen 4 (CTLA4) (24, 35, 94, 95), as well as orphan nuclear receptor 4A (NR4A) and thymocyte selection-associated high mobility group box (TOX) family members, which act to impose exhaustion (96, 97), further inhibiting TCF1 expression (Figure 3C) (24, 35). These events eventually drive CD8+ T cell exhaustion and limit the development of TCF1+ memory-like T cells and anti-tumor activity (Figure 3A). This chromatin binding imbalance due to the accumulation of IRF4/AP1 TF complexes was also found in CAR-T cell therapy (89).
Fortunately, Lynn et al. (89) found that ectopic overexpression of c-Jun in exhausted CAR-T cells can effectively rescue exhaustion and restore anti-tumor activity by disrupting and/or displacing immunosuppressive transcriptional complexes containing IRF4 and AP1 family members (89). Moreover, based on the overexpression of BATF and IRF4 in exhausted T cells (89, 98), knockdown of BATF or IRF4 could remarkably enhance the tumor-killing ability of CAR-T cells by reversing their exhaustion and prolonging their persistence (89, 90). Likewise, Seo et al. (25) found that overexpressed BATF in BATF-transduced CAR-T cells could cooperate with appropriate amount of IRF4 to counteract exhaustion, promoting the expansion of CD8+ CAR-T cells and increasing their effector cytokine production. Nevertheless, inhibiting the interaction between BATF and IRF4 will greatly weaken the tumor control ability of BATF-overexpressing CAR-T cells (25).
Collectively, these findings show that persistent overexpression of IRF4 drives T cell exhaustion depending on the specific microenvironment and the amount and functional status of its binding partners. Therefore, manipulating the formation of IRF4/AP1 complexes may be an inspiring therapeutic strategy to overcome T cell exhaustion. Yet, the core transcriptional network of IRF4 involved in these two opposing programs still needs to be further elucidated.
Various immunosuppressive cells and multiple soluble chemokines and cytokines in the TME interact to not only establish an immunosuppressive TME but also directly or indirectly inhibit the proliferation and activation of CD8+ T cells (99, 100), which may cause chemoresistance and failure of immunotherapy and facilitate tumor growth and metastasis (101–103). IRF4 plays important and complicated roles in the development and function of immunosuppressive cells and their interaction with T cells (Figure 4) (104, 105).
There are two types of macrophages: M1 (anti-tumor activity) (106, 107) and M2 (pro-tumor activity) (108). Generally, TAMs mainly refer to M2 macrophages, which are characterized by high expression of arginase1 (Arg1), chitinase-like 3 (Ym1/Chil3), found in inflammatory zone 1 (Fizz1) and mannose receptor (MR) (109, 110). IRFs play a key role in macrophage maturation and phenotypic polarization. Of the nine IRFs, IRF1, IRF5 and IRF8 are involved in the commitment of M1 macrophages, whereas IRF3 and IRF4 are crucial for M2 macrophage polarization through regulating the expression of Arg1 and Ym1, which further sufficiently produces Th2 and directly suppresses effector T cell proliferation (111–113).
In addition, it has been reported that Jumonji domain containing 3 (Jmjd3) is essential for M2 macrophage polarization, and IRF4 is a Jmjd3 target gene (110, 114). Phosphatidylserine released by apoptotic tumor cells could induce the polarization and accumulation of M2 macrophages via a STAT3-Jmjd3-IRF4 signaling axis (115); therefore, down-regulation of Jmjd3 by targeting the STAT3-Jmjd3-IRF4 axis may be a candidate approach for inhibiting the accumulation of M2 macrophages in tumor sites and remodeling the TME. Moreover, some miRNAs have been found to promote the transformation of macrophages from M2 to M1 by targeting IRF4 to activate IRF5 (116, 117). Given that IRF4 promotes the polarization of M2 macrophages, targeting IRF4 to reprogram TAM polarization in the TME appears to be a promising therapy for tumors.
DCs, known as professional antigen presenting cells, play a major role in orchestrating immune responses, and can be mainly divided into three subtypes: plasmacytoid DCs (pDCs), classical DCs (cDCs, including cDC1 and cDC2), and monocyte-derived DCs (mo-DCs) (9, 118, 119). However, the differentiation and maturation of DCs are often impaired by the immunosuppressive TME, which leads to DC dysfunction and induces tolerance to tumor cells (8, 9, 118, 119). For instance, mature pDCs exert immunostimulatory function, which is characterized by the production of large amounts of type I IFNs. Whereas, in the TME, pDCs with reduced production of type I IFNs favor the development of Treg cell, exert immunosuppressive effects on CTLs and promote tumor progression (8, 9, 120, 121). Several studies have indicated a role for IRF4 in development of monocytes, pDCs, and cDCs (122–124). IRF4 contributes to the differentiation of pDCs (122). In addition, IRF4 plays a key role in the development of cDC2 and promotes their survival and migration to lymph nodes and is essential TF for cDC2-mediated Th2 induction (122). By contrast, inhibition of IRF4 in DCs represses Th2 and promotes Th17 responses (123).
The monocytes in the TME can prioritize differentiation into monocyte-derived macrophages (mo-Macs) rather than mo-DCs (3, 105). The presence of mo-DCs has been correlated with CD8+ T cell activation and successful anti-tumor therapy (125). IRF4 is essential for human mo-DC differentiation and efficient antigen cross-presentation, whereas IRF4-deficent monocytes are phone to differentiation into mo-Macs (124). Devalaraja et al. found that the TME induces tumor cells to produce retinoic acid (RA) in murine sarcoma models, which drives intratumor monocyte polarization to mo-Macs instead of mo-DCs by inhibiting IRF4 (3). Interestingly, overexpression of IRF4 in human monocytes can sufficiently block RA-mediated mo-Mac differentiation (3, 124). Collectively, these results suggest that IRF4 plays critical and complicated roles in the maturation and differentiation of DCs in the TME.
MDSCs are immature myeloid cells that do not differentiate into mature myeloid cells, and this is a major obstacle to achieving successful immunotherapy in tumors (126, 127). Two major subpopulations, monocytic (M) MDSCs and polymorphonuclear (PMN)-MDSCs, have an immune suppressive function. IRF4 plays a role in the lymphoid cell development. However, IRF4 expression is decreased in immature myeloid cells, such as MDSCs in tumor-bearing mice and chronic myeloid leukemia cells (104, 128). Accordingly, IRF4 deficiency further favors the generation of MDSCs in the TME, and increases the expansion of M-MDSCs and the infiltration of PMN-MDSCs with a strong suppressive capacity, which inhibits the proliferation of CD8+ T cells through IL10 and ROS generation and promotes tumor growth (104, 129). By contrast, an increase in the IRF4 expression in MDSCs from bone marrow cells inhibits the numbers of MDSCs through induction differentiation, and further damages the immunosuppressive function of MDSCs (104). Unfortunately, IRF4 expression is remarkably suppressed during the development of MDSCs and tumor formation in the TME (104).
Altogether, these data show that IRF4 plays a critical role in preventing the generation of MDSCs; nevertheless, IRF4 expression is limited by tumors and MDSCs, which may in turn boost the accumulation and suppressive activity of MDSCs to accelerate the generation of an immunosuppressive TME. Thus far, the exact mechanisms regulating IRF4 in the differentiation of MDSCs remains largely unknown.
IRF4 plays key roles in the development of various immunosuppressive cells in the TME. More importantly, this TF is also indispensable in the differentiation and function of effector T cells, particularly memory T cells in the secondary response (32, 64, 78). Notably, the amount and duration of IRF4 expression determines CD8+ T cell differentiation into effector T cells or exhausted T cells, depending on the specific microenvironment and states of its binding partners (24, 34, 35, 91). Thus far, the dual regulatory mechanism of IRF4 in T cell immunity is not completely clear. Given the imbalance between the activating and immunoregulatory IRF4/AP1 complexes induced by persistent high expression of IRF4 and AP1 family members in specific contexts, manipulating the composition of the IRF4/AP1 complexes may be a novel therapeutic strategy for overcoming T cell exhaustion and improving anti-tumor potency.
Recently, several studies have reported exciting findings, including the regulation of the physical interaction between IRF4 and its binding partners, the formation of ternary complexes through overexpression of BATF or c-Jun, and the regulation of the amount of IRF4 or BATF, which are essential for rescuing exhaustion and improving anti-tumor potency in tumor-specific CAR-T cells (25, 89, 90). In addition, several recent studies have focused on targeting Roquin and Regnase1, negative regulators of T cell activation and differentiation, to enhance the proliferation and persistence of tumor-antigen-specific CD8+ T cells or CAR-T cells and effectively inhibit tumor growth (71, 130–132). In fact, the beneficial effects of the regulation of these targets are caused not only by loss of function of a single gene, but likely also caused by the cooperative regulation of multiple targets. For instance, the promotion of the survival and proliferation of tumor-antigen-specific CD8+ T cells by inactivating Roquin1 is highly dependent on the expression of IRF4 (71). Similarly, Regnase1 deficiency contributed to CAR-T cell survival and proliferation, which also specifically required BATF (130), further enhancing recall responses by increasing TCF1+ CAR-T cell population (131). By coincidence, proper reduction of IRF4 contributes to the generation of TCF1+ memory T cells that control tumor recurrence (25). Together, these findings point to promising new targets for improving immunotherapy.
Taken together, based on the close cooperation and regulatory relationships between IRF4, BATF, TCF1 and Roquin or Regnase1, targeting IRF4 or IRF4-based multi-target combination is an important direction for regulating human anti-tumor T cell immunity and the TME to improve therapeutic efficacy in the future.
JL reviewed the literature and wrote the manuscript. TL and PL contributed to literature collection and manuscript revision. QY designed the review, and wrote and revised the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by Bethune · Blood Scientific Research Capacity Building Project (J202201E023), the Medical Science and Technology Research Project of Henan Province (LHGJ20220185) and National Natural Science Foundation of China (82200222).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Fu C, Jiang A. Dendritic cells and CD8 T cell immunity in tumor microenvironment. Front Immunol (2018) 9:3059. doi: 10.3389/fimmu.2018.03059
2. Nakamura K, Smyth MJ. Myeloid immunosuppression and immune checkpoints in the tumor microenvironment. Cell Mol Immunol (2020) 17(1):1–12. doi: 10.1038/s41423-019-0306-1
3. Devalaraja S, To TKJ, Folkert IW, Natesan R, Alam MZ, Li M, et al. Tumor-derived retinoic acid regulates intratumoral monocyte differentiation to promote immune suppression. Cell (2020) 180(6):1098–114.e16. doi: 10.1016/j.cell.2020.02.042
4. Mougiakakos D, Bach C, Bottcher M, Beier F, Röhner L, Stoll A, et al. The IKZF1-IRF4/IRF5 axis controls polarization of myeloma-associated macrophages. Cancer Immunol Res (2021) 9(3):265–78. doi: 10.1158/2326-6066.CIR-20-0555
5. Carbó JM, León TE, Font-Díaz J, de la Rosa JV, Castrillo A, Picard FR, et al. Pharmacologic activation of LXR alters the expression profile of tumor-associated macrophages and the abundance of regulatory T cells in the tumor microenvironment. Cancer Res (2021) 81(4):968–85. doi: 10.1158/0008-5472.CAN-19-3360
6. Li K, Shi H, Zhang B, Ou X, Ma Q, Chen Y, et al. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transd Target Ther (2021) 6(1):362. doi: 10.1038/s41392-021-00670-9
7. Yang Q, Xie H, Li X, Feng Y, Xie S, Qu J, et al. Interferon regulatory factor 4 regulates the development of polymorphonuclear myeloid-derived suppressor cells through the transcription of c-myc in cancer. Front Immunol (2021) 12:627072. doi: 10.3389/fimmu.2021.627072
8. Verneau J, Sautés-Fridman C, Sun CM. Dendritic cells in the tumor microenvironment: Prognostic and theranostic impact. Semin Immunol (2020) 48:101410. doi: 10.1016/j.smim.2020.101410
9. Subtil B, Cambi A, Tauriello DVF, de Vries IJM. The therapeutic potential of tackling tumor-induced dendritic cell dysfunction in colorectal cancer. Front Immunol (2021) 12:724883. doi: 10.3389/fimmu.2021.724883
10. Lu C, Rong D, Zhang B, Zheng W, Wang X, Chen Z, et al. Current perspectives on the immunosuppressive tumor microenvironment in hepatocellular carcinoma: Challenges and opportunities. Mol Cancer (2019) 18(1):130. doi: 10.1186/s12943-019-1047-6
11. Xiao Y, Yu D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol Ther (2021) 221:107753. doi: 10.1016/j.pharmthera.2020.107753
12. Liu Y, Song Y, Yin Q. Effects of ibrutinib on T-cell immunity in patients with chronic lymphocytic leukemia. Front Immunol (2022) 13:962552. doi: 10.3389/fimmu.2022.962552
13. Hanna BS, Yazdanparast H, Demerdash Y, Roessner PM, Schulz R, Lichter P, et al. Combining ibrutinib and checkpoint blockade improves CD8+ T-cell function and control of chronic lymphocytic leukemia in em-TCL1 mice. Haematologica (2021) 106(4):968–77. doi: 10.3324/haematol.2019.238154
14. Lejeune M, Köse MC, Duray E, Einsele H, Beguin Y, Caers J. Bispecific, T-cell-recruiting antibodies in b-cell malignancies. Front Immunol (2020) 11:762. doi: 10.3389/fimmu.2020.00762
15. Mehta PH, Fiorenza S, Koldej RM, Jaworowski A, Ritchie DS, Quinn KM. T Cell fitness and autologous CAR T cell therapy in haematologic malignancy. Front Immunol (2021) 12:780442. doi: 10.3389/fimmu.2021.780442
16. Zhang ZZ, Wang T, Wang XF, Zhang YQ, Song SX, Ma CQ. Improving the ability of CAR-T cells to hit solid tumors: Challenges and strategies. Pharmacol Res (2022) 175:106036. doi: 10.1016/j.phrs.2021.106036
17. Zhang M, Kim JA, Huang AY. Optimizing tumor microenvironment for cancer immunotherapy: Beta-glucan-based nanoparticles. Front Immunol (2018) 9:341. doi: 10.3389/fimmu.2018.00341
18. Barnestein R, Galland L, Kalfeist L, Ghiringhelli F, Ladoire S, Limagne E. Immunosuppressive tumor microenvironment modulation by chemotherapies and targeted therapies to enhance immunotherapy effectiveness. Oncoimmunology (2022) 11(1):2120676. doi: 10.1080/2162402X.2022.2120676
19. Wong RWJ, Ong JZL, Theardy MS, Sanda T. IRF4 as an oncogenic master transcription factor. Cancers (2022) 14(17):4314. doi: 10.3390/cancers14174314
20. Sundararaj S, Seneviratne S, Williams SJ, Enders A, Casarotto MG. The molecular basis for the development of adult T-cell leukemia/lymphoma in patients with an IRF4(K59R) mutation. Protein Sci (2022) 31(4):787–96. doi: 10.1002/pro.4260
21. Maffei R, Fiorcari S, Benatti S, Atene CG, Martinelli S, Zucchini P, et al. IRF4 modulates the response to BCR activation in chronic lymphocytic leukemia regulating IKAROS and SYK. Leukemia (2021) 35(5):1330–43. doi: 10.1038/s41375-021-01178-5
22. Nam S, Lim JS. Essential role of interferon regulatory factor 4 (IRF4) in immune cell development. Arch Pharm Res (2016) 39(11):1548–55. doi: 10.1007/s12272-016-0854-1
23. Huber M, Lohoff M. IRF4 at the crossroads of effector T-cell fate decision. Eur J Immunol (2014) 44(7):1886–95. doi: 10.1002/eji.201344279
24. Man K, Gabriel SS, Liao Y, Gloury R, Preston S, Henstridge DC, et al. Transcription factor IRF4 promotes CD8(+) T cell exhaustion and limits the development of memory-like T cells during chronic infection. Immunity (2017) 47(6):1129–41.e5. doi: 10.1016/j.immuni.2017.11.021
25. Seo H, González-Avalos E, Zhang W, Ramchandani P, Yang C, Lio CJ, et al. BATF and IRF4 cooperate to counter exhaustion in tumor-infiltrating CAR T cells. Nat Immunol (2021) 22(8):983–95. doi: 10.1038/s41590-021-00964-8
26. Krishnamoorthy V, Kannanganat S, Maienschein-Cline M, Cook SL, Chen J, Bahroos N, et al. The IRF4 gene regulatory module functions as a read-write integrator to dynamically coordinate T helper cell fate. Immunity (2017) 47(3):481–97.e7. doi: 10.1016/j.immuni.2017.09.001
27. Sundararaj S, Seneviratne S, Williams SJ, Enders A, Casarotto MG. Structural determinants of the IRF4/DNA homodimeric complex. Nucleic Acids Res (2021) 49(4):2255–65. doi: 10.1093/nar/gkaa1287
28. Remesh SG, Santosh V, Escalante CR. Structural studies of IRF4 reveal a flexible autoinhibitory region and a compact linker domain. J Biol Chem (2015) 290(46):27779–90. doi: 10.1074/jbc.M115.678789
29. Li P, Spolski R, Liao W, Leonard WJ. Complex interactions of transcription factors in mediating cytokine biology in T cells. Immunol Rev (2014) 261(1):141–56. doi: 10.1111/imr.12199
30. Li P, Spolski R, Liao W, Wang L, Murphy TL, Murphy KM, et al. BATF-JUN is critical for IRF4-mediated transcription in T cells. Nature (2012) 490(7421):543–6. doi: 10.1038/nature11530
31. Murphy TL, Tussiwand R, Murphy KM. Specificity through cooperation: BATF-IRF interactions control immune-regulatory networks. Nat Rev Immunol (2013) 13(7):499–509. doi: 10.1038/nri3470
32. Harberts A, Schmidt C, Schmid J, Reimers D, Koch-Nolte F, Mittrücker HW, et al. Interferon regulatory factor 4 controls effector functions of CD8(+) memory T cells. Proc Natl Acad Sci U.S.A. (2021) 118(16):e2014553118. doi: 10.1073/pnas.2014553118
33. Glasmacher E, Agrawal S, Chang AB, Murphy TL, Zeng W, Vander Lugt B, et al. A genomic regulatory element that directs assembly and function of immune-specific AP-1-IRF complexes. Science (2012) 338(6109):975–80. doi: 10.1126/science.1228309
34. Papavassiliou AG, Musti AM. The multifaceted output of c-jun biological activity: Focus at the junction of CD8 T cell activation and exhaustion. Cells (2020) 9(11):2470. doi: 10.3390/cells9112470
35. Seo W, Jerin C, Nishikawa H. Transcriptional regulatory network for the establishment of CD8(+) T cell exhaustion. Exp Mol Med (2021) 53(2):202–9. doi: 10.1038/s12276-021-00568-0
36. Hombach AA, Abken H. Most do, but some do not: CD4(+)CD25(-) T cells, but not CD4(+)CD25(+) treg cells, are cytolytic when redirected by a chimeric antigen receptor (CAR). Cancers (2017) 9(9):112. doi: 10.3390/cancers9090112
37. Brown CY, Sadlon T, Hope CM, Wong YY, Wong S, Liu N, et al. Molecular insights into regulatory T-cell adaptation to self, environment, and host tissues: Plasticity or loss of function in autoimmune disease. Front Immunol (2020) 11:1269. doi: 10.3389/fimmu.2020.01269
38. Alvisi G, Brummelman J, Puccio S, Mazza EM, Tomada EP, Losurdo A, et al. IRF4 instructs effector treg differentiation and immune suppression in human cancer. J Clin Invest (2020) 130(6):3137–50. doi: 10.1172/JCI130426
39. Bollig N, Brüstle A, Kellner K, Ackermann W, Abass E, Raifer H, et al. Transcription factor IRF4 determines germinal center formation through follicular T-helper cell differentiation. Proc Natl Acad Sci U.S.A. (2012) 109(22):8664–9. doi: 10.1073/pnas.1205834109
40. Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, et al. Bcl6 and blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science (2009) 325(5943):1006–10. doi: 10.1126/science.1175870
41. Kwon H, Thierry-Mieg D, Thierry-Mieg J, Kim HP, Oh J, Tunyaplin C, et al. Analysis of interleukin-21-induced Prdm1 gene regulation reveals functional cooperation of STAT3 and IRF4 transcription factors. Immunity (2009) 31(6):941–52. doi: 10.1016/j.immuni.2009.10.008
42. Wu H, Deng Y, Zhao M, Zhang J, Zheng M, Chen G, et al. Molecular control of follicular helper T cell development and differentiation. Front Immunol (2018) 9:2470. doi: 10.3389/fimmu.2018.02470
43. Capone A, Volpe E. Transcriptional regulators of T helper 17 cell differentiation in health and autoimmune diseases. Front Immunol (2020) 11:348. doi: 10.3389/fimmu.2020.00348
44. Bunte K, Beikler T. Th17 cells and the IL-23/IL-17 axis in the pathogenesis of periodontitis and immune-mediated inflammatory diseases. Int J Mol Sci (2019) 20(14):3394. doi: 10.3390/ijms20143394
45. Brüstle A, Heink S, Huber M, Rosenplänter C, Stadelmann C, Yu P, et al. The development of inflammatory T(H)-17 cells requires interferon-regulatory factor 4. Nat Immunol (2007) 8(9):958–66. doi: 10.1038/ni1500
46. Sha Y, Markovic-Plese S. Activated IL-1RI signaling pathway induces Th17 cell differentiation via interferon regulatory factor 4 signaling in patients with relapsing-remitting multiple sclerosis. Front Immunol (2016) 7:543. doi: 10.3389/fimmu.2016.00543
47. Lorenz G, Moschovaki-Filippidou F, Würf V, Metzger P, Steiger S, Batz F, et al. IFN regulatory factor 4 controls post-ischemic inflammation and prevents chronic kidney disease. Front Immunol (2019) 10:2162. doi: 10.3389/fimmu.2019.02162
48. Tominaga N, Ohkusu-Tsukada K, Udono H, Abe R, Matsuyama T, Yui K. Development of Th1 and not Th2 immune responses in mice lacking IFN-regulatory factor-4. Int Immunol (2003) 15(1):1–10. doi: 10.1093/intimm/dxg001
49. Xu WD, Pan HF, Ye DQ, Xu Y. Targeting IRF4 in autoimmune diseases. Autoimmun Rev (2012) 11(12):918–24. doi: 10.1016/j.autrev.2012.08.011
50. Schumann K, Raju SS, Lauber M, Kolb S, Shifrut E, Cortez JT, et al. Functional CRISPR dissection of gene networks controlling human regulatory T cell identity. Nat Immunol (2020) 21(11):1456–66. doi: 10.1038/s41590-020-0784-4
51. Aramini B, Masciale V, Samarelli AV, Dubini A, Gaudio M, Stella F, et al. Phenotypic, functional, and metabolic heterogeneity of immune cells infiltrating non-small cell lung cancer. Front Immunol (2022) 13:959114. doi: 10.3389/fimmu.2022.959114
52. Saleh R, Elkord E. FoxP3(+) T regulatory cells in cancer: Prognostic biomarkers and therapeutic targets. Cancer Lett (2020) 490:174–85. doi: 10.1016/j.canlet.2020.07.022
53. van der Veeken J, Glasner A, Zhong Y, Hu W, Wang ZM, Bou-Puerto R, et al. The transcription factor Foxp3 shapes regulatory T cell identity by tuning the activity of trans-acting intermediaries. Immunity (2020) 53(5):971–84.e5. doi: 10.1016/j.immuni.2020.10.010
54. Arnold PR, Wen M, Zhang L, Ying Y, Xiao X, Chu X, et al. Suppression of FOXP3 expression by the AP-1 family transcription factor BATF3 requires partnering with IRF4. Front Immunol (2022) 13:966364. doi: 10.3389/fimmu.2022.966364
55. Zheng Y, Chaudhry A, Kas A, deRoos P, Kim JM, Chu TT, et al. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature (2009) 458(7236):351–6. doi: 10.1038/nature07674
56. Koizumi SI, Ishikawa H. Transcriptional regulation of differentiation and functions of effector T regulatory cells. Cells (2019) 8(8):939. doi: 10.3390/cells8080939
57. Cretney E, Leung PS, Trezise S, Newman DM, Rankin LC, Teh CE, et al. Characterization of blimp-1 function in effector regulatory T cells. J Autoimmun (2018) 91:73–82. doi: 10.1016/j.jaut.2018.04.003
58. Cretney E, Xin A, Shi W, Minnich M, Masson F, Miasari M, et al. The transcription factors blimp-1 and IRF4 jointly control the differentiation and function of effector regulatory T cells. Nat Immunol (2011) 12(4):304–11. doi: 10.1038/ni.2006
59. Van Damme H, Dombrecht B, Kiss M, Roose H, Allen E, Van Overmeire E, et al. Therapeutic depletion of CCR8(+) tumor-infiltrating regulatory T cells elicits antitumor immunity and synergizes with anti-PD-1 therapy. J Immunother Cancer (2021) 9(2):e001749. doi: 10.1136/jitc-2020-001749
60. Stéphan P, Lautraite R, Voisin A, Grinberg-Bleyer Y. Transcriptional control of regulatory T cells in cancer: Toward therapeutic targeting? Cancers (2020) 12(11):3194. doi: 10.3390/cancers12113194
61. Swatler J, Turos-Korgul L, Kozlowska E, Piwocka K. Immunosuppressive cell subsets and factors in myeloid leukemias. Cancers (2021) 13(6):1203. doi: 10.3390/cancers13061203
62. Kavazović I, Polić B, Wensveen FM. Cheating the hunger games; mechanisms controlling clonal diversity of CD8 effector and memory populations. Front Immunol (2018) 9:2831. doi: 10.3389/fimmu.2018.02831
63. Reina-Campos M, Scharping NE, Goldrath AW. CD8(+) T cell metabolism in infection and cancer. Nat Rev Immunol (2021) 21(11):718–38. doi: 10.1038/s41577-021-00537-8
64. Nayar R, Schutten E, Bautista B, Daniels K, Prince AL, Enos M, et al. Graded levels of IRF4 regulate CD8+ T cell differentiation and expansion, but not attrition, in response to acute virus infection. J Immunol (2014) 192(12):5881–93. doi: 10.4049/jimmunol.1303187
65. Richard AC. Divide and conquer: Phenotypic and temporal heterogeneity within CD8(+) T cell responses. Front Immunol (2022) 13:949423. doi: 10.3389/fimmu.2022.949423
66. Gallagher MP, Conley JM, Vangala P, Garber M, Reboldi A, Berg LJ. Hierarchy of signaling thresholds downstream of the T cell receptor and the tec kinase ITK. Proc Natl Acad Sci U.S.A. (2021) 118(35):e2025825118. doi: 10.1073/pnas.2025825118
67. Pritzl CJ, Daniels MA, Teixeiro E. Interplay of inflammatory, antigen and tissue-derived signals in the development of resident CD8 memory T cells. Front Immunol (2021) 12:636240. doi: 10.3389/fimmu.2021.636240
68. Miyakoda M, Honma K, Kimura D, Akbari M, Kimura K, Matsuyama T, et al. Differential requirements for IRF4 in the clonal expansion and homeostatic proliferation of naive and memory murine CD8(+) T cells. Eur J Immunol (2018) 48(8):1319–28. doi: 10.1002/eji.201747120
69. Yao S, Buzo BF, Pham D, Jiang L, Taparowsky EJ, Kaplan MH, et al. Interferon regulatory factor 4 sustains CD8(+) T cell expansion and effector differentiation. Immunity (2013) 39(5):833–45. doi: 10.1016/j.immuni.2013.10.007
70. Wu H, Witzl A, Ueno H. Assessment of TCR signal strength of antigen-specific memory CD8(+) T cells in human blood. Blood Adv (2019) 3(14):2153–63. doi: 10.1182/bloodadvances.2019000292
71. Zhao H, Liu Y, Wang L, Jin G, Zhao X, Xu J, et al. Genome-wide fitness gene identification reveals roquin as a potent suppressor of CD8 T cell expansion and anti-tumor immunity. Cell Rep (2021) 37(10):110083. doi: 10.1016/j.celrep.2021.110083
72. Grusdat M, McIlwain DR, Xu HC, Pozdeev VI, Knievel J, Crome SQ, et al. IRF4 and BATF are critical for CD8(+) T-cell function following infection with LCMV. Cell Death Differ (2014) 21(7):1050–60. doi: 10.1038/cdd.2014.19
73. Raczkowski F, Ritter J, Heesch K, Schumacher V, Guralnik A, Höcker L, et al. The transcription factor interferon regulatory factor 4 is required for the generation of protective effector CD8+ T cells. Proc Natl Acad Sci U.S.A. (2013) 110(37):15019–24. doi: 10.1073/pnas.1309378110
74. Zou D, Fu J, Guo Z, Chen W. Interferon regulatory factor 4 deficiency in CD8(+) T cells abrogates terminal effector differentiation and promotes transplant acceptance. Immunology (2020) 161(4):364–79. doi: 10.1111/imm.13258
75. Behrens G, Heissmeyer V. Cooperation of RNA-binding proteins - a focus on roquin function in T cells. Front Immunol (2022) 13:839762. doi: 10.3389/fimmu.2022.839762
76. Finlay D. IRF4 links antigen affinity to CD8+ T-cell metabolism. Immunol Cell Biol (2014) 92(1):6–7. doi: 10.1038/icb.2013.72
77. Man K, Miasari M, Shi W, Xin A, Henstridge DC, Preston S, et al. The transcription factor IRF4 is essential for TCR affinity-mediated metabolic programming and clonal expansion of T cells. Nat Immunol (2013) 14(11):1155–65. doi: 10.1038/ni.2710
78. Man K, Kallies A. Synchronizing transcriptional control of T cell metabolism and function. Nat Rev Immunol (2015) 15(9):574–84. doi: 10.1038/nri3874
79. Lugli E, Brummelman J, Pilipow K, Roychoudhuri R. Paths to expansion: Differential requirements of IRF4 in CD8(+) T-cell expansion driven by antigen and homeostatic cytokines. Eur J Immunol (2018) 48(8):1281–4. doi: 10.1002/eji.201847727
80. Parga-Vidal L, van Gisbergen KPJM. Area under immunosurveillance: Dedicated roles of memory CD8 T-cell subsets. Cold Spring Harb Perspect Biol (2020) 12(11):a037796. doi: 10.1101/cshperspect.a037796
81. Abou-Daya KI, Tieu R, Zhao D, Rammal R, Sacirbegovic F, Williams AL, et al. Resident memory T cells form during persistent antigen exposure leading to allograft rejection. Sci Immunol (2021) 6(57):eabc8122. doi: 10.1126/sciimmunol.abc8122
82. Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, et al. Intratumoral Tcf1(+)PD-1(+)CD8(+) T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity (2019) 50(1):195–211.e10. doi: 10.1016/j.immuni.2018.12.021
83. Pagliarulo F, Cheng PF, Brugger L, van Dijk N, van den Heijden M, Levesque MP, et al. Molecular, immunological, and clinical features associated with lymphoid neogenesis in muscle invasive bladder cancer. Front Immunol (2022) 12:793992. doi: 10.3389/fimmu.2021.793992
84. Liang T, Wang X, Liu Y, Ai H, Wang Q, Wang X, et al. Decreased TCF1 and BCL11B expression predicts poor prognosis for patients with chronic lymphocytic leukemia. Front Immunol (2022) 13:985280. doi: 10.3389/fimmu.2022.985280
85. Zhang J, Lyu T, Cao Y, Feng H. Role of TCF-1 in differentiation, exhaustion, and memory of CD8(+) T cells: A review. FASEB J (2021) 35(5):e21549. doi: 10.1096/fj.202002566R
86. Jiang W, He Y, He W, Wu G, Zhou X, Sheng Q, et al. Exhausted CD8+T cells in the tumor immune microenvironment: New pathways to therapy. Front Immunol (2021) 11:622509. doi: 10.3389/fimmu.2020.622509
87. Kim C, Jin J, Weyand CM, Goronzy JJ. The transcription factor TCF1 in T cell differentiation and aging. Int J Mol Sci (2020) 21(18):6497. doi: 10.3390/ijms21186497
88. Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity (2007) 27(4):670–84. doi: 10.1016/j.immuni.2007.09.006
89. Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, et al. C-jun overexpression in CAR T cells induces exhaustion resistance. Nature (2019) 576(7786):293–300. doi: 10.1038/s41586-019-1805-z
90. Jiang P, Zhang Z, Hu Y, Liang Z, Han Y, Li X, et al. Single-cell ATAC-seq maps the comprehensive and dynamic chromatin accessibility landscape of CAR-T cell dysfunction. Leukemia (2022) 36(11):2656–68. doi: 10.1038/s41375-022-01676-0
91. Kurachi M, Barnitz RA, Yosef N, Odorizzi PM, DiIorio MA, Lemieux ME, et al. The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8+ T cells. Nat Immunol (2014) 15(4):373–83. doi: 10.1038/ni.2834
92. Chennupati V, Held W. Feeling exhausted? tuning Irf4 energizes dysfunctional T cells. Immunity (2017) 47(6):1009–11. doi: 10.1016/j.immuni.2017.11.028
93. Martinez GJ, Pereira RM, Äijö T, Kim EY, Marangoni F, Pipkin ME, et al. The transcription factor NFAT promotes exhaustion of activated CD8(+) T cells. Immunity (2015) 42(2):265–78. doi: 10.1016/j.immuni.2015.01.006
94. Mognol GP, Spreafico R, Wong V, Scott-Browne JP, Togher S, Hoffmann A, et al. Exhaustion-associated regulatory regions in CD8(+) tumor-infiltrating T cells. Proc Natl Acad Sci U.S.A. (2017) 114(13):E2776–85. doi: 10.1073/pnas.1620498114
95. Chen J, López-Moyado IF, Seo H, Lio CJ, Hempleman LJ, Sekiya T, et al. NR4A transcription factors limit CAR T cell function in solid tumours. Nature (2019) 567(7749):530–4. doi: 10.1038/s41586-019-0985-x
96. Seo H, Chen J, González-Avalos E, Samaniego-Castruita D, Das A, Wang YH, et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc Natl Acad Sci U.S.A. (2019) 116(25):12410–5. doi: 10.1073/pnas.1905675116
97. Scott AC, Dündar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature (2019) 571(7764):270–4. doi: 10.1038/s41586-019-1324-y
98. Doering TA, Crawford A, Angelosanto JM, Paley MA, Ziegler CG, Wherry EJ. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity (2012) 37(6):1130–44. doi: 10.1016/j.immuni.2012.08.021
99. Takahashi K, Kurashina K, Yamaguchi H, Kanamaru R, Ohzawa H, Miyato H, et al. Altered intraperitoneal immune microenvironment in patients with peritoneal metastases from gastric cancer. Front Immunol (2022) 13:969468. doi: 10.3389/fimmu.2022.969468
100. Almeida-Nunes DL, Mendes-Frias A, Silvestre R, Dinis-Oliveira RJ, Ricardo S. Immune tumor microenvironment in ovarian cancer ascites. Int J Mol Sci (2022) 23(18):10692. doi: 10.3390/ijms231810692
101. Salmaninejad A, Valilou SF, Soltani A, Ahmadi S, Abarghan YJ, Rosengren RJ, et al. Tumor-associated macrophages: role in cancer development and therapeutic implications. Cell Oncol (Dordr) (2019) 42(5):591–608. doi: 10.1007/s13402-019-00453-z
102. Wu Y, Yi M, Niu M, Mei Q, Wu K. Myeloid-derived suppressor cells: an emerging target for anticancer immunotherapy. Mol Cancer (2022) 21(1):184. doi: 10.1186/s12943-022-01657-y
103. Fujimura T, Aiba S. Significance of immunosuppressive cells as a target for immunotherapies in melanoma and non-melanoma skin cancers. Biomolecules (2020) 10(8):1087. doi: 10.3390/biom10081087
104. Nam S, Kang K, Cha JS, Kim JW, Lee HG, Kim Y, et al. Interferon regulatory factor 4 (IRF4) controls myeloid-derived suppressor cell (MDSC) differentiation and function. J Leukoc Biol (2016) 100(6):1273–84. doi: 10.1189/jlb.1A0215-068RR
105. Jahchan NS, Mujal AM, Pollack JL, Binnewies M, Sriram V, Reyno L, et al. Tuning the tumor myeloid microenvironment to fight cancer. Front Immunol (2019) 10:1611. doi: 10.3389/fimmu.2019.01611
106. Wang C, Lin Y, Zhu H, Zhou Y, Mao F, Huang X, et al. The prognostic and clinical value of tumor-associated macrophages in patients with breast cancer: A systematic review and meta-analysis. Front Oncol (2022) 12:905846. doi: 10.3389/fonc.2022.905846
107. Zhang SY, Song XY, Li Y, Ye LL, Zhou Q, Yang WB. Tumor-associated macrophages: A promising target for a cancer immunotherapeutic strategy. Pharmacol Res (2020) 161:105111. doi: 10.1016/j.phrs.2020.105111
108. Cao H, Huang T, Dai M, Kong X, Liu H, Zheng Z, et al. Tumor microenvironment and its implications for antitumor immunity in cholangiocarcinoma: Future perspectives for novel therapies. Int J Biol Sci (2022) 18(14):5369–90. doi: 10.7150/ijbs.73949
109. Wang LX, Zhang SX, Wu HJ, Rong XL, Guo J. M2b macrophage polarization and its roles in diseases. J Leukoc Biol (2019) 106(2):345–58. doi: 10.1002/JLB.3RU1018-378RR
110. Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol (2010) 11(10):936–44. doi: 10.1038/ni.1920
111. El Chartouni C, Schwarzfischer L, Rehli M. Interleukin-4 induced interferon regulatory factor (Irf) 4 participates in the regulation of alternative macrophage priming. Immunobiology (2010) 215(9–10):821–5. doi: 10.1016/j.imbio.2010.05.031
112. Oishi S, Takano R, Tamura S, Tani S, Iwaizumi M, Hamaya Y, et al. M2 polarization of murine peritoneal macrophages induces regulatory cytokine production and suppresses T-cell proliferation. Immunology (2016) 149(3):320–8. doi: 10.1111/imm.12647
113. Al Mamun A, Chauhan A, Qi S, Ngwa C, Xu Y, Sharmeen R, et al. Microglial IRF5-IRF4 regulatory axis regulates neuroinflammation after cerebral ischemia and impacts stroke outcomes. Proc Natl Acad Sci U.S.A. (2020) 117(3):1742–52. doi: 10.1073/pnas.1914742117
114. Liang H, Liu B, Gao Y, Nie J, Feng S, Yu W, et al. Jmjd3/IRF4 axis aggravates myeloid fibroblast activation and M2 macrophage to myofibroblast transition in renal fibrosis. Front Immunol (2022) 13:978262. doi: 10.3389/fimmu.2022.978262
115. Liang X, Luo M, Shao B, Yang JY, Tong A, Wang RB, et al. Phosphatidylserine released from apoptotic cells in tumor induces M2-like macrophage polarization through the PSR-STAT3-JMJD3 axis. Cancer Commun (2022) 42(3):205–22. doi: 10.1002/cac2.12272
116. Xing Y, Ruan G, Ni H, Qin H, Chen S, Gu X, et al. Tumor immune microenvironment and its related miRNAs in tumor progression. Front Immunol (2021) 12:624725. doi: 10.3389/fimmu.2021.624725
117. Hu A, Chen X, Bi Q, Xiang Y, Jin R, Ai H, et al. A parallel and cascade control system: magnetofection of miR125b for synergistic tumor-association macrophage polarization regulation and tumor cell suppression in breast cancer treatment. Nanoscale (2020) 12(44):22615–27. doi: 10.1039/d0nr06060g
118. Gardner A, Ruffell B. Dendritic cells and cancer immunity. Trends Immunol (2016) 37(12):855–65. doi: 10.1016/j.it.2016.09.006
119. Laoui D, Keirsse J, Morias Y, Van Overmeire E, Geeraerts X, Elkrim Y, et al. The tumour microenvironment harbours ontogenically distinct dendritic cell populations with opposing effects on tumour immunity. Nat Commun (2016) 7:13720. doi: 10.1038/ncomms13720
120. Conrad C, Gregorio J, Wang YH, Ito T, Meller S, Hanabuchi S, et al. Plasmacytoid dendritic cells promote immunosuppression in ovarian cancer via ICOS costimulation of Foxp3(+) T-regulatory cells. Cancer Res (2012) 72(20):5240–9. doi: 10.1158/0008-5472.CAN-12-2271
121. Caro AA, Deschoemaeker S, Allonsius L, Coosemans A, Laoui D. Dendritic cell vaccines: A promising approach in the fight against ovarian cancer. Cancers (2022) 14(16):4037. doi: 10.3390/cancers14164037
122. Sichien D, Scott CL, Martens L, Vanderkerken M, Van Gassen S, Plantinga M, et al. IRF8 transcription factor controls survival and function of terminally differentiated conventional and plasmacytoid dendritic cells, respectively. Immunity (2016) 45(3):626–40. doi: 10.1016/j.immuni.2016.08.013
123. Lee J, Zhang J, Chung YJ, Kim JH, Kook CM, González-Navajas JM, et al. Inhibition of IRF4 in dendritic cells by PRR-independent and -dependent signals inhibit Th2 and promote Th17 responses. Elife (2020) 9:e49416. doi: 10.7554/eLife.49416
124. Briseño CG, Haldar M, Kretzer NM, Wu X, Theisen DJ, Kc W, et al. Distinct transcriptional programs control cross-priming in classical and monocyte-derived dendritic cells. Cell Rep (2016) 15(11):2462–74. doi: 10.1016/j.celrep.2016.05.025
125. Kuhn S, Yang J, Ronchese F. Monocyte-derived dendritic cells are essential for CD8(+) T cell activation and antitumor responses after local immunotherapy. Front Immunol (2015) 6:584. doi: 10.3389/fimmu.2015.00584
126. Bruger AM, Dorhoi A, Esendagli G, Barczyk-Kahlert K, van der Bruggen P, Lipoldova M, et al. How to measure the immunosuppressive activity of MDSC: Assays, problems and potential solutions. Cancer Immunol Immunother (2019) 68(4):631–44. doi: 10.1007/s00262-018-2170-8
127. Sanaei MJ, Salimzadeh L, Bagheri N. Crosstalk between myeloid-derived suppressor cells and the immune system in prostate cancer: MDSCs and immune system in prostate cancer. J Leukoc Biol (2020) 107(1):43–56. doi: 10.1002/JLB.4RU0819-150RR
128. Cumbo C, Tarantini F, Anelli L, Zagaria A, Redavid I, Minervini CF, et al. IRF4 expression is low in Philadelphia negative myeloproliferative neoplasms and is associated with a worse prognosis. Exp Hematol Oncol (2021) 10(1):58. doi: 10.1186/s40164-021-00253-y
129. Metzger P, Kirchleitner SV, Boehmer DFR, Hörth C, Eisele A, Ormanns S, et al. Systemic but not MDSC-specific IRF4 deficiency promotes an immunosuppressed tumor microenvironment in a murine pancreatic cancer model. Cancer Immunol Immunother (2020) 69(10):2101–12. doi: 10.1007/s00262-020-02605-9
130. Wei J, Long L, Zheng W, Dhungana Y, Lim SA, Guy C, et al. Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature (2019) 576(7787):471–6. doi: 10.1038/s41586-019-1821-z
131. Zheng W, Wei J, Zebley CC, Jones LL, Dhungana Y, Wang YD, et al. Regnase-1 suppresses TCF-1+ precursor exhausted T-cell formation to limit CAR-t-cell responses against ALL. Blood (2021) 138(2):122–35. doi: 10.1182/blood.2020009309
Keywords: IRF4, tumor microenvironment, immunosuppressive cells, T cell exhaustion, immunoregulation
Citation: Lu J, Liang T, Li P and Yin Q (2023) Regulatory effects of IRF4 on immune cells in the tumor microenvironment. Front. Immunol. 14:1086803. doi: 10.3389/fimmu.2023.1086803
Received: 01 November 2022; Accepted: 18 January 2023;
Published: 06 February 2023.
Edited by:
Mazdak Ganjalikhani Hakemi, Isfahan University of Medical Sciences, IranReviewed by:
Lionel Franz Poulin, INSERM U1003 Laboratoire de Physiologie Cellulaire, FranceCopyright © 2023 Lu, Liang, Li and Yin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qingsong Yin, am55aW5xaW5nc29uZ0AxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.