 Haijiao Jing
Haijiao Jing Xiaoming Wu1
Xiaoming Wu1 Jialan Shi
Jialan Shi- 1Department of Hematology, The First Hospital, Harbin Medical University, Harbin, China
- 2Department of Research, VA Boston Healthcare System, Harvard Medical School, Boston, MA, United States
- 3Department of Medical Oncology, Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA, United States
COVID-19 patients have a high incidence of thrombosis, and thromboembolic complications are associated with severe COVID-19 and high mortality. COVID-19 disease is associated with a hyper-inflammatory response (cytokine storm) mediated by the immune system. However, the role of the inflammatory response in thrombosis remains incompletely understood. In this review, we investigate the crosstalk between inflammation and thrombosis in the context of COVID-19, focusing on the contributions of inflammation to the pathogenesis of thrombosis, and propose combined use of anti-inflammatory and anticoagulant therapeutics. Under inflammatory conditions, the interactions between neutrophils and platelets, platelet activation, monocyte tissue factor expression, microparticle release, and phosphatidylserine (PS) externalization as well as complement activation are collectively involved in immune-thrombosis. Inflammation results in the activation and apoptosis of blood cells, leading to microparticle release and PS externalization on blood cells and microparticles, which significantly enhances the catalytic efficiency of the tenase and prothrombinase complexes, and promotes thrombin-mediated fibrin generation and local blood clot formation. Given the risk of thrombosis in the COVID-19, the importance of antithrombotic therapies has been generally recognized, but certain deficiencies and treatment gaps in remain. Antiplatelet drugs are not in combination with anticoagulant treatments, thus fail to dampen platelet procoagulant activity. Current treatments also do not propose an optimal time for anticoagulation. The efficacy of anticoagulant treatments depends on the time of therapy initiation. The best time for antithrombotic therapy is as early as possible after diagnosis, ideally in the early stage of the disease. We also elaborate on the possible mechanisms of long COVID thromboembolic complications, including persistent inflammation, endothelial injury and dysfunction, and coagulation abnormalities. The above-mentioned contents provide therapeutic strategies for COVID-19 patients and further improve patient outcomes.
Introduction
Recent studies demonstrate that the incidences of venous thromboembolism (VTE) and pulmonary embolism in hospitalized patients with COVID-19 are 17% and 7.1%, respectively, while the incidence of major bleeding is only 3.9% (1, 2). Systematic reviews have shown a higher incidence of VTE at 28% in COVID-19 patients admitted to the ICU (3, 4). Autopsy studies on COVID-19 patients have reported thrombi composed of platelets and fibrin in pulmonary vascular (5, 6). COVID-19 patients exhibit unique coagulopathy, characterized by thrombus formation with elevated levels of cytokines, including tumor necrosis factor (TNF)-α, interleukin (IL)-8, and IL-6, among others (7). Inflammatory molecules and immune cells are considered the main drivers for thrombosis in COVID-19 patients. Transient increases of thrombotic risk induced by viruses (including SARS-CoV-2) indicates that subsequent host immune response is a major trigger of vascular thromboembolic events, rather than the infection itself, independent of the pathogen species (8). Even with effective antithrombotic drugs, thrombotic events cannot be completely prevented, which suggests the presence of other unresolved mechanisms that result in under-treatment, such as inflammation. The above evidence supports the hypothesis that COVID-19-associated thrombosis is immunothrombosis triggered by inflammation. Immunothrombosis is described as the immune system triggering coagulation to form thrombosis that prevents pathogen transmission, with immune cells (neutrophils, etc.), NETs, and platelets playing important roles in this process (9, 10). Molecular interactions between inflammation and coagulation have been gradually confirmed. Inflammation and the coagulation cascade form complex networks that work to counterbalance the body’s response to pathogen invasion and tissue damage, and maintain homeostasis (11). Inflammation is a defense mechanism triggered by the immune system in response to injury or infection and is characterized by increased local blood flow, leukocyte recruitment, and the release of cytokines and chemokines, which can promote pathogen clearance. Inflammation also induces increased expression of adhesion molecules on endothelium, the subsequent interactions between leukocytes and endothelial cells (ECs) can lead to immune cell activation, thereby forming immunothrombosis at the injured area (12–14). Immunothrombosis can further limit systemic transmission of pathogens via the bloodstream and is an emergent mechanism for the host to inhibit infection (15). However, in COVID-19, thrombosis is related to disease progression and is associated with increased mortality (16–18). Therefore, to improve prognosis, early prevention and treatment of thrombosis is critical for COVID-19 patients.
Mechanisms of inflammation in COVID-19
Severe COVID-19 is the result of intense interactions between SARS-CoV-2 and the host, causing progressive damage to tissues and organs (19). Spike (S) protein in the viral envelope exists as homotrimers, including the receptor-binding S1 subunit and the membrane fusion S2 subunit. The receptor-binding domain (RBD) of the virus S1 subunit binds to ACE2 on the epithelium of the nasopharynx. S1/S2 or S2 site hydrolysis by transmembrane protease serine 2 (TMPRSS2) is required for the activation of RBD, causing a confirmation change in the S protein which catalyzes virus-host membrane fusion (20, 21). This is followed by viral RNA replication, particle assembly and release (22). SARS-CoV-2 viral RNA is recognized by the innate immune cells via pattern recognition receptors (PRRs), such as Toll-like receptors (TLR, including TLR3, 7, 8), as well as the NOD-like receptors (NLR). Then, the activated down-stream transcription factor, NF-κB, induces the generation of pro-inflammatory cytokines. These cytokines contribute to the formation of the cytokine storm. Additionally, SARS-CoV-2 has been reported to exacerbate the inflammatory response of myeloid cells through C-type lectin receptors and Tweety family members (23). This non-TLR pathway contributes to virus-induced cytokine storm. C-type lectins not only contribute to cytokine storm. Among the members of the human C-type lectin family, dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN), L-SIGN, LSECtin and Syk-coupled C-type lectin member 5A (CLEC5A) and CLEC2 are critical in virus-induced NET formation (24). Dengue virus (DV) activates platelets to release extracellular vesicles (EVs) by CLEC2, including exosomes (EXOs) and microvesicles or microparticles (MVs or MPs). DV induced EXOs (DV-EXOs) and MVs (DV-MVs) further activate neutrophils and macrophages by CLEC5A and TLR2, thereby inducing NET formation and pro-inflammatory cytokine release. Consequently, the blockade of C-type lectins is a promising strategy to attenuate virus-induced NETs formation (25). In addition to DV, influenza A virus (IAV, H5N1) can also activate platelets to release EVs, thereby enhancing NET formation. A recent study shows that SARS-CoV-2 viral proteins induce NET formation via a Syk-dependent pathway of downstream signaling molecules of C-type lectin receptors (26). Further study shows that EVs released from SARS-CoV-2-activated platelets induce robust formation of NET via CLEC5A and TLR2, which in turn enhance thrombosis (27).
Interferon regulatory factor 3/7 (IRF3/7) is activated to enhance the transcription of interferon, which contributes to the host antiviral response. Growing evidence indicates protective type I interferon responses of COVID-19 patients are significantly reduced, which further leads to disease progression (19). Siglecs (sialic acid binding immunoglobulin-like lectins) are receptors of immune cell membranes and can interact with sialoglycans on viruses. Typically, most siglecs with structural domains containing the immunoreceptor tyrosine-based inhibitory motif (ITIM) mainly exert a negative regulatory role to prevent excessive immune responses. Sialoglycans on the SARS-CoV-2 envelope interact with siglecs on host cells to downregulate host immunity to clear virus, thereby aggravating viral infection (28). In the lung parenchyma, the virus damages the alveolar epithelial cells, releasing damage-associated molecular patterns (DAMPs), including nucleosomes, histone, and human high mobility group protein 1 (HMGB1), which activate innate immune cells to release chemokines and cytokines, such as TNF-α, IL-6, IL-1β, etc. Then, these cytokines and chemokines recruit more innate immune cells (neutrophils, monocytes, macrophages, natural killer cells) and activate adaptive immune cells (CD4+, CD8+ T) to also produce cytokines, ultimately leading to a cytokine storm (28, 29). The build-up of cytokines aggravates the damage, necrosis, and even sloughing of the alveolar epithelium, and forms a positive feedback loop of epithelial damage. Myeloid cells are the main source of inflammatory dysregulation in the lung tissue of COVID-19 patients (30).
Inflammatory markers in COVID-19 patients
Increased levels of IL-8, IL-6, monocyte chemoattractant protein-1, and TNF-α are observed in the plasma of COVID-19 patients. Elevated TNF-α, in particular, is a marker of poor prognosis and may lead to endothelial dysfunction in patients with COVID-19 (31, 32). IL-6 plays an essential role in driving pathological inflammatory responses, and is also associated with more severe outcomes. In a study of 2782 COVID-19 patients, high levels of C reactive protein (CRP) correlate with progression to severe cases, acute kidney injury, and all-cause mortality (33, 34). CRP levels display a positive relationship with COVID-19 severity and have been demonstrated to predict the risk of thrombosis (33, 35).
The normal physiological function of endothelium
Virchow’s triad describes the contributing factors to thrombus formation: damaged vessel walls, alterations in blood flow, and changed blood composition (hypercoagulability), and is fundamental to understanding the pathophysiology of venous thrombosis (36, 37). Damage to and subsequent activation of ECs are the first and critical steps for thrombosis. Intact ECs maintain blood fluidity and vascular homeostasis primarily through anti-inflammatory and anticoagulant effects.
Endothelial barrier function is dependent on stable cell-cell junctions, including adherens and tight junctions (38). ACE2 on the endothelium generates angiotensin (1–7), which binds to Mas1 receptor, modulating anti-inflammatory and antithrombotic signaling pathways (39, 40). Heparan sulfate within the glycocalyx binds to and activates antithrombin, and thus limits thrombosis on the endothelium. Additionally, ECs also support natural anticoagulant and antifibrinolytic substances, including thrombomodulin (TM), endothelial protein C receptor (EPCR), and tissue factor pathway inhibitor (TFPI) (41, 42). TM on the endothelial surface binds to thrombin, and then forms a complex with ERCP, and converts protein C to its active form (activated protein C, APC). APC and its cofactor protein S inactivate FVa and FVIIIa, thereby reducing thrombin generation (43, 44). TFPI, an endogenous serine protease inhibitor, can directly inhibit FXa and FVIIa/TF complex, and thereby exert anticoagulant effects via the extrinsic pathway of coagulation. ECs can secrete tissue plasminogen activator (t-PA) and urokinase-type plasminogen activator (u-PA), express the u-PA receptor (uPAR), and provide potent fibrinolytic ability. The inhibitors of complement cascade can protect quiescent ECs from complement deposition. Quiescent endothelium can also prevent platelet adhesion by the expression of CD39 and the release of prostaglandin I2 (PGI2) and nitric oxide (NO), thereby dampening platelet activation (45, 46). NO and PGI2 also exert other cytoprotective effects. NO can suppress the recruitment of leukocytes to the vessel wall through reducing the expression of chemokines and the transcription of adhesion molecules on the endothelial surface. PGI2 can reduce leucocyte adhesion, activation, and extravasation to attenuate inflammatory responses (47).
Inflammation-driven immunothrombosis
Inflammatory stimuli can overwhelm the delicate antithrombotic balance described above, promote inflammatory environments, and eventually lead to thrombus formation (48, 49). Cytokine signaling, hypoxia, tissue damage, and DAMPs trigger the transition of ECs from an anticoagulant to a pro-coagulant phenotype (11). Under inflammatory conditions, the loss of vascular integrity, reduced expression of antithrombotic molecules, the interactions between neutrophils and platelets, platelet activation, TF expression on monocytes, microparticle release, and inhibition of fibrinolysis lead to immunothrombosis (50, 51).
The interaction between platelet and neutrophil
DAMPs and cytokines induce an increased release of vWF (von Willebrand factor) from Weibel-Palade bodies to ECs surface. Platelets bind to vWF on the ECs through glycoprotein Ib (GP Ib). ADAMTS13 (a disintegrin and metalloprotease with thrombospondin type-1 repeats, member-13) regulates the size of vWF multimers and inhibits platelet-rich thrombosis by cleavage of vWF (52, 53). Plasma ADAMTS13 levels are decreased in patients with severe COVID-19 compared to those with mild disease, and reduced ADAMTS13 levels are associated with increased mortality rates (52). The elevated vWF and decreased ADAMTS13 levels are the potent predictors of adverse outcomes in severe COVID-19 patients (51, 54). ECs perturbation, increased release of vWF, and relatively insufficient vWF cleavage owing to the deficiency of ADAMTS13 are responsible for increased interactions between platelets and the vessel wall to cause thrombotic microangiopathies.
Platelets in COVID-19 patients are characterized by hyperreactivity (increased aggregation, increased expression of CD40 and p-selectin). In posthoc analyses of 300 patients with COVID-19, plasma p-selectin levels at admission were strongly associated with the subsequent diagnosis of venous thromboembolism (55). Mean platelet volume (a biomarker of platelet hyperactivity) was related to the occurrence of thrombosis and disease severity (56). In a meta-analysis of 15 studies, the presence of larger, more immature platelets was associated with an increased risk of thrombosis and all-cause mortality (57). Additionally, Analyses of tissue and blood from COVID-19 patients revealed that SARS-CoV-2 viral virions entered megakaryocytes and platelets, which was related to alterations in the platelet transcriptome and activation profile (56).
Cytokines result in increased expression of adhesion molecules on the surface of ECs including P-selectin and E-selectin, which favor rapid recruitment of leukocytes to the site of injury and inflammation (11). Although leukocytes are not typical parts of the coagulation system, they are essential for immunothrombosis, and contribute to the initiation of coagulation by ECs. Neutrophils bind to chemokine (C-X-C motif) ligand 1 and P-selectin on ECs by (C-X-C motif) chemokine receptor 2 and P-selectin glycoprotein ligand-1, respectively, whereas platelets attach to EC-bound vWF. Bound platelets are then activated by SARS-CoV-2 and release HMGB1-bearing extracellular vesicles, inducing neutrophils to generate neutrophil extracellular traps (NETs), web-like structures of which entrap blood cells and coagulation factors (58). In turn, active substances such as antimicrobial peptides released by NETs can further activate platelets. In this way, NETs provide scaffolds for thrombosis. This creates a positive feedback loop, and the crosslinking of platelets with NETs components results in immunothrombosis. The interactions of activated platelets with immune cells stimulate the coagulation system, forming an intertwined process bridging thrombotic and inflammatory pathways (59). Indeed, studies have shown that the levels of neutrophil–platelet aggregates in the blood of COVID-19 patients are elevated (60). Additionally, among COVID-19 patients requiring hospitalization, those admitted to the ICU have the highest levels of platelet-leukocyte aggregation, suggesting that platelet-leukocyte aggregation may be a surrogate marker of COVID-19 severity (60, 61).
NETs are an important interface between inflammation and thrombosis. NETs provide scaffolds for the activated coagulation system and link the immune and coagulation systems by immunothrombosis. NETs can bind to TF, thereby promoting the generation of fibrin. The cell-free DNA released in NETs provides a negatively charged surface to allow FXII binding and activation of the contact (intrinsic) coagulation pathway, resulting in microvascular occlusion (62, 63). Additionally, neutrophil elastase (NE) and myeloperoxidase, two other components of NETs, can cleave and inactivate natural anticoagulants (TFPI and TM), thereby leading to a procoagulant shift of the hemostatic balance and local clot formation (54, 64).
The generation of NETs contributes to adverse coagulation function and immunothrombosis. The components of NETs were observed in serum, plasma, and post-mortem specimens of COVID-19 patients (15, 65). The levels of DNA, myeloperoxidase (MPO)-DNA complexes and citrullinated histones (H3Cit) increased two-fold in COVID-19 patients compared to healthy controls (65). Plasma levels of MPO-DNA complexes and H3Cit correlated with COVID-19 not only with thrombotic events but also with disease severity (66, 67). A prospective cohort study of the autopsy showed that 10 of 21 COVID-19 patients presented with neutrophil embolism (68). Histopathology of the lung and other organs consistently indicated that aggregated NETs led to microvascular obstruction associated with the disruption of the endothelium (33). These observations highlight the potential relationship between NETs and persistent COVID-19 thrombosis (Figure 1).
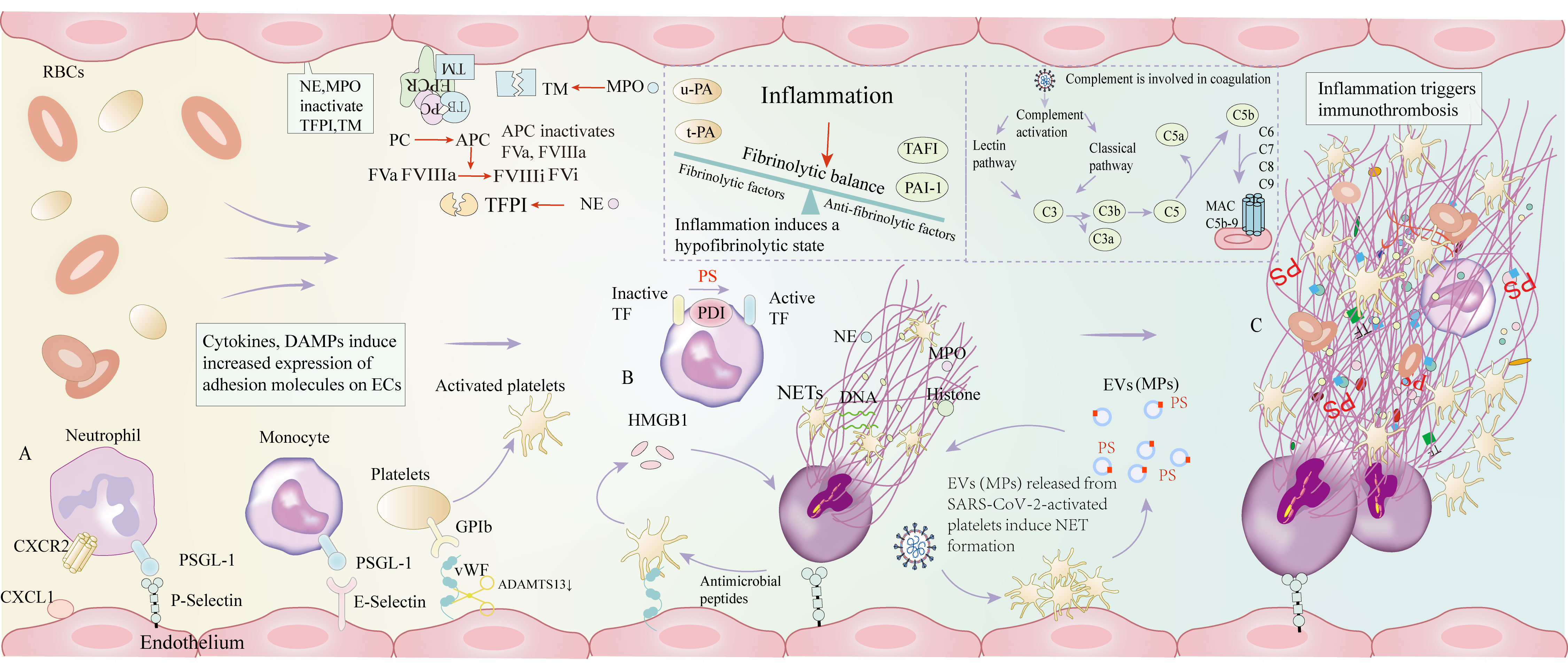
Figure 1 The mechanisms of immunothrombosis in COVID-19 patients. (A) Inflammation induces immune cell recruitment and increased expression of adhesion molecules, including P-selectin, E-selectin, and vWF. (B) Activated platelets activate neutrophils and generate NETs through the release of HMGB1, and the components of NETs, antimicrobial peptides, further induce platelet activation, forming a positive feedback activation. NE and MPO can inactivate TFPI and TM, thereby inhibiting anticoagulant function. Monocytes exert pro-coagulant effects by converting inactive TF to active TF, which is required for PS and PDI. EVs (MPs) are mainly derived from SARS-CoV-2-activated platelets. Platelet-derived EVs (MPs) are responsible for robust NET formation, which plays a critical role in COVID-19 thrombosis. Activated platelets and platelet-derived EVs are markers and central mediators of COVID-19 immunothrombosis. Under inflammatory settings, elevated levels of thrombin-activatable fibrinolysis inhibitor and PAI-1 surpass the role of u-PA and t-PA, thereby resulting in inhibition of fibrinolysis. SARS-CoV-2 induces complement activation, which is involved in coagulation. (C) The above processes eventually cause immunothrombosis. vWF: von Willebrand factor; NETs: Neutrophil extracellular traps; HMGB1: human high mobility group protein 1; NE: Neutrophil elastase; MPO: Myeloperoxidase; TFPI: tissue factor pathway inhibitor; TM: thrombomodulin; TF: tissue factor; PS: Phosphatidylserine; PDI: Protein disulfide isomerase; EVs: Extracellular vesicles; MPs: Microparticles; PAI-1: plasminogen activator inhibitor; u-PA: urokinase-type plasminogen activator; t-PA: tissue plasminogen activator; SARS-CoV-2: severe acute respiratory syndrome coronavirus 2; CXCL-1: chemokine (C-X-C motif) ligand 1; CXCR2: C-X-C motif chemokine receptor 2; PSGL-1: P-selectin glycoprotein ligand-1.
Monocytes TF expression
Inflammation can induce the recruitment of monocytes, which bind to ECs through E-selectin. Monocytes initiate coagulation by the expression of TF and enhance thromboinflammation by the activation of the inflammasome. TF expression is triggered by pyroptosis, which is a cell death process dependent on caspase-1, involving the formation of pores in the cell membrane and the release of inflammation mediators (IL-1β and IL-18). TF is the primary activator of the extrinsic coagulation pathway that functions by forming a complex with circulating FVIIa and enhancing the activation of FIX and FX, which goes on to initiate coagulation (10, 69). Thus, TF exposure induces increased thrombin generation, which leads to the formation of fibrin clots. Protein disulfide isomerase (PDI) is an important regulator of extracellular disulfide-exchange and is associated with TF coagulant activity on the cell surface (70). PDI released at the site of vascular lesions contributes to TF activation by converting TF from a nonfunctional encrypted state to its active form (71, 72). The release of PDI is tightly regulated to prevent continuous thrombosis under physiological conditions. Adherent platelets and impaired vascular wall can promote thromboinflammation by releasing PDI. Besides PDI, phosphatidylserine (PS) exposure (externalization) can also directly promote TF-dependent fibrin formation, as explained below (Figure 1).
EVS release and PS exposure
Activated platelets and platelet-derived extracellular vesicles (EVs) are markers and central mediators of COVID-19 immunothrombosis. The levels of platelet-derived EVs were higher in COVID-19 patients requiring hospitalization than in those not requiring hospitalization (73). Platelet-derived as well as endothelium-derived EVs are responsible for NET formation and endothelial cell death (10, 74). Robust formation of NET induced by platelet-derived EVs further enhances immunothrombosis (59). Additionally, increased levels of TF+ EVs in the plasma of COVID-19 patients are related to thrombin generation, the severity of respiratory condition and mortality risk (75, 76).
MPs (a kind of EVs) are enveloped by a phospholipid bilayer membrane and released from apoptotic or activated cells. Cytokines within the microvasculature induce the apoptosis of blood cells and ECs, causing the release of MPs, which exert pro-coagulant effects. Additionally, platelets activated by SARS-CoV-2 are the primary source of pro-coagulant MPs. Several studies have mentioned that the MPs released from virus-activated platelets cause robust NET formation and inflammatory reactions (25, 74). Strong evidence suggests that MPs are procoagulant due to the exposure of PS. Type IV P-type ATPases (P4 ATPases) are flippases that transport specific lipids (PS) from the inner to the outer leaflet of the membrane bilayer. Transmembrane protein 16F (TMEM16F) and Xk-related protein 8 (Xkr8) are two scramblases involved in the process of PS transport (77, 78). PS on MPs moves to the outside layers through caspase-dependent Xkr8 activation and calcium-dependent P4-ATPase inactivation, which is referred to as PS externalization. PS provides binding sites for coagulation factors V and VIII. TMEM16F-mediated PS externalization on the surface of monocytes triggers the caspase-11 and gasdermin D pathways, which regulates the coagulation activity of TF and promotes the release of MPs bearing TF (79) (Figure 2). MPs release and PS externalization are the main drivers of COVID-19-associated pulmonary coagulopathy. First, as the severity of the disease worsens, more and a larger diversity of apoptotic cells release MPs, including red blood cells, platelets, monocytes/macrophages, and ECs. PS on MPs is a potent modulator of the human blood-clotting system. Secondly, PS initiates the extrinsic coagulation pathway by decrypting TF. PS enhances the catalytic efficiency of the tenase and prothrombinase complexes by providing scaffolds for these coagulation factors (80, 81) (Figure 2). Elevated platelet-derived MPs in circulation correlate with an increased risk of thromboembolic complications, leading to increased severity and mortality of COVID-19 (82, 83). The number of PS+ peripheral blood mononuclear cells (PBMCs) in blood samples from COVID-19 patients is abnormally high, and becomes a new parameter that correlates strongly with disease severity. Compared to those without thrombosis, COVID-19 ICU patients with thromboembolic events had significantly increased platelet PS externalization, an apoptotic marker, suggesting the link between venous thromboembolism and PS externalization (84).
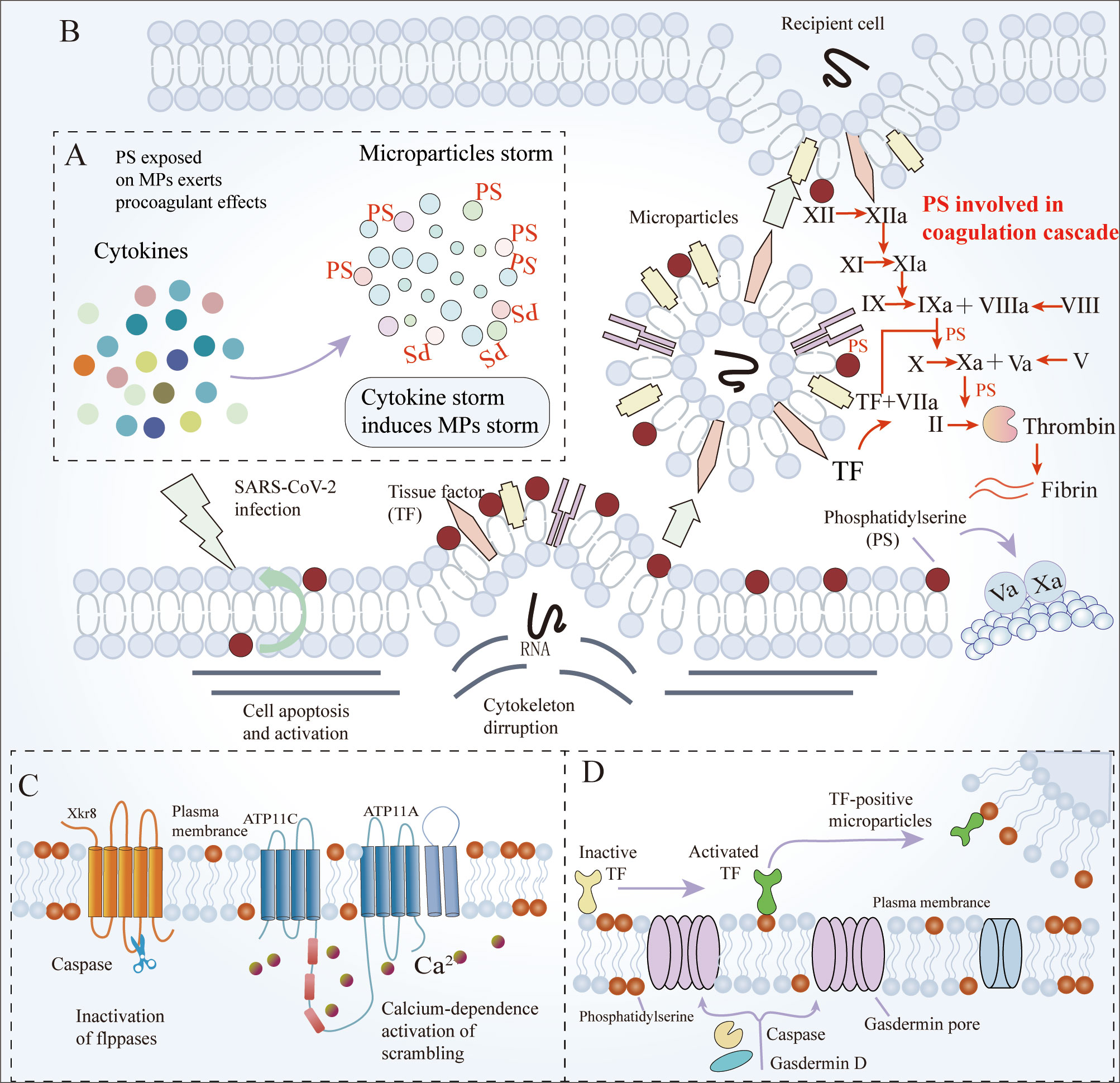
Figure 2 The mechanisms of MPs and PS involved in coagulation. (A) High levels of cytokines induce the apoptosis of blood cells, and ECs, release MPs (mainly derived from platelets), and exert pro-coagulant effects. Additionally, platelet activation is the primary source of pro-coagulant MPs. (B) PS is exposed on MPs in case of cell apoptosis and activation, and then is involved in coagulation by the coagulation cascade (C) Caspase-dependent Xkr8 activation and calcium-dependent P4-ATPase inactivation are responsible for PS externalization. (D) Coagulation activity of TF and the release of MPs bearing TF require caspase-dependent gasdermin D-mediated PS externalization. ECs, Endothelial cells; MPs, microparticles; PS, phosphatidylserine; Xkr8, Xk-related protein 8; TF, Tissue factor.
Complement activation
SARS-CoV-2 activates the complement cascade by the classical and the lectin pathways (85). The complement system is a major contributor to inflammation and thrombosis. C3a and C5a recruit and activate neutrophils and monocytes to release pro-inflammatory cytokines (IL-6, IL-8). C5a can upregulate the expression of TF on neutrophils and ECs, which in turn exerts a procoagulant effect (86). C5a can also inhibit fibrinolysis by increased release of plasminogen activator inhibitor 1 (PAI-1) from mast cells and basophils (86, 87). In an in vitro system, the plasma of COVID-19 patients induces complement activation to trigger NETosis in a C5a-dependent manner (88). Membrane attack complex (MAC) induces ECs to actively secrete vWF and assemble the prothrombinase complex (89). Multiple complement-centric interconnections between inflammatory pathways and prothrombotic mechanisms act synergistically to suppress pathogens. Complement factors (C1q and C3), and MAC can activate platelets. In turn, platelets provide a surface for complement activation, and enhance inflammatory function of neutrophils by binding to complement (90, 91). Furthermore, platelets capture pathogens in a C3- and GP Ibα-dependent manner, transport them to antigen-presenting dendritic cells, and thereby initiate adaptive immune responses (92). Complement factors (C5b-9 and C4b) and MASP2 are shown in pulmonary microvascular of severe COVID-19 patients (15, 93). According to autopsy outcomes, complement is related to microvascular occlusion in the context of severe COVID-19 (Figure 1).
Fibrinolytic inhibition
When thrombus occurs in the vascular intima, ECs can express plasminogen activators including t-PA and u-PA, thereby contributing to the generation of endogenous fibrinolytic substances. Activated ECs can also produce PAI-1, which antagonizes u-PA and t-PA, thereby maintaining fibrinolytic balance. But inflammation can result in a hypo-fibrinolytic state in COVID-19 patients. Studies show that low levels of plasminogen are significantly related to inflammatory markers (CRP, procalcitonin, and IL-6) (94, 95). Inflammation itself can promote the local release of t-PA and PAI-1 from ECs, suggesting that increased PAI-1 levels may affect fibrinolysis more than elevated t-PA levels which are simultaneously presented in COVID-19 patients (96, 97). In addition, elevated levels of thrombin-activatable fibrinolysis inhibitor (TAFI) and PAI-1 surpass the role of u-PA and t-PA, thereby resulting in inhibition of fibrinolytic function (98) (Figure 1).
The crosstalk between inflammation and coagulation
The crosstalk between inflammation and coagulation mainly depends on three coagulation factors: TF, thrombin, and FXII. In addition to promoting coagulation, TF also initiates inflammatory pathways by G protein-coupled protease-activated receptors (PARs), which are differentially expressed on ECs, platelets, and leukocytes (99). The activation of PARs initiates multiple inflammatory signals including cytokines and growth factors, which induce the expression of adhesion molecules. TF is the pivotal trigger of coagulation activation, and the initial events leading to its exposure in circulating blood propel an escalating feedback loop between inflammation and coagulation. If thromboinflammation loses its natural brakes, it may result in overwhelming thrombotic and inflammatory injury. The partial pro-inflammatory effects of thrombin are mediated by ECs stimulation. Thrombin activates ECs primarily by PAR1 proteolysis, and induces increased P-selectin expression and mobilization of Weibel-Palade bodies (41, 100). Thrombin cleaves PARs on platelets, triggers the release of granular contents, such as ADP, CD40 ligand, and multiple pro-inflammatory mediators (101, 102). Thrombin also stimulates platelets, which appear to enhance rapid and efficient recruitment of neutrophils to localized endothelial injury sites (103). The activation of the contact system of coagulation can also release pro-inflammatory mediators including bradykinin (BK) (104). Proteins of the contact system (FXII, high molecular weight kininogen (HK) and plasma kallikrein (PK)) can assemble on the cell surface to form a complex, which binds to soluble contact activator (including poly P) carried by the cell surface, resulting in FXII activation. FXIIa, in turn, triggers thrombus formation by the intrinsic coagulation pathway. FXIIa can also activate PK, and generate BK from HK. BK and its active metabolite Des-Arg 9-BK exert pro-inflammatory effects.
Long COVID
Long COVID or post-acute COVID-19 syndrome (PACS) is an emerging syndrome worldwide, characterized by the persistence of signs and symptoms of COVID-19 for more than four weeks after infection (105). The symptoms of long COVID mainly include fatigue, dyspnea, reduced exercise tolerance, chest pain, and thrombotic complications (106, 107). About 10% of COVID-19 patients develop persistent or relapsing and remitting symptoms beyond 4 to 12 weeks following infection (106). Studies show that 4.5%-36.6% of COVID-19 patients remain symptomatic greater than 3 months after infection (108–110). This percentage is as high as 76% among patients requiring hospitalization during the infection (111, 112). A single-center report of 163 patients who did not undergo post-discharge thromboprophylaxis found that the cumulative incidence of thrombosis 30 days after discharge was 2.5%, including pulmonary embolism and ischemic stroke, etc (113). Prospective studies of 6 weeks post-discharge follow up assessing d-dimer levels and venous ultrasound showed 8% of COVID-19 patients receiving thromboprophylaxis after hospital discharge (114, 115). Currently, vaccination is the most effective preventative measure for severe COVID-19 symptoms. Since the rollout of COVID-19 vaccines, a global vaccination campaign has been carried out. Studies have shown that receiving at least one dose of COVID-19 vaccine significantly reduces the risk of ICU stay, and hypoxemia, but not some outcomes of long COVID. Receiving two doses of vaccine (i.e. BNT162b2 Pfizer/BioNTech, mRNA-1273 Moderna, or Ad26.COV2. S Janssen) is associated with a lower risk of most outcomes (116, 117). Some studies also suggest that vaccine administered pre-infection can only provide partial protection during the acute stage of the disease, but not reduce the long-term consequences of SARS-CoV-2 infection (which may include thrombosis) (118, 119). Therefore, we will focus on the thromboembolic complications of Long COVID, which can lead to serious consequences for COVID-19 patients.
Despite the increasing awareness of Long-COVID, the potential pathophysiological mechanisms of it remain poorly characterized, and may correlate with several factors (120, 121). Proposed causes include lingering viruses, chronic inflammation, or microscopic thrombi (122, 123). Long COVID may be caused by a combination of multiple factors (124–126). In the following section, we summarize the possible mechanisms of long COVID thrombosis, including endothelial lesions, coagulation abnormalities and persistent inflammation.
In the acute phase of COVID-19, apart from mediating inflammatory immune responses, SARS-CoV-2 can directly damage ECs by binding to AEC2 (127). The damage of ECs leads to endothelial activation, loss of barrier function, and increased permeability followed by microvascular leakage (128–130). Endothelial damage can induce diffuse and systemic endothelial dysfunction, and activate multiple immune-mediated inflammatory pathways and thrombus formation, thereby resulting in severe multi-organ involvement and subsequent increased mortality (131, 132). SARS-CoV-2 has also been detected in ECs, and elevated levels of circulating endothelial cells (CECs, a biomarker of vascular injury) can be found in patients admitted for COVID-19 (124, 125, 133). However, some pathological processes persist even when SARS-CoV-2 is no longer detectable. Elevated markers of endothelial lesions and coagulation are observed in a significant proportion of convalescent patients, suggesting infection may induce chronic coagulopathy, endotheliitis, and microvascular lesions, accompanied by microthrombi formation (126, 134).
Endothelial damage and dysfunction
In studies of COVID-19 patients enrolled in medical wards or intensive care unit (ICU), the marked endothelial dysfunction gradually improved at the 6 month follow-up, but remained impaired compared to healthy controls (135, 136). The levels of CECs in patients recovered from COVID-19 were elevated, which may be driven by activated T lymphocyte-related cytokines. Furthermore, increased expression of ICAM1, SELP, and CX3CL1 on CECs may indicate that ECs from the site of vascular injury were in pro-inflammatory and procoagulant states (137). In a study of 50 patients with SARS-CoV-2 infection, the highest level of soluble thrombomodulin (14.4 ng/ml) was observed in a patient who did not require hospitalization, indicating persistent endothelial lesions in convalescence were not exclusive to those who exhibited severe COVID-19 (138). During the post-infection phase, flow-mediated peripheral arterial dilation was significantly decreased in COVID-19 patients, suggesting the presence of endothelial dysfunction (139). Cross-sectional studies assessing vascular function 3-4 weeks following SARS-CoV-2 infection showed a significant decrease in systemic vascular function and increased arterial stiffness in SARS-CoV-2-positive participants (135, 140). Overall, results from these studies show persistent endothelial lesions are common in the convalescent phase of COVID-19. The normal endothelium plays an important role in preventing thrombosis by expressing antiplatelet and anticoagulant substances to block platelet aggregation and fibrin clot formation as well as inhibiting the adhesion of coagulation proteins (128, 141). It has been previously demonstrated that endothelial dysfunction is associated with an increased risk of thrombosis (142, 143). Consequently, long COVID thrombosis may be secondary to endothelial damage and subsequent endothelial dysfunction.
Endothelial activation and coagulation abnormalities
Recent studies have suggested that persistent endothelial activation is commonly observed in convalescent COVID-19 patients (138, 139). Especially, several groups have reported consistently increased levels of endogenous thrombin potential (ETP) and D-dimer in COVID-19 patients 4-12 months following acute infection (138, 144, 145). Significantly elevated peak thrombin, and a shorter time to reach the peak of thrombin formation are also seen in convalescent COVID-19 patients (138, 146). Interestingly, the percentage of intermediate monocytes is associated with elevated ETP and peak thrombin (146). In a cohort of convalescent patients, plasma FVIII: C levels were still significantly higher than in controls. In the same study, reduced ADAMTS13 as well as elevated vWF: Ag levels mean that the vWF: Ag/ADAMTS13 ratio dramatically increased in patients recovered from COVID-19 (107). Studies indicated that markers of platelet activation returned to normal at 2-3 months post-discharge (147, 148). Notably, circulating NETs biomarkers reverted towards normal levels around 4 months following infection compared to healthy controls (149, 150). A series of complex events in the vasculature including endothelial activation, NET formation, vWF secretion, and blood cell adhesion and aggregation collectively mediate immunothrombosis. Therefore, persistent EC activation and increased pro-coagulant substances in convalescence may be linked to long COVID thromboembolic complications.
Persistent inflammation
Researchers have found the evidence of SARS-CoV-2 in peripheral organs of the body, especially the intestine, for several months following viral clearance from the respiratory airways (151). Weeks to months after the disappearance of acute symptoms, viral RNA and protein antigens can be detected in the central nervous system, intestines, and secondary lymphoid organs (152, 153). Residual antigens may activate T- and B-lymphocytes, thereby resulting in persistent inflammation. The levels of pro-inflammatory markers in patients with long COVID remain elevated. Markers that are elevated during the acute phase including CRP and IL, may remain high 6 months or longer after disease onset (126, 154, 155). In the early recovery phase of COVID-19, patients who continued to develop PACS generally had higher levels of inflammatory biomarkers including TNF-α and interferon-inducible protein 10 (IP-10). PACS patients had an inflammatory state characterized by the up-regulation of TNF-α, IL-6, and IL-13, with decreased levels of IP-10 (156, 157). The levels of IL-17 and IL-2 were higher in the long COVID patient group, while higher levels of IL-10, IL-6, and IL-4 were shown in patients without sequelae (158, 159). Clinical manifestations of PACS correlate with the persistent pro-inflammatory phenotype induced by SARS-CoV-2 infection. We have elaborated on how inflammation triggers the activation of the coagulation cascade, eventually leading to thrombin-mediated fibrin generation and obstructive clot formation in the acute phase of COVID-19. Therefore, systemic inflammation may also be involved in Long COVID thrombosis. The presence of long-term systemic inflammation indicates that anti-inflammatory drugs could be useful not only during the acute phase of infection, but also in long COVID.
Treatment strategy
Antiviral therapy
A hyper-inflammatory response triggered by SARS-CoV-2 leads to immunothrombosis, thus early effective antiviral therapies are necessary for COVID-19 patients to prevent thrombus formation. Since its discovery, Omicron variant has rapidly increased in prevalence. Compared with Delta, Omicron has a significantly reduced risk of severe outcomes, with a higher reduction in the risk of serious consequences at the more severe endpoints (160). The antiviral monoclonal antibodies currently under investigation include molnupiravir, sotrovimab, and casirivimab-imdevimab. Molnupiravir is effective against Alpha and Beta variants in hamster and human cell models (in vivo), and against Delta and Omicron in studies in vitro (no in vivo data) (161). Sotrovimab retains its activity on Omicron, but requires higher concentrations to neutralize the virus. Casirivimab-imdevimab lacks efficacy against Omicron ba1variant (162, 163). Consequently, casirivimab-imdevimab is no longer recommended for COVID-19 treatment, unless infection with an earlier SARS-CoV-2 variant is confirmed (164, 165).
Anticoagulant therapy
A large-scale observational study has shown that prophylactic doses of enoxaparin can improve the intubation rate and survival of hospitalized patients with COVID-19 (1). In large randomized controlled trials of hospitalized patients with COVID-19, the incidence of VTE in patients given prophylactic doses of anticoagulant therapy ranges from 6% to10%, and an incidence of 4-8% with therapeutic doses of anticoagulant therapy (166–168). In a prospective study of 803 hospitalized patients with COVID-19, increased doses of anticoagulant agents determined by the combination of disease severity, body weight, and D-dimer levels is associated with reduced mortality (169). In another observational study of 4,389 COVID-19 patients, analyses suggest more pronounced benefit with therapeutic compared to prophylactic dosing in anticoagulant therapy (170). In non-critically ill COVID-19 patients, therapeutic-dose anticoagulant therapy with heparin increases the probability of survival to hospital discharge and reduces the use of cardiovascular or respiratory organ support compared with thromboprophylaxis (1). These evidences indicate that therapeutic-dose anticoagulant therapy is superior to prophylactic-dose anticoagulant therapy in increasing the probability of hospital discharge and reducing mortality. However, recent studies do not favor the use of high dose of anticoagulant therapy (therapeutic-dose or full-dose anticoagulant therapy) in critically ill patients with COVID-19 (171, 172). The efficacy of anticoagulant treatment depends on the time of therapy initiation. As the disease progresses, anticoagulant therapeutic effect may be not satisfactory owing to the interactions of inflammation with the coagulation cascade. Full-dose anticoagulant therapy cannot reverse established disease process (thrombus formation) at the advanced stage (173, 174). Additionally, thrombosis leads to the consumption of coagulation factors, and full-dose anticoagulant therapy conversely worsens bleeding risk. Therefore, early administration of therapeutic-dose anticoagulant therapy can effectively reduce the incidence of thrombosis. Prophylactic anticoagulant therapy for COVID-19 patients after discharge remains controversial. However, post-discharge prophylactic anticoagulant treatment is beneficial to patients with a high risk of VTE. In patients at high risk of VTE discharged after hospitalisation due to COVID-19, thromboprophylaxis with rivaroxaban 10 mg/day for 35 days improved clinical outcomes compared with unextended thromboprophylaxis (175).
Thrombotic events are considered to be major causes of COVID-19 mortality, and current guidelines recommend the use of heparin for thromboprophylaxis and anticoagulant therapy (176). Heparin, including unfractionated heparin and low-molecular-weight heparin (LMWH), can bind to antithrombin (AT), and enhance inhibitory effects of AT on FXa and thrombin (177, 178). Accumulating evidence suggests that heparin can regulate the inflammatory response in multiple ways, including inhibition of both selectin-induced neutrophil adhesion and the release of inflammatory mediators (179, 180). Recent studies address the relationship between heparin and SARS-CoV-2, considering whether heparin directly binds to the S protein of SARS-CoV-2, and exerts direct antiviral effects (181).
In COVID-19, FXII is an attractive and rational therapeutic target. FXII can induce BK activation, which leads to the activation of downstream complement and the production of inflammatory cytokines (182). FXII inhibitors block the NETs-mediated contact activation (intrinsic) pathway of coagulation, and have anti-inflammatory effects (183). The most advanced FXII inhibitors are fully human or humanized monoclonal antibodies (including 3F7). Another potential therapeutic target is FXI, and FXI inhibitors include IONIS416858, bay1213790, MAA868, etc (184, 185). High doses of IONIS416858 (an ASO targeting FXI) used to treat venous thrombosis outperformed enoxaparin without increased bleeding risk. Direct oral anticoagulants (DOACs) directly inhibit thrombin or FXa to achieve an anticoagulant effect. FXa inhibitor rivaroxaban has been shown to prevent ischemic events in patients with cardiovascular diseases, and also slow down the progression of established atherosclerosis (186, 187). In patients with atrial fibrillation, oral rivaroxaban anticoagulation reduced plasma IL-6 and CRP levels (188). Compared to standard prophylactic anticoagulant therapy, rivaroxaban for 30 days improved clinical outcomes in hospitalized patients with COVID-19 and elevated d-dimer levels (189). Promising results have also been found using FXa inhibitors (primarily apixaban) in preventing thrombotic complications and reducing bleeding risks in COVID-19 patients compared with LMWH (190). Relatively few studies of the thrombin inhibitor dabigatran for anticoagulant therapy in COVID-19 have been reported. Compared with warfarin, DOACs have a broader therapeutic index, and do not require regular monitoring. DOACs are associated with a lower risk of systemic thromboembolic events and intracranial hemorrhage. Additionally, in the case of excessive anticoagulation, DOACs-induced bleeding can be reversed (191, 192). However, DOACs are not a universally accepted approach because of their high price. Considering the role PS plays in COVID-19 thrombosis, it may be an effective target for emerging therapeutics. The milk-fat globule protein lactadherin has been shown to compete with FV and FVIII for PS binding sites and reduce procoagulant activity in vitro and clotting in mouse bleeding models, and may therefore be a potential therapy to suppress thrombus formation.
t-PA is used for thrombolysis (generally after the failure of heparin treatment), rescue of severely ill patients, or for the prevention of possible thrombosis (193, 194). Considering there have been few adverse events of fibrinolytic therapy, early and local application of tPA, combined with accurate anti-inflammatory interventions targeting cytokines and chemokines, can improve survival in severe COVID-19 patients with ARDS (194). For COVID-19 patients without ARDS or other severe complications, fibrinolytic therapy can significantly improve the prognosis (194, 195). Currently, case reports and case series show that t-PA has significant efficacy for severe COVID-19 ARDS patients, with improved PaO2/FiO2 and no bleeding complications (196, 197). In a recent group of cases, five COVID-19 patients with severe hypoxemia (PaO2 <80 mmHg) and d-dimer greater than 1.5 μg/mL, receiving thrombolytic therapy (25 mg t-PA intravenous injection for 2 hours, 25 mg continuous infusion for the next 22 hours) and subsequent continuous infusion of heparin, showed improvement in oxygen demand, and three patients avoided mechanical ventilation after t-PA injection (196). In another study, three severe COVID-19 patients with respiratory failure undergoing the treatment of t-PA had a transient improvement in respiratory status, and one patient achieved a durable response (197).
Antiplatelet therapy
Activated platelets are one of the important sources of MPs, and their essential role in COVID-19 coagulation have gradually been confirmed. Three classes of drugs can be used as antiplatelet therapy (198). The first category is aspirin, which irreversibly inactivates platelet cyclooxygenase-1 (COX-1), thus effectively preventing the production of the platelet agonist thromboxane A2 (TXA2). The second class of drugs that inhibit platelet activation is P2Y12 receptor antagonists (including clopidogrel, etc). P2Y12 is a major ADP receptor on platelets, and ADP binds to the P2Y12 receptor on platelets, promoting platelet aggregation and activation (199). The last category of drugs is the FDA-approved PAR1 antagonist (vorapaxar). Besides its antithrombotic activity due to inhibition of platelet aggregation, dipyridamole has also been shown to have antiviral, anti-inflammatory, and antioxidant properties. A clinical trial is currently underway to assess the effects of dipyridamole in COVID-19 patients (200, 201).
Observational studies showed that the mortality of hospitalized COVID-19 patients who received antiplatelet therapy before hospitalization was lower (202, 203). In a multivariate analysis of a propensity-matched population of COVID-19 patients including age, sex, hypertension, diabetes, renal failure, and invasive ventilation, administration of aspirin was associated with a lower risk of in-hospital mortality (204). Antiplatelet therapy during COVID-19 in-hospital stays reduced the risk of mortality, and shortened the duration of mechanical ventilation without simultaneously increasing the risk of bleeding (205, 206). Antiplatelet therapy also improved the ventilation/perfusion ratio in COVID-19 patients with severe respiratory failure, the mechanisms of which were the regulation of megakaryocyte function and platelet adhesion, and the prevention and intervention of pulmonary capillary thrombosis (207).
Targeting nets
Targeting components of NETs are considered as potential interventions for COVID-19, the best known of which is Cl-amidine, an inhibitor of protein-arginine deiminase 4 and the NE inhibitor sivelestat (ONO-5046) (208). Cl-amidine was shown to inhibit NET release in both SARS-CoV-2- infected healthy neutrophils and blood neutrophils of COVID-19 patients (66). Sivelestat can improve lung function and oxygen saturation in patients with ARDS (209). A new generation of NE inhibitors (Lonodelestat, Alvelestat, CHF6333 and Elafin) is currently in clinical trials. Exogenous recombinant deoxyribonuclease 1 has been shown to reduce the concentrations of cell-free DNA and NETs in plasma in vitro and is a promising treatment option. The treatment of recombinant human (rh)DNase 1 was associated with a reduction in pulmonary DNA-MPO complexes as well as improved oxygenation.
Beyond NETs as a treatment target, platelets can promote NET formation, and platelet-derived HMGB1 is considered as a key molecule in arterial and venous thrombosis (210–212). Inhibiting HMGB1 with BoxA reduced thrombus burden deep venous thrombosis and improved the outcome of ischemic stroke (210, 212). Additionally, EVs from virus-activated platelets are potent endogenous danger signals, and the blockade of C-type lectins is a promising strategy to attenuate virus-induced NETs formation and coagulopathy. Targeting platelet-neutrophil interactions with antibodies against P-selectin or PSGL-1 may be an interesting approach regarding platelet-mediated NET formation (213). Crizanlizumab is a monoclonal antibody against p-selectin that is currently being investigated for the treatment of COVID-19-related vascular disease (214). More research into the mechanisms of NET formation in COVID-19 may pave the way for novel therapeutic approaches.
Anti-inflammatory treatment
Anticoagulant therapy alone cannot resolve inflammation-driven thrombosis. Therefore, combination therapies are often required for COVID-19 patients. Additionally, targeting inflammation to prevent thrombosis does not affect the hemostatic balance, and avoids bleeding risk with existing treatments. Anakinra is a recombinant human IL-1 receptor antagonist. In a study of approximately 600 COVID-19 pneumonia patients at risk of respiratory failure, anakinra significantly improved the survival rate and shortened the length of hospital stay (215). Recent meta- analyses have suggested that COVID-19 patients the C-reactive protein serum concentrations greater than 100 mg/L benefited most from anakinra treatment (216, 217). In other trials, the level of soluble urokinase plasminogen activator receptor (suPAR) was used to identify the risk of severe COVID-19 at early stages, and anakinra was found to substantially reduce progression to severe respiratory failure and 30-day mortality (218, 219). Tocilizumab, a humanized monoclonal antibody targeting IL-6R, is an immunosuppressive drug commonly used in the treatment of chronic rheumatoid arthritis (220, 221). In hospitalized COVID-19 patients, treatment with IL-6 receptor antagonist (IL-6RA) reduced all-cause mortality. The most evident beneficiaries from IL-6RA were COVID-19 patients on respiratory support, including both non-invasive ventilation and invasive mechanical ventilation (221). Anti-TNF-α therapy is associated with lower hospitalization rates and a reduction in severe COVID-19 (admitted to critical care units or death) (222). However, these inflammatory mediators fulfill a crucial role in host defense. Therefore, the risk must be weighed between the potential benefits of suppressing cytokine storm and reducing defenses against pathogens.
Anti-complement therapy
Elevated levels of several complement factors (C3 and C5) are observed in the plasma of COVID-19 patients and these factors are correlated to disease severity (223, 224). In a non-randomized clinical trial, eculizumab (a C5 inhibitor) significantly improved 15-day survival and oxygenation in severe COVID-19 patients (225). C3 inhibitor AMY-101 has been used for COVID-19 patients and is currently being evaluated in several clinical trials. Narsoplimab (an antibody against MASP-2), inhibits the lectin pathway, and has shown efficacy in a small study of COVID-19 patients (226) (Table 1).
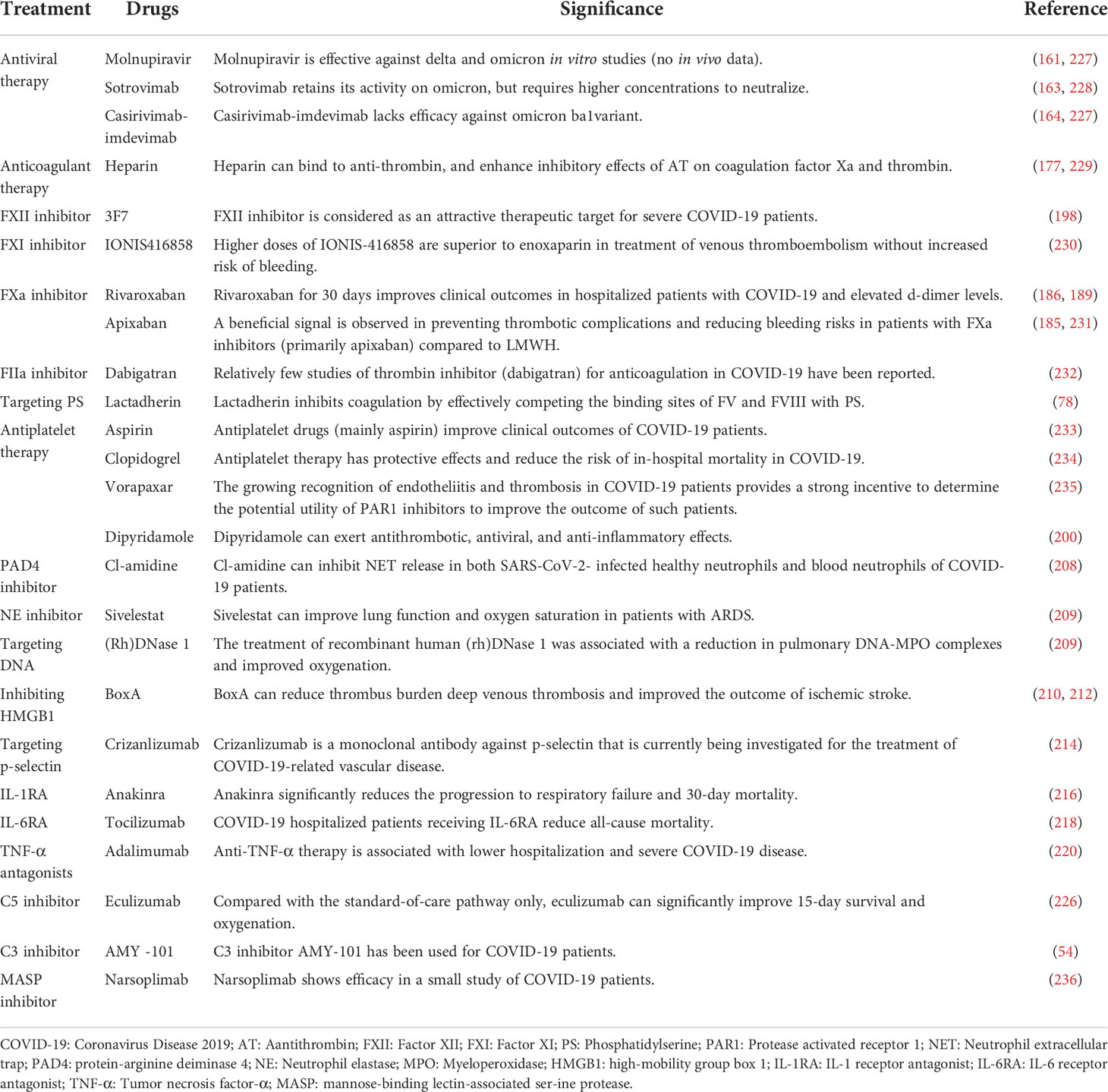
Table 1 Prevention and treatment strategies for COVID-19 patients.
Conclusion
The COVID-19 pandemic has brought unprecedented challenges. This review summarizes the mechanisms of how inflammation triggers the coagulation cascade, with a more nuanced understanding of interactions between coagulation and inflammatory system, inflammation can be targeted for the prevention and treatment of VTE and to blunt hyper-inflammatory and pro-thrombotic states. Accumulating evidence suggests that anti-inflammatory therapy plays a beneficial role in COVID-19. However, an important consideration in anti-inflammatory therapy is to balance the benefits of mitigating cytokine storm and the risk of immunosuppression hampering viral clearance. In advanced critically ill patients, the positive feedback loop between inflammation and the coagulation cascade leads to thrombosis and depletion of coagulation factors, which anti-thrombotic therapy cannot reverse. Consequently, early anti-thrombotic therapy can reduce adverse outcomes and improve the prognosis of COVID-19 patients.
Author contributions
HJ and JS wrote the draft. XW, MX, and LL reviewed the manuscript, and JS and VN revised the manuscript and JS provided funding support. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Farkouh ME, Stone GW, Lala A, Bagiella E, Moreno PR, Nadkarni GN, et al. Anticoagulation in patients with COVID-19: JACC review topic of the week. J Am Coll Cardiol (2022) 79(9):917–28. doi: 10.1016/j.jacc.2021.12.023
2. Jiménez D, García-Sanchez A, Rali P, Muriel A, Bikdeli B, Ruiz-Artacho P, et al. Incidence of VTE and bleeding among 468Hospitalized patients with coronavirus disease 2019: A systematic review and meta-analysis. Chest (2021) 159(3):1182–4691196. doi: 10.1016/j.chest.2020.11.005
3. Mansory EM, Srigunapalan S, Lazo-Langner A. Venous thromboembolism in hospitalized critical and noncritical COVID-19 patients: A systematic review and meta-analysis. TH Open (2021) 5(3):e286–94. doi: 10.1055/s-0041-1730967
4. Nopp S, Moik F, Jilma B, Pabinger I, Ay C. Risk of venous thromboembolism in patients with COVID-19: A systematic review and meta-analysis. Res Pract Thromb Haemost (2020) 4(7):1178–91. doi: 10.1002/rth2.12439
5. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in covid-19. N Engl J Med (2020) 383(2):120–8. doi: 10.1056/NEJMoa2015432
6. O'Donnell JS, Peyvandi F, Martin-Loeches I. Pulmonary immuno-thrombosis in COVID-19 ARDS pathogenesis. Intensive Care Med (2021) 47(8):899–902. doi: 10.1007/s00134-021-06419-w
7. Pitt B, Tate AM, Gluck D, Rosenson RS, Goonewardena SN. Repurposing low-dose naltrexone for the prevention and treatment of immunothrombosis in COVID-19. Eur Heart J Cardiovasc Pharmacother (2022) 8(4):402–5. doi: 10.1093/ehjcvp/pvac014
8. Corrales-Medina VF, Musher DM, Wells GA, Chirinos JA, Chen L, Fine MJ. Cardiac complications in patients with community-acquired pneumonia: Incidence, timing, risk factors, and association with short-term mortality. Circulation (2012) 125(6):773–81. doi: 10.1161/CIRCULATIONAHA.111.040766
9. Palankar R, Greinacher A. Challenging the concept of immunothrombosis. Blood (2019) 133(6):508–9. doi: 10.1182/blood-2018-11-886267
10. Wienkamp AK, Erpenbeck L, Rossaint J. Platelets in the NETworks interweaving inflammation and thrombosis. Front Immunol (2022) 3:953129. doi: 10.3389/fimmu.2022.953129
11. Colling ME, Tourdot BE, Kanthi Y. Inflammation, infection and venous thromboembolism. Circ Res (2021) 128(12):2017–36. doi: 10.1161/CIRCRESAHA.121.318225
12. Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol (2013) 13(1):34–45. doi: 10.1038/nri3345
13. Bovill EG, van der Vliet A. Venous valvular stasis-associated hypoxia and thrombosis: What is the link? Annu Rev Physiol (2011) 73:527–45. doi: 10.1146/annurev-physiol-012110-142305
14. Jian D, Wang Y, Jian L, Tang H, Rao L, Chen K, et al. METTL14 aggravates endothelial inflammation and atherosclerosis by increasing FOXO1 N6-methyladeosine modifications. Theranostics (2020) 10(20):8939–56. doi: 10.7150/thno.45178
15. Stark K, Massberg S. Interplay between inflammation and thrombosis in cardiovascular pathology. Nat Rev Cardiol (2021) 18(9):666–82. doi: 10.1038/s41569-021-00552-1
16. Wagner DD, Heger LA. Thromboinflammation: from atherosclerosis to COVID-19. Arterioscler Thromb Vasc Biol (2022) 42(9):1103–12. doi: 10.1161/ATVBAHA.122.317162
17. Ng H, Havervall S, Rosell A, Aguilera K, Parv K, von Meijenfeldt FA, et al. Circulating markers of neutrophil extracellular traps are of prognostic value in patients with COVID-19. Arterioscler Thromb Vasc Biol (2021) 41(2):988–94. doi: 10.1161/ATVBAHA.120.315267
18. Iba T, Levy JH, Connors JM, Warkentin TE, Thachil J, Levi M. The unique characteristics of COVID-19 coagulopathy. Crit Care (2020) 24(1):360. doi: 10.1186/s13054-020-03077-0
19. Lowery SA, Sariol A, Perlman S. Innate immune and inflammatory responses to SARS-CoV-2: Implications for COVID-19. Cell Host Microbe (2021) 29(7):1052–62. doi: 10.1016/j.chom.2021.05.004
20. Nagaraja S, Jain D, Kesavardhana S. Inflammasome regulation in driving COVID-19 severity in humans and immune tolerance in bats. J Leukoc Biol (2022) 111(2):497–508. doi: 10.1002/JLB.4COVHR0221-093RR
21. Zhou YW, Xie Y, Tang LS, Pu D, Zhu YJ, Liu JY, et al. Therapeutic targets and interventional strategies in COVID-19: Mechanisms and clinical studies. Signal Transduct Target Ther (2021) 6(1):317. doi: 10.1038/s41392-021-00733-x
22. Islamuddin M, Mustfa SA, Ullah SNMN, Omer U, Kato K, Parveen S. Innate immune response and inflammasome activation during SARS-CoV-2 infection. Inflammation (2022) 45(5):1849–63. doi: 10.1007/s10753-022-01651-y
23. Lu Q, Liu J, Zhao S, Gomez Castro MF, Laurent-Rolle M, Dong J, et al. SARS-CoV-2 exacerbates proinflammatory responses in myeloid cells through c-type lectin receptors and tweety family member 2. Immunity (2021) 54(6):1304–1319.e9. doi: 10.1016/j.immuni.2021.05.006
24. Sung PS, Hsieh SL. C-type lectins and extracellular vesicles in virus-induced NETosis. . J BioMed Sci (2021) 28(1):46. doi: 10.1186/s12929-021-00741-7
25. Sung PS, Huang TF, Hsieh SL. Extracellular vesicles from CLEC2-activated platelets enhance dengue virus-induced lethality via CLEC5A/TLR2. Nat Commun (2019) 10(1):2402. doi: 10.1038/s41467-019-10360-4
26. Youn YJ, Lee YB, Kim SH, Jin HK, Bae JS, Hong CW. Nucleocapsid and spike proteins of SARS-CoV-2 drive neutrophil extracellular trap formation. Immune Netw (2021) 21(2):e16. doi: 10.4110/in.2021.21.e16
27. Sung PS, Yang SP, Peng YC, Sun CP, Tao MH, Hsieh SL. CLEC5A and TLR2 are critical in SARS-CoV-2-induced NET formation and lung inflammation. J BioMed Sci (2022) 29(1):52. doi: 10.1186/s12929-022-00832-z
28. Wielgat P, Narejko K, Car H. SARS-CoV-2 attacks in the brain: Focus on the sialome. Cells (2022) 11(9):1458. doi: 10.3390/cells11091458
29. Tang Y, Liu J, Zhang D, Xu Z, Ji J, Wen C. Cytokine storm in COVID-19: The current evidence and treatment strategies. Front Immunol (2020) 11:1708. doi: 10.3389/fimmu.2020.01708
30. Melms JC, Biermann J, Huang H, Wang Y, Nair A, Tagore S, et al. A molecular single-cell lung atlas of lethal COVID-19. Nature (2021) 595(7865):114–9. doi: 10.1038/s41586-021-03569-1
31. Ruan Q, Yang K, Wang W, Jiang L, Song J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from wuhan, China. Intensive Care Med (2020) 46(5):846–8. doi: 10.1007/s00134-020-05991-x
32. Teuwen LA, Geldhof V, Pasut A, Carmeliet P. COVID-19: the vasculature unleashed. Nat Rev Immunol (2020) 20(7):389–91. doi: 10.1038/s41577-020-0343-0
33. Gorog DA, Storey RF, Gurbel PA, Tantry US, Berger JS, Chan MY, et al. Current and novel biomarkers of thrombotic risk in COVID-19: a consensus statement from the international COVID-19 thrombosis biomarkers colloquium. Nat Rev Cardiol (2022) 19(7):475–95. doi: 10.1038/s41569-021-00665-7
34. Del Valle DM, Kim-Schulze S, Huang HH, Beckmann ND, Nirenberg S, Wang B, et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat Med (2020) 26(10):1636–43. doi: 10.1038/s41591-020-1051-9
35. Smilowitz NR, Kunichoff D, Garshick M, Shah B, Pillinger M, Hochman JS, et al. C-reactive protein and clinical outcomes in patients with COVID-19. Eur Heart J (2021) 42(23):2270–9. doi: 10.1093/eurheartj/ehaa1103
36. Chernysh IN, Nagaswami C, Kosolapova S, Peshkova AD, Cuker A, Cines DB, et al. The distinctive structure and composition of arterial and venous thrombi and pulmonary emboli. Sci Rep (2020) 10(1):5112. doi: 10.1038/s41598-020-59526-x
37. Wolberg AS, Aleman MM, Leiderman K, Machlus KR. Procoagulant activity in hemostasis and thrombosis: Virchow's triad revisited. Anesth Analg (2012) 114(2):275–85. doi: 10.1213/ANE.0b013e31823a088c
38. Wallez Y, Huber P. Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochim Biophys Acta (2008) 1778(3):794–809. doi: 10.1016/j.bbamem.2007.09.003
39. Ni W, Yang X, Yang D, Bao J, Li R, Xiao Y, et al. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit Care (2020) 24(1):422. doi: 10.1186/s13054-020-03120-0
40. Fraga-Silva RA, Da Silva DG, Montecucco F, Mach F, Stergiopulos N, da Silva RF, et al. The angiotensin-converting enzyme 2/Angiotensin-(1-7)/Mas receptor axis: a potential target for treating thrombotic diseases. Thromb Haemost (2012) 108(6):1089–96. doi: 10.1160/TH12-06-0396
41. Jackson SP, Darbousset R, Schoenwaelder SM. Thromboinflammation: Challenges of therapeutically targeting coagulation and other host defense mechanisms. Blood (2019) 133(9):906–18. doi: 10.1182/blood-2018-11-882993
42. Esmon CT. Coagulation inhibitors in inflammation. Biochem Soc Trans (2005) 33(Pt 2):401–5. doi: 10.1042/BST0330401
43. Martin FA, Murphy RP, Cummins PM. Thrombomodulin and the vascular endothelium: Insights into functional, regulatory, and therapeutic aspects. Am J Physiol Heart Circ Physiol (2013) 304(12):H1585–97. doi: 10.1152/ajpheart.00096.2013
44. Ito T, Thachil J, Asakura H, Levy JH, Iba T. Thrombomodulin in disseminated intravascular coagulation and other critical conditions-a multi-faceted anticoagulant protein with therapeutic potential. Crit Care (2019) 23(1):280. doi: 10.1186/s13054-019-2552-0
45. Jin RC, Voetsch B, Loscalzo J. Endogenous mechanisms of inhibition of platelet function. Microcirculation (2005) 12(3):247–58. doi: 10.1080/10739680590925493
46. Libby P, Lüscher T. COVID-19 is, in the end, an endothelial disease. Eur Heart J (2020) 41(32):3038–44. doi: 10.1093/eurheartj/ehaa623
47. Levi M, van der Poll T. Inflammation and coagulation. Crit Care Med (2010) 38(2Suppl):S26–34. doi: 10.1097/CCM.0b013e3181c98d21
48. Welsh JD, Hoofnagle MH, Bamezai S, Oxendine M, Lim L, Hall JD, et al. Hemodynamic regulation of perivalvular endothelial gene expression prevents deep venous thrombosis. J Clin Invest (2019) 129(12):5489–500. doi: 10.1172/JCI124791
49. Brooks EG, Trotman W, Wadsworth MP, Taatjes DJ, Evans MF, Ittleman FP, et al. Valves of the deep venous system: an overlooked risk factor. Blood (2009) 114(6):1276–9. doi: 10.1182/blood-2009-03-209981
50. Radermecker C, Detrembleur N, Guiot J, Cavalier E, Henket M, d'Emal C, et al. Neutrophil extracellular traps infiltrate the lung airway, interstitial, and vascular compartments in severe COVID-19. J Exp Med (2020) 217(12):e20201012. doi: 10.1084/jem.20201012
51. Flaumenhaft R, Enjyoji K, Schmaier AA. Vasculopathy in COVID-19. Blood (2022) 140(3):222–35. doi: 10.1182/blood.2021012250
52. Mancini I, Baronciani L, Artoni A, Colpani P, Biganzoli M, Cozzi G, et al. The ADAMTS13-von willebrand factor axis in COVID-19 patients. J Thromb Haemost (2021) 19(2):513–21. doi: 10.1111/jth.15191
53. Martinelli N, Montagnana M, Pizzolo F, Friso S, Salvagno GL, Forni GL, et al. A relative ADAMTS13 deficiency supports the presence of a secondary microangiopathy in COVID 19. Thromb Res (2020) 193:170–2. doi: 10.1016/j.thromres.2020.07.034
54. Favaloro EJ, Henry BM, Lippi G. Increased VWF and decreased ADAMTS-13 in COVID-19: Creating a milieu for (Micro) thrombosis. Semin Thromb Hemost (2021) 47(4):400–18. doi: 10.1055/s-0041-1727282
55. Fenyves BG, Mehta A, Kays KR. MGH COVID-19 collection & processing team, Goldberg MB, hacohen n, filbin MR. plasma p-selectin is an early marker of thromboembolism in COVID-19. Am J Hematol (2021). doi: 10.1101/2021.07.10.21260293
56. Barrett TJ, Bilaloglu S, Cornwell M, Burgess HM, Virginio VW, Drenkova K, et al. Platelets contribute to disease severity in COVID-19. J Thromb Haemost (2021) 19(12):3139–53. doi: 10.1111/jth.15534
57. Barrett TJ, Lee AH, Xia Y, Lin LH, Black M, Cotzia P, et al. Platelet and vascular biomarkers associate with thrombosis and death in coronavirus disease. Circ Res (2020) 127(7):945–7. doi: 10.1161/CIRCRESAHA.120.317803
58. Stark K, Philippi V, Stockhausen S, Busse J, Antonelli A, Miller M, et al. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood (2016) 128(20):2435–49. doi: 10.1182/blood-2016-04-710632
59. Ding J, Song B, Xie X, Li X, Chen Z, Wang Z, et al. Inflammation in cerebral venous thrombosis. Front Immunol (2022) 13:833490. doi: 10.3389/fimmu.2022.833490
60. Manne BK, Denorme F, Middleton EA, Portier I, Rowley JW, Stubben C, et al. Platelet gene expression and function in patients with COVID-19. Blood (2020) 136(11):1317–29. doi: 10.1182/blood
61. Hottz ED, Azevedo-Quintanilha IG, Palhinha L, Teixeira L, Barreto EA, Pão CRR, et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood (2020) 136(11):1330–41. doi: 10.1182/blood.2020007252
62. Nieuwdorp M, van Haeften TW, Gouverneur MC, Mooij HL, van Lieshout MH, Levi M, et al. Loss of endothelial glycocalyx during acute hyperglycemia coincides with endothelial dysfunction and coagulation activation in vivo. Diabetes (2006) 55(2):480–6. doi: 10.2337/diabetes.55.02.06.db05-1103
63. Noubouossie DF, Whelihan MF, Yu YB, Sparkenbaugh E, Pawlinski R, Monroe DM, et al. In vitro activation of coagulation by human neutrophil DNA and histone proteins but not neutrophil extracellular traps. Blood (2017) 129(8):1021–9. doi: 10.1182/blood-2016-06-722298
64. Blanch-Ruiz MA, Ortega-Luna R, Gómez-García G, Martínez-Cuesta MÁ, Álvarez Á. Role of neutrophil extracellular traps in COVID-19 progression: An insight for effective treatment. Biomedicines (2021) 10(1):31. doi: 10.3390/biomedicines10010031
65. Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, et al. Neutrophil extracellular traps in COVID-19. JCI Insight (2020) 5(11):e138999. doi: 10.1172/jci.insight.138999
66. Veras FP, Pontelli MC, Silva CM, Toller-Kawahisa JE, de Lima M, Nascimento DC, et al. SARS-CoV-2-Triggered neutrophil extracellular traps mediate COVID-19 pathology. J Exp Med (2020) 217(12):e20201129. doi: 10.1084/jem.20201129
67. Guéant JL, Fromonot J, Guéant-Rodriguez RM, Lacolley P, Guieu R, Regnault V. Blood myeloperoxidase-DNA, a biomarker of early response to SARS-CoV-2 infection? Allergy (2021) 76(3):892–6. doi: 10.1111/all.14533
68. Schurink B, Roos E, Radonic T, Barbe E, Bouman CSC, de Boer HH, et al. Viral presence and immunopathology in patients with lethal COVID-19: a prospective autopsy cohort study. Lancet Microbe (2020) 1(7):e290–9. doi: 10.1016/S2666-5247(20)30144-0
69. Mackman N. Triggers, targets and treatments for thrombosis. Nature (2008) 451(7181):914–8. doi: 10.1038/nature06797
70. Ruf W. Role of thiol pathways in TF procoagulant regulation. Thromb Res (2012) 129 Suppl 2(Suppl 2):S11–2. doi: 10.1016/j.thromres.2012.02.020
71. Popescu NI, Lupu C, Lupu F. Extracellular protein disulfide isomerase regulates coagulation on endothelial cells through modulation of phosphatidylserine exposure. Blood (2010) 116(6):993–1001. doi: 10.1182/blood-2009-10-249607
72. Beckmann L, Rolling CC, Voigtländer M, Mäder J, Klingler F, Schulenkorf A, et al. Bacitracin and rutin regulate tissue factor production in inflammatory monocytes and acute myeloid leukemia blasts. Cancers (Basel) (2021) 13(16):3941. doi: 10.3390/cancers13163941
73. Cappellano G, Raineri D, Rolla R, Giordano M, Puricelli C, Vilardo B, et al. Circulating platelet-derived extracellular vesicles are a hallmark of sars-Cov-2 infection. Cells (2021) 10(1):85. doi: 10.3390/cells10010085
74. Garnier Y, Claude L, Hermand P, Sachou E, Claes A, Desplan K, et al. Plasma microparticles of intubated COVID-19 patients cause endothelial cell death, neutrophil adhesion and netosis, in a phosphatidylserine-dependent manner. Br J Haematol (2022) 196(5):1159–69. doi: 10.1111/bjh.18019
75. Guervilly C, Bonifay A, Burtey S, Sabatier F, Cauchois R, Abdili E, et al. Dissemination of extreme levels of extracellular vesicles: Tissue factor activity in patients with severe COVID-19. Blood Adv (2021) 5(3):628–34. doi: 10.1182/bloodadvances.2020003308
76. Middleton EA, He XY, Denorme F, Campbell RA, Ng D, Salvatore SP, et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood (2020) 136(10):1169–79. doi: 10.1182/blood.2020007008
77. Bevers EM, Williamson PL. Getting to the outer leaflet: Physiology of phosphatidylserine exposure at the plasma membrane. Physiol Rev (2016) 96(2):605–45. doi: 10.1152/physrev.00020.2015
78. Shin HW, Takatsu H. Phosphatidylserine exposure in living cells. Crit Rev Biochem Mol Biol (2020) 55(2):166–78. doi: 10.1080/10409238.2020.1758624
79. Yang X, Cheng X, Tang Y, Qiu X, Wang Y, Kang H, et al. Bacterial endotoxin activates the coagulation cascade through gasdermin d-dependent phosphatidylserine exposure. Immunity (2019) 51(6):983–996.e6. doi: 10.1016/j.immuni.2019.11.005
80. Tripisciano C, Weiss R, Karuthedom George S, Fischer MB, Weber V. Extracellular vesicles derived from platelets, red blood cells, and monocyte-like cells differ regarding their ability to induce factor XII-dependent thrombin generation. Front Cell Dev Biol (2020) 8:298. doi: 10.3389/fcell.2020.00298
81. Millington-Burgess SL, Bonna AM, Rahman T, Harper MT. Ethaninidothioic acid (R5421) is not a selective inhibitor of platelet phospholipid scramblase activity. Br J Pharmacol (2020) 177(17):4007–20. doi: 10.1111/bph.15152
82. Rausch L, Lutz K, Schifferer M, Winheim E, Gruber R, Oesterhaus EF, et al. Binding of phosphatidylserine-positive microparticles by PBMCs classifies disease severity in COVID-19 patients. J Extracell Vesicles (2021) 10(14):e12173. doi: 10.1002/jev2.12173
83. Wang C, Yu C, Novakovic VA, Xie R, Shi J. Circulating microparticles in the pathogenesis and early anticoagulation of thrombosis in COVID-19 with kidney injury. Front Cell Dev Biol (2022) 9:784505. doi: 10.3389/fcell.2021.784505
84. Althaus K, Marini I, Zlamal J, Pelzl L, Singh A, Häberle H, et al. Antibody-induced procoagulant platelets in severe COVID-19 infection. Blood (2021) 137(8):1061–71. doi: 10.1182/blood.2020008762
85. Lim EHT, van Amstel RBE, de Boer VV, van Vught LA, de Bruin S, Brouwer MC, et al. Complement activation in COVID-19 and targeted therapeutic options: A scoping review. Blood Rev (2022) 100995. doi: 10.1016/j.blre.2022.100995
86. Lim MS, Mcrae S. COVID-19 and immunothrombosis: Pathophysiology and therapeutic implications. Crit Rev Oncol Hematol (2021) 168:103529. doi: 10.1016/j.critrevonc.2021.103529
87. Esmon CT. Inflammation and the activated protein c anticoagulant pathway. Semin Thromb Hemost (2006) 32 Suppl 1:49–60. doi: 10.1055/s-2006-939554
88. Skendros P, Mitsios A, Chrysanthopoulou A, Mastellos DC, Metallidis S, Rafailidis P, et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J Clin Invest (2020) 130(11):6151–7. doi: 10.1172/JCI141374
89. Hamilton KK, Hattori R, Esmon CT, Sims PJ. Complement proteins C5b-9 induce vesiculation of the endothelial plasma membrane and expose catalytic surface for assembly of the prothrombinase enzyme complex. J Biol Chem (1990) 265(7):3809–14. doi: 10.1016/S0021-9258(19)39666-8
90. Peerschke EI, Yin W, Ghebrehiwet B. Complement activation on platelets: Implications for vascular inflammation and thrombosis. Mol Immunol (2010) 47(13):2170–5. doi: 10.1016/j.molimm.2010.05.009
91. Polley MJ, Nachman R. The human complement system in thrombin-mediated platelet function. J Exp Med (1978) 147(6):1713–26. doi: 10.1084/jem.147.6.1713
92. Verschoor A, Neuenhahn M, Navarini AA, Graef P, Plaumann A, Seidlmeier A, et al. A platelet-mediated system for shuttling blood-borne bacteria to CD8α+ dendritic cells depends on glycoprotein GPIb and complement C3. Nat Immunol (2011) 12(12):1194–201. doi: 10.1038/ni.2140
93. Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl Res (2020) 220:1–13. doi: 10.1016/j.trsl.2020.04.007
94. Della-Morte D, Pacifici F, Ricordi C, Massoud R, Rovella V, Proietti S, et al. Low level of plasminogen increases risk for mortality in COVID-19 patients. Cell Death Dis (2021) 12(8):773. doi: 10.1038/s41419-021-04070-3
95. Gacche RN, Gacche RA, Chen J, Li H, Li G. Predictors of morbidity and mortality in COVID-19. Eur Rev Med Pharmacol Sci (2021) 25(3):1684–707. doi: 10.26355/eurrev_202102_24880
96. Bachler M, Bösch J, Stürzel DP, Hell T, Giebl A, Ströhle M, et al. Impaired fibrinolysis in critically ill COVID-19 patients. Br J Anaesth (2021) 126(3):590–8. doi: 10.1016/j.bja.2020.12.010
97. Meltzer ME, Lisman T, de Groot PG, Meijers JC, le Cessie S, Doggen CJ, et al. Venous thrombosis risk associated with plasma hypofibrinolysis is explained by elevated plasma levels of TAFI and PAI-1. Blood (2010) 116(1):113–21. doi: 10.1182/blood-2010-02-267740
98. Susen S, Tacquard CA, Godon A, Mansour A, Garrigue D, Nguyen P, et al. Prevention of thrombotic risk in hospitalized patients with COVID-19 and hemostasis monitoring. Crit Care (2020) 24(1):364. doi: 10.1186/s13054-020-03000-7
99. Heuberger DM, Schuepbach RA. Protease-activated receptors (PARs): Mechanisms of action and potential therapeutic modulators in PAR-driven inflammatory diseases. Thromb J (2019) 17:4. doi: 10.1186/s12959-019-0194-8
100. Tull SP, Bevins A, Kuravi SJ, Satchell SC, Al-Ani B, Young SP, et al. PR3 and elastase alter PAR1 signaling and trigger vWF release via a calcium-independent mechanism from glomerular endothelial cells. PloS One (2012) 7(8):e43916. doi: 10.1371/journal.pone.0043916
101. Coppinger JA, Cagney G, Toomey S, Kislinger T, Belton O, McRedmond JP, et al. Characterization of the proteins released from activated platelets leads to localization of novel platelet proteins in human atherosclerotic lesions. Blood (2004) 103(6):2096–104. doi: 10.1182/blood-2003-08-2804
102. Coughlin SR. How the protease thrombin talks to cells. Proc Natl Acad Sci U.S.A. (1999) 96(20):11023–7. doi: 10.1073/pnas.96.20.11023
103. Kaplan ZS, Zarpellon A, Alwis I, Yuan Y, McFadyen J, Ghasemzadeh M, et al. Thrombin-dependent intravascular leukocyte trafficking regulated by fibrin and the platelet receptors GPIb and PAR4. Nat Commun (2015) 6:7835. doi: 10.1038/ncomms8835
104. Busch MH, Timmermans SAMEG, Nagy M, Visser M, Huckriede J, Aendekerk JP, et al. Neutrophils and contact activation of coagulation as potential drivers of COVID-19. Circulation (2020) 142(18):1787–90. doi: 10.1161/CIRCULATIONAHA.120.050656
105. Wiech M, Chroscicki P, Swatler J, Stepnik D, De Biasi S, Hampel M, et al. Remodeling of T cell dynamics during long COVID is dependent on severity of SARS-CoV-2 infection. Front Immunol (2022) 13:886431. doi: 10.3389/fimmu.2022.886431
106. Subramanian A, Nirantharakumar K, Hughes S, Myles P, Williams T, Gokhale KM, et al. Symptoms and risk factors for long COVID in non-hospitalized adults. Nat Med (2022) 28(8):1706–14. doi: 10.1038/s41591-022-01909-w
107. Prasannan N, Heightman M, Hillman T, Wall E, Bell R, Kessler A, et al. Impaired exercise capacity in post-COVID-19 syndrome: the role of VWF-ADAMTS13 axis. Blood Adv (2022) 6(13):4041–8. doi: 10.1182/bloodadvances
108. Sudre CH, Murray B, Varsavsky T, Graham MS, Penfold RS, Bowyer RC, et al. Attributes and predictors of long COVID. Nat Med (2021) 27(4):626–31. doi: 10.1038/s41591-021-01292-y
109. Blomberg B, Mohn KG, Brokstad KA, Zhou F, Linchausen DW, Hansen BA, et al. Long COVID in a prospective cohort of home-isolated patients. Nat Med (2021) 27(9):1607–13. doi: 10.1038/s41591-021-01433-3
110. Honigsbaum M, Krishnan L. Taking pandemic sequelae seriously: from the Russian influenza to COVID-19 long-haulers. Lancet (2020) 396(10260):1389–91. doi: 10.1016/S0140-6736(20)32134-6
111. Huang C, Huang L, Wang Y, Li X, Ren L, Gu X, et al. 6-month consequences of COVID-19 in patients discharged from hospital: a cohort study. Lancet (2021) 397(10270):220–32. doi: 10.1016/S0140-6736(20)32656-8
112. Heesakkers H, van der Hoeven JG, Corsten S, Janssen I, Ewalds E, Simons KS, et al. Clinical outcomes among patients with 1-year survival following intensive care unit treatment for COVID-19. JAMA (2022) 327(6):559–65. doi: 10.1001/jama.2022.0040
113. Patell R, Bogue T, Koshy A, Bindal P, Merrill M, Aird WC, et al. Postdischarge thrombosis and hemorrhage in patients with COVID-19. Blood (2020) 136(11):1342–6. doi: 10.1182/blood.2020007938
114. Engelen MM, Vandenbriele C, Balthazar T, Claeys E, Gunst J, Guler I, et al. Venous thromboembolism in patients discharged after COVID-19 hospitalization. Semin Thromb Hemost (2021) 47(4):362–71. doi: 10.1055/s-0041-1727284
115. Montani D, Savale L, Noel N, Meyrignac O, Colle R, Gasnier M, et al. Post-acute COVID-19 syndrome. Eur Respir Rev (2022) 31(163):210185. doi: 10.1183/16000617.0185-2021
116. Taquet M, Dercon Q, Harrison PJ. Six-month sequelae of post-vaccination SARS-CoV-2 infection: A retrospective cohort study of 10,024 breakthrough infections. Brain Behav Immun (2022) 103:154–62. doi: 10.1016/j.bbi.2022.04.013
117. Butt AA, Nafady-Hego H, Chemaitelly H, Abou-Samra AB, Khal AA, Coyle PV, et al. Outcomes among patients with breakthrough SARS-CoV-2 infection after vaccination. Int J Infect Dis (2021) 110:353–8. doi: 10.1016/j.ijid.2021.08.008
118. Al-Aly Z, Bowe B, Xie Y. Long COVID after breakthrough SARS-CoV-2 infection. Nat Med (2022) 28(7):1461–7. doi: 10.1038/s41591-022-01840-0
119. Flemming A. Vaccines only partially protect against long COVID. Nat Rev Immunol (2022) 22(7):410. doi: 10.1038/s41577-022-00749-6
120. Newell KL, Waickman AT. Inflammation, immunity, and antigen persistence in post-acute sequelae of SARS-CoV-2 infectionimmunity and inflammaion in post-acute sequelae of SARS-CoV-2 infection. Curr Opin Immunol (2022) 77:102228. doi: 10.1016/j.coi.2022.102228
121. Leppkes M, Neurath MF. Rear window-what can the gut tell us about long-COVID? Gastroenterology (2022) 163(2):376–8. doi: 10.1053/j.gastro.2022.05.044
122. Couzin-Frankel J. Clues to long COVID. Science (2022) 376(6599):1261–5. doi: 10.1126/science.add4297
123. Ledford H. Can drugs reduce the risk of long COVID? what scientists know so far. Nature (2022) 604(7904):20–1. doi: 10.1038/d41586-022-00823-y
124. Colmenero I, Santonja C, Alonso-Riaño M, Noguera-Morel L, Hernández-Martín A, Andina D, et al. SARS-CoV-2 endothelial infection causes COVID-19 chilblains: Histopathological, immunohistochemical and ultrastructural study of seven paediatric cases. Br J Dermatol (2020) 183(4):729–37. doi: 10.1111/bjd.19327
125. Khider L, Gendron N, Goudot G, Chocron R, Hauw-Berlemont C, Cheng C, et al. Curative anticoagulation prevents endothelial lesion in COVID-19 patients. J Thromb Haemost (2020) 18(9):2391–9. doi: 10.1111/jth.14968
126. Gyöngyösi M, Alcaide P, Asselbergs FW, Brundel BJJM, Camici GG, da Costa Martins P, et al. Long COVID and the cardiovascular system - elucidating causes and cellular mechanisms in order to develop targeted diagnostic and therapeutic strategies: A joint scientific statement of the ESC working groups on cellular biology of the heart and myocardial & pericardial diseases. Cardiovasc Res (2022). doi: 10.1093/cvr/cvac115
127. Targosz-Korecka M, Kubisiak A, Kloska D, Kopacz A, Grochot-Przeczek A, Szymonski M. Endothelial glycocalyx shields the interaction of SARS-CoV-2 spike protein with ACE2 receptors. Sci Rep (2021) 11(1):12157. doi: 10.1038/s41598-021-91231-1
128. Prasad M, Leon M, Lerman LO, Lerman A. Viral endothelial dysfunction: A unifying mechanism for COVID-19. Mayo Clin Proc (2021) 96(12):3099–108. doi: 10.1016/j.mayocp.2021.06.027
129. De Lorenzo A, Escobar S, Tibiriçá E. Systemic endothelial dysfunction: A common pathway for COVID-19, cardiovascular and metabolic diseases. Nutr Metab Cardiovasc Dis (2020) 30(8):1401–2. doi: 10.1016/j.numecd.2020.05.007
130. Escher R, Breakey N, Lämmle B. Severe COVID-19 infection associated with endothelial activation. Thromb Res (2020) 190:62. doi: 10.1016/j.thromres.2020.04.014
131. Armstrong SM, Darwish I, Lee WL. Endothelial activation and dysfunction in the pathogenesis of influenza a virus infection. Virulence (2013) 4(6):537–42. doi: 10.4161/viru.25779
132. Krautkrämer E, Zeier M, Plyusnin A. Hantavirus infection: an emerging infectious disease causing acute renal failure. Kidney Int (2013) 83(1):23–7. doi: 10.1038/ki.2012.360
133. Evans PC, Rainger GE, Mason JC, Guzik TJ, Osto E, Stamataki Z, et al. Endothelial dysfunction in COVID-19: a position paper of the ESC working group for atherosclerosis and vascular biology, and the ESC council of basic cardiovascular science. Cardiovasc Res (2020) 116(14):2177–84. doi: 10.1093/cvr/cvaa230
134. Xie Y, Xu E, Bowe B, Al-Aly Z. Long-term cardiovascular outcomes of COVID-19. Nat Med (2022) 28(3):583–90. doi: 10.1038/s41591-022-01689-3
135. Izzo R, Trimarco V, Mone P, Aloè T, MC M, Diana A, et al. Combining l-arginine with vitamin c improves long-COVID symptoms: The nationwide multicenter LINCOLN study. Pharmacol Res (2022) 106360. doi: 10.1016/j.phrs.2022.106360
136. Oikonomou E, Souvaliotis N, Lampsas S, Siasos G, Poulakou G, Theofilis P, et al. Endothelial dysfunction in acute and long standing COVID-19: A prospective cohort study. Vascul Pharmacol (2022) 144:106975. doi: 10.1016/j.vph.2022.106975
137. Chioh FW, Fong SW, Young BE, Wu KX, Siau A, Krishnan S, et al. Convalescent COVID-19 patients are susceptible to endothelial dysfunction due to persistent immune activation. Elife (2021) 10:e64909. doi: 10.7554/eLife.64909
138. Fogarty H, Townsend L, Morrin H, Ahmad A, Comerford C, Karampini E, et al. Persistent endotheliopathy in the pathogenesis of long COVID syndrome. J Thromb Haemost (2021) 19(10):2546–53. doi: 10.1111/jth.15490
139. Mejia-Renteria H, Travieso A, Sagir A, Martínez-Gómez E, Carrascosa-Granada A, Toya T, et al. In-vivo evidence of systemic endothelial vascular dysfunction in COVID-19. Int J Cardiol (2021) 345:153–5. doi: 10.1016/j.ijcard.2021.10.140
140. Szeghy RE, Province VM, Stute NL, Augenreich MA, Koontz LK, Stickford JL, et al. Carotid stiffness, intima-media thickness and aortic augmentation index among adults with SARS-CoV-2. Exp Physiol (2022) 107(7):694–707. doi: 10.1113/EP089481
141. Gaitzsch E, Czermak T, Ribeiro A, Heun Y, Bohmer M, Merkle M, et al. Double-stranded DNA induces a prothrombotic phenotype in the vascular endothelium. Sci Rep (2017) 7(1):1112. doi: 10.1038/s41598-017-01148-x
142. Loscalzo J. Oxidative stress in endothelial cell dysfunction and thrombosis. Pathophysiol Haemost Thromb (2002) 32(5-6):359–60. doi: 10.1159/000073600
143. Kakar P, Lip GY. Hypertension: Endothelial dysfunction, the prothrombotic state and antithrombotic therapy. Expert Rev Cardiovasc Ther (2007) 5(3):441–50. doi: 10.1586/14779072.5.3.441
144. Townsend L, Fogarty H, Dyer A, Martin-Loeches I, Bannan C, Nadarajan P, et al. Prolonged elevation of d-dimer levels in convalescent COVID-19 patients is independent of the acute phase response. J Thromb Haemost (2021) 19(4):1064–70. doi: 10.1111/jth.15267
145. Fan BE, Wong SW, Sum CLL, Lim GH, Leung BP, Tan CW, et al. Hypercoagulability, endotheliopathy, and inflammation approximating 1 year after recovery: Assessing the long-term outcomes in COVID-19 patients. Am J Hematol (2022) 97(7):915–23. doi: 10.1002/ajh.26575
146. Fogarty H, Ward SE, Townsend L, Karampini E, Elliott S, Conlon N, et al. Sustained VWF-ADAMTS-13 axis imbalance and endotheliopathy in long COVID syndrome is related to immune dysfunction. J Thromb Haemost (2022) 20(10):2429–38. doi: 10.1111/jth.15830
147. Petito E, Falcinelli E, Paliani U, Cesari E, Vaudo G, Sebastiano M, et al. Association of neutrophil activation, more than platelet activation, with thrombotic complications in coronavirus disease 2019. J Infect Dis (2021) 223(6):933–44. doi: 10.1093/infdis/jiaa756
148. Zhou Y, Nishikawa M, Kanno H, Yang R, Ibayashi Y, Xiao TH, et al. Long-term effects of pfizer-BioNTech COVID-19 vaccinations on platelets. Cytometry A (2022). doi: 10.1002/cyto.a.24677
149. Zhu Y, Chen X, Liu X. NETosis and neutrophil extracellular traps in COVID-19: Immunothrombosis and beyond. Front Immunol (2022) 13:838011. doi: 10.3389/fimmu.2022.838011
150. Lou M, Yuan D, Liao S, Tong L, Li J. Potential mechanisms of cerebrovascular diseases in COVID-19 patients. J Neurovirol (2021) 27(1):35–51. doi: 10.1007/s13365-021-00948-2
151. Dryden M, Mudara C, Vika C, Blumberg L, Mayet N, Cohen C, et al. Post-COVID-19 condition 3 months after hospitalisation with SARS-CoV-2 in south Africa: A prospective cohort study. Lancet Glob Health (2022) 10(9):e1247–56. doi: 10.1016/S2214-109X(22)00286-8
152. Van Cleemput J, van Snippenberg W, Lambrechts L, Dendooven A, D'Onofrio V, Couck L, et al. Organ-specific genome diversity of replication-competent SARS-CoV-2. Nat Commun (2021) 12(1):6612. doi: 10.1038/s41467-021-26884-7
153. Gaebler C, Wang Z, Lorenzi JCC, Muecksch F, Finkin S, Tokuyama M, et al. Evolution of antibody immunity to SARS-CoV-2. Nature (2021) 591(7851):639–44. doi: 10.1038/s41586-021-03207-w
154. Laing AG, Lorenc A, Del Molino Del Barrio I, Das A, Fish M, Monin L, et al. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat Med (2020) 26(10):1623–35. doi: 10.1038/s41591-020-1038-6
155. Lopez-Leon S, Wegman-Ostrosky T, Perelman C, Sepulveda R, Rebolledo PA, Cuapio A, et al. More than 50 long-term effects of COVID-19: a systematic review and meta-analysis. Sci Rep (2021) 11(1):16144. doi: 10.1038/s41598-021-95565-8
156. Acosta-Ampudia Y, Monsalve DM, Rojas M, Rodríguez Y, Zapata E, Ramírez-Santana C, et al. Persistent autoimmune activation and proinflammatory state in post-coronavirus disease 2019 syndrome. J Infect Dis (2022) 225(12):2155–62. doi: 10.1093/infdis/jiac017
157. Ripani U, Bisaccia M, Meccariello L. Dexamethasone and nutraceutical therapy can reduce the myalgia due to COVID-19 - a systemic review of the active substances that can reduce the expression of interlukin-6. Med Arch (2022) 76(1):66–71. doi: 10.5455/medarh.2022.76.66-71
158. Queiroz MAF, Neves PFMD, Lima SS, Lopes JDC, Torres MKDS, Vallinoto IMVC, et al. Cytokine profiles associated with acute COVID-19 and long COVID-19 syndrome. Front Cell Infect Microbiol (2022) 12:922422. doi: 10.3389/fcimb.2022.922422
159. Shuwa HA, Shaw TN, Knight SB, Wemyss K, McClure FA, Pearmain L, et al. Alterations in T and b cell function persist in convalescent COVID-19 patients. Med (NY) (2021) 2(6):720–735.e4. doi: 10.1016/j.medj.2021.03.013
160. Muecksch F, Wang Z, Cho A, Gaebler C, Ben Tanfous T, DaSilva J, et al. Increased memory b cell potency and breadth after a SARS-CoV-2 mRNA boost. Nature (2022) 607(7917):128–34. doi: 10.1038/s41586-022-04778-y
161. Pitre T, Van Alstine R, Chick G, Leung G, Mikhail D, Cusano E, et al. Antiviral drug treatment for nonsevere COVID-19: a systematic review and network meta-analysis. CMAJ (2022) 194(28):E969–80. doi: 10.1503/cmaj.220471
162. Piccicacco N, Zeitler K, Ing A, Montero J, Faughn J, Silbert S, et al. Real-world effectiveness of early remdesivir and sotrovimab in the highest-risk COVID-19 outpatients during the omicron surge. J Antimicrob Chemother (2022) 77(10):2693–700. doi: 10.1093/jac/dkac256
163. Tao K, Tzou PL, Kosakovsky Pond SL, Ioannidis JPA, Shafer RW. Susceptibility of SARS-CoV-2 omicron variants to therapeutic monoclonal antibodies: Systematic review and meta-analysis. Microbiol Spectr (2022) 10(4):e0092622. doi: 10.1128/spectrum.00926-22
164. Huang DT, McCreary EK, Bariola JR, Minnier TE, Wadas RJ, Shovel JA, et al. Effectiveness of casirivimab-imdevimab and sotrovimab during a SARS-CoV-2 delta variant surge: A cohort study and randomized comparative effectiveness trial. JAMA Netw Open (2022) 5(7):e2220957. doi: 10.1001/jamanetworkopen
165. Hirsch C, Park YS, Piechotta V, Chai KL, Estcourt LJ, Monsef I, et al. SARS-CoV-2-neutralising monoclonal antibodies to prevent COVID-19. Cochrane Database Syst Rev (2022) 6(6):CD014945. doi: 10.1002/14651858
166. Lopes RD, de Barros E Silva PGM, Furtado RHM, Macedo AVS, Bronhara B, Damiani LP, et al. Therapeutic versus prophylactic anticoagulation for patients admitted to hospital with COVID-19 and elevated d-dimer concentration (ACTION): an open-label, multicentre, randomised, controlled trial. Lancet (2021) 397(10291):2253–63. doi: 10.1016/S0140-6736(21)01203-4
167. REMAP-CAP Investigators, ACTIV-4a Investigators, ATTACC Investigators, Goligher EC, Bradbury CA, McVerry BJ, et al. ;Therapeutic anticoagulation with heparin in critically ill patients with covid-19. N Engl J Med (2021) 385(9):777–89. doi: 10.1056/NEJMoa2103417
168. ATTACC Investigators, ACTIV-4a Investigators, REMAP-CAP Investigators, Lawler PR, Goligher EC, Berger JS, et al. ;Therapeutic anticoagulation with heparin in noncritically ill patients with covid-19. N Engl J Med (2021) 385(9):790–802. doi: 10.1056/NEJMoa2105911
169. Farrar JE, Trujillo TC, Mueller SW, Beltran L, Nguyen C, Hassell K, et al. Evaluation of a patient specific, targeted-intensity pharmacologic thromboprophylaxis protocol in hospitalized patients with COVID-19. J Thromb Thrombolysis (2022) 53(2):446–53. doi: 10.1007/s11239-021-02552-x
170. Nadkarni GN, Lala A, Bagiella E, Chang HL, Moreno PR, Pujadas E, et al. Anticoagulation, bleeding, mortality, and pathology in hospitalized patients with COVID-19. J Am Coll Cardiol (2020) 76(16):1815–26. doi: 10.1016/j.jacc.2020.08.041
171. Hoogenboom WS, Lu JQ, Musheyev B, Borg L, Janowicz R, Pamlayne S, et al. Prophylactic versus therapeutic dose anticoagulation effects on survival among critically ill patients with COVID-19. PloS One (2022) 17(1):e0262811. doi: 10.1371/journal.pone.0262811
172. Yamashita Y, Yachi S, Takeyama M, Nishimoto Y, Tsujino I, Nakamura J, et al. Therapeutic-dose vs. prophylactic-dose anticoagulation therapy for critically ill patients with COVID-19 in a practice-based observational study. Circ J (2022) 86(7):1137–42. doi: 10.1253/circj.CJ-22-0209
173. Sofia R, Carbone M, Landoni G, Zangrillo A, Dagna L. Anticoagulation as secondary prevention of massive lung thromboses in hospitalized patients with COVID-19. Eur J Intern Med (2022) 100:21–4. doi: 10.1016/j.ejim.2022.04.009
174. Spyropoulos AC, Goldin M, Giannis D, Diab W, Wang J, Khanijo S, et al. Efficacy and safety of therapeutic-dose heparin vs standard prophylactic or intermediate-dose heparins for thromboprophylaxis in high-risk hospitalized patients with COVID-19: The HEP-COVID randomized clinical trial. JAMA Intern Med (2021) 181(12):1612–20. doi: 10.1001/jamainternmed.2021.6203
175. Ramacciotti E, Barile Agati L, Calderaro D, Aguiar VCR, Spyropoulos AC, de Oliveira CCC, et al. Rivaroxaban versus no anticoagulation for post-discharge thromboprophylaxis after hospitalisation for COVID-19 (MICHELLE): an open-label, multicentre, randomised, controlled trial. Lancet (2022) 399(10319):50–9. doi: 10.1016/S0140-6736(21)02392-8
176. Cuker A, Tseng EK, Nieuwlaat R, Angchaisuksiri P, Blair C, Dane K, et al. American Society of hematology living guidelines on the use of anticoagulation for thromboprophylaxis in patients with COVID-19: May 2021 update on the use of intermediate-intensity anticoagulation in critically ill patients. Blood Adv (2021) 5(20):3951–9. doi: 10.1182/bloodadvances.2021005493
177. Fragkou PC, Palaiodimou L, Stefanou MI, Katsanos AH, Lambadiari V, Paraskevis D, et al. Effects of low molecular weight heparin and fondaparinux on mortality, hemorrhagic and thrombotic complications in COVID-19 patients. Ther Adv Neurol Disord (2022) 15:17562864221099472. doi: 10.1177/17562864221099472
178. Leentjens J, van Haaps TF, Wessels PF, Schutgens REG, Middeldorp S. COVID-19-associated coagulopathy and antithrombotic agents-lessons after 1 year. Lancet Haematol (2021) 8(7):e524–33. doi: 10.1016/S2352-3026(21)00105-8
179. Hong L, Chen G, Cai Z, Liu H, Zhang C, Wang F, et al. Balancing microthrombosis and inflammation via injectable protein hydrogel for inflammatory bowel disease. Adv Sci (Weinh) (2022) 9(20):e2200281. doi: 10.1002/advs.202200281
180. Patel B, Rashid J, Gupta N, Ahsan F. Low-Molecular-Weight heparin-coated and montelukast-filled inhalable particles: A dual-drug delivery system for combination therapy in asthma. J Pharm Sci (2017) 106(4):1124–35. doi: 10.1016/j.xphs.2016.12.025
181. McFadyen JD, Stevens H, Peter K. The emerging threat of (Micro) thrombosis in COVID-19 and its therapeutic implications. Circ Res (2020) 127(4):571–87. doi: 10.1161/CIRCRESAHA.120.317447
182. Renné T, Stavrou EX. Roles of factor XII in innate immunity. Front Immunol (2019) 10:2011. doi: 10.3389/fimmu.2019.02011
183. Mailer RK, Rangaswamy C, Konrath S, Emsley J, Renné T. An update on factor XII-driven vascular inflammation. Biochim Biophys Acta Mol Cell Res (2022) 1869(1):119166. doi: 10.1016/j.bbamcr.2021.119166
184. Eikelboom J, Floege J, Thadhani R, Weitz JI, Winkelmayer WC. Anticoagulation in patients with kidney failure on dialysis: Factor XI as a therapeutic target. Kidney Int (2021) 100(6):1199–207. doi: 10.1016/j.kint.2021.08.028
185. Nopp S, Kraemmer D, Ay C. Factor XI inhibitors for prevention and treatment of venous thromboembolism: A review on the rationale and update on current evidence. Front Cardiovasc Med (2022) 9:903029. doi: 10.3389/fcvm.2022.903029
186. Eikelboom JW, Connolly SJ, Bosch J, Dagenais GR, Hart RG, Shestakovska O, et al. Rivaroxaban with or without aspirin in stable cardiovascular disease. N Engl J Med (2017) 377(14):1319–30. doi: 10.1056/NEJMoa1709118
187. Branch KR, Probstfield JL, Eikelboom JW, Bosch J, Maggioni AP, Cheng RK, et al. Rivaroxaban with or without aspirin in patients with heart failure and chronic coronary or peripheral artery disease. Circulation (2019) 140(7):529–37. doi: 10.1161/CIRCULATIONAHA.119.039609
188. Kirchhof P, Ezekowitz MD, Purmah Y, Schiffer S, Meng IL, Camm AJ, et al. Effects of rivaroxaban on biomarkers of coagulation and inflammation: A Post hoc analysis of the X-VeRT trial. TH Open (2020) 4(1):e20–32. doi: 10.1055/s-0040-1701206
189. Lopes RD, de Barros E Silva PGM, Furtado RHM, Macedo AVS, Ramacciotti E, Damini LP, et al. Randomized clinical trial to evaluate a routine full anticoagulation strategy in patients with coronavirus infection (SARS-CoV2) admitted to hospital: Rationale and design of the ACTION (Anticoagulation coronavirus)-coalition IV trial. Am Heart J (2021) 238:1–11. doi: 10.1016/j.ahj.2021.04.005
190. Sholzberg M, Tang GH, Negri E, Rahhal H, Kreuziger LB, Pompilio CE, et al. Coagulopathy of hospitalised COVID-19: A pragmatic randomised controlled trial of therapeutic anticoagulation versus standard care as a rapid response to the COVID-19 pandemic (RAPID COVID COAG - RAPID Trial): A structured summary of a study protocol for a randomised controlled trial. Trials (2021) 22(1):202. doi: 10.1186/s13063-021-05076-0
191. Prasannan N, Heightman M, Hillman T, Wall E, Bell R, Kessler A, et al. Impaired exercise capacity in post-COVID syndrome: The role of VWF-ADAMTS13 axis. Blood Adv (2022) 6(13):4041–8. doi: 10.1182/bloodadvances.2021006944
192. Ricottini E, Nusca A, Ussia GP, Grigioni F. Antithrombotic treatment for valve prostheses: Which drug, which dose, and when? Prog Cardiovasc Dis (2022) 72:4–14. doi: 10.1016/j.pcad.2022.05.008
193. Douin DJ, Shaefi S, Brenner SK, Gupta S, Park I, Wright FL, et al. Tissue plasminogen activator in critically ill adults with COVID-19. Ann Am Thorac Soc (2021) 18(11):1917–21. doi: 10.1513/AnnalsATS.202102-127RL
194. Ji HL, Dai Y, Zhao R. Fibrinolytic therapy for COVID-19: a review of case series. Acta Pharmacol Sin (2022) 43(8):2168–70. doi: 10.1038/s41401-021-00827-w
195. Ji HL, Zhao R, Matalon S, Matthay MA. Elevated plasmin (ogen) as a common risk factor for COVID-19 susceptibility. Physiol Rev (2020) 100(3):1065–75. doi: 10.1152/physrev.00013.2020
196. Barrett CD, Oren-Grinberg A, Chao E, Moraco AH, Martin MJ, Reddy SH, et al. Rescue therapy for severe COVID-19-Associated acute respiratory distress syndrome with tissue plasminogen activator: A case series. J Trauma Acute Care Surg (2020) 89(3):453–7. doi: 10.1097/TA.0000000000002786
197. Wang J, Hajizadeh N, Moore EE, McIntyre RC, Moore PK, Veress LA, et al. Tissue plasminogen activator (tPA) treatment for COVID-19 associated acute respiratory distress syndrome (ARDS): A case series. J Thromb Haemost (2020) 18(7):1752–5. doi: 10.1111/jth.14828
198. Mackman N, Bergmeier W, Stouffer GA, Weitz JI. Therapeutic strategies for thrombosis: New targets and approaches. Nat Rev Drug Discovery (2020) 19(5):333–52. doi: 10.1038/s41573-020-0061-0
199. Bikdeli B, Madhavan MV, Jimenez D, Chuich T, Dreyfus I, Driggin E, et al. COVID-19 and thrombotic or thromboembolic disease: Implications for prevention, antithrombotic therapy, and follow-up: JACC state-of-the-Art review. J Am Coll Cardiol (2020) 75(23):2950–73. doi: 10.1016/j.jacc.2020.04.031
200. Aliter KF, Al-Horani RA. Potential therapeutic benefits of dipyridamole in COVID-19 patients. Curr Pharm Des (2021) 27(6):866–75. doi: 10.2174/1381612826666201001125604
201. Nappi F, Giacinto O, Ellouze O, Nenna A, Avtaar Singh SS, Chello M, et al. Association between COVID-19 diagnosis and coronary artery thrombosis: A narrative review. Biomedicines (2022) 10(3):702. doi: 10.3390/biomedicines10030702
202. Spaetgens B, Nagy M, Ten Cate H. Antiplatelet therapy in patients with COVID-19-More is less? JAMA (2022) 327(3):223–4. doi: 10.1001/jama.2021.23866
203. Haji Aghajani M, Moradi O, Amini H, Azhdari Tehrani H, Pourheidar E, Rabiei MM, et al. Decreased in-hospital mortality associated with aspirin administration in hospitalized patients due to severe COVID-19. J Med Virol (2021) 93(9):5390–5. doi: 10.1002/jmv.27053
204. Santoro F, Núñez-Gil IJ, Vitale E, Viana-Llamas MC, Romero R, Maroun Eid C, et al. Aspirin therapy on prophylactic anticoagulation for patients hospitalized with COVID-19: A propensity score-matched cohort analysis of the HOPE-COVID-19 registry. J Am Heart Assoc (2022) 11(13):e024530. doi: 10.1161/JAHA.121.024530
205. Santoro F, Nuñez-Gil IJ, Vitale E, Viana-Llamas MC, Reche-Martinez B, Romero-Pareja R, et al. Antiplatelet therapy and outcome in COVID-19: the health outcome predictive evaluation registry. Heart (2022) 108(2):130–6. doi: 10.1136/heartjnl-2021-319552
206. Meizlish ML, Goshua G, Liu Y, Fine R, Amin K, Chang E, et al. Intermediate-dose anticoagulation, aspirin, and in-hospital mortality in COVID-19: A propensity score-matched analysis. Am J Hematol (2021) 96(4):471–9. doi: 10.1002/ajh.26102
207. Viecca M, Radovanovic D, Forleo GB, Santus P. Enhanced platelet inhibition treatment improves hypoxemia in patients with severe COVID-19 and hypercoagulability. a case control, proof of concept study. Pharmacol Res (2020) 158:104950. doi: 10.1016/j.phrs.2020.104950
208. Barnes BJ, Adrover JM, Baxter-Stoltzfus A, Borczuk A, Cools-Lartigue J, Crawford JM, et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J Exp Med (2020) 217(6):e20200652. doi: 10.1084/jem.20200652
209. Pastorek M, Dúbrava M, Celec P. On the origin of neutrophil extracellular traps in COVID-19. Front Immunol (2022) 13:821007. doi: 10.3389/fimmu.2022.821007
210. von Brühl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med (2012) 209(4):819–35. doi: 10.1084/jem.20112322
211. Dyer MR, Chen Q, Haldeman S, Yazdani H, Hoffman R, Loughran P, et al. Deep vein thrombosis in mice is regulated by platelet HMGB1 through release of neutrophil-extracellular traps and DNA. Sci Rep (2018) 8(1):2068. doi: 10.1038/s41598-018-20479-x
212. Denorme F, Portier I, Rustad JL, Cody MJ, de Araujo CV, Hoki C, et al. Neutrophil extracellular traps regulate ischemic stroke brain injury. J Clin Invest (2022) 132(10):e154225. doi: 10.1172/JCI154225
213. Kaiser R, Escaig R, Erber J, Nicolai L. Neutrophil-platelet interactions as novel treatment targets in cardiovascular disease. Front Cardiovasc Med (2022) 8:824112. doi: 10.3389/fcvm.2021.824112
214. Lee YY, Park HH, Park W, Kim H, Jang JG, Hong KS, et al. Long-acting nanoparticulate DNase-1 for effective suppression of SARS-CoV-2-mediated neutrophil activities and cytokine storm. Biomaterials (2021) 267:120389. doi: 10.1016/j.biomaterials.2020.120389
215. Franzetti M, Forastieri A, Borsa N, Pandolfo A, Molteni C, Borghesi L, et al. IL-1 receptor antagonist anakinra in the treatment of COVID-19 acute respiratory distress syndrome: A retrospective, observational study. J Immunol (2021) 206(7):1569–75. doi: 10.4049/jimmunol.2001126
216. Cavalli G, Dagna L. The course of action for effective anti-cytokine treatment in COVID-19. Lancet Respir Med (2021) 9(12):1353–4. doi: 10.1016/S2213-2600(21)00405-7
217. Kyriazopoulou E, Huet T, Cavalli G, Gori A, Kyprianou M, Pickkers P, et al. Effect of anakinra on mortality in patients with COVID-19: a systematic review and patient-level meta-analysis. Lancet Rheumatol (2021) 3(10):e690–7. doi: 10.1016/S2665-9913(21)00216-2
218. Vora SM, Lieberman J, Wu H. Inflammasome activation at the crux of severe COVID-19. Nat Rev Immunol (2021) 21(11):694–703. doi: 10.1038/s41577-021-00588-x
219. Cron RQ, Caricchio R, Chatham WW. Calming the cytokine storm in COVID-19. Nat Med (2021) 27(10):1674–5. doi: 10.1038/s41591-021-01500-9
220. Zizzo G, Tamburello A, Castelnovo L, Laria A, Mumoli N, Faggioli PM, et al. Immunotherapy of COVID-19: Inside and beyond IL-6 signalling. Front Immunol (2022) 13:795315. doi: 10.3389/fimmu.2022.795315
221. Matthay MA, Luetkemeyer AF. IL-6 receptor antagonist therapy for patients hospitalized for COVID-19: Who, when, and how? JAMA (2021) 326(6):483–5. doi: 10.1001/jama.2021.11121
222. Li X, Shao M, Zeng X, Qian P, Huang H. Signaling pathways in the regulation of cytokine release syndrome in human diseases and intervention therapy. Signal Transduct Target Ther (2021) 6(1):367. doi: 10.1038/s41392-021-00764-4
223. Siggins MK, Davies K, Fellows R, Thwaites RS, Baillie JK, Semple MG, et al. Alternative pathway dysregulation in tissues drives sustained complement activation and predicts outcome across the disease course in COVID-19. Immunology (2022). doi: 10.1111/imm.13585
224. Cugno M, Meroni PL, Gualtierotti R, Griffini S, Grovetti E, Torri A, et al. Complement activation and endothelial perturbation parallel COVID-19 severity and activity. J Autoimmun (2021) 116:102560. doi: 10.1016/j.jaut.2020
225. Rambaldi A, Gritti G, Micò MC, Frigeni M, Borleri G, Salvi A, et al. Endothelial injury and thrombotic microangiopathy in COVID-19: Treatment with the lectin-pathway inhibitor narsoplimab. Immunobiology (2020) 225(6):152001. doi: 10.1016/j.imbio.2020.152001
226. Annane D, Heming N, Grimaldi-Bensouda L, Frémeaux-Bacchi V, Vigan M, Roux AL, et al. Eculizumab as an emergency treatment for adult patients with severe COVID-19 in the intensive care unit: A proof-of-concept study. EClinicalMedicine (2020) 28:100590. doi: 10.1016/j.eclinm.2020.100590
227. Agarwal A, Rochwerg B, Lamontagne F, Siemieniuk RA, Agoritsas T, Askie L, et al. A living WHO guideline on drugs for covid-19. BMJ (2020) 370:m3379. doi: 10.1136/bmj.m3379
228. Drożdżal S, Rosik J, Lechowicz K, Machaj F, Szostak B, Przybyciński J, et al. An update on drugs with therapeutic potential for SARS-CoV-2 (COVID-19) treatment. Drug Resist Update (2021) 59:100794. doi: 10.1016/j.drup.2021.100794
229. Lund LC, Hedberg P, Andreasen AH, Petersen J, Petersen TS, Pottegård A, et al. Prophylactic anticoagulation with low molecular weight heparin in COVID-19: Cohort studies in Denmark and Sweden. Clin Microbiol Infect (2022) 28(9):1291.e1–5. doi: 10.1016/j.cmi.2022.03.006
230. Büller HR, Bethune C, Bhanot S, Gailani D, Monia BP, Raskob GE, et al. Factor XI antisense oligonucleotide for prevention of venous thrombosis. N Engl J Med (2015) 372(3):232–40. doi: 10.1056/NEJMoa1405760
231. Pekrul I, Spannagl M, Nitschmann S. Antikoagulation bei patienten mit COVID-19 [Anticoagulation in patients with COVID-19]. Internist (Berl) (2021) 62(11):1253–5. doi: 10.1007/s00108-021-01190-y
232. Open SAFELY Collaborative, Wong AYS, LA T, JP B, Elson W, AJ W, et al. Association between warfarin and COVID-19-related outcomes compared with direct oral anticoagulants: Population-based cohort study. J Hematol Oncol (2021) 14(1):172. doi: 10.1186/s13045-021-01185-0
233. Attaway AH, Scheraga RG, Bhimraj A, Biehl M, Hatipoğlu U. Severe covid-19 pneumonia: Pathogenesis and clinical management. BMJ (2021) 372:n436. doi: 10.1136/bmj.n436
234. Chow JH, Yin Y, Yamane DP, Davison D, Keneally RJ, Hawkins K, et al. Association of prehospital antiplatelet therapy with survival in patients hospitalized with COVID-19: A propensity score-matched analysis. J Thromb Haemost (2021) 19(11):2814–24. doi: 10.1111/jth.15517
235. Sriram K, Insel PA. Proteinase-activated receptor 1: A target for repurposing in the treatment of COVID-19? Br J Pharmacol (2020) 177(21):4971–4. doi: 10.1111/bph.15194
Keywords: inflammation, immunothrombosis, anti-inflammatory treatment, antithrombotic therapy, long COVID-19
Citation: Jing H, Wu X, Xiang M, Liu L, Novakovic VA and Shi J (2022) Pathophysiological mechanisms of thrombosis in acute and long COVID-19. Front. Immunol. 13:992384. doi: 10.3389/fimmu.2022.992384
Received: 12 July 2022; Accepted: 27 October 2022;
Published: 16 November 2022.
Edited by:
Robert Lindsay Medcalf, Monash University, AustraliaReviewed by:
Emmanuel Favaloro, Westmead Hospital, AustraliaShie-Liang Hsieh, Academia Sinica, Taiwan
Copyright © 2022 Jing, Wu, Xiang, Liu, Novakovic and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jialan Shi, amlhbGFuX3NoaUBkZmNpLmhhcnZhcmQuZWR1; c2hpNzM2NjFAZ21haWwuY29t