 Arailym Sarsembayeva1†
Arailym Sarsembayeva1† Melanie Kienzl1†Eva Gruden1†
Melanie Kienzl1†Eva Gruden1† Dusica Ristic1†
Dusica Ristic1† Kathrin Maitz1
Kathrin Maitz1 Paulina Valadez-Cosmes1†Ana Santiso1†Carina Hasenoehrl1
Paulina Valadez-Cosmes1†Ana Santiso1†Carina Hasenoehrl1 Luka Brcic2†
Luka Brcic2† Jörg Lindenmann3†
Jörg Lindenmann3† Julia Kargl1†
Julia Kargl1† Rudolf Schicho1,4*†
Rudolf Schicho1,4*†- 1Division of Pharmacology, Otto Loewi Research Center, Medical University of Graz, Graz, Austria
- 2Diagnostic and Research Institute of Pathology, Medical University of Graz, Graz, Austria
- 3Division of Thoracic and Hyperbaric Surgery, Department of Surgery, Medical University of Graz, Graz, Austria
- 4BioTechMed, Graz, Austria
Cannabinoid (CB) receptors (CB1 and CB2) are expressed on cancer cells and their expression influences carcinogenesis in various tumor entities. Cells of the tumor microenvironment (TME) also express CB receptors, however, their role in tumor development is still unclear. We, therefore, investigated the role of TME-derived CB1 and CB2 receptors in a model of non-small cell lung cancer (NSCLC). Leukocytes in the TME of mouse and human NSCLC express CB receptors, with CB2 showing higher expression than CB1. In the tumor model, using CB1- (CB1-/-) and CB2-knockout (CB2-/-) mice, only deficiency of CB2, but not of CB1, resulted in reduction of tumor burden vs. wild type (WT) littermates. This was accompanied by increased accumulation and tumoricidal activity of CD8+ T and natural killer cells, as well as increased expression of programmed death-1 (PD-1) and its ligand on lymphoid and myeloid cells, respectively. CB2-/- mice responded significantly better to anti-PD-1 therapy than WT mice. The treatment further increased infiltration of cytotoxic lymphocytes into the TME of CB2-/- mice. Our findings demonstrate that TME-derived CB2 dictates the immune cell recruitment into tumors and the responsiveness to anti-PD-1 therapy in a model of NSCLC. CB2 could serve as an adjuvant target for immunotherapy.
Introduction
Cannabinoid (CB) receptors CB1 and CB2 are widely found in human tumor tissue and are well-known to influence the growth of tumor cells (1). However, whether they act as tumor promotors or suppressors, and whether CB receptors located in cancer cells or/and in immune cells of the tumor microenvironment (TME) are involved in tumor progression, is less clear. In particular, CB receptors could significantly influence the development of lung cancer, as suggested by previous studies of non-small cell lung cancer (NSCLC) (2, 3). Some studies show that agonists of CB1 and/or CB2 attenuate the carcinogenic potential in lung cancer cells (2, 4–6), and reduce tumor growth in immunodeficient (7) and FVB/N mice (8), however, other studies report the opposite. For instance, CB1/CB2 agonist tetrahydrocannabinol (THC) may promote proliferation of lung cancer cells (9) and the growth of breast cancer in vivo (10). In addition, silencing of CB2 in lung cancer cells reportedly decreases their proliferation, migration, and invasion (3). A number of studies on the prognostic value of CB expression revealed discrepant findings based on the cancer type (reviewed in (11)). While some articles described high expression of CB1/CB2 receptors in human samples of NSCLC correlating with prolonged survival (2), others described a positive correlation of CB2 expression with increased tumor size and pathological grading of NSCLC (3), indicating a complex and still unclear role of CB receptors in NSCLC.
CB1 and CB2 receptors are part of the endocannabinoid system (ECS), acting in concert with their endogenous ligands (endocannabinoids) and enzymes for synthesis and degradation of these ligands (12, 13). CB1 is abundantly expressed in the central nervous system (14), but is also detectable in peripheral tissues including the immune system (15, 16). The majority of immune cells express CB1 at low levels, and its expression is generally affected by the activation status and cell type, as well as the presence of immune stimuli and endocannabinoids (17). In contrast, CB2 is highly expressed in immune cells, and controls functions such as proliferation, migration, activity, cytokine release, antigen presentation, and antibody production (15, 18). The receptor has previously been described for its immunosuppressive behavior (15, 19). For instance, in plaque-forming cell assays in mouse splenocytes (which measure the capacity of the spleen cells to mount a primary antibody response to sheep red blood cells), THC could directly inhibit the cells via CB2 (20). In addition, the endocannabinoid anandamide suppresses release of pro-inflammatory cytokines like IL-2, TNF-α and IFN-γ from activated human peripheral T-lymphocytes, acting primarily through CB2 (21). These effects can be mimicked by the CB2 agonist JWH-015, and blocked by the CB2 antagonist SR144528 (22). Cannabinoids have been reported to reduce natural killer cell (NK) activity, thus, in vivo administration of THC in male Swiss mice results in inhibition of splenic NK cytolytic activity without altering proliferation of splenocytes (23). Also, in human NK cells, THC has been demonstrated to reduce cytolytic activity (reviewed in Braile et al. (24)). CB2 has previously been suggested to play a key role in suppressing immune activity in cancer, a concept supported by Zhu et al., who showed that CB2 controls tumor immunity of lung cancer by increasing the levels of Th2 cytokines like IL-10 and TGF, and by downregulating the Th1 cytokine IFN-γ (10).
Based on their well-described impact on immune cells, CB receptors could significantly influence immune cell behavior and regulatory components of immune activity, including inhibitory checkpoint proteins like programmed death-1 (PD-1) and its ligand PD-L1, within the TME. PD-1 is an inhibitory receptor expressed on T cells after antigen stimulation, while PD-L1 is found on tumor cells and antigen presenting cells (25). Particularly, in NSCLC, the PD-1/PD-L1 axis has emerged as a successful target for the use of immune checkpoint inhibitors (ICI). However, limited response rates and resistance have hampered their success (26), warranting the discovery of new targets to boost ICI therapy. In this regard, clinical trials using combination therapies of ICIs with anti-angiogenic agents, chemotherapy, ataxia telangiectasia and Rad3-related (ATR) kinase and mitogen-activated protein kinase kinase (MEK) inhibitors, have been conducted or are still ongoing (reviewed in Blach et al. (26)).
In the present study, we investigated whether CB receptors located in the TME control tumor growth and influence susceptibility to ICI treatment. To investigate our hypothesis, we used a mouse model of NSCLC, in which immunocompetent wild type (WT) and CB1-knockout (CB1-/-) or CB2-knockout (CB2-/-) mice received a subcutaneous (s.c.) injection of syngeneic lung adenocarcinoma cells (KP cells (27)), thus creating a tumor model with TME cells that either express or lack CB receptor. We report that tumors in CB2-/- mice are smaller than in their WT littermates, and that CB2-/- mice respond better to anti-PD-1 therapy, indicating that CB2 expression in the TME is a critical determinant of immune suppression in this NSCLC model.
Results
Tumor and TME cells express CB receptors in situ, and blockade of CB2, and not CB1, inhibits tumor growth in a murine NSCLC model
As the role of TME-derived CB receptors in lung cancer has not yet been investigated, we aimed to identify whether TME host cells lacking CB1 or CB2 would influence primary tumor growth. After injecting KP cells s.c. into the flanks of CB1-/-, CB2-/-, and WT mice, ex vivo measurement of tumor weight and volume demonstrated that tumor burden of CB1-/- mice did not differ from WTs in our mouse model (Figure 1A). In contrast, mice devoid of CB2 showed more than 50% reduction in both tumor weight and volume, as compared to WT littermates (Figure 1B). We then investigated whether pharmacological blockade of CB receptors in tumor-bearing C57BL/6J mice could replicate findings obtained in knockout mice using previously tested doses of CB1 antagonist SR141716 (28, 29) and CB2 antagonist SR144528 (29, 30). As a result, treatment with CB1 antagonist SR141716 had no effect on both tumor weight and volume (Figure 1C), whereas tumor-bearing C57BL/6J mice treated with CB2 antagonist SR144528 showed a significant reduction in tumor weight and volume as compared to vehicle-treated animals (Figure 1D).
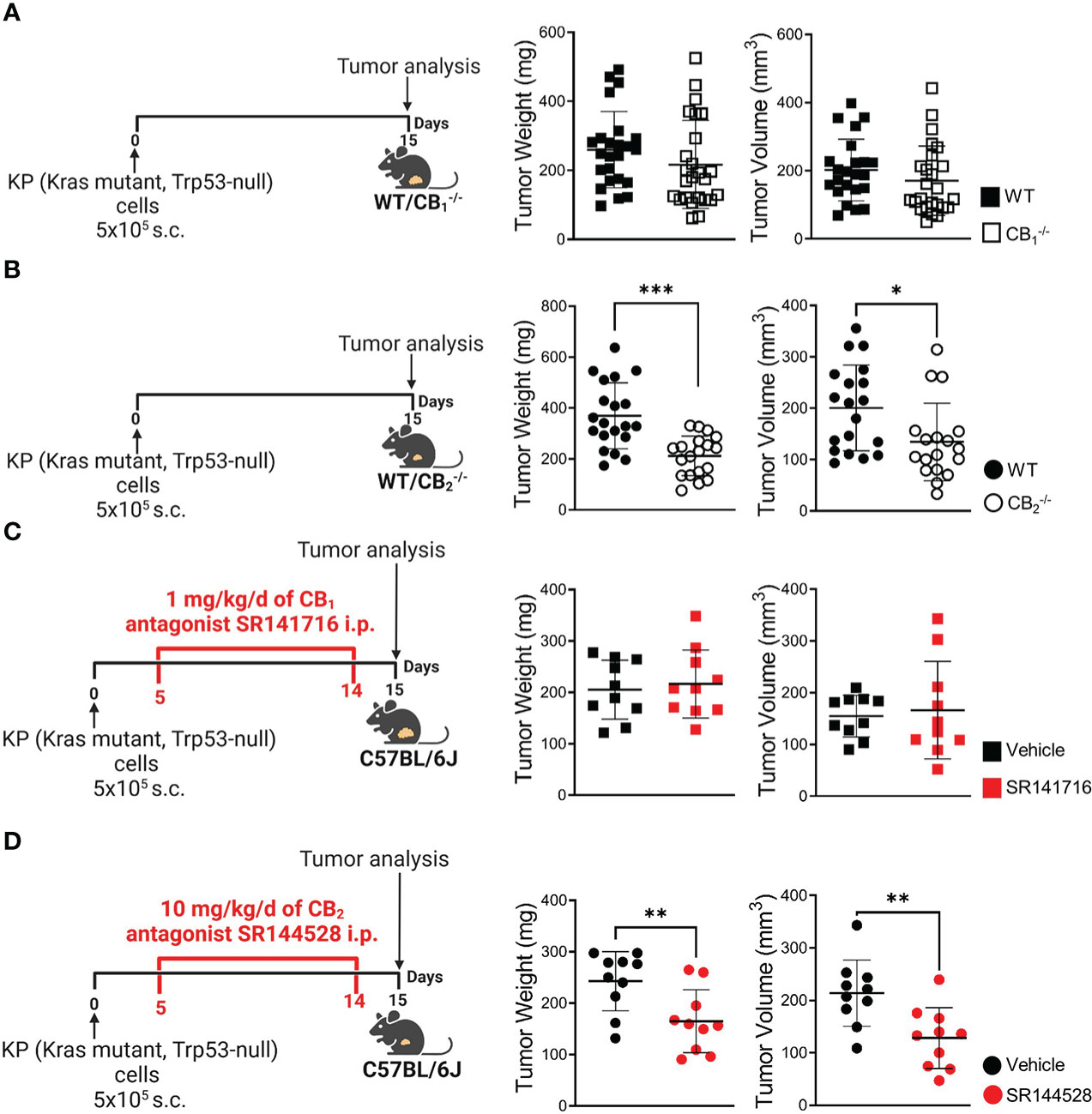
Figure 1 Blockade of CB2, but not CB1, inhibits tumor growth in a mouse model of NSCLC. (A) Experimental design: CB1-/- mice and wild type (WT) littermates were subcutaneously (s.c.) injected with 5x105 KP (Kras mutant, Trp53-null) lung adenocarcinoma cells on day 0. On day 15, tumors were measured ex vivo and harvested for analysis. Data indicate mean values ± SD from three pooled independent experiments. n= 23-25. (B) Experimental design: CB2-/- mice and WT littermates were s.c. injected with 5x105 KP lung adenocarcinoma cells on day 0. On day 15, tumors were measured ex vivo and collected for analysis. Data indicate mean values ± SD from two pooled independent experiments. n= 18-20. (C, D) Experimental design: C57BL/6J WT mice were s.c. injected with 5x105 KP lung adenocarcinoma cells on day 0. Five-days post-inoculation, KP cell tumor-bearing mice started receiving intraperitoneal (i.p.) injections of either (C) 1 mg/kg/d of CB1 antagonist SR141716 or (D) 10 mg/kg/d of CB2 antagonist SR144528 (or vehicle). On day 15, tumor weight and volume were measured ex vivo. One representative experiment is shown. Data indicate mean values ± SD, n= 9-10. All statistical differences were evaluated by using unpaired student`s t-test (A–D). *p < .05; **p < .01; ***p < .001. NSCLC, non-small cell lung cancer.
To further investigate the role of CB receptors in the TME, we identified mRNA expression of these receptors in tumor cells and infiltrating immune cells in situ. We used in situ hybridization (ISH) technique with specific probes against CB1 and CB2 mRNA in combination with immunofluorescence (IF). Dual ISH-IF analysis displayed CB1 expression in cancer cells as well as immune cells of the TME, but to a far lesser extent than expression of CB2 (Figure 2A). Around 25% of tumor cells (which positively stained for cytokeratin) co-localized with CB2 mRNA (Figure 2B). Within the TME, we detected CB2 mRNA expression in CD3+ T cells, CD8+ T cells, NKp46/NCR1+ cells, CD163+ or F4/80+ macrophages, and CD11b+ cells. Co-localizations ranged between ~20-40% (Figure 2B).
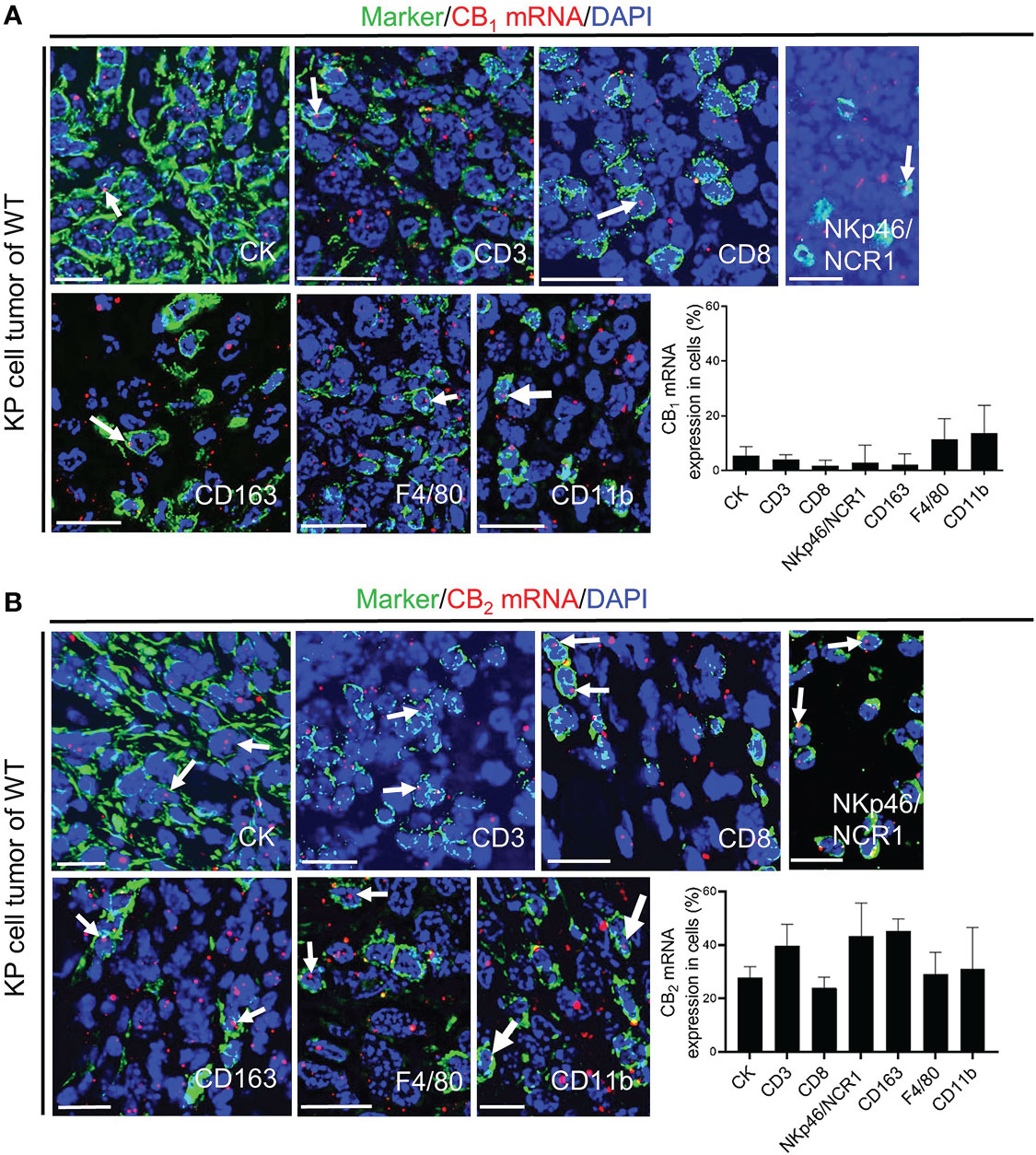
Figure 2 CB1 and CB2 mRNA in tumor cells and immune cells of the TME. (A, B) In situ hybridization (ISH)/immunofluorescence (IF) of tumor/immune cells in KP cell tumor sections from wild type mice. (A) The graph demonstrates the percentages of co-localization of CB1 mRNA positive signals with tumor cells (cytokeratin-stained, CK+ cells; ~ 5%) and leukocytes of the TME, such as CD3+ T cells (~ 4%), CD8+ T cells (~ 3%), NKp46/NCR1+ cells (natural killer, NK cells; ~ 14%), CD163+ M2 macrophages (~ 7%), F4/80+ M1 and M2 macrophages (~ 11%), and CD11b+ myeloid cells (~ 14%). (B) The graph shows the percentages of co-localization of CB2 mRNA signals with tumor cells (~ 25%) and tumor-infiltrating immune cells, including CD3+ T cells (~ 39%), CD8+ T cells (~ 24%), NKp46/NCR1+ NK cells (~ 43%), CD163+ M2 macrophages (~ 43%), F4/80+ M1 and M2 macrophages (~ 29%), and CD11b+ myeloid cells (~ 29%). Arrows denote CB1 or CB2 ISH mRNA signals within tumor and immune cells. Calibration bars=20 μm. Data indicate mean values +SD; n=3 (sections from three different tumors, 30-150 cells counted per section). TME, tumor microenvironment.
Since several studies described CB receptor expression in tumors of NSCLC patients (2, 3, 7), we stained sections of human lung cancer tissues to assess the distribution of CB1 and CB2 receptors in tumor cells and infiltrating immune cells, and also applied flow cytometry in freshly resected NSCLC tissues. In line with our mouse data, CB1 and CB2 expression were not only seen in lung cancer cells, but also in infiltrated immune cells, such as CD3+ T and CD8+ T cells, NKp46/NCR1+or CD56+ NK cells, and CD163+ macrophages. Expression of CB2 was generally higher than that of CB1 (Figures 3A, B, S2A).
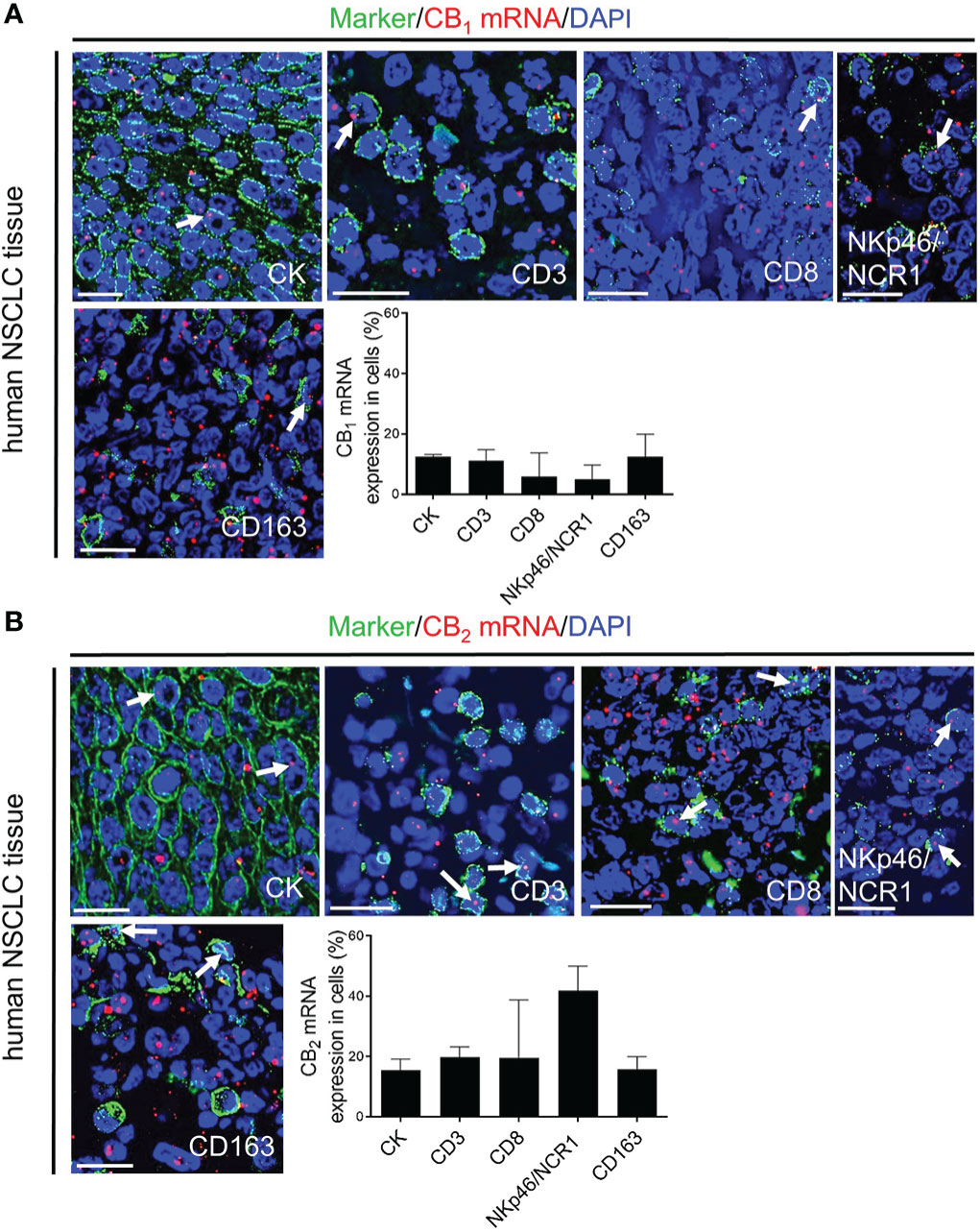
Figure 3 In situ hybridization (ISH)/immunofluorescence (IF) of human NSCLC tissue sections (A, B) Representative fluorescence microscopy images of human NSCLC tissue sections. The graphs show the percentages of co-localization of CB1 and CB2 mRNA signals with tumor cells (cytokeratin-stained, CK+ cells) as well as tumor-infiltrating immune cells (CD3+ T cells, CD8+ T cells, NKp46/NCR1+ NK cells, and CD163+ M2 macrophages). Arrows indicate CB1 and CB2 ISH signals within tumor and immune cells of the TME. Calibration bars = 20 μm. Data indicate mean values +SD. n=3 (tumor sections from three different patients with NSCLC were used for quantification, 30-150 cells counted per section). NSCLC, non-small cell lung cancer; NK, natural killer cells; TME, tumor microenvironment.
These results indicate that CB1 and CB2 is expressed in both tumor and tumor-infiltrated immune cells, however, only deletion of CB2 on host cells or systemic blockade of CB2, but not of CB1, results in a reduction of tumor burden. To validate our results from the KP cell tumor model, we used Lewis lung carcinoma (LLC1) cells in CB2-/- vs. WT mice and identified that tumor burden was significantly reduced in CB2-/- mice when compared to WT mice (Figure S2C).
Tumor reduction exclusively relies on deletion of CB2 in TME host cells
According to dual ISH-IF, we found that besides immune cells, around 20-25% of tumor cells in human NSCLC (Figures 3B, S2A) and mouse tumor (Figures 4A, S2D) tissue co-localized with CB2 mRNA. According to RT-qPCR, tumors of WT mice showed higher levels of CB2 mRNA than those from CB2-/- mice, because host cells, such as immune cells infiltrating the TME in CB2-/- mice, are devoid of CB2 expression (Figure 4B). KP cells in culture cells expressed minimal levels of CB2 (Figures 4B, S2D). We confirmed the specificity of our CB2 PCR primers by absence of CB2 mRNA expression in spleen tissue of CB2-/- mice in comparison to WT mice (Figure S2E).
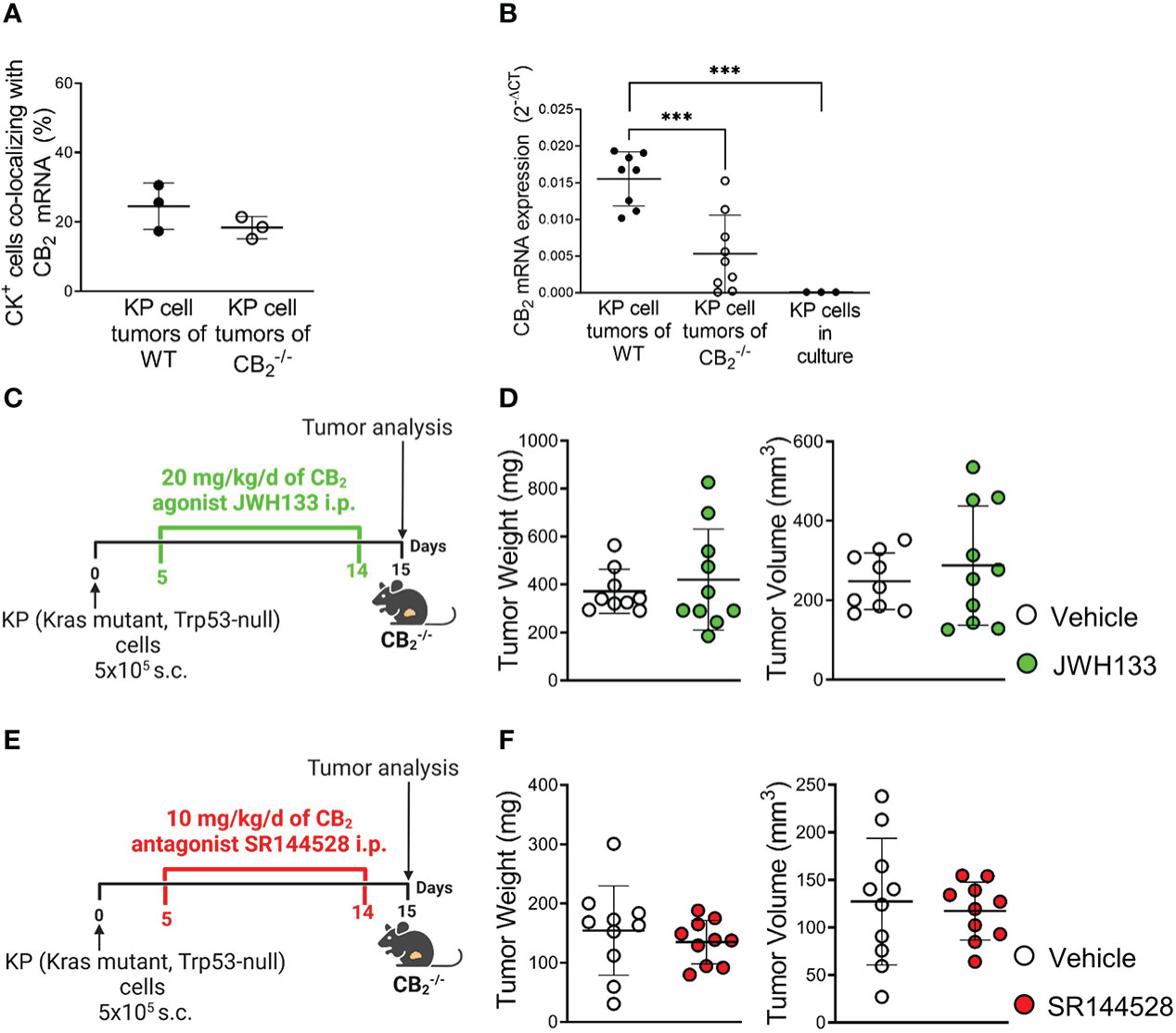
Figure 4 Tumor reduction exclusively relies on deletion of CB2 in TME host cells. (A) The graph depicts the percentage of CB2 mRNA positive cells co-localizing with cytokeratin-stained (CK+) tumor cells in mouse KP cell tumors, as evaluated by ISH-IF. Data indicate mean values ± SD. n=3/group (sections from three different tumors, 75-150 cells counted per section). (B) Relative CB2 mRNA expression as measured by qPCR in lysates from KP cell tumors from WT and CB2-/- mice, as well as KP cells in culture. Data indicate mean values ± SD. n≥8/group; n=3 (consecutive passages of KP cells). (C–F) Experimental design: CB2-/- mice were subcutaneously (s.c.) injected with 5x105 KP (Kras mutant, Trp53-null) lung adenocarcinoma cells on day 0. For ten days, CB2-/- mice were treated intraperitoneally (i.p.) with either (C) 20 mg/kg/d of CB2 agonist JWH133 or (E) 10mg/kg/d of CB2 antagonist SR144528 (or vehicle). Tumor weight and volume were measured at the end of the experiment ex vivo on day 15. One representative experiment is shown. Data indicate mean values ± SD. n≥9. Statistical differences were evaluated by using unpaired student`s t-test (A, D, F) or one-way ANOVA with Tukey’s multiple comparison test (B). ***p<.001. TME, tumor microenvironment; ISH/IF, in situ hybridization and immunofluorescence; WT, wild type.
To address the role of CB2-expressing KP cells on tumor growth in situ, we pharmacologically activated or blocked CB2 in tumor-bearing CB2-/- mice using a CB2 agonist (JWH133) (Figure 4C) or CB2 antagonist (SR144528) (Figure 4E) at previously published doses (29, 31). The results revealed that activation or inhibition of CB2 in tumor cells of tumor-bearing CB2-/- mice had no effect on tumor weight and volume (Figures 4D, F), indicating that the tumor reduction we observed in the CB2-/- mice solely depended on CB2, expressed in cells of the TME.
Knockout of CB2 in cells of the TME favors an anti-carcinogenic immune cell profile and enhances CD8+ T and NK cell activity
To determine the immune cell profile in tumors of CB2-/- and WT mice, we used flow cytometry and identified changes in infiltration of immune cells and their subtypes, observing a significant shift of lymphoid cell populations in CB2-/- as compared to WT mice (gating strategies shown in Figures S1A–C). There were no significant differences in the infiltration of CD45+ leukocytes and myeloid cells between tumors of CB2-/- and WT mice (Figures 5A, B, S3A). We, however, observed an increased infiltration of T cells (CD3+), NK cells (NKp46+), and CD8+ T cells (Figures 5C, D, S3B–D), but no differences in infiltration of CD4+ T and regulatory T cells (Tregs) into tumors of CB2-/- mice vs. WTs (Figure 5D). Within the CD8+ T cell population, the number of effector CD8+ T cells increased while naïve CD8+ T cells decreased (Figures 5E, S3E), indicating that CD8+ T cells from CB2-/-, but not from WT mice, were primed to become effector cells. Percentages of infiltrating CD8+ T (Figure 5F) as well as NK cells (Figure 5G) negatively correlated with tumor weight in CB2-/- mice. Furthermore, no significant changes in lymphoid immune cell composition, including T, B, NK, and NKT cells were seen in the spleens and lungs of healthy CB2-/- and WT mice (Figures S3F, G).
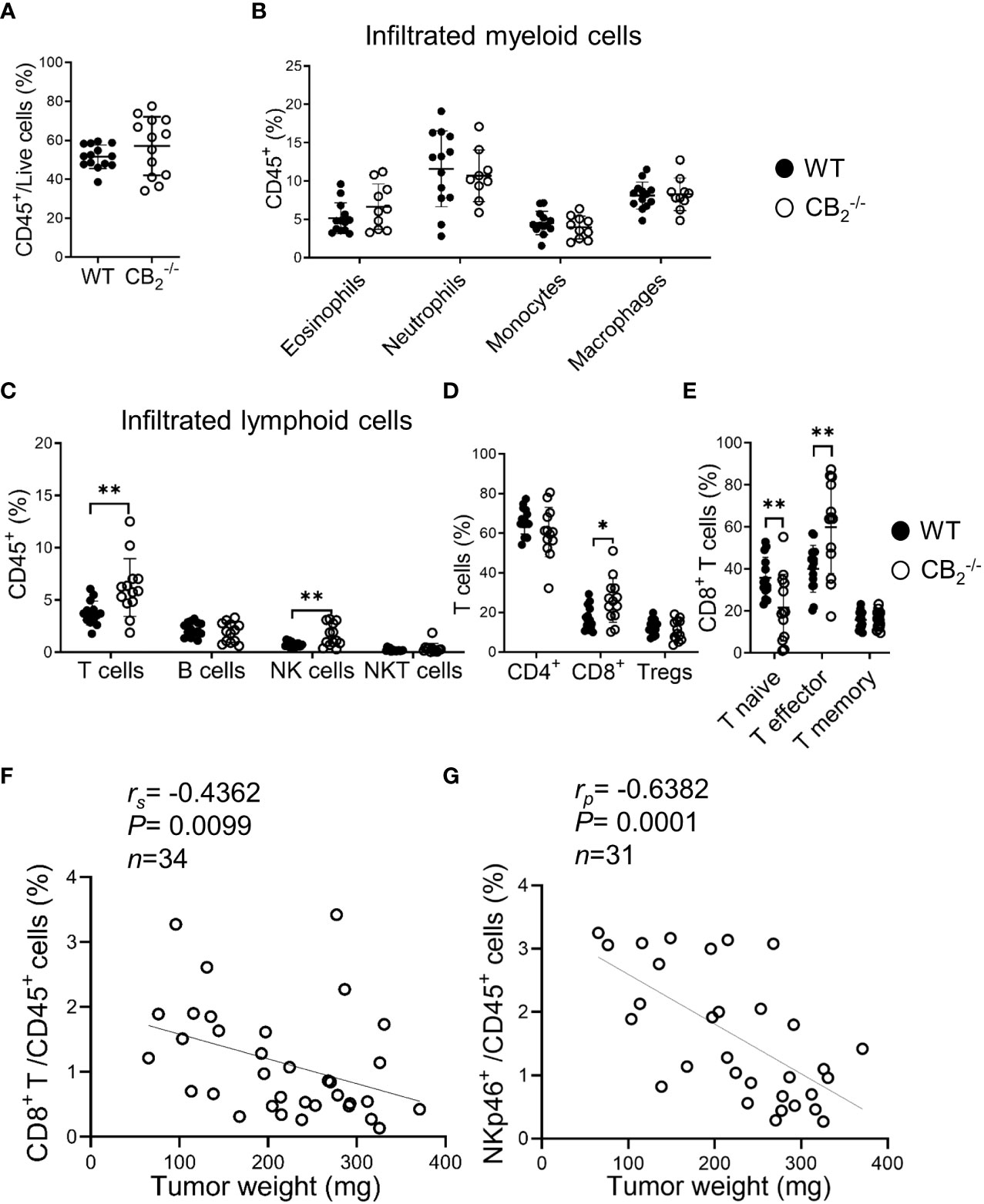
Figure 5 Knockout of CB2 in cells of the TME favors an anti-carcinogenic immune cell profile. (A–E) Flow cytometric analysis of single cell suspensions from KP cell tumors. Data indicate mean values ± SD from two pooled independent experiments. n≥10. Detailed information on immune cell markers is provided in Figure S1. Statistical differences were evaluated by using unpaired student`s t-test (A), multiple t-tests (B–E). (F, G) The percentages of tumor-infiltrating CD8+ T (CD45+/CD3+/CD8+) and NK (CD45+/CD3-/CD19-/NKp46+) cells (out of CD45+ cells) were plotted against tumor weights from CB2-/- mice. Data were pooled from four independent experiments. n=31-34. Correlation of samples was assessed using Spearman (rs) and Pearson (rp) correlation coefficients after testing for normality. *p < .05; **p < .01. NK, natural killer cells, NKT, natural killer T cells; TME, tumor microenvironment; Tregs, regulatory T cells; WT, wild type.
To identify underlying mechanisms of the tumor reduction in CB2-/- mice, we checked for apoptosis and proliferation rates of tumor cells (CD45-) and infiltrating immune cells (CD45+). Flow cytometric analysis and cleaved-caspase-3/caspase-3 immunoblotting of tumors from CB2-/- and WT mice showed no significant differences in apoptosis rates (Figures S4B–D). Similarly, in vivo and in vitro cell proliferation in tumor cells and infiltrating immune cells from CB2-/- mice using bromodeoxyuridine (BrdU) incorporation assay and Ki-67 immunofluorescence did not differ from WT mice (Figures S5B, C). To test whether cytotoxic immune cells were more activated in the CB2-/- mice, we stimulated tumor-infiltrating CD8+ T and NK cells from CB2-/- and WT mice ex vivo with PMA/Iono and assessed the activity of these cells using flow cytometry. In comparison to WT mice, tumors of CB2-/- mice showed increased expression levels of IFN-γ on CD8+ T cells (Figure 6A), and CD107a on NK cells (Figures 6B, C), signifying a local activation and enhanced tumoricidal activity of CD8+ T and NK cells. Therefore, a deficiency of CB2 in the TME leads to a higher number as well as to an increased activity of cytotoxic lymphocytes in the tumor.
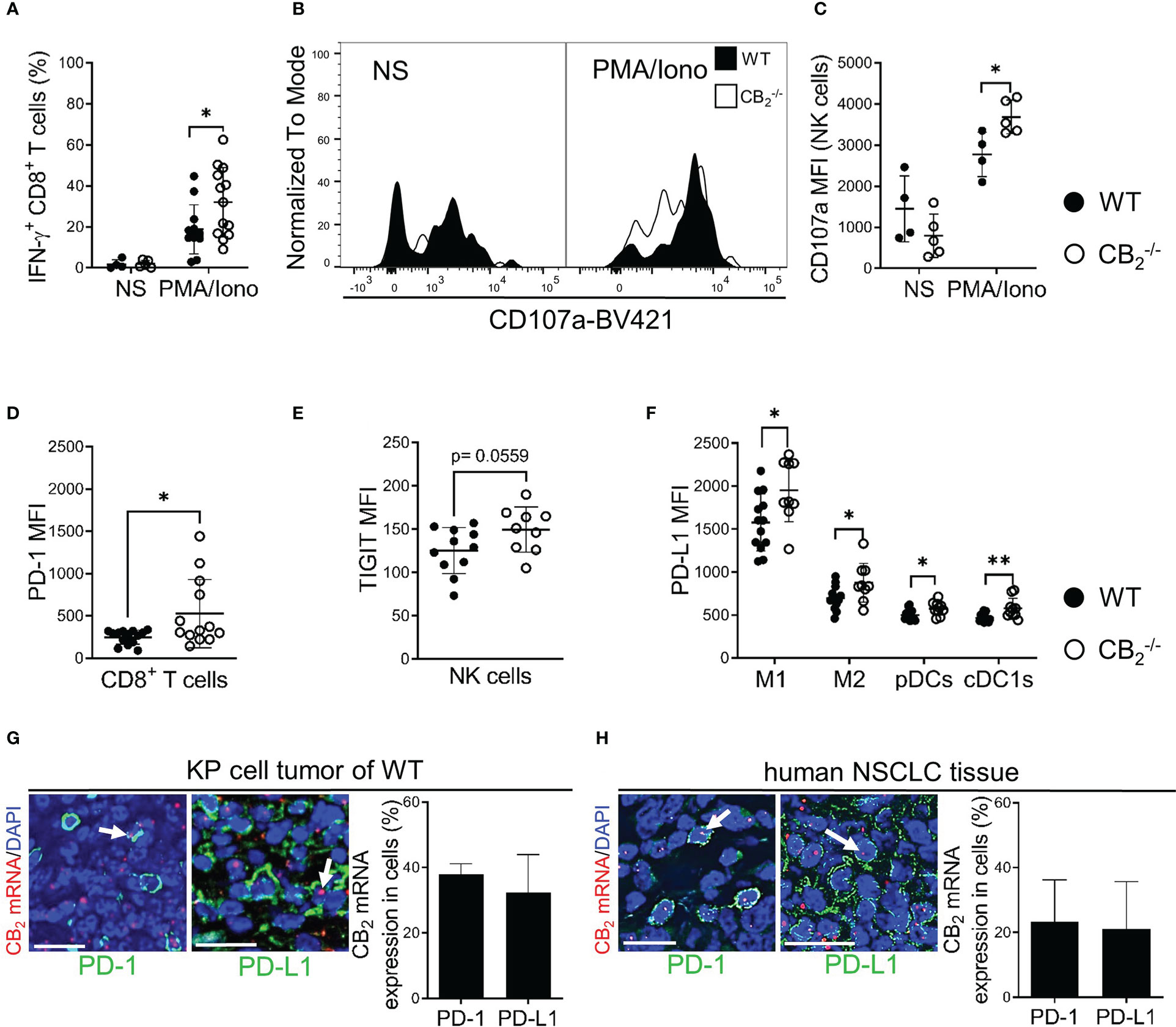
Figure 6 A CB2 deficient TME stimulates activity of CD8+ T and NK cells and alters expression of immune checkpoint proteins. (A) IFN-γ production of intratumoral CD8+ T (CD45+/CD3+/CD8+) cells prior to (non-stimulated, NS) and after ex vivo stimulation with phorbol myristate acetate/ionomycin (PMA/Iono). Data indicate mean values ± SD from two pooled independent experiments. n=4 (for NS); n≥11 (for PMA/Iono).(B, C) Degranulation capacity of tumor-infiltrated NK (CD45+/CD3-/NKp46+) cells before (NS) and after ex vivo stimulation with PMA/Iono. The graph depicts MFI of CD107a on NK cells. One representative experiment is shown. Data indicate mean values ± SD. n≥4. (D) MFI of PD-1 on tumor-infiltrated CD8+ T cells is shown. Data indicate mean values ± SD from two pooled independent experiments. n=13-14. (E) MFI of TIGIT on tumor-infiltrated NK cells. Data indicate mean values ± SD from two pooled independent experiments. n=13-14. (F) MFI of PD-L1 on tumor-infiltrated myeloid cells. Data indicate mean values ± SD from two pooled independent experiments. n=10-13. Detailed information on immune cell markers is provided in Figure S1. (G) ISH-IF analysis of KP cell tumors. Co-localization of PD-1/PD-L1 positively stained cells with CB2 mRNA is shown. Data indicate mean values +SD. n=3 (sections from three different tumors, 30-150 cells counted per section). (H) ISH-IF staining of human NSCLC tissue sections. The graph depicts co-localization of PD-1+/PD-L1+ stained cells with CB2 mRNA. Data indicate mean values +SD. n=3 (tumor sections from three different patients with NSCLC were used for quantification, 30-150 cells counted per section). Arrows indicate co-localization of CB2 mRNA with cells positive for PD-1/PD-L1. Calibration bars=20 μm. Statistical differences were evaluated by using unpaired student`s t-test (D, E), multiple t-tests (A, C, F). *p<.05; **p<.01. IFN-γ, interferon-gamma; NK, natural killer cells; MFI, median fluorescence intensity; PD-1, programmed death-1; TIGIT, T cell immunoglobulin and ITIM domain; PD-L1, programmed death-ligand 1; M1, M1 macrophages; M2, M2 macrophages, pDCs, plasmacytoid dendritic cells; cDC1, type 1 conventional dendritic cells, ISH-IF, in situ hybridization and immunofluorescence; WT, wild type; NSCLC, non-small cell lung cancer.
A CB2 deficient TME leads to a higher expression of immune checkpoint proteins and an enhanced responsiveness to PD-1 blocking antibodies
We next aimed to identify possible immune-based therapeutic strategies that could augment tumor reduction and hypothesized that a CB2 deficiency in the TME would have a favorable effect on immune checkpoint blockade. Thus, we first measured surface expression of different immune checkpoint proteins on immune cells. Results show that PD-1 expression was increased on tumor-infiltrating CD8+ T cells, but not on NK cells in CB2-/- vs. WT mice. On NK cells, only TIGIT (T cell immunoglobulin and ITIM domain) showed higher expression (Figures 6D, E, S6A). We also detected enhanced expression of PD-L1 on myeloid cells (macrophages and DCs) of CB2-/- vs. WT mice (Figure 6F). Regarding the other immune checkpoint proteins, no significant differences were detected for CTLA-4 (cytotoxic T-lymphocyte antigen-4), TIM-3 (T cell immunoglobulin and mucin domain-containing protein-3), and LAG-3 (lymphocyte activation gene-3) on NK and CD8+ T cells (Figures S6B–H). Dual ISH-IF revealed that approximately 40% of PD-1+ and PD-L1+ cells co-localized with CB2 mRNA in the KP cell tumors (Figure 6G). In human lung cancer, about 20% of PD-1+ and PD-L1+ cells co-localized with CB2 mRNA (Figure 6H).
Based on these findings, we treated CB2-/- mice with anti-PD-1 to boost immune cell activity (Figure 7A). Deficiency of CB2 on host cells augmented the responsiveness to PD-1 antibody treatment, resulting in an enhanced reduction of tumor growth in the CB2-/- mice (Figures 7B, C).
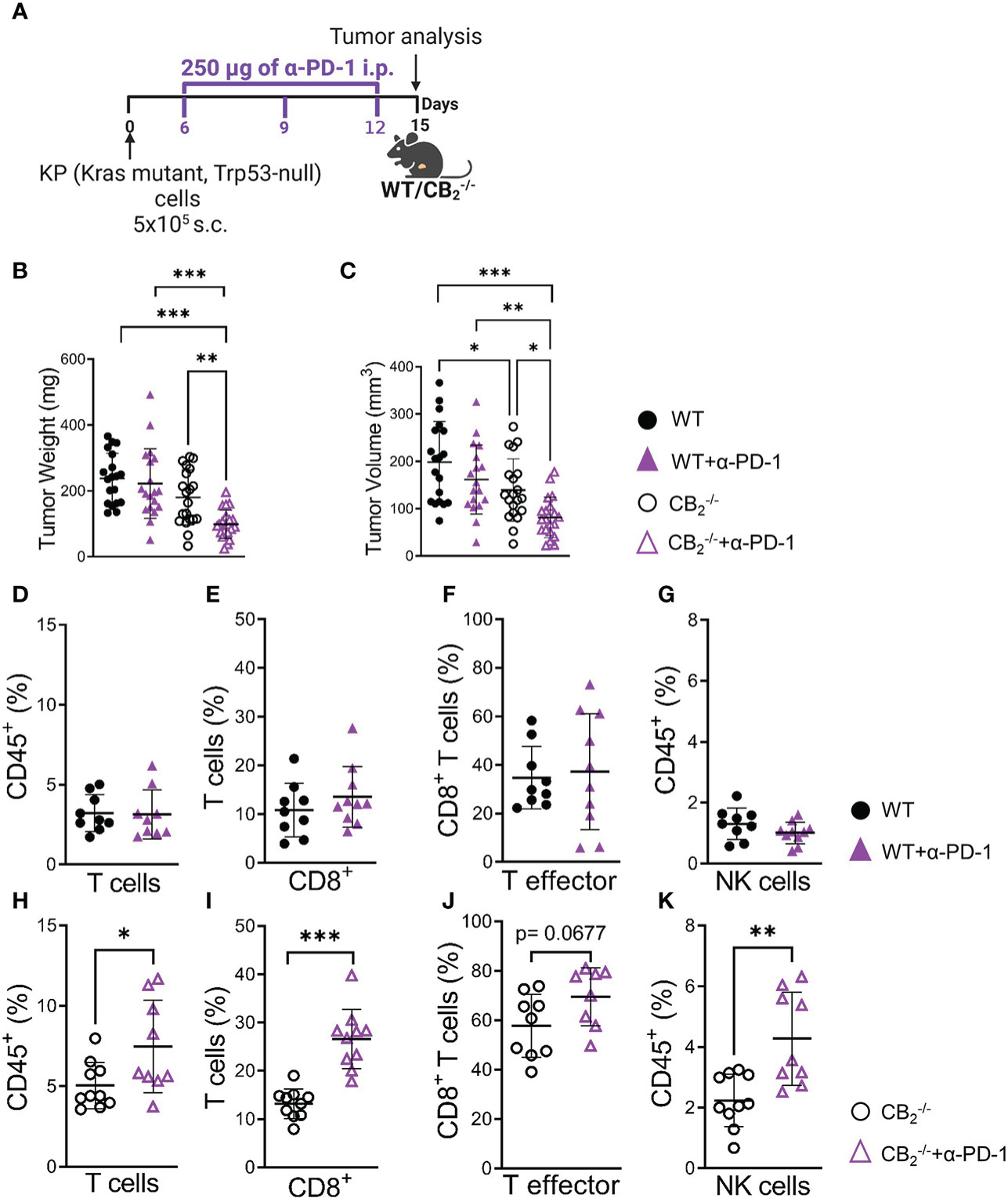
Figure 7 CB2-/- mice are more responsive to anti-PD-1 antibody treatment than their wild type littermates. (A) Experimental design: CB2-/- mice and WT littermates were subcutaneously (s.c.) injected with 5x105 KP (Kras mutant, Trp53-null) lung adenocarcinoma cells on day 0. On days 6, 9, and 12, mice were treated with 250 µg of anti-PD-1 (α-PD-1) antibodies (or isotype control). (B, C) Tumor weight and volume were measured at the end of the experiment on day 15 ex vivo. Data indicate mean values ± SD from two pooled independent experiments. n=19-21. (D–K) Flow cytometric analysis was performed on single cell suspensions from KP cell tumors of CB2-/- and WT α-PD-1 (or isotype control) treated mice. Detailed information on immune cell markers is provided in Figure S1. Data indicate mean values ± SD. One representative experiment is shown. n≥8. Statistical differences were evaluated by using one-way ANOVA, Tukey’s multiple comparison test (B, C), unpaired student`s t-test (D–K). *p < .05; **p < .01; ***p < .001; WT, wild type; NK, natural killer cells.
Flow cytometric analysis showed that PD-1 antibody therapy potentiated an increase in the number of CD8+ T and NK cells in tumors of CB2-/- mice (Figures 7H–K), but not in WT (Figures 7D-G), indicating that the deletion of CB2 in the TME favors an enhanced responsiveness to PD-1 therapy and causes a reduction in tumor burden.
Discussion
For many decades, the concept that cancer development is mainly driven by genetic mutations within tumor cells, has been studied in detail. However, cancer progression is additionally regulated by the surrounding niche, called the TME, which may deliver vital factors that promote cancer development or escape from host immune surveillance (32). A number of studies have identified the significance of immune cells of the TME in tumor development and as targets in immunotherapy. As such, cytotoxic lymphocytes like CD8+ T and NK cells are important prerequisites for successful immunotherapy (33–37).
CB1 and CB2 are over-expressed in various types of cancer, such as skin (38), breast (39) and NSCLC (4), and they have long been implicated in cancer progression (2, 3, 11, 38, 39). However, in addition to tumor cells, CB1 and CB2 are expressed in immune cells that can potentially populate the TME, where they could play a pro- or anti-tumorigenic role (27). A number of studies have focused on CB receptor/ligand interactions in tumor cells and how this axis influences tumor growth in vitro and in vivo (40), including studies in lung cancer cells and models of lung cancer (3, 4, 8). In contrast, little has been described on CB receptors in immune cells of the TME and how TME-derived CB receptors shape the immune cell profile and the response to immunotherapy. In our current study, we demonstrated that deficiency of CB2 in the TME host cells contributes to a reduction in tumor burden in a mouse model of NSCLC (summarized in Figure 8).
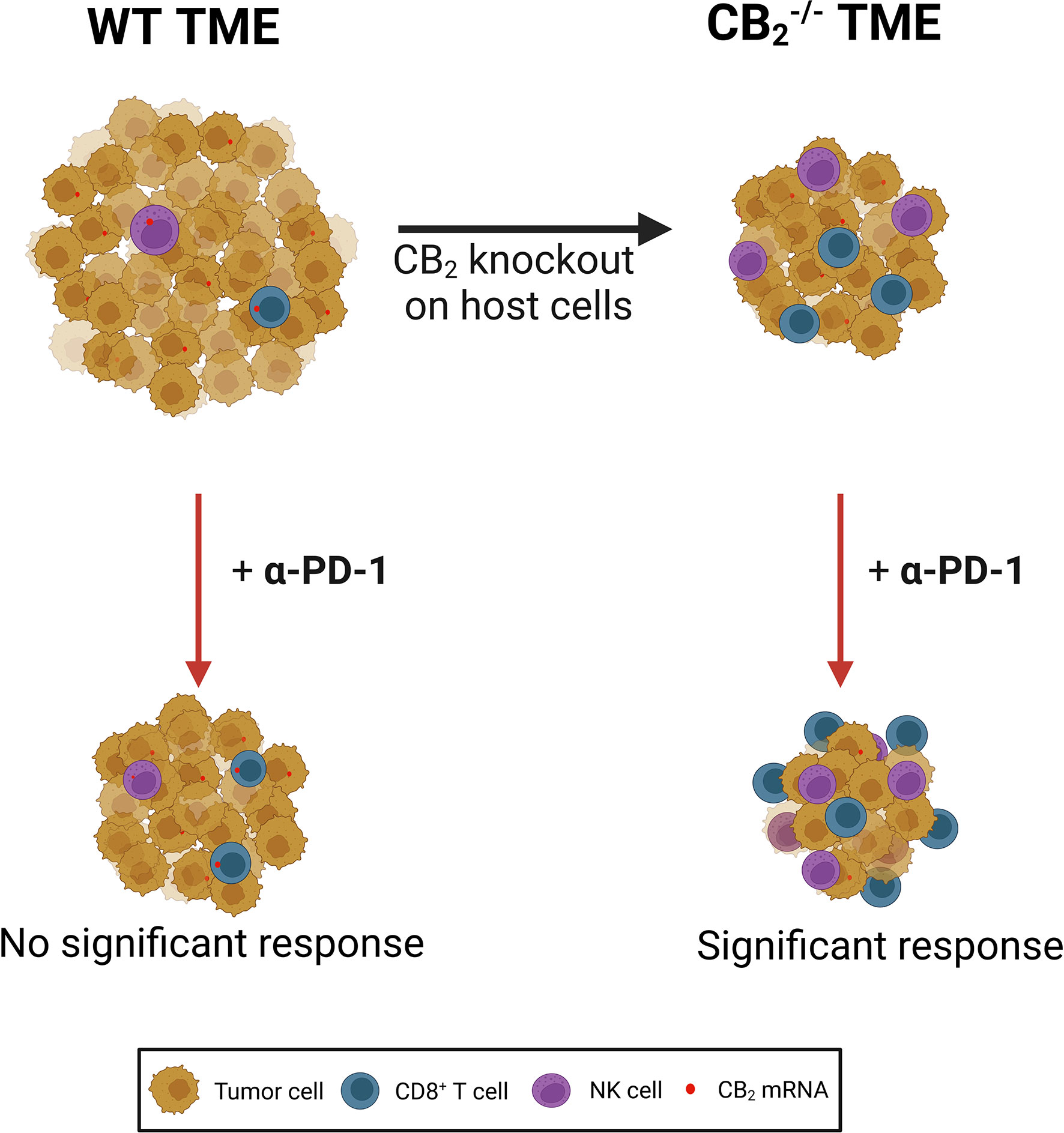
Figure 8 CB2 expressed on tumor microenvironment (TME) cells creates a pro-tumorigenic microenvironment by limiting the activity of cytotoxic lymphocytes in a mouse model of NSCLC. Deletion of the gene encoding CB2 on host cells results in a reduction of tumor growth as well as increased infiltration and local tumoricidal activity of CD8+ T and natural killer (NK) cells. CB2 knockout (CB2-/-) mice responded significantly better to anti-PD-1 (α-PD-1) therapy than wild type (WT) mice. α-PD-1 therapy further increased accumulation of CD8+ T and NK cells in the TME of CB2-/- mice. NSCLC, non-small cell lung cancer.
CB receptors are present in tumor cells and immune cells in situ
Using dual ISH-IF analysis of mouse and human lung cancer sections, we revealed that tumor cells as well as tumor-infiltrating immune cells, such as CD8+ T, NK cells, and macrophages express CB2 at much higher levels than CB1. ISH-IF showed co-localization of CB2 mRNA in around 20-40% of immune cells, and 25% in KP tumor cells, suggesting TME cell-mediated and/or possible direct effects on tumor cells by CB2. Pharmacological activation or inhibition of CB2 in CB2-/- mice (i.e., targeting only CB2-expressing KP tumor cells) revealed no influence of tumor cell-derived CB2 on tumor growth, indicating that only CB2 expressed in TME cells was responsible for the diminished tumor growth. The conflicting findings of CB2 in lung cancer (2–4), therefore, suggest a heterogeneous role for CB2 in lung carcinogenesis, which most likely depends not only on CB2 expressing tumor cells, but also on the type of TME-infiltrating immune cells expressing CB2.
TME-derived CB2 controls immune cell infiltrates to the tumor
Cannabinoid ligands are known to suppress phagocytosis, antigen presentation, and other features of immune cells that are essential for regulation of immune activity in the TME (16). As we detected widespread CB2 expression in immune cells of the TME, we assessed the immune cell profile of the tumors.
Our flow cytometric analyses demonstrated that the immune cell landscape was altered in the absence of CB2 in the TME. Although there was no shift in the myeloid cell profile, we observed a significant infiltration of cytotoxic lymphocytes, mainly of cytotoxic CD8+ T and NK cells into the TME of CB2-/- as compared to WT mice. We also found a negative correlation between the percentages of infiltrated CD8+ T and NK cells into the TME and the tumor weights in CB2 deficient mice, suggesting an involvement of CD8+ T and NK cells in the reduction of tumor growth. A more detailed investigation of these cells revealed that tumor-infiltrating CD8+ T and NK cells of CB2-/- mice possessed higher cytotoxic activity (higher levels of IFN-γ and CD107a). These data are fully consistent with studies describing that an increased infiltration of the cytotoxic lymphocytes into the TME is associated with a good prognosis (41–43). Particularly in NSCLC, activity of CD8+ T and NK cells may be hampered: NK cells can overexpress inhibitory receptors (44), additionally they have been shown to poorly infiltrate NSCLC tumors, and are found more frequently in normal lung than neoplastic tissues (45). Moreover, a reduced number of cytotoxic T cells along with a reduction in IFN-γ expression was observed in NSCLC patients (46, 47). Hence, CB2 deficiency reversed the low infiltration of NK and CD8+ T cells in our model and boosted their activity, likely contributing to a reduction in tumor size.
CB2-/- mice are highly susceptible to PD-1 checkpoint inhibitor treatment
Immunotherapies using checkpoint inhibitors have been demonstrated to increase survival of patients in a number of cancer types, including melanoma and lung cancer (48, 49). Among all known checkpoints, the most prominent target for treatment is the PD-1/PD-L1 axis, owing to its proven efficacy in several types of cancers (48–50). Previous studies found that one of the critical requirements for ICIs to work is a sufficient infiltration of lymphocytes, including CD8+ T cells, at tumor sites (33, 51). A major finding of our study is that tumor-bearing CB2-/- mice responded significantly better to anti-PD-1 treatment than the WT mice (as demonstrated by the significant reduction in tumor burden). In addition, we noticed increased PD-1 expression on CD8+ T cells in tumors of CB2-/- mice, an indication of high T cell activity against tumor antigens as well as a possible prediction of anti-PD-1 therapy response (25, 34, 52). Our data also revealed increased PD-L1 expression on tumor-infiltrating myeloid cells in CB2-/- mice, another important finding that the tumor might respond favorably to anti-PD-1 therapy (53–55). Cytotoxic CD8+ T cells are often the main focus of interest in terms of improving immune checkpoint blockade therapies, but other immune cells, such as NK cells may provide an important contribution to the efficacy of checkpoint inhibitors (reviewed in (56)). Thus, the presence of intratumoral cytotoxic NK cells promotes a positive response to immunotherapies, by also targeting the PD-1/PD-L1 axis (35, 36). Recent studies found that the number of NK cells correlated with the responsiveness to anti-PD-1 treatment, and improved overall survival in melanoma and metastatic melanoma patients (37, 43). Zhang et al. identified that the presence of NK cells provided an enhanced clinical benefit of PD-L1 as well as TIGIT-based immunotherapies, as NK cells improved the functional role of CD8+ T cells and/or inhibited their exhaustion (57). The TME of CB2-/- mice had a significantly higher number of NK cells than WTs, and their presence, therefore, may enhance the susceptibility to immunotherapy with anti-PD-1.
To further assess susceptibility to checkpoint blockade, we determined other proteins that inhibit T and NK cells activity/proliferation, such as CTLA-4, TIM-3, TIGIT, and LAG-3 (58–62). Except for increased expression of TIGIT on NK cells, there were no significant differences between CB2-/- and WT mice littermates as to the expression rates of these proteins on CD8+ T and NK cells. Collectively, our data suggest that CD8+ T and NK cells in CB2-/- mice were in an active, non-exhausted state (high levels of IFN-γ and PD-1 on CD8+ T cells, and of CD107a on NK cells).
Deficiency of CB2 in the TME increases the PD-1 antibody-induced effect on CD8+ T and NK cells
The effect of an anti-PD-1/PD-L1 therapy on the immune cell composition has often been associated with the restoration of effector CD8+ T cell activity to kill tumor cells (63). Other cytotoxic lymphocytes, including NK cells, also contribute to the response to immunotherapy (reviewed in (64)): Lee et al. demonstrated increased frequency of intratumoral and peritumoral NK cells in melanoma patients who responded well to PD-1 blockade (37). Hsu et al. also identified that, in addition to T cells, NK cells can mediate the effect of anti-PD-1/PD-L1 therapy (35). In our study, the anti-PD-1 therapy further increased the number of CD8+ T and NK cells at the tumor site of CB2-/- as compared to WT mice. This supports the concept that a successful anti-PD-1 therapy is inherently linked to the presence of CD8+ T and NK cells in the TME. It should be mentioned that PD-1 expression in tumor-infiltrating NK cells of CB2-/- mice was not different from WT mice, and that PD-1 expression was lower on NK than CD8+ T cells. This calls into question whether there is a direct effect of anti-PD-1 antibodies on NK cells, as the checkpoint blockade may have indirectly modulated anti-cancer NK cell functions via the crosstalk with other immune cell populations, as previously described (65, 66). While this manuscript was in preparation, a study was published, describing that THC and exogenous cannabinoids (approved for the treatment of chemotherapy-induced nausea) reduced the effect of anti-PD-1 therapy (67), reconfirming our own observations. Cannabis is well-known for its immunosuppressive effects (68), which is also supported by a recent observation that the use of cannabis during treatment with PD-1 checkpoint inhibitor nivolumab in cancer patients lowered their response rate (69). With our study, we highlight a possible mechanism for a lower response, which includes CB2, CD8+ T and NK cells.
Conclusion
Our results demonstrate that the CB2 receptor in the TME of NSCLC tumors may act as an immunosuppressor that impedes CD8+ T and NK cell activity, thereby promoting tumor growth. Deletion of CB2 in the TME releases the immunosuppressive break rendering tumors to be more susceptible to PD-1 inhibitor treatment. The findings also suggest that the use of cannabis or cannabinoid-based medicine during immunotherapy may lead to a low treatment response. Altogether, the CB2 receptor maybe an interesting adjuvant target for ICI therapy.
Materials and methods
Cancer cell lines and mice
The mouse KP cell line (a generous gift by Dr. McGarry Houghton from the Fred Hutchinson Cancer Center, Seattle, USA) was isolated from a lung adenocarcinoma, grown in a Kras mutant/Trp53-null (KrasLSL-G12D/p53fl/fl) mouse following intratracheal administration of adenoviral Cre recombinase, as described before (70). Briefly, pieces of mechanically disintegrated lung tumor were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with FBS (10%), penicillin (100units/mL) and streptomycin (100μg/mL). Clonal cells were derived by single-cell dilution into 96 well plates (70). Lewis lung carcinoma (LLC1) cell line was purchased from ATCC (Rockville, Maryland, USA). Both cell lines were maintained in DMEM media containing 10% fetal bovine serum (FBS, Life Technologies) and 1% penicillin/streptomycin (P/S, PAA Laboratories) and kept in a humidified incubator (5% CO2) at 37°C and passaged every 48 hrs. The cell lines were mycoplasma free.
All animals were bred and maintained in the animal facilities of the Medical University of Graz. Wild type C57BL/6J (B6) mice were purchased from Charles River, Germany. CB1-/- mice on B6 background were obtained from Dr. Andreas Zimmer, University of Bonn, Germany. CB2-/- mice (B6.129P2-Cnr2tm1Dgen/J on B6 background) were obtained from Jackson Laboratories (Bar Harbor, ME, USA). Experiments were performed on 6-14-week-old mice. All procedures were granted by the Austrian Federal Ministry of Science and Research (protocol number: BMBWF-66.010/0041-V/3b/2018). Subcutaneous (s.c.) injections of KP or LLC1 cells were performed under inhaled isoflurane anaesthesia. To generate s.c. tumors, KP or LLC1 cells (5×105) suspended in 450 µL Dulbecco’s Phosphate Buffered Saline (PBS, Gibco) were injected s.c. into the lower flanks of mice on day 0. Tumors were harvested at the experimental endpoint (day 15 for KP cell tumor model; day 21 for LLC1 lung tumor model) and were subsequently weighed, measured with a digital caliper ex vivo, and collected for analysis. Tumor volume was calculated based on the following formula: v = length x width x height x π/6 (71).
Pharmacology
To pharmacologically block CB1 receptors, tumor-bearing C57BL/6J WT mice were intraperitoneally (i.p.) treated with 1 mg/kg/d SR141716 (28, 29) (CB1 antagonist, Cayman Chemical, Ann Arbor, MI). For pharmacological activation of CB2 receptors, tumor-bearing CB2-/- mice were i.p. treated with 20 mg/kg/d JWH-133 (31) (CB2 agonist, Axon Medchem, Groningen, NL). To pharmacologically block CB2 receptors, tumor-bearing CB2-/- mice and C57BL/6J WT mice were i.p. treated with 10 mg/kg/d SR144528 (29, 30) (CB2 antagonist, Cayman Chemical, Ann Arbor, MI) or vehicle (ethanol). The treatment period for all mentioned interventions was ten days, starting from day 5 when the s.c. tumors were palpable, until day 14. For inhibition of PD-1, tumor-bearing CB2-/- mice and WT littermates were injected i.p. with 250 μg of rat monoclonal anti-mouse PD-1 antibody (72) (clone 29F.1A12, BioXCell, Lebanon, NH) or rat IgG2a isotype control (clone 2A3, BioXCell, Lebanon, NH) on days 6, 9, and 12.
Single-cell suspensions
Single cell suspensions of dissected mouse KP cell tumors were prepared as previously described (71). Briefly, using surgical scissors, tumors were cut into small pieces, and afterwards digested with DNase I (160 U/ml; Worthington) and collagenase (4.5 U/ml; Worthington) for 20 min at 37°C, while rotating at 800-1000 rpm. The tissue was then passed through a 40 μm cell strainer, washed in staining buffer (SB, PBS+2% FBS), suspended in PBS, counted, and used for surface, intracellular and nuclear antigen staining.
Flow cytometry of dissected KP cell tumors
To exclude dead cells, single cell suspensions were initially incubated for 20 min in Fixable Viability Dye (FVD) eFluor™ 780 (eBioscience) at 4°C in the dark. Prior to staining with surface, intracellular and nuclear antibodies, single cell suspensions were incubated in 1 μg TruStain FcX™ (BioLegend, # 101320) for 10 min at 4°C. Immunostaining was performed for 30 min at 4°C (protected from light) using the pre-mixed antibody panels (Table S1). To detect FoxP3 nuclear antigen within the cells, surface stained cells were permeabilized and fixed with Transcription Factor Buffer Set (BD Biosciences, # 562574) before staining with FoxP3 antibody (Table S1). To detect expression of IFN-γ and CD107a, single-cell suspensions of the tumors (2×106 cells per well) were suspended in RPMI media supplemented with 10% FBS, 1% P/S, and GolgiStop (1.5 μl/ml, BD Biosciences), seeded into 96- well U-bottomed plates, and incubated for 4 hrs at 37°C (5% CO2). During incubation time, CD107a was added, and cells were stimulated with phorbol myristate acetate (PMA) (100 ng/ml, Sigma Aldrich) and ionomycin (Iono) (1 μg/ml, Sigma Aldrich), or used unstimulated (73, 74). Afterwards, surface and intracellular stainings (BD Cytofix/Cytoperm™ Kit) were performed with the pre-mixed antibody panel (Table S1). Cells were then washed and fixed in eBioscience™ IC Fixation Buffer (ThermoFisher Scientific, # 00-8222-49) for 10 min at 4°C. Fixed cells were either acquired on a BD LSR Fortessa™ or a BD Canto™ flow cytometer connected to FACSDiva software (BD Biosciences). FlowJo software (Treestar) was used for analysis and compensation. Fluorescence minus-one-samples were used to define gates (Figures S1A-D).
RNA extraction and RT-qPCR
RNA was extracted from tissue and KP cells using Trizol (Life Technologies) and RNeasy Kit (Qiagen), respectively. Samples were treated either with a DNA-free™ DNA Removal Kit (Invitrogen) or RNase-Free DNase set (Qiagen). The quality and concentration of RNA were determined using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific). Reverse transcription of purified RNA (1 µg) was performed by High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Gene expression was assessed by reverse transcription-quantitative polymerase chain reaction (RT-qPCR) using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad). Primers were acquired from Eurofins (Table S2) and Bio-Rad (Table S3). Relative gene expression was calculated using 2-ΔΔCT methods (75).
In situ hybridization and immunofluorescence
Mouse and human NSCLC tissue samples
Tumors from mice were fixed in acid-free phosphate-buffered 10% formaldehyde solution (Roti®- Histofix 10%, pH 7) for 24-48 hrs at room temperature with gentle shaking. Tissue was further processed for paraffin embedding based on standard procedures. Human NSCLC tissue samples (formalin-fixed and paraffin-embedded) were acquired from the Biobank of the Medical University of Graz. Ethical approval was obtained from the Institutional Review Board of the Medical University of Graz (EK-numbers: 30-105 ex 17/18). All procedures involving clinical samples followed the ethical standards of the institutional and/or national research committee and the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. All patients participated in the study gave informed consent.
ISH probes used to detect CB1 and CB2 mRNAs in mouse tumor and human NSCLC tissue were purchased from Advanced Cell Diagnostics (ACD, Newark, USA) (Table S4). ISH was performed using RNAscope® 2.5 HD red kit according to manufacturer’s instructions. Briefly, tumor tissue sections were first treated with H2O2 at room temperature for 10 min and target retrieval was performed using the Brown FS3000 food steamer at 95°C for 15 min. Then, the sections were digested with protease IV in HybEZ™ II oven (ACD, Newark, USA) at 40°C for 20 min, washed in distilled water, followed by incubation with the corresponding probes at 40°C for 2 hrs and stained with Fast Red. To compare tissue samples from CB1-/- or CB2-/- and WT mice, sections were placed on a single slide. The specificity of the mouse CB1 and CB2 probes was previously verified in CB1-/- and CB2-/- mice (76). Immunofluorescence of tumor cells and infiltrated immune cells of the TME was conducted using primary antibodies listed in Table S5. Alexa Fluor® 488-labeled goat anti-rabbit IgG (1:500, Jackson Immuno Research, #111-546-144) and Alexa Fluor® 488-labelled bovine anti-goat IgG (H+L) (1:500, Jackson Immuno Research, # 805-545-180) were used as secondary antibodies. In parallel, sections were processed in the absence of primary antibody as a negative control. Then, sections were mounted with Vectashield® (containing DAPI) (Vector Laboratories) and images were taken using an Olympus IX73 fluorescence microscope (Olympus) connected with a Hamamatsu ORCA-ER digital camera (Hamamatsu Photonics K.K., Japan). Images were processed with an Olympus CellSens® 1.17 imaging software containing a deconvolution program (Olympus). ImageJ software was used to quantify expression and co-localization with the corresponding probes.
Statistical analysis
Data are presented as means ± standard deviation (SD) or standard error of means (SEM) and analyzed using Prism v.9.3.1 (GraphPad Software, La Jolla, CA, USA). Differences between experimental groups were assessed by unpaired student’s t-tests, multiple t-tests or two-way analysis of variance (ANOVA) with the indicated post hoc test for corrections of multiple comparisons, whereas for multiple comparisons with three or more experimental groups, a one-way ANOVA was applied with the indicated post hoc test for corrections of multiple comparisons. Shapiro-Wilk and Kolmogorov-Smirnov tests were used to test a normal distribution. Correlations between tumor weight and infiltration of CD8+ T and NK cells in the TME was determined using Pearson’s correlation coefficient (rp) and Spearman’s correlation coefficient rho (rs).
In all cases, a p-value <0.05 was considered significant and represented with one, two or three asterisks when lower than 0.05, 0.01, or 0.001, respectively.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the Medical University of Graz. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by the Austrian Federal Ministry of Education, Science and Research.
Author contributions
ArS, MK, JK, CH and RS contributed to the conception and design of the study. ArS, MK, EG, DR, CH, KM, AnS and PVC performed experiments and acquired data. ArS, MK, EG, JK and RS contributed to the analysis and interpretation of the data. ArS and RS participated in the writing of the manuscript. LB and JL provided the human lung cancer samples. All authors contributed to the article and approved the submitted version.
Funding
Ph.D. candidate ArS received funding from the Medical University of Graz (PhD program: Molecular Medicine). Work in the lab of JK is funded by the Austrian Science Fund (FWF grant P-35294-B). RS is funded by the Austrian Science Fund (FWF; grants P33325 and KLI887).
Acknowledgments
PhD candidates AnS, DR and PVC were trained within the frame of the PhD programs Molecular Medicine and DK-MOLIN of the Medical University of Graz and the Austrian Science Fund. We are grateful to Veronika Pommer and Sabine Kern for their excellent technical assistance; we thank Sabine Donner for animal care and Marah Runtsch for carefully reading the manuscript. Experimental design diagrams and the overview of the study were created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.997115/full#supplementary-material
References
1. Fraguas-Sánchez AI, Martín-Sabroso C, Torres-Suárez AI. Insights into the effects of the endocannabinoid system in cancer: A review. Br J Pharmacol (2018) 175:2566–80. doi: 10.1111/bph.14331
2. Milian L, Mata M, Alcacer J, Oliver M, Sancho-Tello M, de Llano JJM, et al. Cannabinoid receptor expression in non-small cell lung cancer. effectiveness of tetrahydrocannabinol and cannabidiol inhibiting cell proliferation and epithelial-mesenchymal transition in vitro. PLoS One (2020) 15:e0228909. doi: 10.1371/journal.pone.0228909
3. Xu S, Ma H, Bo Y, Shao M. The oncogenic role of CB2 in the progression of non-small-cell lung cancer. BioMed Pharmacother (2019) 117:109080. doi: 10.1016/j.biopha.2019.109080
4. Preet A, Qamri Z, Nasser MW, Prasad A, Shilo K, Zou X, et al. Cannabinoid receptors, CB1 and CB2, as novel targets for inhibition of non-small cell lung cancer growth and metastasis. Cancer Prev Res (2011) 4:65–75. doi: 10.1158/1940-6207.CAPR-10-0181
5. Boyacıoğlu Ö, Bilgiç E, Varan C, Bilensoy E, Nemutlu E, Sevim D, et al. ACPA decreases non-small cell lung cancer line growth through Akt/PI3K and JNK pathways in vitro. Cell Death Dis (2021) 12:56. doi: 10.1038/s41419-020-03274-3
6. Vidinský B, Gál P, Pilátová M, Vidová Z, Solár P, Varinská L, et al. Anti-proliferative and anti-angiogenic effects of CB2 r agonist (JWH-133) in non-small lung cancer cells (A549) and human umbilical vein endothelial cells: an in vitro investigation. CzechRepublic Folia Biol (2012) 58:75–80.
7. Preet A, Ganju RK, Groopman JE. Δ9-tetrahydrocannabinol inhibits epithelial growth factor-induced lung cancer cell migration in vitro as well as its growth and metastasis in vivo. Oncogene (2008) 27:339–46. doi: 10.1038/sj.onc.1210641
8. Ravi J, Elbaz M, Wani NA, Nasser MW, Ganju RK. Cannabinoid receptor-2 agonist inhibits macrophage induced EMT in non-small cell lung cancer by downregulation of EGFR pathway. Mol Carcinog (2016) 55:2063–76. doi: 10.1002/mc.22451
9. Hart S, Fischer OM, Ullrich A. Cannabinoids induce cancer cell proliferation via tumor necrosis factor α-converting enzyme (TACE/ADAM17)-mediated transactivation of the epidermal growth factor receptor. Cancer Res (2004) 64:1943–50. doi: 10.1158/0008-5472.CAN-03-3720
10. Zhu LX, Sharma S, Stolina M, Gardner B, Roth MD, Tashkin DP, et al. Δ-9-Tetrahydrocannabinol inhibits antitumor immunity by a CB2 receptor-mediated, cytokine-dependent pathway. J Immunol (2000) 165:373–80. doi: 10.4049/jimmunol.165.1.373
11. Pyszniak M, Tabarkiewicz J, Łuszczki JJ. Endocannabinoid system as a regulator of tumor cell malignancy – biological pathways and clinical significance. Onco Targets Ther (2016) 9:4323–36. doi: 10.2147/OTT.S106944
12. Cristino L, Bisogno T, Di Marzo V. Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nat Rev Neurol (2020) 16:9–29. doi: 10.1038/s41582-019-0284-z
13. Lu HC, Mackie K. Review of the endocannabinoid system. Biol Psychiatry Cognit Neurosci Neuroimaging (2021) 6:607–15. doi: 10.1016/j.bpsc.2020.07.016
14. Tsou K, Brown S, Sañudo-Peña MC, K Mackie JW. Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience (1998) 83:393–411. doi: 10.1016/s0306-4522(97)00436-3
15. Basu S, Dittel BN. Unraveling the complexities of cannabinoid receptor 2 (CB2) immune regulation in health and disease. Immunol Res (2011) 51:26–38. doi: 10.1007/s12026-011-8210-5
16. Kienzl M, Kargl J, Schicho R. The immune endocannabinoid system of the tumor microenvironment. Int J Mol Sci (2020) 21:1–25. doi: 10.3390/ijms21238929
17. Kaplan BLF. The role of CB1 in immune modulation by cannabinoids. Pharmacol Ther (2013) 137:365–74. doi: 10.1016/j.pharmthera.2012.12.004
18. Klein TW, Newton C, Friedman H. Cannabinoid receptors and immunity. Immunol Today (1998) 19:373–81. doi: 10.1016/S0167-5699(98)01300-0
19. Eisenstein TK, Meissler JJ. Effects of cannabinoids on T-cell function and resistance to infection. J Neuroimmune Pharmacol (2015) 10:204–16. doi: 10.1007/s11481-015-9603-3
20. Eisenstein TK, Meissler JJ, Wilson Q, Gaughan JP, Adler MW. Anandamide and Δ9-tetrahydrocannabinol directly inhibit cells of the immune system via CB2 receptors. J Neuroimmunol (2007) 189:17–22. doi: 10.1016/j.jneuroim.2007.06.001
21. Cencioni MT, Chiurchiù V, Catanzaro G, Borsellino G, Bernardi G, Battistini L, et al. Anandamide suppresses proliferation and cytokine release from primary human T-lymphocytes mainly via CB2 receptors. PloS One (2010) 5:e8688. doi: 10.1371/journal.pone.0008688
22. Montecucco F, Burger F, Mach F, Steffens S. CB2 cannabinoid receptor agonist JWH-015 modulates human monocyte migration through defined intracellular signaling pathways. Am J Physiol Hear Circ Physiol (2008) 294:H1145–55. doi: 10.1152/ajpheart.01328.2007
23. Massi P, Fuzio D, Vigano D, Sacerdote P, Parolaro D. Relative involvement of cannabinoid CB and CB receptors in the Δ9-tetrahydrocannabinol-induced inhibition of natural killer activity. Eur J Pharmacol (2000) 387:343–7. doi: 10.1016/S0014-2999(99)00860-2
24. Braile M, Marcella S, Marone G, Galdiero MR, Varricchi G, Loffredo S. The interplay between the immune and the endocannabinoid systems in cancer. Cells (2021) 10:1282. doi: 10.3390/cells10061282
25. Simon S, Labarriere N. PD-1 expression on tumor-specific T cells: Friend or foe for immunotherapy? Oncoimmunology (2018) 7:e1364828. doi: 10.1080/2162402X.2017.1364828
26. Błach J, Wojas-Krawczyk K, Nicoś M, Krawczyk P. Failure of immunotherapy–the molecular and immunological origin of immunotherapy resistance in lung cancer. Int J Mol Sci (2021) 22:9030. doi: 10.3390/ijms22169030
27. Kienzl M, Hasenoehrl C, Maitz K, Sarsembayeva A, Taschler U, Valadez-Cosmes P, et al. Monoacylglycerol lipase deficiency in the tumor microenvironment slows tumor growth in non-small cell lung cancer. Oncoimmunology (2021) 10:e1965319. doi: 10.1080/2162402X.2021.1965319
28. Rinaldi-Carmona M, Barth F, Hkaulme M, Shireb D, Calandrab B, Congy C, et al. SR1417 16A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett (1994) 350:240–4. doi: 10.1016/0014-5793(94)00773-x
29. Di Marzo V. Targeting the endocannabinoid system: To enhance or reduce? Nat Rev Drug Discovery (2008) 7:438–55. doi: 10.1038/nrd2553
30. Rinaldi-Carmona M, Barth F, Millan J, Derocq J-M, Casellas P, Congy C, et al. SR144528, the first potent and selective antagonist of the CB2 cannabinoid receptor. J Pharmacol Exp Ther (1998) 284:644–50.
31. Storr MA, Keenan CM, Zhang H, Patel KD, Makriyannis A, Sharkey KA. Activation of the cannabinoid 2 receptor (CB2) protects against experimental colitis. Inflamm Bowel Dis (2009) 15:1678–85. doi: 10.1002/ibd.20960
32. Hanahan D. Hallmarks of cancer: New dimensions. Cancer Discov (2022) 12:31–46. doi: 10.1158/2159-8290.CD-21-1059
33. Farhood B, Najafi M, Mortezaee K. CD8+ cytotoxic T lymphocytes in cancer immunotherapy: A review. J Cell Physiol (2019) 234:8509–21. doi: 10.1002/jcp.27782
34. Thommen DS, Koelzer VH, Herzig P, Roller A, Trefny M, Dimeloe S, et al. A transcriptionally and functionally distinct PD-1+ CD8+ T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat Med (2018) 24:994–1004. doi: 10.1038/s41591-018-0057-z
35. Hsu J, Hodgins JJ, Marathe M, Nicolai CJ, Bourgeois-Daigneault MC, Trevino TN, et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J Clin Invest (2018) 128:4654–68. doi: 10.1172/JCI99317
36. Dong W, Wu X, Ma S, Wang Y, Nalin AP, Zhu Z, et al. The mechanism of anti–PD-L1 antibody efficacy against PD-L1–negative tumors identifies NK cells expressing PD-L1 as a cytolytic effector. Cancer Discov (2019) 9:1422–37. doi: 10.1158/2159-8290.CD-18-1259
37. Lee H, Quek C, Silva I, Tasker A, Batten M, Rizos H, et al. Integrated molecular and immunophenotypic analysis of NK cells in anti-PD-1 treated metastatic melanoma patients. Oncoimmunology (2019) 8:e1537581. doi: 10.1080/2162402X.2018.1537581
38. Llanos Casanova M, Blázquez C, Martínez-Palacio J, Villanueva C, Fernández-Aceñero MJ, Huffman JW, et al. Inhibition of skin tumor growth and angiogenesis in vivo by activation of cannabinoid receptors. J Clin Invest (2003) 111:43–50. doi: 10.1172/JCI200316116
39. Caffarel MM, Sarrió D, Palacios J, Guzmán M, Sánchez C. Δ9-tetrahydrocannabinol inhibits cell cycle progression in human breast cancer cells through Cdc2 regulation. Cancer Res (2006) 66:6615–21. doi: 10.1158/0008-5472.CAN-05-4566
40. Laezza C, Pagano C, Navarra G, Pastorino O, Proto MC, Fiore D, et al. The endocannabinoid system: A target for cancer treatment. Int J Mol Sci (2020) 21:747. doi: 10.3390/ijms21030747
41. Martínez-Lostao L, Anel A, Pardo J. How do cytotoxic lymphocytes kill cancer cells? Clin Cancer Res (2015) 21:5047–56. doi: 10.1158/1078-0432.CCR-15-0685
42. Takanami I, Takeuchi K, Giga M. The prognostic value of natural killer cell infiltration in resected pulmonary adenocarcinoma. J Thorac Cardiovasc Surg (2001) 121:1058–63. doi: 10.1067/mtc.2001.113026
43. Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, et al. A natural killer–dendritic cell axis defines checkpoint therapy–responsive tumor microenvironments. Nat Med (2018) 24:1178–91. doi: 10.1038/s41591-018-0085-8
44. Jin S, Deng Y, Hao JW, Li Y, Liu B, Yu Y, et al. NK cell phenotypic modulation in lung cancer environment. PloS One (2014) 9:e109976. doi: 10.1371/journal.pone.0109976
45. Stankovic B, Bjørhovde HAK, Skarshaug R, Aamodt H, Frafjord A, Müller E, et al. Immune cell composition in human non-small cell lung cancer. Front Immunol (2019) 9:3101. doi: 10.3389/fimmu.2018.03101
46. Prado-Garcia H, Romero-Garcia S, Aguilar-Cazares D, Meneses-Flores M, Lopez-Gonzalez JS. Tumor-induced CD8+ T-cell dysfunction in lung cancer patients. Clin Dev Immunol (2012) 2012:741741. doi: 10.1155/2012/741741
47. Sheng SY, Gu Y, Lu CG, Zou JY, Hong H, Wang RF. The distribution and function of human memory T cell subsets in lung cancer. Immunol Res (2017) 65:639–50. doi: 10.1007/s12026-016-8882-y
48. Amaria RN, Reddy SM, Tawbi HA, Davies MA, Ross MI, Glitza IC, et al. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat Med (2018) 24:1649–54. doi: 10.1038/s41591-018-0197-1
49. Suresh K, Naidoo J, Lin CT, Danoff S. Immune checkpoint immunotherapy for non-small cell lung cancer: Benefits and pulmonary toxicities. Chest (2018) 154:1416–23. doi: 10.1016/j.chest.2018.08.1048
50. Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD-L1 checkpoint. Immunity (2018) 48:434–52. doi: 10.1016/j.immuni.2018.03.014
51. Leclerc M, Voilin E, Gros G, Corgnac S, de Montpréville V, Validire P, et al. Regulation of antitumour CD8 T-cell immunity and checkpoint blockade immunotherapy by neuropilin-1. Nat Commun (2019) 10:3345. doi: 10.1038/s41467-019-11280-z
52. Legat A, Speiser DE, Pircher H, Zehn D, Fuertes Marraco SA. Inhibitory receptor expression depends more dominantly on differentiation and activation than “exhaustion” of human CD8 T cells. Front Immunol (2013) 4:455. doi: 10.3389/fimmu.2013.00455
53. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N Engl J Med (2012) 366:2443–54. doi: 10.1056/nejmoa1200690
54. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature (2014) 515:563–7. doi: 10.1038/nature14011
55. Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res (2014) 20:5064–74. doi: 10.1158/1078-0432.CCR-13-3271
56. Shaver KA, Croom-Perez TJ, Copik AJ. Natural killer cells: The linchpin for successful cancer immunotherapy. Front Immunol (2021) 12:679117. doi: 10.3389/fimmu.2021.679117
57. Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol (2018) 19:723–32. doi: 10.1038/s41590-018-0132-0
58. Pansy K, Uhl B, Krstic J, Szmyra M, Fechter K, Santiso A, et al. Immune regulatory processes of the tumor microenvironment under malignant conditions. Int J Mol Sci (2021) 22:13311. doi: 10.3390/ijms222413311
59. Marin-Acevedo JA, Dholaria B, Soyano AE, Knutson KL, Chumsri S, Lou Y. Next generation of immune checkpoint therapy in cancer: New developments and challenges. J Hematol Oncol (2018) 11:39. doi: 10.1186/s13045-018-0582-8
60. Chauvin JM, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C, et al. TIGIT and PD-1 impair tumor antigen-specific CD8+ T cells in melanoma patients. J Clin Invest (2015) 125:2046–58. doi: 10.1172/JCI80445
61. Lanitis E, Dangaj D, Irving M, Coukos G. Mechanisms regulating T-cell infiltration and activity in solid tumors. Ann Oncol (2017) 28:xii18–32. doi: 10.1093/annonc/mdx238
62. Harjunpää H, Guillerey C. TIGIT as an emerging immune checkpoint. Clin Exp Immunol (2020) 200:108–19. doi: 10.1111/cei.13407
63. Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, et al. T-Cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature (2017) 545:60–5. doi: 10.1038/nature22079
64. Pesce S, Greppi M, Grossi F, Del Zotto G, Moretta L, Sivori S, et al. PD/1-PD-Ls checkpoint: Insight on the potential role of NK cells. Front Immunol (2019) 10:1242. doi: 10.3389/fimmu.2019.01242
65. Alvarez M, Simonetta F, Baker J, Morrison AR, Wenokur AS, Pierini A, et al. Indirect impact of PD-1/PD-L1 blockade on a murine model of NK cell exhaustion. Front Immunol (2020) 11:7. doi: 10.3389/fimmu.2020.00007
66. Oyer JL, Gitto SB, Altomare DA, Copik AJ. PD-L1 blockade enhances anti-tumor efficacy of NK cells. Oncoimmunology (2018) 7:e1509819. doi: 10.1080/2162402X.2018.1509819
67. Xiong X, Chen S, Shen J, You H, Yang H, Yan C, et al. Cannabis suppresses antitumor immunity by inhibiting JAK/STAT signaling in T cells through CNR2. Signal Transduct Target Ther (2022) 7:99. doi: 10.1038/s41392-022-00918-y
68. Danial Yahyal M, Watson RR. Immunomodulation by morphine and marijuana. Life Sci (1987) 41:2503–10. doi: 10.1016/0024-3205(87)90434-6
69. Taha T, Meiri D, Talhamy S, Wollner M, Peer A, Bar-Sela G. Cannabis impacts tumor response rate to nivolumab in patients with advanced malignancies. Oncologist (2019) 24:549–54. doi: 10.1634/theoncologist.2018-0383
70. Mathsyaraja H, Catchpole J, Freie B, Eastwood E, Babaeva E, Geuenich M, et al. Loss of MGA repression mediated by an atypical polycomb complex promotes tumor progression and invasiveness. Elife (2021) 10:e64212. doi: 10.7554/eLife.64212
71. Kienzl M, Hasenoehrl C, Valadez-Cosmes P, Maitz K, Sarsembayeva A, Sturm E, et al. IL-33 reduces tumor growth in models of colorectal cancer with the help of eosinophils. Oncoimmunology (2020) 9:1–12. doi: 10.1080/2162402X.2020.1776059
72. Strauss L, Mahmoud MAA, Weaver JD, Tijaro-Ovalle NM, Christofides A, Wang Q, et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci Immunol (2020) 5:1–27. doi: 10.1126/sciimmunol.aay1863
73. Lucarini V, Ziccheddu G, Macchia I, La Sorsa V, Peschiaroli F, Buccione C, et al. IL-33 restricts tumor growth and inhibits pulmonary metastasis in melanoma-bearing mice through eosinophils. Oncoimmunology (2017) 6:e1317420. doi: 10.1080/2162402X.2017.1317420
74. Alter G, Malenfant JM, Altfeld M. CD107a as a functional marker for the identification of natural killer cell activity. J Immunol Methods (2004) 294:15–22. doi: 10.1016/j.jim.2004.08.008
75. Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res (2001) 29:e45. doi: 10.1093/nar/29.9.e45
76. Grill M, Hasenoehrl C, Kienzl M, Kargl J, Schicho R. Cellular localization and regulation of receptors and enzymes of the endocannabinoid system in intestinal and systemic inflammation. Histochem Cell Biol (2019) 151:5–20. doi: 10.1007/s00418-018-1719-0
Glossary
Keywords: CB1, CB2, cannabinoid receptors, non-small cell lung cancer, tumor microenvironment, CD8+ T cells, NK cells, immunotherapy
Citation: Sarsembayeva A, Kienzl M, Gruden E, Ristic D, Maitz K, Valadez-Cosmes P, Santiso A, Hasenoehrl C, Brcic L, Lindenmann J, Kargl J and Schicho R (2023) Cannabinoid receptor 2 plays a pro-tumorigenic role in non-small cell lung cancer by limiting anti-tumor activity of CD8+ T and NK cells. Front. Immunol. 13:997115. doi: 10.3389/fimmu.2022.997115
Received: 18 July 2022; Accepted: 07 December 2022;
Published: 09 January 2023.
Edited by:
Gulderen Yanikkaya Demirel, Yeditepe University, TurkeyReviewed by:
Fernando Guimaraes, Diamantina Institute, The University of Queensland, AustraliaMaite Alvarez, University of Navarra, Spain
Copyright © 2023 Sarsembayeva, Kienzl, Gruden, Ristic, Maitz, Valadez-Cosmes, Santiso, Hasenoehrl, Brcic, Lindenmann, Kargl and Schicho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rudolf Schicho, cnVkb2xmLnNjaGljaG9AbWVkdW5pZ3Jhei5hdA==
†ORCID: Arailym Sarsembayeva, orcid.org/0000-0001-9793-0789
Melanie Kienzl, orcid.org/0000-0001-6508-176X
Eva Gruden, orcid.org/0000-0001-6376-5842
Dusica Ristic, orcid.org/0000-0001-7821-5595
Paulina Valadez-Cosmes, orcid.org/0000-0002-8693-1521
Ana Santiso, orcid.org/0000-0003-3436-8121
Luka Brcic, orcid.org/0000-0002-9098-8416
Jörg Lindenmann, orcid.org/0000-0003-1276-7666
Julia Kargl, orcid.org/0000-0002-0870-0816
Rudolf Schicho, orcid.org/0000-0002-5726-4731