 Dabin Huang
Dabin Huang Ping Wang
Ping Wang Juncao Chen1
Juncao Chen1 Mingwei Zhu
Mingwei Zhu Yaping Tang
Yaping Tang Wei Zhou
Wei Zhou- 1Department of Neonatology, Guangzhou Women and Children’s Medical Center, Guangzhou Medical University, Guangzhou, China
- 2Department of Pediatrics, Guangdong Second Provincial General Hospital, Guangzhou, China
- 3Guangzhou Institute of Pediatrics, Guangzhou Women and Children’s Medical Center, Guangzhou Medical University, Guangzhou, China
Neonatal necrotizing enterocolitis (NEC) is an inflammatory disease that occurs in premature infants and has a high mortality rate; however, the mechanisms behind this disease remain unclear. The TLR4 signaling pathway in intestinal epithelial cells, mediated by TLR4, is important for the activation of the inflammatory storm in NEC infants. Myeloid differentiation protein 2 (MD2) is a key auxiliary component of the TLR4 signaling pathway. In this study, MD2 was found to be significantly increased in intestinal tissues of NEC patients at the acute stage. We further confirmed that MD2 was upregulated in NEC rats. MD2 inhibitor (MI) pretreatment reduced the occurrence and severity of NEC in neonatal rats, inhibited the activation of NF-κB and the release of inflammatory molecules (TNF-α and IL-6), and reduced the severity of intestinal injury. MI pretreatment significantly reduced enterocyte apoptosis while also maintaining tight junction proteins, including occludin and claudin-1, and protecting intestinal mucosal permeability in NEC rats. In addition, an NEC in vitro model was established by stimulating IEC-6 enterocytes with LPS. MD2 overexpression in IEC-6 enterocytes significantly activated NF-κB. Further, both MD2 silencing and MI pretreatment inhibited the inflammatory response. Overexpression of MD2 increased damage to the IEC-6 monolayer cell barrier, while both MD2 silencing and MI pretreatment played a protective role. In conclusion, MD2 triggers an inflammatory response through the TLR4 signaling pathway, leading to intestinal mucosal injury in NEC. In addition, MI alleviates inflammation and reduces intestinal mucosal injury caused by the inflammatory response by blocking the TLR4-MD2/NF-κB signaling axis. These results suggest that inhibiting MD2 may be an important way to prevent NEC.
Introduction
Neonatal necrotizing enterocolitis (NEC) is a common and serious intestinal inflammatory disease occurring in the neonatal period and is considered a destructive gastrointestinal disorder leading to the death of premature infants (1–3). A cohort study of 9,552 premature infants with gestational age less than 32 weeks found that the incidence of NEC was 4.9% based on data collected from 57 tertiary hospitals in 25 provinces of China in 2019 (4). The common long-term and severe complications of the NEC survivors included delayed neuropsychomotor development, short bowel syndrome, cholestasis, intestinal failure and intestinal stenosis (5–7). Although NEC etiology and pathogenesis are not clearly understood, studies have shown that exaggerated inflammatory signal imbalance in the intestinal mucosa and intestinal injury work together to induce NEC in premature infants (8). Pattern recognition receptors are widely expressed in various cells of the intestinal epithelium; these receptors recognize pathogenic molecules and immunogenic factors and initiate a series of signal cascade reactions. Thus, pattern recognition receptors are the key component of intestinal innate immunity and play a central role in the first line of intestinal immunity defense (9, 10). Toll-like receptors (TLRs) are a class of typical pattern recognition receptors (8–10). As microbial ligands stimulate intestinal mucosa, TLR inflammatory signaling pathways are activated to begin the inflammatory cascade. In particular, TLR4, which is expressed in intestinal epithelial cells, is the receptor for lipopolysaccharide (LPS), and is associated with the occurrence and development of NEC (11–14). Exaggerated proinflammatory signal transduction of the TLR4 signaling pathway promotes mucosal injury in the intestine and enhances the destruction of the intestinal barrier (15). TLR4-induced epithelial cell death in the intestine is the foundation for intestinal mucosal injury in NEC (16–18). Targeting the TLR4 signaling pathway might be an attractive approach for treating NEC based on studies linking TLR4 and NEC.
As a coreceptor of TLR4, myeloid differentiation protein 2 (MD2) is required in order to recognize LPS and subsequently activate the TLR4 signaling pathway (19, 20). When LPS binds to MD2, the LPS-MD2-TLR4 complex is dimerized and further recruits myeloid differentiation factor 88 (MyD88). This event leads to activation of downstream signaling pathways, especially the nuclear factor kappa-B (NF-κB) pathway, which results in the expression of proinflammatory molecules (20). A clinical study found that the transcription level of the MD2 gene in acute-phase intestinal tissues of NEC patients was approximately 8 times higher than that in recovery-phase tissues (21). A study of 42 newborns with NEC showed that a polymorphic site in the promoter region of MD2 (rs11465996) was closely associated with the occurrence and severity of NEC (22). Previous studies have shown that human milk oligosaccharides can reduce the severity of NEC in mice and piglets, and computer molecular modeling suggested that human milk oligosaccharides might compete for the LPS binding sites on the MD2-TLR4 complex (23). Therefore, MD-2 may be related to the development of NEC, and its abnormal expression, structural abnormalities, or functional abnormalities may affect the expression of the MD2-TLR4/NF-κB signaling pathway and cause an abnormal immune response. The LPS-induced inflammatory response may be significantly diminished by targeting MD2 (24), thereby decreasing the likelihood of adverse events including sepsis (25), cardiac/kidney injuries (26, 27), cancer (28), colitis (29) and neuroinflammation (30). These findings indicate that targeting MD2 to inhibit the excessive activation of TLR4 signaling may be an effective strategy for the treatment of NEC.
At present, the role of MD2 in NEC is unknown. Our clinical samples showed that MD2 expression was significantly higher in acute-phase intestinal tissue of NEC patients than in tissue collected in the recovery period. We hypothesized that injury to intestinal epithelial cells induced by the TLR4 signaling pathway is dependent on MD2. In this study, an MD2 inhibitor (MI) was applied to an NEC rat model, and different MD2 intervention strategies (MD2 overexpression, MD2 knockout and MI pretreatment) were used in IEC-6 enterocytes to test this hypothesis. Overall, we elucidated the mechanism of MD2 in the occurrence and development of NEC. Further, the results suggested that MD2 may be an effective target for reducing the incidence and severity of NEC by early diagnosis and intervention.
Materials and methods
NEC patient selection and sample collection
According to the Declaration of Helsinki, after informed consent from the patients’ parents in Department of Neonatology in Guangzhou Women and Children’s Medical Center and approval by the Medical Ethics Review Committee were obtained (Ethical approval number: [2022] 059A01), the clinical intestinal tissues of NEC neonates and control neonates were collected. The intestinal tissue of NEC neonates came from patients who underwent intestinal resection due to severe intestinal necrosis. The severity of NEC was assessed in clinics based on Bell’s classification, which identifies the disease as stage I, II, or III (31). In our study, we collected samples from 4 infants with Bell stage II and III NEC, 2 convalescent infants with NEC and 2 infants with congenital intestinal atresia as controls. The inclusion criteria for the subjects in the control group were based on previous studies (32).
Animal experiments
All animal experiments were approved by the Institutional Animal Care and Use Committee of Guangzhou Medical University and performed according to the institutional guidelines. Forty-eight newborn male or female Sprague–Dawley (SD) rats (6–8 g) were collected from pregnant SD rats (Beijing Vital River Laboratory Animal Technology Co., Ltd., Beijing, China). Neonatal rats were separated from maternal rats immediately after birth and randomly divided into 4 groups with 12 rats in each group. The overall experimental scheme, including grouping, NEC protocol, treatment and sampling times, is summarized in sFigure 1.
The NEC model was achieved by formula feeding and the asphyxia/cold stress method with reference to previous studies (33, 34). Briefly, a newborn incubator was used to warm and humidify neonatal rats at 37°C. After stabilization, rats were hand-fed with formula gavage (Abbott Similac infant formula: Esbilac canine milk replacer 2:1) six times per day. A slow increase in feeding was introduced at the onset, starting at 0.10 mL every four hours and gradually increasing to 0.2–0.3 mL every four hours. In addition, every 12 hours, the neonatal rats were subjected to hypoxia (95% nitrogen for 90 seconds) and hypothermia (4 °C for 10 min). In the treatment group, rats were given MI (20 mg/kg/d) by gavage (35). MI (MD2-IN-1, Selleck, Shanghai, China) was dissolved in 1% sodium carboxymethyl cellulose (CMC-Na) for in vivo administration. MD2-IN-1 is a specific MD2 inhibitor that blocks the LPS-induced activation of TLR4/MD2-downstream proinflammatory MAPK/NF-κB signaling pathways by binding to the hydrophobic pocket of MD2 (35). In the control group, rats were normally breast fed. As a vehicle control, animals were gavaged with 1:100 dilutions of CMC-Na. Throughout the experiment, the clinical signs of NEC, such as abdominal distension, stool traits, gastric retention, apnea and lethargy, were monitored every 6 hours in the animals. Animals were euthanized after 72 hours or when clinical symptoms of NEC appeared.
Animal sampling
On the 7th day after birth, the rats were euthanized after fasting for 12 hours, and the intestines were collected and evaluated macroscopically for signs of NEC (gas accumulation, hemorrhage and necrosis). Upon sacrifice, the ileum was taken from the ileocecum junction approximately 1 cm away, fixed with 4% paraformaldehyde and embedded in paraffin for histological examination or stored at -80° for protein collection and cytokine measurement.
Histopathological examination and grading
Paraffin ileal tissue sections (4 mm) of humans and rats were prepared and stained with hematoxylin and eosin after dewaxing and hydration. The stained sections were observed by optical microscopy. The intestinal injury of rats was evaluated according to the histopathological scoring system of Nadler (36), and the pathological grades were as follows: grade 0 indicates normal intestine; grade 1 indicates exfoliation or separation of epithelial cells; grade 2 indicates moderate submucosal separation and local villous necrosis; grade 3 indicates severe submucosal separation and villous necrosis; and grade 4 indicates transmural necrosis. Samples graded 2 or higher were classified as NEC positive after histopathologic evaluation by two blinded pathologists.
Western blotting and immunoprecipitation
The total protein from the ileal tissues was extracted with lysis buffer (Beyotime, China). A Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime, China) was used for the preparation of nuclear and cytoplasmic fractions. The concentration of protein was determined by BCA assay (Invitrogen, USA). The protein was separated by SDS−PAGE and transferred to polyvinylidene fluoride membranes to further incubate with 5% skim milk in TBS/0.05% Tween-20 for 1 hour at room temperature. Following overnight incubation with primary antibodies at 4°C, membranes were incubated with HRP-conjugated secondary antibodies for 1 hour. These were the primary antibodies used: anti-MD2 (1:500, ab24182, Abcam), anti-MyD88 (1:1000, NB100-56698, Novus), anti-TLR4 (1:1000, sc-293072, Santa Cruz), anti- inhibitor of IκB alpha (IκBα) (1:1000, 10268-1-AP, Proteintech), anti-Phospho-IκBα (Ser32) (1:1000, 2859, Cellsignal), anti-NF-κB p65 (1:1000, 10745-1-AP, Proteintech), anti-phospho-NF-κB p65 (Ser536) (1:1000, 3033, Cellsignal), anti-high-mobility group box 1 (HMGB1) (1:1000, 10829-1-AP, Proteintech), anti-caspase-3 (1:1000, 19677-1-AP, Proteintech), anti-cleaved-caspase-3 (1:1000, 9661, Cellsignal), anti-occludin (1:1000, 66378-1-Ig, Proteintech), anti-claudin-1 (1:1000, 13050-1-AP, Proteintech), anti-GAPDH (1:2000, 2118, Cellsignal), anti-β-actin (1:2000, 4970, Cellsignal) and anti-Lamin-B1 (1:1000, 12987-1-AP, Proteintech). Immunoreactive bands were detected by enhanced chemiluminescence reagents (Millipore, USA). ImageJ software (National Institutes of Health, Bethesda, MD) was used to calculate relative protein levels.
For coimmunoprecipitation assays, intestinal tissue lysates (300–500 μg) were subjected to an overnight incubation with primary TLR4 antibody at 4°C and then immunoprecipitated using Protein A/G Sepharose beads. The bound protein was eluted in denatured SDS loading buffer and detected by western blotting.
Immunofluorescence staining
For immunofluorescence, sections of paraffin ileum tissue (4 mm) were prepared and fixed with 4% PFA. Then, 5% goat serum was added for 1 hour at room temperature, and the primary antibodies were applied overnight in a dark chamber at 4°C. After incubating with the second antibody at room temperature for 1 hour, the sections were mounted with VECTASHIELD Antifade Mounting Medium with DAPI to stain the nucleus. Using a Leica SP8 inverted fluorescence microscope, immunofluorescent images were acquired. ImageJ software was used to calculate the average fluorescence intensity of MD2 and occludin.
The following was the primary antibodies and their concentrations: rabbit anti-rat MD2 (ab24182, Abcam) (1:50), mouse anti-rat occludin (66378-1-Ig, Proteintech) (1:200). The secondary antibodies and their concentrations were as follows: goat anti-mouse or rabbit IgG H&L-Alexa Fluor 555 (ab150114 or ab150078, Abcam) (1:1000).
ELISA
According to the manufacturer’s instructions, ELISA kits (CUSABIO, Wuhan, China) were used to measure IL-6 and TNF-α concentrations in rat intestinal homogenates. The BCA method was used to measure the total protein content.
Intestinal permeability measurement
After the NEC protocol was performed, rats were gavaged with fluorescein isothiocyanate-dextran (FITC-D, Sigma; MW 10,000; 40 mg FITC-D/100 g) 4 hours before being sacrificed. After collecting the serum of rats, the fluorescence density was measured by a Multiskan Go plate reader (Thermo Fisher, USA). Additionally, the serum FITC-D concentration was determined.
TUNEL staining
According to the manufacturer’s protocol, the TUNEL assay was performed by using the In Situ Cell Death Detection Kit TMR Red (12156792910, Roche). Enterocyte apoptosis in vivo was determined by measuring the number of intestinal epithelial cells that were TUNEL+ by confocal microscopy per high-power field, and three high-power visual fields were randomly selected for each sample. The area of intestinal tissue in each high-power field was calculated by ImageJ software. To calculate the number of apoptotic cells per square millimeter, we used the following formula: number of apoptotic cells per square millimeter (number/mm2) = TUNEL+ cell number/area mm2.
Cell culture and treatment
IEC-6 enterocytes were purchased from iCell Bioscience Inc. (Shanghai, China) and cultured according to the manufacturer’s recommendations. DMEM-high glucose medium (GIBCO, USA) containing 10% fetal bovine serum and 1% antibiotics (100 U/mL penicillin and 0.1 mg/mL streptomycin) was used for cell culture. LPS (L2880, Sigma) was added to serum-free DMEM according to the experimental design. MI was dissolved in DMSO for in vitro experiments, and cells in the vehicle control were treated with DMSO at a 1:1000 dilution.
Lentivirus transduction and small interfering RNA transfection
Lentiviral vector particles (Genechem, Shanghai, China) containing MD2 short hairpin RNA (shRNA) were transfected into intestinal cells to generate MD2-overexpressing IEC-6 enterocytes. The shRNA sequence is as follows: 5′-GAG GAT CCC CGG GTA CCG GTC GCC ACC ATG TTG CCA TTT TTT CTC TTT TC-3′. Cell lines overexpressing MD2 were screened using medium containing puromycin (3 μg/mL). An siRNA specific for MD2 (5′-CAU GUU GAG UUC AUU CCA ATT-3′) was designed and synthesized by Tsingke (Beijing, China). According to the manufacturer’s protocol, this siRNA was transfected into cells with Lipofectamine 3000 transfection reagent (Invitrogen, Waltham, MA, USA). MD2 protein and mRNA expression levels were detected by real-time quantitative PCR (RT–qPCR) and western blot in each group after overexpression or knockdown of MD2.
RT–qPCR
TRIzol reagent (Invitrogen) was used to extract total RNA from enterocytes, after which a PrimeScript RT kit (RR047A, Takara, Japan) was used to reverse transcribe the RNA to cDNA. RT–qPCR was performed using a SYBR Premix Ex Taq II kit (RR820A, Takara, Japan) according to the manufacturer’s protocol. The following is the information of the primers: rat MD2 (forward: 5′-CTG AAA GGG GAG GCT GTC AAC AC-3′; reverse: 5′-GCA ATG GCT TCT GCA ACA CAT CTG-3′); rat TNF-α (forward: 5′-ATG GGC TCC CTC TCA TCA GTT CC-3′; reverse: 5′-GCT CCT CCG CTT GGT GGT TTG-3′); rat IL-6 (forward: 5′-ACT TCC AGC CAG TTG CCT TCT TG-3′; reverse: 5′-TGG TCT GTT GTG GGT GGT ATC CTC-3′); rat beta-actin (forward: 5′-CTG AGA GGG AAA TCG TGC GTG AC-3′; reverse: 5′-AGG AAG AGG ATG CGG CAG TGG-3′).
Determination of transepithelial electrical resistance and FITC-D permeability measurement
The Transwell cells were inoculated with 5 × 104 cells per well (200 μL) in the upper compartment, and 600 μL culture medium was added to the lower compartment. Transepithelial electrical resistance (TEER) was measured by a Millicell-ERS voltmeter after changing the culture medium and balancing the incubator for 30 minutes. In short, the two electrode probes of the resistance instrument were placed in the upper and lower compartment of the Transwell chamber, and the average TEER values of three different points were measured and calculated as the actual TEER values. The assay was carried out in triplicate. Two Transwell chambers with only culture medium were used as blank controls. To calculate the standard TEER value, we used the formula TEER value = (measured value − blank control value)/0.33 cm2.
After relevant intervention, FITC-D (100 μL, 1 mg/mL) was added to the upper compartment of the Transwell chamber, and 600 μL serum-free medium was added to the lower compartment. Then, the Transwell chamber was incubated at 37°C for 1 hour. The medium in the lower compartment was collected, and the fluorescence intensity was measured by a Multiskan Go plate reader (ThermoFisher, USA) (emission wavelength, 525 nm; excitation wavelength, 488 nm). According to the standard curve, the fluorescence intensity of each sample was converted into the FITC-D concentration. To measure FITC-D permeability, the following equation was used: FITC-D permeability (%/h/cm2) = (FITC-D concentration in the lower compartment/initial concentration of FITC-D upper compartment)/1 hr/0.33 cm2 × 100%.
Statistical analysis
Data analysis was performed using SPSS 25.0 software (SPSS, Inc., Chicago, IL, USA) and GraphPad Prism 8 (GraphPad Software Inc., USA). The comparison of multiple groups was performed using one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test. A two-tailed unpaired t test was used for comparisons between two groups. The pathological scores among multiple groups were compared by the Kruskal–Wallis test. All data are presented as the mean ± standard deviation. P<0.05 was considered a statistically significant difference.
Results
Clinical characteristics and MD2 expression levels in intestinal tissues of NEC patients
We collected samples from 4 neonates with Bell stage III NEC, 2 neonates with congenital intestinal atresia and 2 convalescent neonates with NEC who served as the controls. Among the NEC cases, infants often developed NEC after 4 weeks of hospitalization, and their birth weight was less than 1500 g (Table 1). Serum hypersensitive C-reactive protein (hs-CRP), which is an inflammatory cascade biomarker, was significantly increased in patients with acute NEC. Of note, 1 NEC neonate had intestinal failure, and 3 NEC neonates had neonatal cholestasis (Table 1). Next, we investigated whether MD2 plays a role in NEC. Compared with that in the control samples, MD2 expression was significantly increased in clinical samples from NEC neonates according to immunofluorescence analysis (P<0.05) (Figures 1A, B).
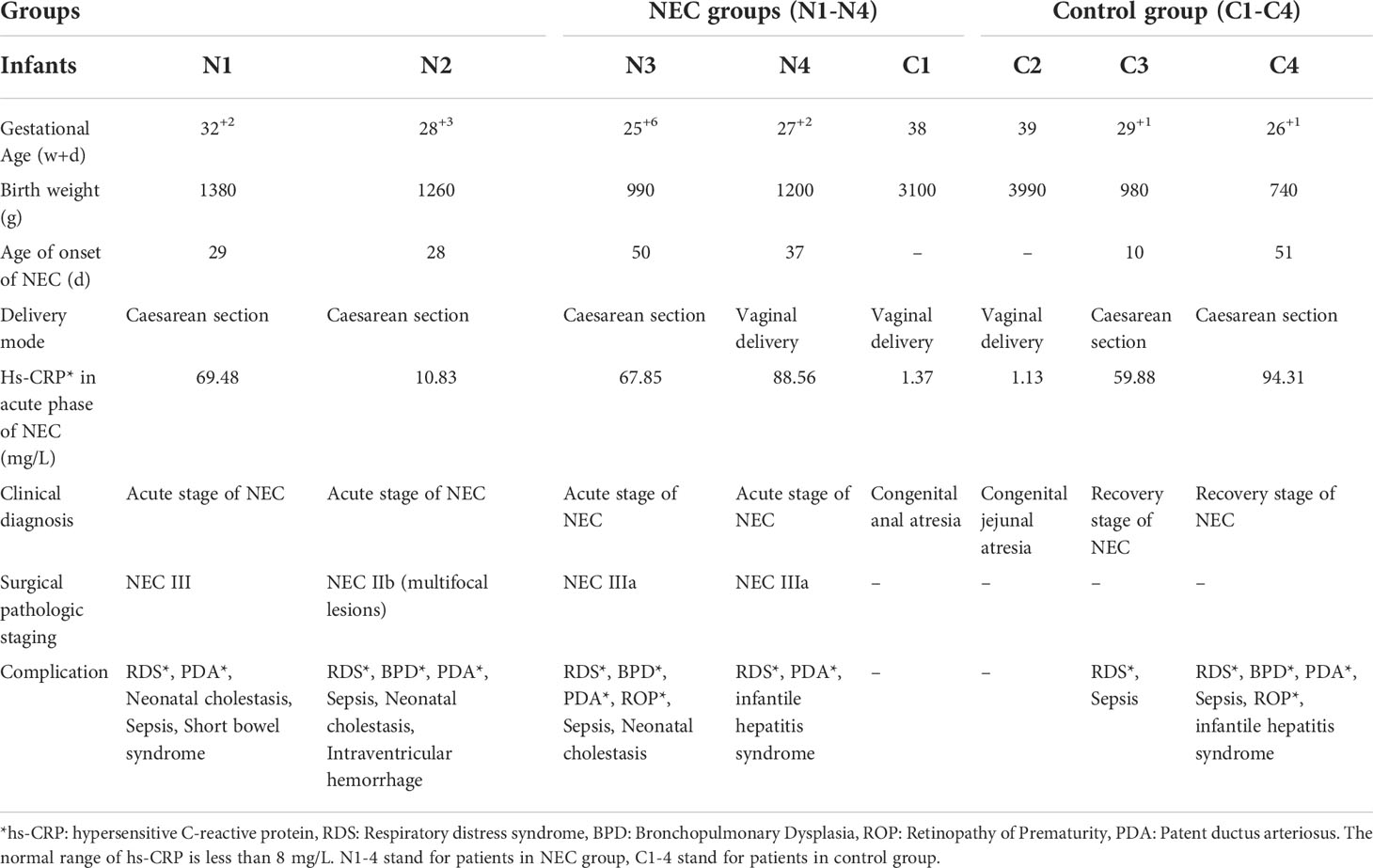
Table 1 Clinical and laboratory data of NEC infants and control volunteers (n=8).
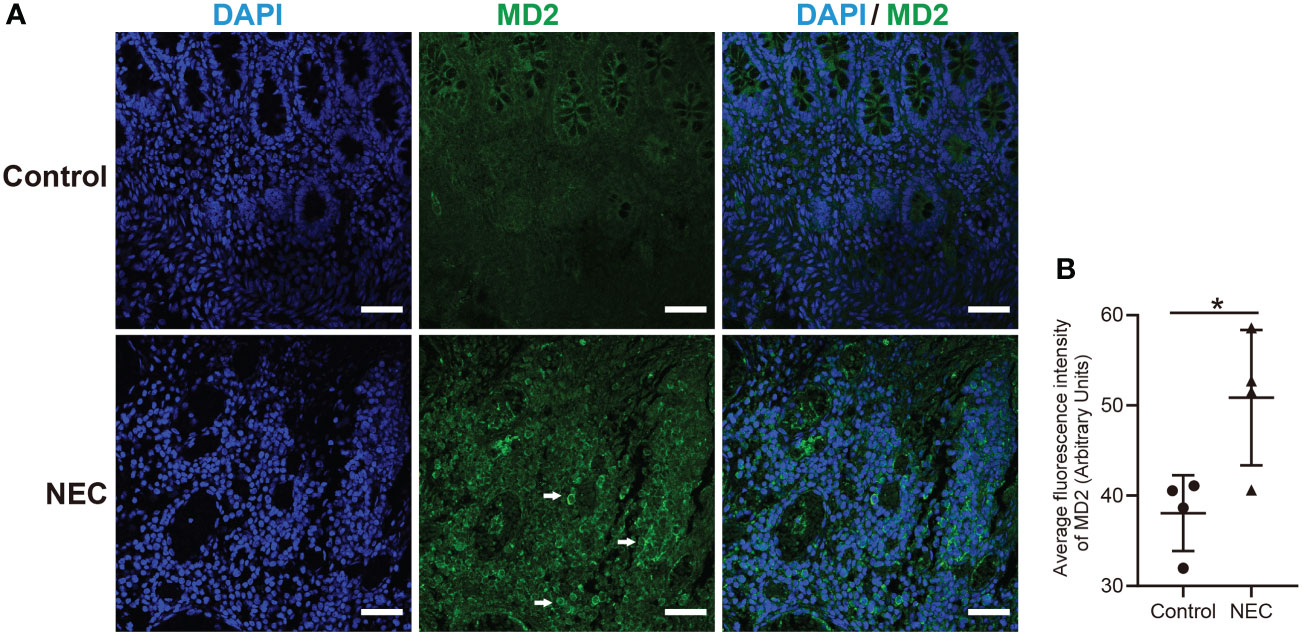
Figure 1 Clinical characteristics and MD2 expression levels in intestinal tissues of NEC patients. (A) MD2 expression in intestinal tissue of patients. MD2 (green) and DAPI staining of nuclei (blue) in intestinal tissue of the control and NEC groups. Scale bar, 50 μm. (B) Average fluorescence intensity of MD2. Data are presented as the mean ± SD (n=4). The scatter plots represent the mean value of the average intensity of fluorescence in 6 random fields in each section of the quantification diagram. *p value < 0.05 for each group comparison using a two-tailed Student’s t test.
Evaluation of the general state, pathological alterations and MD2 protein expression in NEC rats
During the experiment, NEC neonatal rats gradually developed abdominal distension, diarrhea and yellow mucous stool excretion, and their activity decreased as determined by poor response and drowsiness. In the treatment group, neonatal rats also gradually developed abdominal distension, diarrhea and poor response. In contrast, the treatment group had later occurrence and a lower degree of disease than the NEC group. The general condition of the control and vehicle-treated rats was good, with normal behavior and feces. First, we evaluated the expression of MD2 in the intestinal tissue of NEC rats. As predicted, results from western blot analysis (Figures 2A, B) showed that MD2 expression was higher in experimental NEC rats than in control rats. In the NEC group samples, the intestinal tract showed extensive edema, dim coloration with intestinal dilatation, and air within the bowel wall (known as pneumatosis intestinalis), and in severe cases, these effects were accompanied by beaded changes. The intestinal dilatation and pneumatosis of rats treated with MI were mild (Figure 2C). Histological examination showed edema in the submucosa and muscular layer, separation of the submucosa and extensive exfoliation of villi in the NEC intestines (Figure 2D). Compared with the control rats, the NEC rats presented with a significantly higher grade of histological injury (Figure 2E). However, the rats in the treatment group showed a significant decrease in histological injury grade compared to those in the NEC group (Figure 2E).
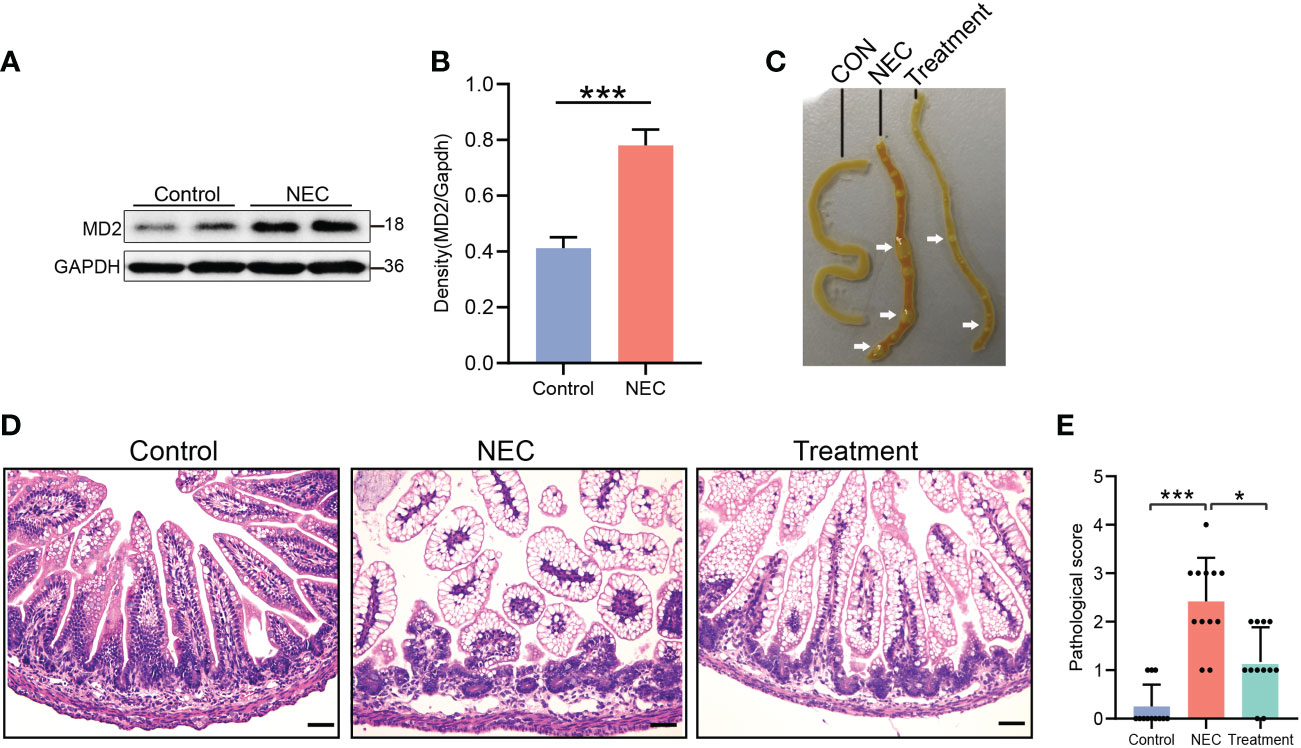
Figure 2 Evaluation of pathological condition, histological score and MD2 protein expression in NEC rats. (A, B) The expression of MD2 in the intestinal tissue of NEC neonatal rats was detected by western blot analysis (n=4). The p value for each group comparison using a two-tailed Student’s t test. ***p < 0.001. (C) Representative images showing the gross intestinal morphology. (D, E) Histological H&E staining of the transverse section of ileal tissue and histological NEC score in each group. Scores of 2 or more were regarded as NEC positive. Scale bar, 50 μm. Data are presented as the mean ± SD (n=12) and analyzed by the Kruskal–Wallis test. *p < 0.05, ***p < 0.001.
Blocking MD2 inhibits NF-κB and inflammatory responses in NEC rats
We determined the expression level of the MD2-TLR4 complex by coimmunoprecipitation of rat intestinal tissue. As expected, the level of the MD2-TLR4 complex in the NEC rats was 4 times higher than in the control rats, but the increase was not observed in the treatment group rats (Figures 3A, B). The expression of TLR4 and MyD88 in the NEC group was higher than that in the control group, while expression in the treatment group was lower (Figures 3C, D). We next evaluated the phosphorylation of IκBα, a step that is necessary for translocation of the NF-κB p65 subunit and activation of NF-κB. The levels of IκBα phosphorylation and NF-κB translocation in the intestinal tissue of the NEC group were significantly increased, while pretreatment with MI significantly inhibited the activation of NF-κB (Figures 3C, D). The activation of MD2-TLR4/NF-κB signal transduction prompts the production of proinflammatory cytokines, leading to an inflammatory cascade reaction. ELISA analysis of inflammatory molecules in intestinal tissue showed that the expression of TNF-α (Figure 3E) and IL-6 (Figure 3F) was upregulated in both the NEC group and the treatment group compared to the control group; however, the levels in the treatment group were lower than those in the NEC group. These results showed that MD2 blockade prevents the activation of MD2-TLR4 signaling and inhibits the production of proinflammatory factors.
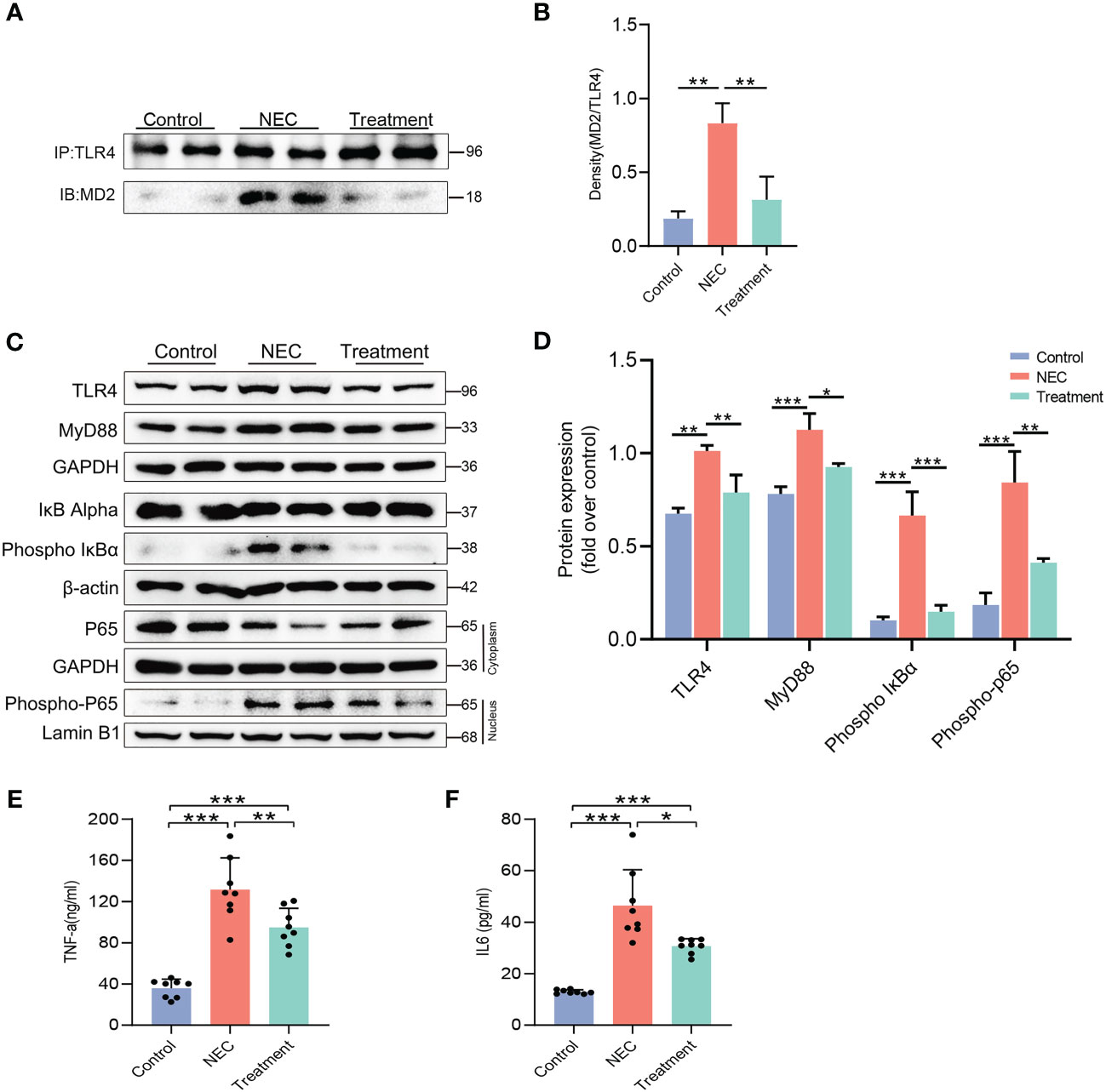
Figure 3 Blocking MD2 inhibits NF-κB and inflammatory responses in NEC rats. (A, B) Immunoprecipitation assays showed that MD2 inhibitor pretreatment significantly reduced the formation of the TLR4–MD2 complex in rat intestinal tissue. IB indicates immunoblotting antibody, and IP indicates precipitating antibody (n=3). (C, D) Western blot analysis of TLR4 signaling-related proteins, NF-κB activation and nuclear translocation of the p65 subunit of NF-κB (n=3). (E, F) TNF-α and IL-6 levels in intestinal tissue lysates were detected by ELISA, and values were normalized to the total protein (n=8). The p value was analyzed by one-way ANOVA with multiple comparisons, followed by Tukey’s post hoc test. *p < 0.05, **p < 0.01, ***p < 0.001.
MD2 inhibition attenuates enterocyte apoptosis and intestinal barrier dysfunction in NEC rats
Next, we further studied the protective mechanism of MI. Enterocyte apoptosis induced by the TLR4 inflammatory cascade is the central pathway leading to NEC-induced mucosal inflammation. TUNEL staining revealed prominent NEC enterocyte apoptosis, which was significantly attenuated by MI (Figures 4A, B). Caspase-3 is a marker for apoptosis, and its degree of cleavage can reflect the level of apoptosis. The increased expression of the protein HMGB1 can reflect an increase in tissue or cell injury. As shown in Figures 4C–E, the expression levels of cleaved caspase-3 and HMGB1 were highly increased in NEC, while the expression levels of cleaved caspase-3 and HMGB1 were markedly suppressed by MI.
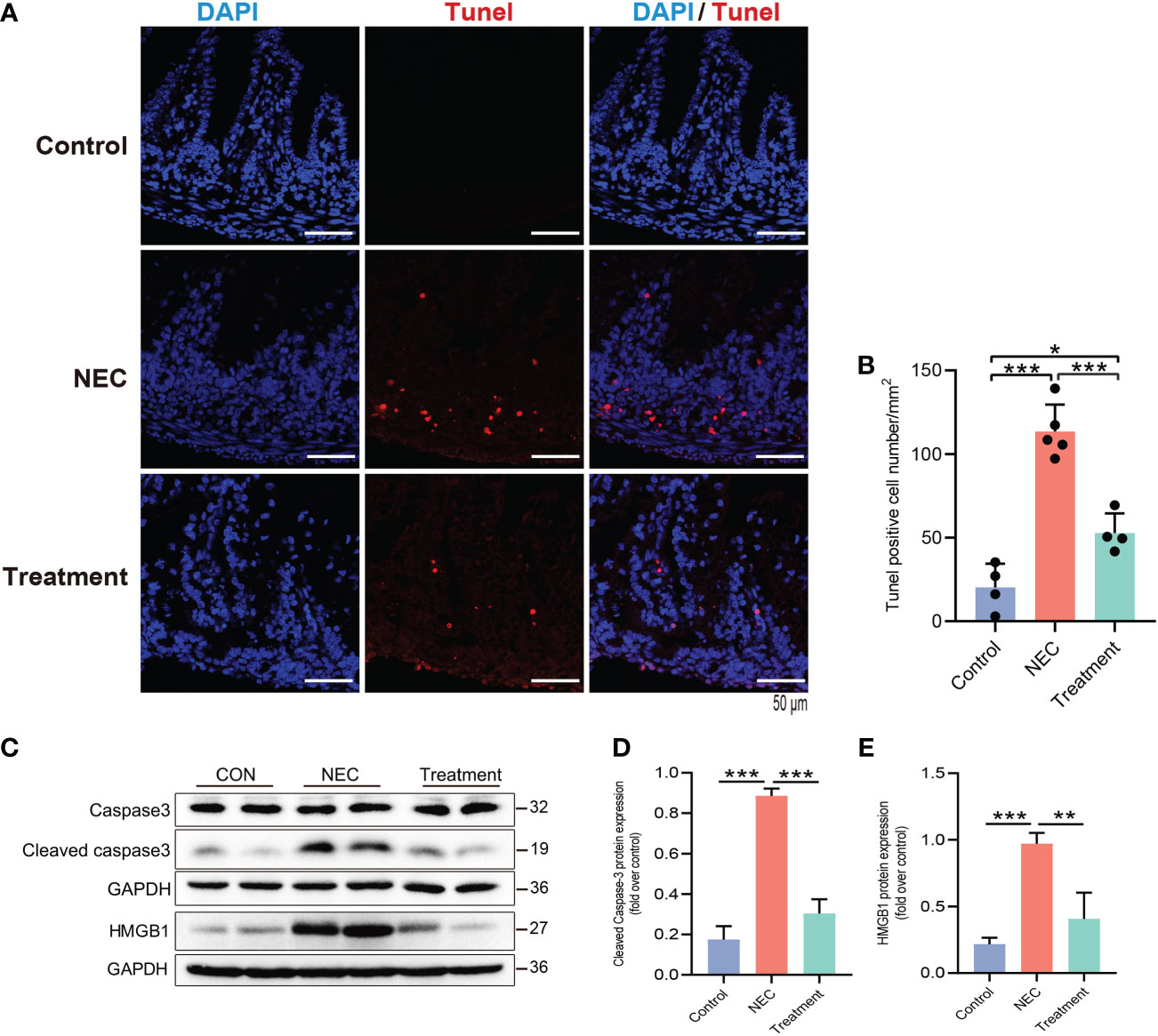
Figure 4 MD2 blockade attenuates apoptosis of intestinal epithelial cells in NEC rats. (A, B) Detection of intestinal cell apoptosis in each group of neonatal rats; red represents TUNEL+ cells. NEC significantly increased TUNEL+ cells, while pretreatment with MD2 inhibitor significantly blocked this effect (n=4 in the control group, n=4 in the treatment group and n=5 in the NEC group). Scale bar, 50 μm. (C–E) The protein expression of cleaved caspase-3 and HMGB1 in the intestines of the three groups was analyzed by Western blotting (n=3). The p value was analyzed by one-way ANOVA with multiple comparisons, followed by Tukey’s post hoc test. *p < 0.05, **p < 0.01, ***p < 0.001.
We further evaluated the intestinal barrier function of neonatal rats. The expression of occludin protein in the NEC group was decreased, while the expression of occludin protein in the control and treatment groups was relatively high and evenly distributed (Figures 5A, B). In the NEC group, the levels of tight junction (TJ) proteins, including claudin-1 and occludin, were decreased (Figures 5C, D), thereby leading to impairment of the intestinal barrier function. MD2 blockade protected against this loss of function caused by NEC (Figures 5A–D). The permeability of the intestinal mucosa was detected by FITC-D. The intestinal permeability of FITC-D was significantly enhanced in NEC rats compared to control rats, while the leakage of FITC-D into circulation was reduced in rats pretreated with MI (Figure 5E).
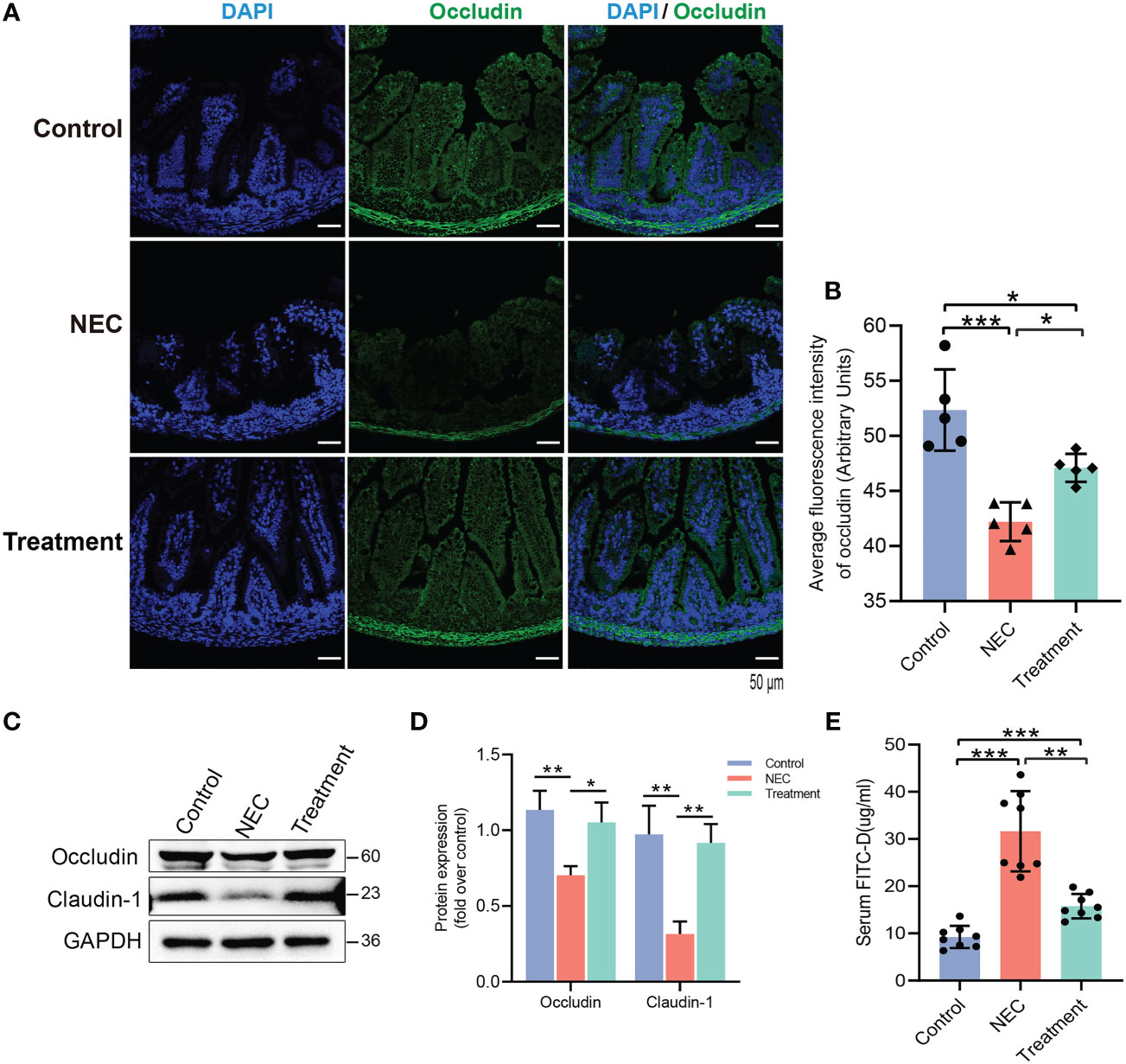
Figure 5 Evaluation of the effect of MD2 on intestinal mucosal barrier function in NEC rats. (A, B) Occludin expression in the intestinal tissue of rats. Occludin (green) and DAPI staining of nuclei (blue). The scatter plots represent the mean value of the average optical density in 3 random fields in each section of the quantification diagram. Scale bar, 50 μm, n=5. (C, D) The protein expression of occludin and claudin-1 in the intestines of the three groups was analyzed by Western blotting (n=3). (E) The intestinal permeability was detected by FITC-D (n=8). The p value was analyzed by one-way ANOVA with multiple comparisons, followed by Tukey’s post hoc test. *p < 0.05, **p < 0.01, ***p < 0.001.
Efficiency of MD2 overexpression or knockdown and effects of LPS on NF-κB and the inflammatory response in IEC-6 enterocytes
We next used a lentivirus-mediated stable MD2 overexpression cell line (IEC-6), with the MD2 gene silenced by siRNA, to further explore the effect of MD2 on IEC-6 enterocytes stimulated by LPS. Transfection of the lentiviral plasmid expressing MD2 shRNA resulted in significantly higher MD2 protein and mRNA levels in IEC-6MD2-Over cells than in IEC-6-CON cells (Figures 6A–C). After transfection with siRNA, the mRNA and protein levels of MD2 in IEC-6MD2-Si cells were significantly downregulated compared to those in nontarget-treated cells, and the effective silencing of MD2 expression was 80.5% (Figures 6D–F).
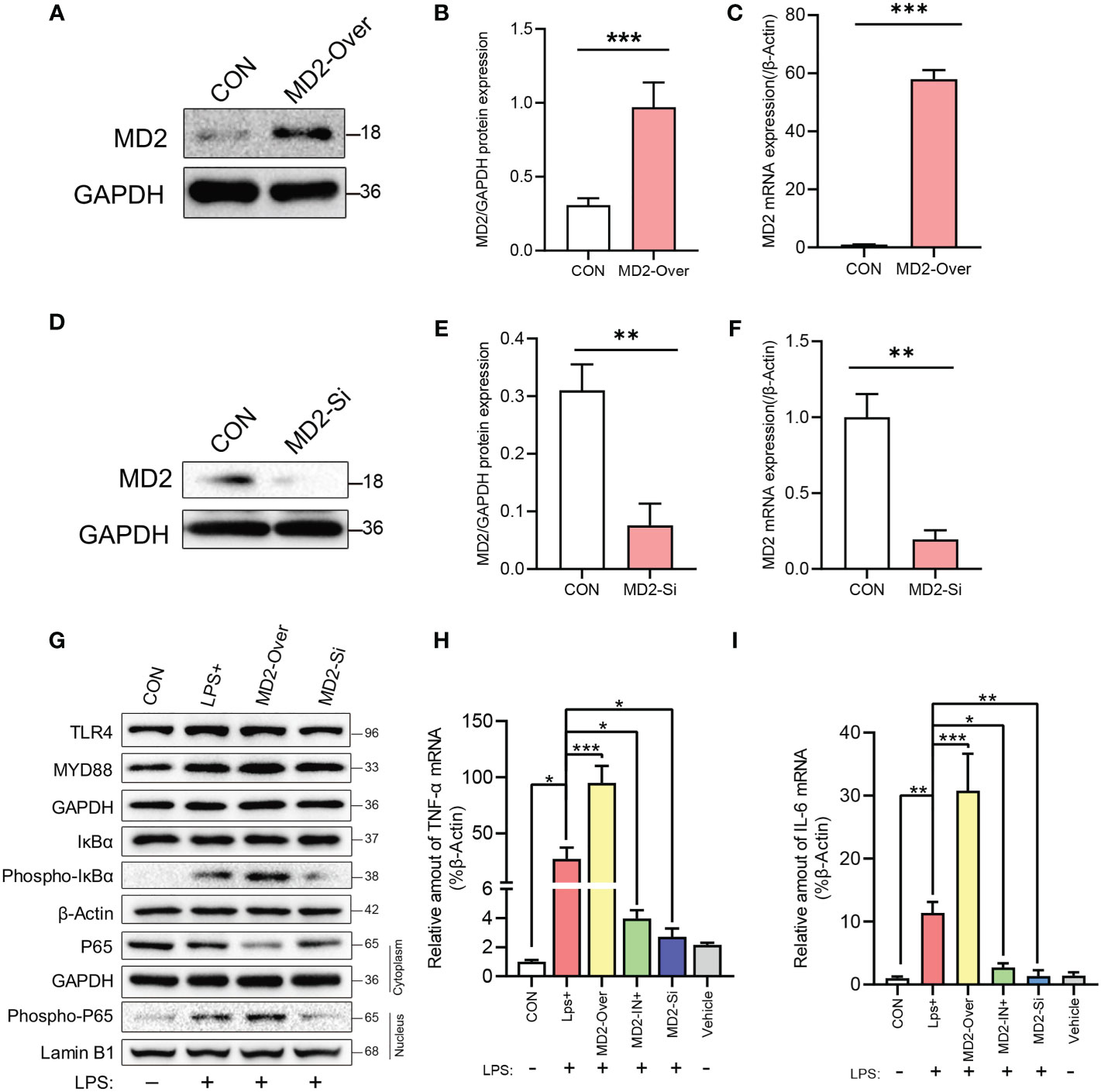
Figure 6 Efficiency of MD2 overexpression or knockdown and the effects of LPS on NF-κB and the inflammatory response in IEC-6 enterocytes. (A–C) Evaluation of MD2 overexpression efficiency by western blot analysis (n=4) and RT–qPCR analysis (n=3). (D–F) Evaluation of MD2 silencing efficiency (n=3). (G) Effects of LPS on TLR4 signaling-related proteins, NF-κB activation and nuclear translocation of the p65 subunit of NF-κB in IEC-6 enterocytes (n=3). (H, I) The effect of LPS on proinflammatory cytokines in IEC-6 enterocytes was analyzed by RT–qPCR (n=3). LPS+, IEC-6 + LPS; MD2-Over, MD2-overexpressing IEC-6 + LPS; MD2-IN-1+, IEC-6 + MD2 inhibitor + LPS; MD2-Si, MD2-deficient IEC-6 + LPS; Vehicle, IEC-6 treated with DMSO at a 1:1000 dilution. (B–F) p value for each group comparison using a two-tailed Student’s t test. **p < 0.01, ***p < 0.001. (H, I) The p value was analyzed by one-way ANOVA with multiple comparisons, followed by Tukey’s post hoc test. *p < 0.05, **p < 0.01, ***p < 0.001.
After LPS induction, the levels of IκBα phosphorylation and NF-κB P65 nuclear translocation in IEC-6MD2-Over cells were upregulated compared to those in control cells, while the activation of NF-κB was decreased in IEC-6MD2-Si cells (Figure 6G). RT–qPCR analysis indicated that LPS significantly increased the expression of TNF-α and IL-6 mRNA in IEC-6MD2-Over cells compared to IEC-6-CON cells, and this increase in TNF-α and IL-6 mRNA expression was prevented by MD2 gene silencing and MI pretreatment (Figures 6H, I).
Effects of MD2 on inflammatory injury and barrier function in IEC-6 enterocytes
As shown in Figure 7A, LPS stimulation highly activated the inflammatory injury-related protein HMGB1 and the apoptosis-related protein caspase-3 in IEC-6 enterocytes. Moreover, MD2 overexpression further increased this activation, whereas MD2 silencing reduced this activation. After LPS-induced injury, the expression of claudin-1 and occludin in IEC-6MD2-Over cells was significantly decreased compared to IEC-6-CON cells and IEC-6MD2-Si cells (Figure 7A). TEER is a common method to detect monolayer cell barriers and evaluate the integrity of cell−cell tight junctions. The TEER assay showed that the TEER value of monolayer cells decreased after IEC-6 cells were injured by LPS. The most severe decrease in TEER value was seen in IEC-6MD2-Over cells, while pretreatment with MI suppressed the decrease in TEER value (Figure 7B). Notably, the decrease in TEER value was accompanied by an increase in FITC-D fluorescence intensity. FITC-D detection showed that the cell permeability of IEC-6MD2-Over cells was higher than that of IEC-6 cells with or without LPS intervention, while the cell permeability of IEC-6 cells pretreated with MI was diminished (Figure 7C).
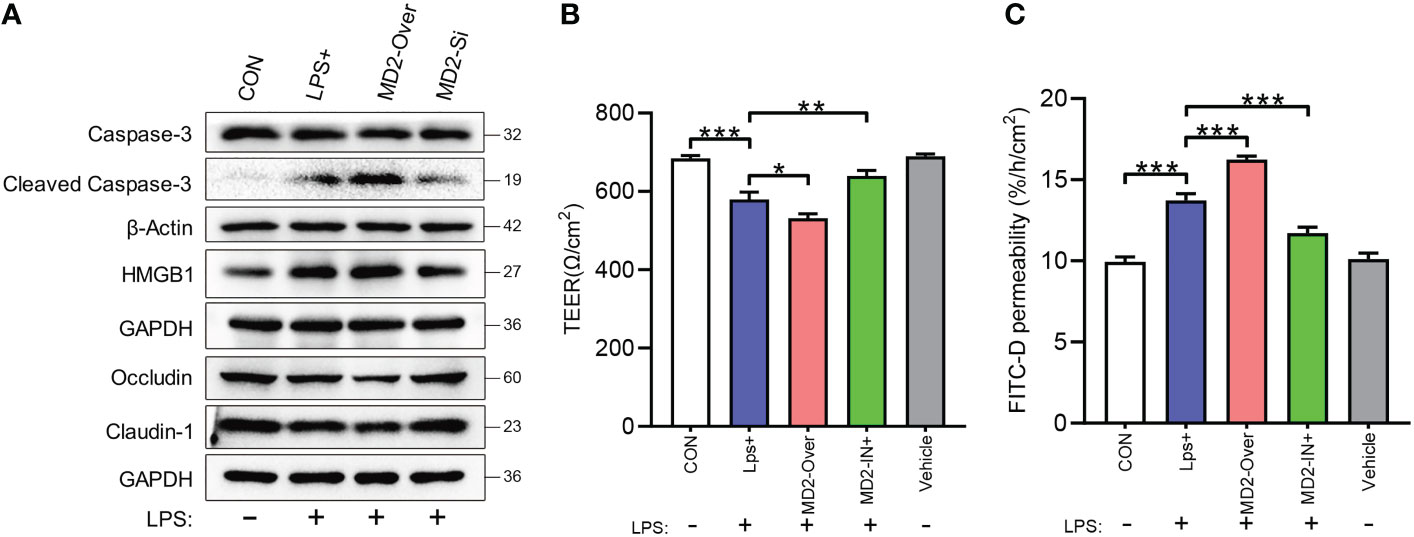
Figure 7 Effects of MD2 on inflammatory injury and barrier function in IEC-6 enterocytes. (A) The expression of HMGB1, caspase-3, cleaved caspase-3, occludin and claudin-1 was measured by western blot analysis (n=3). (B) Cell monolayer permeability was detected using TEER. (C) Determination of FITC-D permeability (n=3). LPS+, IEC-6 + LPS; MD2-Over, MD2-overexpressing IEC-6 + LPS; MD2-IN-1+, IEC-6 + MD2 inhibitor + LPS; Vehicle, IEC-6 treated with DMSO at a 1:1000 dilution. p value was analyzed by one-way ANOVA with multiple comparisons, followed by Tukey’s post hoc test. *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
NEC is a destructive and inflammatory intestinal disease that affects the life of premature infants and is characterized by multiple intestinal injuries ranging from epithelial injury to transmural involvement and perforation (2, 37). The role of TLR4 in the pathogenesis of neonatal NEC has become increasingly important. Excessive stimulation of TLR4 in the intestinal epithelium leads to NEC by initiating the signal cascade reaction and further leading to breakdown of the intestinal barrier (15, 37–39). Activated TLR4 triggers the activation of downstream signaling pathways and leads to the translocation of NF-κB, leading to the overexpression of proinflammatory molecules (40). Excessive inflammatory mediator damage and necrotic intestinal tissue destroy the mucosal barrier, resulting in the development of NEC (41). Of note, apoptosis of enterocytes is the precursor of intestinal mucosal injury in NEC (42).
MD2 can directly recognize and bind to the LPS lipid A domain with the activation of TLR4, which is an indispensable auxiliary protein (43). MyD88 is the main downstream effector of the TLR4-MD2-LPS complex following binding to LPS (44). The present study found that MD2 is highly correlated with NEC in preterm infants. Previous studies have shown that the transcription level of the MD2 gene in acute-phase intestinal tissues of NEC patients was increased (21). Similarly, we confirmed that the MD2 protein was upregulated in the acute phase of neonatal NEC. Formula feeding, hypothermia, and asphyxia have been identified as risk factors for the development of NEC (45–47). In this work, the neonatal rat NEC model was constructed by targeting multiple factors in the pathogenesis of NEC using formula feeding and hypoxia-cold stimulation with reference to previous studies (48–51). This model has been proven to be an ideal NEC modeling method that reproduces the typical pathophysiological changes in NEC by combining a variety of pathogenic factors. In this experiment, NEC rats had NEC-like lesions similar to those observed in clinical NEC patients, including abdominal distension, gastric retention, diarrhea, intestinal dilatation, inflammatory leukocyte infiltration, necrosis and perforation. Pretreatment with MI reduced the incidence and severity of NEC and prevented intestinal inflammatory injury caused by hypoxia/cold stress, determined by the decrease in NEC scores and the levels of TNF-α and IL-6 in the ileum. A large amount of evidence shows that there are high concentrations of LPS and TLR/MyD88-associated proinflammatory cytokines in the serum of NEC patients, including IL-1β, IL-6 and TNF-α (52, 53). MI administration in experimental NEC rats preserved the intestinal villous architecture, maintained barrier function and significantly decreased apoptosis levels. During initiation of the inflammatory cascade reaction, rapid degradation of IκB-α in the cytoplasm and downstream translocation of phosphorylated NF-κB p65 are necessary to activate NF-κB. In this study, both of these processes were attenuated by pretreatment with MI. Thus, the findings presented here showed that MI prevented NEC-related intestinal injury in neonatal rats by targeting the MD2-TLR4/NF-κB inflammatory pathway.
Barrier dysfunction is another important pathophysiological basis of NEC. During the early stages of NEC development, abnormal cell death and epithelial barrier disruption are necessary conditions for further development (11, 15, 16). Numerous studies have demonstrated that the intestinal barrier dysfunction observed in human NEC is characterized by the downregulation of mucins and TJ proteins, such as zonula occludens-1, occludin and claudin-4 (54–56). In this study, we found that the leakage of FITC-D into the circulation increased and the expression of claudin-1 and occludin decreased in NEC samples, suggesting severe barrier dysfunction. The intestinal epithelial (enterocyte) cell line IEC-6 is often used as an NEC model system in vitro (17, 57–59). IEC-6 cells are nontransformed intestinal cells that have abundant expression of TLR4 and respond to LPS. These cells are derived from rat small intestine with epithelioid morphological characteristics and an immature crypt-like phenotype (60). The IEC-6 model induced by LPS in vitro showed that overexpression of MD2 stimulated damage to the single-layer cell barrier, while both MD2 silencing and MI pretreatment were protective. Previous studies have shown that suppression of the TLR4 signaling pathway protects the intestinal barrier function of NEC (61–63). The barrier-protective effect of MD2 has been previously reported in mice with chronic colitis (29). Barrier dysfunction caused by severe intestinal epithelial cell death is the basis of intestinal mucosal injury in NEC, in which TLR4 plays a key role (17, 64). In the present study, we found that enterocyte apoptosis increased and that caspase-3 and HMGB1 were highly activated in NEC rats, and these effects were reversed by pretreatment with MI. HMGB1 is an important proinflammatory cytokine and inflammatory transmitter (65–67). The increased expression of HMGB1 leads to enhancement of the inflammatory response in the tissue and promotes the development of NEC (68, 69).
The present study showed that targeting MD2 to block the activation of TLR4 reduces excessive inflammatory storms, intestinal barrier function injury and intestinal cell apoptosis in NEC patients. The present study had several limitations. The number of human samples was small, and the direct effect of MD2 on TJs was not explored in depth. In addition, genetically engineered rats and human intestinal epithelial cell lines were not used to further explore the mechanism. The specificity of the treatment window that was explored is another limitation of this study. In future work, we will further explore the appropriate time course of treatment.
Conclusion
In summary, we found that the MD2 protein is upregulated in NEC infants. MD2 leads to overactive inflammation through the MD2-TLR4/NF-κB signaling axis and promotes enterocyte apoptosis and barrier dysfunction, thereby exacerbating NEC. Because TLR4 activation is a common pathogenic alteration of NEC, inhibiting MD2 is a potential therapeutic target for the treatment of NEC.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Materials. Further inquiries can be directed to the corresponding author.
Ethics statement
All the studies followed the guidelines for the ethical treatment of human specimens and were approved by the Ethics Committee of Guangzhou Women and Children's Medical Center (Ethical approval number: [2022] 059A01). According to the Declaration of Helsinki, written informed consent was obtained from the parents of all the neonates. All animal experiments were approved by the Institutional Animal Care and Use Committee of Guangzhou Medical University and performed according to the institutional guidelines.
Author contributions
DH, WZ, YT and MZ contributed to the study design. DH, YL and JC performed experiments. DH and PW analyzed data. WZ performed and/or contributed critically to all experiments. DH and PW drafted manuscript. PW and WZ edited and revised the manuscript. All the authors have read and approved the final manuscript.
Funding
Guangzhou Science and Technology Plan Project (No. 201904010484 to WZ, No. 202102080247 to PW). Basic and Applied Basic Research Foundation of Guangdong Province (No. 2022A1515012354 to PW).
Acknowledgments
We thank Professors WZ and YT for providing reagents and help. The authors also thank all the staff of the Yaping Tang Experimental Research Group Unit of the Institute of Pediatrics, Guangzhou Women and Children’s Medical Center.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.995791/full#supplementary-material
References
1. Eaton S, Rees CM, Hall NJ. Current research on the epidemiology, pathogenesis, and management of necrotizing enterocolitis. Neonatology (2017) 111(4):423–30. doi: 10.1159/000458462
2. Alganabi M, Lee C, Bindi E, Li B, Pierro A. Recent advances in understanding necrotizing enterocolitis. F1000Res (2019) 8:F1000 Faculty Rev-107. doi: 10.12688/f1000research.17228.1
3. Jiang S, Yan W, Li S, Zhang L, Zhang Y, Shah PS, et al. Mortality and morbidity in infants <34 weeks' gestation in 25 NICUs in China: A prospective cohort study. Front Pediatr (2020) 8:33. doi: 10.3389/fped.2020.00033
4. Cao Y, Jiang S, Sun J, Hei M, Wang L, Zhang H, et al. Assessment of neonatal intensive care unit practices, morbidity, and mortality among very preterm infants in china. JAMA Netw Open (2021) 4(8):e2118904. doi: 10.1001/jamanetworkopen.2021.18904
5. Hau EM, Meyer SC, Berger S, Goutaki M, Kordasz M, Kessler U. Gastrointestinal sequelae after surgery for necrotising enterocolitis: A systematic review and meta-analysis. Arch Dis Child Fetal Neonatal Ed (2019) 104(3):F265–73. doi: 10.1136/archdischild-2017-314435
6. Jones IH, Hall NJ. Contemporary outcomes for infants with necrotizing enterocolitis-a systematic review. J Pediatr (2020) 220:86–92. doi: 10.1016/j.jpeds.2019.11.011
7. Imren C, Vlug LE, de Koning B, Diertens T, Snel HE, Suurland J, et al. Necrotizing enterocolitis in a dutch cohort of very preterm infants: Prevalence, mortality, and long-term outcomes. Eur J Pediatr Surg (2022) 32(1):111–9. doi: 10.1055/s-0041-1741544
8. Hackam DJ, Sodhi CP. Toll-like receptor-mediated intestinal inflammatory imbalance in the pathogenesis of necrotizing enterocolitis. Cell Mol Gastroenterol Hepatol (2018) 6(2):229–38. doi: 10.1016/j.jcmgh.2018.04.001
9. Mara MA, Good M, Weitkamp JH. Innate and adaptive immunity in necrotizing enterocolitis. Semin Fetal Neonatal Med (2018) 23(6):394–9. doi: 10.1016/j.siny.2018.08.002
10. Fitzgerald KA, Kagan JC. Toll-like receptors and the control of immunity. Cell. (2020) 180(6):1044–66. doi: 10.1016/j.cell.2020.02.041
11. Sodhi CP, Neal MD, Siggers R, Sho S, Ma C, Branca MF, et al. Intestinal epithelial toll-like receptor 4 regulates goblet cell development and is required for necrotizing enterocolitis in mice. Gastroenterology (2012) 143(3):708–18. doi: 10.1053/j.gastro.2012.05.053
12. Yazji I, Sodhi CP, Lee EK, Good M, Egan CE, Afrazi A, et al. Endothelial TLR4 activation impairs intestinal microcirculatory perfusion in necrotizing enterocolitis via eNOS-NO-nitrite signaling. Proc Natl Acad Sci U S A. (2013) 110(23):9451–6. doi: 10.1073/pnas.1219997110
13. Egan CE, Sodhi CP, Good M, Lin J, Jia H, Yamaguchi Y, et al. Toll-like receptor 4-mediated lymphocyte influx induces neonatal necrotizing enterocolitis. J Clin Invest. (2016) 126(2):495–508. doi: 10.1172/JCI83356
14. Mihi B, Good M. Impact of toll-like receptor 4 signaling in necrotizing enterocolitis: The state of the science. Clin Perinatol (2019) 46(1):145–57. doi: 10.1016/j.clp.2018.09.007
15. Leaphart CL, Cavallo J, Gribar SC, Cetin S, Li J, Branca MF, et al. A critical role for TLR4 in the pathogenesis of necrotizing enterocolitis by modulating intestinal injury and repair. J Immunol (2007) 179(7):4808–20. doi: 10.4049/jimmunol.179.7.4808
16. Werts AD, Fulton WB, Ladd MR, Saad-Eldin A, Chen YX, Kovler ML, et al. A novel role for necroptosis in the pathogenesis of necrotizing enterocolitis. Cell Mol Gastroenterol Hepatol (2020) 9(3):403–23. doi: 10.1016/j.jcmgh.2019.11.002
17. Zhou Y, Li Y, Zhou B, Chen K, Lyv Z, Huang D, et al. Inflammation and apoptosis: Dual mediator role for toll-like receptor 4 in the development of necrotizing enterocolitis. Inflamm Bowel Dis (2017) 23(1):44–56. doi: 10.1097/MIB.0000000000000961
18. Liu T, Zong H, Chen X, Li S, Liu Z, Cui X, et al. Toll-like receptor 4-mediated necroptosis in the development of necrotizing enterocolitis. Pediatr Res (2022) 91(1):73–82. doi: 10.1038/s41390-021-01457-y
19. Schromm AB, Lien E, Henneke P, Chow JC, Yoshimura A, Heine H, et al. Molecular genetic analysis of an endotoxin nonresponder mutant cell line: A point mutation in a conserved region of MD-2 abolishes endotoxin-induced signaling. J Exp Med (2001) 194(1):79–88. doi: 10.1084/jem.194.1.79
20. Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. (2009) 458(7242):1191–5. doi: 10.1038/nature07830
21. Chan KL, Wong KF, Luk JM. Role of LPS/CD14/TLR4-mediated inflammation in necrotizing enterocolitis: Pathogenesis and therapeutic implications. World J Gastroenterol (2009) 15(38):4745–52. doi: 10.3748/wjg.15.4745
22. Zhou W, Yuan W, Huang L, Wang P, Rong X, Tang J. Association of neonatal necrotizing enterocolitis with myeloid differentiation-2 and GM2 activator protein genetic polymorphisms. Mol Med Rep (2015) 12(1):974–80. doi: 10.3892/mmr.2015.3499
23. Sodhi CP, Wipf P, Yamaguchi Y, Fulton WB, Kovler M, Nino DF, et al. The human milk oligosaccharides 2'-fucosyllactose and 6'-sialyllactose protect against the development of necrotizing enterocolitis by inhibiting toll-like receptor 4 signaling. Pediatr Res (2021) 89(1):91–101. doi: 10.1038/s41390-020-0852-3
24. Chen L, Fu W, Zheng L, Wang Y, Liang G. Recent progress in the discovery of myeloid differentiation 2 (MD2) modulators for inflammatory diseases. Drug Discov Today (2018) 23(6):1187–202. doi: 10.1016/j.drudis.2018.01.015
25. Duan G, Zhu J, Xu J, Liu Y. Targeting myeloid differentiation 2 for treatment of sepsis. Front Biosci (Landmark Ed). (2014) 19:904–15. doi: 10.2741/4256
26. Wang Y, Qian Y, Fang Q, Zhong P, Li W, Wang L, et al. Saturated palmitic acid induces myocardial inflammatory injuries through direct binding to TLR4 accessory protein MD2. Nat Commun (2017) 8:13997. doi: 10.1038/ncomms13997
27. Xu Z, Li W, Han J, Zou C, Huang W, Yu W, et al. Angiotensin II induces kidney inflammatory injury and fibrosis through binding to myeloid differentiation protein-2 (MD2). Sci Rep (2017) 7:44911. doi: 10.1038/srep44911
28. Rajamanickam V, Yan T, Xu S, Hui J, Xu X, Ren L, et al. Selective targeting of the TLR4 co-receptor, MD2, prevents colon cancer growth and lung metastasis. Int J Biol Sci (2020) 16(8):1288–302. doi: 10.7150/ijbs.39098
29. Han C, Guan Q, Guo L, Yang Y, Ruan S, Zhang X. The effects and mechanisms of myeloid differentiation protein 2 on intestinal mucosal permeability in mice with chronic colitis. J Cell Physiol (2019) 234(11):21089–99. doi: 10.1002/jcp.28711
30. Oo TT, Pratchayasakul W, Chattipakorn N, Chattipakorn SC. Potential roles of myeloid differentiation factor 2 on neuroinflammation and its possible interventions. Mol Neurobiol (2020) 57(11):4825–44. doi: 10.1007/s12035-020-02066-2
31. Bell MJ, Ternberg JL, Feigin RD, Keating JP, Marshall R, Barton L, et al. Neonatal necrotizing enterocolitis. therapeutic decisions based upon clinical staging. Ann Surg (1978) 187(1):1–7. doi: 10.1097/00000658-197801000-00001
32. Wei J, Tang D, Lu C, Yang J, Lu Y, Wang Y, et al. Irf5 deficiency in myeloid cells prevents necrotizing enterocolitis by inhibiting M1 macrophage polarization. Mucosal Immunol (2019) 12(4):888–96. doi: 10.1038/s41385-019-0169-x
33. Wei J, Zhou Y, Besner GE. Heparin-binding EGF-like growth factor and enteric neural stem cell transplantation in the prevention of experimental necrotizing enterocolitis in mice. Pediatr Res (2015) 78(1):29–37. doi: 10.1038/pr.2015.63
34. Kovler ML, Sodhi CP, Hackam DJ. Precision-based modeling approaches for necrotizing enterocolitis. Dis Model Mech (2020) 13(6):dmm044388. doi: 10.1242/dmm.044388
35. Zhang Y, Wu J, Ying S, Chen G, Wu B, Xu T, et al. Discovery of new MD2 inhibitor from chalcone derivatives with anti-inflammatory effects in LPS-induced acute lung injury. Sci Rep (2016) 6:25130. doi: 10.1038/srep25130
36. Nadler EP, Dickinson E, Knisely A, Zhang XR, Boyle P, Beer-Stolz D, et al. Expression of inducible nitric oxide synthase and interleukin-12 in experimental necrotizing enterocolitis. J Surg Res (2000) 92(1):71–7. doi: 10.1006/jsre.2000.5877
37. Neu J, Walker WA. Necrotizing enterocolitis. N Engl J Med (2011) 364(3):255–64. doi: 10.1056/NEJMra1005408
38. Nanthakumar N, Meng D, Goldstein AM, Zhu W, Lu L, Uauy R, et al. The mechanism of excessive intestinal inflammation in necrotizing enterocolitis: An immature innate immune response. PloS One (2011) 6(3):e17776. doi: 10.1371/journal.pone.0017776
39. Hodzic Z, Bolock AM, Good M. The role of mucosal immunity in the pathogenesis of necrotizing enterocolitis. Front Pediatr (2017) 5:40. doi: 10.3389/fped.2017.00040
40. Tanner SM, Berryhill TF, Ellenburg JL, Jilling T, Cleveland DS, Lorenz RG, et al. Pathogenesis of necrotizing enterocolitis: Modeling the innate immune response. Am J Pathol (2015) 185(1):4–16. doi: 10.1016/j.ajpath.2014.08.028
41. Satoh T, Izumi H, Iwabuchi N, Odamaki T, Namba K, Abe F, et al. Bifidobacterium breve prevents necrotising enterocolitis by suppressing inflammatory responses in a preterm rat model. Benef. Microbes (2016) 7(1):75–82. doi: 10.3920/BM2015.0035
42. Liu Y, Zhu L, Fatheree NY, Liu X, Pacheco SE, Tatevian N, et al. Changes in intestinal toll-like receptors and cytokines precede histological injury in a rat model of necrotizing enterocolitis. Am J Physiol Gastrointest Liver Physiol (2009) 297(3):G442–50. doi: 10.1152/ajpgi.00182.2009
43. Ryu JK, Kim SJ, Rah SH, Kang JI, Jung HE, Lee D, et al. Reconstruction of LPS transfer cascade reveals structural determinants within LBP, CD14, and TLR4-MD2 for efficient LPS recognition and transfer. Immunity. (2017) 46(1):38–50. doi: 10.1016/j.immuni.2016.11.007
44. Park BS, Lee JO. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp Mol Med (2013) 45:e66. doi: 10.1038/emm.2013.97
45. Nino DF, Sodhi CP, Hackam DJ. Necrotizing enterocolitis: New insights into pathogenesis and mechanisms. Nat Rev Gastroenterol Hepatol (2016) 13(10):590–600. doi: 10.1038/nrgastro.2016.119
46. Isani MA, Delaplain PT, Grishin A, Ford HR. Evolving understanding of neonatal necrotizing enterocolitis. curr. opin. Pediatr (2018) 30(3):417–23. doi: 10.1097/MOP.0000000000000629
47. Rose AT, Patel RM. A critical analysis of risk factors for necrotizing enterocolitis. Semin Fetal Neonatal Med (2018) 23(6):374–9. doi: 10.1016/j.siny.2018.07.005
48. Hou Y, Lu X, Zhang Y. IRAK inhibitor protects the intestinal tract of necrotizing enterocolitis by inhibiting the toll-like receptor (TLR) inflammatory signaling pathway in rats. Med Sci Monit (2018) 24:3366–73. doi: 10.12659/MSM.910327
49. Zhang D, Wen J, Zhou J, Cai W, Qian L. Milk fat globule membrane ameliorates necrotizing enterocolitis in neonatal rats and suppresses lipopolysaccharide-induced inflammatory response in IEC-6 enterocytes. JPEN J Parenter Enteral Nutr (2019) 43(7):863–73. doi: 10.1002/jpen.1496
50. Sun L. Low-dose cyclooxygenase-2 (COX-2) inhibitor celecoxib plays a protective role in the rat model of neonatal necrotizing enterocolitis. Bioengineered. (2021) 12(1):7234–45. doi: 10.1080/21655979.2021.1980646
51. Bell RL, Withers GS, Kuypers FA, Stehr W, Bhargava A. Stress and corticotropin releasing factor (CRF) promote necrotizing enterocolitis in a formula-fed neonatal rat model. PloS One (2021) 16(6):e246412. doi: 10.1371/journal.pone.0246412
52. Sharma R, Tepas JR, Hudak ML, Mollitt DL, Wludyka PS, Teng RJ, et al. Neonatal gut barrier and multiple organ failure: Role of endotoxin and proinflammatory cytokines in sepsis and necrotizing enterocolitis. J Pediatr Surg (2007) 42(3):454–61. doi: 10.1016/j.jpedsurg.2006.10.038
53. Li Z, Sheng L. Significance of dynamic evolution of TNF-alpha, IL-6 and intestinal fatty acid-binding protein levels in neonatal necrotizing enterocolitis. Exp Ther Med (2018) 15(2):1289–92. doi: 10.3892/etm.2017.5532
54. Moore SA, Nighot P, Reyes C, Rawat M, McKee J, Lemon D, et al. Intestinal barrier dysfunction in human necrotizing enterocolitis. J Pediatr Surg (2016) 51(12):1907–13. doi: 10.1016/j.jpedsurg.2016.09.011
55. Bein A, Eventov-Friedman S, Arbell D, Schwartz B. Intestinal tight junctions are severely altered in NEC preterm neonates. Pediatr Neonatol (2018) 59(5):464–73. doi: 10.1016/j.pedneo.2017.11.018
56. Liu D, Xu Y, Feng J, Yu J, Huang J, Li Z. Mucins and tight junctions are severely altered in necrotizing enterocolitis neonates. Am J Perinatol. (2021) 38(11):1174–80. doi: 10.1055/s-0040-1710558
57. Qureshi FG, Leaphart C, Cetin S, Li J, Grishin A, Watkins S, et al. Increased expression and function of integrins in enterocytes by endotoxin impairs epithelial restitution. Gastroenterology. (2005) 128(4):1012–22. doi: 10.1053/j.gastro.2005.01.052
58. Subramanian S, Geng H, Tan XD. Cell death of intestinal epithelial cells in intestinal diseases. Sheng Li Xue Bao (2020) 72(3):308–24. doi: 10.13294/j.aps.2020.0039
59. Yuan X, Xiong Z, Liu W, Li Y, Li H, Zhang X, et al. Novel therapeutic targeting of CCL3-CCR4 axis mediated apoptotic intesitnal injury in necrotizing enterocolitis. Front Immunol (2022) 13:859398. doi: 10.3389/fimmu.2022.859398
60. Cetin S, Ford HR, Sysko LR, Agarwal C, Wang J, Neal MD, et al. Endotoxin inhibits intestinal epithelial restitution through activation of rho-GTPase and increased focal adhesions. J Biol Chem (2004) 279(23):24592–600. doi: 10.1074/jbc.M313620200
61. Yang G, Bao P, Zhang L, Lyu Z, Zhou B, Chen K, et al. Critical role of myeloid differentiation factor 88 in necrotizing enterocolitis. Pediatr Res (2014) 75(6):707–15. doi: 10.1038/pr.2014.39
62. Huang K, Mukherjee S, DesMarais V, Albanese JM, Rafti E, Draghi IA, et al. Targeting the PXR-TLR4 signaling pathway to reduce intestinal inflammation in an experimental model of necrotizing enterocolitis. Pediatr Res (2018) 83(5):1031–40. doi: 10.1038/pr.2018.14
63. Shi Y, Liu T, Zhao X, Yao L, Hou A, Fu J, et al. Vitamin d ameliorates neonatal necrotizing enterocolitis via suppressing TLR4 in a murine model. Pediatr Res (2018) 83(5):1024–30. doi: 10.1038/pr.2017.329
64. Jilling T, Lu J, Jackson M, Caplan MS. Intestinal epithelial apoptosis initiates gross bowel necrosis in an experimental rat model of neonatal necrotizing enterocolitis. Pediatr Res (2004) 55(4):622–9. doi: 10.1203/01.PDR.0000113463.70435.74
65. Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature (2002) 418(6894):191–5. doi: 10.1038/nature00858
66. Bell CW, Jiang W, Reich CR, Pisetsky DS. The extracellular release of HMGB1 during apoptotic cell death. Am J Physiol Cell Physiol (2006) 291(6):C1318–25. doi: 10.1152/ajpcell.00616.2005
67. Yang H, Wang H, Andersson U. Targeting inflammation driven by HMGB1. Front Immunol (2020) 11:484. doi: 10.3389/fimmu.2020.00484
68. Vitali R, Terrin G, Palone F, Laudadio I, Cucchiara S, Boscarino G, et al. Fecal high-mobility group box 1 as a marker of early stage of necrotizing enterocolitis in preterm neonates. Front Pediatr (2021) 9:672131. doi: 10.3389/fped.2021.672131
Keywords: NEC, MD2, TLR4, NF-κB, intestinal barrier
Citation: Huang D, Wang P, Chen J, Li Y, Zhu M, Tang Y and Zhou W (2022) Selective targeting of MD2 attenuates intestinal inflammation and prevents neonatal necrotizing enterocolitis by suppressing TLR4 signaling. Front. Immunol. 13:995791. doi: 10.3389/fimmu.2022.995791
Received: 02 August 2022; Accepted: 14 October 2022;
Published: 01 November 2022.
Edited by:
Guirong Wang, Upstate Medical University, United StatesReviewed by:
Qinghe Meng, Upstate Medical University, United StatesXinlin Hou, First Hospital, Peking University, China
Copyright © 2022 Huang, Wang, Chen, Li, Zhu, Tang and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Zhou, emhvdXdlaV9wdTAwMkAxMjYuY29t