 Gerard Badia-Bringué
Gerard Badia-Bringué María Canive
María Canive Marta Alonso-Hearn
Marta Alonso-Hearn- 1Department of Animal Health, NEIKER-Basque Institute for Agricultural Research and Development, Basque Research and Technology Alliance (BRTA), Derio, Bizkaia, Spain
- 2Doctoral Program in Molecular Biology and Biomedicine, Universidad del País Vasco/Euskal Herriko Unibertsitatea (UPV/EHU), Leioa, Bizkaia, Spain
The genetic loci influencing individual resistance to Mycobacterium avium subsp. paratuberculosis (MAP) infection are still largely unknown. In the current study, we searched for genetic loci associated with resistance to MAP infection by evaluating the performance of monocyte-derived macrophages (MDMs) isolated from the peripheral blood of 75 healthy Holsteins cows and infected ex vivo with MAP. Bacterial load (log colony-forming units, log CFUs) within MDMs was quantified at 2 h and 7 days p. i. using a BACTEC MGIT 960 instrument. In addition, the expression levels of some genes with important roles in the innate immune response including epiregulin (EREG), complement component C3 (C3), galectin-9 (Gal9), and nitric oxide (NO-) were measured in the supernatant of the infected cells. DNA from peripheral blood samples of the animals included in the study was isolated and genotyped with the EuroG MD bead Chip (44,779 single nucleotide-polymorphisms, SNPs). Linear mixed models were used to calculate the heritability (h2) estimates for each indicator of MDM performance, MAP load within MDMs and EREG, C3, Gal9, and NO-expression. After performing a genome-wide association study, the only phenotypes that showed SNPs with a significant association were the bacterial load within MDMs at 2 h (h2 = 0. 87) and 7 days (h2 = 0.83) p.i. A total of 6 SNPs, 5 candidate genes, and one microRNA on the Bos taurus chromosomes BTA2, BTA17, BTA18, and BTA21 were associated with MAP load at 2 h p.i. Overlap was seen in two SNPs associated with the log CFUs at 2 h and 7 d p.i. The identified SNPs had negative regression coefficients, and were, therefore, associated with a low bacterial load within MDMs. Some of the identified SNPs were located within QTLs previously associated with longevity, reproductive, and udder health traits. Some of the identified candidate genes; Oxysterol Binding Protein Like 6, Cysteine and Serine Rich Nuclear Protein 3, and the Coiled-Coil Domain Containing 92 regulate cellular cholesterol trafficking and efflux, apoptosis, and interferon production, respectively. Taken together, our results define a heritable and distinct immunogenetic profile in MAP-infected macrophages designed to limit bacterial load early after infection.
1 Introduction
Bovine paratuberculosis (PTB) or Johne´s disease (JD) is a chronic enteritis of domestic and wild ruminants caused by Mycobacterium avium subsp. paratuberculosis (MAP). PTB is a major problem for animal health that compromises animal welfare and causes important economic losses to the dairy industry (1). In Europe and North America, PTB is endemic in dairy cattle, with herd prevalence estimates higher than 50% (2). PTB-associated losses include increased susceptibility to other diseases, increased somatic cell counts, increased incidence of clinical mastitis, reduced fertility, reduced milk production, costs of testing, and involuntary culling of cows (3). Animals are infected early in life through the fecal-oral route but clinical onset appears when animals are 18 months or older (4). Once ingested, MAP reaches the jejune and ileum and crosses the intestinal mucosa by binding to fibronectin β1 receptors present on M cells located in Peyer’s patches (5, 6). In the submucosa, MAP is phagocytized by sub-epithelial macrophages. Within infected macrophages, MAP has developed several survival mechanisms such as preventing the maturation and acidification of the phagosome and its fusion with the lysosome and preventing the presentation of antigens to the immune system (7). MAP has a zoonotic potential and it has been detected in samples of patients with Crohn´s disease (CD), ulcerative colitis, and idiopathic inflammatory bowel disease (IBD)-associated colorectal cancer (8–11). Inactivated vaccines against MAP interfere with diagnostic tests for bovine tuberculosis and therefore are not allowed in many countries. One solution to control this disease is the identification of genetic loci determining variation in immune-related traits that could be used to select PTB-resistant animals.
PTB is a multifactorial disease that arises as the result of the interaction of genetic, environmental, and microbial factors which affect the various disease outcomes. According to their extension in the intestine, cellular infiltrate, and amount of MAP; PTB-associated lesions were classified as focal, multifocal, and diffuse (diffuse paucibacillary or lymphoplasmacytic, diffuse intermediate, and diffuse multibacillary or histiocytic) (12, 13). Our research group has explored very recently the associations between PTB-associated pathology and host genetics (14). Overall, a total of 380 SNPs were found associated (FDR ≤ 0.05; P ≤ 5 10-7) with ante-mortem (serum ELISA) and post-mortem (tissue PCR and culture and histopathology) PTB diagnostic definitions in a common set of Spanish Holstein cattle (N = 983) using whole-genome sequence data (WGS) data (14–16). However, we were unable to identify SNPs associated with resistance to MAP infection.
Reductionist approaches investigate a host biological subsystem such as a key cellular function whose performance is the criterion for the classification of the population (17). Recently, a strong effect of host genetics on the in vitro nitric oxide (NO-) production of monocyte-derived macrophages (MDMs) in response to two common bacterial pathogens of dairy cattle, Escherichia coli and Staphylococcus aureus, has been described (18). Macrophages are important host defense cells against MAP infection. Since MAP-infected macrophages’ performance can be better analyzed under a controlled environment, the probability of identifying resistant animals is much higher using ex vivo macrophage models in comparison to holistic approaches based on field studies. In addition, it is generally hypothesized that mycobacteria growth in macrophages in vitro predicts the in vivo risk of disease or infection (19, 20). Moreover, the direct mycobacteria growth inhibition assay (MGIA) has demonstrated cross-species potential (21). Recently, we have demonstrated that MAP-infected MDMs constitute a very useful model of infection to validate the functional impact of specific expression quantitative trait loci (cis-eQTLs) potentially implicated in susceptibility to MAP infection (22). Cis-eQTLs are genetic variants typically located in gene regulatory regions that alter gene expression in an allele-specific manner. Cis-eQTLs are functional links between genomic variants, gene expression, and ultimately phenotype. In the current study, we hypothesize that MAP-infected MDMs might help to identify resistant individuals that respond stronger than others to MAP infection and, as result, reduce the intracellular MAP burden significantly.
The measure of indicators of MDMs’ performance in response to MAP infection is another strategy for the identification of resistant cattle. To date, however, no serum biomarkers have been associated with resistance to MAP infection. In our previous study (22), the integration of gene expression data (RNA-Seq) and genotypes (54,609 SNPs per animal) from a cohort of cows naturally exposed to MAP allowed the identification of 192 cis-eQTLs associated with the expression of 145 genes in peripheral blood samples. Four of these cis-eQTLs regulated the expression of several genes with important roles in the innate immune response; the complement component C3 (C3), epiregulin (EREG), galectin-9 (Gal9), and nitric oxide synthase (NOS1). In the current study, we tested whether the measurement of MAP load within infected MDMs and the expression in the supernatants of MAP-infected MDMs of several genes with important roles in the innate immune response could be used as correlates of MDMs performance. Our objective was to identify genetic loci associated with resistance to MAP infection using ex vivo MAP-infected MDMs from Spanish Holstein cows. For this purpose, we performed a genome-wide association study (GWAS) to identify SNPs and candidate genes associated with the MAP load within infected MDMs and with the expression of Gal9, ERG, C3, and NO- in the supernatants of infected MDMs. Since there is evidence that some allelic variants may contribute to resistance to multiple pathogens, the identified SNPs and candidate genes were compared with QTLs and candidate genes for other bovine diseases and health and reproductive traits. In addition, the candidate genes identified in our study were compared with human candidate genes previously identified for CD, IBD, and colorectal cancer. The workflow of the study is presented in Figure 1.
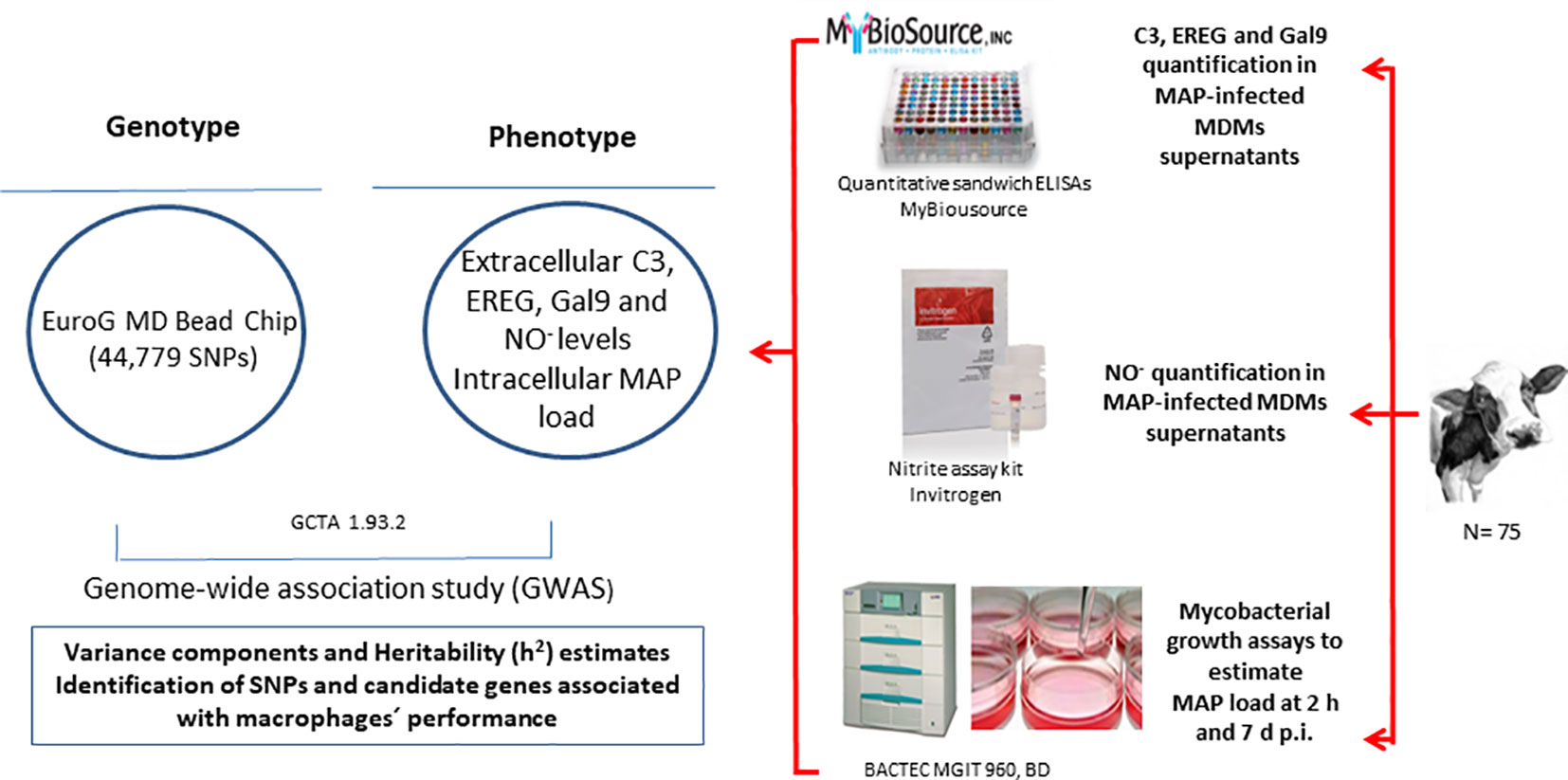
Figure 1 Study design. The approach starts from the measures of MAP load within MAP infected-MDMs and the C3, EREG, Gal9, and NO- expression levels in the supernatants of MAP-infected-MDMs. These phenotypes are combined with the genotypes of the animals in a GWAS study for the calculation of variance components and heritability (h2) estimates and the identification of associated SNPs and candidate genes.
2 Materials and methods
2.1 Ethical statement
Animals used in this study were not submitted to any in vivo experimentation. Blood sampling was conducted with the approval of the Ethics Committee of Animal Experimentation of NEIKER (OEBA-2022-010), following current legal regulations and in particular, according to pertinent Basque (Basque Government Decree 454/1994), Spanish (Spanish Government Law 32/2007/7 and Royal decree 53/2013/1), and European (Community Council Animal Welfare Guidelines 2010/63/EU) regulations.
2.2 Animals and blood samples
The reference population consisted of 75 Spanish Holstein cows from a single farm located in the Basque Country. In 2022, only five cows from this herd had an ELISA positive result and all the animals had a negative fecal PCR result. Only adult cows (2 years or older, 4.5 years mean age) were included in the study. All the animals included in this study were registered with the Spanish Federation of Holstein cattle (CONAFE; www.conafe.com.) and had negative fecal PCR results when blood samples were collected and in subsequent annual samplings. In addition, the animals used in this study were not diagnosed with any other pathogen. Blood samples were collected in groups of 16 per sampling day. The validation population consisted of 16 cows from the same farm.
2.3 Mycobacterial growth inhibition assays
MGIA were performed as previously described (22–24). Briefly, fifteen milliliters of peripheral blood were drawn from the tail vein of healthy Holstein cows into heparinized Vacutainer tubes (Becton, Dickinson and Company, Sparks, MD, USA) and diluted 1:2 in Hanks balanced salt solution (HBSS). Leucosep tubes were filled with 15 ml of Ficoll-Paque (1.084 g/cm3) (GE Healthcare, Uppsala, Sweden) and centrifuged at 1,000 rpm for 30 seconds at room temperature. Subsequently, the diluted blood was overlaid on the top of the Ficoll-Paque and centrifuged at 800 g for 15 minutes at room temperature. The plasma layer was removed and the cell interphase containing peripheral blood mononuclear cells (PBMCs) was collected and transferred to a clean tube. PBMCs were washed twice in HBSS and centrifuged at 400 g for 10 minutes to remove platelets. Supernatants were aspirated and the purified PBMCs were resuspended in RPMI-1640 supplemented with 20 mM L-glutamine, 10% heat-inactivated bovine serum (Lonza, Spain), 100 U ml-1 penicillin G, and 100 mg ml-1 streptomycin sulfate (Lonza, Spain). PBMCs were cultured at a concentration of 1 x 106 cells/ml into 24-well plates and incubated at 37°C in a humidified 5% CO2 incubator for 2 h. Non-adherent cells were removed by washing, and adherent cells were incubated in fresh medium for 7 days at 37°C to allow differentiation to MDMs. Differentiated MDMs were inoculated in triplicate with a single-cell suspension of MAP K10 strain at a multiplicity of infection (MOI) of 10:1 (bacteria:cells). After 2 h, the supernatant was removed, and the cells were washed twice with HBSS to remove extracellular bacteria. Infected MDMs were lysed at this time (2 h p. i.) or were cultured at 37°C for 7 days in fresh medium. At each time point, the supernatant was aspirated and infected MDMs were lysed by vigorous pipetting with 0.5 ml of 0.1% Triton X-100 (Sigma-Aldrich) in sterile water for 10 min.
2.4 Assessment of viable intracellular MAP using the BACTEC MGIT 960 system.
MGIA assessment using the BACTEC MGIT 960 system was performed as previously described (23–25). Briefly, supplemented Mycobacteria Growth Indicator tubes (MGIT) (Becton, Dickinson, and Company, Sparks, MD) were inoculated with 0.5 ml of the cell lysates. The concentration of the initial suspension of the MAP K10 strain used to inoculate the MDMs was validated in the BACTEC MGIT 960. The tubes were incubated at 37 ± 2°C for up to 42 days in a BACTEC MGIT 960 instrument (Becton, Dickinson, and Company). The earliest instrumental indicator of positivity (i.e., time to detection [TTD]) for each tube was recorded. The predicted number of bacteria in each positive tube was calculated using standard curves that relate TTD (in days) to the estimated log CFUs (25). For the standard curves, a ten-fold dilution series of a MAP K10 strain cellular suspension (McFarland= 1) were prepared and the TTDs of 100 µl of each dilution in the BACTEC MGIT 960 system were detected. MAP load (log CFUs) were plotted versus TTDs and the mathematical equation was used to determine the estimated log CFUs for each sample. The log CFU ratios were calculated by dividing the estimated log CFUs at day 7 by that at 2 h p. i. MAP survival indexes were calculated according to Martinez et al. (26), and Price et al. (27), as the square root CFU ratio percentage at 7 days p. i. with respect to 2 h p. i. Lower values of the MAP survival index reflect higher resistance to the infection.
2.5 NO- quantification
NO- levels were measured in the supernatants collected from the MAP-infected MDMs after 2 h of infection with the Measure-iT ™ High-Sensitivity Nitrite Assay Kit (Invitrogen, Paisley, UK) according to the manufacturer's instructions. The assay has an optimal range of 20–500 pmol nitrite, making it up to 50 times more sensitive than colorimetric methods utilizing the Griess reagent. Nitrates are analyzed after quantitative conversion to nitrites through enzymatic reduction; used in this manner, the assay provides effective quantitation of NO-. Briefly, the supernatants from the MAP-infected MDMs after 2 h of infection (40 µl) were brought to a volume of 50 µl by adding 10 µl of H2O and placed into 96-well black plates in triplicate. Subsequently, 100 µl of quantitation reagent were added and mixed by pipetting. The plate was incubated for 10 minutes at room temperature and 5 µl of quantitation developer were added to each well and gently mixed by pipetting. Right after quantitation developer addition, fluorescence was measured using a multimodal microplate reader Synergy™ HTX (Biotek, Winooski, Vermont, US) at 365/450 nm (ex/em). A standard curve was prepared with the Measure-iT™ nitrite quantitation reagent concentrate included in the kit (0, 2.75, 5.5, 11, 22, 33, 44, 55 µM). Assays were conducted in duplicate in a final volume of 110 µl by adding 100 µl of quantification agent, fluorescence was measured, and plotted versus picomoles of NO-. The equation was used to determine the NO- concentration for each sample.
2.6 Quantification of EREG, Gal9, and C3 protein levels
The EREG, Gal9, and C3 expression was assessed in the supernatants of MAP-infected MDMs collected at 7 days p. i. using quantitative sandwich ELISAs according to the manufacturer’s instructions (MyBioSource, San Diego, US). The sensitivity of the EREG, Gal9, and C3 ELISA kits is 5 pg/ml (detection range, 31.2-1.000 pg/ml), 0.1 ng/ml (detection range, 0.625-20 ng/ml), and 2 µg/ml (detection range, 15.6 μg/ml-500 μg/ml), respectively. Briefly, standards and samples (50 µl) were added in duplicate into a Microelisa Stripplate provided with each kit. One hundred microliters of horseradish peroxidase-conjugated antibody were added to each well. After incubation for 60 min at 37°C in the dark, the plate was washed four times with 350 µl of wash solution and incubated with 50 µl of 3, 3′, 5, 5′-Tetramethylbenzidine for 15 min at 37°C in the dark. After adding 50 µl of stop solution into each well, the OD values were measured in an ELISA reader at 450 nm (Thermo Scientific Multiskan, US). We average the duplicate readings for each standard and sample and subtract the average OD of the blank. A standard curve was generated by plotting the mean OD values of each standard on the vertical axis and the corresponding concentration on the horizontal axis. The levels of EREG, Gal9, and C3 in each sample were interpolated from the standard curve. The correlations between the intracellular MAP load within MDMs at 2 h and 7 d p. i., and the quantification of EREG, Gal9, and C3 protein levels were analyzed using the Spearman’s rank correlation coefficient (ρ) implemented in R 4.1.2. considering a coefficient with a P-value less than or equal to 0.05 as significant.
2.7 Genotyping
Peripheral blood samples were collected in EDTA tubes and DNA was extracted using the QIAmp DNA Blood Mini Kit according to the manufacturer’s instructions (Qiagen, Hilden, Germany). Purified genomic DNA was genotyped with the EuroG MD Bead Chip at the molecular genetic laboratory service of the Spanish Federation of Holstein Cattle (CONAFE). The InfiniumTM iScan software (Illumina, San Diego, CA) was used for allele assignation. All the SNPs passed a call rate > 0.80. After filtering out SNPs with minimum allele frequency (MAF)< 0.01, the final marker set included 44,779 SNPs.
2.8 Variance components and heritability (h2) estimates
The intracellular MAP load at 2 h and 7 d p. i. and the expression of NO-, EREG, Gal9, and C3 were the quantitative phenotypes analyzed. The variance components and h2 explained by all the SNPs were calculated using the genome-wide complex trait analysis (GCTA) software 1.93.2, according to the following formula
where Is the variance explained by all the SNPs and is the residual variance (28).
2.9 Genome-wide association study
Genotypes and the quantitative phenotypes were analyzed using the mixed linear model association analysis of the GCTA 1.93.2 software. Briefly, the model is y = a + bx + g + e, where y is the phenotype, a is the mean term, b is the additive effect (fixed effect) of the candidate SNP to be tested for association, x is the SNP genotype indicator variable coded as 0, 1 or 2, g is the polygenic effect (random effect) assumed to be distributed as N~(0 ),and e is the residual assumed to be distributed as N~(0 ). Age was included as a covariate in the analysis. To account for multiple testing, a 5% genome-wide false discovery rate (FDR) was used (29). The inflation factor (λ) and quantile-quantile plots were used to compare the observed distributions of –log (P-values) to the expected distribution under the no-association model. λ values close to 1 suggest appropriate adjustment for potential substructure and λ > 1.2 suggest population stratification. The regression coefficients (b-values) were calculated using the GCTA 1.93.2 software.
2.10 Identification of SNPs and candidate genes
The EuroG MD Bead chip is based on the University of Maryland UMD 3.1 Bos taurus reference genome; hence, the coordinates of the GWAS-identified SNPs were converted to the ARS-UCD1.2 genome by using liftOver software (https://genome.ucsc.edu). The location of the identified SNPs (e.g. upstream or downstream of a transcript, in the coding sequence, in non-coding RNA, in regulatory regions) in the ARS-UCD1.2 genome was determined using the Ensembl Variant Effect predictor (VEP). None of the identified SNPs were within 500,000 base pairs of each other or on linkage disequilibrium. The candidate genes located within 50,000 base pairs to each side of the SNPs were identified using Ensembl (https://www.ensembl.org). The function of all the identified genes was searched in GeneCards (http://www.genecards.org) by searching their gene symbol. Since there is evidence that some allelic variants may contribute to resistance to multiple pathogens, the identified SNPs and candidate genes were compared with QTLs and candidate genes previously associated with other bovine diseases, longevity, and reproductive and health traits (http://www.animalgenome.org). In addition, the identified candidate genes were also compared with human candidate genes previously identified for CD, IBD, and colorectal cancer (http://www.ebi.ac.uk/gwas).
2.11 Genomic estimated breeding values for MAP survival indexes within MDMs and validation of the genomic predictions
gEBVs for MAP survival indexes within MDMs were calculated for each animal in the reference population using the gBLUP model of GCTA 1.93.2 (30). Subsequently, gEBVs for MAP survival indexes within MDMs were predicted in a validation population. PBMCs were isolated from animals with high (N = 8) and low (N = 8) gEBVs, differentiated to MDMs, infected ex vivo with MAP, and at 2 h and 7 d p. i. the intracellular load was quantified in the BACTEC MGIT system as described in point 2.3. An unpaired student t-test with the Welch-Satterthwaite correction was used to compare the experimental MAP survival indexes calculated for the animals with high and low gEBVs (GraphPad Prism 8, San Diego, CA, US). Differences were considered significant when P-values were ≤ 0.05. Correlations between the gEBVs and experimental MAP survival indexes within infected MDMs were calculated with the Pearson correlation implemented in R 4.1.2.
3 Results
3.1 Functional phenotyping of bovine MDMs in response to MAP infection
As indicators of bovine MDMs performance, intracellular MAP load within MDMs at 2 h and 7 d p. i. and NO−, EREG, Gal9, and C3 secretion were measured in MAP-infected MDMs isolated from 75 cows (Figure 2). The NO- secretion was measured in the supernatants of MAP-infected MDMs at 2 h p.i. while EREG, Gal9, and C3 were measured at 7 d p. i. To estimate the technical reproducibility of each phenotype, the technical coefficients of variation (CVs) were calculated for each phenotype. The technical CV calculated using the duplicate measures within one experiment for C3, EREG, Gal9, NO-, and MAP load with macrophages at 2 h and 7 d p. i. were 6.47% (0% - 17.54%), 4.76% (0% - 10.92%), 4.08% (0% - 9.48%), 14.07% (0.89% - 76.69%), 2.00% (0% - 7.12%), and 7.11% (0% - 33.89%), respectively. To estimate the variation within the population, the CV were calculated for the measures of each phenotype within the population. The CV of Gal9, C3, EREG, NO-, and MAP load within MDMs at 2 h and 7 d p. i. were 7, 9, 14, 41, 12, and 31%, respectively. The biological reproducibility of the MAP load within macrophages at 2 h and 7 d p. i. was previously assessed by calculating the CVs of biological triplicates within three different experiments; 9.40 and 10.72%, respectively (24). The biological CV considers the precision of the quantification of MAP load within macrophages to correctly characterize the cow or the fidelity of the reading of the phenotype. The correlations between the six phenotypes were analyzed using Spearman’s rank correlation (Table 1). The strongest positive correlation (ρ = 0.80) was found between the intracellular MAP load at 2 h and 7 d p. i., while the highest negative correlation (ρ = - 0.64) was observed between the secretion of NO- measured at 2 h p. i. and the intracellular MAP load at 7 d p. i. The NO- measured at 2 h p. i. also correlated negatively with the intracellular MAP load at 2 h p. i. (ρ = - 0.29). The secretion of C3 was positively correlated with the MAP intracellular load at 2 h and 7 d p.i.; ρ = 0.34 and ρ= 0.48, respectively. On the other hand, negative correlations were found between NO- and Gal9, and NO- and EREG expression; ρ = - 0.24 and ρ = - 0.35, respectively. Positive correlations were found between Gal9 land EREG, and Gal9 and C3 expression; ρ = 0.28 and ρ = 0.44, respectively.
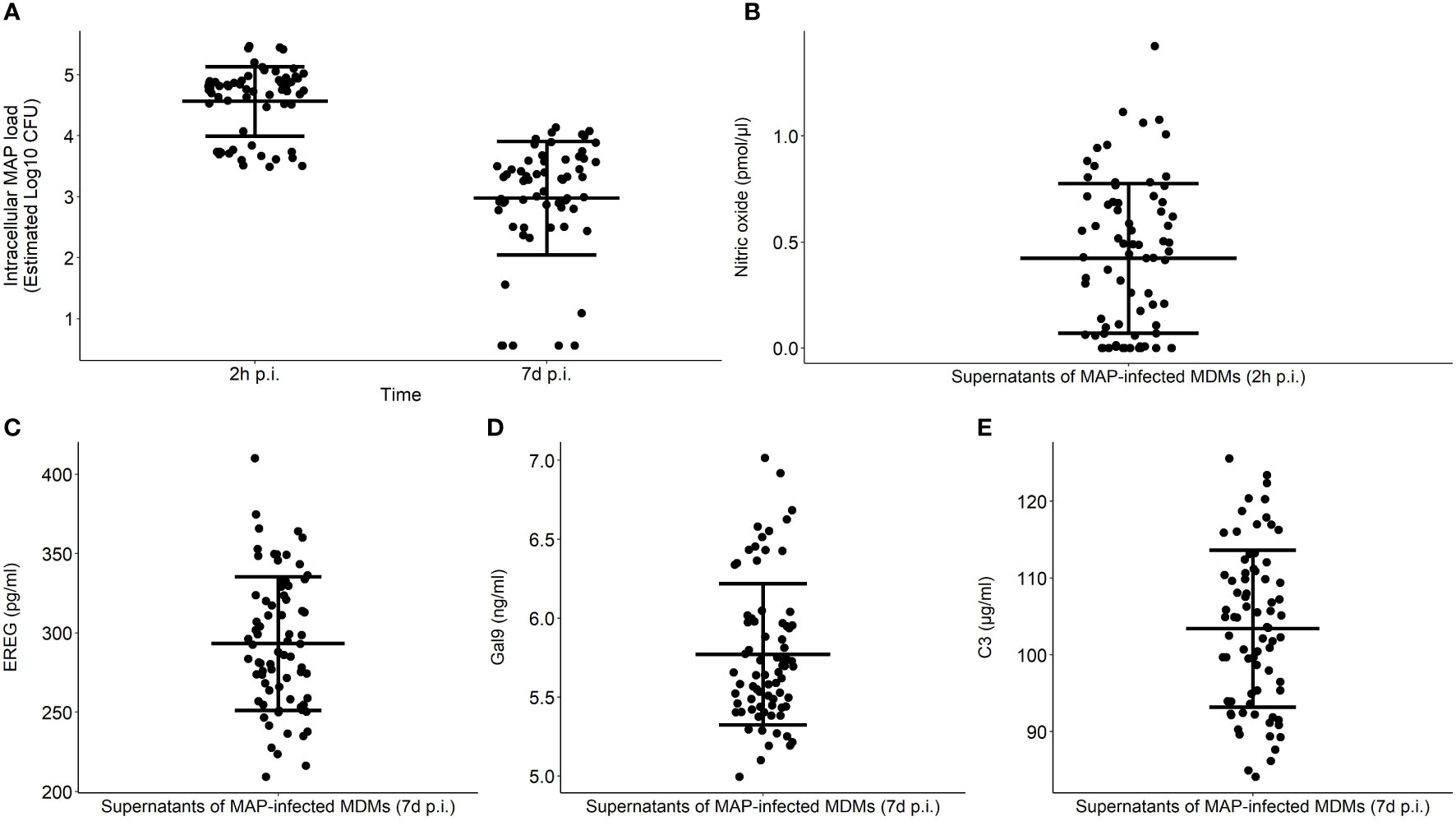
Figure 2 Functional phenotyping of bovine MDMs in response to MAP infection. MDMs performance was determined by measuring the intracellular MAP load at 2 h and 7 d p. i. (A) and extracellular NO- (B), EREG (C), Gal9 (D), and C3 production (E) in response to MAP infection. Extracellular NO- levels were measured in the supernatants of MAP-infected MDMs at 2 h p. i. while EREG, Gal9, and C3 production were tested at 7 d p. i. Each dot represents one individual response, the middle line represents the average in each group, and the error bars represent the standard deviation from the mean in each group.
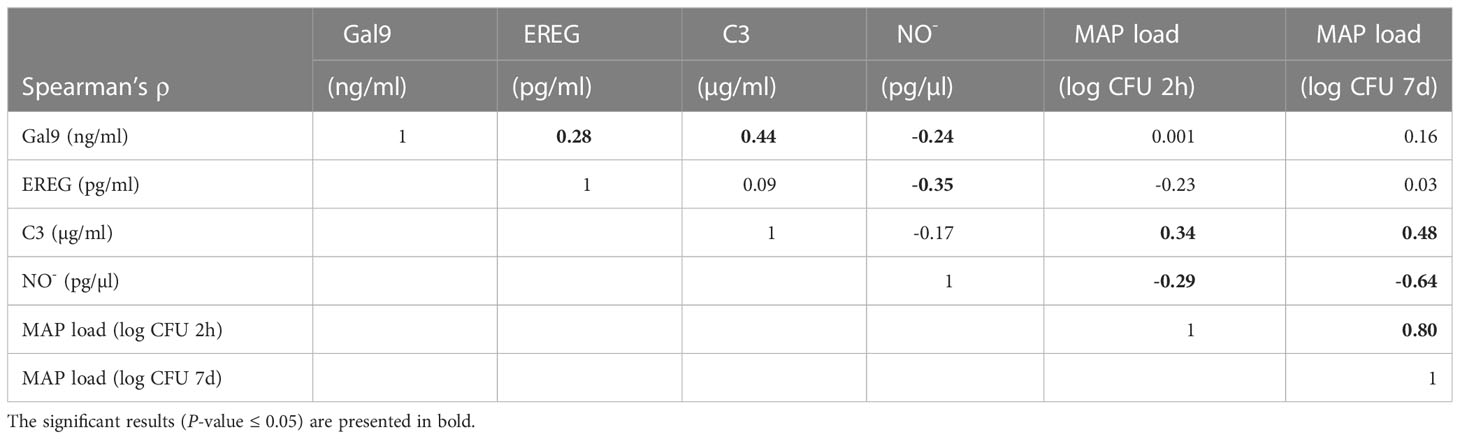
Table 1 Spearman’s rank correlation coefficient (ρ) between the intracellular MAP load within MDMs at 2 h and 7 d p. i., and the extracellular Gal9, EREG, C3, and NO- levels.
3.2 Variance components and heritability (h2) estimates
The intracellular MAP load at 2 h and 7 d p. i. and the extracellular NO-, EREG, Gal9, and C3 levels were the quantitative phenotypes analyzed. The variance components, additive genetic variance () and residual variance (), and h2 explained by all the SNPs were calculated using the genome-wide complex trait analysis (GCTA) software 1.93.2. Table 2 summarizes the variance components and h2 estimates for each phenotype. The highest h2 estimates were obtained for the MAP load within MDMs at 2 h (h2 = 0.87) and 7 days p. i. (h2 = 0.83). The estimated h2 for C3 (h2 = 0.78), EREG (h2 = 0.74), NO- (h2 = 0.44) and Gal9 (h2 = 0.13) levels were also calculated. While the h2 estimates for intracellular MAP load, C3 (h2 = 0.78), and EREG (h2 = 0.74) were high; NO- (h2 = 0.44) and Gal9 (h2 = 0.13) estimates were moderate to low.
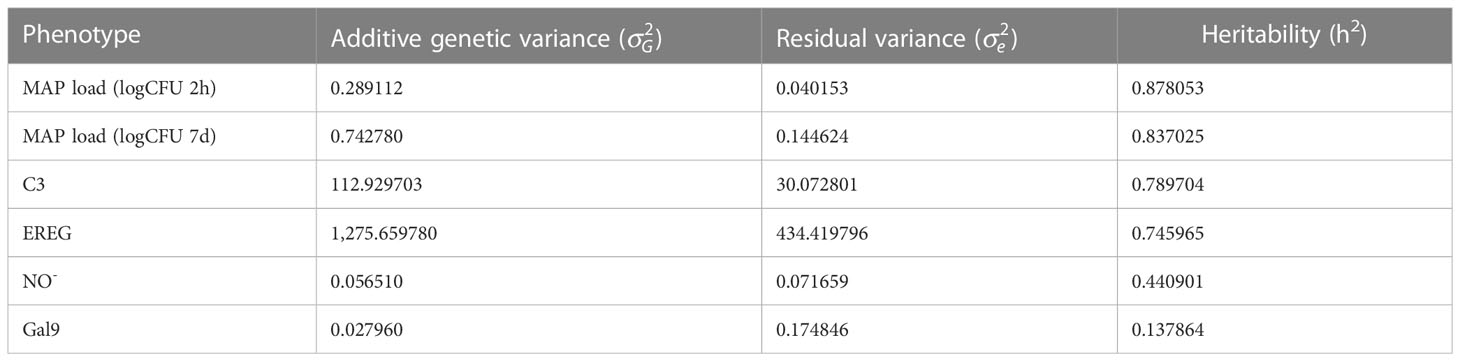
Table 2 Variance components and h2 estimates for the intracellular MAP load within MDMs at 2 h and 7 d p. i., and the extracellular Gal9, EREG, C3, and NO- levels.
3.3 GWAS
To identify associations between genetic variants and phenotypes, a GWAS was performed for each phenotype. Only the bacterial load within MDMs at 2 h and 7 d p. i. showed SNPs with a statistically significant association. Six SNPs surpassed the PFDR correction for the MAP load at 2 h p. i. and two for the MAP load at 7 d p. i. Overlap was seen in two SNPs associated with MAP load within MDMs at 2 h and 7 d p. i. These two SNPs were located on BTA17 and BTA18. Manhattan plots showing –log10 (P-values) of association between each SNP and MAP load within MDMs at 2 h and 7 d p. i. are presented in Figures 3A, B, respectively. Quantile-quantile plots comparing the observed distribution of –log (P-values) to the expected values under the null hypothesis for the MAP load within MDMs at 2 h and 7 d p. i. are presented in Figures 3C, D, respectively. The plots showed a distribution close to the expected line; λmedian= 1.00 and λmedian= 1.01 for MAP load at 2 h and 7 d p. i., respectively. This indicates that significant values were not overestimated due to population stratification or cryptic relatedness.
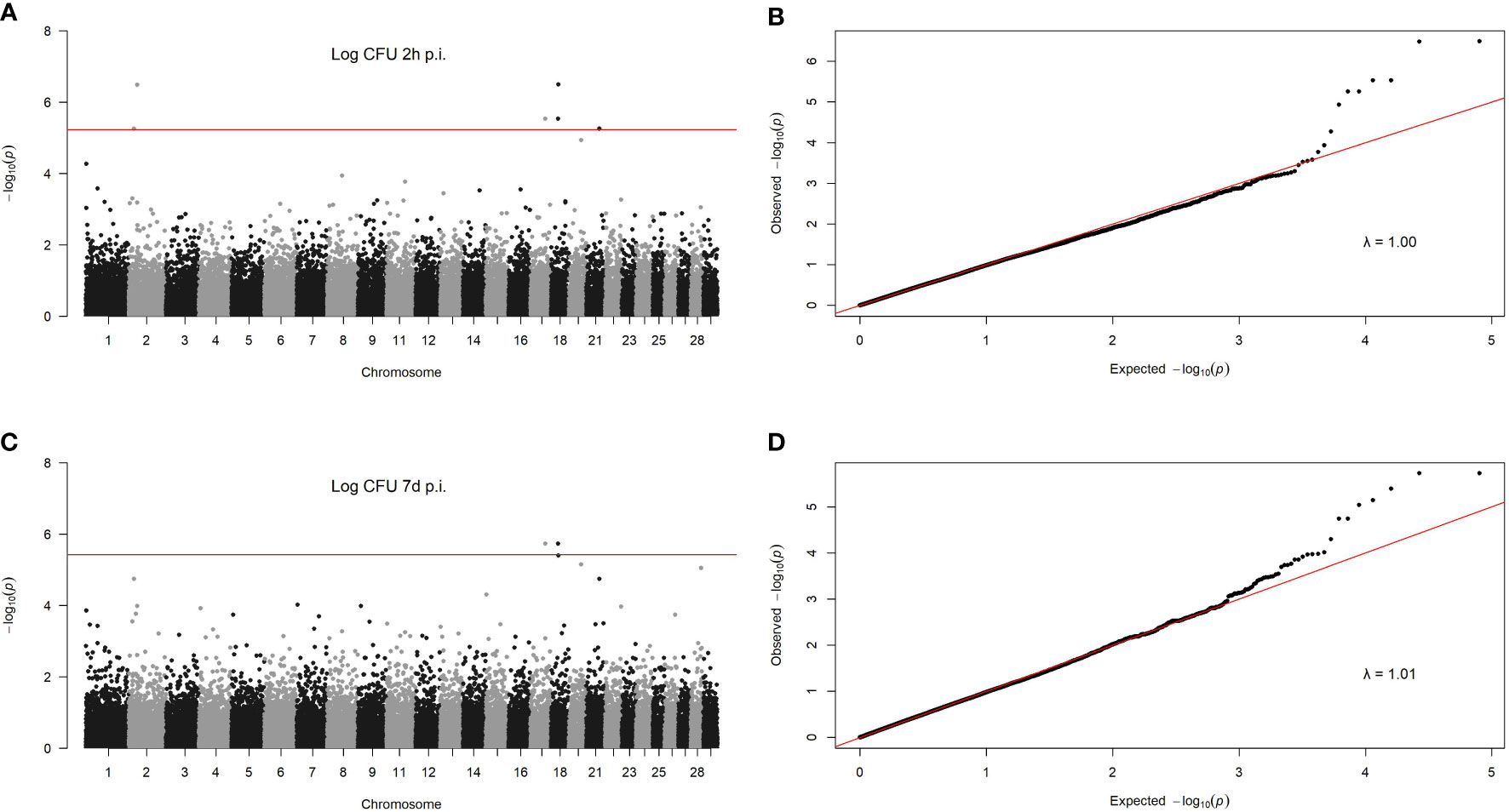
Figure 3 GWAS for MAP load within infected MDMs. Manhattan plots showing –log10 (P-values) of association between every single SNP and intracelllar MAP load within MDMs at 2 h (A) and 7 d p. i. (B) and 7 d p. i. (C). Each dot represents the result from the test association for a single SNP. The chromosomes´ locationof the SNPs is indicated on the x-axis. The horizontal red line represents the genome-wide significance threshold –log10 (PFDR). Quantile-quantileplots comparing the observed distribution of –log (P-values) for the intracellular MAP load within MDMs at 2 h (D) to the expected values under the null hypothesis.
3.4 SNPs associated with MAP load within MDMs and candidate genes identification
The SNPs surpassing the significance threshold (PFDR ≤ 0.05), P-values, and candidate genes are presented in Table 3. The six identified SNPs were located on BTA2, BTA17, BTA18, and BTA21. Three of the SNPs were in intergenic regions, two in introns, and one is a downstream variant. The most genome-wide significantly associated SNP (P = 3.28 × 10-7) was located on BTA2. The regression coefficients (b-values) of the six identified SNPs were all negative suggesting that all the SNPs were associated with a low bacterial load within MDMs. None of the six SNPs identified by GWAS was in strong linkage disequilibrium. A total of five candidate genes and one miRNA were identified within 50,000 bp upstream and downstream of the identified SNPs including the Cysteine and Serine Rich Nuclear Protein 3 (CSRNP3), U6 spliceosomal RNA (U6), Oxysterol Binding Protein Like 6 (OSBPL6), Coiled-Coil Domain Containing 92 (CCDC92), ENSBTAG00000042858, and bta-mir-2365. Two of the six identified SNPs were in intergenic regions and showed no evidence of a candidate gene within 100,000 bp distance.
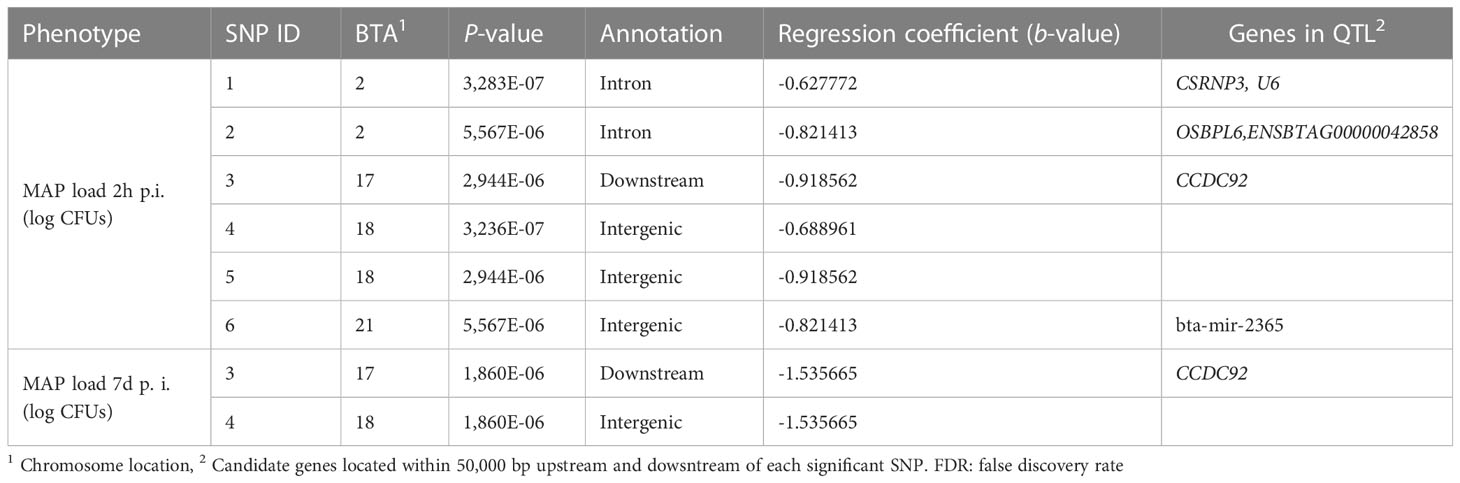
Table 3 SNPs surpassing the significance threshold (PFDR ≤ 0.05) for evidence of an association with MAP load within infected MDMs at 2 h and 7 d p. i.
3.5 Comparison of the identified SNPs and candidate genes to previously reported QTLs and candidate genes
The SNPs and candidate genes that were identified in the current study had not been associated with bovine PTB, tuberculosis, or clinical mastitis before. By examining the available cattle QTL database (Table 4), we observed that two of the identified SNPs were located on BTA18 within QTLs previously associated with the somatic cell score (QTL:3556), length of productive life (QTL:3555), and reproductive traits such as stillbirth (QTL:11362), birth index (QTL: 30537), and dystocia (QTL:2707) (31–34). In addition, the SNP located on BTA21 was located within QTLs associated with IgG levels (QTL:66212), somatic cell score (QTL:5446 and QTL:10190), and calving ease (QTL:11126) (35–38). Finally, the SNP on BTA17 was located within a QTL associated with calving ease (36). The identified candidate genes have not been associated with PTB or with other diseases in cattle or in other animal species.
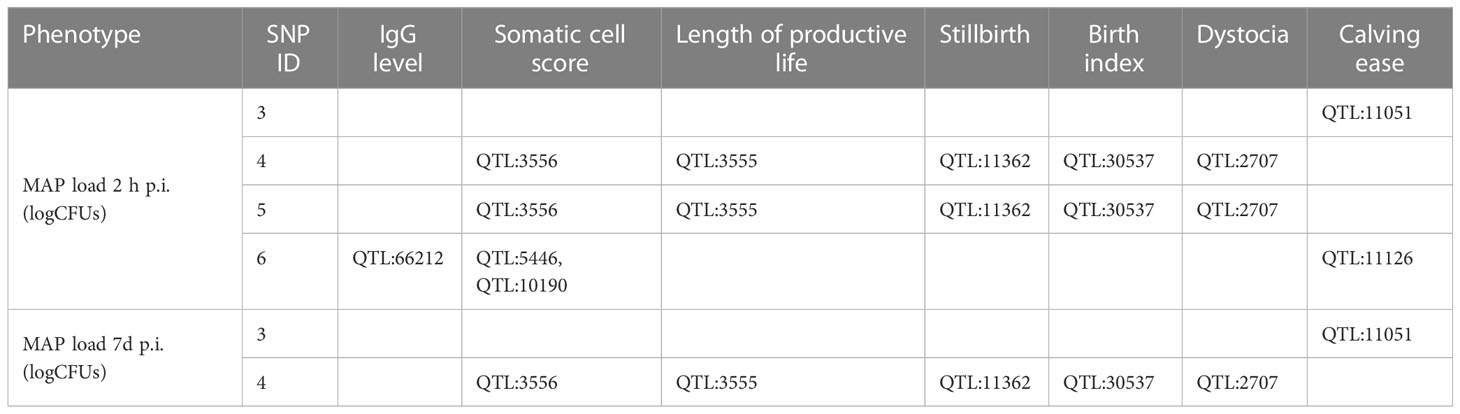
Table 4 Overlapping of the identified SNPs with QTLs associated with other health and reproductive traits.
3.6 Validation of the genomic predictions
The gBLUP model (30) was used to estimate gEBVs of each individual in the study population based on the effect of the six SNPs with evidence of association with the survival indexes of MAP within MDMs and their genomic relationships. Subsequently, the six SNPs with evidence of association with the phenotype were used to predict the gEBVs of the validation population. For the validation of the genomic predictions, 16 animals with high (N = 8) and low (N = 8) gEBVs were selected. The distribution of the gEBVs is presented in Figure 4A. MDMs of the selected animals were isolated and infected ex vivo with MAP as previously described in Materials and Methods. Intracellular bacterial load was quantified at 2 h and 7 d p. i. and the experimental MAP survival indexes were calculated for the animals with low (mean = 75.7) and high gEBVs (mean = 84.1). There were significant differences in the killing capacity of macrophages from cows with low and high gEBVs (P-value = 0.0006) and the data demonstrated a tighter grouping of the macrophages from cows with low gEBVs having superior killing capacity (Figure 4B). Pearson correlation coefficient between the gEBVs and experimental MAP survival indexes was positive (0.78) (P-value = 0.0003). These results demonstrated a significant effect of the six identified SNPs on MAP survival within macrophages and confirmed the existence of a strong genetic effect on macrophages’ performance in response to MAP infection.
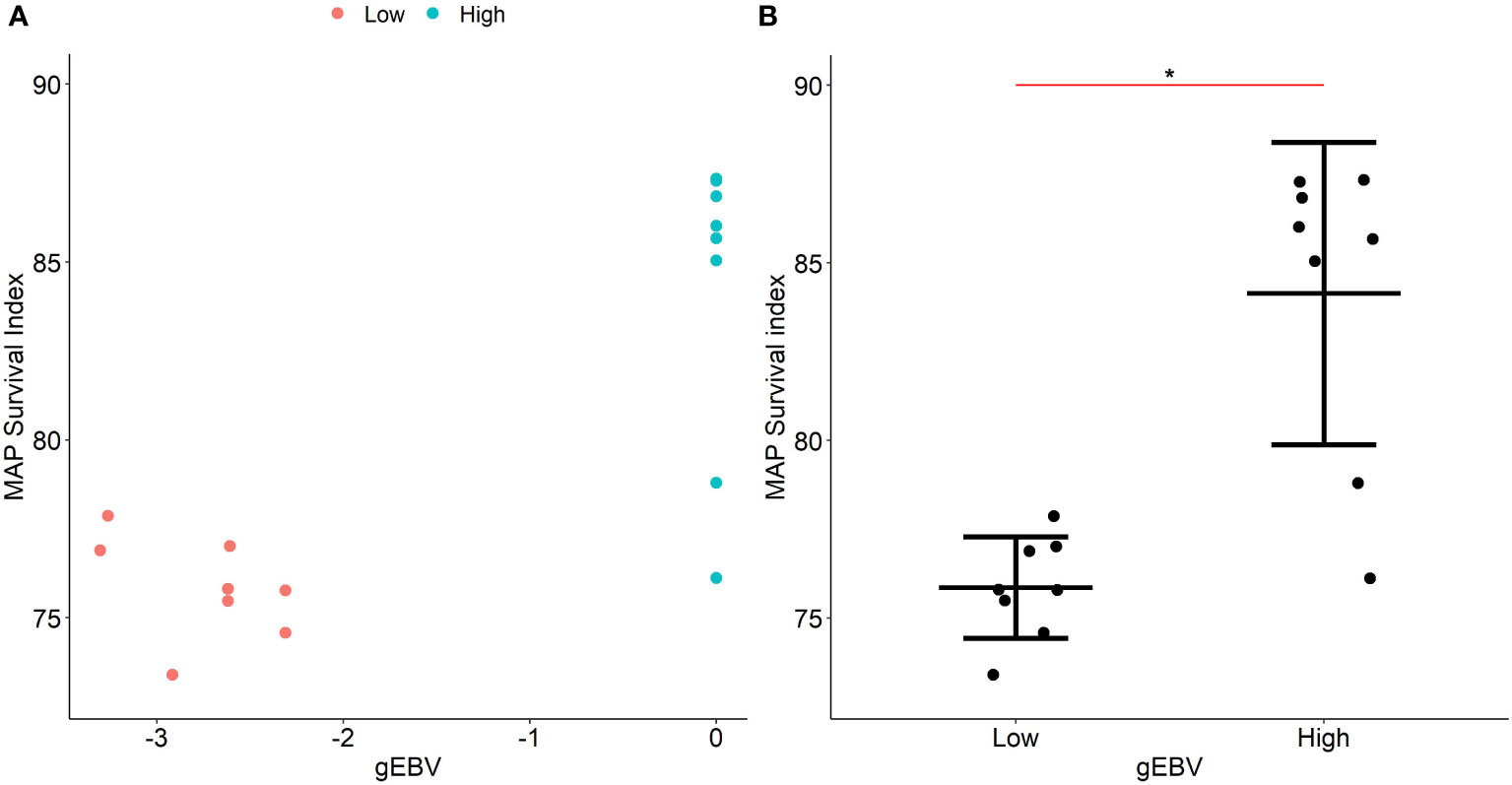
Figure 4 Validation of the gEBVs for MAP survival indexes within infected MDMs. (A). Scatter plots showing the distribution of the gEBVs for MAP survival within infected MDMs for the validation population. gEBVs for MAP survival indexes within MDMs were calculated for each animal in the study population using a gBLUP model and the genomic predictions were validated in a validation population consisting of animals with high (N= 8) and low (N= 8) gEBVs. (B). Experimental MAP survival indexes within MDMs from the validation population. MDMs from animals with high (N= 8) and low (N= 8) gEBVs were isolated, infected with MAP for 2 h and 7 d p. i., and the MAP survival indexes were calculated. Differences in the MAP survival indexes between animals with high and low gEBVs were statistically significant (P< 0.0006). In the validation population, the correlations between the experimental MAP survival indexes within MDMs and the gEBVs were positive (Pearson r = 0.78, P = 0.0003). The asterisk indicates statistically significant differences.
4 Discussion
Macrophages are important antigen presenter cells that participate in the destruction of pathogens by producing antimicrobial components after phagocytosis (39–44). Some animals might be able to induce a strong early innate immune response and limit MAP load within macrophages better than others. A considerable proportion of this inter-individual variability might be genetic (16). Defining the relevant phenotype is the main challenge in identifying resistant animals and the genetic profile of resistance against MAP infection. Since ex vivo assays measure the capacity of a cell population to respond to a challenge, functional testing represents the best way to evaluate resistance and vaccine protection (45). MGIA assays using bovine MDMs were previously used to assess macrophages´ s capacity to clear MAP strains isolated from different hosts (23, 24), to test novel therapeutic approaches for the treatment of MAP-infected animals (46), and to test vaccines’ efficacy and protection (47). Our study is the first in using a quantitative MGIA as an unbiased measure of the bovine MDMs´ ability to control MAP survival in vitro and as a correlate of resistance to MAP infection. Since the MGIA models MAP infection, it reveals immune parameters induced by in vivo infection and contributes to understanding the control of intracellular MAP load. In addition, evidence exists that the ability of the macrophages to produce an antimicrobial profile early after infection can cause differences in disease susceptibility or resistance in connection to their cellular responses (48–50). Previously, next-generation RNA (RNA-Seq) sequencing was used to study the transcriptomic response to MAP infection of the macrophages from cows that were naturally infected and identified as positive for JD (JD (+); n = 22) or negative for JD (healthy/resistant, JD (−); n = 28) (51). In addition to identifying genetic variants from RNA-seq data, SNP variants were also identified using the Bovine SNP50 DNA chip. Significant variants from JD (+/-) macrophages were identified by a genome-wide association study in a case-control approach and revealed two novel eQTLs on BTA4 and 11 (P< 5 × 10-7). This study was conducted using MDMs from cattle that were naturally infected with MAP. In our study, however, we used MDMs obtained from cattle that were negative by PCR for the detection of MAP DNA in fecal samples before blood collection and in subsequent annual samplings. Our study is the first in using a quantitative MGIA as an unbiased measure of the bovine MDMs´ ability to control MAP load and as a correlate of resistance to MAP infection. In our previous study (22), the integration of RNA-Seq data and genotypes from a cohort of cows naturally exposed to MAP allowed the identification of four cis-eQTLs regulating the expression of genes with important roles in the induction of the innate immune response early after infection; the C3, EREG, Gal9, and NOS1 genes. As shown in Supplementary Figure 1, none of the animals with the highest levels of expression of these genes showed diffuse lesions, which suggested that the measure of the levels of expression of these genes might be used to evaluate macrophages performance and for the identification of cattle able to limit MAP infection. The goal of the current study was to identify genetic loci associated with resistance to MAP infection by combining genotypes and phenotypes including the intracellular MAP load within infected MDMs and the extracellular expression of NO-, Gal9, C3, and EREG.
NOS1 belongs to the family of NO- synthases which synthesize NO- from L-arginine. NO- is one of the major macrophage-killing mechanisms (52). It was previously demonstrated that MAP interferes with NO- synthase expression early after infection (53). EREG belongs to the Epidermal Growth factor (EGF) family together with the Heparin Binding EGF-like Growth factor (HBEGF); acting both as mitogenic stimulators via binding to EGF receptors (EGFRs). EREG overexpression leads to downstream signaling events including Mitogen-Activated Protein Kinase 8 (MAPK8) phosphorylation and activation which results in apoptosis induction. Using RNA-Seq transcriptomic profiling, we previously detected down-regulation of EREG in peripheral blood and ileocecal valve from cows with diffuse lesions versus controls which may favor MAP survival (54). Gal9 is an S-type lectin with antimicrobial activity in infected macrophages by causing macrophage activation, Interleukin 1ß (IL1ß) secretion, and pathogen clearance (55, 56). Higher C3 levels may allow increased opsonophagocytosis and effective bacterial clearance of several microorganisms including mycobacteria (47, 57). In our study, negative correlations were observed between the extracellular NO- and the MAP load within infected MDMs at 2 h and 7 d p. i. The correlations between EREG and Gal9 and intracellular MAP load within MDMs were not statistically significant. On the other hand, positive correlations were found between MAP load within macrophages and C3 expression at 2 h and 7 d p. i.
One of the critical factors that influence the power of detection of SNP (s)-associated with a trait is how much of the phenotypic variance is explained by the associated SNP (s). The highest the h2 estimate, the chance of detecting SNPs increases. The h2 estimates obtained in our study for the MAP load (log CFUs) within infected MDMs at 2 h and 7 d p. i. were 0.87 and 0.83, respectively. These findings suggest a strong influence of host genetics on MAP load within infected macrophages. To our knowledge, the h2 estimate for the macrophages´ performance assessed by measuring MAP load within macrophages had not been estimated before and is higher than h2 estimates calculated for other immunocompetence traits. For instance, the analysis of MDMs phenotypic variation revealed an h2 = 0.77 for the NO- production by MDMs in response to E.coli (18). After performing the GWAS, we did not identify SNPs significantly associated with the NO-, Gal9, EREG, and C3 expression. However, six SNPs located on BTA2, BTA17, BTA18, and BTA21 were significantly associated with a lower MAP load within MDMs after 2 h p. i. This discordance can be explained because the MGIA is a functional assay that assesses the combined effects of a range of immune and metabolic mechanisms that influence mycobacteria growth in vitro, which is one of its advantages over measuring single predefined immune markers. It is important to highlight that for the assessment of viable intracellular MAP, we used the BACTEC MGIT 960 system, a fully automated solution for mycobacterial liquid culture that provides accurate measures of MAP load.
We identified six SNPs associated with low MAP load within MDMs in dairy cattle. Interestingly, the two identified SNPs located on BTA18 overlapped with QTLs associated with the somatic cell score (QTL:3556), length of productive life (QTL:3555), and reproductive traits such as stillbirth (QTL:11362), birth Index (QTL:30537), and dystocia (QTL:2707) highlighting the important role of this region (23.5-31.4 Mbp) on chromosome 18 in health, reproduction, and longevity (31–34, 58). This suggests that an animal that can limit MAP infection has also improved udder health, fewer reproductive problems, and, consequently, a longer productive life. In addition, the SNP located on BTA21 was located within QTLs associated with IgG levels (QTL:66212), somatic cell score (QTL:5446 and QTL:10190), and calving ease (QTL:11126) (35–38). This finding suggests that there might be genetic loci that impact both the primary innate immune response and the secondary production of IgG which is activated later if innate mechanisms are not able to eliminate the pathogen. Finally, the identified SNP on BTA17 was located within a QTL associated with calving ease (36). The identified SNPs on BTA17, BTA18, and BTA21 overlapped with QTLs for longevity, udder health, and reproductive traits. The identified markers could be useful for marker-assisted selection in dairy cattle breeding programs for PTB resistance in combination with other strategies like management and biosecurity.
Five candidate genes and one miRNA were identified 50,000 bp upstream and downstream of some of the identified SNPs; Cysteine and Serine Rich Nuclear Protein 3 (CSRNP3), the Coiled-Coil Domain Containing 92 (CCDC92), Oxysterol Binding Protein like 6 (OSBPL6), U6, a non-characterized protein (ENSBTAG0000004285), and the bta-mir-2365. Specific functions of the four identified candidate genes are summarized in Supplementary Table 2. CSRNP3, also named TGF-Beta Induced Apoptosis Protein 2, is a transcriptional activator involved in positive apoptosis regulation. Apoptotic clearance of pathogens including MAP plays a critical function in the resolution of the infection by the removal of the infected macrophages along with the ingested organisms. CCDC92 is an interferon-γ stimulated protein that plays an important role in the regulation of innate immunity and attenuation of microbial infections (59). The OSBPL6 is an intracellular lipid receptor that contributes to the maintenance of cholesterol homeostasis by regulating cellular cholesterol trafficking and efflux. Previously, genetic variants in candidate genes involved in cholesterol metabolism (bta04979) such as Angiopoietin-like 4 and 8 (ANGPTL4 and ANGPTL8), Low Lipropotein Receptor (LDRL), and Neutral cholesterol Ester Hydrolase 1 (NCEH1) appeared enriched in animals with PTB-associated diffuse lesions (22). Consequently, reduced levels of plasma cholesterol were detected in cattle with diffuse lesions. Moreover, we observed that the cholesterol route (bta04977) was dysregulated in the ileocecal valve of cows with diffuse lesions with four upregulated Apoliprotein genes (APOA1, APOC3, APOA4, APOB) matching this route (54). Recent data suggest that MAP-infected macrophages accumulate intracellular cholesterol droplets and depict a foam phenotype during infection providing an enriched environment for MAP survival (60, 61). In addition to the observations that increased lipid biogenesis and transport to the place of the infection may favor MAP growth by the modulation of host immune response leading to an anti-inflammatory milieu, MAP may potentially mobilize lipid body content as a source of nutrients to enhance their survival and replication in host cells (62). In an opposite manner, our study suggests that animals with genetic variants in candidate genes favoring cholesterol homeostasis such as the OSBPL6 might be able to limit MAP infection by reducing the lipid body content within infected MDMs. The U6 spliceosomal RNA and the bta-miR-2365 were also identified as candidate genes in our study. The U6 is a small non-coding RNA involved in the post-transcriptional regulation of gene expression by affecting both the stability and translation of mRNAs. Using miRmap, we predicted target genes for the bta-miR-2365. When we used ClusterProfiler with the predicted target genes, an enrichment in genes associated with the endocytosis (bta04144) and the Hippo signaling pathway (bta04390) was found. The Hippo signaling pathway controls organ size by coordinately regulating cell growth, proliferation, and apoptosis (63). Emerging evidence has suggested that the innate immune response was extensively regulated by multiple core components of the Hippo cascade while the activation of innate immune signaling conversely led to substantial modulations of the Hippo pathway (64). In most cases, the Hippo signaling potentiates the innate immune response. In addition, the Hippo pathway-mediated processes are interconnected with those of other key signaling cascades, such as those mediated by TGF-beta and Wnt growth factors. Therefore, the bta-miR-2365 along with the other candidate genes identified in our study could modulate MAP’s entry and survival within macrophages. The candidate genes identified in our study were also compared to candidate genes previously identified in CD, IBD, and colorectal cancer to determine if there was any overlap. We found that CSRNP3 was previously found to be associated with colorectal cancer (65). In clear cell renal cell carcinoma (ccRCC), the CSRNP3 may serve as a prognostic biomarker to predict the overall survival of patients (66). CSRNP3 might impact the immune environment of ccRCC through immunocyte infiltration of natural killer cells and plasmacytoid dendritic cells.
The sample size used in the current study could be considered small in comparison to traditional GWAS analysis using a larger population. However, the assessment of the macrophages´ performance by measuring MAP load within infected MDMs is a functional and controlled trait that can be measured in the absence of environmental and physiological effects. Other phenotypes, such as the NO- response of bovine MDMs in response to E.coli or S. aureus, used approximately 60 samples to identify associated cis-SNPs (18). Further studies are needed to assess the capacity of macrophages to kill MAP as a phenotype for resistance to MAP infection in other cattle populations and ruminant breeds.
5 Conclusions
We report a new phenotype, the control of MAP early survival within infected macrophages, for evaluating resistance to MAP infection in dairy cows. This resistance is genetically determined and is mediated at the level of macrophages´ function. Considering how difficult it is to assess resistance to MAP infection, our approach represents a major advance in the field. The SNPs and candidate genes identified here revealed the association between MAP load within macrophages and genetics, which helps fill the knowledge gap between genetics, innate immune responses, and resistance to MAP infection in cattle. Our results define a heritable and distinct immunogenetic profile in MAP-infected macrophages associated with cholesterol homeostasis regulation and with the induction of apoptosis and IFNγ production. A strong effect of host genetics in limiting bacteria load within MDMs very early after infection was observed (h2 estimate = 0.8) which opens the possibility of ranking Holstein cows based on predicted MAP survival variation within infected MDMs. The identified SNPs might be used to develop genetic evaluations for immunocompetence in the Spanish breeding program which would allow producers to select cattle more resistant to MAP infection and likely to other intracellular pathogens as well; ultimately reducing the prevalence of diseases and the dependence on antimicrobials, preventing economic losses, increasing the length of cattle productive life, and improving food safety.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was reviewed and approved by the Ethics Committee of Animal Experimentation of NEIKER.
Author contributions
MC measured intracellular MAP load within MDMs, extracellular EREG, C3, Gal9, and NO- levels, and performed the GWAS. GB-B performed the gBLUP model and contributed to the statistical analysis of the data. MC and GB-B performed the validation of the genomic predictions. MA-H was the principal investigator of the project and participated in project management, experimental design, and data analysis. All authors contributed to the article and approved the submitted version.
Funding
Funding support was provided by grants (RTI2018-094192-R-C21 and PID2021-122197OR-C21) funded by the Spanish Ministry of Science and Innovation (MCIN/AEI/10.13039/501100011033) and by European Regional Funds (FEDER, “Una manera de hacer Europa”). GB-B has been awarded a fellowship from MCIN/AEI/10.13039/501100011033 and “FSE Invierte en tu futuro”; grant PRE2019-090562.
Acknowledgments
This work has been possible thanks to the support of the computing infrastructure of the i2BASQUE Research and Academic Network. The authors want to express their gratitude to Dr. Almudena Fernández and Dr. Oscar González-Recio from the Animal Breeding Department, Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria, CSIC, Madrid, Spain, for useful comments and suggestions. We gratefully acknowledge Kyle P. Hearn for proofreading the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1042638/full#supplementary-material
References
1. Rasmussen P, Barkema HW, Mason S, Beaulieu E, Hall DC. Economic losses due to johne’s disease (paratuberculosis) in dairy cattle. J Dairy Sci (2021) 104:3123–43. doi: 10.3168/jds.2020-19381
2. Nielsen SS, Toft N. A review of prevalences of paratuberculosis in farmed animals in Europe. Prev Vet Med (2009) 88:1–14. doi: 10.1016/j.prevetmed.2008.07.003
3. Whittington R, Donat K, Weber MF, Kelton D, Nielsen SS, Eisenberg S, et al. Control of paratuberculosis: Who, why and how. a review of 48 countries. BMC Vet Res (2019) 15:1–29. doi: 10.1186/s12917-019-1943-4
4. Hailat N, Hemida H, Hananeh W, Stabel J, Rezig FE, Jaradat S, et al. Investigation on the occurrence and pathology of paratuberculosis (Johne’s disease) in apparently healthy cattle in Jordan. Comp Clin Pathol (2011) 21:879–88. doi: 10.1007/s00580-011-1192-9
5. Momotani E, Whipple DL, Thiermann AB, Cheville NF. Role of m cells and macrophages in the entrance of Mycobacterium paratuberculosis into domes of ileal peyer’s patches in calves. Vet Pathol (1988) 25:131–7. doi: 10.1177/030098588802500205
6. Sweeney RW. Pathogenesis of paratuberculosis. Vet Clin North Am - Food Anim Pract (2011) 27:537–46. doi: 10.1016/j.cvfa.2011.07.001
7. Bermudez LE, Petrofsky M, Sommer S, Barletta RG. Peyer’s patch-deficient mice demonstrate that Mycobacterium avium subsp. paratuberculosis translocates across the mucosal barrier via both m cells and enterocytes but has inefficient dissemination. Infect Immun (2010) 78:3570–7. doi: 10.1128/IAI.01411-09
8. Feller M, Huwiler K, Stephan R, Altpeter E, Shang A, Furrer H, et al. Mycobacterium avium subspecies paratuberculosis and crohn’s disease: a systematic review and meta-analysis. Lancet Infect Dis (2007) 7:607–13. doi: 10.1016/S1473-3099(07)70211-6
9. Juste RA, Elguezabal N, Garrido JM, Pavon A, Geijo MV, Sevilla I, et al. On the prevalence of m. avium subspecies paratuberculosis DNA in the blood of healthy individuals and patients with inflammatory bowel disease. PloS One (2008) 3:3–8. doi: 10.1371/journal.pone.0002537
10. Jeyanathan M, Boutros-Tadros O, Radhi J, Semret M, Bitton A, Behr MA. Visualization of Mycobacterium avium in crohn’s tissue by oil-immersion microscopy. Microbes Infect (2007) 9:1567–73. doi: 10.1016/j.micinf.2007.09.001
11. Pierce ES. Could Mycobacterium avium subspecies paratuberculosis cause crohn’s disease, ulcerative colitis and colorectal cancer? Infect Agent Cancer (2018) 13:1–6. doi: 10.1186/s13027-017-0172-3
12. González J, Geijo MV, García-Pariente C, Verna A, Corpa JM, Reyes LE, et al. Histopathological classification of lesions associated with natural paratuberculosis infection in cattle. J Comp Pathol (2005) 133:184–96. doi: 10.1016/j.jcpa.2005.04.007
13. Balseiro A, Perez V, Juste RA. Chronic regional intestinal inflammatory disease: A trans-species slow infection? Comp Immunol Microbiol Infect Dis (2019) 62:88–100. doi: 10.1016/j.cimid.2018.12.001
14. Canive M, Badia-Bringué G, Vázquez P, González-Recio O, Fernández A, Garrido JM, et al. Identification of loci associated with pathological outcomes in Holstein cattle infected with Mycobacterium avium subsp. paratuberculosis using whole-genome sequence data. Sci Rep (2021) 11:1–13. doi: 10.1038/s41598-021-99672-4
15. Canive M, González-Recio O, Fernández A, Vázquez P, Badia-Bringué G, Lavín JL, et al. Identification of loci associated with susceptibility to Mycobacterium avium subsp. paratuberculosis infection in Holstein cattle using combinations of diagnostic tests and imputed whole-genome sequence data. PloS One (2021) 16:1–17. doi: 10.1371/journal.pone.0256091
16. Alonso-Hearn M, Badia-Bringué G, Canive M. Genome-wide association studies for the identification of cattle susceptible and resilient to paratuberculosis. Front Vet Sci (2022) 9:935133. doi: 10.3389/fvets.2022.935133
17. Ko DC, Shukla KP, Fong C, Wasnick M, Brittnacher MJ, Wurfel MM, et al. A genome-wide In vitro bacterial-infection screen reveals human variation in the host response associated with inflammatory disease. Am J Hum Genet (2009) 85:214–27. doi: 10.1016/j.ajhg.2009.07.012
18. Emam M, Tabatabaei S, Sargolzaei M, Sharif S, Schenkel F, Mallard B. The effect of host genetics on in vitro performance of bovine monocyte-derived macrophages. J Dairy Sci (2019) 102:9107–16. doi: 10.3168/jds.2018-15960
19. Tanner R, Satti I, Harris SA, O’Shea MK, Cizmeci D, O’Connor D, et al. Tools for assessing the protective efficacy of TB vaccines in humans: in vitro mycobacterial growth inhibition predicts outcome of in vivo mycobacterial infection. Front Immunol (2020) 10:2983. doi: 10.3389/fimmu.2019.02983
20. Bok J, Hofland RW, Evans CA. Whole blood mycobacterial growth assays for assessing human tuberculosis susceptibility: A systematic review and meta-analysis. Front Immunol (2021) 12:641082. doi: 10.3389/fimmu.2021.641082
21. Brennan MJ, Tanner R, Morris S, Scriba TJ, Achkar JM, Zelmer A, et al. The cross-species mycobacterial growth inhibition assay (MGIA) project, 2010–2014. Clin Vaccine Immunol (2017) 24:2010–4. doi: 10.1128/CVI.00142-17
22. Canive M, Fernandez-Jimenez N, Casais R, Vázquez P, Lavín JL, Bilbao JR, et al. Identification of loci associated with susceptibility to bovine paratuberculosis and with the dysregulation of the MECOM, eEF1A2, and U1 spliceosomal RNA expression. Sci Rep (2021) 11:1–14. doi: 10.1038/s41598-020-79619-x
23. Abendaño N, Sevilla IA, Prieto JM, Garrido JM, Juste RA, Alonso-Hearn M. Mycobacterium avium subspecies paratuberculosis isolates from sheep and goats show reduced persistence in bovine macrophages than cattle, bison, deer and wild boar strains regardless of genotype. Vet Microbiol (2013) 163:325–34. doi: 10.1016/j.vetmic.2012.12.042
24. Alonso-Hearn M, Magombedze G, Abendaño N, Landin M, Juste RAJ. Deciphering the virulence of Mycobacterium avium subsp. paratuberculosis isolates in animal macrophages using mathematical models. Theor Biol (2019) 468:82–91. doi: 10.1016/j.jtbi.2019.01.040
25. Abendaño N, Sevilla I, Prieto JM, Garrido JM, Juste RA, Alonso-Hearn M. Quantification of Mycobacterium avium subsp. paratuberculosis strains representing distinct genotypes and isolated from domestic and wildlife animal species by use of an automatic liquid culture system. J Clin Microbiol (2012) 50:2609–17. doi: 10.1128/JCM.00441-1
26. Martinez R, Toro R, Montoya F, Burbano M, Tobón J, Gallego J, et al. Bovine SLC11A1 3’ UTR SSCP genotype evaluated by a macrophage in vitro killing assay employing a brucella abortus strain. Anim Breed Genet (2008) 125:271–9. doi: 10.1111/j.1439-0388.2008.00727.x
27. Price RE, Templeton JW, Smith R, Adams LG. Ability of mononuclear phagocytes from cattle naturally resistant or susceptible to brucellosis to control in vitro intracellular survival of brucella abortus. Infect Immun (1990) 58:879–86. doi: 10.1128/iai.58.4.879-886.1990
28. Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: A tool for genome-wide complex trait analysis. Am J Hum Genet (2011) 88:76–82. doi: 10.1016/j.ajhg.2010.11.011
29. Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc Ser B (1995) 57:289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
30. VanRaden PM, Sullivan PG. International genomic evaluation methods for dairy cattle. Genet Sel Evol (2010) 42:1–9. doi: 10.1186/1297-9686-42-7
31. Brand B, Baes C, Mayer M, Reinsch N, Kühn C. Identification of a two-marker-haplotype on bos taurus autosome 18 associated with somatic cell score in German Holstein cattle. BMC Genet (2009) 10:50. doi: 10.1186/1471-2156-10-50
32. Höglund JK, Guldbrandtsen B, Su G, Thomsen B, Lund MS. Genome scan detects quantitative trait loci affecting female fertility traits in Danish and Swedish Holstein cattle. J Dairy Sci (2009) 92:2136–43. doi: 10.3168/jds.2008-1104
33. Kühn C, Bennewitz J, Reinsch N, Xu N, Thomsen H, Looft C, et al. Quantitative trait loci mapping of functional traits in the german Holstein cattle population. J Dairy Sci (2003) 86:360–8. doi: 10.3168/jds.S0022-0302(03)73614-5
34. Muncie SA, Cassady JP, Ashwell MS. Refinement of quantitative trait loci on bovine chromosome 18 affecting health and reproduction in US holsteins. Anim Genet (2006) 37:273–5. doi: 10.1111/j.1365-2052.2006.01425.x
35. Leach RJ, Craigmile SC, Knott SA, Williams JL, Glass EJ. Quantitative trait loci for variation in immune response to a foot-and-Mouth disease virus peptide. BMC Genet (2010) 11:1–14. doi: 10.1186/1471-2156-11-107
36. McClure MC, Morsci NS, Schnabel RD, Kim JW, Yao P, Rolf MM, et al. A genome scan for quantitative trait loci influencing carcass, post-natal growth and reproductive traits in commercial Angus cattle. Anim Genet (2010) 41:597–607. doi: 10.1111/j.1365-2052.2010.02063.x
37. Rodriguez-Zas SL, Southey BR, Heyen DW, Lewin HA. Interval and composite interval mapping of somatic cell score, yield, and components of milk in dairy cattle. J Dairy Sci (2002) 85:3081–91. doi: 10.3168/jds.S0022-0302(02)74395-6
38. Schulman NF, Viitala SM, De Koning DJ, Virta J, Mäki-Tanila A, Vilkki JH. Quantitative trait loci for health traits in Finnish Ayrshire cattle. J Dairy Sci (2004) 87:443–9. doi: 10.3168/jds.S0022-0302(04)73183-5
39. Gordon S. Pattern recognition receptors: doubling up for the innate immune response. Cell (2002) 111:927–30. doi: 10.1016/s0092-8674(02)01201-1
40. Rabinovich GA, Riera CM, Sotomayor CE. Galectin-1, an alternative signal for T cell death, is increased in activated macrophages. Braz J Med Biol Res (1999) 32:557–67. doi: 10.1590/s0100-879x1999000500009
41. Silva MT. Macrophage phagocytosis of neutrophils at inflammatory/infectious foci: a cooperative mechanism in the control of infection and infectious inflammation. J Leukoc Biol (2011) 89:675–83. doi: 10.1189/jlb.0910536
42. Gordon S. Elie metchnikoff, the man and the myth. J Innate Immun (2016) 8:223–7. doi: 10.1159/000443331
43. Lavin Y, Mortha A, Rahman A, Merad M. Regulation of macrophage development and function in peripheral tissues. Nat Rev Immunol (2015) 15:731–44. doi: 10.1038/nri3920
44. Hirayama D, Iida T, Nakase H. The phagocytic function of macrophage-enforcing innate immunity and tissue homeostasis. Int J Mol Sci (2017) 19:92. doi: 10.3390/ijms19010092
45. Albert-Vega C, Tawfik DM, Trouillet-Assant S, Vachot L, Mallet F, Textoris J. Immune functional assays, from custom to standardized tests for precision medicine. Front Immunol (2018) 9:2367. doi: 10.3389/fimmu.2018.02367
46. Canive M, Badia-Bringué G, Alonso-Hearn M. The upregulation of Cathepsin g is associated with resistance to bovine paratuberculosis. Animals (2022) 12(21):3038. doi: 10.3390/ani12213038
47. Juste RA, Alonso-Hearn M, Garrido JM, Abendaño N, Sevilla IA, Gortazar C, et al. Increased lytic efficiency of bovine macrophages trained with killed mycobacteria. PloS One (2016) 11:1–12. doi: 10.1371/journal.pone.0165607
48. Cheers C, Ho M. Resistance and susceptibility of mice to bacterial infection. IV. functional specificity in natural resistance to facultative intracellular bacteria. J Reticuloendothel Soc (1983) 34:299–309.
49. Morissette C, Francoeur C, Darmond-Zwaig C, Gervais F. Lung phagocyte bactericidal function in strains of mice resistant and susceptible to pseudomonas aeruginosa. Infect Immun (1996) 64:4984–92. doi: 10.1128/iai.64.12.4984-4992
50. Gross WB, Siegel PB. Environment-genetic influences on immunocompetence. J Anim Sci (1988) 66:2091–4. doi: 10.2527/jas1988.6682091x
51. Ariel O, Brouard JS, Marete A, Miglior F, Ibeagha-Awemu E, Bissonnette N. Genome-wide association analysis identified both RNA-seq and DNA variants associated to paratuberculosis in Canadian Holstein cattle ‘in vitro’ experimentally infected macrophages. BMC Genomics (2021) 22:162. doi: 10.1186/s12864-021-07487-4
52. Weiss G, Schaible UE. Macrophage defense mechanisms against intracellular bacteria. Immunol Rev (2015) 264:182–203. doi: 10.1111/imr.12266
53. Sommer S, Pudrith CB, Colvin CJ, Coussens PM. Mycobacterium avium subspecies paratuberculosis suppresses expression of IL-12p40 and iNOS genes induced by signalling through CD40 in bovine monocyte-derived macrophages. Vet Immunol Immunopathol (2009) 128:44–52. doi: 10.1016/j.vetimm.2008.10.294
54. Alonso-Hearn M, Canive M, Blanco-Vazquez C, Torremocha R, Balseiro A, Amado J, et al. RNA-Seq analysis of ileocecal valve and peripheral blood from Holstein cattle infected with Mycobacterium avium subsp. paratuberculosis revealed dysregulation of the CXCL8/IL8 signaling pathway. Sci Rep (2019) 9:1–17. doi: 10.1038/s41598-019-51328-0
55. Sada-Ovalle I, Chávez-Galán L, Torre-Bouscoulet L, Nava-Gamiño L, Barrera L, Jayaraman P, et al. The Tim3–galectin 9 pathway induces antibacterial activity in human macrophages infected with Mycobacterium tuberculosis. J Immunol (2012) 189:5896–902. doi: 10.4049/jimmunol.1200990
56. Jayaraman P, Sada-Ovalle I, Nishimura T, Anderson AC, Kuchroo VK, Remold H, et al. IL-1β promotes antimicrobial immunity in macrophages by regulating TNFR signaling and caspase-3 activation. J Immunol (2013) 109(8):4196–204. doi: 10.4049/jimmunol.1202688
57. De La Fuente J, Gortázar C, Juste R. Complement component 3: A new paradigm in tuberculosis vaccine. Expert Rev Vaccines (2016) 15:275–7. doi: 10.1586/14760584.2016.1125294
58. Höglund JK, Guldbrandtsen B, Lund MS, Sahana G. Analyzes of genome-wide association follow-up study for calving traits in dairy cattle. BMC Genet (2012) 13:71. doi: 10.1186/1471-2156-13-71
59. Kuroda M, Halfmann PJ, Hill-Batorski L, Ozawa M, Lopes TJS, Neumann G, et al. Identification of interferon-stimulated genes that attenuate Ebola virus infection. Nat Commun (2020) 11:2953. doi: 10.1038/s41467-020-16768-7
60. Johansen MD, de Silva K, Plain KM, Whittington RJ, Purdie AC. Mycobacterium avium subspecies paratuberculosis is able to manipulate host lipid metabolism and accumulate cholesterol within macrophages. Microb Pathog (2019) 130:44–53. doi: 10.1016/j.micpath.2019.02.031
61. Ariel O, Gendron D, Dudemaine PL, Gévry N, Ibeagha-Awemu EM, Bissonnette N. Transcriptome profiling of bovine macrophages infected by Mycobacterium avium spp. paratuberculosis depicts foam cell and innate immune tolerance phenotypes. Front Immunol (2020) 10:2874. doi: 10.3389/fimmu.2019.02874
62. Wright K, Mizzi R, Plain KM, Purdie AC, de Silva K. Mycobacterium avium subsp. paratuberculosis exploits miRNA expression to modulate lipid metabolism and macrophage polarisation pathways during infection. Sci Rep (2022) 12:1–14. doi: 10.1038/s41598-022-13503-8
63. Pan D. Hippo signaling in organ size control. Genes Dev (2007) 21:886–97. doi: 10.1101/gad.1536007.
64. Wang S, Zhou L, Ling L, Meng X, Chu F, Zhang S, et al. The crosstalk between hippo-YAP pathway and innate immunity. Front Immunol (2020) 11:323. doi: 10.3389/fimmu.2020.00323
65. Cheng THT, Thompson D, Painter J, Omara T, Gorman M, Martin L, et al. Meta-analysis of genome-wide association studies identifies common susceptibility polymorphisms for colorectal and endometrial cancer near SH2B3 and TSHZ1. Sci Rep (2015) 5:1–12. doi: 10.1038/srep17369
Keywords: paratuberculosis, mycobacterial growth inhibition assay (MGIA), host resistance, macrophages, innate immune response, phagocytosis, immunoprofiling
Citation: Badia-Bringué G, Canive M and Alonso-Hearn M (2023) Control of Mycobacterium avium subsp. paratuberculosis load within infected bovine monocyte-derived macrophages is associated with host genetics. Front. Immunol. 14:1042638. doi: 10.3389/fimmu.2023.1042638
Received: 12 September 2022; Accepted: 09 February 2023;
Published: 22 February 2023.
Edited by:
Felix Ngosa Toka, Ross University School of Veterinary Medicine, Saint Kitts and NevisReviewed by:
Thomas Hall, University College Dublin, IrelandNathalie Bissonnette, Agriculture and Agri-Food Canada (AAFC), Canada
Copyright © 2023 Badia-Bringué, Canive and Alonso-Hearn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marta Alonso-Hearn, bWFsb25zb0BuZWlrZXIuZXVz