 Sijia He
Sijia He Qian Huang
Qian Huang Jin Cheng
Jin Cheng
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 13 March 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1073561
This article is part of the Research Topic Endogenous Danger Signals in Cancer Immunology and Immunotherapy View all 7 articles
Generally, the demise of cancer cells in different ways enables the body to clear these harmful cells. However, cancer cells obtain unlimited replication and immortality from successful circumvention of cell death via various mechanisms. Some evidence suggests that treatment-induced dying tumor cells even promote cancer progression. Notably, therapeutic interventions to harness the immune system against tumor cells have shown complicated influences in clinics. Herein, there is an urgent need to clarify the underlying mechanisms that influence the outcome and regulation of the immune system during cancer treatment. In this review, we provide an account on the cell death modes and the relationship between dying tumor cells with tumor immune microenvironment during cancer treatment, focusing on immunotherapy, from mechanistic standpoint to emerging limitations and future directions.
Cancer is characterized by uncontrolled cell proliferation and lack of cell death, which has become a major threat to human health. Usually, treatment options for patients with cancers depend on the stage and type of cancer, which include surgical removal, chemotherapy, radiotherapy, targeted therapy and immunotherapy. It is worth noting that immunotherapy has changed the landscape of cancer treatment. Immunotherapy is based on the knowledge of tumor escape mechanisms and aims to manipulate the immune system to reactivate the antitumor immune response and surmount the signaling pathways leading to escape. Early approaches to immunotherapy were focused on targeting cytokines that affected immune cell function. Nowadays, therapeutic alternatives are investigated to maneuver many facets of the immune system, such as immune checkpoint inhibitors (ICIs), adoptive cell therapy (ACT), oncolytic viruses, and cancer vaccines. Although, there are various approaches to tumor treatment, one of the major reasons for the unsatisfactory efficacy is that cancer cells, including those that are about to undergo death or are dying, can sculpt their surrounding microenvironment via secreting cytokines, chemokines, and other factors. An increasing number of researchers are beginning to investigate the diversity of cell death outputs and how they coordinate with the immune system. The immunogenic potential of dying tumor cells relies on their antigenicity and capacity to generate adjuvant signals commonly known as damage-associated molecular patterns (DAMPs) (1), and dying tumor cells that activate adaptive immune responses in immunocompetent hosts is also referred to as immunogenic cell death (ICD) (2). DAMPs emitted in the course of ICD, such as calreticulin (CALR), heat-shock proteins (HSPs), high mobility group box 1 (HMGB1), annexin A1 (ANXA1), secreted ATP, and type I interferons (IFNs), can be recognized by both the innate and adaptive immune systems via distinct pattern recognition receptors (PRRs) actuating chemoattraction, homing, activation, maturation, ultimately leading to the cross-presentation of tumor antigens to CD8+ cytotoxic T lymphocytes (CTLs) in the context of robust immune-stimulation (1). A tremendous amount of evidence suggests that when immune cells are present in the tumor microenvironment (TME), they promote tumor progression. Crosstalk between cancer cells and the proximal immune cells ultimately establishes soils that fosters tumor growth and recurrence. In this review, we will enumerate the current cell death occurred during cancer treatment, laying special emphasis on the link between different cell death modalities and tumor immune microenvironment (TIME), in order to figure out the underlying mechanisms that affect the results and regulation of immune system during cancer treatment.
Previous forms of cell death were classified into three main types based on macroscopic morphological changes: apoptosis (type I cell death), autophagy (type II cell death) and necrosis (type III cell death) (3). Since 2005, the cell death category system has been revamped by the Nomenclature Committee on Cell Death (NCCD), an organization formulating guidelines for the definition and interpretation of cell death from morphological, biochemical, and functional perspectives (2, 4). Fundamentally, cell death can be classified into accidental cell death (ACD) and regulated cell death (RCD) (2). ACD is uncontrolled and is a process caused by a biologically uncontrolled and harmful stimulus. Thus, ACD cannot be blocked by any medicinal or genetic interferences. However, cells undergoing ACD can release products toxic to surrounding cells, which may extend primary injury. In this context, the releasing molecules from cells undergoing ACD can be inhibited by particular Interventions, which may have a beneficial effect on prolonged disease outcomes. RCD is governed by a genetically encoded molecular machinery (2). Therefore, targeting RCD has promising potential in treating cancer. Although the forms of RCD are constantly evolving, only a few have been confirmed in the context of cancer. Hereby, we concentrate on the most pertinent and salient RCD patterns, namely apoptosis, necroptosis, pyroptosis, and ferroptosis (Figure 1). These four lytic forms of RCD have been discovered to trigger immune responses under the right circumstances, which are also known as ICD. Since our perception of cell death has radically changed, it is impractical to associate ICD to a specific type of RCD (2). Other cell death modalities have been extensively reviewed elsewhere (2).
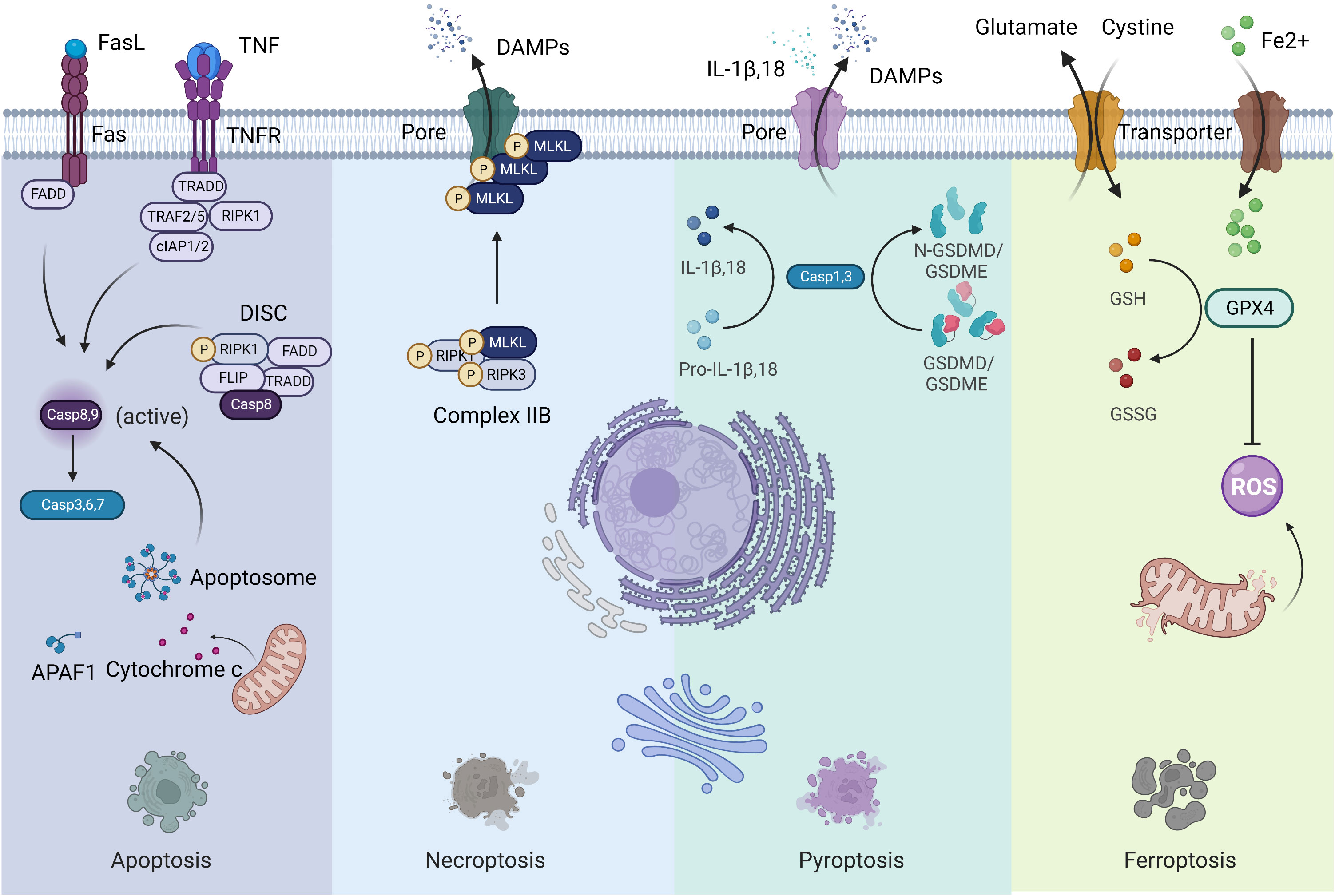
Figure 1 Four forms of regulated cell death. Left: Apoptosis; Middle to the left: Necroptosis; Middle to the right: Pyroptosis; Right: Ferroptosis.
Apoptosis is the most extensively studied form of RCD characterized by morphological alterations of cell rounding, cytoplasmic shrinkage, plasma membrane blebbing and chromatin condensation, that participates in various physiological and pathophysiological processes (2). Apoptosis is usually divided into two forms: the intrinsic pathway (mitochondria) and the extrinsic pathway (death receptors). The intrinsic pathway is initiated by various internal traumas that eventuate in mitochondrial outer membrane permeabilization (MOMP), cytochrome c being released from the intermembrane space, and caspase 9 being activated by the formation of the apoptosome complex (2). The extrinsic pathway is mediated by death receptors, mainly Fas cell surface death receptor and TNF receptor superfamily members. When binding with ligands, the intracellular structural domains of these death receptors interact with adaptor proteins through the interplay of death structural and death effector domains, resulting in the activation of caspase 8 in a cytosolic complex called the death-inducing signaling complex (DISC). Both caspase 8 and caspase 9 are initiator caspases that can cleave and activate executioner caspases such as caspase 3, 6 and 7, leading to apoptosis. Surface-exposed calreticulin (ecto-CRT) and secreted ATP are crucial DAMPs for immunogenic apoptosis. It was reported that dying tumor cells induced by photodynamic therapy (PDT) underwent immunogenic apoptosis characterized by phenotypic maturation and functional stimulation of dendritic cells (DCs) as well as induction of a protective anticancer immune response (5). Early after PDT, cancer cells exhibited ecto-CRT and secreted ATP before displaying a biochemical feature of apoptosis, via a novel PERK-dependent subroutine that necessitate a competent secretory pathway and PI3K-mediated plasma membrane/extracellular trafficking (5).
Necroptosis is considered as a regulated necrotic cell death modality driven by receptor-interacting kinase protein 1 (RIPK1) via forming complex IIB (necrotic body), requiring RIPK3 activity and being executed by mixed lineage kinase domain-like pseudokinase (MLKL) protein (6). Cancer cells undergoing necroptosis are characterized by cell membrane perforation and increased intracellular osmotic pressure, morphologically being cell rounding and swelling, and explosive rupture of the plasma membrane, resulting in releasing cell contents into extracellular space and exacerbating inflammatory response (7). Natural compounds, kinase inhibitors, irradiation and chemotherapy can induce necroptosis (8). Shikonin, an effective anticancer constituent, has been reported to inhibit both primary and metastatic osteosarcoma by inducing necroptosis (9). Necroptosis has been reported to promote or suppress cancer progression, contextualized in different types of cancers (8). Necroptotic pancreatic cancer cells can facilitate their migration and invasive abilities via CXCL5-CXCR2 axis (10). Necroptosis acts as an inflammatory mode of cell death with several signaling pathways activation, such as NF-κB and MAPK pathways, favoring cancer development (11).
Pyroptosis is an inflammatory caspase-dependent cell death modality that requires members of the gasdermin protein family, characterized by pore-formation, cell swelling and lysis, and release of proinflammatory mediators, including IL-1β, IL-18, ATP, and HMGB1 (12). Chemotherapy drugs can trigger tumor cell pyroptosis through caspase 3 cleavage of gasdermin E (GSDME) (13). Metformin treatment induces gasdermin D (GSDMD)-mediated pyroptosis of esophageal squamous cell carcinoma (ESCC) by impacting miR-497/proline-, glutamic acid- and leucine-rich protein-1 (PELP1) axis (14). The involvement of pyroptosis in cancer development appears to be contingent on the types of cancer, genetics, and the duration of pyroptosis induction (15). As the activation of pyroptosis is often accompanied by an inflammatory microenvironment, it is evident that pyroptosis contribute to cancer progression under some circumstances (15).
Ferroptosis is a newly defined form of RCD characterized by iron overload, lipid reactive oxygen species (ROS) accumulation, and lipid peroxidation (16). When cells undergo ferroptosis, mitochondria are smaller than normal, with a reduction or loss of mitochondrial cristae and rupture of the outer mitochondrial membrane, but the nuclear size is still normal, without chromatin condensation (17). Ferroptosis can be regulated via extrinsic and intrinsic pathways. The extrinsic pathway (transporter-dependent pathway) is triggered by repression of cell membrane transporters such as the cystine/glutamate antiporter system or by initiation of the iron transporters serotransferrin and lactotransferrin. The intrinsic pathway (enzyme-regulated pathway) is activated through blockade of intracellular antioxidant enzymes such as glutathione peroxidase 4 (GPX4) (18). However, the effector molecules of ferroptosis remain to be determined (19). The role of ferroptosis in tumorigenesis depends on the release of DAMPs and the activation of immune response within the TME. Many antitumor therapies have been reported to induce ferroptosis (17–19). Cisplatin induces ferroptosis through depleting intracellular glutathione (GSH) and inhibiting GPXs (20). The dysregulation of ferroptosis contributes to antitumor therapy resistance and failure.
Notably in 2022, Tsvetkov et al. discovered that excess intracellular copper could cause a proteotoxic stress response and ultimately cell death through direct binding to lipid acylated components of the tricarboxylic acid (TCA) cycle, leading to abnormal aggregation of lipid acylated proteins and loss of iron-sulfur (Fe-S) cluster proteins in the respiratory chain complex. The team found that this cell death is not blocked by apoptosis, necroptosis, pyroptosis and ferroptosis-related inhibitors, and therefore, is considered as a novel cell death mode and named as cuproptosis (21). This study shows that ferredoxin 1 (FDX1, a reductase that reduces Cu2+ to the more toxic Cu1+) and protein lipoylation are pivotal modulators of cuproptosis (21). However, there are few studies related to cuproptosis, and its overall regulatory mechanism in cancer remains to be further investigated.
Antitumor therapy achieves its ultimate therapeutic goal by causing cell death, but many studies have now found that tumor cell death can also promote tumor recurrence or enhance the therapeutic resistance of tumor cells by affecting the TME. The TME consists of immune cells, stromal cells, blood vessels, lymphatic vessels, nerve terminals and extracellular matrix (ECM) immunomodulating the microenvironment through various signaling molecules that continuously remodel local immunity. The composition of the TME is a key factor in tumor-host interactions. The tumor immune microenvironment (TIME), on the other hand, emphasizes the different cell populations of the immune system and their interactions in the TME, and is of interest for its key role in carcinogenesis, cancer progression and response to therapy (22). In this section, we will present how cell death affects the TIME, both directly and indirectly (Figure 2).
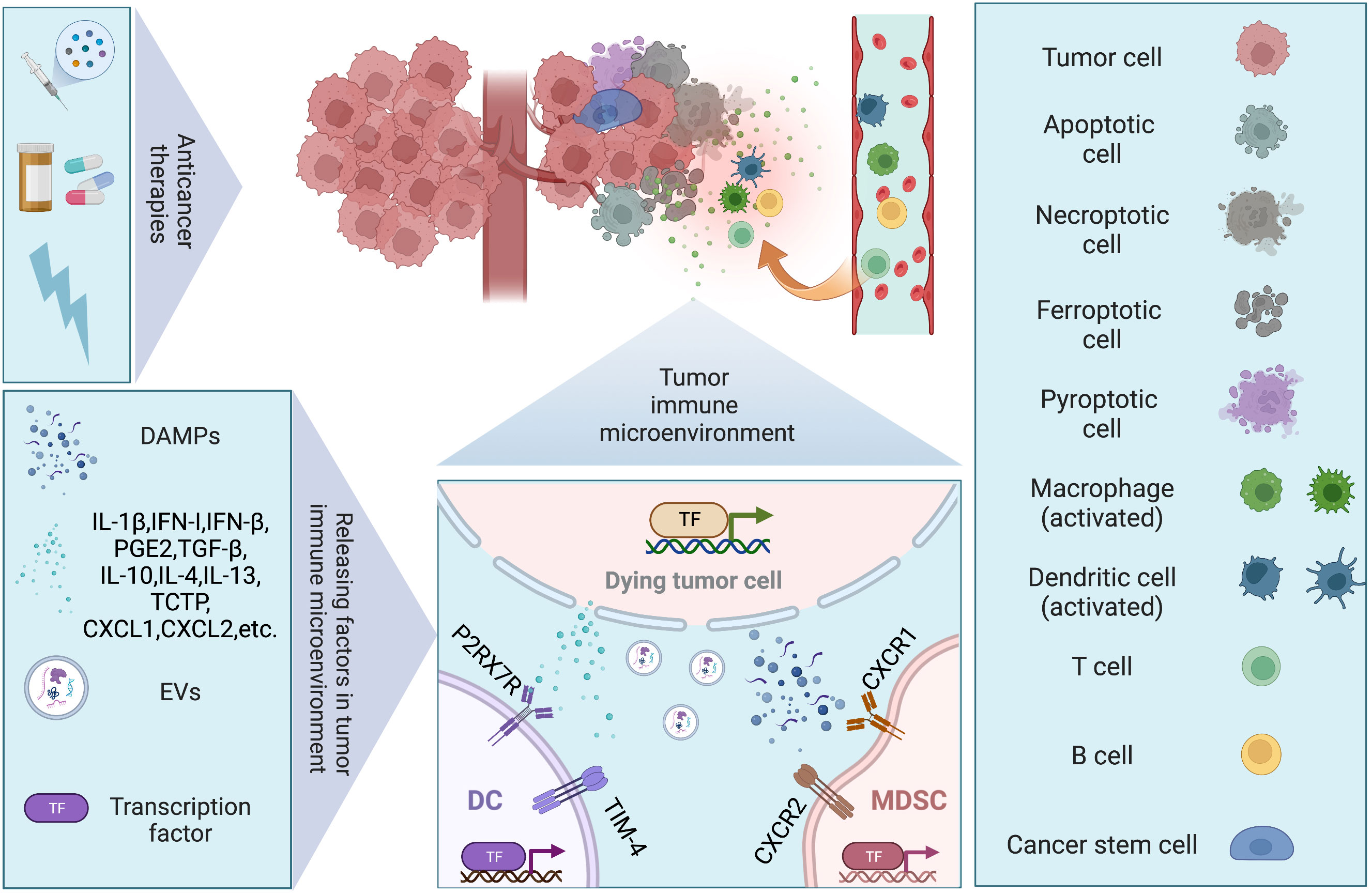
Figure 2 Dying tumor cells and tumor immune microenvironment. Conventional therapies (chemotherapy, targeted therapy, and radiotherapy) remain the mainstay of management in solid tumor, inducing different modalities of cell death. Regulated cell death (RCD), such as apoptosis, necroptosis, pyroptosis, and ferroptosis, have been discovered to trigger immune responses under the right circumstances, which are also known as immunogenic cell death (ICD). Under stresses, dying tumor cells release several factors including damage-associated molecular patterns (DAMPs), chemokines, cytokines, and extracellular vesicles (EVs), to modulate immune system via facilitating cancer stem cell repopulation, governing inflammation, promoting angiogenesis, reprogramming metabolites.
Phagocytosis of dying tumor cells by DCs results in cross-presentation of cell-associated antigen, and the priming of CD8+ T cells, which has been implicated in the maintenance of immune homeostasis. When mice were immunized with cells that experienced RIPK3-mediated necroptosis, CD8+ T cells cross-priming was significantly higher compared to immunization with caspase-8-mediated apoptotic cells (23). Both RIPK1 signaling and NF-κB activation within the dying tumor cells are required for efficient cross-priming and antitumor immunity (23). It was demonstrated that ATP released by dying cancer cells activated P2RX7 receptors on DCs and induced the nucleotide-binding oligomerization domain (NOD)-like receptor family, pyrin domain containing-3 protein (NLRP3) inflammasome, linking the innate and adaptive immune responses against dying cancer cells via secretion of IL-1β (24). Cytosolic DNA released by dying tumor cells induces the production of type I IFN (IFN-I) by DCs through cGAS-STING pathway, contributing to antitumor immunity (25). However, it is also reported that DAMPs released from dying tumor cells upregulate T cell immunoglobulin and mucin domain protein-4 (TIM-4) expression levels on TAMs and DCs, which activating autophagy and impeding anti-tumor immunity via TIM-4-AMPKα1 interaction (26). Conventional type 1 dendritic cells (cDC1) are vital for antitumor immunity and the accumulation of cDC1 relies on chemoattractants CCL5 and XCL1 produced by NK cells (27). Dying tumor cells produced prostaglandin E2 (PGE2) to evade from the NK cell-cDC1 axis by reducing NK cell viability and chemokine production, as well as by downregulating chemokine receptor expression in cDC1 (27). MDSCs are a heterogenous group of myeloid cells that are highly suppressive to antitumor lymphocyte function and trafficking to tumor (28). Two major subsets of MDSCs have been identified in humans: polymorphonuclear MDSCs (PMN-MDSCs) and monocytic MDSCs (M-MDSCs) (28, 29). Translationally controlled tumor protein (TCTP) released by dying tumor cells induces expression of CXCL1 and CXCL2, which then recruits PMN-MDSC population to TIME and exerts immunosuppressive function (29). The resultant accumulation of PMN-MDSCs also strongly attenuates the activation and aggregation of CD8+ T and NK cells in the TIME (29–31). Inhibition of TCTP averts PMN-MDSC accumulation and tumor growth (29). Additionally, the immunomodulatory activity of irradiation-induced cell death varies. High non-fractionated radiation doses above the threshold (ranging from 12 to 18 Gy in different cancer cells) induce the expression of the DNA exonuclease Trex1, which degrades cytoplasmic DNA and mitigates the production of IFN-I and the immunostimulatory effects on DCs (32, 33). In contrast, radiation given in fractionated doses below the dose threshold for Trex1 induction stimulates dying tumor cells to release IFN-β cytokine, which recruits Batf3-dependent DCs and activates anti-tumor CD8+ T cells (33).
Emerging evidence demonstrates that cancer development and progression are associated with a tumor subpopulation, termed cancer stem cells (CSCs). CSCs not only show treatment resistance, but also regulate the immune system in order to avoid being eliminated by immune cells (34). CSCs have been proved to facilitate macrophage differentiation toward an immunosuppressive M2 phenotype by secreting macrophage-colony stimulating factor (M-CSF), which depends on the constitutive activation of interferon-regulatory factor 5 (IRF5) (35). CSCs also promote the production of IL-6 and IL-1β to mediate Th17 cell differentiation, as well as IL-8 and TNF-α to stimulate neutrophil tumor infiltration and angiogenesis (35–37). In turn, TAMs promote the expansion of CSCs via IL-6 and STAT3 signaling, which stimulate further cytokine production, generating a positive feedback loop for CSC self-renewal (38, 39). Moreover, CSCs secrete several cytokines to exert inhibitory effects on immune cells in TME, including TGF-β, IL-10, IL-4 and IL-13 (40). Although chemotherapy effectively elicited apoptosis, associated PGE2 released by these dying tumor cells could paradoxically facilitate neighboring CSC repopulation (41). Blockade of PGE2 production using cyclooxygenase-2 (COX2) inhibitors abrogated this effect (41).
Several studies have highlighted the interconnections between cell death and inflammatory signal transduction (42). Inflammasomes are cytoplasmic multimeric protein complex that provide molecular platforms for activation of caspase-1, contributing to pyroptosis (43). The formation of inflammasome requires cytosolic sensing of pathogen-associated molecular patterns (PAMPs) or DAMPs by the nucleotide-binding domain and leucine-rich repeat receptors (NLRs) or absent in melanoma 2 (AIM2)-like receptors (ALRs) (43). Various innate immune receptors also assemble inflammasome complexes (42). It is widely believed that inflammasomes either favor or suppress carcinogenesis in a highly context-dependent manner. Among NLRs, NLRP3 senses the broadest array of stimuli and participates in adaptive immunity. It is testified that NLRP3 inflammasome drives a T-cell response towards dying tumor cells (24). However, the detrimental effect of NLRP3 is observed when mice are treated with DC vaccination against the poorly immunogenic melanoma cell line B16-F10, via enhancing the accumulation of MDSCs (44). NLRP3 is also an important suppressor of NK cell-mediated responses to carcinogen-induced tumors and metastases (45).
A functional vascular network delivers continued oxygen and nutrients to cells, however, the vasculature of tumor is structurally and functionally abnormal, such as poorly organized vasculature, tortuous and dilated vessels with uneven diameter, excessive branches and shunts (46). The impaired vessel perfusion and high vascular permeability increase tissue hypoxia, acidosis, and necrosis, which could reprogram the resident macrophages into a pro-tumorigenic and immunosuppressive phenotype (47, 48). Also, the hypoxic and acidic microenvironment could up-regulate programmed cell death ligand 1(PD-L1) on MDSCs, DCs and cancer cells, polarize TAMs to the immune inhibitory M2-like phenotype to suppress T-effector cell function. Chronic hypoxia and microenvironment acidity contribute to immune evasion by inducing T cells differentiation into immunosuppressive Tregs (49, 50). Actually, hypoxia is often found in solid tumors due to the rapid growth of cancer cells (51). Hypoxia could influence monocytic cells with respect to morphology, antigen expression, and cytokine secretion, and to induce various genes including vascular endothelial growth factor (VEGF), fibroblast growth factor-3 (FGF-3), BCL2-interacting killer (BIK), and MMPs (52–54). Hypoxia-inducible factors (HIFs) are major transcription factors for cancer cells to adapt hypoxic stress and the activation of HIF pathway results in angiogenesis (55). VEGF family members play important roles in HIF induced angiogenesis. There is mounting evidence to support the perspective that angiogenesis and inflammation are interdependent (56, 57). It is well established that cancer cells are able to secrete pro-angiogenic factors as well as inflammatory mediators (37). Our previous studies have shown that apoptotic tumor cells mediate proangiogenic response via caspase 3/NF-κB/COX2/PGE2 and caspase 3/p-eIF4E/VEGF-A pathways after irradiation (58). Additionally, normalizing blood vessels with anti-angiogenic therapy normalizes tumor vasculature, creates a more uniform distribution of perfused tumor vessels, and promotes infiltration of T effector cells while inhibiting the accumulation of MDSC (56, 57). Normalized vasculature also alleviates the hypoxia and acidity in TIME, which in term polarizing TAMs to an immunostimulatory M1-like phenotype (56, 57).
Emerging evidence has pointed out that the immune response is correlated with dramatic modifications in tissue metabolism, such as nutrients exhaustion, oxygen depletion, and increased accumulation of reactive nitrogen and oxygen intermediates (59). Given their coexistence, cancer cells and immune cells are in the competition for the local resources available in the microenvironment. When cancer cells underwent dying process, the nutrients competition would still exist. The dying tumor cells either utilized the nutrients around themselves to survival or against demise, or sacrificed themselves to support remnant tumor cells proliferation or escape from immune clearance. It has been reported that apoptotic cells can release metabolites to activate specific programs in neighboring cells (60). Cancer cell death leads to a local ionic imbalance within the TIME as well. In necrotic areas, dying tumor cells release a high number of cations, specifically potassium ions, causing functional starvation of the tumor-infiltrating T cells in parallel to suppression of AKT/mTOR phosphorylation (61). Overabundance of potassium in the TIME also limited T cell effector function by promoting autophagy and metabolic reprogramming, while decreased histone acetylation at effector and exhaustion loci, which in turn generating T cell stemness with improved persistence, engraftment, self-renewal, multipotency, and tumor clearance (62). Lactic acid, a metabolic product of cancer cells, is reported to act as an epigenetic regulator and elicit M2 macrophage polarization through epigenetic reprogramming (63). It binds straight to the histone lysine lactylation (Kla) sites and the transcription of downstream genes is stimulated, which then invokes M2 polarization and reinforces inflammation-independent biological pathways (63). G protein-coupled receptor 81 (GPR81) is a lactic acid receptor found on both immune cells and cancer cells (64). Its activation in cancer cells promotes proliferation, therapy resistance, and enhanced expression of PD-L1 (65–67). Its activation on DCs is associated with decreased cAMP, IL-6, IL-12, and prevents tumor-specific antigen presentation (67). The depletion of glutamine, arginine, and tryptophan also show immunosuppressive effects. It is demonstrated that cancer cells could express indoleamine 2,3-dioxygenase (IDO), an enzyme that catalyzing tryptophan (TRP) and suppresses immune response within the TME (68). Gut microbiota impacts the amenability of some tumor types to therapies by influencing regulatory aspects of the immune response. More clearly, the microbiome has been reported to play a critical role in determining DC functionality (69, 70).
Extracellular vesicles (EVs) are a heterogeneous class of cell-derived membranous structures produced by all cells that contain proteins, lipids and genetic material (71). EVs are essential mediators of cell-cell communication and can be broadly divided into two distinct classes: exosomes and microvesicles (MVs) (71). Tumor-derived exosomes are involved in many stages of tumor progression, including angiogenesis (72), therapy resistance (73), invasion and metastasis (74), and immune evasion (75). Dying tumor cells treated by antitumor agent topotecan (TPT) could release exosomes that contain DNA to activate DCs via a STING-dependent pathway (76). Exosomes derived from tunicamycin (TM)-treated cancer cells contain abundant miR-23a-3p, which inhibit PTEN expression and subsequently elevated phosphorylated AKT and PD-L1 expression in macrophages, thus helping cancer cells to escape from antitumor immunity (77). MVs arise via direct outward budding and fission of the plasma membrane and participate in cell-to-cell communication, immune regulation, and tumor progression (71). Dying tumor cells could release MVs loaded with PD-L1 to suppress the function of T cells and induce M2 phenotype macrophages via TBK1/STAT6 and AKT/mTOR signals, contributing to the suppressive TIME (78).
While conventional therapies remain the mainstay of management in solid tumor, immunotherapy is rapidly being incorporated with these therapies to prolong patient survival (79). Immunotherapies are currently confined to those antibodies targeting PD-1/PD-L1 and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4). However, these ICIs often show transient efficacy, especially in microsatellite stable tumors which often present minor genetic abnormalities and possess limited antigenic capacity (80). The immunosuppressive TME also contributes to the restricted effects of ICIs. It is reported that the response rate to single agent PD-1 blockade ranges from 40% to 70% in cancers such as melanoma or Hodgkin’s lymphoma, while limited to 10-25% in most other cancer types (81–83). Moreover, these patients who initially respond to ICIs can eventually have disease progression. Thus, strategies to improve efficacy are urgently needed. Combination strategies have been formulated to confront the challenge of immunotherapy resistance and broaden the responding population (Table 1). For instance, immunotherapy has been combined with chemotherapy in lung, breast and gastric cancer, with TKIs in RCC and bladder cancer (83). In the CheckMate 816 study (NCT02998528), neoadjuvant immunotherapy combined with chemotherapy was associated with significantly longer event-free survival than chemotherapy alone in patients with resectable NSCLC (31.6 months vs. 20.8 months) and a higher proportion of patients with pathologic complete responses (24% vs. 2.2%). The addition of immunotherapy to neoadjuvant chemotherapy did not increase the incidence of adverse events or impede the feasibility of surgery (84). Ceralasertib has excellent antitumor activity as an oral kinase inhibitor in combination with immunotherapy (NCT03780608), with a durable response in patients with advanced gastric cancer (85). During study treatment, investigators also found an upregulation of cytoplasmic DNA-induced innate immune responses, activation of intratumoral lymphocytes and expansion of circulating tumor-reactive CD8+ T cell clones in responders (85). Of course, some clinical studies have concluded that combination therapy does not improve the effectiveness of immunotherapy very well. A multicenter randomized phase II clinical study found that radiotherapy did not increase the response to PD-L1 plus CTLA-4 combination inhibition in NSCLC patients resistant to PD(L)-1 therapy (NCT02888743) (86). Thus, clinically, the effectiveness of immunotherapy needs to be further clarified, and the mechanism of limited effect of combined immunotherapy and non-response to immunotherapy may be related to TIME, and more basic research on molecular mechanism is expected to follow.
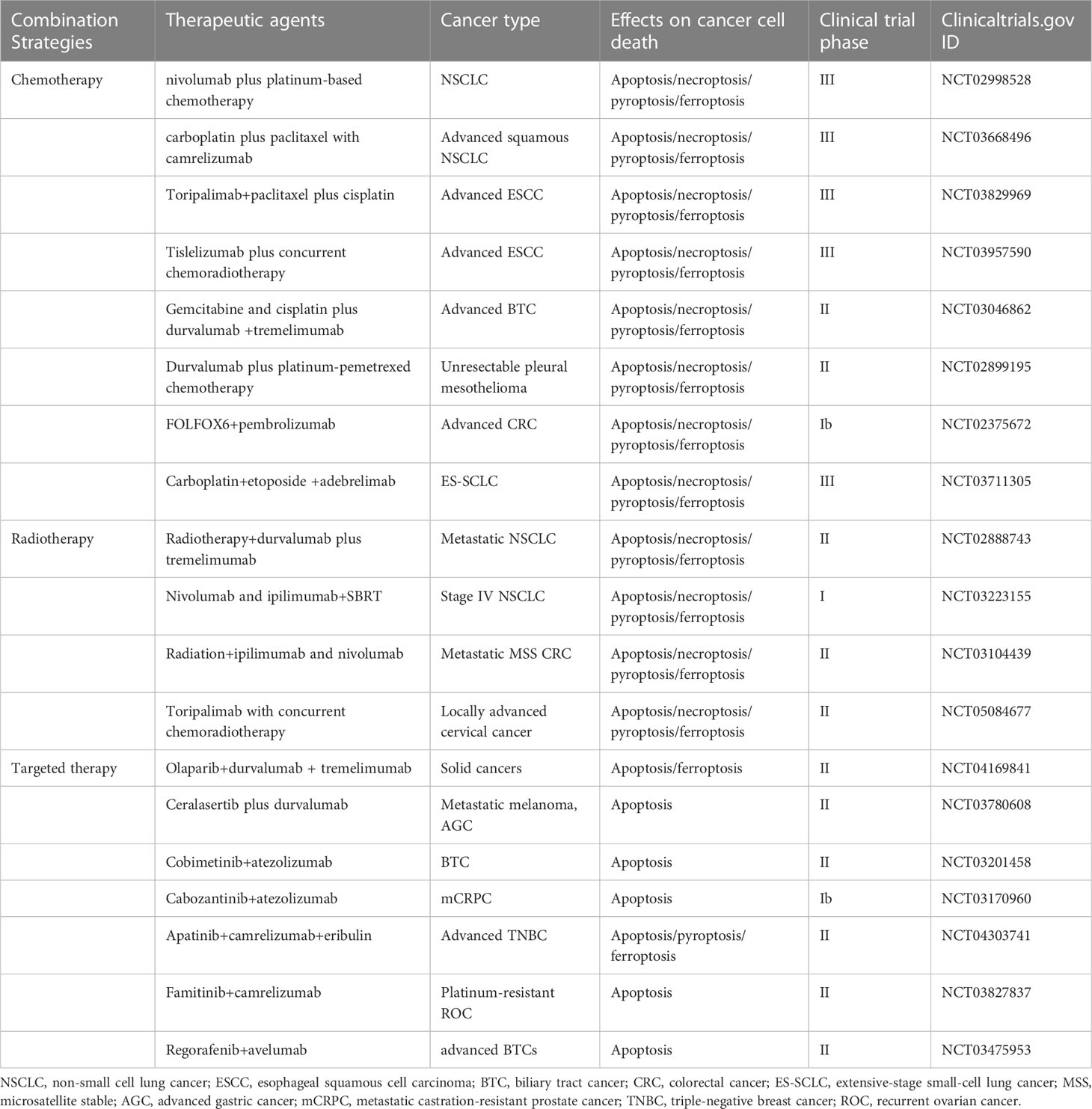
Table 1 Summary some of the clinical trials testing combinations of immunotherapy with therapeutic agents that may target apoptosis, necroptosis, pyroptosis, and ferroptosis.
Effective elimination of cancer cells has always been the mainstay and goal of clinical cancer treatment. However, no matter what treatment options are adopted, there are still residual cancer cells surviving and even proliferating to form new tumor masses. In this process, TIME has been proven to play an important role. And there are multiple molecular interactions between cancer cell death and TIME, which affect the efficacy of immunotherapy to an extent. Thus, investigation and targeting of molecules at the crossroad of dying tumor cells and host immune system may provide new therapeutic opportunities in the area of cancer treatment especially immunotherapy.
SJH and JC conceived the manuscript. SJH wrote the manuscript and designed the figures and tables. JC and QH reviewed and revised the manuscript. All the authors contributed to the manuscript have read and agreed to the published version.
This work was funded by the National Natural Science Foundation of China (No. 81972887 to JC, No. 81903117 to SJH), the Natural Science Foundation of Shanghai (No. 21ZR1451100 to JC), the Shanghai Pujiang Program (No. 2021PJD056 to JC) and the Shanghai “Rising Stars of Medical Talents” Youth Development Program (No. SHWRS (2021) _099 to JC).
All figures were created with BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Fucikova J, Kepp O, Kasikova L, Petroni G, Yamazaki T, Liu P, et al. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis (2020) 11(11):1013. doi: 10.1038/s41419-020-03221-2
2. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: Recommendations of the nomenclature committee on cell death 2018. Cell Death Diff (2018) 25(3):486–541. doi: 10.1038/s41418-017-0012-4
3. Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zitvogel L, et al. Cell death modalities: Classification and pathophysiological implications. Cell Death Diff (2007) 14(7):1237–43. doi: 10.1038/sj.cdd.4402148
4. Kroemer G, El-Deiry WS, Golstein P, Peter ME, Vaux D, Vandenabeele P, et al. Classification of cell death: Recommendations of the nomenclature committee on cell death. Cell Death Diff (2005) 12 Suppl 2:1463–7. doi: 10.1038/sj.cdd.4401724
5. Garg AD, Krysko DV, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T, et al. A novel pathway combining calreticulin exposure and atp secretion in immunogenic cancer cell death. EMBO J (2012) 31(5):1062–79. doi: 10.1038/emboj.2011.497
6. Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol (2017) 17(2):97–111. doi: 10.1038/nri.2016.107
7. Aaes TL, Kaczmarek A, Delvaeye T, De Craene B, De Koker S, Heyndrickx L, et al. Vaccination with necroptotic cancer cells induces efficient anti-tumor immunity. Cell Rep (2016) 15(2):274–87. doi: 10.1016/j.celrep.2016.03.037
8. Gong Y, Fan Z, Luo G, Yang C, Huang Q, Fan K, et al. The role of necroptosis in cancer biology and therapy. Mol Cancer (2019) 18(1):100. doi: 10.1186/s12943-019-1029-8
9. Fu Z, Deng B, Liao Y, Shan L, Yin F, Wang Z, et al. The anti-tumor effect of shikonin on osteosarcoma by inducing Rip1 and Rip3 dependent necroptosis. BMC Cancer (2013) 13:580. doi: 10.1186/1471-2407-13-580
10. Ando Y, Ohuchida K, Otsubo Y, Kibe S, Takesue S, Abe T, et al. Necroptosis in pancreatic cancer promotes cancer cell migration and invasion by release of Cxcl5. PloS One (2020) 15(1):e0228015. doi: 10.1371/journal.pone.0228015
11. Qin X, Ma D, Tan YX, Wang HY, Cai Z. The role of necroptosis in cancer: A double-edged sword? Biochim Biophys Acta Rev Cancer (2019) 1871(2):259–66. doi: 10.1016/j.bbcan.2019.01.006
12. Xia X, Wang X, Cheng Z, Qin W, Lei L, Jiang J, et al. The role of pyroptosis in cancer: Pro-cancer or pro-"Host"? Cell Death Dis (2019) 10(9):650. doi: 10.1038/s41419-019-1883-8
13. Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature (2017) 547(7661):99–103. doi: 10.1038/nature22393
14. Wang L, Li K, Lin X, Yao Z, Wang S, Xiong X, et al. Metformin induces human esophageal carcinoma cell pyroptosis by targeting the mir-497/Pelp1 axis. Cancer Lett (2019) 450:22–31. doi: 10.1016/j.canlet.2019.02.014
15. Qi S, Wang Q, Zhang J, Liu Q, Li C. Pyroptosis and its role in the modulation of cancer progression and antitumor immunity. Int J Mol Sci (2022) 23(18):10494. doi: 10.3390/ijms231810494
16. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
17. Wang H, Cheng Y, Mao C, Liu S, Xiao D, Huang J, et al. Emerging mechanisms and targeted therapy of ferroptosis in cancer. Mol Ther (2021) 29(7):2185–208. doi: 10.1016/j.ymthe.2021.03.022
18. Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: Molecular mechanisms and health implications. Cell Res (2021) 31(2):107–25. doi: 10.1038/s41422-020-00441-1
19. Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: The role of ferroptosis in cancer. Nat Rev Clin Oncol (2021) 18(5):280–96. doi: 10.1038/s41571-020-00462-0
20. Guo J, Xu B, Han Q, Zhou H, Xia Y, Gong C, et al. Ferroptosis: A novel anti-tumor action for cisplatin. Cancer Res Treat (2018) 50(2):445–60. doi: 10.4143/crt.2016.572
21. Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M, et al. Copper induces cell death by targeting lipoylated tca cycle proteins. Science (2022) 375(6586):1254–61. doi: 10.1126/science.abf0529
22. Xu L, Zou C, Zhang S, Chu TSM, Zhang Y, Chen W, et al. Reshaping the systemic tumor immune environment (Stie) and tumor immune microenvironment (Time) to enhance immunotherapy efficacy in solid tumors. J Hematol Oncol (2022) 15(1):87. doi: 10.1186/s13045-022-01307-2
23. Yatim N, Jusforgues-Saklani H, Orozco S, Schulz O, Barreira da Silva R, Reis e Sousa C, et al. Ripk1 and nf-kappab signaling in dying cells determines cross-priming of Cd8(+) T cells. Science (2015) 350(6258):328–34. doi: 10.1126/science.aad0395
24. Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the Nlrp3 inflammasome in dendritic cells induces il-1beta-Dependent adaptive immunity against tumors. Nat Med (2009) 15(10):1170–8. doi: 10.1038/nm.2028
25. Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, et al. Sting-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity (2014) 41(5):843–52. doi: 10.1016/j.immuni.2014.10.019
26. Baghdadi M, Yoneda A, Yamashina T, Nagao H, Komohara Y, Nagai S, et al. Tim-4 glycoprotein-mediated degradation of dying tumor cells by autophagy leads to reduced antigen presentation and increased immune tolerance. Immunity (2013) 39(6):1070–81. doi: 10.1016/j.immuni.2013.09.014
27. Bottcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. Nk cells stimulate recruitment of Cdc1 into the tumor microenvironment promoting cancer immune control. Cell (2018) 172(5):1022–37 e14. doi: 10.1016/j.cell.2018.01.004
28. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol (2016) 37(3):208–20. doi: 10.1016/j.it.2016.01.004
29. Hangai S, Kawamura T, Kimura Y, Chang CY, Hibino S, Yamamoto D, et al. Orchestration of myeloid-derived suppressor cells in the tumor microenvironment by ubiquitous cellular protein tctp released by tumor cells. Nat Immunol (2021) 22(8):947–57. doi: 10.1038/s41590-021-00967-5
30. Mao Y, Sarhan D, Steven A, Seliger B, Kiessling R, Lundqvist A. Inhibition of tumor-derived prostaglandin-E2 blocks the induction of myeloid-derived suppressor cells and recovers natural killer cell activity. Clin Cancer Res (2014) 20(15):4096–106. doi: 10.1158/1078-0432.CCR-14-0635
31. Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med (2011) 208(10):1949–62. doi: 10.1084/jem.20101956
32. Cai X, Chiu YH, Chen ZJ. The cgas-Cgamp-Sting pathway of cytosolic DNA sensing and signaling. Mol Cell (2014) 54(2):289–96. doi: 10.1016/j.molcel.2014.03.040
33. Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, et al. DNA Exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun (2017) 8:15618. doi: 10.1038/ncomms15618
34. Liu S, Wicha MS. Targeting breast cancer stem cells. J Clin Oncol (2010) 28(25):4006–12. doi: 10.1200/JCO.2009.27.5388
35. Yamashina T, Baghdadi M, Yoneda A, Kinoshita I, Suzu S, Dosaka-Akita H, et al. Cancer stem-like cells derived from chemoresistant tumors have a unique capacity to prime tumorigenic myeloid cells. Cancer Res (2014) 74(10):2698–709. doi: 10.1158/0008-5472.CAN-13-2169
36. Jinushi M. Role of cancer stem cell-associated inflammation in creating pro-inflammatory tumorigenic microenvironments. Oncoimmunology (2014) 3:e28862. doi: 10.4161/onci.28862
37. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature (2008) 454(7203):436–44. doi: 10.1038/nature07205
38. Wan S, Zhao E, Kryczek I, Vatan L, Sadovskaya A, Ludema G, et al. Tumor-associated macrophages produce interleukin 6 and signal Via Stat3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology (2014) 147(6):1393–404. doi: 10.1053/j.gastro.2014.08.039
39. Korkaya H, Liu S, Wicha MS. Regulation of cancer stem cells by cytokine networks: Attacking cancer's inflammatory roots. Clin Cancer Res (2011) 17(19):6125–9. doi: 10.1158/1078-0432.CCR-10-2743
40. Clara JA, Monge C, Yang Y, Takebe N. Targeting signalling pathways and the immune microenvironment of cancer stem cells - a clinical update. Nat Rev Clin Oncol (2020) 17(4):204–32. doi: 10.1038/s41571-019-0293-2
41. Kurtova AV, Xiao J, Mo Q, Pazhanisamy S, Krasnow R, Lerner SP, et al. Blocking Pge2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature (2015) 517(7533):209–13. doi: 10.1038/nature14034
42. Kesavardhana S, Malireddi RKS, Kanneganti TD. Caspases in cell death, inflammation, and pyroptosis. Annu Rev Immunol (2020) 38:567–95. doi: 10.1146/annurev-immunol-073119-095439
43. Man SM, Kanneganti TD. Regulation of inflammasome activation. Immunol Rev (2015) 265(1):6–21. doi: 10.1111/imr.12296
44. van Deventer HW, Burgents JE, Wu QP, Woodford RM, Brickey WJ, Allen IC, et al. The inflammasome component Nlrp3 impairs antitumor vaccine by enhancing the accumulation of tumor-associated myeloid-derived suppressor cells. Cancer Res (2010) 70(24):10161–9. doi: 10.1158/0008-5472.CAN-10-1921
45. Chow MT, Sceneay J, Paget C, Wong CS, Duret H, Tschopp J, et al. Nlrp3 suppresses nk cell-mediated responses to carcinogen-induced tumors and metastases. Cancer Res (2012) 72(22):5721–32. doi: 10.1158/0008-5472.CAN-12-0509
46. Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature (2000) 407(6801):249–57. doi: 10.1038/35025220
47. Palazon A, Aragones J, Morales-Kastresana A, de Landazuri MO, Melero I. Molecular pathways: Hypoxia response in immune cells fighting or promoting cancer. Clin Cancer Res (2012) 18(5):1207–13. doi: 10.1158/1078-0432.CCR-11-1591
48. Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer (2011) 11(6):393–410. doi: 10.1038/nrc3064
49. Clever D, Roychoudhuri R, Constantinides MG, Askenase MH, Sukumar M, Klebanoff CA, et al. Oxygen sensing by T cells establishes an immunologically tolerant metastatic niche. Cell (2016) 166(5):1117–31 e14. doi: 10.1016/j.cell.2016.07.032
50. Huber V, Camisaschi C, Berzi A, Ferro S, Lugini L, Triulzi T, et al. Cancer acidity: An ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin Cancer Biol (2017) 43:74–89. doi: 10.1016/j.semcancer.2017.03.001
51. McKeown SR. Defining normoxia, physoxia and hypoxia in tumours-implications for treatment response. Br J Radiol (2014) 87(1035):20130676. doi: 10.1259/bjr.20130676
52. Lahat N, Rahat MA, Ballan M, Weiss-Cerem L, Engelmayer M, Bitterman H. Hypoxia reduces Cd80 expression on monocytes but enhances their lps-stimulated tnf-alpha secretion. J Leukoc Biol (2003) 74(2):197–205. doi: 10.1189/jlb.0303105
53. Mizukami Y, Li J, Zhang X, Zimmer MA, Iliopoulos O, Chung DC. Hypoxia-inducible factor-1-Independent regulation of vascular endothelial growth factor by hypoxia in colon cancer. Cancer Res (2004) 64(5):1765–72. doi: 10.1158/0008-5472.can-03-3017
54. Koong AC, Denko NC, Hudson KM, Schindler C, Swiersz L, Koch C, et al. Candidate genes for the hypoxic tumor phenotype. Cancer Res (2000) 60(4):883–7. doi: 10.1016/S0360-3016(99)90122-0
55. Krock BL, Skuli N, Simon MC. Hypoxia-induced angiogenesis: Good and evil. Genes Cancer (2011) 2(12):1117–33. doi: 10.1177/1947601911423654
56. Ribatti D, Crivellato E. Immune cells and angiogenesis. J Cell Mol Med (2009) 13(9A):2822–33. doi: 10.1111/j.1582-4934.2009.00810.x
57. Wigner P, Grebowski R, Bijak M, Saluk-Bijak J, Szemraj J. The interplay between oxidative stress, inflammation and angiogenesis in bladder cancer development. Int J Mol Sci (2021) 22(9):4483. doi: 10.3390/ijms22094483
58. Feng X, Yu Y, He S, Cheng J, Gong Y, Zhang Z, et al. Dying glioma cells establish a proangiogenic microenvironment through a caspase 3 dependent mechanism. Cancer Lett (2017) 385:12–20. doi: 10.1016/j.canlet.2016.10.042
59. Guerra L, Bonetti L, Brenner D. Metabolic modulation of immunity: A new concept in cancer immunotherapy. Cell Rep (2020) 32(1):107848. doi: 10.1016/j.celrep.2020.107848
60. Medina CB, Mehrotra P, Arandjelovic S, Perry JSA, Guo Y, Morioka S, et al. Metabolites released from apoptotic cells act as tissue messengers. Nature (2020) 580(7801):130–5. doi: 10.1038/s41586-020-2121-3
61. Eil R, Vodnala SK, Clever D, Klebanoff CA, Sukumar M, Pan JH, et al. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature (2016) 537(7621):539–43. doi: 10.1038/nature19364
62. Vodnala SK, Eil R, Kishton RJ, Sukumar M, Yamamoto TN, Ha NH, et al. T Cell stemness and dysfunction in tumors are triggered by a common mechanism. Science (2019) 363(6434):eaau0135. doi: 10.1126/science.aau0135
63. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature (2019) 574(7779):575–80. doi: 10.1038/s41586-019-1678-1
64. Brown TP, Ganapathy V. Lactate/Gpr81 signaling and proton motive force in cancer: Role in angiogenesis, immune escape, nutrition, and warburg phenomenon. Pharmacol Ther (2020) 206:107451. doi: 10.1016/j.pharmthera.2019.107451
65. Feng J, Yang H, Zhang Y, Wei H, Zhu Z, Zhu B, et al. Tumor cell-derived lactate induces taz-dependent upregulation of pd-L1 through Gpr81 in human lung cancer cells. Oncogene (2017) 36(42):5829–39. doi: 10.1038/onc.2017.188
66. Yang X, Lu Y, Hang J, Zhang J, Zhang T, Huo Y, et al. Lactate-modulated immunosuppression of myeloid-derived suppressor cells contributes to the radioresistance of pancreatic cancer. Cancer Immunol Res (2020) 8(11):1440–51. doi: 10.1158/2326-6066.CIR-20-0111
67. Brown TP, Bhattacharjee P, Ramachandran S, Sivaprakasam S, Ristic B, Sikder MOF, et al. The lactate receptor Gpr81 promotes breast cancer growth Via a paracrine mechanism involving antigen-presenting cells in the tumor microenvironment. Oncogene (2020) 39(16):3292–304. doi: 10.1038/s41388-020-1216-5
68. Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol (2013) 34(3):137–43. doi: 10.1016/j.it.2012.10.001
69. Vetizou M, Pitt JM, Daillere R, Lepage P, Waldschmitt N, Flament C, et al. Anticancer immunotherapy by ctla-4 blockade relies on the gut microbiota. Science (2015) 350(6264):1079–84. doi: 10.1126/science.aad1329
70. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (Time) for effective therapy. Nat Med (2018) 24(5):541–50. doi: 10.1038/s41591-018-0014-x
71. van Niel G, D'Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol (2018) 19(4):213–28. doi: 10.1038/nrm.2017.125
72. Olejarz W, Kubiak-Tomaszewska G, Chrzanowska A, Lorenc T. Exosomes in angiogenesis and anti-angiogenic therapy in cancers. Int J Mol Sci (2020) 21(16):5840. doi: 10.3390/ijms21165840
73. Wang X, Zhang H, Yang H, Bai M, Ning T, Deng T, et al. Exosome-delivered circrna promotes glycolysis to induce chemoresistance through the mir-122-Pkm2 axis in colorectal cancer. Mol Oncol (2020) 14(3):539–55. doi: 10.1002/1878-0261.12629
74. Zhang C, Ji Q, Yang Y, Li Q, Wang Z. Exosome: Function and role in cancer metastasis and drug resistance. Technol Cancer Res Treat (2018) 17:1533033818763450. doi: 10.1177/1533033818763450
75. Kim DH, Kim H, Choi YJ, Kim SY, Lee JE, Sung KJ, et al. Exosomal pd-L1 promotes tumor growth through immune escape in non-small cell lung cancer. Exp Mol Med (2019) 51(8):1–13. doi: 10.1038/s12276-019-0295-2
76. Kitai Y, Kawasaki T, Sueyoshi T, Kobiyama K, Ishii KJ, Zou J, et al. DNA-Containing exosomes derived from cancer cells treated with topotecan activate a sting-dependent pathway and reinforce antitumor immunity. J Immunol (2017) 198(4):1649–59. doi: 10.4049/jimmunol.1601694
77. Liu J, Fan L, Yu H, Zhang J, He Y, Feng D, et al. Endoplasmic reticulum stress causes liver cancer cells to release exosomal mir-23a-3p and up-regulate programmed death ligand 1 expression in macrophages. Hepatology (2019) 70(1):241–58. doi: 10.1002/hep.30607
78. Li C, Qiu S, Jin K, Zheng X, Zhou X, Jin D, et al. Tumor-derived microparticles promote the progression of triple-negative breast cancer Via pd-L1-Associated immune suppression. Cancer Lett (2021) 523:43–56. doi: 10.1016/j.canlet.2021.09.039
79. Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell (2018) 175(4):998–1013 e20. doi: 10.1016/j.cell.2018.10.038
80. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. Pd-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med (2015) 372(26):2509–20. doi: 10.1056/NEJMoa1500596
81. Chen R, Zinzani PL, Fanale MA, Armand P, Johnson NA, Brice P, et al. Phase ii study of the efficacy and safety of pembrolizumab for Relapsed/Refractory classic Hodgkin lymphoma. J Clin Oncol (2017) 35(19):2125–32. doi: 10.1200/JCO.2016.72.1316
82. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med (2019) 381(16):1535–46. doi: 10.1056/NEJMoa1910836
83. Schoenfeld AJ, Hellmann MD. Acquired resistance to immune checkpoint inhibitors. Cancer Cell (2020) 37(4):443–55. doi: 10.1016/j.ccell.2020.03.017
84. Forde PM, Spicer J, Lu S, Provencio M, Mitsudomi T, Awad MM, et al. Neoadjuvant nivolumab plus chemotherapy in resectable lung cancer. N Engl J Med (2022) 386(21):1973–85. doi: 10.1056/NEJMoa2202170
85. Kwon M, Kim G, Kim R, Kim KT, Kim ST, Smith S, et al. Phase ii study of ceralasertib (Azd6738) in combination with durvalumab in patients with advanced gastric cancer. J Immunother Cancer (2022) 10(7):e005041. doi: 10.1136/jitc-2022-005041
86. Schoenfeld JD, Giobbie-Hurder A, Ranasinghe S, Kao KZ, Lako A, Tsuji J, et al. Durvalumab plus tremelimumab alone or in combination with low-dose or hypofractionated radiotherapy in metastatic non-Small-Cell lung cancer refractory to previous Pd(L)-1 therapy: An open-label, multicentre, randomised, phase 2 trial. Lancet Oncol (2022) 23(2):279–91. doi: 10.1016/S1470-2045(21)00658-6
Keywords: cancer, cell death, tumor immune microenvironment, cancer treatment, immunotherapy
Citation: He S, Huang Q and Cheng J (2023) The unfolding story of dying tumor cells during cancer treatment. Front. Immunol. 14:1073561. doi: 10.3389/fimmu.2023.1073561
Received: 18 October 2022; Accepted: 27 February 2023;
Published: 13 March 2023.
Edited by:
Na Li, Shandong Cancer Hospital, ChinaReviewed by:
Scott McComb, National Research Council Canada (NRC), CanadaCopyright © 2023 He, Huang and Cheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jin Cheng, amluLmNoZW5nQHNoZ2guY24=; Qian Huang, aHVhbmdxaWFuX3NqdHVAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.