 Fan Jiang1†Mingjie Xia2†Yanan Zhang1,3†Jie Chang4Jiang Cao4Zhongkai Zhang5*Zhanyang Qian6*Lei Yang1,7*
Fan Jiang1†Mingjie Xia2†Yanan Zhang1,3†Jie Chang4Jiang Cao4Zhongkai Zhang5*Zhanyang Qian6*Lei Yang1,7*- 1Department of Orthopedics, Taizhou People’s Hospital, Nanjing Medical University, Taizhou, China
- 2Department of Orthopedics, Nanjing First Hospital, Nanjing Medical University, Nanjing, China
- 3Postgraduate School, Dalian Medical University, Dalian, China
- 4Department of Orthopedics, Affiliated First Hospital of Nanjing Medical University, Nanjing, China
- 5Department of Orthopedics, Shandong Provincial Hospital Affiliated to Shandong First Medical University, Jinan, China
- 6Department of Orthopedics, Zhongda Hospital, Nanjing, China
- 7School of Bioinformatics Engineering, Nanjing Medical University, Nanjing, China
Background: Neuroinflammation following spinal cord injury (SCI) results in prolonged neurological damage and locomotor dysfunction. Polarization of microglia is vital to regulation of neuroinflammation, although the underlying mechanisms have not yet been elucidated. Endocannabinoid receptor subtype 2 (CB2R) is reported to ameliorate neurodegeneration via immunomodulation activities. However, the underlying machinery in the context of SCI remains unclear.
Methods: A lipopolysaccharide-induced microglia inflammation model and a mouse model of SCI were employed to investigate the regulatory role of CB2R in the polarization of microglia in response to excess neuroinflammation. Markers of inflammation and autophagy were measured by Western blot analysis, immunofluorescence, flow cytometry, and enzyme-linked immunosorbent assays. Histological staining with hematoxylin and eosin, Nissl, and Luxol® fast blue was conducted using commercial kits. The locomotor function of the hindlimbs of the experimental mice was evaluated with the Basso Mouse Scale, Louisville Swim Scale, and footprint assay.
Results: The results showed that CB2R promoted M2 differentiation, increased interleukin (IL)-10 expression, and inhibited M1 differentiation with decreased expression of IL-1β and IL-6. CB2R activation also increased ubiquitination of the NLRP3 inflammasome and interacted with the autophagy-related proteins p62 and microtubule-associated proteins 1B light chain 3. Treatment with the CB2R activator JWH-133 reduced loss of myelin, apoptosis of neurons, and glial scarring, leading to improved functional recovery of the hindlimbs, while the CB2R antagonist AM630 produced opposite results.
Conclusion: Taken together, these results suggested that CB2R activation attenuated neuroinflammation targeting microglial polarization by promoting NLRP3 clearance, thereby facilitating functional recovery post-SCI.
Introduction
Spinal cord injury (SCI) is a deteriorative neuropathy that results in irreversible neurological dysfunction and severe physio-psychological damage to the patient (1, 2). However, the pathogenesis of SCI remains unclear, as available reports have mostly focused on either the induction of neuroinflammation, accumulation of reactive oxygen species (ROS), or development of neurotoxicity (3). Microglia are indispensable components of pathological events (4). Damage to the spinal cord initiates various inflammatory responses via immunoregulation of microglial injury and subsequent activation of damage-associated molecular patterns, which promote activation of microglia in the resting state via multiple molecular signaling pathways, leading to increased differentiation of M1 microglia that release destructive pro-inflammatory mediators and induce ROS generation and a lack of M2 microglia, thereby limiting excess inflammation, but fueling neuroinflammation (5–7). The cross-kingdom efficacy of microglial polarization as an immune-inflammatory junction suggests a potential target as a defensive mechanism, although the specific components remain unclear.
The NLRP3 (NOD-, LRR-, and pyrin domain-containing 3) inflammasome, an intracellular signaling multiprotein complex, is reported to be activated in M1 microglia. It promotes production of numerous inflammatory amplifiers and has been implicated in the pathogenesis of various non-infectious diseases (8–10). As the most characteristic inflammatory sensor molecule, NLRP3 triggers activation of capase-1 and maturation of interleukin (IL)-1β and IL-18 (11, 12). Moreover, autophagy is a scavenging mechanism that delivers damaged organelles as well as excess proteins to lysosomes. Multiple lines of evidence show that accumulation of the NLRP3 inflammasome induces autophagosome formation, which limits inflammasome activity (13–15). However, the therapeutic effect of the destruction of the NLRP3 inflammasome via regulation of autophagy to alleviate secondary injury post-SCI remains unclear.
Previous studies have suggested that activated microglia produce endocannabinoids and express cannabinoid receptor subtypes that suppress neuroinflammation (16). In addition, endocannabinoids were reported to be an effective adjuvant to inhibit activation of NLRP3 inflammasome (17). The endocannabinoid system is mainly composed of endocannabinoid receptor subtype 1 (CB1R), endocannabinoid receptor subtype 2 (CB2R), and endocannabinoids like 2-arachidonoyl glycerol, anandamide, and other structurally related lipids, as well as related enzymes for transportation, synthesis, and degradation, which are involved in a variety of physiological functions and pathological processes (18–20). Of interest, activation of CB2R in the central nervous system plays a protective role against neurodegenerative diseases attributed to immune regulation (21–25). Previous reports suggest that targeting CB2R manipulates M1/M2 transition of macrophages in response to inflammation and regulates autophagy-related signaling (26, 27). However, the unique mechanisms underlying regulation of neuroinflammation post-SCI via CB2R and autophagy remain elusive.
In the present study, a model of lipopolysaccharide (LPS)-induced inflammation of microglia and a mouse model of contusion-induced SCI were employed to explore the role of CB2R in SCI-induced neuroinflammation in vitro and in vivo, respectively. The results showed that activation of CB2R facilitated phenotype transition of microglia to the M2 state, rather than M1, restrained neuronal apoptosis, and protected neurological functions, suggesting that CB2R attenuated SCI-induced neuroinflammation by promoting ubiquitination and clearance of NLRP3 through autophagy in microglia.
Materials and methods
Study approval
The animal study protocol was approved by the Institutional Animal Care and Use Committee of Nanjing Medical University (Nanjing, China) and conducted in accordance with the guidelines described in “Animal Research: Reporting of In Vivo Experiments”.
Antibodies
The antibodies (Abs) mentioned in this study are listed in Table 1.
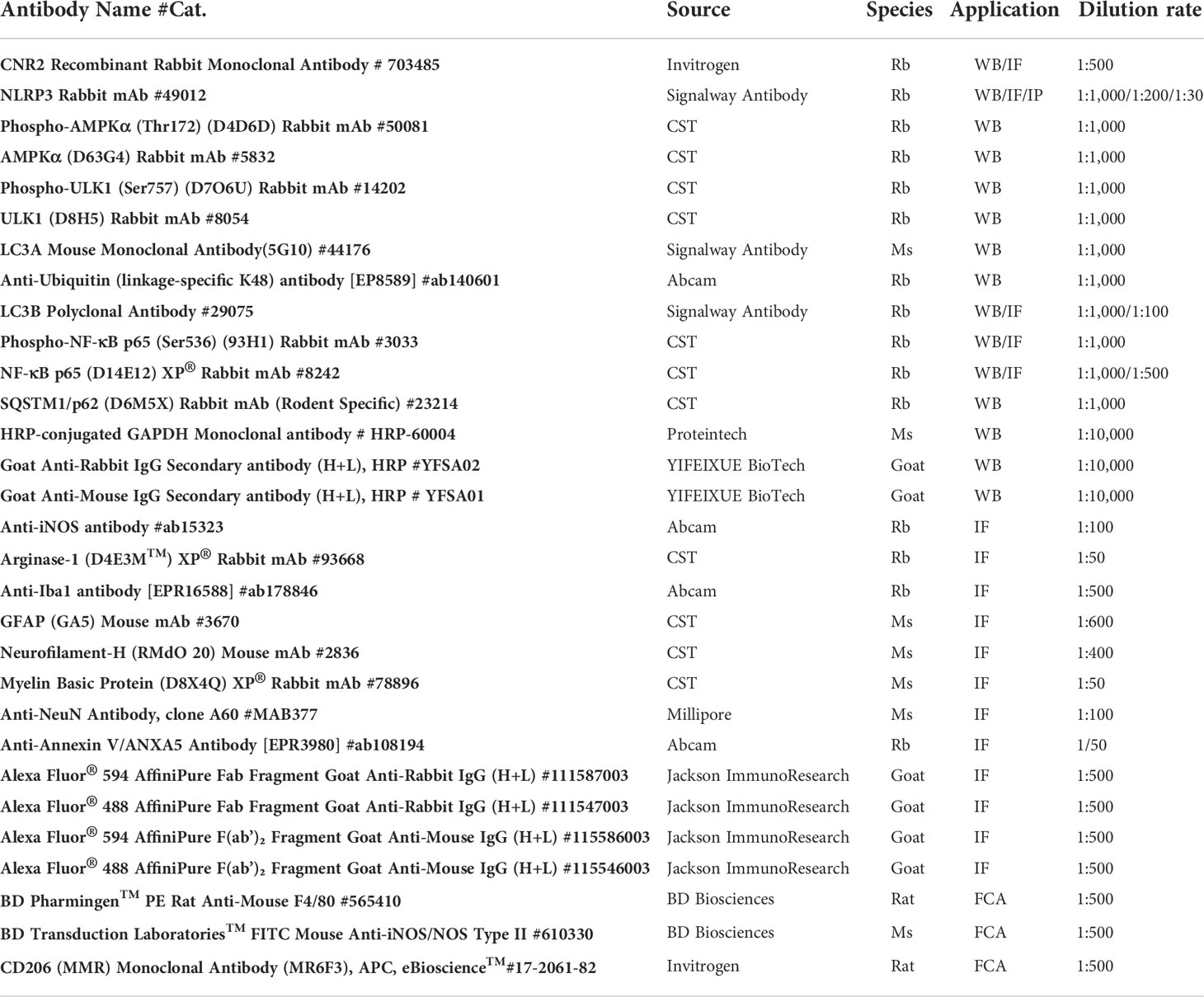
Table 1 The information of Abs
Cell culture
BV2 microglia and human embryonic kidney (HEK)-293T cells were obtained from the Institute of Cell Research, Chinese Academy of Medical Sciences (Shanghai, China) and cultured in Dulbecco’s modified Eagle medium (KGM12800-500; Nanjing KeyGen Biotech. Co. Ltd., Nanjing, China) supplemented with 10% fetal bovine serum (Gibco, Grand Island, NY, USA). At 80% confluence, the microglia and HEK-293T cells were pretreated for 24 h with the CB2R activator JWH-133 (10 nM; B7941; APExBIO Technology LLC, Houston, TX, USA) or antagonist AM630 (40 nM; A3168; APExBIO Technology LLC) dissolved in 0.1% dimethyl sulfoxide (KGT5131; Nanjing KeyGen Biotech. Co. Ltd.). Inflammation was stimulated by treatment with 1 μg/ml of LPS (Sigma-Aldrich, St. Louis, MO, USA) for 24 h.
Establishment of a mouse model of SCI
Male C57BL/6J mice, aged 8 weeks, were obtained from the Experimental Animal Center of Nanjing Medical University and randomly assigned to one of the following four groups: sham group (laminectomy and daily intraperitoneal [i.p.] administration of a vehicle solution composed of 5% ethanol, 5% dimethyl sulfoxide, 5% Tween 20, and 85% normal saline), SCI group (SCI and i.p. administration of the vehicle solution), SCI+J group (SCI and i.p. administration of JWH-133 at 2 mg/kg per day), or SCI+A group (SCI and i.p. administration of AM630 at 2 mg/kg per day). JWH-133 and AM630 were initially administered 30 min prior to SCI and continued for 3 days. For SCI modeling, mice were anesthetized with ketamine (80 mg/kg) and xylazine (4 mg/kg) prior to laminectomy. A moderate contusion to the spinal cord at T10 was created with a 5-g impactor dropped from a height of 5 cm (28). The bladder of all mice with SCI was manually emptied once per day until the development of incontinence.
Western blot analysis
Proteins were extracted from cells and tissues using a total protein extraction kit (KGP2100; Nanjing KeyGen Biotech. Co. Ltd.) in accordance with the manufacturer’s protocol. Following quantification using an enhanced bicinchoninic acid kit (P0010; Beyotime Institute of Biotechnology, Shanghai, China), equal amounts of protein from each group were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and then transferred to polyvinylidene fluoride membranes (thickness, 0.45 μm; EMD Millipore Corporation, Billerica, MA, USA), which were blocked with 5% skim milk diluted in tris-buffered saline-Tween-20 for 1 h at room temperature and then incubated with primary Abs overnight at 4°C, followed by secondary Abs for 1 h at room temperature. Afterward, the membrane was visualized using a chemiluminescence system (4600; Tanon Science and Technology Co., Ltd., Shanghai, China) and analyzed with ImageJ software (https://imagej.nih.gov/ij/).
Enzyme-linked immunosorbent assay
The supernatant of the cell culture of each group was collected by centrifugation at 3,000 rpm for 10 min and stored at −80°C. Expression levels of inflammatory markers (IL-1β, IL-6, and IL-10) were measured using appropriate ELISA kits [EK201B, EK206, EK210; Multisciences (Lianke) Biotech Co., Ltd., Hangzhou, China] in accordance with the manufacturer’s protocols. Absorbance was determined at 550 nm using a multifunctional microplate reader (Synergy™ HT; BioTek Instruments, Winooski, VT, USA).
Flow cytometry
Polarization of microglia was induced by incubation with phycoerythrin (PE)-conjugated F4/80 Ab plus fluorescein isothiocyanate-conjugated Ab against inducible nitric oxide synthase (iNOS) or PE-conjugated F4/80 Ab plus allophycocyanin-conjugated Ab against CD206 at 4°C for 30 min. Then, the proportions of polarized microglia in each group were determined using a BD FACSVerse™ flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA). The generated dataset was analyzed using FlowJo software (version 8.0; https://www.flowjo.com/).
Terminal-deoxynucleotidyl transferase mediated nick end labeling assay
Cell death in injured spinal cord tissue was determined using a one-step terminal-deoxynucleotidyl transferase mediated nick end labeling (TUNEL) apoptosis kit (KGA7062; Nanjing KeyGen Biotech. Co. Ltd.) in accordance with the manufacturer’s instructions. Briefly, tissue sections were incubated with a working buffer containing proteinase K at 37°C for 30 min, then washed three times with PBS for 5 min and incubated in DNase I solution at 37°C for 30 min. Afterward, the tissue sections were probed with an enzyme working buffer at 37°C for 1 h, then labeled with tetramethylrhodamine-conjugated streptavidin in the dark at 37°C for 30 min. The nuclei were counterstained with Fluoromount-G®, a water-soluble compound containing 4´,6-diamidino-2-phenylindole (0100; SouthernBiotech, Birmingham, AL, USA). The tissue sections were imaged with a fluorescence microscope (BX51; Olympus Corporation, Tokyo, Japan).
Immunoprecipitation
BV2 and HEK-293T cells were lysed with protein lysis buffer on ice for 30 min and centrifugated at 12,000 × g and 4°C for 10 min. Then, 50 μl of the supernatant containing protein was collected as the “input” group, while the remaining supernatant was probed with Protein G Sepharose® 4 Fast Flow beads (GE Healthcare Sverige AB, Stockholm, Sweden) coated with an Ab against NLRP3 at 60 rpm and 4°C overnight using a rotary table. The beads were boiled twice in 2× loading buffer at 100°C for 5 min and then washed at 120 rpm and 4°C.
Immunofluorescence staining
Microglia were deparaffinated, rehydrated, and fixed with 4% paraformaldehyde (P0099; Beyotime Institute of Biotechnology) for 15 min for antigen retrieval and blocked using Immunol Staining Blocking Buffer (P0102; Beyotime Institute of Biotechnology) at room temperature for 1 h. The samples were probed with primary Abs overnight at 4°C, followed by secondary fluorescent Abs at room temperature for 1 h. The nuclei were counterstained with Fluoromount-G® and the samples were observed using a fluorescence microscope.
Histological staining
Histological staining with hematoxylin and eosin (H&E), Nissl, and Luxol® fast blue (LFB) was conducted using commercial kits (G1120, G1436, G3240; Beijing Solarbio Science and Technology Co., Ltd., Beijing, China) in accordance with the manufacturer’s protocols. Sections were mounted with neutral balsam (G8590; Beijing Solarbio Science and Technology Co., Ltd.) and then photographed using a fluorescence microscope.
Behavioral assessment
Locomotor function of the hindlimbs of the experimental mice was evaluated by two blinded researchers in accordance with the Basso Mouse Scale, the Louisville Swim Scale, and a footprint assay as previously described (29, 30).
Statistical analysis
Statistical analysis was performed with Prism 8.3.0 software (GraphPad Software, Inc., San Diego, CA, USA) and the data are presented as the mean ± standard deviation. Comparisons between two groups were conducted using the unpaired t-test and among three or more groups using one-way or two-way analysis of variance followed by Tukey’s post hoc test. A probability (p) value < 0.05 was considered statistically significant.
Results
Expression patterns of CB2R in cord tissue post-SCI
CB2R protein expression was measured from day 3 to week 6 post-SCI (Figure 1A). Post-SCI, CB2R protein expression significantly increased and peaked by 1 week, then gradually decreased to week 6 (Figure 1B), which was consistent with the IF staining results (Figure 1C). Furthermore, IF staining on day 7 post-SCI showed that the distribution of CB2R expression largely overlapped with the neuron-specific marker NeuN and microglia-specific marker IBA-1 in both the uninjured and injured mice. In contrast, astrocytes positive for expression of glial fibrillary acidic protein (GFAP) had expressed relatively low levels of CB2R (Figures 1D, E). Hence, microglia were selected for further studies.
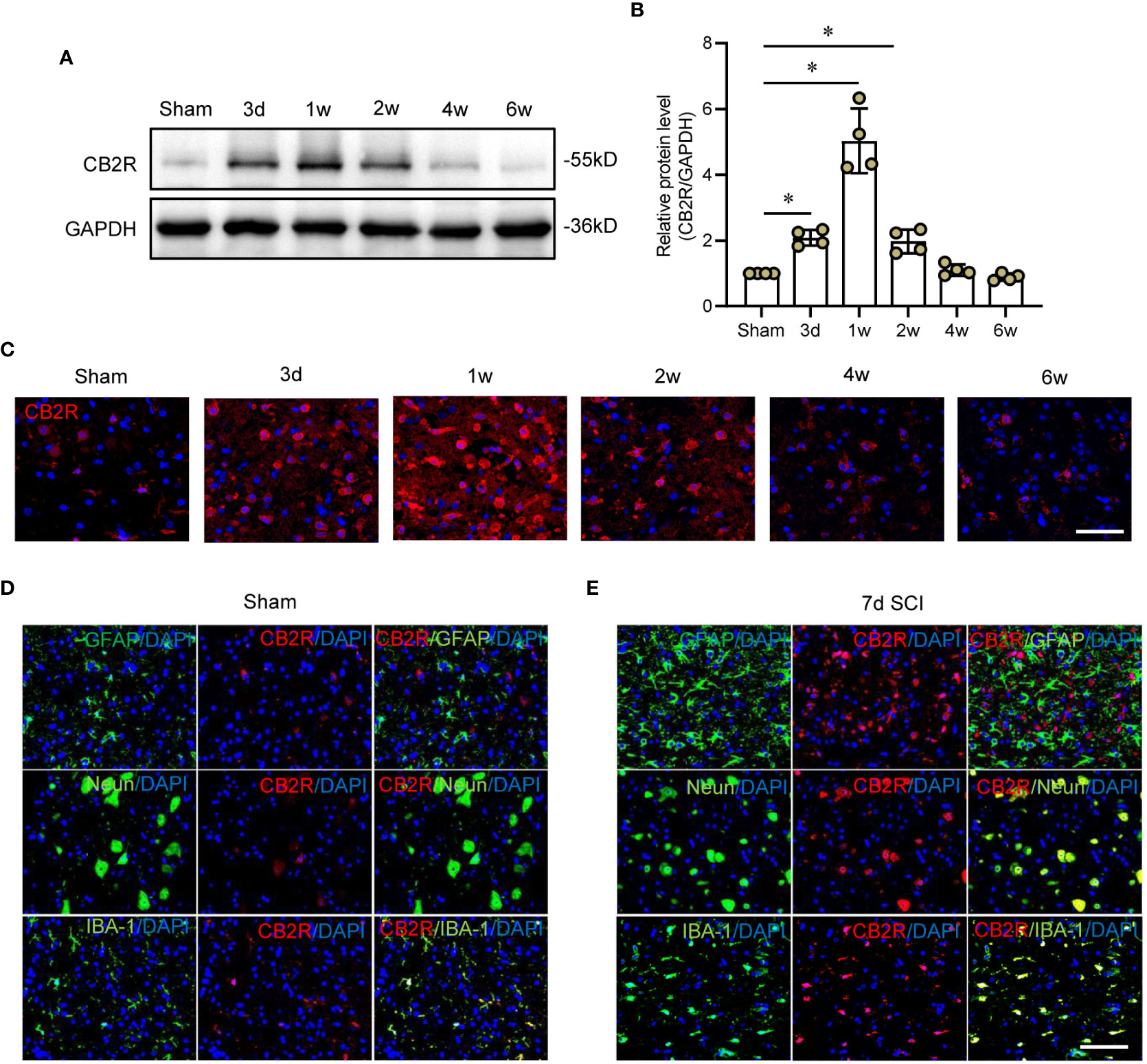
Figure 1 Expression patterns of CB2R in cord tissues post-SCI. (A) Representative protein band of CB2R on day 3 and weeks 1, 2, 4, and 6 post-SCI. (B) Quantitative analysis showing that CB2R expression was significantly increased on day 3 and weeks 1 and 2 post-SCI. (C) IF staining showing that CB2R (red) in cords of mice within 6W post-SCI. (D) IF staining of CB2R (red) with GFAP, Neun, or IBA-1 (green) in cords of sham mice. (E) IF staining of CB2R (red) with GFAP, Neun, or IBA-1 (green) in SCI mice on day 7 post-SCI. Scale bar = 100 μm. *p < 0.05.
CB2R regulates inflammation-mediated polarization of microglia
The CB2R activator JWH-133 and antagonist AM630 were employed to determine whether CB2R participates in regulation of the immunophenotype of LPS-treated microglia (31, 32). Specifically, the expression patterns of the M1 subtype marker iNOS and M2 subtype marker ARG-1 were determined by IF staining (Figure 2A). The results showed that LPS markedly increased expression of iNOS, but reduced that of ARG-1 in microglia. Subsequent treatment with JWH-133 decreased LPS-stimulated upregulated expression of iNOS, but rescued pull-down of ARG-1, while treatment with AM630 aggravated LPS-stimulated upregulation of iNOS and inhibited expression of ARG-1 (Figures 2B, C). In addition, the proportion of polarized cells was determined by flow cytometry. The results showed that the proportion of iNOS+ M1 microglia was significantly increased after LPS stimulation, and activation of CB2R by JWH-133 remarkably reduced such increase, while inhibition of CB2R by AM630 further intensified this effect (Figures 2D, E). In contrast, LPS stimulation remarkably reduced the proportion of CD206+ M2 microglia, which was further decreased by treatment with AM630 but increased by treatment with JWH-133 (Figures 2F, G). LPS stimulation increased expression of IL-1β, IL-6, and IL-10, whereas JWH-133 decreased expression of IL-1β and IL-6, but increased that of IL-10. In contrast, AM630 increased expression of IL-1β and IL-6, but decreased that of IL-10 (Figures 2H–J).
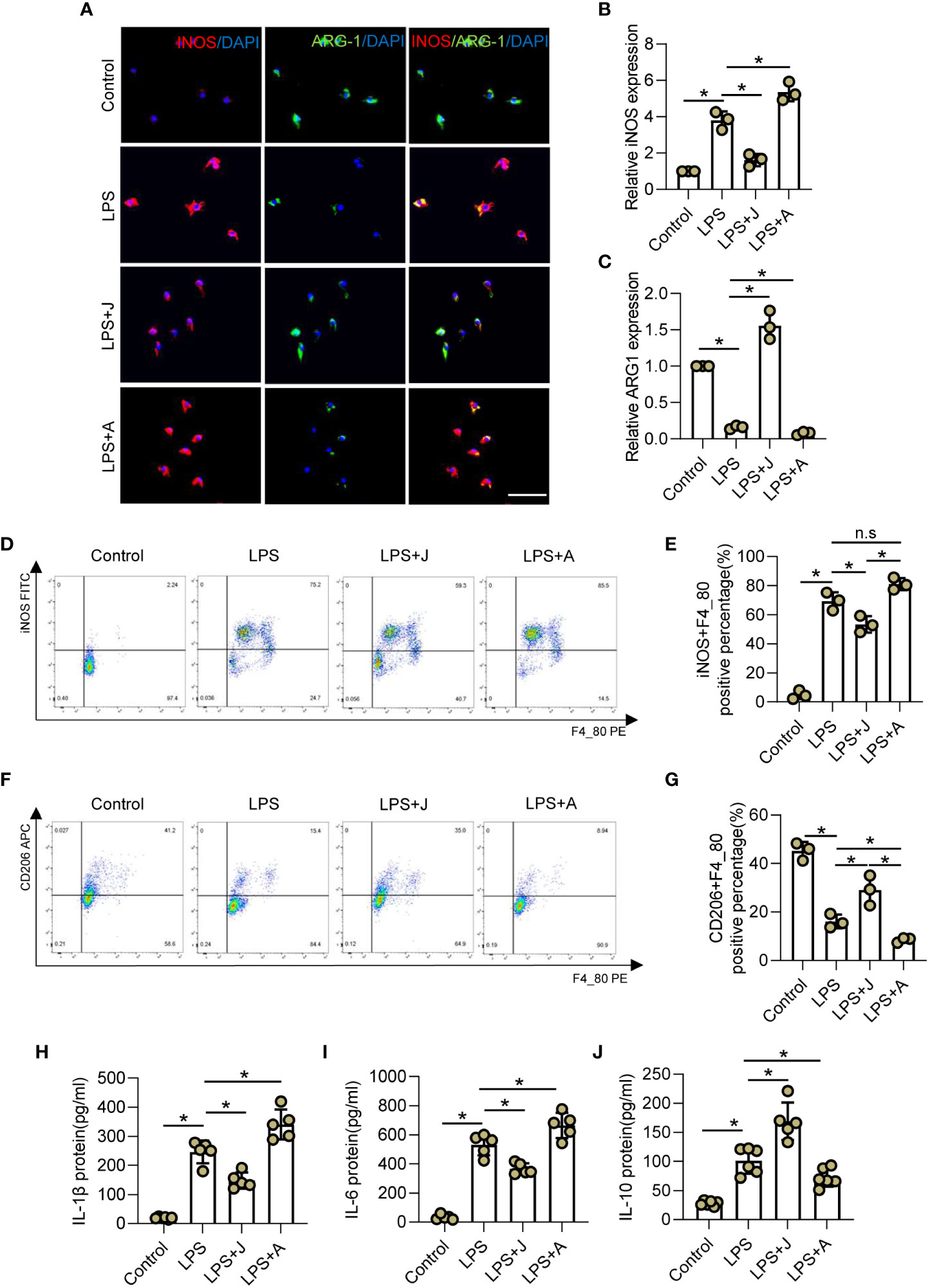
Figure 2 CB2R regulates inflammation-mediated polarization of microglia. (A) IF staining of iNOS (red) and ARG-1 (green) in BV2 microglia after LPS insult for 24 h with CB2R activation by JWH-133 (J) or inactivation by AM630 (A). Scale bar = 50 μm. (B) Quantitative analysis indicating that CB2R significantly altered expression of the M1 marker iNOS during inflammation. (C) Quantitative analysis indicating that CB2R significantly altered expression of the M2 marker ARG-1 after LPS stimulation. (D) Flow cytometry results showing the relative proportion of M1 microglia (red) after LPS insult with and without CB2R activation or inactivation. (E) Quantitative analysis of M1 microglia. (F) Flow cytometry results showing the relative proportion of M2 microglia (red) after LPS insult with and without CB2R activation or inactivation. (G) Quantitative analysis of M2 microglia. ELISA results of IL-1β (H), IL-6 (I), and IL-10 (J) in the control, LPS, LPS+J (JWH-133), and LPS+A (AM630) groups. *p < 0.05. n.s means no significance.
CB2R promoted ubiquitination and autophagy-induced degradation of NLRP3 via the adenosine monophosphate-activated protein kinase/unc-51-like autophagy activating kinase signaling pathway
To elucidate the specific mechanism underlying CB2R-regulated polarization of microglia, the relationship between NLRP3 and CB2R was determined by IF staining. As shown in Figure 3A, stimulation with LPS increased expression of CB2R and NLRP3, while treatment with JWH-133 increased expression of CB2R, but decreased that of NLRP3, and treatment with AM630 had opposite effects (Figures 3B, C). Furthermore, the expression levels of the autophagy-related proteins adenosine monophosphate-activated protein kinase (AMPK), unc-51-like autophagy activating kinase 1 (ULK1), and microtubule-associated proteins 1A/1B light chain 3 (LC3A/B) were determined by WB analysis. The results showed that treatment with JWH-133 promoted phosphorylation of AMPK and ULK-1, leading to significantly increased expression of LC3B, which was reversed following CB2R inhibition by AM630 (Figures 3D–G). Moreover, activation of CB2R by JWH-133 decreased LPS-induced upregulation of NLRP3 and significantly reduced expression of p62, while treatment with AM630 had opposite effects (Figures 3H, J, K). Moreover, activation of CB2R inhibited phosphorylation of p65, while inhibition of CB2R fueled phosphorylation and nuclear expression of p65 (Figures 3H, I, L). The results of IP analysis showed that p62 was pulled down by NLRP3, indicating a direct interaction between NLRP3 and p62 (Figure 3M). Furthermore, in LPS-stimulated HEK-293T cells, treatment with JWH-133 increased binding of ubiquitin linkage-specific K48 to NLRP3, while treatment with AM630 had opposite effects (Figure 3N). These findings indicated that CB2R might also regulate ubiquitination of NLRP3 in LPS-stimulated microglia.
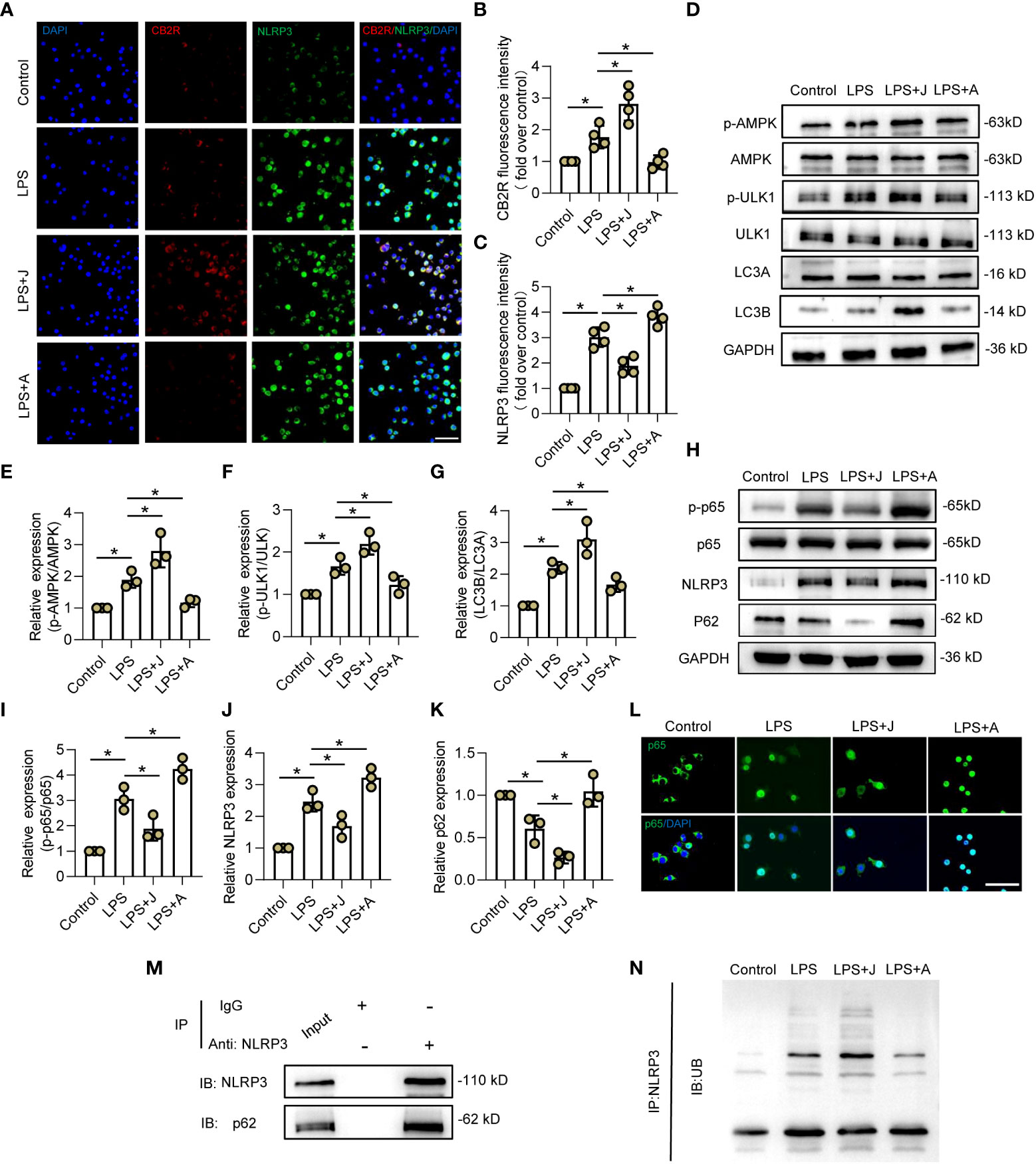
Figure 3 CB2R promoted ubiquitination and autophagy-induced degradation of NLRP3 via the AMPK/ULK signaling pathway. (A) IF staining of CB2R (red) and NLRP3 (green) after LPS stimuli for 24 h with CB2R activation by JWH-133 (J) or inactivation by AM630 (A). Scale bar = 50 μm. (B) Quantitative analysis of CB2R. (C) Quantitative analysis of NLRP3. (D) Representative protein bands of p-AMPK, AMPK, p-ULK1, ULK1, LC3A, and LC3B in each group after 24 h of LPS insult. (E) Quantitative analysis of the p-AMPK/AMPK ratio. (F) Quantitative analysis of the p-ULK1/ULK1 ratio. (G) Quantitative analysis of the LC3B/LC3A ratio. (H) Representative protein bands of p-p65, p65, NLRP3, and p62 after 24 h of LPS stimulation. (I) Quantitative analysis of the p-p65/p65 ratio. (J) Quantitative analysis of NLRP3. (K) Quantitative analysis of p62. (L) IF staining of CB2R (red) and NLRP3 (green) after LPS stimulation for 24 h with CB2R activation by JWH-133 or CB2R inactivation by AM630. Scale bar = 50 μm. (M) Representative protein bands of NLRP3 and p62 in BV2 microglia after IP assay using an Ab against NLRP3. (N) Representative protein bands of ubiquitin linkage-specific K48 bound to NLRP3. *p < 0.05.
CB2R alleviated inflammation by promoting NLRP3-autophagosome formation in injured cords post-SCI
In vivo, the microglial polarization markers iNOS and ARG-1 with IBA-1 were measured. IF staining showed that the proportion of iNOS+ microglia was increased, while that of ARG-1+ microglia was decreased post-SCI. However, administration of JWH-133 markedly reduced the proportion of M1 microglia and increased that of the M2 subtype, while treatment with AM630 had opposite effects (Figures 4A–D). The WB results showed that JWH-133 treatment significantly increased phosphorylation of APMK and ULK1 as well as the LC3B/LC3A ratio, but markedly decreased NLRP3 expression, while treatment with AM630 had opposite effects (Supplementary Figures 2A–E). In addition, increased NLRP3 expression was mitigated by JWH-133-induced increased LC3B expression on day 3 post-SCI, while treatment with AM630 increased expression of NLRP3 and LC3B (Figure 4E). These results suggested that activated CB2R might protect against neuroinflammation by promoting the formation of the NLRP3-autophagosome.
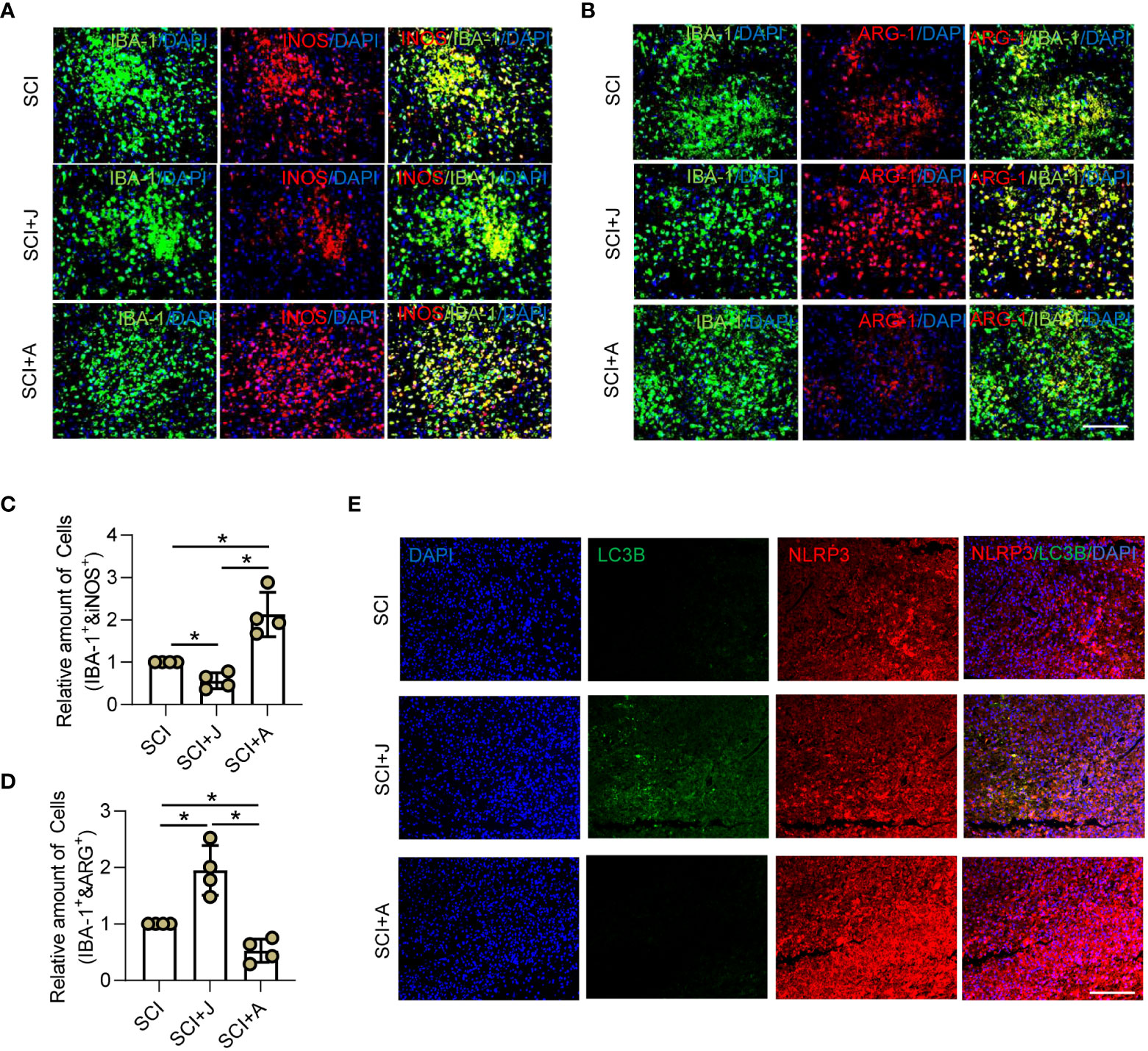
Figure 4 CB2R alleviated inflammation by promoting NLRP3-autophagosome formation in injured cords post-SCI. On day 3 post-SCI with CB2R activation by JWH-133 or inactivation by AM630, (A) IF staining of iNOS (red) and IBA-1 (green); (B) IF staining of ARG-1 (red) and IBA-1 (green). Scale bar = 100 μm. (C) Quantitative analysis of the number of iNOS+ microglia. (D) Quantitative analysis of the number of ARG-1+ microglia. (E) IF staining of NLRP3 (red) and LC3B (green) on day 3 post-SCI. Scale bar = 100 μm. *p < 0.05.
CB2R ameliorated secondary neuronal apoptosis and demyelination post-SCI
Given that CB2R was found to play a protective role in secondary injury, the LFB staining profiles of the injured spinal cord on day 7 post-SCI were compared among the groups. The results showed that treatment with JWH-133 resulted in a larger area of myelin as compared to the SCI mice, while treatment with AM630 caused significant myelin loss (Figures 5A, B), which was consistent with IF staining of myelin basic protein (Figures 5C, E). IF staining showed that treatment with JWH-133 increased the amount of neurofilaments, while treatment with AM630 further aggravated loss of neurofilaments as compared to the SCI mice (Figures 5C, D). TUNEL staining revealed that treatment with JWH-133 significantly reduced the amount of dead cells, while treatment with AM630 significantly increased cell death, as compared to the SCI mice (Figures 5F, G). Moreover, the number of apoptotic neurons was determined by staining with Annexin V, a marker of early apoptosis, IF analysis, and Nissl staining. The results showed that, as compared to the SCI mice, administration of JWH-133 resulted in fewer apoptotic neurons and a larger number of viable neurons, while treatment with AM630 produced opposite effects (Figures 5H–K).
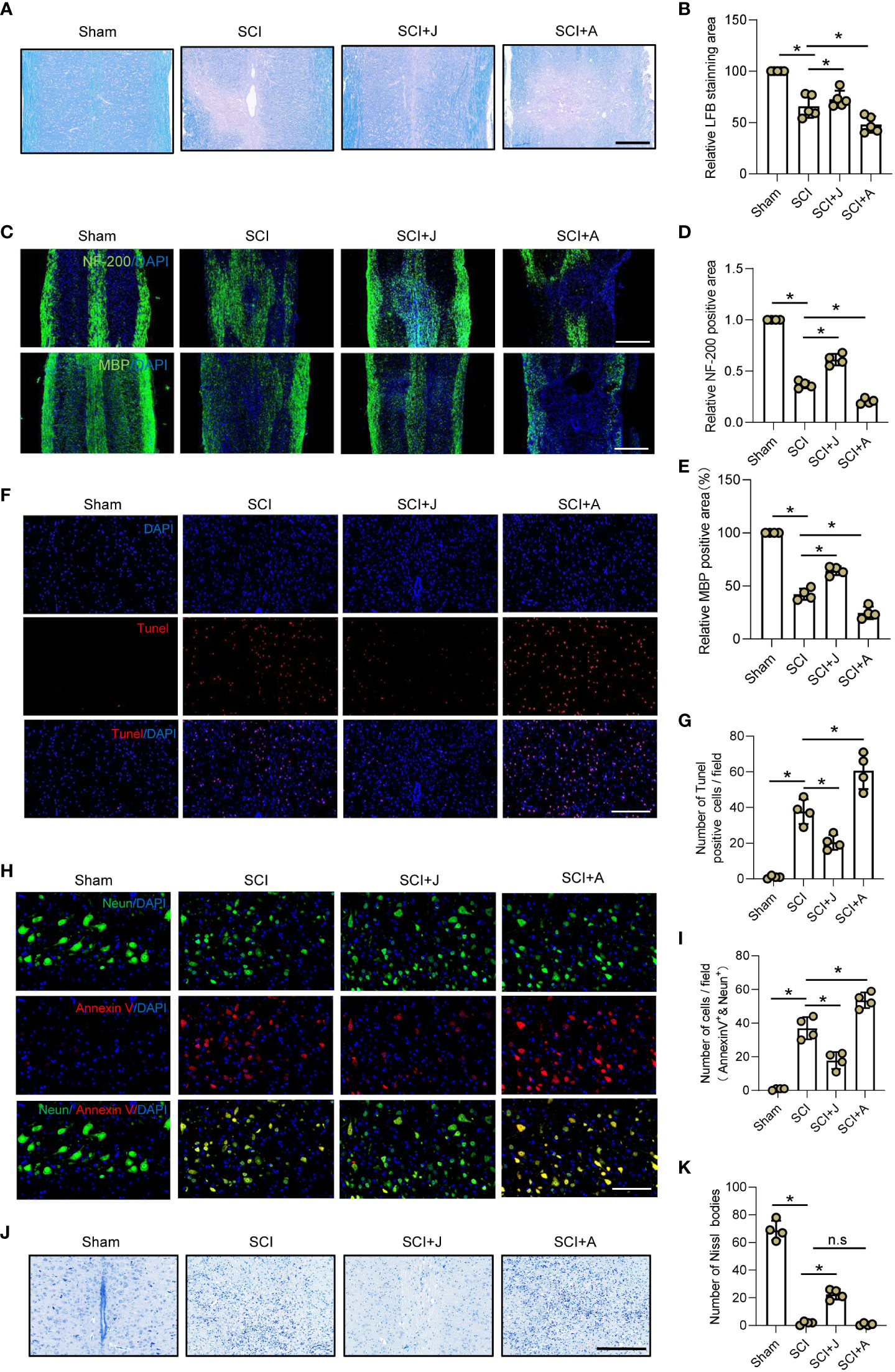
Figure 5 CB2R ameliorated secondary neuronal apoptosis and demyelination post-SCI. (A) LFB staining of spinal cords on day 7 post-SCI. Scale bar = 200 μm. (B) Quantitative analysis of LFB-labeled area. (C) IF staining of NF200 or myelin basic protein (green) on day 7 post-SCI. Scale bar = 500 μm. (D) Quantitative analysis of NF200. (E) Quantitative analysis of myelin basic protein. (F) TUNEL staining of spinal cords on day 7 post-SCI. Scale bar = 100 μm. (G) Quantitative analysis of TUNEL-positive cells. (H) IF staining of Neun (green) and Annexin V (red) on day 7 post-SCI. Scale bar = 100 μm. (I) Quantitative analysis of Annexin V-positive neurons. (J) Nissl staining of spinal cords on day 7 post-SCI. Scale bar = 200 μm. (K) Quantitative analysis of the number of neurons. *p < 0.05. n.s means no significance.
CB2R mitigated expansion of neuronal damage and formation of glial scarring post-SCI
Since glial scarring was initiated on day 7 and completed on day 28 post-SCI, IF staining of the injured foci on days 7 and 28 post-SCI was compared. IF staining on day 7 post-SCI showed that JWH-133 administration significantly reduced the extent of neuronal damage in areas occupied by microglia and astrocytes, as compared to the SCI mice, while administration of AM630 had opposite effects (Figures 6A–D). Assessment of these indices showed consistent trends on days 7 and 28 post-SCI during the period of spontaneous healing (Figures 6E–H).
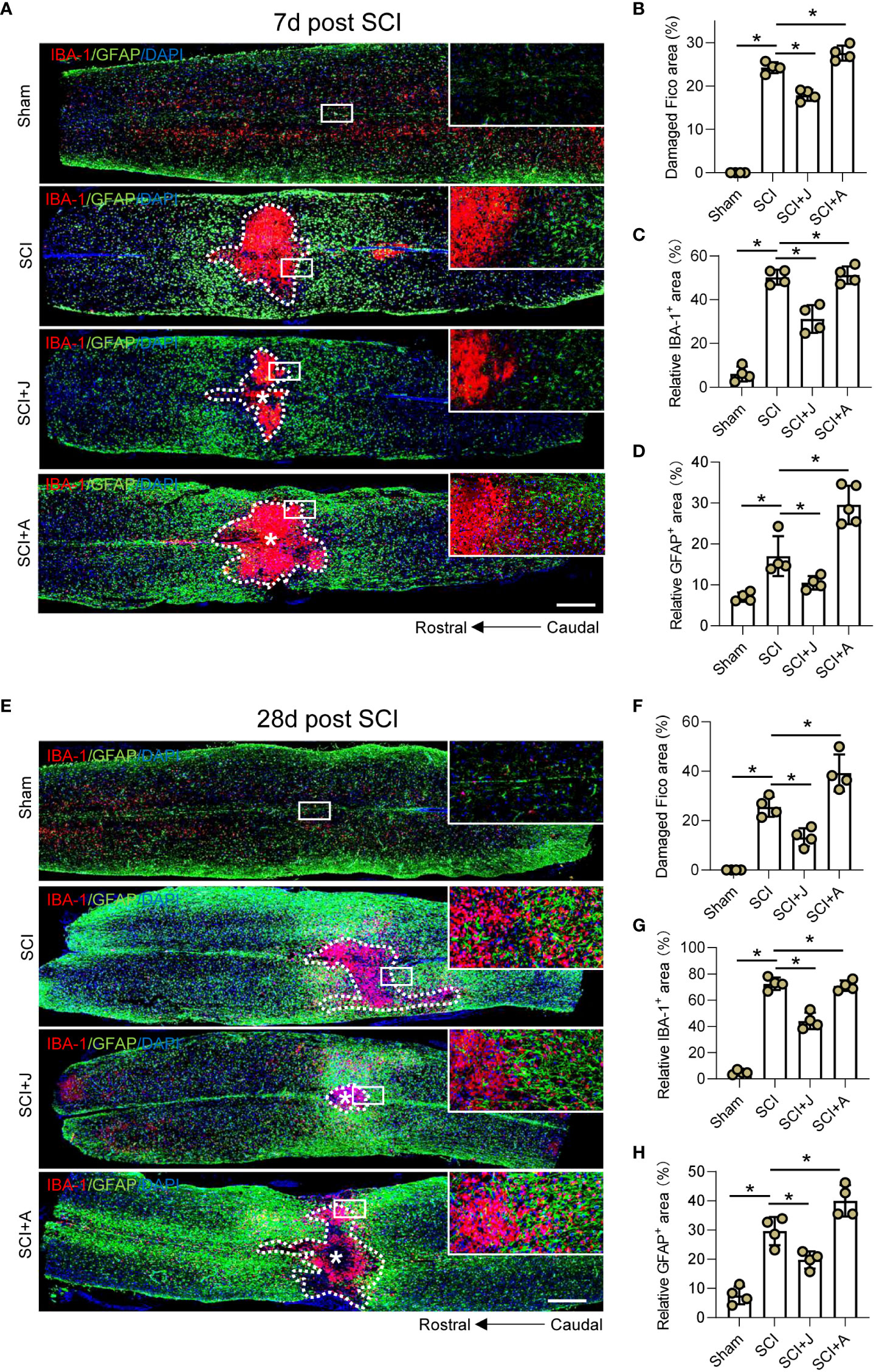
Figure 6 CB2R mitigated expansion of neuronal damage and formation of glial scarring post-SCI. (A) IF staining of GFAP (green) and IBA-1 (red) on day 7 post-SCI. Scale bar = 500 μm. Quantitative analysis of the area of injured foci inside the white dotted line (B), microglia-occupied area, (C) and astrocyte-occupied area (D) on day 7 post-SCI. (E) IF staining of GFAP (green) and IBA-1 (red) on day 28 post-SCI. Scale bar = 500 μm. Quantitative analysis of the area of injured foci inside the white dotted line (F), microglia-occupied area, (G) and astrocyte-occupied area (H) on day 28 post-SCI. *p < 0.05.
CB2R attenuated histological and functional damage post-SCI
H&E staining showed that as compared to the SCI group on day 3 post-SCI, administration of JWH-133 reduced the areas of hemorrhage and damage of the injured cord tissues, while administration of AM630 increased the areas of hemorrhage and significantly increased the damaged areas (Figures 7A, B; Supplementary Figure 1A). Furthermore, H&E staining on day 28 post-SCI showed that treatment with JWH-133 prevented further histological damage to the injured foci, while treatment with AM630 exacerbated tissue damage, as compared to the SCI mice (Figures 7C, D; Supplementary Figure 1B). The results of the footprint assay showed that treatment with JWH-133 lowered the frequency of dorsiflexion of the hindlimbs with longer stride length and width, while treatment with AM630 was associated with lower crawling indices, as compared to the SCI mice (Figures 7E–G). The results of the swimming test showed that JWH-133-treated mice exhibited a smaller angle ranging from the trunk to the water surface with a higher frequency of hindlimb movements, while AM630-treated mice seldom exhibited any evident difference, as compared to the SCI mice on day 28 post-SCI (Figure 7H). Therefore, the Louisville Swim Scale scores of the JWH-133-treated mice were higher than those of the SCI mice and AM630-treated mice beginning on day 14 post-SCI (Figure 7I). The Basso Mouse Scale scores also showed that, as compared to the SCI mice, treatment with JWH-133 significantly improved locomotion, while treatment with AM630 significantly decreased locomotion beginning on day 7 post-SCI (Figure 7J).
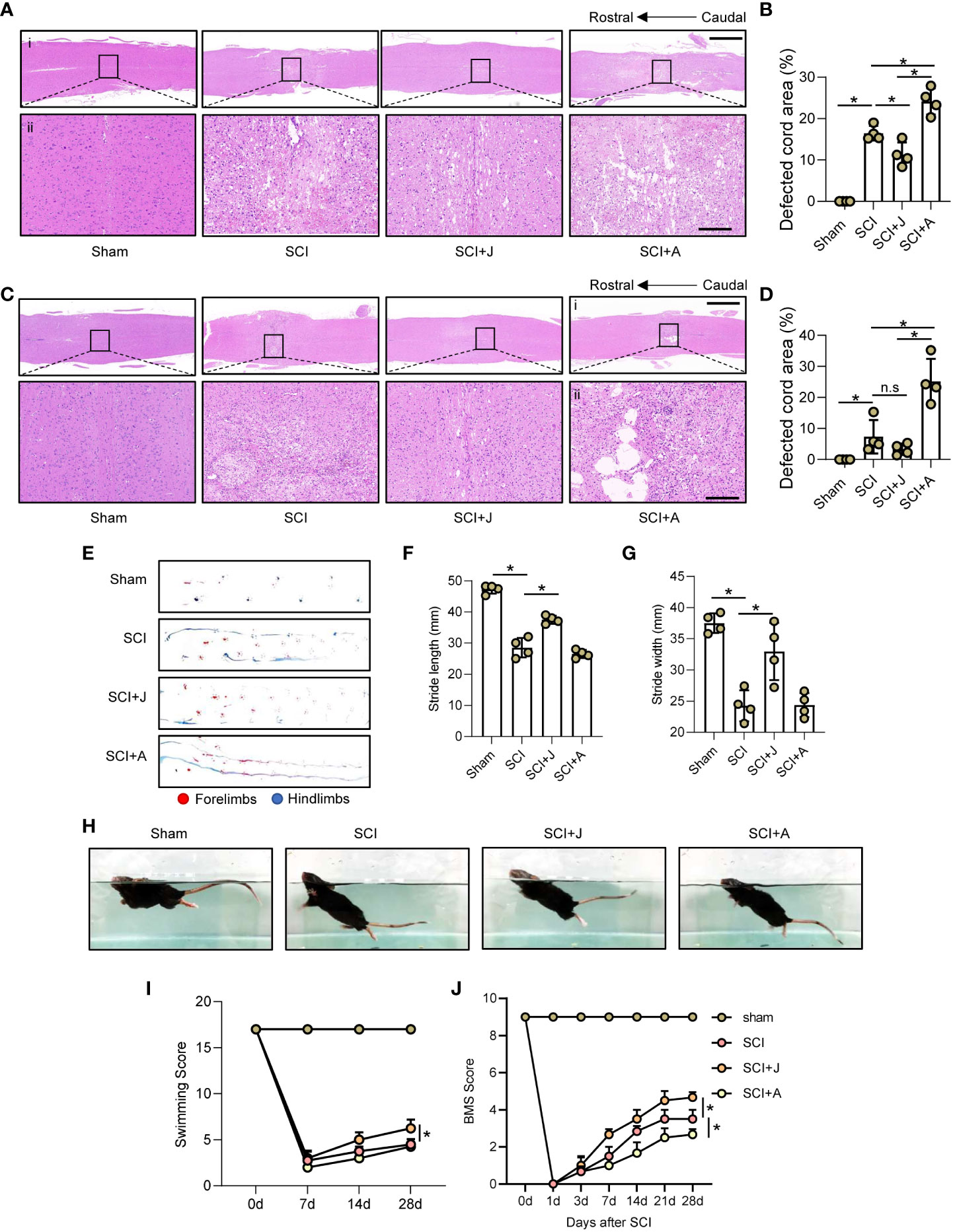
Figure 7 CB2R attenuated histological and functional damage post-SCI. (A) Representative images of H&E staining of spinal cords on day 3 post-SCI (injured cords in black frames). Scale bar (i) = 600 μm (ii) = 100 μm. (B) Quantitative analysis of defected area by H&E staining. (C) H&E staining of spinal cords on day 3 post-SCI (injured cords in black frames). Scale bar: (i) = 600 μm, (ii) = 100 μm. (D) Quantitative analysis of defected area by H&E staining. (E) Mouse footprint assay on day 28 post-SCI. Quantitative analysis of stride length (F) and width (G) of the hindlimbs of mice post-SCI. (H) Representative images of a mouse swimming on day 28 post-SCI. Statistical analysis of the Louisville Swim Scale score (I) and Basso Mouse Scale (J) over a 28-day period. *p < 0.05. n.s means no significance.
Discussion
The findings of the present study suggested that the neuroprotective effects of CB2R activation in SCI were partly regulated by two main mechanisms: (i) attenuation of neuroinflammatory responses and reversed microglial polarization through inactivation of NLRP3 via AMPK/ULK1 signaling-induced autophagy and (ii) CB2R mitigation of neuroinflammation-mediated secondary neuropathology due to myelin loss, neuron apoptosis, and glial scar accumulation post-SCI.
LPS provokes inflammatory responses in multiple immune cells, including macrophages, T cells, and microglia (33, 34). Hence, LPS is often used to establish in vitro aseptic inflammatory models to mimic the pathogenesis of various neuro-immune diseases, such as autoimmune encephalomyelitis, multiple sclerosis, and depression (35–37). Of note, LPS is also widely used to induce microglial inflammation in mimetic models of SCI (38–40). LPS binds to Toll-like receptor 4 and promotes activation of p65, which then translocates to the nucleus and triggers the transcription of pro-inflammatory molecules (41, 42). The results of the current study confirmed that CB2R expression was significantly increased both in the cord tissues of SCI mice and in LPS-stimulated microglia, which then inhibited activation of p65, leading to decreased translocation to the nucleus. A previous report also suggested that CB2R activation could mitigate LPS-induced neuroinflammation (43), but the underlying molecular mechanism regulated by CB2R has not yet been elucidated. The results of the current study showed that pharmacological activation of CB2R by JWH-133 inhibited LPS-induced proinflammatory transition of the M1 subtype and increased the proportion of immunoregulatory M2 microglia in vitro, which was reversed by treatment with the CB2R antagonist AM630. These findings indicated that inhibition of neuroinflammation via CB2R potentially occurred through unidentified signaling pathways related to immunoregulation.
NLRP3, a classic biomarker of inflammation, responds to trauma via regulating activation of caspase-1 and maturation of IL-1β, thereby promoting inflammation-dominated neuropathology post-SCI (44, 45). Accumulating evidence indicates that either suppression of NLRP3 expression or promotion of cell autophagy ameliorates neuroinflammation-induced neurological injury, although the underlying mechanisms remain unclear. Numerous mechanisms have been proposed to explain the interactions between the NLRP3 inflammasome and autophagy, including autophagy activation by inflammatory signaling via limited IL-1β production and targeting ubiquitinated inflammasomes (13). In neurological diseases, non-coding RNA, bio-enzymes, and small molecular compounds have been implicated in the interplay between ubiquitination and autophagy of NLRP3 (46–48). In LPS-stimulated microglia, CB2R activation decreased NLRP3 expression associated with formation of autophagosomes, resulting in accumulation of LC3B and exhaustion of p62. Consistently, Shao et al. (49) demonstrated that alleviation of experimental autoimmune encephalomyelitis by CB2R activation was linked to activation of autophagosomes and inhibition of the NLRP3 inflammasome. In contrast, the results of the present study suggest that CB2R regulates NLRP3 ubiquitination by ubiquitin linkage-specific K48 and promotes binding of NLRP3 with p62, an autophagy substrate, and LC3B, an autophagy activator. K48-linked polyubiquitination of NLRP3 mediates self-degradation and reduces cellular NLRP3 levels, which are essential for inactivation of the NLRP3 inflammasome (50) and p62-dependent selective autophagy also inhibited the capacity of NLRP3 inflammasome-related proteins to attenuate inflammation-related pathology (51, 52). These findings confirmed that CB2R activation indeed mitigated neuroinflammation targeting polarization of microglia by regulating autophagy- and ubiquitination-mediated degradation of the NLRP3 inflammasome. AMPK, a nutrient and energy sensor that controls mitochondrial homeostasis, is activated by multiple physio-pathological insults, resulting in inflammation (53, 54), and subsequent phosphorylation of ULK1, resulting in activation of autophagosomes (55). Strikingly, CB2R has been implicated in upregulation of AMPK expression in models of aging, inflammation, and hyperglycemia (55–57), implying that CB2R might also participate in the regulation of autophagy. Interestingly, the results of the present study showed that activation of CB2R increased AMPK/ULK1 phosphorylation, leading to increased expression of LC3B, indicating that CB2R activation in a state of neuroinflammation facilitated autophagy via activation of the AMPK/ULK1 axis.
An increase in the proportion of pro-inflammatory M1 microglia that express iNOS with a decrease in the proportion of anti-inflammatory M2 microglia expressing ARG-1 in the early stage of SCI plays a crucial role in neuroinflammation (58, 59). Therefore, upregulation of iNOS and downregulation of ARG-1 promote deteriorative secondary neuropathy and more intense neuroinflammation in the injured cord tissues post-SCI. The results of the in vivo experiments found that pro-inflammatory M1 microglia predominantly expressing iNOS and penuriously expressing ARG-1 had accumulated in the vicinity of the injured cord, while CB2R activation via administration of JWH-133 resulted in increased expression of ARG-1 with reduced levels of iNOS in microglia around the injury. In contrast, treatment with the CB2R antagonist AM630 promoted the formation of M1, rather than M2, microglia, suggesting that CB2R activation limited inflammation by manipulating polarization of microglia in the injured spinal cord. Accordingly, CB2R activation reduced secondary pathology like neuron loss, demyelination, and glial scarring, thereby improving functional recovery. However, these corrections were neutralized in AM630-treated mice, suggesting that CB2R may play a potent neuroprotective role in addition to anti-inflammatory activities. In summary, CB2R attenuated inflammatory responses via targeting microglial differentiation by facilitating autophagy- and ubiquitination-induced degradation of the NLRP3 inflammasome. CB2R expression in SCI mice reduced the area of glial scarring, rescued the deficiency of neurons and myelin, and improved recovery of locomotor function, in part, by inhibiting neuroinflammation.
These preliminary findings verified the role of CB2R in post-SCI neuroinflammation and subsequent neuropathology. Hence, further research is warranted to explore therapeutic strategies targeting CB2R and other functions of CB2R in neurons to reveal their potential immunotherapeutic value for SCI.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics statement
This study was reviewed and approved by the Institutional Animal Care and Use Committee of Nanjing Medical University.
Author contributions
FJ and ZZ conceived and designed the experiments. FJ performed the cell experiments. MX and YZ performed the animal experiments. JCh and JCa analyzed the data. ZQ wrote the paper. LY funded and supervised the research. All authors contributed to the article and approved the submitted version.
Funding
This study was supproted by 2022 Lifting Project for Young Scientific and Technological Talents Funded by Jiangsu Association of Science and Technology (To LY Grant No#:TJ-2022-033).
Acknowledgments
We thank the Key Laboratory of Taizhou People’s Hospital for technological support and the use of equipment.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.993168/full#supplementary-material
Supplementary Figure 1 | Photos of 4% paraformaldehyde-fixed spinal cords on days 3 (A) and 28 (B) post-SCI in each group.
Supplementary Figure 2 | (A) Representative protein bands including of p-AMPK, AMPK, p-ULK1, ULK1, LC3A, LC3B, and NLPR3 in each group 3 days after SCI. (B) Quantitative analysis of ratio of the p-AMPK/AMPK ratio. (C) Quantitative analysis of the p-ULK1/ULK1 ratio. (D) Quantitative analysis of the LC3B/LC3A ratio. (E) Quantitative analysis of NLRP3 expression.
References
1. Zipser CM, Cragg JJ, Guest JD, Fehlings MG, Jutzeler CR, Anderson AJ, et al. Cell-based and stem-cell-based treatments for spinal cord injury: Evidence from clinical trials. Lancet Neurol (2022) 21(7):659–70. doi: 10.1016/S1474-4422(21)00464-6
2. Slater PG, Dominguez-Romero ME, Villarreal M, Eisner V, Larrain J. Mitochondrial function in spinal cord injury and regeneration. Cell Mol Life Sci (2022) 79:239. doi: 10.1007/s00018-022-04261-x
3. Li Y, Ritzel RM, He J, Cao T, Sabirzhanov B, Li H, et al. The voltage-gated proton channel Hv1 plays a detrimental role in contusion spinal cord injury via extracellular acidosis-mediated neuroinflammation. Brain Behav Immun (2021) 91:267–83. doi: 10.1016/j.bbi.2020.10.005
4. Tan W, Su PP, Leff J, Gao X, Chen J, Guan AK, et al. Distinct phases of adult microglia proliferation: A myc-mediated early phase and a Tnfaip3-mediated late phase. Cell Discovery (2022) 8:34. doi: 10.1038/s41421-022-00377-3
5. Xu GY, Xu S, Zhang YX, Yu ZY, Zou F, Ma XS, et al. Cell-free extracts from human fat tissue with a hyaluronan-based hydrogel attenuate inflammation in a spinal cord injury model through M2 microglia/microphage polarization. Small (2022) 18:e2107838. doi: 10.1002/smll.202107838
6. Zhai X, Chen K, Yang H, Li B, Zhou T, Wang H, et al. Extracellular vesicles derived from CD73 modified human umbilical cord mesenchymal stem cells ameliorate inflammation after spinal cord injury. J Nanobiotechnology (2021) 19:274. doi: 10.1186/s12951-021-01022-z
7. Wang C, Xu T, Lachance BB, Zhong X, Shen G, Xu T, et al. Critical roles of sphingosine kinase 1 in the regulation of neuroinflammation and neuronal injury after spinal cord injury. J Neuroinflamm (2021) 18:50. doi: 10.1186/s12974-021-02092-4
8. Coll RC, Schroder K, Pelegrin P. NLRP3 and pyroptosis blockers for treating inflammatory diseases. Trends Pharmacol Sci (2022) 43(8):653–68. doi: 10.1016/j.tips.2022.04.003
9. Gangopadhyay A, Devi S, Tenguria S, Carriere J, Nguyen H, Jager E, et al. NLRP3 licenses NLRP11 for inflammasome activation in human macrophages. Nat Immunol (2022) 23(6):892–903. doi: 10.1038/s41590-022-01220-3
10. Xu Z, Li D, Qu W, Yin Y, Qiao S, Zhu Y, et al. Card9 protects sepsis by regulating Ripk2-mediated activation of NLRP3 inflammasome in macrophages. Cell Death Dis (2022) 13:502. doi: 10.1038/s41419-022-04938-y
11. Zhang Z, Guo P, Huang S, Jia Z, Chen T, Liu X, et al. Inhibiting microglia-derived NLRP3 alleviates subependymal edema and cognitive dysfunction in posthemorrhagic hydrocephalus after intracerebral hemorrhage via AMPK/Beclin-1 pathway. Oxid Med Cell Longev (2022) 2022:4177317. doi: 10.1155/2022/4177317
12. Zhang H, Wang J, Ruan C, Gao Z, Zhu Q, Li S. Co-Exposure of chronic stress and alumina nanoparticles aggravates hippocampal microglia pyroptosis by activating cathepsin B/NLRP3 signaling pathway. J Hazard Mater (2022) 436:129093. doi: 10.1016/j.jhazmat.2022.129093
13. Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, et al. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol (2012) 13:255–63. doi: 10.1038/ni.2215
14. Lin Q, Li S, Jiang N, Shao X, Zhang M, Jin H, et al. PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol (2019) 26:101254. doi: 10.1016/j.redox.2019.101254
15. Cai B, Zhao J, Zhang Y, Liu Y, Ma C, Yi F, et al. USP5 attenuates NLRP3 inflammasome activation by promoting autophagic degradation of NLRP3. Autophagy (2021), 18(5):990–1004. doi: 10.1080/15548627.2021.1965426
16. Young AP, Denovan-Wright EM. Synthetic cannabinoids reduce the inflammatory activity of microglia and subsequently improve neuronal survival. vitro. Brain Behav Immun (2022) 105:29–43. doi: 10.1016/j.bbi.2022.06.011
17. Johnson CT, Bradshaw HB. Modulatory potential of cannabidiol on the opioid-induced inflammatory response. Cannabis Cannabinoid Res (2021) 6:211–20. doi: 10.1089/can.2020.0181
18. Cristino L, Bisogno T, Di Marzo V. Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nat Rev Neurol (2020) 16:9–29. doi: 10.1038/s41582-019-0284-z
19. Chiurchiu V, Maccarrone M. Bioactive lipids as modulators of immunity, inflammation and emotions. Curr Opin Pharmacol (2016) 29:54–62. doi: 10.1016/j.coph.2016.06.005
20. Moreno E, Cavic M, Canela EI. Functional fine-tuning of metabolic pathways by the endocannabinoid system-implications for health and disease. Int J Mol Sci (2021) 22(7):3661. doi: 10.3390/ijms22073661
21. Galve-Roperh I, Chiurchiu V, Diaz-Alonso J, Bari M, Guzman M, Maccarrone M. Cannabinoid receptor signaling in progenitor/stem cell proliferation and differentiation. Prog Lipid Res (2013) 52:633–50. doi: 10.1016/j.plipres.2013.05.004
22. Chiurchiu V, Leuti A, Maccarrone M. Cannabinoid signaling and neuroinflammatory diseases: A melting pot for the regulation of brain immune responses. J Neuroimmune Pharmacol (2015) 10:268–80. doi: 10.1007/s11481-015-9584-2
23. Bisogno T, Oddi S, Piccoli A, Fazio D, Maccarrone M. Type-2 cannabinoid receptors in neurodegeneration. Pharmacol Res (2016) 111:721–30. doi: 10.1016/j.phrs.2016.07.021
24. Chiurchiu V, van der Stelt M, Centonze D, Maccarrone M. The endocannabinoid system and its therapeutic exploitation in multiple sclerosis: Clues for other neuroinflammatory diseases. Prog Neurobiol (2018) 160:82–100. doi: 10.1016/j.pneurobio.2017.10.007
25. Tiberi M, Evron T, Saracini S, Boffa L, Mercuri NB, Chintalacharuvu SR, et al. Potent T cell-mediated anti-inflammatory role of the selective CB2 agonist lenabasum in multiple sclerosis. Neuropathol Appl Neurobiol (2022) 48:e12768. doi: 10.1111/nan.12768
26. Zhou H, Du R, Li G, Bai Z, Ma J, Mao C, et al. Cannabinoid receptor 2 promotes the intracellular degradation of HMGB1 via the autophagy-lysosome pathway in macrophage. Int Immunopharmacol (2020) 78:106007. doi: 10.1016/j.intimp.2019.106007
27. Luo XQ, Li A, Yang X, Xiao X, Hu R, Wang TW, et al. Paeoniflorin exerts neuroprotective effects by modulating the M1/M2 subset polarization of microglia/macrophages in the hippocampal CA1 region of vascular dementia rats via cannabinoid receptor 2. Chin Med (2018) 13:14. doi: 10.1186/s13020-018-0173-1
28. Qian Z, Chen H, Xia M, Chang J, Li X, Ye S, et al. Activation of glucagon-like peptide-1 receptor in microglia attenuates neuroinflammation-induced glial scarring via rescuing arf and rho GAP adapter protein 3 expressions after nerve injury. Int J Biol Sci (2022) 18:1328–46. doi: 10.7150/ijbs.68974
29. Basso DM, Fisher LC, Anderson AJ, Jakeman LB, McTigue DM, Popovich PG. Basso mouse scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J Neurotrauma (2006) 23:635–59. doi: 10.1089/neu.2006.23.635
30. Smith RR, Burke DA, Baldini AD, Shum-Siu A, Baltzley R, Bunger M, et al. The Louisville swim scale: A novel assessment of hindlimb function following spinal cord injury in adult rats. J Neurotrauma (2006) 23:1654–70. doi: 10.1089/neu.2006.23.1654
31. Ghosh K, Zhang GF, Chen H, Chen SR, Pan HL. Cannabinoid CB2 receptors are upregulated via bivalent histone modifications and control primary afferent input to the spinal cord in neuropathic pain. J Biol Chem (2022), 298(6):101999. doi: 10.1016/j.jbc.2022.101999
32. Sophocleous A, Yiallourides M, Zeng F, Pantelas P, Stylianou E, Li B, et al. Association of cannabinoid receptor modulation with normal and abnormal skeletal remodelling: A systematic review and meta-analysis of in vitro, in vivo and human studies. Pharmacol Res (2022) 175:105928. doi: 10.1016/j.phrs.2021.105928
33. Fan H, Chen Z, Tang HB, Shan LQ, Chen ZY, Wang XH, et al. Exosomes derived from olfactory ensheathing cells provided neuroprotection for spinal cord injury by switching the phenotype of macrophages/microglia. Bioeng Transl Med (2022) 7:e10287. doi: 10.1002/btm2.10287
34. Wang C, Xiao Y, Yu L, Tian F, Zhao J, Zhang H, et al. Protective effects of different bacteroides vulgatus strains against lipopolysaccharide-induced acute intestinal injury, and their underlying functional genes. J Adv Res (2022) 36:27–37. doi: 10.1016/j.jare.2021.06.012
35. Arioz BI, Tastan B, Tarakcioglu E, Tufekci KU, Olcum M, Ersoy N, et al. Melatonin attenuates LPS-induced acute depressive-like behaviors and microglial NLRP3 inflammasome activation through the SIRT1/Nrf2 pathway. Front Immunol (2019) 10:1511. doi: 10.3389/fimmu.2019.01511
36. Al-Ghezi ZZ, Busbee PB, Alghetaa H, Nagarkatti PS, Nagarkatti M. Combination of cannabinoids, delta-9-tetrahydrocannabinol (THC) and cannabidiol (CBD), mitigates experimental autoimmune encephalomyelitis (EAE) by altering the gut microbiome. Brain Behav Immun (2019) 82:25–35. doi: 10.1016/j.bbi.2019.07.028
37. Li Y, Zhou D, Ren Y, Zhang Z, Guo X, Ma M, et al. Mir223 restrains autophagy and promotes CNS inflammation by targeting ATG16L1. Autophagy (2019) 15:478–92. doi: 10.1080/15548627.2018.1522467
38. Xu S, Wang J, Zhong J, Shao M, Jiang J, Song J, et al. CD73 alleviates GSDMD-mediated microglia pyroptosis in spinal cord injury through PI3K/AKT/Foxo1 signaling. Clin Transl Med (2021) 11:e269. doi: 10.1002/ctm2.269
39. Liu Z, Yao X, Sun B, Jiang W, Liao C, Dai X, et al. Pretreatment with kaempferol attenuates microglia-mediate neuroinflammation by inhibiting MAPKs-NF-kappaB signaling pathway and pyroptosis after secondary spinal cord injury. Free Radic Biol Med (2021) 168:142–54. doi: 10.1016/j.freeradbiomed.2021.03.037
40. Zhang L, Zhuang X, Kotitalo P, Keller T, Krzyczmonik A, Haaparanta-Solin M, et al. Intravenous transplantation of olfactory ensheathing cells reduces neuroinflammation after spinal cord injury via interleukin-1 receptor antagonist. Theranostics (2021) 11:1147–61. doi: 10.7150/thno.52197
41. Li T, Sun H, Li Y, Su L, Jiang J, Liu Y, et al. Downregulation of macrophage migration inhibitory factor attenuates NLRP3 inflammasome mediated pyroptosis in sepsis-induced AKI. Cell Death Discovery (2022) 8:61. doi: 10.1038/s41420-022-00859-z
42. Yang D, Dai X, Xing Y, Tang X, Yang G, Harrison AG, et al. Intrinsic cardiac adrenergic cells contribute to LPS-induced myocardial dysfunction. Commun Biol (2022) 5:96. doi: 10.1038/s42003-022-03007-6
43. Sahu P, Mudgal J, Arora D, Kinra M, Mallik SB, Rao CM, et al. Cannabinoid receptor 2 activation mitigates lipopolysaccharide-induced neuroinflammation and sickness behavior in mice. Psychopharmacol (Berl) (2019) 236:1829–38. doi: 10.1007/s00213-019-5166-y
44. Xue S, Cao ZX, Wang JN, Zhao QX, Han J, Yang WJ, et al. Receptor-interacting protein kinase 3 inhibition relieves mechanical allodynia and suppresses NLRP3 inflammasome and NF-kappaB in a rat model of spinal cord injury. Front Mol Neurosci (2022) 15:861312. doi: 10.3389/fnmol.2022.861312
45. Mi J, Yang Y, Yao H, Huan Z, Xu C, Ren Z, et al. Inhibition of heat shock protein family a member 8 attenuates spinal cord ischemia-reperfusion injury via astrocyte NF-kappaB/NLRP3 inflammasome pathway: HSPA8 inhibition protects spinal ischemia-reperfusion injury. J Neuroinflamm (2021) 18:170. doi: 10.1186/s12974-021-02220-0
46. Xue Z, Zhang Z, Liu H, Li W, Guo X, Zhang Z, et al. lincRNA-Cox2 regulates NLRP3 inflammasome and autophagy mediated neuroinflammation. Cell Death Differ (2019) 26:130–45. doi: 10.1038/s41418-018-0105-8
47. Han X, Sun S, Sun Y, Song Q, Zhu J, Song N, et al. Small molecule-driven NLRP3 inflammation inhibition via interplay between ubiquitination and autophagy: Implications for Parkinson disease. Autophagy (2019) 15:1860–81. doi: 10.1080/15548627.2019.1596481
48. Chang P, Li H, Hu H, Li Y, Wang T. The role of HDAC6 in autophagy and NLRP3 inflammasome. Front Immunol (2021) 12:763831. doi: 10.3389/fimmu.2021.763831
49. Shao BZ, Wei W, Ke P, Xu ZQ, Zhou JX, Liu C. Activating cannabinoid receptor 2 alleviates pathogenesis of experimental autoimmune encephalomyelitis via activation of autophagy and inhibiting NLRP3 inflammasome. CNS Neurosci Ther (2014) 20:1021–8. doi: 10.1111/cns.12349
50. Song H, Zhao C, Yu Z, Li Q, Yan R, Qin Y, et al. UAF1 deubiquitinase complexes facilitate NLRP3 inflammasome activation by promoting NLRP3 expression. Nat Commun (2020) 11:6042. doi: 10.1038/s41467-020-19939-8
51. Cheng Y, Pan X, Wang J, Li X, Yang S, Yin R, et al. Fucoidan inhibits NLRP3 inflammasome activation by enhancing p62/SQSTM1-dependent selective autophagy to alleviate atherosclerosis. Oxid Med Cell Longev (2020) 2020:3186306. doi: 10.1155/2020/3186306
52. Su SH, Wu YF, Lin Q, Wang DP, Hai J. URB597 protects against NLRP3 inflammasome activation by inhibiting autophagy dysfunction in a rat model of chronic cerebral hypoperfusion. J Neuroinflamm (2019) 16:260. doi: 10.1186/s12974-019-1668-0
53. Herzig S, Shaw RJ. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol (2018) 19:121–35. doi: 10.1038/nrm.2017.95
54. Garcia D, Shaw RJ. AMPK: Mechanisms of cellular energy sensing and restoration of metabolic balance. Mol Cell (2017) 66:789–800. doi: 10.1016/j.molcel.2017.05.032
55. Du J, Xu M, Kong F, Zhu P, Mao Y, Liu Y, et al. CB2R attenuates intervertebral disc degeneration by delaying nucleus pulposus cell senescence through AMPK/GSK3beta pathway. Aging Dis (2022) 13:552–67. doi: 10.1016/j.molcel.2017.05.032
56. Choi IY, Ju C, Anthony JA, Lee DI, Prather PL, Kim WK. Activation of cannabinoid CB2 receptor-mediated AMPK/CREB pathway reduces cerebral ischemic injury. Am J Pathol (2013) 182:928–39. doi: 10.1016/j.ajpath.2012.11.024
57. Wu A, Hu P, Lin J, Xia W, Zhang R. Activating cannabinoid receptor 2 protects against diabetic cardiomyopathy through autophagy induction. Front Pharmacol (2018) 9:1292. doi: 10.3389/fphar.2018.01292
58. Hernandez J, Francos-Quijorna I, Redondo-Castro E, Lopez-Vales R, Navarro X. Microglia stimulation by protein extract of injured rat spinal cord. A novel in vitro model for studying activated microglia. Front Mol Neurosci (2021) 14:582497. doi: 10.3389/fnmol.2021.582497
Keywords: cannabinoid receptor-2, neuroinflammation, NLRP3, autophagy, spinal cord injury
Citation: Jiang F, Xia M, Zhang Y, Chang J, Cao J, Zhang Z, Qian Z and Yang L (2022) Cannabinoid receptor-2 attenuates neuroinflammation by promoting autophagy-mediated degradation of the NLRP3 inflammasome post spinal cord injury. Front. Immunol. 13:993168. doi: 10.3389/fimmu.2022.993168
Received: 13 July 2022; Accepted: 06 September 2022;
Published: 26 September 2022.
Edited by:
Bo Li, Sun Yat-sen University, ChinaReviewed by:
Shiqing Feng, Tianjin Medical University General Hospital, ChinaValerio Chiurchiù, National Research Council (CNR), Italy
Copyright © 2022 Jiang, Xia, Zhang, Chang, Cao, Zhang, Qian and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lei Yang, leiyang@njmu.edu.cn; Zhanyang Qian, spineqzy@126.com; Zhongkai Zhang, zhangzhongkai@sdfmu.edu.cn
†These authors have contributed equally to this work