 Yuehan Xiong
Yuehan Xiong Menghua Cai
Menghua Cai Yi Xu1
Yi Xu1 Peng Dong
Peng Dong Hui Chen
Hui Chen Jianmin Zhang
Jianmin Zhang- 1Department of Immunology, Chinese Academy of Medical Sciences (CAMS) Key Laboratory of T Cell and Cancer Immunotherapy, State Key Laboratory of Medical Molecular Biology, Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences (CAMS) and School of Basic Medicine, Peking Union Medical College, Beijing, China
- 2Changzhou Xitaihu Institute for Frontier Technology of Cell Therapy, Changzhou, China
Spondyloarthritis (SpA) refers to a group of diseases with inflammation in joints and spines. In this family, ankylosing spondylitis (AS) is a rare but classic form that mainly involves the spine and sacroiliac joint, leading to the loss of flexibility and fusion of the spine. Compared to other diseases in SpA, AS has a very distinct hereditary disposition and pattern of involvement, and several hypotheses about its etiopathogenesis have been proposed. In spite of significant advances made in Th17 dynamics and AS treatment, the underlying mechanism remains concealed. To this end, we covered several topics, including the nature of the immune response, the microenvironment in the articulation that is behind the disease’s progression, and the split between the hypotheses and the evidence on how the intestine affects arthritis. In this review, we describe the current findings of AS and SpA, with the aim of providing an integrated view of the initiation of inflammation and the development of the disease.
Introduction
Ankylosing spondylitis (AS) is a classic type of inflammatory disease that starts usually with an inflammation in the sacroiliac (SI) joint and ends with the fusion of the spine with a pathognomonic feature called the “bamboo spine”. It belongs to a group of diseases named spondyloarthritis (SpA), featuring inflammation flaring up in the spine, peripheral joints, ligaments, and tendons.
Along with the rapid advancement in medicine, the classification of SpA keeps changing, which in turn inspires as well as limits the way we study it. Initially, AS was recognized as a kind of disease related to rheumatoid arthritis (RA) or rheumatoid spondylitis (1). RA has the hallmark of persistent symmetrical peripheral polyarthritis, including in the hands and feet, and it can affect the cervical spine, causing neck pain and stiffness, while AS usually begins with inflammation in the bilateral lumbosacral joints and less frequently involves peripheral joint inflammation; though, AS was not clearly distinguished from rheumatoid disorders until the discovery of the rheumatoid factor and anti-citrullinated protein antibody (ACPA) (2). In 1974, Moll and Wright grouped non-RA inflammatory diseases into an inter-related family named ‘seronegative spondyarthrides’, including AS, psoriatic arthritis (PsA), reactive arthritis (also known as Reiter’s disease), inflammatory bowel disease (IBD), etc (3). This classification scheme was not widely accepted, yet it did mark off diseases that have considerable comorbidity rates and share signaling cellular pathways in hindsight (4, 5). The classification criteria launched by the Assessment of Spondyloarthritis International Society (ASAS) in 2009 is regarded as a landmark in AS classification, recognizing axSpA as the only entity in comparison to peripheral SpA (pSpA), and the term ‘ankylosing spondylitis’ was fundamentally replaced by ‘radiographical axSpA (r-axSpA)’ (6). These criteria were modified for better classification of the patients and recognition of the medical demand for axSpA that presents unreadable or mild radiographical change (non-radiographical axSpA, or nr-axSpA). However, as a practical standard in classifying patients and avoiding people with unprovoked inflammation in their back but no MRI sign, the term axSpA limits efficient discussion on more closely inter-related constellation of features, including SI damage, HLA-B27, and anterior uveitis that is much more prevalent in former defined AS patients than the other kinds of axSpA or r-axSpA, such as PsA (7). Therefore, AS, as a concept that may or may not manifest on individuals simultaneously, still affords us a useful model to explore the mechanism of pathogenesis.
The pathogenic mechanism of AS remains obscure, but several hypotheses about the initiating process have been proposed. The first one is a direct inference based on ‘self-nonself’ immunology that autoinflammation should be attributed to specific arthritogenic peptides, i.e., aiming to discover a molecular mimicry between the foreign and self-peptide. Despite growing evidence of T-cell clonal expansion in patients, the existence of conclusive common antigens is still challenging to verify (8). Some other theories involve the conformational plasticity of the most critical risk gene, HLA-B27, which has a high prevalence of genetic variants among AS patients (9), as the cause of AS. A series of researches highlights the unfolded protein response (UPR), which is activated during the biosynthesis process of HLA-B27 alleles in the endoplasmic reticulum (ER), due to its consequences of inflammation and autophagy. An error-prone folding process of specific HLA-B27 alleles can instigate endoplasmic reticulum stress (ERS), which activates the UPR and leads to necrotic death and cytokine secretion. Surface-expressed HLA-B27 molecules with unconventional conformation may activate the immune system through the intrinsic HLA monitoring receptor, the killer immunoglobulin-like receptors (KIRs) expressed by CD4+ T cells, and natural killer (NK) cells.
Deep understanding of T cell-mediated inflammation has spawned the cytokine blockade strategy, but little is known why this strategy leads to overreaction and underreaction in different situations. In this review, we present organized evidence about how pathophysiological factors contribute to AS development, analyze the consistency among different hypotheses, and make an attempt to reconcile immunological understanding in other fields with findings in AS.
Cause of AS: Infection or danger?
The danger theory and AS
A successful immune response would be a quick strong response targeting invading foreign organisms by recognizing its antigenic components via immunoglobulin receptors, as the prototypical ‘self-nonself’ theory indicates. Two major modified versions of the theory have been raised to describe the nature of the ligands, known as Charles Janeway’s ‘infectious-nonself’ theory and Polly Matzinger’s ‘danger model’ (10, 11). An adaptive immune response is initiated by the activation of nonclonally rearranged receptors, for instance, the interaction between LPS and Toll-like receptors (TLRs). Janeway proposed these ligands possessing an exogenous nature related to infectious pathogens, termed ‘pathogen-associated molecular patterns (PAMPs)’, while Matzinger held the view that the immune system is more concerned with dangerous conditions, in which chemical substances, named by ‘danger-associated molecular patterns (DAMPs)’, are recognized by the immune system.
With growing knowledge of stimulatory and inhibitory signaling pathways of immunocytes, the canonical model has been expanded. It was known that the activation of naïve T cells requires a primary response when PAMP/DAMPs or cytokine-activated antigen-presenting cells (APCs) simultaneously deliver antigens to TCR (signal one) and costimulatory signals (signal two); while memory T cells only react after re-encountering the same antigen (epitope). However, the bystander activation process is not dependent on antigen encountering. Early in the 90s, Tough et al. have already confirmed that both CD4 and CD8 T cells can be activated with bacterial/viral PAMP but TCR signaling, namely, LPS and poly(I:C) (12, 13). Recent discoveries in animal models such as experimental autoimmune encephalomyelitis (EAE), a model induced by autoantigen to mimic human multiple sclerosis (MS), confirmed that most of the infiltrating T cells are not provided with autoantigen-specificity, while the severity of inflammation could be rescued by knocking down TLRs (14).
Perhaps two suppositions could be made on the initial stage of AS: 1) the production of autoantigen has a feature of tissue-specific expression, incurring attack directed by adaptive immunity; or 2) there is a persistent damaged tissue in the axis, releasing signals and priming and bystander-activating immune cells. These signals, either DAMPs or pathogen-associated molecular patterns (PAMPs), can work to offset peripheral tolerance and allow the immune system to attack itself (Figure 1).
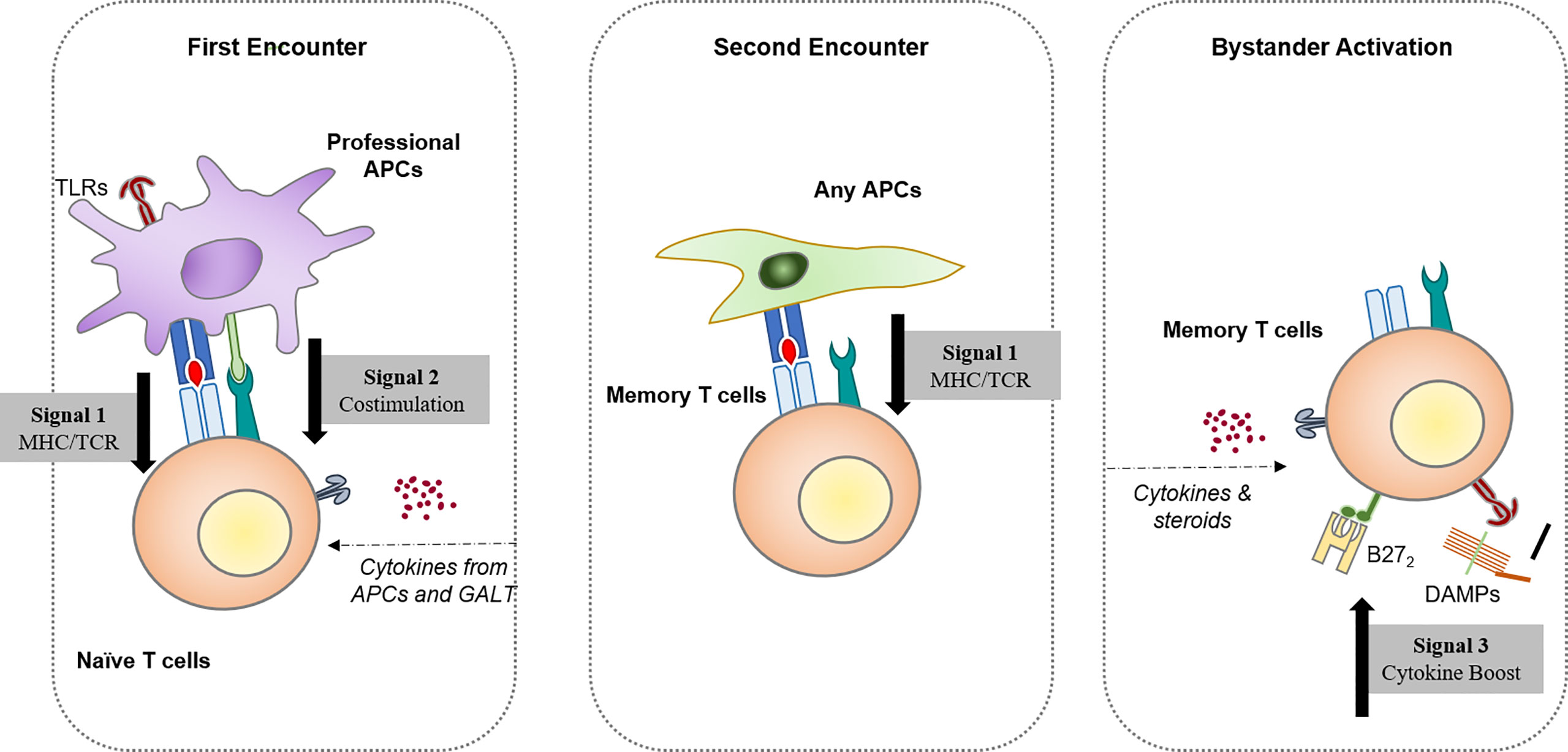
Figure 1 Three different ways resting/naïve T lymphocytes become activated. Activation by the first encounter requires naïve lymphocytes associated with professional antigen-presenting cells, namely, dendritic cells. The second encounter with the antigen is enough to generate a fast, strong response for memory lymphocytes in the resting state. Hypotheses based on the canonical first/second encounter models led to an exploration of potential cross-reactive antigens that elicit spondyloarthritis after pathogen infection. With improved knowledge of the immune regulatory network, the antigen-independent activation of T cells has been identified, which provides a possible pathway for autoreactive T cells that bypasses central tolerogenesis mechanisms.
Searching for antigens
Molecular mimicry is a common etiological mechanism caused by similar antigenic epitopes shared between self and foreign antigens. Hence, an infection-driven immune response would result in a self-reactive immune system and unresolved inflammation. Early results suggested Klebsiella pneumoniae, a Gram-negative bacterium causing pneumonia and sepsis, as a potential candidate. It was first related to AS development due to its fecal carriage and increased serum antibody levels compared to those in HLA-B27-negative patients (15). Then members from Enterobacteriaceae including Yersinia, Salmonella, and Shigella are considered as potential candidates for causing typhoid fever and sequelae of arthritis. The cross-reactivity between HLA-B27 and K. pneumoniae was well-studied (16, 17), and underlying molecular mimicry was found, including a homologous hexa- or octapeptide from K. pneumoniae nitrogenase (residues 188-193) and HLA-B*2701 (residues 72-77) (18, 19), a tetrapeptide from bacterial pullulanase and collagens (20), and a dodecapeptide from bacterial dipeptidase and collagens (21). The same situation was revealed for Candida albicans infection, a dimorphic fungus colonizing the intestinal mucosa, where some research suggests a positive correlation among patients (22) but others did not (23, 24). These conflicting results might be an outcome of the complexity of the intestinal microbiome, where either commensal or pathogenic bacteria generate antibody response, and their function as facultatively causal, consequential, and bystanders to diseases requires arduous work to confirm.
A macroscopic demonstration was given by a recent meta-analysis incorporating 1.3 million cases of AS and 7.6 million healthy controls, which found that there was no significant contribution from bacterial infection (RR [95% CI] = 0.70 [0.10–4.78]), even though previous infections increase the AS risk (25). In comparison, viral infection significantly contributes to AS (RR [95% CI] = 1.43 [1.22–1.66]) (25). HLA-B27 can recognize and present peptides of various viral origins, including influenza A, human immunodeficiency virus (HIV), hepatitis C virus (HCV) (26–29), etc. At the same time, a follow-up study showed that former HPV infection increases the risk of AS (30). Two cohort studies suggested an inverse effect of HIV infection on AS development, suggesting the protective role of an overactivated immune system against viral infection (31, 32). However, even if viral infections increase the susceptibility to AS, it elicits another question about the role of HLA-B27 molecule during this process. Since most peptide-presenting function is inherent to HLA-B27 allotypes (33), these observations suggest that the risk of persistent viral infection is not dependent on a specific antigen but on the pathogenic induction of immune response (unless an epitope expansion occurs).
Recent advances
Both computational biology and transcriptomics have made great progress in recent years. A host of studies have been conducted with 16S rRNA sequencing and shotgun metagenomic sequencing in search of specific intestinal microorganisms enriched in AS patients. Some bacterial peptides derived from the enriched population are found to match HLA-B27 epitopes according to the prediction tool (the immune epitope database, IEDB) (34, 35). One peptide from Bacteroides fragilis mimicking human type II collagen is suggested to be able to interact with HLA-B27 and stimulate the IFN-γ production of PBMC (34). However, it is uncertain whether the pro-inflammatory effect of that peptide depends on carrying the HLA-B27 allotype or heterogeneous inflammatory state among AS patients and healthy controls, the direct binding between HLA-B27 and the peptide appear unlikely in the original study (36). Besides, few consensuses have been reached among other studies: some research confirmed the difference of both microbiota diversity and abundance of specific species, associated with HLA-B27 (37, 38); one suggested that HLA-B27 is not involved in the shift of microbiota during the disease (39), and some discovered an insignificant difference in α-diversity between the AS and HC groups, although specific species correlate with disease activity (40–42). Clonal expansion of CD4+ and/or CD8+ T cells is also analyzed with immune repertoire sequencing, implying specific antigen-TCR binding in AS (43–45). However, inferring the CDR3 sequences to antigen epitopes is still challenging. Schittenhelm et al. utilized mass spectrometry to enrich HLA-B27 binding peptides with different affinities among HLA-B27 allotypes, but there was only a slight change in abundance (46). Nonetheless, research combining the peptidome and TCR motif brought about some consensual outcomes that disease-related HLA-B27 serotypes bind peptides with C-terminal elongation and specific amino acids are enriched (43, 47, 48). As some studies suggest, this could either be fundamental for the cross-reactive immune response or a consequence of a malfunctioning MHC assembly (48, 49).
In brief, although many research studies suggested a role of HLA-B27 as a direct antigen or mediator of pathogen entry, there are limited clues supporting the causal relation that AS is a sequel to this molecular mimicry process. Instead, HLA-B27 has a potential in providing immunostimulation according to recent investigations for its intercellular and intracellular function. For instance, HLA is under the surveillance of the immunoglobulin-like receptor (KIR) family by the killer cell. KIRs have extraordinary polymorphism and the capability to ‘license’ killer cell function during sensing altered HLA expression and conformers (50). Moreover, it is noteworthy that HLA-B27-related hypotheses could be overrepresented, since only a small percentage of HLA-B27-positive individuals are prone to develop the disease (51); AS patients could be likewise misdiagnosed for carrying HLA-B27, according to a recent study in Spain (52).
From injury to inflammation
Enthesitis and synovitis
Our lumbar spine and lower extremities experience high mechanical stress as upright standing species. In the attachment of the tendon or ligament to the bone, the connective part (the enthesis) is easily damaged by stress. This could be one reason why inflammation of entheses, or enthesitis, usually occurs at these sites, particularly in AxSpA (53). Entheses have a similar structure to the growth plate (epiphyseal plate), in which expansion and differentiation of chondrocytes make up a continuous gradient from the uncalcified tendon to the calcified bone. They and their adjacent tissue synovium, named ‘synovio-entheseal complexes (SECs)’ (54), are likely to represent a highly vulnerable part to inflammation. In 1971, John Ball made a very insightful conclusion based on pathological evidence that AS has less destructive erosive synovitis than rheumatoid disease, with unique inflammatory enthesopathy (55). A more modern view of enthesopathy today might be that the repetitive microtrauma as a consequence of body movement could provide a very early signal to initiate SpA and be regarded as a prodromal symptom of it (56, 57). By unloading physical stress in the hind limbs of TNF-overexpressing mice, Jacques and colleagues proved that enthesitis and osteophytes develop only under adequate mechanical stimulation, which was later confirmed in both collagen-induced arthritis (CIA) and collagen antibody-induced arthritis (CAIA) models (58, 59). A discrepancy in body weight bearing leads to different levels of inflammation in different parts of the tarsal and metatarsal bones, reminding us of how AS erodes large joints. However, this model may elucidate only one piece of the genuine disease development in humans. Although the site of enthesitis is found to be connected with bone erosion in humans (60), a cohort covering HLA-B27+ subclinical SpA patients revealed that most individuals reported arthralgia, 19% reported inflammatory back pain, and only 5% reported enthesitis (61). The difference between the hypothesis and the findings highlights that enthesitis may be needed for bone lesions rather than inflammation and encourages more planned comparative studies to determine what happens in the early stages of the disease.
Nonantigenic stimuli from cartilage
Modeled after human RA, the phenotype discovered in both CIA and CAIA mice may still offer some insights into the mechanism of AS development, as efforts to screen cartilage biomarkers for diagnostic and prognostic utility found that peptides derived from type II collagens are correlated with tissue destruction and AS severity (62, 63). It was suspected that type II collagen, the major component of hyaline cartilage, is a potential target antigen during the disease process (64). Similarly, Zou and his colleagues demonstrated that the G1 domain of aggrecan, a large proteoglycan in articular cartilage, is a targetable epitope in AS and RA patients (65, 66). They found that G1 peptide-specific CD8+ T cells exist in more than half of the patients, which unfortunately elicited no further exploration. In retrospect, CD8+ T cells, even in terms of adaptive immunity, have a questionable role in disease initiation. As shown by the HLA-B27/huβ2m rat model, CD8+ T-cell deficiency does not influence the process of SpA initiation (67, 68). Moreover, the pathological imbalance in CD8+ T-cell frequency is not influenced by TNFα inhibition therapy, which has proven its efficacy in treating AS patients (69). It was not until Plow and Kollias developed CIA in Rag1-/- immune-deficient mice that the nonantigenic function of type II collagen became obvious (70). Lambert et al. identified a specific fragment derived from type II collagen, named Coll2-1, which can activate synoviocytes to secrete IL-8 (CXCL8) in a TLR-4-dependent manner (71).
In addition to collagen, hyaluronan is an essential component of the extracellular matrix (ECM) and synovial fluid (SF) which is degraded rapidly with aging and inflammation (72). In AS patients, serum hyaluronic acid is also slightly elevated (p = 0.04) and is correlated with some clinical features, such as c-reactive protein (CRP) test, Schober’s test, and the finger-to-floor distance (73). The effect of hyaluronan in inflammation is related to its molecular weight; lower-molecular-weight hyaluronic acid has been reported to facilitate the immune response by binding TLR2 and TLR4 (74, 75), while higher-molecular-weight hyaluronic acid has been demonstrated to be anti-inflammatory through the CD44 signaling pathway (76). Parallel to these findings, it has been shown that TLR1 and TLR2 are strongly expressed in primary human chondrocytes (77). The expression of TNFα and TLR1/2 could even be drastically upregulated by TNFα or TLR1/2 stimuli. Moreover, chondrocytes are able to worsen cartilage degradation by the upregulation of matrix metalloproteinases (MMPs), cathepsin B, and L, which antagonizes the MMP inhibitor (TIMP-1/2) and downregulates ECM proteins (78–80). These findings collectively suggest a very proinflammatory function of chondrocytes during AS development.
Landscape of innate immunity in AS
Generally, AS is characterized by neutrophil and macrophage/monocyte expansion and synovial infiltration. The hemogram of AS patients shows an elevated neutrophil to lymphocyte ratio (NLR), platelet to lymphocyte ratio (PLR), and monocyte to lymphocyte ratio (MLR) compared to healthy individuals (81–83). All of these features are positively correlated with ESR and CRP, wherein MLR is believed to be a better diagnostic parameter than the others. Immunohistology also demonstrated shared features among non-RA SpA, including AS, PsA, ReA, and JIA, and CD163+ M2 macrophage and neutrophil counts were greater in synovial biopsies than in RA and HC (84–86). This specific myelopoiesis pattern could be attributed to different causes, including enhanced monopoiesis/neutropoiesis in the bone marrow (BM) and extramedullary sites, noncanonical precursor differentiation, and the pro-survival effect of cytokines.
In addition to the commonly discussed effect of elevated inflammatory cytokines such as GM-CSF or M-CSF, it has been suggested that monocytes can be generated with an alternative protocol in AS. A traditional paradigm of monocyte differentiation involves sequential binary decisions from granulocyte and monocyte progenitors (GMPs) to monocyte/DC progenitors (MDPs) and finally to common monocyte precursors (CMPs). Since the identification and nomenclature of myeloid cells in the published literature may face a problem of consistency (87), detailed cell identities will not be overly emphasized. It has been proposed by Yáñez et al. that GMP and MDP are derived independently from GMP and are both able to generate so-called Ly6Chi ‘classical’ monocytes in mice, which stands for different means of emergency monopoiesis (88). Two diverse microbial components have been demonstrated to induce different responses. Lipopolysaccharide (LPS), which is conventionally recognized by TLR4 and TLR2 to a degree, stimulates both neutrophil and (neutrophil-like) monocyte differentiation from GMPs, while unmethylated CpG, recognized by TLR9, stimulates monocyte and conventional DC production (88). In line with their observations, increasing evidence suggests a similar process in AS myelopoiesis. In curdlan-injected SKG mice, myelopoiesis is skewed toward GMP, and extramedullary proliferation of GMP is induced by GM-CSF secreted by CD4+ T cells and mast cells (89). In humans, monocytes from HLA-B27+ AS patients show an adaptation toward a GMP-driven neutrophil-like phenotype when challenged by LPS and cytokines (90).
Moreover, the adaptation of innate immunity, mediated by epigenetic reprogramming, could last a long time. This so-called ‘trained immunity’ has been proven in vaccine- or adjuvant-treated models; for instance, mice treated with β-glucan (a PAMP that activates dectin-1) show increased myeloid cell expansion and enhanced proinflammatory cytokine production from monocytes (91, 92). Mitroulis et al. have demonstrated that hematopoietic stem cells (HSC) from mice treated with β-glucan can preserve a biased differentiation pattern that prefers GMP differentiation after being transferred to irradiated recipient mice for 12 weeks (93). Since AS patients have TLR stimulants including hyaluronan, collagen fragments, and TNFα floating in the SF and peripheral blood (PB), this novel mechanism of innate immunity training can be regarded as a theoretical underpinning of cellular pathogenesis.
It has been well studied that damaged synovium/entheses are able to produce various kinds of inflammatory cytokines and chemokines, engaging in myeloid cell recruitment, activation, polarization, and even osteoclast (trans-) differentiation. Entheseal mesenchymal cells from a mouse model have been demonstrated to secrete CXCL1 and CCL2 (MCP1), which bind CXCR2 and CCR2/4, respectively, in response to mechanical stress (59). Ex vivo mesenchymal stem cells (MSCs) from AS patients also show increased CCL2 production (94). Correspondingly, CCR2+ M2 macrophages (CD163+) are largely increased in PB and synovial biopsies of SpA patients and are correlated with AS disease activity (86, 95–97). This axis participates not only in chemotaxis but also shapes the polarization of macrophages, as CCR2 blockade polarizes macrophages toward an inflammatory M1 phenotype (98, 99). Neutrophil infiltration may also benefit from CCL2/CCR2, as high CCR2 expression and responsiveness are observed in RA patients and an antigen-induced arthritis (AIA) mouse model (100). Research on the neutrophil-specific chemokine CXCL8 shows controversial outcomes, as synovial CXCL8 has been suggested to be elevated only in the SF of RA, aside from other SpA (101, 102), while serum CXCL8 is higher in PsA and AS than in RA (103). These results further suggest a stepwise recruitment protocol of AS wherein neutrophils are recruited by endogenous chemoattractants through vessels and are eventually assembled by TLR stimuli and/or complement components in articulate tissues (104).
The IL-23/IL-17 axis
The findings of nonantigenic immune activation do not necessarily cancel the importance of adaptive immunity. In contrast, the reduction in the severity of disease in immune-deficient mice confirms a harmful influence of activated lymphocytes, when the IL-23/IL-17 axis has been long suspected to play important role in AS development. In the canonical model of the IL-23/IL-17 axis, the inflammatory IL-17 family as well as other cytokines such as IL-22, TNFα, IL-6 is secreted by Th17 cells, which is induced and maintained by IL-23. A general activation of this axis in AS is proven by an elevated level of IL-23 and IL-17 found in a wide range of AS patients (105–107). Other than Th17 cells, tissue-resident innate immunocytes, such as γδT17 cells and ILC3s, possess a comparable ability in secreting IL-17 and IL-22 (108–110). These cells are the innate counterpart of Th17 cells and have been proven to drive SpA development in IL-23-overexpressing mouse models.
However, after achieving a favorable response rate in PsA and CD, IL-23 blockers failed to show efficacy in treating AS (111–113). Despite evidence from animal models that IL-23-driven IL-17 can stir up enthesitis and SpA (108–110), its causality to AS development has never been settled. According to current evidence, the indeterminate relationship between IL-23 and IL-17 can be explained by redundant IL-17-inducing pathways. Cuthbert et al. firstly reported that the Vδ1 subset of γδT lacking IL-23R is able to secrete IL-17 and IL-22 under anti-CD3/CD28 (mimicking TCR stimulation) or phorbol myristate acetate (PMA) stimulation (114), which provides a substitutional mechanism for IL-23/IL-17 axis. Another potential IL-23-independent activator is prostaglandin E2 (PGE2), a principal mediator of inflammation, which is widely observed to be elevated among AS patients that made the response rate to NSAIDs that inhibits the biosynthesis of PGE2 a criterion for AS diagnosis. Early studies reveal its capability to stimulate IL-17 production and proliferation synergistically with IL-23 in vitro (115, 116), while later an IL-23-independent manner is found on Th17 cells from methylated bovine serum albumin (mBSA)-induced RA model mice as well as RA patients (117, 118). PGE2 is transduced through the prostaglandin E2 receptor 2 subtype (EP2) and the 4 subtype (EP4) expressed on the T cell surface. Under normal conditions, the signaling pathway from PGE2 to the Th17 effector function is regulated in a negative feedback manner, where RAR-related orphan receptor-γ (RORC), the lineage-defining transcriptional factor of Th17, silences EP2 expression by directly binding to its protein coding gene PTGER2 (119). However, the pathogenic Th17 from MS patients shows impaired binding to PTGER2, resulting in an unsuppressed EP2 level and stronger PGE2 signaling. Considering strengthened PGE2 signaling in AS patients compared to HC (120), it remains to be seen that the lacking regulatory capacity of PGE2 signaling could promote AS development by activating Th17 cells. Likewise, a heterogeneity in inflammatory signaling has also been identified in PsA, in that patients with higher skin scores have lower arachidonic acid-derived oxylipins (such as PGE2); on the contrary, those with higher oxylipins are associated with enthesitis (121). This is corroborated by better cutaneous response and limited osteoarticular response to IL-23 inhibitors observed in PsA patients (122). The parallel pro-inflammatory role of IL-23 and PGE2 in both diseases is worthy of further examination.
Searching an inhibitor for IL-17-producing cells brings us to a deeper understanding of IL-17 function. Interesting results are found in screened RORC inhibitors, in that ideally suppressing Th17 function should rescue animal models from developing inflammation. Guendisch et al. first reported that the RORC inhibitor cpd 1 efficiently reduces major Th17 cytokines, including IL-17A, IL-17F, and IL-22, resulting in attenuation in mBSA-immunized antigen-induced arthritis rats (123). Another RORC antagonist, BIX119, has been reported to ablate IL-17 production selectively while sparing IL-22-producing subsets in human SpA- and CD-derived cells (124, 125). Van Tok and colleagues found that using the same compound in vivo reduced serum IL-17A, IL-17F, and IL-22 but accelerated the mean onset and worsened the severity in the HLA-B27/huβ2m rat model (126). This paradoxical effect may easily remind us of anti-IL17 treatment in IBD patients (127), but no intestinal inflammation or weight loss was detected in these rats. This counterintuitive problem brings about a deeper examination of the capacity of Th17, in that if it is not a consequence of an imbalanced intestinal immunity, the loss of IL-17 or IL-22 should contribute to the pathogenicity in another way. Chong et al. revealed that the neutralization of IL-17A with a monoantibody abolished the expression of IL-24 and enhanced IL-17F and GM-CSF secretion in autoimmune uveitis and its model (128). They suggested that autocrine IL-17 activates NFκB, leading to IL-24 production, which in turn represses IL-17F and GM-CSF production via the SOCS1/3 pathway. This discovery was later supported by a study in EAE mice, where Rorc-/- increases GM-CSF production in CD4+ T cells and splenocytes (129). These two elegant studies describe the positive and negative feedback in Th17-cell functioning, where GM-CSF could stimulate macrophages to produce more IL-23 to skew Th17 polarization and increase GM-CSF production, which is suspended when a high concentration of IL-17A activates SOCS1/3 and blocks the expression of GM-CSF (Figure 2).
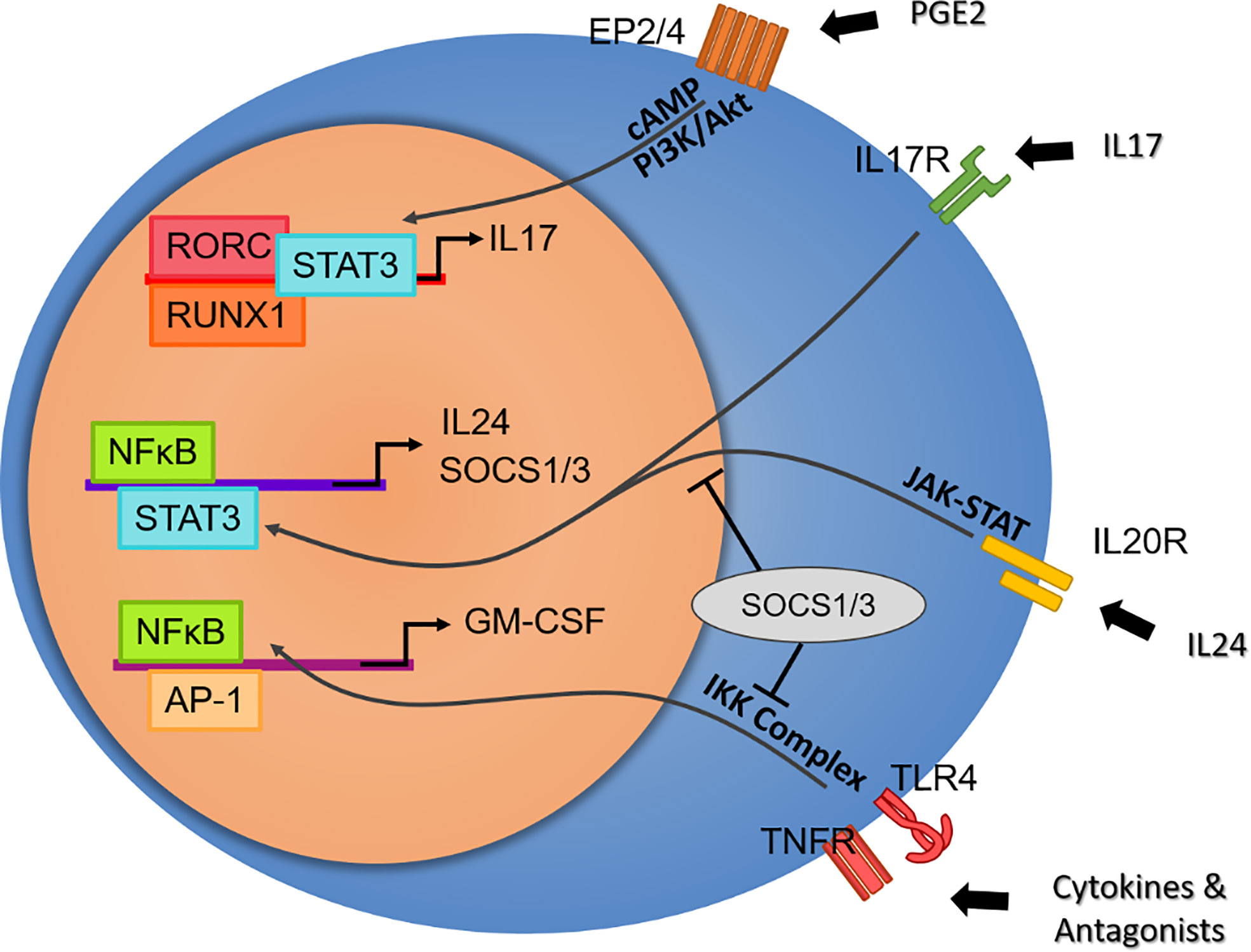
Figure 2 Potential effector axis of Th17 cells independent of IL-23 stimulation. From top to bottom: Prostaglandin E2 plays a critical role in ankylosing spondylitis by activating Th17 cells in an RORC-dependent pathway. Despite its multifaceted proinflammatory effects, IL-17 may overrepresent Th17 function. The paradoxical inflammation induced by the RORC blockade elicits the discovery of the intrinsic negative feedback of IL-17, where the IL-24 autocrine induced by IL-17 regulates cytokine production by SOCS1/3. In contrast, GM-CSF, a secret weapon of Th17, is not under the regulation of RORC. GM-CSF secretion by T cells or nonimmune cells may represent a protocol of the early stage in response to a broad range of tissue damage and DAMPs via TLR and TNFR signaling, as well as a particular pathogenic pathway in autoimmune disease.
Human Th17 cells are long-lived inflammatory cells, with abundant antiapoptotic gene expression as well as a stem-cell-like phenotype (130). Th17 population is more likely to be self-maintained after differentiation, considering their remarkable capacity to proliferate in resistance to immunosuppression compared with Th0/1/2 (131). In an HLA-B27/Huβ2m mouse model, van Tok et al. showed that anti-IL23R treatment only has a prophylactic protective effect, while anti-IL17A is able to halt inflammation and block new bone formation (132). This may answer the question of why the IL23R polymorphism is associated with AS occurrence rather than its severity (133).
Taken together, disintegrated connective tissues accumulated during the mechanical trauma of the synovium-entheses or cartilage provide enough danger signals to stimulate both innate and adaptive immunity. As a chronic disease, the innate arm can act quickly to injured articulation and sustain a proinflammatory effect by alternating the differentiation program; although there lacks direct evidence of bystander activation in AS, there are alternative ways for the adaptive arm to activate and be activated, while stimulating innate immunity.
HLA-B27 tuned immunity
Non-conventional conformers of HLA-B27
In addition to mechanical stress, Polachek et al. discovered an increased risk of enthesitis in PsA with HLA-B27-positive results among 225 patients (134). A similar effect of HLA-B27 occurs in juvenile idiopathic arthritis (JIA) (135). It seems that HLA-B27 alone is capable of perpetuating enthesitis or arthritis in the lower extremities. In the first part of this article, we mentioned that misfolded HLA-B27 is supposed to play a dual role in AS pathogenesis. Conventional MHC molecules are composed of one HLA molecule and a β2m molecule to form a functional MHC heterodimer. The unconventional conformations of HLA-B27 may exist as a free heavy chain (FHC) on the surface or form a homodimer (B272) via a disulfide bond on Cys67 (136). The presence of unconventional HLA-B27 has been reported in the intestine and joints of SpA patients (133, 137, 138). They can bind or be recognized by a set of receptors related to the KIR family, including KIR3DL2, leukocyte immunoglobulin-like receptor LILRB2, and the homologous paired immunoglobulin-like receptors (PIRs) of mice (139, 140). Samples from AS patients showed proliferative KIR3DL2+ CD4+ T cells and NK cells producing significantly more IL-17 than KIR3DL2- cells, which could be reproduced ex vivo by coculturing KIR3DL2+ cells with B272- expressing APCs (141, 142). This reaction could even be rescued by a B272- binding monoclonal antibody, which inhibits the proliferation and survival of KIR3DL2+ cells as well as the production of cytokines in vitro (133, 143). Along with these findings, the idea of HLA-B27 as a KIR stimulant is consistent with the GWAS results, in that the proinflammatory function of KIR-bearing cells is suppressed in people carrying protective HLA-B27 variants or the ERAP1 variant that causes less non-conventional HLA-B27 surface expression (144–146).
Unfolded protein and endoplasmic reticulum
The formation of an unconventional HLA-B27 conformation is also considered to be inflammatory as a trigger of the unfolded protein response (UPR). MHC class I molecules are assembled in the ER and bind endogenous proteins originating from the cytosol (147). Perturbation during this process may cause protein accumulation and impaired ER function. The conformational change of HLA-B27 is commonly detected via conformation-specific antibodies such as ME-1 and W6/32, which can detect folded HLA-B27, and HC10, which is reactive against unfolded or partially folded proteins (148). It has been reported since 2002 that after HLA-B27 heavy chain (HC) synthesis, there can be HC10-reactive peptide accumulation in the ER, while hours later, W6/32-reactive peptides emerge (149). This could be worsened without sufficient β2m or tapasin, a binding protein of ABC transporter TAP. A recent study precisely measured the timing-consumption of different alleles. It takes 3.5 h for susceptible HLA-B*2705 to construct the MHC-peptide complex and 30–90 min for nonsusceptible B*2706 and B*2709 (150). It has also been hypothesized that this prolonged time course may increase the possibility of oxidation at Cys67 and promote B272 formation (149, 151).
UPR orchestration involves one sensor protein, BiP, and three main pathways: inositol requiring 1 (IRE1), protein kinase R-like ER kinase (PERK), and activating transcription factor 6 (ATF6). Finally, the mammalian target of rapamycin (mTOR) and the c-Jun N-terminal kinase (JNK) pathways are initiated and maintain proteostasis through a reduction in protein synthesis, induction of chaperone molecules, and degradation of severely misfolded proteins via the ER-associated degradation (ERAD) pathway (152). In HLA-B27/huβ2m rats, enhanced UPR has been detected in intestinal macrophages with increased inflammatory cytokines, including IFNγ and IL-23 (153, 154). A similar difference has been observed in AS patients. Samples from peripheral blood (PB), BM, and SF demonstrated that the expansion of proinflammatory plasmacytoid dendritic cells (pDCs) is accompanied by the upregulation of the PERK pathway (155). Macrophages from PB or SF show the upregulation of UPR as measured by mRNA differential expression, enabling MHC surface expression and the secretion of cytokines, including TNFα and IFNγ (156–158).
In contrast, Ambarus and colleagues found no evidence of UPR in B27-positive macrophages from PB and SF using the same markers as in previous studies (BiP, CHOP, ERdj4) (159). It is contended by Ciccia that autophagy (MAP1LC3A, ATG5/12) but not UPR is activated in AS (160). They investigated intestinal biopsies from AS and Crohn’s disease (CD) patients with both immunohistochemistry and RT-qPCR, and only ERAD ubiquitin ligase SYVN1 was colocalized with HLA-B27 FHCs. In contrast, the findings from the team of Rik Lories support the downregulation of autophagy-related genes (ATG16L1, IRGM, HSP90AA1) in PB (161). Further research revealed autophagy dysfunction with the downregulation of MAP1LC3A and ATG5/12/16L1 in PBMCs, covering all markers used in the previous study (162). This dysfunction is attributed to a decrease in the autophagy mediator lncRNA GAS5.
The function of autophagy is highly contextual, and both increased and decreased autophagy may be involved in disease (163). Autophagy can improve the clearance of pathogenic proteins (164), inhibit apoptotic cell death (165), and obstruct the NLRP3 inflammasome via the autophagic removal of NLRP3 activators (166). In the context of autoinflammatory disease, autophagy seems to be more protective. Rapamycin could induce autophagy by blocking the mTOR pathway. Treating HLA-B27/huβ2m rats with rapamycin is able to reduce misfolded proteins by 50% and the severity of disease (167, 168). In humans, it can downregulate the IL-17 and TNFα secretion by PBMCs and inhibit the osteogenic differentiation of fibroblast-like synoviocytes ex vivo (169). With misfolded HLA-B27 accumulating in the ER, autophagy could be induced as a compensatory method for survival but could eventually be disrupted, leading to a remodeled inflammatory phenotype and cell death. This is very likely to occur in antigen-presenting cells, as they are both MHC-bearers and cytokine-producers. In other inflammatory conditions, it has been proven that imbalanced autophagy in dendritic cells and macrophages is extremely pathogenic (170). In contrast, autophagy promotes the production of lysophosphatidylcholine (LPC), an apoptotic inducer and chemotaxin, and the well-known ‘eat-me’ signal phosphatidylserine (PtdSer) (67). These signals promote the scavenging of inflammatory or even antigenic cellular contents. It can be expected that researchers will make better use of autophagy in treating AS. Recent attempts to induce autophagy and apoptotic death in synovial fibroblasts have identified emodin (171) and a combined TNF antagonist and ferroptosis inducer treatment to exert an anti-inflammatory effect (172).
HLA-B27-related proteins
Beyond HLA-B27, ERAP1, a peptide-trimming enzyme in the ER, is the second strongest risk gene for AS (173). Causing a shift in the peptidome presented by the peptide-MHC complex (174), the pathophysiological effect of ERAP1 is tightly bound to the abnormal assembly of the peptide HLA-B27 (175, 176). In 2013, Kirino et al. discovered that HLA-B51-related BD is associated with ERAP1 alleles in a recessive model based on Turkish and Japanese patients (rs17482078, combined OR [95% CI] = 4.56 [2.88–7.22]), strongly suggesting an independent effect of ERAP1 (177). Following this study, additional SNPs of ERAP1, whether protective or detrimental, were identified among Han Chinese and Turks (178, 179). Kuiper et al. also found that birdshot uveitis, which is prone to affect the HLA-A29+ population, was significantly associated with ERAP1, including rs10044354 (OR [95% CI] = 2.07 [1.58–2.71]) and rs2287987 (OR [95% CI] = 2.01 [1.51–2.67]) (180). Given the association with MHC-I risk genes, these diseases were classified by McGonagle et al. into a unified group of ‘MHC-I-opathy’, which provided a good reason for us to explore the commonality in ER dynamics other than the particularity of HLA-B27 (181). It has also been suggested that ERAP1 could intervene in the cleavage or shedding of cell surface receptors of inflammatory cytokines, which after cleavage bind to cytokines without inducing intracellular signals and thus exhibit an inhibitory function. Cui et al. first found that the level of membrane-associated TNF receptor 1 (TNFRI) is negatively correlated with ERAP1 expression without direct protein–protein interactions, suggesting that ERAP1 may assist TNFRI sheddase function (182). That sheddase was later identified as the tumor necrosis factor-alpha-converting enzyme (TACE, or ADAM17) (183). However, several clinical studies verified an increased level of soluble TNFRI in both AS and RA, correlating with ESR and CRP, while it decreased after infliximab or etanercept treatment (169, 184). This makes ERAP1 unlikely to be detrimental by reducing decoy receptors to amplify inflammatory signals.
Collectively, mutations in ERAP1 and HLA-B27 seem to be constantly monitored by a proteostasis network composed of the intracellular UPR, which is meant to correct abnormal folding but could ultimately become apoptotic and proinflammatory, and be subjected to intercellular surveillance by NK cells via the surface MHC sensor and KIR family. All of these aspects place accumulative stress on the entheseal or synovial tissue and are probably shared by other MHC-I-related diseases. This presumed mechanism may also evoke an idea of AS treatment by downregulating MHC-I expression in the opposite way as the intrinsic ability of immune escape in tumor cells.
The potentiality and actuality of gut-joint migration
Comparison between IBD and AS
Up to 50% of AS patients have subclinical intestinal inflammation, while approximately 10% develop overt IBD, which remarkably resembles CD (185). CD and AS share indisputable similarities. They share many gene variants that are either protective or susceptible, including IL-23R, ERAP1, NOD2, CARD15, etc. Some bacteria, such as Ruminococcus gnavus, have been confirmed to be involved in the dysbiosis of both of them (186–188). Enhanced T-cell maturation occurs even in the noninflamed part of the intestinal mucosa from SpA patients, with an increase in the number of lymphoid follicles, CD11c+ dendritic cells, CD68+ macrophages, and CD11a+ pan-lymphoid cells. The pathological features of the AS intestine resemble the early phase of chronic CD, including mixing infiltrating cells and villous atrophy (189–191). However, they differ in several aspects. Recruited CD14+ macrophages accumulate in the LP (lamina propria) of CD, where the proinflammatory microenvironment polarizes half or more macrophages toward the M1 phenotype (192–195). At the same time, macrophages in the AS intestine are mainly tissue-resident and undergo M2 polarization (95, 193). What’s more, CD has an increase in regulatory T cells (Tregs) in the intestinal mucosa but a huge decrease in the PB, suggesting a decompensation in the immunotolerogenesis of CD (196, 197), while AS brings no significant change of Tregs count of PB, but an upregulation in LP comparing to HC (198, 199). As a result, IL-17-producing cells controlled by Tregs are more polarized and proliferative in CD than in AS (199–202), leading to the impaired barrier function reflected by Paneth cell malfunction. Overall, CD and AS are not the same diseases even in the local intestine.
The gastrointestinal system is highly complicated, and it could be problematic to expect a stimulus to penetrate the intestinal homeostasis and affect specific aspects of articulation. Taurog et al. reported that rats cultured in germ-free conditions cannot develop arthritis or colitis until the gut microbiome is reimported (203). However, considering that even pulse dosing of antibiotics could impact the expansion and development of intestinal immunocytes (196, 197, 204), it could be deduced that a germ-free environment impedes the maturation and proliferation of intestinal lymphocytes, comprising up to 20% of total lymphocytes in the body (205). Many studies have implied that intestinal microbiota leads to local immune dysregulation through its metabolites and the breakdown of the intestinal barrier, of which the stimulus is transduced thoroughly to articulation through the immune system.
Gut-joint chemotaxis
The migration hypothesis holds the same rationale as the idea of bacterial infection-driven AS, in that there is spatial proximity between the sacroiliac joint and the draining lymph nodes located in the lower gastrointestinal tract and the pelvic floor. Cellular identification provides much evidence that cells expressing gut-specific markers are present in inflamed joints. For example, Ciccia et al. identified a group of classic monocytes (CD14++CD16+) in the synovium expressing CCR9, which directs gut homing under homeostatic conditions, suggesting that gut-derived monocytes participate in AS development (206). The gut could be the major source of circulating antigen-experienced T cells that could be activated in the joint, and CCL20 could be the most important chemokine in this process. Ridley and colleagues reported that KIR3DL2+ CD4+ Th17 cells expressing gut-homing CCR9 are expanded in the PB in patients with AS (142). They predominantly express CCR6 (207), and its only ligand, CCL20, has been shown to be elevated, particularly in the SF, in AS and RA patients (155, 208). CD14+ myeloid cells isolated from human enthesis tissues and adjacent bones have been shown to be the primary CCL20 producer after ex vivo induction by LPS and IFNγ (209). In addition, tendon stromal cells also have the capacity to secrete CCL20 after IL-23 overexpression (210). The migration of mucosal-associated invariant T (MAIT) cells, which travel around the PB and barrier tissue, is also largely dependent on the CCL20–CCR6 interaction (211–213). As a consequence, Gracey et al. found a reduced frequency of MAIT cells in blood but an increased number of IL-17+ MAIT cells in the SF (214), while Toussirot et al. observed that IFN-γ+/IL-17A+ MAIT cells were increased in the PB of AS patients (215). Interestingly, resident memory CD8+ T cells (named CD8+ TRM and marked by CD8+CD69+CD103+) were also found to expand in both the inflamed mucosa and PB of HLA-B27+ SpA patients, secreting IFNγ (216). They were believed to never leave the tissue in which they reside; however, newly reported evidence suggests that they have the potential to leave their resident tissues, which is called ‘retrograde migration’, and produce circulating effector cells (217). Since few studies concern their migration in response to inflammation, their migration could be CCL20-CCR6 dependent based on discovery in tumor-infiltrating CD8+ TRM, and the cytokine pattern in articulation as hereinafter described (218, 219). TCR repertoire sequencing from AS patients has already identified an oligoclonal expansion shared by the intestine and joints, expressing cytokines such as IFNγ, IL-10, and TNFα upon ex vivo stimulation (220). A more direct and decisive model is needed, similar to that established by Duc and colleagues in the MS model, whereby disrupting gut homing, they prevented inflammatory cells from getting primed and activated, therefore protecting the mice against developing EAE (221).
Joint retention of cells from the gut
Chemotaxis is only one aspect of cell recruitment; the other aspects of adhesion and retention require the interaction of adhesion molecules expressed on high endothelial venules (HEVs) or synovial tissues during this step (222). By blocking the binding between integrin and adhesion molecules, anti-integrin biologics prevent immune cells from extravasating and reaching inflammatory sites, to the benefit of both clinical treatment and research. Natalizumab and vedolizumab are two popular anti-integrin biologics used in treating IBD: vedolizumab selectively antagonizes integrin α4β7 while natalizumab blocks α4β1 and α4β7 by binding the α4 subunit (223, 224). With a similar mechanism of action, vedolizumab but not natalizumab is associated with SpA, causing sacroiliac arthritis or arthralgias, particularly as de novo cases (225–227). This leads to the conjecture that the additional blockade of the α4β1 of vedolizumab, which plays an important role in ileal homing (228), deflects immunocytes towards articulation. Inflamed joints have abundant ligands for α4β1-binding, including VCAM-1, ICAM-1, and fibronectin. VCAM-1 and ICAM-1 are already proven to be elevated significantly under the inflammatory situation in humans and mice (229–233). The RGD motif containing the three amino acids Arg-Gly-Asp presented in the classical integrin ligand is also found in several ECM proteins, including unraveled fibronectin fragments from the inflamed cartilage (234). In OA patients, fibronectin fragments have been confirmed to interact with chondrocytes, as chondrocytes from the inflamed tissue specifically express α2β1, α4β1, and α6β1 to bind these ECM proteins (235). Fibronectin controlling T cell recruitment is also revealed in the human dermis recently (236).
Besides integrin α4β1, α4β7 is also shared by joint-homing and gut-homing. Mucosal vascular addressing cell adhesion molecule 1 (MAdCAM-1) preferentially interacts with integrin α4β7, directing lymphocyte traffic to intestine. Previous studies shows a putative role for MAdCAM-1 and α4β7 in mediating the BM-homing of hematopoietic cells (237). Ciccia et al. discovered α4β7-expressing ILC3 existing in the intestine, PB, and BM from AS patients (238). A recent finding brings a new perspective to this problem, suggesting that the conformer switch of α4β7 is controlled by CCL25-CXCL10 (239). The CXCL10-activated integrin selectively binds to VCAM-1, while CCL25 induces an extended conformation that binds MAdCAM-1. Correspondingly, the expression level of CCL25 and CXCL10 shifts between the intestinal epithelium and synovium (101, 240). This novel mechanism may further strengthen the ability for T cells to switch their target from gut to joint, utilizing the same integrin binding to different adhesion molecules (Figure 3). This deduction is partially confirmed by new findings. For example, Qaiyum et al. discovered that mature CD8+ T cells enriched in the SF of AS have a distinct expression pattern of integrins, including α1, αE (CD103), β1, and β4 (of course, they did not bring the universally expressed α4 into the comparison) (241).
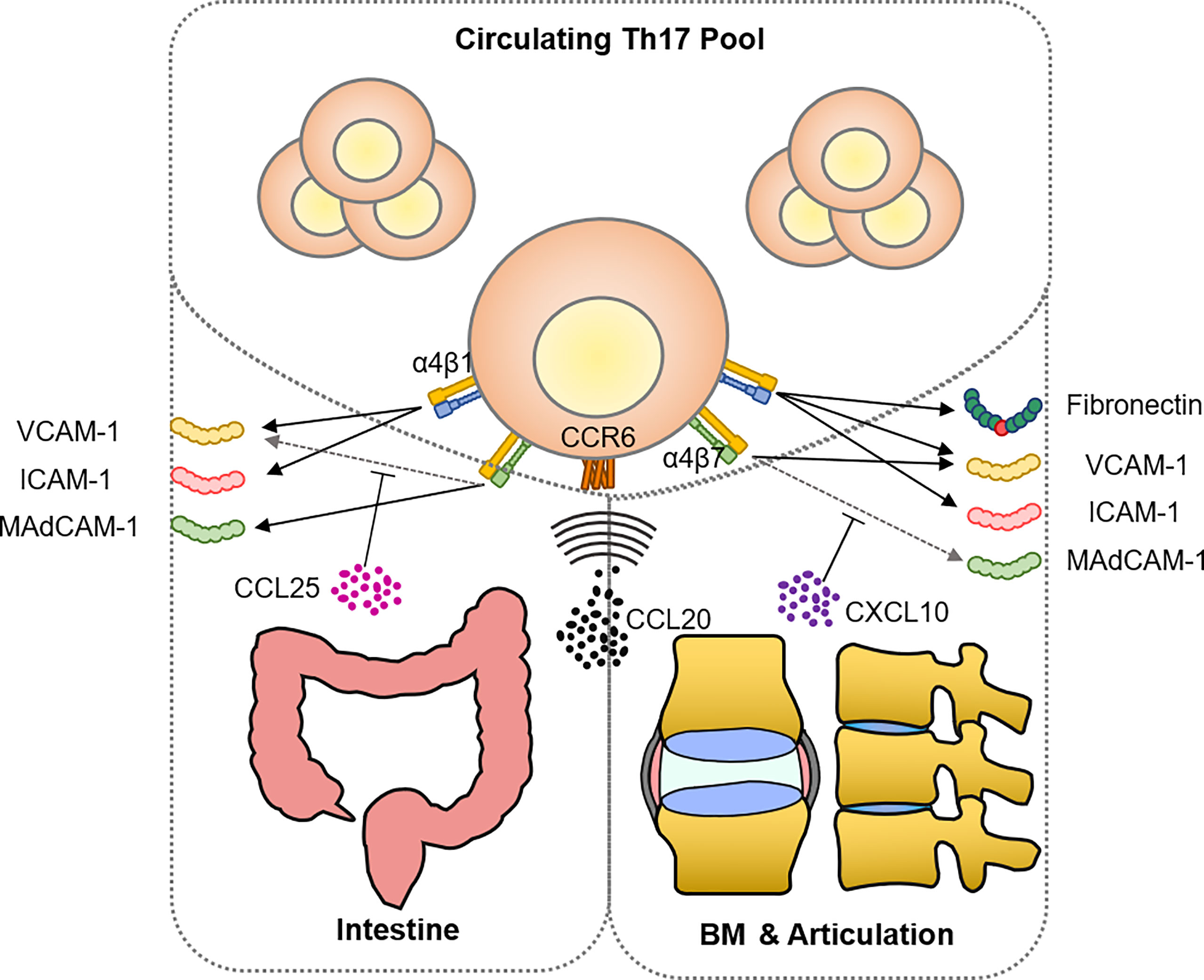
Figure 3 Shared mechanism of recruitment between intestine and articulation in AS. The ligand-receptor pair CCL20-CCR6 constitutes the predominant chemoattraction route of Th17 targeting both intestine and inflamed articulation. Emerging evidence has shown that both tissues express compatible adhesion molecules including VCAM-1, ICAM-1, and MAdCAM-1 for integrin binding, allowing Th17 cells to enter the inflammatory site. Mechanical stress remodeled ECM provides not only T cell stimulant, as the aforementioned hyaluronan and collagen fragment, but also fibronectin fragments that are able to bind and stimulate Th17 cells via integrin α4β1.
Combining clinical evidence and characteristics of immunocytes plus inflamed tissues, a growing body of evidence has provided a framework of the cytological gut-joint axis, which strengthens the idea of gut microbiota-driven disease. Though the synergism and antagonism that exist between intestinal bacteria and the crosstalk with the intestinal immune system remain difficult to investigate, it can be expected that the maneuver of gut-derived cells through recruitment and retention would become a promising target for AS intervention, as it has shown potential in the treatment of MS (221, 242).
Conclusion
In summary, evidence suggests that there is a radial network in AS pathogenesis, where IL-17-producing T cells play a central role in sensing danger signals and amplifying immune response. On the one hand, the mechanical damage of articulation provides abundant T cell stimulants that may bypass antigen encountering to bystander-activate immune response, yet further investigation on bystander activation in AS is much required. On the other hand, the molecular mechanism of HLA-B27 misfolding and non-conventional presentation becomes apparently associated with immune activation. Although it could still be inaccurate to assert that the AS is not driven directly by any infection, no certain arthritogenic peptide nor molecular mimicry matches have been identified from benchside to bedside. We already know that inflamed joints have a large number of stimuli of TLR and other innate receptors, and that innate immune cells are not only able to respond but also maintain an inflammatory immune memory, which gives weight to the role of non-immune and innate immune cells in autoinflammatory disease. Similarly, alternative pathways have been identified by researchers besides the IL-23/IL-17 axis, where non-immune and innate immune cells are involved through the upstream inducer PGE2 and downstream effector GM-CSF. Furthermore, the transportation between the gut and the joint, through chemotaxis and adhesion, is very clear and shall play an important role in relaying influence from microbiota to autoinflammatory disease.
Nevertheless, we are still trapped in a framework dominated by adaptive immunity, while various pivotal aspects of this protocol remain to be discussed. For example, peripheral tolerogenesis is not emphasized as an equal measure, which could be more important in explaining why many HLA-B27-positive people remain healthy. In addition, bone homeostasis, which regulates the balance of osteoporosis and ectopic ossification, is rarely mentioned, along with other adjacent tissues, since every part of our bodies should be considered as one piece of the immune system. With scientists’ endeavors to decipher this disease over the years, we are finally brought to the stage where the great majority of issues have been identified, although they are not fully understood. This enables us to join every piece of knowledge together to better assist patients and embrace the nature of the complexity of how the body works.
Author contributions
YHX: Drafting the manuscript. MC: Conception and design of study. YX, HC, and PD: Revising the manuscript critically for important intellectual content. JZ and WH: Supervision, Writing-Reviewing, and Editing. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (82071791, 31970843, 81972866 and U20A20374), the CAMS Initiative for Innovative Medicine (2021-1-I2M-005 and 2021-1-I2M-035), and the CAMS Central Public Welfare Scientific Research Institute Basal Research Expenses (2018PT32004, 2018PT31052 and 3332020035).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ashrafi M, Ermann J, Weisman MH. Spondyloarthritis evolution: what is in your history? Curr Opin Rheumatol (2020) 32(4):321–9. doi: 10.1097/bor.0000000000000712
2. Aletaha D, Neogi T, Silman AJ, Funovits J, Felson D. T, Bingham C. O, et al. 2010 Rheumatoid arthritis classification criteria: an American college of Rheumatology/European league against rheumatism collaborative initiative. Ann Rheum Dis (2010) 69(9):1580–8. doi: 10.1136/ard.2010.138461
3. Moll JM, Haslock I, Macrae IF, Wright V. Associations between ankylosing spondylitis, psoriatic arthritis, reiter's disease, the intestinal arthropathies, and behcet's syndrome. Med (Baltimore) (1974) 53(5):343–64. doi: 10.1097/00005792-197409000-00002
4. Wang S, Tsou HK, Chiou JY, Wang YH, Zhang Z, Wei JC. Increased risk of inflammatory bowel disease among patients with ankylosing spondylitis: A 13-year population-based cohort study. Front Immunol (2020) 11:578732. doi: 10.3389/fimmu.2020.578732
5. Gupta S, Syrimi Z, Hughes DM, Zhao SS. Comorbidities in psoriatic arthritis: a systematic review and meta-analysis. Rheumatol Int (2021) 41(2):275–84. doi: 10.1007/s00296-020-04775-2
6. Sieper J, Rudwaleit M, Baraliakos X, Brandt J, Braun J, Burgos-Vargas R, et al. The assessment of SpondyloArthritis international society (ASAS) handbook: a guide to assess spondyloarthritis. Ann Rheum Dis (2009) 68 Suppl 2:ii1–44. doi: 10.1136/ard.2008.104018
7. Rudwaleit M, Khan MA, Sieper J. The challenge of diagnosis and classification in early ankylosing spondylitis: do we need new criteria? Arthritis Rheumatol (2005) 52(4):1000–8. doi: 10.1002/art.20990
8. Mauro D, Thomas R, Guggino G, Lories R, Brown MA, Ciccia F. Ankylosing spondylitis: an autoimmune or autoinflammatory disease? Nat Rev Rheumatol (2021) 17(7):387–404. doi: 10.1038/s41584-021-00625-y
9. Schlosstein L, Terasaki PI, Bluestone R, Pearson CM. High association of an HL-a antigen, W27, with ankylosing spondylitis. N Engl J Med (1973) 288(14):704–6. doi: 10.1056/nejm197304052881403
10. Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol (1994) 12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015
11. Janeway CA Jr. The immune system evolved to discriminate infectious nonself from noninfectious self. Immunol Today (1992) 13(1):11–6. doi: 10.1016/0167-5699(92)90198-g
12. Tough DF, Borrow P, Sprent J. Induction of bystander T cell proliferation by viruses and type I interferon in vivo. Science. (1996) 272(5270):1947–50. doi: 10.1126/science.272.5270.1947
13. Tough DF, Sun S, Sprent J. T Cell stimulation in vivo by lipopolysaccharide (LPS). J Exp Med (1997) 185(12):2089–94. doi: 10.1084/jem.185.12.2089
14. Shim CH, Cho S, Shin YM, Choi JM. Emerging role of bystander T cell activation in autoimmune diseases. BMB Rep (2022) 55(2):57–64. doi: 10.5483/BMBRep.2022.55.2.183
15. Long F, Wang T, Li Q, Xiong Y, Zeng Y. Association between klebsiella pneumoniae and ankylosing spondylitis: A systematic review and meta-analysis. Int J Rheum Dis (2022) 25(4):422–32. doi: 10.1111/1756-185x.14283
16. Seager K, Bashir HV, Geczy AF, Edmonds J, de Vere-Tyndall A. Evidence for a specific B27-associated cell surface marker on lymphocytes of patients with ankylosing spondylitis. Nature. (1979) 277(5691):68–70. doi: 10.1038/277068a0
17. Avakian H, Welsh J, Ebringer A, Entwistle CC. Ankylosing spondylitis, HLA-B27 and klebsiella. II Cross-reactivity Stud Hum Tissue typing sera. Br J Exp Pathol (1980) 61(1):92–6.
18. Schwimmbeck PL, Oldstone MB. Molecular mimicry between human leukocyte antigen B27 and klebsiella. consequences for spondyloarthropathies. Am J Med (1988) 85(6a):51–3. doi: 10.1016/0002-9343(88)90385-3
19. Ewing C, Ebringer R, Tribbick G, Geysen HM. Antibody activity in ankylosing spondylitis sera to two sites on HLA B27.1 at the MHC groove region (within sequence 65-85), and to a klebsiella pneumoniae nitrogenase reductase peptide (within sequence 181-199). J Exp Med (1990) 171(5):1635–47. doi: 10.1084/jem.171.5.1635
20. Fielder M, Pirt SJ, Tarpey I, Wilson C, Cunningham P, Ettelaie C, et al. Molecular mimicry and ankylosing spondylitis: possible role of a novel sequence in pullulanase of klebsiella pneumoniae. FEBS Lett (1995) 369(2-3):243–8. doi: 10.1016/0014-5793(95)00760-7
21. Puccetti A, Dolcino M, Tinazzi E, Moretta F, D'Angelo S, Olivieri I, et al. Antibodies directed against a peptide epitope of a klebsiella pneumoniae-derived protein are present in ankylosing spondylitis. PloS One (2017) 12(1):e0171073. doi: 10.1371/journal.pone.0171073
22. Wei JC, Chou MC, Huang JY, Chang R, Hung YM. The association between candida infection and ankylosing spondylitis: a population-based matched cohort study. Curr Med Res Opin (2020) 36(12):2063–9. doi: 10.1080/03007995.2020.1838460
23. Trull AK, Ebringer R, Panayi GS, Colthorpe D, James DC, Ebringer A. IgA antibodies to klebsiella pneumoniae in ankylosing spondylitis. Scand J Rheumatol (1983) 12(3):249–53. doi: 10.3109/03009748309098543
24. Höhler T, Hug R, Schneider PM, Krummenauer F, Gripenberg-Lerche C, Granfors K, et al. Ankylosing spondylitis in monozygotic twins: studies on immunological parameters. Ann Rheum Dis (1999) 58(7):435–40. doi: 10.1136/ard.58.7.435
25. Zhang X, Sun Z, Zhou A, Tao L, Chen Y, Shi X, et al. Association between infections and risk of ankylosing spondylitis: A systematic review and meta-analysis. Front Immunol (2021) 12:768741. doi: 10.3389/fimmu.2021.768741
26. McMichael A, Bowness P. HLA-B27: natural function and pathogenic role in spondyloarthritis. Arthritis Res (2002) 4 Suppl 3(Suppl 3):S153–8. doi: 10.1186/ar571
27. Neumann-Haefelin C, McKiernan S, Ward S, Viazov S, Spangenberg H. C, Killinger T, et al. Dominant influence of an HLA-B27 restricted CD8+ T cell response in mediating HCV clearance and evolution. Hepatology. (2006) 43(3):563–72. doi: 10.1002/hep.21049
28. Dazert E, Neumann-Haefelin C, Bressanelli S, Fitzmaurice K, Kort J, Timm J, et al. Loss of viral fitness and cross-recognition by CD8+ T cells limit HCV escape from a protective HLA-B27-restricted human immune response. J Clin Invest (2009) 119(2):376–86. doi: 10.1172/jci36587
29. Goulder PJ, Phillips RE, Colbert RA, McAdam S, Ogg G, Nowak MA, et al. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat Med (1997) 3(2):212–7. doi: 10.1038/nm0297-212
30. Wei CY, Lin JY, Wang YT, Huang JY, Wei JC, Chiou JY. Risk of ankylosing spondylitis following human papillomavirus infection: A nationwide, population-based, cohort study. J Autoimmun (2020) 113:102482. doi: 10.1016/j.jaut.2020.102482
31. Yen YF, Chuang PH, Jen IA, Chen M, Lan YC, Liu YL, et al. Incidence of autoimmune diseases in a nationwide HIV/AIDS patient cohort in Taiwan, 2000-2012. Ann Rheum Dis (2017) 76(4):661–5. doi: 10.1136/annrheumdis-2016-209815
32. Damba JJ, Laskine M, Jin Y, Sinyavskaya L, Durand M. Incidence of autoimmune diseases in people living with HIV compared to a matched population: a cohort study. Clin Rheumatol (2021) 40(6):2439–45. doi: 10.1007/s10067-020-05500-x
33. Schittenhelm RB, Sian TC, Wilmann PG, Dudek NL, Purcell AW. Revisiting the arthritogenic peptide theory: quantitative not qualitative changes in the peptide repertoire of HLA-B27 allotypes. Arthritis Rheumatol (2015) 67(3):702–13. doi: 10.1002/art.38963
34. Zhou C, Zhao H, Xiao XY, Chen BD, Guo R J, Wang Q, et al. Metagenomic profiling of the pro-inflammatory gut microbiota in ankylosing spondylitis. J Autoimmun (2020) 107:102360. doi: 10.1016/j.jaut.2019.102360
35. Yin J, Sternes PR, Wang M, Song J, Morrison M, Li T, et al. Shotgun metagenomics reveals an enrichment of potentially cross-reactive bacterial epitopes in ankylosing spondylitis patients, as well as the effects of TNFi therapy upon microbiome composition. Ann Rheum Dis (2020) 79(1):132–40. doi: 10.1136/annrheumdis-2019-215763
36. Ziegler A, Loll B, Biesiadka J, Saenger W, Kellermann T, Misselwitz R, et al. A cartilage-derived self peptide presented by HLA-B27 molecules? comment on the article by atagunduz et al. Arthritis Rheumatol (2005) 52(8):2581–2. doi: 10.1002/art.21281
37. Asquith M, Sternes PR, Costello ME, Karstens L, Diamond S, Martin, et al. HLA alleles associated with risk of ankylosing spondylitis and rheumatoid arthritis influence the gut microbiome. Arthritis Rheumatol (2019) 71(10):1642–50. doi: 10.1002/art.40917
38. Berland M, Meslier V, Berreira Ibraim S, Le Chatelier E, Pons N, Maziers N, et al. Both disease activity and HLA-B27 determine gut microbiome dysbiosis in spondyloarthritis. Arthritis Rheumatol (2022). doi: 10.1002/art.42289
39. Stoll ML, DeQuattro K, Li Z, Sawhney H, Weiss PF, Nigrovic PA, et al. Impact of HLA-B27 and disease status on the gut microbiome of the offspring of ankylosing spondylitis patients. Children (Basel) (2022) 9(4):569. doi: 10.3390/children9040569
40. Zhang L, Han R, Zhang X, Fang G, Chen J, Li J, et al. Fecal microbiota in patients with ankylosing spondylitis: Correlation with dietary factors and disease activity. Clin Chim Acta (2019) 497:189–96. doi: 10.1016/j.cca.2019.07.038
41. Liu G, Hao Y, Yang Q, Deng S. The association of fecal microbiota in ankylosing spondylitis cases with c-reactive protein and erythrocyte sedimentation rate. Mediators Inflamm (2020) 2020:8884324. doi: 10.1155/2020/8884324
42. Berlinberg AJ, Regner EH, Stahly A, Brar A, Reisz JA, Gerich ME, et al. Multi 'Omics analysis of intestinal tissue in ankylosing spondylitis identifies alterations in the tryptophan metabolism pathway. Front Immunol (2021) 12:587119. doi: 10.3389/fimmu.2021.587119
43. Faham M, Carlton V, Moorhead M, Zheng J, Klinger M, Pepin F, et al. Discovery of T cell receptor β motifs specific to HLA-B27-Positive ankylosing spondylitis by deep repertoire sequence analysis. Arthritis Rheumatol (2017) 69(4):774–84. doi: 10.1002/art.40028
44. Zheng M, Zhang X, Zhou Y, Tang J, Han Q, Zhang Y, et al. TCR repertoire and CDR3 motif analyses depict the role of αβ T cells in ankylosing spondylitis. EBioMedicine. (2019) 47:414–26. doi: 10.1016/j.ebiom.2019.07.032
45. Hanson AL, Nel HJ, Bradbury L, Thomas R, Kenna TJ, Brown MA, et al. Altered repertoire diversity and disease-associated clonal expansions revealed by T cell receptor immunosequencing in ankylosing spondylitis patients. Arthritis Rheumatol (2020) 72(8):1289–302. doi: 10.1002/art.41252
46. Schittenhelm RB, Sivaneswaran S, Lim Kam Sian TC, Croft NP, Purcell AW. Human leukocyte antigen (HLA) B27 allotype-specific binding and candidate arthritogenic peptides revealed through heuristic clustering of data-independent acquisition mass spectrometry (DIA-MS) data. Mol Cell Proteomics (2016) 15(6):1867–76. doi: 10.1074/mcp.M115.056358
47. García-Medel N, Sanz-Bravo A, Alvarez-Navarro C, C, Gómez-Molina P, Barnea E, Marcilla, et al. Peptide handling by HLA-B27 subtypes influences their biological behavior, association with ankylosing spondylitis and susceptibility to endoplasmic reticulum aminopeptidase 1 (ERAP1). Mol Cell Proteomics (2014) 13(12):3367–80. doi: 10.1074/mcp.M114.039214
48. Barnea E, Melamed Kadosh D, Haimovich Y, Satumtira N, Dorris ML, Nguyen MT, et al. The human leukocyte antigen (HLA)-B27 peptidome in vivo, in spondyloarthritis-susceptible HLA-B27 transgenic rats and the effect of Erap1 deletion. Mol Cell Proteomics (2017) 16(4):642–62. doi: 10.1074/mcp.M116.066241
49. Tedeschi V, Paldino G, Paladini F, Mattorre B, Tuosto L, Sorrentino R, et al. The impact of the 'Mis-peptidome' on HLA class I-mediated diseases: Contribution of ERAP1 and ERAP2 and effects on the immune response. Int J Mol Sci (2020) 21(24):9608. doi: 10.3390/ijms21249608
50. Pende D, Falco M, Vitale M, Cantoni C, Vitale C, Munari E, et al. Killer ig-like receptors (KIRs): Their role in NK cell modulation and developments leading to their clinical exploitation. Front Immunol (2019) 10:1179. doi: 10.3389/fimmu.2019.01179
51. Brown MA, Kennedy LG, MacGregor AJ, Darke C, Duncan E, Shatford JL, et al. Susceptibility to ankylosing spondylitis in twins: the role of genes, HLA, and the environment. Arthritis Rheumatol (1997) 40(10):1823–8. doi: 10.1002/art.1780401015
52. Arévalo M, Gratacós Masmitjà J, Moreno M, Calvet J, Orellana C, Ruiz D, et al. Influence of HLA-B27 on the ankylosing spondylitis phenotype: results from the REGISPONSER database. Arthritis Res Ther (2018) 20(1):221. doi: 10.1186/s13075-018-1724-7
53. McGonagle D, Aydin SZ, Marzo-Ortega H, Eder L, Ciurtin C. Hidden in plain sight: Is there a crucial role for enthesitis assessment in the treatment and monitoring of axial spondyloarthritis? Semin Arthritis Rheumatol (2021) 51(6):1147–61. doi: 10.1016/j.semarthrit.2021.07.011
54. McGonagle D, Aydin SZ, Tan AL. The synovio-entheseal complex and its role in tendon and capsular associated inflammation. J Rheumatol Suppl. (2012) 89:11–4. doi: 10.3899/jrheum.120233
55. Ball J. Enthesopathy of rheumatoid and ankylosing spondylitis. Ann Rheum Dis (1971) 30(3):213–23. doi: 10.1136/ard.30.3.213
56. Carter S, Braem K, Lories RJ. The role of bone morphogenetic proteins in ankylosing spondylitis. Ther Adv Musculoskelet Dis (2012) 4(4):293–9. doi: 10.1177/1759720x12444175
57. Perrotta FM, Lories R, Lubrano E. To move or not to move: the paradoxical effect of physical exercise in axial spondyloarthritis. RMD Open (2021) 7(1):e001480. doi: 10.1136/rmdopen-2020-001480
58. Jacques P, Lambrecht S, Verheugen E, Pauwels E, Kollias G, Armaka M, et al. Proof of concept: enthesitis and new bone formation in spondyloarthritis are driven by mechanical strain and stromal cells. Ann Rheum Dis (2014) 73(2):437–45. doi: 10.1136/annrheumdis-2013-203643
59. Cambré I, Gaublomme D, Burssens A, Jacques P, Schryvers N, De Muynck A, et al. Mechanical strain determines the site-specific localization of inflammation and tissue damage in arthritis. Nat Commun (2018) 9(1):4613. doi: 10.1038/s41467-018-06933-4
60. McGonagle D, Wakefield RJ, Tan AL, D'Agostino MA, Toumi H, Hayashi K, et al. Distinct topography of erosion and new bone formation in achilles tendon enthesitis: implications for understanding the link between inflammation and bone formation in spondylarthritis. Arthritis Rheumatol (2008) 58(9):2694–9. doi: 10.1002/art.23755
61. de Jong HMY, de Winter JJH, van der Horst-Bruinsma IE, van Schaardenburg DJ, van Gaalen FA, van Tubergen AM, et al. Progression from subclinical inflammation to overt SpA in first degree relatives of SpA patients is associated with HLA-B27: the pre-SpA cohort. Arthritis Care Res (Hoboken) (2021). doi: 10.1002/acr.24743
62. Kim TH, Stone M, Payne U, Zhang X, Ionescu M, Lobanok T, et al. Cartilage biomarkers in ankylosing spondylitis: relationship to clinical variables and treatment response. Arthritis Rheumatol (2005) 52(3):885–91. doi: 10.1002/art.20870
63. Vosse D, Landewé R, Garnero P, van der Heijde D, van der Linden S, Geusens P. Association of markers of bone- and cartilage-degradation with radiological changes at baseline and after 2 years follow-up in patients with ankylosing spondylitis. Rheumatol (Oxford) (2008) 47(8):1219–22. doi: 10.1093/rheumatology/ken148
64. Lee S, Khare SD, Griffiths MM, Luthra HS, David CS. HLA-B27 transgenic mice are susceptible to collagen-induced arthritis: type II collagen as a potential target in human disease. Hum Immunol (2000) 61(2):140–7. doi: 10.1016/s0198-8859(99)00148-2
65. Zou J, Zhang Y, Thiel A, Rudwaleit M, Shi SL, Radbruch A, et al. Predominant cellular immune response to the cartilage autoantigenic G1 aggrecan in ankylosing spondylitis and rheumatoid arthritis. Rheumatol (Oxford) (2003) 42(7):846–55. doi: 10.1093/rheumatology/keg230
66. Zou J, Appel H, Rudwaleit M, Thiel A, Sieper J. Analysis of the CD8+ T cell response to the G1 domain of aggrecan in ankylosing spondylitis. Ann Rheum Dis (2005) 64(5):722–9. doi: 10.1136/ard.2004.024455
67. May E, Dorris ML, Satumtira N, Iqbal I, Rehman MI, Lightfoot E, et al. CD8 alpha beta T cells are not essential to the pathogenesis of arthritis or colitis in HLA-B27 transgenic rats. J Immunol (2003) 170(2):1099–105. doi: 10.4049/jimmunol.170.2.1099
68. Taurog JD, Dorris ML, Satumtira N, Tran TM, Sharma R, Dressel R, et al. Spondylarthritis in HLA-B27/human beta2-microglobulin-transgenic rats is not prevented by lack of CD8. Arthritis Rheumatol (2009) 60(7):1977–84. doi: 10.1002/art.24599
69. Yang M, Lv Q, Wei Q, Jiang Y, Qi J, Xiao M, et al. TNF-α inhibitor therapy can improve the immune imbalance of CD4+ T cells and negative regulatory cells but not CD8+ T cells in ankylosing spondylitis. Arthritis Res Ther (2020) 22(1):149. doi: 10.1186/s13075-020-02226-8
70. Plows D, Kontogeorgos G, Kollias G. Mice lacking mature T and b lymphocytes develop arthritic lesions after immunization with type II collagen. J Immunol (1999) 162(2):1018–23.
71. Lambert C, Borderie D, Dubuc JE, Rannou F, Henrotin Y. Type II collagen peptide Coll2-1 is an actor of synovitis. Osteoarthritis Cartilage. (2019) 27(11):1680–91. doi: 10.1016/j.joca.2019.07.009
72. Gupta RC, Lall R, Srivastava A, Sinha A. Hyaluronic acid: Molecular mechanisms and therapeutic trajectory. Front Vet Sci (2019) 6:192. doi: 10.3389/fvets.2019.00192
73. Duruöz MT, Turan Y, Cerrahoglu L, Isbilen B. Serum hyaluronic acid levels in patients with ankylosing spondylitis. Clin Rheumatol (2008) 27(5):621–6. doi: 10.1007/s10067-007-0757-0
74. Scheibner KA, Lutz MA, Boodoo S, Fenton MJ, Powell JD, Horton MR. Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. J Immunol (2006) 177(2):1272–81. doi: 10.4049/jimmunol.177.2.1272
75. Gariboldi S, Palazzo M, Zanobbio L, Selleri S, Sommariva M, Sfondrini L, et al. Low molecular weight hyaluronic acid increases the self-defense of skin epithelium by induction of beta-defensin 2 via TLR2 and TLR4. J Immunol (2008) 181(3):2103–10. doi: 10.4049/jimmunol.181.3.2103
76. Hashizume M, Mihara M. High molecular weight hyaluronic acid inhibits IL-6-induced MMP production from human chondrocytes by up-regulating the ERK inhibitor, MKP-1. Biochem Biophys Res Commun (2010) 403(2):184–9. doi: 10.1016/j.bbrc.2010.10.135
77. Sillat T, Barreto G, Clarijs P, Soininen A, Ainola M, Pajarinen J, et al. Toll-like receptors in human chondrocytes and osteoarthritic cartilage. Acta Orthop (2013) 84(6):585–92. doi: 10.3109/17453674.2013.854666
78. Cunnane G, FitzGerald O, Hummel KM, Gay RE, Gay S, Bresnihan B. Collagenase, cathepsin b and cathepsin l gene expression in the synovial membrane of patients with early inflammatory arthritis. Rheumatol (Oxford) (1999) 38(1):34–42. doi: 10.1093/rheumatology/38.1.34
79. Kostoulas G, Lang A, Nagase H, Baici A. Stimulation of angiogenesis through cathepsin b inactivation of the tissue inhibitors of matrix metalloproteinases. FEBS Lett (1999) 455(3):286–90. doi: 10.1016/s0014-5793(99)00897-2
80. Kapoor M, Martel-Pelletier J, Lajeunesse D, Pelletier JP, Fahmi H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol (2011) 7(1):33–42. doi: 10.1038/nrrheum.2010.196
81. Xu S, Ma Y, Wu M, Zhang X, Yang J, Deng J, et al. Neutrophil lymphocyte ratio in patients with ankylosing spondylitis: A systematic review and meta-analysis. Mod Rheumatol (2020) 30(1):141–8. doi: 10.1080/14397595.2018.1564165
82. Al-Osami MH, Awadh NI, Khalid KB, Awadh AI. Neutrophil/lymphocyte and platelet/lymphocyte ratios as potential markers of disease activity in patients with ankylosing spondylitis: a case-control study. Adv Rheumatol (2020) 60(1):13. doi: 10.1186/s42358-020-0113-5
83. Huang Y, Deng W, Zheng S, Feng F, Huang Z, Huang Q, et al. Relationship between monocytes to lymphocytes ratio and axial spondyloarthritis. Int Immunopharmacol (2018) 57:43–6. doi: 10.1016/j.intimp.2018.02.008
84. Kruithof E, Baeten D, De Rycke L, Vandooren B, Foell D, Roth J, et al. Synovial histopathology of psoriatic arthritis, both oligo- and polyarticular, resembles spondyloarthropathy more than it does rheumatoid arthritis. Arthritis Res Ther (2005) 7(3):R569–80. doi: 10.1186/ar1698
85. Kruithof E, Van den Bossche V, De Rycke L, Vandooren B, Joos R, Cañete JD, et al. Distinct synovial immunopathologic characteristics of juvenile-onset spondylarthritis and other forms of juvenile idiopathic arthritis. Arthritis Rheumatol (2006) 54(8):2594–604. doi: 10.1002/art.22024
86. Baeten D, Kruithof E, De Rycke L, Boots AM, Mielants H, Veys EM, et al. Infiltration of the synovial membrane with macrophage subsets and polymorphonuclear cells reflects global disease activity in spondyloarthropathy. Arthritis Res Ther (2005) 7(2):R359–69. doi: 10.1186/ar1501
87. Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, et al. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol (2014) 14(8):571–8. doi: 10.1038/nri3712
88. Yáñez A, Coetzee SG, Olsson A, Muench DE, Berman BP, Hazelett DJ, et al. Granulocyte-monocyte progenitors and monocyte-dendritic cell progenitors independently produce functionally distinct monocytes. Immunity. (2017) 47(5):890–902.e4. doi: 10.1016/j.immuni.2017.10.021
89. Regan-Komito D, Swann JW, Demetriou P, Cohen ES, Horwood NJ, Sansom SN, et al. GM-CSF drives dysregulated hematopoietic stem cell activity and pathogenic extramedullary myelopoiesis in experimental spondyloarthritis. Nat Commun (2020) 11(1):155. doi: 10.1038/s41467-019-13853-4
90. Karow F, Smiljanovic B, Grün JR, Poddubnyy D, Proft F, Talpin A, et al. Monocyte transcriptomes from patients with axial spondyloarthritis reveal dysregulated monocytopoiesis and a distinct inflammatory imprint. Arthritis Res Ther (2021) 23(1):246. doi: 10.1186/s13075-021-02623-7
91. Moorlag S, Khan N, Novakovic B, Kaufmann E, Jansen T, van Crevel R, et al. β-glucan induces protective trained immunity against mycobacterium tuberculosis infection: A key role for IL-1. Cell Rep (2020) 31(7):107634. doi: 10.1016/j.celrep.2020.107634
92. Dos Santos JC, Barroso de Figueiredo AM, Teodoro Silva MV, Cirovic B, de Bree LCJ, Damen M, et al. β-Glucan-Induced trained immunity protects against leishmania braziliensis infection: a crucial role for IL-32. Cell Rep (2019) 28(10):2659–2672.e6. doi: 10.1016/j.celrep.2019.08.004
93. Mitroulis I, Ruppova K, Wang B, Chen LS, Grzybek M, Grinenko T, et al. Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell. (2018) 172(1-2):147–161.e12. doi: 10.1016/j.cell.2017.11.034
94. Xie Z, Wang P, Li J, Li Y, Wang S, Wu X, et al. MCP1 triggers monocyte dysfunctions during abnormal osteogenic differentiation of mesenchymal stem cells in ankylosing spondylitis. J Mol Med (Berl) (2017) 95(2):143–54. doi: 10.1007/s00109-016-1489-x
95. Zhao J, Yuan W, Tao C, Sun P, Yang Z, Xu W. M2 polarization of monocytes in ankylosing spondylitis and relationship with inflammation and structural damage. Apmis. (2017) 125(12):1070–5. doi: 10.1111/apm.12757
96. De Rycke L, Vandooren B, Kruithof E, De Keyser F, Veys EM, Baeten D. Tumor necrosis factor alpha blockade treatment down-modulates the increased systemic and local expression of toll-like receptor 2 and toll-like receptor 4 in spondylarthropathy. Arthritis Rheumatol (2005) 52(7):2146–58. doi: 10.1002/art.21155
97. Solmaz D, Tekinalp A, Avci O, Turgut B. Ccr2 expression was increased in patients with ankylosing spondylitis independent from disease activity. Ann Rheumatic Dis (2015) 74:495–6. doi: 10.1136/annrheumdis-2015-eular.2492
98. Sierra-Filardi E, Nieto C, Domínguez-Soto A, Barroso R, Sánchez-Mateos P, Puig-Kroger A, et al. CCL2 shapes macrophage polarization by GM-CSF and m-CSF: identification of CCL2/CCR2-dependent gene expression profile. J Immunol (2014) 192(8):3858–67. doi: 10.4049/jimmunol.1302821
99. Deci MB, Ferguson SW, Scatigno SL, Nguyen J. Modulating macrophage polarization through CCR2 inhibition and multivalent engagement. Mol Pharm (2018) 15(7):2721–31. doi: 10.1021/acs.molpharmaceut.8b00237
100. Talbot J, Bianchini FJ, Nascimento DC, Oliveira RD, Souto FO, Pinto LG, et al. CCR2 expression in neutrophils plays a critical role in their migration into the joints in rheumatoid arthritis. Arthritis Rheumatol (2015) 67(7):1751–9. doi: 10.1002/art.39117
101. Proost P, Struyf S, Loos T, Gouwy M, Schutyser E, Conings R, et al. Coexpression and interaction of CXCL10 and CD26 in mesenchymal cells by synergising inflammatory cytokines: CXCL8 and CXCL10 are discriminative markers for autoimmune arthropathies. Arthritis Res Ther (2006) 8(4):R107. doi: 10.1186/ar1997
102. Aleksandrova E, Novikov A, Kulakova P, Dorofeev A, Savenkova N, Volnukhin E, et al. Serum levels of il-6 and il-8 in ankylosing spondylitis patients: Associations with disease activity. Ann Rheumatic Dis (2020) 79:880–1. doi: 10.1136/annrheumdis-2020-eular.3082
103. Hueber W, Tomooka BH, Zhao X, Kidd BA, Drijfhout JW, Fries JF, et al. Proteomic analysis of secreted proteins in early rheumatoid arthritis: anti-citrulline autoreactivity is associated with up regulation of proinflammatory cytokines. Ann Rheum Dis (2007) 66(6):712–9. doi: 10.1136/ard.2006.054924
104. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol (2013) 13(3):159–75. doi: 10.1038/nri3399
105. Milanez FM, Saad CG, Viana VT, Moraes JC, Périco GV, Sampaio-Barros PD, et al. IL-23/Th17 axis is not influenced by TNF-blocking agents in ankylosing spondylitis patients. Arthritis Res Ther (2016) 18:52. doi: 10.1186/s13075-016-0949-6
106. Andersen T, Rasmussen TK, Hvid M, Holm CK, Madsen KJ, Jurik AG, et al. Increased plasma levels of IL-21 and IL-23 in spondyloarthritis are not associated with clinical and MRI findings. Rheumatol Int (2012) 32(2):387–93. doi: 10.1007/s00296-010-1655-3
107. Chen WS, Chang YS, Lin KC, Lai CC, Wang SH, Hsiao KH, et al. Association of serum interleukin-17 and interleukin-23 levels with disease activity in Chinese patients with ankylosing spondylitis. J Chin Med Assoc (2012) 75(7):303–8. doi: 10.1016/j.jcma.2012.05.006
108. Sherlock JP, Joyce-Shaikh B, Turner SP, Chao CC, Sathe M, Grein J, et al. IL-23 induces spondyloarthropathy by acting on ROR-γt+ CD3+CD4-CD8- entheseal resident T cells. Nat Med (2012) 18(7):1069–76. doi: 10.1038/nm.2817
109. Reinhardt A, Yevsa T, Worbs T, Lienenklaus S, Sandrock I, Oberdörfer L, et al. Interleukin-23-Dependent γ/δ T cells produce interleukin-17 and accumulate in the enthesis, aortic valve, and ciliary body in mice. Arthritis Rheumatol (2016) 68(10):2476–86. doi: 10.1002/art.39732
110. Cuthbert RJ, Fragkakis EM, Dunsmuir R, Li Z, Coles M, Marzo-Ortega H, et al. Brief report: Group 3 innate lymphoid cells in human enthesis. Arthritis Rheumatol (2017) 69(9):1816–22. doi: 10.1002/art.40150
111. Yin Y, Wang M, Liu M, Zhou E, Ren T, Chang X, et al. Efficacy and safety of IL-17 inhibitors for the treatment of ankylosing spondylitis: a systematic review and meta-analysis. Arthritis Res Ther (2020) 22(1):111. doi: 10.1186/s13075-020-02208-w
112. Baeten D, Adamopoulos IE. IL-23 inhibition in ankylosing spondylitis: Where did it go wrong? Front Immunol (2020) 11:623874. doi: 10.3389/fimmu.2020.623874
113. McGonagle D, Watad A, Sharif K, Bridgewood C. Why inhibition of IL-23 lacked efficacy in ankylosing spondylitis. Front Immunol (2021) 12:614255. doi: 10.3389/fimmu.2021.614255
114. Cuthbert RJ, Watad A, Fragkakis EM, Dunsmuir R, Loughenbury P, Khan A, et al. Evidence that tissue resident human enthesis γδT-cells can produce IL-17A independently of IL-23R transcript expression. Ann Rheum Dis (2019) 78(11):1559–65. doi: 10.1136/annrheumdis-2019-215210
115. Chizzolini C, Chicheportiche R, Alvarez M, de Rham C, Roux-Lombard P, Ferrari-Lacraz S, et al. Prostaglandin E2 synergistically with interleukin-23 favors human Th17 expansion. Blood. (2008) 112(9):3696–703. doi: 10.1182/blood-2008-05-155408
116. Yao C, Sakata D, Esaki Y, Li Y, Matsuoka T, Kuroiwa K, et al. Prostaglandin E2-EP4 signaling promotes immune inflammation through Th1 cell differentiation and Th17 cell expansion. Nat Med (2009) 15(6):633–40. doi: 10.1038/nm.1968
117. Paulissen SM, van Hamburg JP, Davelaar N, Asmawidjaja PS, Hazes JM, Lubberts E. Synovial fibroblasts directly induce Th17 pathogenicity via the cyclooxygenase/prostaglandin E2 pathway, independent of IL-23. J Immunol (2013) 191(3):1364–72. doi: 10.4049/jimmunol.1300274
118. Lemos HP, Grespan R, Vieira SM, Cunha TM, Verri WA Jr, Fernandes KS, et al. Prostaglandin mediates IL-23/IL-17-induced neutrophil migration in inflammation by inhibiting IL-12 and IFNgamma production. Proc Natl Acad Sci USA (2009) 106(14):5954–9. doi: 10.1073/pnas.0812782106
119. Kofler DM, Marson A, Dominguez-Villar M, Xiao S, Kuchroo VK, Hafler DA. Decreased RORC-dependent silencing of prostaglandin receptor EP2 induces autoimmune Th17 cells. J Clin Invest (2014) 124(6):2513–22. doi: 10.1172/jci72973
120. Klasen C, Meyer A, Wittekind PS, Waqué I, Nabhani S, Kofler DM. Prostaglandin receptor EP4 expression by Th17 cells is associated with high disease activity in ankylosing spondylitis. Arthritis Res Ther (2019) 21(1):159. doi: 10.1186/s13075-019-1948-1
121. Coras R, Kavanaugh A, Kluzniak A, Holt D, Weilgosz A, Aaron A, et al. Differences in oxylipin profile in psoriasis versus psoriatic arthritis. Arthritis Res Ther (2021) 23(1):200. doi: 10.1186/s13075-021-02575-y
122. Nerviani A, Boutet MA, Tan WSG, Goldmann K, Purkayastha N, Lajtos TA, et al. IL-23 skin and joint profiling in psoriatic arthritis: novel perspectives in understanding clinical responses to IL-23 inhibitors. Ann Rheum Dis (2021) 80(5):591–7. doi: 10.1136/annrheumdis-2020-218186
123. Guendisch U, Weiss J, Ecoeur F, Riker JC, Kaupmann K, Kallen J, et al. Pharmacological inhibition of RORγt suppresses the Th17 pathway and alleviates arthritis in vivo. PloS One (2017) 12(11):e0188391. doi: 10.1371/journal.pone.0188391
124. Venken K, Jacques P, Mortier C, Labadia ME, Decruy T, Coudenys, et al. RORγt inhibition selectively targets IL-17 producing iNKT and γδ-T cells enriched in spondyloarthritis patients. Nat Commun (2019) 10(1):9. doi: 10.1038/s41467-018-07911-6
125. Bassolas-Molina H, Raymond E, Labadia M, Wahle J, Ferrer-Picón E, Panzenbeck M, et al. An RORγt oral inhibitor modulates IL-17 responses in peripheral blood and intestinal mucosa of crohn's disease patients. Front Immunol (2018) 9:2307. doi: 10.3389/fimmu.2018.02307
126. van Tok MN, Mandour M, Wahle J, Labadia ME, van de Sande MGH, Nabozny G., et al. Paradoxical augmentation of experimental spondyloarthritis by RORC inhibition in HLA-B27 transgenic rats. Front Immunol (2021) 12:699987. doi: 10.3389/fimmu.2021.699987
127. Fauny M, Moulin D, D'Amico F, Netter P, Petitpain N, Arnone D, et al. Paradoxical gastrointestinal effects of interleukin-17 blockers. Ann Rheum Dis (2020) 79(9):1132–8. doi: 10.1136/annrheumdis-2020-217927
128. Chong WP, Mattapallil MJ, Raychaudhuri K, Bing SJ, Wu S, Zhong Y, et al. The cytokine IL-17A limits Th17 pathogenicity via a negative feedback loop driven by autocrine induction of IL-24. Immunity. (2020) 53(2):384–397.e5. doi: 10.1016/j.immuni.2020.06.022
129. El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol (2011) 12(6):568–75. doi: 10.1038/ni.2031
130. Kryczek I, Zhao E, Liu Y, Wang Y, Vatan L, Szeliga W, et al. Human TH17 cells are long-lived effector memory cells. Sci Transl Med (2011) 3(104):104ra100. doi: 10.1126/scitranslmed.3002949
131. Crawford MP, Sinha S, Renavikar PS, Borcherding N, Karandikar NJ. CD4 T cell-intrinsic role for the T helper 17 signature cytokine IL-17: Effector resistance to immune suppression. Proc Natl Acad Sci U S A. (2020) 117(32):19408–14. doi: 10.1073/pnas.2005010117
132. van Tok MN, Na S, Lao CR, Alvi M, Pots D, van de Sande MGH, et al. The initiation, but not the persistence, of experimental spondyloarthritis is dependent on interleukin-23 signaling. Front Immunol (2018) 9:1550. doi: 10.3389/fimmu.2018.01550
133. Payeli SK, Kollnberger S, Marroquin Belaunzaran O, Thiel M, McHugh K, Giles J, et al. Inhibiting HLA-B27 homodimer-driven immune cell inflammation in spondylarthritis. Arthritis Rheumatol (2012) 64(10):3139–49. doi: 10.1002/art.34538
134. Polachek A, Cook R, Chandran V, Abji F, Gladman D, Eder L. The association between HLA genetic susceptibility markers and sonographic enthesitis in psoriatic arthritis. Arthritis Rheumatol (2018) 70(5):756–62. doi: 10.1002/art.40423
135. Berntson L, Damgård M, Andersson-Gäre B, et al. HLA-B27 predicts a more extended disease with increasing age at onset in boys with juvenile idiopathic arthritis. J Rheumatol (2008) 35(10):2055–61.
136. Allen RL, O'Callaghan CA, McMichael AJ, Bowness P. Cutting edge: HLA-B27 can form a novel beta 2-microglobulin-free heavy chain homodimer structure. J Immunol (1999) 162(9):5045–8.
137. Rysnik O, McHugh K, van Duivenvoorde L, et al. Non-conventional forms of HLA-B27 are expressed in spondyloarthritis joints and gut tissue. J Autoimmun (2016) 70:12–21. doi: 10.1016/j.jaut.2016.03.009
138. McHugh K, Rysnik O, Kollnberger S, Shaw J, Utriainen L, Al-Mossawi MH, et al. Expression of aberrant HLA-B27 molecules is dependent on B27 dosage and peptide supply. Ann Rheum Dis (2014) 73(4):763–70. doi: 10.1136/annrheumdis-2012-203080
139. Kollnberger S, Chan A, Sun MY, Chen LY, Wright C, di Gleria K, et al. Interaction of HLA-B27 homodimers with KIR3DL1 and KIR3DL2, unlike HLA-B27 heterotrimers, is independent of the sequence of bound peptide. Eur J Immunol (2007) 37(5):1313–22. doi: 10.1002/eji.200635997
140. Giles J, Shaw J, Piper C, Wong-Baeza I, McHugh K, Ridley A, et al. HLA-B27 homodimers and free h chains are stronger ligands for leukocyte ig-like receptor B2 than classical HLA class I. J Immunol (2012) 188(12):6184–93. doi: 10.4049/jimmunol.1102711
141. Bowness P, Ridley A, Shaw J, Chan AT, Wong-Baeza I, Fleming M, et al. Th17 cells expressing KIR3DL2+ and responsive to HLA-B27 homodimers are increased in ankylosing spondylitis. J Immunol (2011) 186(4):2672–80. doi: 10.4049/jimmunol.1002653
142. Ridley A, Hatano H, Wong-Baeza I, Shaw J, Matthews KK, Al-Mossawi H, et al. Activation-induced killer cell immunoglobulin-like receptor 3DL2 binding to HLA-B27 licenses pathogenic T cell differentiation in spondyloarthritis. Arthritis Rheumatol (2016) 68(4):901–14. doi: 10.1002/art.39515
143. Marroquin Belaunzaran O, Kleber S, Schauer S, Hausmann M, Nicholls F, Van den Broek M, et al. HLA-B27-Homodimer-Specific antibody modulates the expansion of pro-inflammatory T-cells in HLA-B27 transgenic rats. PloS One (2015) 10(6):e0130811. doi: 10.1371/journal.pone.0130811
144. Wong-Baeza I, Ridley A, Shaw J, Hatano H, Rysnik O, McHugh K, et al. KIR3DL2 binds to HLA-B27 dimers and free h chains more strongly than other HLA class I and promotes the expansion of T cells in ankylosing spondylitis. J Immunol (2013) 190(7):3216–24. doi: 10.4049/jimmunol.1202926
145. Cauli A, Shaw J, Giles J, Hatano H, Rysnik O, Payeli S, et al. The arthritis-associated HLA-B*27:05 allele forms more cell surface B27 dimer and free heavy chain ligands for KIR3DL2 than HLA-B*27:09. Rheumatol (Oxford) (2013) 52(11):1952–62. doi: 10.1093/rheumatology/ket219
146. Chen L, Ridley A, Hammitzsch A, Al-Mossawi MH, Bunting H, Georgiadis D, et al. Silencing or inhibition of endoplasmic reticulum aminopeptidase 1 (ERAP1) suppresses free heavy chain expression and Th17 responses in ankylosing spondylitis. Ann Rheum Dis (2016) 75(5):916–23. doi: 10.1136/annrheumdis-2014-206996
147. Adiko AC, Babdor J, Gutiérrez-Martínez E, Guermonprez P, Saveanu L. Intracellular transport routes for MHC I and their relevance for antigen cross-presentation. Front Immunol (2015) 6:335. doi: 10.3389/fimmu.2015.00335
148. Powis SJ, Antoniou AN. Measuring synthesis and degradation of MHC class I molecules. Methods Mol Biol (2019) 1988:83–100. doi: 10.1007/978-1-4939-9450-2_7
149. Dangoria NS, DeLay ML, Kingsbury DJ, Mear JP, Uchanska-Ziegler B, Ziegler A, et al. HLA-B27 misfolding is associated with aberrant intermolecular disulfide bond formation (dimerization) in the endoplasmic reticulum. J Biol Chem (2002) 277(26):23459–68. doi: 10.1074/jbc.M110336200
150. Guiliano DB, North H, Panayoitou E, Campbell EC, McHugh K, Cooke FG, et al. Polymorphisms in the f pocket of HLA-B27 subtypes strongly affect assembly, chaperone interactions, and heavy-chain misfolding. Arthritis Rheumatol (2017) 69(3):610–21. doi: 10.1002/art.39948
151. Mear JP, Schreiber KL, Münz C, Zhu X, Stevanović S, Rammensee HG, et al. Misfolding of HLA-B27 as a result of its b pocket suggests a novel mechanism for its role in susceptibility to spondyloarthropathies. J Immunol (1999) 163(12):6665–70.
152. Guiliano DB, Fussell H, Lenart I, Tsao E, Nesbeth D, Fletcher AJ, et al. Endoplasmic reticulum degradation-enhancing α-mannosidase-like protein 1 targets misfolded HLA-B27 dimers for endoplasmic reticulum-associated degradation. Arthritis Rheumatol (2014) 66(11):2976–88. doi: 10.1002/art.38809
153. DeLay ML, Turner MJ, Klenk EI, Smith JA, Sowders DP, Colbert RA. HLA-B27 misfolding and the unfolded protein response augment interleukin-23 production and are associated with Th17 activation in transgenic rats. Arthritis Rheumatol (2009) 60(9):2633–43. doi: 10.1002/art.24763
154. Turner MJ, Sowders DP, DeLay ML, Mohapatra R, Bai S, Smith JA, et al. HLA-B27 misfolding in transgenic rats is associated with activation of the unfolded protein response. J Immunol (2005) 175(4):2438–48. doi: 10.4049/jimmunol.175.4.2438
155. Liu CH, Chou CT, Chen CH, Yang SY, Ko YA, Wu YT, et al. Aberrant distribution and function of plasmacytoid dendritic cells in patients with ankylosing spondylitis are associated with unfolded protein response. Kaohsiung J Med Sci (2020) 36(6):441–9. doi: 10.1002/kjm2.12184
156. Feng Y, Ding J, Fan CM, Zhu P. Interferon-γ contributes to HLA-B27-associated unfolded protein response in spondyloarthropathies. J Rheumatol (2012) 39(3):574–82. doi: 10.3899/jrheum.101257
157. Rezaiemanesh A, Mahmoudi M, Amirzargar AA, Vojdanian M, Jamshidi AR, Nicknam MH. Ankylosing spondylitis m-CSF-derived macrophages are undergoing unfolded protein response (UPR) and express higher levels of interleukin-23. Mod Rheumatol (2017) 27(5):862–7. doi: 10.1080/14397595.2016.1259716
158. Zeng L, Lindstrom MJ, Smith JA. Ankylosing spondylitis macrophage production of higher levels of interleukin-23 in response to lipopolysaccharide without induction of a significant unfolded protein response. Arthritis Rheumatol (2011) 63(12):3807–17. doi: 10.1002/art.30593
159. Ambarus CA, Yeremenko N, Baeten DL. Altered cytokine expression by macrophages from HLA-B27-positive spondyloarthritis patients without evidence of endoplasmic reticulum stress. Rheumatol Adv Pract (2018) 2(1):rky014. doi: 10.1093/rap/rky014
160. Ciccia F, Accardo-Palumbo A, Rizzo A, Guggino G, Raimondo S, Giardina A, et al. Evidence that autophagy, but not the unfolded protein response, regulates the expression of IL-23 in the gut of patients with ankylosing spondylitis and subclinical gut inflammation. Ann Rheum Dis (2014) 73(8):1566–74. doi: 10.1136/annrheumdis-2012-202925
161. Neerinckx B, Carter S, Lories R. IL-23 expression and activation of autophagy in synovium and PBMCs of HLA-B27 positive patients with ankylosing spondylitis. response to: 'Evidence that autophagy, but not the unfolded protein response, regulates the expression of IL-23 in the gut of patients with ankylosing spondylitis and subclinical gut inflammation' by ciccia et al. Ann Rheum Dis (2014) 73(11):e68. doi: 10.1136/annrheumdis-2014-206277
162. Tan M, Zhang QB, Liu TH, Yang YY, Zheng JX, Zhou WJ, et al. Autophagy dysfunction may be involved in the pathogenesis of ankylosing spondylitis. Exp Ther Med (2020) 20(4):3578–86. doi: 10.3892/etm.2020.9116
163. Thorburn A. Autophagy and disease. J Biol Chem (2018) 293(15):5425–30. doi: 10.1074/jbc.R117.810739
164. Lin F, Qin ZH. Degradation of misfolded proteins by autophagy: is it a strategy for huntington's disease treatment? J Huntingtons Dis (2013) 2(2):149–57. doi: 10.3233/jhd-130052
165. Yonekawa T, Thorburn A. Autophagy and cell death. Essays Biochem (2013) 55:105–17. doi: 10.1042/bse0550105
166. Biasizzo M, Kopitar-Jerala N. Interplay between NLRP3 inflammasome and autophagy. Front Immunol (2020) 11:591803. doi: 10.3389/fimmu.2020.591803
167. Navid F, Layh-Schmitt G, Sikora KA, Cougnoux A, Colbert RA. The role of autophagy in the degradation of misfolded HLA-B27 heavy chains. Arthritis Rheumatol (2018) 70(5):746–55. doi: 10.1002/art.40414
168. Chen S, van Tok MN, Knaup VL, Kraal L, Pots D, Bartels L, Gravallese EM, et al. mTOR blockade by rapamycin in spondyloarthritis: Impact on inflammation and new bone formation in vitro and in vivo. Front Immunol (2019) 10:2344. doi: 10.3389/fimmu.2019.02344
169. Haroon N, Tsui FW, Chiu B, Tsui HW, Inman RD. Serum cytokine receptors in ankylosing spondylitis: relationship to inflammatory markers and endoplasmic reticulum aminopeptidase polymorphisms. J Rheumatol (2010) 37(9):1907–10. doi: 10.3899/jrheum.100019
170. Painter JD, Galle-Treger L, Akbari O. Role of autophagy in lung inflammation. Front Immunol (2020) 11:1337. doi: 10.3389/fimmu.2020.01337
171. Ma C, Wen B, Zhang Q, et al. Emodin induces apoptosis and autophagy of fibroblasts obtained from patient with ankylosing spondylitis. Drug Des Devel Ther (2019) 13:601–9. doi: 10.2147/dddt.S182087
172. Wu J, Feng Z, Chen L, Li Y, Bian H, Geng J, et al. TNF antagonist sensitizes synovial fibroblasts to ferroptotic cell death in collagen-induced arthritis mouse models. Nat Commun (2022) 13(1):676. doi: 10.1038/s41467-021-27948-4
173. Haroon N, Inman RD. Endoplasmic reticulum aminopeptidases: Biology and pathogenic potential. Nat Rev Rheumatol (2010) 6(8):461–7. doi: 10.1038/nrrheum.2010.85
174. Serwold T, Gonzalez F, Kim J, Jacob R, Shastri N. ERAAP customizes peptides for MHC class I molecules in the endoplasmic reticulum. Nature. (2002) 419(6906):480–3. doi: 10.1038/nature01074
175. York IA, Chang SC, Saric T, Keys JA, Favreau JM, Goldberg AL, et al. The ER aminopeptidase ERAP1 enhances or limits antigen presentation by trimming epitopes to 8-9 residues. Nat Immunol (2002) 3(12):1177–84. doi: 10.1038/ni860
176. Kanaseki T, Shastri N. Endoplasmic reticulum aminopeptidase associated with antigen processing regulates quality of processed peptides presented by MHC class I molecules. J Immunol (2008) 181(9):6275–82. doi: 10.4049/jimmunol.181.9.6275
177. Kirino Y, Bertsias G, Ishigatsubo Y, Mizuki N, Tugal-Tutkun I, Seyahi E, et al. Genome-wide association analysis identifies new susceptibility loci for behçet's disease and epistasis between HLA-B*51 and ERAP1. Nat Genet (2013) 45(2):202–7. doi: 10.1038/ng.2520
178. Zhang L, Yu H, Zheng M, Li H, Liu Y, Kijlstra A, et al. Association of ERAP1 gene polymorphisms with behçet's disease in han Chinese. Invest Ophthalmol Vis Sci (2015) 56(10):6029–35. doi: 10.1167/iovs.15-17544
179. Takeuchi M, Ombrello MJ, Kirino Y, Erer B, Tugal-Tutkun I, Seyahi E, et al. A single endoplasmic reticulum aminopeptidase-1 protein allotype is a strong risk factor for behçet's disease in HLA-B*51 carriers. Ann Rheum Dis (2016) 75(12):2208–11. doi: 10.1136/annrheumdis-2015-209059
180. Kuiper JJW, Setten JV, Devall M, Cretu-Stancu M, Hiddingh S, Ophoff RA, et al. Functionally distinct ERAP1 and ERAP2 are a hallmark of HLA-A29-(Birdshot) uveitis. Hum Mol Genet (2018) 27(24):4333–43. doi: 10.1093/hmg/ddy319
181. McGonagle D, Aydin SZ, Gül A, Mahr A, Direskeneli H. 'MHC-i-opathy'-unified concept for spondyloarthritis and behçet disease. Nat Rev Rheumatol (2015) 11(12):731–40. doi: 10.1038/nrrheum.2015.147
182. Cui X, Hawari F, Alsaaty S, Lawrence M, Combs CA, Geng W, et al. Identification of ARTS-1 as a novel TNFR1-binding protein that promotes TNFR1 ectodomain shedding. J Clin Invest (2002) 110(4):515–26. doi: 10.1172/jci13847
183. Bell JH, Herrera AH, Li Y, Walcheck B. Role of ADAM17 in the ectodomain shedding of TNF-alpha and its receptors by neutrophils and macrophages. J Leukoc Biol (2007) 82(1):173–6. doi: 10.1189/jlb.0307193
184. Schulz M, Dotzlaw H, Neeck G. Ankylosing spondylitis and rheumatoid arthritis: serum levels of TNF-α and its soluble receptors during the course of therapy with etanercept and infliximab. BioMed Res Int (2014) 2014:675108. doi: 10.1155/2014/675108
185. Ciccia F, Rizzo A, Triolo G. Subclinical gut inflammation in ankylosing spondylitis. Curr Opin Rheumatol (2016) 28(1):89–96. doi: 10.1097/bor.0000000000000239
186. Tito RY, Cypers H, Joossens M, Varkas G, Van Praet L, Glorieus E, et al. Brief report: Dialister as a microbial marker of disease activity in spondyloarthritis. Arthritis Rheumatol (2017) 69(1):114–21. doi: 10.1002/art.39802
187. Breban M, Tap J, Leboime A, Said-Nahal R, Langella P, Chiocchia G, et al. Faecal microbiota study reveals specific dysbiosis in spondyloarthritis. Ann Rheum Dis (2017) 76(9):1614–22. doi: 10.1136/annrheumdis-2016-211064
188. Costello ME, Ciccia F, Willner D, Warrington N, Robinson PC, Gardiner B, et al. Brief report: Intestinal dysbiosis in ankylosing spondylitis. Arthritis Rheumatol (2015) 67(3):686–91. doi: 10.1002/art.38967
189. Bjarnason I, Helgason KO, Geirsson AJ, Sigthorsson G, Reynisdottir I, Gudbjartsson D, et al. Subclinical intestinal inflammation and sacroiliac changes in relatives of patients with ankylosing spondylitis. Gastroenterology. (2003) 125(6):1598–605. doi: 10.1053/j.gastro.2003.08.035
190. Cuvelier C, Barbatis C, Mielants H, De Vos M, Roels H, Veys E. Histopathology of intestinal inflammation related to reactive arthritis. Gut. (1987) 28(4):394–401. doi: 10.1136/gut.28.4.394
191. Ciccia F, Guggino G, Rizzo A, Alessandro R, Luchetti MM, Milling S, et al. Dysbiosis and zonulin upregulation alter gut epithelial and vascular barriers in patients with ankylosing spondylitis. Ann Rheum Dis (2017) 76(6):1123–32. doi: 10.1136/annrheumdis-2016-210000
192. Kamada N, Hisamatsu T, Okamoto S, Chinen H, Kobayashi T, Sato T, et al. Unique CD14 intestinal macrophages contribute to the pathogenesis of crohn disease via IL-23/IFN-gamma axis. J Clin Invest (2008) 118(6):2269–80. doi: 10.1172/jci34610
193. Ciccia F, Alessandro R, Rizzo A, Accardo-Palumbo A, Raimondo S, Raiata F, et al. Macrophage phenotype in the subclinical gut inflammation of patients with ankylosing spondylitis. Rheumatol (Oxford) (2014) 53(1):104–13. doi: 10.1093/rheumatology/ket323
194. Du Y, Rong L, Cong Y, Shen L, Zhang N, Wang B. Macrophage polarization: an effective approach to targeted therapy of inflammatory bowel disease. Expert Opin Ther Targets (2021) 25(3):191–209. doi: 10.1080/14728222.2021.1901079
195. Isidro RA, Appleyard CB. Colonic macrophage polarization in homeostasis, inflammation, and cancer. Am J Physiol Gastrointest Liver Physiol (2016) 311(1):G59–73. doi: 10.1152/ajpgi.00123.2016
196. Maul J, Loddenkemper C, Mundt P, Berg E, Giese T, Stallmach A, et al. Peripheral and intestinal regulatory CD4+ CD25(high) T cells in inflammatory bowel disease. Gastroenterology. (2005) 128(7):1868–78. doi: 10.1053/j.gastro.2005.03.043
197. Saruta M, Yu QT, Fleshner PR, Mantel PY, Schmidt-Weber CB, Banham AH, et al. Characterization of FOXP3+CD4+ regulatory T cells in crohn's disease. Clin Immunol (2007) 125(3):281–90. doi: 10.1016/j.clim.2007.08.003
198. Lai NL, Zhang SX, Wang J, Zhang JQ, Wang CH, Gao C, et al. The proportion of regulatory T cells in patients with ankylosing spondylitis: A meta-analysis. J Immunol Res (2019) 2019:1058738. doi: 10.1155/2019/1058738
199. Ciccia F, Accardo-Palumbo A, Giardina A, Di Maggio P, Principato A, Bombardieri M, et al. Expansion of intestinal CD4+CD25(high) treg cells in patients with ankylosing spondylitis: a putative role for interleukin-10 in preventing intestinal Th17 response. Arthritis Rheumatol (2010) 62(12):3625–34. doi: 10.1002/art.27699
200. Chaudhry A, Samstein RM, Treuting P, Liang Y, Pils MC, Heinrich JM, et al. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity. (2011) 34(4):566–78. doi: 10.1016/j.immuni.2011.03.018
201. Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. (2003) 52(1):65–70. doi: 10.1136/gut.52.1.65
202. Ciccia F, Bombardieri M, Principato A, Giardina A, Tripodo C, Porcasi R, et al. Overexpression of interleukin-23, but not interleukin-17, as an immunologic signature of subclinical intestinal inflammation in ankylosing spondylitis. Arthritis Rheumatol (2009) 60(4):955–65. doi: 10.1002/art.24389
203. Taurog JD, Richardson JA, Croft JT, Simmons WA, Zhou M, Fernández-Sueiro JL, et al. The germfree state prevents development of gut and joint inflammatory disease in HLA-B27 transgenic rats. J Exp Med (1994) 180(6):2359–64. doi: 10.1084/jem.180.6.2359
204. Tupin E, Kinjo Y, Kronenberg M. The unique role of natural killer T cells in the response to microorganisms. Nat Rev Microbiol (2007) 5(6):405–17. doi: 10.1038/nrmicro1657
205. Ganusov VV, De Boer RJ. Do most lymphocytes in humans really reside in the gut? Trends Immunol (2007) 28(12):514–8. doi: 10.1016/j.it.2007.08.009
206. Ciccia F, Guggino G, Zeng M, Thomas R, Ranganathan V, Rahman A, et al. Proinflammatory CX3CR1+CD59+Tumor necrosis factor-like molecule 1A+Interleukin-23+ monocytes are expanded in patients with ankylosing spondylitis and modulate innate lymphoid cell 3 immune functions. Arthritis Rheumatol (2018) 70(12):2003–13. doi: 10.1002/art.40582
207. Wang C, Kang SG, Lee J, Sun Z, Kim CH. The roles of CCR6 in migration of Th17 cells and regulation of effector T-cell balance in the gut. Mucosal Immunol (2009) 2(2):173–83. doi: 10.1038/mi.2008.84
208. Melis L, Vandooren B, Kruithof E, Jacques P, De Vos M, Mielants H, et al. Systemic levels of IL-23 are strongly associated with disease activity in rheumatoid arthritis but not spondyloarthritis. Ann Rheum Dis (2010) 69(3):618–23. doi: 10.1136/ard.2009.107649
209. Bridgewood C, Watad A, Russell T, Palmer TM, Marzo-Ortega H, Khan A, et al. Identification of myeloid cells in the human enthesis as the main source of local IL-23 production. Ann Rheum Dis (2019) 78(7):929–33. doi: 10.1136/annrheumdis-2018-214944
210. Shi Z, Garcia-Melchor E, Wu X, Getschman AE, Nguyen M, Rowland DJ, et al. Targeting the CCR6/CCL20 axis in entheseal and cutaneous inflammation. Arthritis Rheumatol (2021) 73(12):2271–81. doi: 10.1002/art.41882
211. Solders M, Gorchs L, Tiblad E, Gidlöf S, Leeansyah E, Dias J, et al. Recruitment of MAIT cells to the intervillous space of the placenta by placenta-derived chemokines. Front Immunol (2019) 10:1300. doi: 10.3389/fimmu.2019.01300
212. Melo AM, O'Brien AM, Phelan JJ, Kennedy SA, Wood NAW, Veerapen N, et al. Mucosal-associated invariant T cells display diminished effector capacity in oesophageal adenocarcinoma. Front Immunol (2019) 10:1580. doi: 10.3389/fimmu.2019.01580
213. Ling L, Lin Y, Zheng W, Hong S, Tang X, Zhao P, et al. Circulating and tumor-infiltrating mucosal associated invariant T (MAIT) cells in colorectal cancer patients. Sci Rep (2016) 6:20358. doi: 10.1038/srep20358
214. Gracey E, Qaiyum Z, Almaghlouth I, Lawson D, Karki S, Avvaru N, et al. IL-7 primes IL-17 in mucosal-associated invariant T (MAIT) cells, which contribute to the Th17-axis in ankylosing spondylitis. Ann Rheum Dis (2016) 75(12):2124–32. doi: 10.1136/annrheumdis-2015-208902
215. Toussirot É, Laheurte C, Gaugler B, Gabriel D, Saas P. Increased IL-22- and IL-17A-Producing mucosal-associated invariant T cells in the peripheral blood of patients with ankylosing spondylitis. Front Immunol (2018) 9:1610. doi: 10.3389/fimmu.2018.01610
216. Guggino G, Rizzo A, Mauro D, Macaluso F, Ciccia F. Gut-derived CD8(+) tissue-resident memory T cells are expanded in the peripheral blood and synovia of SpA patients. Ann Rheum Dis (2021) 80(11):e174. doi: 10.1136/annrheumdis-2019-216456
217. Kok L, Masopust D, Schumacher TN. The precursors of CD8(+) tissue resident memory T cells: from lymphoid organs to infected tissues. Nat Rev Immunol (2021) 22(5):283–93. doi: 10.1038/s41577-021-00590-3
218. Djenidi F, Adam J, Goubar A, Durgeau A, Meurice G, de Montpréville V, et al. CD8+CD103+ tumor-infiltrating lymphocytes are tumor-specific tissue-resident memory T cells and a prognostic factor for survival in lung cancer patients. J Immunol (2015) 194(7):3475–86. doi: 10.4049/jimmunol.1402711
219. Corgnac S, Malenica I, Mezquita L, Auclin E, Voilin E, Kacher J, et al. CD103(+)CD8(+) T(RM) cells accumulate in tumors of anti-PD-1-Responder lung cancer patients and are tumor-reactive lymphocytes enriched with Tc17. Cell Rep Med (2020) 1(7):100127. doi: 10.1016/j.xcrm.2020.100127
220. May E, Märker-Hermann E, Wittig BM, Zeitz M, Meyer zum Büschenfelde KH, Duchmann R. Identical T-cell expansions in the colon mucosa and the synovium of a patient with enterogenic spondyloarthropathy. Gastroenterology. (2000) 119(6):1745–55. doi: 10.1053/gast.2000.20173
221. Duc D, Vigne S, Bernier-Latmani J, Yersin Y, Ruiz F, Gaïa N, et al. Disrupting myelin-specific Th17 cell gut homing confers protection in an adoptive transfer experimental autoimmune encephalomyelitis. Cell Rep (2019) 29(2):378–390.e4. doi: 10.1016/j.celrep.2019.09.002
222. Salmi M, Andrew DP, Butcher EC, Jalkanen S. Dual binding capacity of mucosal immunoblasts to mucosal and synovial endothelium in humans: dissection of the molecular mechanisms. J Exp Med (1995) 181(1):137–49. doi: 10.1084/jem.181.1.137
223. Battat R, Dulai PS, Jairath V, Vande Casteele N. A product review of vedolizumab in inflammatory bowel disease. Hum Vaccin Immunother (2019) 15(10):2482–90. doi: 10.1080/21645515.2019.1591139
224. Targan SR, Feagan BG, Fedorak RN, Lashner BA, Panaccione R, Present DH, et al. Natalizumab for the treatment of active crohn's disease: results of the ENCORE trial. Gastroenterology. (2007) 132(5):1672–83. doi: 10.1053/j.gastro.2007.03.024
225. Varkas G, Thevissen K, De Brabanter G, Van Praet L, Czul-Gurdian F, Cypers H, et al. An induction or flare of arthritis and/or sacroiliitis by vedolizumab in inflammatory bowel disease: a case series. Ann Rheum Dis (2017) 76(5):878–81. doi: 10.1136/annrheumdis-2016-210233
226. Dubash S, Marianayagam T, Tinazzi I, Al-Araimi T, Pagnoux C, Weizman AV, et al. Emergence of severe spondyloarthropathy-related entheseal pathology following successful vedolizumab therapy for inflammatory bowel disease. Rheumatol (Oxford) (2019) 58(6):963–8. doi: 10.1093/rheumatology/key267
227. Diaz LI, Keihanian T, Schwartz I, Bin Kim S, Calmet F, Alejandra Quintero M, et al. Vedolizumab-induced De novo extraintestinal manifestations. Gastroenterol Hepatol (N Y) (2020) 16(2):75–81.
228. Zundler S, Fischer A, Schillinger D, Binder MT, Atreya R, Rath T, et al. The α4β1 homing pathway is essential for ileal homing of crohn's disease effector T cells in vivo. Inflammation Bowel Dis (2017) 23(3):379–91. doi: 10.1097/mib.0000000000001029
229. Salmi M, Rajala P, Jalkanen S. Homing of mucosal leukocytes to joints. distinct endothelial ligands in synovium mediate leukocyte-subtype specific adhesion. J Clin Invest (1997) 99(9):2165–72. doi: 10.1172/jci119389
230. Carter RA, Campbell IK, O'Donnel KL, Wicks IP. Vascular cell adhesion molecule-1 (VCAM-1) blockade in collagen-induced arthritis reduces joint involvement and alters b cell trafficking. Clin Exp Immunol (2002) 128(1):44–51. doi: 10.1046/j.1365-2249.2002.01794.x
231. Silverman MD, Haas CS, Rad AM, Arbab AS, Koch AE. The role of vascular cell adhesion molecule 1/ very late activation antigen 4 in endothelial progenitor cell recruitment to rheumatoid arthritis synovium. Arthritis Rheumatol (2007) 56(6):1817–26. doi: 10.1002/art.22706
232. Kong DH, Kim YK, Kim MR, Jang JH, Lee S. Emerging roles of vascular cell adhesion molecule-1 (VCAM-1) in immunological disorders and cancer. Int J Mol Sci (2018) 19(4):1057. doi: 10.3390/ijms19041057
233. Lin CC, Pan CS, Wang CY, Liu SW, Hsiao LD, Yang CM. Tumor necrosis factor-alpha induces VCAM-1-mediated inflammation via c-src-dependent transactivation of EGF receptors in human cardiac fibroblasts. J BioMed Sci (2015) 22(1):53. doi: 10.1186/s12929-015-0165-8
234. Jin H, Jiang S, Wang R, Zhang Y, Dong J, Li Y. Mechanistic insight into the roles of integrins in osteoarthritis. Front Cell Dev Biol (2021) 9:693484. doi: 10.3389/fcell.2021.693484
235. Loeser RF. Integrins and chondrocyte-matrix interactions in articular cartilage. Matrix Biol (2014) 39:11–6. doi: 10.1016/j.matbio.2014.08.007
236. Fernandes NRJ, Reilly NS, Schrock DC, Hocking DC, Oakes PW, Fowell DJ. CD4(+) T cell interstitial migration controlled by fibronectin in the inflamed skin. Front Immunol (2020) 11:1501. doi: 10.3389/fimmu.2020.01501
237. Tada T, Inoue N, Widayati DT, Fukuta K. Role of MAdCAM-1 and its ligand on the homing of transplanted hematopoietic cells in irradiated mice. Exp Anim. (2008) 57(4):347–56. doi: 10.1538/expanim.57.347
238. Ciccia F, Guggino G, Rizzo A, Saieva L, Peralta S, Giardina A, et al. Type 3 innate lymphoid cells producing IL-17 and IL-22 are expanded in the gut, in the peripheral blood, synovial fluid and bone marrow of patients with ankylosing spondylitis. Ann Rheum Dis (2015) 74(9):1739–47. doi: 10.1136/annrheumdis-2014-206323
239. Wang S, Wu C, Zhang Y, Zhong Q, Sun H, Cao W, et al. Integrin α4β7 switches its ligand specificity via distinct conformer-specific activation. J Cell Biol (2018) 217(8):2799–812. doi: 10.1083/jcb.201710022
240. Ericsson A, Kotarsky K, Svensson M, Sigvardsson M, Agace W. Functional characterization of the CCL25 promoter in small intestinal epithelial cells suggests a regulatory role for caudal-related homeobox (Cdx) transcription factors. J Immunol (2006) 176(6):3642–51. doi: 10.4049/jimmunol.176.6.3642
241. Qaiyum Z, Gracey E, Yao Y, Inman RD. Integrin and transcriptomic profiles identify a distinctive synovial CD8+ T cell subpopulation in spondyloarthritis. Ann Rheum Dis (2019) 78:1566–1575. doi: 10.1136/annrheumdis-2019-215349
Keywords: axial spondyloarthritis, hereditary autoinflammatory diseases, Th17 cells, etiology, genetics
Citation: Xiong Y, Cai M, Xu Y, Dong P, Chen H, He W and Zhang J (2022) Joint together: The etiology and pathogenesis of ankylosing spondylitis. Front. Immunol. 13:996103. doi: 10.3389/fimmu.2022.996103
Received: 17 July 2022; Accepted: 28 September 2022;
Published: 17 October 2022.
Edited by:
Michele Maria Luchetti Gentiloni, Marche Polytechnic University, ItalyReviewed by:
Patricia Ruiz Limón, Universidad de Málaga, SpainTom Macleod, University of Leeds, United Kingdom
Weibin Du, Zhejiang Chinese Medical University, China
Copyright © 2022 Xiong, Cai, Xu, Dong, Chen, He and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianmin Zhang, anpoYW5nQGlibXMucHVtYy5lZHUuY24=; Wei He, aGV3ZWluZ2RAMTI2LmNvbQ==