 Xi Liu1
Xi Liu1
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 03 October 2022
Sec. Parasite Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.999412
This article is part of the Research TopicA year in review: Discussions in Parasite ImmunologyView all 4 articles
Non-alcoholic fatty liver disease (NAFLD) is closely related to obesity, diabetes, and metabolic syndrome (MetS), and it has become the most common chronic liver disease. Helminths have co-evolved with humans, inducing multiple immunomodulatory mechanisms to modulate the host’s immune system. By using their immunomodulatory ability, helminths and their products exhibit protection against various autoimmune and inflammatory diseases, including obesity, diabetes, and MetS, which are closely associated with NAFLD. Here, we review the pathogenesis of NAFLD from abnormal glycolipid metabolism, inflammation, and gut dysbiosis. Correspondingly, helminths and their products can treat or relieve these NAFLD-related diseases, including obesity, diabetes, and MetS, by promoting glycolipid metabolism homeostasis, regulating inflammation, and restoring the balance of gut microbiota. Considering that a large number of clinical trials have been carried out on helminths and their products for the treatment of inflammatory diseases with promising results, the treatment of NAFLD and obesity-related diseases by helminths is also a novel direction and strategy.
With the improvement of living standards in modern society, obesity and related metabolic syndrome have become a global epidemic, especially in developed western countries and wealthy regions in other countries. Non-alcoholic fatty liver disease (NAFLD) has become an important cause of modern chronic liver disease (1, 2). NAFLD has a global prevalence of 25.8% (3, 4), and the population with NAFLD is expected to increase by 21% by 2030 (2). Moreover, NAFLD is being diagnosed in a growing number of obese children and adolescents (5). Despite the increasing prevalence of NAFLD, which imposes a health burden on society and a huge economic burden on the medical industry, no medicines are available yet for the treatment of NAFLD (6, 7). Therefore, new therapeutic strategies for NAFLD should be explored.
Since the 1980s, the incidence of allergic diseases and autoimmune diseases has increased significantly in western countries and most modern countries because of the improvement of people’s living environment and the decrease in the incidence of major infectious and parasitic diseases. Accordingly, experts have coined the term “hygiene hypothesis” (8, 9). A consensus among the general public states that helminths are harmful to humans. However, “hygiene hypothesis” suggests that helminth infections can, in some cases, have beneficial effects on the host and their products may be potential therapeutic modalities (10, 11). Parasite can prevent inflammatory, autoimmune, and metabolic diseases through their excretory/secretory products (ESPs), such as extracellular vesicles, glycans, proteins, and microRNAs (10, 12–18). Parasitic infection can induce the type 2 immune response, effectively control the host inflammatory response, promote wound healing, and regulate tissue repair (19, 20). A recent epidemiological investigation manifested that previous schistosome infection is negatively associated with fatty liver and coronary heart disease (CHD) (21). Schistosoma infection may provide new direction for the prevention and treatment of fatty liver and CHD (21). Therefore, the ability of worm infection and/or its products to intervene with NAFLD needs to be determined. In this review, the pathogenesis of NAFLD is discussed, and the effect of helminth infection and their products on the pathogenesis of NAFLD for improved NAFLD was determined.
NAFLD is a progressive disease that describes a continuum of clinical liver abnormalities, showing changes from non-alcoholic fatty liver disease (NAFL) to non-alcoholic steatohepatitis (NASH). In this process, liver fibrosis and cirrhosis gradually progress. Patients with NASH are at high risk for eventual end-stage liver disease and hepatocellular carcinoma (2, 3, 22). NAFLD is histologically defined as more than 5% of hepatocytes, excluding hepatocyte damage caused by alcohol and other specific factors (23). The pathogenesis of NAFLD is complex, and NAFLD is related to metabolic dysfunction. Considering the heterogeneity of NAFLD, experts have introduced the term “metabolism-related fatty liver disease (MAFLD)”, which is considered to be a more accurate expression of the current understanding of NAFLD, that is, hepatic manifestations in the systemic metabolic disorders (24, 25). NAFLD is a disease caused by multiple factors, including overnutrition, obesity, type 2 diabetes mellitus (T2DM), metabolic syndrome (MetS), and genetics (26). These conditions can cause various injuries, such as lipid accumulation in the liver, insulin resistance (IR), glycolipid metabolism disorder, oxidative stress, release of inflammatory cytokines, and changes in the gut–liver axis, affect the occurrence and development of NAFLD (26, 27). Here, the pathogenesis of NAFLD is discussed from the aspects of glucose and lipid metabolism, inflammation, and gut microbiota.
T2DM is substantially associated with NAFLD (28). NAFLD, T2DM, and MetS are frequently coexisting. Generally, MetS is a major risk factor for T2DM. Additionally, among the risk factors that induce NAFLD and NASH, MetS is the strongest (29). MetS is affected by various environmental factors mainly because of high-fat and high-carbohydrate diet, resulting in hyperglycemia, hypertension, dyslipidemia, and a high incidence of fatty liver disease. The main feature of MetS is IR, indicating that insulin’s ability to make use of glucose is declining, particularly in non-hepatic tissues such as adipose and muscular tissues. Hyperinsulinemia, a direct manifestation of IR, is caused by increased blood glucose levels caused by decreased glucose utilization, followed by increased insulin compensatory activity (30, 31). Thus, IR is a pathogenic factor for NAFLD.
Considering that obesity is common characteristic in patients with NAFLD (4), obesity along with overnutrition may contribute to various injuries and thus lead to lipid accumulation in the liver (32). Therefore, the mechanisms of hepatic steatosis development driven by excessive hepatic lipid accumulation need to be understood. The main components of lipids or triglycerides (TG) accumulated in the human liver are primarily derived from circulating free fatty acids (FFAs) in adipose tissue (33, 34). De novo lipogenesis (DNL) and dietary fat intake are two important pathways for hepatic lipid accumulation (27, 32, 34). Lipolysis is a process in which fat is hydrolyzed into glycerol and fatty acid (FA) under the action of lipase, thus providing energy for the body (32). The lipase that catalyzes TG is a rate-limiting enzyme and is governed by a large number of hormones. When the lipid content in the body exceeds its processing capacity, the synthesis and utilization of TG are out of balance, leading to the accumulation of TG in the liver, which may be the first inducing factor of NAFLD (35). Liver lipid metabolism homeostasis is regulated by multiple mechanisms, such as hormones, metabolic pathways, and signaling pathways, such as phosphatidylinositol 3-kinase(PI3K)/AKT/PTEN pathway in hepatocytes (36), among which insulin signaling plays a crucial role (37). In general, plasma circulating FFA concentration increases during fasting because of insulin signaling and declines after feeding because of the inhibition of lipolysis. By contrast, in the presence of IR, the decomposition of fat is not restricted, thus increasing the degree of steatosis, thus remarkably increasing the level of circulating FFA, leading to hepatic steatosis (27, 32).
The incidence of liver DNL was substantially increased in patients with NAFLD (38, 39). DNL is the process by which excess carbohydrates are converted into FFAs. In this process, the acetyl-coenzyme A (acetyl-CoA) produced by glycolysis is initially converted to malonyl-coenzyme A (malonyl-CoA) through acetyl-CoA carboxylase (ACC) and ultimately to palmitate, a saturated fatty acid. These FFAs can be converted to TG for storage along with glycerol (39, 40). DNL in patients with NAFLD is regulated by insulin and glucose at the transcriptional level and is negatively correlated with insulin sensitivity (41). Two key transcription factors are involved in the regulation of DNL, including the sterol regulatory element binding protein 1C (SREBP1c), also named as sterol regulatory element binding transcription factor 1 (SREBF1), and the carbohydrate regulatory element binding protein (ChREBP). The activation of SREBP1c is mediated by insulin and liver X receptor (LXR), while ChREBP is activated by carbohydrate metabolites (32, 40–43). Hyperinsulinemia caused by insulin resistance increases the activity of SREBP1c. SREBP1c activation results in the transcription of lipogenic genes such as stearoyl-CoA desaturase 1 (SCD1) and stimulates elevated liver DNL. Hyperglycemia stimulates ChREBP and further induces the transcription of pyruvate kinase, contributing to the conversion of phosphoenolpyruvate and ADP into ATP and pyruvate, and the decarboxylation of pyruvate into CoA, which is then used in DNL, a synthetic pathway (44, 45). Based on mouse studies, the presence of excess lipids enhance liver DNL, creating a vicious cycle (46).
A diet in high calories, fat, and sucrose is closely associated with the occurrence of NAFLD and can promote the synthesis of FAs (4). Sucrose can be decomposed into fructose and glucose in the gut. Fructose is essential because of its ability to activate the key transcription factor SREBP1c and ChREBP in liver DNL (47). Although both glucose and fructose are metabolically regulated in the liver, fructose metabolism is more detrimental, because fructose can be directly extracted from the portal vein circulation and then transported to the liver. As a result, hepatocytes are exposed to higher concentrations of fructose than other tissues (47–49). Fructose activates ChREBP by increasing the intracellular concentration of fructose-1-phosphate and enhancing glycolysis flux. ChREBP, together with SREBP1c, induces the increased expression of acetyl-CoA carboxylase (ACC) and fatty acid synthase (FAS), thus increasing the very low-density lipoprotein (VLDL) TG secretion and DNL (35, 50–52). Therefore, abnormal glucose lipid metabolism can lead to NAFLD (Figure 1).
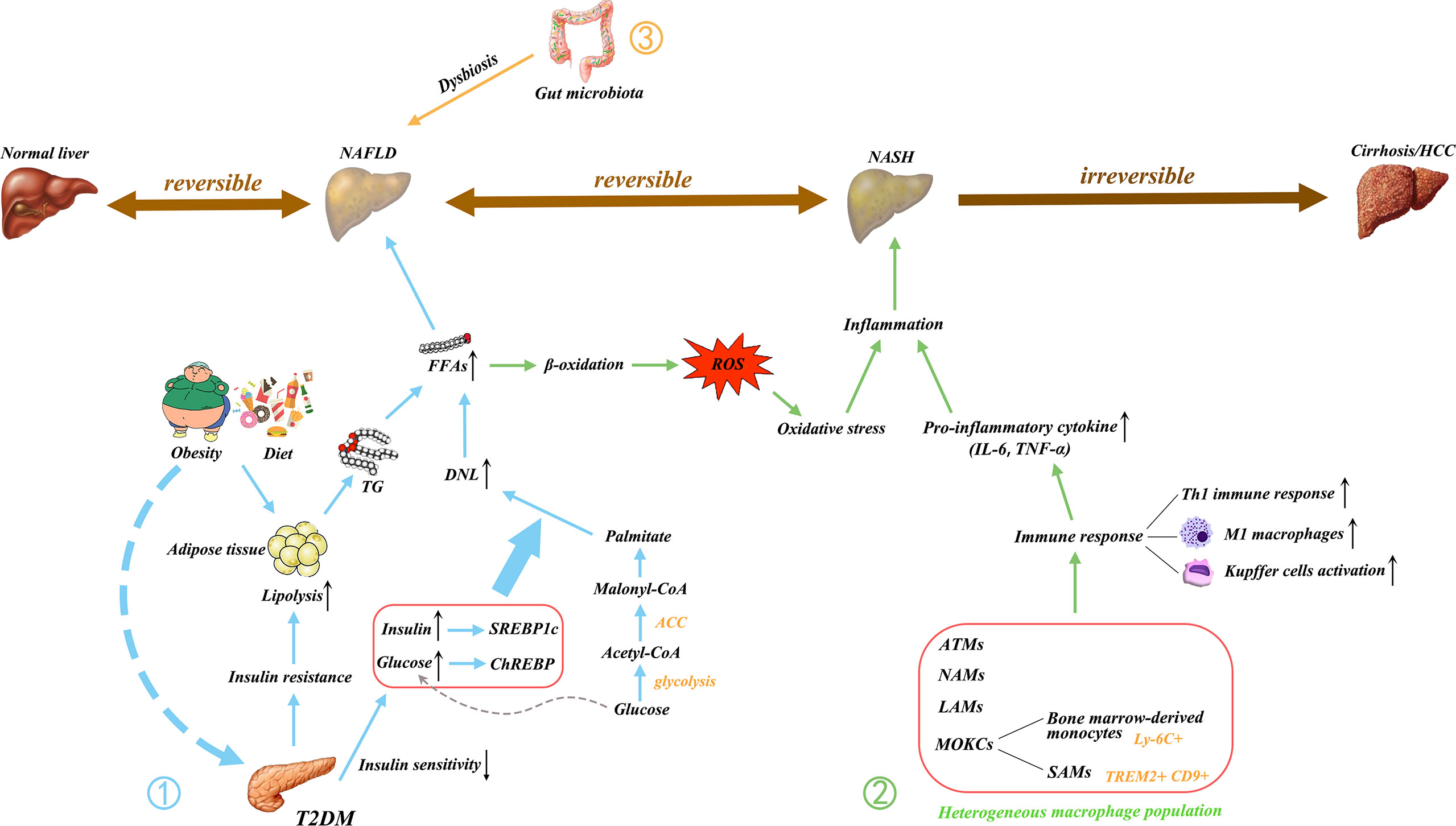
Figure 1 Main pathogenic mechanism of NAFLD. NAFLD is a multifactorial disease; Obesity, diet, and insulin resistance are linked to pathogenesis. ① The increase in free fatty acids (FFAs) is essential to the development of NAFLD. The FFAs produced by the lipolysis of TG in adipose tissue are delivered to the liver, resulting in hepatic steatosis. T2DM causes IR and decreases insulin sensitivity, thus triggering an increase in DNL, which is another major contributor to the increase in FFAs. The excessive delivery of FFAs to the liver upregulates β-oxidation and promotes lipotoxicity, leading to oxidative stress. ② In addition, the activated immune cells secrete pro-inflammatory cytokines. These processes initiate hepatic inflammation and contribute to the transition from NAFLD to NASH. ③ Gut microbiota dysregulation has also been implicated in the pathogenesis of NAFLD. Gut dysbiosis disrupt the intercellular tight junctions, thus allowing the entry of bacterial lipopolysaccharide into the systemic circulation and increasing liver input. Ultimately, it results in liver exposure to inflammatory mediators. NAFLD, nonalcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis; HCC, hepatocellular carcinoma; FFAs, free fatty acids; TG, triglyceride; T2DM, type 2 diabetes mellitus; IR, insulin resistance; DNL, de novo lipogenesis; LPS, lipopolysaccharide; ROS, reactive oxygen species; IL, interleukin; TNF, tumor necrosis factor; Th1, T helper type 1; M1, classically activated macrophage; ATMs, adipose tissue macrophages; NAMs, NASH-associated macrophages; LAMs, lipid-associated macrophages; MOKC, monocyte-derived KCs; SAMs, scar-associated macrophage; TREM2, triggering receptors expressed on myeloid cells 2; CD, cluster of differentiation; CoA, coenzyme A; ACC, acetyl-CoA carboxylase; SREBP1c, sterol regulatory element binding protein 1C (SREBP1c); ChREBP, carbohydrate regulatory element binding protein.
In NAFLD, inflammation may be chronic, and liver biopsy may be ignored to some extent, but nearly one-third of patients will develop from simple fatty liver to NASH (35). Excess lipids are ectopically deposited in multiple organs and tissues and impair their function because of overnutrition. In NASH, lipotoxicity caused by excess FFAs delivered to the liver can upregulate mitochondria β oxidation, thus increasing reactive oxygen species (ROS) generation, and inducing oxidative stress. Oxidative stress is a prominent driver of hepatic inflammation (53–55).
The inflammatory response generally occurs after the appearance of tissue damage, and after liver injury in patients with NAFLD, a large number of immune cells (e.g., resident and recruited macrophages, dendritic cells, neutrophils, monocytes, innate lymphoid cells, and mast cells), endothelial cells, and liver parenchymal cells participate in the inflammatory response. Resident liver macrophages such as Kupffer cells (KC), endothelial cells, and liver parenchymal cells have surface receptors that recognize DAMPs and PAMPs, which can bind the receptors and induce the synthesis and release of inflammatory mediators, leading to the recruitment of inflammatory immune cells into the damaged liver tissue (26, 56–59).
The innate immune response triggers hepatic inflammation, thus inducing disease progression toward NASH. Macrophages play a central role. Macrophages that are stimulated by different environmental factors are activated to transform into two distinct functional phenotypes, namely, the classically activated macrophages (M1) and the alternatively activated macrophages (M2) (60). However, this M1/M2 paradigm is only suitable for in vitro research (61). Instead of fixed phenotypes in vivo, macrophages exhibit high plasticity and can adopt different activation states according to the environment (62, 63). Adipose tissue macrophages (ATMs) accumulate in obese individuals, causing chronic low-grade inflammation. Long-term chronic inflammation leads to insulin resistance and metabolic imbalances. Kratz and colleague found that ATMs from obese humans converts “metabolically activated” phenotype, which is distinct from M1 or M2 markers. These metabolically activated macrophages regulate the balance between cytokine production and lipid metabolism and are driven by independent pro-inflammatory and anti-inflammatory pathways (64). Metabolic disease-specific macrophage phenotypes are also found in NAFLD. Xiong and colleague conducted single-cell RNA sequencing analysis and found that NASH-associated macrophages (NAMs) are highly expressive of triggering receptors expressed on myeloid cells 2 (Trem2) (65). These NAMs are also associated with disease severity and are highly responsive to drug and dietary interventions (65). KCs are replaced by bone marrow-derived macrophages and thus reduced in MAFLD. These recruited macrophages in the liver of MAFLD have two different activation states, one similar to homeostatic KCs and the other to lipid-associated macrophages (LAMs) from obese adipose tissues (66). These LAMs express NASH patient marker osteopontin, which is associated with fibrosis development (66). Compared with the traditional M1/M2 phenotype, the macrophage pool has considerable heterogeneity and requires more specific macrophage-targeting strategies in MAFLD. Tran and colleague also found that liver-resident KCs are replaced by Ly-6C+ monocytes during NASH (67). These monocyte-derived KCs (MoKCs) reduce hepatic triglyceride storage, increase inflammation, promote liver damage, and are linked to disease progression during NASH (67). Seidman et al. (68) also identified a subset of MoKCs that resembles scar-associated macrophage (SAM) phenotype. These SAMs express Trem2 and CD9 and are also profibrogenic in liver cirrhosis (69). A dual C-C chemokine receptor type 2 and 5 dual antagonist (named cenicriviroc) can inhibit macrophage infiltration and antifibrosis in NASH animal models. Cenicriviroc entered phase 2b trial in the CENTAUR and improved antifibrosis compared with that in the placebo group (70). Thus, targeting macrophages as inflammation mediators can improve disease progression in NASH or NAFLD. The inflammatory response in the liver promotes the progress in NAFLD (Figure 1).
Another well-recognized risk factor in the progression of NAFLD is the gut microbiota. Extensive research has confirmed that the gut microbiome of patients with NASH and normal subjects have different characteristics. The liver and the gut are inextricably linked to some extent through the “gut-liver axis”, which is an important two-way communication pathway between the gut and the liver. The products of the liver, such as bile acids, can influence the composition of the intestinal flora, the integrity of the intestinal barrier, and intestinal permeability; in turn, the intestinal microbiota influences bile acid synthesis and glycolipid metabolism in the liver (71, 72). Changes in the abundance and diversity of gut microbes are closely related to the progression of NAFLD. Each stage of NAFLD has its special gut microbiota signature (73, 74). Notably, the microbes, bacterial lipopolysaccharides (LPS), and the metabolites of the microorganisms, can affect the function of the liver (75). Thus, the enterohepatic axis plays a non-negligible role in NAFLD and is an effective and important target for the prevention and treatment of NAFLD in the future.
NAFLD is associated with dysbiosis and intestinal bacteria overgrowth (76, 77). Dysbiosis occurs when the beneficial and harmful bacteria in the gut are in imbalance qualitatively and quantitatively, generating a pathological combination. Many researchers speculated that differences in gut bacterial composition cause obese individuals to develop NAFLD. For example, obesity was replicated by transferring the gut microbiota from obese mice or humans to germ-free mice (78). In NAFLD, Firmicutes and Proteobacteria were increased, and Bacteroidetes were reduced at the phylum level. Ruminococcaceae and Rikenellaceae were decreased, and Enterobacteriaceae was increased at the family level. Anaerosporobacter, Faecalibacterium, Eubacterium, Prevotella, and Coprococcus were reduced, and Peptoniphilus, Dorea, and Escherichia were increased at the genera level (79). Therefore, fecal microbiome transplantation (FMT) that regulates the gut microbiome can be used to treat NAFLD. Xue and colleague found that FMT can improve intestinal microbiota disorders, thereby reducing fat aggregation and relieving fatty liver. Moreover, FMT improves the reconstruction of gut microbiota in lean NAFLD compared with that in obese patients with NAFLD (80). However, Craven et al. (81) found that allogenic or autologous FMT did not improve IR and hepatic proton density fat fraction but allogenic FMT decreased the small intestinal permeability in patients with NAFLD. Indeed, altered intestinal permeability is associated with liver disease (81). The intestinal mucosal barrier prevents the invasion of bacteria and the absorption of toxins. The mechanical barrier, also known as the physical barrier, is of utmost importance among the intestinal mucosal barriers and rests on the physiological structure of the mucosal epithelium, the lamina propria, and the mucosal base, where the intestinal epithelial cells are tightly arranged by cell junctions. Tight junction protein (TJP), including Zonula Occludens (ZOs), occluding, and claudin, connect the intestinal epithelial cells and maintain the integrity of the intestinal barrier (82). The dysbiosis of the gut microbiota leads to impaired TJP and the disruption of the tight junction between intestinal cells, thus increasing intestinal permeability (83). Under such circumstances, the LPS of gram-negative bacilli is translocated from the intestine to the portal system, and bacterial translocation leads to the exposure of the liver to inflammatory mediators (84, 85). HFD can induce proinflammatory signal, increase intestinal permeability, and cause the development of severe steatohepatitis (86). In comparison with healthy controls, patients with NAFLD increase intestinal permeability. Inflammation and early liver damage alter intestinal permeability in patients with NAFLD (87). Gut dysbiosis caused by inflammasome deficiency leads to the abnormal accumulation of bacterial metabolites in the portal circulation. Liver exposure to high concentrations of portal system products, especially pre-conditioned by lipid accumulation in hepatocytes by excessive nutrient, make it vulnerable to the development of NAFLD/NASH (88). The dysregulation of gut microbiota involved in NAFLD pathogenesis in shown in Figure 1.
It is worth noted that white adipose tissue (WAT) is an inflammatory organ where adipocytes interact with immune cells to maintain tissue homeostasis. In general, IL-10, IL-4, and other anti-inflammatory cytokines secreted from T regulatory cells (Tregs) and eosinophils polarize ATMs towards M2 or alternatively activated macrophages, thereby maintaining a tolerogenic phenotype (89, 90). However, excessive nutrition leads to WAT amplification and fat cell hypoxia, followed by chemokine production to induce immune cell infiltration and IL-6, TNF-α, and IL-1β release, resulting in a low-grade inflammatory response in obese individuals (91). Moreover, obesity, T2DM, and other environmental factors can alter intestinal permeability, causing gut-derived endotoxins to penetrate into the circulatory system, affecting liver lipid deposition, and accelerating liver inflammation and fibrosis processes in NAFLD (92).
Helminth infections can antagonize allergic and autoimmune disorders, as suggested by the hygiene hypothesis (8, 93, 94). However, helminth-mediated protection is not limited to autoimmune and allergic diseases. Recent studies have found that chronic helminth infection or helminth-derived products have beneficial effects on host metabolism and improve insulin resistance and T2DM (10). NAFLD is closely related to insulin resistance, T2DM, and MetS. Therefore, helminth may provide the protection against NAFLD.
Considering that NAFLD is closely related to disorders of glycolipid metabolism, the effective intervention of metabolic dysfunction can further influence the development of NAFLD. Numerous mouse experiments and several human studies have confirmed that helminth infection and its derived molecules attenuate obesity, improve IR, glucose tolerance, and MetS (15, 95). Nippostrongylus brasiliensis (N. brasiliensis) infection decreases weight gain and adipose tissue mass, relieves hepatic steatosis, reduces the expression of key lipogenic enzymes, and improves glucose metabolism in HFD mice (96). Schistosoma japonicum (S. japonicum) infection upregulates glycolysis-related genes, such as Ldha, Glut4, Pkm2, Glut1, Pfkfb3, Aldoc, HK2, and Pfk, and downregulates gluconeogenesis gene G6pc in mouse liver. Furthermore, S. japonicum infection downregulates FA synthesis genes and lipid uptake gene and upregulates FA oxidation-related gene. SEA-stimulated macrophages showed increased gene expression related to glycolysis and FA oxidation but decreased gene expression related to gluconeogenesis, FA synthesis, and lipid uptake. Therefore, S. japonicum infection promotes the catabolism of glycolipids and inhibits their anabolic metabolism in mouse livers, possibly via the AMPK, AKT, and mTORC1 pathways in macrophages (97). The mice in S. japonicum infection reduces miR-802 and lipid metabolism. The reduced miR-802 decreases hepatic lipogenesis by AMPK phosphorylation. Sjp40, a main component of SEA from S. japonicum binding with CD36 on hepatocytes to suppress miR-802, leading to the activation of AMPK pathway and mitigation of lipogenesis in liver. Sjp40 shows therapeutic potential in treating obesity-related fatty liver (14). da Silva Filomeno CE et al. demonstrated that Schistosoma mansoni (S. mansoni) infection improves glucose tolerance, body mass and liver steatosis in HFD mice but aggravates pathological damage to the liver in mice (98). Similarly, Taenia pisiformis infection decreases body weight and cholesterol level in obese rabbits and changes the metabolic features in rabbits (99). Bhargava et al. further demonstrated that lacto-N-fucopentaose III (LNFPIII) or soluble egg antigen (SEA) from S. mansoni improves glucose tolerance and insulin sensitivity, suppresses lipogenesis in the liver, and provides a strong protective effect against HFD-induced hepatosteatosis in mice. The upregulation of IL-10 in activated macrophages and dendritic cells and extracellular signal-regulated kinase (Erk)-Ap1 pathway in liver mediate the effects of LNFPIII on glucose and lipid metabolic pathway, respectively (15). The synthetic small molecule analogues (SMAs) of ES-62 from Acanthocheilonema viteae (A.viteae) exhibit antimetabolic disorder activity in mice on a high calorie diet (HCD) by reducing fasting glucose levels in male mice and mitigating ileal villus length and liver fibrosis caused by HCD in female mice. Thus, SMAs have the potential to prevent or treat metabolic disorders associated with obesity (100).
Human studies also found that helminth infection improves glycolipid metabolism. Rennie C et al. found that helminth infection induces lower fasting blood glucose, HbA1c levels, prevalence of MetS, and T2DM than without infection people by using meta-analysis (101). Helminth infections generally improve metabolic function, but different species have different effects, and S. mansoni infection remarkably improves metabolic outcomes (101). Zinsou JF et al. found that individuals infected by Schistosoma haematobium (S. haematobium) exhibit decreased serum triglyceride (TG), total cholesterol (TC), and high-density lipoprotein (HDL)-C, especially in overweight/obese individuals. TC, HDL-C, LDL-C, and TG levels in serum are inversely correlated with the intensity of S. haematobium infection in overweight/obese individuals. S. haematobium infection can improve lipid profile in overweight/obese individuals and contribute to the decreased risk of cardiometabolic diseases caused by hyperlipidemia (102). Thus, helminth infection or helminth-derived molecules can improve NAFLD by regulating glycolipid metabolism (Figure 2).
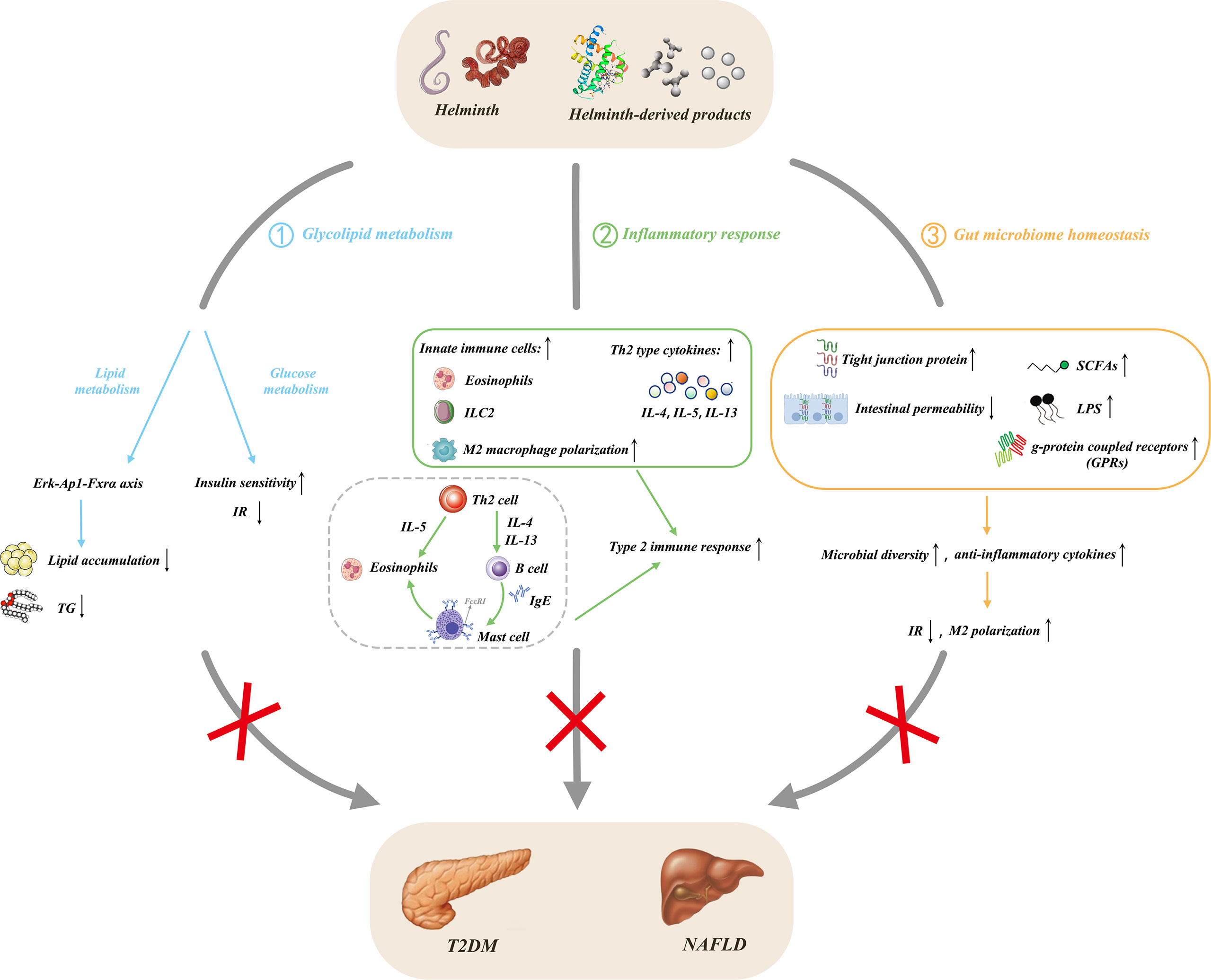
Figure 2 Protective effects of helminths and their products in NAFLD. ① Helminth infection and its derived products regulate glucose metabolism, alleviate insulin resistance, and improve insulin sensitivity; they also affect lipid metabolism and reduce lipogenesis through the Erk-Ap1-Fxrα axis; ②secrete anti-inflammatory cytokines by inducing type 2 immunity and M2 macrophage polarization; ③ influence the composition of gut microbiota through the liver-gut axis, increase the level of fecal SCFAs and upregulate the expression of its main receptors, GPRs, thereby coordinating multiple signaling pathways to prevent obesity. Meanwhile, the increase of tight junction proteins leads to the decrease of intestinal permeability and the expansion of the dominant bacteria, causing the increase of intestinal microbial abundance and diversity. NAFLD, nonalcoholic fatty liver disease; T2DM, type 2 diabetes mellitus; TG, triglyceride; IR, insulin resistance; IL, interleukin; LPS, lipopolysaccharide; Erk, extracellular signal-regulated kinase; AP-1, activator protein-1; Fxrα, farnesoid X receptor alpha; Th2, T helper type 2; M2, alternatively activated macrophage; ILC2, type 2 innate lymphoid cell; Ig, immunoglobulin; SCFAs, short chain fatty acids; GRPs, g-protein coupled receptors.
Obesity is a chronic low-grade inflammation, also named meta-inflammation, which easily alters glycolipid metabolism and insulin resistance and ultimately results in the development of T2DM (103). Various innate and adaptive immune cells are activated in metabolic organs, especially adipose tissue and liver, which produce pro-inflammatory cytokines, such as IL-6 and TNF-α, resulting in insulin resistance and metabolic dysregulation (104). Helminth infection potently induces type 2 immune responses characterized by Th2 phenotype polarization, secretion of IL-4, IL-5, and IL-13, and type 2 innate lymphoid cells (ILC2s). Helminth infection also induces immune tolerance, which is characterized by the abundance of IL-10 and TGF-β secreted by regulatory T cells (Tregs), regulatory B cells (Bregs), tolerogenic dendritic cells (DCs), and M2 macrophages (105–107). Thus, the tolerable response to helminth infection can inhibit the inflammation of allergic and autoimmune diseases (10) and the inflammation of obesity, T2DM, MetS, and NAFLD (108).
Helminth infection and their products can decrease inflammation in adipose tissue and improve glucose tolerance and weight gain in obese individuals and mice (10). Litomosoides sigmodontis (L. sigmodontis) infection resulted in the recruitment of eosinophil and M2 macrophages in epididymal adipose tissue (EAT) and promoted glucose homeostasis in HFD mice. L. sigmodontis antigen (LsAg) treatment also improved glucose tolerance, reduced inflammatory responses, and promoted insulin signaling in EAT. However, LsAg-mediated glucose homeostasis was independent of Foxp3 Tregs (109). Heligmosomoides polygyrus (H. polygyrus) infection improved insulin sensitivity and reduced fat accumulation in the liver and obesity-related inflammation in HFD mice. Treg frequency and suppressor function in adipose tissue increased after H. polygyrus infection. H. polygyrus contributed to improve weight gain and MetS by regulating adipose tissue Tregs in obese mice (110). N. brasiliensis infection increased IL-5 and IL-13 levels in visceral adipose tissue (VAT) and promoted insulin sensitivity in HFD mice. IL-33 promoted ILC2s, produced IL-5 and IL-13, promoted eosinophils, and alternatively activated macrophages in VAT, thus promoting metabolic homeostasis (111). Similarly, S. mansoni infection increased eosinophils and M2 macrophages in white adipose tissue (WAT), enhanced insulin sensitivity, and decreased weight gain, fat mass, and adipocyte size. SEA injection also induced type 2 immune responses in WAT and liver and promoted whole-body metabolic homeostasis (112). Individuals infected with Strongyloides stercoralis infection (Ss+) showed a decrease in various pro-inflammatory cytokines and chemokine levels in plasma, but anthelmintic treatment restored the reduced inflammatogenic cytokines and chemokines (113). Peroxisome proliferator-activated receptor-γ (PPAR-γ), a key master transcription factor for adipogenesis, stimulates lipogenic enzyme expression, thereby promoting lipid storage in adipose tissue (114). PPAR-γ suppress inflammation, resulting in lower TG/HDL cholesterol ratio when activated by its agonist pioglitazone in diabetic patients (115, 116). Lysophosphatidylcholine (LPC) from S. mansoni induces M2 macrophage polarization by increasing PPARγ expression in macrophages (117). The T2 ribonuclease omega-1 (ω1) is an RNase that degrades host RNA. ω1 from S. mansoni treatment improves glycolipid metabolism, and this phenomenon is associated with the induction of type 2 immunity in HFD mice. Th2 cells, eosinophils, and M2 macrophages are increased in the WAT of mice after ω1 injection. Furthermore, ω1 induced type 2 immunity, thus promoting whole-body metabolic homeostasis by suppressing food intake via signal transducer and activator of transcription 6 (STAT6)-independent mechanism in obese mice (118). Surendar and colleagues demonstrated that HFD increased the numbers of CD4+ and CD8+ T cells and production of IFN-γ and IL-17 in the adipose tissue. Adiponectin can inhibit the differentiation of Th1 and Th17 cells and decrease the level of IFN-γ and IL-17 in HFD mice. LsAg from L. sigmodontis treatment increased adiponectin level and decreased Th1 and Th17 cell frequencies in the adipose tissue of mice, thereby improving obesity and insulin resistance in obese mice (119). Similarly, LNFPIII from S. mansoni improved glucose homeostasis, and this process is partially mediated by IL-10 secreted from DCs and macrophages (15). Notably, SJMHE1, an HSP60-derived peptide from S. japonicum, could induce the CD4+CD25+ Treg amplification in vivo and in vitro (120). Furthermore, SJMHE1 suppresses delayed-type hypersensitivity and collagen-induced arthritis, asthma, and colitis by regulating the balance of Th cells (16–18, 121, 122); it also promotes peripheral nerve repair through inducing M2 macrophages (123). Considering that that NAFLD progression is caused by lipid accumulation in liver to steatohepatitis characterized by inflammation, whether SJMHE1 could be potential to intervene in NAFLD needs to be evaluated. Helminths and their products would be promising to contribute to innovative therapy for obesity-related fatty liver by regulating inflammation (Figure 2).
Obesity, diabetes, and NAFLD have dysregulation of the gut microbiota (124, 125). Reducing the diversity and abundance of gut microbiota is often associated with obesity, IR, MetS, and NAFLD (79, 126, 127). Moreover, helminth infection can change the composition and function of gut microbiota, improve host metabolism, reduce systemic inflammation, and promote insulin sensitivity (128, 129). Khudhair and colleagues found that N. brasiliensis infection remarkably alters the composition in gut microbiota at both the phylum and order level, induces type 2 immune responses in adipose tissue, liver, and gut, and promotes glucose homeostasis, thereby preventing T2D (129). N. brasiliensis infection decreases the abundance of Verrucomicrobia and TM7 phyla and increases the richness of Proteobacteria at the phylum level in mice fed with high glycemic index (HGI) diet. N. brasiliensis infection increases the abundance of Clostridiales and Desufovibrionales at the order level in high-fat (HF) mice. Furthermore, N. brasiliensis infection elevates short chain fatty acid (SCFA) levels, which can be beneficial for regulating inflammation and promoting insulin sensitivity (129). SCFAs can maintain gut integrity, immune, and metabolism homeostasis, and it can regulate appetite, weight gain, and glycolipid metabolism (130, 131). Pace et al. found that Strongyloides venezuelensis infection modified gut microbiota, most notably by increasing Lactobacillus spp in HDF mice. This alteration of microbiota increased anti-inflammatory cytokine production, induced M2 macrophage polarization in adipose tissue, increased tight junction protein expression in intestinal cells, and reduced LPS level in serum, thereby promoting glucose metabolism homeostasis (128). ESPs from N. brasiliensis treatment improve glucose tolerance, and decrease weight gain, and induce type 2 immune response in HGI mice. N. brasiliensis ESPs also alter the composition of gut microbiota in mice fed with HGI diet at the phylum and order levels. N. brasiliensis ESP treatment increases the abundance of Bacteroidetes and Patescibacteria phylum, reduces the abundance of Actinobacteria phylum, which is related to the HGI group. ESPs from N. brasiliensis adult worm treatment also increase the abundance of Lactobacillales and Saccharimonadales but decrease Coriobacteriales abundance. N. brasiliensis ESPs induces immune response and gut microbiota changes, which confer protection against abnormal glucose metabolism in mice (132). Similarly, ESPs from the larval Echinococcus granulosus (E. granulosus) treatment can mitigate damage to the intestinal barrier caused by high-fat (HF) diet, increase the expression of zonula occludens-1 (ZO-1), relieve the translocation of bacterial endotoxins and gut inflammation, and attenuate HF diet-induced microbiota dysbiosis, thereby antagonizing obesity-related neurodegenerative diseases (133). ES-62 from A. viteae treatment improved a range of inflammatory, metabolic, and microbiome parameters of aging in HCD-accelerated aging mice (134). A phase 1b clinical trial with Necator americanus (N.americanus) inoculated obesity and MetS individuals is ongoing. The evaluation of the safety and tolerability of infection with N. americanus, metabolic and immunological parameters, and the composition of fecal microbiome in this phase 1b clinical trial will provide valuable information about the use of N. americanus for the treatment of metabolic diseases (135). Helminths can affect host immune function by secreting various molecules or indirectly by altering the gut microbiota (136). The interactions between helminth and microbota can shape the homeostasis of the immune system, thereby improving metabolic homeostasis and immune balance in obesity, MetS, and NAFLD (Figure 2).
Although helminths and their molecules have beneficial outcomes in obesity, MetS, T2DM, and NAFLD, many challenges remain to be considered. First, helminth infection can cause a series of pathological effects. For example, S. mansoni infection can cause anemia, malnutrition, growth stunting, progressive liver fibrosis, and portal hypertension (137). Furthermore, liver flukes Opisthorchis viverrini (O. viverrini) and Clonorchis sinensis infection can induce cholangiocarcinoma (138, 139), and S. haematobium infection can cause squamous cell carcinoma of the bladder (140). In addition to the above harmful effects, parasitic infections can also weaken vaccination effects and increase susceptibility to other pathogens (141, 142). Although O. viverrini infection improved insulin resistance and liver lipid accumulation in high-fructose diet hamsters, the animals showed severe NAFLD as indicated by histopathological analysis (143). Helminths or their products that induce type 2 immunity may increase the incidence of asthma. Caraballo et al. reported that Ascaris lumbricoides tropomyosin has strong allergenic activity (144). Furthermore, the incidence of asthma increases in obese patients (145). Therefore, this issue should be considered in helminth molecules for the treatment of obesity-related diseases, including NAFLD. In addition, the pharmacokinetics of helminth molecules and the mechanism in which they reach target tissues are also challenges. Helminth proteins are only present for a few hours (10). Similar to most active molecules, nanocarriers for the delivery of helminth molecules may increase their half-life and improve their activity in vivo compared with free molecules (146).
Recently, extracellular vesicles (EVs) released by helminths at various life stages can package and deliver immune modulators to host target cells, thereby manipulating the host immune response and exerting immunomodulatory effects (147). Thus, helminth EVs have anti-inflammatory therapeutic potential. Fasciola hepatica EVs prevent DSS-induced ulcerative colitis (148), and Trichinella spiralis EVs decrease the severity of DSS- and TNBS-induced colitis (149, 150). Whether the immunomodulatory propensity of helminth-derived EVs can be used for the treatment of obesity, T2DM, MetS, and NAFLD must be explored in the future. Current research mainly focuses on animal experiments, but the immunology and gene expression patterns in inflammatory diseases greatly differ between human patients and animal models (151, 152). Thus, future research must focus on helminth molecules to intervene in NAFLD-related diseases in human clinical trials.
Despite these challenges, helminth molecules for evolutionary stress selection are effective and safe, especially for NAFLD treatments without effective drugs. Many nature-inspired drugs, such as venom from insects and reptiles, have been used for the treatment of various diseases for millennia (153). Helminths have been included in this list as other nature-inspired drugs and warrant further studies. Most reports on helminth regulation of NAFLD-related diseases, including obesity, T2DM, and MetS, focused on nematodes and trematodes, such as N. brasiliensis, H. polygyrus, L. sigmodontis, S. venezuelensis, S. mansoni, and S. japonicum (95). In the case of cestodes, only T. pisiformis interventions were seen in obese rabbits (99). In addition, ESPs from larval E. granulosus can reverse HF-induced gut barrier dysfunction and microbiota dysbiosis, thereby protecting against obesity-associated neurodegenerative diseases (133). Helminths and their molecules regulated NAFLD-related diseases as shown in Table 1. Only one study reported that ESPs from adult worms and infective third-stage larvae of N. brasiliensis can improve glucose tolerance and attenuate body weight gain in HGI mice (132). Nevertheless, helminth intervention for NAFLD-related diseases is still in its infancy, and whether helminths can treat metabolic disorders in obesity, T2DM, MetS, and NAFLD remains unclear. However, studies on controlled human infection (CHI) with N. americanus and single-sex S. mansoni are ongoing (154–156). Clinical trials using CHI or helminth molecules to treat inflammatory metabolic diseases, such as obesity, T2DM, NASH and NAFLD, will provide new insights and key messages in the future. Clinical trials of the porcine whipworm Trichuris suis ova (TSO) in the treatment of Crohn’s disease demonstrated that patients received a single dose of 7500 TSO still showed tolerance and did not display any short- or long-term treatment-related side effects (157). Clinical trials of experimental helminths in the treatment of inflammatory diseases have been comprehensively reviewed and have yielded promising results (10). Recently, a phase 1b clinical trial by using the larvae III stage of N. americanus in the treatment of obesity and MetS is underway to evaluate the safety and tolerability of hookworm (135). These clinical trials lay the foundation for the development of next generation of therapies against obesity-related diseases, including NAFLD.
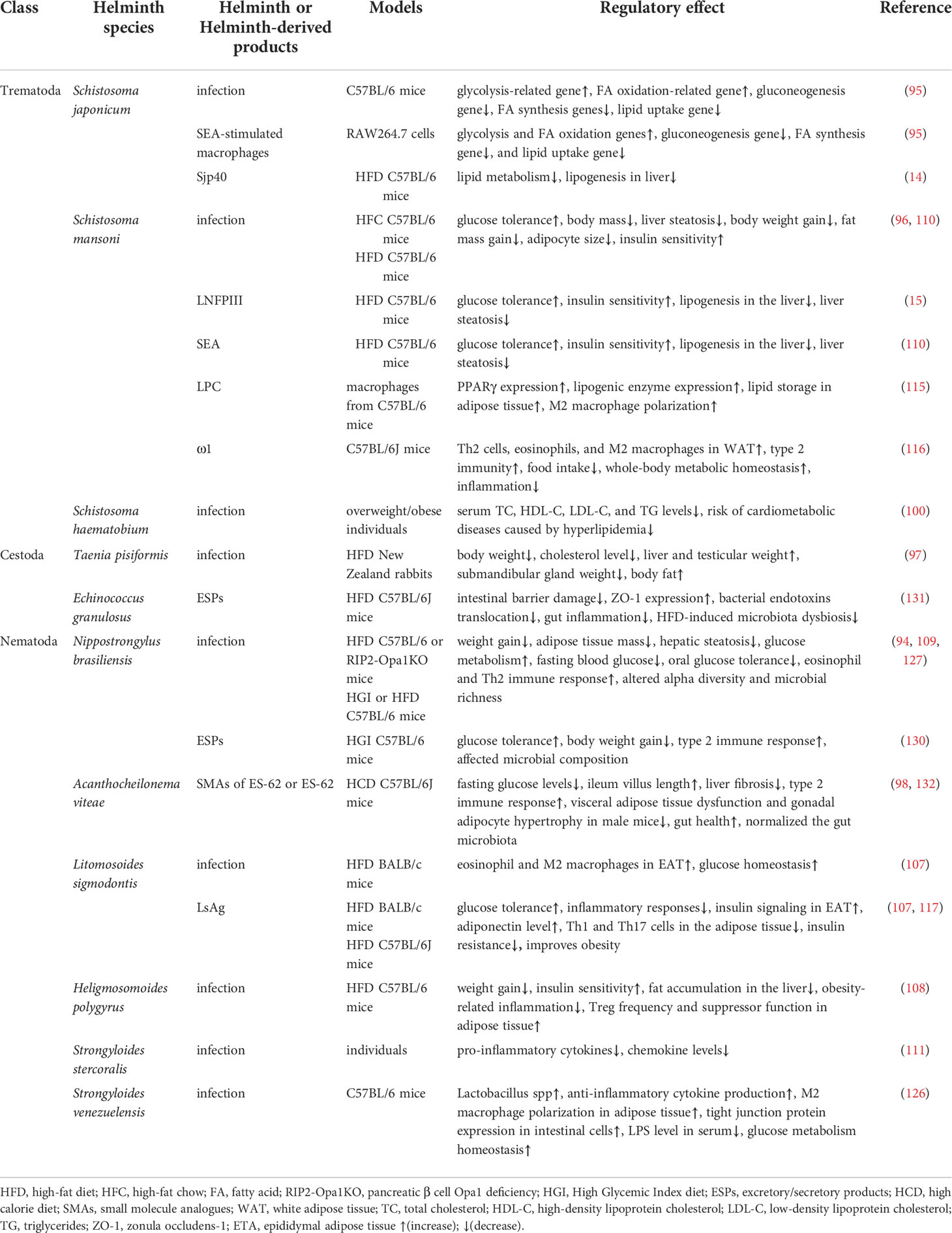
Table 1 Helminths or helminth-derived products suppress obesity, T2DM, and NAFLD-related diseases.
XL, YJ, JY, and XW wrote the manuscript. XL researched the literatures and drew Figures. XW revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by a grant from the National Natural Science Foundation of China (81871243), the key research and development programs of Jiangsu Province (BE2017697), the Six Talent Peaks of Jiangsu Province (WSN-009), and Zhenjiang Clinical Research Center of Gynecological Diseases of Traditional Chinese Medicine (SS202204-KFB02).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Loomba R, Sanyal AJ. The global nafld epidemic. Nat Rev Gastroenterol Hepatol (2013) 10(11):686–90. doi: 10.1038/nrgastro.2013.171
2. Estes C, Razavi H, Loomba R, Younossi Z, Sanyal AJ. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology (2018) 67(1):123–33. doi: 10.1002/hep.29466
3. Monelli F, Venturelli F, Bonilauri L, Manicardi E, Manicardi V, Rossi PG, et al. Systematic review of existing guidelines for nafld assessment. Hepatoma Res (2021) 7:25. doi: 10.20517/2394-5079.2021.03
4. Cotter TG, Rinella M. Nonalcoholic fatty liver disease 2020: The state of the disease. Gastroenterology (2020) 158(7):1851–64. doi: 10.1053/j.gastro.2020.01.052
5. Trandafir LM, Frasinariu OE, Leon-Constantin MM, Chiriac S, Trandafirescu MF, Miron IC, et al. Pediatric nonalcoholic fatty liver disease - a changing diagnostic paradigm. Rom J Morphol Embryol (2020) 61(4):1023–31. doi: 10.47162/RJME.61.4.04
6. Yoneda M, Honda Y, Saito S, Nakajima A. What considerations are there for the pharmacotherapeutic management of nonalcoholic steatohepatitis? Expert Opin Pharmacother (2021) 22(10):1217–20. doi: 10.1080/14656566.2021.1912014
7. Prikhodko VA, Bezborodkina NN, Okovityi SV. Pharmacotherapy for non-alcoholic fatty liver disease: Emerging targets and drug candidates. Biomedicines (2022) 10(2):274. doi: 10.3390/biomedicines10020274
8. Bach JF. The hygiene hypothesis in autoimmunity: The role of pathogens and commensals. Nat Rev Immunol (2018) 18(2):105–20. doi: 10.1038/nri.2017.111
9. Murdaca G, Greco M, Borro M, Gangemi S. Hygiene hypothesis and autoimmune diseases: A narrative review of clinical evidences and mechanisms. Autoimmun Rev (2021) 20(7):102845. doi: 10.1016/j.autrev.2021.102845
10. Ryan SM, Eichenberger RM, Ruscher R, Giacomin PR, Loukas A. Harnessing helminth-driven immunoregulation in the search for novel therapeutic modalities. PloS Pathog (2020) 16(5):e1008508. doi: 10.1371/journal.ppat.1008508
11. Zhang B, Gems D. Gross ways to live long: Parasitic worms as an anti-inflammaging therapy? Elife (2021) 10:e65180. doi: 10.7554/eLife.65180
12. Arai T, Lopes F. Potential of human helminth therapy for resolution of inflammatory bowel disease: The future ahead. Exp Parasitol (2022) 232:108189. doi: 10.1016/j.exppara.2021.108189
13. Gao YR, Sun XZ, Li R, Tang CL, Zhang RH, Zhu YW, et al. The effect of regulatory T cells in schistosoma-mediated protection against type 2 diabetes. Acta Trop (2021) 224:106073. doi: 10.1016/j.actatropica.2021.106073
14. Ni Y, Xu Z, Li C, Zhu Y, Liu R, Zhang F, et al. Therapeutic inhibition of mir-802 protects against obesity through ampk-mediated regulation of hepatic lipid metabolism. Theranostics (2021) 11(3):1079–99. doi: 10.7150/thno.49354
15. Bhargava P, Li C, Stanya KJ, Jacobi D, Dai L, Liu S, et al. Immunomodulatory glycan lnfpiii alleviates hepatosteatosis and insulin resistance through direct and indirect control of metabolic pathways. Nat Med (2012) 18(11):1665–72. doi: 10.1038/nm.2962
16. Zhang W, Li L, Zheng Y, Xue F, Yu M, Ma Y, et al. Schistosoma japonicum peptide Sjmhe1 suppresses airway inflammation of allergic asthma in mice. J Cell Mol Med (2019) 23(11):7819–29. doi: 10.1111/jcmm.14661
17. Shan W, Zhang W, Xue F, Ma Y, Dong L, Wang T, et al. Schistosoma japonicum peptide Sjmhe1 inhibits acute and chronic colitis induced by dextran sulfate sodium in mice. Parasit Vectors (2021) 14(1):455. doi: 10.1186/s13071-021-04977-y
18. Li L, Shan W, Zhu H, Xue F, Ma Y, Dong L, et al. Sjmhe1 peptide from schistosoma japonicum inhibits asthma in mice by regulating Th17/Treg cell balance Via mir-155. J Inflammation Res (2021) 14:5305–18. doi: 10.2147/JIR.S334636
19. Harris NL, Loke P. Recent advances in type-2-Cell-Mediated immunity: Insights from helminth infection. Immunity (2017) 47(6):1024–36. doi: 10.1016/j.immuni.2017.11.015
20. Cortes-Selva D, Fairfax K. Schistosome and intestinal helminth modulation of macrophage immunometabolism. Immunology (2021) 162(2):123–34. doi: 10.1111/imm.13231
21. Zou ZQ, Liu M, Zhong HQ, Guan GY. Association of previous schistosome infection with fatty liver and coronary heart disease: A cross-sectional study in China. Parasite Immunol (2021) 43(4):e12822. doi: 10.1111/pim.12822
22. Simon TG, Roelstraete B, Khalili H, Hagstrom H, Ludvigsson JF. Mortality in biopsy-confirmed nonalcoholic fatty liver disease: Results from a nationwide cohort. Gut (2021) 70(7):1375–82. doi: 10.1136/gutjnl-2020-322786
23. European Association for the Study of the L, European Association for the Study of D, European Association for the Study of O. Easl-Easd-Easo clinical practice guidelines for the management of non-alcoholic fatty liver disease. J Hepatol (2016) 64(6):1388–402. doi: 10.1016/j.jhep.2015.11.004
24. Eslam M, Sanyal AJ, George J, International Consensus P. Mafld: A consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology (2020) 158(7):1999–2014.e1. doi: 10.1053/j.gastro.2019.11.312
25. Eslam M, Newsome PN, Sarin SK, Anstee QM, Targher G, Romero-Gomez M, et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J Hepatol (2020) 73(1):202–9. doi: 10.1016/j.jhep.2020.03.039
26. Tacke F, Weiskirchen R. Non-alcoholic fatty liver disease (Nafld)/Non-alcoholic steatohepatitis (Nash)-related liver fibrosis: Mechanisms, treatment and prevention. Ann Transl Med (2021) 9(8):729. doi: 10.21037/atm-20-4354
27. Bence KK, Birnbaum MJ. Metabolic drivers of non-alcoholic fatty liver disease. Mol Metab (2021) 50:101143. doi: 10.1016/j.molmet.2020.101143
28. Loosen SH, Demir M, Kunstein A, Jordens M, Qvarskhava N, Luedde M, et al. Variables associated with increased incidence of non-alcoholic fatty liver disease (Nafld) in patients with type 2 diabetes. BMJ Open Diabetes Res Care (2021) 9(1):e002243. doi: 10.1136/bmjdrc-2021-002243
29. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of nafld development and therapeutic strategies. Nat Med (2018) 24(7):908–22. doi: 10.1038/s41591-018-0104-9
30. Hudish LI, Reusch JE, Sussel L. B cell dysfunction during progression of metabolic syndrome to type 2 diabetes. J Clin Invest (2019) 129(10):4001–8. doi: 10.1172/jci129188
31. Fahed G, Aoun L, Bou Zerdan M, Allam S, Bou Zerdan M, Bouferraa Y, et al. Metabolic syndrome: Updates on pathophysiology and management in 2021. Int J Mol Sci (2022) 23(2):786. doi: 10.3390/ijms23020786
32. Dorairaj V, Sulaiman SA, Abu N, Abdul Murad NA. Nonalcoholic fatty liver disease (Nafld): Pathogenesis and noninvasive diagnosis. Biomedicines (2021) 10(1):15. doi: 10.3390/biomedicines10010015
33. Alves-Bezerra M, Cohen DE. Triglyceride metabolism in the liver. Compr Physiol (2017) 8(1):1–8. doi: 10.1002/cphy.c170012
34. Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted Via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest (2005) 115(5):1343–51. doi: 10.1172/jci23621
35. Arab JP, Arrese M, Trauner M. Recent insights into the pathogenesis of nonalcoholic fatty liver disease. Annu Rev Pathol (2018) 13:321–50. doi: 10.1146/annurev-pathol-020117-043617
36. Matsuda S, Kobayashi M, Kitagishi Y. Roles for Pi3k/Akt/Pten pathway in cell signaling of nonalcoholic fatty liver disease. ISRN Endocrinol (2013) 2013:472432. doi: 10.1155/2013/472432
37. Hardy T, Oakley F, Anstee QM, Day CP. Nonalcoholic fatty liver disease: Pathogenesis and disease spectrum. Annu Rev Pathol (2016) 11:451–96. doi: 10.1146/annurev-pathol-012615-044224
38. Smith GI, Shankaran M, Yoshino M, Schweitzer GG, Chondronikola M, Beals JW, et al. Insulin resistance drives hepatic De novo lipogenesis in nonalcoholic fatty liver disease. J Clin Invest (2020) 130(3):1453–60. doi: 10.1172/JCI134165
39. Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased De novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology (2014) 146(3):726–35. doi: 10.1053/j.gastro.2013.11.049
40. Sanders FW, Griffin JL. De novo lipogenesis in the liver in health and disease: More than just a shunting yard for glucose. Biol Rev Camb Philos Soc (2016) 91(2):452–68. doi: 10.1111/brv.12178
41. Pafili K, Roden M. Nonalcoholic fatty liver disease (Nafld) from pathogenesis to treatment concepts in humans. Mol Metab (2021) 50:101122. doi: 10.1016/j.molmet.2020.101122
42. Shimano H, Sato R. Srebp-regulated lipid metabolism: Convergent physiology — divergent pathophysiology. Nat Rev Endocrinol (2017) 13(12):710–30. doi: 10.1038/nrendo.2017.91
43. Uyeda K, Repa JJ. Carbohydrate response element binding protein, chrebp, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab (2006) 4(2):107–10. doi: 10.1016/j.cmet.2006.06.008
44. Dentin R, Girard J, Postic C. Carbohydrate responsive element binding protein (Chrebp) and sterol regulatory element binding protein-1c (Srebp-1c): Two key regulators of glucose metabolism and lipid synthesis in liver. Biochimie (2005) 87(1):81–6. doi: 10.1016/j.biochi.2004.11.008
45. Perla FM, Prelati M, Lavorato M, Visicchio D, Anania C. The role of lipid and lipoprotein metabolism in non-alcoholic fatty liver disease. Children (Basel) (2017) 4(6):46. doi: 10.3390/children4060046
46. Roden M, Shulman GI. The integrative biology of type 2 diabetes. Nature (2019) 576(7785):51–60. doi: 10.1038/s41586-019-1797-8
47. Softic S, Cohen DE, Kahn CR. Role of dietary fructose and hepatic De novo lipogenesis in fatty liver disease. Dig Dis Sci (2016) 61(5):1282–93. doi: 10.1007/s10620-016-4054-0
48. Softic S, Gupta MK, Wang GX, Fujisaka S, O'Neill BT, Rao TN, et al. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J Clin Invest (2018) 128(3):1199. doi: 10.1172/jci99009
49. Jensen T, Abdelmalek MF, Sullivan S, Nadeau KJ, Green M, Roncal C, et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J Hepatol (2018) 68(5):1063–75. doi: 10.1016/j.jhep.2018.01.019
50. Hannou SA, Haslam DE, McKeown NM, Herman MA. Fructose metabolism and metabolic disease. J Clin Invest (2018) 128(2):545–55. doi: 10.1172/jci96702
51. Kim MS, Krawczyk SA, Doridot L, Fowler AJ, Wang JX, Trauger SA, et al. Chrebp regulates fructose-induced glucose production independently of insulin signaling. J Clin Invest (2016) 126(11):4372–86. doi: 10.1172/jci81993
52. Merino B, Fernández-Díaz CM, Cózar-Castellano I, Perdomo G. Intestinal fructose and glucose metabolism in health and disease. Nutrients (2019) 12(1):94. doi: 10.3390/nu12010094
53. Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology (2010) 52(2):774–88. doi: 10.1002/hep.23719
54. Yesilova Z, Yaman H, Oktenli C, Ozcan A, Uygun A, Cakir E, et al. Systemic markers of lipid peroxidation and antioxidants in patients with nonalcoholic fatty liver disease. Am J Gastroenterol (2005) 100(4):850–5. doi: 10.1111/j.1572-0241.2005.41500.x
55. Sumida Y, Niki E, Naito Y, Yoshikawa T. Involvement of free radicals and oxidative stress in Nafld/Nash. Free Radic Res (2013) 47(11):869–80. doi: 10.3109/10715762.2013.837577
56. Cai J, Zhang XJ, Li H. The role of innate immune cells in nonalcoholic steatohepatitis. Hepatology (2019) 70(3):1026–37. doi: 10.1002/hep.30506
57. Lefere S, Tacke F. Macrophages in obesity and non-alcoholic fatty liver disease: Crosstalk with metabolism. JHEP Rep (2019) 1(1):30–43. doi: 10.1016/j.jhepr.2019.02.004
58. Sutti S, Albano E. Adaptive immunity: An emerging player in the progression of nafld. Nat Rev Gastroenterol Hepatol (2020) 17(2):81–92. doi: 10.1038/s41575-019-0210-2
59. Hammoutene A, Rautou PE. Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease. J Hepatol (2019) 70(6):1278–91. doi: 10.1016/j.jhep.2019.02.012
60. Murray PJ. Macrophage polarization. Annu Rev Physiol (2017) 79:541–66. doi: 10.1146/annurev-physiol-022516-034339
61. Gessain G, Blériot C, Ginhoux F. Non-genetic heterogeneity of macrophages in diseases-a medical perspective. Front Cell Dev Biol (2020) 8:613116. doi: 10.3389/fcell.2020.613116
62. Watanabe S, Alexander M, Misharin AV, Budinger GRS. The role of macrophages in the resolution of inflammation. J Clin Invest (2019) 129(7):2619–28. doi: 10.1172/jci124615
63. Rückerl D, Allen JE. Macrophage proliferation, provenance, and plasticity in macroparasite infection. Immunol Rev (2014) 262(1):113–33. doi: 10.1111/imr.12221
64. Kratz M, Coats BR, Hisert KB, Hagman D, Mutskov V, Peris E, et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab (2014) 20(4):614–25. doi: 10.1016/j.cmet.2014.08.010
65. Xiong X, Kuang H, Ansari S, Liu T, Gong J, Wang S, et al. Landscape of intercellular crosstalk in healthy and Nash liver revealed by single-cell secretome gene analysis. Mol Cell (2019) 75(3):644–60.e5. doi: 10.1016/j.molcel.2019.07.028
66. Remmerie A, Martens L, Thoné T, Castoldi A, Seurinck R, Pavie B, et al. Osteopontin expression identifies a subset of recruited macrophages distinct from kupffer cells in the fatty liver. Immunity (2020) 53(3):641–57.e14. doi: 10.1016/j.immuni.2020.08.004
67. Tran S, Baba I, Poupel L, Dussaud S, Moreau M, Gélineau A, et al. Impaired kupffer cell self-renewal alters the liver response to lipid overload during non-alcoholic steatohepatitis. Immunity (2020) 53(3):627–40.e5. doi: 10.1016/j.immuni.2020.06.003
68. Seidman JS, Troutman TD, Sakai M, Gola A, Spann NJ, Bennett H, et al. Niche-specific reprogramming of epigenetic landscapes drives myeloid cell diversity in nonalcoholic steatohepatitis. Immunity (2020) 52(6):1057–74.e7. doi: 10.1016/j.immuni.2020.04.001
69. Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature (2019) 575(7783):512–8. doi: 10.1038/s41586-019-1631-3
70. Friedman SL, Ratziu V, Harrison SA, Abdelmalek MF, Aithal GP, Caballeria J, et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology (2018) 67(5):1754–67. doi: 10.1002/hep.29477
71. Han H, Jiang Y, Wang M, Melaku M, Liu L, Zhao Y, et al. Intestinal dysbiosis in nonalcoholic fatty liver disease (Nafld): Focusing on the gut-liver axis. Crit Rev Food Sci Nutr (2021), 1–18. doi: 10.1080/10408398.2021.1966738
72. Albillos A, de Gottardi A, Rescigno M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J Hepatol (2020) 72(3):558–77. doi: 10.1016/j.jhep.2019.10.003
73. Albhaisi SAM, Bajaj JS, Sanyal AJ. Role of gut microbiota in liver disease. Am J Physiol Gastrointest Liver Physiol (2020) 318(1):G84–98. doi: 10.1152/ajpgi.00118.2019
74. Loomba R, Seguritan V, Li W, Long T, Klitgord N, Bhatt A, et al. Gut microbiome-based metagenomic signature for non-invasive detection of advanced fibrosis in human nonalcoholic fatty liver disease. Cell Metab (2017) 25(5):1054–62.e5. doi: 10.1016/j.cmet.2017.04.001
75. Carpino G, Del Ben M, Pastori D, Carnevale R, Baratta F, Overi D, et al. Increased liver localization of lipopolysaccharides in human and experimental nafld. Hepatology (2020) 72(2):470–85. doi: 10.1002/hep.31056
76. Schnabl B, Brenner DA. Interactions between the intestinal microbiome and liver diseases. Gastroenterology (2014) 146(6):1513–24. doi: 10.1053/j.gastro.2014.01.020
77. de Faria Ghetti F, Oliveira DG, de Oliveira JM, de Castro Ferreira L, Cesar DE, Moreira APB. Influence of gut microbiota on the development and progression of nonalcoholic steatohepatitis. Eur J Nutr (2018) 57(3):861–76. doi: 10.1007/s00394-017-1524-x
78. Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science (2013) 341(6150):1241214. doi: 10.1126/science.1241214
79. Aron-Wisnewsky J, Vigliotti C, Witjes J, Le P, Holleboom AG, Verheij J, et al. Gut microbiota and human nafld: Disentangling microbial signatures from metabolic disorders. Nat Rev Gastroenterol Hepatol (2020) 17(5):279–97. doi: 10.1038/s41575-020-0269-9
80. Xue L, Deng Z, Luo W, He X, Chen Y. Effect of fecal microbiota transplantation on non-alcoholic fatty liver disease: A randomized clinical trial. Front Cell Infect Microbiol (2022) 12:759306. doi: 10.3389/fcimb.2022.759306
81. Leung C, Rivera L, Furness JB, Angus PW. The role of the gut microbiota in nafld. Nat Rev Gastroenterol Hepatol (2016) 13(7):412–25. doi: 10.1038/nrgastro.2016.85
82. Slifer ZM, Blikslager AT. The integral role of tight junction proteins in the repair of injured intestinal epithelium. Int J Mol Sci (2020) 21(3):972. doi: 10.3390/ijms21030972
83. Allam-Ndoul B, Castonguay-Paradis S, Veilleux A. Gut microbiota and intestinal trans-epithelial permeability. Int J Mol Sci (2020) 21(17):6402. doi: 10.3390/ijms21176402
84. Vancamelbeke M, Vermeire S. The intestinal barrier: A fundamental role in health and disease. Expert Rev Gastroenterol Hepatol (2017) 11(9):821–34. doi: 10.1080/17474124.2017.1343143
85. Mao JW, Tang HY, Zhao T, Tan XY, Bi J, Wang BY, et al. Intestinal mucosal barrier dysfunction participates in the progress of nonalcoholic fatty liver disease. Int J Clin Exp Pathol (2015) 8(4):3648–58.
86. Rohr MW, Narasimhulu CA, Rudeski-Rohr TA, Parthasarathy S. Negative effects of a high-fat diet on intestinal permeability: A review. Adv Nutr (2020) 11(1):77–91. doi: 10.1093/advances/nmz061
87. Luther J, Garber JJ, Khalili H, Dave M, Bale SS, Jindal R, et al. Hepatic injury in nonalcoholic steatohepatitis contributes to altered intestinal permeability. Cell Mol Gastroenterol Hepatol (2015) 1(2):222–32. doi: 10.1016/j.jcmgh.2015.01.001
88. Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, et al. Inflammasome-mediated dysbiosis regulates progression of nafld and obesity. Nature (2012) 482(7384):179–85. doi: 10.1038/nature10809
89. Exley MA, Hand L, O'Shea D, Lynch L. Interplay between the immune system and adipose tissue in obesity. J Endocrinol (2014) 223(2):R41–8. doi: 10.1530/joe-13-0516
90. Huh JY, Park YJ, Ham M, Kim JB. Crosstalk between adipocytes and immune cells in adipose tissue inflammation and metabolic dysregulation in obesity. Mol Cells (2014) 37(5):365–71. doi: 10.14348/molcells.2014.0074
91. Francisco V, Pino J, Gonzalez-Gay MA, Mera A, Lago F, Gómez R, et al. Adipokines and inflammation: Is it a question of weight? Br J Pharmacol (2018) 175(10):1569–79. doi: 10.1111/bph.14181
92. Kim H, Lee DS, An TH, Park HJ, Kim WK, Bae KH, et al. Metabolic spectrum of liver failure in type 2 diabetes and obesity: From nafld to Nash to hcc. Int J Mol Sci (2021) 22(9):4495. doi: 10.3390/ijms22094495
93. Mu Y, McManus DP, Hou N, Cai P. Schistosome infection and schistosome-derived products as modulators for the prevention and alleviation of immunological disorders. Front Immunol (2021) 12:619776. doi: 10.3389/fimmu.2021.619776
94. Berbudi A, Ajendra J, Wardani AP, Hoerauf A, Hübner MP. Parasitic helminths and their beneficial impact on type 1 and type 2 diabetes. Diabetes Metab Res Rev (2016) 32(3):238–50. doi: 10.1002/dmrr.2673
95. van der Zande HJP, Zawistowska-Deniziak A, Guigas B. Immune regulation of metabolic homeostasis by helminths and their molecules. Trends Parasitol (2019) 35(10):795–808. doi: 10.1016/j.pt.2019.07.014
96. Yang Z, Grinchuk V, Smith A, Qin B, Bohl JA, Sun R, et al. Parasitic nematode-induced modulation of body weight and associated metabolic dysfunction in mouse models of obesity. Infect Immun (2013) 81(6):1905–14. doi: 10.1128/iai.00053-13
97. Xu ZP, Chang H, Ni YY, Li C, Chen L, Hou M, et al. Schistosoma japonicum infection causes a reprogramming of glycolipid metabolism in the liver. Parasit Vectors (2019) 12(1):388. doi: 10.1186/s13071-019-3621-6
98. da Silva Filomeno CE, Costa-Silva M, Correa CL, Neves RH, Mandarim-de-Lacerda CA, Machado-Silva JR. The acute schistosomiasis mansoni ameliorates metabolic syndrome in the C57bl/6 mouse model. Exp Parasitol (2020) 212:107889. doi: 10.1016/j.exppara.2020.107889
99. Arias-Hernández D, García-Jiménez S, Domínguez-Roldan R, Murcia-Mejía C, Báez-Saldaña A, Hallal-Calleros C, et al. Effects of taenia pisiformis infection and obesity on clinical parameters, organometry and fat distribution in Male rabbits. Pathogens (2020) 9(11):861. doi: 10.3390/pathogens9110861
100. Lumb FE, Crowe J, Doonan J, Suckling CJ, Selman C, Harnett MM, et al. Synthetic small molecule analogues of the immunomodulatory acanthocheilonema viteae product Es-62 promote metabolic homeostasis during obesity in a mouse model. Mol Biochem Parasitol (2019) 234:111232. doi: 10.1016/j.molbiopara.2019.111232
101. Rennie C, Fernandez R, Donnelly S, McGrath KC. The impact of helminth infection on the incidence of metabolic syndrome: A systematic review and meta-analysis. Front Endocrinol (Lausanne) (2021) 12:728396. doi: 10.3389/fendo.2021.728396
102. Zinsou JF, Janse JJ, Honpkehedji YY, Dejon-Agobe JC, Garcia-Tardon N, Hoekstra PT, et al. Schistosoma haematobium infection is associated with lower serum cholesterol levels and improved lipid profile in Overweight/Obese individuals. PloS Negl Trop Dis (2020) 14(7):e0008464. doi: 10.1371/journal.pntd.0008464
103. Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature (2017) 542(7640):177–85. doi: 10.1038/nature21363
104. Lackey DE, Olefsky JM. Regulation of metabolism by the innate immune system. Nat Rev Endocrinol (2016) 12(1):15–28. doi: 10.1038/nrendo.2015.189
105. Pearce EJ, C MK, Sun J JJT, McKee AS, Cervi L. Th2 response polarization during infection with the helminth parasite schistosoma mansoni. Immunol Rev (2004) 201:117–26. doi: 10.1111/j.0105-2896.2004.00187.x
106. Acharya S, Da'dara AA, Skelly PJ. Schistosome immunomodulators. PloS Pathog (2021) 17(12):e1010064. doi: 10.1371/journal.ppat.1010064
107. Shi W, Xu N, Wang X, Vallée I, Liu M, Liu X. Helminth therapy for immune-mediated inflammatory diseases: Current and future perspectives. J Inflammation Res (2022) 15:475–91. doi: 10.2147/jir.S348079
108. Dai M, Yang X, Yu Y, Pan W. Helminth and host crosstalk: New insight into treatment of obesity and its associated metabolic syndromes. Front Immunol (2022) 13:827486. doi: 10.3389/fimmu.2022.827486
109. Berbudi A, Surendar J, Ajendra J, Gondorf F, Schmidt D, Neumann AL, et al. Filarial infection or antigen administration improves glucose tolerance in diet-induced obese mice. J Innate Immun (2016) 8(6):601–16. doi: 10.1159/000448401
110. Queiroz-Glauss CP, Vieira MS, Gonçalves-Pereira MH, Almeida SS, Freire RH, Gomes MA, et al. Helminth infection modulates number and function of adipose tissue tregs in high fat diet-induced obesity. PloS Negl Trop Dis (2022) 16(5):e0010105. doi: 10.1371/journal.pntd.0010105
111. Molofsky AB, Nussbaum JC, Liang HE, Van Dyken SJ, Cheng LE, Mohapatra A, et al. Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J Exp Med (2013) 210(3):535–49. doi: 10.1084/jem.20121964
112. Hussaarts L, Garcia-Tardon N, van Beek L, Heemskerk MM, Haeberlein S, van der Zon GC, et al. Chronic helminth infection and helminth-derived egg antigens promote adipose tissue M2 macrophages and improve insulin sensitivity in obese mice. FASEB J (2015) 29(7):3027–39. doi: 10.1096/fj.14-266239
113. Rajamanickam A, Munisankar S, Dolla C, Menon PA, Thiruvengadam K, Nutman TB, et al. Helminth infection modulates systemic pro-inflammatory cytokines and chemokines implicated in type 2 diabetes mellitus pathogenesis. PloS Negl Trop Dis (2020) 14(3):e0008101. doi: 10.1371/journal.pntd.0008101
114. Gao H, Li H, Li W, Shen X, Di B. Pioglitazone attenuates atherosclerosis in diabetic mice by inhibition of receptor for advanced glycation end-product (Rage) signaling. Med Sci Monit (2017) 23:6121–31. doi: 10.12659/msm.907401
115. Nicholls SJ, Tuzcu EM, Wolski K, Bayturan O, Lavoie A, Uno K, et al. Lowering the Triglyceride/High-density lipoprotein cholesterol ratio is associated with the beneficial impact of pioglitazone on progression of coronary atherosclerosis in diabetic patients: Insights from the periscope (Pioglitazone effect on regression of intravascular sonographic coronary obstruction prospective evaluation) study. J Am Coll Cardiol (2011) 57(2):153–9. doi: 10.1016/j.jacc.2010.06.055
116. Mizoguchi M, Tahara N, Tahara A, Nitta Y, Kodama N, Oba T, et al. Pioglitazone attenuates atherosclerotic plaque inflammation in patients with impaired glucose tolerance or diabetes a prospective, randomized, comparator-controlled study using serial fdg Pet/Ct imaging study of carotid artery and ascending aorta. JACC Cardiovasc Imaging (2011) 4(10):1110–8. doi: 10.1016/j.jcmg.2011.08.007
117. Assunção LS, Magalhães KG, Carneiro AB, Molinaro R, Almeida PE, Atella GC, et al. Schistosomal-derived lysophosphatidylcholine triggers M2 polarization of macrophages through pparγ dependent mechanisms. Biochim Biophys Acta Mol Cell Biol Lipids (2017) 1862(2):246–54. doi: 10.1016/j.bbalip.2016.11.006
118. van der Zande HJP, Gonzalez MA, de Ruiter K, Wilbers RHP, García-Tardón N, van Huizen M, et al. The helminth glycoprotein omega-1 improves metabolic homeostasis in obese mice through type 2 immunity-independent inhibition of food intake. FASEB J (2021) 35(2):e21331. doi: 10.1096/fj.202001973R
119. Surendar J, Frohberger SJ, Karunakaran I, Schmitt V, Stamminger W, Neumann AL, et al. Adiponectin limits ifn-Γ and il-17 producing Cd4 T cells in obesity by restraining cell intrinsic glycolysis. Front Immunol (2019) 10:2555. doi: 10.3389/fimmu.2019.02555
120. Wang X, Zhou S, Chi Y, Wen X, Hoellwarth J, He L, et al. Cd4+Cd25+ treg induction by an Hsp60-derived peptide Sjmhe1 from schistosoma japonicum is Tlr2 dependent. Eur J Immunol (2009) 39(11):3052–65. doi: 10.1002/eji.200939335
121. Wang X, Wang J, Liang Y, Ni H, Shi L, Xu C, et al. Schistosoma japonicum Hsp60-derived peptide Sjmhe1 suppresses delayed-type hypersensitivity in a murine model. Parasit Vectors (2016) 9:147. doi: 10.1186/s13071-016-1434-4
122. Wang X, Li L, Wang J, Dong L, Shu Y, Liang Y, et al. Inhibition of cytokine response to tlr stimulation and alleviation of collagen-induced arthritis in mice by schistosoma japonicum peptide Sjmhe1. J Cell Mol Med (2017) 21(3):475–86. doi: 10.1111/jcmm.12991
123. Ma Y, Wei C, Qi X, Pu Y, Dong L, Xu L, et al. Schistosoma japonicum-derived peptide Sjmhe1 promotes peripheral nerve repair through a macrophage-dependent mechanism. Am J Transl Res (2021) 13(3):1290–306.
124. Chen J, Vitetta L. Bile acids and butyrate in the effects of Probiotics/Synbiotics on nonalcoholic fatty liver disease. Eur J Gastroenterol Hepatol (2019) 31(11):1475–6. doi: 10.1097/meg.0000000000001506
125. Muñoz-Garach A, Diaz-Perdigones C, Tinahones FJ. Gut microbiota and type 2 diabetes mellitus. Endocrinol Nutr (2016) 63(10):560–8. doi: 10.1016/j.endonu.2016.07.008
126. Harsch IA, Konturek PC. The role of gut microbiota in obesity and type 2 and type 1 diabetes mellitus: New insights into "Old" diseases. Med Sci (2018) 6(2):32. doi: 10.3390/medsci6020032
127. Saad MJ, Santos A, Prada PO. Linking gut microbiota and inflammation to obesity and insulin resistance. Physiol (Bethesda) (2016) 31(4):283–93. doi: 10.1152/physiol.00041.2015
128. Pace F, Carvalho BM, Zanotto TM, Santos A, Guadagnini D, Silva KLC, et al. Helminth infection in mice improves insulin sensitivity Via modulation of gut microbiota and fatty acid metabolism. Pharmacol Res (2018) 132:33–46. doi: 10.1016/j.phrs.2018.04.008
129. Khudhair Z, Alhallaf R, Eichenberger RM, Whan J, Kupz A, Field M, et al. Gastrointestinal helminth infection improves insulin sensitivity, decreases systemic inflammation, and alters the composition of gut microbiota in distinct mouse models of type 2 diabetes. Front Endocrinol (Lausanne) (2020) 11:606530. doi: 10.3389/fendo.2020.606530
130. Vinolo MA, Rodrigues HG, Nachbar RT, Curi R. Regulation of inflammation by short chain fatty acids. Nutrients (2011) 3(10):858–76. doi: 10.3390/nu3100858
131. Morrison DJ, Preston T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes (2016) 7(3):189–200. doi: 10.1080/19490976.2015.1134082
132. Khudhair Z, Alhallaf R, Eichenberger RM, Field M, Krause L, Sotillo J, et al. Administration of hookworm Excretory/Secretory proteins improves glucose tolerance in a mouse model of type 2 diabetes. Biomolecules (2022) 12(5):637. doi: 10.3390/biom12050637
133. Wu J, Zhu Y, Zhou L, Lu Y, Feng T, Dai M, et al. Parasite-derived excretory-secretory products alleviate gut microbiota dysbiosis and improve cognitive impairment induced by a high-fat diet. Front Immunol (2021) 12:710513. doi: 10.3389/fimmu.2021.710513
134. Crowe J, Lumb FE, Doonan J, Broussard M, Tarafdar A, Pineda MA, et al. The parasitic worm product Es-62 promotes health- and life-span in a high calorie diet-accelerated mouse model of ageing. PloS Pathog (2020) 16(3):e1008391. doi: 10.1371/journal.ppat.1008391
135. Pierce D, Merone L, Lewis C, Rahman T, Croese J, Loukas A, et al. Safety and tolerability of experimental hookworm infection in humans with metabolic disease: Study protocol for a phase 1b randomised controlled clinical trial. BMC Endocr Disord (2019) 19(1):136. doi: 10.1186/s12902-019-0461-5
136. Gause WC, Maizels RM. Macrobiota - helminths as active participants and partners of the microbiota in host intestinal homeostasis. Curr Opin Microbiol (2016) 32:14–8. doi: 10.1016/j.mib.2016.04.004
137. Wilson MS, Mentink-Kane MM, Pesce JT, Ramalingam TR, Thompson R, Wynn TA. Immunopathology of schistosomiasis. Immunol Cell Biol (2007) 85(2):148–54. doi: 10.1038/sj.icb.7100014
138. de Martel C, Ferlay J, Franceschi S, Vignat J, Bray F, Forman D, et al. Global burden of cancers attributable to infections in 2008: A review and synthetic analysis. Lancet Oncol (2012) 13(6):607–15. doi: 10.1016/s1470-2045(12)70137-7
139. Brindley PJ, Loukas A. Helminth infection-induced malignancy. PloS Pathog (2017) 13(7):e1006393. doi: 10.1371/journal.ppat.1006393
140. Brindley PJ, da Costa JM, Sripa B. Why does infection with some helminths cause cancer? Trends Cancer (2015) 1(3):174–82. doi: 10.1016/j.trecan.2015.08.011
141. Hartmann W, Brunn ML, Stetter N, Gabriel G, Breloer M. Pre-existing helminth infection impairs the efficacy of adjuvanted influenza vaccination in mice. PloS One (2022) 17(3):e0266456. doi: 10.1371/journal.pone.0266456
142. Su C, Su L, Li Y, Long SR, Chang J, Zhang W, et al. Helminth-induced alterations of the gut microbiota exacerbate bacterial colitis. Mucosal Immunol (2018) 11(1):144–57. doi: 10.1038/mi.2017.20
143. Chaidee A, Onsurathum S, Intuyod K, Haonon O, Pannangpetch P, Pongchaiyakul C, et al. Opisthorchis viverrini infection augments the severity of nonalcoholic fatty liver disease in high-Fat/High-Fructose diet-fed hamsters. Am J Trop Med Hyg (2019) 101(5):1161–9. doi: 10.4269/ajtmh.19-0442
144. Caraballo L, Coronado S. Parasite allergens. Mol Immunol (2018) 100:113–9. doi: 10.1016/j.molimm.2018.03.014
145. Ali Z, Ulrik CS. Obesity and asthma: A coincidence or a causal relationship? a systematic review. Respir Med (2013) 107(9):1287–300. doi: 10.1016/j.rmed.2013.03.019
146. Chariou PL, Ortega-Rivera OA, Steinmetz NF. Nanocarriers for the delivery of medical, veterinary, and agricultural active ingredients. ACS Nano (2020) 14(3):2678–701. doi: 10.1021/acsnano.0c00173
147. Drurey C, Maizels RM. Helminth extracellular vesicles: Interactions with the host immune system. Mol Immunol (2021) 137:124–33. doi: 10.1016/j.molimm.2021.06.017
148. Roig J, Saiz ML, Galiano A, Trelis M, Cantalapiedra F, Monteagudo C, et al. Extracellular vesicles from the helminth fasciola hepatica prevent dss-induced acute ulcerative colitis in a T-lymphocyte independent mode. Front Microbiol (2018) 9:1036. doi: 10.3389/fmicb.2018.01036
149. Yang Y, Liu L, Liu X, Zhang Y, Shi H, Jia W, et al. Extracellular vesicles derived from trichinella spiralis muscle larvae ameliorate tnbs-induced colitis in mice. Front Immunol (2020) 11:1174. doi: 10.3389/fimmu.2020.01174
150. Gao X, Yang Y, Liu X, Wang Y, Yang Y, Boireau P, et al. Extracellular vesicles derived from trichinella spiralis prevent colitis by inhibiting M1 macrophage polarization. Acta Trop (2021) 213:105761. doi: 10.1016/j.actatropica.2020.105761
151. Mestas J, Hughes CC. Of mice and not men: Differences between mouse and human immunology. J Immunol (2004) 172(5):2731–8. doi: 10.4049/jimmunol.172.5.2731
152. Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U.S.A. (2013) 110(9):3507–12. doi: 10.1073/pnas.1222878110
153. Pennington MW, Czerwinski A, Norton RS. Peptide therapeutics from venom: Current status and potential. Bioorg Med Chem (2018) 26(10):2738–58. doi: 10.1016/j.bmc.2017.09.029
154. Roestenberg M, Hoogerwerf MA, Ferreira DM, Mordmüller B, Yazdanbakhsh M. Experimental infection of human volunteers. Lancet Infect Dis (2018) 18(10):e312–e22. doi: 10.1016/s1473-3099(18)30177-4
155. Egesa M, Ssali A, Tumwesige E, Kizza M, Driciru E, Luboga F, et al. Ethical and practical considerations arising from community consultation on implementing controlled human infection studies using schistosoma mansoni in Uganda. Glob Bioeth (2022) 33(1):78–102. doi: 10.1080/11287462.2022.2091503
156. Loukas A, Hotez PJ, Diemert D, Yazdanbakhsh M, McCarthy JS, Correa-Oliveira R, et al. Hookworm infection. Nat Rev Dis Primers (2016) 2:16088. doi: 10.1038/nrdp.2016.88
Keywords: helminth infection, helminth-derived products, NAFLD, glycolipid metabolism, inflammation, gut dysbiosis
Citation: Liu X, Jiang Y, Ye J and Wang X (2022) Helminth infection and helminth-derived products: A novel therapeutic option for non-alcoholic fatty liver disease. Front. Immunol. 13:999412. doi: 10.3389/fimmu.2022.999412
Received: 21 July 2022; Accepted: 15 September 2022;
Published: 03 October 2022.
Edited by:
Fabrizio Bruschi, University of Pisa, ItalyReviewed by:
Jesuthas Ajendra, University Hospital Bonn, GermanyCopyright © 2022 Liu, Jiang, Ye and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuefeng Wang, xuefengwang@ujs.edu.cn
†ORCID: Xuefeng Wang, 0000-0002-5980-6530
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.