 Teng Zhang
Teng Zhang Li Yu-Jing
Li Yu-Jing Tao Ma
Tao Ma
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 09 March 2023
Sec. Microbial Immunology
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1029438
This article is part of the Research TopicA year in review: Discussions in Microbial ImmunologyView all 7 articles
Long term immunosuppression is problematic during sepsis. The PD-1 and PD-L1 immune checkpoint proteins have potent immunosuppressive functions. Recent studies have revealed several features of PD-1 and PD-L1 and their roles in sepsis. Here, we summarize the overall findings of PD-1 and PD-L1 by first reviewing the biological features of PD-1 and PD-L1 and then discussing the mechanisms that control the expression of PD-1 and PD-L1. We then review the functions of PD-1 and PD-L1 in physiological settings and further discuss PD-1 and PD-L1 in sepsis, including their involvement in several sepsis-related processes and their potential therapeutic relevance in sepsis. In general, PD-1 and PD-L1 have critical roles in sepsis, indicating that their regulation may be a potential therapeutic target for sepsis.
Sepsis is a severe illness caused by an aberrant host response to infections, and it is associated with acute organ failure and a high mortality risk (1). Although there has been a global improvement in clinical outcomes as a result of improved treatment practices resulting from the dissemination and implementation of the Surviving Sepsis Campaign guidelines (2) over the preceding decades (3), mortality rates remain unacceptably high, ranging from 25 to 30 percent for sepsis and 40 to 50 percent in cases of septic shock, with country-specific variations (4–6). Moreover, many sepsis survivors have long-term physical and cognitive impairments as well as higher death rates than the general population (7–11).
Years ago, it was believed that sepsis mortality and morbidity resulted from an excessive systemic inflammatory response, but medications designed to reduce this response did not enhance survival (12, 13). Several investigations have shown that sepsis is not only characterized by early acute inflammation but is also a concomitant immunosuppressed condition that may last for months after the original episode of sepsis (14, 15). Immune suppression during sepsis makes it harder to eliminate the underlying infection and increases the chance of subsequent infections (16, 17). Importantly, the chronic immunosuppressed states generated by defective innate and adaptive immune responses are responsible for reduced immunity, multi-organ damage, protracted hospital stays, and mortality (17–21). To properly treat this condition, it is essential to understand how sepsis produces immunosuppression.
Immune checkpoint pathways are endogenous immune system components that govern the immune response under normal physiological conditions (22). The programmed cell death protein 1 (PD-1) and programmed death ligand 1 (PD-L1) immune checkpoint is an important regulator that inhibits T cell receptor-induced activation signals (23). In addition, interaction between PD-L1 and PD-1 suppresses the immune system systemically in many cells (24, 25). Multiple clinical studies have established a correlation between PD-1 or PD-L1 expression and sepsis mortality (26–28). Immunotherapy for sepsis using anti-PD-1 and anti-PD-L1 antibodies has shown benefit in animal studies (29–32) while clinical trials in humans have not given the direct benefit evidence of PD-1 or PD-L1 blockade (33, 34). The purpose of this review is to describe the biological properties of PD-1 and PD-L1 and their functions in physiological conditions, focusing on the mechanisms that regulate PD-1 and PD-L1 expression and the roles of the PD-1/PD-L1 axis in sepsis. In general, PD-1 and PD-L1 have critical roles in sepsis, indicating that regulation of their expression may be a potential therapeutic target for sepsis.
PD-1, also known as CD279, is one of the co-inhibitory receptors initially found on the surface of antigen-activated T lymphocytes (35). A small percentage of lymph node, spleen, and bone marrow cells, as well as immature CD4+CD8+ thymocytes has been reported to express the PD-1 protein (36). The presence of PD-1 (mRNA or protein) is seldom detected and appears only after a period of stimulation (37). Activation of lymphocyte B cell receptors or T cell receptors is often associated with an increase in PD-1 expression (38, 39).
PD-L1 (CD274) and PD-L2 (CD273) are the two ligands for PD-1 (CD279). PD-L1 expression can be found on hematopoietic cells, such as T lymphocytes, B lymphocytes, macrophages and dendritic cells (DCs), as well as non-hematopoietic healthy tissue cells, such as vascular endothelial cells, keratinocytes, pancreatic islet cells, astrocytes, corneal epithelial cells, and endothelial cells (40). It has been reported that macrophages, DCs, and mast cells express PD-L2 (41). Binding to PD-L1 is the major mechanism of PD-1 function in sepsis (42). Moreover, PD-L1 gene deficiency improves sepsis survival, while PD-L2 gene deficiency does not show a survival benefit for sepsis (43).
Both PD-1 and PD-L1 are type I transmembrane immunoglobulin (Ig) superfamily members (41). PD-1 contains a cytoplasmic domain that comprises two tyrosine-based signaling motifs and an extracellular domain that mimics Ig-V as well as a transmembrane domain (40). PD-L1 has an Ig-V extracellular domain, an Ig-C-like extracellular domain, a transmembrane domain, and a short cytoplasmic tail devoid of conventional signaling patterns (44). It is possible for the extracellular domains of PD-L1 and PD-1 to interact, causing PD-1 to alter its shape, which allows Src family kinases to phosphorylate the immuno-receptor tyrosine-based inhibitory motif (ITIM) and immuno-receptor tyrosine-based switch motif (ITSM) (45) (Figure 1). These phosphorylated tyrosine patterns attract the SHP-2 and SHP-1 protein tyrosine phosphatases, which suppress the activation of T lymphocytes (46). When the PD-1 receptor is ligated, SHP-2 inhibits the Akt and ERK/MAPK signaling pathways by dephosphorylating PI3K (47). In the absence of SHP-2-induced T cell exhaustion, SHP-1 plays a compensatory role (48). In addition, the SHP2 phosphatase is capable of dephosphorylating the CD28 costimulatory receptor (44).
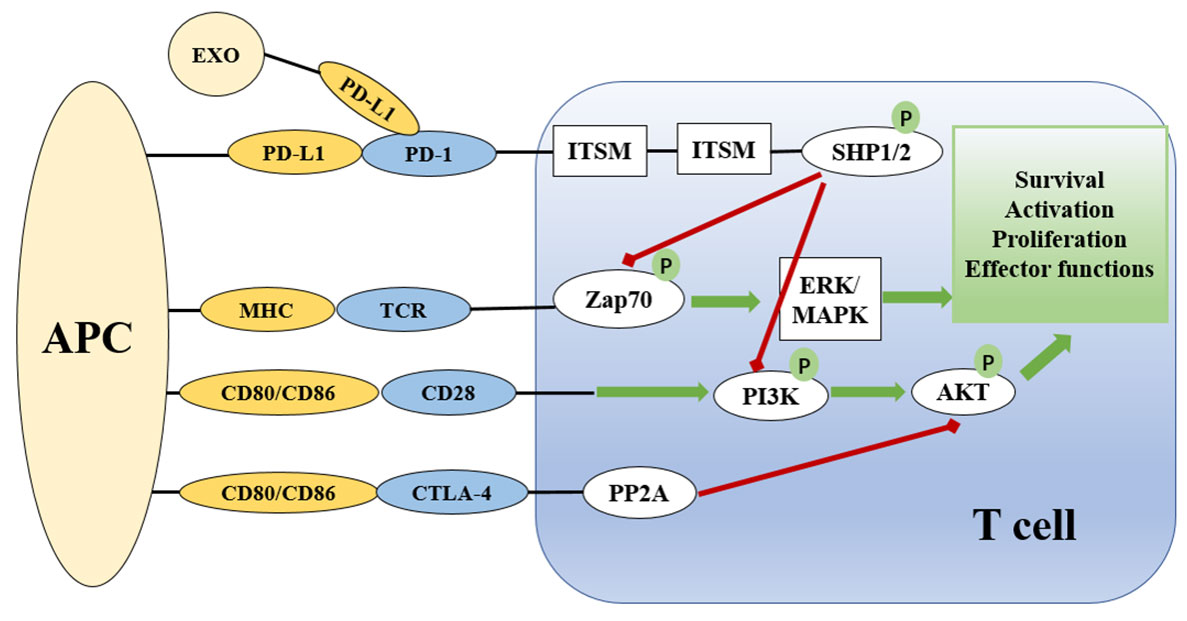
Figure 1 Overview of the PD-1 and PD-L1 checkpoints and signaling pathways associated with them. The presentation of antigen by MHC on APCs to the TCR complex on T cells activates T cells via the Zap70 and ERK/MAPK signaling pathways. CD28 on T cells binds to CD80/86 on APCs to provide co-stimulatory signals. PD-1/PD-L and CTLA-4 signaling suppress the AKT signaling pathway to limit T cell activation. CTLA-4 suppresses the AKT pathway directly by recruiting PP2A, while PD-1 signaling includes SHP-mediated regulation of Zap20 and the PI3K/AKT signaling pathway. Green lines indicate stimulatory messages, while red lines represent inhibitory ones. ITSM and ITIM are intracellular domains of immunological checkpoints that are responsible for intracellular signaling. APC, antigen presenting cell; TCR, T cell receptor; MHC, major histocompatibility complex; PD-1, programmed death-1; CTLA4, cytotoxic T lymphocyte antigen-4; ZAP70, zeta chain of T cell receptor associated protein kinase 70; PI3K, phosphoinositide 3 kinase; PP2A, protein phosphatase 2A; ERK, extracellular signal-regulated kinase; MAPK, mitogen activated protein kinase; AKT, protein kinase B; ITIM, immunoreceptor tyrosine-based inhibition motif; ITSM, immunoreceptor tyrosine-based motif; SHP, Src homology region 2 domain-containing phosphatase.
The mechanisms that regulate PD-1 expression in T cells are well known. PD-1 is barely detectable on naive T cells, but PD-1 surface expression rapidly increases on all T cells upon first antigen-mediated activation through the T cell receptor (TCR) (49). When the activating antigen is rapidly eliminated, PD-1 expression levels on responding T cells decrease (50, 51). If the antigen is not eliminated, such as during persistent infections and malignancies, PD-1 expression persists at a high level (50–52). PD-1 expression on antigen-activated T cells is controlled by many transcription factors, including nuclear factor of activated T cells (NFAT), cytoplasmic 1, fork head box protein O1 (FOXO1), T-bet, and B lymphocyte-induced maturation protein 1 (Blimp-1) (40, 53), as well as the serine–threonine kinase glycogen synthase kinase 3 (GSK3) (54). Although TCR activation is the most essential factor in controlling T cell PD-1 expression, other factors independent of TCR activation also play a role. For instance, in chronic infection, PD-1 expression may be sustained even after antigen clearance (55–57). The following re-expansion of exhausted CD8+ T cell populations under infection also persistently express PD-1 (56). There are dynamic patterns of DNA methylation at the Pdcd1 gene that correspond with PD-1 expression during T cell development (58). Using assay for transposase-accessible chromatin with sequencing (ATAC-seq), researchers identified a distinct pattern of accessibility of the Pdcd1 gene in fatigued T cells (57, 59), and ablation of a regulatory region 23 kb upstream of the transcriptional start site decreases PD-1 expression (59). This 23 kb upstream region in mouse T cells is essential for regulating PD-1 expression (59).
In contrast to PD-1, PD-L1 is ubiquitously expressed by several kinds of cells and regulated by more factors in an inflammation environment. There are three major regulatory mechanisms of PD-L1 expression (Figure 2) as follows: 1) proinflammatory signals promote the expression of PD-L1; 2) microRNAs control the post-transcriptional regulation of PD-L1 gene expression; 3) protein circulation, ubiquitination, and glycosylation all influence PD-L1 levels. The regulators of PD-L1 expression have been listed in Table 1.
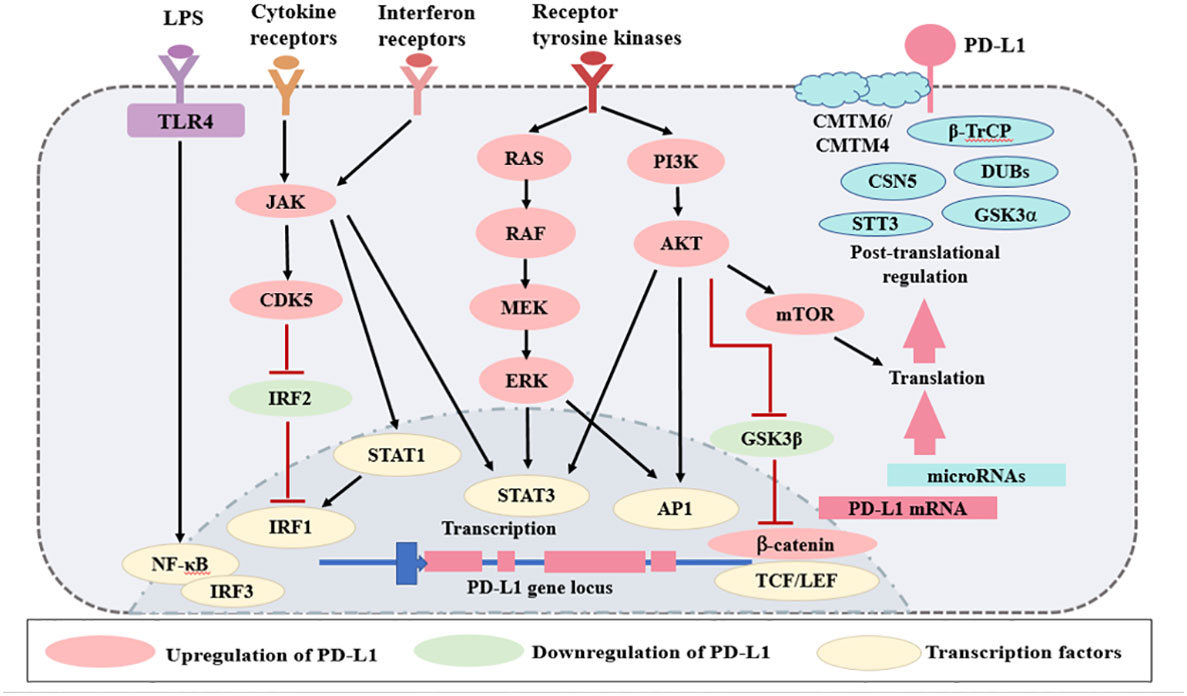
Figure 2 Regulation of PD-L1 expression. The expression of PD-L1 is regulated by several mechanisms. Various signaling pathways, such as TLRs, interferon receptors, cytokine receptors, and receptor tyrosine kinases, increase PD-L1 expression. There are several microRNAs that regulate PD-L1 mRNA transcription. Finally, PD-L1 is regulated at the protein level by protein circulation, ubiquitination, and glycosylation. Red lines represent inhibitory signals, and black lines represent stimulatory signals. AP1, activator protein 1; LPS, lipopolysaccharides; LEF, lymphoid enhancer- binding factor; TLRs, Toll-like receptors; NF- κB, nuclear factor κB; IRF, interferon regulatory factor; STAT, signal transducer and activator of transcription; TCF, T cell- specific transcription factor; GSK3, glycogen synthase kinase 3; JAK, Janus kinase; CDK5, cyclin dependent kinase 5; CMTM, CKLF-like MARVEL transmembrane domain containing; DUBs, deubiquitinating enzymes; STT3, subunit of the oligosaccharyltransferase complex; GSK3α, glycogen synthase kinase 3α; β-TrCP, β-tranducin repeat-containing protein.
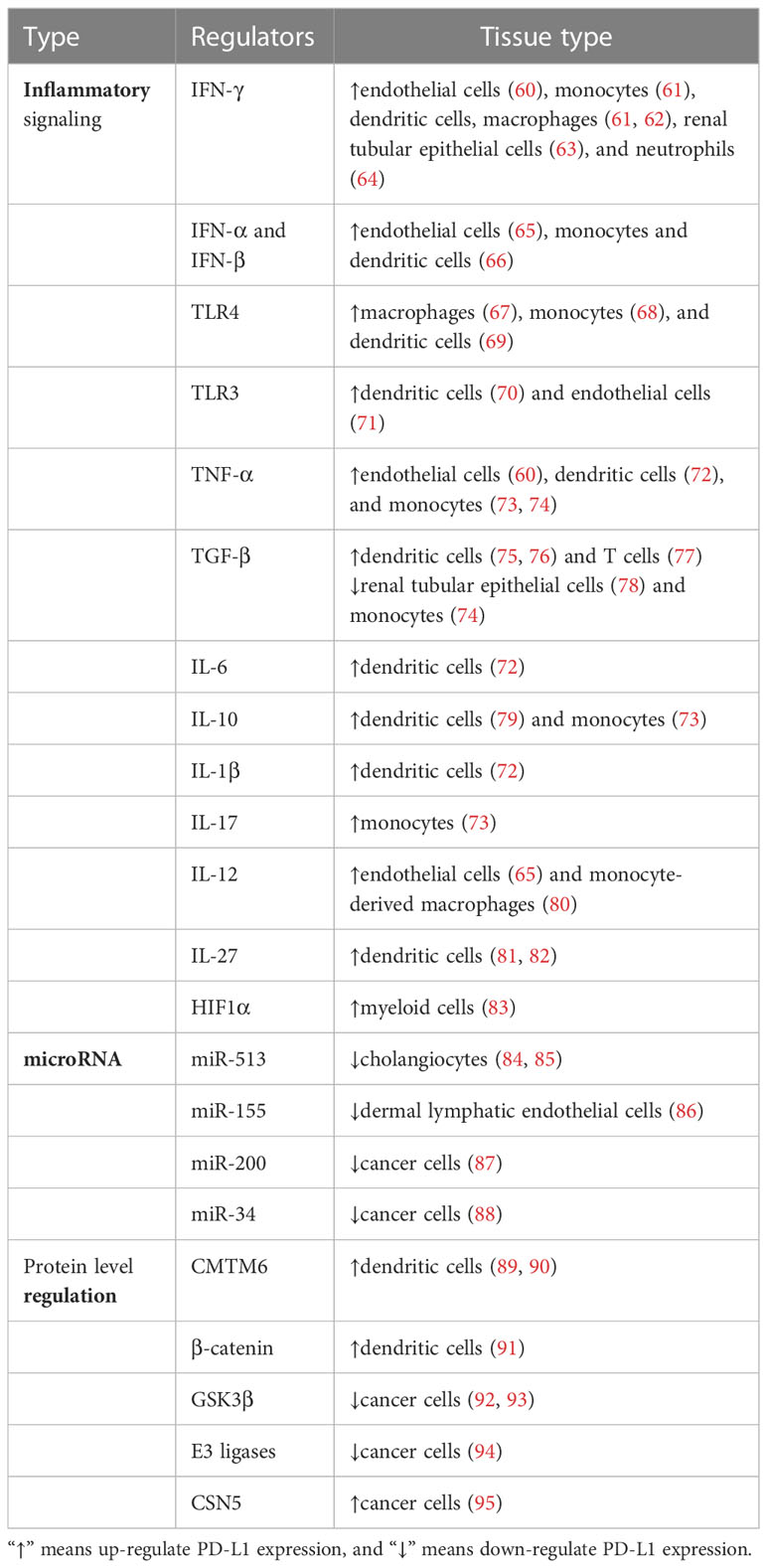
Table 1 Regulators of PD-L1 in normal cells.
PD-L1 gene expression is linked to inflammation, which is consistent with its role in preventing T cell activation (96). Numerous soluble chemicals generated by immune cells have recently been discovered as PD-L1-inducing agents. IFN-γ is traditionally considered to be the most potent soluble inducer of PD-L1, indicating that PD-L1 expression may be a crude indicator of IFN-γ signaling and T cell activation in the majority of instances (97). In sepsis, activated T cells produce significant levels of the IFN-γ proinflammatory cytokine (98). Upon binding to its receptor, IFN-γ activates the JAK-STAT pathway, which in turn activates the STAT1 protein (99), resulting in an increase in a group of transcription factors known as interferon-responsive factors (100), which increase the induction of PD-L1 (101). In addition to IFN-γ, type I interferons (IFN-α and IFN-β) may stimulate PD-L1 expression in endothelial cells, monocytes, and DCs in vitro (65, 66). Type I and type II interferons may activate the AKT-mTOR cascade, which regulates interferon-dependent mRNA translation (102), indicating that the interferon receptor signaling pathway and the AKT-mTOR signaling pathway interact (103). The phosphatidylinositol 3-kinase (PI3K) signaling pathway influences cell growth and survival (104), and pharmacological inhibition of PI3K-AKT signaling inhibits IFN-induced PD-L1 expression (100). Additionally, the PI3K-AKT pathway may regulate PD-L1 expression in an IFN-independent manner, and it has been proposed that at least a part of this regulation occurs via altering PD-L1 mRNA levels by mTOR (105).
Lipopolysaccharide (LPS) is the principal component of the outer membrane of Gram-negative bacteria, contributing significantly to the structural integrity of the bacterium and protecting it against certain forms of chemical attack (106). LPS treatment of macrophages (67), monocytes (68), and primary bone marrow-derived DCs (69) results in enhanced PD-L1 expression. LPS signals via Toll-like receptor 4 (TLR4), and activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) leads to the generation of type I interferons (107, 108). In addition, RELA, an NF-kB subunit, assembles into a complex with the PD-L1 promoter called RELA-MUC1-C, which in turn increases transcription of PD-L1 (109). Polyinosinic: polycytidylic acid [poly(I:C)] is an immunostimulant that is used to simulate viral infections (110). Poly(I:C) induces TLR3 activation on DCs and endothelial cells, thereby increasing PD-L1 expression (111), and this process requires PI3K signaling for the increase of PD-L1 expression (112).
Hypoxia is a critical feature of sepsis, as impaired lung function and drastic inflammation often outgrows the oxygen supply. Immune cells respond to this oxygen deficiency by activating a series of hypoxia-inducible factors (HIFs) (113). Both HIF-1a and HIF-2a have been shown to physically interact with the hypoxia response element (HRE) in the promoter region of PD-L1 (83, 114).
Furthermore, it has been shown that the expression of PD-L1 may be regulated by other stimulators. In vitro cultivated monocytes and tubular epithelial cells express less PD-L1 when treated with transforming growth factor (TGF)-β (115). PD-L1 expression in endothelial cells may be stimulated by IL-12 (116) and tumor necrosis factor (TNF) (117). When stimulated with IL-2, IL-17, IL-15 (118), IL-12 (116), IL-4, and granulocyte-macrophage colony-stimulating factor (GM-CSF) (119), monocytes and macrophages display higher amounts of surface PD-L1expression. DCs treated with IL-1, IL-6, IL-10, IL-27 (120), and TNF (117) exhibit elevated levels of PD-L1. Despite the fact that the aforementioned findings reveal that a large range of inflammatory mediators may regulate PD-L1 expression, it remains unclear in many situations whether this control occurs indirectly, such as through influencing IFN production.
MicroRNAs play an important role in normal physiology as posttranscriptional gene expression regulators by controlling the degradation of target mRNA and/or inhibiting translation. A recent study has demonstrated the role of microRNAs in the control of PD-L1 expression (121), which may take place either directly by binding to PD-L1 mRNA or indirectly by regulating the expression of other PD-L1 regulators. Traditionally, miR-513 and miR-155 are mechanisms for fine-tuning PD-L1 expression in response to IFN-signaling. Both miR-513 and miR-155 suppress PD-L1 at the translational level by direct binding to the 3’ UTR of PD-L1 mRNA (84, 122). IFN-γ suppresses miR-513 expression while reinforcing PD-L1 expression, whereas IFN-γ induces miR-155 while suppressing PD-L1 expression (86). In addition to these direct effects, miRNAs may also indirectly affect PD-L1 expression by influencing the expression of other PD-L1 regulators, such as by repressing PTEN (a tumor-suppressor gene that negatively regulates PI3K-AKT signaling) to increase PD-L1 expression, or by inhibiting PD-L1 expression via its direct action on the STAT3 transcription factor (123, 124).
Expression of PD-L1 is ultimately controlled by posttranslational regulation. Autophagy and endocytosis require lysosomal breakdown to recycle cytoplasmic proteins, organelles, extracellular proteins, and cell surface receptors. CKLF-like MARVEL transmembrane domain-containing 6 (CMTM6) is a transmembrane protein that interacts with the PD-L1 protein on the cell surface (89). CMTM6 binds to PD-L1 and extends its half-life by blocking ubiquitination and lysosomal degradation during protein recycling (89). These effects increase and sustain elevated levels of PD-L1 on the cell surface (90). Inhibition of CMTM6 expression reduces PD-L1 protein synthesis, but it has little effect on PD-L1 mRNA levels (69).
PD-L1 has four residues, namely, N35, N192, N200, and N219, which are attached by an oligosaccharide (92). This N-linked glycosylation is essential for PD-L1 stability and PD-L1 binding ability (92, 125). β-catenin enhances PD-L1 glycosylation and stabilization by increasing transcription of the STT3 subunit (N-glycosyltransferase component) of the oligosaccharyltransferase complex (126, 127). Unglycosylated PD-L1 is a fragile protein (92). GSK3β phosphorylates residues T180 and S184 of PD-L1, which are subsequently bound by the β-tranducin repeats containing protein (β-TrCP) E3 ubiquitin ligase and then targeted for ubiquitin-dependent degradation by the 26S proteasome (92, 93). Glycosylation at N192, N200, and N219 impairs the interaction with GSK3β and stabilizes PD-L1 as a result (92). The phosphorylation and degradation of PD-L1 by GSK3β is a crucial mechanism for decreasing PD-L1 levels.
E3 ligases perform a critical function by binding ubiquitin chains to their targets, thereby designating them for degradation (128). There are several different E3 ligases that can degrade PD-L1 in both normal and diseased states (94). Deubiquitinating enzymes (DUBs) prevent substrate protein ubiquitination by removing ubiquitin chains, therefore stabilizing the protein (95). Deubiquitination mediated by COP9 signalosome 5 (CSN5) leads to TNF-induced activation of PD-L1 (95). Further, there are many other protein regulatory mechanisms of PD-L1 that have been reviewed in other articles (96).
PD-1 and PD-L1 are important to maintain a healthy body (129). In the absence of PD-1, excessive immune-mediated tissue damage may have catastrophic effects on the host. Different genetic backgrounds of PD-1-deficient animals are susceptible to developing lupus-like autoimmune disease (130, 131) or catastrophic autoimmune cardiomyopathy (132). PD-1 inhibition, whether genetic or antibody-based, has also been shown to accelerate the onset of diabetes in individuals who are neither obese nor diabetic (133). Other findings include the defect of T-cell training in the thymus in PD-1 deficient mice (131) and the impairment of maternal tolerance in fetuses and their mothers as a result of PD-L1 inhibition (134).
The PD-1 pathway plays an important role in limiting immunopathological responses in host tissues by promoting inflammatory response downregulation and return to immune system balance (135). If CD8+ T cell responses are not well regulated, significant immunopathology may occur from the production of proinflammatory cytokines, such as IFN-γ and TNF. Lethal immunopathology occurs in PD-1deficient or PD-L1deficient animals after infection with strains of lymphocytic choriomeningitis virus (LCMV) that produce chronic infection, illustrating the critical function of the PD-1 pathway in regulating immune-mediated tissue damage (50, 136, 137). This deadly immunopathology is based on CD8+ T cells and may involve the perforin-dependent destruction of vascular endothelial cells (136). The PD-1 pathway also regulates proatherogenic inflammatory responses because animals defective in the low-density lipoprotein receptor develop more atherosclerotic lesions if they lack PD-L1 (138). The reduced vascular integrity that occurs in the absence of PD-1 signaling provides a significant hurdle for PD-1 immunotherapy because inhibiting PD-1 may increase the risk of heart attacks, strokes, and edema by altering the permeability of the vascular barrier (138).
The PD-1 pathway also affects the development and responses of memory T cells. Compared to wild-type T cells, PD-1-deficient T cells isolated from vaccinia virus-infected mixed bone marrow chimera mice display greater amounts of CC-chemokine receptor 7 (CCR7) and CD62L, and they are skewed toward a central memory T cell phenotype (139). Experimentally induced deletion of PD-1 results in a higher proliferation of memory T cells when they are transplanted into wild-type recipients and then challenged with another strain of vaccine virus (139). Experiments using vaccinia virus infection have indicated that PD-1 inhibition during secondary challenge may repair deficiencies in CD8+ T cell responses in the absence of CD4+ T cell assistance (140). Secondary PD-1 blockage after viral lung infection significantly improved CD8+ T cell activities (141). The amount and quality of memory T cell responses may be affected by PD-1 inhibition during primary versus secondary challenges, and this may be dependent on the illness environment.
Sepsis is a lethal uncontrolled host reaction to infection. Clinically, sepsis is currently defined as having an infection and a sudden change in how an organ works, as measured by the Sequential Organ Failure Assessment score (142). We still don’t know all the details of how cell damage and organ malfunction result from sepsis. Reduced T lymphocyte function, impaired myeloid cell activity, and non-immune cell death have been described as the pathophysiological features of sepsis (143).
The number of cells that express the PD-1 and PD-L1 genes is increased during sepsis (144). In the CLP model, CD4+ T cells increase PD-1 expression within 24 hours, CD8+ T cells increase PD-1 expression at a time of 3 days to 7 days, and myeloid cells increase PD-L1 expression within 24 hours (144). A previous study has reported that endothelial cells in splenic capillaries of individuals who died of sepsis had a higher level of PD-L1 than endothelial cells in the spleens of individuals with brain death or injury necessitating immediate splenectomy (27). Individuals with sepsis have increased surface PD-1 expression on T lymphocytes and increased surface PD-L1 expression on myeloid cells (28). Loss of PD-1 signaling often enhances immunological control of numerous forms of infection, such as viral, fungal, and bacterial infections (136, 141, 145–147). Numerous studies have connected PD-1/PD-L1 axis to altered immune cell activity in sepsis (Figure 3).
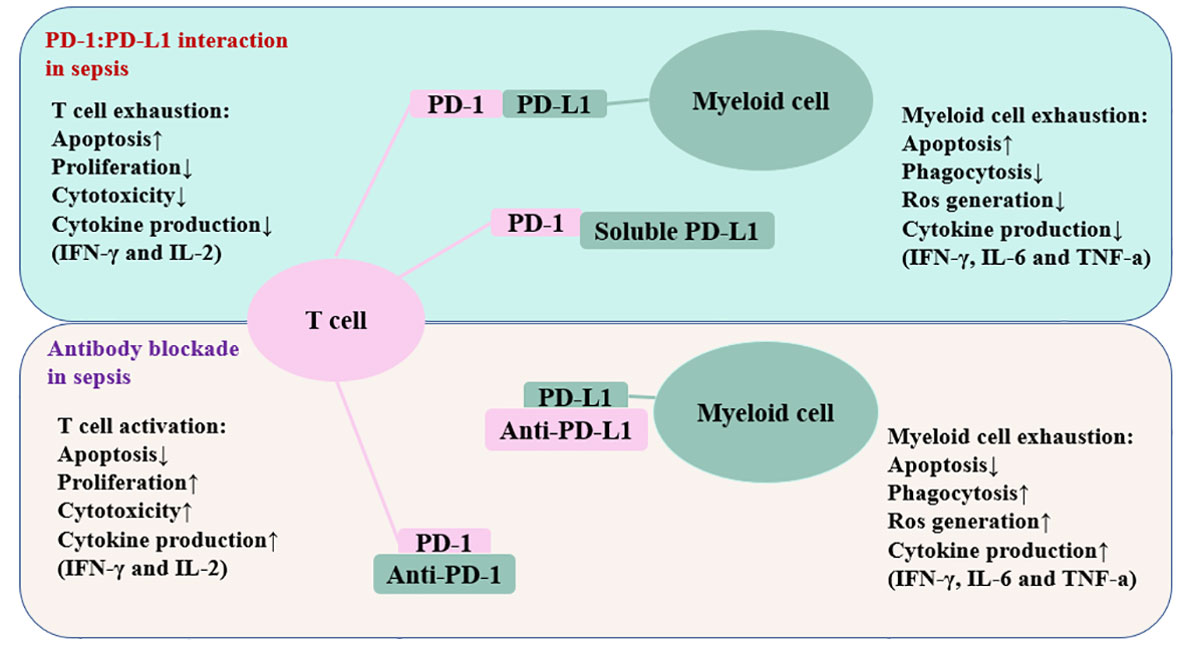
Figure 3 Schematic depiction of the PD-1/PD-L1 related immune cell dysfunction. The interaction between PD-1 and PD-L1 impairs T cell function and myeloid cell function. Antibodies against these inhibitory molecules restore the immune system’s function and boost resistance to infection in patients suffering from sepsis. The up arrow represents an increase, whereas the down arrow denotes a decrease. PD-1, programmed cell death-1; PD-L1, programmed cell death ligand-1; IFN-γ, interferon-gamma; IL-2, interleukin-2; IL-6, inerleukin-6.
High levels of PD-1 on T cells have been detected in patients with sepsis (148). Higher PD-1 expression on T cells has been associated with lymphopenia, T cell death, increased mortality (27, 28, 149, 150), and subsequent nosocomial infections (151). By suppressing T cell proliferation, survival, cytokine generation, and other effector activities, engagement of PD-1 by PD-L1 changes the activity of T cells in a number of ways (79, 152, 153). Binding of PD-L1 to effector T cells that express PD-1 reduces costimulatory signaling, resulting in depletion of the T cell response capacity, characterized by diminished co-stimulatory receptor expression (such as CD28), activation of inhibitory immunological checkpoints, and metabolic derangements, leading to impaired synthesis of effector cytokines, poor proliferation, and increased susceptibility to apoptotic cell death (154). In sepsis, blocking the binding of PD-1 on T cells with PD-L1 prevents T cell depletion and is associated with increased microbial clearance and a decreased mortality rate (27, 155–159).
Sepsis inhibits the functionality of myeloid cells via the PD-1/PD-L1 axis (144). In sepsis patients, an increase in PD-L1 expression on monocytes and neutrophils is accompanied with a decrease in their phagocytic capabilities (158). It has been demonstrated that cecal ligation and puncture (CLP)-induced murine sepsis increases PD-1 expression on liver Kupffer cells and that deleting PD-1 enhances their phagocytic activity (160). In a sepsis CLP model, the expression of PD-L1 on circulating neutrophils is correlated with both pro- and anti-inflammatory cytokine levels and mortality (161). Patients with septic shock who have elevated PD-L1 levels on their circulating monocytes are more likely to die during the first 28 days of their illness (162). Recent research has shown that natural killer (NK) cell PD-L1 expression within 24 h of ICU admission is related to increased sepsis severity (163). Patients with sepsis have elevated PD-L1 levels in their peripheral blood monocytes, and binding with PD-1 impairs cell survival and function. In addition, anti-PD-1 antibody therapy restores monocyte production of critical cytokines, including IFN-γ and IL-2 (158).
PD-1/PD-L1 signaling has been associated with organ damage induced by sepsis (158). In addition to leukocytes, non-immune cells, such as the lung, liver, kidney, colon, small intestine, and tissue endothelial cells express PD-L1 (40, 43, 164). CLP-induced PD-L1 defective sepsis mice have lower levels of serum bilirubin, alanine aminotransferase (ALT), and aspartate aminotransferase (AST), and their endothelial permeability barrier is unaltered, with greater systemic bacteria clearance and better survival (43). Deletion of PD-L2 enhances systemic bacterial clearance but does not protect against the increase in hepatocellular damage markers, such as serum bilirubin and AST, and it does not influence the survival of CLP model (43). Both PD-1 and PD-L1 are increased on liver sinusoidal endothelial cells in a CLP model of sepsis, and deleting PD-L1 protects against sepsis-induced increases in hepatic vascular leakage, edema, and endothelial cell mortality (165). Patients with severe sepsis have PD-L1 expression in their postmortem lung tissue (27). Pulmonary endothelial cell permeability and lung damage are reduced when PD-1 or PD-1 is deleted from lung endothelial or parenchymal cells during sepsis (166, 167). Neutrophil infiltration and sepsis-induced lung damage may both be reduced by intravenous delivery of siRNAs that specifically target PD-L1 expression, indicating that this protein may be a therapeutic target for the prevention of sepsis-induced lung injury (166). PD-L1 also controls the intestinal damage caused by sepsis. Deletion or treatment with an antibody of PD-L1 lowers the degree of intestinal damage resulting from sepsis (168). In the early phase of systemic LCMV infection, PD-L1 deficiency on endothelial cell leads to increased vascular permeability and ultimately to circulatory collapse (136). It is important to note that shock or systemic inflammatory syndromes related to different pathogens might respond differently to checkpoint blockade or inhibition.
PD-1/PD-L1 signaling is a potential therapeutic target of sepsis. Patients with sepsis have been integrated using the Persistent Inflammation, Immunosuppression, and Catabolism Syndrome (PICS) (142). The inflammatory and immune-suppressing phases were considered to happen at the same time in sepsis (143). At initially, several anti-inflammatory strategies were tested since sepsis was thought to be fundamentally hyperinflammation. Yet, none of these sepsis therapy methods have shown any evidence of success (142). Therefore, immunological stimulation is a novel approach to combating sepsis, particularly its Immunoparalysis component. In CLP-induced sepsis, PD-1 knockout animals have a greater likelihood of survival than wild-type mice (169). Anti-PD-L1 antibody treatment in mouse models of CLP-induced sepsis reduces T cell apoptosis, increases bacterial clearance, and minimizes organ damage (31). Anti-PD-L1 antibody treatment 24h after fungal sepsis caused by Candida albicans protects T cell function and enhances survival (30). The two-hit model is another type of sepsis model. It has a higher death rate than the CLP model and a more severe immune suppression than the CLP model. Using a two-pronged attack, first CLP-induced sepsis and then fungal sepsis using C. albicans, researchers created a model of sepsis and found that the novel PD-L1 blocking peptide, compound 8, decreases mortality by half (170). Similarly, a recent study has reported that treating CLP-induced sepsis with the novel LD01 peptide, which suppresses PD-1 signaling, enhances macrophage phagocytosis, T cell activity, and survival (171). No single study showed benefit of PD-1/PD-L1 blockage in sepsis caused by pulmonary infection, and the majority of pre-clinical models examined did not include antibiotic treatment, which is a critical part of therapy for sepsis (172). Anti-PD-L1 therapy did not alter the survival of sepsis model caused by Staphylococcus aureus pneumonia (173). It should be noted that different organisms, different sites of infection, and timing of therapy all may have an effect on the outcomes of anti-PD-1 or anti-PD-L1 antibody treatment in sepsis.
For humans, most studies compared PD-1 or PD-L1 expression between sepsis patients and healthy volunteers but not critically ill patients. The effectiveness of PD-1 or PD-L1 blockade on human cells has just been tested in vitro blockade assays (174). Ex vivo administration with an anti-PD-1 antibody decreases apoptosis and increases IFN-γ production in CD8 T cells collected from septic patients (150) as well as reverses sepsis-induced T cell dysfunction and enhances neutrophil and monocyte phagocytic activity in circulating blood cells collected from septic patients (158). The anti-PD-1 antibody, nivolumab, has been licensed for use in cancer patients (175, 176). Clinical trials for nivolumab in sepsis have also made progress. A 2019 Phase 1b clinical trial investigating the safety and tolerability of nivolumab in septic patients reported no side effects, such as a cytokine storm (33). Similarly, another 2019 Phase 1b clinical trial investigating the safety of the anti-PD-L1 antibody, BMS-936559, in sepsis patients validated the antibody’s safety with no instances of hypercytokinemia (34). All studies gave evidence that there were no safety concerns with PD-1/PD-L1 blockade in ICU-bound sepsis patients at high risk for mortality and no indication of a “cytokine storm”. No evidence has shown the benefit of immunotherapy (PD-1 and PD-L1 blockade) in clinical trials in humans for sepsis (33, 34). Interesting, recent studies have indicated that the anti-PD-1/PD-L1 axis may be safe for use in sepsis (33, 34). However, cancer research studies have revealed a wide variety of major side effects related to immune checkpoint inhibition, including liver damage, thrombocytopenia, pneumonitis, colitis, thyroiditis, and vasculitis (177). Additional clinical trials will provide better knowledge of the blockade of the PD-1/PD-L1 axis in sepsis.
PD-1 and PD-L1 are potent immune checkpoint proteins in several cells, and they are upregulated by several inflammation signals. In sepsis, PD-1 and PD-L1 expression is corelated with the mortality rate, and blockade of PD-1 or PD-L1 protects against sepsis. In current state, the blockade of PD-1/PD-L1 would be able to correct the immunosuppression in sepsis, as in oncology treatment. However, many unknowns remain. First, it is unknown how PD-1 and PD-L1 differ from other inhibitory receptors in terms of their specific and shared functions. Inhibitory receptors are not known to act in a hierarchical manner; if one receptor is destroyed, other receptors may compensate. These signaling pathways are still undefined. For example, it remains unknown how blocking one receptor affects the ability of another to communicate. Second, methods are needed to inhibit PD-1 or PD-L1 expression increases in sepsis. The blockade of PD-1/PD-L1 can cause the immune system to attack healthy cells, leading to a range of immune-related adverse events such as rash, colitis, pneumonitis, and hepatitis (178). In sepsis, PD-L1 expression is increased as a result of immune dysregulation. Restoring PD-L1 expression levels to normal by inhibiting PD-L1 expression upregulation is a potential therapeutic modality capable of avoiding autoimmune damage due to PD-L1 blockade. In summary, we believe that regulating the PD-1/PD-L1 pathway will be a potent weapon to protect against sepsis in the future.
TZ contributed to collection of references and manuscript preparation. TM and LY-J contributed to manuscript modifications. All authors contributed to the article and approved the submitted version.
This work was supported by grants from National Natural Science Foundation of China (82172122 to TM), Tianjin Municipal Health Commission (KJ20145 to TZ). This work was also funded by Tianjin Key Medical Discipline (Specialty) Construction Project.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Evans L, Rhodes A, Alhazzani W, Antonelli M, Coopersmith CM, French C, et al. Surviving sepsis campaign: International guidelines for management of sepsis and septic shock 2021. Intensive Care Med (2021) 47(11):1181–247. doi: 10.1007/s00134-021-06506-y
2. Rhodes A, Evans LE, Alhazzani W, Levy MM, Antonelli M, Ferrer R, et al. Surviving sepsis campaign: International guidelines for management of sepsis and septic shock: 2016. Intensive Care Med (2017) 43(3):304–77. doi: 10.1007/s00134-017-4683-6
3. Kaukonen KM, Bailey M, Suzuki S, Pilcher D, Bellomo R. Mortality related to severe sepsis and septic shock among critically ill patients in Australia and new Zealand, 2000-2012. JAMA (2014) 311(13):1308–16. doi: 10.1001/jama.2014.2637
4. Schinkel M, Nanayakkara PWB, Wiersinga WJ. Sepsis performance improvement programs: From evidence toward clinical implementation. Crit Care (2022) 26(1):77. doi: 10.1186/s13054-022-03917-1
5. Vincent JL, Marshall JC, Namendys-Silva SA, Francois B, Martin-Loeches I, Lipman J, et al. Assessment of the worldwide burden of critical illness: The intensive care over nations (Icon) audit. Lancet Respir Med (2014) 2(5):380–6. doi: 10.1016/S2213-2600(14)70061-X
6. Vincent JL, Sakr Y, Sprung CL, Ranieri VM, Reinhart K, Gerlach H, et al. Sepsis in European intensive care units: Results of the soap study. Crit Care Med (2006) 34(2):344–53. doi: 10.1097/01.ccm.0000194725.48928.3a
7. Iwashyna TJ, Ely EW, Smith DM, Langa KM. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA (2010) 304(16):1787–94. doi: 10.1001/jama.2010.1553
8. Prescott HC, Osterholzer JJ, Langa KM, Angus DC, Iwashyna TJ. Late mortality after sepsis: Propensity matched cohort study. BMJ (2016) 353:i2375. doi: 10.1136/bmj.i2375
9. Mayr FB, Yende S, Angus DC. Epidemiology of severe sepsis. Virulence (2014) 5(1):4–11. doi: 10.4161/viru.27372
10. Pandharipande PP, Girard TD, Ely EW. Long-term cognitive impairment after critical illness. N Engl J Med (2014) 370(2):185–6. doi: 10.1056/NEJMc1313886
11. Angus DC, Opal S. Immunosuppression and secondary infection in sepsis: Part, not all, of the story. JAMA (2016) 315(14):1457–9. doi: 10.1001/jama.2016.2762
12. Abraham E. Why immunomodulatory therapies have not worked in sepsis. Intensive Care Med (1999) 25(6):556–66. doi: 10.1007/s001340050903
13. Marshall JC. Why have clinical trials in sepsis failed? Trends Mol Med (2014) 20(4):195–203. doi: 10.1016/j.molmed.2014.01.007
14. Schorr CA, Dellinger RP. The surviving sepsis campaign: Past, present and future. Trends Mol Med (2014) 20(4):192–4. doi: 10.1016/j.molmed.2014.02.001
15. Torres LK, Pickkers P, van der Poll T. Sepsis-induced immunosuppression. Annu Rev Physiol (2022) 84:157–81. doi: 10.1146/annurev-physiol-061121-040214
16. Hotchkiss RS, Coopersmith CM, McDunn JE, Ferguson TA. The sepsis seesaw: Tilting toward immunosuppression. Nat Med (2009) 15(5):496–7. doi: 10.1038/nm0509-496
17. Patil NK, Bohannon JK, Sherwood ER. Immunotherapy: A promising approach to reverse sepsis-induced immunosuppression. Pharmacol Res (2016) 111:688–702. doi: 10.1016/j.phrs.2016.07.019
18. Xiao W, Mindrinos MN, Seok J, Cuschieri J, Cuenca AG, Gao H, et al. A genomic storm in critically injured humans. J Exp Med (2011) 208(13):2581–90. doi: 10.1084/jem.20111354
19. Delano MJ, Ward PA. Sepsis-induced immune dysfunction: Can immune therapies reduce mortality? J Clin Invest (2016) 126(1):23–31. doi: 10.1172/JCI82224
20. Tang BM, Huang SJ, McLean AS. Genome-wide transcription profiling of human sepsis: A systematic review. Crit Care (2010) 14(6):R237. doi: 10.1186/cc9392
21. Boomer JS, Green JM, Hotchkiss RS. The changing immune system in sepsis: Is individualized immuno-modulatory therapy the answer? Virulence (2014) 5(1):45–56. doi: 10.4161/viru.26516
22. Sharma P, Allison JP. The future of immune checkpoint therapy. Science (2015) 348(6230):56–61. doi: 10.1126/science.aaa8172
23. Bretscher P, Cohn M. A Theory Self-Nonself Discrimination. Science (1970) 169(3950):1042–9. doi: 10.1126/science.169.3950.1042
24. Yi M, Zheng X, Niu M, Zhu S, Ge H, Wu K. Combination strategies with pd-1/Pd-L1 blockade: Current advances and future directions. Mol Cancer (2022) 21(1):28. doi: 10.1186/s12943-021-01489-2
25. Budimir N, Thomas GD, Dolina JS, Salek-Ardakani S. Reversing T-cell exhaustion in cancer: Lessons learned from pd-1/Pd-L1 immune checkpoint blockade. Cancer Immunol Res (2022) 10(2):146–53. doi: 10.1158/2326-6066.CIR-21-0515
26. Jiang W, Li X, Ding H, Wang K, Liu X, Wang Q, et al. Pd-1 in tregs predicts the survival in sepsis patients using sepsis-3 criteria: A prospective, two-stage study. Int Immunopharmacol (2020) 89(Pt A):107175. doi: 10.1016/j.intimp.2020.107175
27. Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA (2011) 306(23):2594–605. doi: 10.1001/jama.2011.1829
28. Zhang Y, Li J, Lou J, Zhou Y, Bo L, Zhu J, et al. Upregulation of programmed death-1 on T cells and programmed death ligand-1 on monocytes in septic shock patients. Crit Care (2011) 15(1):R70. doi: 10.1186/cc10059
29. Zhang Y, Zhou Y, Lou J, Li J, Bo L, Zhu K, et al. Pd-L1 blockade improves survival in experimental sepsis by inhibiting lymphocyte apoptosis and reversing monocyte dysfunction. Crit Care (2010) 14(6):R220. doi: 10.1186/cc9354
30. Chang KC, Burnham CA, Compton SM, Rasche DP, Mazuski RJ, McDonough JS, et al. Blockade of the negative Co-stimulatory molecules pd-1 and ctla-4 improves survival in primary and secondary fungal sepsis. Crit Care (2013) 17(3):R85. doi: 10.1186/cc12711
31. Brahmamdam P, Inoue S, Unsinger J, Chang KC, McDunn JE, Hotchkiss RS. Delayed administration of anti-Pd-1 antibody reverses immune dysfunction and improves survival during sepsis. J Leukoc Biol (2010) 88(2):233–40. doi: 10.1189/jlb.0110037
32. Triantafyllou E, Gudd CL, Mawhin MA, Husbyn HC, Trovato FM, Siggins MK, et al. Pd-1 blockade improves kupffer cell bacterial clearance in acute liver injury. J Clin Invest (2021) 131(4):e140196. doi: 10.1172/JCI140196
33. Hotchkiss RS, Colston E, Yende S, Crouser ED, Martin GS, Albertson T, et al. Immune checkpoint inhibition in sepsis: A phase 1b randomized study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of nivolumab. Intensive Care Med (2019) 45(10):1360–71. doi: 10.1007/s00134-019-05704-z
34. Hotchkiss RS, Colston E, Yende S, Angus DC, Moldawer LL, Crouser ED, et al. Immune checkpoint inhibition in sepsis: A phase 1b randomized, placebo-controlled, single ascending dose study of antiprogrammed cell death-ligand 1 antibody (Bms-936559). Crit Care Med (2019) 47(5):632–42. doi: 10.1097/CCM.0000000000003685
35. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of pd-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J (1992) 11(11):3887–95. doi: 10.1002/j.1460-2075.1992.tb05481.x
36. Gianchecchi E, Delfino DV, Fierabracci A. Recent insights into the role of the pd-1/Pd-L1 pathway in immunological tolerance and autoimmunity. Autoimmun Rev (2013) 12(11):1091–100. doi: 10.1016/j.autrev.2013.05.003
37. Qin W, Hu L, Zhang X, Jiang S, Li J, Zhang Z, et al. The diverse function of pd-1/Pd-L pathway beyond cancer. Front Immunol (2019) 10:2298. doi: 10.3389/fimmu.2019.02298
38. Haas KM, Poe JC, Tedder TF. Cd21/35 promotes protective immunity to streptococcus pneumoniae through a complement-independent but Cd19-dependent pathway that regulates pd-1 expression. J Immunol (2009) 183(6):3661–71. doi: 10.4049/jimmunol.0901218
39. Liu MY, Klement JD, Langan CJ, van Riggelen J, Liu K. Expression regulation and function of pd-1 and pd-L1 in T lymphoma cells. Cell Immunol (2021) 366:104397. doi: 10.1016/j.cellimm.2021.104397
40. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. Pd-1 and its ligands in tolerance and immunity. Annu Rev Immunol (2008) 26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331
41. Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol (2005) 23:515–48. doi: 10.1146/annurev.immunol.23.021704.115611
42. Wang JF, Wang YP, Xie J, Zhao ZZ, Gupta S, Guo Y, et al. Upregulated pd-L1 delays human neutrophil apoptosis and promotes lung injury in an experimental mouse model of sepsis. Blood (2021) 138(9):806–10. doi: 10.1182/blood.2020009417
43. Rossi AL, Le M, Chung CS, Chen Y, Fallon EA, Matoso A, et al. A novel role for programmed cell death receptor ligand 2 in sepsis-induced hepatic dysfunction. Am J Physiol Gastrointest Liver Physiol (2019) 316(1):G106–G14. doi: 10.1152/ajpgi.00204.2018
44. Doroshow DB, Bhalla S, Beasley MB, Sholl LM, Kerr KM, Gnjatic S, et al. Pd-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat Rev Clin Oncol (2021) 18(6):345–62. doi: 10.1038/s41571-021-00473-5
45. Fernandes RA, Su L, Nishiga Y, Ren J, Bhuiyan AM, Cheng N, et al. Immune receptor inhibition through enforced phosphatase recruitment. Nature (2020) 586(7831):779–84. doi: 10.1038/s41586-020-2851-2
46. Marasco M, Berteotti A, Weyershaeuser J, Thorausch N, Sikorska J, Krausze J, et al. Molecular mechanism of Shp2 activation by pd-1 stimulation. Sci Adv (2020) 6(5)::eaay4458. doi: 10.1126/sciadv.aay4458
47. Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol (2007) 8(3):239–45. doi: 10.1038/ni1443
48. Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. Shp-1 and shp-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol (2004) 173(2):945–54. doi: 10.4049/jimmunol.173.2.945
49. Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, et al. Expression of the pd-1 antigen on the surface of stimulated mouse T and b lymphocytes. Int Immunol (1996) 8(5):765–72. doi: 10.1093/intimm/8.5.765
50. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted Cd8 T cells during chronic viral infection. Nature (2006) 439(7077):682–7. doi: 10.1038/nature04444
51. Crawford A, Angelosanto JM, Kao C, Doering TA, Odorizzi PM, Barnett BE, et al. Molecular and transcriptional basis of Cd4(+) T cell dysfunction during chronic infection. Immunity (2014) 40(2):289–302. doi: 10.1016/j.immuni.2014.01.005
52. Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol (2015) 36(4):265–76. doi: 10.1016/j.it.2015.02.008
53. Schildberg FA, Klein SR, Freeman GJ, Sharpe AH. Coinhibitory pathways in the B7-Cd28 ligand-receptor family. Immunity (2016) 44(5):955–72. doi: 10.1016/j.immuni.2016.05.002
54. Taylor A, Harker JA, Chanthong K, Stevenson PG, Zuniga EI, Rudd CE. Glycogen synthase kinase 3 inactivation drives T-Bet-Mediated downregulation of Co-receptor pd-1 to enhance Cd8(+) cytolytic T cell responses. Immunity (2016) 44(2):274–86. doi: 10.1016/j.immuni.2016.01.018
55. Angelosanto JM, Blackburn SD, Crawford A, Wherry EJ. Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection. J Virol (2012) 86(15):8161–70. doi: 10.1128/JVI.00889-12
56. Utzschneider DT, Legat A, Fuertes Marraco SA, Carrie L, Luescher I, Speiser DE, et al. T Cells maintain an exhausted phenotype after antigen withdrawal and population reexpansion. Nat Immunol (2013) 14(6):603–10. doi: 10.1038/ni.2606
57. Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by pd-1 blockade. Science (2016) 354(6316):1160–5. doi: 10.1126/science.aaf2807
58. Youngblood B, Oestreich KJ, Ha SJ, Duraiswamy J, Akondy RS, West EE, et al. Chronic virus infection enforces demethylation of the locus that encodes pd-1 in antigen-specific Cd8(+) T cells. Immunity (2011) 35(3):400–12. doi: 10.1016/j.immuni.2011.06.015
59. Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, et al. The epigenetic landscape of T cell exhaustion. Science (2016) 354(6316):1165–9. doi: 10.1126/science.aae0491
60. Mazanet MM, Hughes CC. B7-H1 is expressed by human endothelial cells and suppresses T cell cytokine synthesis. J Immunol (2002) 169(7):3581–8. doi: 10.4049/jimmunol.169.7.3581
61. Brown JA, Dorfman DM, Ma FR, Sullivan EL, Munoz O, Wood CR, et al. Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J Immunol (2003) 170(3):1257–66. doi: 10.4049/jimmunol.170.3.1257
62. Kryczek I, Wei S, Gong W, Shu X, Szeliga W, Vatan L, et al. Cutting edge: Ifn-gamma enables apc to promote memory Th17 and abate Th1 cell development. J Immunol (2008) 181(9):5842–6. doi: 10.4049/jimmunol.181.9.5842
63. Schoop R, Wahl P, Le Hir M, Heemann U, Wang M, Wuthrich RP. Suppressed T-cell activation by ifn-Gamma-Induced expression of pd-L1 on renal tubular epithelial cells. Nephrol Dial Transplant (2004) 19(11):2713–20. doi: 10.1093/ndt/gfh423
64. de Kleijn S, Langereis JD, Leentjens J, Kox M, Netea MG, Koenderman L, et al. Ifn-Gamma-Stimulated neutrophils suppress lymphocyte proliferation through expression of pd-L1. PloS One (2013) 8(8):e72249. doi: 10.1371/journal.pone.0072249
65. Eppihimer MJ, Gunn J, Freeman GJ, Greenfield EA, Chernova T, Erickson J, et al. Expression and regulation of the pd-L1 immunoinhibitory molecule on microvascular endothelial cells. Microcirculation (2002) 9(2):133–45. doi: 10.1038/sj/mn/7800123
66. Schreiner B, Mitsdoerffer M, Kieseier BC, Chen L, Hartung HP, Weller M, et al. Interferon-beta enhances monocyte and dendritic cell expression of B7-H1 (Pd-L1), a strong inhibitor of autologous T-cell activation: Relevance for the immune modulatory effect in multiple sclerosis. J Neuroimmunol (2004) 155(1-2):172–82. doi: 10.1016/j.jneuroim.2004.06.013
67. Loke P, Allison JP. Pd-L1 and pd-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci U.S.A. (2003) 100(9):5336–41. doi: 10.1073/pnas.0931259100
68. Huang G, Wen Q, Zhao Y, Gao Q, Bai Y. Nf-kappab plays a key role in inducing Cd274 expression in human monocytes after lipopolysaccharide treatment. PloS One (2013) 8(4):e61602. doi: 10.1371/journal.pone.0061602
69. Mezzadra R, Sun C, Jae LT, Gomez-Eerland R, de Vries E, Wu W, et al. Identification of Cmtm6 and Cmtm4 as pd-L1 protein regulators. Nature (2017) 549(7670):106–10. doi: 10.1038/nature23669
70. Pulko V, Liu X, Krco CJ, Harris KJ, Frigola X, Kwon ED, et al. Tlr3-stimulated dendritic cells up-regulate B7-H1 expression and influence the magnitude of Cd8 T cell responses to tumor vaccination. J Immunol (2009) 183(6):3634–41. doi: 10.4049/jimmunol.0900974
71. Cole JE, Navin TJ, Cross AJ, Goddard ME, Alexopoulou L, Mitra AT, et al. Unexpected protective role for toll-like receptor 3 in the arterial wall. Proc Natl Acad Sci U.S.A. (2011) 108(6):2372–7. doi: 10.1073/pnas.1018515108
72. Karakhanova S, Meisel S, Ring S, Mahnke K, Enk AH. Erk/P38 map-kinases and Pi3k are involved in the differential regulation of B7-H1 expression in dc subsets. Eur J Immunol (2010) 40(1):254–66. doi: 10.1002/eji.200939289
73. Zhao Q, Xiao X, Wu Y, Wei Y, Zhu LY, Zhou J, et al. Interleukin-17-Educated monocytes suppress cytotoxic T-cell function through B7-H1 in hepatocellular carcinoma patients. Eur J Immunol (2011) 41(8):2314–22. doi: 10.1002/eji.201041282
74. Ou JN, Wiedeman AE, Stevens AM. Tnf-alpha and tgf-beta counter-regulate pd-L1 expression on monocytes in systemic lupus erythematosus. Sci Rep (2012) 2:295. doi: 10.1038/srep00295
75. Ni XY, Sui HX, Liu Y, Ke SZ, Wang YN, Gao FG. Tgf-beta of lung cancer microenvironment upregulates B7h1 and gitrl expression in dendritic cells and is associated with regulatory T cell generation. Oncol Rep (2012) 28(2):615–21. doi: 10.3892/or.2012.1822
76. Song S, Yuan P, Wu H, Chen J, Fu J, Li P, et al. Dendritic cells with an increased pd-L1 by tgf-beta induce T cell anergy for the cytotoxicity of hepatocellular carcinoma cells. Int Immunopharmacol (2014) 20(1):117–23. doi: 10.1016/j.intimp.2014.02.027
77. Baas M, Besancon A, Goncalves T, Valette F, Yagita H, Sawitzki B, et al. Tgfbeta-dependent expression of pd-1 and pd-L1 controls Cd8(+) T cell anergy in transplant tolerance. Elife (2016) 5:e08133. doi: 10.7554/eLife.08133
78. Starke A, Wuthrich RP, Waeckerle-Men Y. Tgf-beta treatment modulates pd-L1 and Cd40 expression in proximal renal tubular epithelial cells and enhances Cd8 cytotoxic T-cell responses. Nephron Exp Nephrol (2007) 107(1):e22–9. doi: 10.1159/000106506
79. Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med (2003) 9(5):562–7. doi: 10.1038/nm863
80. Xiong HY, Ma TT, Wu BT, Lin Y, Tu ZG. Il-12 regulates B7-H1 expression in ovarian cancer-associated macrophages by effects on nf-kappab signalling. Asian Pac J Cancer Prev (2014) 15(14):5767–72. doi: 10.7314/apjcp.2014.15.14.5767
81. Karakhanova S, Bedke T, Enk AH, Mahnke K. Il-27 renders dc immunosuppressive by induction of B7-H1. J Leukoc Biol (2011) 89(6):837–45. doi: 10.1189/jlb.1209788
82. Matta BM, Raimondi G, Rosborough BR, Sumpter TL, Thomson AW. Il-27 production and Stat3-dependent upregulation of B7-H1 mediate immune regulatory functions of liver plasmacytoid dendritic cells. J Immunol (2012) 188(11):5227–37. doi: 10.4049/jimmunol.1103382
83. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. Pd-L1 is a novel direct target of hif-1alpha, and its blockade under hypoxia enhanced mdsc-mediated T cell activation. J Exp Med (2014) 211(5):781–90. doi: 10.1084/jem.20131916
84. Gong AY, Zhou R, Hu G, Li X, Splinter PL, O'Hara SP, et al. Microrna-513 regulates B7-H1 translation and is involved in ifn-Gamma-Induced B7-H1 expression in cholangiocytes. J Immunol (2009) 182(3):1325–33. doi: 10.4049/jimmunol.182.3.1325
85. Gong AY, Zhou R, Hu G, Liu J, Sosnowska D, Drescher KM, et al. Cryptosporidium parvum induces B7-H1 expression in cholangiocytes by down-regulating microrna-513. J Infect Dis (2010) 201(1):160–9. doi: 10.1086/648589
86. Yee D, Shah KM, Coles MC, Sharp TV, Lagos D. Microrna-155 induction Via tnf-alpha and ifn-gamma suppresses expression of programmed death ligand-1 (Pd-L1) in human primary cells. J Biol Chem (2017) 292(50):20683–93. doi: 10.1074/jbc.M117.809053
87. Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn YH, Byers LA, et al. Metastasis is regulated Via microrna-200/Zeb1 axis control of tumour cell pd-L1 expression and intratumoral immunosuppression. Nat Commun (2014) 5:5241. doi: 10.1038/ncomms6241
88. Cortez MA, Ivan C, Valdecanas D, Wang X, Peltier HJ, Ye Y, et al. Pdl1 regulation by P53 Via mir-34. J Natl Cancer Inst (2016) 108(1):djv303. doi: 10.1093/jnci/djv303
90. Burr ML, Sparbier CE, Chan YC, Williamson JC, Woods K, Beavis PA, et al. Cmtm6 maintains the expression of pd-L1 and regulates anti-tumour immunity. Nature (2017) 549(7670):101–5. doi: 10.1038/nature23643
91. Karnam A, Bonam SR, Rambabu N, Wong SSW, Aimanianda V, Bayry J. Wnt-Beta-Catenin signaling in human dendritic cells mediates regulatory T-cell responses to fungi Via the pd-L1 pathway. mBio (2021) 12(6):e0282421. doi: 10.1128/mBio.02824-21
92. Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun (2016) 7:12632. doi: 10.1038/ncomms12632
93. Wu Y, Zhang C, Liu X, He Z, Shan B, Zeng Q, et al. Arih1 signaling promotes anti-tumor immunity by targeting pd-L1 for proteasomal degradation. Nat Commun (2021) 12(1):2346. doi: 10.1038/s41467-021-22467-8
94. Yang Z, Wang Y, Liu S, Deng W, Lomeli SH, Moriceau G, et al. Enhancing pd-L1 degradation by itch during mapk inhibitor therapy suppresses acquired resistance. Cancer Discovery (2022) 12(8):1942–59. doi: 10.1158/2159-8290.CD-21-1463
95. Lim SO, Li CW, Xia W, Cha JH, Chan LC, Wu Y, et al. Deubiquitination and stabilization of pd-L1 by Csn5. Cancer Cell (2016) 30(6):925–39. doi: 10.1016/j.ccell.2016.10.010
96. Yamaguchi H, Hsu JM, Yang WH, Hung MC. Mechanisms regulating pd-L1 expression in cancers and associated opportunities for novel small-molecule therapeutics. Nat Rev Clin Oncol (2022) 19(5):287–305. doi: 10.1038/s41571-022-00601-9
97. Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon receptor signaling pathways regulating pd-L1 and pd-L2 expression. Cell Rep (2017) 19(6):1189–201. doi: 10.1016/j.celrep.2017.04.031
98. Hokama LT, Veiga ADM, Menezes MCS, Sardinha Pinto AA, de Lima TM, Ariga SKK, et al. Endothelial injury in covid-19 and septic patients. Microvasc Res (2022) 140:104303. doi: 10.1016/j.mvr.2021.104303
99. Liu C, Yao Z, Wang J, Zhang W, Yang Y, Zhang Y, et al. Macrophage-derived Ccl5 facilitates immune escape of colorectal cancer cells Via the P65/Stat3-Csn5-Pd-L1 pathway. Cell Death Differ (2020) 27(6):1765–81. doi: 10.1038/s41418-019-0460-0
100. Gao Y, Yang J, Cai Y, Fu S, Zhang N, Fu X, et al. Ifn-Gamma-Mediated inhibition of lung cancer correlates with pd-L1 expression and is regulated by Pi3k-akt signaling. Int J Cancer (2018) 143(4):931–43. doi: 10.1002/ijc.31357
101. Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, et al. Exosomal pd-L1 contributes to immunosuppression and is associated with anti-Pd-1 response. Nature (2018) 560(7718):382–6. doi: 10.1038/s41586-018-0392-8
102. Weichhart T, Saemann MD. The Pi3k/Akt/Mtor pathway in innate immune cells: Emerging therapeutic applications. Ann Rheum Dis (2008) 67 Suppl 3:iii70–4. doi: 10.1136/ard.2008.098459
103. Zhao R, Song Y, Wang Y, Huang Y, Li Z, Cui Y, et al. Pd-1/Pd-L1 blockade rescue exhausted Cd8+ T cells in gastrointestinal stromal tumours Via the Pi3k/Akt/Mtor signalling pathway. Cell Prolif (2019) 52(3):e12571. doi: 10.1111/cpr.12571
104. Vasan N, Cantley LC. At A crossroads: How to translate the roles of Pi3k in oncogenic and metabolic signalling into improvements in cancer therapy. Nat Rev Clin Oncol (2022) 19(7):471–85. doi: 10.1038/s41571-022-00633-1
105. Alsuliman A, Colak D, Al-Harazi O, Fitwi H, Tulbah A, Al-Tweigeri T, et al. Bidirectional crosstalk between pd-L1 expression and epithelial to mesenchymal transition: Significance in claudin-low breast cancer cells. Mol Cancer (2015) 14:149. doi: 10.1186/s12943-015-0421-2
106. Kumar V, Barrett JE. Toll-like receptors (Tlrs) in health and disease: An overview. Handb Exp Pharmacol (2022) 276:1–21. doi: 10.1007/164_2021_568
107. Li H, Xia JQ, Zhu FS, Xi ZH, Pan CY, Gu LM, et al. Lps promotes the expression of pd-L1 in gastric cancer cells through nf-kappab activation. J Cell Biochem (2018) 119(12):9997–10004. doi: 10.1002/jcb.27329
108. Kang X, Li P, Zhang C, Zhao Y, Hu H, Wen G. The Tlr4/Erk/Pdl1 axis may contribute to nsclc initiation. Int J Oncol (2020) 57(2):456–65. doi: 10.3892/ijo.2020.5068
109. Witt DA, Donson AM, Amani V, Moreira DC, Sanford B, Hoffman LM, et al. Specific expression of pd-L1 in rela-fusion supratentorial ependymoma: Implications for pd-1-Targeted therapy. Pediatr Blood Cancer (2018) 65(5):e26960. doi: 10.1002/pbc.26960
110. Fortier ME, Kent S, Ashdown H, Poole S, Boksa P, Luheshi GN. The viral mimic, Polyinosinic:Polycytidylic acid, induces fever in rats Via an interleukin-1-Dependent mechanism. Am J Physiol Regul Integr Comp Physiol (2004) 287(4):R759–66. doi: 10.1152/ajpregu.00293.2004
111. Boes M, Meyer-Wentrup F. Tlr3 triggering regulates pd-L1 (Cd274) expression in human neuroblastoma cells. Cancer Lett (2015) 361(1):49–56. doi: 10.1016/j.canlet.2015.02.027
112. Ogawa T, Kan OK, Shiota A, Fujita A, Ishii Y, Fukuyama S, et al. Inhibition of Pi3kdelta differentially regulates poly I:C- and human metapneumovirus-induced pd-L1 and pd-L2 expression in human bronchial epithelial cells. Front Immunol (2021) 12:767666. doi: 10.3389/fimmu.2021.767666
113. Schodel J, Ratcliffe PJ. Mechanisms of hypoxia signalling: New implications for nephrology. Nat Rev Nephrol (2019) 15(10):641–59. doi: 10.1038/s41581-019-0182-z
114. Donskov F, Pinto CA, Predoiu R, Fox C, Georgsen JB, Skaarup K, et al. Molecular analysis and favorable clinical outcomes in real-world patients with metastatic renal cell carcinoma. Acta Oncol (2022) 61(10):1268–77. doi: 10.1080/0284186X.2022.2119100
115. Hussain SM, Kansal RG, Alvarez MA, Hollingsworth TJ, Elahi A, Miranda-Carboni G, et al. Role of tgf-beta in pancreatic ductal adenocarcinoma progression and pd-L1 expression. Cell Oncol (Dordr) (2021) 44(3):673–87. doi: 10.1007/s13402-021-00594-0
116. Cheng X, Zhao Z, Ventura E, Gran B, Shindler KS, Rostami A. The pd-1/Pd-L pathway is up-regulated during il-12-Induced suppression of eae mediated by ifn-gamma. J Neuroimmunol (2007) 185(1-2):75–86. doi: 10.1016/j.jneuroim.2007.01.012
117. Wang X, Yang L, Huang F, Zhang Q, Liu S, Ma L, et al. Inflammatory cytokines il-17 and tnf-alpha up-regulate pd-L1 expression in human prostate and colon cancer cells. Immunol Lett (2017) 184:7–14. doi: 10.1016/j.imlet.2017.02.006
118. Chen S, Crabill GA, Pritchard TS, McMiller TL, Wei P, Pardoll DM, et al. Mechanisms regulating pd-L1 expression on tumor and immune cells. J Immunother Cancer (2019) 7(1):305. doi: 10.1186/s40425-019-0770-2
119. Liang CL, Jiang H, Feng W, Liu H, Han L, Chen Y, et al. Total glucosides of paeony ameliorate pristane-induced lupus nephritis by inducing pd-1 ligands(+) macrophages Via activating il-4/Stat6/Pd-L2 signaling. Front Immunol (2021) 12:683249. doi: 10.3389/fimmu.2021.683249
120. Peng Q, Qiu X, Zhang Z, Zhang S, Zhang Y, Liang Y, et al. Pd-L1 on dendritic cells attenuates T cell activation and regulates response to immune checkpoint blockade. Nat Commun (2020) 11(1):4835. doi: 10.1038/s41467-020-18570-x
121. Sun C, Mezzadra R, Schumacher TN. Regulation and function of the pd-L1 checkpoint. Immunity (2018) 48(3):434–52. doi: 10.1016/j.immuni.2018.03.014
122. Li X, Wang S, Mu W, Barry J, Han A, Carpenter RL, et al. Reactive oxygen species reprogram macrophages to suppress antitumor immune response through the exosomal mir-155-5p/Pd-L1 pathway. J Exp Clin Cancer Res (2022) 41(1):41. doi: 10.1186/s13046-022-02244-1
123. Fujita Y, Yagishita S, Hagiwara K, Yoshioka Y, Kosaka N, Takeshita F, et al. The clinical relevance of the mir-197/Cks1b/Stat3-Mediated pd-L1 network in chemoresistant non-Small-Cell lung cancer. Mol Ther (2015) 23(4):717–27. doi: 10.1038/mt.2015.10
124. Zhu J, Chen L, Zou L, Yang P, Wu R, Mao Y, et al. Mir-20b, -21, and -130b inhibit pten expression resulting in B7-H1 over-expression in advanced colorectal cancer. Hum Immunol (2014) 75(4):348–53. doi: 10.1016/j.humimm.2014.01.006
125. Li CW, Lim SO, Chung EM, Kim YS, Park AH, Yao J, et al. Eradication of triple-negative breast cancer cells by targeting glycosylated pd-L1. Cancer Cell (2018) 33(2):187–201 e10. doi: 10.1016/j.ccell.2018.01.009
126. Hsu JM, Xia W, Hsu YH, Chan LC, Yu WH, Cha JH, et al. Stt3-dependent pd-L1 accumulation on cancer stem cells promotes immune evasion. Nat Commun (2018) 9(1):1908. doi: 10.1038/s41467-018-04313-6
127. Ruan Z, Liang M, Lai M, Shang L, Deng X, Su X. Kya1797k down-regulates pd-L1 in colon cancer stem cells to block immune evasion by suppressing the beta-Catenin/Stt3 signaling pathway. Int Immunopharmacol (2020) 78:106003. doi: 10.1016/j.intimp.2019.106003
128. Wang L, Wu J, Li J, Yang H, Tang T, Liang H, et al. Host-mediated ubiquitination of a mycobacterial protein suppresses immunity. Nature (2020) 577(7792):682–8. doi: 10.1038/s41586-019-1915-7
129. Merelli B, Massi D, Cattaneo L, Mandala M. Targeting the Pd1/Pd-L1 axis in melanoma: Biological rationale, clinical challenges and opportunities. Crit Rev Oncol Hematol (2014) 89(1):140–65. doi: 10.1016/j.critrevonc.2013.08.002
130. Chua KH, Lian LH, Sim XJ, Cheah TE, Lau TP. Association between Pdcd1 gene polymorphisms and risk of systemic lupus erythematosus in three main ethnic groups of the Malaysian population. Int J Mol Sci (2015) 16(5):9794–803. doi: 10.3390/ijms16059794
131. Nishimura H, Honjo T, Minato N. Facilitation of beta selection and modification of positive selection in the thymus of pd-1-Deficient mice. J Exp Med (2000) 191(5):891–8. doi: 10.1084/jem.191.5.891
132. Wang J, Okazaki IM, Yoshida T, Chikuma S, Kato Y, Nakaki F, et al. Pd-1 deficiency results in the development of fatal myocarditis in mrl mice. Int Immunol (2010) 22(6):443–52. doi: 10.1093/intimm/dxq026
133. Godoy GJ, Olivera C, Paira DA, Salazar FC, Ana Y, Stempin CC, et al. T Regulatory cells from non-obese diabetic mice show low responsiveness to il-2 stimulation and exhibit differential expression of anergy-related and ubiquitination factors. Front Immunol (2019) 10:2665. doi: 10.3389/fimmu.2019.02665
134. Morita K, Tsuda S, Kobayashi E, Hamana H, Tsuda K, Shima T, et al. Analysis of tcr repertoire and pd-1 expression in decidual and peripheral Cd8(+) T cells reveals distinct immune mechanisms in miscarriage and preeclampsia. Front Immunol (2020) 11:1082. doi: 10.3389/fimmu.2020.01082
135. Fife BT, Bluestone JA. Control of peripheral T-cell tolerance and autoimmunity Via the ctla-4 and pd-1 pathways. Immunol Rev (2008) 224:166–82. doi: 10.1111/j.1600-065X.2008.00662.x
136. Frebel H, Nindl V, Schuepbach RA, Braunschweiler T, Richter K, Vogel J, et al. Programmed death 1 protects from fatal circulatory failure during systemic virus infection of mice. J Exp Med (2012) 209(13):2485–99. doi: 10.1084/jem.20121015
137. Mueller SN, Vanguri VK, Ha SJ, West EE, Keir ME, Glickman JN, et al. Pd-L1 has distinct functions in hematopoietic and nonhematopoietic cells in regulating T cell responses during chronic infection in mice. J Clin Invest (2010) 120(7):2508–15. doi: 10.1172/JCI40040
138. Gotsman I, Grabie N, Dacosta R, Sukhova G, Sharpe A, Lichtman AH. Proatherogenic immune responses are regulated by the pd-1/Pd-L pathway in mice. J Clin Invest (2007) 117(10):2974–82. doi: 10.1172/JCI31344
139. Allie SR, Zhang W, Fuse S, Usherwood EJ. Programmed death 1 regulates development of central memory Cd8 T cells after acute viral infection. J Immunol (2011) 186(11):6280–6. doi: 10.4049/jimmunol.1003870
140. Fuse S, Tsai CY, Molloy MJ, Allie SR, Zhang W, Yagita H, et al. Recall responses by helpless memory Cd8+ T cells are restricted by the up-regulation of pd-1. J Immunol (2009) 182(7):4244–54. doi: 10.4049/jimmunol.0802041
141. Erickson JJ, Gilchuk P, Hastings AK, Tollefson SJ, Johnson M, Downing MB, et al. Viral acute lower respiratory infections impair Cd8+ T cells through pd-1. J Clin Invest (2012) 122(8):2967–82. doi: 10.1172/JCI62860
142. Cecconi M, Evans L, Levy M, Rhodes A. Sepsis and septic shock. Lancet (2018) 392(10141):75–87. doi: 10.1016/S0140-6736(18)30696-2
143. van der Poll T, Shankar-Hari M, Wiersinga WJ. The immunology of sepsis. Immunity (2021) 54(11):2450–64. doi: 10.1016/j.immuni.2021.10.012
144. Ruan WS, Feng MX, Xu J, Xu YG, Song CY, Lin LY, et al. Early activation of myeloid-derived suppressor cells participate in sepsis-induced immune suppression Via pd-L1/Pd-1 axis. Front Immunol (2020) 11:1299. doi: 10.3389/fimmu.2020.01299
145. Lazar-Molnar E, Gacser A, Freeman GJ, Almo SC, Nathenson SG, Nosanchuk JD. The pd-1/Pd-L costimulatory pathway critically affects host resistance to the pathogenic fungus histoplasma capsulatum. Proc Natl Acad Sci U.S.A. (2008) 105(7):2658–63. doi: 10.1073/pnas.0711918105
146. Phares TW, Ramakrishna C, Parra GI, Epstein A, Chen L, Atkinson R, et al. Target-dependent B7-H1 regulation contributes to clearance of central nervous system infection and dampens morbidity. J Immunol (2009) 182(9):5430–8. doi: 10.4049/jimmunol.0803557
147. Yao S, Wang S, Zhu Y, Luo L, Zhu G, Flies S, et al. Pd-1 on dendritic cells impedes innate immunity against bacterial infection. Blood (2009) 113(23):5811–8. doi: 10.1182/blood-2009-02-203141
148. Spec A, Shindo Y, Burnham CA, Wilson S, Ablordeppey EA, Beiter ER, et al. T Cells from patients with candida sepsis display a suppressive immunophenotype. Crit Care (2016) 20:15. doi: 10.1186/s13054-016-1182-z
149. Guignant C, Lepape A, Huang X, Kherouf H, Denis L, Poitevin F, et al. Programmed death-1 levels correlate with increased mortality, nosocomial infection and immune dysfunctions in septic shock patients. Crit Care (2011) 15(2):R99. doi: 10.1186/cc10112
150. Chang K, Svabek C, Vazquez-Guillamet C, Sato B, Rasche D, Wilson S, et al. Targeting the programmed cell death 1: Programmed cell death ligand 1 pathway reverses T cell exhaustion in patients with sepsis. Crit Care (2014) 18(1):R3. doi: 10.1186/cc13176
151. Wilson JK, Zhao Y, Singer M, Spencer J, Shankar-Hari M. Lymphocyte subset expression and serum concentrations of pd-1/Pd-L1 in sepsis - pilot study. Crit Care (2018) 22(1):95. doi: 10.1186/s13054-018-2020-2
152. Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity (2007) 27(1):111–22. doi: 10.1016/j.immuni.2007.05.016
153. Latchman YE, Liang SC, Wu Y, Chernova T, Sobel RA, Klemm M, et al. Pd-L1-Deficient mice show that pd-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc Natl Acad Sci U.S.A. (2004) 101(29):10691–6. doi: 10.1073/pnas.0307252101
154. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol (2015) 15(8):486–99. doi: 10.1038/nri3862
155. Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: A novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis (2013) 13(3):260–8. doi: 10.1016/S1473-3099(13)70001-X
156. Patil NK, Guo Y, Luan L, Sherwood ER. Targeting immune cell checkpoints during sepsis. Int J Mol Sci (2017) 18(11):2413. doi: 10.3390/ijms18112413
157. McNab FW, Berry MP, Graham CM, Bloch SA, Oni T, Wilkinson KA, et al. Programmed death ligand 1 is over-expressed by neutrophils in the blood of patients with active tuberculosis. Eur J Immunol (2011) 41(7):1941–7. doi: 10.1002/eji.201141421
158. Patera AC, Drewry AM, Chang K, Beiter ER, Osborne D, Hotchkiss RS. Frontline science: Defects in immune function in patients with sepsis are associated with pd-1 or pd-L1 expression and can be restored by antibodies targeting pd-1 or pd-L1. J Leukoc Biol (2016) 100(6):1239–54. doi: 10.1189/jlb.4HI0616-255R
159. Patil NK, Luan L, Bohannon JK, Hernandez A, Guo Y, Sherwood ER. Frontline science: Anti-Pd-L1 protects against infection with common bacterial pathogens after burn injury. J Leukoc Biol (2018) 103(1):23–33. doi: 10.1002/JLB.5HI0917-360R
160. Wang F, Huang X, Chung CS, Chen Y, Hutchins NA, Ayala A. Contribution of programmed cell death receptor (Pd)-1 to kupffer cell dysfunction in murine polymicrobial sepsis. Am J Physiol Gastrointest Liver Physiol (2016) 311(2):G237–45. doi: 10.1152/ajpgi.00371.2015
161. Huang X, Chen Y, Chung CS, Yuan Z, Monaghan SF, Wang F, et al. Identification of B7-H1 as a novel mediator of the innate Immune/Proinflammatory response as well as a possible myeloid cell prognostic biomarker in sepsis. J Immunol (2014) 192(3):1091–9. doi: 10.4049/jimmunol.1302252
162. Shao R, Fang Y, Yu H, Zhao L, Jiang Z, Li CS. Monocyte programmed death ligand-1 expression after 3-4 days of sepsis is associated with risk stratification and mortality in septic patients: A prospective cohort study. Crit Care (2016) 20(1):124. doi: 10.1186/s13054-016-1301-x
163. Jiang W, Li X, Wen M, Liu X, Wang K, Wang Q, et al. Increased percentage of pd-L1(+) natural killer cells predicts poor prognosis in sepsis patients: A prospective observational cohort study. Crit Care (2020) 24(1):617. doi: 10.1186/s13054-020-03329-z
164. Collins M, Ling V, Carreno BM. The B7 family of immune-regulatory ligands. Genome Biol (2005) 6(6):223. doi: 10.1186/gb-2005-6-6-223
165. Hutchins NA, Wang F, Wang Y, Chung CS, Ayala A. Kupffer cells potentiate liver sinusoidal endothelial cell injury in sepsis by ligating programmed cell death ligand-1. J Leukoc Biol (2013) 94(5):963–70. doi: 10.1189/jlb.0113051
166. Xu S, Yang Q, Bai J, Tao T, Tang L, Chen Y, et al. Blockade of endothelial, but not epithelial, cell expression of pd-L1 following severe shock attenuates the development of indirect acute lung injury in mice. Am J Physiol Lung Cell Mol Physiol (2020) 318(4):L801–L12. doi: 10.1152/ajplung.00108.2019
167. Lomas-Neira J, Monaghan SF, Huang X, Fallon EA, Chung CS, Ayala A. Novel role for pd-1:Pd-L1 as mediator of pulmonary vascular endothelial cell functions in pathogenesis of indirect Ards in mice. Front Immunol (2018) 9:3030. doi: 10.3389/fimmu.2018.03030
168. Wu Y, Chung CS, Chen Y, Monaghan SF, Patel S, Huang X, et al. A novel role for programmed cell death receptor ligand-1 (Pd-L1) in sepsis-induced intestinal dysfunction. Mol Med (2017) 22:830–40. doi: 10.2119/molmed.2016.00150
169. Huang X, Venet F, Wang YL, Lepape A, Yuan Z, Chen Y, et al. Pd-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc Natl Acad Sci U.S.A. (2009) 106(15):6303–8. doi: 10.1073/pnas.0809422106
170. Shindo Y, McDonough JS, Chang KC, Ramachandra M, Sasikumar PG, Hotchkiss RS. Anti-Pd-L1 peptide improves survival in sepsis. J Surg Res (2017) 208:33–9. doi: 10.1016/j.jss.2016.08.099
171. Phares TW, Kotraiah V, Chung CS, Unsinger J, Mazer M, Remy KE, et al. A peptide-based checkpoint immunomodulator alleviates immune dysfunction in murine polymicrobial sepsis. Shock (2021) 55(6):806–15. doi: 10.1097/SHK.0000000000001682
172. Busch LM, Sun J, Cui X, Eichacker PQ, Torabi-Parizi P. Checkpoint inhibitor therapy in preclinical sepsis models: A systematic review and meta-analysis. Intensive Care Med Exp (2020) 8(1):7. doi: 10.1186/s40635-019-0290-x
173. Curran CS, Busch LM, Li Y, Xizhong C, Sun J, Eichacker PQ, et al. Anti-Pd-L1 therapy does not improve survival in a murine model of lethal staphylococcus aureus pneumonia. J Infect Dis (2021) 224(12):2073–84. doi: 10.1093/infdis/jiab274
174. Busch LM, Sun J, Eichacker PQ, Torabi-Parizi P. Inhibitory immune checkpoint molecule expression in clinical sepsis studies: A systematic review. Crit Care Med (2020) 48(9):1365–74. doi: 10.1097/CCM.0000000000004496
175. Motzer RJ, Powles T, Burotto M, Escudier B, Bourlon MT, Shah AY, et al. Nivolumab plus cabozantinib versus sunitinib in first-line treatment for advanced renal cell carcinoma (Checkmate 9er): Long-term follow-up results from an open-label, randomised, phase 3 trial. Lancet Oncol (2022) 23(7):888–98. doi: 10.1016/S1470-2045(22)00290-X
176. Provencio M, Serna-Blasco R, Nadal E, Insa A, Garcia-Campelo MR, Casal Rubio J, et al. Overall survival and biomarker analysis of neoadjuvant nivolumab plus chemotherapy in operable stage iiia non-Small-Cell lung cancer (Nadim phase ii trial). J Clin Oncol (2022) 40(25):2924–33. doi: 10.1200/JCO.21.02660
177. Dolladille C, Ederhy S, Sassier M, Cautela J, Thuny F, Cohen AA, et al. Immune checkpoint inhibitor rechallenge after immune-related adverse events in patients with cancer. JAMA Oncol (2020) 6(6):865–71. doi: 10.1001/jamaoncol.2020.0726
Keywords: PD-1, PD-L1, sepsis, immune cells, immunology
Citation: Zhang T, Yu-Jing L and Ma T (2023) Role of regulation of PD-1 and PD-L1 expression in sepsis. Front. Immunol. 14:1029438. doi: 10.3389/fimmu.2023.1029438
Received: 27 August 2022; Accepted: 27 February 2023;
Published: 09 March 2023.
Edited by:
Ian Marriott, University of North Carolina at Charlotte, United StatesReviewed by:
Parizad Torabi-Parizi, National Institutes of Health, United StatesCopyright © 2023 Zhang, Yu-Jing and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tao Ma, dGFvbWFAdG11LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.