
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 23 January 2023
Sec. B Cell Biology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1067459
 Amit Singh1†
Amit Singh1† Mary Kaileh1†
Mary Kaileh1† Supriyo De2
Supriyo De2 Krystyna Mazan-Mamczarz2
Krystyna Mazan-Mamczarz2 Dashzeveg Bayarsaihan3
Dashzeveg Bayarsaihan3 Ranjan Sen1
Ranjan Sen1 Ananda L. Roy1*
Ananda L. Roy1*The ubiquitously expressed transcription factor TFII-I is a multifunctional protein with pleiotropic roles in gene regulation. TFII-I associated polymorphisms are implicated in Sjögren’s syndrome and Lupus in humans and, germline deletion of the Gtf2i gene in mice leads to embryonic lethality. Here we report a unique role for TFII-I in homeostasis of innate properties of B lymphocytes. Loss of Gtf2i in murine B lineage cells leads to an alteration in transcriptome, chromatin landscape and associated transcription factor binding sites, which exhibits myeloid-like features and coincides with enhanced sensitivity to LPS induced gene expression. TFII-I deficient B cells also show increased switching to IgG3, a phenotype associated with inflammation. These results demonstrate a role for TFII-I in maintaining immune homeostasis and provide clues for GTF2I polymorphisms associated with B cell dominated autoimmune diseases in humans.
TFII-I was discovered as a transcription factor that bound to the adenovirus major late core promoter Initiator (Inr) element. TFII-I also interacts with a sequence-specific DNA element (the E-box element) together with the upstream stimulatory factor (USF) in cell-free systems (1). Subsequent studies have suggested a broader, multifunctional role for TFII-I in a variety of cell types perhaps by virtue of its interaction with cell type-specific proteins and signaling intermediates (2). TFII-I is shown to be phosphorylated in response to several cell surface receptor signaling pathways, including growth factor receptor and immune receptors (3), further suggesting its role in these pathways. Recent studies also indicate its involvement in DNA-damage and response pathways (4).
TFII-I is vertebrate-specific and encoded by the GTF2I gene in humans and Gtf2i in mice (5). The locus comprises 36 exons that give rise to several alternatively spliced isoforms (6). Various disease pathologies are associated with either gene dosage effects or mutations or gene fusion events involving GTF2I (7). Most notably, GTF2I haploinsufficiency is associated with Williams-Beuren Syndrome (WBS) a neurodevelopmental disorder with specific craniofacial features (7–9). GTF2I gene dosage is further ascribed to autism spectrum disorder (ASD) (10) and single nucleotide polymorphisms in GTF2I loci are associated with autoimmune disorders like SLE, RA and Sjogren syndrome (11, 12), and reviewed in (3). Finally, a single point mutation in GTF2I is associated with thymic epithelial tumors as well as GTF2I gene fusions are known to occur in various forms of cancers (13, 14). Collectively, these studies clearly point to an essential and important role for TFII-I in various human diseases. Consistent with pleiotropic roles of TFII-I, germline deletion of the gene Gtf2i in mice leads to early embryonic lethality likely due to severe defects in vasculogenesis and angiogenesis (15, 16).
Although ubiquitously expressed in most if not all cell types, a series of studies showed that TFII-I biochemically interacts with Bruton’s tyrosine kinase (Btk) in B cells and corresponding kinase Itk in T cells, suggesting a role in immune cell type specific functions (17). Moreover, biochemically TFII-I interacts with the B-cell specific co-activator, OCA-B (18) and B cell transcription factor Bright (19) and regulates Immunoglobulin gene expression in cell-based assays (18). Intrigued by TFII-I’s potential involvement in the immune system, we conditionally deleted the Gtf2i gene in mice using a CD19-Cre driver to test its potential role in mature B cells.
Murine naïve B cells are generally classified into three distinct subsets, B-1 B cells of peritoneal origin, follicular (FO) B cells, and marginal zone (MZ) B cells (20). A large fraction of FO B cells is IgDhiIgMlowCD21mid cells (termed as follicular type I B cells), while a smaller fraction are IgDhiIgMhiCD21mid B cells (termed as follicular type II B cells) (21). In contrast, only a minor population of splenic B cells are MZ cells expressing high levels of IgM, CD21 and CD1d. MZ B cells are generated as naive B cells that intrinsically have some properties resembling those of memory cells. MZ B cells are also considered to be innate-like cells that can be induced to differentiate into short-lived plasma cells in the absence of BCR ligation (22). These properties allow the MZ B cells to crossover between adaptive and innate immunity (23).
We show here that ablation of TFII-I in B cell lineage selectively reduces murine MZ B cells. Further, transcriptomic and chromatin studies show that the surviving MZ B cells as well as FO B cells exhibit enhanced innate cell-like molecular features. Consistent with this notion, the splenic B cells in TFII-I deleted mice exhibit a noticeable increase in lipopolysaccharide (LPS) sensitivity, once again resembling features that are characteristic of innate immune cells. These results demonstrate that TFII-I is critical for maintaining B cell homeostasis and its absence accentuates innate properties of B lymphocytes.
To interrogate the role of TFII-I in B cell function we generated a “conditional knock-out” mouse model by breeding ‘floxed’ alleles of Gtf2i (24) with B cell-specific CD19-driver Cre. The B cell specific deletion of Gtf2i at genomic, mRNA and protein level were confirmed (Supp Figure 1A, B). Total splenocytes and B cell numbers were unaffected upon Gtf2i ablation (Supp Figure 1C). Various splenic B cells subsets (transitional, FO and MZ) were identified by previously described flow cytometry gating strategy (20) (Figure 1A), the percentages of total B, FO, MZ and transitional B (T1, T2) cells from 18 - 20 independent experiments are shown in (Figures 1B-E) and the cell numbers from 4 independent experiments are shown in (Supp Figure 1D). Both percentages and numbers of MZ B cells were significantly reduced in Gtf2i cKO (homozygous, HO) mice compared to control (wild type, WT and heterozygous, Het) mice (Figure 1C and Supp Figure 1D). However, total B cells as well as the FO and transitional B cell percentage and numbers were comparable among control and cKO mice (Figures 1B, D, E and Supp Figure 1D).
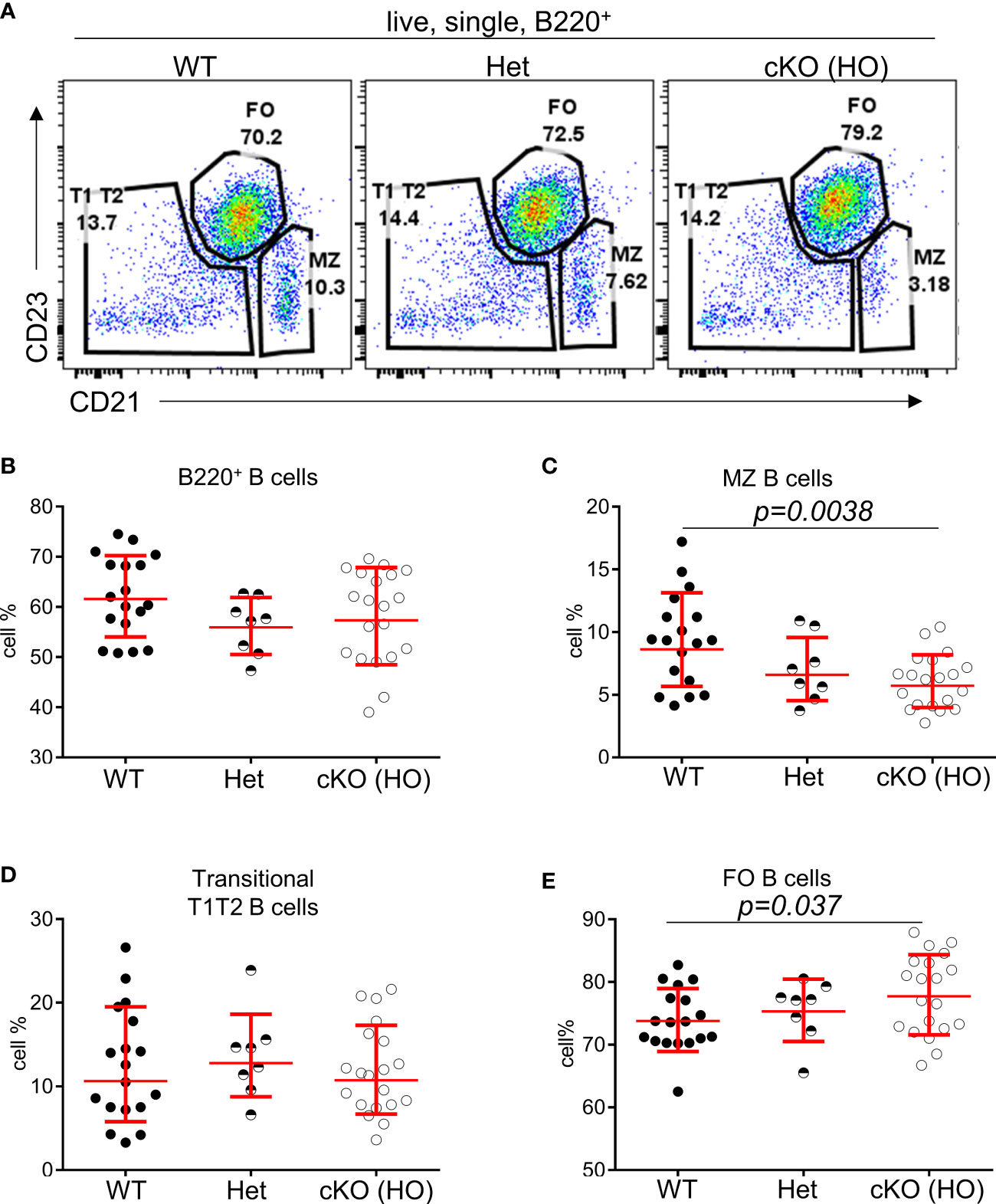
Figure 1 Reduced percentages of MZ B cells in Gtf2i cKO mice. (A) Representative plots of splenic B cell immunophenotyping of WT, Gtf2ifl/+CD19-Cre+ (Het) and Gtf2ifl/flCD19-Cre+ (HO) mice. FACS dot plots of CD21 (x axis) and CD23 (y axis) expression in B cells show CD21loCD23hi Follicular (FO) B cells, CD21hiCD23lo Marginal Zone (MZ) B cells and CD21loCD23lo Transitional (T1T2) B cells. (B) Splenocytes of wild type (WT), heterozygous (Het) and homozygous null (HO) mice were stained with B220 antibody and the percentages of B220+ total B cells are shown. (C–E) Primary splenic B cells were isolated from WT, Het and HO mice using EasySep magnetic purification techniques by negative selection. About 1 million B cells were stained with anti CD21-FITC and anti CD23-PE for 35 minutes, then washed and analyzed at the BD FACSAria Fusion. The percentages of FO, MZ and T1T2 B cells from 18 - 20 independent experiments are shown. Significance (P values) were calculated using paired student’s t test in Graphpad Prism.
We further investigated the tonic signaling (low level of antigen independent BCR signaling responsible for B cell survival) (25) in total splenic B cells in absence of TFII-I, as it can have Btk mediated B cell survival effects (26, 27). B cells were cultured ex vivo with or without B cell activating factor (BAFF) for the indicated times (Supp Figure 1E) and viability was checked using flow cytometry. However, there was no appreciable difference in the viability of B cells with or without BAFF among the WT, Het, and HO mice, suggesting no obvious role of TFII-I in B cell survival under these experimental conditions. Taken together these results indicate that although B cell specific ablation of Gtf2i does not interfere broadly with peripheral B cell homeostasis and survival, it selectively reduces MZ B cells in the spleen.
To understand the possible mechanisms of peripheral differences in B cell subsets, we first analyzed the transcriptome using bulk RNA-seq. Total RNA was prepared from FACS sorted FO and MZ B cells of WT and cKO mice spleens. We first identified MZ B cells differentially expressed genes (DEGs) between Gtf2i cKO and WT mice via DESeq2 algorithm taking ≥1.5 fold and FDR≤ 0.05 as cutoff (Figure 2 and Supp Figure 2A). DEG numbers were low, 64 DEGs were unique to WT and 40 genes were specific to Gtf2i cKO MZ B cells (Figure 2A). Of these small number of genes, we noticed Zfp36l2 was selectively downregulated in cKO MZ B cells. Zfp36l2 belongs to RNA binding protein (RBP) family which is involved in MZ B cell maintenance (28) and regulation of immune inflammation (29). This may partially provide an explanation as to why the cKO mice have reduced numbers of MZ B cells. Whether Zfp36l2 is a target of TFII-I is presently unknown. Next, we similarly identified FO B cells DEGs between the cKO and WT mice and only 6 DEGs were WT specific and 22 were cKO specific (Supp Figure 2A, B).
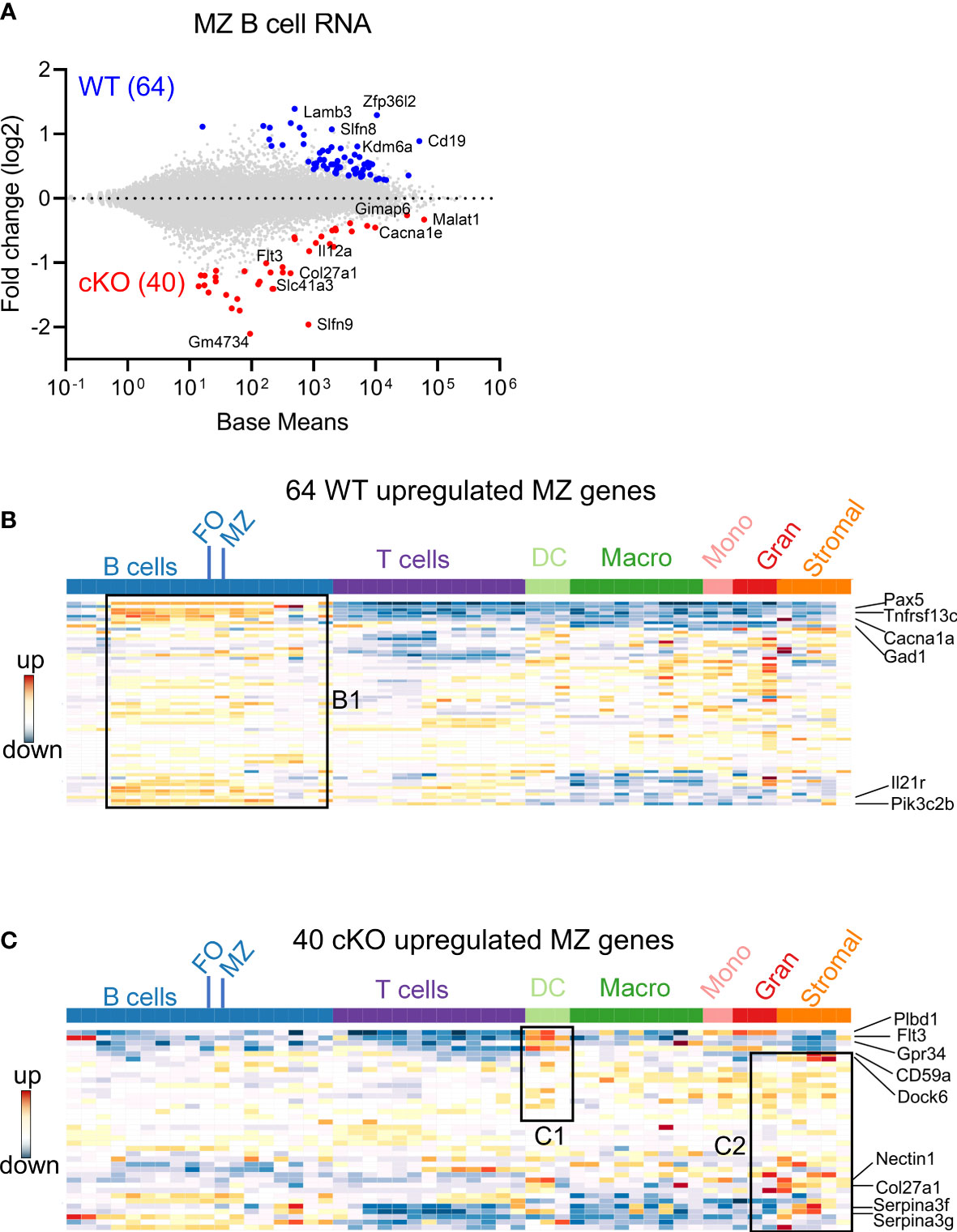
Figure 2 Differentially expressed genes in FO and MZ B cells derived from Gtf2i cKO and WT mice. Total RNA was extracted from FO and MZ B cells of WT and cKO mice. The RNA was used for bar-coded library preparations and sequencing. (A) MA plot displaying the log fold-change compared with mean expression and showing the DEGs of the WT MZ B cells (64 genes) and of the cKO MZ B cells (40 genes). (B) 64 WT MZ specific genes were queried against the ImmGen-Database and the heatmap shows the expression levels of these genes in B and other immune cells. Box B1 shows genes that are significantly enriched in the different B cell compartments. (C) Heatmap shows the expression levels of the 40 cKO MZ specific genes when they were queried against the ImmGen-Database. Genes that are enriched in DC compartment are shown in box C1 while genes that are enriched in stromal and granulocytes are shown in box C2.
Overlapping FO and MZ DEGs of cKO B cells identified TFII-I target genes, although at this stage we don’t know whether these are direct or indirect targets. Out of 62 cKO DEGs (40 MZ and 22 FO), 12 genes were shared by both FO and MZ subsets, 10 were FO specific and 28 were MZ specific genes (Supp Figure 2C). We noticed that these cKO-specific genes further exhibited specific innate immune related features (Supp Figure 2C, boxes bold fonts). This observation led us to further analyze MZ DEGs for presence of potential broad immune cell features using Immunological Genome Database (ImmGen-Database) (30). Genes which were up-regulated in the absence of TFII-I (40 genes) demonstrated enrichment only in MZ B cells (Supp Figure 2D, E, box A), while genes which were downregulated in the absence of TFII-I (64 genes) were enriched in the MZ subset as well as in other peripheral B cell subsets, which included mainly the transitional B cells genes like Pax5, Tnfrsf13c, Cacna1, Gad1, Il21r and Pik3c2b (Figure 2B box B1 and Supp Figure 2D, box A). Interestingly, ImmGen database comparison indicated that some of these 40 cKO genes are also highly expressed in two splenic dendritic cell subsets of bone marrow origin (CD4+ and CD8+ DCs) genes like Plbd1, Flt3 and Gpr34 (31, 32) and normally not expressed in splenic B cells (Figure 2C box C1 and Supp Figure 2D, box B). Further, they exhibited genes such as Cd59a, Dock6, Nectin1, Col27a1, Serpina3f and Serpina3g (Figure 2C box C2 and Supp Figure 2D, box C) which come from stromal subsets of Thymic Medullary Epithelial Cells “TEC”, fibroblastic reticular cells “FRC”, lymphatic endothelial cells “LEC” and also represent genes involved in myeloid development (33–35). Together, these observations indicate that although Gtf2i deletion in B cells doesn’t make significant transcriptome alteration, it leads both FO and MZ subsets to upregulate selective genes that are of innate and stromal cell origin.
Both follicular and marginal zone B cells develop from transitional B cells and attain their signature genes (20). Since marginal zone cells were reduced in the absence of TFII-I we tested whether TFII-I could alter the signatures of FO and MZ defining gene expression. Using DeSeq2 algorithm, we first identified DEGs between our WT (C57BL6) FO and MZ B cells (Supp Figure 3A) and then compared with Immgen-Database C57BL6 FO-MZ DEGs. The analysis of our WT B cells identified 328 upregulated genes in FO B cells and 714 genes in MZ B cells (Figure 3A). These numbers were similar to ImmGen-Database (30) when FO-MZ DEGs were compared, indicating a robustness in our analysis (Supp Figure 3B). We next assessed the FO and MZ signature DEGs in the absence of TFII-I using cKO mice (Supp Figure 3A). Gtf2i ablation shows slightly more DEGs between FO and MZ B cells comparable to the WT; 380 genes in the cKO FO cells, while 827 genes in the cKO MZ cells (Figure 3B). To determine the features associated with these cKO-specific genes, they were compared to WT using Venn overlap in both FO and MZ subsets.
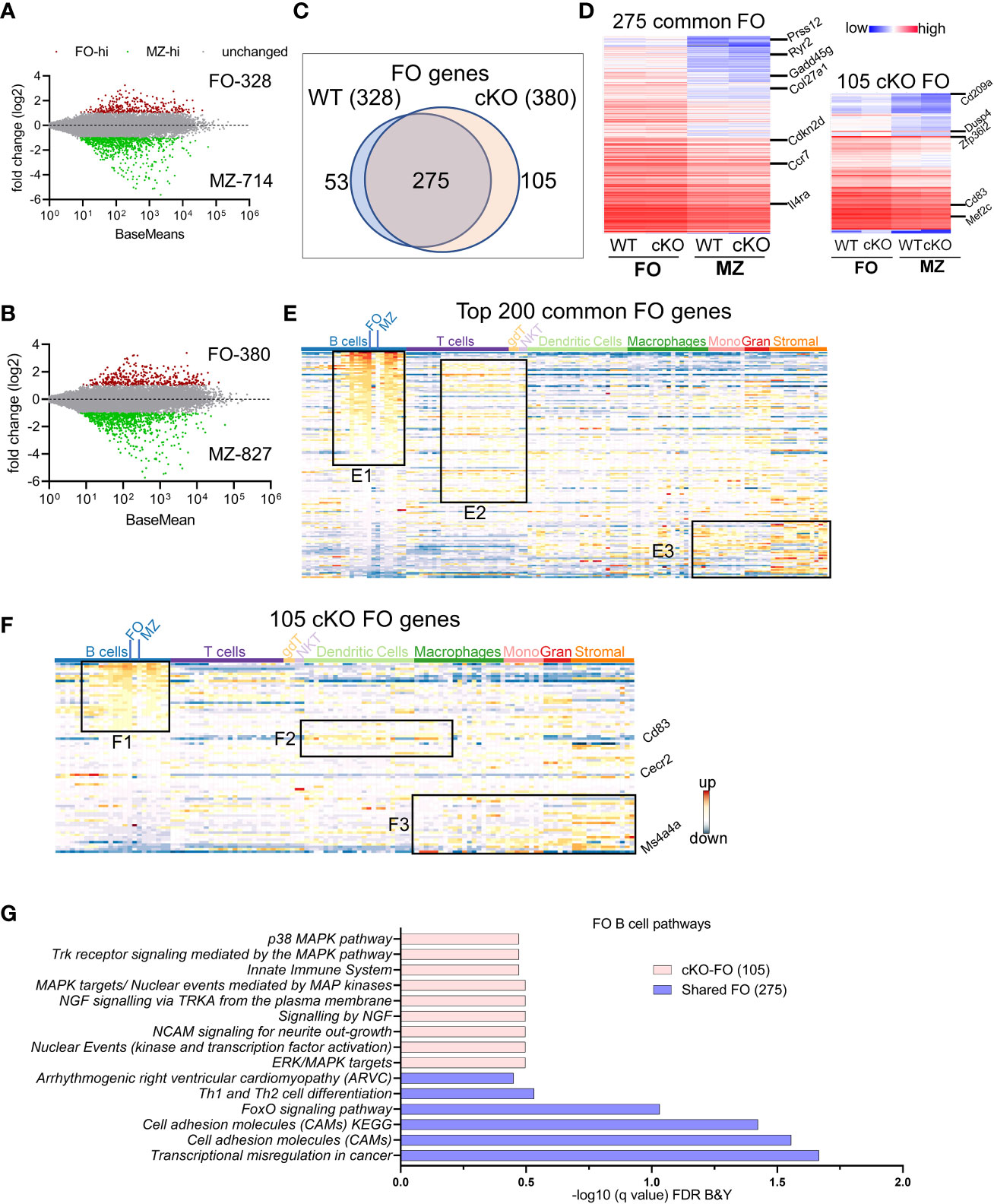
Figure 3 FO and MZ B cell defining transcriptome in WT and cKO mice. Total RNA was extracted from FO and MZ B cells of WT and cKO mice. The RNA was used for bar-coded library preparations and sequencing. (A, B) MA plot displaying the log fold-change compared with mean expression and showing the differentially expressed genes (DEGs) in FO and MZ B cells of the WT (A) and of the cKO (B) mice. (C) Venn diagram showing the genes that are common verses the ones unique to FO B cells between the WT and the cKO mice. (D) Heatmap demonstrating average counts (CPM, n=2) normalized by DESeq2 of the 275 FO common genes (left panel) and of the 105 unique cKO genes (right panel). (E) 200 common FO genes were queried against the ImmGen-Database to identify their enrichment in the different immune cell populations. Heatmap representing the expression of these genes in all immune cells. Genes enriched in the B cell compartments are shown in box E1, genes enriched in T cells compartments are shown in E2 and genes enriched in monocytes, granulocytes and stromal cells are shown in E3. (F) 105 cKO FO specific genes were queried against the ImmGen-Database as in E and the genes enriched in B cell compartments are shown in box F1, genes enriched in the Dendritic and Macrophage cells are shown in box F2 while genes that are enriched in the myeloid and stromal lineages are shown in box F3. (G) Gene Ontologies (GO) analysis using ToppGene suite, the analysis was carried out using either the shared (shared between WT and cKO) FO or the cKO specific FO B cell genes.
FO B cell genes: For FO specific genes, majority (275) were commonly upregulated (unaltered by Gtf2i absence) in both groups (Figure 3C) with a similar expression level (Figure 3D left panel and Supp Table 1). Of these genes Prss12, Ryr2, Il4ra, and Gadd45g are known to be FO specific (36). Top 200 common FO genes were analyzed using ImmGen-Database for immune-related transcriptomic signatures, including B cell, T cell, dendritic cells (DC), macrophages (Macro), monocytes (Mono), granulocytes (Gran) and stromal cells (Supp Figure 3C). These genes demonstrated features consistent with FO B cell specific genes (Supp Figure 3C upper panel red arrow). Interestingly, nearly half of these common FO genes were also significantly enriched in other peripheral subsets of B cells except MZ, germinal center (GC) and peritoneal cavity B cells (Figure 3E, box E1 and Supp Figure 3D). This analysis reiterates that transcriptionally FO B cells are significantly far from MZ, and GC B cells compare to transitional B cells. A subset of these common genes was expressed to a lower extent in mature T cell subsets (Figure 3E, box E2 and Supp Figure 3D), while the genes that are expressed at lower levels in FO B cells were upregulated in some subsets of macrophages, monocytes, and stromal cell (Figure 3E, box E3 and Supp Figure 3D). However, in the absence of Gtf2i, 105 genes were upregulated in FO B cells, including Zfp36l2, Dusp4, Cd83, Cd209a and Mef2c (Figure 3D right panel and Supp Table 2). Similarly, 53 genes were downregulated in Gtf2i ablated FO B cells (upregulated in WT FO B cells) including Dab2ip and Abca8b. Immgen-Data base comparison of these 53 genes shows that they were also expressed in other B cell subsets (Supp Figure 3E, box SE1), including Vpreb3 (a gene expressed during B cell development) (37). The poorly expressed FO WT genes were expressed in other myeloid and stromal cell compartments (Supp Figure 3E, box SE2), a feature observed in the common set of FO genes (Figure 3E, box E3). Because these genes were expressed in the absence of Gtf2i we concluded that these could be TFII-I (direct or indirect) regulated genes.
Genes upregulated in FO B cells in the absence of Gtf2i (105 cKO) were also significantly expressed (Supp Figure 3D) in other B cell subsets (Figure 3F, box F1). For instance, a Pax5 target gene Cecr2, which is expressed during B cells development (38) is still expressed in FO B cells in the absence of Gtf2i (Figure 3F). In contrast and unlike common and WT FO genes, the FO genes that are specific to the cKO group showed some enrichment of dendritic and macrophage subset genes such as Cd83 (Figure 3F, box F2). Further, the genes that are expressed at low levels in the cKO FO subset were also expressed in other myeloid and stromal cell lineages (Figure 3F, box F3). The enriched Gene Ontologies (GO) of the 275 common FO genes were identified using ToppGene suite and demonstrate mostly cell adhesion features (Figure 3G). cKO specific 105 FO genes show enrichment of multiple MAPK pathways and innate immune system GOs (Figure 3G). These observations indicate that in the absence of TFII-I, and in comparison, to the MZ B cells, FO B cells maintain expression of genes that are highly expressed during B cell developmental stages (Vpreb3 and Cecr2) and genes which mark GC B cells or innate immune cells like Cd83 and Cd209a.
MZ-specific genes: We next compared the transcriptomic features of MZ B cell genes that are enriched when compared to FO B cells in WT and in cKO. The overlapping of 714 upregulated MZ specific genes in WT with 827 genes in Gtf2i cKO resulted in 626 common set of genes (Figure 4A) that also exhibited comparable expression levels (Figure 4B, left panel and Supp Table 3). When analyzed for gene features of other immune cells, top 200 common MZ specific genes showed significant (Supp Figure 3F) dominating MZ B cell features (Figure 4C, box C1). These common genes also recapitulated features of innate immune cell genes (36) (Figure 4C, box C2 and Supp Figure 3F) and shared features characteristic of granulocytes and stromal cell compartments (Figure 4C, box C3). While 201 MZ genes were uniquely upregulated in the absence of Gtf2i (Gtf2i cKO MZ cell genes) when compared to cKO FO B cells, which include Mpo, Gm4734, Mest, Gpr34 and Asns (Figure 4B, right panel and Supp table 4). Likewise, 88 MZ genes were downregulated in Gtf2i lacking B cells (upregulated in WT MZ cell genes) including Zfp385a, Gap43, and E2f7. WT MZ-specific genes (88 genes) didn’t demonstrate any significant features for other immune cell types (Supp Figure 3G). However, the 201 genes derived from the MZ compartment of cKO demonstrated significant enrichment of genes belonging to developing stages of both B and T lymphocytes (Figure 4D, box D1 and Supp Figure 3H). These genes were generally poorly expressed in mature peripheral lymphocytes of WT mice (Figure 4D, box D1, middle region dotted lines) and are mainly cell cycle related genes (Supp Table 4). Remarkably, in the absence of Gtf2i, MZ high expressing genes further exhibited features resembling innate immune compartments (Figure 4D, box D2 and Supp Figure 3H). Additionally, cKO MZ genes also showed commonality with granulocyte and stromal cell genes, which are generally not expressed in WT MZ B cells (Figure 4D, box D3). Like FO B cells, these observations too suggest that there is an alteration of MZ defining basal transcriptome in the absence of TFII-I. The enriched GOs of the 626 common MZ genes identified mostly cell cycle enriched pathways (Figure 4E). While GO analysis for Gtf2i cKO specific 201 MZ genes included the PLK1 signaling that has a role in the progression of mitosis, the ATM and ATR signaling pathways that are crucial for DNA damage response and maintains genomic integrity (Figure 4E).
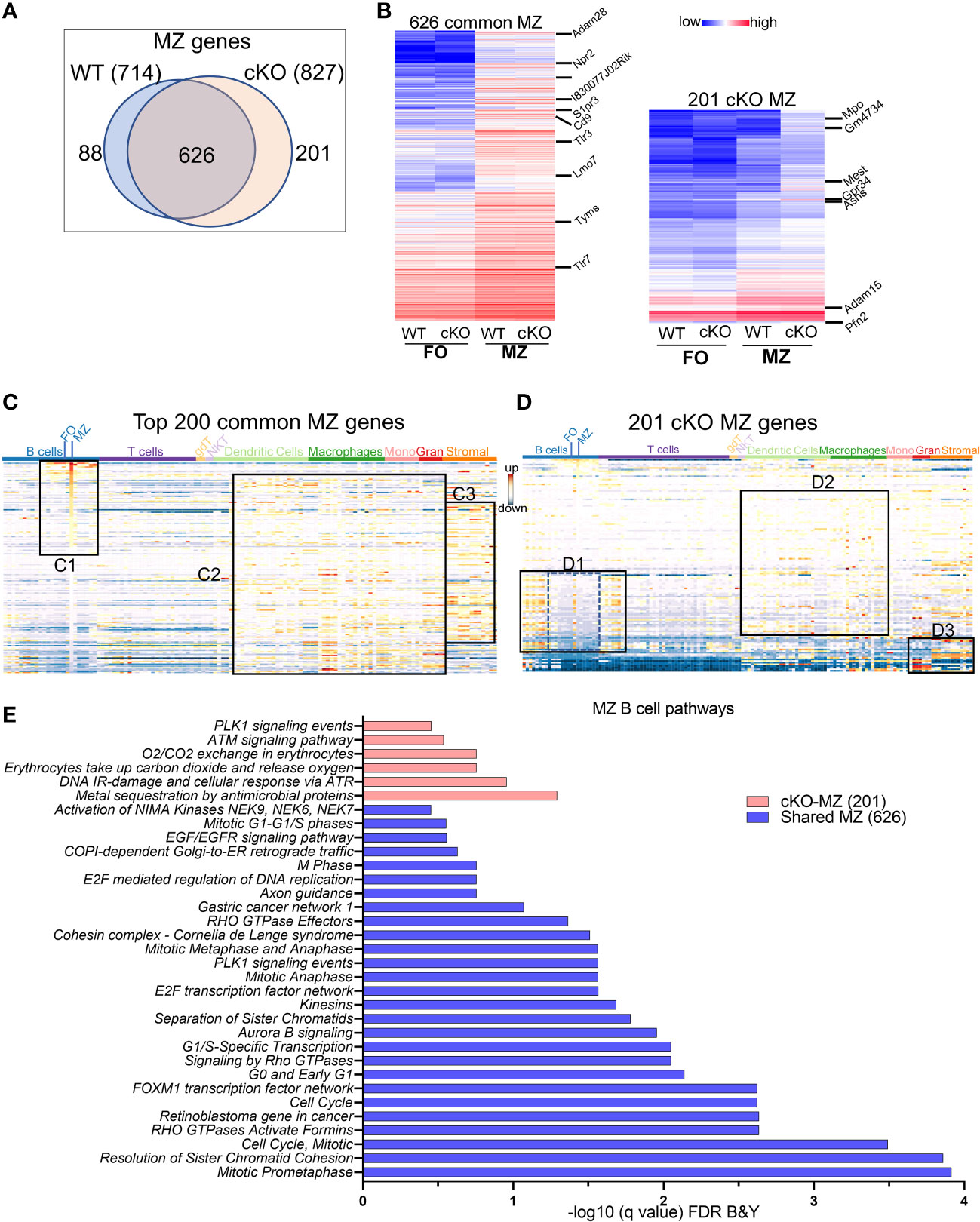
Figure 4 MZ B cell defining transcriptome in WT and cKO mice. Total RNA was extracted from MZ B cells of WT and cKO mice. The RNA was used for bar-coded library preparations and sequencing (A) Venn diagram showing the genes that are common verses the ones that are unique to MZ B cells between the WT and the cKO mice. (B) Heatmap demonstrating average counts (CPM, n=2) normalized by DESeq2 of the 626 MZ common genes (right panel) and of the 201 unique cKO genes (left panel). (C) Top 200 common MZ genes were queried against the ImmGen-Database and the heatmap representing the expression of these genes in all immune cells. Box C1 shows the genes that are enriched mostly in the MZ compartment, box C2 shows the genes that are enriched in the innate compartment and box C3 shows the genes that are enriched in the granulocyte and stromal cell compartments. (D) 201 cKO MZ genes were queried against the ImmGen-Database as in (C) and the genes that are enriched in B and T cells compartments are shown in box D1, while the genes that are enriched in Dendritic cells and Macrophages are shown in box D2 and the genes that are enriched in myeloid and stromal lineages are shown in box D3. (E) Gene Ontologies (GO) analysis using ToppGene suite, the analysis was carried out using either the shared (shared between WT and cKO) MZ or the cKO specific MZ B cell genes.
In summary, these data demonstrate that although in the absence of TFII-I the overall transcriptomic features distinguishing FO and MZ are largely retained, FO B cells lacking TFII-I exhibit signatures that skew them towards DC and macrophage subsets and have some features of GC B cells. In contrast, MZ B cells gained expression of cell cycle-related genes from developing stages of lymphocytes and further exhibit enhanced signatures characteristic of innate cells.
Although TFII-I is a transcription factor that functions via cis-regulatory elements and expected to impact chromatin accessibility, this has not been directly explored. Open chromatin impressions are associated with cellular identities (39). Thus, to address whether absence of TFII-I would alter chromatin landscape of FO and MZ B cells, we performed Assay for Transposase-Accessible Chromatin with high-throughput sequencing (ATAC-seq) (40), using sorted FO and MZ B cells from WT and cKO spleens. After quality checks and alignment to the mouse genome, open chromatin regions (peaks) were called using MACS2 algorithm. We observed substantially fewer peaks in FO B cells in both WT and cKO (44,293 peaks in WT FO and 56,871 peaks in cKO FO) compared to MZ B cells (93,887 peaks in WT MZ and 104,543 peaks in cKO MZ) (Figure 5A). Furthermore, we also noted that both FO and MZ B cell subsets of cKO have more open chromatin peaks than WT counterparts, (12,578 and 10,656) respectively. These observations interestingly coincided with the master regulator of chromatin architecture, Ctcf mRNA expression levels in FO and MZ B cells. More accessible chromatin of MZ B cells have lower Ctcf mRNA compared to FO B cells and similarly cKO FO and MZ B cells also have relatively low expression of Ctcf (Supp Figure 4A). This result may partially explain the enhanced accessible chromatin peaks observed in cKO B cells (41). Interestingly, a direct interaction between CTCF and TFII-I to regulate gene expression has been observed (42).
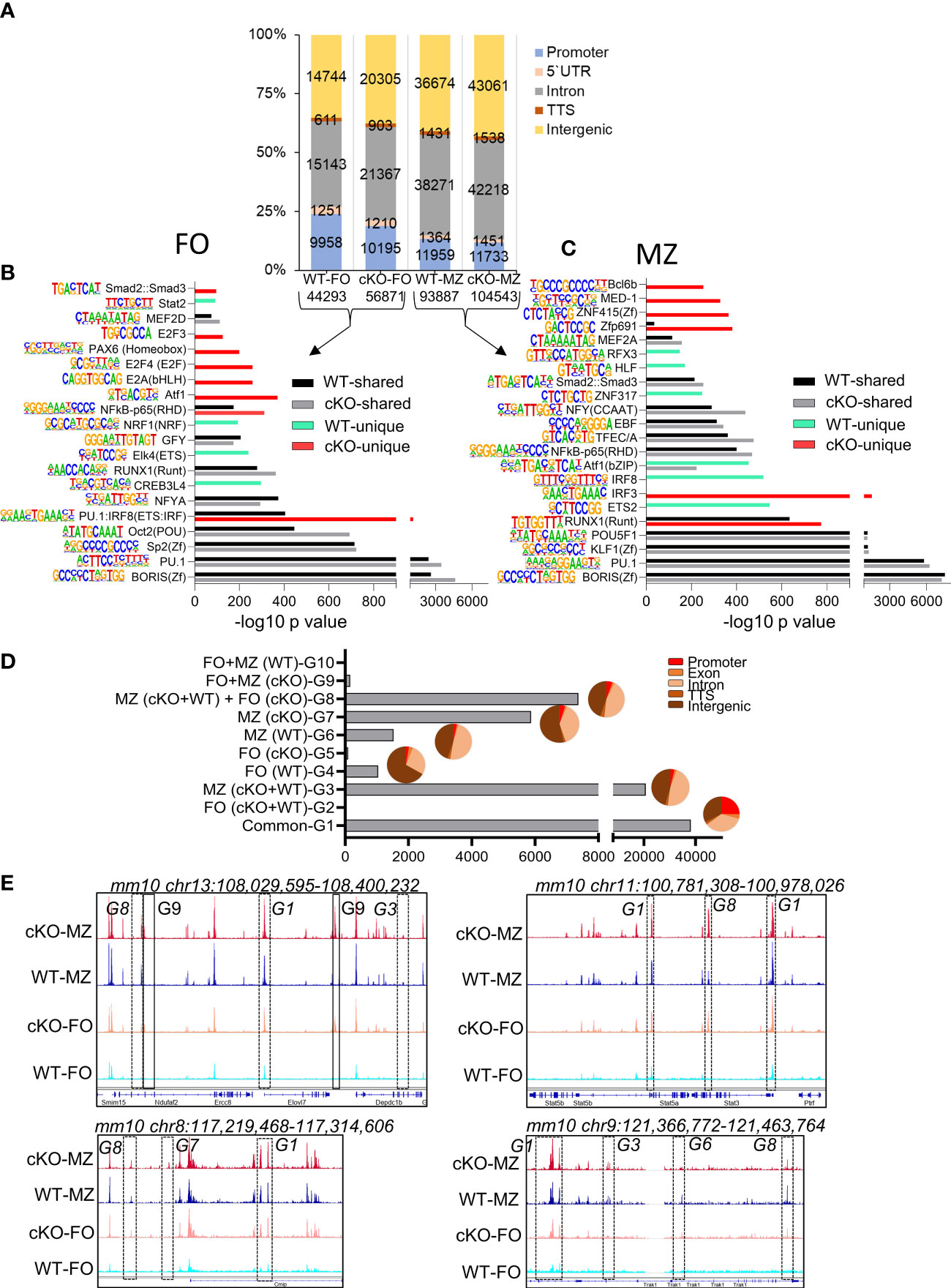
Figure 5 Chromatin landscape of FO and MZ B cells of WT and cKO mice. Quantitative description of chromatin landscape of four different splenic B cell populations: WT FO and MZ B cells and Gtf2i cKO FO and MZ B cells. (A) Peaks from FO and MZ B cells (numbers of peaks on bottom) from the indicated mouse strain and the annotations regarding their location in the genome. The peak numbers per region are shown inside each region (B, C) Presence of transcription factor motifs in open chromatin of WT and cKO FO (B) and MZ B cells (C), bars represent the -log10 p-value. cKO selective transcription factor motifs are marked by red color columns while WT selective transcription factor motifs are marked by light green color columns. Motif logos are listed next to each transcription factor. (D) Overlapping peaks from all four populations were identified using mergePeaks.pl suite (with -d 50) in HOMER. Ten different clustered groups (G1 – G10) of the overlapping peaks are shown with their indicated properties on the y axis. The pie charts show the peaks distribution in the genomic regions for the specified groups. (E) Genomic tracks of open chromatin (snapshot from IGV genome track viewer) from indicated B cell populations demonstrating open chromatin regions of chromosome 13, 11, 8 and 9. Group specific peaks are outlined by boxes.
To determine where these peaks are located in the genome, we annotated the open chromatin peaks to five distinct genomic regions: promoter, 5`UTR, intron, TTS, and intergenic areas with the number of peaks per region as shown in Figure 5A. In the absence of TFII-I the percentage of open promoter regions were reduced in cKO FO B cells with a concomitant gain of open chromatin peaks in the intergenic and intronic regions. Genomic distribution of open chromatin locations in MZ B cells was similar between WT and cKO. We concluded that in the absence of TFII-I the chromatin is more permissive in both FO and MZ splenic B cells and that accessible chromatin is preferentially localized to the intergenic and intronic regions.
We next analyzed RNA corresponding to the accessible chromatin in B cell subsets. Open chromatin peaks were annotated to their associated genes using annotatePeaks.pl algorithm of HOMER. Maximum number of RNA corresponding to the open chromatin (14611) were shared by both FO and MZ subsets of WT and cKO (Supp Figure 4B). While MZ B cell subset of both WT and cKO shared next highest number (2476) of open chromatin related genes. Interestingly, there were 1481 open chromatin corresponding genes shared by MZ of both WT and cKO and with FO of cKO (Supp Figure 4B). This analysis indicates the possibility that TFII-I might skew cKO FO B cell chromatin accessibility resembling chromatin landscape in MZ B cells.
Open chromatin is often associated with presence or absence of specific transcription factor (TF) or families of TFs binding sites (43). To investigate the specific TF binding sites associated with ATAC-seq peaks, we analyzed the open chromatin of FO and MZ B cells identified in Figure 5A using findMotifsGenome.pl algorithm. Similar TF motifs were observed with many open chromatin features associated with WT and cKO derived FO and MZ B cells (Figures 5B, C). However, our de novo analysis demonstrated IRF8(IRF) motif, which is mainly associated with myeloid and B lymphocyte differentiation (44), was relatively more enriched in FO B cells of cKO as a composite of PU.1:IRF8 (ETS : IRF) shared motif (Figure 5B). Interestingly, similar pattern was also observed in MZ B cells of WT (Figure 5C). Motifs like IRF8 enriched in WT MZ open chromatin and subtly different motif of IRF3 enriched in cKO MZ open chromatin. Furthermore, RUNX1 motif was enriched in both MZ of cKO and WT with higher enrichment in the cKO MZ than in the WT MZ (Figure 5C). Presence of BORIS and PU.1 motifs was quite evident in both FO and MZ cell open chromatin from both strains. The cKO FO and MZ B cell accessible regions additionally exhibited presence of Atf1, E2A, E2F4, NF-κB, Zfp691, ZNF415(Zf), MED-1, and Bcl6b motifs (Figures 5B, C). We concluded that though there is more permissible chromatin in the absence of TFII-I, the enrichment of TF-motifs was comparable to WT B cells. However, cKO accessible regions show specific enrichment of E2A and E2F motifs in FO B cells, which can make these cells poised towards GC B cells in the absence of TFII-I (45).
To further understand the global effects of Gtf2i ablation on open chromatin landscape across both B cell subsets, we identified common and unique peaks using mergePeaks.pl from HOMER algorithm (46). To reduce redundancy, we considered peaks across the cell types (FO or MZ) or strains (WT or cKO) as same if their start and end sites were within a span of 50 bp. This method provided 10 clustered groups with substantial number of peaks as defined in Figure 5D. As expected, the largest number of peaks were common to both cell types (FO and MZ) and strains (WT and Gtf2i cKO) as defined in group G1. We hypothesized that these are the open chromatin features of mature splenic B cells. Second largest number of peaks was present in MZ B cells for both strains and is represented by group G3 (20694 peaks). These peaks represented distinct features of MZ cells compared to FO cells regardless of the strain. Furthermore, substantial number of unique peaks (5871) were present in cKO MZ (group G7), suggesting permissive chromatin of MZ B cells in the absence of TFII-I. Surprisingly, apart from these three major groups, 7370 unique peaks were also present in group G8 which is defined as a group consisting of peaks common to both FO and MZ of cKO and MZ of WT. We inferred that even FO B cells partly exhibit open chromatin features like that of MZ B cells in the absence of TFII-I.
Annotation of the common and unique peaks of these groups to the genomic regions is shown in the pie charts in Figure 5D. The peaks in group G1 which represent mature B cell open chromatin are almost equally distributed between the promoter (25%), intronic (35%) and intergenic (33%) regions. However, the peaks in the MZ B cells specific group G3 fall mostly in the intronic and intergenic regions with less opening at the promoter region compared to G1. Like G3, the peaks of cKO MZ specific G7 and WT MZ specific G6 fall also mostly in the intronic and intergenic regions. Group G8 which denotes MZ like chromatin of FO B cells in the absence of TFII-I, shows open chromatin mostly in the intronic and intergenic regions, suggesting a role for intronic and intergenic region in regulating the chromatin landscape in these cells. Representative genome browser tracks demonstrating open chromatin peaks from various groups are shown in (Figure 5E). Although these open chromatin features suggest that majority of chromatin landscape is not altered by ablation of TFII-I in both FO and MZ B cells, striking differences in selective peaks (as represented by G8) indicate that in the absence of TFII-I, a part of FO B cell open chromatin landscape features begin to skew towards chromatin features resembling that of MZ B cells.
After identifying basal state properties of open chromatin of FO and MZ B cell in the absence of TFII-I, we next analyzed FO and MZ defining open chromatin features in WT and cKO B cells by using DiffBind algorithm (47). Significantly more chromatin peaks were open in MZ B cells compared to FO B cells of both strains, WT (39444 vs 983) and cKO (33222 vs 378) respectively (Figure 6A). We further noticed >2fold decrease in differentially open chromatin (DOC) peak numbers in cKO FO B cells when compared to WT FO peaks (378 cKO FO vs 983 WT FO peaks) (Figure 6A). When examined for their genomic location annotations, Gtf2i cKO FO and MZ specific peaks were reduced at the promoter and 5` UTR region compared to their WT counterpart while there was more cKO FO peaks at the intronic and intergenic region compared to WT FO peaks (Figure 6B, top). Relative to the murine genome (mm10) these enrichments were significant for 5` UTR and promoter in both WT and cKO B cells (Figure 6B bottom). However, relative to the murine genome, FO B cell, intron and intergenic peak enrichments were less in WT compared to cKO, while MZ B cells appeared to have similar distribution in intron and intergenic region DOCs (Figure 6B, bottom). These results indicate that TFII-I ablation also leads to reduced chromatin landscape differences between FO and MZ B cells.
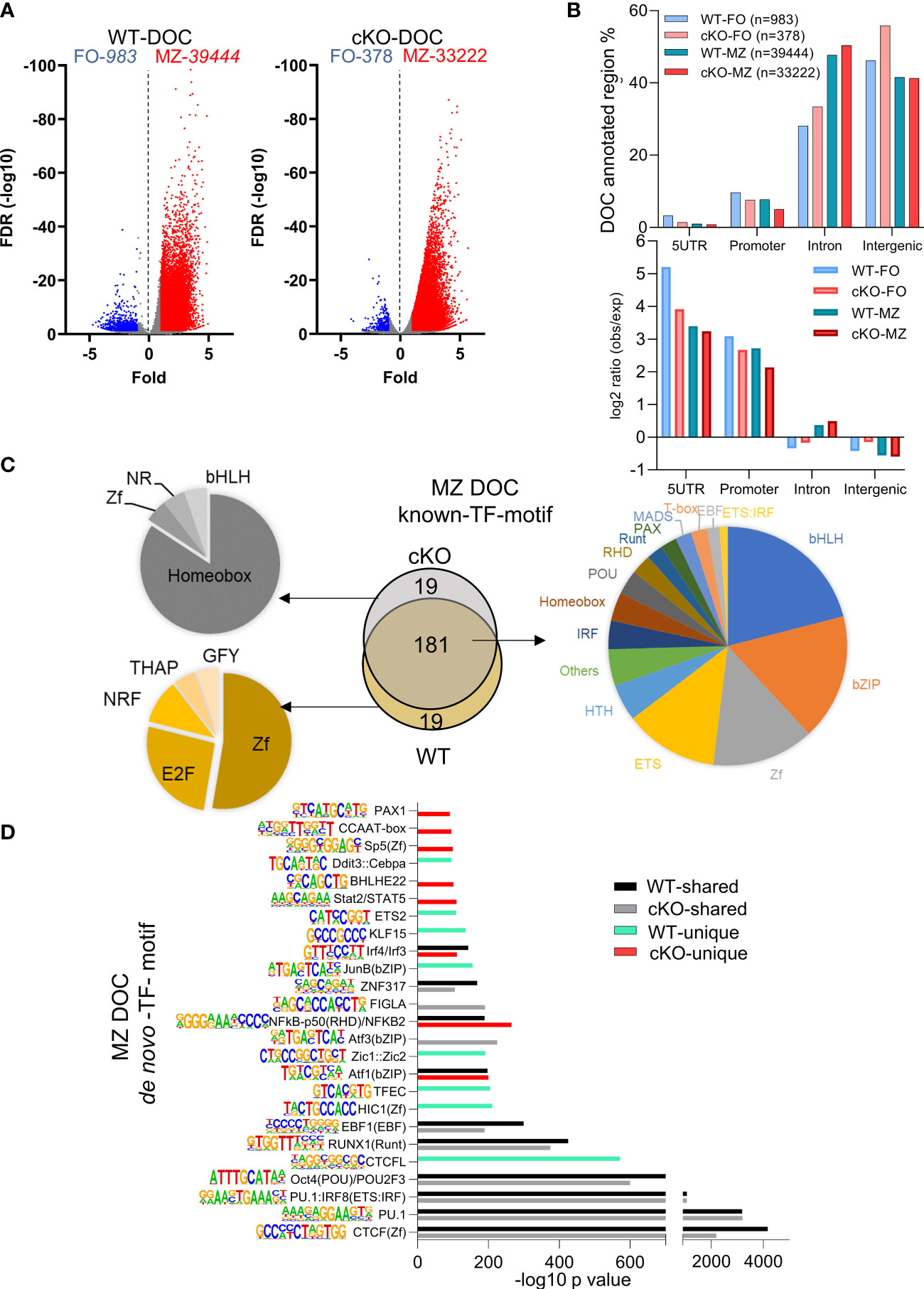
Figure 6 Transcription factors binding motifs in open chromatin of FO and MZ B cells. (A) Volcano plot showing the differentially open chromatin (DOC) from FO and MZ B cells of WT (left) and cKO (right); numbers of DOCs per cell type for the WT and cKO are shown on top. (B) The DOCs were annotated to the 4 genomic regions (5UTR, promoter, intron, intergenic) and the percentages of these DOC annotated regions are shown for the four cell types. Their observed vs expected values relative to genomic background is shown in bottom. (C, D) The top 200 transcription factor (TF) motifs of DOC of MZ from WT and cKO B cells identified by known (C) and de novo (D) TF motif algorithm of HOMER. (C) Venn diagram (middle) shows the number of predicted common and unique TF binding motifs. Common motifs are represented by the pie chart on the right while the unique ones are represented on the left side. (D) bars represent the -log10 p-value. cKO selective transcription factor motifs are marked by red color columns while WT selective transcription factor motifs are marked by light green color columns. Motif logos are listed next to each transcription factor.
Because of higher number of DOCs, we next focused on MZ specific regions for presence of DEGs association and transcription factor motifs using both known and de novo algorithms of HOMER suite. MZ B cell DOCs were annotated to their respective genes and were correlated with DEGs in these cells (Supp Figure 4C). Around half of the DEGs (414 genes) have shared DOCs, while the remaining of the genes cannot be associated with differentially open chromatin (Supp Figure 4C). By comparing the top 200 transcription factor motifs (identified by known motifs algorithm) of MZ DOC between WT and cKO, 90% (181) of motifs were shared between both genotypes. We also noticed significantly enriched transcription factor motifs of basic-helix-loop-helix (bHLH), basic-leucine zipper (bZIP), zinc finger (Zf), and ETS family in this analysis (Figure 6C). In the absence of Gtf2i, 19 motifs under MZ DOC were dominated by Homeobox TF motifs while the WT MZ DOC were enriched for Zf and E2F motifs (Figure 6C).
In our de novo TF motif algorithm analysis (which ranks the dominating TF motifs/TF-family), we found that PU.1, CTCF, POU2F and RUNX1(Runt) motifs were present in open chromatin of both WT and cKO MZ B cells (Figure 6D). Interestingly, CTCFL, EBF1, Jun, TFEC and HIC1 motifs were selectively enriched in WT MZ specific open chromatin, while Aft3, Bhleh22, PAX1 and Stat2/STAT5 motifs dominated in the cKO MZ specific open chromatin (Figure 6D). Gene expression levels of many of these transcription factors are shown in Supp Figure 4D. For example, although Stat family factors show that FO B cells of WT and cKO have similar expression levels, they are higher in the FOs compared to the MZs. On the other hand, Irf4 shows higher expression in the cKO FO and MZ B cells, while Irf8 has higher expression levels in FO B cells of WT and cKO. These selective patterns of activated cell and innate like TF-motifs enrichment in differentially open chromatin suggest that the altered chromatin landscape may stem from FO B cell stages itself and the absence of TFII-I may influence all mature B cells.
It is well-established that murine B cells are less responsive to bacterial endotoxin LPS compared to the innate immune cells (e.g., macrophages and dendritic cells) (48). Given the clear indications from transcriptome and chromatin landscape analysis that removal of TFII-I accentuates innate-like features in B cells, we next aimed to test this notion by challenging B cells from WT and cKO in vitro with various dosages of LPS and assay for gene expression at various time points using Toll-like Receptor Signaling Pathway RT2 Profiler PCR Array (Figure 7A). Naïve splenic B cells from WT and cKO mice were stimulated with 1 and 5 µg/ml of LPS for 0, 2, 8 and 24 hours. After RNA extraction and cDNA preparation, samples were subjected to the PCR array analysis. Differential gene expression analysis showed that 36% of the genes (30 genes) of the Toll-like receptor signaling pathway (84 genes) were expressed at higher levels in cKO B cells at basal level itself (Figure 7B, red), while three out of the 84 genes exhibited higher expression in WT B cells (Figure 7B, blue). These results show that at the basal level most of the Toll-like receptor pathway genes are upregulated in the cKO B cells compared to WT B cells.
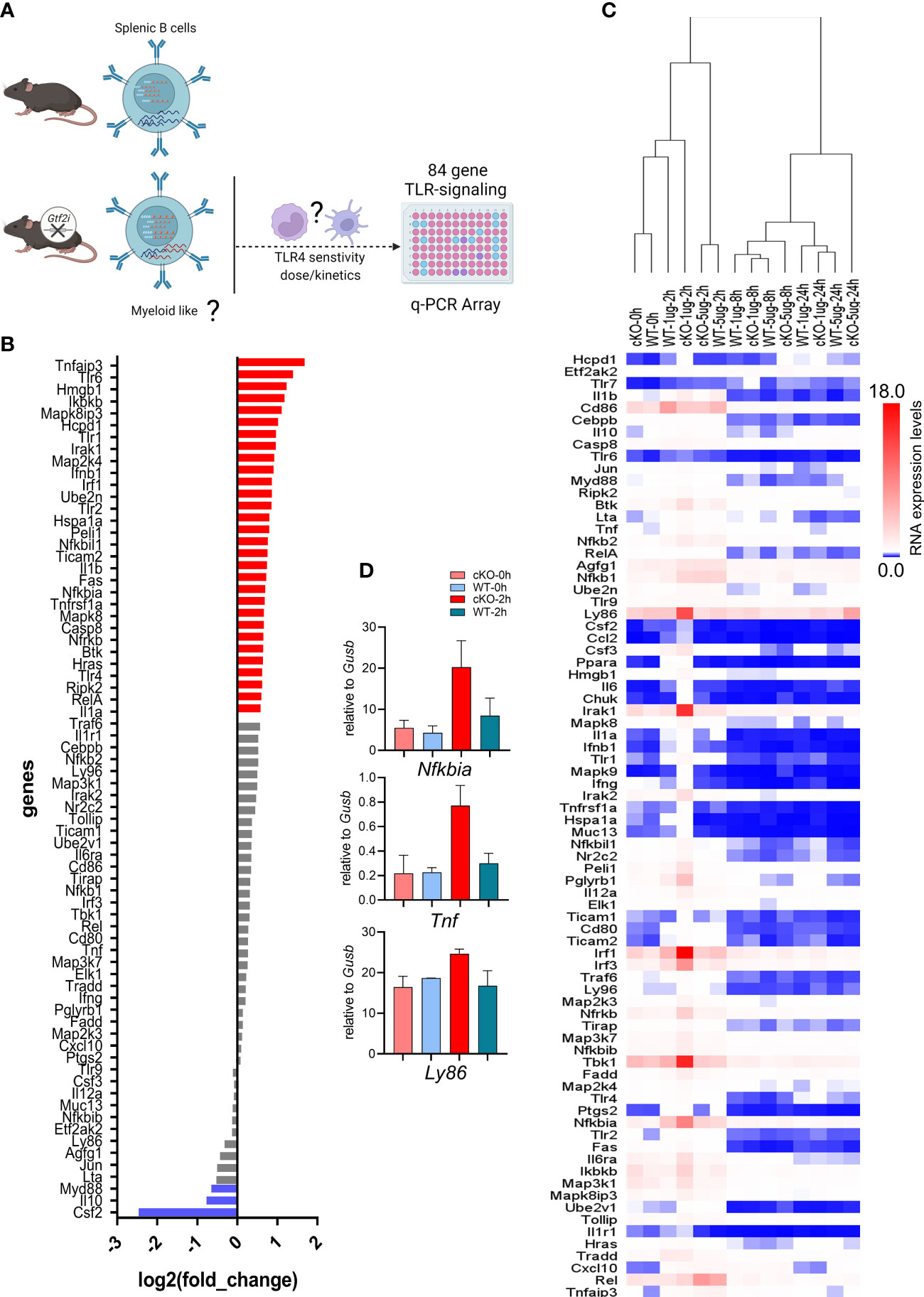
Figure 7 Transcriptome of LPS responsiveness splenic B cells upon Gtf2i ablation. (A) Scheme representing (created using BioRender.com) the identified differences of transcriptome and open chromatin in B cells upon Gtf2i ablation. These differences indicate cKO B cells to possess myeloid like features, which are tested for TLR4 sensitivity by activation with LPS (5 and 1µg/ml) at early, mid and late time points (2, 8 and 24 hours) using TLR-signaling array. (B) Basal level expression of genes downstream of TLR-signaling in WT and cKO B cells. Red bars represent 30 genes highly expressed in cKO and blue are highly expressed in WT by at least 1.5-folds (n=4). (C) Heatmap representing the RNA expression levels of 84 TLR-responsive genes from LPS high (5µg/ml) and low (1µg/ml) dose at early (2h), mid (8h) and late (24h) time points (n=3). Relative expression of target genes was calculated to geometric mean of Ct values of Gusb and Hsp90a. (D) Nfkbia, Tnf and Ly86 mRNA levels were determined from the Mouse Toll-Like Receptor Signaling Pathway” RT2 Profiler PCR Array as in (C), the average of 2 independent experiments is shown. Error bars represent standard deviation.
Interestingly, the suboptimal dose of LPS (1μg/ml) for B cells shows striking differences at early time point (2h) (Fig 7C - D). In hierarchal clustering the WT B cells (2h, 1µg/ml) clustered with unstimulated B cells (0h) while cKO B cells with the LPS 1μg/ml dose clustered away from WT (1µg/ml) and the unstimulated samples but closer to LPS 5µg/ml dose. We noticed with 1μg/ml dose activation at 2h in cKO B cells some genes, Ly86, Irak1, Irf1, Tbk1 and Nfkbia were induced even higher than 5µg/ml dose. However, the response to optimal dose of LPS (5μg/ml) was similar between the cKO and WT B cells at earlier and late time points (2 and 24 hours) while mid time point (8h) shows some differences (Figure 7C). Consistent with the RNA-seq and ATAC-seq analysis, these results together suggest that in the absence of TFII-I, the LPS sensitivity in B cells is increased, further indicating enhancement of their innate and/or lower threshold towards activation.
In the absence of TFII-I in B cells, our transcriptome and chromatin landscape demonstrated skewed properties towards innate like features. Even, FO B cells exhibited enhanced MZ-like molecular features. We thus tested the consequences of such alteration by analyzing ex vivo proliferative properties of B cells after T-independent and dependent (antigen) activation accomplished by LPS and anti-IgM Fab`2 stimulation respectively (Figure 8). B cells from Gtf2i cKO demonstrated higher proliferation capacities upon LPS-mediated/T-independent stimulation in a dose dependent fashion (1.25, 2.5, 5 and 10ug/ml) particularly at 72h (Figure 8A and quantified Figure 8B) while there was no difference in proliferation in response to anti-IgM F(ab)`2/T-dependent stimulation (Figure 8A). We also noticed that significantly more quiescent B cells progressed into cell cycle and completed first mitotic division in cKO B cells compared to WT B cells upon LPS activation (Figure 8B, G0 population). Furthermore, the percentages of cells that proceed to G2, G3 and G4 were significantly higher in cKO B cells compared to the WT (Figure 8B). This increased proliferative property after LPS stimulation was further demonstrated by higher division index of cKO B cells at 72h (Figure 8C).
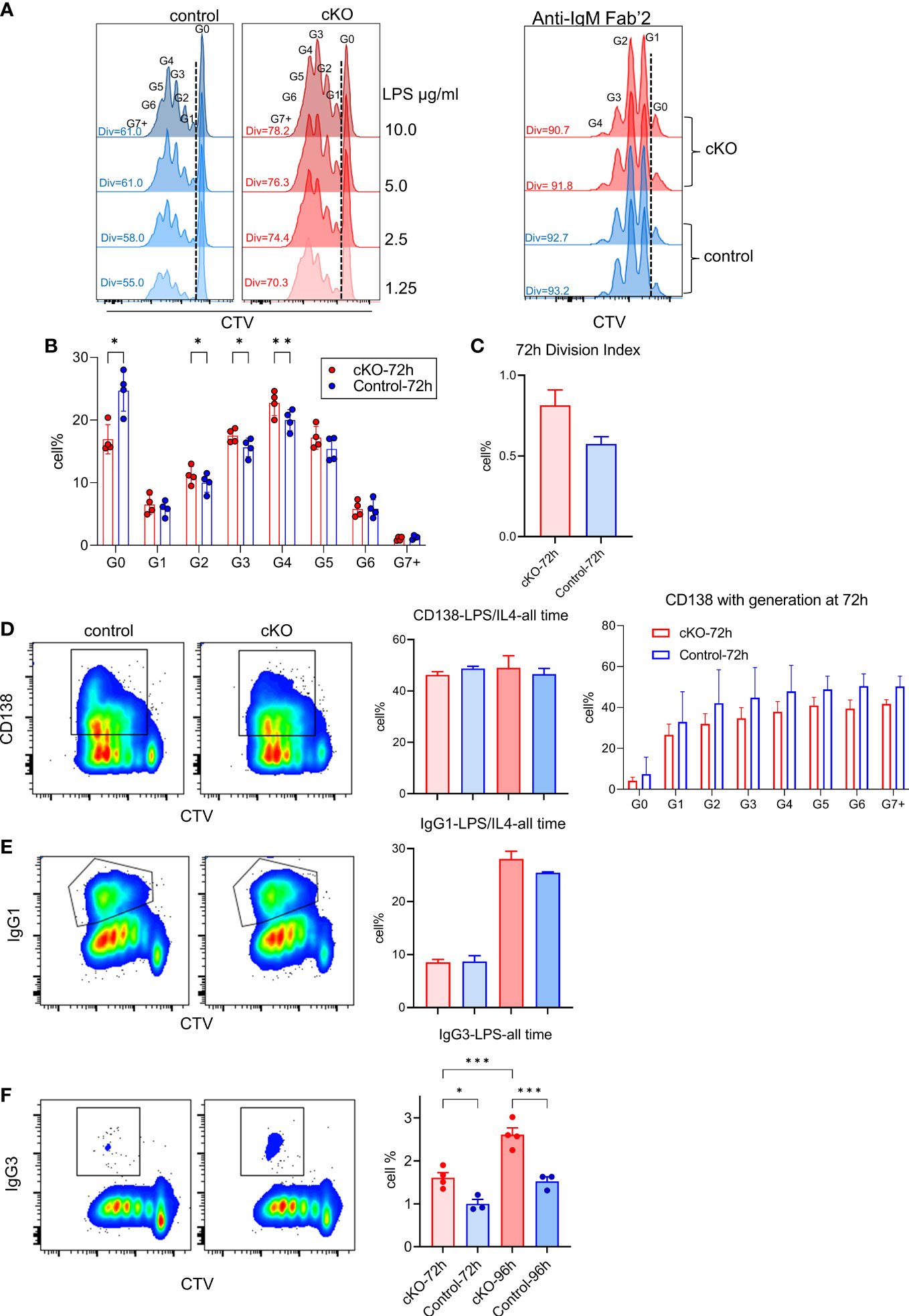
Figure 8 Functional characterization of splenic B cells from WT and cKO derived mice. Splenic B cells from WT and cKO mice were activated with LPS (1.25, 2.5, 5 and 10µg/ml), F(ab`)2 anti-IgM (10µg/ml) or LPS (10µg/ml) plus IL-4 (20ng/ml) for the indicated time points followed by flow cytometry analysis. (A) Representative flow cytometry plot of Cell Trace Violet (CTV) dilution after 72 hours of LPS (10µg/ml n = 4, other concentrations n = 2) or anti-IgM (n = 2) activation. The Div numbers indicate the percentages of divided cells for the indicated mouse strain. Numbers above the CTV peaks refer to the cell generation (G0 – G7+) (B) Cell percentages at each cell division after 72 hours of LPS activation. Bars represent the mean +/- SEM obtained from 4 independent experiments and p values are shown for paired t-test corrected for multiple comparisons. (C) Division Index was calculated for cKO and WT B cells after 72 hours using FlowJo proliferation platform. (D, E) Representative flow cytometry plot of CD138 (D) or IgG1 (E) versus CTV for WT and cKO (left) after 96 hours of LPS plus IL-4 activation. Gating of CD138+ and IgG1+ cells are indicated. The bar plot on the right shows the average of the CD138+ (and the CD138+ cell percentages per cell division) or IgG1+ cell percentages at the indicated times. (F) Representative flow cytometry plot of IgG3 versus CTV after 96 hours of LPS activation and the bar plot shows the IgG3+ cell percentages at the indicated time points. Data represent 4 independent experiments *p≤0.05, **p≤0.01, ***p≤0.001.
To further characterize these B cells functionally, LPS and IL4 treated splenic B cells from WT and cKO mice were tested for CD138+ plasma cells differentiation and IgG1 class switching. Although, at total population level CD138+ cells were similar between WT and cKO (Figure 8D, left), we noticed the trend of reduced plasma cell differentiation at each division in LPS+IL4 treated cKO B cells (Figure 8D, right). Similarly, we did not observe any significant difference in IgG1 class switching (Figure 8E). Only LPS treated B cells, however, demonstrated switching to inflammation associated IgG3 and was significantly higher in cKO B cells (Figure 8F). We concluded that in the absence of Gtf2i, splenic B cells become more responsive to innate signals like LPS activation that is reflected in enhanced proliferation and become biased towards class switching to a potently proinflammatory antibody, IgG3 isotype.
Immune responses to pathogens often involve a two-step process: first a nonspecific but rapid defense mechanism of the innate immune system is triggered and subsequently, a specific but slow defense mechanism of the adaptive immune system kicks in. While the innate arm is conserved from drosophila to man, the adaptive arm is exclusively present in vertebrates. These two arms evolved different surface receptors and specialized cells to respond distinctly to different cellular threats and environmental insults via triggering distinct intracellular signaling pathways (49). Although there is a boundary between these two arms with distinct cells and signaling pathways mediating such immune responses, murine MZ B cells are ideally located as sentinels at the interface between the circulation and lymphoid tissue to respond to blood-borne pathogens, thereby adopting a hybrid immune strategy that crosses over such boundaries (22). Thus, together with B-1 B cells, MZ B cells are endowed with a “natural memory” that provides a bridge between innate and adaptive immune responses (23). Consistent with this notion, MZ B cells are intrinsically capable of very rapidly maturing into plasmablasts, which may be due, in part, to the ability of MZ B cells to respond quickly than follicular B cells, to both B cell receptor (BCR) and to Toll-like receptors (TLRs) (23). In addition, MZ B cells also express high levels of TLRs (comparable to innate cells like DCs, macrophages and granulocytes). These properties allow the MZ B cells to crossover between the two arms of the immune system and rapidly respond to pathogens both via T-independent as well as T-dependent pathways (20, 23).
In this study, we determined that conditional ablation of Gtf2i in B cells leads to significant reduction in MZ B cells (Fig 1), without altering the number of splenocytes or B cells. Moreover, there was no appreciable difference in the viability of B cells with or without BAFF between control and Gtf2i cKO mice. More surprising is the fact that the remaining MZ B cells as well as the FO B cells derived from Gtf2i cKO mice acquired accentuated molecular features of the innate-like properties of MZ B cells. This is reflected in total transcriptomic/RNA-seq (Figs 2-4) as well as chromatin landscape/ATAC-seq analyses (Figs 5 and 6). Although the exact molecular mechanisms for this function of TFII-I is currently unclear, there are several indications from our results. One key molecule may be the RNA binding protein (RBP) family gene Zfp36l2 that was selectively upregulated in WT MZ B cells, but not in Gtf2i cKO MZ B cells (Fig 2A). ZFP36L1 protein post-transcriptionally represses expression of the transcription factors KLF2 and IRF8, which are known to favor the FO B cell phenotype. Given this established role of Zfp36 family in MZ B cell maintenance (28), downregulation of Zfp36l2 might provide a rationale as to why the cKO mice have reduced numbers of MZ B cells. Consistent with this notion, we also noted that genes like Cd83 which play crucial role in B cell activation and GC B cell responses (50) was upregulated FO cKO B cells. This indicates that in absence of TFII-I B cells may be poised towards activation and can have different kinetics of GC responses. IRF8 motif was significantly more enriched in the open chromatin of the cKO FO B cells (Figure 5). Given that TFII-I has been implicated in regulating homeotic genes like Dlx5 and Dlx6 (51) our observation that loss of TFII-I results in the open chromatin of homeobox containing motifs (Figure 6) also suggests that regulation of homeobox genes in B cell function could be an important role of TFII-I. Further, it can also substantiate the association of homeobox transcription factors with MZ B cell lymphomagenesis (52) thereby indicating a potential role for TFII-I in this process. We noted that differences in B cell chromatin landscape observed upon removal of TFII-I in basal state is far more pronounced than the differences noted with our transcriptomic/gene expression analysis. The incongruence between gene expression and chromatin features has been noted and could be due to a number of factors, including but not limited to the fact that the ATAC-seq assay provides a more dynamic change reflecting the cellular state versus the transcriptomic analysis which is more of a static snapshot (53). But it is also possible that a far more robust difference in gene expression and correlation with open chromatin peaks might be more readily observed in the absence of TFII-I upon antigenic stimulation or infection in vivo.
B cell response to LPS is far less robust compared to innate cells, requiring greatly increased dose of LPS to elicit a comparable downstream effect (48). We rationalized that if Gtf2i cKO B cells acquired enhanced innate-like features, they might respond to a lower dose of LPS. We found that LPS sensitivity of these cells was also altered (Figure 7). In particular, a suboptimal dose of LPS at early time points elicited a robust response in cKO ex vivo splenic B cells, compared to the WT control B cells. To our surprise, 5 genes (Ly86, Irak1, Irf1, Tbk1 and Nfkbia) were even more induced with the lower dose of LPS compared to higher dose. This apparently paradoxical regulatory mechanism may be due to time kinetics and/or dose dependent activation of stimulus specific downstream genes via enhancer-based mechanisms (54). Further specific studies are required to understand this interesting phenomenon.
To further distinguish functional differences between cKO and WT B cells and considering the fact that Gtf2i cKO MZ B cells exhibit enhanced expression of cell cycle genes like developing lymphocytes, we analyzed their proliferative potentials in response to LPS. B cells from Gtf2i cKO demonstrated higher proliferation capacities at 72h both at suboptimal and optimal dose of LPS (Fig 8A), while no such appreciable differences were noted when these cells were stimulated using anti-IgM Fab`2. Consistent with this observation, we also noted a far greater number of cells progressed into cell cycle in cKO B cells compare to WT B (Figure 8B, G0 population) as demonstrated by higher division index of cKO B cells at 72h (Figure 8C). Thus, a TFII-I deficiency in an adaptive immune cell such as B lymphocytes leads to a better proliferation capacity in response to innate signals. Differences in proliferative responses to TLR4 stimulation versus BCR crosslinking also indicate the possibility that TFII-I may play a critical role during T-independent B cell response rather than a T-dependent one. Further, this gained cell number can potentially generate increased immune response, which could be beneficial or deleterious if not regulated properly. Paradoxically, TFII-I is noted to be an oncogene and a gain of function mutation is involved in thymomas and exhibits pro-proliferative capabilities in in vitro experiments (14), although proliferative potentials of TFII-I in B cells have never been tested before. Given the distinct and often opposing roles of TFII-I and its isoforms and its different family members in various cell types, further work needs to be done to clearly delineate the function of TFII-I in proliferation in different cell types.
Functional studies of CD138+ plasma cell differentiation and Ig class switching upon LPS and IL4 or only LPS stimulation ex vivo (Fig 8) further confirm that in the absence of Gtf2i, splenic B cells exhibit enhanced responsiveness to LPS by undergoing increased proliferation (a characteristic feature of innate cells), without significantly altering their in vitro plasma differentiation properties. But interestingly, Gtf2i cKO B cells are clearly biased towards class switching to a potent proinflammatory antibody, IgG3 (55), further suggesting a role for TFII-I in altering B cell differentiation properties. We anticipate that these B cell-specific functions of TFII-I observed under basal conditions will be more pronounced in vivo particularly upon T-independent antigenic challenge.
In these studies, we uncovered an unexpected cell-type specific role for TFII-I in murine B cell homeostasis through its conditional ablation. Given that the gain-of-function of TFII-I results in tumorigenic properties, we also conjecture that gain of TFII-I function could lead to B cell specific tumors. We further speculate that the B cell specific functions of TFII-I uncovered here might be related to its known role in autoimmune disorders, like Sjogren syndrome and Lupus. Especially in light of the fact that serum IgG levels (including IgG3) are elevated in Sjogren syndrome and B cell subsets are altered (56), association of these features with lack of TFII-I could explain some aspects of the disease. Finally, although typical WBS syndrome clinical features do not include immune dysregulation, Kimura et al. recently showed differential expression of genes related to B cell activation in WBS patients relative to controls (57). We anticipate future work will address the precise role of TFII-I in autoimmunity and WBS.
Gtf2ifl/+CD19-Cre+ (Het) and Gtf2ifl/flCD19-Cre+ (HO) were generated by crossing Gtf2ifl/fl (24) with Cd19-cre (006785, The Jackson Laboratory). BL6 (WT), Gtf2ifl/+CD19-Cre+ (Het) and Gtf2ifl/flCD19-Cre+ (HO) mice were maintained in the animal facility of the National Institute on Aging. Eight to 12 weeks old mice were used for all experiments. The studies were carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals (NRC 2011). Mice were euthanized with carbon dioxide and spleens were harvested for analysis. The protocol was approved by the Animal Care and Use Committee of the NIA Intramural Research Program, NIH. This program is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC) (File 000401), registered by the United States Department of Agriculture (51-F-0016) and maintains an assurance with the Public Health Service (D16-00602).
Primary B lymphocytes were isolated from spleens of WT, Het and HO mice using EasySep B cell kits by negative selection according to the manufacturer’s instructions (Stemcell Technologies, Canada). B cells were stained with anti CD21-FITC and anti CD23-PE antibodies (Biolegend, San Diego, CA) and sorted at the BD FACS-Aria Fusion. Follicular (FO) B cells were sorted as CD21loCD23hi, and Marginal Zone (MZ) B cells were sorted as CD21hiCD23lo. Cells were cultured in RPMI 1640 medium (Invitrogen, MA) containing 10% heat inactivated FBS (Gemini Bioproducts, CA), 50μM β-mercaptoethanol (Sigma-Aldrich, MO), 1% L-glutamine and 1% penicillin-streptomycin solution (Gibco, MA). B cell purity was > 95% based on flow cytometric analysis following staining with anti-CD19 (Biolegend, San Diego, CA). FO and MZ B cells were >90% pure based on the criteria that they were sorted on.
Total RNA was extracted from FO and MZ B cells using the RNeasy Mini Kit (Qiagen, Valencia, CA) according to the manufacturer’s protocol. Total RNA was sequenced at the Johns Hopkins Deep Sequencing and Microarray Core using standard protocol for NextSeq 500 sequencer. Ribosomal RNA was depleted, barcoded libraries were made, and 50bp single end reads were generated for each sample. Reads were analyzed using Galaxy RNA-seq pipeline (usegalaxy.org) and adapter trimmed sequences were aligned to mouse genome (mm10) using HISAT2. FeatureCounts and htseq-count were used for differential gene expression analysis using DESeq2. Using “MyGeneset” suite we identified our gene set expression across ImmGen cell-types in immgen.org database (58). Gene ontologies and Immgen-cell type enrichment scores were calculated using ToppFun suite at https://toppgene.cchmc.org/.
FO and MZ splenic B cells were FACS sorted and ATAC-seq libraries were made as previously described (Buenrostro, Giresi et al., 2013) with minor modifications. Briefly, 100,000 cells were lysed in 100 ul of lysis buffer (10 mM Tris-Cl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% NP-40). After centrifuging at 500g for 10 min at 4°C, pelleted nuclei were resuspended with 50 ul of transposition mix (1X Tagment DNA buffer, Tn5 Transposase, nuclease-free H2O) and incubated for 30 min at 37°C in a thermomixer with slow revolutions. Transposed DNA was purified using MinElute columns (28004, QIAGEN) and subsequently amplified with Nextera sequencing primers and NEB high fidelity 2X PCR master mix (New England Biolabs) for 11 cycles. PCR-amplified DNA libraries were size selected after fractioning on high melting agarose gel electrophoresis. Library was pooled and sequenced using the Illumina NextSeq sequencer with paired end reads of 75 bases. After sequencing the ATAC-seq analysis was performed on Galaxy server (usegalaxy.org). Reads were run for quality control and treated accordingly for trimming and adapter removing.
QC passed reads, were aligned to mouse mm10 genome using Bowtie2 and peak calling was done by MACS2 peak caller algoritm. HOMER (http://homer.ucsd.edu/homer) software was used for analysis of the open chromatin peaks, mergePeaks.pl (with 50bp overlap to remove redundancy) was used to identify overlapping and unique peaks between various cell subsets. Similarly, within the open chromatin regions findMotifsGenome.pl algorithm was used to identify transcription factor binding motifs. From HOMER suite annotatePeaks.pl algorithm identified genes associated with the peak, location in the genome and observed/expected values of the annotated regions. Differential open chromatin was identified using DiffBind algorithm.
Splenic B cells were isolated from WT and cKO mice and cultured in presence of LPS (Invivogen, San Diego, CA) 5µg/ml or 1µg/ml for 2, 8 and 24 h time points. Total RNA was extracted from B cells using RNeasy Mini Kit (Qiagen, Valencia, CA). 100ng RNA was reverse transcribed using SuperScript IV VILO Master Mix (Thermofisher Scientific, MA) and the cDNA was loaded onto the “Mouse Toll-Like Receptor Signaling Pathway” RT2 Profiler PCR Array according to the manufacturer instructions (Qiagen, Valencia, CA). Expression of the target genes were normalized to the geometric mean of Ct values of the two housekeeping genes Gusb and Hsp90a using the ddCt method (n = 3). Basal level (0 h) fold change was calculated by dividing the gene expression of cKO B cells on the gene expression of WT B cells (n = 4).
Splenic B cells were labeled with Cell Trace Violet (CTV) (Invitrogen, MA) according to the manufacturer instructions. After washing cells were activated with LPS 1.25, 2.5, 5 and 10 µg/ml (10µg/ml n = 4, other concentrations n =2) or with F(ab`)2 anti-IgM 10µg/ml (n = 2) for 72 h followed by flow cytometry analysis for CTV dilution. Each division generation are labeled from G0 – G6. Proliferation plugin of FlowJo (TreeStar BD Biosciences) was used to calculate division and expansion index.
For plasma cell differentiation and IgG1 class switching analysis, LPS/IL4 activated B cells were co-stained with anti-CD138-BV711 and anti-IgG1-PE respectively at 72 and 96 h time points. For IgG3 class switching analysis, LPS activated B cells were stained with anti-IgG3-FITC at 72 and 96 h. Flow acquisition was performed on BD Symphony A5 system and analysis was done on FloJo (TreeStar BD Bioscience).
Graphad Prism (v9.4.1) was used to perform statistical tests as well as generating graphs for the data. Students t-test was performed as indicated for paired or unpaired tests.
The data presented in the study are deposited in the GEO repository, accession number GSE222325.
The animal study was reviewed and approved by National Institute on Aging and Animal Care and Use Committee of the NIA Intramural Research Program, NIH.
AS and MK designed, performed and analyzed experiments. AS and MK share equal first authorship. SD and KM-M analysed RNA-seq and ATAC-seq data. DB generated mouse strain. AS, MK, RS, and AR wrote manuscript. RS and AR secured intramural funding. All authors contributed to the article and approved the submitted version.
This work is generously supported by the Division of Program Coordination, Planning, and Strategic Initiatives, Office of the Director and the Intramural Research Program of the National Institute on Aging, National Institutes of Health.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1067459/full#supplementary-material
1. Roy AL, Meisterernst M, Pognonec P, Roeder RG. Cooperative interaction of an initiator-binding transcription initiation factor and the helix-loop-helix activator USF. Nature (1991) 354(6350):245–8. doi: 10.1038/354245a0
2. Grueneberg DA, Henry RW, Brauer A, Novina CD, Cheriyath V, Roy AL, et al. A multifunctional DNA-binding protein that promotes the formation of serum response factor/homeodomain complexes: identity to TFII-I. Genes Dev (1997) 11(19):2482–93. doi: 10.1101/gad.11.19.2482
3. Roy AL. Pathophysiology of TFII-I: Old guard wearing new hats. Trends Mol Med (2017) 23(6):501–11. doi: 10.1016/j.molmed.2017.04.002
4. Roy AL. Role of the multifunctional transcription factor TFII-I in DNA damage repair. DNA Repair (Amst) (2021) 106:103175. doi: 10.1016/j.dnarep.2021.103175
5. Roy AL. Biochemistry and biology of the inducible multifunctional transcription factor TFII-I: 10 years later. Gene (2012) 492(1):32–41. doi: 10.1016/j.gene.2011.10.030
6. Hakre S, Tussie-Luna MI, Ashworth T, Novina CD, Settleman J, Sharp PA, et al. Opposing functions of TFII-I spliced isoforms in growth factor-induced gene expression. Mol Cell (2006) 24(2):301–8. doi: 10.1016/j.molcel.2006.09.005
7. Morris CA, Mervis CB, Hobart HH, Gregg RG, Bertrand J, Ensing GJ, et al. GTF2I hemizygosity implicated in mental retardation in williams syndrome: genotype-phenotype analysis of five families with deletions in the williams syndrome region. Am J Med Genet A (2003) 123A(1):45–59. doi: 10.1002/ajmg.a.20496
8. Perez Jurado LA, Wang YK, Peoples R, Coloma A, Cruces J, Francke U. A duplicated gene in the breakpoint regions of the 7q11.23 williams-beuren syndrome deletion encodes the initiator binding protein TFII-I and BAP-135, a phosphorylation target of BTK. Hum Mol Genet (1998) 7(3):325–34. doi: 10.1093/hmg/7.3.325
9. Kozel BA, Barak B, Kim CA, Mervis CB, Osborne LR, Porter M, et al. Williams syndrome. Nat Rev Dis Primers (2021) 7(1):42. doi: 10.1038/s41572-021-00276-z
10. Barak B, Feng G. Neurobiology of social behavior abnormalities in autism and williams syndrome. Nat Neurosci (2016) 19(6):647–55. doi: 10.1038/nn.4276
11. Li Y, Zhang K, Chen H, Sun F, Xu J, Wu Z, et al. A genome-wide association study in han Chinese identifies a susceptibility locus for primary sjogren's syndrome at 7q11.23. Nat Genet (2013) 45(11):1361–5. doi: 10.1038/ng.2779
12. Kim K, Bang SY, Ikari K, Yoo DH, Cho SK, Choi CB, et al. Association-heterogeneity mapping identifies an Asian-specific association of the GTF2I locus with rheumatoid arthritis. Sci Rep (2016) 6:27563. doi: 10.1038/srep27563
13. Kim IK, Rao G, Zhao X, Fan R, Avantaggiati ML, Wang Y, et al. Mutant GTF2I induces cell transformation and metabolic alterations in thymic epithelial cells. Cell Death Differ (2020) 27(7):2263–79. doi: 10.1038/s41418-020-0502-7
14. Petrini I, Meltzer PS, Kim IK, Lucchi M, Park KS, Fontanini G, et al. A specific missense mutation in GTF2I occurs at high frequency in thymic epithelial tumors. Nat Genet (2014) 46(8):844–9. doi: 10.1038/ng.3016
15. Mammoto A, Connor KM, Mammoto T, Yung CW, Huh D, Aderman CM, et al. A mechanosensitive transcriptional mechanism that controls angiogenesis. Nature (2009) 457(7233):1103–8. doi: 10.1038/nature07765
16. Enkhmandakh B, Makeyev AV, Erdenechimeg L, Ruddle FH, Chimge NO, Tussie-Luna MI, et al. Essential functions of the williams-beuren syndrome-associated TFII-I genes in embryonic development. Proc Natl Acad Sci U S A. (2009) 106(1):181–6. doi: 10.1073/pnas.0811531106
17. Yang W, Desiderio S. BAP-135, a target for bruton's tyrosine kinase in response to b cell receptor engagement. Proc Natl Acad Sci U S A. (1997) 94(2):604–9. doi: 10.1073/pnas.94.2.604
18. Ren X, Siegel R, Kim U, Roeder RG. Direct interactions of OCA-b and TFII-I regulate immunoglobulin heavy-chain gene transcription by facilitating enhancer-promoter communication. Mol Cell (2011) 42(3):342–55. doi: 10.1016/j.molcel.2011.04.011
19. Schmidt C, Kim D, Ippolito GC, Naqvi HR, Probst L, Mathur S, et al. Signalling of the BCR is regulated by a lipid rafts-localised transcription factor, bright. EMBO J (2009) 28(6):711–24. doi: 10.1038/emboj.2009.20
20. Allman D, Pillai S. Peripheral b cell subsets. Curr Opin Immunol (2008) 20(2):149–57. doi: 10.1016/j.coi.2008.03.014
21. Pillai S, Cariappa A. The follicular versus marginal zone b lymphocyte cell fate decision. Nat Rev Immunol (2009) 9(11):767–77. doi: 10.1038/nri2656
22. Pillai S, Cariappa A, Moran ST. Marginal zone b cells. Annu Rev Immunol (2005) 23:161–96. doi: 10.1146/annurev.immunol.23.021704.115728
23. Cerutti A, Cols M, Puga I. Marginal zone b cells: virtues of innate-like antibody-producing lymphocytes. Nat Rev Immunol (2013) 13(2):118–32. doi: 10.1038/nri3383
24. Enkhmandakh B, Stoddard C, Mack K, He W, Kaback D, Yee SP, et al. Generation of a mouse model for a conditional inactivation of Gtf2i allele. Genesis (2016) 54(7):407–12. doi: 10.1002/dvg.22948
25. Stadanlick JE, Kaileh M, Karnell FG, Scholz JL, Miller JP, Quinn WJ 3rd, et al. Tonic b cell antigen receptor signals supply an NF-kappaB substrate for prosurvival BLyS signaling. Nat Immunol (2008) 9(12):1379–87. doi: 10.1038/ni.1666
26. Novina CD, Kumar S, Bajpai U, Cheriyath V, Zhang K, Pillai S, et al. Regulation of nuclear localization and transcriptional activity of TFII-I by bruton's tyrosine kinase. Mol Cell Biol (1999) 19(7):5014–24. doi: 10.1128/MCB.19.7.5014
27. Mohammad DK, Nore BF, Hussain A, Gustafsson MO, Mohamed AJ, Smith CI. Dual phosphorylation of btk by akt/protein kinase b provides docking for 14-3-3zeta, regulates shuttling, and attenuates both tonic and induced signaling in b cells. Mol Cell Biol (2013) 33(16):3214–26. doi: 10.1128/MCB.00247-13
28. Newman R, Ahlfors H, Saveliev A, Galloway A, Hodson DJ, Williams R, et al. Maintenance of the marginal-zone b cell compartment specifically requires the RNA-binding protein ZFP36L1. Nat Immunol (2017) 18(6):683–93. doi: 10.1038/ni.3724
29. Makita S, Takatori H, Nakajima H. Post-transcriptional regulation of immune responses and inflammatory diseases by RNA-binding ZFP36 family proteins. Front Immunol (2021) 12:711633. doi: 10.3389/fimmu.2021.711633
30. Heng TS, Painter MW. Immunological genome project c. the immunological genome project: networks of gene expression in immune cells. Nat Immunol (2008) 9(10):1091–4. doi: 10.1038/ni1008-1091
31. Miller JC, Brown BD, Shay T, Gautier EL, Jojic V, Cohain A, et al. Deciphering the transcriptional network of the dendritic cell lineage. Nat Immunol (2012) 13(9):888–99. doi: 10.1038/ni.2370
32. Jager E, Schulz A, Lede V, Lin CC, Schoneberg T, Le Duc D. Dendritic cells regulate GPR34 through mitogenic signals and undergo apoptosis in its absence. J Immunol (2016) 196(6):2504–13. doi: 10.4049/jimmunol.1501326
33. Sladowski D, Wasiutynski A, Wilczynski G, Grabska-Liberek I, Coecke S, Kinsner A, et al. Expression of the membrane complement regulatory proteins (CD55 and CD59) in human thymus. Folia Histochem Cytobiol (2006) 44(4):263–7.
34. Malhotra D, Fletcher AL, Astarita J, Lukacs-Kornek V, Tayalia P, Gonzalez SF, et al. Transcriptional profiling of stroma from inflamed and resting lymph nodes defines immunological hallmarks. Nat Immunol (2012) 13(5):499–510. doi: 10.1038/ni.2262
35. Missen MA, Haylock D, Whitty G, Medcalf RL, Coughlin PB. Stage specific gene expression of serpins and their cognate proteases during myeloid differentiation. Br J Haematol (2006) 135(5):715–24. doi: 10.1111/j.1365-2141.2006.06360.x
36. Yoshida H, Lareau CA, Ramirez RN, Rose SA, Maier B, Wroblewska A, et al. The cis-regulatory atlas of the mouse immune system. Cell (2019) 176(4):897–912 e20. doi: 10.1016/j.cell.2018.12.036
37. Kitamura D, Kudo A, Schaal S, Muller W, Melchers F, Rajewsky K. A critical role of lambda 5 protein in b cell development. Cell (1992) 69(5):823–31. doi: 10.1016/0092-8674(92)90293-L
38. Pridans C, Holmes ML, Polli M, Wettenhall JM, Dakic A, Corcoran LM, et al. Identification of Pax5 target genes in early b cell differentiation. J Immunol (2008) 180(3):1719–28. doi: 10.4049/jimmunol.180.3.1719
39. Fernando N, Sciume G, O'Shea JJ, Shih HY. Multi-dimensional gene regulation in innate and adaptive lymphocytes: A view from regulomes. Front Immunol (2021) 12:655590. doi: 10.3389/fimmu.2021.655590
40. Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods (2013) 10(12):1213–8. doi: 10.1038/nmeth.2688
41. Xu B, Wang H, Wright S, Hyle J, Zhang Y, Shao Y, et al. Acute depletion of CTCF rewires genome-wide chromatin accessibility. Genome Biol (2021) 22(1):244. doi: 10.1186/s13059-021-02466-0
42. Pena-Hernandez R, Marques M, Hilmi K, Zhao T, Saad A, Alaoui-Jamali MA, et al. Genome-wide targeting of the epigenetic regulatory protein CTCF to gene promoters by the transcription factor TFII-I. Proc Natl Acad Sci U.S.A. (2015) 112(7):E677–86. doi: 10.1073/pnas.1416674112
43. Klemm SL, Shipony Z, Greenleaf WJ. Chromatin accessibility and the regulatory epigenome. Nat Rev Genet (2019) 20(4):207–20. doi: 10.1038/s41576-018-0089-8
44. Wang H, Morse HC 3rd. IRF8 regulates myeloid and b lymphoid lineage diversification. Immunol Res (2009) 43(1-3):109–17. doi: 10.1007/s12026-008-8055-8
45. Wohner M, Tagoh H, Bilic I, Jaritz M, Poliakova DK, Fischer M, et al. Molecular functions of the transcription factors E2A and E2-2 in controlling germinal center b cell and plasma cell development. J Exp Med (2016) 213(7):1201–21. doi: 10.1084/jem.20152002
46. Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and b cell identities. Mol Cell (2010) 38(4):576–89. doi: 10.1016/j.molcel.2010.05.004
47. Stark R, Brown G. DiffBind: differential binding analysis of ChIP-seq peak data. r package version, (Cancer Research UK : Cambridge Institute, University of Cambridge) Vol. 100. (2011).
48. Guery JC, Ria F, Galbiati F, Adorini L. Normal b cells fail to secrete interleukin-12. Eur J Immunol (1997) 27(7):1632–9. doi: 10.1002/eji.1830270707
49. Luecke S, Sheu KM, Hoffmann A. Stimulus-specific responses in innate immunity: Multilayered regulatory circuits. Immunity (2021) 54(9):1915–32. doi: 10.1016/j.immuni.2021.08.018
50. Krzyzak L, Seitz C, Urbat A, Hutzler S, Ostalecki C, Glasner J, et al. CD83 modulates b cell activation and germinal center responses. J Immunol (2016) 196(9):3581–94. doi: 10.4049/jimmunol.1502163
51. Poitras L, Yu M, Lesage-Pelletier C, Macdonald RB, Gagne JP, Hatch G, et al. An SNP in an ultraconserved regulatory element affects Dlx5/Dlx6 regulation in the forebrain. Development (2010) 137(18):3089–97. doi: 10.1242/dev.051052
52. Robles EF, Mena-Varas M, Barrio L, Merino-Cortes SV, Balogh P, Du MQ, et al. Homeobox NKX2-3 promotes marginal-zone lymphomagenesis by activating b-cell receptor signalling and shaping lymphocyte dynamics. Nat Commun (2016) 7:11889. doi: 10.1038/ncomms11889
53. Cao J, Cusanovich DA, Ramani V, Aghamirzaie D, Pliner HA, Hill AJ, et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science (2018) 361(6409):1380–5. doi: 10.1126/science.aau0730
54. Wibisana JN, Inaba T, Shinohara H, Yumoto N, Hayashi T, Umeda M, et al. Enhanced transcriptional heterogeneity mediated by NF-kappaB super-enhancers. PloS Genet (2022) 18(6):e1010235. doi: 10.1371/journal.pgen.1010235
55. Damelang T, Rogerson SJ, Kent SJ, Chung AW. Role of IgG3 in infectious diseases. Trends Immunol (2019) 40(3):197–211. doi: 10.1016/j.it.2019.01.005
56. Zhang H, Li P, Wu D, Xu D, Hou Y, Wang Q, et al. Serum IgG subclasses in autoimmune diseases. Med (Baltimore) (2015) 94(2):e387. doi: 10.1097/MD.0000000000000387
57. Kimura R, Ishii Y, Tomiwa K, Awaya T, Nakata M, Kato T, et al. Williams-beuren syndrome as a potential risk factor for burkitt lymphoma. Front Genet (2018) 9:368. doi: 10.3389/fgene.2018.00368
Keywords: B cell, Gtf2i, FO and MZ B cells, innate and adaptive immunity, chromatin accessibility
Citation: Singh A, Kaileh M, De S, Mazan-Mamczarz K, Bayarsaihan D, Sen R and Roy AL (2023) Transcription factor TFII-I fine tunes innate properties of B lymphocytes. Front. Immunol. 14:1067459. doi: 10.3389/fimmu.2023.1067459
Received: 11 October 2022; Accepted: 04 January 2023;
Published: 23 January 2023.
Edited by:
Hermann Eibel, University of Freiburg Medical Center, GermanyReviewed by:
Reuben Matthew Tooze, University of Leeds, United KingdomCopyright © 2023 Singh, Kaileh, De, Mazan-Mamczarz, Bayarsaihan, Sen and Roy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ananda L. Roy, YW5hbmRhLnJveUBuaWguZ292
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.