
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 17 February 2023
Sec. Vaccines and Molecular Therapeutics
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1036563
 Jing Pan1,2
Jing Pan1,2 Donghai Xiong1,2
Donghai Xiong1,2 Qi Zhang1,2Katie Palen3
Qi Zhang1,2Katie Palen3 Robert H. Shoemaker4
Robert H. Shoemaker4 Bryon Johnson3
Bryon Johnson3 Shizuko Sei4Yian Wang1,2
Shizuko Sei4Yian Wang1,2 Ming You1,2*
Ming You1,2*Epidermal growth factor receptor (EGFR) mutations occur in about 50% of lung adenocarcinomas in Asia and about 15% in the US. EGFR mutation-specific inhibitors have been developed and made significant contributions to controlling EGFR mutated non-small cell lung cancer. However, resistance frequently develops within 1 to 2 years due to acquired mutations. No effective approaches that target mutant EGFR have been developed to treat relapse following tyrosine kinase inhibitor (TKI) treatment. Vaccination against mutant EGFR is one area of active exploration. In this study, we identified immunogenic epitopes for the common EGFR mutations in humans and formulated a multi-peptide vaccine (Emut Vax) targeting the EGFR L858R, T790M, and Del19 mutations. The efficacy of the Emut Vax was evaluated in both syngeneic and genetic engineered EGFR mutation-driven murine lung tumor models with prophylactic settings, where the vaccinations were given before the onset of the tumor induction. The multi-peptide Emut Vax effectively prevented the onset of EGFR mutation-driven lung tumorigenesis in both syngeneic and genetically engineered mouse models (GEMMs). Flow cytometry and single-cell RNA sequencing were conducted to investigate the impact of Emut Vax on immune modulation. Emut Vax significantly enhanced Th1 responses in the tumor microenvironment and decreased suppressive Tregs to enhance anti-tumor efficacy. Our results show that multi-peptide Emut Vax is effective in preventing common EGFR mutation-driven lung tumorigenesis, and the vaccine elicits broad immune responses that are not limited to anti-tumor Th1 response.
Lung cancer is the second most common cancer and the leading cancer killer, accounting for 25% of all cancer-related deaths (1). The majority of lung cancers (about 85%) are non-small cell lung cancer (NSCLC), and epidermal growth factor receptor (EGFR) mutations are the most frequent driver mutation in lung cancer, occur in around 50% of Asia-Pacific patients with NSCLC and 15% of Western patients (2). Although EGFR mutations occur more frequently in females, Asians, and never smokers, they are not restricted to patients with these characteristics. More than 50% of patients with EGFR mutations in the PIONEER study were not female non-smokers (2). These findings strongly support EGFR mutation testing in all patients with NSCLC and suggest that targeting EGFR for prevention will have a significant impact on controlling this disease.
Clinically approved therapeutics for NSCLC include small molecule tyrosine kinase inhibitors (TKIs) and monoclonal antibodies such as Cetuximab and Panitumumab (3–5). Advanced NSCLC patients harboring mutations in the EGFR gene show initially good responses to TKIs but ultimately relapse (6). The monoclonal antibodies are also limited in clinical use due to limited efficacy, toxicity, and the emergence of resistance (7, 8). While immune checkpoint blockade has shown encouraging effects in subtypes of NSCLC patients, relatively poor response in EGFR mutant NSCLC patients means that those patients who develop resistance to EGFR inhibitors have no effective treatment options. Thus, there is an urgent need to develop new strategies for safer and more efficacious therapeutics.
Key concepts in lung cancer disease control are prevention of lung cancer progression in patients with premalignant lesions and prevention of lung cancer recurrence in previously treated patients. Previous research indicates that EGFR mutations are not only an “early event” occurring during the initiation of lung cancer (9), but they are also the molecular driver of NSCLC (10, 11). The most common EGFR mutations (>90%) are exon 21 mutations resulting in L858R substitutions (~30–45%), and in-frame deletions in exon 19 (~40–60%). EGFR mutation-specific inhibitors have been developed for controlling NSCLC with EGFR mutations. However, second mutations (e.g., T790M) are a common mechanism of resistance. Therefore, alternative approaches to prevent tumor progression in EGFR mutated patients are needed.
Tumor-specific antigens (TSA) can be processed and presented by cell surface major histocompatibility complex (MHC) proteins as tumor mutation-encoding neoantigen (NeoAg) epitopes, which can serve as targets for tumor-specific T cell-mediated killing (12, 13). Several NeoAg vaccines have been developed from preclinical models with significant antitumor efficacy, and recent clinical trials using peptide-based or RNA-based NeoAg vaccine approaches have demonstrated successful induction of NeoAg-specific T cells response with improved clinical outcomes in melanoma patients (14–18). Furthermore, personalized NeoAg peptide vaccine trials for glioblastoma patients were shown to successfully induce antigen specific CD4+ and CD8+ T cell responses and increased T cell infiltration in tumors. Multiple epitopes specific to mutant EGFR were identified, including epitopes specific to L858R and T790M, and vaccines incorporating these epitopes induced immune responses, with clinical responses observed in some patients (19). These findings suggest that targeting neoantigens derived from EGFR mutations could be a useful treatment strategy for NSCLC patients with EGFR-TKI resistance. While the majority of these epitopes are MHC I specific, MHC II epitopes can be designed to target multiple HLA-DR alleles and are thus applicable for broader populations of cancer patients (20–24).
Induction of Th1 helper CD4+ T cell immunity is critical for immunomodulation-mediated cancer eradication (23). MHC II-restricted peptide vaccines can elicit NeoAg-specific Th1 immunity that orchestrates tumor-reactive CD8+ cytotoxic T lymphocyte (CTL) responses, for example, by increasing the number of antigens available to cytotoxic CD8+ T cells through the restoration of MHC class I expression on tumor and immune cells, providing cytokines to facilitate CD8+ responses, and by facilitating the generation of robust immunologic memory. Recently, Th1 cytotoxic T cell function has also been observed in various immune settings. Th1 Cytotoxic CD4+ T cells can directly kill cancer cells in an MHC II-restricted fashion through the secretion of granzyme B and perforin (12, 25, 26). We previously reported an MHC II-restricted EGFR multi-peptide vaccine that targets the wild-type EGFR protein and reduces EGFR-driven lung tumor burdens in a preclinical murine model (22). However, this vaccine is not specific to the mutant forms of EGFR; therefore, in this study, we extended our MHC II-restricted multi-peptide EGFR vaccine to include peptides for common EGFR mutations including the L746–S750 deletion, (Del19) T790M and L858R. We identified the most immunogenic epitopes specific to these mutations and tested this mutation-specific, multi-peptide vaccine, Emut Vax, in both syngeneic and genetically engineered mouse models harboring mutant forms of EGFR.
For the syngeneic tumor model, we used the UN-SCC680 (UN680) lung cancer cell line that expresses the L858R/T790M or Del19 (L746-S750) mutant human EGFR protein. pBabe_EGFR(L858R/T790M) and pBabe_ EGFR(Del19) were gifts from Dr. Matthew Meyerson.
(Addgene plasmid # 32073 and #32062; http://n2t.net/addgene:32062; RRID : Addgene_32062 http://n2t.net/addgene:32073; RRID : Addgene_32073). Lentivirus particles were generated by co-transfecting pBabe_EGFR(L858R/T790M) and pBabe_EGFR(Del19) with packaging plasmids (pVSVG) into HEK293T cells. To overexpress EGFR L858R/T790M or Del19 mutations, UN680 cells were transduced with pBabe_EGFR(L858R/T790M) and pBabe_EGFR (Del19) lentivirus particles and then were selected in complete growth medium contains puromycin (1~2µg/ml). The protein expression of L858R/T790M, Del19, and phosphorylated EGFR was confirmed by western blot analysis.
The mouse NSCLC UN680 cells derived from the A/J background were a generous gift from Dr. Jackeline Agorreta. UN680-derivatives UN680L858R/T790M and UN680Del746-750 were established to represent NSCLC harboring common EGFR mutations. Five- to six-week-old female A/J mice were purchased from the Jackson Laboratory. After one week, they were grouped according to similar body weights into two groups: (1) adjuvant control mice receiving CFA/IFA; (2) Emut Vax group receiving EGFR mutant-specific peptides suspended in adjuvant once per week for a total of four doses. For the syngraft model, UN680L858R/T790M or UN680Del746-750 cells (5 × 106 cells in 0.1 ml of PBS) were inoculated subcutaneously into the flanks of experimental and control mice one week after the fourth vaccine (Day 0). The growth of UN680L858R/T790M or UN680Del746-750 tumors was measured every four to six days by a digital caliper. Tumor volumes were calculated using the formula: V = (length × width × width)/2 (27).
To further test Emut Vax in the prophylaxis setting, an inducible genetically engineered mouse model was used. Transgenic mice harboring human EGFR containing both L858R and T790M mutations (EGFRL858R/T790M mice) on a C57BL/6 background were requested from the NCI mouse repository. EGFRL858R/T790M mice were bred with CCSP mice that expresses the Tet-on Clara Cell Secreted Protein (CCSP) on the C57BL/6 background to generated double transgenic mice (CCSP-EGFRL858R/T790M), which will have lung-specific expression of the transgene EGFRL858R/T790M upon doxycycline induction. All mice were housed in the Biomedical Resource Center at the Medical College of Wisconsin, Milwaukee, WI. All procedures were approved by the institutional animal care and use committee (IACUC). For primary lung cancer experiments, 4-5 week-old CCSP-EGFRL858R/T790M mice were divided into the following two groups: (1) adjuvant control mice that received complete Freund’s adjuvant (CFA) and incomplete Freund’s adjuvant (IFA); (2) Emut Vax treated mice that received the EGFR mutants specific peptides suspended in adjuvant. Doxycycline-containing diets (TD. 01306, Envigo Teklad) were administered as food pellets starting one week after initial 4-weekly vaccinations.
To predict HLA class II epitopes for each mutant EGFR of interest in our study, we used a newly developed algorithm, MixMHC2pred (28). Briefly, each mutation was represented by a sequence that was centered by the mutation site with 14 amino acids extended from both sides, such that the typical sequence length is 29 amino acids (14 + mutation site + 14). We then took 15-amino-acid sliding windows along these sequences and generate an average of 9 different peptides for each mutation, then ran each peptide (mutant form or wild type form) with the most common human HLA alleles to generate a MixMHC2pred score, % Rank_best, which corresponds to a percentile rank (best score is about 0, worst score is 100) of the given peptide expected to be presented by the alleles.
EGFR mutation-specific peptides (Supplementary Table 1) that are 100% identical between humans and mice were synthesized with >95% purity by Genemeds, and their identity was confirmed by HPLC. Peptides were dissolved in 10% DMSO/PBS at 10mg/ml. CFA and IFA were used as adjuvants throughout the study. The vaccine was prepared by mixing an equal volume of peptides in PBS with CFA or IFA till a uniform water-in-oil emulsion formed. Each mouse received 50 μg of each peptide for one vaccine injection. The total vaccine volume was 100 μL/mouse. Mice were injected subcutaneously in the right flank with the first vaccination prepared in CFA starting at 5 weeks of age, followed by 4 weekly booster vaccines prepared in IFA, and then additional boosters administered every 4 weeks thereafter in the GEMM as shown in Figures 1A, 2A.
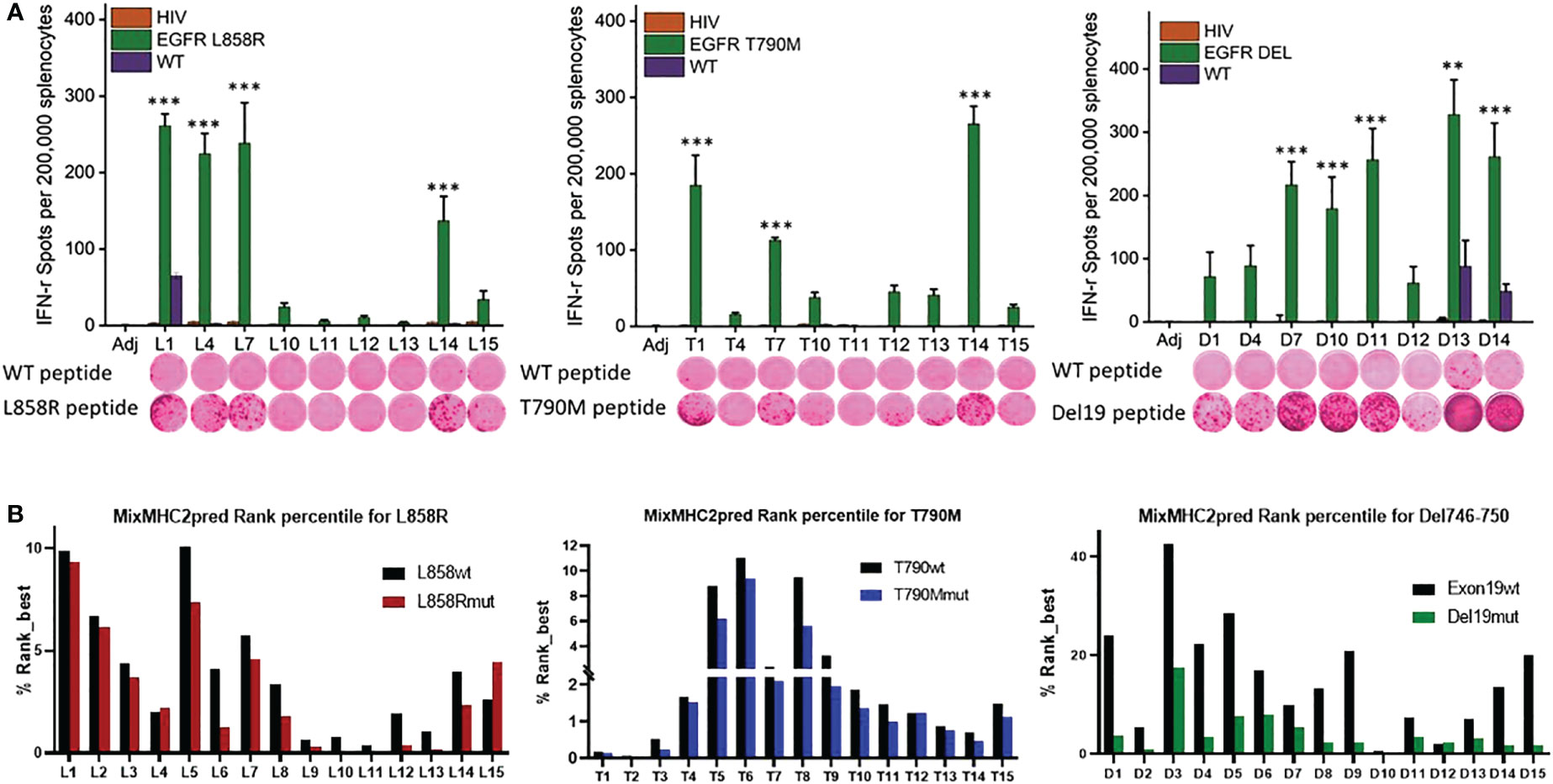
Figure 1 Identification of immunogenic epitopes specific to common EGFR mutations. (A) Quantified (bars in panels) and representative (inset images) ELISPOT results showing T cell responses from naïve mouse splenocytes stimulated with wild-type peptides (first row images) or mutation-specific peptides (second row images). Mice were vaccinated 4 times weekly with individual Emut peptides, splenocytes were collected 7 days after the last vaccination, and the splenocytes pulsed with negative HIV control peptide, each EGFR wild type or mutant peptide. After 72h of incubation, ELISPOT assays were performed, plates were scanned, and spot numbers were statistically analyzed. Data are shown as the mean ± SE of three replicate wells per group, n=5, *P ≤ 0.05, **P ≤ 0.01, ***P<0.001. (B) MixMHC2pred prediction for best-ranked peptides specific to L858R (top panel), T790M (middle panel) and Del19 (bottom panel) EGFR mutations, the general score (%Rank_best) was predicted with the default algorithm setting that includes over 50 most common human MHCII molecules, and the score represents the percentile rank of the peptide expected to be presented and immunogenic with these common alleles.
Spleens were mechanically mashed through a 40 μm cell strainer (BD). Red blood cells were lysed in ACK lysis buffer for 2 min, neutralized with complete RPMI1640 medium, centrifuged at 1000g for 3 min, then re-suspended in T cell medium (RPMI1640 plus 10% FBS, 1% pen/strep and 0.05 mM β-ME). 100 µl of splenocytes (3.0x105 cells/ml) were seeded onto a MAIPS4510 Multiscreen 96-well plate that was coated with anti-IFN-γ capture antibody. The cells were stimulated with mutation-specific peptides, concanavalin A (positive control), an HIV peptide (negative control), and wild-type peptides (negative control) for 72hr. The plates were then washed four times with PBS with 0.5% Tween-20 and the anti-IFN-γ detection antibody (BD) was added for overnight culture at 4°C. After washing with PBS four times, HRP-streptavidin was added for 1hr incubation, and then developed by adding AEC substrate following the manufacturer’s instruction. Plates were scanned and quantified by an automated plate reader system (CTL Technologies).
Splenocytes isolated from ADJ or Emut Vax group of mice were stimulated with Emut peptides for 72 hr, then the supernatant was harvested and sent to EVE’s Technologies (Calgary, AB Canada) for analysis with Mouse Cytokine Array (MD31, https://www.evetechnologies.com/product/mouse-cytokine-array-chemokine-array-31-plex/).
To monitor lung tumor development, CCSP-EGFRL858R/T790M mice were imaged by a 9.4T MRI (Bruker, Billerica, MA) equipped with a customized birdcage style quadrature coil. Animals were anesthetized with isoflurane (2% for induction and ~1.5% for maintenance). Mouse body temperature, heart rate, and respiratory rate were continuously monitored throughout the imaging procedure. Both respiratory and cardiac gating using an electrocardiogram was used to ensure that images were acquired during latent periods of the respiratory cycle and at a consistent point during the cardiac cycle. Tumors were scanned using a multi-slice, multi-echo acquisition (MSME). The following parameters were used to acquire the images: TE = 8.07 ms, TR ≥ 400 ms (variable), matrix = 128 × 128, on average, 20 axial slices. The Dicom viewer was used to process the coronal slice images.
Mouse lungs were inflated and formalin-fixed overnight and transferred to 70% ethanol. Fixed lung tissues were then paraffin-embedded (Sakura Tissue Tek VIP5), sectioned at 5 μm for H&E staining. The NanoZoomer slide scanner was used to scan and measure.
H&E slides of lungs were also scored according to a grading scheme that was previously developed for this same GEMM (29). Briefly, 0 indicates no noticeable lesions; 1 indicates minor abnormalities, the majority of the lung is intact with rare tiny atypical adenomas and multifocal mild atypical type II pneumocyte hyperplasia; 2 indicates minor lesions: small areas of mild atypical hyperplasia or atypical adenomas, large regions are unaffected; 3 indicates moderate lesions: large regions of atypical hyperplasia; occasional well differentiated and demarcated adenocarcinomas with no lymphovascular invasion; 4 indicates moderately severe lesions: multifocal to coalescing adenocarcinomas and atypical adenomas that affect the majority of a lung lobe; 5 is for severe lesions: diffuse involvement, adenocarcinoma is invasive with lymph vascular invasion.
To analyze the tumor-infiltrating T cells, tumors were first minced into 1-2 mm3 pieces and then processed into single cell suspensions using mouse tumor dissociation kits (Miltenyi Biotec, CA) according to the manufacturer’s instructions. Tumor-infiltrating leukocytes (TILs) were enriched by centrifugation on 40–70% Percoll (GE) gradients. Cells were first stained with fixable live/dead dye in PBS, after wash then followed with surface staining buffer containing fluorochrome-conjugated anti-CD45, anti-CD4, anti-CD8a, anti-CD44, and anti-CD62L antibodies. For intracellular cytokine staining (ICS), cells were stimulated for 4 h in T medium (RPMI1640, 10% FBS, 2 mM L-glutamine, 50 μM 2-mercaptoethanol, 1% penicillin-streptavidin) containing KRas peptides (10ug/ml), 1x monensin, 1x Brefeldin A (Thermofisher Sci). Stimulated cells were then stained with fixable live/dead dye in PBS, followed by cell surface staining buffer containing anti-CD45, anti-CD4, and anti-CD8 antibodies. After washing, the cells were fixed with 2% paraformaldehyde at room temperature for 10min, permeabilized with FOXP3 Fix/Perm Buffer Set (Biolegend), and stained with 1x Perm buffer containing anti-granzyme B, anti-IFN-γ and anti-TNFα antibody, and analyzed by flow cytometry. T cells stained with isotype control antibody were used as negative controls. Flow cytometry was analyzed using an LSR-II flow cytometer (Becton Dickinson). Data were processed with FlowJo software (Tree Star).
Lungs from transgenic CCSP-EGFRL858R/T790M mice were perfused with PBS through the right ventricle letting the blood flow out of the premade incision at the left ventricle till the lungs turned white, then tumors were microdissected from each mouse in the adjuvant or Emut Vax group at the end of the efficacy study. scRNA-seq and data analysis was conducted as previously reported (30).
GraphPad Prism 9.0 software was used for statistical analysis.
Two-tailed Student’s t-test or one-way ANOVA were used for multiple columns or group analyses. *P< 0.05, **P< 0.01, or ***P< 0.001 were considered as significant.
To identify the most immunogenic epitopes, each somatic mutation was centered by a sequence with 14 amino acids upstream and downstream of the mutation site, such that the sequence length was 29 amino acids. 15-amino-acid sliding windows along these sequences were taken (Supplementary Table 1) and every single peptide was evaluated in naïve A/J wild-type mice. Mice were first given 4 weekly vaccinations with each different single epitope, and splenocytes were harvested 7 days after the last vaccination for testing in IFN-γ ELISPOT assays. Splenocytes were re-stimulated with either the vaccinated mutant forms of epitopes, the corresponding wildtype peptides, or a nonspecific HIV peptide. As shown in Figure 1A, L858R peptides L1, L4, L7 and L14 induced strong immune responses specific to the mutant forms of the vaccine peptides. In contrast, all vaccinated mice showed no response to nonspecific HIV epitope stimulation. Of note, L1 elicited a relatively weak response to the wild-type form of the EGFR peptide, implying L1 vaccination may lead to potential side effects by targeting wildtype EGFR. For the T790M mutation, T1, T7, and T14 exhibited strong immune responses to the specific mutant forms of the peptides; none of these peptides induced immune recognition of the wildtype EGFR or nonspecific HIV peptides. For Del746-750, almost all the mutant epitopes generated a strong immune response except for D1, D4 and D12 in naïve mice, and these immune responses were specific to the mutant epitopes but not to nonspecific HIV or wildtype EGFR epitopes, except for D13 and D14, which induced weak responses to the wildtype EGFR (Figure 1A).
Using the MHC II prediction algorithm MixMHC2pred, which predicts MHC II binding, epitope processing, cleavage, and antigen presentation, we compared the MixMHC2pred score between wildtype and mutated epitopes with the most common HLA-II alleles in humans. The MixMHC2pred general score (%Rank_best) represents the percentile rank of the peptide expected to be presented and immunogenic with these common alleles, the best score is about 0, and the worst score is 100, the lower score represents a better predicted immunogenicity. The mutations of EGFR shifted these epitopes towards a more immunogenic state. As shown in Figure 1B lower panel, Del19mut epitopes had lower %Rank_Best scores from this algorithm versus wildtype epitopes, which indicates better binding and presentation and is in line with our findings that the majority of the Del746-750 epitopes demonstrated strong and specific immune responses when tested in naïve mice. Mice immunized with adjuvant didn’t develop antigen-specific IFN-γ response (P<0.001, Figure 1A). To include peptides specific against mutant forms of EGFR, peptides L4, L7 and L14 (Figure 1A upper left panel), T1, T7, T14 (Figure 1A lower panel), and D7, D10 and D13 (Figure 1A upper right panel) were chosen to formulate a multi-peptide Emut Vax in the preventive efficacy study.
To test the preventive efficacy of the newly formulated Emut Vax in immunocompetent NSCLC mouse models, we first used a syngeneic lung tumor, UN680, a mouse NSCLC cell line developed in the A/J mouse background after chronic chemical carcinogenesis, which is responsive to multiple immunomodulatory agents including anti-PD1 (31). The UN680 cells were engineered to overexpress either the L858R/T790M double mutation or Del746-750 mutation, and expression of the EGFR mutant forms was confirmed by western blotting (Figure 2A). The mutant lines also express higher levels of phosphorylated EGFR (data not shown). In this model, Emut Vax was given subcutaneously weekly for four weeks, and the tumor cells were inoculated one week later. Compared to the adjuvant control group, Emut Vax decreased tumor load by ~70% (Figure 2B) for both EGFR mutant tumor cell lines, indicating a strong protective effect. IFN-γ ELISpot assay of splenocytes showed that vaccinated animals significantly expressed IFN-γ T cell response upon peptide re-stimulation (Figure 2C). The reactivity to the EGFR peptides was highly specific to this mutant EGFR, which indicated by the lack of response present in non-vaccinated but tumor-bearing animals, suggesting that the immune responses were induced by vaccination but not the boosting effect promoted by tumor inoculation. Although the peptides chosen for the Emut Vax were all immunogenic when tested alone in naïve mice, when combined into one vaccine, the ELISPOT immune response was strongest with T790M and Del746-750 specific peptides.
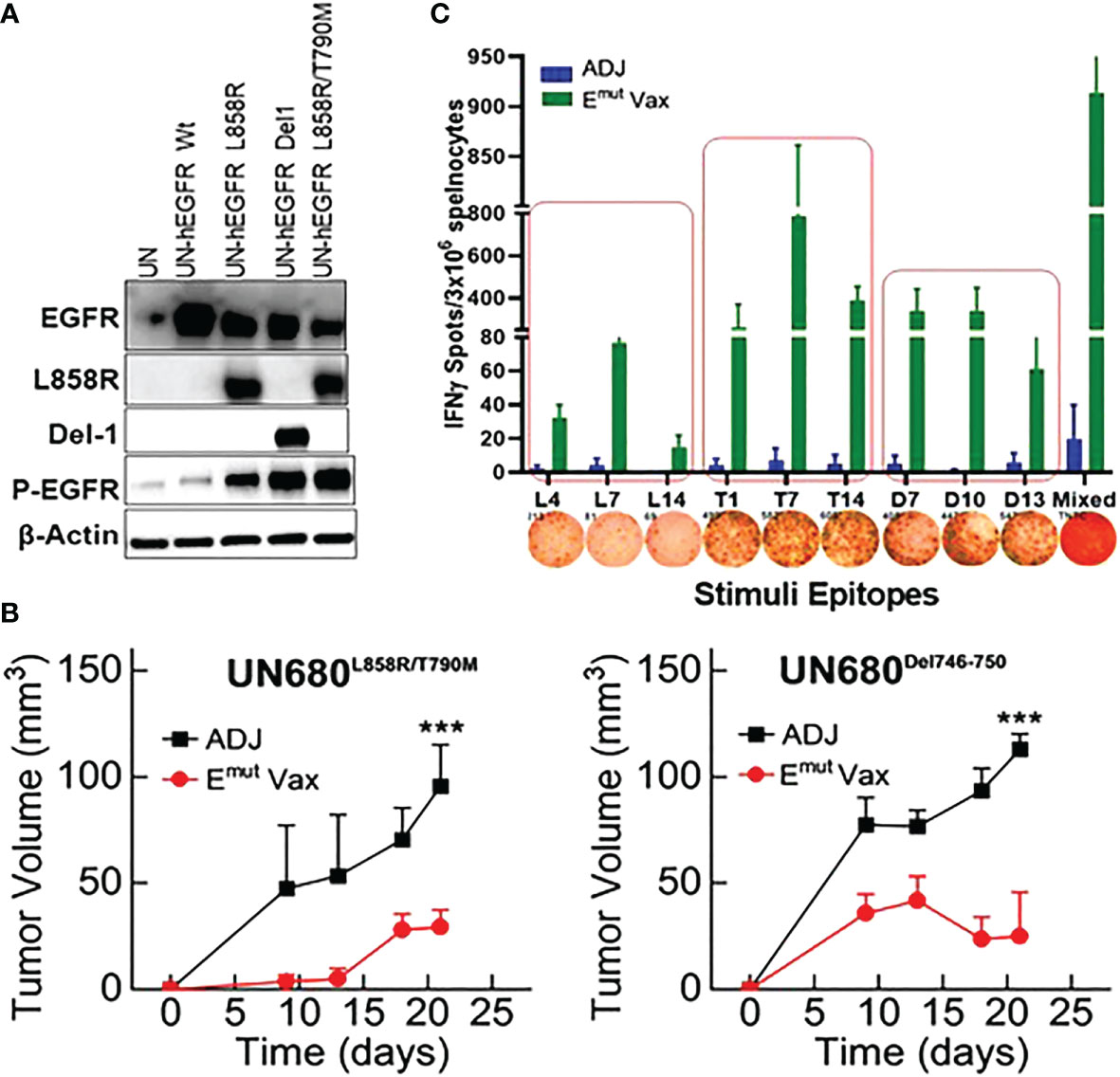
Figure 2 Emut Vax inhibits lung tumor progression in syngraft models. (A) Western blots to confirm expression of different mutant forms of EGFR in UN680 cells. (B) Tumor volume quantitation over time in the L858R/T790M resistant UN680 syngraft tumor model (left panel) or UN680 Del19 syngraft model (right panel). (C) Quantified and representative (insets) ELISPOT results from the spleens harvested at the endpoint of the efficacy study from the UN680 syngraft models. Data were combined from both L858R/T790M resistant or Del19 syngraft models. Data are the mean ± SE, n=10, ***P ≤ 0.001 vs Adj Ctrl.
Flow cytometry analysis from splenocytes harvested at the endpoint revealed that overall percentages of CD4+ and CD8+ cells of vaccinated animals were not significantly different from adjuvant control groups. However, the frequencies of IFN-γ-secreting CD4+ and CD8+ T cells was significantly increased in splenocytes harvested from the vaccinated mice (Figure 3). In the meantime, the frequencies of CD4+ T cells secreting granzyme B and CD8+ T cells secreting TNF-α were also showed a trend of increase (Figure 3). According to these findings, Emut Vax-induced T cell responses are specific to the mutant peptides and aid in the development of an adaptive anti-tumor response. Since none of the animals responded to an irrelevant antigen (HIV peptides), this further confirming the idea that the immune response seen in the vaccinated group was highly specific to mutant EGFR.
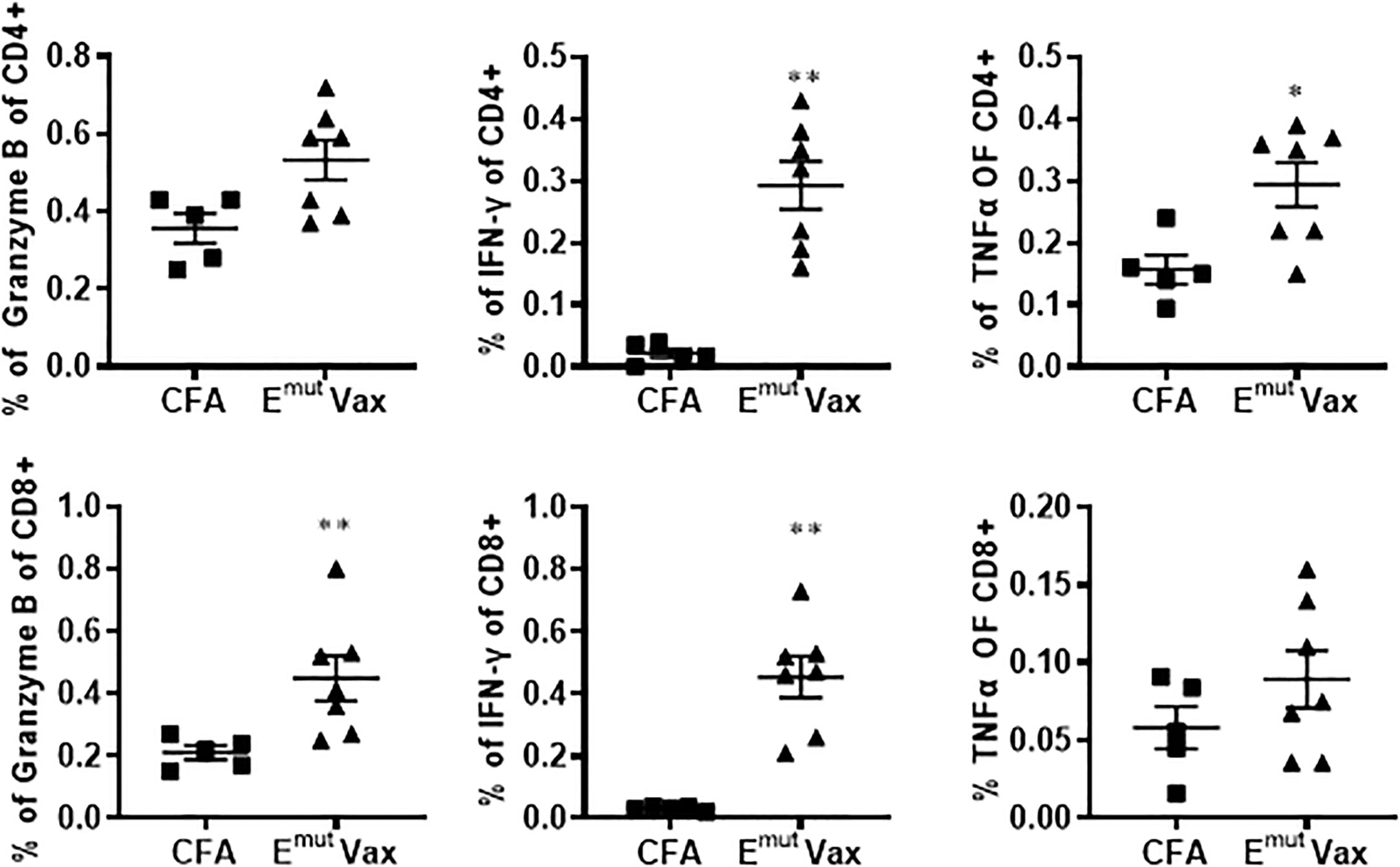
Figure 3 Emut Vax increased Th1 immune responses in a tumor-bearing syngraft model. Spleens were harvested from mice in the UN680 Del19 syngraft model, restimulated with Emut multi-peptides, and frequencies of Th1 cytokine and cytolytic granule-producing CD4+ and CD8+ were analyzed. Data are shown as the mean ± SE of three replicate samples per group, n=5 in ADJ and n=7 in Emut VAX, *P ≤ 0.05, **P ≤ 0.01 vs ADJ Ctrl.
We employed mice designed to express full-length human EGFR with the L858R and T790M mutations to assess Emut Vax. This animal model can develop lung adenocarcinomas quickly, are resistant to TKI treatment, and represent patients who have undergone first line TKI standard of care therapy with no further treatment options. Mice were vaccinated according to the treatment schedule in Figure 4A, and doxycycline administration was started one week following the last vaccine. Representative MRI imaging showed that while the non-vaccinated animals receiving adjuvant treatment displayed hypercellular lung parenchyma with few air spaces, the vaccine group displayed normal alveolar structures with little evidence of lung disease but some evidence of aggregates with a lymphoid character (Figure 4B).
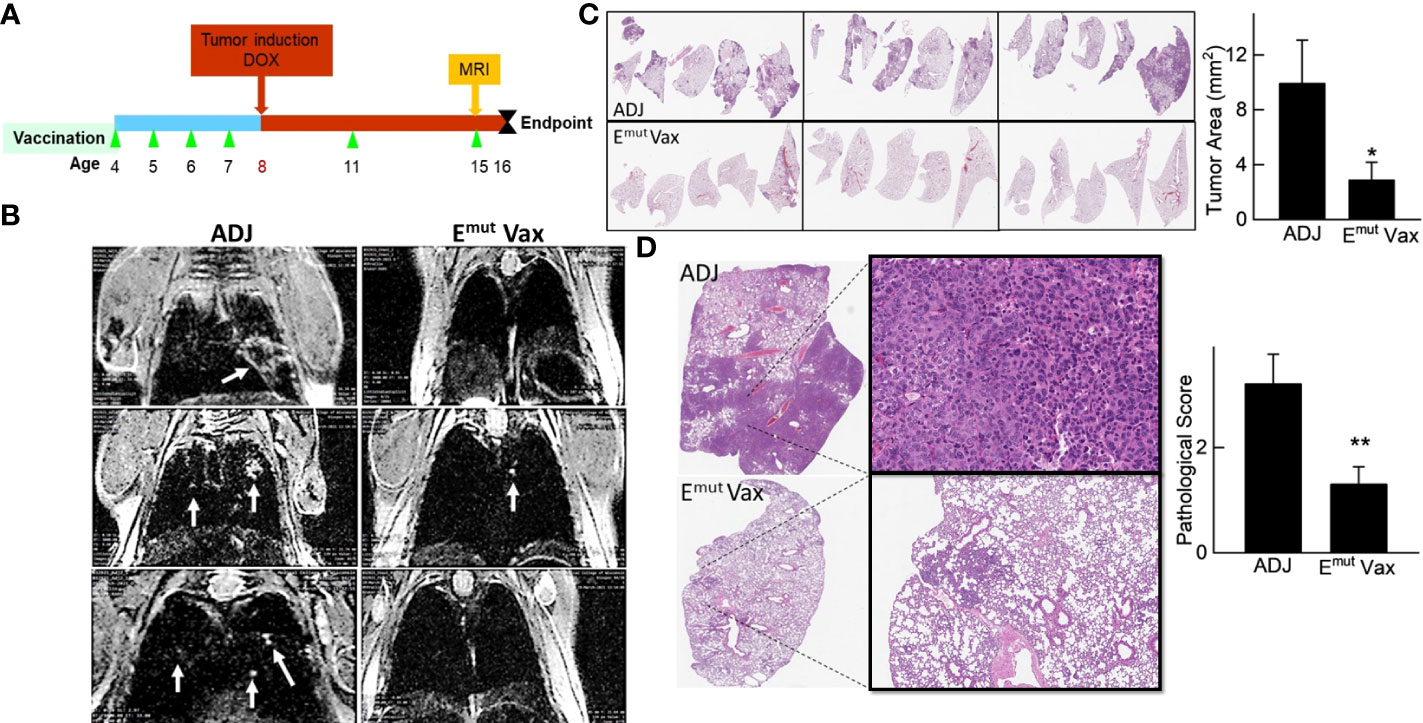
Figure 4 Emut Vax inhibits lung tumor progression in a genetically engineered mouse model. (A) Experimental design outlining the timing of vaccine administration and tumor induction with DOX diet, the time point for MRI monitoring, and the experimental endpoint. (B) Representative MRI images from mice injected with either adjuvant control or Emut Vax. (C) Representative lung lobes (left panel) and quantification of tumors (right panel) from each group. (D) Representative lung lobes (left panel) and pathological scores (right panel) from each group. Data are shown as the mean ± SE, n=10, *P ≤ 0.05, **P≤0.01.
As previously reported and outlined in the Methods section, tumors in the transgenic animals were located within the lung parenchyma, these tumors were hard to be quantified on the surface of the lung. Slides from different anatomic regions were analyzed. The tumor load within each animal was averaged to evaluate changes. Vaccination decreased tumor burden after induction of the EGFR transgene. Vaccinated animals demonstrated an average of 2.5 mm2 tumor area, versus an average of 10.76 mm2 tumor area found in unvaccinated animals, equating to a 75% reduction in tumor burden (Figure 4C; P = 0.0014). To quantify the histopathology response to Emut, the morphology and extent in the tumors were graded, with scores ranging from 0 (no lesions) to 5 (multifocal to consolidating adenocarcinoma) based on previously described criteria (29). The histopathologic severity grades were mostly 4 and up in the adjuvant group, and the lung lobes were affected with moderately severe lesions, consisting of multifocal to coalescing adenocarcinomas, atypical adenomas and adenocarcinomas. Emut Vax prevented tumor progression and decreased pathological score from 3.2 in the adjuvant group to 1.3 (Figure 4D); most tumors were grade 2, which is defined as occasional atypical adenomas with rare borderline/early adenocarcinoma.
To reveal the specific changes in tumor infiltrating lymphocyte compositions and gene expression in Emut VAX treated animals, scRNA-seq was performed on lung tumors harvested from transgenic CCSP-EGFRL858R/T790M mice in the adjuvant or Emut Vax group at the end of the efficacy study. We utilized the Seurat package (32, 33) to perform fine clustering of RNA sequenced single cells in mouse lung tumor samples from the adjuvant and Emut VAX treatment groups. Single-cell gene expression data were aligned and projected in 2 dimensions through UMAP (34). The gene expression patterns of canonical markers were analyzed to characterize different immune cell populations in the tumor samples. Nine different immune cell populations were detected in the lung tumor samples including CD4+ T cells, Treg, Tgd (gamma/delta T cells), CD8+ T cells, neutrophils, B cells, natural killer (NK) cells, macrophages, and dendritic cells (DC), etc. (Supplementary Figures 1A, B). Compared to the adjuvant control group, Emut VAX treatment increased CD4+ T, CD8+ T, Tgd cells, B cells, and DC, and decreased macrophages, neutrophils, and Treg cells (Figure 5). These changes suggest that overall compositional changes in immune cells towards those exhibiting anti-tumor reactivity occur after Emut VAX treatment.
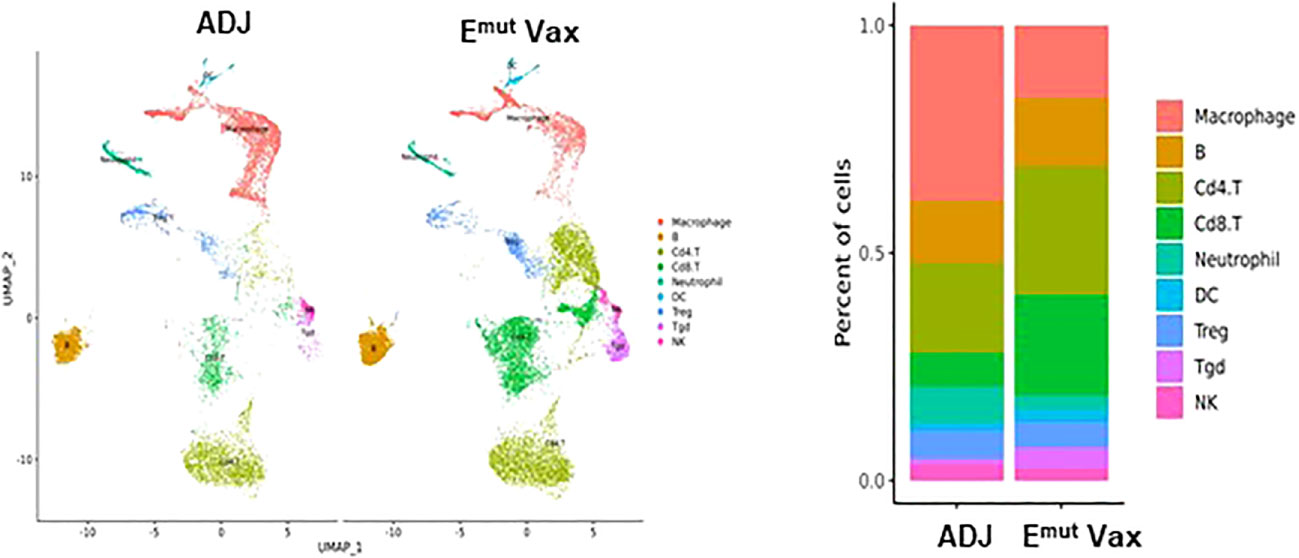
Figure 5 Clustering analysis of scRNA-seq data from tumor-infiltrating cells in adjuvant controls and Emut Vax treated mice. Left panel: The overall immune landscape across adjuvant control and Emut Vax treatment groups. Right panel: The percentage changes of each immune cell population between the adjuvant control and Emut Vax treatment groups. Emut Vax versus ADJ group cell percentage comparison: Macrophage: P = 0.008; B cells: P = 0.09; Cd4+ T cells: P = 0.02; Cd8+ T cells: P = 0.03; Neutrophil: P = 0.05; DC: P = 0.043; Treg: P = 0.11; Tgd: P = 0.052; NK: P = 0.36.
We analyzed all CD4+ T cells in mouse lung tumors from the control and treatment groups (Figure 6A) and identified five types of CD4+ T cell subsets, i.e., CD4CM (CD4+ central-memory T cells), CD4TH1 (Th1 cells), CD4TH9 (Th9 cells), CD4TH17 (Th17 cells) and Treg cell subpopulations (Figures 6B, C). Emut Vax treatment significantly increased proportions of anti-tumor CD4+ Th1, Th9 and Th17 cells in the lung tumors, while the vaccine decreased the abundance of Treg cells (Figure 6D). Cytokine analysis from Emut Vax treated spleens also confirmed the upregulated IFN-Ɣ secretion, as well as IL-1β, IL-2, IL-4, IL-9 and IL-17 upregulation (Figure 6E). IL-1β together with IL-4 could induce antitumor Th9 cell differentiation even in the absence of TGFβ. IL-2 signaling is important for the development of both Tregs and Th9 cells, while the enhanced IL-6 is critical for the inhibition of Foxp3+ Tregs and favors Th17 differentiation. Our results indicate that the anti-tumor function of the CD4+ T cells, especially CD4+ Th1 cells, was significantly enhanced by Emut Vax, together with the complex cytokine network that may control the transdifferentiation of different T helper lineages.
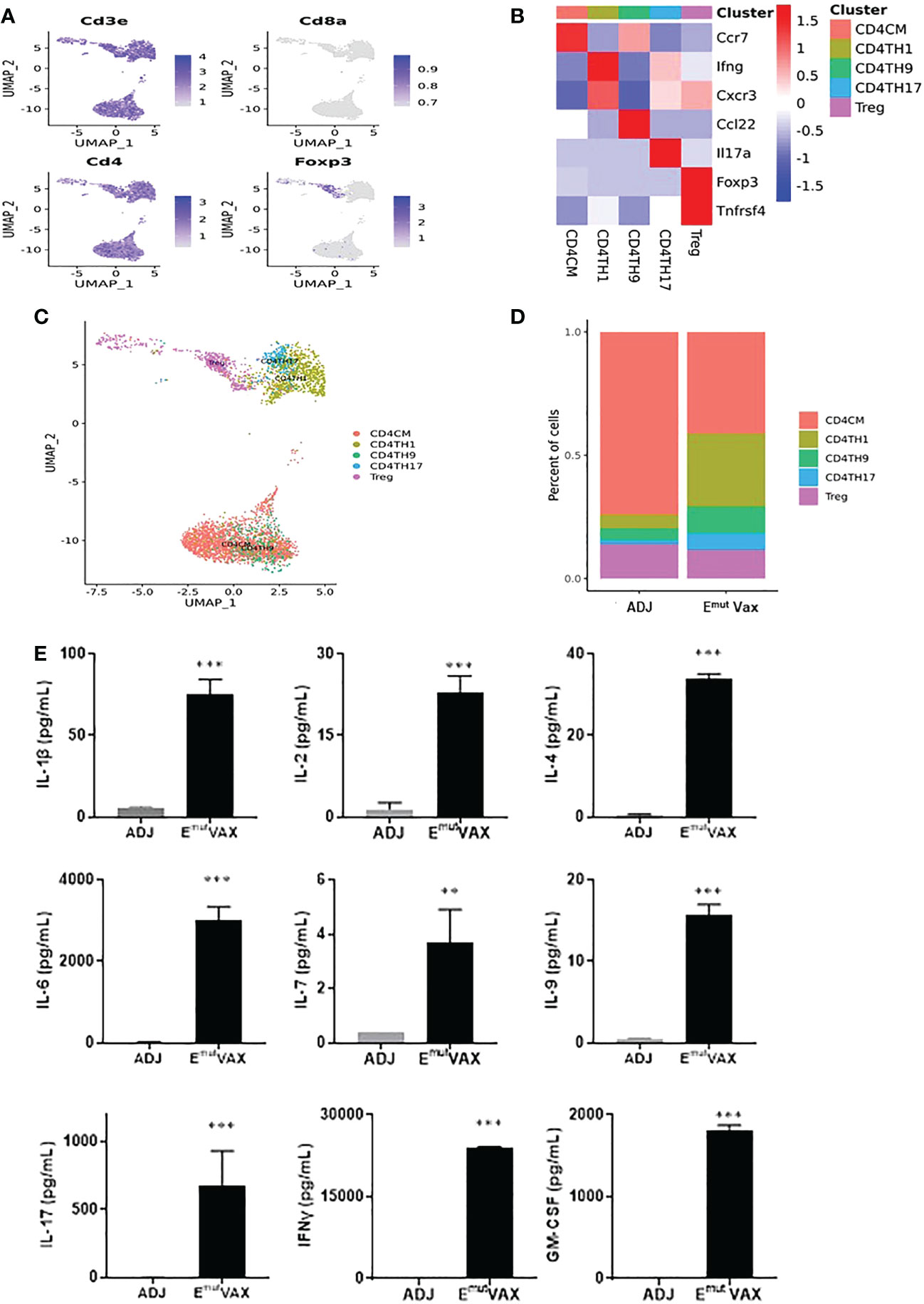
Figure 6 Emut Vax treatment stimulated anti-tumor CD4+ effector T cells within tumor-bearing lungs. (A) Canonical CD4+ T cell marker expression based on scRNA-seq data. (B) Heatmap of the expression markers for each CD4+ T cell subset. (C) The distribution of each CD4+ T cell subset. (D) Percent changes of the CD4+ T cell subsets across control and Emut Vax treatment groups. Data are shown as the mean ± SE, n=5, CD4CM: P = 0.032; CD4TH1: P = 0.016; CD4TH9: P = 0.037; CD4TH17: P = 0.046; Treg: P = 0.12. (E) Cytokine analysis from Emut Vax treated mice vs ADJ mice. Splenocytes were harvested at the endpoint, restimulated with Emut peptide vaccines for 72hr, then supernatant was collected for the cytokine analysis. Data are shown as the mean ± SE, n=5, **P ≤ 0.01, ***P ≤ 0.001 vs ADJ control.
To determine the effects of Emut Vax treatment on different subsets of CD8+ T cells in mouse lung tumors, deep clustering of the CD8+ T cells was performed. Identification of CD8+ T cell subsets was performed by using canonical markers (Figure 7A). The TILPRED program was used for unsupervised clustering of CD8+ T cells (35) and three CD8+ T cells subsets with distinct transcriptomic profiles including effector memory (EM)-like, memory-like, and naïve cells were identified (Figures 7B, C). In the lung tumor microenvironment, Emut Vax treatment greatly increased the abundance of the anti-tumor EM-like CD8+ T cells (Figure 7D). The frequency of memory-like CD8+ T cells was also increased, while that of naive CD8+ T cells was decreased by Emut Vax treatment. These results suggest that the Emut Vax improves the overall composition of antitumor CD8+ T cells in the lung tumor microenvironment.
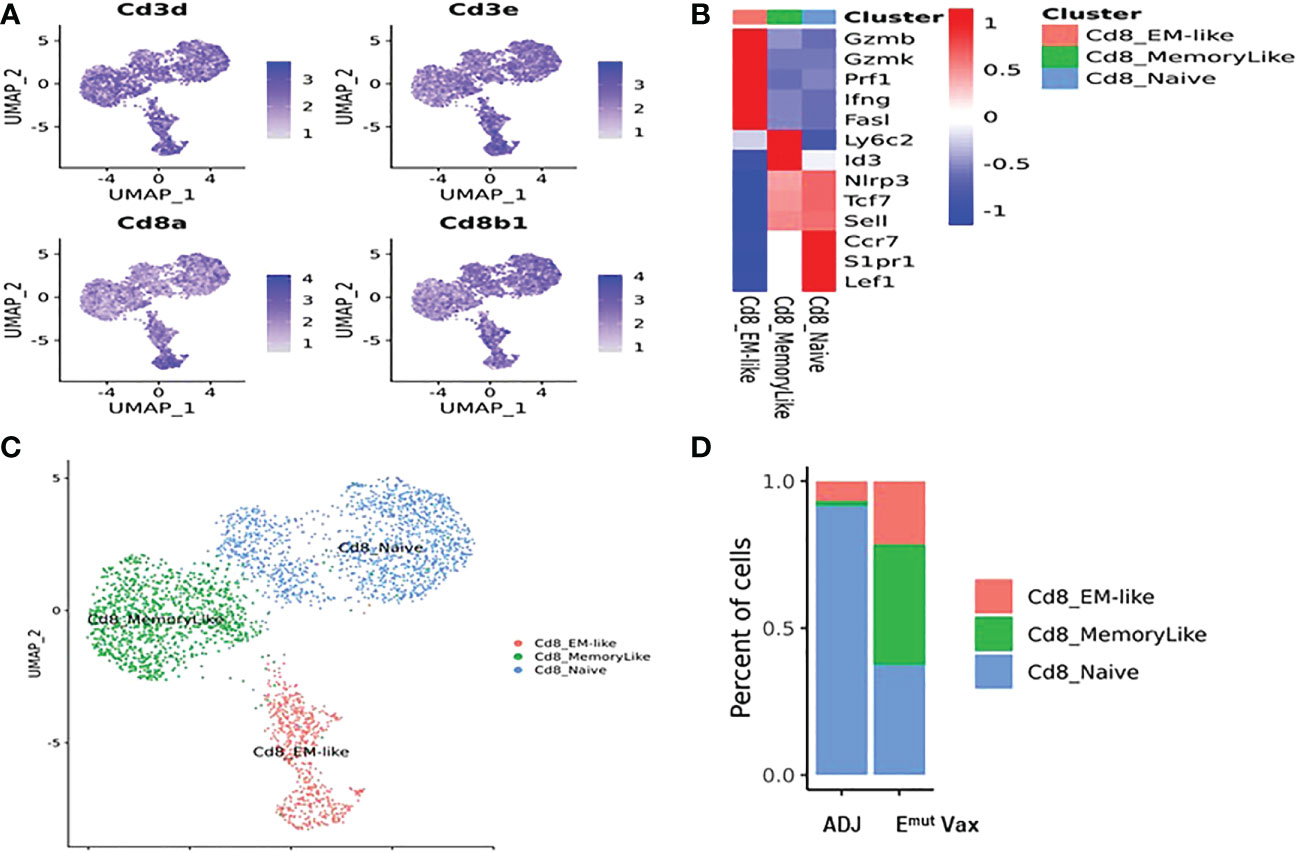
Figure 7 Emut Vax treatment increased anti-tumor CD8+ cytotoxic T cells within tumor-bearing lungs. (A) Canonical CD8+ T cell marker expression based on scRNA-seq data. (B) Heatmap of the expression markers for each CD8+ T cell subset. (C) Distribution of each CD8+ T cell subset; (D) Percent changes of the CD8+ T cells subsets across adjuvant control and Emut Vax treatment groups. CD8_EM-like: P = 0.013; CD8_MemoryLike: P = 0.007; CD8_Naive: P = 0.009.
We further analyzed the other immune cell populations in the mice lung TME for their association with the Emut VAX treatment. Macrophages were subdivided into the M1 and M2 macrophages according to the marker gene expression (Supplementary Figures 1B, 2). It was found that the abundance of the anti-tumor M1 macrophages increased and the pro-tumor M2 macrophages decreased greatly in the Emut VAX treatment groups compared to the control group (Supplementary Figure 2D). These indicated that the anti-tumor response by Emut VAX treatment may also be mediated via macrophages. We also identified two subpopulations of the NK cells: the CD27-CD11b+ NK cells corresponding to the human CD56dim NK cells which have strong cytotoxic activity and the CD27+CD11b- NK cells corresponding to the human CD56bright NK cells which are pre-mature and less cytotoxic (8). The total NK cells were identified by the expression of the Ncr1 gene (Supplementary Figure 3A) and the respective signature scores were used to locate each of these two NK cell subtypes (Supplementary Figures 3B, 3C). Under the Emut VAX treatment, the proportion of the cytotoxic CD27-CD11b+ NK cells increased significantly while that of the pre-mature CD27+CD11b- NK cells decreased (Supplementary Figure 3D), suggesting the involvement of the cytotoxic NK cells in the anti-tumor function of the Emut VAX.
EGFR is one of the most frequently mutated oncogene families that drives human NSCLC development (23). EGFR mutations are not only an “early event” occurring during the initiation of lung cancer (9), they are the molecular driver of NSCLC (10, 11). Therefore, approaches to target EGFR mutation to prevent tumor progression in EGFR mutated populations are an urgent need. Several EGFR inhibitors have been demonstrated to be highly effective in cancer patients with EGFR mutants. However, these inhibitors mostly do not show a significant benefit on overall survival due to secondary mutations that result in resistance to TKIs (18). Few strategies are available once patients become resistant to TKI treatments.
Deletions in exon 19 OR a point mutation in exon 21 L858R are the most common EGFR alterations associated with TKI sensitivity. About 90% of all EGFR alterations are these two alterations (36), which can serve as ideal targets for cancer immunoprevention. Here, we provided the first proof of principle study showing that a multi-peptide vaccine specific to common EGFR mutations (Emut Vax) elicits a strong immune response and provides protection against lung tumorigenesis when given in the preventive setting. We selected peptides that induced a relatively strong Th1 response for the final vaccine formulation. Emut Vax was very effective to prevent tumor development, which showed a 75% decrease in tumor volume in a highly aggressive transgenic model that recapitulates salient features of human EGFR lung cancers. The amino acid peptides we used are 100% identical between humans and mice. Furthermore, we used the newly developed MixMHC2 to predict the most common human HLA class II proteins and found the mutated epitopes generated better MHC-II binding and presentation for human HLA class II. Therefore, the peptides formulated Emut Vax not only had strong immunogenicity in mice but also have a high likelihood of human class II binding, making it possible to be directly translated to clinical studies. EGFR mutant-specific immune responses were not seen in diseased, non-vaccinated adjuvant control mice, indicating that the mice are tolerant to the transgene product and that prophylactic vaccination with Emut Vax can break this immune tolerance resulting in inhibition of tumor development.
Therapeutic cancer peptide vaccines have shown limited efficacy in clinical trials, most likely due to the profound immune suppression that develops in subjects with advanced tumors (37). The role of peptide vaccines has not been extensively studied as prevention prior to the establishment of tumor-induced immune suppression. Personalized vaccines have also been developed against multiple neo-Ags, however, most have focused on specific MHC I epitopes (12, 14, 38, 39). MHC II epitopes can be designed to bind to multiple HLA-DR alleles and are thus applicable to broader populations of at-risk or cancer patients. We tested a multi-peptide EGFR vaccine against multiple mutant epitopes in an EGFR mutant transgenic lung adenocarcinoma mouse model. The prophylactic administration of this multi-valent vaccine can prevent tumorigenesis driven by mutant EGFR. The high incidence of EGFR mutant tumors in the East Asian population supports the concept of performing a true prevention trial with this vaccine. Because our vaccine is directed against specific mutations, vaccination may be a powerful adjuvant treatment to prevent the spread of treatment-resistant disease. The low levels of immune suppressive tumor microenvironment mediated by EGFR inhibitor treatment may create a window of opportunity, in which vaccination can more effectively generate anti-tumor immunity for secondary prevention.
In the current study, Emut Vax predominantly elicited a Th1 T cell response without significant induction of a Th2 response. Consistent with our previous findings, Emut Vax significantly increased tumor infiltrating CD4+ T cells. Interestingly, vaccination also markedly enhanced tumor infiltrating CD8+ T cells and boosted both the central memory (TCM) and effector memory (TEM) CD8+ T cell frequencies and function in CD4+ and CD8+ T cell subsets. Both TCM and TEM are antigen-experienced T cells, with TEM functions as sentinels for immediate protection, while TCM represents long term memory population that provide protection from a systemic challenge and can generate a second wave of effector cells. The enhanced TEM and TCM together might confer the profound host protection during tumorigenesis. The proliferation and recruitment of CD8+ T cells can be promoted by cytokines into the tumor microenvironment. IFN-γ can reverse functional defects in antigen presentation by increasing the expression of MHC I on tumor cells or immune cells (40). other epitope based IFN-γ stimulating vaccines study also showed a similar role for CD8+ T cells in tumor eradication (41). Cross-presentation is also one of the potential major mechanisms for MHC-II restricted epitopes to induce CD8+ CTL responses, macro-autophagy of the exogenous antigens in tumor cells is essential process for antigen sequestration and delivery to dendritic cells for cross-presentation (42), CD4+ Th cells itself can also modify the APC, convert it into an effective stimulator for the successful cross-priming (43).
One of the most important T-helper cell types in anticancer immunity is Th1 cells. and vaccines that were designed to stimulate tumor antigen-specific T cells, particularly MHC II directed vaccines that promote IFN-γ secretion, could uniquely modulate the tumor microenvironment. Our scRNA-seq studies from tumor-infiltrating T lymphocytes revealed that the Emut Vax not only promotes the generation of Th1 helper CD4+ cells with enhanced function but also induces Th9 and Th17 cells, further cytokine analysis also confirmed the upregulated IL1, IL-4, IL9 and IL17 etc. after Emut peptide restimulation. In our study, peptides were formulated with CFA/IFA adjuvant, which is known to induce a Th1-dominated response in company with Th2 and Th17 responses. No toxicity related to the observed increase in Th17 cells was found. Therefore, more research is needed to determine the role of Th17 in neoepitope-induced immune responses. Additionally, CFA also contains ligands for Toll-like receptors, which is known to stimulate cytokines, such as IL-1β secretion. IL-1β together with IL-4 could induce antitumor Th9 cell differentiation even in the absence of TGFb (44). It is reported that Th9 cells were effective in the elimination of solid tumors (45). Th17 cells may play a dual role in antitumor immunity (37), which are a key player in inflammation and tissue destruction (38). IL-6 appears critical for inhibition of Foxp3+ Treg differentiation and favors the induction of Th17 cells. In this study, we observed IL-6 induction and Tregs downregulation. IL-6 signaling plays a complex role in inflammation and cancer (46). The pro-tumor functions of IL-6 are mostly through IL6-STAT3 axis which triggers up-regulation of target genes responsible for tumor cell survival, the activation of STAT3 could also block the maturation of dendritic cells (DCs) and inhibit T cell function (47). However, increasing evidence supports that IL-6 trans-signaling is a key player in anti-tumor T cell responses. IL6-STAT3 is also known to downregulate Foxp3 expression and promote Tregs to Th17 cells conversion (47), which inhibits the immune suppression in the tumor microenvironment. The plasticity and differential roles of Th17 cells in tumorigenesis have been reported by the previous studies. For example, Th17 cells differentiated by IL-6, IL-23 and IL-1β in the absence of TGF-β express Th1 master regulator T-bet encoding gene Tbx21, secrete IFNƔ, and promote CD8 effector T cell function and tumor regression, whereas Th17 cells generated by IL-6 in the presence of TGF-β secreted protumorigenic IL-10 and expressed CD39 and CD73, promoting the release of immunosuppressive adenosine (48–51). In addition, IL-6 regulates the priming in tumors and promotes lymphocyte proliferation and trafficking, especially for CD8+ lymphocytes (52).
Emut Vax regulated T cells and reshaped other immune cells in the tumor microenvironment towards anti-tumor reactivity without any signs of autoimmunity. Emut Vax skewed macrophages towards the anti-tumor M1 macrophage subset (Supplementary Figure 2) and enhanced the proportion of cytotoxic CD27-CD11b+ NK cells while decreasing the immature CD27+CD11b- NK cells (Supplementary Figure 3). It is possible that the skewing of these innate immune cells helps to influence or promote the generation of the CD4+ and CD8+ T cell anti-tumor effector cells.
In summary, the current work provides the first proof-of-principle study for the development of an MHC II multi-peptide vaccine against EGFR neoantigens for lung cancer interception. EGFR mutations induce an un-inflamed TME with less T cell infiltration and increased numbers of Tregs, which results in a more suppressive immune environment (53). EGFR inhibition through TKI treatment is known to enhance MHC class I and II antigen presentation, induce more robust infiltration by immune cells and increases local proliferation of T cells in tumors, which leads to increased activation of immune cells and T-cell mediated tumor killing (54, 55). Thus, TKI treatment before (or at the same time) with Emut VAX may potentially offer a better clinical benefit than treatment with TKI after Emut VAX. With further development, this form of precision immunointerception may have the potential to enhance the precision medicine approach to treatment. We demonstrated that prophylactic vaccination can be an efficacious strategy to control disease occurrence and progression, and it has the potential advantage of inducing and mobilizing multiple anti-tumoral immune cell populations in patients with EGFR mutations that are resistant to EGFR TKI treatments.
The raw processed single-cell RNA-seq data used in the study are deposited in the NCBI repository, which can be accessed via the BioProject ID PRJNA933186.
The animal study was reviewed and approved by All procedures were approved by the institutional animal care and use committee (IACUC). All mice were housed in the Biomedical Resource Center at the Medical College of Wisconsin, Milwaukee, (AA00001807). Written informed consent was obtained from the owners for the participation of their animals in this study.
MY, JP were responsible for the overall experimental design with input by KP, QZ and BJ. The project was supervised by MY, JP, QZ, BJ, YW, SS, RS, and KP and JP. conducted flow cytometry analysis, JP and QZ assessed anti-cancer efficacy in animal models. DX did the scRNA-seq analysis. The following were responsible for writing, reviewing, and editing the manuscript: JP, QZ, RS, SS, YW, and MY. All authors contributed to the article and approved the submitted version.
This work was supported from the National Cancer Institute Prevent Program (75N91019D00020 and HHSN‐261201500037I) and grants from the National Institutes of Health (R01CA223804 (MY); R01CA232433 (MY); NIH R01CA208648 (MY); NIH CA205633 (MY).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2023.1036563/full#supplementary-material
Supplementary Figure 1 | Clustering analysis of scRNA-seq data from tumor-infiltrating immune cells in adjuvant control and Emut Vax treated mice. A. Expression of the marker genes for CD8+ T, CD4+ T, Treg and Tgd cell. B. Expression of the marker genes for NK cells, neutrophils, B cells, macrophages, and dendritic cells.
Supplementary Figure 2 | Effects of the Emut Vax treatment on macrophages within tumor-bearing lungs. (A). Canonical macrophage marker expression based on scRNA-seq data. (B). Heatmap of gene expression for the indicated macrophage subsets – M1 (anti-tumor) and M2 (pro-tumor) macrophages. (C). Distribution of tumor-infiltrating M1 and M2 macrophages. (D). Percent changes of the M1 and M2 macrophage subsets across the control and Emut Vax treatment groups.
Supplementary Figure 3 | The effects of Emut Vax treatment on NK cells within tumor-bearing lungs. (A). The total NK cells were identified based on the expression of the Ncr1 gene. (B). Expression of the CD27-CD11b+ NK gene signature and the CD27+CD11b- NK gene signature in total NK cells. (C). Clustering of the total NK cells into CD27-CD11b+ NK and CD27+CD11b- NK cell subsets. (D). Percent changes of CD27-CD11b+ NK and CD27+CD11b- NK cells across the control and Emut Vax treatment groups.
Supplementary Table 1 | Peptide sequences target different EGFR mutations.
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
2. Midha A, Dearden S, McCormack R. EGFR mutation incidence in non-small-cell lung cancer of adenocarcinoma histology: A systematic review and global map by ethnicity (mutMapII). Am J Cancer Res (2015) 5(9):2892–911.
3. Saavedra D, Neninger E, Rodriguez C, Viada C, Mazorra Z, Lage A, et al. CIMAvax-EGF: Toward long-term survival of advanced NSCLC. Semin Oncol (2018) 45(1-2):34–40. doi: 10.1053/j.seminoncol.2018.04.009
4. Cohenuram M, Saif MW. Epidermal growth factor receptor inhibition strategies in pancreatic cancer: Past, present and the future. JOP (2007) 8(1):4–15.
5. Harari PM. Epidermal growth factor receptor inhibition strategies in oncology. Endocr Relat Cancer. (2004) 11(4):689–708. doi: 10.1677/erc.1.00600
6. Wu L, Ke L, Zhang Z, Yu J, Meng X. Development of EGFR TKIs and options to manage resistance of third-generation EGFR TKI osimertinib: Conventional ways and immune checkpoint inhibitors. Front Oncol (2020) 10:602762. doi: 10.3389/fonc.2020.602762
7. Herbst RS, Baas P, Kim DW, Felip E, Perez-Gracia JL, Han JY, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet (2016) 387(10027):1540–50. doi: 10.1016/S0140-6736(15)01281-7
8. Rittmeyer A, Barlesi F, Waterkamp D. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet (2017) 389(10077):E5–E. doi: 10.1016/S0140-6736(16)32517-X
9. McGranahan N, Swanton C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell (2015) 27(1):15–26. doi: 10.1016/j.ccell.2014.12.001
10. Farnsworth DA, Chen YT, de Rappard Yuswack G, Lockwood WW. Emerging molecular dependencies of mutant EGFR-driven non-small cell lung cancer. Cells (2021) 10(12). doi: 10.3390/cells10123553
11. Politi K, Zakowski MF, Fan PD, Schonfeld EA, Pao W, Varmus HE. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev (2006) 20(11):1496–510. doi: 10.1101/gad.1417406
12. Hu Z, Leet DE, Allesoe RL, Oliveira G, Li S, Luoma AM, et al. Personal neoantigen vaccines induce persistent memory t cell responses and epitope spreading in patients with melanoma. Nat Med (2021) 27(3):515–25. doi: 10.1038/s41591-020-01206-4
13. Chen F, Zou Z, Du J, Su S, Shao J, Meng F, et al. Neoantigen identification strategies enable personalized immunotherapy in refractory solid tumors. J Clin Invest. (2019) 129(5):2056–70. doi: 10.1172/JCI99538
14. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature (2017) 547(7662):217–21. doi: 10.1038/nature22991
15. Hailemichael Y, Dai Z, Jaffarzad N, Ye Y, Medina MA, Huang XF, et al. Persistent antigen at vaccination sites induces tumor-specific CD8(+) t cell sequestration, dysfunction and deletion. Nat Med (2013) 19(4):465–72. doi: 10.1038/nm.3105
16. Li L, Goedegebuure SP, Gillanders WE. Preclinical and clinical development of neoantigen vaccines. Ann Oncol (2017) 28(suppl_12):xii11–xii7. doi: 10.1093/annonc/mdx681
17. Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature (2017) 547(7662):222–6. doi: 10.1038/nature23003
18. Kreiter S, Vormehr M, van de Roemer N, Diken M, Lower M, Diekmann J, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature (2015) 520(7549):692–6. doi: 10.1038/nature14426
19. Li F, Deng L, Jackson KR, Talukder AH, Katailiha AS, Bradley SD, et al. Neoantigen vaccination induces clinical and immunologic responses in non-small cell lung cancer patients harboring EGFR mutations. J Immunother Cancer (2021) 9(7). doi: 10.1136/jitc-2021-002531corr1
20. Pan J, Zhang Q, Sei S, Shoemaker RH, Lubet RA, Wang Y, et al. Immunoprevention of KRAS-driven lung adenocarcinoma by a multipeptide vaccine. Oncotarget (2017) 8(47):82689–99. doi: 10.18632/oncotarget.19831
21. Pan J, Zhang Q, Palen K, Wang L, Qiao L, Johnson B, et al. Potentiation of kras peptide cancer vaccine by avasimibe, a cholesterol modulator. EBioMedicine (2019) 49:72–81. doi: 10.1016/j.ebiom.2019.10.044
22. Ebben JD, Lubet RA, Gad E, Disis ML, You M. Epidermal growth factor receptor derived peptide vaccination to prevent lung adenocarcinoma formation: An in vivo study in a murine model of EGFR mutant lung cancer. Mol Carcinog. (2016) 55(11):1517–25. doi: 10.1002/mc.22405
23. Disis ML. Immune regulation of cancer. J Clin Oncol (2010) 28(29):4531–8. doi: 10.1200/JCO.2009.27.2146
24. Disis ML, Gad E, Herendeen DR, Lai VP, Park KH, Cecil DL, et al. A multiantigen vaccine targeting neu, IGFBP-2, and IGF-IR prevents tumor progression in mice with preinvasive breast disease. Cancer Prev Res (Phila). (2013) 6(12):1273–82. doi: 10.1158/1940-6207.CAPR-13-0182
25. Takeuchi A, Saito T. CD4 CTL, a cytotoxic subset of CD4(+) t cells, their differentiation and function. Front Immunol (2017) 8:194. doi: 10.3389/fimmu.2017.00194
26. Ishizaki K, Yamada A, Yoh K, Nakano T, Shimohata H, Maeda A, et al. Th1 and type 1 cytotoxic t cells dominate responses in t-bet overexpression transgenic mice that develop contact dermatitis. J Immunol (2007) 178(1):605–12. doi: 10.4049/jimmunol.178.1.605
27. Huang M, Xiong D, Pan J, Zhang Q, Sei S, Shoemaker RH, et al. Targeting glutamine metabolism to enhance immunoprevention of EGFR-driven lung cancer. Adv Sci (Weinh) (2022), e2105885. doi: 10.1002/advs.202105885
28. Racle J, Michaux J, Rockinger GA, Arnaud M, Bobisse S, Chong C, et al. Robust prediction of HLA class II epitopes by deep motif deconvolution of immunopeptidomes. Nat Biotechnol (2019) 37(11):1283–6. doi: 10.1038/s41587-019-0289-6
29. Weaver Z, Difilippantonio S, Carretero J, Martin PL, El Meskini R, Iacovelli AJ, et al. Temporal molecular and biological assessment of an erlotinib-resistant lung adenocarcinoma model reveals markers of tumor progression and treatment response. Cancer Res (2012) 72(22):5921–33. doi: 10.1158/0008-5472.CAN-12-0736
30. Pan J, Chen Y, Zhang Q, Khatun A, Palen K, Xin G, et al. Inhibition of lung tumorigenesis by a small molecule CA170 targeting the immune checkpoint protein VISTA. Commun Biol (2021) 4(1):906. doi: 10.1038/s42003-021-02381-x
31. Azpilikueta A, Agorreta J, Labiano S, Perez-Gracia JL, Sanchez-Paulete AR, Aznar MA, et al. Successful immunotherapy against a transplantable mouse squamous lung carcinoma with anti-PD-1 and anti-CD137 monoclonal antibodies. J Thorac Oncol (2016) 11(4):524–36. doi: 10.1016/j.jtho.2016.01.013
32. Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol (2018) 36(5):411–20. doi: 10.1038/nbt.4096
33. Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, et al. Comprehensive integration of single-cell data. Cell (2019) 177(7):1888–902e21. doi: 10.1016/j.cell.2019.05.031
34. Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol (2018). doi: 10.1038/nbt.4314
35. Carmona SJ, Siddiqui I, Bilous M, Held W, Gfeller D. Deciphering the transcriptomic landscape of tumor-infiltrating CD8 lymphocytes in B16 melanoma tumors with single-cell RNA-seq. Oncoimmunology (2020) 9(1):1737369. doi: 10.1080/2162402X.2020.1737369
36. Truini A, Starrett JH, Stewart T, Ashtekar K, Walther Z, Wurtz A, et al. The EGFR exon 19 mutant L747-A750>P exhibits distinct sensitivity to tyrosine kinase inhibitors in lung adenocarcinoma. Clin Cancer Res (2019) 25(21):6382–91. doi: 10.1158/1078-0432.CCR-19-0780
37. Mougel A, Terme M, Tanchot C. Therapeutic cancer vaccine and combinations with antiangiogenic therapies and immune checkpoint blockade. Front Immunol (2019) 10:467. doi: 10.3389/fimmu.2019.00467
38. van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, et al. Tumor exome analysis reveals neoantigen-specific t-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol (2013) 31(32):e439–42. doi: 10.1200/JCO.2012.47.7521
39. Hollingsworth RE, Jansen K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines (2019) 4:7. doi: 10.1038/s41541-019-0103-y
40. Balasubramanian A, John T, Asselin-Labat ML. Regulation of the antigen presentation machinery in cancer and its implication for immune surveillance. Biochem Soc Trans (2022) 50(2):825–37. doi: 10.1042/BST20210961
41. Ohkuri T, Kosaka A, Ikeura M, Salazar AM, Okada H. IFN-gamma- and IL-17-producing CD8(+) t (Tc17-1) cells in combination with poly-ICLC and peptide vaccine exhibit antiglioma activity. J Immunother Cancer. (2021) 9(6). doi: 10.1136/jitc-2021-002426
42. Li Y, Wang LX, Yang G, Hao F, Urba WJ, Hu HM. Efficient cross-presentation depends on autophagy in tumor cells. Cancer Res (2008) 68(17):6889–95. doi: 10.1158/0008-5472.CAN-08-0161
43. Bennett SR, Carbone FR, Karamalis F, Miller JF, Heath WR. Induction of a CD8+ cytotoxic t lymphocyte response by cross-priming requires cognate CD4+ t cell help. J Exp Med (1997) 186(1):65–70. doi: 10.1084/jem.186.1.65
44. Xue G, Jin G, Fang J, Lu Y. IL-4 together with IL-1beta induces antitumor Th9 cell differentiation in the absence of TGF-beta signaling. Nat Commun (2019) 10(1):1376.
45. Chen T, Guo J, Cai Z, Li B, Sun L, Shen Y, et al. Th9 cell differentiation and its dual effects in tumor development. Front Immunol (2020) 11:1026. doi: 10.3389/fimmu.2020.01026
46. Maccio A, Madeddu C. Blocking inflammation to improve immunotherapy of advanced cancer. Immunology (2020) 159(4):357–64. doi: 10.1111/imm.13164
47. Chonov DC, Ignatova MMK, Ananiev JR, Gulubova MV. IL-6 activities in the tumour microenvironment. part 1. Open Access Maced J Med Sci (2019) 7(14):2391–8. doi: 10.3889/oamjms.2019.589
48. Chalmin F, Mignot G, Bruchard M, Chevriaux A, Vegran F, Hichami A, et al. Stat3 and gfi-1 transcription factors control Th17 cell immunosuppressive activity via the regulation of ectonucleotidase expression. Immunity (2012) 36(3):362–73. doi: 10.1016/j.immuni.2011.12.019
49. McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, et al. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by t cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol (2007) 8(12):1390–7. doi: 10.1038/ni1539
50. Muranski P, Borman ZA, Kerkar SP, Klebanoff CA, Ji Y, Sanchez-Perez L, et al. Th17 cells are long lived and retain a stem cell-like molecular signature. Immunity (2011) 35(6):972–85. doi: 10.1016/j.immuni.2011.09.019
51. Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature (2010) 467(7318):967–71. doi: 10.1038/nature09447
52. Fisher DT, Appenheimer MM, Evans SS. The two faces of IL-6 in the tumor microenvironment. Semin Immunol (2014) 26(1):38–47. doi: 10.1016/j.smim.2014.01.008
53. Madeddu C, Donisi C, Liscia N, Lai E, Scartozzi M, Maccio A. EGFR-mutated non-small cell lung cancer and resistance to immunotherapy: Role of the tumor microenvironment. Int J Mol Sci (2022) 23(12). doi: 10.3390/ijms23126489
54. Selenz C, Compes A, Nill M, Borchmann S, Odenthal M, Florin A, et al. EGFR inhibition strongly modulates the tumour immune microenvironment in EGFR-driven non-Small-Cell lung cancer. Cancers (Basel) (2022) 14(16). doi: 10.3390/cancers14163943
Keywords: precision immunoprevention, immunointerception, EGFR mutation, peptide vaccine, MHC class II, T cell
Citation: Pan J, Xiong D, Zhang Q, Palen K, Shoemaker RH, Johnson B, Sei S, Wang Y and You M (2023) Precision immunointerception of EGFR-driven tumorigenesis for lung cancer prevention. Front. Immunol. 14:1036563. doi: 10.3389/fimmu.2023.1036563
Received: 04 September 2022; Accepted: 16 January 2023;
Published: 17 February 2023.
Edited by:
Esther M. Lafuente, Complutense University of Madrid (UCM), SpainReviewed by:
Pablo Sarobe, University of Navarra, SpainCopyright © 2023 Pan, Xiong, Zhang, Palen, Shoemaker, Johnson, Sei, Wang and You. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ming You, bXlvdUBob3VzdG9ubWV0aG9kaXN0Lm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.