 Naoki Iwamoto
Naoki Iwamoto Atsushi Kawakami
Atsushi Kawakami
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 12 September 2022
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.998554
This article is part of the Research Topic Novel Insights into the Pathology of Rheumatoid Arthritis: Emerging Role of Macrophage & T Cell Immunity View all 6 articles
Rheumatoid arthritis (RA) is an autoimmune disease characterized by joint inflammation leading to joint destruction and deformity. The crucial role of osteoclasts in the bone erosion in RA has been demonstrated. Deregulated osteoclastogenesis which is affected by environmental factors including the inflammatory state, as well as genetic and epigenetic factors, is one of hallmarks of RA pathogenesis. An enhanced-monocyte-to-osteoclast transition plays an important role in osteoclast upregulation in RA because under specific stimuli, circulating monocytes might migrate to a specific location in the bones and fuse with each other to become mature multinucleated osteoclasts. To understand the mechanism of bone damage in RA and to develop novel treatments targeting osteoclast upregulation, it is important to clarify our understanding of the monocyte-to-osteoclast transition in RA. Several potential targets which inhibit both inflammation and osteoclastogenesis, as well as regulators that affect the monocyte-to-osteoclast transition have been revealed by recent studies. Here, we review the factors affecting osteoclastogenesis in RA, summarize the anti-osteoclastogenic effects of current RA treatments, and identify promising therapeutic targets relating to both inflammation and osteoclastogenesis.
Rheumatoid arthritis is a chronic inflammatory autoimmune disease affecting approximately 1% of the world’s population, caused by multiple genetic, epigenetic and environment factors. RA is systemic disease characterized by chronic arthritis that ultimately results in joint destruction (1). Advanced osteoarticular destruction and deformation can cause irreversible loss of function, in turn leading to physical disabilities that negatively impact quality of life. Therefore, prevention of joint destruction by early diagnosis and early therapeutic intervention is critical in the management of RA (2). Osteoclasts play a central role in bone destruction. Osteoclasts are multinuclear giant cells derived from the monocyte-macrophage lineage and are responsible for bone resorption. Under specific stimuli, circulating monocytes migrate to a specific location in the bones and then fuse with each other to become mature multinucleated osteoclasts (3, 4). Current therapies for RA are not effective in all patients, recently several studies reported that approximately 3~10% of RA patients have been estimated as “difficult-to-treat” RA which defined by multiple biologic disease modifying anti rheumatic drugs (bDMARDs)/targeted synthetic DMARDs failure, rapid progressive bone destruction, moderate disease activity despite appropriate treatment and so on (5–7). Moreover, not all patients can be treated due to the high cost of medication and the occurrence of adverse events such as infection. Therefore, development of novel treatments targeting specific pathogenic cells such as osteoclasts are needed.
Many research groups have investigated how osteoclastogenesis is enhanced in RA or arthritic conditions in vivo. Elucidating such dysregulation in RA might help not only reveal the pathogenesis of RA but also point toward new therapeutic targets. Inflammation is closely related to enhanced osteoclastogenesis in RA; however, other osteoclastogenesis-enhancing mechanisms exist in the pathogenesis of RA.
In this review, we focus on the pathological enhancement of the monocyte-to-osteoclast transition in RA mainly independent of cytokines. Additionally, we summarize the osteoclastogenesis-inhibiting effect of current RA treatments and promising therapeutic targets related to both inflammation and osteoclastogenesis in RA.
Increases in both the number of circulating osteoclast precursor cells/monocyte and in the bone resorption ability of osteoclasts have been reported in RA. Osteoclasts generated from peripheral blood mononuclear cells (PBMCs) including osteoclast precursor cells/monocytes from RA patients showed higher bone resorption compared with those from ankylosing spondylitis patients (8). Mononuclear cells from RA patients with infliximab treatment showed reduction of bone resorption ability. A study conducted by Ikics et al. clearly revealed data showing increased osteoclastogenic potential in RA (9). When similar numbers of PBMCs from RA patients and healthy controls were cultured in osteoclastogenic conditions (cultured under the presence of the receptor activator of nuclear factor-κB ligand (RANKL) with macrophage colony-stimulating factor (M-CSF)), more osteoclasts formed in the RA than the control culture. Spontaneous osteoclast differentiation has also been suspected in RA (10). Hydrochloric acid released by mature osteoclasts close to the ruffled border of osteoclasts solubilizes calcium from the bone matrix and produces matrix metalloproteinases (MMPs) and cathepsin K, which degrade the remaining bone matrix. Mononuclear cells from RA synovial fluid have been shown to express these genes after 21 days’ culture without addition of RANKL, indicating spontaneous osteoclast formation.
As mentioned previously, in RA patients the osteoclastogenic potential of PBMCs is increased; moreover, elevated osteoclast formation in RA has been shown to be correlated with bone loss (11). The number of osteoclasts generated from RA mononuclear cells was correlated with the patients’ sharp score (the joint destruction score evaluated using X-ray) and the lumbar T-score. Also in this experiment, the osteoclast formation of mononuclear cells from RA patients was significantly higher than that from healthy donors. In addition to enhanced osteoclastogenesis, RA affects the longevity of osteoclasts; that is, the number of osteoclasts undergoing apoptosis after osteoclast formation was significantly lower in RA patients than in healthy donors (12).
Joint inflammation contributes to osteoclast-causing bone destruction; thus, in the inflammatory environment of RA, circulating monocytes might continue to transfer to the joint cavity and differentiate into osteoclasts. In a murine model, osteoclasts in the pannus originated from circulating bone marrow-derived cells, not from locally resident macrophages (13). These arthritis-associated osteoclastogenic macrophages (CX3CR1+HLA-DRhiCD11c+CD80−CD86+ cells) are present in the inflamed synovitis and are distinct from conventional osteoclast precursors in homeostatic bone remodeling. Moreover, these arthritis-associated macrophages (CX3CR1+HLA-DRhiCD11c+CD80−CD86+ cells) were detected in synovial samples from human patients with RA.
Classification of monocyte subsets using CD14 and CD16 expression have been a major focus in the analysis of monocyte function. Depending on their expressions of CD14 and CD16, monocytes are defined as classical monocytes (CD14++CD16-), intermediate monocytes (CD14++CD16+) and non-classical monocytes (CD14+CD16++) (14). The expression of CD14 and CD16 is upregulated in the monocytes of RA patients (15, 16); among these three subsets, the intermediate monocytes are dominant in the peripheral blood and synovial tissue of RA (17, 18). Intermediate monocytes secrete pro-inflammatory cytokines such as tumor necrosis factor-α (TNF- α), interleukin-1β (IL-1 β) and IL-6, and they can differentiate into inflammatory macrophages (M1 macrophages) (19, 20). Moreover, the intermediate monocytes in RA are characterized by an increased expression of HLA-DR, and these HLA-DR-positive intermediate monocytes express a high level of costimulatory molecules (CD80 and C86), which promote the induction of IL17+CD14+ T-cells (17, 21, 22). The relative compositions of classical and non-classical monocytes in RA have been controversial. Some reports have described increased proportions of classical monocytes in RA compared with healthy subjects whereas other reports have shown increased proportions of non-classical monocytes (22, 23). From the view of osteoclastogenesis, the classical monocytes are the most important of the three subsets. Analysis of osteoclastogenic function focusing on CD14/CD16 expression has shown that the monocytes that can differentiate into osteoclasts are CD14-positive but CD16-negative, i.e., CD14+CD16- monocytes are the precursors of circulating osteoclasts (24).
Compared with monocytes from healthy donors, those from the peripheral blood of RA patients showed upregulation of the osteoclast-associated receptor (OSCAR), which is an activating receptor expressed by human myeloid cells, for which both collagen type I (COLI) and collagen type II (COLII) serve as ligands (25, 26). This OSCAR-collagen interaction stimulates RANKL-dependent osteoclastogenesis. Interestingly, OSCAR expression is correlated with disease activity and acute-phase reactant concentrations. OSCAR-collagen interactions are also involved in cytokine production; namely, COL2 stimulates the release of proinflammatory cytokines by monocytes whereas this effect was completely blocked in the presence of OSCAR-antibody.
The M1/M2 subsets of macrophages are well-known for being involved in disease pathogenesis including that of RA. Interestingly, M1-like and M2-like subsets are present in macrophage precursor cells, i.e., monocytes. They were first reported to be associated with diabetes mellitus, then also discovered in hypercholesterolemia (27, 28). M1 monocytes are defined as positive for CD14, CD68, and CCR2, and M2 monocyte are defined as positive for CD14, CX3CR1, and CD163 or CD206. In RA, the M1/M2 ratio of monocytes is correlated with osteoclastogenesis and cytokine production from monocytes (29). Specifically, the number of osteoclasts differentiated from the monocytes of RA patients with high M1/M2 ratios was significantly greater than that from patients whose monocytes had low M1/M2 ratios. M1-dominant monocytes were also found to secrete more IL-6 compared with M2 monocytes.
Although the presence of RANKL-independent pathway in inflammatory-osteoclastogenesis pathway is controversial because, for example, there is the fact that the TNF-α overexpression failed to induce bone destruction in RANKL-deficient mice (30), Yokota et al. recently reported that PBMC can differentiate into osteoclasts by a combination of TNF and IL-6 stimulation without RANKL (31). The behavior of osteoclasts generated by this combination was different from that of those induced by RANKL. Expression levels of IL-1β, TNF, IL12p40 and MMP-3 were significant increased in the TNF-/IL-6-induced osteoclasts, but not in RANKL-induced osteoclasts. And the joint destruction defined by total sharp score correlated with the number of TNF- and IL-6-induced osteoclasts, but not with the number of RANKL-induced osteoclasts. By contrast, the bone mineral density of the whole body was correlated with RANKL-induced osteoclasts, but not with TNF- or IL-6-induced osteoclasts, indicating that “inflammatory cytokine-generated osteoclasts” may contribute to the pathology of RA but not to whole-body osteoporosis.
Monocyte-derived dendritic cells (Mo-DC), generated from monocytes, were first reported by Steinman et al. on the basis of their unique morphology, which distinguished them from macrophages and dendritic cells, the other source of osteoclast cells in RA (32, 33). The first study conducted by Rivollier et al. demonstrated that Mo-DC, generated by stimulation with granulocyte macrophage-colony stimulating factor (GM-CSF) and IL-4, differentiated into osteoclasts in cultivation with RANKL and M-CSF, and this osteoclastogenesis was greatly enhanced by adding synovial fluid from RA patients (34). After this study, the detailed mechanism of RA synovial fluid-enhanced osteoclastogenesis of Mo-DC was reported (35). The Mo-DC-to-osteoclast transition was correlated with peptidyl arginase deiminase (PAD) activity and protein citrullination. PAD enzymes govern the citrullination process, which is dysregulated in RA and contributes to both the production and maintenance of anti-citrullinated protein antibody (ACPA). Purified ACPA was shown to enhance OC differentiation from Mo-DC, and this enhancement might be caused by targeting citrullinated actin and vimentin deposited on the Mo-DC surface, colocalizing with ACPAs binding to the cells. This study suggested that protein citrullination and ACPA binding to immature DCs might one of the mechanisms of bone erosion in ACPA-positive RA.
MicroRNAs (miRNAs) are evolutionarily conserved small non-coding RNAs (length is 18-25 nucleotides) that regulate gene expression at the post-transcriptional level. There is increasing interest in the involvement of miRNAs in autoimmune diseases including RA. MiRNAs modulates cell proliferation, apoptosis and cell differentiation including both osteogenesis and osteoclastogenesis (36–38). Several miRNAs have been reported in relation with the monocyte-to-osteoclast transition in RA (Table 1). Although miR-146a was found to be upregulated in PBMC, synovial fluid and synovial fibroblasts in RA (45), miR-146a behaved as a negative regulator of inflammation (46). Regarding osteoclastogenesis, miR-146a also showed a protective role in inflammatory arthritis: overexpression of miR-146a significantly reduced the osteoclast formation of PBMCs from RA patients, and administration of double-stranded miR-146a prevented joint destruction in collagen-induced arthritis (CIA) mice (47). Interestingly, a recent study revealed that the cells playing a central role in this effect are monocytes: specifically, miR-146a overexpression in LY6Chi monocytes decreased joint destruction in CIA mice (48). MiR-125a has been proposed as an important regulator of the innate immune and inflammatory responses in various inflammatory diseases including RA (49, 50). Plasma levels of miR-125a were found to be elevated in RA, suggesting that this miR might be useful as another biomarker for RA (39, 40). MiR-125a has been founded to be deeply related with monocytes/macrophages, especially macrophage polarization (41, 42). In addition, miR-125a is important for osteoclastogenesis, upregulated during the monocyte-to-osteoclast transitio (41, 43). Overexpression of miR-125a inhibited TNF receptor superfamily member 1B gene (TNFRSF1B) protein expression and promoted osteoclast differentiation whereas inhibition of miR-125a showed an opposite result; furthermore, it was shown that mir-125a promotes osteoclastogenesis by targeting TNFRSF1B (44). MiR-223 has also been thought to be related to osteoclastogenesis in RA although the exact effect on osteoclastogenesis is controversial. One study indicated that miR-223 was highly expressed in the RA synovium, leading to the in vitro inhibition of osteoclastogenesis (51) whereas another study indicated miR-223 upregulation in the RA synovium, but knockout rather than overexpression of miR-223 decreased osteoclastogenesis (52). Another possible miR related to osteoclastogenesis in RA is miR-124. Expression of miR-124a was decreased in RA synovial fibroblasts, and in a rat model of arthritis (adjuvant-induced), rats with overexpression of miR-124 (rat analogue of human miR-124a) showed reduced osteoclastogenesis and the expression levels of RANKL, integrin β1 and nuclear factor of activated T cells cytoplasmic 1 (NFATc1) (53, 54).

Table 1 Summary of microRNA related to osteoclastogenesis in RA.
The available disease modifying anti rheumatic drugs (DMARDs) are subdivided into conventional synthetic DMARDs (csDMARDs: methotrexate (MTX), salazosulfapyridine, etc), bDMARDs and targeted synthetic DMARDs. The effect on osteoclastogenesis by these DMARDs have been reported (Table 2). In the current treatment strategy, after the diagnosis of RA, MTX is suggested as a first-line therapy of RA (phase1). In case MTX is contraindicated, another csDMARD is used. The patient moves to phase 2 if treatment by phase 1 agents shows inadequate response (2).
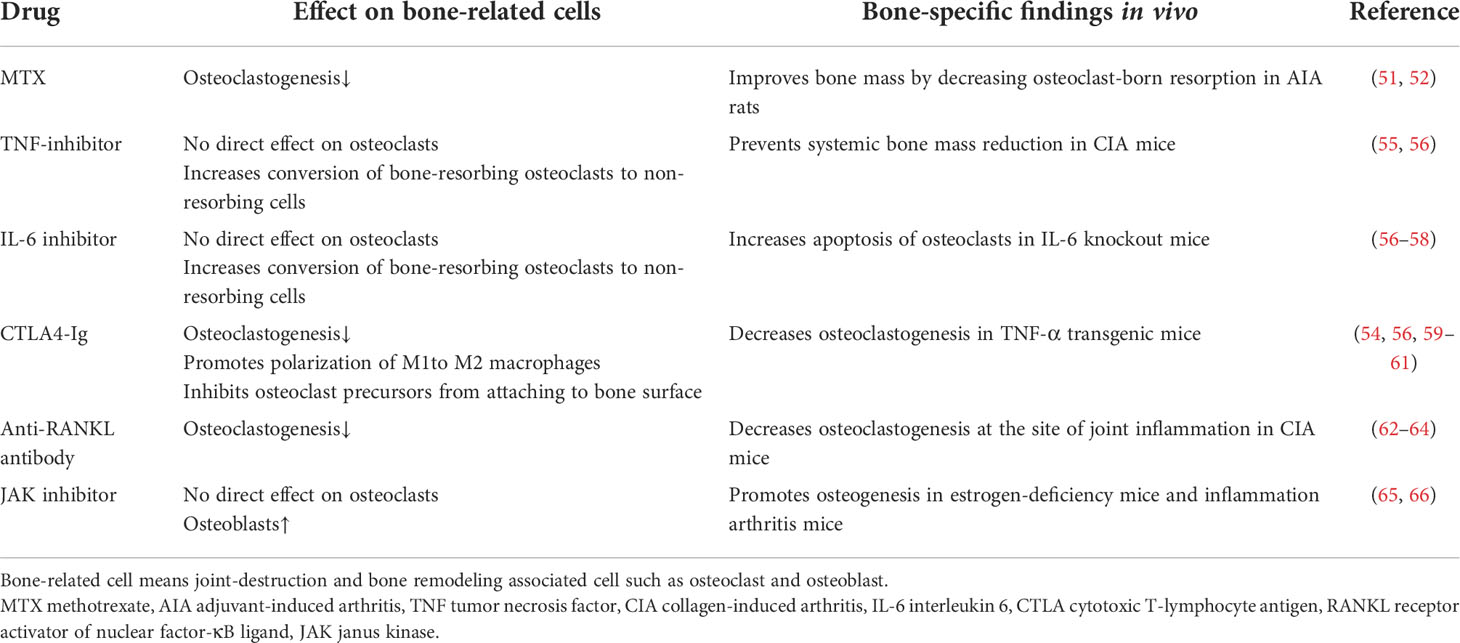
Table 2 The effect of current RA treatments on bone-related cells.
The direct effects of MTX on osteoclastogenesis were reported early on. Specifically, Segawa et al. reported that MTX improved bone mass by preventing decreased osteogenesis and increased bone resorption in adjuvant-induced arthritis rats (59). After that, Kanagawa et al. reported that MTX significantly inhibited osteoclastogenesis through inhibiting RANK-dependent calcium influx into osteoclast progenitors (60).
bDMARDs are used in phase 2 in the treatment strategy, in case MTX/cs DMARDs do not result in an adequate response. In this group of medications, TNF-inhibitors, IL-6 inhibitors and CTLA4-Ig are widely used for treatment of RA. Protection of joint destruction by suppressing inflammation is the main effect of bDMARDs, but inflammation-independent effects of bDMARDs also exist (61). Among the bDMARDs, the one most deeply explored regarding inhibition of osteoclastogenesis is CTLA4-Ig. The binding of CTLA4-Ig to CD80/86 induced activation of the enzyme indoleamine 2,3-dioxygenase (IDO) in osteoclast precursor cells, leading to degradation of tryptophan and the promotion of apoptosis (55, 67). This direct osteoclastogenesis-inhibitory effect of CTLA4-Ig was further analyzed recently (57). Experiments using mice bone marrow macrophages revealed that CTLA4-Ig directly inhibited osteoclastogenesis by interfering with intracellular calcium oscillations. Moreover, CTLA4-Ig treatment promotes M1-to-M2 macrophage polarization, M2 macrophage play anti-osteoclastogenic role as compared with M1 macrophage (58), on monocyte-derived macrophages from RA patients (56). Regarding TNF inhibitor and IL-6 inhibitor, there is no evidence for direct inhibition of osteoclastogenesis; however, TNF-α inhibition has been shown to decrease systemic bone mass reduction in the collagen-induced arthritis model; in IL-6 knockout mice, osteoclast apoptosis is promoted (62–64). In addition to the effect on osteoclastogenesis, the effects on bone reportion by these 3 classes of bDMARDs were recently revealed by Matsuura et al. using in vivo visualization of osteoclast behavior (68). In that study, TNF inhibitor and IL-6 inhibitor affected mature osteoclasts and switched bone-resorbing osteoclasts to non-resorbing cells; on the other hand, CTLA4-Ig had no action on mature osteoclasts but affected osteoclast precursors by decreasing their firm attachment to the bone surface, thereby preventing bone resorption.
The anti-RANKL antibody denosumab is another biologic approved in Japan to inhibit the progression of bone erosion associated with RA. Because the RANK-RANKL pathway is essential for osteoclastogenesis (69), denosumab, through its selective binding to RANKL, inhibits the RANK-RANKL interaction, inhibiting osteoclastogenesis. Phase 2 and 3 clinical trials revealed that denosumab did not inhibit inflammation of RA, clinically defined as the disease activity score, but did inhibit bone erosion; this might come from the mechanism by which denosumab only inhibits osteoclasts but not other cells that play a critical role in RA inflammation such as synovial cells and macrophages (65, 66).
Small molecules are playing an increasing role in the treatment of RA (70); in particular, JAK inhibitors are small-molecule drugs that inhibit the JAK-STAT pathway activity, which is involved in many biologic functions including the activation of immune cells and is associated with several cytokines that are closely related to the pathogenesis of RA (71).In vitro studies have revealed that JAK inhibitors have no direct effects on osteoclasts, but they do suppress osteoclastogenesis via inhibiting RANKL expression on mesenchymal cells such as osteoblasts (72, 73). Interestingly, JAK inhibitors promote osteogenesis (72). In experiments using two different bone-loss model mice (estrogen-deficiency and inflammation arthritis model), the JAK inhibitors tofacitinib and baricitinib showed no direct effects on osteoclasts, but the miRNA sequencing and ingenuity pathway analysis of osteoblasts showed robust up-regulation of Wnt1 and β-catenin; i.e., JAK inhibitors promote osteogenesis by activation of the canonical Wnt signaling pathway.
Research is ongoing into inhibiting osteoclastogenesis as a treatment option for RA (Table 3). One such candidate is the targeting C-X3-C motif chemokine ligand 1 (CX3CL1)- C-X3-C motif chemokine receptor 1 (CX3CR1) axis. CX3CR1, a unique receptor for CX3CL1, is expressed in monocytes/macrophages, dendritic cells and osteoclast precursor cells (86–89). In RA synovial tissue, the expression of CX3CL1 is high and the serum level of soluble CX3CL1 correlates with the disease activity of RA (74, 90). As mentioned above, CX3CL1 plays an important role in the pathogenesis of RA, and a clinical trial using an anti-CX3CL1 monoclonal antibody have been done (75–77). Since CX3CL1 promotes osteoclastogenesis, anti-CX3CL1 therapy has the potential to prevent joint destruction by targeting osteoclast in RA (86).

Table 3 Potential therapeutic targets for treatment of RA by targeting osteoclastogenesis.
The contribution of GM-CSF to the pathogenesis of RA, for example, its elevation in synovial effusion, its genetic level and its crucial role in experimental inflammatory arthritis models, have been well documented by several reports (78, 91–94). GM-CSF induces fusion of prefusion osteoclasts to form bone-resorbing osteoclasts and thereby promotes bone erosion (79). Based on these findings, a GM-CSF inhibitor is now being investigated in a clinical trial (80–82). Molecules that inhibit osteoclastogenesis as well as inflammation are a promising therapeutic option for RA. Semaphorin 3A (Sema3A) has been recently identified as an essential player in bone homeostasis, and Sema3A suppresses osteoclastogenesis and increases osteoblastic bone formation (83, 95). Sema3A levels are decreased in RA serum as well as synovial fluid, and administration of sema3A was shown not only to ameliorate inflammation and bone erosion but also to increase bone formation in a serum transfer-induced arthritis model, which indicated that sema3A is both an immunosuppressive factor and osteoprotective factor (96). Semaphorin 4D (Sema4D) is also known to be an osteoimmune molecule in the semaphorin family. In contrast to the effect of Sema3A, Sema4D promotes inflammation and inhibits osteoblastic bone formation (97, 98). Sema4D levels are increased in both RA serum and RA synovial fluid. In a CIA model, treatment with anti-Sema4D antibody suppressed arthritis and reduced proinflammatory cytokine production (98).
Recently, Koga et al. reported the role of calcium/calmodulin-dependent protein kinase IV (CaMK4) in bone destruction under arthritic conditions (84). CaMK4-deficient mice subjected to CIA showed improved arthritis and decreased Th17 cells. This dual effect on Th17 cells and joint destruction was confirmed in cells from RA patients. CaMK4 inhibition suppressed IL-17 production by CD4+ cells from RA patients and suppressed osteoclastogenesis of monocytes from RA patients.
Notch signaling has an important role in RA, Specifically, Notch signal expression and activation can stimulate synovial cells, thereby accelerating the production of proinflammatory cytokines in RA, and Notch signal are highly expressed in RA synovial tissues related to proliferation and invasion of synovial fibroblasts (85, 99–101). The Notch signaling pathway also can affect osteoclastic differentiation and bone balance; thus, Notch inhibition increases bone volume by enhancing osteoblastic bone formation (102, 103).
Abnormal upregulation of osteoclastogenesis is an important part of the pathogenesis of RA. The monocyte-to-osteoclast transition is crucial for joint destruction although osteoclast precursor cells other than monocytes exist, and osteogenic dysregulation is also important for bone destruction. Numerous studies have shown the environmental, genetic and epigenetic factors that combine to upregulate osteoclastogenesis and modulate osteoclast precursor cells in RA; recent findings regarding these factors such as microRNAs are presented in this review (Figure 1). Many other rheumatic diseases such as Sjogren syndrome, systemic lupus erythematosus and inflammatory myositis cause arthritis but no destructive arthritis. Several parts of pathological condition such as abundant production of cytokines, activation of immune cells share with RA, while the enhancing of monocyte-to-osteoclast transition in these rheumatic disease have been rarely reported. Considering these, abnormal monocyte priming by miRNAs, cytokines and so on, monocyte characteristic property such as M1/M2 imbalance might contribute to joint destruction in RA as combined effect. And, though it should be interpreted with caution because in vitro/vivo effects of anti-osteoclastogenesis do not always reflect clinical effects of ant-bone destruction in RA, recent studies have revealed potential targets contributing to inhibition of both inflammation and osteoclastogenesis. Although much work remains to be performed to gain an integrated overview of osteoclastogenesis in RA, such research may lead to the discovery of novel therapeutic approaches to RA.
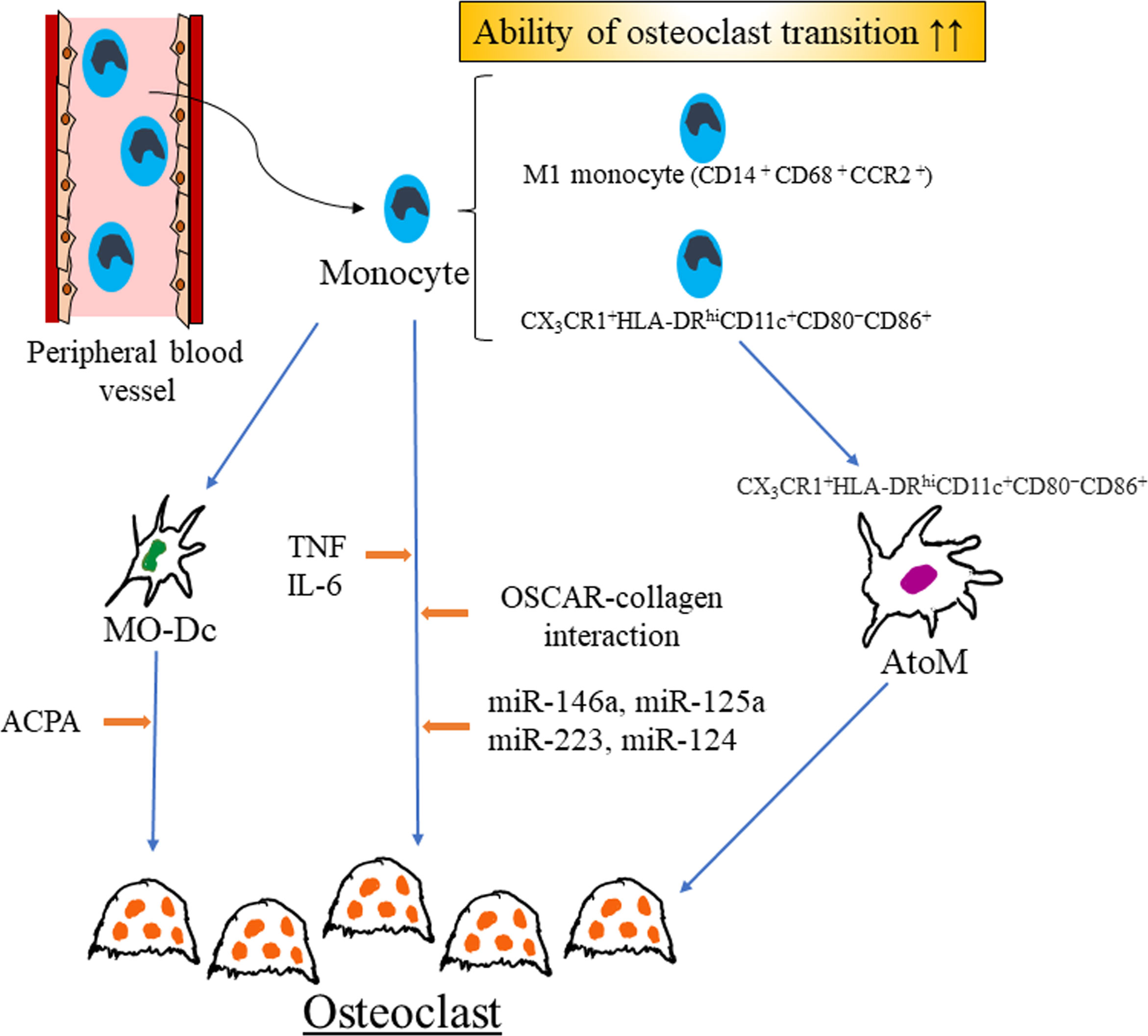
Figure 1 Recent findings of regulators of monocyte-to-osteoclast transition in rheumatoid arthritis. M1 monocyte might have enhanced ability of osteoclast transition as compared with M2 monocyte. Several microRNAs, combination of cytokines and OSCAR-collagen interaction promote osteoclast differentiation in RA. Mo-DCs are generated from monocytes and ACPA enhance osteoclastogenesis of Mo-DCs. In rheumatoid arthritis, arthritis-associated osteoclastogenic macrophages (AtoM) are generated from CX3CR1+HLA-DRhiCD11c+CD80−CD86+ monocyte and differentiated into osteoclast. RA rheumatoid arthritis, Mo-Dc monocyte-derived dendritic cells, MO-Dc monocyte-derived dendritic cells, ACPA anti-citrullinated protein antibody, TNF tumor necrosis factor, IL-6 interleukin-6, OSCAR osteoclast-associated receptor, AtoM arthritis-associated osteoclastogenic macrophages.
NI: wrote the manuscript. NI and AK: reviewed and edited the manuscript. Both authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet (2010) 376(9746):1094–108. doi: 10.1016/S0140-6736(10)60826-4
2. Smolen JS, Landewe RBM, Bijlsma JWJ, Burmester GR, Dougados M, Kerschbaumer A, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann Rheum Dis (2020) 79(6):685–99. doi: 10.1136/annrheumdis-2019-216655
3. Takayanagi H. RANKL as the master regulator of osteoclast differentiation. J Bone Miner Metab (2021) 39(1):13–8. doi: 10.1007/s00774-020-01191-1
4. Parfitt AM. High bone turnover is intrinsically harmful: Two paths to a similar conclusion. The parfitt view. J Bone Miner Res (2002) 17(8):1558–9. doi: 10.1359/jbmr.2002.17.8.1558
5. de Hair MJH, Jacobs JWG, Schoneveld JLM, van Laar JM. Difficult-to-treat rheumatoid arthritis: an area of unmet clinical need. Rheumatol (Oxford) (2018) 57(7):1135–44. doi: 10.1093/rheumatology/kex349
6. Nagy G, Roodenrijs NMT, Welsing PM, Kedves M, Hamar A, van der Goes MC, et al. EULAR definition of difficult-to-treat rheumatoid arthritis. Ann Rheum Dis (2021) 80(1):31–5. doi: 10.1136/annrheumdis-2020-217344
7. Watanabe R, Hashimoto M, Murata K, Murakami K, Tanaka M, Ohmura K, et al. Prevalence and predictive factors of difficult-to-treat rheumatoid arthritis: The KURAMA cohort. Immunol Med (2022) 45(1):35–44. doi: 10.1080/25785826.2021.1928383
8. Gengenbacher M, Sebald HJ, Villiger PM, Hofstetter W, Seitz M. Infliximab inhibits bone resorption by circulating osteoclast precursor cells in patients with rheumatoid arthritis and ankylosing spondylitis. Ann Rheum Dis (2008) 67(5):620–4. doi: 10.1136/ard.2007.076711
9. Ikic M, Jajic Z, Lazic E, Ivcevic S, Grubisic F, Marusic A, et al. Association of systemic and intra-articular osteoclastogenic potential, pro-inflammatory mediators and disease activity with the form of inflammatory arthritis. Int Orthop (2014) 38(1):183–92. doi: 10.1007/s00264-013-2121-0
10. Greisen SR, Einarsson HB, Hvid M, Hauge EM, Deleuran B, Kragstrup TW. Spontaneous generation of functional osteoclasts from synovial fluid mononuclear cells as a model of inflammatory osteoclastogenesis. APMIS (2015) 123(9):779–86. doi: 10.1111/apm.12416
11. Shang W, Zhao LJ, Dong XL, Zhao ZM, Li J, Zhang BB, et al. Curcumin inhibits osteoclastogenic potential in PBMCs from rheumatoid arthritis patients via the suppression of MAPK/RANK/c-Fos/NFATc1 signaling pathways. Mol Med Rep (2016) 14(4):3620–6. doi: 10.3892/mmr.2016.5674
12. Durand M, Boire G, Komarova SV, Dixon SJ, Sims SM, Harrison RE, et al. The increased in vitro osteoclastogenesis in patients with rheumatoid arthritis is due to increased percentage of precursors and decreased apoptosis - the In vitro osteoclast differentiation in arthritis (IODA) study. Bone (2011) 48(3):588–96. doi: 10.1016/j.bone.2010.10.167
13. Hasegawa T, Kikuta J, Sudo T, Matsuura Y, Matsui T, Simmons S, et al. Identification of a novel arthritis-associated osteoclast precursor macrophage regulated by FoxM1. Nat Immunol (2019) 20(12):1631–43. doi: 10.1038/s41590-019-0526-7
14. Ziegler-Heitbrock L, Ancuta P, Crowe S, Dalod M, Grau V, Hart DN, et al. Nomenclature of monocytes and dendritic cells in blood. Blood (2010) 116(16):e74–80. doi: 10.1182/blood-2010-02-258558
15. Liote F, Boval-Boizard B, Weill D, Kuntz D, Wautier JL. Blood monocyte activation in rheumatoid arthritis: increased monocyte adhesiveness, integrin expression, and cytokine release. Clin Exp Immunol (1996) 106(1):13–9. doi: 10.1046/j.1365-2249.1996.d01-820.x
16. Yano R, Yamamura M, Sunahori K, Takasugi K, Yamana J, Kawashima M, et al. Recruitment of CD16+ monocytes into synovial tissues is mediated by fractalkine and CX3CR1 in rheumatoid arthritis patients. Acta Med Okayama (2007) 61(2):89–98. doi: 10.18926/AMO/32882
17. Rossol M, Kraus S, Pierer M, Baerwald C, Wagner U. The CD14(bright) CD16+ monocyte subset is expanded in rheumatoid arthritis and promotes expansion of the Th17 cell population. Arthritis Rheum (2012) 64(3):671–7. doi: 10.1002/art.33418
18. Kawanaka N, Yamamura M, Aita T, Morita Y, Okamoto A, Kawashima M, et al. CD14+,CD16+ blood monocytes and joint inflammation in rheumatoid arthritis. Arthritis Rheum (2002) 46(10):2578–86. doi: 10.1002/art.10545
19. Yoon BR, Yoo SJ, Choi Y, Chung YH, Kim J, Yoo IS, et al. Functional phenotype of synovial monocytes modulating inflammatory T-cell responses in rheumatoid arthritis (RA). PloS One (2014) 9(10):e109775. doi: 10.1371/journal.pone.0109775
20. Boyette LB, Macedo C, Hadi K, Elinoff BD, Walters JT, Ramaswami B, et al. Phenotype, function, and differentiation potential of human monocyte subsets. PloS One (2017) 12(4):e0176460. doi: 10.1371/journal.pone.0176460
21. Rana AK, Li Y, Dang Q, Yang F. Monocytes in rheumatoid arthritis: Circulating precursors of macrophages and osteoclasts and, their heterogeneity and plasticity role in RA pathogenesis. Int Immunopharmacol (2018) 65:348–59. doi: 10.1016/j.intimp.2018.10.016
22. Weldon AJ, Moldovan I, Cabling MG, Hernandez EA, Hsu S, Gonzalez J, et al. Surface APRIL is elevated on myeloid cells and is associated with disease activity in patients with rheumatoid arthritis. J Rheumatol (2015) 42(5):749–59. doi: 10.3899/jrheum.140630
23. Klimek E, Mikolajczyk T, Sulicka J, Kwasny-Krochin B, Korkosz M, Osmenda G, et al. Blood monocyte subsets and selected cardiovascular risk markers in rheumatoid arthritis of short duration in relation to disease activity. BioMed Res Int (2014) 2014:736853. doi: 10.1155/2014/736853
24. Komano Y, Nanki T, Hayashida K, Taniguchi K, Miyasaka N. Identification of a human peripheral blood monocyte subset that differentiates into osteoclasts. Arthritis Res Ther (2006) 8(5):R152. doi: 10.1186/ar2046
25. Herman S, Muller RB, Kronke G, Zwerina J, Redlich K, Hueber AJ, et al. Induction of osteoclast-associated receptor, a key osteoclast costimulation molecule, in rheumatoid arthritis. Arthritis Rheum (2008) 58(10):3041–50. doi: 10.1002/art.23943
26. Schultz HS, Guo L, Keller P, Fleetwood AJ, Sun M, Guo W, et al. OSCAR-collagen signaling in monocytes plays a proinflammatory role and may contribute to the pathogenesis of rheumatoid arthritis. Eur J Immunol (2016) 46(4):952–63. doi: 10.1002/eji.201545986
27. Fadini GP, de Kreutzenberg SV, Boscaro E, Albiero M, Cappellari R, Krankel N, et al. An unbalanced monocyte polarisation in peripheral blood and bone marrow of patients with type 2 diabetes has an impact on microangiopathy. Diabetologia (2013) 56(8):1856–66. doi: 10.1007/s00125-013-2918-9
28. Fadini GP, Simoni F, Cappellari R, Vitturi N, Galasso S, Vigili de Kreutzenberg S, et al. Pro-inflammatory monocyte-macrophage polarization imbalance in human hypercholesterolemia and atherosclerosis. Atherosclerosis (2014) 237(2):805–8. doi: 10.1016/j.atherosclerosis.2014.10.106
29. Fukui S, Iwamoto N, Takatani A, Igawa T, Shimizu T, Umeda M, et al. M1 and M2 monocytes in rheumatoid arthritis: A contribution of imbalance of M1/M2 monocytes to osteoclastogenesis. Front Immunol (2017) 8:1958. doi: 10.3389/fimmu.2017.01958
30. Pettit AR, Ji H, von Stechow D, Muller R, Goldring SR, Choi Y, et al. TRANCE/RANKL knockout mice are protected from bone erosion in a serum transfer model of arthritis. Am J Pathol (2001) 159(5):1689–99. doi: 10.1016/S0002-9440(10)63016-7
31. Yokota K, Sato K, Miyazaki T, Aizaki Y, Tanaka S, Sekikawa M, et al. Characterization and function of tumor necrosis factor and interleukin-6-Induced osteoclasts in rheumatoid arthritis. Arthritis Rheumatol (2021) 73(7):1145–54. doi: 10.1002/art.41666
32. Laperine O, Blin-Wakkach C, Guicheux J, Beck-Cormier S, Lesclous P. Dendritic-cell-derived osteoclasts: A new game changer in bone-resorption-associated diseases. Drug Discovery Today (2016) 21(9):1345–54. doi: 10.1016/j.drudis.2016.04.022
33. Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. i. morphology, quantitation, tissue distribution. J Exp Med (1973) 137(5):1142–62. doi: 10.1084/jem.137.5.1142
34. Rivollier A, Mazzorana M, Tebib J, Piperno M, Aitsiselmi T, Rabourdin-Combe C, et al. Immature dendritic cell transdifferentiation into osteoclasts: A novel pathway sustained by the rheumatoid arthritis microenvironment. Blood (2004) 104(13):4029–37. doi: 10.1182/blood-2004-01-0041
35. Krishnamurthy A, Ytterberg AJ, Sun M, Sakuraba K, Steen J, Joshua V, et al. Citrullination controls dendritic cell transdifferentiation into osteoclasts. J Immunol (2019) 202(11):3143–50. doi: 10.4049/jimmunol.1800534
36. Iwamoto N, Fukui S, Takatani A, Shimizu T, Umeda M, Nishino A, et al. Osteogenic differentiation of fibroblast-like synovial cells in rheumatoid arthritis is induced by microRNA-218 through a ROBO/Slit pathway. Arthritis Res Ther (2018) 20(1):189. doi: 10.1186/s13075-018-1703-z
37. Ji L, Li X, He S, Chen S. Regulation of osteoclast-mediated bone resorption by microRNA. Cell Mol Life Sci (2022) 79(6):287. doi: 10.1007/s00018-022-04298-y
38. He X, Wu F, Zhou Y, Wu J, Long Y, Zhou X. miR-4463 regulates hypoxia-induced autophagy and apoptosis by targeting ULK1 in endothelial cells. Front Biosci (Landmark Ed) (2022) 27(6):175. doi: 10.31083/j.fbl2706175
39. Duroux-Richard I, Pers YM, Fabre S, Ammari M, Baeten D, Cartron G, et al. Circulating miRNA-125b is a potential biomarker predicting response to rituximab in rheumatoid arthritis. Mediators Inflammation (2014) 2014:342524. doi: 10.1155/2014/342524
40. Cheng P, Wang J. The potential of circulating microRNA-125a and microRNA-125b as markers for inflammation and clinical response to infliximab in rheumatoid arthritis patients. J Clin Lab Anal (2020) 34(8):e23329. doi: 10.1002/jcla.23329
41. Banerjee S, Cui H, Xie N, Tan Z, Yang S, Icyuz M, et al. miR-125a-5p regulates differential activation of macrophages and inflammation. J Biol Chem (2013) 288(49):35428–36. doi: 10.1074/jbc.M112.426866
42. Zhao JL, Huang F, He F, Gao CC, Liang SQ, Ma PF, et al. Forced activation of notch in macrophages represses tumor growth by upregulating miR-125a and disabling tumor-associated macrophages. Cancer Res (2016) 76(6):1403–15. doi: 10.1158/0008-5472.CAN-15-2019
43. de la Rica L, Garcia-Gomez A, Comet NR, Rodriguez-Ubreva J, Ciudad L, Vento-Tormo R, et al. NF-kappaB-direct activation of microRNAs with repressive effects on monocyte-specific genes is critical for osteoclast differentiation. Genome Biol (2015) 16:2. doi: 10.1186/s13059-014-0561-5
44. Sun L, Lian JX, Meng S. MiR-125a-5p promotes osteoclastogenesis by targeting TNFRSF1B. Cell Mol Biol Lett (2019) 24:23. doi: 10.1186/s11658-019-0146-0
45. Stanczyk J, Pedrioli DM, Brentano F, Sanchez-Pernaute O, Kolling C, Gay RE, et al. Altered expression of MicroRNA in synovial fibroblasts and synovial tissue in rheumatoid arthritis. Arthritis Rheum (2008) 58(4):1001–9. doi: 10.1002/art.23386
46. Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA (2006) 103(33):12481–6. doi: 10.1073/pnas.0605298103
47. Nakasa T, Shibuya H, Nagata Y, Niimoto T, Ochi M. The inhibitory effect of microRNA-146a expression on bone destruction in collagen-induced arthritis. Arthritis Rheum (2011) 63(6):1582–90. doi: 10.1002/art.30321
48. Ammari M, Presumey J, Ponsolles C, Roussignol G, Roubert C, Escriou V, et al. Delivery of miR-146a to Ly6C(high) monocytes inhibits pathogenic bone erosion in inflammatory arthritis. Theranostics (2018) 8(21):5972–85. doi: 10.7150/thno.29313
49. Lee HM, Kim TS, Jo EK. MiR-146 and miR-125 in the regulation of innate immunity and inflammation. BMB Rep (2016) 49(6):311–8. doi: 10.5483/BMBRep.2016.49.6.056
50. Zhang B, Wang LS, Zhou YH. RETRACTED: Elevated microRNA-125b promotes inflammation in rheumatoid arthritis by activation of NF-kappaB pathway. BioMed Pharmacother (2017) 93:1151–7. doi: 10.1016/j.biopha.2017.07.042
51. Shibuya H, Nakasa T, Adachi N, Nagata Y, Ishikawa M, Deie M, et al. Overexpression of microRNA-223 in rheumatoid arthritis synovium controls osteoclast differentiation. Mod Rheumatol (2013) 23(4):674–85. doi: 10.3109/s10165-012-0710-1
52. Li YT, Chen SY, Wang CR, Liu MF, Lin CC, Jou IM, et al. Brief report: amelioration of collagen-induced arthritis in mice by lentivirus-mediated silencing of microRNA-223. Arthritis Rheum (2012) 64(10):3240–5. doi: 10.1002/art.34550
53. Nakamachi Y, Kawano S, Takenokuchi M, Nishimura K, Sakai Y, Chin T, et al. MicroRNA-124a is a key regulator of proliferation and monocyte chemoattractant protein 1 secretion in fibroblast-like synoviocytes from patients with rheumatoid arthritis. Arthritis Rheum (2009) 60(5):1294–304. doi: 10.1002/art.24475
54. Nakamachi Y, Ohnuma K, Uto K, Noguchi Y, Saegusa J, Kawano S. MicroRNA-124 inhibits the progression of adjuvant-induced arthritis in rats. Ann Rheum Dis (2016) 75(3):601–8. doi: 10.1136/annrheumdis-2014-206417
55. Axmann R, Herman S, Zaiss M, Franz S, Polzer K, Zwerina J, et al. CTLA-4 directly inhibits osteoclast formation. Ann Rheum Dis (2008) 67(11):1603–9. doi: 10.1136/ard.2007.080713
56. Cutolo M, Soldano S, Gotelli E, Montagna P, Campitiello R, Paolino S, et al. CTLA4-ig treatment induces M1-M2 shift in cultured monocyte-derived macrophages from healthy subjects and rheumatoid arthritis patients. Arthritis Res Ther (2021) 23(1):306. doi: 10.1186/s13075-021-02691-9
57. Okada H, Kajiya H, Omata Y, Matsumoto T, Sato Y, Kobayashi T, et al. CTLA4-ig directly inhibits osteoclastogenesis by interfering with intracellular calcium oscillations in bone marrow macrophages. J Bone Miner Res (2019) 34(9):1744–52. doi: 10.1002/jbmr.3754
58. Sun Y, Li J, Xie X, Gu F, Sui Z, Zhang K, et al. Macrophage-osteoclast associations: Origin, polarization, and subgroups. Front Immunol (2021) 12:778078. doi: 10.3389/fimmu.2021.778078
59. Segawa Y, Yamaura M, Aota S, Omata T, Tuzuike N, Itokazu Y, et al. Methotrexate maintains bone mass by preventing both a decrease in bone formation and an increase in bone resorption in adjuvant-induced arthritic rats. Bone (1997) 20(5):457–64. doi: 10.1016/S8756-3282(97)00023-9
60. Kanagawa H, Masuyama R, Morita M, Sato Y, Niki Y, Kobayashi T, et al. Methotrexate inhibits osteoclastogenesis by decreasing RANKL-induced calcium influx into osteoclast progenitors. J Bone Miner Metab (2016) 34(5):526–31. doi: 10.1007/s00774-015-0702-2
61. Smolen JS, Han C, Bala M, Maini RN, Kalden JR, van der Heijde D, et al. Evidence of radiographic benefit of treatment with infliximab plus methotrexate in rheumatoid arthritis patients who had no clinical improvement: a detailed subanalysis of data from the anti-tumor necrosis factor trial in rheumatoid arthritis with concomitant therapy study. Arthritis Rheum (2005) 52(4):1020–30. doi: 10.1002/art.20982
62. Saidenberg-Kermanac’h N, Corrado A, Lemeiter D, deVernejoul MC, Boissier MC, Cohen-Solal ME. TNF-alpha antibodies and osteoprotegerin decrease systemic bone loss associated with inflammation through distinct mechanisms in collagen-induced arthritis. Bone (2004) 35(5):1200–7. doi: 10.1016/j.bone.2004.07.004
63. Takeuchi T, Yoshida H, Tanaka S. Role of interleukin-6 in bone destruction and bone repair in rheumatoid arthritis. Autoimmun Rev (2021) 20(9):102884. doi: 10.1016/j.autrev.2021.102884
64. Liu H, Feng W, Yimin Cui J, Lv S, Hasegawa T, Sun B, et al. Histological evidence of increased osteoclast cell number and asymmetric bone resorption activity in the tibiae of interleukin-6-Deficient mice. J Histochem Cytochem (2014) 62(8):556–64. doi: 10.1369/0022155414537830
65. Cohen SB, Dore RK, Lane NE, Ory PA, Peterfy CG, Sharp JT, et al. Denosumab treatment effects on structural damage, bone mineral density, and bone turnover in rheumatoid arthritis: a twelve-month, multicenter, randomized, double-blind, placebo-controlled, phase II clinical trial. Arthritis Rheum (2008) 58(5):1299–309. doi: 10.1002/art.23417
66. Takeuchi T, Tanaka Y, Soen S, Yamanaka H, Yoneda T, Tanaka S, et al. Effects of the anti-RANKL antibody denosumab on joint structural damage in patients with rheumatoid arthritis treated with conventional synthetic disease-modifying antirheumatic drugs (DESIRABLE study): a randomised, double-blind, placebo-controlled phase 3 trial. Ann Rheum Dis (2019) 78(7):899–907. doi: 10.1136/annrheumdis-2018-214827
67. Bozec A, Zaiss MM, Kagwiria R, Voll R, Rauh M, Chen Z, et al. T Cell costimulation molecules CD80/86 inhibit osteoclast differentiation by inducing the IDO/tryptophan pathway. Sci Transl Med (2014) 6(235):235ra60. doi: 10.1126/scitranslmed.3007764
68. Matsuura Y, Kikuta J, Kishi Y, Hasegawa T, Okuzaki D, Hirano T, et al. In vivo visualisation of different modes of action of biological DMARDs inhibiting osteoclastic bone resorption. Ann Rheum Dis (2018) 77(8):1219–25. doi: 10.1136/annrheumdis-2017-212880
69. Kamijo S, Nakajima A, Ikeda K, Aoki K, Ohya K, Akiba H, et al. Amelioration of bone loss in collagen-induced arthritis by neutralizing anti-RANKL monoclonal antibody. Biochem Biophys Res Commun (2006) 347(1):124–32. doi: 10.1016/j.bbrc.2006.06.098
70. Massalska M, Maslinski W, Ciechomska M. Small molecule inhibitors in the treatment of rheumatoid arthritis and beyond: Latest updates and potential strategy for fighting COVID-19. Cells (2020) 9(8):1876. doi: 10.3390/cells9081876
71. Schwartz DM, Bonelli M, Gadina M, O’Shea JJ. Type I/II cytokines, JAKs, and new strategies for treating autoimmune diseases. Nat Rev Rheumatol (2016) 12(1):25–36. doi: 10.1038/nrrheum.2015.167
72. Adam S, Simon N, Steffen U, Andes FT, Scholtysek C, Muller DIH, et al. JAK inhibition increases bone mass in steady-state conditions and ameliorates pathological bone loss by stimulating osteoblast function. Sci Transl Med (2020) 12(530):eaay4447. doi: 10.1126/scitranslmed.aay4447
73. Murakami K, Kobayashi Y, Uehara S, Suzuki T, Koide M, Yamashita T, et al. A Jak1/2 inhibitor, baricitinib, inhibits osteoclastogenesis by suppressing RANKL expression in osteoblasts. Vitro PloS One (2017) 12(7):e0181126. doi: 10.1371/journal.pone.0181126
74. Volin MV, Huynh N, Klosowska K, Chong KK, Woods JM. Fractalkine is a novel chemoattractant for rheumatoid arthritis fibroblast-like synoviocyte signaling through MAP kinases and akt. Arthritis Rheum (2007) 56(8):2512–22. doi: 10.1002/art.22806
75. Tanaka Y, Takeuchi T, Umehara H, Nanki T, Yasuda N, Tago F, et al. Safety, pharmacokinetics, and efficacy of E6011, an antifractalkine monoclonal antibody, in a first-in-patient phase 1/2 study on rheumatoid arthritis. Mod Rheumatol (2018) 28(1):58–65. doi: 10.1080/14397595.2017.1337056
76. Tanaka Y, Takeuchi T, Yamanaka H, Nanki T, Umehara H, Yasuda N, et al. Efficacy and safety of E6011, an anti-fractalkine monoclonal antibody, in patients with active rheumatoid arthritis with inadequate response to methotrexate: Results of a randomized, double-blind, placebo-controlled phase II study. Arthritis Rheumatol (2021) 73(4):587–95. doi: 10.1002/art.41555
77. Tanaka Y, Takeuchi T, Yamanaka H, Nanki T, Umehara H, Yasuda N, et al. A phase 2 study of E6011, an anti-fractalkine monoclonal antibody, in patients with rheumatoid arthritis inadequately responding to biological disease-modifying antirheumatic drugs. Mod Rheumatol (2021) 31(4):783–9. doi: 10.1080/14397595.2020.1868675
78. Louis C, Souza-Fonseca-Guimaraes F, Yang Y, D'Silva D, Kratina T, Dagley L, et al. NK cell-derived GM-CSF potentiates inflammatory arthritis and is negatively regulated by CIS. J Exp Med (2020) 217(5):e20191421. doi: 10.1084/jem.20191421
79. Lee MS, Kim HS, Yeon JT, Choi SW, Chun CH, Kwak HB, et al. GM-CSF regulates fusion of mononuclear osteoclasts into bone-resorbing osteoclasts by activating the Ras/ERK pathway. J Immunol (2009) 183(5):3390–9. doi: 10.4049/jimmunol.0804314
80. Behrens F, Tak PP, Ostergaard M, Stoilov R, Wiland P, Huizinga TW, et al. MOR103, a human monoclonal antibody to granulocyte-macrophage colony-stimulating factor, in the treatment of patients with moderate rheumatoid arthritis: results of a phase Ib/IIa randomised, double-blind, placebo-controlled, dose-escalation trial. Ann Rheum Dis (2015) 74(6):1058–64. doi: 10.1136/annrheumdis-2013-204816
81. van Nieuwenhuijze A, Koenders M, Roeleveld D, Sleeman MA, van den Berg W, Wicks IP. GM-CSF as a therapeutic target in inflammatory diseases. Mol Immunol (2013) 56(4):675–82. doi: 10.1016/j.molimm.2013.05.002
82. Burmester GR, McInnes IB, Kremer JM, Miranda P, Vencovsky J, Godwood A, et al. Mavrilimumab, a fully human granulocyte-macrophage colony-stimulating factor receptor alpha monoclonal antibody: Long-term safety and efficacy in patients with rheumatoid arthritis. Arthritis Rheumatol (2018) 70(5):679–89. doi: 10.1002/art.40420
83. Hayashi M, Nakashima T, Taniguchi M, Kodama T, Kumanogoh A, Takayanagi H. Osteoprotection by semaphorin 3A. Nature (2012) 485(7396):69–74. doi: 10.1038/nature11000
84. Koga T, Umeda M, Yoshida N, Satyam A, Jha M, Scherlinger M, et al. Inhibition of calcium/calmodulin-dependent protein kinase IV in arthritis: Dual effect on Th17 cell activation and osteoclastogenesis. Rheumatol (Oxford) (2022):keac381. doi: 10.1093/rheumatology/keac381
85. Gao W, Sweeney C, Connolly M, Kennedy A, Ng CT, McCormick J, et al. Notch-1 mediates hypoxia-induced angiogenesis in rheumatoid arthritis. Arthritis Rheum (2012) 64(7):2104–13. doi: 10.1002/art.34397
86. Muraoka S, Kaneko K, Motomura K, Nishio J, Nanki T. CX3CL1/fractalkine regulates the differentiation of human peripheral blood monocytes and monocyte-derived dendritic cells into osteoclasts. Cytokine (2021) 146:155652. doi: 10.1016/j.cyto.2021.155652
87. Kim KW, Vallon-Eberhard A, Zigmond E, Farache J, Shezen E, Shakhar G, et al. In vivo structure/function and expression analysis of the CX3C chemokine fractalkine. Blood (2011) 118(22):e156–67. doi: 10.1182/blood-2011-04-348946
88. Matsuura T, Ichinose S, Akiyama M, Kasahara Y, Tachikawa N, Nakahama KI. Involvement of CX3CL1 in the migration of osteoclast precursors across osteoblast layer stimulated by interleukin-1ss. J Cell Physiol (2017) 232(7):1739–45. doi: 10.1002/jcp.25577
89. Koizumi K, Saitoh Y, Minami T, Takeno N, Tsuneyama K, Miyahara T, et al. Role of CX3CL1/fractalkine in osteoclast differentiation and bone resorption. J Immunol (2009) 183(12):7825–31. doi: 10.4049/jimmunol.0803627
90. Matsunawa M, Isozaki T, Odai T, Yajima N, Takeuchi HT, Negishi M, et al. Increased serum levels of soluble fractalkine (CX3CL1) correlate with disease activity in rheumatoid vasculitis. Arthritis Rheum (2006) 54(11):3408–16. doi: 10.1002/art.22208
91. Xu WD, Firestein GS, Taetle R, Kaushansky K, Zvaifler NJ. Cytokines in chronic inflammatory arthritis. II. granulocyte-macrophage colony-stimulating factor in rheumatoid synovial effusions. J Clin Invest (1989) 83(3):876–82. doi: 10.1172/JCI113971
92. Hartong DT, Dange M, McGee TL, Berson EL, Dryja TP, Colman RF. Insights from retinitis pigmentosa into the roles of isocitrate dehydrogenases in the Krebs cycle. Nat Genet (2008) 40(10):1230–4. doi: 10.1038/ng.223
93. Cook AD, Braine EL, Campbell IK, Rich MJ, Hamilton JA. Blockade of collagen-induced arthritis post-onset by antibody to granulocyte-macrophage colony-stimulating factor (GM-CSF): requirement for GM-CSF in the effector phase of disease. Arthritis Res (2001) 3(5):293–8. doi: 10.1186/ar318
94. Achuthan A, Cook AD, Lee MC, Saleh R, Khiew HW, Chang MW, et al. Granulocyte macrophage colony-stimulating factor induces CCL17 production via IRF4 to mediate inflammation. J Clin Invest (2016) 126(9):3453–66. doi: 10.1172/JCI87828
95. Xu R. Semaphorin 3A: A new player in bone remodeling. Cell Adh Migr (2014) 8(1):5–10. doi: 10.4161/cam.27752
96. Teng Y, Yin Z, Li J, Li K, Li X, Zhang Y. Adenovirus-mediated delivery of Sema3A alleviates rheumatoid arthritis in a serum-transfer induced mouse model. Oncotarget (2017) 8(39):66270–80. doi: 10.18632/oncotarget.19915
97. Negishi-Koga T, Shinohara M, Komatsu N, Bito H, Kodama T, Friedel RH, et al. Suppression of bone formation by osteoclastic expression of semaphorin 4D. Nat Med (2011) 17(11):1473–80. doi: 10.1038/nm.2489
98. Yoshida Y, Ogata A, Kang S, Ebina K, Shi K, Nojima S, et al. Semaphorin 4D contributes to rheumatoid arthritis by inducing inflammatory cytokine production: Pathogenic and therapeutic implications. Arthritis Rheumatol (2015) 67(6):1481–90. doi: 10.1002/art.39086
99. Xu WD, Huang AF. Hypoxia-induced synovial fibroblast activation in inflammatory arthritis and the role of notch-1 and notch-3 signaling: Comment on the article by Chen et al. Arthritis Rheumatol (2021) 73(12):2349–50. doi: 10.1002/art.41909
100. Ando K, Kanazawa S, Tetsuka T, Ohta S, Jiang X, Tada T, et al. Induction of notch signaling by tumor necrosis factor in rheumatoid synovial fibroblasts. Oncogene (2003) 22(49):7796–803. doi: 10.1038/sj.onc.1206965
101. Ishii H, Nakazawa M, Yoshino S, Nakamura H, Nishioka K, Nakajima T. Expression of notch homologues in the synovium of rheumatoid arthritis and osteoarthritis patients. Rheumatol Int (2001) 21(1):10–4. doi: 10.1007/s002960100119
102. Ashley JW, Ahn J, Hankenson KD. Notch signaling promotes osteoclast maturation and resorptive activity. J Cell Biochem (2015) 116(11):2598–609. doi: 10.1002/jcb.25205
Keywords: monocyte, osteoclast, rheumatoid arthritis, disease modifying anti-rheumatic drugs (DMARD), microRNA
Citation: Iwamoto N and Kawakami A (2022) The monocyte-to-osteoclast transition in rheumatoid arthritis: Recent findings. Front. Immunol. 13:998554. doi: 10.3389/fimmu.2022.998554
Received: 20 July 2022; Accepted: 24 August 2022;
Published: 12 September 2022.
Edited by:
Tsutomu Takeuchi, Keio University School of Medicine, JapanReviewed by:
Masaru Ishii, Osaka University, JapanCopyright © 2022 Iwamoto and Kawakami. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Naoki Iwamoto, bmFva2ktaXdhQG5hZ2FzYWtpLXUuYWMuanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.