 Yan Tao
Yan Tao Quanhong Jiang
Quanhong Jiang Qun Wang
Qun Wang- Key Laboratory of Infection and Immunity of Shandong Province, Department of Immunology, School of Basic Medical Sciences, Cheeloo College of Medicine, Shandong University, Jinan, China
Hepatic glucose production (HGP) is fine-regulated via glycogenolysis or gluconeogenesis to maintain physiological concentration of blood glucose during fasting-feeding cycle. Aberrant HGP leads to hyperglycemia in obesity-associated diabetes. Adipose tissue cooperates with the liver to regulate glycolipid metabolism. During these processes, adipose tissue macrophages (ATMs) change their profiles with various physio-pathological settings, producing diverse effects on HGP. Here, we briefly review the distinct phenotypes of ATMs under different nutrition states including feeding, fasting or overnutrition, and detail their effects on HGP. We discuss several pathways by which ATMs regulate hepatic gluconeogenesis or glycogenolysis, leading to favorable or unfavorable metabolic consequences. Furthermore, we summarize emerging therapeutic targets to correct metabolic disorders in morbid obesity or diabetes based on ATM-HGP axis. This review puts forward the importance and flexibility of ATMs in regulating HGP, proposing ATM-based HGP modulation as a potential therapeutic approach for obesity-associated metabolic dysfunction.
Introduction
Glucose is an essential energy source for most tissue cells in mammals. The circulation levels of glucose are strictly controlled within a stable range in healthy individuals. Abnormally low level of blood glucose, known as hypoglycemia, produces deleterious effects on physiological functions of various tissues and organs. Severe hypoglycemia substantially reduces glucose supply to the brain, resulting in central nervous system damage with consequent seizures, coma and death. In contrast, chronic hyperglycemia, a state of long-term high blood glucose usually in diabetes mellitus, may lead to dysfunction and/or failure of various organs (1). Thus, glucose homeostasis is critical for physiological fasting-feeding cycle particularly in overnutrition state.
During the fed state, dietary intake supplies exogenous glucose to provide energy; while under fasting conditions, endogenous glucose production becomes an indispensable way to maintain blood glucose levels. The liver produces endogenous glucose that accounts for the largest amount of glucose output. As such, the capacity for hepatic glucose production (HGP) plays a crucial role in maintaining blood glucose during fasting. Contrarily, aberrant elevations of HGP lead to hyperglycemia and associated metabolic disorders, particularly in individuals with morbid obesity and associated diabetes mellitus (2, 3). Thus, HGP modulation is a pivotal approach for the treatment of obesity-associated diabetes.
Adipose tissue closely interacts with the liver to regulate energy metabolism. In the fed state, triglycerides synthesized in hepatocytes are packed into VLDL and delivered into adipose tissue for storage (4). In the fasted state, the lipolysis of adipose tissue supplies the liver with non-esterified fatty acids (NEFAs) and glycerol that serve as the precursors of HGP (4–6). Besides metabolic products, adipose tissue communicates with the liver by exchanging diverse information including adipokines and extracellular vesicles (EVs) (5, 7). During these interactions, macrophages exhibit high heterogeneity in dynamic adipose tissue niches, playing flexible roles in metabolic and immune processes. Accumulating studies are unraveling the involvement of adipose tissue macrophages (ATMs) in hepatic glycolipid metabolism. Here, we review their emerging roles in modulating HGP in the context of physiological fasting-feeding cycle as well as obesity or diabetes. Related ATM profiles and regulatory mechanisms for hepatic gluconeogenesis are discussed.
HGP
Hepatocytes produce endogenous glucose relying on gluconeogenesis and glycogenolysis. The former generates glucose from non-carbohydrate substrates, while the latter produces glucose through glycogen breakdown. Both glycogenolysis and gluconeogenesis contribute to HGP during fasting period, whereas glycogenolysis only exists during early fasting stages due to limited glycogen storage (3, 4, 8, 9).
Hepatic gluconeogenesis
Gluconeogenesis is a biological process of glucose generation from non-carbohydrate precursors such as glycerol, lactate, pyruvate and glucogenic amino acids (8, 10). As part of the reverse reaction of glycolysis, gluconeogenesis requires four critical enzymes to bypass the irreversible steps. Pyruvate carboxylase (PC) carboxylates pyruvate derived from glycolysis into oxaloacetate, which is decarboxylated by phosphoenolpyruvate carboxykinase (PEPCK) to form phosphoenolpyruvate. Fructose-1,6-bisphosphatase catalyzes fructose 1,6-bisphosphate into fructose 6-phosphate; while glucose 6-phosphatase (G6Pase) dephosphorylates glucose-6-phosphate (G6P) into glucose (8, 11, 12). Hepatic gluconeogenesis is strictly regulated by hormones in different nutrition states. These hormones regulate gluconeogenic gene expression by promoting or suppressing the binding of specific transcription factors with their promoters. To date, multiple transcription factors including CREB, PGC-1α, FOXO1 have been identified to promote PEPCK and/or G6Pase transactivation (13). In fasting conditions, glucagon secretion is increased whilst insulin levels decreased. Upon binding to their receptors on hepatocytes, glucagon activates cAMP-PKA signaling to mediate CREB activation, thereby promoting PEPCK and G6Pase transcriptions. CREB-mediated hepatic gluconeogenesis can be further enhanced by coactivator p300/CBP and CRTC2. In addition, CREB stimulates PGC-1α expression, another coactivator to cooperate with FOXO1 in promoting PEPCK and G6Pase transcriptions. During the fed state, insulin secretion is stimulated whilst glucagon production is inhibited. Insulin signaling mediates downstream AKT activation that phosphorylates FOXO1 for degradation, thereby reducing PEPCK and G6Pase transactivation (4, 9, 14–16). Fasting-induced glucocorticoid also contributes to hepatic gluconeogenesis through direct transactivation of PEPCK or G6Pase (17, 18). In case of hormone dysregulation, for instance, insulin resistance in human or animals with type 2 diabetes leads to the failure of FOXO1 degradation, causing hyperglycemia via abnormal gluconeogenesis (16, 19).
Hepatic glycogenolysis
Glycogenolysis is a process of breaking down glycogen into glucose. Glycogen phosphorylase breaks down glycogen into glucos-1-phosphate, and the latter is converted into G6P by phosphoglucomutase, which is hydrolyzed into glucose by G6Pase. Glycogenolysis also indirectly contributes to hepatic gluconeogenesis by supplying glycolytic intermediates. A very recent study on mouse models provides evidence that glycogen in liver and muscle tissue supplies glycolytic intermediate lactic acid, hence contributing to gluconeogenesis even in the fed state (20).
ATMs and their influences on HGP
There are two kinds of typical adipose tissue in mammals, white adipose tissue (WAT) responsible for energy storge and brown adipose tissue (BAT) responsible for energy dissipation (21). WAT has a close crosstalk with the liver to regulate metabolism and immune reactions. Under different pathophysiological conditions, heterogenous ATMs constitute special immune microenvironments, producing broad impacts on both adipose tissues and liver. Based on phenotypic characteristics, ATMs can be classified into M1 macrophages expressing high levels of proinflammatory cytokines like TNF-α, IL-1, IL-6, iNOS, and M2 macrophages with high levels of anti-inflammatory IL-10 and arginase 1. M2 macrophages are predominant in lean WAT to maintain immune hemostasis in healthy individuals, whereas M1 macrophages are accumulated in obese WAT and mediate adipose tissue inflammation and insulin resistance in metabolically unhealthy individuals. However, during metabolic switch from homeostasis to disorders, some phenotype markers may be overlapped between M1 and M2 macrophages and some potential markers have not been identified or included. There are still some difficulties to depict full and precise ATM profiles based on this simple classification, we therefore specify the phenotypic characteristics of ATMs related to HGP modulation here.
Cytokines from ATMs on HGP Modulation
IL-10 in concert with insulin action: Physiological control of HGP
Insulin is considered as the primary contributor to HGP suppression in the fed state. A recent study adds ATM-derived IL-10 to the list of HGP suppression, providing an elaborate explanation for feeding-induced HGP reduction. An earlier study showed that in vitro treatment with IL-10 plus IL-1β inhibited glucose production in primary rat hepatocytes through reducing PEPCK expression, but the underlying mechanisms remained unclear (22). Using different myeloid-specific gene knockout mouse models, Toda et al. demonstrated that macrophages, in response to postprandial signals LPS and insulin, produced IL-10 to suppress gluconeogenic gene expression and hepatocyte glucose production. This inhibitory effect was mediated by IL-10-stimulated Stat3 activation. Feeding induced IL-10 from ATMs, whereas obesity markedly decreased IL-10-producing macrophages in epidydimal WAT. Thus, ATM-derived IL-10 contributes to HGP suppression in cooperation with insulin in fed state (Figure 1). Upon obesity, ATMs with low IL-10 production failed to inhibit hepatic gluconeogenesis, resulting in high HGP and hyperglycemia. Importantly, IL-10 restoration successfully lowered the plasma glucose in obese mice, accompanied by the suppression of gluconeogenic genes in the liver (23, 24). Therefore, ATM-derived IL-10 may be a promising indicator to monitor HGP homeostasis during fasting-feeding cycle and a potential therapeutic target for obesity-induced metabolic dysfunction.
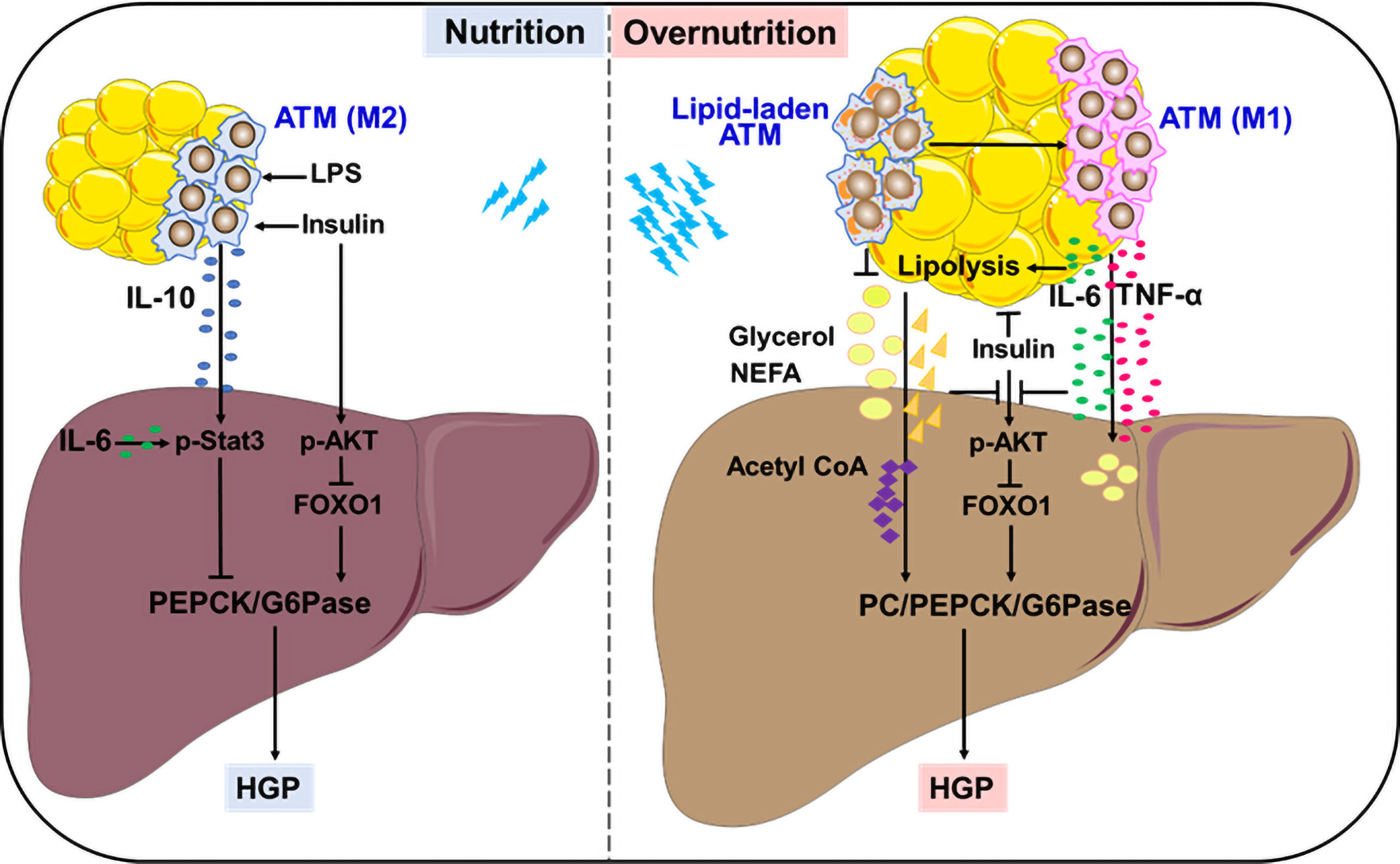
Figure 1 Remote modulation on HGP by ATMs. In the fed state, ATMs in response to insulin and LPS produce IL-10 to control gluconeogenesis-mediated HGP. In overnutrition state, ATMs secret large amounts of IL-6 and TNF-α, which link WAT inflammation with lipolysis, resulting in exacerbated HGP through interfering with insulin action or supplying precursors to stimulate aberrant gluconeogenesis. Lipid-associated ATMs play either protective or detrimental roles in controlling aberrant HGP.
TNF-α in association with insulin resistance: Pathological stimulation of HGP
Insulin resistance is the major cause and typical feature of type 2 diabetes, which is closely associated with abnormal hepatocyte gluconeogenesis that contributes to enhanced HGP (16, 25, 26). An early study provided direct evidence that mice received constant infusion of TNF-α showed significant elevation in HGP together with decrease in insulin action and increase in glucagon secretion (27). Later, it was verified that TNF-α expression was significantly upregulated in WAT from rodents and human with obesity or diabetes, and closely associated with systemic insulin resistance. Importantly, TNF-α deficiency provided protection from obesity-induced insulin resistance in mice (28–30). ATMs are remarkably increased in obese WAT, which produce large amounts of TNF-α to drive insulin resistance and interfere with hepatic glycolipid metabolism (31, 32). TNF-α impairs insulin signaling by blocking tyrosine phosphorylation of insulin receptor substrate-1, the failure of AKT activation may facilitate HGP via glycogenolysis and gluconeogenesis. On the other hand, TNF-α may induce lipid overload in the liver, either from de novo lipogenesis through insulin-independent SREBP1 activation, or from excessive lipolysis of WAT elicited by insulin resistance (31, 33–35). Excessive lipolysis supplies lots of gluconeogenetic precursors like glycerol, NEFAs to promote aberrant HGP. Notably, more lipotoxic intermediators such as saturated palmitic acid and diacylglycerol are generated in these processes, which induce hepatic endoplasmic reticulum stress, inflammation and ensuing insulin resistance (5, 36–39). Therefore, TNF-α from obese ATMs may serve as a strong stimulator for abnormal HGP in obesity and associated diabetes mellitus (Figure 1).
IL-6 in cooperation with lipolysis: Indirect regulation of HGP
Besides TNF-α, IL-6 is another critical cytokine secreted from ATMs in obese condition. But differently, IL-6 regulates HGP in a more complex and flexible manner. To a large extent, the diversity of IL-6 modulation depends on its distinct target organs in different settings, which is more likely influenced by its sources and amounts. In fact, when directly acting on liver or primary hepatocytes, IL-6 at physiological concentrations elicited a suppression in glucagon-stimulated HGP and gluconeogenic genes (40, 41). In line with this, hepatic IL-6 that was induced by insulin action in the brain showed an inhibition on HGP through promoting Stat3 activation and reducing hepatocyte gluconeogenesis via PEPCK and G6pase suppression (42–44). Upon targeting adipocytes, IL-6 plays an indirect role in driving HGP process. In response to exercise or early fasting, skeletal muscle-derived IL-6 promotes lipolysis in WAT, thereby providing materials to support HGP (45–47). Of note, in case of morbid obesity, ATMs produce large amounts of IL-6 to stimulate excess WAT lipolysis, which abrogates insulin action on HGP suppression, resulting in hyperglycemia in rodents or adolescents (48, 49). Using in vivo metabolomics approach in combination with myeloid-specific JNK-deficient mice, and IL-6 neutralization or infusion treatment, Perry et al. provided evidence that insulin failed to suppress hepatic acetyl CoA, PC activity/flux and subsequent HGP in morbidly obese condition. During this process, IL-6 from ATMs played a key role in potentiating abnormal hepatic gluconeogenesis by increasing WAT lipolysis, leading to uncontrolled increases in hepatic acetyl CoA content and PC activity (Figure 1). These findings provide an alternative explanation for ATM-regulated, lipolysis-dependent HGP in diet-induced obesity, and propose a novel therapeutic target based on ATM-derived IL-6 (49). Moreover, the role of IL-6 in suppressing insulin signaling via SOCS3 induction in the liver should also be considered (48, 50, 51), which will not be detailed here.
ATMs on the bridge linking WAT inflammation with hepatic inflammation
As noted above, ATMs make pivotal contribution to hepatic insulin resistance through releasing proinflammatory cytokines. More interestingly, WAT inflammation also triggers hepatic inflammation, producing an overlapped interference on glycolipid homeostasis including HGP process. Through intraperitoneally transplanting visceral WAT from obese mice, Bijnen et al. established evident hepatic inflammation characterized by neutrophil and macrophage accumulation in receipt mice, which contributed to the development of non-alcoholic steatohepatitis (52). When ATMs were deleted from WAT prior to transplantation, this effect could be reversed. This study reveals the key position of ATMs in linking WAT inflammation and hepatic inflammation, in which CD11c+ ATMs that highly express neutrophil chemotactic genes are considered to recruit neutrophils followed by macrophages into the liver (52). In support of this, two earlier studies demonstrated an association of omental ATMs with hepatic inflammation and steatosis in insulin-resistant subjects, as well as the WAT inflammation prior to hepatic inflammation in mice during obesity development (53, 54).
Lipid-laden ATMs on HGP Modulation
Besides the anti- or pro-inflammatory ATMs in lean or obese conditions, a peculiar population of lipid-laden ATMs intersecting metabolism and immunity is significantly upregulated by obesity in human and mouse WAT, particularly visceral WAT (55, 56). These lipid-laden ATMs not only showed an association with human body mass index, but also had an inhibitory effect on insulin signaling of fat explants from lean mice (56). Through analyzing the lipid profiles of ATMs in genetically obese (ob/ob) mice that were treated with or without rosiglitazone, Prieur et al. proposed that the cytotoxic lipid species including free cholesterol and saturated triglyceride could induce the switch of ATMs from M2 toward M1 phenotype, thereby resulting in obesity-induced inflammation and insulin resistance (55). In agreement with this, obese mice that received diet switching from high-fat diet to normal chow diet had a rapid normalization of metabolic parameters including reduced hepatic steatosis and gluconeogenesis-based HPG, accompanied by an obvious reduction in lipid-laden ATMs (57). These findings imply a close linkage of lipid-laden ATMs with liver metabolism. Notably, however, accumulating evidences demonstrated that some lipid-associated ATMs could improve glucose metabolic homeostasis by taking up and sequestering excessive lipids to avoid their damage on both adipose tissue and liver. Silence or deficiency of lipoprotein lipase or Trem2 associated with lipid metabolism abrogated the lipid uptake and storage in ATMs, resulting in insulin resistance and aggravating abnormal HGP via gluconeogenesis in obese mice (58, 59). As such, the lipid-associated ATMs may undergo dynamic changes with the development of obesity and have bidirectional regulation on HGP. It is very likely that ATMs assist in handling lipid during the metabolic adaptation to energy surplus, achieving a fine control of HGP through reducing lipid overload and toxicity on adipocytes and hepatocytes. By contrast, in case of lipid overload, toxic lipid species may stimulate ATMs to induce inflammation, insulin resistance and aberrant HPG, thereby producing deleterious effects on metabolic health (60, 61) (Figure 1). In agreement with this, an exosome-dependent transfer of neutral lipid from adipocytes to macrophages has recently been revealed (62, 63), further elucidation on lipid composition and amounts will help to elucidate their protective or detrimental effects on HGP modulation and consequent metabolic health.
Potential ATM-based Modulators for HGP
Given the diversity of ATMs in different nutrition states, emerging ATM-associated modulators for HGP have been identified, and several potential targets are briefly summarized here.
Osteopontin (OPN)
OPN is a secreted matrix glycoprotein significantly upregulated in human and mouse adipose tissue upon obesity, and ATMs are its important source. OPN contributes to ATM accumulation via recruitment or proliferation in obese WAT, which is therefore closely associated with obesity-induced WAT inflammation, insulin resistance and HGP. Genetic deletion or antibody neutralization of OPN successfully alleviated ATM infiltration, WAT and hepatic inflammation in obese mice, thus improving insulin sensitivity accompanied by normalization of HGP markers. Of note, FOXO1 decrease mediated by AKT activation and Stat3 activation have been involved in HGP correction, confirming that OPN plays critical role in stimulating hepatic gluconeogenesis, though its direct or indirect effects remain to be carefully investigated (64–67). Interestingly, obese mice with selectively silenced OPN in epididymal ATMs showed an improvement in systemic glucose tolerance, further implying the contribution of ATM-derived OPN to the dysregulation of glucose metabolism. In addition, it should be noted that a population of recruited hepatic macrophages expressing OPN has recently been found to promote the development of non-alcohol fatty liver disease (68), which is warranted to be further studied regarding its regulation on HGP.
Dipeptidyl peptidase 4 (DPP4)
DPP4 is a glycoprotein ubiquitously expressed by many cell types. Given its multiple roles in immune activation and metabolic influence, DPP4 has been considered as a potential therapeutic target for type 2 diabetes. DPP4 has a direct role in enzymatic cleavage of incretin hormone glucagon-like peptide-1, thereby interfering with normal glucose homeostasis. In addition, DPP4 is elevated in obese WAT where both adipocytes and ATMs are counted. Factors related to metabolic stress such as glucocorticoids and oxidized LDL have been reported to induce DPP4 upregulation on macrophages, which can stimulate macrophage inflammation and T cell activation, hence potentiating WAT inflammation and insulin resistance (69–72). As DPP4 can function in either soluble or membrane form, and hepatocytes also express DPP4, detailed functions of DPP4 including HGP modulation are yet to be determined.
Retinol binding protein 4 (RBP4)
RBP4 is a serum retinol transporter secreted by the liver and fat. In obese mice, RBP4 stimulates ATM activation dependent on toll-like receptor 2/4, leading to production of proinflammatory cytokines like TNF-α, IL-12, IL-6, IL-1β, and subsequent T cell activation. This action plays a critical role in eliciting obesity-induced WAT inflammation and systemic insulin resistance (73–75). Interestingly, EVs from visceral WAT of obese mice were found to carry RBP4 that possessed the capacity to activate macrophages and elicit insulin resistance (76). More recently, a study on women with obesity linked RBP4 with HGP relying on its stimulation on adipocyte lipolysis. Direct stimulation on basal lipolysis as well as increased lipolysis by indirectly activating ATMs were both included (77). Therefore, RBP4 may act as another promising candidate for ATM-based HGP modulation, particularly in obesity-related glucose dysregulation.
Concluding remarks and perspectives
Fine-regulation of HGP is critical to maintain blood glucose homeostasis in various nutrition states. HGP dysregulation contributes to hyperglycemia in obesity-associated diabetes. During fasting-feeding cycle or metabolic stress like overnutrition, ATMs change their cytokine or lipid profiles to remotely modulate HGP. Feeding induces IL-10 from ATMs, which controls HGP by inhibiting gluconeogenesis in concert with insulin action. While exacerbated HGP usually coincides with large amounts of IL-6 and TNF-α from ATMs, which link WAT inflammation with lipolysis, further interfering with insulin action or supplying precursors to stimulate gluconeogenesis. In response to overnutrition, lipid-associated ATMs play either protective or detrimental roles in controlling aberrant HGP (Figure 1). Therefore, ATM-based HGP modulation may be the promising strategies to treat obesity-associated diabetes.
Regarding ATM-based HGP modulation, more regulatory modes and targets remain to be unraveled, whilst some regulators of macrophages, such as a E3 ubiquitin ligase TRIM29 that inhibits macrophage activation and proinflammatory cytokine production (78), would be the promising candidates. Besides secretary molecules, EVs may be another indispensable remoter for fat-liver crosstalk. It is therefore interesting to explore whether and how EVs regulate the interactions between ATMs, adipocytes and hepatocytes, and their influences on HGP homeostasis. Upon challenged with energy surplus, WAT undergoes the process from metabolic adaptation to maladaptation, in which HGP-related ATM phenotypes and functions need to be carefully determined due to their complexity and flexibility. Given that gluconeogenesis is precisely regulated by circadian clocks, it is yet open but interesting to explore the changes of ATMs in this process. Furthermore, possible HGP regulation by BAT macrophages is also attractive, due to their involvement in energy metabolism (79, 80).
Author contributions
YT and QJ drafted the manuscript. YT designed and created the figure. QW wrote and revised the manuscript, designed the figure. All authors approved the submitted version of the manuscript.
Funding
This work is supported by the National Natural Science Foundation of China (81970733, 81770838, 81471065).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. American Diabetes A. Diagnosis and classification of diabetes mellitus. Diabetes Care (2009) 32(Suppl 1):S62–7. doi: 10.2337/dc13-S067.
2. Gerich JE. Role of the kidney in normal glucose homeostasis and in the hyperglycaemia of diabetes mellitus: Therapeutic implications. Diabetes Med (2010) 27(2):136–42. doi: 10.1111/j.1464-5491.2009.02894.x
3. Petersen MC, Vatner DF, Shulman GI. Regulation of hepatic glucose metabolism in health and disease. Nat Rev Endocrinol (2017) 13(10):572–87. doi: 10.1038/nrendo.2017.80
4. Rui L. Energy metabolism in the liver. Compr Physiol (2014) 4(1):177–97. doi: 10.1002/cphy.c130024
5. Targher G, Corey KE, Byrne CD, Roden M. The complex link between NAFLD and type 2 diabetes mellitus - mechanisms and treatments. Nat Rev Gastroenterol Hepatol (2021) 18(9):599–612. doi: 10.1038/s41575-021-00448-y
6. Kranendonk ME, Visseren FL, van Herwaarden JA, Nolte-'t Hoen EN, de Jager W, Wauben MH, et al. Effect of extracellular vesicles of human adipose tissue on insulin signaling in liver and muscle cells. Obes (Silver Spring) (2014) 22(10):2216–23. doi: 10.1002/oby.20847
7. Fuchs A, Samovski D, Smith GI, Cifarelli V, Farabi SS, Yoshino J, et al. Associations among adipose tissue immunology, inflammation, exosomes and insulin sensitivity in people with obesity and nonalcoholic fatty liver disease. Gastroenterology (2021) 161(3):968–81.e12. doi: 10.1053/j.gastro.2021.05.008
8. Nuttall FQ, Ngo A, Gannon MC. Regulation of hepatic glucose production and the role of gluconeogenesis in humans: Is the rate of gluconeogenesis constant? Diabetes Metab Res Rev (2008) 24(6):438–58. doi: 10.1002/dmrr.863
9. Cui A, Ding D, Li Y. Regulation of hepatic metabolism and cell growth by the atf/creb family of transcription factors. Diabetes (2021) 70(3):653–64. doi: 10.2337/dbi20-0006
10. McCommis KS, Finck BN. Mitochondrial pyruvate transport: A historical perspective and future research directions. Biochem J (2015) 466(3):443–54. doi: 10.1042/BJ20141171
11. Beale EG, Hammer RE, Antoine B, Forest C. Disregulated glyceroneogenesis: PCK1 as a candidate diabetes and obesity gene. Trends Endocrinol Metab (2004) 15(3):129–35. doi: 10.1016/j.tem.2004.02.006
12. Hutton JC, O’Brien RM. Glucose-6-phosphatase catalytic subunit gene family. J Biol Chem (2009) 284(43):29241–5. doi: 10.1074/jbc.R109.025544
13. Oh KJ, Han HS, Kim MJ, Koo SH. CREB and FoxO1: Two transcription factors for the regulation of hepatic gluconeogenesis. BMB Rep (2013) 46(12):567–74. doi: 10.5483/BMBRep.2013.46.12.248
14. Nakae J, Kitamura T, Silver DL, Accili D. The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J Clin Invest (2001) 108(9):1359–67. doi: 10.1172/JCI200112876
15. Lu M, Wan M, Leavens KF, Chu Q, Monks BF, Fernandez S, et al. Insulin regulates liver metabolism in vivo in the absence of hepatic akt and Foxo1. Nat Med (2012) 18(3):388–95. doi: 10.1038/nm.2686
16. Hatting M, Tavares CDJ, Sharabi K, Rines AK, Puigserver P. Insulin regulation of gluconeogenesis. Ann N Y Acad Sci (2018) 1411(1):21–35. doi: 10.1111/nyas.13435
17. Korenfeld N, Finkel M, Buchshtab N, Bar-Shimon M, Charni-Natan M, Goldstein I. Fasting hormones synergistically induce amino acid catabolism genes to promote gluconeogenesis. Cell Mol Gastroenterol Hepatol (2021) 12(3):1021–36. doi: 10.1016/j.jcmgh.2021.04.017
18. Xu S, Liu Y, Hu R, Wang M, Stöhr O, Xiong Y, et al. TAZ inhibits glucocorticoid receptor and coordinates hepatic glucose homeostasis in normal physiological states. Elife (2021) 10:e57462. doi: 10.7554/eLife.57462.
19. Magnusson I, Rothman DL, Katz LD, Shulman RG, Shulman GI. Increased rate of gluconeogenesis in type-ii diabetes-mellitus - a c-13 nuclear-magnetic-resonance study. J Clin Invest (1992) 90(4):1323–7. doi: 10.1172/JCI115997
20. TeSlaa T, Bartman CR, Jankowski CSR, Zhang Z, Xu X, Xing X, et al. The source of glycolytic intermediates in mammalian tissues. Cell Metab (2021) 33(2):367–78.e5. doi: 10.1016/j.cmet.2020.12.020
21. Peirce V, Carobbio S, Vidal-Puig A. The different shades of fat. Nature (2014) 510(7503):76–83. doi: 10.1038/nature13477
22. Yerkovich ST, Rigby PJ, Fournier PA, Olynyk JK, Yeoh GC. Kupffer cell cytokines interleukin-1beta and interleukin-10 combine to inhibit phosphoenolpyruvate carboxykinase and gluconeogenesis in cultured hepatocytes. Int J Biochem Cell Biol (2004) 36(8):1462–72. doi: 10.1016/j.biocel2003.10.022.
23. Toda G, Soeda K, Okazaki Y, Kobayashi N, Masuda Y, Arakawa N, et al. Insulin- and lipopolysaccharide-mediated signaling in adipose tissue macrophages regulates postprandial glycemia through akt-mtor activation. Mol Cell (2020) 79(1):43–53.e4. doi: 10.1016/j.molcel.2020.04.033
24. Sulen A, Aouadi M. Fed macrophages hit the liver’s sweet spot with il-10. Mol Cell (2020) 79(1):1–3. doi: 10.1016/j.molcel.2020.06.016.
25. Kubota N, Kubota T, Kajiwara E, Iwamura T, Kumagai H, Watanabe t, et al. Differential hepatic distribution of insulin receptor substrates causes selective insulin resistance in diabetes and obesity. Nat Commun (2016) 7:12977. doi: 10.1038/ncomms12977
26. Yan H, Yang W, Zhou F, Li X, Pan Q, Shen Z, et al. Estrogen improves insulin sensitivity and suppresses gluconeogenesis via the transcription factor foxo1. Diabetes (2019) 68(2):291–304. doi: 10.2337/db18-0638
27. Lang CH, Dobrescu C, Bagby GJ. Tumor necrosis factor impairs insulin action on peripheral glucose disposal and hepatic glucose output. Endocrinology (1992) 130(1):43–52. doi: 10.1210/endo.130.1.1727716
28. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science (1993) 259(5091):87–91. doi: 10.1126/science.7678183
29. Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest (1995) 95(5):2409–15. doi: 10.1172/JCI117936
30. Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature (1997) 389(6651):610–4. doi: 10.1038/39335
31. Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest (2006) 116(7):1793–801. doi: 10.1172/JCI29069
32. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest (2003) 112(12):1796–808. doi: 10.1172/JCI200319246
33. Yu YH, Ginsberg HN. Adipocyte signaling and lipid homeostasis: sequelae of insulin-resistant adipose tissue. Circ Res (2005) 96(10):1042–52. doi: 10.1161/01.RES.0000165803.47776.38
34. Leclercq IA, Da Silva Morais A, Schroyen B, Van Hul N, Geerts A. Insulin resistance in hepatocytes and sinusoidal liver cells: mechanisms and consequences. J Hepatol (2007) 47(1):142–56. doi: 10.1016/j.jhep.2007.04.002
35. Rui L, Aguirre V, Kim JK, Shulman GI, Lee A, Corbould A, et al. Insulin/IGF-1 and TNF-alpha stimulate phosphorylation of IRS-1 at inhibitory Ser307 via distinct pathways. J Clin Invest (2001) 107(2):181–9. doi: 10.1172/JCI10934
36. Stefan N, Cusi K. A global view of the interplay between non-alcoholic fatty liver disease and diabetes. Lancet Diabetes Endocrinol (2022) 10(4):284–96. doi: 10.1016/S2213-8587(22)00003-1
37. Stefan N. Causes, consequences, and treatment of metabolically unhealthy fat distribution. Lancet Diabetes Endocrinol (2020) 8(7):616–27. doi: 10.1016/S2213-8587(20)30110-8
38. Steensels S, Qiao J, Zhang Y, Maner-Smith KM, Kika N, Holman CD, et al. Acyl-coenzyme a thioesterase 9 traffics mitochondrial short-chain fatty acids toward de novo lipogenesis and glucose production in the liver. Hepatology (2020) 72(3):857–72. doi: 10.1002/hep.31409
39. Mashek DG. Hepatic lipid droplets: A balancing act between energy storage and metabolic dysfunction in NAFLD. Mol Metab (2021) 50:101115. doi: 10.1016/j.molmet.2020.101115
40. Dent JR, Chowdhury MK, Tchijov S, Dulson D, Smith G. Interleukin-6 is a negative regulator of hepatic glucose production in the isolated rat liver. Arch Physiol Biochem (2016) 122(2):103–9. doi: 10.3109/13813455.2016.1146773
41. Christ B, Yazici E, Nath A. Phosphatidylinositol 3-kinase and protein kinase c contribute to the inhibition by interleukin 6 of phosphoenolpyruvate carboxykinase gene expression in cultured rat hepatocytes. Hepatology (2000) 31(2):461–8. doi: 10.1002/hep.510310228
42. Ramadoss P, Unger-Smith NE, Lam FS, Hollenberg AN. STAT3 targets the regulatory regions of gluconeogenic genes. in vivo Mol Endocrinol (2009) 23(6):827–37. doi: 10.1210/me.2008-0264
43. Inoue H, Ogawa W, Ozaki M, Haga S, Matsumoto M, Furukawa K, et al. Role of STAT-3 in regulation of hepatic gluconeogenic genes and carbohydrate metabolism. in vivo Nat Med (2004) 10(2):168–74. doi: 10.1038/nm980
44. Inoue H, Ogawa W, Asakawa A, Okamoto Y, Nishizawa A, Matsumoto M, et al. Role of hepatic STAT3 in brain-insulin action on hepatic glucose production. Cell Metab (2006) 3(4):267–75. doi: 10.1016/j.cmet.2006.02.009
45. Wueest S, Item F, Boyle CN, Jirkof P, Cesarovic N, Ellingsgaard H, et al. Interleukin-6 contributes to early fasting-induced free fatty acid mobilization in mice. Am J Physiol Regul Integr Comp Physiol (2014) 306(11):R861–7. doi: 10.1152/ajpregu.00533.2013
46. Kistner TM, Pedersen BK, Lieberman DE. Interleukin 6 as an energy allocator in muscle tissue. Nat Metab (2022) 4(2):170–9. doi: 10.1038/s42255-022-00538-4
47. Wedell-Neergaard AS, Lang Lehrskov L, Christensen RH, Legaard DE, Dorph E, Larsen MK, et al. Exercise-induced changes in visceral adipose tissue mass are regulated by il-6 signaling: a randomized controlled trial. Cell Metab (2019) 29(4):844–55.e3. doi: 10.1016/j.cmet.2018.12.007
48. Hoene M, Weigert C. The role of interleukin-6 in insulin resistance, body fat distribution and energy balance. Obes Rev (2008) 9(1):20–9.doi: 10.1111/j.1467-789X.2007.00410.x.
49. Perry RJ, Camporez JG, Kursawe R, Titchenell PM, Zhang D, et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell (2015) 160(4):745–58. doi: 10.1016/j.cell.2015.01.012
50. Senn JJ, Klover PJ, Nowak JJ, Zimmers TA, Koniaris LG, Furlanetto RW, et al. Suppressor of cytokine signaling-3 (SOCS-3), a potential mediator of interleukin-6-dependent insulin resistance in hepatocytes. J Biol Chem (2003) 278(16):13740–6. doi: 10.1074/jbc.M210689200
51. Kim JH, Kim JE, Liu HY, Cao W, Chen J. Regulation of interleukin-6-induced hepatic insulin resistance by mammalian target of rapamycin through the STAT3-SOCS3 pathway. J Biol Chem (2008) 283(2):708–15. doi: 10.1074/jbc.M708568200
52. Bijnen M, et al. Adipose tissue macrophages induce hepatic neutrophil recruitment and macrophage accumulation in mice. Gut (2018) 67(7):1317–27. doi: 10.1136/gutjnl-2016-313654
53. van der Heijden RA, Sheedfar F, Morrison MC, Hommelberg PP, Kor D, Kloosterhuis NJ, et al. High-fat diet induced obesity primes inflammation in adipose tissue prior to liver in C57BL/6j mice. Aging (Albany NY) (2015) 7(4):256–68. doi: 10.18632/aging.100738
54. Tordjman J, Poitou C, Hugol D, Bouillot JL, Basdevant A, Bedossa P, et al. Association between omental adipose tissue macrophages and liver histopathology in morbid obesity: Influence of glycemic status. J Hepatol (2009) 51(2):354–62. doi: 10.1016/j.jhep.2009.02.031
55. Prieur X, Mok CY, Velagapudi VR, Núñez V, Fuentes L, Montaner D, et al. Differential lipid partitioning between adipocytes and tissue macrophages modulates macrophage lipotoxicity and M2/M1 polarization in obese mice. Diabetes (2011) 60(3):797–809. doi: 10.2337/db10-0705
56. Shapiro H, Pecht T, Shaco-Levy R, Harman-Boehm I, Kirshtein B, Kuperman Y, et al. Adipose tissue foam cells are present in human obesity. J Clin Endocrinol Metab (2013) 98(3):1173–81. doi: 10.1210/jc.2012-2745
57. Vatarescu M, Bechor S, Haim Y, Pecht T, Tarnovscki T, Slutsky N, et al. Adipose tissue supports normalization of macrophage and liver lipid handling in obesity reversal. J Endocrinol (2017) 233(3):293–305. doi: 10.1530/JOE-17-0007
58. Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, et al. Lipid-associated macrophages control metabolic homeostasis in a trem2-dependent manner. Cell (2019) 178(3):686–98.e14. doi: 10.1016/j.cell.2019.05.054
59. Aouadi M, Vangala P, Yawe JC, Tencerova M, Nicoloro SM, Cohen JL, et al. Lipid storage by adipose tissue macrophages regulates systemic glucose tolerance. Am J Physiol Endocrinol Metab (2014) 307(4):E374–83. doi: 10.1152/ajpendo.00187.2014
60. Ertunc ME, Hotamisligil GS. Lipid signaling and lipotoxicity in metaflammation: indications for metabolic disease pathogenesis and treatment. J Lipid Res (2016) 57(12):2099–114. doi: 10.1194/jlr.R066514
61. Caslin HL, Bhanot M, Bolus WR, Hasty AH. Adipose tissue macrophages: Unique polarization and bioenergetics in obesity. Immunol Rev (2020) 295(1):101–13. doi: 10.1111/imr.12853
62. Flaherty SE, Grijalva A, Xu X, Ables E, Nomani A, Ferrante AW Jr. A lipase-independent pathway of lipid release and immune modulation by adipocytes. Science (2019) 363(6430):989–93. doi: 10.1126/science.aaw2586
63. Antonyak MA, Lukey MJ, Cerione RA. Lipid-filled vesicles modulate macrophages. Science (2019) 363(6430):931–2. doi: 10.1126/science.aaw6765
64. Nomiyama T, Perez-Tilve D, Ogawa D, Gizard F, Zhao Y, Heywood EB, et al. Osteopontin mediates obesity-induced adipose tissue macrophage infiltration and insulin resistance in mice. J Clin Invest (2007) 117(10):2877–88. doi: 10.1172/JCI31986
65. Kiefer FW, Zeyda M, Gollinger K, Pfau B, Neuhofer A, Weichhart T, et al. Neutralization of osteopontin inhibits obesity-induced inflammation and insulin resistance. Diabetes (2010) 59(4):935–46. doi: 10.2337/db09-0404
66. Kiefer FW, Neschen S, Pfau B, Legerer B, Neuhofer A, Kahle M, et al. Osteopontin deficiency protects against obesity-induced hepatic steatosis and attenuates glucose production in mice. Diabetologia (2011) 54(8):2132–42. doi: 10.1007/s00125-011-2170-0
67. Tardelli M, Zeyda K, Moreno-Viedma V, Wanko B, Grün NG, Staffler G, et al. Osteopontin is a key player for local adipose tissue macrophage proliferation in obesity. Mol Metab (2016) 5(11):1131–7. doi: 10.1016/j.molmet.2016.09.003
68. Remmerie A, Martens L, Thoné T, Castoldi A, Seurinck R, Pavie B, et al. Osteopontin expression identifies a subset of recruited macrophages distinct from kupffer cells in the fatty liver. Immunity (2020) 53(3):641–57.e14. doi: 10.1016/j.immuni.2020.08.004
69. Diaz-Jimenez D, Petrillo MG, Busada JT, Hermoso MA, Cidlowski JA. Glucocorticoids mobilize macrophages by transcriptionally up-regulating the exopeptidase DPP4. J Biol Chem (2020) 295(10):3213–27. doi: 10.1074/jbc.RA119.010894
70. Deacon CF. Physiology and pharmacology of DPP-4 in glucose homeostasis and the treatment of type 2 diabetes. Front Endocrinol (Lausanne) (2019) 10:80. doi: 10.3389/fendo.2019.00080
71. Rao X, Zhao S, Braunstein Z, Mao H, Razavi M, Duan L, et al. Oxidized LDL upregulates macrophage DPP4 expression via TLR4/TRIF/CD36 pathways. EBioMedicine (2019) 41:50–61. doi: 10.1016/j.ebiom.2019.01.065
72. Zillessen P, Celner J, Kretschmann A, Pfeifer A, Racké K, Mayer P. Metabolic role of dipeptidyl peptidase 4 (DPP4) in primary human (pre)adipocytes. Sci Rep (2016) 6:23074. doi: 10.1038/srep23074
73. Moraes-Vieira PM, Yore MM, Sontheimer-Phelps A, Castoldi A, Norseen J, Aryal P, et al. Retinol binding protein 4 primes the NLRP3 inflammasome by signaling through toll-like receptors 2 and 4. Proc Natl Acad Sci USA (2020) 117(49):31309–18. doi: 10.1073/pnas.2013877117
74. Moraes-Vieira PM, Yore MM, Sontheimer-Phelps A, Castoldi A, Norseen J, Aryal P, et al. RBP4 activates antigen-presenting cells, leading to adipose tissue inflammation and systemic insulin resistance. Cell Metab (2014) 19(3):512–26. doi: 10.1016/j.cmet.2014.01.018
75. Moraes-Vieira PM, Castoldi A, Aryal P, Wellenstein K, Peroni OD, Kahn BB. Antigen presentation and t-cell activation are critical for rbp4-induced insulin resistance. Diabetes (2016) 65(5):1317–27. doi: 10.2337/db15-1696
76. Deng ZB, Poliakov A, Hardy RW, Clements R, Liu C, Liu Y, et al. Adipose tissue exosome-like vesicles mediate activation of macrophage-induced insulin resistance. Diabetes (2009) 58(11):2498–505. doi: 10.2337/db09-0216
77. Kilicarslan M, de Weijer BA, Simonyté Sjödin K, Aryal P, Ter Horst KW, Cakir H, et al. RBP4 increases lipolysis in human adipocytes and is associated with increased lipolysis and hepatic insulin resistance in obese women. FASEB J (2020) 34(5):6099–110. doi: 10.1096/fj.201901979RR
78. Xing J, Weng L, Yuan B, Wang Z, Jia L, Jin R, et al. Identification of a role for TRIM29 in the control of innate immunity in the respiratory tract. Nat Immunol (2016) 17(12):1373–80. doi: 10.1038/ni.3580
79. Wolf Y, Boura-Halfon S, Cortese N, Haimon Z, Sar Shalom H, Kuperman Y, et al. Brown-adipose-tissue macrophages control tissue innervation and homeostatic energy expenditure. Nat Immunol (2017) 18(6):665–74. doi: 10.1038/ni.3746
Keywords: hepatic glucose production, adipose tissue, macrophage, liver, gluconeogenesis
Citation: Tao Y, Jiang Q and Wang Q (2022) Adipose tissue macrophages in remote modulation of hepatic glucose production. Front. Immunol. 13:998947. doi: 10.3389/fimmu.2022.998947
Received: 20 July 2022; Accepted: 03 August 2022;
Published: 24 August 2022.
Edited by:
Junji Xing, Houston Methodist Research Institute, United StatesReviewed by:
Manuel Gutierrez-Aguilar, National Autonomous University of Mexico, MexicoCopyright © 2022 Tao, Jiang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qun Wang, d2FuZ3F1bkBzZHUuZWR1LmNu