 Dara McCreary1
Dara McCreary1 Ebun Omoyinmi1,2
Ebun Omoyinmi1,2 Ying Hong1
Ying Hong1 Barbara Jensen1
Barbara Jensen1 Alice Burleigh1,3
Alice Burleigh1,3 Fiona Price-Kuehne1*
Fiona Price-Kuehne1* Kimberly Gilmour4
Kimberly Gilmour4 Despina Eleftheriou1,3,5
Despina Eleftheriou1,3,5 Paul Brogan1,5
Paul Brogan1,5- 1Inflammation and Rheumatology Section, University College London Great Ormond Street Institute of Child Health, London, United Kingdom
- 2National Amyloidosis Centre, Royal Free Hospital, London, United Kingdom
- 3Centre for Adolescent Rheumatology, University College London, London, United Kingdom
- 4Camelia Botnar Laboratory, Great Ormond Street Hospital National Health Service (NHS) Foundation Trust, London, United Kingdom
- 5Rheumatology Department, Great Ormond Street Hospital National Health Service (NHS) Foundation Trust, London, United Kingdom
There is an important unmet clinical need for fast turnaround next generation sequencing (NGS) to aid genetic diagnosis of patients with acute and sometimes catastrophic inflammatory presentations. This is imperative for patients who require precise and targeted treatment to prevent irreparable organ damage or even death. Acute and severe hyper- inflammation may be caused by primary immunodeficiency (PID) with immune dysregulation, or more typical autoinflammatory diseases in the absence of obvious immunodeficiency. Infectious triggers may be present in either immunodeficiency or autoinflammation. We compiled a list of 25 genes causing monogenetic immunological diseases that are notorious for their acute first presentation with fulminant inflammation and which may be amenable to specific treatment, including hemophagocytic lymphohistiocytosis (HLH); and autoinflammatory diseases that can present with early-onset stroke or other irreversible neurological inflammatory complications. We designed and validated a pipeline that enabled return of clinically actionable results in hours rather than weeks: the Rapid Autoinflammation Panel (RAP). We demonstrated accuracy of this new pipeline, with 100% sensitivity and 100% specificity. Return of results to clinicians was achieved within 48-hours from receiving the patient’s blood or saliva sample. This approach demonstrates the potential significant diagnostic impact of NGS in acute medicine to facilitate precision medicine and save “life or limb” in these critical situations.
Introduction
In recent years there has been increased adoption of next generation sequencing (NGS) to facilitate molecular diagnoses and routine clinical care. The impact of this has been particularly significant for patients with immunological diseases, notably those with primary immune deficiency (PID) or autoinflammation (1–3). NGS has facilitated the discovery and classification of many new immunological diseases, sometimes providing insights into pathogenesis and resulting in discovery and implementation of targeted treatments (4). A major challenge for the implementation of NGS into routine clinical practice, however, is the time it takes to receive results. Availability of NGS remains patchy within individual countries and throughout Europe, a challenge that the European reference network for rare immunological and autoinflammatory diseases (ERN-RITA) seeks to address (5); but even in countries and centers where this is available, turnaround time (TAT) is usually weeks to months, rather than hours to days. For instance, in the South-East of England, TAT for a modest (24-gene) autoinflammatory panel is 84 days from receipt of a DNA sample. Additional delays incurred by extraction of DNA and transportation to the central genomics laboratory hub may add a further 2-4 weeks. Clinicians, and most importantly patients with chronic inflammatory diseases, are increasingly frustrated by this slow TAT; an issue that becomes especially critical for patients with acute fulminant hyper-inflammatory presentations, who may accrue irreparable organ injury, significant glucocorticoid toxicity (6, 7), or even die during the pre-diagnostic phase of their illness.
The Great Ormond Street Hospital Autoinflammation Centre of Excellence (GOSH-ACE) receives acute referrals nationally in the UK, and internationally. We increasingly noted a crucial unmet clinical need in our service regarding slow TAT of genetic results for acutely unwell pediatric patients. This particularly related to patients presenting for the first time with acute hyper-inflammation, and included various immunological diseases such as hemophagocytic lymphohistiocytosis (HLH) (6, 8, 9); and autoinflammatory diseases presenting with acute vasculitic features such as arterial stroke, digital gangrene, or other life and organ threatening vasculitic injury (e.g. deficiency of adenosine deaminase 2, DADA2; or STING associated vasculopathy with onset in infancy, SAVI, amongst several others). We concluded that there was a need for a bespoke NGS targeted gene panel that would screen for a select number of genetic immunological diseases that in our experience would be important to identify acutely, since targeted treatment might be available.
Previously our group had designed and implemented two diagnostic targeted gene panels which we now deploy for routine patient care: the vasculitis and inflammation panel (2); and the neuroinflammation panel (1). We hypothesized that we could adapt these pipelines to achieve a TAT of 48 to 72 hours. We defined TAT as the time from receipt of whole blood or saliva to the delivery of a formal written report to the clinician. We included 25 monogenetic immunological diseases (the rapid autoinflammation panel, RAP) that in our experience could present acutely with fulminant hyper-inflammation (with or without infection), where rapid deployment of specific or targeted treatment might improve the prognosis (Table 1). The aims of this study were therefore to: 1. design a pathway with potential TAT of 48-72h; 2. assess the accuracy of the readout in healthy controls, and disease controls with known genetic diagnoses; and 3. apply this to consecutive newly presenting patients referred to our service to ascertain the feasibility of this new pipeline in an acute clinical setting.
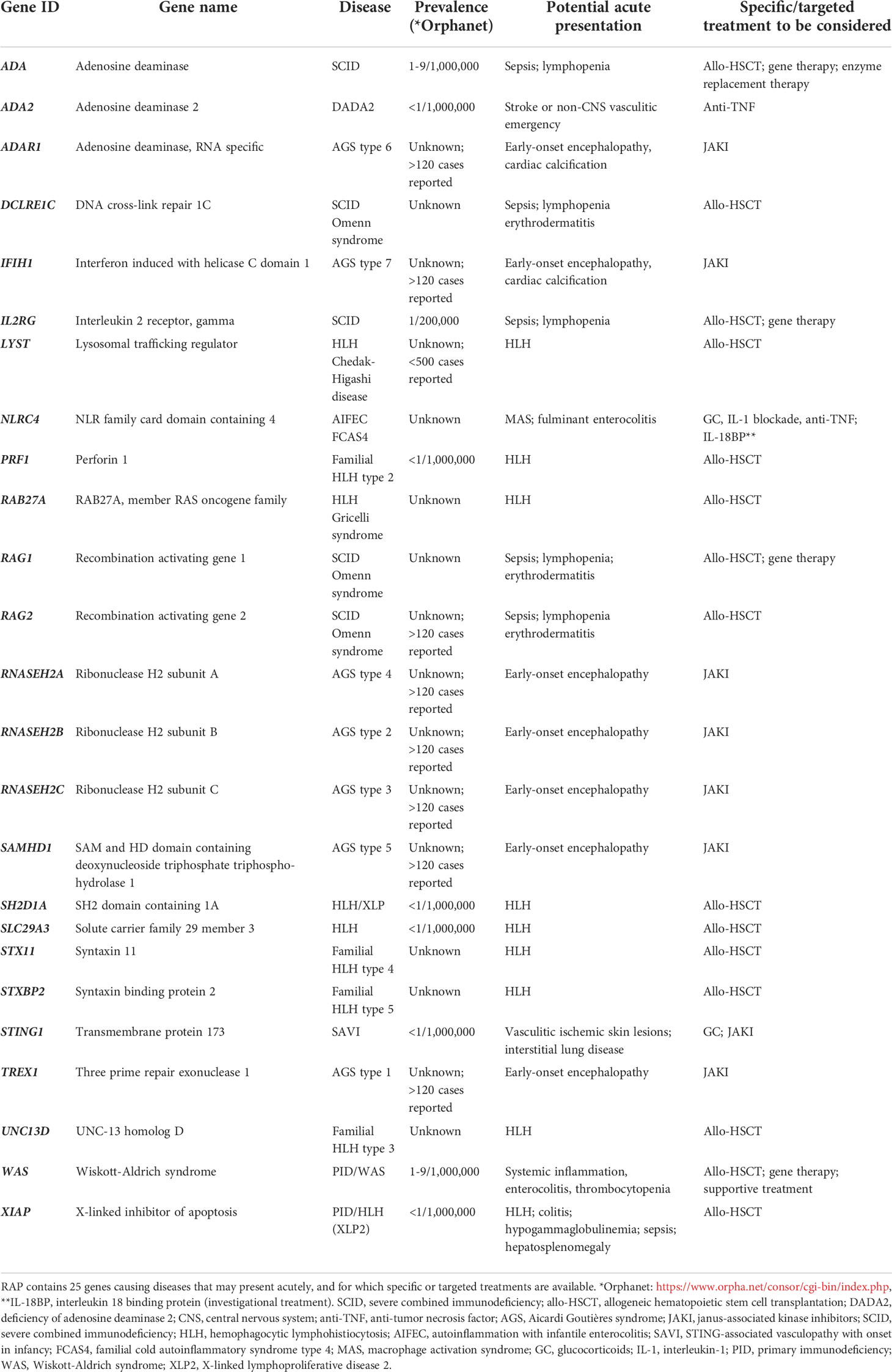
Table 1 The Rapid Autoinflammation Panel (RAP).
Materials and methods
Ethical approval
This study was approved by the National Research Ethics Service Committee (research ethics number 08H071382). All participants and parents provided written consent or assent as appropriate.
Controls
We recruited patients referred to our tertiary referral center (GOSH-ACE). Patients with known genotypes (n=15) from our database served as disease controls to assess accuracy of the new panel. We included 14 samples from patients who were known to have a variant in at least one of the genes included in the RAP; and a single patient with acute disseminated encephalomyelitis (ADEM) known to be without any such variants (Supplemental Table 1). These variants had previously been identified through either Sanger sequencing or NGS performed as part of their routine care. Genome in a Bottle, a DNA reference material, was also used to further evaluate the performance of the RAP, as described previously (10, 11). We sequenced the Genome in a Bottle sample 4 times on 3 separate panel runs to confirm adequate capture of all regions in the panel designed (data not shown).
Prospective patients
Genetic sequencing using the RAP was undertaken in 15 consecutive cases presenting acutely between December 1, 2019, and December 22, 2021, where there was clinical concern for a genetic cause of disease. For each case, in addition to the RAP pipeline, we concurrently sequenced each case using our validated routine larger targeted panels, modified over time to include new genes discovered since we originally published on these panels (1, 2) as the clinical gold-standard used to check the accuracy of the RAP. Table 2 summarizes the clinical indication for requesting genetic testing.
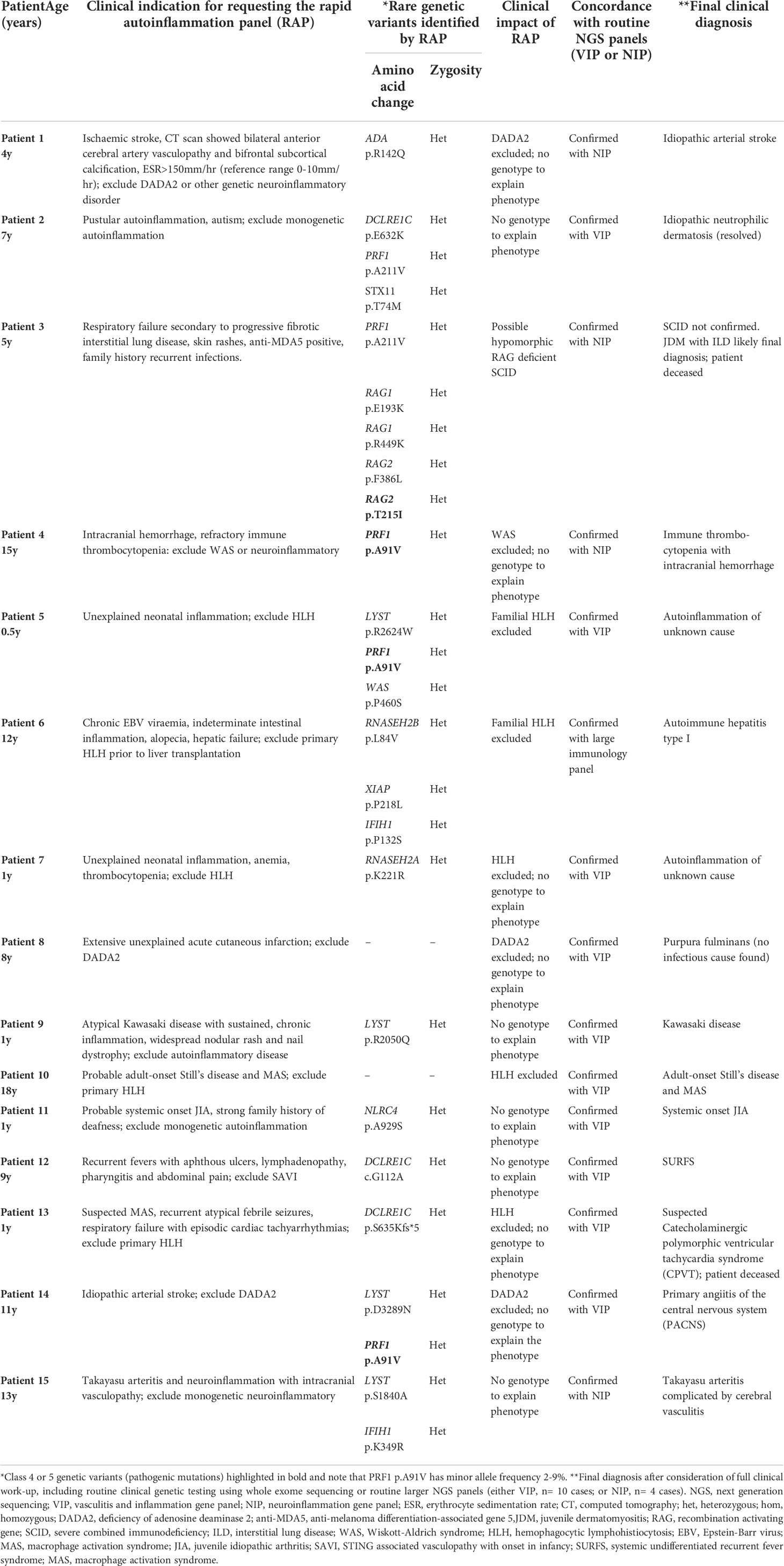
Table 2 Prospective patients, clinical indication for requesting the RAP and final diagnosis.
Workflow design for the RAP
We considered each component step of the workflow sequence from first contact from the requesting clinician, to return of formal genetic report. We next determined the fastest feasible time to complete each step, to achieve a TAT of within 48h, and allocated resources as required to achieve that, as summarized in Figure 1.
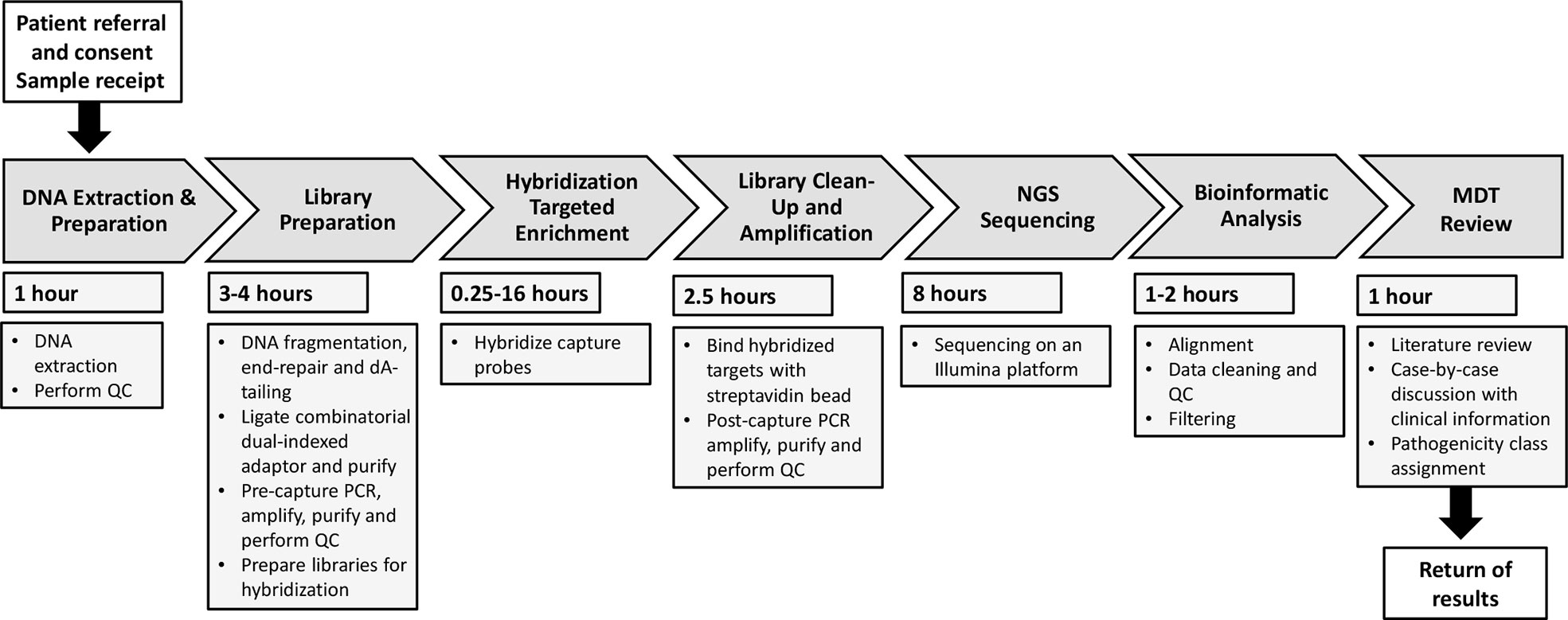
Figure 1 Overall workflow for the Rapid Autoinflammation Panel (RAP). This flow diagram represents the seven stages of the RAP workflow demonstrating how return of results is achieved <48 hours from receiving a patient sample. QC, quality control; PCR, polymerase chain reaction; NGS, next-generation sequencing; MDT, multidisciplinary team.
DNA preparation
Upon receipt of the request (by email) from the clinician, we immediately arranged obtaining a sample for extraction of DNA. We extracted DNA from either blood (EDTA sample) or saliva. One of the most critical time-saving steps was getting the sample to the research team as quickly as possible, achieved by a combination of direct delivery from the clinician, or direct collection by the researcher (DM). DNA was extracted from the EDTA blood sample using the Gentra Puregene extraction kit (Qiagen); DNA from saliva was extracted using the prepIT L2P kit (Genotek).
Library preparation and targeted enrichment
The design and development of the capture library was done with Twist Bioscience (https://www.twistbioscience.com) and was designed to work with the Illumina sequencing kit. The library capture was performed as described in the Twist Bioscience library preparation protocol. First, enzymatic fragmentation was performed before undergoing the Twist Universal Adaptor System step. This was then followed by the Twist Fast Hybridization Target Enrichment Protocol. Once the DNA was extracted it underwent quality control (QC) and was then diluted to 5ng/μl prior to starting the fragmentation. Briefly, the genomic DNA was fragmented by enzymatic reaction, then the fragments end-repaired and dA-tailed. The Twist universal adaptors were then ligated to the dA-tailed DNA fragments before purifying to generate indexed gDNA libraries. The indexed gDNA library was amplified and purified before the QC of the sample was performed. This was performed using TapeStation 4200 Bioanalyzer (Agilent) to assess the fragment size of the indexed library.
Library clean-up and amplification
The indexed ligated libraries were amplified and hybridized to our customized Twist gene panel (Table 1) before being amplified via polymerase chain reaction (PCR). The protocol enabled samples to be processed as a single sample, or multiplex. The hybridization step was incubated for a minimum of 15 minutes; or left to run overnight (depending on the time of receiving the patient sample and the urgency of results). Once the hybridization was complete, the samples were cleaned using purification beads before undergoing a final amplification step.
Sequencing
The captured and indexed library was then sequenced as a single sample using the Illumina MiSeq sequencer with either MiSeq Reagent Kit v2 (500-cycles) MS-102-2003 or the Micro Kit V2 (300-cycles) MS-103-1002. A region was considered a low-coverage region if any single nucleotide in the exon had a coverage less than 30x. After the first eight samples were sequenced, additional baits were added to a total of 8 regions to improve coverage of 4 genes. Subsequently, overall coverage was high and reproducible between the different sequencing runs.
Bioinformatics
Read alignment, variant calling, and annotation were performed using the web-based Galaxy (12). The method uses paired end reads from Illumina MiSeq instruments that were mapped to the human reference genome (GRCh37) using Burrows-Wheeler Aligner–MEM software. The output variant call format (VCF) file from USEGALAXY was annotated using wANNOVAR, the web-based user-interfaced ANNOVAR tool from Wang Genomic Labs, which provided allele frequencies from public databases and in-silico predictions of pathogenicity (13). Identified variants were evaluated for coverage using the Integrative Genomics Viewer (14). We used public databases to search for the frequency of variants: the 1000 Genome Project, Exome Variant Server, Exome Aggregation Consortium, and Genome Aggregation database (15–18). We filtered out synonymous variants and then excluded common polymorphic variants with minor allele frequency of 1% or greater. The only exception to this was the relatively common pathogenic PRF1 p.Ala91Val variant (19). Pathogenicity assessment of identified variants was done from the downloaded wANNOVAR files, and predicted functional impact of any variants using SIFT, PolyPhen-2 and MutationTaster (20–22).
Report writing and return of results
The final step in the RAP workflow was a formal multidisciplinary team (MDT) reporting meeting involving clinicians (PB and DE) and scientists (DM and EO). At that meeting, the clinical phenotype was reviewed in the context of any rare genetic variants identified and a literature review was conducted to inform the pathogenicity assessment. The identified variants were individually assessed and classified into pathogenicity groups (class 1, clearly not pathogenic; class 2, unlikely to be pathogenic; class 3, unknown significance; class 4, likely to be pathogenic; and class 5, clearly pathogenic) according to the Association for Clinical Genetic Science 2013 practice guidelines (23). Each meeting typically took between 30 and 60 minutes per patient. Signed return of results reports were then emailed securely to the lead clinician caring for the patient within 48 hours of sample receipt.
Statistical analysis
Continuous variables were summarized as median and range. Categorical variables were presented as percentages and frequencies. Sensitivity and specificity were calculated using SPSS statistical software version 21 (IBM).
Results
Turnaround time for the RAP
The time taken from receipt of sample for DNA separation to return of report for the 15 prospective cases studied was under 48 hours, confirming our methodological workflow time-based estimate.
Accuracy of the RAP in disease controls
The RAP correctly identified all 16 variants known to be present in the 15 disease controls shown in Supplemental Table 1 and did not falsely call any other rare variants. One disease control sample was known to have no rare variants in any of the genes covered and served as a negative control; this was also confirmed by the RAP. Thus, the RAP was both highly sensitive (100%) and specific (100%) for detection of these known rare variants. In addition, two of the disease control samples ADA2 (p.G47R) and ADA2 (p.P251L) were run on separate runs to check reproducibility of the pipeline, with 100% concordance (data not shown).
Performance of the RAP in prospective patients with unknown diagnoses
Between December 1, 2020, and December 22, 2021, the RAP was requested to be performed on 15 patients referred to our service who required urgent diagnostic genetic testing; 10 males; median age, 7.2 years (range 0.5-18 years). The RAP had actionable clinical impact in all 15 patients by excluding the specific genetic cause queried by the clinician in 14/15 cases, and in one case (patient 3) the RAP identified a non-confirmatory genotype suggestive of hypomorphic recombination-activating gene (RAG) severe combined immunodeficiency (SCID). A total of 25 variants (22 rare variants and the three PRF1 variants) were identified in these 15 patients (median 1, range 0-5 variants per patient). A summary of the phenotypes, genotypes, and final diagnoses of these 15 patients is provided in Table 2.
Pathogenic variants (Class 4 or Class 5)
Class 4 or 5 pathogenic variants are highlighted in bold in Table 2. Patient 3 was a 5-year-old boy from consanguineous parents of Bangladeshi origin. There was a positive family history of a paternal uncle who died of infection at age 3; and a second paternal uncle with recurrent infections now aged 35 years. The proband had two younger siblings with no significant past medical history. He was admitted to the pediatric intensive care unit with a 4-to-6-week history of progressive respiratory failure, with negative infection screen for SARS-CoV-2. Cutaneous features were reminiscent of juvenile dermatomyositis (JDM); and anti-melanoma differentiation associated gene 5 (MDA5) antibodies were positive. Cytomegalovirus (CMV) viremia and CMV were detected in bronchoalveolar lavage raising the possibility of CMV pneumonitis as a contributing cause of the respiratory failure. Immunophenotyping demonstrated low T, normal B, almost absent NK cells, and low naïve CD4 cells. Serum immunoglobulin levels were normal. The RAP identified 2 rare variants in RAG1; 2 variants in RAG2; and a single PRF1 variant (Table 2). These results suggested a possible (but unconfirmed) diagnosis of severe combined immunodeficiency (SCID). The RAG2 (p.T215I) variant has been described previously and associated with typical SCID (24). The RAG2 (p.F386L) variant results in wild-type activity (National Institute of Health, personal communication, data not shown) and is therefore likely benign; both RAG1 variants were also classified as benign. All other common SCID mutations were excluded in this case using our wider genetic panels (IL2RG, JAK3, RAG1, DCLRE1C, PRKDC, LIG4, NHEJ1, ADA, AK2, IL7R) (1). Carrier status for the PRF1 (p.A211V) variant was an incidental finding. The patient died, thus precluding further immunophenotyping for hypomorphic RAG deficient SCID. We concluded that the most likely diagnosis was that of JDM with interstitial lung disease as the cause of death. Patient 4, 5 and 14 (Table 2) were carriers for PRF1 (p.A91V) (19), again deemed incidental to the phenotype.
Variants of unknown significance (Class 3)
A total of 21 unique variants of unknown significance (VUS) in 12 genes were found in 13 patients. Two patients (13.3%) were found to have no rare variants in any of the 25 genes covered in the RAP (Table 2).
Discussion
The diagnostic utility of next-generation sequencing (NGS) has undoubtedly transformed medicine in virtually every specialty. A major limitation, however, is the turnaround time (TAT) for clinically actionable results. Increasingly, molecular diagnoses guide therapeutic decisions, and these decisions are particularly critical for acutely sick patients presenting with suspected monogenetic immunological diseases (4, 6). In this study, we designed and validated a modest targeted panel to aid the diagnosis of patients presenting with fulminant inflammation. We based the choice of the 25 genes included in the RAP (Table 1) purely on the needs of our service as a tertiary referral center for autoinflammation, based on our collective clinical experience of caring for patients with autoinflammation or immunodeficiency. We devised and delivered a workflow that enabled a TAT of less than 48 hours, defined as the time taken from receipt of a sample for DNA extraction to return of an actionable report to the clinician. We validated the accuracy of RAP by comparing it to our routine clinical NGS pipelines and found 100% concordance with these established pipelines.
We found that there were three major resource requirements required to achieve a TAT of less than 48 hours: 1. a dedicated skilled scientist to deliver the panel, including collecting samples from the requesting clinician in real time; 2. access to a sequencer in real time with no notice or need for prior booking; and 3. availability of skilled personnel (including scientists and clinician scientists) to deliver a multidisciplinary team (MDT) report. In summary, these results suggest that it is possible to deliver NGS genetic test results within 48 hours if there is adequate skilled manpower, and availability of a sequencer. The choice of genes included on such rapid return panels can be modified for different clinical settings.
The Genomic Medicine Service (GMS) within the NHS in England (https://www.england.nhs.uk/publication/national-genomic-test-directories/) offers a rapid sequencing service for acutely unwell babies and children with a potentially monogenic disorder, initially applying a targeted virtual gene panel to whole exome/genome sequencing data, with a TAT of 14 days for negative/preliminary results; and 21 days for a final report confirming a clinically relevant finding. Other laudable attempts to shorten the TAT for results include a Canadian study with mean time to preliminary report of 7.2 days (25); pilot studies in the US achieving a mean TAT between 14 and 16.3 days for a formal written report (26, 27); and a Chinese group that managed to deliver a 24-hour rapid trio exome in critically unwell children (28). However, these studies were in research settings, and have not yet translated into routine clinical care. Perhaps more relevant to our study was a recent report from Australia that demonstrated the feasibility of ultra-rapid exome sequencing in a public healthcare system, achieving a median of 3 days from sample receipt to clinical report (29). The cost of this service, however, was not described.
We recognize that rapid return of whole exome/genome sequencing results most likely not be achieved for years to come, although as the cost and speed of NGS continues to decline, we anticipate this will be available in the foreseeable future. However, until this is achieved, panel sequencing remains the mainstay for rapid genetic testing in a clinical setting. The RAP was not designed to replace larger gene panel or clinical whole-exome sequencing that are increasingly part of routine clinical care. Rather, it is designed to be implemented at the beginning of the diagnostic workup for patients with severe inflammatory presentations. We anticipate that modifications of the selection of the genes may be required to further optimize the diagnostic yield. It is unsurprising that none of the 15 prospective patients we studied turned out to conclusively have any of the genetic diseases included in the RAP (nor were any genetic diagnoses identified with subsequent larger panel or WES in these patients). This does not deter from the clinical impact, since a negative test result using a test with a very high negative predictive value is very useful for clinicians, as indicated in Table 2 for each of the 15 patients we studied. Clinicians are increasingly aware that genetic testing for ultra-rare diseases usually returns negative results, and that this allows rapid exclusion of these rare but treatable diseases early in the patient pathway (30).
Several practical factors can adversely affect the TAT. Lessons learned to mitigate against barriers to rapid TAT include the aforementioned availability of a dedicated and skilled workforce; and open access to a sequencer. Other practical considerations are co-location of the sequencing laboratory and personnel close to the requesting clinical site; clear lines of communication between requesting clinicians and scientists; and close working relationships between scientists and clinician scientists to generate reports. Consideration of maintaining patient confidentiality must always be remembered when returning reports to clinicians via email. Another practical consideration regarding implementation is cost; in the UK, the current cost of a gene panel is in the region of £750 to £1030 (National Health Service North Thames genomics laboratory Hub, personal communication). An estimate of the cost of the RAP per patient (excluding the potentially expensive input of the MDT to generate reports) was approximately £1000.
While targeted gene panels can also offer benefit for detecting somatic mutations, they must be designed for this purpose, i.e., careful selection of a restricted number of genes and ensuring high read depth by using a sequencing technology with greater output. Our study chose a read depth of a minimum of 30x, which is commonly accepted for diagnostic purposes (31), but higher coverage would be required for detection of somatic mutations. Furthermore, somatic mutations in the 25 RAP genes we included are not known to cause disease at low levels of somatic mosaicism.
In conclusion, we have devised and validated a workflow that successfully offers rapid return of genetic test results within 48 hours. The panel was designed to aid the diagnosis of patients presenting with fulminant inflammation but could be modified for virtually any clinical setting where rapid return of results may modify treatment and improve patient outcomes. The main resources required to deliver this workflow included a dedicated skilled scientist, open access to a sequencer, and availability of a rapid response skilled clinical/scientific MDT to generate reports. We suggest that our study brings us ever closer to the ultimate desirable goal of rapid return of genetic results at the point of patient care.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by National Research Ethics Committee: research ethics number 08H071382. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
DM, EO, DE, KG, and PB conceptualized the study; DM, FP-K, and PB wrote the first draft of the manuscript; DM, EO, AB, YH, BJ and FP-K collected and analyzed the data. All authors contributed to the final article and approved the submitted version.
Funding
This study was funded in whole from a peer-reviewed grant from Rosetrees Trust (grant number CF1\100009). All research at Great Ormond Street Hospital NHS Foundation Trust and UCL Great Ormond Street Institute of Child Health is supported by the NIHR Great Ormond Street Hospital Biomedical Research Centre.
Acknowledgments
We thank all families who agreed to take part in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.998967/full#supplementary-material
References
1. McCreary D, Omoyinmi E, Hong Y, Mulhern C, Papadopoulou C, Casimir M, et al. Development and validation of a targeted next-generation sequencing gene panel for children with neuroinflammation. JAMA Network Open (2019) 2(10):e1914274. doi: 10.1001/jamanetworkopen.2019.14274
2. Omoyinmi E, Standing A, Keylock A, Price-Kuehne F, Melo Gomes S, Rowczenio D, et al. Clinical impact of a targeted next-generation sequencing gene panel for autoinflammation and vasculitis. PloS One (2017) 12(7):e0181874. doi: 10.1371/journal.pone.0181874
3. Haimel M, Pazmandi J, Heredia RJ, Dmytrus J, Bal SK, Zoghi S, et al. Curation and expansion of human phenotype ontology for defined groups of inborn errors of immunity. J Allergy Clin Immunol (2022) 149(1):369–78. doi: 10.1016/j.jaci.2021.04.033
4. Stoffels M, Kastner DL. Old dogs, new tricks: monogenic autoinflammatory disease unleashed. Annu Rev Genomics Hum Genet (2016) 17(1):245–72. doi: 10.1146/annurev-genom-090413-025334
5. Dolezalova P, Anton J, Avcin T, Beresford MW, Brogan PA, Constantin T, et al. The European network for care of children with paediatric rheumatic diseases: care across borders. Rheumatology. (2019) 58(7):1188–95. doi: 10.1093/rheumatology/key439
6. Kul Cinar O, Putland A, Wynne K, Eleftheriou D, Brogan PA. Hereditary systemic autoinflammatory diseases: therapeutic stratification. Front Pediatr (2022) 10:867679. doi: 10.3389/fped.2022.867679
7. Garg S, Wynne K, Omoyinmi E, Eleftheriou D, Brogan P. Efficacy and safety of anakinra for undifferentiated autoinflammatory diseases in children: a retrospective case review. Rheumatol Adv Pract (2019) 3:rkz004. doi: 10.1093/rap/rkz004
8. Murphy C, Nanthapisal S, Gilmour K, Laurent S, D'Arco F, Hemingway C, et al. Progressive neurologic disorder: Initial manifestation of hemophagocytic lymphohistiocytosis. Neurology. (2016) 86(22):2109–11. doi: 10.1212/WNL.0000000000002729
9. Cruikshank M, Anoop P, Nikolajeva O, Rao A, Rao K, Gilmour K, et al. Screening assays for primary haemophagocytic lymphohistiocytosis in children presenting with suspected macrophage activation syndrome. Pediatr Rheumatol Online J (2015) 12(Suppl 1):48. doi: 10.1186/s12969-015-0043-7
10. Cornish A, Guda C. A comparison of variant calling pipelines using genome in a bottle as a reference. BioMed Res Int (2015) 2015:456479. doi: 10.1155/2015/456479
11. Zook JM, Catoe D, McDaniel J, Vang L, Spies N, Sidow A, et al. Extensive sequencing of seven human genomes to characterize benchmark reference materials. Sci Data (2016) 3(1):160025. doi: 10.1038/sdata.2016.25
12. Afgan E, Baker D, Batut B, van den Beek Marius, Bouvier D, Čech M, et al. The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res (2018) 46(W1):W537–W44. doi: 10.1093/nar/gky379
13. Yang H, Wang K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat Protoc (2015) 10(10):1556–66. doi: 10.1038/nprot.2015.105
14. Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol (2011) 29(1):24–6. doi: 10.1038/nbt.1754
15. Auton A, Abecasis GR, Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, et al. A global reference for human genetic variation. Nature. (2015) 526(7571):68–74. doi: 10.1038/nature15393
16. Karczewski KJ, Weisburd B, Thomas B, Solomonson M, Ruderfer DM, Kavanagh D, et al. The ExAC browser: displaying reference data information from over 60 000 exomes. Nucleic Acids Res (2017) 45(D1):D840–D5. doi: 10.1093/nar/gkw971
17. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. (2020) 581(7809):434–43. doi: 10.1038/s41586-020-2308-7
18. Exome variant server, NHLBI GO exome sequencing project . Available at: http://evs.gs.washington.edu/EVS/ (Accessed October 2021).
19. House IG, Thia K, Brennan AJ, Tothill R, Dobrovic A, Yeh WZ, et al. Heterozygosity for the common perforin mutation, p.A91V, impairs the cytotoxicity of primary natural killer cells from healthy individuals. Immunol Cell Biol (2015) 93(6):575–80. doi: 10.1038/icb.2015.1
20. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc (2009) 4(7):1073–81. doi: 10.1038/nprot.2009.86
21. Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet (2013) 76(1):7.20.1–7.41. doi: 10.1002/0471142905.hg0720s76
22. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods (2014) 11(4):361–2. doi: 10.1038/nmeth.2890
23. Wallis Y, Payne S, McAnulty C, Bodmer D, Sistermans E, Robertson K, et al. Practice guidelines for the evaluation of pathogenicity and the reporting of sequence variants in clinical molecular genetics. ACGS & VGKL (2013). Available at: https://www.acgs.uk.com/media/10791/evaluation_and_reporting_of_sequence_variants_bpgs_june_2013_-_finalpdf.pdf.
24. Meshaal SS, El Hawary RE, Abd Elaziz DS, Eldash A, Alkady R, Lotfy S, et al. Phenotypical heterogeneity in RAG-deficient patients from a highly consanguineous population. Clin Exp Immunol (2019) 195(2):202–12. doi: 10.1111/cei.13222
25. Elliott AM, Du Souich C, Lehman A, Guella I, Evans DM, Candido T, et al. RAPIDOMICS: rapid genome-wide sequencing in a neonatal intensive care unit–successes and challenges. Eur J Pediatrics (2019) 178(8):1207–18. doi: 10.1007/s00431-019-03399-4
26. Brunelli L, Jenkins SM, Gudgeon JM, Bleyl SB, Miller CE, Tvrdik T, et al. Targeted gene panel sequencing for the rapid diagnosis of acutely ill infants. Mol Genet Genomic Med (2019) 7(7):e796. doi: 10.1002/mgg3.796
27. Carey AS, Schacht JP, Umandap C, Fasel D, Weng C, Cappell J, et al. Rapid exome sequencing in PICU patients with new-onset metabolic or neurological disorders. Pediatr Res (2020) 88(5):761–8. doi: 10.1038/s41390-020-0858-x
28. Wang H, Qian Y, Lu Y, Qin Q, Lu G, Cheng G, et al. Clinical utility of 24-h rapid trio-exome sequencing for critically ill infants. Nat Genomic Med (2020) 5(1). doi: 10.1038/s41525-020-0129-0
29. Lunke S, Eggers S, Wilson M, Patel C, Barnett CP, Pinner J, et al. Feasibility of ultra-rapid exome sequencing in critically ill infants and children with suspected monogenic conditions in the Australian public health care system. JAMA. (2020) 323(24):2503. doi: 10.1097/01.ogx.0000722040.32795.04
30. Thaventhiran JED, Lango Allen H, Burren OS, Rae W, Greene D, Staples E, et al. Whole-genome sequencing of a sporadic primary immunodeficiency cohort. Nature. (2020) 583(7814):90–5. doi: 10.1038/s41586-020-2265-1
Keywords: genetics, autoinflammation, immunodeficiency, hyper-inflammation, turnaround time (TAT)
Citation: McCreary D, Omoyinmi E, Hong Y, Jensen B, Burleigh A, Price-Kuehne F, Gilmour K, Eleftheriou D and Brogan P (2022) A rapid turnaround gene panel for severe autoinflammation: Genetic results within 48 hours. Front. Immunol. 13:998967. doi: 10.3389/fimmu.2022.998967
Received: 20 July 2022; Accepted: 05 September 2022;
Published: 20 September 2022.
Edited by:
Sinisa Savic, University of Leeds, United KingdomReviewed by:
Tadej Avcin, University Medical Centre Ljubljana, SloveniaSeth Lucian Masters, The University of Melbourne, Australia
Copyright © 2022 McCreary, Omoyinmi, Hong, Jensen, Burleigh, Price-Kuehne, Gilmour, Eleftheriou and Brogan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fiona Price-Kuehne, ZmlvbmEucHJpY2Uta3VlaG5lQHVjbC5hYy51aw==