 Xuejiao Cui
Xuejiao Cui Futao Wang2
Futao Wang2- 1Department of Endocrinology, Shengjing Hospital of China Medical University, Shenyang, China
- 2Department of Endocrinology, Changchun Central Hospital, Changchun, China
Graves’ orbitopathy (GO) is an organ-specific autoimmune disease, but its pathogenesis remains unclear. There are few review articles on GO research from the perspective of target cells and target antigens. A systematic search of PubMed was performed, focusing mainly on studies published after 2015 that involve the role of target cells, orbital fibroblasts (OFs) and orbital adipocytes (OAs), target antigens, thyrotropin receptor (TSHR) and insulin-like growth factor-1 receptor (IGF-1R), and their corresponding antibodies, TSHR antibodies (TRAbs) and IGF-1R antibodies (IGF-1R Abs), in GO pathogenesis and the potentially effective therapies that target TSHR and IGF-1R. Based on the results, OFs may be derived from bone marrow-derived CD34+ fibrocytes. In addition to CD34+ OFs, CD34- OFs are important in the pathogenesis of GO and may be involved in hyaluronan formation. CD34- OFs expressing Slit2 suppress the phenotype of CD34+ OFs. β-arrestin 1 can be involved in TSHR/IGF-1R crosstalk as a scaffold. Research on TRAbs has gradually shifted to TSAbs, TBAbs and the titre of TRAbs. However, the existence and role of IGF-1R Abs are still unknown and deserve further study. Basic and clinical trials of TSHR-inhibiting therapies are increasing, and TSHR is an expected therapeutic target. Teprotumumab has become the latest second-line treatment for GO. This review aims to effectively describe the pathogenesis of GO from the perspective of target cells and target antigens and provide ideas for its fundamental treatment.
Introduction
Graves’ orbitopathy (GO), also known as thyroid eye disease and thyroid-associated ophthalmopathy, is relatively rare, with an incidence of 0.54-0.9 cases/100,000/year in males and 2.67-3.3 cases/100,000/year in females (1). Approximately 25%-50% of patients with Graves’ disease (GD) have GO, with the incidence varying based on the testing tool used to diagnose GO, such as clinical signs and symptoms, physical examination, and imaging studies. Common symptoms of GO include eye pain, photophobia, blurred vision, excessive tear production and double vision. GO can cause periorbital oedema, exophthalmos, eyelid recession and changes in eye movement, affecting one or both eyes. Approximately 5–6% of patients with GO have severe disease with compressive optic neuropathy or sight-threatening corneal ulceration, and these patients may experience vision loss. GO occurs at any age, but women 30-50 years of age are most commonly affected (2). Nevertheless, the severity of GO tends to be worse in men and in patients who are first diagnosed when they are older than 50 years old (3). Traditional risk factors for GO include smoking, hypercholesterolaemia, thyroid dysfunction, radioiodine therapy, high thyrotropin receptor antibodies (TRAbs), and low thyroid peroxidase antibodies (TPOAbs) and thyroglobulin antibodies (TgAbs). New risk factors for GO include high thyroglobulin (Tg) (4), selenium deficiency, intestinal flora imbalance, and increased levels of both Yersinia enterocolitica and Escherichia coli in the digestive tract (5).
GO is characterized by inflammation in retrobulbar tissues, increased adipogenesis, and accumulation of extraocular intramuscular glycosaminoglycans (GAGs), which result in expansion and remodelling of the orbital contents (6, 7). The pathogenesis of GO is unclear. Orbital fibroblasts (OFs) and orbital adipocytes (OAs) are important cells that are targeted by the autoimmune response, and thyrotropin receptor (TSHR) and insulin-like growth factor-1 receptor (IGF-1R) are key target antigens. Moreover, increased production of TRAbs is associated with the prevalence and severity of GO. This review describes recent advances in GO research related to primary target cells (OFs and OAs), primary target antigens (TSHR and IGF-1R) and associated antibodies, and targeted antigen-specific therapies for GO. Data acquisition was based on PubMed search strategies, with a particular focus on papers published after 2015.
Immune cells and GO
Both cellular and humoral immunity play important roles in the pathogenesis of GO. High expression of HLA-DR and adhesion molecules on the vascular endothelium of the orbital tissues of GO patients leads to strong infiltration of orbital immune cells, such as dendritic cells (DCs), macrophages, mast cells, B lymphocytes and T lymphocytes. Most of the current evidence indicates that the level of T and B lymphocyte infiltration correlates with GO disease activity (8, 9). Antigen-presenting cells (APCs), such as DCs and macrophages, present TSHR to CD4+ T lymphocytes in the context of MHC-II molecules. Subsequently, CD4+ T lymphocytes release cytokines to activate CD8+ T lymphocytes or autoantibody-producing B lymphocytes, and a cascade of effects amplifies the immune response and promotes the autoimmune process. CD4+ T lymphocytes are the main cells dominating orbital inflammatory infiltration and are classified into various subtypes, including T helper (Th) 1, Th2, Th17, and Treg cells (10). Different subgroups of T lymphocytes play dominant roles in different stages of GO. Th1 cells, which produce interleukin (IL)-1β, IL-2, TNF-α, and IFN-γ, induce a cell-mediated immune response in the early stage, whereas Th2 cells, which release IL-4, IL-5, IL-10, and IL-13, activate humoral reactions and promote the production of IgG in the late stage. A recent study showed that increased concentrations of Th2 chemokines (CCL2) in plasma from patients with GO may reflect disease activity (11). Additionally, Fang et al. (12) reported that compared with healthy control individuals, patients with GO have significantly higher levels of IL-17A-producing T lymphocytes and recruitment of both CD4+ and CD8+ T lymphocytes in the orbits. Furthermore, orbital tissues from patients with GO express more IL-17A receptor, IL-17A and its related cytokines, with severe fibrotic changes, compared with healthy control individuals. Another study by Fang et al. (13) showed that IFN-γ- and IL-22-expressing Th17 cells are increased in patients with GO, which was positively related to the clinical activity score (CAS), and that IL-17A promotes TGF-β-induced fibrosis in CD90+ OFs from patients with GO (GO-OFs). These findings suggest the potential pathogenic role of Th17 cells in the inflammatory response and fibrosis associated with GO. However, uncertainty remains as to whether Treg cell levels correlate with the severity and activity, clinical course, or treatment response of GO (14, 15).
Target cells of GO
Orbital fibroblasts
OFs are target cells of the autoimmune response in GO. They participate in proliferation, adipogenesis and overproduction of extracellular matrix GAGs, including hyaluronan (HA), which are involved in GO pathogenesis (16). Recently, reported proteomics and DNA methylation data indicate that OFs from patients with active GO are involved in inflammation, adipogenesis, and GAG production, and OFs from patients with inactive GO are more inclined to play an active role in extracellular matrix remodelling (17). MicroRNAs have been reported to be involved in HA production. The results of one study indicated that overexpression of miR-146a reduces the production of HA and collagen I in GO-OFs, and it was demonstrated that miR-146a downregulates the secretion of HA and collagen I in GO-OFs in vitro (18). A recent study showed that the LPAL2/miR-1287-5p axis modulates TGF-β1-induced increases in cell adhesion factor levels and GO-OF activation via EGFR/AKT signalling (19). OFs express MHC-II molecules (20, 21), and the study showed that GO-OFs present their own antigens to T lymphocytes via MHC-II molecules and employ CD40-CD40 L signalling, leading to activation of T lymphocytes and further stimulating fibroblast proliferation. According to the literature, most OFs are positive for CD90 (Thy-1) expression and negative for CD45 expression (22); these proteins are positive and negative markers of mesenchymal stem cells, respectively. Activated OFs differentiate according to the expression of Thy-1 on their cell surface. Thy-1+ OFs are mainly present in extraocular muscles; they differentiate into myofibroblasts and overproduce HA. This leads to extraocular muscle oedema and enlargement. A recent study showed that simvastatin inhibits TGF-β-induced myofibroblast differentiation by inhibiting the RhoA/ROCK/ERK and p38 MAPK signalling pathways, suggesting that simvastatin is a potential therapeutic drug for the prevention and treatment of GO orbital fibrosis (23). Thy-1- OFs, which are called preadipocytes, are mainly present in connective tissue and differentiate into adipocytes (24); Thy-1+ OFs suppress adipocytic differentiation of Thy-1- OFs by producing antiadipocytic factors. Therefore, the balance and relative proportion of Thy-1- and Thy-1+ OFs modulate tissue remodelling in GO (25).
Bone marrow-derived CD34+ fibrocytes are a monocyte subpopulation of peripheral blood mononuclear cells that infiltrate orbital tissues and promote the onset of GO (26). A recent study showed that the concentration of circulating fibrocytes is significantly higher in patients with GO than in patients with GD and healthy control individuals, and in GO patients, these fibrocytes express a significantly higher level of TSHR (27). CD34+ OFs have been reported to express major thyroid autoantigens, including TSHR, thyroperoxidase (TPO), thyroglobulin (Tg) and sodium-iodide symporter (NIS) (28). In response to inflammatory factors, CD34+ OFs further differentiate into myofibroblasts or adipocytes (29).
CD34- OFs coexist with residential CD34+ OFs, but little was previously known about the role of CD34- OFs until CD34- OFs and CD34+ OFs were recently sorted separately by cytometry. The expression of cytokines and HA synthases (HASs) differs in these cell subsets. HAS1, HAS2, and HAS3 are differentially inducible in various cell types by several cytokines and growth factors (30). These enzymes differ from each other in catalytic activities (HAS3 > HAS2 > HAS1) and the sizes of their final products. HAS1 and HAS2 polymerize long stretches of GlcAGlcNAc disaccharide chains, whereas HAS3 polymerizes relatively short stretches (<300 kDa) (31). IL-12p35 is mainly expressed in CD34- OFs, and IL-23p19, IL-6 and TNF-α levels are higher in CD34+ OFs (32, 33). Basal and TSH-induced HAS1 expression occurs in CD34+ OFs, and HAS2 and UDP-glucose dehydrogenase are mainly expressed by CD34- OFs (33). HAS2 appears to be responsible for HA synthesis in both cell subpopulations, and the relatively low levels of HAS2 in CD34+ OFs appear to represent the basis for the substantially higher levels of HA synthesized in CD34− OFs (33). Slit2, an axon guidance glycoprotein, is expressed and released by CD34- OFs and dampens the inflammatory phenotype of CD34+ OFs. Specifically, rhSlit2 dramatically represses the expression of TPO, Tg, TSHR, AIRE (Autoimmune regulator) and NIS and represses that of TNF-α, IL-6, and IL-23 induced by TSH in CD34+ OFs (33, 34). Slit2 knockdown enhances HAS1 expression in CD34- OFs but reduces basal and TSH-dependent HAS2 expression (33). Hence, Slit2 may represent a factor that was previously unrecognized for its capacity to modulate immune responses and HA synthesis in human tissues.
Orbital adipocytes
Adipogenesis contributes to orbital adipose tissue expansion. A fibroblast has an approximate diameter of 30 microns; the diameter of a mature adipocyte is approximately 150 microns, which is 5 times larger. According to the literature, increased expression of TSHR during adipogenesis and binding of TRAb to TSHR on adipocytes leads to upregulation of HAS gene expression and excessive production of HA (35). Signalling pathways, including those for cAMP, PI3K-AKT, AGE-RAGE, lipolysis regulation, and thyroid hormone, are enriched in orbital fat isolated from patients with GO. The IGF-1R and Wnt signalling pathways appear to be enriched early in adipogenesis (36), and Wnt signalling inhibits adipogenesis in GO-OFs (37). PI3K can be activated by TSH, IGF-1, or multiple cytokine receptors, resulting in AKT activation. Activated AKT inhibits FOXO1 and activates mTOR to promote HA production and adipogenesis via peroxisome proliferator activator gamma (PPARγ) (38). PPARγ belongs to the nuclear receptor family of transcription factors and is expressed in adipocytes; it acts as a transcription factor and regulates the homeostasis of lipids and glucose. A recent study demonstrated that the mRNA level of protein kinase RNA-like endoplasmic reticulum kinase (PERK) is significantly higher in orbital tissues from patients with GO than in those from patients without GO. The expression of PPARγ is downregulated and oxidative stress and adipogenesis are reduced in PERK siRNA-transfected GO-OFs (39). One study showed that the expression of glycogen synthase kinase-3β (GSK-3β) in GO orbital tissues is significantly higher than that in control orbital tissues. During adipocyte differentiation, fibroblasts treated with the GSK-3β inhibitor CHIR 99021 showed decreased lipid droplets and decreased expression of PPARγ and c/EBPα and -β. Moreover, inhibition of Wnt and β-catenin in adipogenesis is reversed by CHIR 99021 (40). MicroRNAs have also been reported to be involved in adipogenesis; miR-130a is upregulated in Thy-1-OFs, inhibits AMPK activation, and promotes lipid accumulation in GO-OFs, leading to excessive fatty tissue accumulation in the orbit (41). Another recent study demonstrated that TGF-β-treated human placental mesenchymal stem cells (hPMSCs) suppress adipogenesis and lipogenesis in GO-OFs and in GO mice; the effects are mediated by the SMAD 2/3 pathways, suggesting that these cells may be a new and safe method to promote the antiadipogenic function of hPMSCs to treat GO (42). HA overproduction and adipogenesis have been observed in fibroblasts from orbital adipose tissue (OAT) but not in those from white adipose tissue (WAT) (43). This finding can be explained by the fact that human OAT is derived from the neural crest in the orbit (44) but that WAT is derived from the mesoderm (45). A schematic of the theoretical pathogenesis of GO is shown in Figure 1.
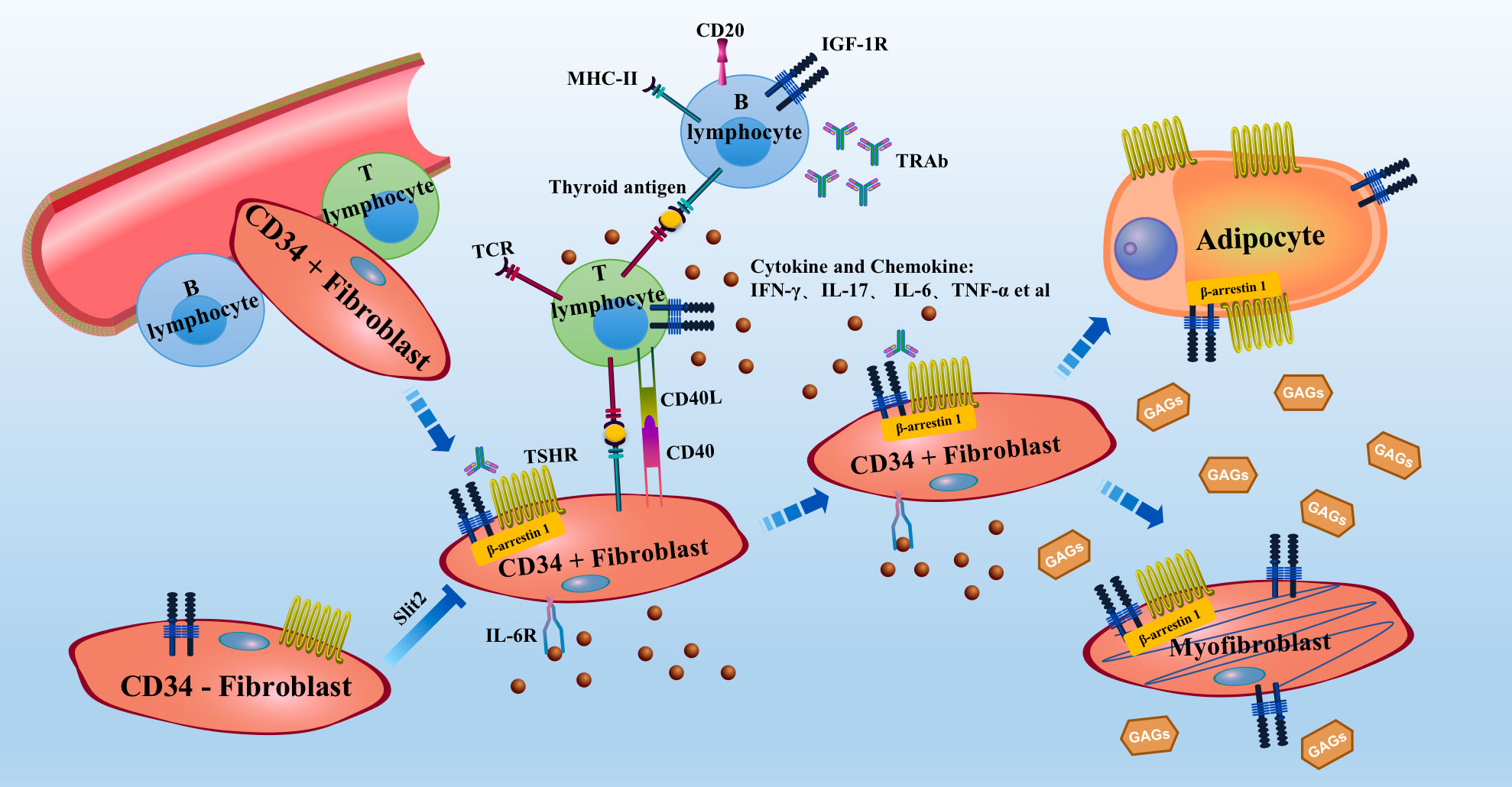
Figure 1 Schematic of the theoretical pathogenesis of GO Bone marrow mononuclear cell-derived CD34+ fibroblasts enter the orbit from the circulation and express low levels of TSHR, thyroglobulin and other thyroid antigens. APCs, such as B lymphocytes or macrophages, present antigens to antigen-specific T lymphocytes and activate them. Activated T lymphocytes release cytokines and chemokines, further causing B lymphocytes to produce antibodies. These processes form an inflammatory microenvironment in the orbit. OFs can also act as APCs. In the inflammatory microenvironment, activated T lymphocytes activate OFs. Activated OFs further secrete cytokines and chemokines and produce excessive levels of GAGs, leading to swelling of the orbital tissue. CD34+ fibroblasts are found in orbit with CD34− fibroblasts. CD34+ fibroblasts further differentiate into myofibroblasts or adipocytes, resulting in thickening of the extraocular muscles and exophthalmos. TSHR expression is increased during adipogenesis. IGF-1R, TSHR, IL-6R, CD20 and TNF-α are current therapeutic targets for GO. APC, antigen-presenting cell; GAG, glycosaminoglycan; GO, Graves’ orbitopathy; OFs, orbital fibroblasts; TSHR, thyrotropin receptor; IGF-1R, insulin-like growth factor-1 receptor.
Autoantigens in the pathogenesis of GO
TSHR
TSHR is a G protein-coupled receptor (GPCR) that contains 3 domains (an ectodomain, transmembrane domain, and intracellular domain). It is composed of an A subunit, comprising a large extracellular domain, and a B subunit, which consists of a short extracellular fragment anchored in the cell membrane and an intracellular part. TSHR is a common target autoantigen of GD and GO (46). A new preclinical model of GO has been established in female BALB/c mice. This mouse model was established via immunization with the human TSHR α subunit by electrotransfection of a plasmid expressing human TSHR. This model exhibits infiltration of a large area around the optic nerve, increased adipogenesis, orbital muscle hypertrophy, extraocular muscle hypertrophy, and conjunctival oedema (47). TSHR is expressed in thyroid cells, which regulate thyroid function, and studies have shown that preadipocyte fibroblasts and myofibroblasts in the orbital tissues of patients with GO also express TSHR (48, 49). TSHR activation of OFs by signalling through cAMP–protein kinase A (PKA) to cAMP response element-binding protein-binding sites in the promoters of HAS1 and HAS2 increases HA production (50). mRNA expression of TSHR in orbital fat/connective tissue in patients with active GO is higher than that in patients with inactive GO, with the levels being directly related to mRNA levels of IL-1β (51). The expression level of TSHR in fibroblasts has been found to be much lower than that in thyroid cells. Indeed, one study showed that TSHR levels are 11-fold higher in GO or control thyrocytes than in fibroblasts (52). The exact mechanism by which TSHR, a specific antigen in thyroid cells, is ectopically expressed in the orbital tissues of patients with GO and becomes the target of the immune response is still unclear. However, this outcome likely occurs because OFs originate from monocyte progenitor cells in the bone marrow and have the ability to express TSHR (49). It has also been shown that preadipocyte fibroblasts express TSHR after stimulation with TSH, TRAb, and IL-6, and the adenylate cyclase/cAMP and PI3K/pAKT pathways become activated. Thus, the differentiation of fibroblasts to adipocytes and HA production are promoted (53). The findings of Woeller et al. (54) also demonstrate that TSHR signalling in GO-OFs stimulates proliferation indirectly through induction of miR-146a and miR-155, reducing the expression of ZNRF3 and PTEN that normally block cell proliferation. The latest interesting study by Draman MS et al. suggests a variant in TSHR that might explain the antigenic role of this receptor in OF even prior to adipogenesis (55). In conclusion, TSHR plays a specific role in GO pathogenesis and has become a therapeutic target.
IGF-1R
Although data show that TSHR plays an important role in GO, IGF-1R may also have a key function. IGF-IR is a membrane-spanning receptor tyrosine kinase (RTK) that binds IGF-I and IGF-II and activates two important downstream signalling pathways, MAPK and PI3K/AKT. Evidence indicates that compared with a normal control, GO-OFs exhibit increased HA synthesis after IGF-1 treatment. Pretreatment with an IGF-1R blocking monoclonal antibody (1H7) or AKT inhibitor significantly decreased the HA concentration. IGF-1 treatment increases the level of pAKT expression; 1H7 and PI3K blockers decrease the expression of PI3K and pAKT protein, and AKT inhibitors decrease the expression of PI3K, AKT and pAKT (56). A study has shown that IGF-IR is overexpressed by OFs and T and B lymphocytes in patients with GO; IGF-1R regulates HA synthesis and adipogenesis in the orbit and defines the phenotype and function of T and B lymphocytes (57). A recent study indicated that 1H7 inhibits autophagy and induces apoptosis by suppressing IGF-1R signalling. This leads to retro-OF/OA death, which may be the main mechanism by which teprotumumab (a human monoclonal IGF-1R blocking antibody) reduces ocular protrusion in patients with GO (58). Teprotumumab has also been reported to attenuate constitutive expression and induction by TSH of MHC-II and B7 family members, including CD80, CD86, and programmed death-ligand 1 in CD34+ fibrocytes (59). At present, teprotumumab is a second-line treatment for GO, which is discussed in detail below.
TSHR and IGF-1R complex
Although TSHR and IGF-1R are two independent receptors, there is a certain connection.
A study in an animal model showed that mice can develop a hyperthyroid state with inflammation and fibrosis of the orbital tissue after being transfected with plasmids expressing IGF-1Rα and TSHR (60). Tsui et al. (52) found that TSHR and IGF-1R colocalize by performing confocal microscopy of OFs and cultured human thyroid epithelial cells. Simultaneous activation of TSHR and IGF-1R causes rapid, synergistic phosphorylation/activation of ERK1 and ERK2 in primary cultures of GO-OFs and human thyrocytes as well as human embryonic kidney (HEK) 293 cells overexpressing TSHR (61). Tramontano et al. (62) reported a possible interaction between IGF-1 and TSH and that IGF-1 enhances the proliferation and DNA synthesis of FRTL-5 thyroid epithelial cells induced by TSH in culture. IGF-1 has also been reported to increase the expression level of TSHR in GO-OFs (63). Conditional knockout of the IGF-1R gene in the thyroid gland significantly reduces the response to TSH (64). Krieger et al. (65) showed that IGF-1R activation by GO immunoglobulins (Igs) occurs via TSHR/IGF-1R crosstalk after binding to TSHR and not through direct binding to IGF-1R. Research has revealed that nuclear FOXO transcription factors serve as convergence points for the IGF-1R and TSHR signalling pathways in GO (66, 67). FOXO1 and FOXO3a act as repressors to prevent excessive fat and HA production in OFs, respectively (66). FOXOs may be an alternative target for nonimmunosuppressive therapy.
Nevertheless, the manner in which TSHR and IGF-1R interact is not clear. They may interact by activating overlapping signalling pathways or physically through direct heterodimerization. Additionally, β-arrestin 1 and β-arrestin 2 have been reported to be key factors in the regulation of TSHR-mediated signalling (68): β-arrestin 2 plays a major role in TSHR desensitization (69), and β-arrestin 1 is primarily involved in activating TSHR signalling (70). Krieger et al. (71) showed that β-arrestin 1 is a member of a preformed complex that includes TSHR and IGF-1R and that its presence is necessary for stability of the complex in GO-OFs, human thyrocytes and U2OS cells. Knockdown of β-arrestin 1 reduces TSHR/IGF-1R colocalization, blocks TSHR/IGF-1R crosstalk and prevents teprotumumab-mediated inhibition of HA production by GO-Igs and M22 (72). These findings support a model of TSHR/IGF-1R crosstalk that may be a general mechanism underlying GPCR/RTK crosstalk that depends on β-arrestin 1 (Figure 2).
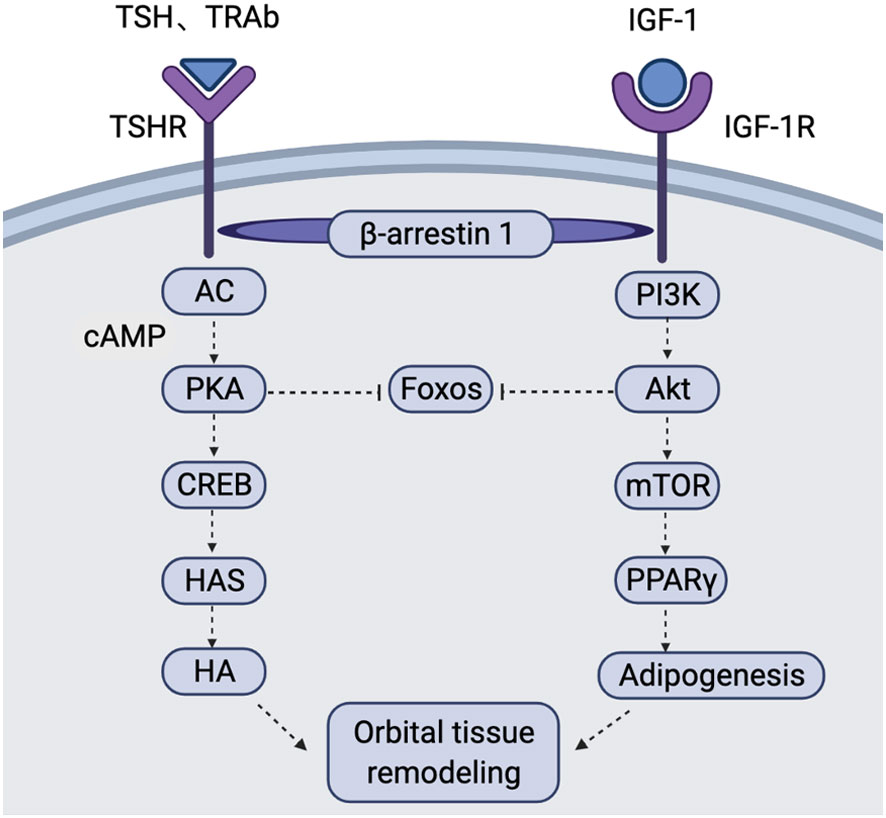
Figure 2 Schematic of the TSHR and IGF-1R signalling pathways in the pathogenesis of GO In OFs and OAs of GO patients, TSH and TRAb bind to TSHR to induce activation of AC. This causes an increase in cAMP production and activates PKA. Activated PKA inhibits FOXOs and activates the transcription factor CREB. CREB further acts on the promoters of HASs to increase HA production. Binding of IGF-1 to IGF-1R activates PI3K, resulting in AKT activation. Activated AKT inhibits FOXOs and activates mTOR to promote adipogenesis via PPARγ. TSHR/IGF-1R crosstalk depends on β-arrestin 1 acting as a scaffold, leading to interaction of the two signalling pathways. This figure is adapted from Draman Mohd Shazli,Zhang Lei,Dayan Colin et al. Front Endocrinol (Lausanne), 2021, 12: 739994. AC, adenylate cyclase; AKT, protein kinase B; cAMP, cyclic adenosine monophosphate; CREB, cAMP response element-binding protein; FOXOs, Forkhead box O; GO, Graves’ orbitopathy; HA, hyaluronan; HASs, hyaluronan synthases; mTOR, mammalian target of rapamycin; OAs, orbital adipocytes; OFs, orbital fibroblasts; PI3K, phosphatidylinositol-3-kinase; PKA, protein kinase A; PPARγ, peroxisome proliferator activator gamma; TRAb, thyrotropin receptor antibody; TSH, thyrotropin; TSHR, thyrotropin receptor.
Autoantibodies in GO pathogenesis
TRAbs
Three types of TRAbs have been identified in recent decades: stimulatory (TSAbs), blocking (TBAbs) and neutral TRAbs (73). Although all three types of TRAbs can be found in patients with GD, TSAbs serve as a marker of the disease (74). Considerable experimental evidence supports the concept that the shedding of the TSHR A subunit in genetically susceptible individuals is a contributing factor to the induction and/or affinity maturation of pathogenic TSAbs, which are the direct cause of GD (75). In recent years, clinical research on TRAbs has gradually moved from general to detailed and now includes research on TSAbs, TBAbs and their titres. A retrospective study confirmed that 70% of euthyroid patients with GO are positive for both TSBAbs and TSAbs at their initial visit (76). A recent study detected TSAb activity in 85 of 91 (93.4%) patients with GO (P < 0.001). The sensitivity rates for differentiating between clinically active versus inactive and mild versus moderate-to-severe GO are both 100% for TSAbs. This finding suggests that TSAbs are highly prevalent in patients with GO and exhibit superior clinical features and predictive potential (77). One study showed a shift from TSAbs to TBAbs in 8 GD patients with/without GO during methimazole treatment that led to remission (78). According to in vitro experiments, TSAbs exhibit selectivity in activating TSHRs, as TSAbs from patients with GO are more effective in stimulating OFs, and TSAbs from patients with GD are more effective in stimulating thyrocytes (79). The study by Kahaly et al. (80) confirmed that TRAb titres, as determined by dilution analysis, significantly differentiate between patients with GD and patients with GO. Although these data are potentially interesting and suggestive of a pathogenic role in GO, these experiments have not been compared with the standard bioassay for TSAbs (CHO cells/cAMP stimulation). Therefore, further confirmation is needed. TRAb binding to TSHR induces GAG production through cAMP and the PI3K/AKT signalling pathway, which overlaps with the signalling pathway downstream of IGF-1R. A recent study found that TSAbs are able to induce IGF-1R phosphorylation and initiate both TSHR and IGF-1R signalling in human and mouse fibroblasts. These findings indicate that TSAbs enhance IGF-1R activity and contribute to retroorbital cellular proliferation and inflammation (58). Kumar et al. (81) showed that treatment of orbital preadipocytes from patients with GO with M22, a human monoclonal TSAb, during adipocyte differentiation results in enhanced mRNA expression levels of IL-6. Furthermore, treatment of orbital cultures with M22 after adipocyte differentiation increases secretion of the IL-6 protein into the medium. These results suggest that M22 increases IL-6 expression in orbital preadipocyte fibroblasts and increases IL-6 release by mature adipocytes and that circulating TRAbs might play a direct role in the clinical activity of GO.
Anti-IGF-1R antibodies
A role for IGF-1R in the pathogenesis of GO has been proposed, but the presence and function of IGF-1R antibodies (IGF-1R Abs) are controversial (82). The findings of Marcus-Samuels et al. (83) demonstrated that knockdown of IGF-1R causes a 6.3-65% decrease in IGF-1-stimulated pAKT but has no effect on GO-Ig stimulation of pAKT, suggesting that GO-Igs contain factors that stimulate pAKT formation but that this factor does not directly activate IGF-1R. It was concluded that there is no evidence of stimulating IGF-R Abs in patients with GO. However, other studies have shown the presence of IGF-1R Abs. A study from Weightman DR et al. first demonstrated that IgG prepared from patients with GD with or without overt ophthalmopathy interacts with IGF-1 binding sites on OFs, suggesting that antibodies may occur in GD that bind to IGF-1R (84). Minich et al. (85) detected IGF-IR Abs in 10 serum samples from control subjects (11%) and 60 serum samples from patients with GO (10%). They found a similar prevalence of IGF-1R Abs in GO patients and controls, with an obvious lack of correlation between IGF-1R Ab concentrations and CAS or NOSPECS status. Therefore, this study suggested that IGF-1R Abs in the blood of patients with GO do not exacerbate the disease and are in fact antagonistic. In addition, this study confirmed that IGF-IR Abs fail to stimulate autophosphorylation of IGF-1R and instead inhibit IGF1-induced signalling in HepG2 hepatocarcinoma cells (85). Another study showed that regardless of the presence of GO, a quarter of patients with GD have IGF-IR Abs in their sera. In patients with GO, there is no relationship between GO severity and IGF-1R Ab levels, although the levels of these antibodies correlate inversely with CAS (86). Lanzolla et al. (87) further designed a cross-sectional investigation to measure IGF-1R Ab levels in patients with GD, with or without GO, who underwent radioiodine therapy followed by glucocorticoid treatment and found higher IGF-1R Ab levels in GD patients without GO than in those with GO. These results suggest a putative protective role for IGF-1R Abs with respect to the development of GO, which is consistent with the beneficial effects of teprotumumab on GO (87). In contrast, Varewijck et al. (88) found that GO patients with high TSAb titres are more likely to have stimulatory IGF-1R Abs. In summary, whether IGF-1R Abs exist and their role deserve further study.
Treatments that target TSHR and IGF-1R
TSHR antagonists
Because fibroblast production of cAMP, pAKT, and HA is activated via TSHR signalling, future inhibitory therapies targeting TSHR may be effective for GO. K1-70, a human monoclonal autoantibody, has considerable promise as a new drug for blocking the actions of thyroid stimulators on TSHR. In a recent case report, K1−70 was injected intramuscularly at 3 weekly intervals in a patient with follicular thyroid cancer, GD and GO, TSAb activity in the serum decreased, and proptosis and inflammation improved (89). In a phase I clinical trial, 18 patients with stable GD that were given antithyroid drug medication received a single intramuscular dose of 25 mg or a single intravenous dose of 50 mg or 150 mg of K1-70. In these patients, the thyroid function, GD and GO clinical manifestations improved after the injection. No immunogenic reaction, death or serious adverse events occurred (90). Currently, several small TSHR antagonists have also been studied in vitro in human samples. For example, in an in vitro human experiment, a small TSHR antagonist (NCGC00229600, also called ANTAG2) inhibited basal cAMP, pAKT, and HA production and that stimulated by TSAb (M22 and MS-1) and bTSH in primary cultures of undifferentiated GO-OFs (91). Another in vitro study found that a low-molecular-weight TSHR antagonist (Org-274179-0) completely blocks cAMP production in differentiated GO-OFs induced by human recombinant GD-IgG, TSH or M22 (92). Marcinkowski et al. (93) showed that a novel, highly selective inhibitor of TSHR (S37a) inhibits TSHR activation not only via TSH but also via stimulatory monoclonal TRAbs M22 (human), KSAb1 (murine) and the allosteric small-molecule agonist C2 in HEK293 cells expressing TSHR in vitro. Moreover, the addition of 50 µM S37a inhibited cAMP accumulation induced by sera from patients with GO by 50‐60% compared to bTSH-treated stable HEK-TSHR cells. In vivo experiments in mice are also being studied and may provide new ideas for treating GO. The findings of in vivo experimental studies by Fassbender et al. (94) demonstrate that novel peptides distinctly reduce serum thyroxine levels, thyroid size, retro-orbital fibrosis and tachycardia in Ad-TSHR289-immunized mice. In immunologically naïve mice, the administration of peptides did not induce any immune response. This evidence also indicates that intravenous administration of TSHR-derived cyclic peptide 19 to Ad-TSHR-immunized mice significantly improves thyroid function, the levels of TRAbs and acidic mucins and the collagen content in orbital tissue, offering a potential novel therapeutic approach for GD and GO (95). A recent study indicated that GO mice intraperitoneally injected with siRNA targeting TSHR exhibited marked improvements in weight loss, serum thyroxine (T4) levels, TSAb levels, TSBAb levels and thyroid uptake of 99mTcO4 (96). Potential novel therapies for GO that directly target TSHR are provided in Table 1.
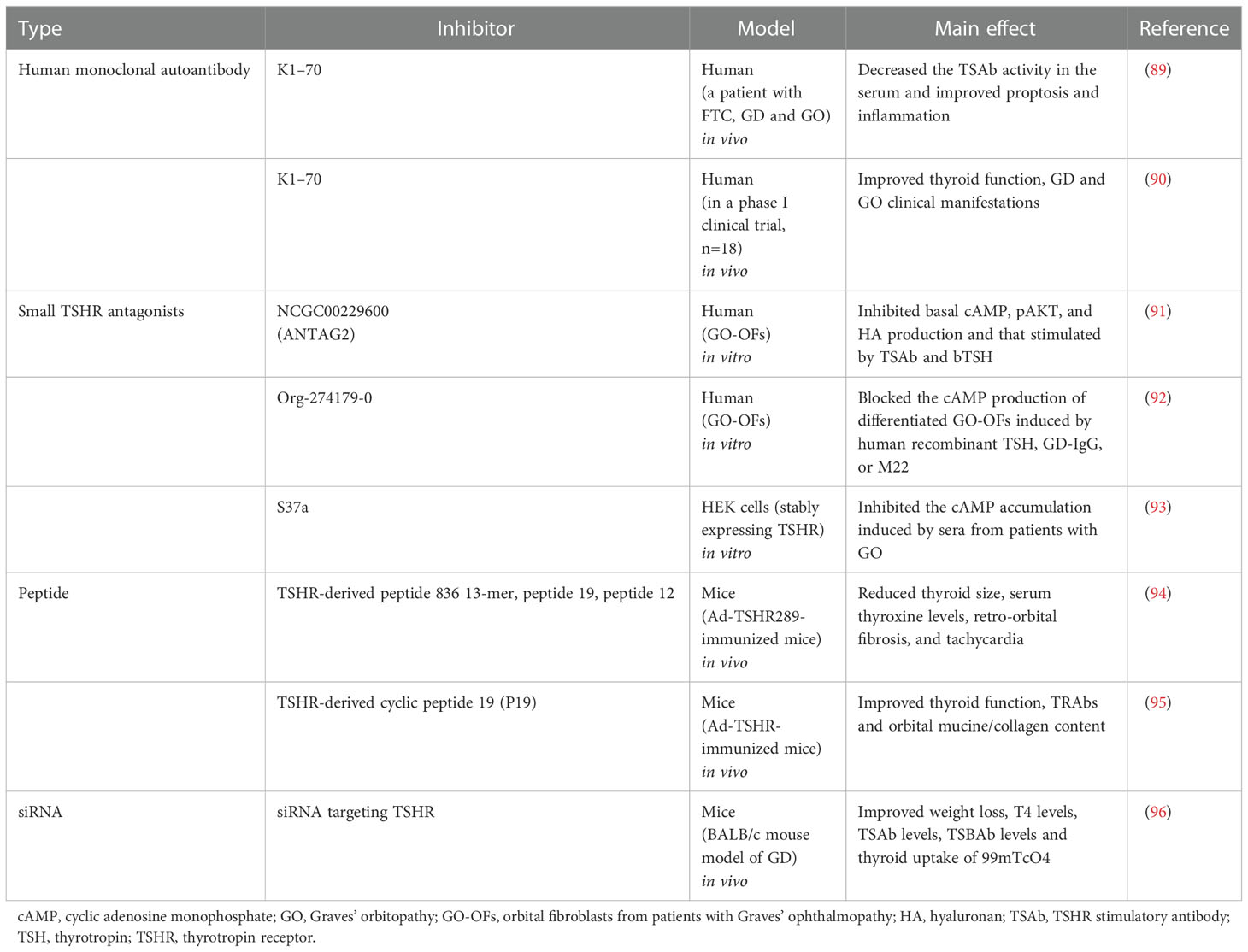
Table 1 Potential novel therapies for GO that directly target TSHR.
IGF-1R blockers
Recently, the FDA-approved drug teprotumumab (a human monoclonal anti-IGF-1R Ab) was identified as a breakthrough drug for treating active GO. Teprotumumab reduces proptosis and the need for orbital decompression surgery. Teprotumumab mainly inhibits the IGF-1R pathway through two mechanisms. First, upon binding to the cysteine-rich domain of human IGF-1R, teprotumumab blocks the binding pocket for both endogenous ligands (IGF-1 and IGF-2) and prevents them from activating the signalling cascade. Second, binding of teprotumumab induces internalization and subsequent degradation of IGF-1R, leading to a 95% reduction in cell surface-accessible IGF-1R (97, 98). Teprotumumab does not bind to TSHRs and inhibits IGF1R-dependent M22-induced HA production, which is mediated by TSHR/IGF-1R crosstalk but not IGF-1R-independent M22 stimulation (72). Fibrocytes from patients with GD display a robust reduction in surface IGF-1R and TSHR expression after teprotumumab treatment (99).
A randomized, placebo-controlled, multicentre, double-masked trial was performed to investigate the efficacy of teprotumumab in patients with active, moderate-to severe GO (100). Eighty-eight patients were randomly assigned to receive the active drug or placebo administered intravenously once every 3 weeks for a total of eight infusions. Compared with patients who received the placebo, those who received teprotumumab showed a response at week 24. The therapeutic effects were rapid; at week 6, 18 of the 43 patients in the teprotumumab group and 2 of the 45 patients in the placebo group showed a response. These data indicate that teprotumumab therapy provides clinical benefits to patients with active, moderate-to-severe GO by reducing proptosis and CAS and by improving quality of life (100).
Another randomized, double-masked, placebo-controlled, phase 3 multicentre trial was carried out in 2020. Forty-one patients were assigned to the teprotumumab group and received intravenous infusions of teprotumumab (10 mg/kg for the first infusion and 20 mg/kg for subsequent infusions). Forty-two patients were assigned to the placebo group and received intravenous infusions of placebo once every 3 weeks for 21 weeks. The last trial visit for this analysis occurred at week 24, at which the percentage of patients showing a proptosis response was higher in the teprotumumab group. All secondary outcomes, including overall response, the proportion of patients with CAS of 0 or 1, the mean change in proptosis, the diplopia response, and the mean change in Graves’ ophthalmopathy-specific quality-of-life overall score, were significantly better after treatment with teprotumumab than with placebo. These findings demonstrate that among patients with active GO, teprotumumab resulted in better outcomes than placebo with respect to proptosis, CAS, diplopia, and quality of life. The most common adverse events (AEs) reported with teprotumumab included muscle spasms, alopecia, nausea, diarrhoea, hearing impairment, fatigue and hyperglycaemia, but they were mild-moderate during treatment. Serious AEs, such as Hashimoto’s encephalopathy, were uncommon (101). A summary of two teprotumumab clinical trials for GO is shown in Table 2.
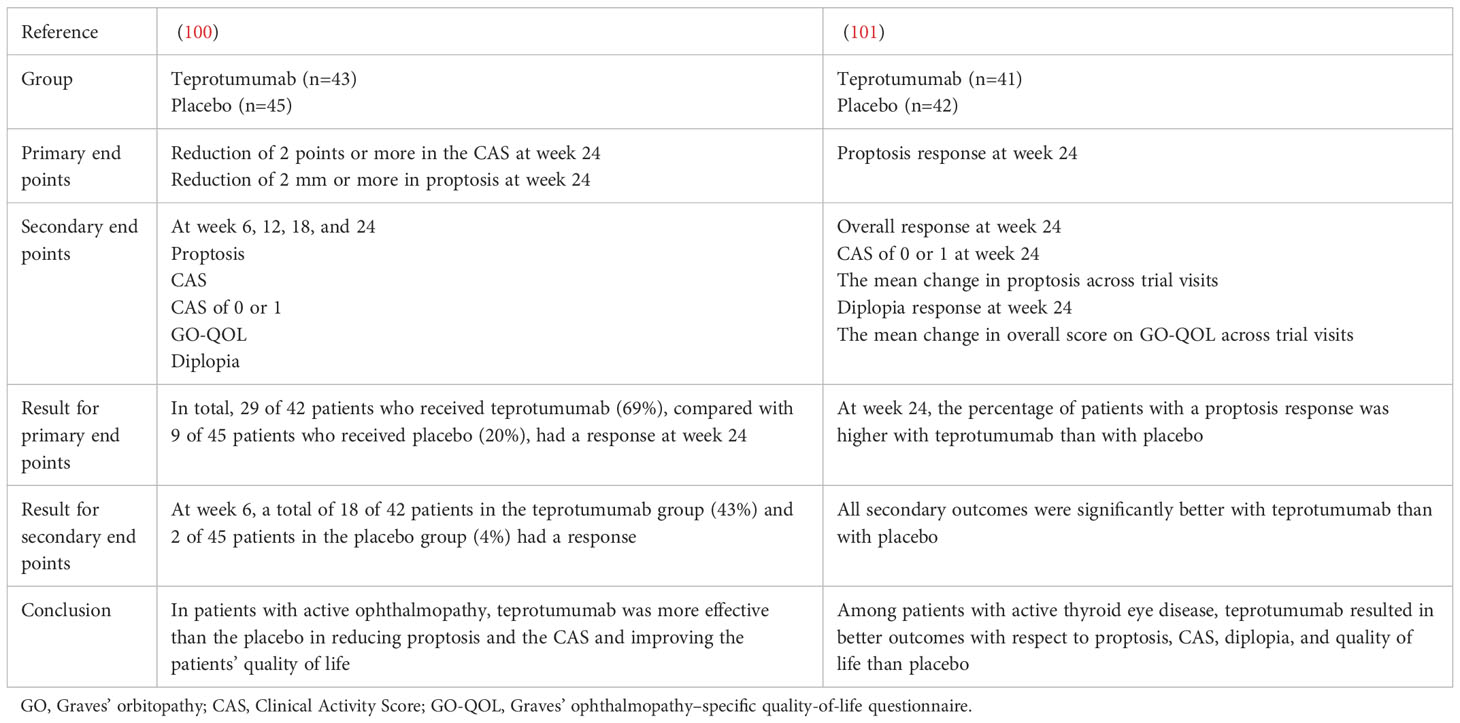
Table 2 Summary of two teprotumumab clinical trials for GO.
Currently, high-dose intravenous methylprednisolone combined with oral mycophenolate is the preferred therapy for moderate-to-severe and active GO, as cited by the 2021 EUGOGO. Other biological agents, such as tocilizumab (a humanized monoclonal antibody against IL-6R) (102–109), rituximab (a monoclonal antibody that targets CD20+ B cells) (108, 110–116), and adalimumab (a TNF-α blocking monoclonal antibody) (117), have proven to be useful and safe therapeutic options as treatments in refractory GO. These agents have produced promising results, including improved proptosis and diplopia and reduced CAS scores. Other biological agents of GO are listed in Table 3.
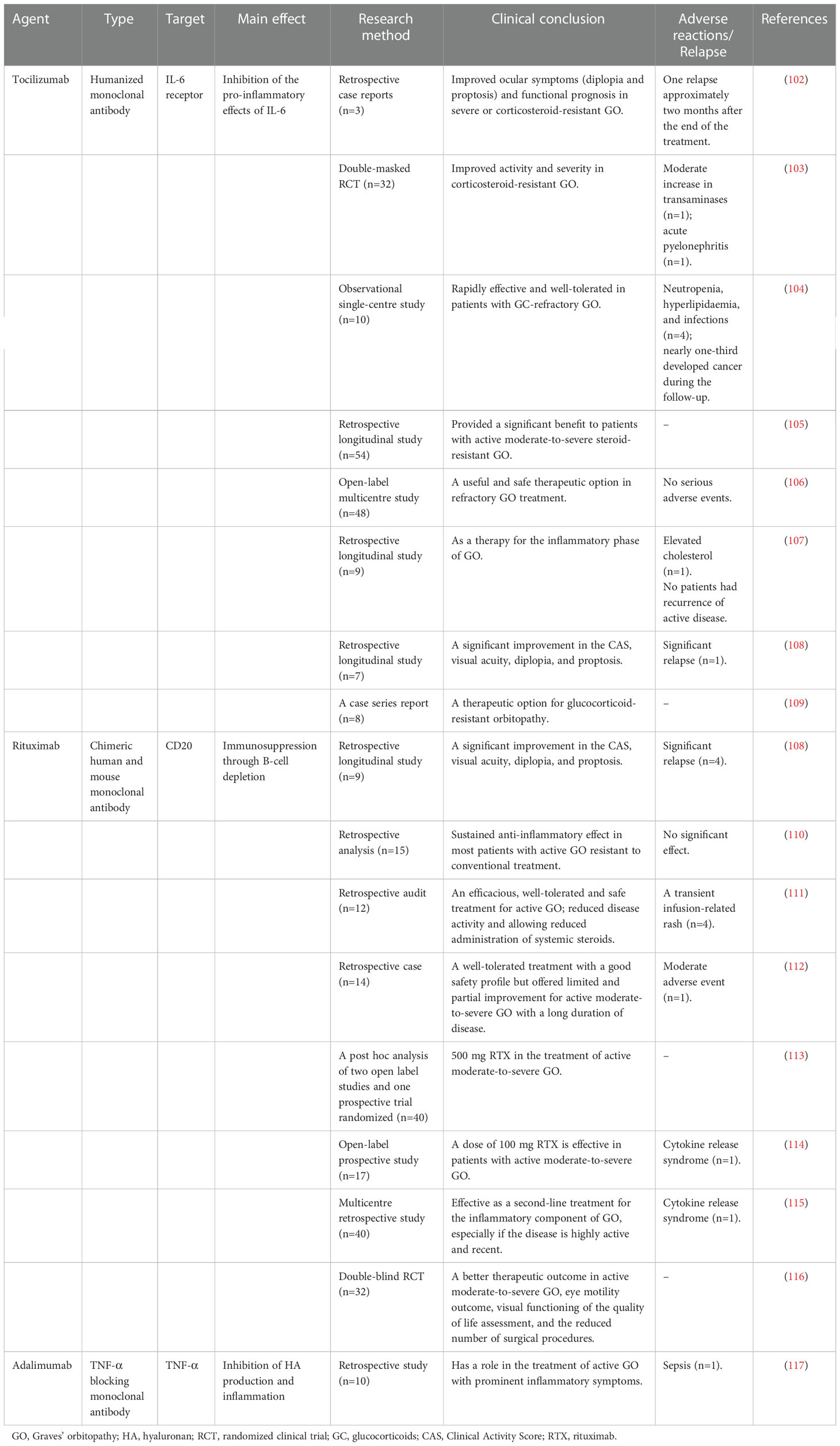
Table 3 Other clinical therapeutic targets for GO.
Conclusions
The following important points were obtained from the review. In addition to CD34+ OFs, CD34- OFs are crucial for GO pathogenesis and may be involved in HA formation. CD34- OFs expressing Slit2 suppress the phenotype of CD34+ OFs, and β-arrestin 1 is involved in TSHR/IGF-1R crosstalk. In recent years, research on TRAbs has gradually shifted to TSAbs and TBAbs, but the existence and role of IGF-1R Abs remain unclear, and further exploration is warranted. The results of clinical trials targeting TSHR inhibition are expected. Teprotumumab has become the latest second-line treatment for GO. Tocilizumab and rituximab hold great promise in the future management of GO and can be useful in cases if intolerance or resistance to standard immunosuppressive treatment.
Author contributions
This review was written by XC. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (Grant no. 82270833) and the Provincial Natural Science Foundation of Liaoning (Grant no.2022-BS-120). This work was also funded by the 345 Talent Project of Shengjing Hospital of China Medical University (Grant no.M1349).
Acknowledgments
We thank the physicians at the Department of Endocrinology of Shengjing Hospital of China Medical University for their advice and assistance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
APCs, Antigen-presenting cells; DCs, dendritic cells; GAGs, glycosaminoglycans; GD, Graves’ disease; GO, Graves’ orbitopathy; GO-OFs, OFs from patients with GO; HA, hyaluronan; IGF-1R, insulin-like growth factor-1 receptor; IGF-1R Abs, IGF-1R antibodies; OAs, orbital adipocytes; OFs:orbital fibroblasts; Tg, thyroglobulin; TgAbs, thyroglobulin antibodies; TPO, thyroperoxidase; TPOAbs, thyroid peroxidase antibodies; TRAbs, TSHR antibodies; TSHR, thyrotropin receptor.
References
1. Bartalena L, Kahaly GJ, Baldeschi L, Dayan CM, Eckstein A, Marcocci C, et al. The 2021 European group on graves’ orbitopathy (EUGOGO) clinical practice guidelines for the medical management of graves’ orbitopathy. Eur J Endocrinol (2021) 185:G43–67. doi: 10.1530/EJE-21-0479
2. Bartalena L. Diagnosis and management of graves disease: a global overview. Nat Rev Endocrinol (2013) 9:724–34. doi: 10.1038/nrendo.2013.193
4. Khamisi S, Lundqvist M, Emadi P, Almby K, Ljunggren O, Karlsson FA. Serum thyroglobulin is associated with orbitopathy in graves’ disease. J Endocrinol Invest (2021) 44:1905–11. doi: 10.1007/s40618-021-01505-8
5. Cao J, Su Y, Chen Z, Ma C, Xiong W. The risk factors for graves’ ophthalmopathy. Graefes Arch Clin Exp Ophthalmol (2022) 260:1043–54. doi: 10.1007/s00417-021-05456-x
6. Smith TJ. TSHR as a therapeutic target in graves’ disease. Expert Opin Ther Targets (2017) 21:427–32. doi: 10.1080/14728222.2017.1288215
7. Krieger CC, Neumann S, Gershengorn MC. TSH/IGF1 receptor crosstalk: mechanism and clinical implications. Pharmacol Ther (2020) 209:107502. doi: 10.1016/j.pharmthera.2020.107502
8. Dottore GR, Torregrossa L, Caturegli P, Ionni I, Sframeli A, Sabini E, et al. Association of T and b cells infiltrating orbital tissues with clinical features of graves orbitopathy. JAMA Ophthalmol (2018) 136:613–9. doi: 10.1001/jamaophthalmol.2018.0806
9. Dottore GR, Torregrossa L, Lanzolla G, Mariotti S, Menconi F, Piaggi P, et al. Role of the mononuclear cell infiltrate in graves’ orbitopathy (GO): results of a large cohort study. J Endocrinol Invest (2022) 45:563–72. doi: 10.1007/s40618-021-01692-4
10. Huang Y, Fang S, Li D, Zhou H, Li B, Fan X. The involvement of T cell pathogenesis in thyroid-associated ophthalmopathy. Eye (Lond) (2019) 33:176–82. doi: 10.1038/s41433-018-0279-9
11. He M, Wang Y, Wang J, Sui J, Ding X, Chen Z, et al. The potential markers involved in newly diagnosed graves’ disease and the development of active graves’ orbitopathy. Cytokine (2020) 127:154998. doi: 10.1016/j.cyto.2020.154998
12. Fang S, Huang Y, Wang S, Zhang Y, Luo X, Liu L, et al. IL-17A exacerbates fibrosis by promoting the proinflammatory and profibrotic function of orbital fibroblasts in TAO. J Clin Endocrinol Metab (2016) 101:2955–65. doi: 10.1210/jc.2016-1882
13. Fang S, Huang Y, Zhong S, Li Y, Zhang Y, Li Y, et al. Regulation of orbital fibrosis and adipogenesis by pathogenic Th17 cells in graves orbitopathy. J Clin Endocrinol Metab (2017) 102:4273–83. doi: 10.1210/jc.2017-01349
14. Siomkajlo M, Mizera L, Szymczak D, Kolackov K, Grzegrzolka J, Bolanowski M, et al. Effect of systemic steroid therapy in graves’ orbitopathy on regulatory T cells and Th17/Treg ratio. J Endocrinol Invest (2021) 44:2475–84. doi: 10.1007/s40618-021-01565-w
15. Matsuzawa K, Izawa S, Okura T, Fujii S, Matsumoto K, Shoji K, et al. Implications of FoxP3-positive and -negative CD4(+) CD25(+) T cells in graves’ ophthalmopathy. Endocr J (2016) 63:755–64. doi: 10.1507/endocrj.EJ16-0108
16. Taylor PN, Zhang L, Lee RWJ, Muller I, Ezra DG, Dayan CM, et al. New insights into the pathogenesis and nonsurgical management of graves orbitopathy. Nat Rev Endocrinol (2020) 16:104–16. doi: 10.1038/s41574-019-0305-4
17. Virakul S, Somparn P, Pisitkun T, van der Spek PJ, Dalm V, Paridaens D, et al. Integrative analysis of proteomics and DNA methylation in orbital fibroblasts from graves’ ophthalmopathy. Front Endocrinol (Lausanne) (2020) 11:619989. doi: 10.3389/fendo.2020.619989
18. Liu W, Ma C, Li HY, Chen L, Yuan SS, Li KJ. MicroRNA-146a downregulates the production of hyaluronic acid and collagen I in graves’ ophthalmopathy orbital fibroblasts. Exp Ther Med (2020) 20:38. doi: 10.3892/etm.2020.9165
19. Wang N, Hou SY, Qi X, Deng M, Cao JM, Tong BD, et al. LncRNA LPAL2/miR-1287-5p/EGFR axis modulates TED-derived orbital fibroblast activation through cell adhesion factors. J Clin Endocrinol Metab (2021) 106:e2866–86. doi: 10.1210/clinem/dgab256
20. Klett ZG, Elner SG, Elner VM. Differential expression of immunoreactive HLA-DR and ICAM-1 in human cultured orbital fibroblasts and orbital tissue. Opht Plast Reconstr Surg (1996) 12:153–62. doi: 10.1097/00002341-199609000-00001
21. Hiromatsu Y, Wang PW, Wosu L, How J, Wall JR. Mechanisms of immune damage in graves’ ophthalmopathy. Horm Res (1987) 26:198–207. doi: 10.1159/000180701
22. Kozdon K, Fitchett C, Rose GE, Ezra DG, Bailly M. Mesenchymal stem cell-like properties of orbital fibroblasts in graves’ orbitopathy. Invest Ophthalmol Vis Sci (2015) 56:5743–50. doi: 10.1167/iovs.15-16580
23. Wei YH, Liao SL, Wang SH, Wang CC, Yang CH. Simvastatin and ROCK inhibitor y-27632 inhibit myofibroblast differentiation of graves’ ophthalmopathy-derived orbital fibroblasts via RhoA-mediated ERK and p38 signaling pathways. Front Endocrinol (Lausanne) (2020) 11:607968. doi: 10.3389/fendo.2020.607968
24. Perros P, Hegedus L, Bartalena L, Marcocci C, Kahaly GJ, Baldeschi L, et al. Graves’ orbitopathy as a rare disease in Europe: a European group on graves’ orbitopathy (EUGOGO) position statement. Orphanet J Rare Dis (2017) 12:72. doi: 10.1186/s13023-017-0625-1
25. Lacheta D, Miskiewicz P, Gluszko A, Nowicka G, Struga M, Kantor I, et al. Immunological aspects of graves’ ophthalmopathy. BioMed Res Int (2019) 2019:7453260. doi: 10.1155/2019/7453260
26. Smith TJ. Potential roles of CD34+ fibrocytes masquerading as orbital fibroblasts in thyroid-associated ophthalmopathy. J Clin Endocrinol Metab (2019) 104:581–94. doi: 10.1210/jc.2018-01493
27. Basak M, Sanyal T, Kar A, Bhattacharjee P, Das M, Chowdhury S. Peripheral blood mononuclear cells - can they provide a clue to the pathogenesis of graves’ orbitopathy? Endocrine (2022) 75:447–55. doi: 10.1007/s12020-021-02865-0
28. Lu Y, Atkins SJ, Fernando R, Trierweiler A, Mester T, Grisolia ABD, et al. CD34- orbital fibroblasts from patients with thyroid-associated ophthalmopathy modulate TNF-alpha expression in CD34+ fibroblasts and fibrocytes. Invest Ophthalmol Vis Sci (2018) 59:2615–22. doi: 10.1167/iovs.18-23951
29. Smith TJ, Hegedus L. Gravesd disease. N Engl J Med (2016) 375:1552–65. doi: 10.1056/NEJMra1510030
30. van Zeijl CJ, Fliers E, van Koppen CJ, Surovtseva OV, de Gooyer ME, Mourits MP, et al. Effects of thyrotropin and thyrotropin-receptor-stimulating graves’ disease immunoglobulin G on cyclic adenosine monophosphate and hyaluronan production in nondifferentiated orbital fibroblasts of graves’ ophthalmopathy patients. Thyroid (2010) 20:535–44. doi: 10.1089/thy.2009.0447
31. Mummert ME. Immunologic roles of hyaluronan. Immunol Res (2005) 31:189–206. doi: 10.1385/IR:31:3:189
32. Fernando R, Atkins SJ, Smith TJ. Slit2 may underlie divergent induction by thyrotropin of IL-23 and IL-12 in human fibrocytes. J Immunol (2020) 204:1724–35. doi: 10.4049/jimmunol.1900434
33. Fernando R, Smith TJ. Slit2 regulates hyaluronan & cytokine synthesis in fibrocytes: potential relevance to thyroid-associated ophthalmopathy. J Clin Endocrinol Metab (2021) 106:e20–33. doi: 10.1210/clinem/dgaa684
34. Fernando R, Grisolia ABD, Lu Y, Atkins S, Smith TJ. Slit2 modulates the inflammatory phenotype of orbit-infiltrating fibrocytes in graves’ disease. J Immunol (2018) 200:3942–9. doi: 10.4049/jimmunol.1800259
35. Wiersinga WM. Autoimmunity in graves’ ophthalmopathy: the result of an unfortunate marriage between TSH receptors and IGF-1 receptors? J Clin Endocrinol Metab (2011) 96:2386–94. doi: 10.1210/jc.2011-0307
36. Kim DW, Taneja K, Hoang T, Santiago CP, McCulley TJ, Merbs SL, et al. Transcriptomic profiling of control and thyroid-associated orbitopathy (TAO) orbital fat and TAO orbital fibroblasts undergoing adipogenesis. Invest Ophthalmol Vis Sci (2021) 62:24. doi: 10.1167/iovs.62.9.24
37. Jung SJ, Choi YJ, Park TK, Woo SE, Kim BY, Yoon JS, et al. Wnt signalling inhibits adipogenesis in orbital fibroblasts from patients with graves’ orbitopathy. Br J Ophthalmol (2022) 106:1019–27. doi: 10.1136/bjophthalmol-2020-316898
38. Ko J, Kim JY, Lee EJ, Yoon JS. Inhibitory effect of idelalisib, a selective phosphatidylinositol 3-kinase delta inhibitor, on adipogenesis in an in vitro model of graves’ orbitopathy. Invest Ophthalmol Vis Sci (2018) 59:4477–85. doi: 10.1167/iovs.18-24509
39. Ko J, Kim JY, Chae MK, Lee EJ, Yoon JS. PERK mediates oxidative stress and adipogenesis in graves’ orbitopathy pathogenesis. J Mol Endocrinol (2021) 66:313–23. doi: 10.1530/JME-21-0057
40. Lee JS, Chae MK, Kikkawa DO, Lee EJ, Yoon JS. Glycogen synthase kinase-3beta mediates proinflammatory cytokine secretion and adipogenesis in orbital fibroblasts from patients with graves’ orbitopathy. Invest Ophthalmol Vis Sci (2020) 61:51. doi: 10.1167/iovs.61.8.51
41. Hammond CL, Roztocil E, Gonzalez MO, Feldon SE, Woeller CF. MicroRNA-130a is elevated in thyroid eye disease and increases lipid accumulation in fibroblasts through the suppression of AMPK. Invest Ophthalmol Vis Sci (2021) 62:29. doi: 10.1167/iovs.62.1.29
42. Shin HA, Park M, Banga JP, Lew H. TGFbeta-treated placenta-derived mesenchymal stem cells selectively promote anti-adipogenesis in thyroid-associated ophthalmopathy. Int J Mol Sci (2022) 23:5603. doi: 10.3390/ijms23105603
43. Zhang L, Grennan-Jones F, Lane C, Rees DA, Dayan CM, Ludgate M. Adipose tissue depot-specific differences in the regulation of hyaluronan production of relevance to graves’ orbitopathy. J Clin Endocrinol Metab (2012) 97:653–62. doi: 10.1210/jc.2011-1299
44. Billon N, Dani C. Developmental origins of the adipocyte lineage: new insights from genetics and genomics studies. Stem Cell Rev Rep (2012) 8:55–66. doi: 10.1007/s12015-011-9242-x
45. Gesta S, Tseng YH, Kahn CR. Developmental origin of fat: tracking obesity to its source. Cell (2007) 131:242–56. doi: 10.1016/j.cell.2007.10.004
46. Ferrari SM, Fallahi P, Elia G, Ragusa F, Camastra S, Paparo SR, et al. Novel therapies for thyroid autoimmune diseases: an update. Best Pract Res Clin Endocrinol Metab (2020) 34:101366. doi: 10.1016/j.beem.2019.101366
47. Xia N, Ye X, Hu X, Song S, Xu H, Niu M, et al. Simultaneous induction of graves’ hyperthyroidism and graves’ ophthalmopathy by TSHR genetic immunization in BALB/c mice. PloS One (2017) 12:e0174260. doi: 10.1371/journal.pone.0174260
48. Choi HY, Jeon H, Yang JW, Ahn JH, Jung JH. Thyroid-stimulating hormone receptor expression on primary cultured human extraocular muscle myoblasts. Curr Eye Res (2018) 43:1484–8. doi: 10.1080/02713683.2018.1501075
49. Smith TJ, Janssen J. Insulin-like growth factor-i receptor and thyroid-associated ophthalmopathy. Endocr Rev (2019) 40:236–67. doi: 10.1210/er.2018-00066
50. Zhang L, Bowen T, Grennan-Jones F, Paddon C, Giles P, Webber J, et al. Thyrotropin receptor activation increases hyaluronan production in preadipocyte fibroblasts: contributory role in hyaluronan accumulation in thyroid dysfunction. J Biol Chem (2009) 284:26447–55. doi: 10.1074/jbc.M109.003616
51. Wakelkamp IM, Bakker O, Baldeschi L, Wiersinga WM, Prummel MF. TSH-r expression and cytokine profile in orbital tissue of active vs. inactive graves’ ophthalmopathy patients. Clin Endocrinol (Oxf) (2003) 58:280–7. doi: 10.1046/j.1365-2265.2003.01708.x
52. Tsui S, Naik V, Hoa N, Hwang CJ, Afifiyan NF, Hikim AS, et al. Evidence for an association between thyroid-stimulating hormone and insulin-like growth factor 1 receptors: a tale of two antigens implicated in graves’ disease. J Immunol (2008) 181:4397–405. doi: 10.4049/jimmunol.181.6.4397
53. Jyonouchi SC, Valyasevi RW, Harteneck DA, Dutton CM, Bahn RS. Interleukin-6 stimulates thyrotropin receptor expression in human orbital preadipocyte fibroblasts from patients with graves’ ophthalmopathy. Thyroid (2001) 11:929–34. doi: 10.1089/105072501753210984
54. Woeller CF, Roztocil E, Hammond C, Feldon SE. TSHR signaling stimulates proliferation through PI3K/Akt and induction of miR-146a and miR-155 in thyroid eye disease orbital fibroblasts. Invest Ophthalmol Vis Sci (2019) 60:4336–45. doi: 10.1167/iovs.19-27865
55. Draman MS, Grennan-Jones F, Taylor P, Muller I, Evans S, Haridas A, et al. Expression of endogenous putative TSH binding protein in orbit. Curr Issues Mol Biol (2021) 43:1794–804. doi: 10.3390/cimb43030126
56. Chen XL, He WM, Li W. Effects and its mechanism of IGF-1R on the synthesis of hyaluronic acid in orbital fibroblasts of thyroid associated ophthalmopathy. Sichuan Da Xue Xue Bao Yi Xue Ban (2017) 48:727–31.
57. Mohyi M, Smith TJ. IGF1 receptor and thyroid-associated ophthalmopathy. J Mol Endocrinol (2018) 61:T29–43. doi: 10.1530/JME-17-0276
58. Morshed SA, Ma R, Latif R, Davies TF. Mechanisms in graves eye disease: apoptosis as the end point of insulin-like growth factor 1 receptor inhibition. Thyroid (2022) 32:429–39. doi: 10.1089/thy.2021.0176
59. Fernando R, Caldera O, Smith TJ. Therapeutic IGF-I receptor inhibition alters fibrocyte immune phenotype in thyroid-associated ophthalmopathy. Proc Natl Acad Sci U.S.A. (2021) 118:e2114244118. doi: 10.1073/pnas.2114244118
60. Zhao SX, Tsui S, Cheung A, Douglas RS, Smith TJ, Banga JP. Orbital fibrosis in a mouse model of graves’ disease induced by genetic immunization of thyrotropin receptor cDNA. J Endocrinol (2011) 210:369–77. doi: 10.1530/JOE-11-0162
61. Krieger CC, Perry JD, Morgan SJ, Kahaly GJ, Gershengorn MC. TSH/IGF-1 receptor cross-talk rapidly activates extracellular signal-regulated kinases in multiple cell types. Endocrinology (2017) 158:3676–83. doi: 10.1210/en.2017-00528
62. Tramontano D, Cushing GW, Moses AC, Ingbar SH. Insulin-like growth factor-I stimulates the growth of rat thyroid cells in culture and synergizes the stimulation of DNA synthesis induced by TSH and graves’-IgG. Endocrinology (1986) 119:940–2. doi: 10.1210/endo-119-2-940
63. Paik JS, Kim SE, Kim JH, Lee JY, Yang SW, Lee SB. Insulin-like growth factor-1 enhances the expression of functional TSH receptor in orbital fibroblasts from thyroid-associated ophthalmopathy. Immunobiology (2020) 225:151902. doi: 10.1016/j.imbio.2019.151902
64. Ock S, Ahn J, Lee SH, Kang H, Offermanns S, Ahn HY, et al. IGF-1 receptor deficiency in thyrocytes impairs thyroid hormone secretion and completely inhibits TSH-stimulated goiter. FASEB J (2013) 27:4899–908. doi: 10.1096/fj.13-231381
65. Krieger CC, Place RF, Bevilacqua C, Marcus-Samuels B, Abel BS, Skarulis MC, et al. TSH/IGF-1 receptor cross talk in graves’ ophthalmopathy pathogenesis. J Clin Endocrinol Metab (2016) 101:2340–7. doi: 10.1210/jc.2016-1315
66. Zhang L, Ji QH, Ruge F, Lane C, Morris D, Tee AR, et al. Reversal of pathological features of graves’ orbitopathy by activation of forkhead transcription factors, FOXOs. J Clin Endocrinol Metab (2016) 101:114–22. doi: 10.1210/jc.2015-2932
67. Kumar S, Coenen M, Iyer S, Bahn RS. Forkhead transcription factor FOXO1 is regulated by both a stimulatory thyrotropin receptor antibody and insulin-like growth factor-1 in orbital fibroblasts from patients with graves’ ophthalmopathy. Thyroid (2015) 25:1145–50. doi: 10.1089/thy.2015.0254
68. Kopp P. Key elements involved in the negative regulation of the TSH receptor: G protein-coupled receptor kinases, arrestin and inducible cAMP early repressor. Eur J Endocrinol (1997) 136:269–70. doi: 10.1530/eje.0.1360269
69. Frenzel R, Voigt C, Paschke R. The human thyrotropin receptor is predominantly internalized by beta-arrestin 2. Endocrinology (2006) 147:3114–22. doi: 10.1210/en.2005-0687
70. Boutin A, Eliseeva E, Gershengorn MC, Neumann S. Beta-Arrestin-1 mediates thyrotropin-enhanced osteoblast differentiation. FASEB J (2014) 28:3446–55. doi: 10.1096/fj.14-251124
71. Krieger CC, Boutin A, Jang D, Morgan SJ, Banga JP, Kahaly GJ, et al. Arrestin-beta-1 physically scaffolds TSH and IGF1 receptors to enable crosstalk. Endocrinology (2019) 160:1468–79. doi: 10.1210/en.2019-00055
72. Krieger CC, Sui X, Kahaly GJ, Neumann S, Gershengorn MC. Inhibition of TSH/IGF-1 receptor crosstalk by teprotumumab as a treatment modality of thyroid eye disease. J Clin Endocrinol Metab (2022) 107:e1653–60. doi: 10.1210/clinem/dgab824
73. Metcalfe R, Jordan N, Watson P, Gullu S, Wiltshire M, Crisp M, et al. Demonstration of immunoglobulin G, a, and e autoantibodies to the human thyrotropin receptor using flow cytometry. J Clin Endocrinol Metab (2002) 87:1754–61. doi: 10.1210/jcem.87.4.8411
74. George A, Diana T, Langericht J, Kahaly GJ. Stimulatory thyrotropin receptor antibodies are a biomarker for graves’ orbitopathy. Front Endocrinol (Lausanne) (2020) 11:629925. doi: 10.3389/fendo.2020.629925
75. Rapoport B, McLachlan SM. TSH receptor cleavage into subunits and shedding of the a-subunit; a molecular and clinical perspective. Endocr Rev (2016) 37:114–34. doi: 10.1210/er.2015-1098
76. Suzuki N, Noh JY, Kameda T, Yoshihara A, Ohye H, Suzuki M, et al. Clinical course of thyroid function and thyroid associated-ophthalmopathy in patients with euthyroid graves’ disease. Clin Ophthalmol (2018) 12:739–46. doi: 10.2147/OPTH.S158967
77. Matutinovic MS, Diana T, Beleslin BN, Ciric J, Zarkovic M, Kahaly GJ, et al. Clinical value of functional thyrotropin receptor antibodies in Serbian patients with graves’ orbitopathy. J Endocrinol Invest (2022) 45:189–97. doi: 10.1007/s40618-021-01652-y
78. Kahaly GJ, Diana T, Kanitz M, Frommer L, Olivo PD. Prospective trial of functional thyrotropin receptor antibodies in graves disease. J Clin Endocrinol Metab (2020) 105:e1006–e14. doi: 10.1210/clinem/dgz292
79. Krieger CC, Kahaly GJ, Azam A, Klubo-Gwiezdzinska J, Neumann S, Gershengorn MC. Graves’ autoantibodies exhibit different stimulating activities in cultures of thyrocytes and orbital fibroblasts not reflected by clinical assays. Thyroid (2022) 32:90–6. doi: 10.1089/thy.2021.0326
80. Kahaly GJ, Wuster C, Olivo PD, Diana T. High titers of thyrotropin receptor antibodies are associated with orbitopathy in patients with graves disease. J Clin Endocrinol Metab (2019) 104:2561–8. doi: 10.1210/jc.2018-02705
81. Kumar S, Schiefer R, Coenen MJ, Bahn RS. A stimulatory thyrotropin receptor antibody (M22) and thyrotropin increase interleukin-6 expression and secretion in graves’ orbital preadipocyte fibroblasts. Thyroid (2010) 20:59–65. doi: 10.1089/thy.2009.0278
82. Krieger CC, Neumann S, Gershengorn MC. Is there evidence for IGF1R-stimulating abs in graves’ orbitopathy pathogenesis? Int J Mol Sci (2020) 21:6561. doi: 10.3390/ijms21186561
83. Marcus-Samuels B, Krieger CC, Boutin A, Kahaly GJ, Neumann S, Gershengorn MC. Evidence that graves’ ophthalmopathy immunoglobulins do not directly activate IGF-1 receptors. Thyroid (2018) 28:650–5. doi: 10.1089/thy.2018.0089
84. Weightman DR, Perros P, Sherif IH, Kendall-Taylor P. Autoantibodies to IGF-1 binding sites in thyroid associated ophthalmopathy. Autoimmunity (1993) 16:251–7. doi: 10.3109/08916939309014643
85. Minich WB, Dehina N, Welsink T, Schwiebert C, Morgenthaler NG, Kohrle J, et al. Autoantibodies to the IGF1 receptor in graves’ orbitopathy. J Clin Endocrinol Metab (2013) 98:752–60. doi: 10.1210/jc.2012-1771
86. Marino M, Dottore GR, Ionni I, Lanzolla G, Sabini E, Ricci D, et al. Serum antibodies against the insulin-like growth factor-1 receptor (IGF-1R) in graves’ disease and graves’ orbitopathy. J Endocrinol Invest (2019) 42:471–80. doi: 10.1007/s40618-018-0943-8
87. Lanzolla G, Ricci D, Nicoli F, Sabini E, Sframeli A, Brancatella A, et al. ‘Putative protective role of autoantibodies against the insulin-like growth factor-1 receptor in graves’ disease: results of a pilot study. J Endocrinol Invest (2020) 43:1759–68. doi: 10.1007/s40618-020-01341-2
88. Varewijck AJ, Boelen A, Lamberts SW, Fliers E, Hofland LJ, Wiersinga WM, et al. Circulating IgGs may modulate IGF-I receptor stimulating activity in a subset of patients with graves’ ophthalmopathy. J Clin Endocrinol Metab (2013) 98:769–76. doi: 10.1210/jc.2012-2270
89. Ryder M, Wentworth M, Algeciras-Schimnich A, Morris JC, Garrity J, Sanders J, et al. Blocking the thyrotropin receptor with K1-70 in a patient with follicular thyroid cancer, graves’ disease, and graves’ ophthalmopathy. Thyroid (2021) 31:1597–602. doi: 10.1089/thy.2021.0053
90. Furmaniak J, Sanders J, Sanders P, Li Y, Rees Smith B. TSH receptor specific monoclonal autoantibody K1-70(TM) targeting of the TSH receptor in subjects with graves’ disease and graves’ orbitopathy-results from a phase I clinical trial. Clin Endocrinol (Oxf) (2022) 96:878–87. doi: 10.1111/cen.14681
91. Turcu AF, Kumar S, Neumann S, Coenen M, Iyer S, Chiriboga P, et al. A small molecule antagonist inhibits thyrotropin receptor antibody-induced orbital fibroblast functions involved in the pathogenesis of graves ophthalmopathy. J Clin Endocrinol Metab (2013) 98:2153–9. doi: 10.1210/jc.2013-1149
92. van Zeijl CJ, van Koppen CJ, Surovtseva OV, de Gooyer ME, Plate R, Conti P, et al. Complete inhibition of rhTSH-, graves’ disease IgG-, and M22-induced cAMP production in differentiated orbital fibroblasts by a low-molecular-weight TSHR antagonist. J Clin Endocrinol Metab (2012) 97:E781–5. doi: 10.1210/jc.2011-2931
93. Marcinkowski P, Hoyer I, Specker E, Furkert J, Rutz C, Neuenschwander M, et al. A new highly thyrotropin receptor-selective small-molecule antagonist with potential for the treatment of graves’ orbitopathy. Thyroid (2019) 29:111–23. doi: 10.1089/thy.2018.0349
94. Fassbender J, Holthoff HP, Li Z, Ungerer M. Therapeutic effects of short cyclic and combined epitope peptides in a long-term model of graves’ disease and orbitopathy. Thyroid (2019) 29:258–67. doi: 10.1089/thy.2018.0326
95. Diana T, Ungerer M, Wuster C, Fassbender J, Li Z, Reimann A, et al. A cyclic peptide significantly improves thyroid function, thyrotropin-receptor antibodies and orbital mucine/collagen content in a long-term graves’ disease mouse model. J Autoimmun (2021) 122:102666. doi: 10.1016/j.jaut.2021.102666
96. Wang X, Liu W, Rui Z, Zheng W, Tan J, Li N, et al. Immunotherapy with a biologically active ICAM-1 mAb and an siRNA targeting TSHR in a BALB/c mouse model of graves’ disease. Endokrynol Pol (2021) 72:592–600. doi: 10.5603/EP.a2021.0087
97. Smith TJ, Janssen JA. Building the case for insulin-like growth factor receptor-i involvement in thyroid-associated ophthalmopathy. Front Endocrinol (Lausanne) (2016) 7:167. doi: 10.3389/fendo.2016.00167
98. Kahaly GJ. Immunotherapies for thyroid eye disease. Curr Opin Endocrinol Diabetes Obes (2019) 26:250–5. doi: 10.1097/MED.0000000000000493
99. Chen H, Mester T, Raychaudhuri N, Kauh CY, Gupta S, Smith TJ, et al. Teprotumumab, an IGF-1R blocking monoclonal antibody inhibits TSH and IGF-1 action in fibrocytes. J Clin Endocrinol Metab (2014) 99:E1635–40. doi: 10.1210/jc.2014-1580
100. Smith TJ, Kahaly GJ, Ezra DG, Fleming JC, Dailey RA, Tang RA, et al. Teprotumumab for thyroid-associated ophthalmopathy. N Engl J Med (2017) 376:1748–61. doi: 10.1056/NEJMoa1614949
101. Douglas RS, Kahaly GJ, Patel A, Sile S, Thompson EHZ, Perdok R, et al. Teprotumumab for the treatment of active thyroid eye disease. N Engl J Med (2020) 382:341–52. doi: 10.1056/NEJMoa1910434
102. Maldiney T, Deschasse C, Bielefeld P. Tocilizumab for the management of corticosteroid-resistant mild to severe graves’ ophthalmopathy, a report of three cases. Ocul Immunol Inflammation (2020) 28:281–4. doi: 10.1080/09273948.2018.1545914
103. Perez-Moreiras JV, Gomez-Reino JJ, Maneiro JR, Perez-Pampin E, Lopez AR, Alvarez FMR, et al. Efficacy of tocilizumab in patients with moderate-to-severe corticosteroid-resistant graves orbitopathy: a randomized clinical trial. Am J Ophthalmol (2018) 195:181–90. doi: 10.1016/j.ajo.2018.07.038
104. Moi L, Hamedani M, Ribi C. Long-term outcomes in corticosteroid-refractory graves’ orbitopathy treated with tocilizumab. Clin Endocrinol (Oxf) (2022) 97:363–70. doi: 10.1111/cen.14655
105. Perez-Moreiras JV, Varela-Agra M, Prada-Sanchez MC, Prada-Ramallal G. Steroid-resistant graves’ orbitopathy treated with tocilizumab in real-world clinical practice: a 9-year single-center experience. J Clin Med (2021) 10:706. doi: 10.3390/jcm10040706
106. Sanchez-Bilbao L, Martinez-Lopez D, Revenga M, Lopez-Vazquez A, Valls-Pascual E, Atienza-Mateo B, et al. Anti-IL-6 receptor tocilizumab in refractory graves’ orbitopathy: national multicenter observational study of 48 patients. J Clin Med (2020) 9:2816. doi: 10.3390/jcm9092816
107. Smith LD, Moscato EE, Seiff SR. Tocilizumab for the management of thyroid-associated orbitopathy. Opht Plast Reconstr Surg (2022) 38:188–92. doi: 10.1097/IOP.0000000000002027
108. Bennedjai A, Bouheraoua N, Gatfosse M, Dupasquier-Fediaevsky L, Errera MH, Tazartes M, et al. Tocilizumab versus rituximab in patients with moderate to severe steroid-resistant graves’ orbitopathy. Ocul Immunol Inflammation (2022) 30:500–5. doi: 10.1080/09273948.2020.1808688
109. Jose JCM, Rivera-Moscoso R, Jorge AFR, Vargas-Sanchez J, Ortega-Gutierrez G, Madriz-Prado R, et al. Tocilizumab in glucocorticoid-resistant graves orbitopathy. a case series report of a mexican population. Ann Endocrinol (Paris) (2020) 81:78–82. doi: 10.1016/j.ando.2020.01.003
110. Du Pasquier-Fediaevsky L, Andrei S, Berche M, Leenhardt L, Heron E, Riviere S. Low-dose rituximab for active moderate to severe graves’ orbitopathy resistant to conventional treatment. Ocul Immunol Inflammation (2019) 27:844–50. doi: 10.1080/09273948.2018.1453078
111. Insull EA, Sipkova Z, David J, Turner HE, Norris JH. Early low-dose rituximab for active thyroid eye disease: an effective and well-tolerated treatment. Clin Endocrinol (Oxf) (2019) 91:179–86. doi: 10.1111/cen.13970
112. Eid L, Coste-Verdier V, Longueville E, Ribeiro E, Nicolescu-Catargi B, Korobelnik JF. The effects of rituximab on graves’orbitopathy: a retrospective study of 14 patients. Eur J Ophthalmol (2020) 30:1008–13. doi: 10.1177/1120672119845224
113. Campi I, Vannucchi G, Muller I, Lazzaroni E, Curro N, Dainese M, et al. Therapy with different dose regimens of rituximab in patients with active moderate-To-Severe graves’ orbitopathy. Front Endocrinol (Lausanne) (2021) 12:790246. doi: 10.3389/fendo.2021.790246
114. Vannucchi G, Campi I, Covelli D, Curro N, Lazzaroni E, Palomba A, et al. Efficacy profile and safety of very low-dose rituximab in patients with graves’ orbitopathy. Thyroid (2021) 31:821–8. doi: 10.1089/thy.2020.0269
115. Deltour JB, Flamen MD, Ladsous M, Giovansili L, Cariou B, Caron P, et al. Efficacy of rituximab in patients with graves’ orbitopathy: a retrospective multicenter nationwide study. Graefes Arch Clin Exp Ophthalmol (2020) 258:2013–21. doi: 10.1007/s00417-020-04651-6
116. Salvi M, Vannucchi G, Curro N, Campi I, Covelli D, Dazzi D, et al. Efficacy of b-cell targeted therapy with rituximab in patients with active moderate to severe graves’ orbitopathy: a randomized controlled study. J Clin Endocrinol Metab (2015) 100:422–31. doi: 10.1210/jc.2014-3014
Keywords: Graves’ orbitopathy, orbital fibroblasts, orbital adipocytes, thyrotropin receptor, insulin-like growth factor-1 receptor
Citation: Cui X, Wang F and Liu C (2023) A review of TSHR- and IGF-1R-related pathogenesis and treatment of Graves’ orbitopathy. Front. Immunol. 14:1062045. doi: 10.3389/fimmu.2023.1062045
Received: 05 October 2022; Accepted: 02 January 2023;
Published: 19 January 2023.
Edited by:
Marian Elizabeth Ludgate, Cardiff University, United KingdomReviewed by:
Mario Salvi, IRCCS Ca ‘Granda Foundation Maggiore Policlinico Hospital, ItalyXiaofan Jia, University of Colorado Anschutz Medical Campus, United States
Copyright © 2023 Cui, Wang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cong Liu, bGl1Y19zamhvc3BpdGFsQDE2My5jb20=