 Tuhina Gupta1†
Tuhina Gupta1† Demba Sarr1†
Demba Sarr1† Kayla Fantone1
Kayla Fantone1 Nuha Milad Ashtiwi1
Nuha Milad Ashtiwi1 Kaori Sakamoto2
Kaori Sakamoto2 Frederick D. Quinn1‡
Frederick D. Quinn1‡ Balázs Rada1*‡
Balázs Rada1*‡- 1Department of Infectious Diseases, College of Veterinary Medicine, University of Georgia, Athens, GA, United States
- 2Department of Pathology, College of Veterinary Medicine, University of Georgia, Athens, GA, United States
Introduction: Mycobacterium tuberculosis (Mtb) is the primary cause of human tuberculosis (TB) and is currently the second most common cause of death due to a singleinfectious agent. The first line of defense against airborne pathogens, including Mtb, is the respiratory epithelium. One of the innate defenses used by respiratory epithelial cells to prevent microbial infection is an oxidative antimicrobial system consisting of the proteins, lactoperoxidase (LPO) and Dual oxidase 1 (Duox1), the thiocyanate anion (SCN-) and hydrogen peroxide (H2O2), which together lead to the generation of antimicrobial hypothiocyanite (OSCN-) in the airway lumen. OSCN- kills bacteria and viruses in vitro, but the role of this Duox1-based system in bacterial infections in vivo remains largely unknown. The goal of this study was to assess whether Duox1 contributes to the immune response against the unique respiratory pathogen, Mtb.
Methods: Duox1-deficient (Duox1 KO) and wild-type (WT) mice were infected with Mtb aerosols and bacterial titers, lung pathology, cytokines and immune cell recruitment were assessed.
Results and discussion: Mtb titers in the lung, spleen and liver were not different 30 days after infection between WT and Duox1 KO mice. Duox1 did not affect lung histology assessed at days 0, 30, and 90 post-Mtb infection. Mtb-infected Duox1 KO animals exhibited enhanced production of certain cytokines and chemokines in the airway; however, this response was not associated with significantly higher numbers of macrophages or neutrophils in the lung. B cell numbers were lower, while apoptosis was higher in the pulmonary lesions of Mtb-infected Duox1 KO mice compared to infected WT animals. Taken together, these data demonstrate that while Duox1 might influence leukocyte recruitment to inflammatory cell aggregates, Duox1 is dispensable for the overall clinical course of Mtb lung infection in a mouse model.
Introduction
Tuberculosis (TB) caused by Mycobacterium tuberculosis (Mtb) continues to be a major global disease annually affecting millions worldwide. An estimated 10 million people developed active TB in 2018, with 1.2 million deaths among HIV-negative people and an additional 251,000 deaths among HIV-positive individuals (1). There are no effective vaccines against pulmonary TB in adults and no biomarkers for protective immunity (2–7). While there are drugs that are effective against Mtb, because of the long-term treatment regimen, patient non-compliance is common leading to development of drug-resistant strains (4).
The earliest encounter between Mtb bacteria and the host occurs at the interface between innate immune cells and the bacilli (8). While innate immunity is critical for early anti-mycobacterial responses, it is also important for the progression of infection and long-term control of Mtb. It has long been recognized that immunity to Mtb can provide both protection and cause tissue damage (8). The pathogenesis of TB begins with inhalation of the bacilli and their interaction with cells in the respiratory tract, and is ultimately mediated by responses of the innate and adaptive immune systems (9, 10). The different steps involve macrophages, dendritic cells, pathogen-associated molecular patterns (PAMPs), pattern recognition receptors (PPRs), cytokines, and T cells, as well as neutrophils and monocytes (8–10).
Additionally, several Mtb gene products are known to interact with airway epithelial cells – heparin-binding hemagglutinin (HBHA) and Rv3351c (11, 12). HBHA is a major adhesin molecule involved in binding of Mtb to the epithelial cells, with ultimate necrosis of these cells likely contributing to extrapulmonary dissemination to various organs. Additionally, our group demonstrated that Mtb Rv3351c is involved in maximally effective entry and trafficking within alveolar epithelial cells (12). However, the role of epithelial cells in innate immunity against Mtb is at an early stage of investigation and the involvement of components of the airway surface liquid (ASL) has mostly been ignored. It is known that upper airway epithelial cells are inert to direct Mtb interaction; however, they release several chemokines and defensins that likely help modify the local environment along with infected alveolar macrophages (13).
Bronchial epithelial cells orchestrate an oxidative extracellular antimicrobial system present in the ASL consisting of the protein lactoperoxidase (LPO), the thiocyanate anion (SCN-), and hydrogen peroxide (H2O2) generated by the NADPH oxidase enzyme Dual oxidase 1 (Duox1) [reviewed by (14)]. LPO oxidizes SCN- into microbicidal hypothiocyanite (OSCN-) in the presence of H2O2 killing a wide variety of viruses, bacteria, fungi and parasites in vitro (15–19). Mtb has not been investigated yet for its in vitro susceptibility to OSCN-. We have recently shown the relevance of Duox1 in antiviral innate immunity in a murine model of influenza infection (20). Mice with global Duox1 deficiency had significantly higher susceptibility to influenza A lung infection than wild-type animals. Our recent in vivo investigations have shown that both SCN- and LPO are present in the airway surface liquid of both Duox1 KO and WT mice (20). This antimicrobial system has long been characterized in vitro, but its relevance in vivo has remained unknown until recently. Because DUOX1 is highly expressed by bronchial epithelial cells and is the main H2O2 provider for the LPO/H2O2/SCN- - antimicrobial system (21–24), we hypothesized that Duox1 KO mice will have higher bacterial loads and more severe lung pathology than wild-type (WT) animals following Mtb infection.
Our data indicate that both Duox1 KO and WT mice have significantly higher inflammation in the lung tissue and airways at 4 weeks post-infection upon Mtb infection compared to mice at day 1 post-Mtb infection. Surprisingly, Duox1 does not have a protective role during Mtb infection in vivo. These data indicate that Duox1 is overall dispensable in Mtb infection in vivo.
Materials and methods
Mice
All mouse procedures were approved by the Animal Care and Use Committee at The University of Georgia (UGA). Duox1 KO and C57BL/6J (WT) mouse colonies were maintained in the Coverdell vivarium at The University of Georgia. The Duox1-deficient mice were established on a B6 background by retroviral-based gene-trapping methodology (25, 26) by Lexicon Pharmaceuticals. We thank the company and Miklós Geiszt (Semmelweiss University, Budapest, Hungary), the sole academic distributor, for providing the Duox1 KO mouse strain. Mice were housed with a standard diet and given water add libitum in a specific pathogen-free facility, according to The University of Georgia Animal Use Protocol Guidelines.
One week before infection, mice were transferred to the Animal Health Research Center, an animal biosafety level 3 (ABSL-3) facility, in order to perform aerosolized Mtb infections. Both male and female mice between the ages of 6 to 12 weeks were used in the study. All animals were housed with 12-hour light/dark cycle, ad libitum mouse chow and water, and provided environmental enrichment. Animals were examined at least daily for signs of distress and discomfort using a scoring system (weight loss 10–20% = 1 point, cyanosis = 1point, mild lethargy = 1 point, hunched back = 1 point, weight loss > 20% = 3 points, dyspnea = 3 points) in which any animal accumulating 3 points is euthanized.
Mtb culture and infection
The seed stocks of Mtb strain Erdman were cultured as pellicles in Proskauer Beck broth medium. Oleic Albumin Dextrose Catalase enrichment was used in the agar plates. The working Mtb stocks used to infect mice were prepared as described previously (27). A stock was used as an inoculum to infect mice with Mtb aerosols generated within a Madison chamber. This chamber has been calibrated to deliver 200 CFU of nebulized bacteria to the lung of the mice during a 15-minute exposure window as previously described (27). The Madison chamber holds 18 cages of mice in a carousel, and all animals per experiment from each genotype (WT and Duox1 KO) were randomly dispersed in the carousels in one single run to control for possible variability of Mtb exposure within the Madison chamber. Each experiment included 15 males and 15 females from each genotype.
Euthanasia and tissue collection
At 1, 30, and 90 days post-infection, mice were euthanized by CO2 asphyxiation, and blood was collected by cardiac puncture. Bronchoalveolar lavage (BAL) fluid was also collected using 1 ml of sterile phosphate-buffered saline (1X PBS). These samples were stored at 4°C until assayed. The lungs were perfused with sterile saline. Whole lungs, spleen and mediastinal lymph nodes (MLN) were collected in RPMI and kept at 4°C until lung leukocytes were isolated and stained. From another set of animals, a portion of the lung (right lobe) was collected (equal number of males and females from each genotype) to determine bacterial load (CFUs) and another piece (left lobe) was fixed with 10% neutral-buffered formalin for histology. Liver and spleen were also collected to determine dissemination and trachea was fixed in 10% buffered formalin for histology and further analysis by immunofluorescence and/or immunohistochemistry.
Enumeration of bacterial load
Lung, liver and spleen from infected mice were aseptically collected in Whirl-Pak bags (Cat# B01062, Nasco, VWR, Radnor, PA) and frozen at -80°C. For plating, the tissues were resuspended in sterile 1X PBS and hand-homogenized. Serial dilutions of the lysates were plated on Middlebrook 7H11 agar plates supplemented with 0.5% glycerol, 10% albumin, dextrose and saline (ADS), 0.05% Tween 80 (7H11gtADS) and incubated at 37°C for 3-6 weeks. The colonies were counted and bacterial load in each organ calculated and graphed on a log10 scale.
Lung single cell suspension
The lungs were chopped in Petri dishes with a sterile scalpel and incubated at 37°C with a digestion medium containing collagenase (Cat#07912, Stemcell Technologies, Cambridge, MA) and DNAse I (Cat# 07900, StemCell Technologies, Cambridge, MA) in a shaking platform. Cell suspensions were passed through a 70 µm nylon mesh strainer before counting and staining with labeled antibodies.
Spleen and mediastinal lymph node single cell preparation
Single cell suspension of the spleen and MLN were prepared by passing hand-homogenized tissues through a 70 µm nylon mesh. The cells were spun at 350 x g for 10 minutes, the cell pellet was resuspended in ACK lysing buffer (Cat# 10-548E; Lonza, Walkersville MO) for 30 seconds. The cells were washed with 1X PBS, resuspended, counted and stained with a viability dye followed by labeled antibody cocktail as above.
Cytokine quantification
The Bio-Plex Pro Mouse Chemokine Panel 31-plex magnetic bead-based assay was used to measure levels of 31 different mouse cytokines and chemokines in Mtb-infected, cell-free BAL fluid (collected on 1 and 30 day post infection, centrifuged, and stored at -80°C as indicated above) using internal standards (Cat#:12009159, Bio-Rad, Hercules, CA) following manufacturer’s instructions and as previously described (20, 28). Samples were read with the Bio-Plex 3D analyzer (Bio-Rad Inc., Hercules, CA) and data were exported to Excel before statistical analysis.
Flow cytometry
The antibodies used here are listed in Table 1. BAL cells, splenocytes, MLN and lung single cell suspensions were washed once in 1X PBS, and leukocytes were resuspended in 1 ml sterile 1X PBS, diluted when necessary and counted with an automatic cell counter Countess TM Automated Cell Counter, Invitrogen. For viability measurement, 1X106 cells were suspended in 100 µl of 1X PBS and stained first with Zombie Aqua™ flexible viability dye (Cat#: 423101; BioLegend, San Diego, CA) at room temperature in the dark for 15 minutes. Following a washing step, the cells were resuspended in 100 µl of 1X PBS containing 1% bovine serum albumin and blocked using TruStain FcX™ anti-mouse (CD16/32) antibody (Cat#: 101320; BioLegend, San Diego, CA) on ice and protected from light for 10 minutes. Multicolor cell surface staining was performed separately using two different antibody cocktails for the characterization of lymphoid (CD45, CD3, CD4, CD8, and NK1.1) and myeloid cells (CD11b, CD11c, F4/80, Ly6G, Ly6C, and CD115) as previously described (20, 28, 29). The cells were subsequently stained with surface antibodies (Table 1) at a 1:100 dilution at the same time to detect the number of cells from each of the myeloid and lymphoid cell subsets following the protocol as described previously (20). Samples were read within 12 hours using the NovoCyte Flow Cytometer (Agilent Technologies, Santa Clara, CA) with the NovoExpress® software at the University of Georgia College of Veterinary Medicine Cytometry Core Facility. Data acquisition was followed by analysis using FlowJo v.10.8.1 (BD Biosciences, San Jose, CA) with high-dimensional data analysis algorithms using FlowJo plugins. Flow data were also analyzed using a previously optimized gating strategy for identification of the cell populations of interest (20). Myeloid cell clustering was also assessed by uniform manifold approximation and projection (UMAP) analysis.
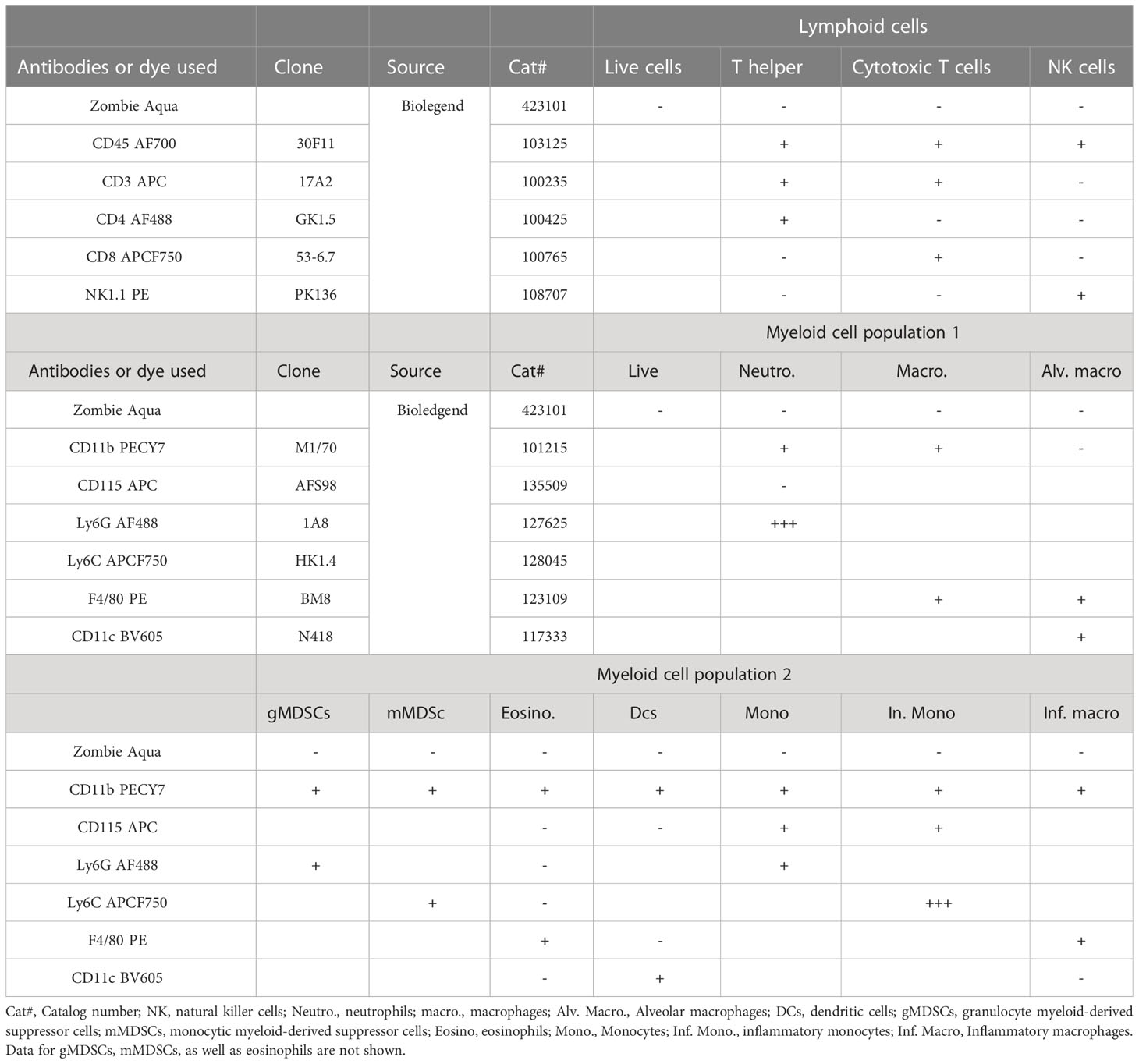
Table 1 Summary table of the antibodies used for multicolor flow cytometry in this study and the gating strategies that were applied for the data analysis.
Histopathology
Duox1 KO and WT mice were sacrificed as indicated above. Lungs were injected with 1 ml of 10% neutral-buffered formalin (NBF) and stored in 10% NBF. Formalin-fixed, paraffin-embedded lung tissue blocks were prepared by the Histology Laboratory at the University of Georgia College of Veterinary Medicine. Lung tissue sections (5-µm thickness) were either left unstained for subsequent immunofluorescence or immunohistochemistry or stained with hematoxylin and eosin (H&E) stains for histopathology. Histopathology was performed by a board-certified, veterinary pathologist who was blinded to the mouse genotype and experimental conditions. In addition to the number of inflammatory cell aggregates (“granulomas”) histologically distinguishable in the section, the percent of affected lung tissue was estimated. Scoring for alveolar infiltrates, alveolar edema, necrosis, and pleuritis was on a scale of 0 to 4 based on distribution (0 = no lesions, 1 = focal, 2 = multifocal, 3 = coalescing, 4 = diffuse). The neutrophil score was based on percentage of neutrophils within alveolar infiltrates (0 = no neutrophils, 1 = up to 25% of cells, 2 = 26-50%, 3 = 51-75%, 4 = 76-100%). The perivascular cuffing (PVC) score was based on thickness of leukocytes around blood vessels (0 = no cuffing, 1 = 1 layer of cells, 2 = 2-5 layers, 3 = 6-10, 4 = greater than 10). The vasculitis score was based on severity of the lesion (0 = no inflammation, 1 = vessel wall infiltrated by cells, 2 = smooth myofiber separation and cellular infiltration, 3 = changes in 2 + fibrinoid change, 4 = effacement of the vessel wall). The interstitial pneumonia score was based on thickness of alveolar septa (0 = no infiltration, 1 = 1 leukocyte thickness, 2 = 2 leukocytes thick, 3 = 3 leukocytes thick, 4 = 4 leukocytes thick).
Immunofluorescence
Unstained lung tissue sections were deparaffinized and rehydrated with a xylene and alcohol gradient. Sections were antigen-retrieved with 0.1 M sodium citrate in a Pascal Pressure Cooker (Agilent Technologies, Santa Clara, CA) for 20 minutes at 95°C, and then permeabilized with 0.1% Triton X-100 while blocking in PBS with 5% BSA and 10% normal horse serum for 1 hour. Primary antibody was used for detection of Mtb bacteria (cat# NR-13820; BEI Resources, Manassas, VA). In some cases, anti-CD45R (Cat#: 550286, BD Biosciences, San Jose, CA) anti-CD68 (Cat#: ab955; Abcam; Waltham MA) and anti-cleaved caspase-3 (Cat#: 9661, Cell Signaling Technologies, Danvers, MA) antibodies were also applied to assess B cells and apoptosis, respectively. In each case, the primary antibody dilution was optimized, and all dilutions and antibody incubations were done in 1X TBST with 0.5% BSA, 1% normal horse serum, and 0.01% Triton X100 overnight at 4°C. Sections were washed the very next morning using 1X TBST, and secondary antibody staining was performed at a 1:1,000 dilution in 1X TBST with 0.05% BSA, 0.1% normal horse serum for 1 hour with horse anti-rabbit IgG antibody (H+L) conjugated with FITC (Vector Laboratories, cat#: FI-2000-1.5). Sections were washed with 1X-TBST, and then Vectashield™ anti-fade mounting medium with DAPI (Cat#: H-1200-10, Vector Laboratories, Burlingame, CA) was applied to sections before adding the coverslip. All digital images were acquired on a Nikon A1R confocal microscope (Nikon Eclipse Ti-E inverted microscope) and examined with NIS Element software (Nikon, Version 6.4). Fluorescence intensity was quantified using ImageJ (NIH, version 1.8.0), and data were exported to Excel and analyzed for statistical significance and plotting using Graph Pad Prism 9.2.0. 3-5 sections per mouse were analyzed.
Immunohistochemistry
The expression pattern of several host proteins in the mouse lung was also assessed by immunohistochemistry using the following primary antibodies: anti-cleaved caspase 3 (Cat# 9661, Cell Signaling, Danvers, MA), anti-myeloperoxidase (Cat# PA5-16672; Invitrogen, Waltham, MA), anti-neutrophil elastase (Cat# PA5-87158; Invitrogen, Waltham, MA), and anti-CD45R (Cat: 550286; BD Pharmingen; San Diego, CA).
The unstained sections of lung tissues from Mtb-infected WT and Duox1 KO mice were deparaffinized and rehydrated as above. After antigen-retrieval, endogenous peroxidase blocking was performed by incubating tissue sections with Dual Endogenous Enzyme Block (Cat# S2003, Agilent Technologies, Santa Clara, CA). Sections were permeabilized in 1X TBST containing 0.5% Triton X-100 for 5 minutes followed by blocking in Rodent Block (Cat# RBM961, Biocare Medical, Pacheco, CA). Tissue sections were then incubated in primary antibodies overnight followed by appropriate Polymer HRP-conjugated secondary antibodies (Cat# D13-18 for rabbit primary antibody or D13-52 for mouse primary antibody, GBI Labs Golden Bridge International, Bothell, WA) followed by a 5-minute 3,3′-Diaminobenzidine (DAB) treatment. Hematoxylin (Cat# H3401, Vector Laboratories, Newark, CA) and Acrytol Mounting Medium (Cat# 13158, EMS, Hatfield, PA)), were used for counterstaining and mounting, respectively. DAB signal quantification was done using ImageJ software (NIH, Bethesda, MA).
Myeloperoxidase and neutrophil elastase ELISA
Using BAL fluid, sera and lung lysates from WT and Duox1 KO mice, we quantified MPO and NE using DuoSet ELISA kits (MPO; Cat# 3667; NE; Cat#4517-05, R&D Systems, Minneapolis, MN) following the manufacturer’s instructions. Primary antibody against MPO (800 ng/ml) or NE (1 μg/ml) was added to 96-well plates overnight at room temperature. Plates were washed, samples and recombinant protein standards (ranging from 250-16,000 pg/ml for MPO and 12.5-800 pg/ml for NE) were added to the plates overnight at 4°C. Plates were again washed and detection antibodies were added for 2 hours at room temperature at concentrations of 50 ng/ml for anti-MPO and 100 ng/ml of anti-NE, respectively. Following incubation, the plates were washed and streptavidin-HRP was added for 20 minutes at room temperature at a 200-fold dilution for MPO and 40-fold dilution for NE. Substrate solution was added at a 1:1 mixture of H2O2 and tetramethylbenzidine. Once the reaction was complete, 1N HCl was added to stop the reaction and absorbance at 450 nm was used to quantify the respective protein in pg/ml.
Neutrophil elastase activity assay
Enzymatic activity of NE was evaluated using the NE activity assay kit (Cat#600610; Cayman Chemical; Ann Arbor, MI) as previously described (28). Neutrophil elastase at 18 mU/ml was used to make a standard curve ranging from 0.156 mU/ml to 10 mU/ml. Standards and samples were added to a black, flat-bottom, 96-well plate. Ten μl of substrate solution consisting of Cell-Based Assay DMF and (Z-Ala-Ala-Ala-Ala)2Rh110 was added to each standard and sample, which is selectively cleaved by elastase to yield the highly fluorescent compound R110. Fluorescence was analyzed with an excitation wavelength of 485 nm and an emission wavelength of 525 nm using a microplate fluorimeter (Varioskan Flash, Thermo Scientific). Kinetic fluorescent measurements were taken every 2 minutes for 2 hours at 37°C. Average fluorescence was taken for each well and quantified in mU/ml with the help of the NE standard.
Statistical analysis
CFU data were log10 transformed before statistical analysis. GraphPad Prism Software 9.2.0 was used to carry out all statistical analysis. Quantified data were either analyzed between groups with unpaired Student’s t test or Mann-Whitney U test as appropriate. In some quantified data, statistical analysis required One-way ANOVA with Tukey’s post-hoc multiple comparison (for comparing three or more groups). Data are expressed as mean ± S.E.M. where *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001 indicates the level of significance. Relevant information, such as test used, the number of animals/samples, and the p values obtained, are given for each figure in the figure legend.
Results
Mtb growth in vivo is Duox1-independent
In our previous study we showed that wild-type mice express Duox1 in their airway epithelium and have elevated H2O2 levels in their bronchoalveolar lavage compared to Duox1-deficient animals (20). To evaluate the effect of a functional Duox1 on Mtb infection, we infected male and female Duox1 KO and C57BL/6J (WT) mice. Following Mtb aerogenic infection, bacterial colony forming units (CFUs) at 1, 30, and 90 days post-infection were measured in the lung, liver and spleen to evaluate bacterial growth and dissemination. Our results show that, compared to WT mice, CFUs in Mtb-infected Duox1 KO mice were not significantly different in the lung, spleen or liver at days 1, 30 or 90 post-infection, respectively (Figures 1A, B). To further evaluate the effects of Duox1 on bacterial growth inside the lung, Mtb-infected WT and Duox1 KO mouse lungs were sectioned and stained with a polyclonal anti-Mtb antibody. Bacteria were not detected inside of the lung tissue at 1 day post-infection, as expected, in both WT and Duox1 KO animals (Figure 2A). However, at 30 days post-infection, bacteria (green) are present in inflammatory cell aggregates (Figure 2A). Quantification of green fluorescence indicative of Mtb burden did not yield significant differences between WT and Duox1 KO mice neither at 1 nor 30 day(s) post-infection (Figures 2B, C). Despite our initial hypothesis, CFU data from the lung, spleen, and liver, and visual quantification of the bacteria inside the lungs suggest that Mtb lung infection does not depend on host Duox1 expression.
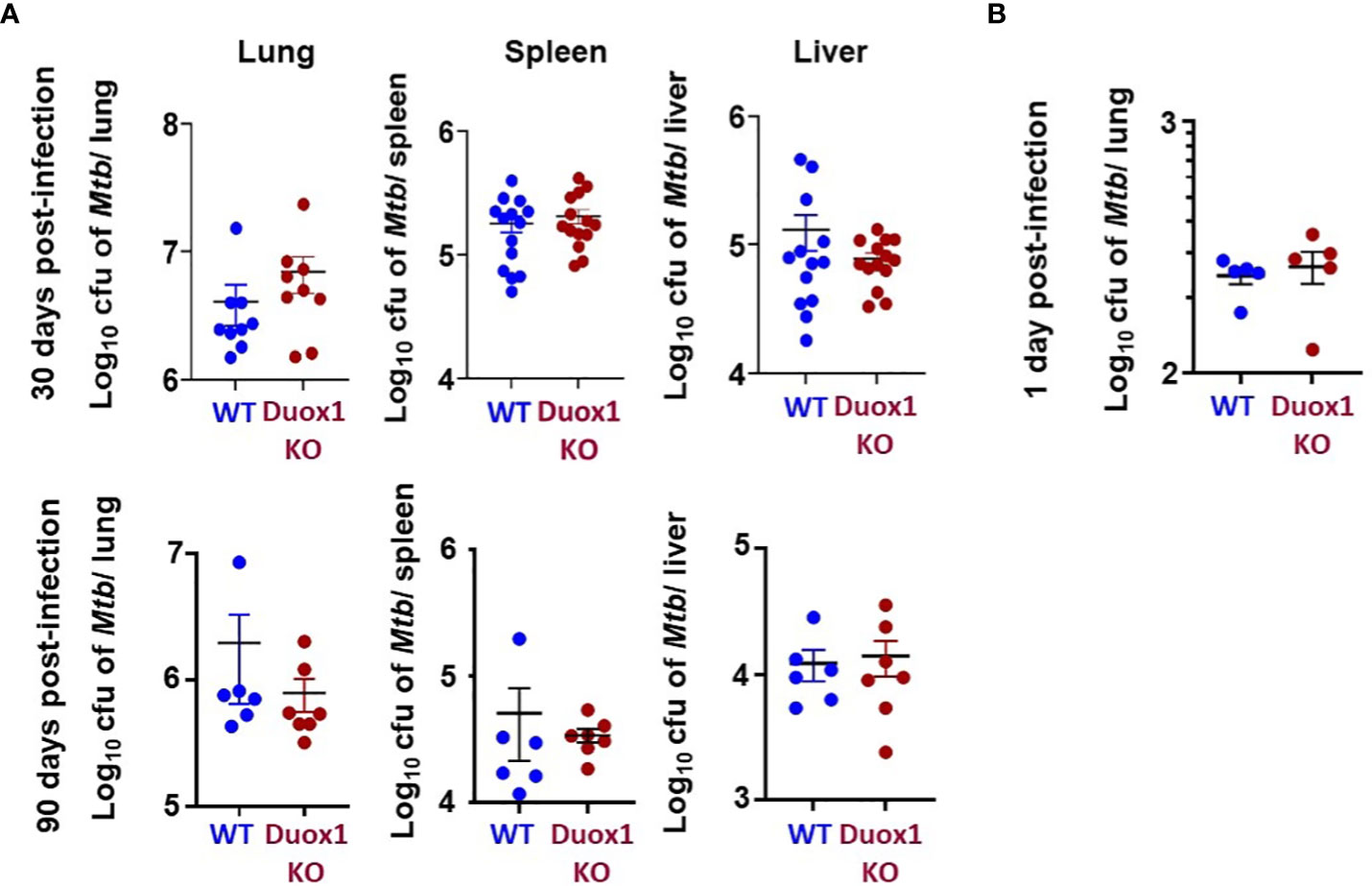
Figure 1 Mtb growth in vivo in Duox1 KO mice. Mature Duox1 KO and WT mice were aerogenically infected with 200 CFU of Mtb (Erdman). (A) On 30 and 90 days post-Mtb infection, mice from each genotype were euthanized and lungs (n=9), spleens (n=14) and livers (n=14) were removed for CFU burden assessment by colony counting. (B) On 1 day post-infection, lungs from mice (n=5) of each genotype were removed for CFU counts. Mann-Whitney U test was used to compare CFUs between animal groups. Mean+/-S.E.M. CFU, colony-forming unit; Duox1 KO: Duox1-deficient; WT, wild-type.
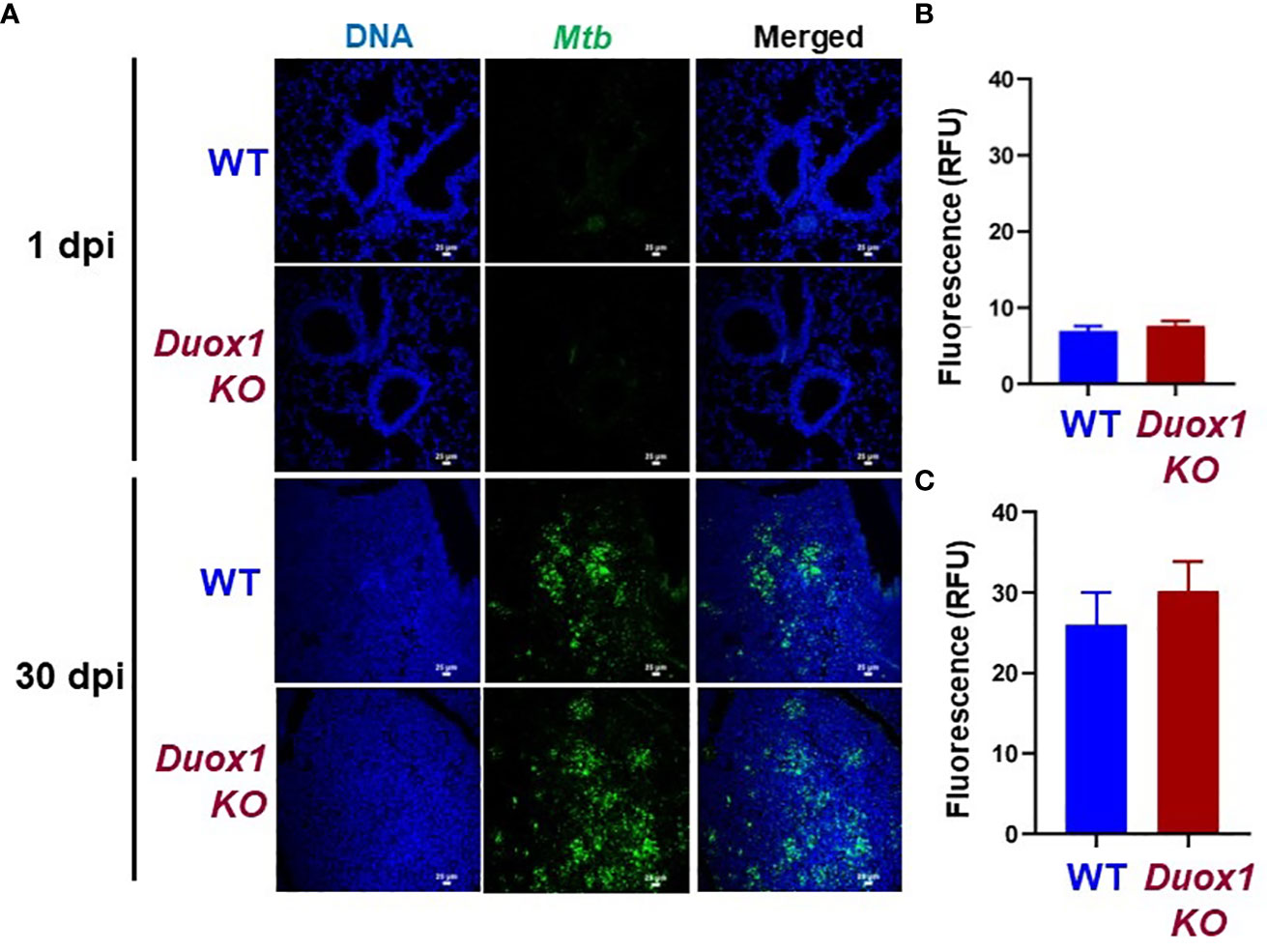
Figure 2 Duox1 does not affect Mtb proliferation in the lung. Lungs from Mtb-infected Duox1 KO and WT mice were harvested at 1 and 30 day(s) post-infection, respectively. Fixed and processed lung tissues were stained with primary anti-Mtb antibody followed by a secondary AF488 antibody. (A) Representative fluorescent images of six similar results are shown. White scale bars indicate 25 μm. Mean green fluorescent intensities (MFI) indicative of Mtb burden were quantified for (B) 1 day post-infection and (C) 30 days post-infection in Duox1 KO (n=6) and WT mice (n=6). Error bars represent ± SEM. Mann Whitney U-test. Duox1 KO: Duox1-deficient; WT, wild-type; RFU, relative fluorescence unit.
Lung histopathology during Mtb infection is not affected by Duox1
Since we hypothesized that Duox1 could affect lung pathology during Mtb infection, lungs of Mtb-infected Duox1 KO and WT mice collected at 1, 30, and 90 day(s) post-infection, were stained with hematoxylin and eosin (Figures 3A-C for WT and Figures 3D-F for Duox1 KO mice) and examined by a board-certified veterinary pathologist blinded to the experimental groups. The percent of lung affected (Figure 3G), the number of “granulomas” or inflammatory cell aggregates (Figure 3H), the extent of alveolar infiltration (Figures 3I, J) and necrosis (Figure 3K), the neutrophil score (Figure 3L), perivascular infiltration (Figure 3M), vasculitis (Figure 3N), interstitial pneumonia (Figure 3O), and pleuritis (Figure 3P) were all not different between Duox1 KO and WT mice on any of the days assessed (1, 30, 90 day(s) post-infection). However, all parameters were significantly higher at 30 or 90 days post-infection compared to 1 day post-infection, suggesting that infection was chronically established in mice, irrespective of their genetic background. Overall, the analysis suggested that Duox1 does not affect lung pathology during Mtb infection in vivo.
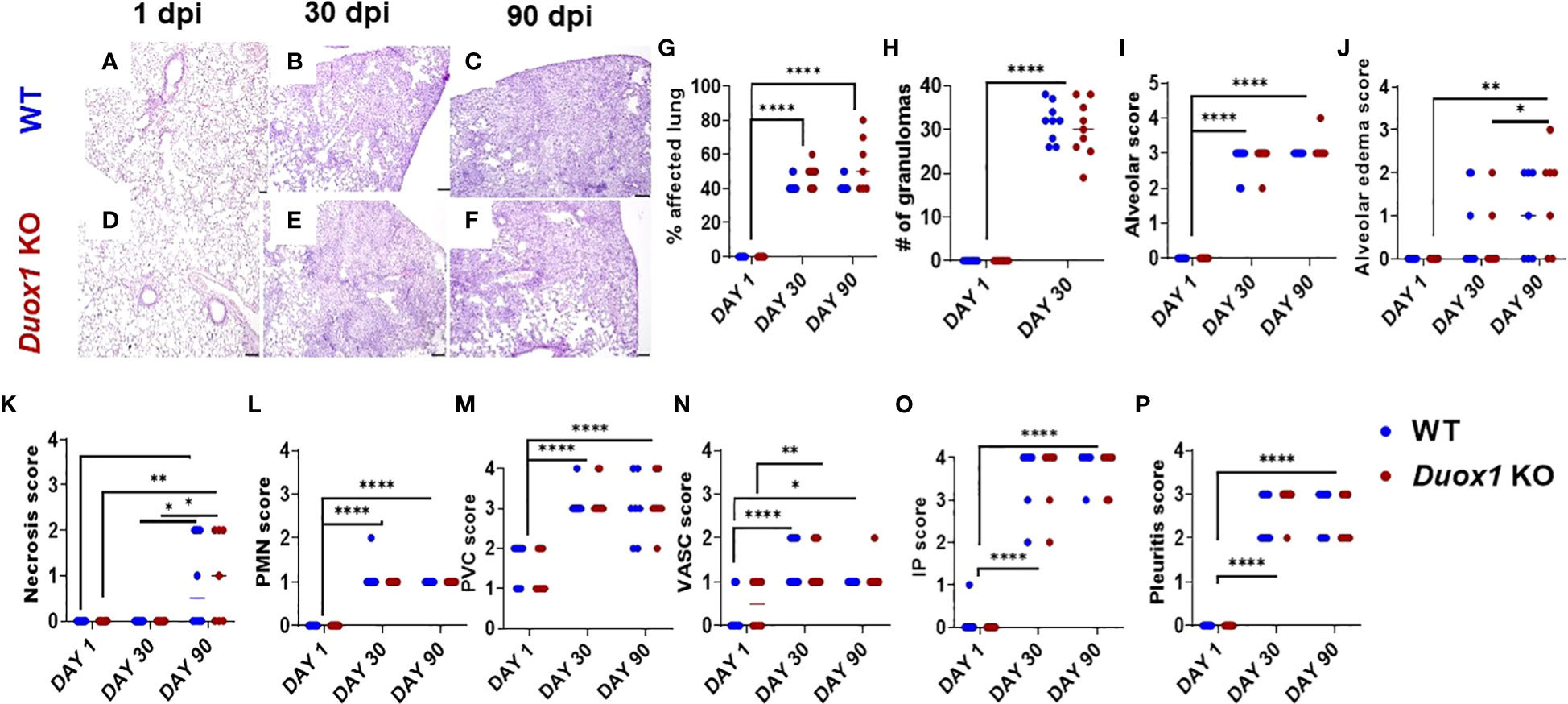
Figure 3 Lung pathology during Mtb infection is independent of Duox1. Mtb-infected lung lobes from both Duox1 KO and WT mice were isolated and fixed in 10% buffered formalin at 1, 30, and 90 day(s) post-infection. Fixed lung tissues were processed for histology, sectioned, and subjected to hematoxylin and eosin staining. Lungs collected at 1 day post-infection from both mouse background (A, D) do not show any evidences of lung pathology while granuloma-like lesions in both groups are visible at 30 (B, E) and 90 (C, F) days post-infection. (G) Comparative percentage of lung tissue affected (0% to 80%). (H) The number of inflammatory aggregates inside the lung tissues (from 0 to 40 maximum). The lung histological score was assessed with a scale from 0 to 4 and included the (I) alveoli, (J) the alveolar edema, (K) necrosis, and (L) polymorphonuclear neutrophils as well as (M) perivascular score. (N) The vascular, (O) IP score and (P) pleuritis are also shown. Sample sizes: n=8 Duox1 KO; n=8 WT mice for histopathological scoring at 1 day post-infection. For 30 days post-infection: n=9 Duox1 KO and n=9 WT mice were used. For 90 days post-infection, n=6 WT and n= 7 Duox1 KO animals were assessed. ANOVA with multiple comparison was used for statistical data analysis. *p<0.05, **p<0.01, ****p<0.0001. IP, intraperitoneal, WT, wild-type; day post infection, days post-infection; PMN, polymorphonuclear neutrophil granulocyte; Duox1 KO, Duox1-deficient; WT, wild-type.
Duox1 influences the cytokine environment in the lung during Mtb infection
A lack of any effect of Duox1 on lung pathology does not mean that Duox1 could not have a role in Mtb infection. To further investigate a possible in vivo role for Duox1 in the immune response to Mtb infection, pro- and anti-inflammatory cytokines/chemokines in the cell-free BAL fluid from both Mtb-infected WT and Duox1 KO mice were quantified using a bioplex beads array. Duox1 expression has been detected by us previously in human macrophages and Duox1 could alter macrophage functions including proinflammatory cytokine release (30). None of the cytokines/chemokines was significantly different between WT and Duox1 KO at 1 day post-infection (Supplementary Figures 1, 2). However, at 30 days post-infection, TNF-α, IL-6, KC, CCL3, CCL2, CCL20, CXCL11, CCL11), CCL27, CXCL5, CXCL12 and CCL5 were all significantly higher in the BAL fluid of Duox1 KO compared to WT mice (Supplementary Figure 1). Only CXCL16 and CCL1 were significantly higher in WT compared to Duox1 KO mice at 30 days post-infection (Supplementary Figure 1), while additional cytokines/chemokines showed no significant differences between WT and Duox1 KO mice (Supplementary Figure 2). Overall, our data suggest a potential role of Duox1 in shaping the localized immune response in Mtb infection in vivo.
Immune cell recruitment to the airways during Mtb infection does not depend on Duox1
Because Duox1 may have a role in the immune responses against Mtb infection in vivo, we next used multicolor flow cytometry to determine the phenotype of immune cells recruited in the airway and the spleen using a previously optimized protocol (20, 28). We determined previously that there are no significant differences in the nature of leukocytes in the airway of uninfected, naïve, WT and Duox1 KO mice (20). The protocol used markers that identifiy lymphoid and myeloid cell subsets. Overall, our data show that there are no significant differences in the percent of cells in the spleen, lung, BAL, and mediastinal lymph node (MLN) for both major lymphoid and myeloid cell subsets in Mtb-infected WT versus Duox1 KO animals (Figures 4A, D; Supplementary Figures 4, 5, 6A, D). To further analyze the multicolor flow data, we used high-dimensional algorithms with FlowJo plugins. All data were analyzed by t-sne analysis (Figure 4 and Supplementary Figures 4-6B, C, E F) while lung myeloid cell populations were also clustered by uniform manifold approximation and projection (UMAP) for comparison (Supplementary Figure 3). Gating overlay of lymphoid cell subsets in t-sne dimension 1 and 2 indicated lower density of CD4 and CD45 clusters in the spleen (Supplementary Figures 4E, F) at 30 days post-infection, while the density of clusters remained identical for WT and Duox1 KO at 1 day post-infection (Supplementary Figures 4B, C). However, the density of CD4+ cells inside the T cell cluster is lower in Duox1 KO at 30 day post infection (Supplementary Figure 3F) compared to WT (Supplementary Figure 4E). Like lymphoid cells, the myeloid cell clusters in the spleen also show different clustering, especially for neutrophils (Supplementary Figures 5E, F). However, these relative differences are not seen in the entire lung single cell suspension at neither 1 (Supplementary Figures 5B, C), nor 30 day(s) post-infection (Supplementary Figures 5E, F). In the BAL and mediastinal lymph node (MLN), no apparent differences between myeloid clusters were observed (Supplementary Figures 6B, C, E, F). Overall, Duox1 does not significantly affect the immune cell composition in the studied organs.
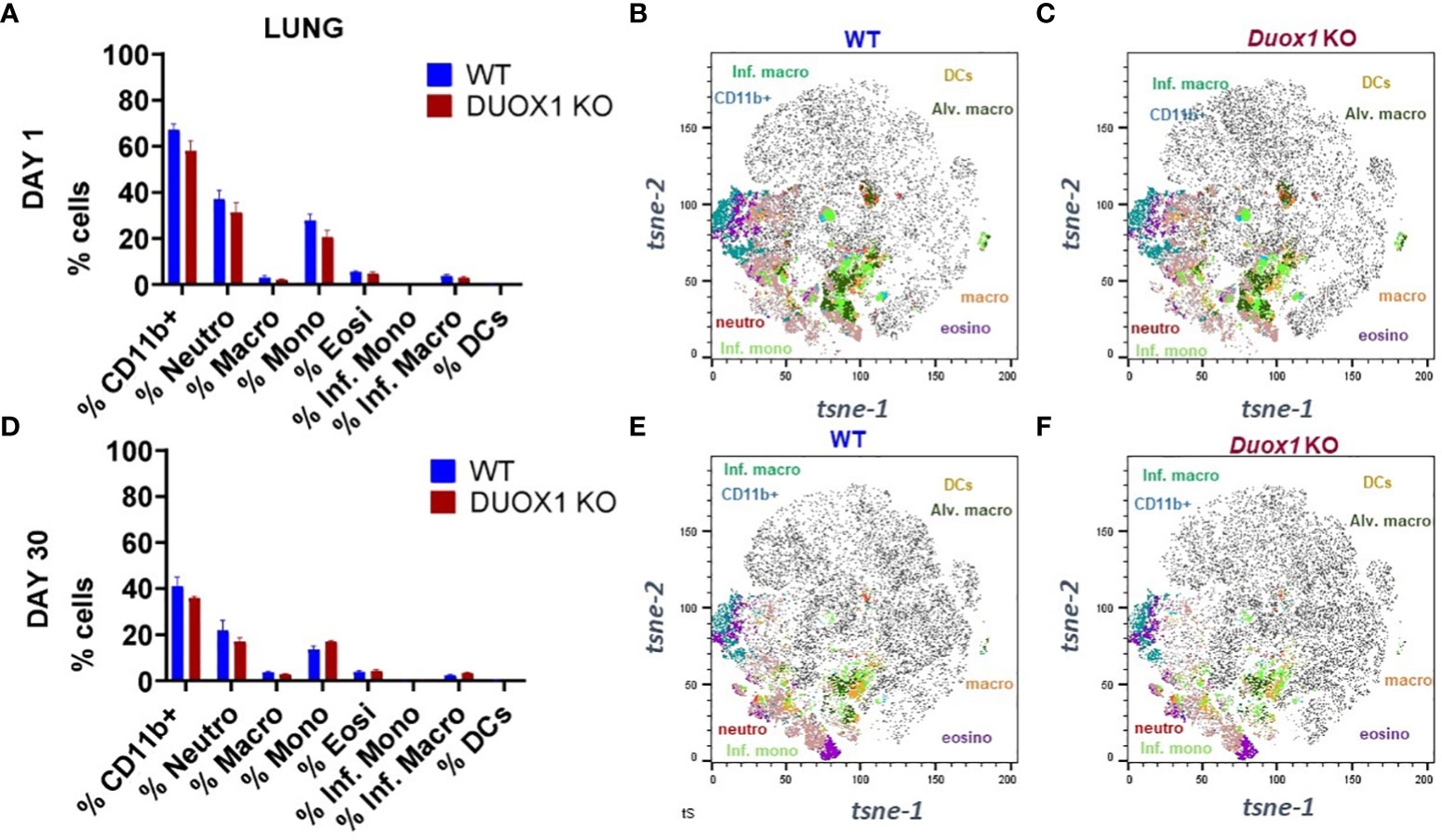
Figure 4 Myeloid cell populations in the lung tissues of Mtb-infected WT and Duox1 KO mice. (A) Mean observed cell proportions determined by flow cytometry in lung lysates from two independent experiments (n= 5 per mouse genotype) of Mtb-infected WT mice at 1 day post-infection. (B) Marker expression by t-sne in Mtb-infected WT mice at 1 day post-infection. (C-F) Marker expressions by t-sne in Mtb-infected Duox1 KO and WT mice at 1 and 30 day(s) post-infection as indicated. Duox1 KO, Duox1-deficient; WT, wild-type.
Airway levels of neutrophil markers increase during Mtb infection in a Duox1-independent fashion
Phagocytic cells (i.e., neutrophils and macrophages) are the predominant “warriors” of Mtb-induced inflammatory responses that play important roles in immune defense but also lung pathology (31, 32). Macrophages act as the first line of defense against Mtb inside the lung (33). The heme protein myeloperoxidase (MPO) is an active major component of neutrophils that can also be found at lower quantities in monocytes and inflammatory macrophages (34, 35). In the presence of H2O2, MPO generates hypochlorous acid (HOCl) using chloride (35). We have shown that naïve, uninfected Duox1 KO mice have significantly lower levels of H2O2 in their BAL fluid than WT mice (20). To assess a possible Duox1-dependent change in MPO levels following Mtb infection, we collected BAL fluids, sera and lung homogenates from Mtb-infected Duox1 KO and WT mice at 1 and 30 day(s) post-infection. Our data show that MPO increases, mainly in the BAL, over time with Mtb infection, but the differences between Mtb-infected WT and Duox1 KO mice were not statistically significant in the BAL (Figure 5A), sera (Figure 5B), or lung lysates (Figure 5C).
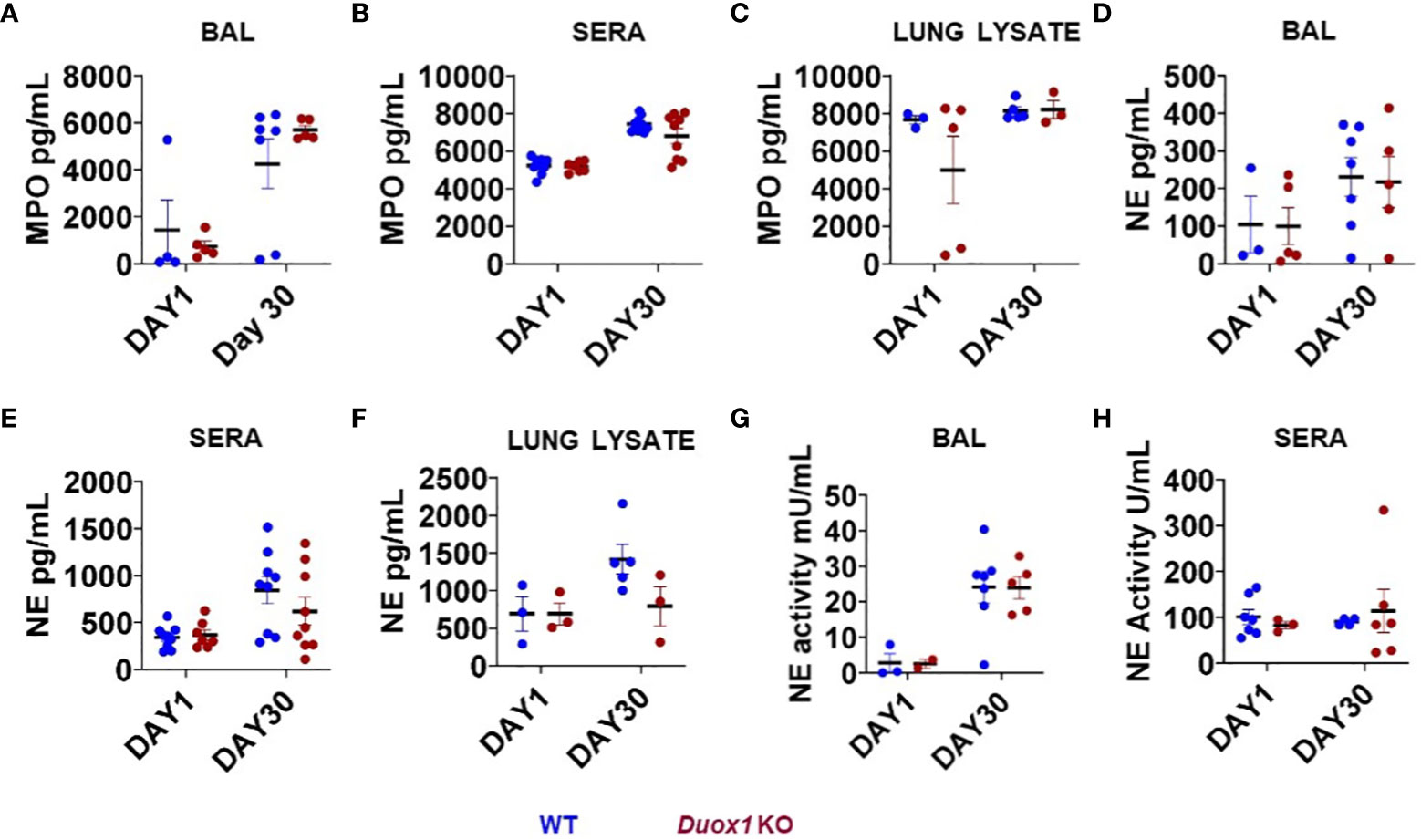
Figure 5 Neutrophil markers in the blood and airways of Mtb-infected Duox1 KO mice. The concentrations of MPO and NE were measured in the BAL fluid, sera and lung lysates from Mtb-infected WT and Duox1 KO mice at 1 and 30 days post-infection. NE enzymatic activities in cell-free BAL fluid and sera are also shown. The following parameters were measured at 1 and 30 day(s) post-infection. (A) BAL MPO, (B) serum MPO, (C) MPO in lung lysates, (D) BAL NE, (E) serum NE, (F) NE in lung lysates, (G) NE activity in BAL and (H) NE activity in sera. Statistical comparisons were done with ANOVA followed by multiple comparisons. No significant differences were found. The concentrations of both MPO and NE are expressed in pg/ml. BAL, bronchoalveolar lavage fluid; Duox1 KO, Duox1-deficient; MPO, myeloperoxidase; NE, neutrophil elastase; WT, wild-type.
Neutrophil elastase (NE) is a major protease released from neutrophils during airway inflammation. A previous study has demonstrated that Duox1 is required for neutrophil recruitment in a mouse model of allergic asthma (36). To address whether NE release inside Mtb-infected airways is affected by Duox1, NE concentrations were assessed by ELISA in the BAL fluid, sera, and lung lysates of Mtb-infected WT and Duox1 KO mice as above (Figures 5D–F). In addition, NE enzymatic activity was measured as previously described (28) and the results show no effect of Duox1 (Figures 5G, H). Additionally, tissue distribution of MPO inside the lung tissue was evaluated by immunohistochemistry (Figure 6). Mtb-infected lungs at 1 day post-infection exhibit undetectable MPO while high levels of MPO are evident inside inflammatory cell aggregates at 30 days post-infection in both WT and Duox1 KO (Figure 6A) mice. Spatial NE expression inside the lung lesions of Mtb-infected mice revealed that NE also accumulates over time following MPO patterns independent of Duox1 (Figure 6B). These data demonstrate that release of the azurophilic granule content from neutrophils is not influenced by Duox1 during Mtb lung infection.
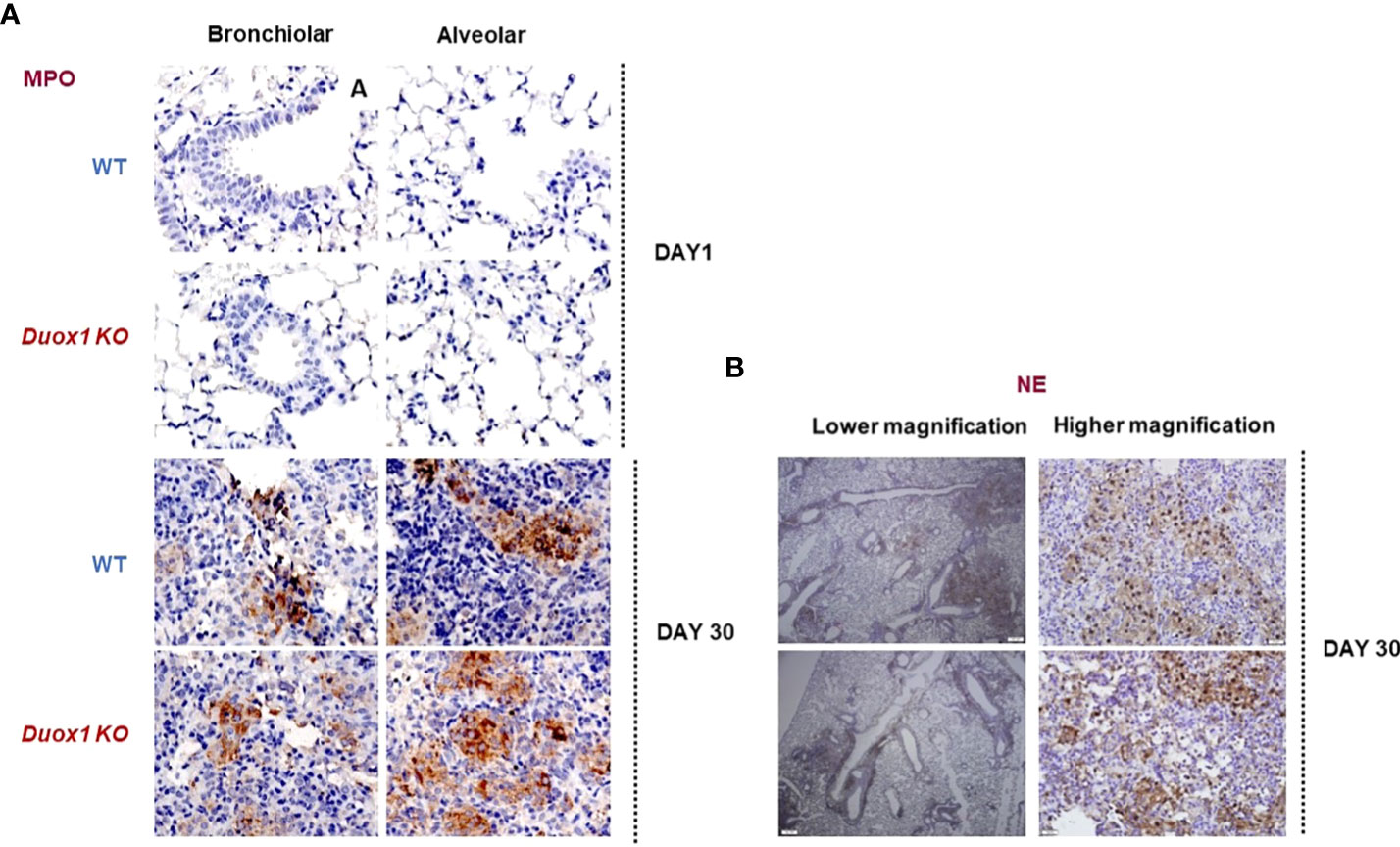
Figure 6 MPO and NE expressions in the lungs of Duox1 KO mice infected with Mtb. Mtb-infected lungs from Duox1 KO and WT mice were harvested at 1 or 30 day(s) post-infection, fixed and processed for staining with rabbit anti-MPO or rabbit anti-NE antibodies followed by anti-rabbit secondary antibody (HRP-linked) and DAB amplification. Sections are counterstained with hematoxylin and representative micrographs are shown in panels. (A) Representative micrograph of MPO staining of Mtb-infected lungs (bronchiolar area) of Duox1 KO and WT mice at 1 and 30 day(s) post-infection. (B) Representative photomicrographs of NE in Mtb-infected lungs at 30 days post-infection, at low (200 µm) and high magnification (20 µm), respectively. Duox1 KO, Duox1-deficient; MPO, myeloperoxidase; NE, neutrophil elastase; WT, wild-type.
Duox1-dependent B cells accumulate in the lung of Mtb-infected WT mice
To further investigate a possible role for Duox1 in the immune responses against Mtb, we harvested the lungs of Mtb-infected WT and Duox1 KO mice at 1 and 30 day(s) post-infection, and proceeded with immunofluorescence staining using a B cell-specific antibody (anti-mouse CD45R). Our results show that B cell numbers in the lung tissues (not BAL) at 1 day post-infection are the same between WT and Duox1 KO mice (Figures 7A, B). However, at 30 days post-infection, the number of B cells is significantly higher in WT mice than in Duox1 KO mice (p≤ 0.05) (Figures 7A, C). In contrast, B cell staining in the spleen at 30 days post-infection is comparable between Mtb-infected Duox1 KO and WT animals (Supplementary Figure 7). When anti-Mtb IgG antibody titers were compared between Mtb-infected Duox1 KO and WT animals in different compartments (BAL, lung lysate, serum), no significant differences were found (Supplementary Figure 8). These results indicate that Duox1 may play a role in B cell trafficking but it is unlikely to affect overall B cell activation or function during Mtb infection in this mouse model.
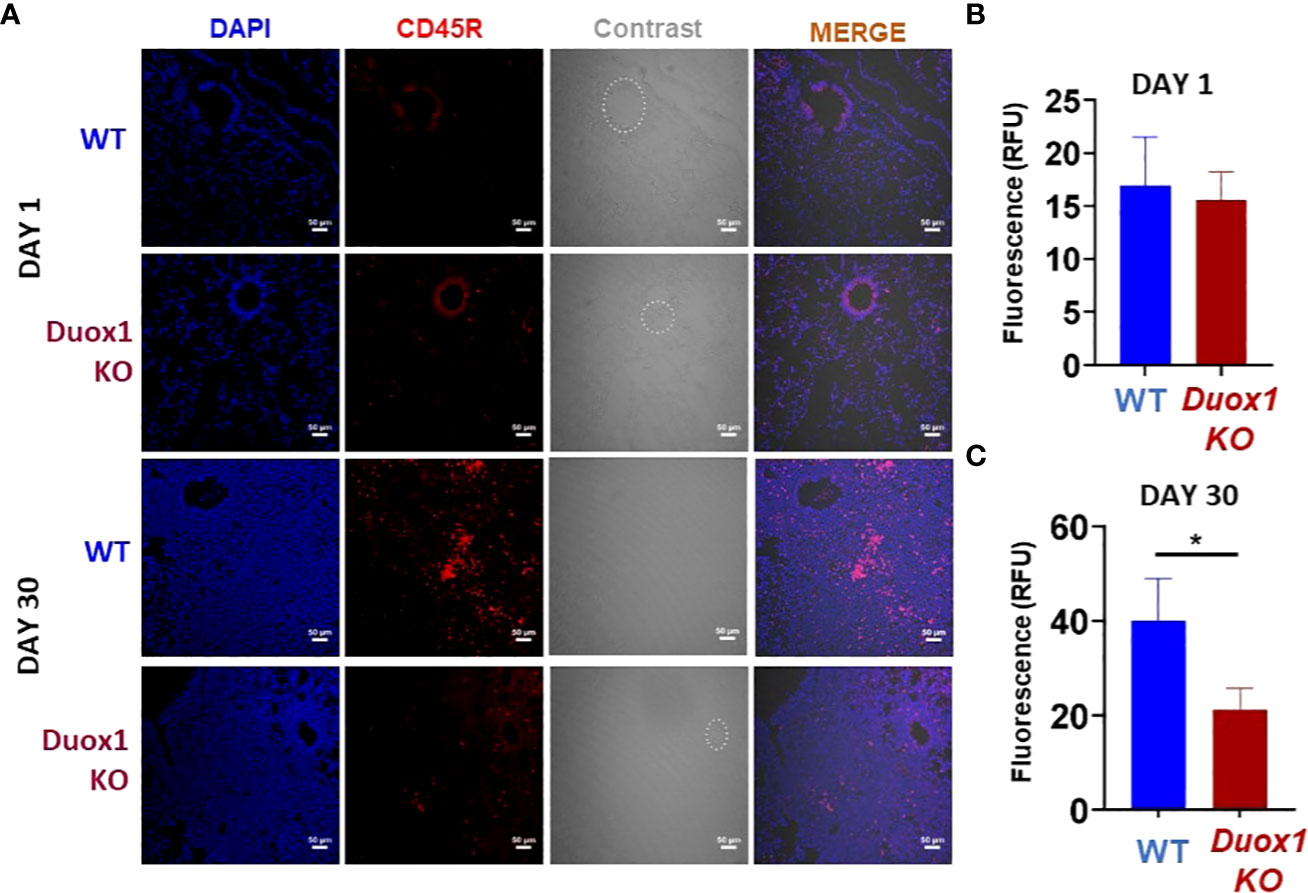
Figure 7 Duox1-depndent B cell accumulation in Mtb-infected lungs. Duox1 KO and WT mice were aerogenically infected with Mtb Erdman (200 CFU). Lung tissues were obtained at 1 and 30 day(s) post-infection, fixed and immunofluorescence staining was performed using anti-mouse CD45R specific for B cells (red). DAPI (blue) was used to depict cellular DNA. (A) Representative images of five similar results (n = 5 WT and n = 5 Duox1 KO mice) are shown. Quantitation of CD45R fluorescence at (B) 1 day post-infection and (C) 30 days post-infection. Scale bars indicate 50 μm. Mean fluorescent intensity (MFI) ± SEM in arbitrary unit (AU) is shown (Mann–Whitney U test). Significance level is indicated as *, p < 0.05. Duox1 KO, Duox1-deficient; WT, wild-type.
Duox1 suppresses caspase-3 activation in the lungs of Mtb-infected mice
Apoptosis plays a vital role in host defenses against pathogens. During Mtb infection, macrophage apoptosis can be beneficial to the host by activating innate and adaptive immune responses (37, 38). Cleaved caspase-3, the major executioner of apoptosis, degrades multiple proteins and is responsible for DNA fragmentation and morphological changes (39). Using immunofluorescence staining [as done previously (20)], we found significantly higher caspase-3 activation/cleavage in Mtb-infected Duox1 KO mice compared to WT counterparts suggesting that Duox1 KO mice are more susceptible to apoptosis while caspase 3 activation was undetectable at day 1 post-Mtb infection in both genotypes (Figure 8A and Supplementary Figure 9). Similar results were obtained using immunohistochemistry showing that cleaved caspase-3 expression is significantly higher in Duox1 KO than in WT mice (p ≤ 0.05) (Figures 8B, C). No Duox1-dependent differences were observed in the tissue expression of the used macrophage marker, CD68 (Figure 8A and Supplementary Figure 9). Overall, these data indicate that Duox1 reduces apoptosis during Mtb lung infection.
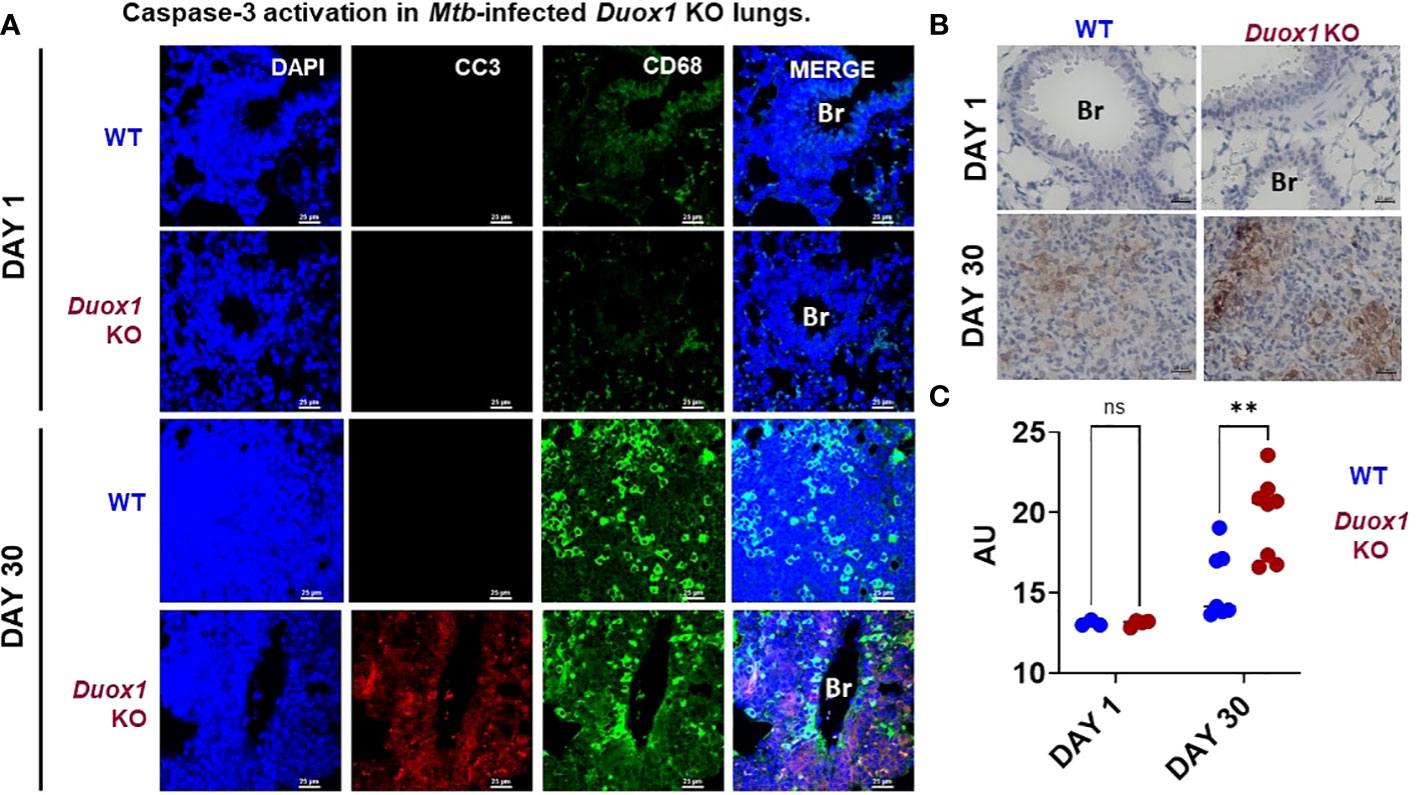
Figure 8 Caspase-3 activation in Mtb-infected Duox1 KO lungs. Mtb-infected lungs of WT and Duox1 KO mice were harvested and fixed at 1 or 30 day(s) post-infection. (A) Representative immunofluorescence staining of cleaved caspase-3 (CC3, red), CD68 (macrophages, green) and DAPI (DNA, blue). Scale bar: 25 µm). (B) Representative results of cleaved caspase-3 immunohistochemistry images at 1 (n=3 per genotype) and 30 days post-infection (n=8 per genotype). Scale bar indicates 25 µm. (C) Comparative non-stochiometric DAB signal quantification with ImageJ shows that the cleaved caspase-3 signal is significantly higher in the lung inflammatory aggregates of Duox1 KO mice compared to WT mice at 1 (n=3) and 30 (n=8) day(s) post-infection. A 2-way Anova with Sidak’s multiple comparison test was used for statistical data analysis. Significance levels are indicated as **P ≤ 0.01. AU, arbitrary unit; Br, bronchus; CC3, cleaved caspase-3; Duox1 KO, Duox1-deficient; WT, wild-type.
Discussion
DUOX1, an NADPH oxidase mainly expressed in bronchial epithelial cells, is the major source of airway epithelium-derived H2O2 (23). Because bronchial epithelial cells are one of the first lines of defense during respiratory infections, including TB, it was hypothesized that Duox1 will be protective against Mtb infection in vivo. Lactoperoxidase (LPO), an antimicrobial enzyme present in the airway, uses H2O2 to oxidize several substrates (15–18). LPO is abundant in the airway surface liquid of naïve mice (20). Its primary substrate, thiocyanate (SCN-) is also abundant in the airway secretions and can be converted by LPO into hypothiocyanite (OSCN-) that possesses antimicrobial properties against several pathogens in vitro (14, 21, 40). DUOX1 has been also shown to be expressed in human and murine macrophages which play a central role in Mtb infection in the lung in both species (30, 41, 42). Our primary goal was to assess whether Duox1 has a protective role against Mtb infection, given that DUOX1 is the main source of H2O2 in the airway. We have previously shown that DUOX1 has an important role in antiviral immunity in vivo by improving airway epithelial influenza viral clearance, shaping the early antiviral innate response, and reducing infection-related morbidity and mortality (20).
To extend these findings to other infectious diseases models, we assessed whether Duox1 prevents or limits Mtb infection, reduces or limits bacterial growth and dissemination, or eliminates or reduces lung pathology in vivo. Using a mouse model of aerosol infection with the strain Mtb Erdman, we addressed these primary objectives. The data show a dispensable role of Duox1 during Mtb infection in vivo. Neither bacterial growth in the lung, nor bacterial dissemination to different organs, such as the spleen and the liver, nor lung pathology were affected by Duox1 in this model. These data indicate that Duox1 does not have a protective role against Mtb, at least for the timeline (90 days post-infection) used as an endpoint in this study. Previous studies using a different strain of Mtb (H37Rv) have shown that mice start to show signs of illness and decreased survival beginning three months after infection (43), the time at which our mice were sacrificed. Additionally, previous studies have shown that Mtb has relatively high resistance to killing with H2O2.
Despite this, Duox1 had some significant effects in our Mtb model at three levels. First, Duox1 KO mice showed significantly higher levels of certain cytokines/chemokines in the BAL fluid at 30 days post-infection as evidenced by higher concentrations of TNF-α, IL6, KC, CCL3, CCL2, CCL20, CXCL11, CCL11, CCL27, CXCL5, CXCL12, and CCL5. A similar, overall role of Duox1 in shaping the early cytokine environment of the airways was established in our prior study using an H1N1 influenza strain (20). While some of the observed differences in cytokine levels between Mtb-infected WT and Duox1 KO mice are of interest, only four mice per group were used, therefore further experiments with more animals are required to confirm these data during Mtb infection. In contrast, our prior influenza study in Duox1 KO mice used 19-20 mice per group for cytokine measurements (20). Duox1 deficiency did not affect the frequencies of lymphoid or myeloid cells at 30 and 90 days post-infection either. None of the neutrophil markers was different between WT and Duox1 KO mice, suggesting that Duox1 is not required for neutrophil recruitment and activation in the lung environment during Mtb infection.
A previous study demonstrated that B cells infiltrate the granulomas of active Mtb patients (44) in accordance with earlier reports on the implication of B cells in TB pathogenesis (45, 46). Mechanistic studies assessing a possible role for B cells in Mtb infection used a mouse model with a moderately high dose of approximately 300 CFUs of the H37Rv strain (43). The study demonstrated that mice with altered B cell populations in the lung were more susceptible to aerosol infection than mice with normal B cells, suggesting that B cells can modulate immunity to Mtb infection (43). A prior study found that Duox1-derived H2O2 negatively regulates in vitro proliferative activity but not Ig isotype production in primary splenic CD19+ B cells in mice (47). Based on the results in our study, Duox1-dependent B cell infiltration in the lung is suggested during Mtb infection in vivo. However, this data was only derived from one method, immunofluorescence staining of a B cell-specific marker and secondary, more quantitative measurements would be required to strengthen the importance of this observation. Using B cell-deficient mice, the role of B cells in TB has been addressed in several studies (48–52). B cells are present in lymphoid clusters in mice (53), non-human primates (54) and human tuberculous granulomas (48, 55, 56). The function of B cells in mice lacking the ability to secrete immunoglobulin was uncovered by a study that supported the hypothesis that B cells can modulate immunity to Mtb in an organ-specific manner (43). Indeed, B cells participate in orchestrating cellular inflammatory aggregate formation during Mtb infection (50, 57). Our data are not sufficient to suggest a clinical or pathological role for B cells in TB in our model (58). Our results also show that the levels of Mtb-specific IgG levels are comparable between WT and Duox1 KO mice. Previous studies have indicated that activation of B cells within the mucosa will generate mainly IgA-secreting plasma cells, while activation in lymph nodes will generate mainly IgG-secreting plasma cells that home preferentially to the bone marrow and provide systemic protection (58). However, recent evidence suggests that IL4-responsive B cells during chronic Mtb infection in mice have a perturbed antibody and inflammatory cytokines profile (59). Previous evidence also suggests that IgA+ B cells produce IL-10 in the gut (60) and during Mtb infection (44) but more detailed investigations are required to assess any relationship between B cells, anti-inflammatory responses, immunoglobulins, and cell death pathways such as apoptosis.
Finally, higher levels of cleaved caspase 3, a marker for apoptosis, in the lung of Duox1 KO compared to WT mice may suggest more apoptotic cell death in Mtb-infected animals. Previous independent studies have shown the presence of apoptotic markers, including cleaved caspase-3, in murine and human lung following virulent Mtb infection (61–65). Apoptosis participates in host defense against Mtb facilitating cross-presentation of bacilli antigens contained in apoptotic bodies in the local lymph nodes (66), but its role in Mtb-infected Duox1 KO mice is not addressed in this study. Previously, we showed a similar trend in an influenza infection model where the Duox1 KO lung tissues (likely airway epithelium) showed higher levels of the same apoptotic marker (20). Unlike in the case of our data here on Mtb, Duox1 affected viral proliferation in the lungs of influenza-infected mice suggesting that Duox1 could play a pathogen-specific role in regulating apoptosis and general respiratory innate immunity.
Study limitations
The study endpoint is short (90 days post-infection) for Mtb infection. Designing future experiments beyond this timeline and after 1 day post-infection may yield a more positive phenotype. Also, using GFP-labelled Mtb bacilli would have helped adding significant values to both the immunostaining on lung tissue sections and the multicolor flow cytometry. The sample size of the bioplex data generated in this study is small (n=4 per genotype per day post infection) and the cell origin of the cytokines/chemokines identified was not addressed. Another limitation is the lack of confirmation of the B cell immunostaining by flow cytometry that shows higher expression in Mtb-infected WT mice at 30 days post-infection compared to Duox1-deficient mice. B cells were originally omitted in the flow cytometry panel because investigating them in the BAL was not our initial goal. It was only later when we did not find significant differences for initial parameters that we assessed the levels of B cells in the lung. In this study, we have not addressed any possible interactions between memory B cells, immunoglobulins, anti-inflammatory cytokines, Duox1, and cell death.
Additionally, previous studies have demonstrated that Mtb bacteria resistant to isoniazid, a treatment for active TB, have deletions in the katG gene, which encodes for the enzyme catalase (67). Several pathogens promote the degradation of H2O2 through the presence of genes that encode for enzymes with catalase activity (68). Recent evidence suggests that Mtb catalase inhibits the formation of mast cell extracellular traps by degrading H2O2 (69). This could be studied further in the context of Duox1. There are differences in lung structure and TB pathogenesis between humans and mice that could not be accounted for in this work solely using a mouse model. Our study was performed in mice with global Duox1 deficiency. Hence, Duox1 deficiency in cells other than airway epithelial cells could have also played a role. While caspase-3 cleavage is widely used as an apoptosis marker, the use of an additional apoptosis assay, such as the TUNEL assay, would be required to confirm the Duox1-dependence of apoptosis during Mtb infection. The cell type with enhanced cleaved caspase-3 signal has not been identified. The cleaved caspase-3 signal largely does not overlap with the CD68 staining indicative of macrophages.
In conclusion, our work unravels that Duox1 does not play a major role in the immune response or pathogenesis of Mtb lung infection in vivo.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was reviewed and approved by Animal Care and Use Committee at the University of Georgia.
Author contributions
BR, TG, and DS designed the experiments. TG, DS, KF, NA performed the experiments. DS, TG, KF, and BR analysed the data. KS performed the pathological evaluation. DS wrote the original draft. DS, TG, KF, NA, KS, FQ, and BR revised and edited the manuscript. FQ provided access to BSL3. BR and FQ received funding and oversaw the project. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Institute of Health (R01AI146857-01A1 and R21AI147097 to BR) and UGA Athletic Association professor endowment funds (FQ).
Acknowledgments
The authors would like to thank Jamie Barber, Julie Nelson, Veronica Obregon-Perko, the FlowJo Application Scientist for the US Southeast at BD Life Sciences for their technical support and assistance in the data analysis of the flow cytometry. Authors would also like to thank Shelly Helms and Dr. Hind Yahyaoui Azami to help run the Madison chamber.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1044703/full#supplementary-material
References
2. Petruccioli E, Scriba TJ, Petrone L, Hatherill M, Cirillo DM, Joosten SA, et al. Correlates of tuberculosis risk: predictive biomarkers for progression to active tuberculosis. Eur Respir J (2016) 48:1751–63. doi: 10.1183/13993003.01012-2016
3. Huang L, Russell DG. Protective immunity against tuberculosis: what does it look like and how do we find it? Curr Opin Immunol (2017) 48:44–50. doi: 10.1016/j.coi.2017.08.001
4. Huang L, Nazarova EV, Russell DG. Mycobacterium tuberculosis: Bacterial fitness within the host macrophage. Microbiol Spectr (2019) 7(2):7.2.04. doi: 10.1128/microbiolspec.BAI-0001-2019
5. Goletti D, Petruccioli E, Joosten SA, Ottenhoff TH. Tuberculosis biomarkers: From diagnosis to protection. Infect Dis Rep (2016) 8:6568. doi: 10.4081/idr.2016.6568
6. Cadena AM, Flynn JL, Fortune SM. The importance of first impressions: Early events in mycobacterium tuberculosis infection influence outcome. mBio (2016) 7:e00342–16. doi: 10.1128/mBio.00342-16
7. Bhatt K, Verma S, Ellner JJ, Salgame P. Quest for correlates of protection against tuberculosis. Clin Vaccine Immunol (2015) 22:258–66. doi: 10.1128/CVI.00721-14
8. Basaraba RJ, Hunter RL. Pathology of tuberculosis: How the pathology of human tuberculosis informs and directs animal models. Microbiol Spectr (2017) 5(3):5.3.17. doi: 10.1128/microbiolspec.TBTB2-0029-2016
9. Heemskerk D, Caws M, Marais B, Farrar J. Tuberculosis in adults and children. London, Springer (2015).
10. Sia JK, Rengarajan J. Immunology of mycobacterium tuberculosis infections. Microbiol Spectr (2019) 7(4):7.4.6. doi: 10.1128/microbiolspec.GPP3-0022-2018
11. Menozzi FD, Rouse JH, Alavi M, Laude-Sharp M, Muller J, Bischoff R, et al. Identification of a heparin-binding hemagglutinin present in mycobacteria. J Exp Med (1996) 184:993–1001. doi: 10.1084/jem.184.3.993
12. Pavlicek RL, Fine-Coulson K, Gupta T, Quinn FD, Posey JE, Willby M, et al. Rv3351c, a mycobacterium tuberculosis gene that affects bacterial growth and alveolar epithelial cell viability. Can J Microbiol (2015) 61:938–47. doi: 10.1139/cjm-2015-0528
13. Reuschl AK, Edwards MR, Parker R, Connell DW, Hoang L, Halliday A, et al. Innate activation of human primary epithelial cells broadens the host response to mycobacterium tuberculosis in the airways. PloS Pathog (2017) 13:e1006577. doi: 10.1371/journal.ppat.1006577
14. Sarr D, Toth E, Gingerich A, Rada B. Antimicrobial actions of dual oxidases and lactoperoxidase. J Microbiol (2018) 56:373–86. doi: 10.1007/s12275-018-7545-1
15. Conner GE, Salathe M, Forteza R. Lactoperoxidase and hydrogen peroxide metabolism in the airway. Am J Respir Crit Care Med (2002) 166:S57–61. doi: 10.1164/rccm.2206018
16. Conner GE, Wijkstrom-Frei C, Randell SH, Fernandez VE, Salathe M. The lactoperoxidase system links anion transport to host defense in cystic fibrosis. FEBS Lett (2007) 581:271–8. doi: 10.1016/j.febslet.2006.12.025
17. Gerson C, Sabater J, Scuri M, Torbati A, Coffey R, Abraham JW, et al. The lactoperoxidase system functions in bacterial clearance of airways. Am J Respir Cell Mol Biol (2000) 22:665–71. doi: 10.1165/ajrcmb.22.6.3980
18. Wijkstrom-Frei C, El-Chemaly S, Ali-Rachedi R, Gerson C, Cobas MA, Forteza R, et al. Lactoperoxidase and human airway host defense. Am J Respir Cell Mol Biol (2003) 29:206–12. doi: 10.1165/rcmb.2002-0152OC
19. Gattas MV, Forteza R, Fragoso MA, Fregien N, Salas P, Salathe M, et al. Oxidative epithelial host defense is regulated by infectious and inflammatory stimuli. Free Radical Biol Med (2009) 47:1450–8. doi: 10.1016/j.freeradbiomed.2009.08.017
20. Sarr D, Gingerich AD, Asthiwi NM, Almutairi F, Sautto GA, Ecker J, et al. Dual oxidase 1 promotes antiviral innate immunity. Proc Natl Acad Sci U.S.A. (2021) 118. doi: 10.1073/pnas.2017130118
21. Rada B, Lekstrom K, Damian S, Dupuy C, Leto TL. The pseudomonas toxin pyocyanin inhibits the dual oxidase-based antimicrobial system as it imposes oxidative stress on airway epithelial cells. J Immunol (2008) 181:4883–93. doi: 10.4049/jimmunol.181.7.4883
22. Forteza R, Salathe M, Miot F, Forteza R, Conner GE. Regulated hydrogen peroxide production by duox in human airway epithelial cells. Am J Respir Cell Mol Biol (2005) 32:462–9. doi: 10.1165/rcmb.2004-0302OC
23. Geiszt M, Witta J, Baffi J, Lekstrom K, Leto TL. Dual oxidases represent novel hydrogen peroxide sources supporting mucosal surface host defense. FASEB J (2003) 17:1502–4. doi: 10.1096/fj.02-1104fje
24. Rada B, Leto TL. Characterization of hydrogen peroxide production by duox in bronchial epithelial cells exposed to pseudomonas aeruginosa. FEBS Lett (2010) 584:917–22. doi: 10.1016/j.febslet.2010.01.025
25. Zambrowicz BP, Friedrich GA, Buxton EC, Lilleberg SL, Person C, Sands AT. Disruption and sequence identification of 2,000 genes in mouse embryonic stem cells. Nature (1998) 392:608–11. doi: 10.1038/33423
26. Donko A, Ruisanchez E, Orient A, Enyedi B, Kapui R, Peterfi Z, et al. Urothelial cells produce hydrogen peroxide through the activation of Duox1. Free Radic Biol Med (2010) 49:2040–8. doi: 10.1016/j.freeradbiomed.2010.09.027
27. Gupta T, LaGatta M, Helms S, Pavlicek RL, Owino SO, Sakamoto K, et al. Evaluation of a temperature-restricted, mucosal tuberculosis vaccine in guinea pigs. Tuberculosis (Edinb) (2018) 113:179–88. doi: 10.1016/j.tube.2018.10.006
28. Almutairi F, Sarr D, Tucker SL, Fantone K, Lee JK, Rada B. RGS10 reduces lethal influenza infection and associated lung inflammation in mice. Front Immunol (2021) 12:772288. doi: 10.3389/fimmu.2021.772288
29. Tucker SL, Sarr D, Rada B. Neutrophil extracellular traps are present in the airways of ENaC-overexpressing mice with cystic fibrosis-like lung disease. BMC Immunol (2021) 22:7. doi: 10.1186/s12865-021-00397-w
30. Rada B, Park JJ, Sil P, Geiszt M, Leto TL. NLRP3 inflammasome activation and interleukin-1beta release in macrophages require calcium but are independent of calcium-activated NADPH oxidases. Inflammation Res (2014) 63:821–30. doi: 10.1007/s00011-014-0756-y
31. O’Garra A, Redford PS, McNab FW, Bloom CI, Wilkinson RJ, Berry MP. The immune response in tuberculosis. Annu Rev Immunol (2013) 31:475–527. doi: 10.1146/annurev-immunol-032712-095939
32. Muefong CN, Sutherland JS. Neutrophils in tuberculosis-associated inflammation and lung pathology. Front Immunol (2020) 11:962. doi: 10.3389/fimmu.2020.00962
33. Bekale RB, Du Plessis SM, Hsu NJ, Sharma JR, Sampson SL, Jacobs M, et al. Mycobacterium tuberculosis and interactions with the host immune system: Opportunities for nanoparticle based immunotherapeutics and vaccines. Pharm Res (2018) 36:8. doi: 10.1007/s11095-018-2528-9
34. Sugiyama S, Okada Y, Sukhova GK, Virmani R, Heinecke JW, Libby P. Macrophage myeloperoxidase regulation by granulocyte macrophage colony-stimulating factor in human atherosclerosis and implications in acute coronary syndromes. Am J Pathol (2001) 158:879–91. doi: 10.1016/S0002-9440(10)64036-9
35. Klebanoff SJ. Myeloperoxidase: friend and foe. J Leukoc Biol (2005) 77:598–625. doi: 10.1189/jlb.1204697
36. Chang S, Linderholm A, Franzi L, Kenyon N, Grasberger H, Harper R. Dual oxidase regulates neutrophil recruitment in allergic airways. Free Radical Biol Med (2013) 65:38–46. doi: 10.1016/j.freeradbiomed.2013.06.012
37. Pan H, Yan BS, Rojas M, Shebzukhov YV, Zhou H, Kobzik L, et al. Ipr1 gene mediates innate immunity to tuberculosis. Nature (2005) 434:767–72. doi: 10.1038/nature03419
38. Malik ZA, Iyer SS, Kusner DJ. Mycobacterium tuberculosis phagosomes exhibit altered calmodulin-dependent signal transduction: contribution to inhibition of phagosome-lysosome fusion and intracellular survival in human macrophages. J Immunol (2001) 166:3392–401. doi: 10.4049/jimmunol.166.5.3392
39. McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol (2013) 5:a008656. doi: 10.1101/cshperspect.a008656
40. Gingerich A, Pang L, Hanson J, Dlugolenski D, Streich R, Lafontaine ER, et al. Hypothiocyanite produced by human and rat respiratory epithelial cells inactivates extracellular H1N2 influenza a virus. Inflammation Res (2016) 65:71–80. doi: 10.1007/s00011-015-0892-z
41. Morris CR, Habibovic A, Dustin CM, Schiffers C, Lin MC, Ather JL, et al. Macrophage-intrinsic DUOX1 contributes to type 2 inflammation and mucus metaplasia during allergic airway disease. Mucosal Immunol (2022) 15:977–89. doi: 10.1038/s41385-022-00530-x
42. Herrera MT, Guzman-Beltran S, Bobadilla K, Santos-Mendoza T, Flores-Valdez MA, Gutierrez-Gonzalez LH, et al. Human pulmonary tuberculosis: Understanding the immune response in the bronchoalveolar system. Biomolecules (2022) 12(8):1148. doi: 10.3390/biom12081148
43. Torrado E, Fountain JJ, Robinson RT, Martino CA, Pearl JE, Rangel-Moreno J, et al. Differential and site specific impact of b cells in the protective immune response to mycobacterium tuberculosis in the mouse. PloS One (2013) 8:e61681. doi: 10.1371/journal.pone.0061681
44. Dai YC, Wang WD, Zhang JA, Chen C, Luo HL, Xu H, et al. MTB driven b cells producing IL-35 and secreting high level of IL-10 in the patients with active pulmonary tuberculosis. Mol Immunol (2019) 112:175–81. doi: 10.1016/j.molimm.2019.05.004
45. Zhang M, Zeng G, Yang Q, Zhang J, Zhu X, Chen Q, et al. Anti-tuberculosis treatment enhances the production of IL-22 through reducing the frequencies of regulatory b cell. Tuberculosis (Edinb) (2014) 94:238–44. doi: 10.1016/j.tube.2013.12.003
46. Zhang M, Zheng X, Zhang J, Zhu Y, Zhu X, Liu H, et al. CD19(+)CD1d(+)CD5(+) b cell frequencies are increased in patients with tuberculosis and suppress Th17 responses. Cell Immunol (2012) 274:89–97. doi: 10.1016/j.cellimm.2012.01.007
47. Sugamata R, Donko A, Murakami Y, Boudreau HE, Qi CF, Kwon J, et al. Duox1 regulates primary b cell function under the influence of IL-4 through BCR-mediated generation of hydrogen peroxide. J Immunol (2019) 202:428–40. doi: 10.4049/jimmunol.1601395
48. Bosio CM, Gardner D, Elkins KL. Infection of b cell-deficient mice with CDC 1551, a clinical isolate of mycobacterium tuberculosis: delay in dissemination and development of lung pathology. J Immunol (2000) 164:6417–25. doi: 10.4049/jimmunol.164.12.6417
49. Johnson CM, Cooper AM, Frank AA, Bonorino CB, Wysoki LJ, Orme IM. Mycobacterium tuberculosis aerogenic rechallenge infections in b cell-deficient mice. Tuber Lung Dis (1997) 78:257–61. doi: 10.1016/S0962-8479(97)90006-X
50. Maglione PJ, Xu J, Chan J. B cells moderate inflammatory progression and enhance bacterial containment upon pulmonary challenge with mycobacterium tuberculosis. J Immunol (2007) 178:7222–34. doi: 10.4049/jimmunol.178.11.7222
51. Turner J, Frank AA, Brooks JV, Gonzalez-Juarrero M, Orme IM. The progression of chronic tuberculosis in the mouse does not require the participation of b lymphocytes or interleukin-4. Exp Gerontol (2001) 36:537–45. doi: 10.1016/S0531-5565(00)00257-6
52. Vordermeier HM, Venkataprasad N, Harris DP, Ivanyi J. Increase of tuberculous infection in the organs of b cell-deficient mice. Clin Exp Immunol (1996) 106:312–6. doi: 10.1046/j.1365-2249.1996.d01-845.x
53. Tsai MC, Chakravarty S, Zhu G, Xu J, Tanaka K, Koch C, et al. Characterization of the tuberculous granuloma in murine and human lungs: cellular composition and relative tissue oxygen tension. Cell Microbiol (2006) 8:218–32. doi: 10.1111/j.1462-5822.2005.00612.x
54. Phuah JY, Mattila JT, Lin PL, Flynn JL. Activated b cells in the granulomas of nonhuman primates infected with mycobacterium tuberculosis. Am J Pathol (2012) 181:508–14. doi: 10.1016/j.ajpath.2012.05.009
55. Ulrichs T, Kosmiadi GA, Jorg S, Pradl L, Titukhina M, Mishenko V, et al. Differential organization of the local immune response in patients with active cavitary tuberculosis or with nonprogressive tuberculoma. J Infect Dis (2005) 192:89–97. doi: 10.1086/430621
56. Ulrichs T, Kosmiadi GA, Trusov V, Jorg S, Pradl L, Titukhina M, et al. Human tuberculous granulomas induce peripheral lymphoid follicle-like structures to orchestrate local host defence in the lung. J Pathol (2004) 204:217–28. doi: 10.1002/path.1628
57. Clatworthy MR. Targeting b cells and antibody in transplantation. Am J Transplant (2011) 11:1359–67. doi: 10.1111/j.1600-6143.2011.03554.x
58. Kato A, Hulse KE, Tan BK, Schleimer RP. B-lymphocyte lineage cells and the respiratory system. J Allergy Clin Immunol (2013) 131:933–57; quiz 958. doi: 10.1016/j.jaci.2013.02.023
59. Parihar SP, Ozturk M, Hoft MA, Chia JE, Guler R, Keeton R, et al. IL-4-Responsive b cells are detrimental during chronic tuberculosis infection in mice. Front Immunol (2021) 12:611673. doi: 10.3389/fimmu.2021.611673
60. Rojas OL, Probstel AK, Porfilio EA, Wang AA, Charabati M, Sun T, et al. Recirculating intestinal IgA-producing cells regulate neuroinflammation via IL-10. Cell (2019) 176:610–624 e18. doi: 10.1016/j.cell.2018.11.035
61. Aguilo JI, Alonso H, Uranga S, Marinova D, Arbues A, de Martino A, et al. ESX-1-induced apoptosis is involved in cell-to-cell spread of mycobacterium tuberculosis. Cell Microbiol (2013) 15:1994–2005. doi: 10.1111/cmi.12169
62. Aguilo N, Marinova D, Martin C, Pardo J. ESX-1-induced apoptosis during mycobacterial infection: To be or not to be, that is the question. Front Cell Infect Microbiol (2013) 3:88. doi: 10.3389/fcimb.2013.00088
63. Blomgran R, Desvignes L, Briken V, Ernst JD. Mycobacterium tuberculosis inhibits neutrophil apoptosis, leading to delayed activation of naive CD4 T cells. Cell Host Microbe (2012) 11:81–90. doi: 10.1016/j.chom.2011.11.012
64. Aporta A, Arbues A, Aguilo JI, Monzon M, Badiola JJ, de Martino A, et al. Attenuated mycobacterium tuberculosis SO2 vaccine candidate is unable to induce cell death. PloS One (2012) 7:e45213. doi: 10.1371/journal.pone.0045213
65. Seimon TA, Kim MJ, Blumenthal A, Koo J, Ehrt S, Wainwright H, et al. Induction of ER stress in macrophages of tuberculosis granulomas. PloS One (2010) 5:e12772. doi: 10.1371/journal.pone.0012772
66. Behar SM, Martin CJ, Booty MG, Nishimura T, Zhao X, Gan HX, et al. Apoptosis is an innate defense function of macrophages against mycobacterium tuberculosis. Mucosal Immunol (2011) 4:279–87. doi: 10.1038/mi.2011.3
67. Zhang Y, Heym B, Allen B, Young D, Cole S. The catalase-peroxidase gene and isoniazid resistance of mycobacterium tuberculosis. Nature (1992) 358:591–3. doi: 10.1038/358591a0
68. Eason MM, Fan X. The role and regulation of catalase in respiratory tract opportunistic bacterial pathogens. Microb Pathog (2014) 74:50–8. doi: 10.1016/j.micpath.2014.07.002
Keywords: Mycobacterium tuberculosis, antimicrobial, Duox1, inflammation, epithelium
Citation: Gupta T, Sarr D, Fantone K, Ashtiwi NM, Sakamoto K, Quinn FD and Rada B (2023) Dual oxidase 1 is dispensable during Mycobacterium tuberculosis infection in mice. Front. Immunol. 14:1044703. doi: 10.3389/fimmu.2023.1044703
Received: 14 September 2022; Accepted: 16 February 2023;
Published: 03 March 2023.
Edited by:
Deepak Kaushal, Southwest National Primate Research Center (SNPRC), United StatesReviewed by:
Laura Ellen Via, National Institute of Allergy and Infectious Diseases (NIH), United StatesStefan Oehlers, Technology and Research (A*STAR), Singapore
Copyright © 2023 Gupta, Sarr, Fantone, Ashtiwi, Sakamoto, Quinn and Rada. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Balázs Rada, cmFkYWJAdWdhLmVkdQ==
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share senior authorship