 Junjie Liu1,2†
Junjie Liu1,2† Haisu Tao3†Tong Yuan1,2†
Haisu Tao3†Tong Yuan1,2† Jiang Li1,2Jian Li1,2
Jiang Li1,2Jian Li1,2 Huifang Liang1,2*
Huifang Liang1,2* Zhiyong Huang1,2*
Zhiyong Huang1,2* Erlei Zhang1,2*
Erlei Zhang1,2*- 1Hepatic Surgery Center, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 2Hubei Key Laboratory of Hepato-Pancreato-Biliary Diseases, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 3Department of Hepatobiliary Surgery, Zhujiang Hospital, Southern Medical University, Guangzhou, China
Anti-PD-1/PD-L1 therapy has shown significant benefits in the treatment of a variety of malignancies. However, not all cancer patients can benefit from this strategy due to drug resistance. Therefore, there is an urgent need for methods that can effectively improve the efficacy of anti-PD-1/PD-L1 therapy. Combining anti-PD-1/PD-L1 therapy with regorafenib has been demonstrated as an effective method to enhance its therapeutic effect in several clinical studies. In this review, we describe common mechanisms of resistance to anti-PD-1/PD-L1 therapy, including lack of tumor immunogenicity, T cell dysfunction, and abnormal expression of PD-L1. Then, we illustrate the role of regorafenib in modifying the tumor microenvironment (TME) from multiple aspects, which is different from other tyrosine kinase inhibitors. Regorafenib not only has immunomodulatory effects on various immune cells, but can also regulate PD-L1 and MHC-I on tumor cells and promote normalization of abnormal blood vessels. Therefore, studies on the synergetic mechanism of the combination therapy may usher in a new era for cancer treatment and help us identify the most appropriate individuals for more precise treatment.
1 Introduction
In recent years, immune checkpoint inhibitors (ICIs) have achieved considerable progress in the immunotherapy of cancer. ICIs act on a suppressed immune system and activate CD8+ T cells to attack tumor cells (1). ICIs are now being researched in clinical studies for a range of malignancies (2, 3). However, there are still some patients who cannot benefit from anti-PD-1/PD-L1 therapy due to the development of resistance in the tumor (4). In past studies, researchers have focused mostly on how immune cells combat tumor cells face-to-face. Nevertheless, it is also essential to consider whether the environment in which this fight takes place is conducive to immune cell activity. Combining anti-PD-1/PD-L1 therapy with other therapeutic medications that help improve the TME may be an effective method to enhance its therapeutic potential.
Regorafenib is a tyrosine kinase inhibitor that is used in patients with colorectal cancer (CRC) (5), gastrointestinal stromal tumor (GIST) (6), and hepatocellular carcinoma (HCC) (7). Regorafenib differs from sorafenib in that a fluorine atom has been added to the central phenyl ring, making it more pharmacologically active than sorafenib (8). Recent studies have shown that regorafenib can optimize the efficacy of anti-PD-1/PD-L1 treatment. Regorafenib combined with nivolumab showed good efficacy and tolerable safety in the treatment of CRC (9). Therefore, regorafenib combined with ICIs therapy may be a promising approach for cancer therapy.
2 The underlying mechanism of resistance to PD-1/PD-L1 blockade
2.1 PD-1 and its ligands
PD-1 (also known as CD279) encodes a 50-55 kDa protein that is a member of the CD28/cytotoxic T-lymphocyte antigen 4 (CTLA4) family (10). PD-1 was originally isolated from the murine T-cell hybridoma cell line 2B4.11 and is thought to play a role in programmed cell death (11). However, subsequent studies revealed that the expression of PD-1 may not be necessary for apoptosis (12). PD-1 deficiency was found to be associated with autoimmune diseases in PD-1-deficient C57BL/6 mice (13). Furthermore, selective upregulation of PD-1 was also observed on exhausted T cells (14). PD-L1 (also known as CD274), which belongs to the B7 gene family (15), is expressed in many immune cells and non-hematopoietic cells, including macrophages, dendritic cells (DCs), B cells, T cells, vascular endothelial cells, hepatocytes, and epithelial cells (16). PD-1 binds to its ligand, PD-L1 or PD-L2, suppresses T-cell receptor (TCR)-mediated phosphorylation of ZAP70 and CD3zeta by recruiting protein tyrosine phosphatase SHP-2, inhibits downstream PI3K/AKT pathways, and transmits negative regulatory antigen receptor signals (17, 18). Similarly, the effect of suppressing B cell receptor (BCR) signaling was observed in B cells (19). In a variety of tumors, the overexpression of PD-1 on tumor-infiltrating immune cells or PD-L1 on tumor cells corresponds with a poor prognosis (20). The binding of PD-1 to PD-L1 is a potential mechanism through which tumors escape the host immune response, and eliminating this interaction can play a pivotal role in tumor immunotherapy (21, 22). Therefore, anti-PD-1/PD-L1 therapy has become an essential cancer therapeutic strategy. Currently, anti-PD-1/PD-L1 therapy has been approved for the treatment of various tumors, including CRC, HCC, non-small cell lung cancer (NSCLC), renal cell carcinoma (RCC), as well as head and neck cancer (HNC), among others (23).
2.2 Resistance to PD-1/PD-L1 blockade
Although anti-PD-1/PD-L1 therapy has great therapeutic prospects in a number of malignancies, there are still many patients who do not benefit from the treatment. In some cancers, primary treatment resistance is observed in more than 60% of patients (24). In non-responsive patients, PD-1/PD-L1 blockade alone is insufficient for cancer treatment. Before utilizing anti-PD-1/PD-L1 therapy, it is vital to evaluate which patients are most suited for this therapy and how to combine it with other treatment strategies. Therefore, it is crucial to understand why drug resistance occurs. Resistance to PD-1/PD-L1 blockade can be classified as primary or acquired treatment resistance (25). Patients who do not initially demonstrate a clinical response to anti-PD-1/PD-L1 therapy are considered to have “primary resistance” to therapy. For example, insufficient antigen immunogenicity and dysfunction of antigen presentation may affect the ability of APCs to present tumor rejection antigens to CD8+ T cells and thus affect the subsequent anti-tumor effects. “Acquired resistance” refers to the phenomenon in which initially immunotherapy-responsive tumor patients acquire treatment resistance or relapse over time. Due to the heterogeneity of tumors, tumor cells with low antigen expression and the upregulation of certain immunosuppressive signaling receptors may result in the development of acquired resistance (25, 26). Mechanistically, resistance to drugs targeting PD-1/PD-L1 can be due to the following aspects: 1) lack of tumor immunogenicity, 2) T cell dysfunction, 3) abnormal expression of PD-L1.
2.2.1 Lack of tumor immunogenicity
Neoantigens produced by tumor cells can activate the immune response of T cells, which is related to stronger immunogenicity and is one of the targets of tumor immunotherapy (27). Firstly, the tumor mutational burden (TMB) can predict the effect of ICIs. A lower TMB tends to be correlated with worse clinical outcomes (28). Secondly, in some mismatch repair deficient cancers, neoantigen expression makes them more sensitive to ICIs (29). Conversely, mismatch repair proficient tumors are insensitive to ICIs. Thirdly, Β2-microglobulin (B2M) mutations lead to the downregulation of MCH-I, triggering the escape of tumor cells from treatment with ICIs (30, 31). All of the above are primary drug resistance. Fourthly, due to the existence of tumor heterogeneity, during the use of ICIs, tumor cells with low antigen expression are selected for continued growth, resulting in the emergence of acquired drug resistance (32).
2.2.2 T cell dysfunction
Multifaceted T cell dysfunction may result in resistance to ICIs. Antigen-presenting cells (APCs) stimulate the activation of tumor antigen-specific effector T cells, whereas a lack of APCs or their inactivation inhibits effector T cell activation (33). In melanoma cells, activation of the WNT/Β-catenin pathway reduces the number of tumor-infiltrating lymphocytes (TILs) because activation of this pathway decreases the expression of chemokine C-C motif ligand 4 (CCL4), which is essential for tumor infiltration by DCs. Insufficient DCs for the activation of T cells results in resistance to ICIs therapy (34). Moreover, Foxp3+ regulatory T cells (Tregs) suppress the immune response to autoantigens to prevent the development of autoimmune diseases and also suppress the anti-tumor immune response (35). CTLA4 blockade can inhibit Tregs, reactivate the repressed T cells in the TME, and overcome the inhibitory immune microenvironment (36). As an immunosuppressive immature myeloid cell, myeloid-derived suppressor cells (MDSCs) also promote tumor invasion and contribute to tumor immune escape (37). The massive accumulation of MDSCs in tumor tissues will limit the inhibitory effect of ICIs on tumors (38). In some solid tumors, tumor-associated macrophages (TAMs) infiltration is strongly correlated with a poorer prognosis (39–41). TAMs play an essential role in strengthening angiogenesis, tumor growth and metastasis, as well as dampening anti-tumor immunity (42). These suppressive immune cells impair the normal function of T cells, leading to primary resistance to ICIs therapy.
In addition to the immune cells that influence the therapeutic efficiency of ICIs, there are also some inhibitory molecules and cytokines in the TME. In epithelial cells, transforming growth factor Β (TGF-Β) has a tumor suppressor effect. In other contexts, however, TGF-Β has the effect of promoting tumor invasion and metastasis. Its tumor-promoting effect is not only limited to tumor cells but also affects the TME (43). Principe et al. reported that TGF-Β reduces T cell infiltration and impairs granzyme B-mediated tumor destruction (44). Moreover, TGF-Β plays an immunosuppressive role independently of Foxp3+Tregs (45). In the hypoxic tumor microenvironment, CD39 and CD73 stimulate the conversion of ATP to adenosine, which weakens antitumor immunity (46, 47). In a preclinical study, researchers observed that treatment targeting CD73 alleviated the suppression of lymphocytes by adenosine and inhibited tumor growth in mice in vivo. Therapy targeted against the adenosinergic pathway combined with anti-PD-1 therapy is more effective than monotherapy (48). In addition, vascular epithelial growth factor (VEGF) (49) and indoleamine 2,3-dioxygenase (IDO) (50) can also induce the function of Tregs, leading to immunosuppression. CD8+T cells in the TME can kill tumor cells by secreting IFN-γ, which exerts its effects through the downstream JAK/STAT pathway (51). Furthermore, IFN-γ can inhibit tumor progression by increasing tumor apoptosis and recruiting other immune cells to limit tumor cell proliferation (52). However, IFN-γ is also a double-edged sword in the anti-tumor immune response, since it promotes the upregulation of PD-L1 in the TME and consequently decreases the activity of CD8+T cells (53). Mutations in IFN-γ pathway-related proteins (such as JAK1/2 and STAT1) can also result in resistance of tumor cells to anti-PD-1/PD-L1 therapy (54, 55).
The expression of other inhibitory receptors can also contribute to T cell depletion, resulting in anti-PD-1/PD-L1 therapy resistance. These include CTLA4 (56), lymphocyte-activation gene 3 (LAG3) (57), T-cell Ig and mucin domain-3 protein (TIM3) (58), as well as B and T lymphocyte attenuator (BTLA) (59). These compensatory inhibitory signals are elevated throughout therapy, which makes the anti-PD-1/PD-L1 treatment unable to achieve the best therapeutic effect, resulting in acquired drug resistance. Therefore, anti-PD-1/PD-L1 therapy can only overcome part of the inhibitory signals in the TME, and there may be more inhibitory axes that play a role in suppressing T cell function in the TME.
2.2.3 Abnormal expression of PD-L1
Low expression of PD-L1 leads to the development of primary resistance, thereby reducing the efficacy of treatment. In a clinical trial of NSCLC, the expression of PD-L1 in more than 50% of tumor cells was related to higher pembrolizumab effectiveness (60). Conversely, an increase in PD-L1 expression may also diminish the effectiveness of anti-PD-1/PD-L1 therapy. Theodoraki et al. demonstrated that patients with high levels of PD-L1+exosomes in their plasma had a worse prognosis (61). IFN-γ stimulates the expression of PD-L1 in exosomes, and these PD-L1+exosomes impair the function of CD8+T cells, promote tumor progression, and trigger immune escape (62). Tumor-infiltrating T cells release IFN-γ, which increases the expression of PD-L1 on the surface of tumor cells or other immune cells infiltrating the tumor. Overexpressed PD-L1 binds to PD-1 on the surface of T cells, hence restricting the effector function of T cells (63). At the same time, several lncRNAs and circular RNAs were found to boost the expression of PD-L1 in tumor cells and promote tumor resistance to anti-PD-1 therapy (64–67). Therefore, the increase or decrease of PD-L1 expression in the tumor microenvironment may affect the efficacy of anti-PD-1/PD-L1 therapy, which can be explained by different immune evasion mechanisms. Furthermore, the expression of PD-L1 in tumor tissue may be heterogeneous, and the results of a single biopsy cannot represent the overall response of the tumor to anti-PD-1/PD-L1 therapy. Therefore, future research should further explore the connection between PD-L1 expression and anti-PD-1 effectiveness.
2.3 Strategies for overcoming drug resistance
As a result of the advancement of cancer research, different therapeutic techniques have been proposed for various causes of drug resistance in tumors. Resistance to anti-PD-1/PD-L1 therapy may emerge at various stages of cancer treatment. Therefore, it is essential to combine anti-PD-1/PD-L1 therapy with other therapeutic techniques. Currently, therapeutic strategies for overcoming drug resistance mainly focus on increasing the recognition of tumor cells by the immune system (68–71), enhancing T cell infiltration in tumor tissue (72, 73), reversing the exhausted state of T cells (74), and improving the immunosuppressive tumor microenvironment (75, 76). Regorafenib has been proven in an increasing number of studies to improve the TME and boost the anti-tumor immune response, hence reducing anti-PD-1/PD-L1 treatment resistance.
3 Immunomodulatory effect of regorafenib
The TME comprises multiple cell types, including immune cells, fibroblasts, neurons, adipocytes, and endothelial cells, as well as extracellular components, including the extracellular matrix, cytokines, growth factors, chemokines, and extracellular vesicles (77, 78). The TME is strongly immunosuppressive, which is why most antitumor therapies targeting tumor cells have limited therapeutic effects. Multiple immunosuppressive cells in the TME facilitate the evasion of anticancer immune responses by tumor cells. TAMs, which are major immunosuppressive cells, can not only promote tumor invasion and metastasis but also promote tumor angiogenesis (79). MDSCs, as a heterogeneous population of immature myeloid cells, are also reported to be involved in resistance to anti-VEGF treatment (80). Tregs in the TME decrease anti-tumor immunity via numerous mechanisms, including directly inhibiting immune cells, interacting with antigen-presenting cells and fibroblasts, secreting cytokines, etc. (81). Moreover, tumor-associated neutrophils (TANs), cancer-associated fibroblasts (CAFs), and tumor-associated endothelial cells (TECs) in the TME all predict poor prognosis in a variety of cancers (82–84). The extracellular matrix also plays an important role in tumor resistance and the promotion of tumor invasion and metastasis. The TME influences the acquisition and maintenance of tumor hallmarks to varying degrees. This dependency of tumor cells on the TME presents a potential for anticancer therapy. Targeting the TME provides additional advantages over traditional therapeutic strategies (78). Transforming a tumor’s immunosuppressive microenvironment into an immunostimulatory microenvironment enables the immune system to eliminate malignancies (85). Stimulation of the antitumor immune response by regorafenib can augment the efficacy of anti-PD-1/PD-L1 treatment and exert a more potent antitumor effect.
Regorafenib targets multiple tyrosine kinases, including vascular endothelial growth factor receptors (VEGFR)1-3, platelet-derived growth factor receptor (PDGFR), fibroblast growth factor receptors (FGFR)1 and 2, tyrosine kinase with immunoglobulin-like and epidermal growth factor-like domains 2 (TIE2), the oncogenic kinases RET and KIT, rapidly accelerated fibrosarcoma (RAF), as well as colony-stimulating factor-1 receptor (CSF1R) (86). These targets have significant roles in tumor angiogenesis, promoting tumor growth and metastasis. Regorafenib alleviates tumor resistance to anti-PD-1/PD-L1 treatment in several aspects through diverse targets, regulates the TME, and reduces tumor progression.
3.1 Effects on natural killer (NK) cells
NK cells are the primary effector cells in innate anticancer immunity and have the ability to autonomously kill target cells. The majority of treatment approaches targeting the tumor microenvironment concentrate on T cell immunity while ignoring the role of NK cells (87). NKG2D, a key receptor on NK cells, can recognize different ligands on tumor cells, including MHC class I polypeptide-related sequence A (MICA), to exert an antitumor effect. The combination of NKG2D and MICA on tumor cells can enhance the tumor-killing activity and cytokine production of NK cells (88). However, several studies have observed that human tumor cells release soluble MICA by proteolytic shedding and that these sMICA molecules result in the downregulation of NKG2D and impede NKG2D-mediated tumor immune surveillance (89–91). ADAM10 and ADAM9 are sheddases that release MICA from the cell membrane of tumor cells. Regorafenib can suppress the transcriptional expression of ADAM10 and ADAM9 and diminish the protein levels of sMICA, thereby improving the function of NK cells in antitumor immunity (92, 93).
In addition, it has been observed that the overexpression of CD24 correlates with the invasiveness and metastasis capacity of malignancies (94), while the activation of STAT3 is related to the expression of CD24 (95, 96), and inhibition of p-STAT3 can limit the expression of CD24 (97). Interestingly, in experiments with liver cancer cell lines and mouse subcutaneous tumors, regorafenib inhibited p-STAT3 by boosting the activity of SHP-1, thereby inducing the apoptosis of tumor cells (98). Siglec-10 is an immunosuppressive receptor expressed on monocytes, eosinophils, and NK cells (99–101). The combination of CD24 and Siglec-10 impairs NK cell function and decreases survival in patients with HCC (102). Therefore, regorafenib may influence the expression of CD24 by modulating the expression of p-STAT3, further affecting the function of NK cells.
3.2 Effects on TAMs
Regorafenib can alleviate the inhibitory tumor microenvironment by suppressing TAMs in the TME through specific mechanisms. There are TIE2-expressing macrophages (TEMs) in tumors, which can accelerate tumor angiogenesis and tumor progression (103). Targeting the ANG2/TIE2 axis has been shown to reduce angiogenesis and tumor growth in preclinical studies (104). Regorafenib can inhibit the infiltration of TAMs by inhibiting the TIE2 pathway (105), while inducing sustained M1 polarization and reversing M2 polarization (106–110). Bai et al. demonstrated that regorafenib not only inhibited the activation of M2 macrophages but also caused M2 inactivation by reducing the production of tumor-suppressive cytokines (111). Moreover, the p38MAPK pathway plays a vital role in tumor development (112). In in vitro experiments, regorafenib promoted antitumor immunity via the p38MAPK/Creb1/Klf4 pathway. In macrophages, regorafenib reduced the phosphorylation of p38MAPK and Creb1, while increasing the expression of Klf4 and M1-related genes (113). In a model of colorectal cancer in mice, regorafenib reduced the abundance of TAMs in a dose-dependent manner through inhibition of CSF1R (114).
Doleschel et al. demonstrated that when treatment was discontinued after 10 days of continuous treatment, the regorafenib combined with anti-PD-1 treatment group still maintained tumor suppression, and the tumor volume increased only slightly until the end of the observation period (108). After withdrawal of treatment, macrophages increased in the group receiving combination treatment, and the combination therapy led to a polarization of tumor-associated macrophages towards the M1 phenotype. These results indicate that the addition of anti-PD-1 therapy to regorafenib therapy can improve the microenvironment of tumors and prevent rapid tumor regeneration (108).
3.3 Effects on CD8+T cells and Tregs
In addition to activating NK cells and affecting the polarization of tumor-associated macrophages, regorafenib also induces CD8+T cell activation and suppresses Tregs. One study showed that regorafenib increased the expression of CXCL10 in HCC cells by inhibiting the activity of STAT3. As a ligand of CXCR3, CXCL10 can mediate the infiltration of T cells into tumor tissues. Furthermore, regorafenib combined with anti-PD-1 treatment increased the number of CXCR3+CD8+T cells in tissues, enhancing the effector role of T cells (115). Although both regorafenib and DC-101 (VEGFR2 receptor inhibitor) inhibit tumor angiogenesis, regorafenib induces tumor infiltration by CD4+ and IFN-γ+CD8+T cells, which is associated with better antitumor efficacy (113). In addition to HCC models, regorafenib combined with ICIs also produces significant antitumor effects in other cancers, such as melanoma (116), CRC (108), and oral squamous cell carcinoma (OSCC) (109). In an orthotopic OSCC model, IFN-γ+IL-2+CD8+T cell infiltration was considerably higher in the regorafenib combined anti-PD-L1 therapy group than in the monotherapy group or the control group. From improving the immunosuppression status to enhancing immune activation, regorafenib further enhanced the therapeutic effect of ICIs in OSCC (109). In addition, through their immunosuppressive effects, CD4+ CD25+ Foxp3+ Tregs play a crucial role in the development and prognosis of HCC (117). The amount of Tregs in the peripheral blood of HCC patients is higher than that of the healthy control group (118). In addition, an increase in Tregs in HCC tumors predicted a poorer outcome (119–121). By secreting cytokines like interleukin-10 (IL-10) and TGF-Β, Tregs in the TME exert immunosuppressive effects (122, 123). Importantly, Tregs express inhibitory receptors like CTLA4, PD-1, and others, making them potential direct targets for improving the TME (117). Regorafenib has been reported to significantly reduce Treg infiltration in the TME (108). Chiang et al. reported that regorafenib combined with anti-PD-L1 treatment decreased the accumulation of Tregs and MDSCs (109). Additionally, suppressive immune cells such as TAMs, MDSCs, and CAFs in the TME promote the accumulation of Tregs (124–126). Therefore, regorafenib may inhibit Tregs by acting on these suppressive immune cells.
3.4 Effects on CAFs
CAFs also play a crucial role in inhibiting antitumor immunity. CAFs recruit inhibitory immune cells to promote tumor growth by secreting inhibitory cytokines. For example, CAFs recruit and activate Tregs by secreting regulators such as CXCL12 and CD73, thereby suppressing the function of effector T cells and promoting the formation of an immunosuppressive microenvironment (125). In addition, CAFs recruit MDSCs and TAMs through the secretion of TGF-Β and complement C5a, thereby boosting their potential to suppress effector T cells and promote tumor growth (127–130). Consequently, suppressing CAFs in the TME is also a method for improving immunotherapy.
Zhang et al. reported that regorafenib suppressed the proliferation and promoted the apoptosis of CAFs by acting on the AKT/Bcl-2/Bax pathway in vitro. Moreover, in vivo experiments revealed that regorafenib reduced TAMs infiltration in tumors and inhibited tumor growth by targeting CAFs (131). In a nude mouse model with co-implanted colon cancer cells and bone marrow mesenchymal stem cells, regorafenib had a more potent antitumor effect than in colon tumor-bearing mice. In both tumor models, pERK1/2 expression was reduced by regorafenib. Studies have shown that regorafenib inhibits not only the interaction between tumor cells and CAFs, but also reduces tumor angiogenesis and lymphangiogenesis, thus decreasing the invasion and metastasis of colon cancer (132).
3.5 Effects on PD-L1 and MCH-I expression
A study evaluating the effects of 161 different kinase inhibitors on melanoma cell lines found that regorafenib is the only drug that reduces the expression of PD-L1 in tumor cells without decreasing the amount of MHC-I on the surface of the cell (116). Regorafenib indirectly inhibits JAK1/2-STAT1 signaling by inhibiting the oncogenic tyrosine kinase RET. Interestingly, RET/Src signaling showed no influence on MHC-I levels, while suppression of RET/Src signaling by regorafenib reduced IFN-γ-induced expression of PD-L1 and IDO1 on tumor cells. The expression of PD-L1 facilitates tumor cell evasion of T-cell effector functions, but regorafenib alleviates immune evasion by inhibiting IFN-γ-induced PD-L1 expression (116). However, in another preclinical investigation, the researchers discovered that in the presence of IFN-γ, regorafenib could upregulate the expression of MCH-I on HCC cells by acting on the IFN-γ/STAT1 signaling pathway (133). At the same time, regorafenib enhanced the expression of MCH-I transcriptional regulators, such as IRF1 and NLRC5. In terms of up-regulating MHC-I, regorafenib was more effective than lenvatinib, sorafenib, and cabozantinib. This may be due to the fact that regorafenib has a more potent inhibitory impact on the MEK/ERK pathway (133). Increasing numbers of studies indicate that regorafenib can impact the expression of PD-L1 and MCH-I. Nevertheless, further efforts are needed to understand the underlying mechanisms.
3.6 Inhibition of angiogenesis
The increase of angiogenic factors such as VEGF and ANG2 in the tumor microenvironment might result in aberrant tumor angiogenesis and structure, as well as facilitate tumor evasion of immunotherapy (134). When tumor blood vessels function abnormally, immune cells in tumor tissue are inhibited by various factors, blocking their normal antitumor effect. Firstly, the hypoxic tumor microenvironment facilitates the infiltration of MDSCs and TAMs and induces the polarization of TAMs toward the M2 type (135). Additionally, the elevated expression of CC-chemokine ligand 28 (CCL28) in a hypoxic environment indirectly results in the accumulation of Tregs in the tumor microenvironment (136). Secondly, the hypoxic environment promotes the expression of CTLA4 and TIM3 on Tregs and stimulates the expression of PD-L1 on tumor cells (137, 138). Therefore, anti-tumor angiogenesis alleviates hypoxia by improving tissue perfusion, enhances the infiltration of the tumor by immune cells, and alleviates the immunosuppressive environment, which is crucial for antitumor therapy.
Regorafenib has been shown to be effective in inhibiting angiogenesis. For example, regorafenib reduced tumor angiogenesis in a mouse orthotopic colon cancer model by targeting the VEGFR2 and TIE2 receptors. The ratios of VEGFR2+/CD31 and TIE2+/CD31 in regorafenib-treated tumors were considerably lower than in the control group (105). Regorafenib also inhibits tumor angiogenesis, which slows the growth of diverse malignancies, including colon cancer (132), breast cancer (139), gastric cancer(GC) (140), and HCC (141). Notably, the original idea was that antiangiogenic drugs could limit oxygen and nutrient delivery to tumors (142). However, excessive inhibition of tumor blood vessels might result in tumor invasion and metastasis by inducing transformation of the tumor into a hypoxia-tolerant phenotype (143). Thus, Jain et al. proposed the notion of normalization of tumor vasculature. Normalization of blood vessels can alleviate hypoxia and enhance the delivery of antitumor medications. Therefore, the application duration and dose of antiangiogenic medicines merit more research (142). In the above-mentioned study of regorafenib combined with anti-PD-1 therapy, researchers discovered that in an orthotopic mouse model of HCC, an intermediate dose (10 mg/kg) of regorafenib combined with anti-PD-1 therapy considerably decreased the region of hypoxia in comparison to other doses of regorafenib. Through the use of 5, 10, and 20 mg/kg of regorafenib in combination with anti-PD-1 therapy in the treatment of the orthotopic mouse model, it was discovered that an intermediate dose of regorafenib can promote the normalization of intratumoral blood vessels, thereby increasing the delivery of therapeutic drugs (115). More research is required to determine the optimal dose and treatment duration of these anti-angiogenic medicines for the treatment of malignancies. (Figure 1)
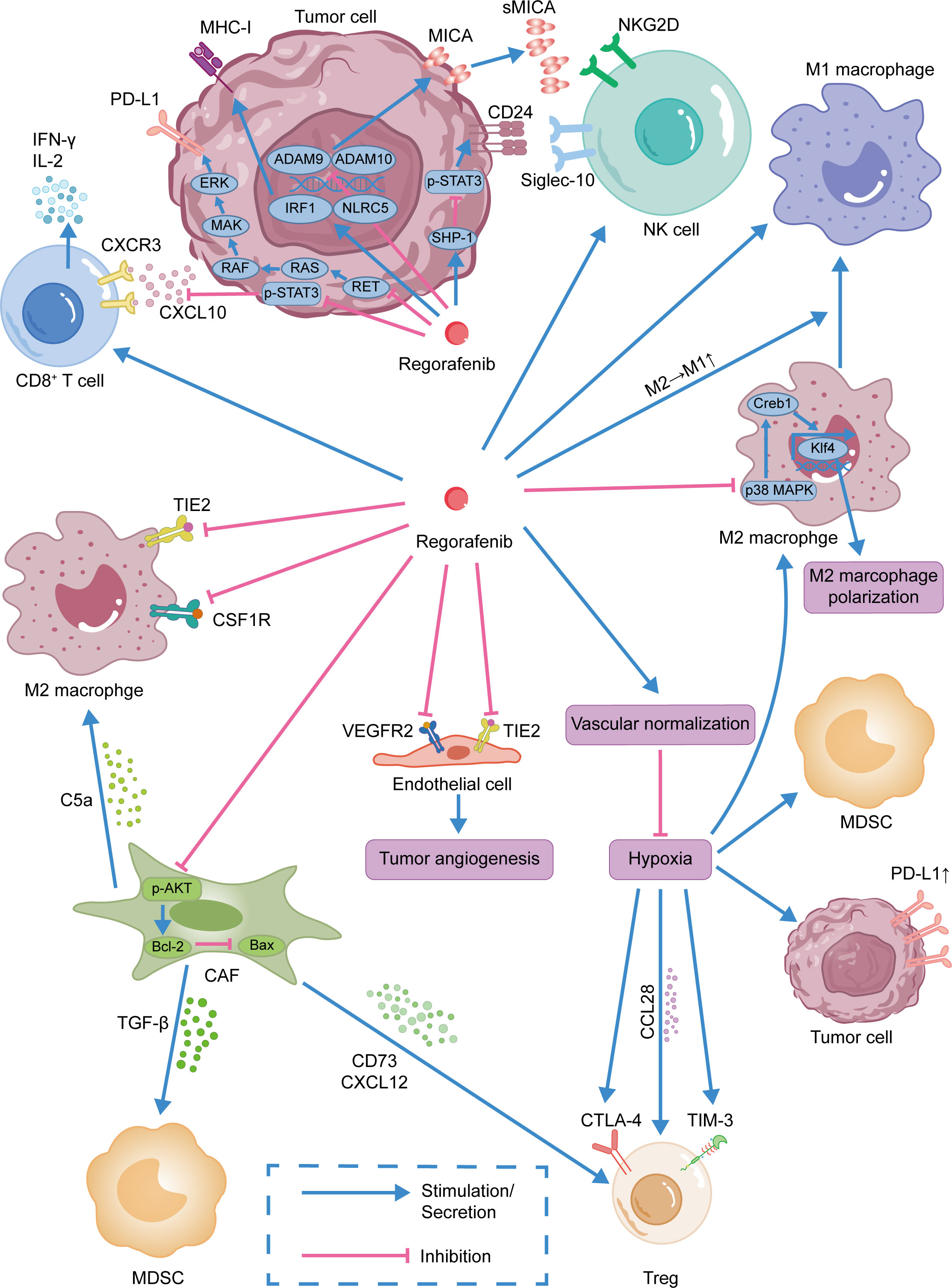
Figure 1 Immunomodulatory effect of regorafenib.Regorafenib was shown to promote the infiltration of CD8+ T cells into tumor tissues and activate NK cells. By normalizing tumor blood vessels, regorafenib improves the immunosuppressive tumor microenvironment. Moreover, regorafenib was also shown to inhibit MDSCs, CAFs, and Tregs and promote the repolarization of TAMs from M2-like to M1-like phenotype.
4 Clinical trials of combined therapy
Regorafenib in combination with anti-PD-1/PD-L1 therapy has been utilized in a growing number of clinical trials. (Table 1) The clinical outcomes of the studies in Table 1 vary to some extent, possibly because the studies are still in the early stages and there are differences in the subjects selected for different studies. Therefore, more clinical trials are needed to verify the efficacy of the combination therapy.
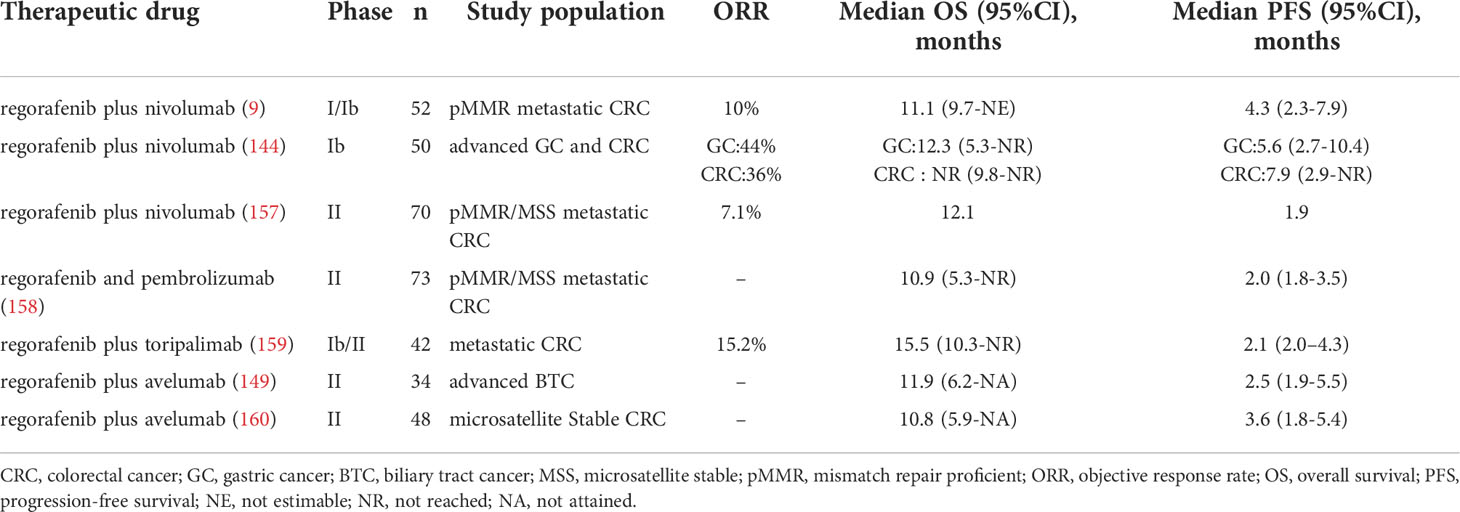
Table 1 Clinical trials of combined therapy.
In REGONIVO trial in CRC patients, the median PFS was 7.9 months (95% confidence interval [CI], 2.9 months-NR), and the objective response rate (ORR) was 36% (95%CI, 18.0%-57.5%) (144). Compared with regorafenib (ORR: 3%) in the INTEGRATE trial (150) or Nivolumab (ORR: 11.9%) in the ATTRACTION-2 trial (151), combination therapy provided greater benefits than monotherapy. In addition, among GC patients, median PFS and median OS were 6.5 months (95% CI, 2.7 months-10.4 months) and 12.3 months (95% CI, 5.3 months-NR), respectively. With regorafenib and nivolumab, three of seven patients with refractory GC who had previously undergone anti-PD-1 monotherapy achieved a response (144). This study demonstrated that regorafenib may overcome anti-PD-1 treatment resistance, and the combination of regorafenib with anti-PD-1 therapy has promising therapeutic potential. In addition, the combination therapy also showed good efficacy and tolerable safety in the treatment of mismatch repair proficient (pMMR) CRC (9). In this phase IB study, 52 patients were recruited to receive the regorafenib and nivolumab combination. The median progression-free survival (PFS) and overall survival (OS) for the 40 evaluable patients were 4.3 and 11.1 months, respectively. Overall, 10 percent of the 40 evaluable patients showed a partial response, and 53 percent attained stable disease. In this clinical experiment, the researchers found that the expression of Treg in tumor tissue was negatively correlated with the FPS of patients (9). The results of the REGONIVO trial mentioned above are better than this clinical trial. This discrepancy may be due to differences in the patients included in the two trials. Therefore, researchers still need to explore more sensitive biomarkers for predicting the efficacy of combination therapy.
Patients with biliary tract cancer (BTC) often have a dismal prognosis, with few therapeutic options available after the failure of first-line therapy, with the limited effectiveness of ICI therapy alone (152, 153). However, research suggests that regorafenib may improve the efficacy of ICIs in BTC patients. In a phase II multicenter, single-arm, open-label trial of regorafenib plus avelumab (anti-PD-L1 antibody), the combination treatment demonstrated promising effects against cholangiocarcinoma. Four (13.8%) of the twenty-nine evaluable patients achieved partial responses, eleven (37.0%) achieved stable disease, and fourteen (48.3%) had progressive disease. The median PFS was 2.5 months (95%CI, 1.9 months-5.5months) and overall survival was 11.9 months (95%CI, 6.2months-NA). In this trial, the researchers also found that patients with higher expression of PD-L1 and IDO1 had better treatment outcomes (148). These results indicate which patients with cholangiocarcinoma might benefit most from regorafenib in combination with ICIs.
In addition to CRC, GC, and BTC, the combination of Regorafenib and anti-PD-1/PD-L1 therapy has also shown promising therapeutic effects in HCC. In a multicenter retrospective study of advanced HCC, researchers evaluated the efficacy of regorafenib plus sintilimab versus regorafenib alone. Combination therapy has longer FPS, higher ORR, and better OS than monotherapy. The median PFS for the combination therapy was 5.6 months (95% CI, 4.2 months–7.0 months) and the median OS was 13.4 months (95% CI, 9.2 months–17.5 months). The ORR of combination therapy was 24.1%, compared with 9.1% for monotherapy (154). In another phase Ib clinical trial of regorafenib plus pembrolizumab for first-line treatment of advanced HCC, the disease control rate (DCR) was 88% in the regorafenib 120 mg group and 91% in the regorafenib 80 mg group (RECIST v1.1) (155).
In conclusion, the combination of regorafenib with anti-PD-1/PD-L1 treatment may have a more potent impact than monotherapy in modifying the tumor microenvironment and reducing angiogenesis. However, further research is necessary to enable an informed choice of the most effective therapy based on the unique features of each patient. Currently, there are many ongoing clinical trials of the combination therapy for the treatment of cancer, including the evaluation of combinations with nivolumab, sintilimab, and toripalimab. (Table 2)
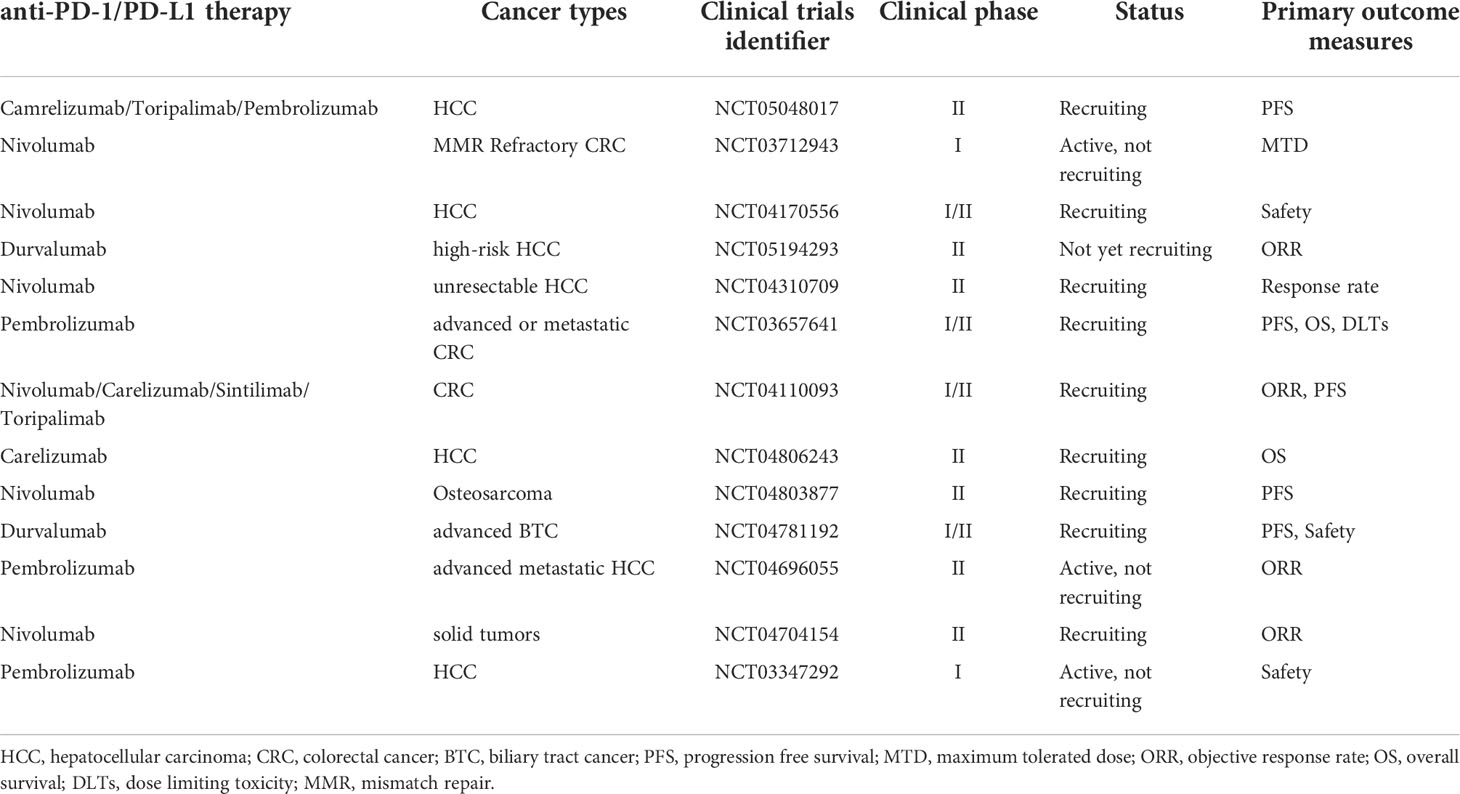
Table 2 Ongoing trials of regorafenib combined with anti-PD-1/PD-L1 therapy.
5 Discussion and future perspectives
Anti-PD-1/PD-L1 therapy has shown significant effects in the treatment of several malignancies. However, the emergence of primary or acquired tumor resistance indicates that not all patients receive therapeutic benefits. As a tyrosine kinase inhibitor, regorafenib has an effect on the TME in addition to its involvement in inhibiting tumor cell invasion and metastasis, as well as promoting tumor cell apoptosis. Firstly, Regorafenib enhances antitumor immunity by leading to a polarization of tumor-associated macrophages towards the M1 phenotype and by increasing the proliferation and activation of CD8+ T cells (105, 113). Secondly, regorafenib decreases the accumulation of Tregs and MDSCs, thereby alleviating the suppressive TME (109). In the TME, TAMs, MDSCs, and CAFs all promote the accumulation of Tregs (124–126). Consequently, regorafenib may inhibit Tregs by acting on these immunosuppressive cells. By inhibiting CAFs’ proliferation and promoting apoptosis, regorafenib also inhibits tumor cell invasion and metastasis (131). Thirdly, regorafenib affects the function of NK cells by inhibiting the expression of sMICA and CD24 on tumor cells (92, 93, 97, 98). Moreover, regorafenib increases MHC-I expression on tumor cells, thus enhancing the immunogenicity of the tumor. Regorafenib also reduces immune evasion by suppressing the expression of PD-L1 on tumor cells (116, 133). Fourthly, by acting on receptors such as VEGFR2 and TIE2, regorafenib promotes intratumoral blood vessel normalization, enhances drug delivery, reduces hypoxia-induced infiltration of suppressive immune cells, and prevents hypoxia-induced PD-L1 upregulation (115, 138). The immunomodulatory actions of regorafenib reduce resistance to anti-PD-1/PD-L1 treatment in multiple ways, which illustrates the therapeutic significance of their combination. (Figure 2)
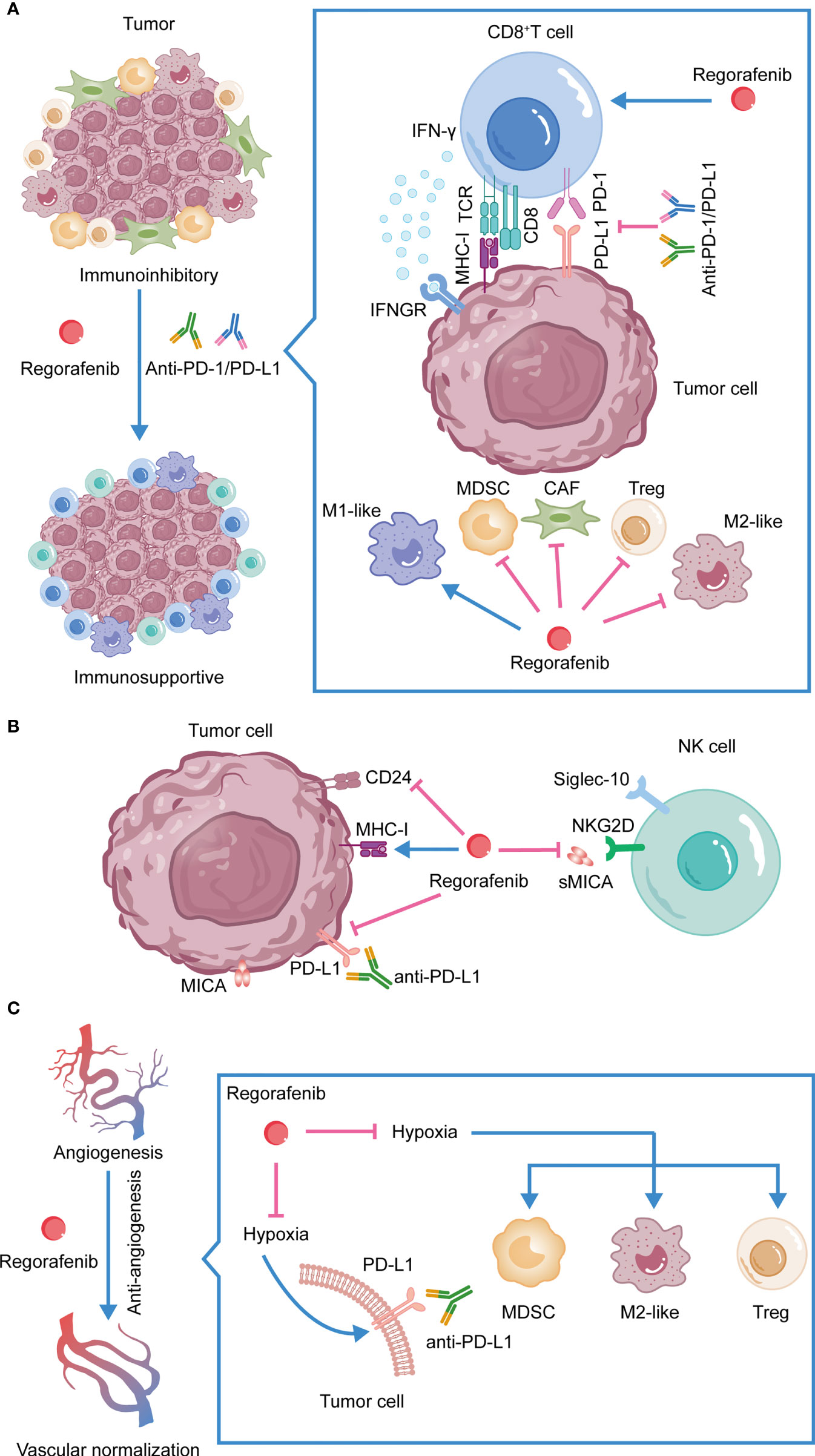
Figure 2 The synergistic antitumor efficacies and mechanisms of anti-PD-1/PD-L1 in combination with regorafenib. (A) Combination therapy reshapes the TME. (B) Regorafenib activates NK cells, inhibits the expression of PD-L1 on tumor cells and promotes the expression of MHC-I. (C) Regorafenib promotes vessel normalization, enhances drug delivery, alleviates hypoxia-induced PD-L1, reduces hypoxia-induced infiltration of suppressive immune cells, and increases the efficacy of anti-PD-1/PD-L1 therapy.
Regorafenib combined with anti-PD-1/PD-L1 therapy is one of the trendiest issues in tumor therapy research in recent years. A patient with lung metastases from HCC who underwent a combination of regorafenib and sintilimab had a complete response of tumor lesions in the liver and lungs, which we previously reported (156). Zhang et al. also reported that a patient with small bowel adenocarcinoma received nivolumab plus regorafenib after chemotherapy and targeted therapy failed, and the symptoms were significantly relieved, and the disease was stable for 15 months (157). In addition, Yong et al. reported that a metastatic rectal adenocarcinoma patient also experienced a complete response after treatment with regorafenib and sintilimab (158). However, the combination therapy also confronts some obstacles. Future research should investigate how to obtain precise treatment, identify specific and extremely sensitive biomarkers, and choose the most suited individuals for combination therapy. A recent study has revealed possible biomarkers for predicting OS in regorafenib-treated patients by analyzing proteins and microRNAs in baseline plasma samples from patients with different responses. Some proteins and microRNAs, such as ANG-1 and MIR122, can be utilized as potential markers to predict the OS of cancer patients (159). Another study demonstrated that a signature composed of five biomarkers (HIF1A, CDKN1A, miR-3607-3p, miR-301a-3p, and miR-93-5p) in glioblastoma patients with regorafenib treatment can be used to identify patients with a survival advantage (160). These biomarkers might effectively predict the response of cancer patients to regorafenib. However, due to the heterogeneity of tumors, the expression of certain proteins may not accurately predict the action of medications on the entire tumor, which will increase the difficulty of finding suitable biomarkers. In any event, the discovery of reliable biomarkers for predicting the efficacy of combination therapy remains the path of future research, which will allow us to identify the most suitable patients for combination therapy.
6 Conclusions
This review outlined the underlying mechanism of resistance to PD-1/PD-L1 blockade and the immunomodulatory effect of regorafenib, providing a theoretical basis for regorafenib combined with anti-PD-1/PD-L1 therapy. Regorafenib was shown to interact with NK cells, TAMs, cytotoxic T cells, Tregs, and CAFs in the TME. Additionally, regorafenib affects PD-L1 and MHC-I on tumor cells and normalizes abnormal blood vessels. From traditional chemotherapy to molecular targeted therapy and combination immunotherapy, the area of anticancer therapy has witnessed several significant developments. There are ever more therapies available to alleviate the patient’s symptoms and improve their prognosis. Regorafenib in combination with anti-PD-1/PD-L1 therapy has shown excellent antitumor efficacy and tolerable safety in the treatment of a variety of malignancies. The study of the synergetic mechanism of combination therapy and the identification of possible biomarkers are worthwhile future research paths that will help us identify the most suitable individual patients for more precise treatment.
Author contributions
All authors contributed to the article and approved the submitted version of the manuscript.
Funding
This project was supported by Hubei provincial special grants for scientific and technical innovation (No.2021BCA115) and the National Natural Science Foundation of China (No. 81902839).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Carlino MS, Larkin J, Long GV. Immune checkpoint inhibitors in melanoma. Lancet (London England) (2021) 398(10304):1002–14. doi: 10.1016/s0140-6736(21)01206-x
2. El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (Checkmate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet (London England) (2017) 389(10088):2492–502. doi: 10.1016/s0140-6736(17)31046-2
3. Apolo AB, Infante JR, Balmanoukian A, Patel MR, Wang D, Kelly K, et al. Avelumab, an anti-programmed death-ligand 1 antibody, in patients with refractory metastatic urothelial carcinoma: Results from a multicenter, phase ib study. J Clin Oncol (2017) 35(19):2117–24. doi: 10.1200/jco.2016.71.6795
4. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell (2017) 168(4):707–23. doi: 10.1016/j.cell.2017.01.017
5. Arai H, Battaglin F, Wang J, Lo JH, Soni S, Zhang W, et al. Molecular insight of regorafenib treatment for colorectal cancer. Cancer Treat Rev (2019) 81:101912. doi: 10.1016/j.ctrv.2019.101912
6. Blay JY, Kang YK, Nishida T, von Mehren M. Gastrointestinal stromal tumours. Nat Rev Dis Primers (2021) 7(1):22. doi: 10.1038/s41572-021-00254-5
7. Llovet JM, Montal R, Sia D, Finn RS. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat Rev Clin Oncol (2018) 15(10):599–616. doi: 10.1038/s41571-018-0073-4
8. Wilhelm SM, Dumas J, Adnane L, Lynch M, Carter CA, Schütz G, et al. Regorafenib (Bay 73-4506): A new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer (2011) 129(1):245–55. doi: 10.1002/ijc.25864
9. Kim RD, Kovari BP, Martinez M, Xie H, Sahin IH, Mehta R, et al. A phase I/Ib study of regorafenib and nivolumab in mismatch repair proficient advanced refractory colorectal cancer. Eur J Cancer (Oxford Engl 1990) (2022) 169:93–102. doi: 10.1016/j.ejca.2022.03.026
10. Okazaki T, Honjo T. Pd-1 and pd-1 ligands: From discovery to clinical application. Int Immunol (2007) 19(7):813–24. doi: 10.1093/intimm/dxm057
11. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of pd-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J (1992) 11(11):3887–95. doi: 10.1002/j.1460-2075.1992.tb05481.x
12. Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, et al. Expression of the pd-1 antigen on the surface of stimulated mouse T and b lymphocytes. Int Immunol (1996) 8(5):765–72. doi: 10.1093/intimm/8.5.765
13. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the pd-1 gene encoding an itim motif-carrying immunoreceptor. Immunity (1999) 11(2):141–51. doi: 10.1016/s1074-7613(00)80089-8
14. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted Cd8 T cells during chronic viral infection. Nature (2006) 439(7077):682–7. doi: 10.1038/nature04444
15. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the pd-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med (2000) 192(7):1027–34. doi: 10.1084/jem.192.7.1027
16. Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol (2007) 8(3):239–45. doi: 10.1038/ni1443
17. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. Ctla-4 and pd-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol (2005) 25(21):9543–53. doi: 10.1128/mcb.25.21.9543-9553.2005
18. Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, et al. Pd-1 inhibits T-cell receptor induced phosphorylation of the Zap70/Cd3zeta signalosome and downstream signaling to pkctheta. FEBS Lett (2004) 574(1-3):37–41. doi: 10.1016/j.febslet.2004.07.083
19. Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. Pd-1 immunoreceptor inhibits b cell receptor-mediated signaling by recruiting src homology 2-Domain-Containing tyrosine phosphatase 2 to phosphotyrosine. Proc Natl Acad Sci United States America (2001) 98(24):13866–71. doi: 10.1073/pnas.231486598
20. Ohaegbulam KC, Assal A, Lazar-Molnar E, Yao Y, Zang X. Human cancer immunotherapy with antibodies to the pd-1 and pd-L1 pathway. Trends Mol Med (2015) 21(1):24–33. doi: 10.1016/j.molmed.2014.10.009
21. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of pd-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by pd-L1 blockade. Proc Natl Acad Sci United States America (2002) 99(19):12293–7. doi: 10.1073/pnas.192461099
22. Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, et al. Blockade of B7-H1 and pd-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res (2005) 65(3):1089–96. doi: 10.1158/0008-5472.1089.65.3
23. Yi M, Zheng X, Niu M, Zhu S, Ge H, Wu K. Combination strategies with pd-1/Pd-L1 blockade: Current advances and future directions. Mol Cancer (2022) 21(1):28. doi: 10.1186/s12943-021-01489-2
24. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-Pd-1 antibody in cancer. N Engl J Med (2012) 366(26):2443–54. doi: 10.1056/NEJMoa1200690
25. Nowicki TS, Hu-Lieskovan S, Ribas A. Mechanisms of resistance to pd-1 and pd-L1 blockade. Cancer J (2018) 24(1):47–53. doi: 10.1097/ppo.0000000000000303
26. Lei Q, Wang D, Sun K, Wang L, Zhang Y. Resistance mechanisms of anti-Pd1/Pdl1 therapy in solid tumors. Front Cell Dev Biol (2020) 8:672. doi: 10.3389/fcell.2020.00672
27. Peng M, Mo Y, Wang Y, Wu P, Zhang Y, Xiong F, et al. Neoantigen vaccine: An emerging tumor immunotherapy. Mol Cancer (2019) 18(1):128. doi: 10.1186/s12943-019-1055-6
28. Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet (2019) 51(2):202–6. doi: 10.1038/s41588-018-0312-8
29. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to pd-1 blockade. Sci (New York NY) (2017) 357(6349):409–13. doi: 10.1126/science.aan6733
30. Yeon Yeon S, Jung SH, Jo YS, Choi EJ, Kim MS, Chung YJ, et al. Immune checkpoint blockade resistance-related B2m hotspot mutations in microsatellite-unstable colorectal carcinoma. Pathology Res Pract (2019) 215(1):209–14. doi: 10.1016/j.prp.2018.11.014
31. Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, et al. Impaired hla class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discovery (2017) 7(12):1420–35. doi: 10.1158/2159-8290.Cd-17-0593
32. Wolf Y, Bartok O, Patkar S, Eli GB, Cohen S, Litchfield K, et al. Uvb-induced tumor heterogeneity diminishes immune response in melanoma. Cell (2019) 179(1):219–35.e21. doi: 10.1016/j.cell.2019.08.032
33. O'Donnell JS, Long GV, Scolyer RA, Teng MW, Smyth MJ. Resistance to Pd1/Pdl1 checkpoint inhibition. Cancer Treat Rev (2017) 52:71–81. doi: 10.1016/j.ctrv.2016.11.007
34. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic Β-catenin signalling prevents anti-tumour immunity. Nature (2015) 523(7559):231–5. doi: 10.1038/nature14404
35. Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res (2017) 27(1):109–18. doi: 10.1038/cr.2016.151
36. Curran MA, Montalvo W, Yagita H, Allison JP. Pd-1 and ctla-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci United States America (2010) 107(9):4275–80. doi: 10.1073/pnas.0915174107
37. Li K, Shi H, Zhang B, Ou X, Ma Q, Chen Y, et al. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal transduction targeted Ther (2021) 6(1):362. doi: 10.1038/s41392-021-00670-9
38. Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, et al. Disruption of Cxcr2-mediated mdsc tumor trafficking enhances anti-Pd1 efficacy. Sci Trans Med (2014) 6(237):237ra67. doi: 10.1126/scitranslmed.3007974
39. Maller O, Drain AP, Barrett AS, Borgquist S, Ruffell B, Zakharevich I, et al. Tumour-associated macrophages drive stromal cell-dependent collagen crosslinking and stiffening to promote breast cancer aggression. Nat materials (2021) 20(4):548–59. doi: 10.1038/s41563-020-00849-5
40. Yang H, Zhang Q, Xu M, Wang L, Chen X, Feng Y, et al. Ccl2-Ccr2 axis recruits tumor associated macrophages to induce immune evasion through pd-1 signaling in esophageal carcinogenesis. Mol Cancer (2020) 19(1):41. doi: 10.1186/s12943-020-01165-x
41. Hofbauer D, Mougiakakos D, Broggini L, Zaiss M, Büttner-Herold M, Bach C, et al. Β(2)-microglobulin triggers Nlrp3 inflammasome activation in tumor-associated macrophages to promote multiple myeloma progression. Immunity (2021) 54(8):1772–87.e9. doi: 10.1016/j.immuni.2021.07.002
42. Cassetta L, Pollard JW. Targeting macrophages: Therapeutic approaches in cancer. Nat Rev Drug Discovery (2018) 17(12):887–904. doi: 10.1038/nrd.2018.169
43. Smith AL, Robin TP, Ford HL. Molecular pathways: Targeting the tgf-Β pathway for cancer therapy. Clin Cancer Res (2012) 18(17):4514–21. doi: 10.1158/1078-0432.Ccr-11-3224
44. Principe DR, DeCant B, Mascariñas E, Wayne EA, Diaz AM, Akagi N, et al. TgfΒ signaling in the pancreatic tumor microenvironment promotes fibrosis and immune evasion to facilitate tumorigenesis. Cancer Res (2016) 76(9):2525–39. doi: 10.1158/0008-5472.Can-15-1293
45. Oh SA, Liu M, Nixon BG, Kang D, Toure A, Bivona M, et al. Foxp3-independent mechanism by which tgf-Β controls peripheral T cell tolerance. Proc Natl Acad Sci United States America (2017) 114(36):E7536–e44. doi: 10.1073/pnas.1706356114
46. Perrot I, Michaud HA, Giraudon-Paoli M, Augier S, Docquier A, Gros L, et al. Blocking antibodies targeting the Cd39/Cd73 immunosuppressive pathway unleash immune responses in combination cancer therapies. Cell Rep (2019) 27(8):2411–25.e9. doi: 10.1016/j.celrep.2019.04.091
47. Young A, Mittal D, Stagg J, Smyth MJ. Targeting cancer-derived adenosine: New therapeutic approaches. Cancer Discovery (2014) 4(8):879–88. doi: 10.1158/2159-8290.Cd-14-0341
48. Hay CM, Sult E, Huang Q, Mulgrew K, Fuhrmann SR, McGlinchey KA, et al. Targeting Cd73 in the tumor microenvironment with Medi9447. Oncoimmunology (2016) 5(8):e1208875. doi: 10.1080/2162402x.2016.1208875
49. Wada J, Suzuki H, Fuchino R, Yamasaki A, Nagai S, Yanai K, et al. The contribution of vascular endothelial growth factor to the induction of regulatory T-cells in malignant effusions. Anticancer Res (2009) 29(3):881–8. doi: 10.1103/PhysRevLett.94.028701
50. Munn DH, Mellor AL. Ido in the tumor microenvironment: Inflammation, counter-regulation, and tolerance. Trends Immunol (2016) 37(3):193–207. doi: 10.1016/j.it.2016.01.002
51. Meraz MA, White JM, Sheehan KC, Bach EA, Rodig SJ, Dighe AS, et al. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the jak-stat signaling pathway. Cell (1996) 84(3):431–42. doi: 10.1016/s0092-8674(00)81288-x
52. Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: An overview of signals, mechanisms and functions. J leukocyte Biol (2004) 75(2):163–89. doi: 10.1189/jlb.0603252
53. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat Med (2002) 8(8):793–800. doi: 10.1038/nm730
54. Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary resistance to pd-1 blockade mediated by Jak1/2 mutations. Cancer Discovery (2017) 7(2):188–201. doi: 10.1158/2159-8290.Cd-16-1223
55. Sucker A, Zhao F, Pieper N, Heeke C, Maltaner R, Stadtler N, et al. Acquired ifnγ resistance impairs anti-tumor immunity and gives rise to T-Cell-Resistant melanoma lesions. Nat Commun (2017) 8:15440. doi: 10.1038/ncomms15440
56. Buchbinder E, Hodi FS. Cytotoxic T lymphocyte antigen-4 and immune checkpoint blockade. J Clin Invest (2015) 125(9):3377–83. doi: 10.1172/jci80012
57. Maruhashi T, Sugiura D, Okazaki IM, Okazaki T. Lag-3: From molecular functions to clinical applications. J immunotherapy Cancer (2020) 8(2):e001014. doi: 10.1136/jitc-2020-001014
58. Liu F, Liu Y, Chen Z. Tim-3 expression and its role in hepatocellular carcinoma. J Hematol Oncol (2018) 11(1):126. doi: 10.1186/s13045-018-0667-4
59. Ning Z, Liu K, Xiong H. Roles of btla in immunity and immune disorders. Front Immunol (2021) 12:654960. doi: 10.3389/fimmu.2021.654960
60. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-Small-Cell lung cancer. N Engl J Med (2015) 372(21):2018–28. doi: 10.1056/NEJMoa1501824
61. Theodoraki MN, Yerneni SS, Hoffmann TK, Gooding WE, Whiteside TL. Clinical significance of pd-L1(+) exosomes in plasma of head and neck cancer patients. Clin Cancer Res (2018) 24(4):896–905. doi: 10.1158/1078-0432.Ccr-17-2664
62. Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, et al. Exosomal pd-L1 contributes to immunosuppression and is associated with anti-Pd-1 response. Nature (2018) 560(7718):382–6. doi: 10.1038/s41586-018-0392-8
63. Restifo NP, Smyth MJ, Snyder A. Acquired resistance to immunotherapy and future challenges. Nat Rev Cancer (2016) 16(2):121–6. doi: 10.1038/nrc.2016.2
64. Huang XY, Zhang PF, Wei CY, Peng R, Lu JC, Gao C, et al. Circular RNA circmet drives immunosuppression and anti-Pd1 therapy resistance in hepatocellular carcinoma Via the mir-30-5p/Snail/Dpp4 axis. Mol Cancer (2020) 19(1):92. doi: 10.1186/s12943-020-01213-6
65. Hong W, Xue M, Jiang J, Zhang Y, Gao X. Circular RNA circ-Cpa4/ let-7 Mirna/Pd-L1 axis regulates cell growth, stemness, drug resistance and immune evasion in non-small cell lung cancer (Nsclc). J Exp Clin Cancer Res CR (2020) 39(1):149. doi: 10.1186/s13046-020-01648-1
66. Zhang H, Zhu C, He Z, Chen S, Li L, Sun C. Lncrna Psmb8-As1 contributes to pancreatic cancer progression Via modulating mir-382-3p/Stat1/Pd-L1 axis. J Exp Clin Cancer Res CR (2020) 39(1):179. doi: 10.1186/s13046-020-01687-8
67. Zhao L, Liu Y, Zhang J, Liu Y, Qi Q. Lncrna Snhg14/Mir-5590-3p/Zeb1 positive feedback loop promoted diffuse Large b cell lymphoma progression and immune evasion through regulating pd-1/Pd-L1 checkpoint. Cell Death Dis (2019) 10(10):731. doi: 10.1038/s41419-019-1886-5
68. Grimaldi A, Cammarata I, Martire C, Focaccetti C, Piconese S, Buccilli M, et al. Combination of chemotherapy and pd-1 blockade induces T cell responses to tumor non-mutated neoantigens. Commun Biol (2020) 3(1):85. doi: 10.1038/s42003-020-0811-x
69. Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature (2015) 520(7547):373–7. doi: 10.1038/nature14292
70. Rassy E, Flippot R, Albiges L. Tyrosine kinase inhibitors and immunotherapy combinations in renal cell carcinoma. Ther Adv Med Oncol (2020) 12:1758835920907504. doi: 10.1177/1758835920907504
71. Chon HJ, Lee WS, Yang H, Kong SJ, Lee NK, Moon ES, et al. Tumor microenvironment remodeling by intratumoral oncolytic vaccinia virus enhances the efficacy of immune-checkpoint blockade. Clin Cancer Res (2019) 25(5):1612–23. doi: 10.1158/1078-0432.Ccr-18-1932
72. Letourneur D, Danlos FX, Marabelle A. Chemokine biology on immune checkpoint-targeted therapies. Eur J Cancer (Oxford Engl 1990) (2020) 137:260–71. doi: 10.1016/j.ejca.2020.06.009
73. Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci United States America (2012) 109(43):17561–6. doi: 10.1073/pnas.1215397109
74. Marin-Acevedo JA, Dholaria B, Soyano AE, Knutson KL, Chumsri S, Lou Y. Next generation of immune checkpoint therapy in cancer: New developments and challenges. J Hematol Oncol (2018) 11(1):39. doi: 10.1186/s13045-018-0582-8
75. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. Csf1/Csf1r blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res (2014) 74(18):5057–69. doi: 10.1158/0008-5472.Can-13-3723
76. Martin CJ, Datta A, Littlefield C, Kalra A, Chapron C, Wawersik S, et al. Selective inhibition of TgfΒ1 activation overcomes primary resistance to checkpoint blockade therapy by altering tumor immune landscape. Sci Trans Med (2020) 12(536). :eaay8456doi: 10.1126/scitranslmed.aay8456
77. Wu T, Dai Y. Tumor microenvironment and therapeutic response. Cancer Lett (2017) 387:61–8. doi: 10.1016/j.canlet.2016.01.043
78. Xiao Y, Yu D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol Ther (2021) 221:107753. doi: 10.1016/j.pharmthera.2020.107753
79. Zhu S, Yi M, Wu Y, Dong B, Wu K. Roles of tumor-associated macrophages in tumor progression: Implications on therapeutic strategies. Exp Hematol Oncol (2021) 10(1):60. doi: 10.1186/s40164-021-00252-z
80. Shojaei F, Wu X, Malik AK, Zhong C, Baldwin ME, Schanz S, et al. Tumor refractoriness to anti-vegf treatment is mediated by Cd11b+Gr1+ myeloid cells. Nat Biotechnol (2007) 25(8):911–20. doi: 10.1038/nbt1323
81. Glasner A, Plitas G. Tumor resident regulatory T cells. Semin Immunol (2021) 52:101476. doi: 10.1016/j.smim.2021.101476
82. Shaul ME, Fridlender ZG. Tumour-associated neutrophils in patients with cancer. Nat Rev Clin Oncol (2019) 16(10):601–20. doi: 10.1038/s41571-019-0222-4
83. Jenkins BH, Buckingham JF, Hanley CJ, Thomas GJ. Targeting cancer-associated fibroblasts: Challenges, opportunities and future directions. Pharmacol Ther (2022) 240:108231. doi: 10.1016/j.pharmthera.2022.108231
84. De Sanctis F, Ugel S, Facciponte J, Facciabene A. The dark side of tumor-associated endothelial cells. Semin Immunol (2018) 35:35–47. doi: 10.1016/j.smim.2018.02.002
85. Locy H, de Mey S, de Mey W, De Ridder M, Thielemans K, Maenhout SK. Immunomodulation of the tumor microenvironment: Turn foe into friend. Front Immunol (2018) 9:2909. doi: 10.3389/fimmu.2018.02909
86. Grothey A, Blay JY, Pavlakis N, Yoshino T, Bruix J. Evolving role of regorafenib for the treatment of advanced cancers. Cancer Treat Rev (2020) 86:101993. doi: 10.1016/j.ctrv.2020.101993
87. Wu SY, Fu T, Jiang YZ, Shao ZM. Natural killer cells in cancer biology and therapy. Mol Cancer (2020) 19(1):120. doi: 10.1186/s12943-020-01238-x
88. Zwirner NW, Fuertes MB, Girart MV, Domaica CI, Rossi LE. Cytokine-driven regulation of nk cell functions in tumor immunity: Role of the mica-Nkg2d system. Cytokine Growth factor Rev (2007) 18(1-2):159–70. doi: 10.1016/j.cytogfr.2007.01.013
89. Salih HR, Rammensee HG, Steinle A. Cutting edge: Down-regulation of mica on human tumors by proteolytic shedding. J Immunol (Baltimore Md 1950) (2002) 169(8):4098–102. doi: 10.4049/jimmunol.169.8.4098
90. Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble mic ligands impair expression of Nkg2d and T-cell activation. Nature (2002) 419(6908):734–8. doi: 10.1038/nature01112
91. Chen D, Gyllensten U. Mica polymorphism: Biology and importance in cancer. Carcinogenesis (2014) 35(12):2633–42. doi: 10.1093/carcin/bgu215
92. Waldhauer I, Goehlsdorf D, Gieseke F, Weinschenk T, Wittenbrink M, Ludwig A, et al. Tumor-associated mica is shed by Adam proteases. Cancer Res (2008) 68(15):6368–76. doi: 10.1158/0008-5472.Can-07-6768
93. Arai J, Goto K, Stephanou A, Tanoue Y, Ito S, Muroyama R, et al. Predominance of regorafenib over sorafenib: Restoration of membrane-bound mica in hepatocellular carcinoma cells. J Gastroenterol Hepatol (2018) 33(5):1075–81. doi: 10.1111/jgh.14029
94. Yang XR, Xu Y, Yu B, Zhou J, Li JC, Qiu SJ, et al. Cd24 is a novel predictor for poor prognosis of hepatocellular carcinoma after surgery. Clin Cancer Res (2009) 15(17):5518–27. doi: 10.1158/1078-0432.Ccr-09-0151
95. Yoshida M, Zhao L, Grigoryan G, Shim H, He P, Yun CC. Deletion of Na+/H+ exchanger regulatory factor 2 represses colon cancer progress by suppression of Stat3 and Cd24. Am J Physiol Gastrointestinal liver Physiol (2016) 310(8):G586–98. doi: 10.1152/ajpgi.00419.2015
96. Lee TK, Castilho A, Cheung VC, Tang KH, Ma S, Ng IO. Cd24(+) liver tumor-initiating cells drive self-renewal and tumor initiation through Stat3-mediated nanog regulation. Cell Stem Cell (2011) 9(1):50–63. doi: 10.1016/j.stem.2011.06.005
97. Hüser L, Altevogt P, Utikal J. Role of Stat3 dependent Sox2 and Cd24 expression in melanoma cell adaptive resistance towards targeted therapies. Oncotarget (2019) 10(18):1662–3. doi: 10.18632/oncotarget.26718
98. Tai WT, Chu PY, Shiau CW, Chen YL, Li YS, Hung MH, et al. Stat3 mediates regorafenib-induced apoptosis in hepatocellular carcinoma. Clin Cancer Res (2014) 20(22):5768–76. doi: 10.1158/1078-0432.Ccr-14-0725
99. Munday J, Kerr S, Ni J, Cornish AL, Zhang JQ, Nicoll G, et al. Identification, characterization and leucocyte expression of siglec-10, a novel human sialic acid-binding receptor. Biochem J (2001) 355(Pt 2):489–97. doi: 10.1042/0264-6021:3550489
100. Whitney G, Wang S, Chang H, Cheng KY, Lu P, Zhou XD, et al. A new siglec family member, siglec-10, is expressed in cells of the immune system and has signaling properties similar to Cd33. Eur J Biochem (2001) 268(23):6083–96. doi: 10.1046/j.0014-2956.2001.02543.x
101. Hoffmann A, Kerr S, Jellusova J, Zhang J, Weisel F, Wellmann U, et al. Siglec-G is a B1 cell-inhibitory receptor that controls expansion and calcium signaling of the B1 cell population. Nat Immunol (2007) 8(7):695–704. doi: 10.1038/ni1480
102. Zhang P, Lu X, Tao K, Shi L, Li W, Wang G, et al. Siglec-10 is associated with survival and natural killer cell dysfunction in hepatocellular carcinoma. J Surg Res (2015) 194(1):107–13. doi: 10.1016/j.jss.2014.09.035
103. Venneri MA, De Palma M, Ponzoni M, Pucci F, Scielzo C, Zonari E, et al. Identification of proangiogenic Tie2-expressing monocytes (Tems) in human peripheral blood and cancer. Blood (2007) 109(12):5276–85. doi: 10.1182/blood-2006-10-053504
104. Huang H, Lai JY, Do J, Liu D, Li L, Del Rosario J, et al. Specifically targeting angiopoietin-2 inhibits angiogenesis, Tie2-expressing monocyte infiltration, and tumor growth. Clin Cancer Res (2011) 17(5):1001–11. doi: 10.1158/1078-0432.Ccr-10-2317
105. Abou-Elkacem L, Arns S, Brix G, Gremse F, Zopf D, Kiessling F, et al. Regorafenib inhibits growth, angiogenesis, and metastasis in a highly aggressive, orthotopic colon cancer model. Mol Cancer Ther (2013) 12(7):1322–31. doi: 10.1158/1535-7163.Mct-12-1162
106. Zhao PF, Yin WM, Wu AH, Tang YS, Wang JY, Pan ZZ, et al. Dual-targeting to cancer cells and M2 macrophages Via biomimetic delivery of mannosylated albumin nanoparticles for drug-resistant cancer therapy. Advanced Funct Materials (2017) 27(44):1700403. doi: 10.1002/adfm.201700403
107. Zhao P, Wang Y, Kang X, Wu A, Yin W, Tang Y, et al. Dual-targeting biomimetic delivery for anti-glioma activity Via remodeling the tumor microenvironment and directing macrophage-mediated immunotherapy. Chem Sci (2018) 9(10):2674–89. doi: 10.1039/c7sc04853j
108. Doleschel D, Hoff S, Koletnik S, Rix A, Zopf D, Kiessling F, et al. Regorafenib enhances anti-Pd1 immunotherapy efficacy in murine colorectal cancers and their combination prevents tumor regrowth. J Exp Clin Cancer Res CR (2021) 40(1):288. doi: 10.1186/s13046-021-02043-0
109. Chiang IT, Lee YH, Tan ZL, Hsu FT, Tu HF. Regorafenib enhances antitumor immune efficacy of anti-Pd-L1 immunotherapy on oral squamous cell carcinoma. Biomedicine pharmacotherapy (2022) 147:112661. doi: 10.1016/j.biopha.2022.112661
110. Li Z, Xu W, Yang J, Wang J, Wang J, Zhu G, et al. A tumor microenvironments-adapted polypeptide Hydrogel/Nanogel composite boosts antitumor molecularly targeted inhibition and immunoactivation. Advanced materials (Deerfield Beach Fla) (2022) 34(21):e2200449. doi: 10.1002/adma.202200449
111. Bai H, Wang J, Phan CU, Chen Q, Hu X, Shao G, et al. Cyclodextrin-based host-guest complexes loaded with regorafenib for colorectal cancer treatment. Nat Commun (2021) 12(1):759. doi: 10.1038/s41467-021-21071-0
112. Martínez-Limón A, Joaquin M, Caballero M, Posas F, de Nadal E. The P38 pathway: From biology to cancer therapy. Int J Mol Sci (2020) 21(6):1913. doi: 10.3390/ijms21061913
113. Ou DL, Chen CW, Hsu CL, Chung CH, Feng ZR, Lee BS, et al. Regorafenib enhances antitumor immunity Via inhibition of P38 Kinase/Creb1/Klf4 axis in tumor-associated macrophages. J immunotherapy Cancer (2021) 9(3):e001657. doi: 10.1136/jitc-2020-001657
114. Hoff S, Grünewald S, Röse L, Zopf D. 1198p - immunomodulation by regorafenib alone and in combination with anti Pd1 antibody on murine models of colorectal cancer. Ann Oncol (2017) 28(5):v423. doi: 10.1093/annonc/mdx376.060
115. Shigeta K, Matsui A, Kikuchi H, Klein S, Mamessier E, Chen IX, et al. Regorafenib combined with Pd1 blockade increases Cd8 T-cell infiltration by inducing Cxcl10 expression in hepatocellular carcinoma. J immunotherapy Cancer (2020) 8(2):e001435. doi: 10.1136/jitc-2020-001435
116. Wu RY, Kong PF, Xia LP, Huang Y, Li ZL, Tang YY, et al. Regorafenib promotes antitumor immunity Via inhibiting pd-L1 and Ido1 expression in melanoma. Clin Cancer Res (2019) 25(14):4530–41. doi: 10.1158/1078-0432.Ccr-18-2840
117. Granito A, Muratori L, Lalanne C, Quarneti C, Ferri S, Guidi M, et al. Hepatocellular carcinoma in viral and autoimmune liver diseases: Role of Cd4+ Cd25+ Foxp3+ regulatory T cells in the immune microenvironment. World J Gastroenterol (2021) 27(22):2994–3009. doi: 10.3748/wjg.v27.i22.2994
118. Zhou W, Deng J, Chen Q, Li R, Xu X, Guan Y, et al. Expression of Cd4+Cd25+Cd127(Low) regulatory T cells and cytokines in peripheral blood of patients with primary liver carcinoma. Int J Med Sci (2020) 17(6):712–9. doi: 10.7150/ijms.44088
119. Kobayashi N, Hiraoka N, Yamagami W, Ojima H, Kanai Y, Kosuge T, et al. Foxp3+ regulatory T cells affect the development and progression of hepatocarcinogenesis. Clinical cancer research : an official journal of the American Association for Cancer Research (2007) 13(3):902–11. doi: 10.1158/1078-0432.Ccr-06-2363
120. Fu J, Xu D, Liu Z, Shi M, Zhao P, Fu B, et al. Increased regulatory T cells correlate with Cd8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology (2007) 132(7):2328–39. doi: 10.1053/j.gastro.2007.03.102
121. Tu JF, Ding YH, Ying XH, Wu FZ, Zhou XM, Zhang DK, et al. Regulatory T cells, especially icos(+) Foxp3(+) regulatory T cells, are increased in the hepatocellular carcinoma microenvironment and predict reduced survival. Sci Rep (2016) 6:35056. doi: 10.1038/srep35056
122. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol (2008) 8(7):523–32. doi: 10.1038/nri2343
123. Yi Y, He HW, Wang JX, Cai XY, Li YW, Zhou J, et al. The functional impairment of hcc-infiltrating Γδ T cells, partially mediated by regulatory T cells in a tgfΒ- and il-10-Dependent manner. J Hepatol (2013) 58(5):977–83. doi: 10.1016/j.jhep.2012.12.015
124. Kos K, Salvagno C, Wellenstein MD, Aslam MA, Meijer DA, Hau CS, et al. Tumor-associated macrophages promote intratumoral conversion of conventional Cd4(+) T cells into regulatory T cells Via pd-1 signalling. Oncoimmunology (2022) 11(1):2063225. doi: 10.1080/2162402x.2022.2063225
125. Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell (2018) 33(3):463–79.e10. doi: 10.1016/j.ccell.2018.01.011
126. Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Krüger C, Manns MP, et al. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces Cd4(+)Cd25(+)Foxp3(+) T cells. Gastroenterology (2008) 135(1):234–43. doi: 10.1053/j.gastro.2008.03.020
127. Chandra Jena B, Sarkar S, Rout L, Mandal M. The transformation of cancer-associated fibroblasts: Current perspectives on the role of tgf-Β in caf mediated tumor progression and therapeutic resistance. Cancer Lett (2021) 520:222–32. doi: 10.1016/j.canlet.2021.08.002
128. Su S, Chen J, Yao H, Liu J, Yu S, Lao L, et al. Cd10(+)Gpr77(+) cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell (2018) 172(4):841–56.e16. doi: 10.1016/j.cell.2018.01.009
129. Li Z, Pang Y, Gara SK, Achyut BR, Heger C, Goldsmith PK, et al. Gr-1+Cd11b+ cells are responsible for tumor promoting effect of tgf-Β in breast cancer progression. Int J Cancer (2012) 131(11):2584–95. doi: 10.1002/ijc.27572
130. Markiewski MM, DeAngelis RA, Benencia F, Ricklin-Lichtsteiner SK, Koutoulaki A, Gerard C, et al. Modulation of the antitumor immune response by complement. Nat Immunol (2008) 9(11):1225–35. doi: 10.1038/ni.1655
131. Zhang M, Li X, Wu W, Gao J, Han Q, Sun Z, et al. Regorafenib induces the apoptosis of gastrointestinal cancer-associated fibroblasts by inhibiting akt phosphorylation. Stem Cells Dev (2022) 31(13-14):383-94. doi: 10.1089/scd.2022.0088
132. Takigawa H, Kitadai Y, Shinagawa K, Yuge R, Higashi Y, Tanaka S, et al. Multikinase inhibitor regorafenib inhibits the growth and metastasis of colon cancer with abundant stroma. Cancer Sci (2016) 107(5):601–8. doi: 10.1111/cas.12907
133. Takahashi A, Umemura A, Yano K, Okishio S, Kataoka S, Okuda K, et al. Tyrosine kinase inhibitors stimulate hla class I expression by augmenting the Ifnγ/Stat1 signaling in hepatocellular carcinoma cells. Front Oncol (2021) 11:707473. doi: 10.3389/fonc.2021.707473
134. Fukumura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat Rev Clin Oncol (2018) 15(5):325–40. doi: 10.1038/nrclinonc.2018.29
135. Huang Y, Kim BYS, Chan CK, Hahn SM, Weissman IL, Jiang W. Improving immune-vascular crosstalk for cancer immunotherapy. Nat Rev Immunol (2018) 18(3):195–203. doi: 10.1038/nri.2017.145
136. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, et al. Tumour hypoxia promotes tolerance and angiogenesis Via Ccl28 and T(Reg) cells. Nature (2011) 475(7355):226–30. doi: 10.1038/nature10169
137. Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, et al. Hypoxia-inducible factors enhance the effector responses of Cd8(+) T cells to persistent antigen. Nat Immunol (2013) 14(11):1173–82. doi: 10.1038/ni.2714
138. Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res (2014) 74(3):665–74. doi: 10.1158/0008-5472.Can-13-0992
139. Su JC, Mar AC, Wu SH, Tai WT, Chu PY, Wu CY, et al. Disrupting vegf-a paracrine and autocrine loops by targeting shp-1 suppresses triple negative breast cancer metastasis. Sci Rep (2016) 6:28888. doi: 10.1038/srep28888
140. Huynh H, Ong R, Zopf D. Antitumor activity of the multikinase inhibitor regorafenib in patient-derived xenograft models of gastric cancer. J Exp Clin Cancer Res CR (2015) 34:132. doi: 10.1186/s13046-015-0243-5
141. Todesca P, Marzi L, Critelli RM, Cuffari B, Caporali C, Turco L, et al. Angiopoietin-2/Tie2 inhibition by regorafenib associates with striking response in a patient with aggressive hepatocellular carcinoma. Hepatol (Baltimore Md) (2019) 70(2):745–7. doi: 10.1002/hep.30588
142. Jain RK. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Sci (New York NY) (2005) 307(5706):58–62. doi: 10.1126/science.1104819
143. Pàez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Viñals F, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell (2009) 15(3):220–31. doi: 10.1016/j.ccr.2009.01.027
144. Fukuoka S, Hara H, Takahashi N, Kojima T, Kawazoe A, Asayama M, et al. Regorafenib plus nivolumab in patients with advanced gastric or colorectal cancer: An open-label, dose-escalation, and dose-expansion phase ib trial (Regonivo, Epoc1603). J Clin Oncol (2020) 38(18):2053–61. doi: 10.1200/jco.19.03296
145. Fakih M, Raghav KPS, Chang DZ, Bendell JC, Larson T, Cohn AL, et al. Single-arm, phase 2 study of regorafenib plus nivolumab in patients with mismatch repair-proficient (Pmmr)/Microsatellite stable (Mss) colorectal cancer (Crc). J Clin Oncol (2021) 39(15):2728–35. doi: 10.1200/JCO.2021.39.15_suppl.3560
146. Barzi A, Azad NS, Yang Y, Tsao-Wei D, Rehman R, Fakih M, et al. Phase I/Ii study of regorafenib (Rego) and pembrolizumab (Pembro) in refractory microsatellite stable colorectal cancer (Msscrc). J Clin Oncol (2022) 40(4):510–9. doi: 10.1200/JCO.2022.40.4_suppl.015
147. Wang F, He MM, Yao YC, Zhao X, Wang ZQ, Jin Y, et al. Regorafenib plus toripalimab in patients with metastatic colorectal cancer: A phase Ib/Ii clinical trial and gut microbiome analysis. Cell Rep Med (2021) 2(9):100383. doi: 10.1016/j.xcrm.2021.100383
148. Cousin S, Cantarel C, Guegan JP, Mazard T, Gomez-Roca C, Metges JP, et al. Regorafenib-avelumab combination in patients with biliary tract cancer (Regomune): A single-arm, open-label, phase ii trial. Eur J Cancer (Oxford Engl 1990) (2022) 162:161–9. doi: 10.1016/j.ejca.2021.11.012
149. Cousin S, Cantarel C, Guegan JP, Gomez-Roca C, Metges JP, Adenis A, et al. Regorafenib-avelumab combination in patients with microsatellite stable colorectal cancer (Regomune): A single-arm, open-label, phase ii trial. Clin Cancer Res (2021) 27(8):2139–47. doi: 10.1158/1078-0432.Ccr-20-3416
150. Pavlakis N, Sjoquist KM, Martin AJ, Tsobanis E, Yip S, Kang YK, et al. Regorafenib for the treatment of advanced gastric cancer (Integrate): A multinational placebo-controlled phase ii trial. J Clin Oncol (2016) 34(23):2728–35. doi: 10.1200/jco.2015.65.1901
151. Chen LT, Satoh T, Ryu MH, Chao Y, Kato K, Chung HC, et al. A phase 3 study of nivolumab in previously treated advanced gastric or gastroesophageal junction cancer (Attraction-2): 2-year update data. Gastric Cancer (2020) 23(3):510–9. doi: 10.1007/s10120-019-01034-7
152. Banales JM, Marin JJG, Lamarca A, Rodrigues PM, Khan SA, Roberts LR, et al. Cholangiocarcinoma 2020: The next horizon in mechanisms and management. Nat Rev Gastroenterol Hepatol (2020) 17(9):557–88. doi: 10.1038/s41575-020-0310-z
153. Piha-Paul SA, Oh DY, Ueno M, Malka D, Chung HC, Nagrial A, et al. Efficacy and safety of pembrolizumab for the treatment of advanced biliary cancer: Results from the keynote-158 and keynote-028 studies. Int J Cancer (2020) 147(8):2190–8. doi: 10.1002/ijc.33013
154. Huang J, Guo Y, Huang W, Hong X, Quan Y, Lin L, et al. Regorafenib combined with pd-1 blockade immunotherapy versus regorafenib as second-line treatment for advanced hepatocellular carcinoma: A multicenter retrospective study. J hepatocellular carcinoma (2022) 9:157–70. doi: 10.2147/jhc.S353956
155. El-Khoueiry AB, Kim RD, Harris WP, Sung MW, Waldschmidt D, Cabrera R, et al. Updated results of a phase 1b study of regorafenib (Reg) 80 Mg/Day or 120 Mg/Day plus pembrolizumab (Pembro) for first-line treatment of advanced hepatocellular carcinoma (Hcc). J Clin Oncol (2021) 39(15):4078. doi: 10.1200/JCO.2021.39.15_suppl.4078
156. Zhang EL, Zhang ZY, Li J, Huang ZY. Complete response to the sequential treatment with regorafenib followed by pd-1 inhibitor in a sorafenib-refractory hepatocellular carcinoma patient. OncoTargets Ther (2020) 13:12477–87. doi: 10.2147/ott.S284092
157. Zhang G, Lin L, Dong D, Qiu H, Liu T, Lian L, et al. Nivolumab plus regorafenib in patients with small bowel adenocarcinoma: A case report. Medicine (2021) 100(4):e24295. doi: 10.1097/md.0000000000024295
158. Zhang Y, Zhang F, Zhao L, Fu X, Shang Y, Gao Q. Long-term survival of a patient with microsatellite-stable refractory colorectal cancer with regorafenib and pd-1 inhibitor sintilimab: A case report and review of literature. BMC Gastroenterol (2021) 21(1):399. doi: 10.1186/s12876-021-01950-y
159. Teufel M, Seidel H, Köchert K, Meinhardt G, Finn RS, Llovet JM, et al. Biomarkers associated with response to regorafenib in patients with hepatocellular carcinoma. Gastroenterology (2019) 156(6):1731–41. doi: 10.1053/j.gastro.2019.01.261
Keywords: regorafenib, PD-1/PD-L1, immunotherapy, resistance, combination therapy, tumor microenvironment, immunomodulatory effects
Citation: Liu J, Tao H, Yuan T, Li J, Li J, Liang H, Huang Z and Zhang E (2022) Immunomodulatory effects of regorafenib: Enhancing the efficacy of anti-PD-1/PD-L1 therapy. Front. Immunol. 13:992611. doi: 10.3389/fimmu.2022.992611
Received: 12 July 2022; Accepted: 18 August 2022;
Published: 02 September 2022.
Edited by:
Simona Kranjc Brezar, Institute of Oncology Ljubljana, SloveniaCopyright © 2022 Liu, Tao, Yuan, Li, Li, Liang, Huang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Erlei Zhang, YmFpeXUxOTg2MTEwNEAxNjMuY29t; Zhiyong Huang, aHVhbmd6eUB0amgudGptdS5lZHUuY24=; Huifang Liang, bGlhbmdodWlmYW5nMTk5N0AxMjYuY29t
†These authors share first authorship