
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 27 September 2022
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.989275
 Zhichao Liu1,2†
Zhichao Liu1,2† Guo Lin1,3†
Guo Lin1,3† Zeping Yan1†
Zeping Yan1† Linduo Li4
Linduo Li4 Xingchen Wu1Jingrong Shi5Jianxing He1Lei Zhao6*
Xingchen Wu1Jingrong Shi5Jianxing He1Lei Zhao6* Hengrui Liang1*Wei Wang1*
Hengrui Liang1*Wei Wang1*Background: Developing prediction tools for immunotherapy approaches is a clinically important and rapidly emerging field. The routinely used prediction biomarker is inaccurate and may not adequately utilize large amounts of medical data. Machine learning is a promising way to predict the benefit of immunotherapy from individual data by individuating the most important features from genomic data and clinical characteristics.
Methods: Machine learning was applied to identify a list of candidate genes that may predict immunotherapy benefits using data from the published cohort of 853 patients with NSCLC. We used XGBoost to capture nonlinear relations among many mutation genes and ICI benefits. The value of the derived machine learning-based mutation signature (ML-signature) on immunotherapy efficacy was evaluated and compared with the tumor mutational burden (TMB) and other clinical characteristics. The predictive power of ML-signature was also evaluated in independent cohorts of patients with NSCLC treated with ICI.
Results: We constructed the ML-signature based on 429 (training/validation = 8/2) patients who received immunotherapy and extracted 88 eligible predictive genes. Additionally, we conducted internal and external validation with the utility of the OAK+POPLAR dataset and independent cohorts, respectively. This ML-signature showed the enrichment in immune-related signaling pathways and compared to TMB, ML-signature was equipped with favorable predictive value and stratification.
Conclusion: Previous studies proposed no predictive difference between original TMB and modified TMB, and original TMB contains some genes with no predictive value. To demonstrate that fewer genetic tests are sufficient to predict immunotherapy efficacy, we used machine learning to screen out gene panels, which are used to calculate TMB. Therefore, we obtained the 88-gene panel, which showed the favorable prediction performance and stratification effect compared to the original TMB.
Immune checkpoint inhibitors (ICI) are an effective treatment for patients with advanced non-small cell lung cancer (NSCLC) (1, 2). The drugs were designed to target programmed death-ligand 1 (PD-L1)/programmed death-1 (PD-1) and disrupt inhibition of the immune response, leading to T-cell activation and restoring anti-tumor immunity (3, 4). Nevertheless, only a minority of patients with advanced NSCLC derive clinical benefits from this treatment (5). Identifying biomarkers and/or prediction models can help inform which patients would be the beneficial candidate for immunotherapy. Emerging predictive biomarkers associated with enhanced response to ICI include microsatellite instability, tumor mutational burden (TMB), PD-L1 expression, and inflammatory gene expression (6, 7). Many efforts are currently being undertaken toward improving the predictive value of different gene mutations (8). However, the routinely used prediction biomarker and/or prediction model is still not accurate enough and may not adequately utilize large amounts of medical data.
With the advent of “era of precision medicine”, the analyses of large-scale molecular data are beneficial for many aspects of oncology research, including the classification of possible subtypes, stages, and treatment of cancer (9). Accurate classification of cancers can greatly help physicians to choose the optimal treatment strategies for patients. To this end, classifying cancer into different groups is regarded as one of the most important issues in cancer therapy (10). Following the explosive growth of huge amounts of biological data, the shift from traditional biostatistical methods to computer-aided means has made machine-learning methods an integral part of today’s cancer diagnostic and prognostic prediction (11). Machine learning would certainly accelerate the progress of prediction for ICI benefits as a data-driven approach by individuating the most important features from genomic data and clinical characteristics in the current practice (12).
In this study, we exploited the potential of machine learning methods to address the issue of identifying NSCLC patients with ICI benefits. With the hypothesis that mutations in certain genes may better predict NSCLC response to ICB treatment, we aimed to develop a machine learning based-mutation signature (ML-signature) to predict ICI clinical benefits effectively.
This retrospective study was approved by our institutional review board (IRB No.202070). The genomic alterations and clinical data for POPLAR (NCT01903993) and OAK (NCT02008227) trials were downloaded from publicly accessible data reported by Gandara et al. (13). Both POPLAR (randomized phase II trial) and OAK (randomized phase III trial) were designed to compare single-agent atezolizumab with docetaxel as second/third-line therapy for patients with advanced NSCLC, who were unselected for PD-L1 status. In this open-label, phase 2/3 randomized controlled trial, patients with NSCLC who progressed on post-chemotherapy, Eastern Cooperative Oncology Group performance status 0 or 1 were recruited. Detailed characteristics were reported in previous reports (14, 15). Relevant data of OAK and POPLAR cohorts were provided in Tables S1 and S2.
Mutations were measured by FoundationOne CDx NGS assay (16), which targets 1.1 Mb of the genomic coding sequence. As per the study protocol of POPLAR and OAK, progress-free survival (PFS) was defined as the time between the date of randomization and the date of first documented disease progression, as assessed by the investigator using RECIST v1.1, or death from any cause, whichever occurs first. Overall survival (OS) was defined as the time between the date of randomization until death from any cause. Objective response was defined as complete response and partial response according to RECIST v1.1 (17).
The ICI dataset was used as a development dataset to determine the gene signature of ICI benefits and consisted of 429 patients treated with ICI from the prospective POPLAR and OAK trials. In the research, we estimated the importance of features (mutation genes) for the ICI-benefit predictive modeling problem using the XGBoost method, which is a gradient boosting decision tree.
Using XGBoost, we developed immunotherapy benefit prediction models based on mutation features (the ML-signature) (18, 19). Gradient boosting decision tree (GBDT) methods employ an ensemble of multiple decision trees to strengthen the classification power. Each decision tree is grown by selecting the most discriminative features from the large feature candidate pool. This process relieves traditional tree-based methods from the onerous feature selection process and allows the classifier to interact directly with the features. The feature selection process of the algorithm facilitates the analysis of the features (mutation genes) that most impact the classifier and thus provides us with a method of investigating the biological mechanisms hidden within the genomic data. The higher the feature importance score of XGBoost is, the more important and effective the corresponding feature (gene) is. We obtain the top-shared ranked features (genes) based on descending order of feature importance to characterize the ML-signature. Details about the XGBoost model and code are shown in Supplemental methods and Supplemental code.
For functional enrichment analysis, all genes in ML-signature were mapped to terms in the Kyoto Encyclopedia of Genes and Genomes database (KEGG) and P < 0.05 as the threshold. KEGG links genomic information with biological functions based on the online platform (https://www.genome.jp/), and KEGG results was visualized by R package ggplot2.
ML-signature was applied to evaluate risk stratification at the individual level. The cut-off points for risk stratification of ML-signature were calculated. The ICI-benefit score was calculated as the number of mutations of the ML-signature found in a patient. We further evaluated the cut-off value for ICI-benefit score to stratify patients into ICI benefit and non-benefit groups with optimal survival stratification. Using the LOWESS smoother fitting curves, we modeled the relationship between PFS-HRs and ICI-benefit score cut-off values (20).
Survival analyses were conducted to compare the predictive performance between ML-signature and TMB. PFS and OS were regarded as endpoints.
To further validate the performance of ML-signature, we evaluated its predictive power on another NSCLC dataset from external cohorts, which can be downloaded from the cBioPortal database (https://www.cbioportal.org).
All analyses were conducted with R software (version 3.5.3) and SAS (version 9.4). A Cox proportional hazards model was used to determine the HR for survival. Kaplan–Meier methodology was used to construct the survival curves and the significances of subgroups were estimated using the log-rank test. A two-sided P <0.05 was considered a statistically significant difference.
To derive the optimal cut-offs of the candidate gene inclusion for ML-signature in the prediction of ICI-benefit/non-benefit stratification, we performed Cox regression analysis to assess the effect of inclusion criteria changes for ML-signature (the number of gene mutations was analyzed as a continuous variable) on PFS in the immunotherapy treatment arm. The curves of Wald statistic of PFS HR of different ML-signature stratification, which was determined at numbers of mutations in ML-signature, were fitted using a locally estimated scatterplot smoothing (LOESS) with a span of 0.80 (21); and structural breakpoints were then determined by Chow test (22). The Spearman’s rank coefficient was used to compare ML-signature (as a continuous variable) with TMB and SLD (sum of the longest diameter of target lesions at baseline).
This study contained two sections (schematic of study design in Figure 1): 1.) ML-signature development, 2.) validation of the performance of ML-signature for immunotherapy efficacy.
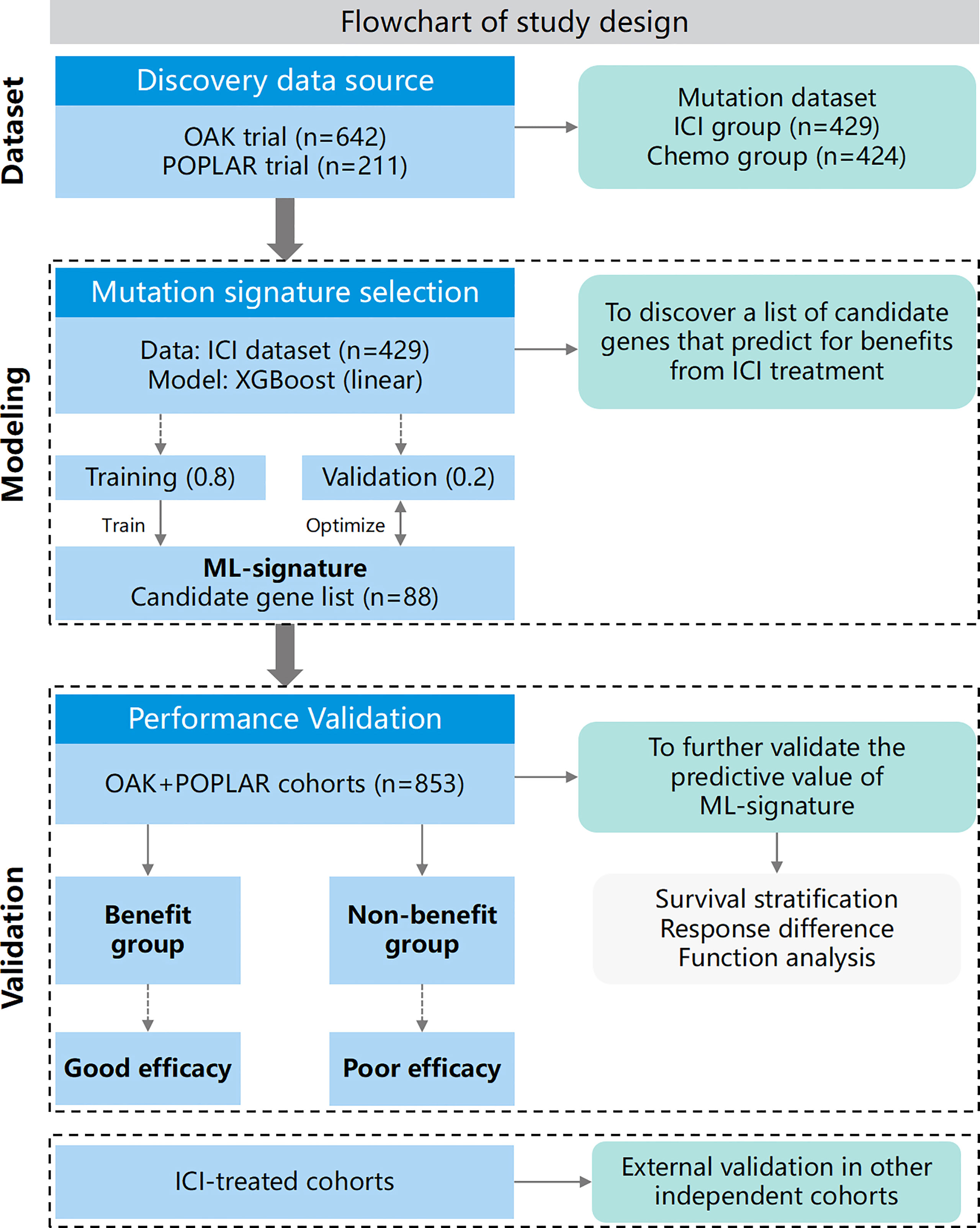
Figure 1 Flowchart of this study.
We hypothesized that gene mutations not only produced neoantigens but also could functionally affect the efficacy of ICI. Based on this hypothesis, we aimed to identify genes whose mutations could positively influence ICI treatment efficacy by investigating NSCLC patients from two large, published cohorts of patients treated with ICI and sequenced with FoundationOne CDx (F1CDx): 391 genes known to be involved in cancer development.
The ML-signature development is shown in Figure 2. XGBoost classification method was used to capture the importance of mutation genes for the ICI benefits. The XGBoost algorithm generates a regression model based on an ensemble of decision trees. The mutation genes are ranked based on the permutation importance method in the XGboost model. After 350 repetitions of modeling in this study, the top-ranked gene intersection between models stabilized at 88 genes. Applying these criteria to the targeted sequencing gene panel used in the study, we obtained an 88-gene panel (Table S3).
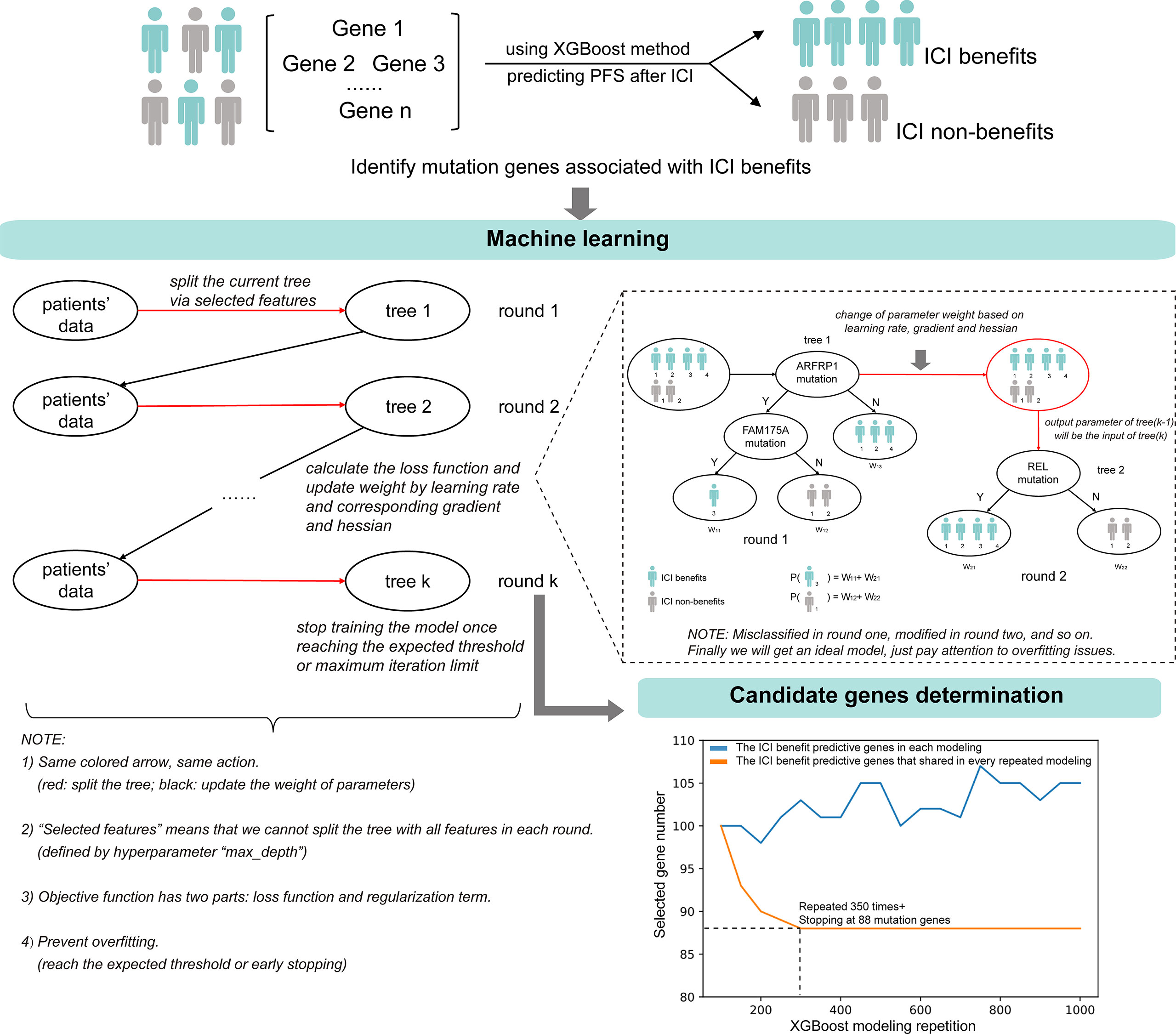
Figure 2 The identification of candidate mutation genes using XGBoost feature selection and the structural diagram of the machine learning model.
We examined the biological functions and signal transduction pathway associated with the ICI-benefits related ML-signature. Functional enrichment (Figures S1A, S2) showed the immune-related pathways were significantly enriched in the ML-signature compared with those excluded genes. This result is a biological reasonableness check for the ICI-benefits gene identification using the gradient boosting decision tree analysis. Among the 853 patients with NSCLC, 467 had mutations in at least one of the 88 genes, and 243 had mutations in at least two of the 88 genes (Figure S1B). There was no significant difference in the distribution of mutations in 88 genes between the immunotherapy and chemotherapy groups (P>0.05).
We conducted survival analyses to decide whether the ICI-benefit score could be a predictor for immunotherapy. The ICI-benefit score based on 88 genes was significant associated with favorable PFS (HR 0.755, 95%CI, 0.696-0.820, P<0.001) and OS (HR 0.849, 95%CI, 0.778-0.926, P<0.001) in patients receiving immunotherapy; while the tumor mutational burden (TMB) score was not (PFS: HR 0.991, 95%CI, 0.982-1.0, P=0.056; OS: HR 1.003, 95%CI, 0.993-1.013, P=0.572). In patients receiving chemotherapy, the ICI-benefit score was associated with poor PFS (HR, 1.061, 95%CI, 1.011-1.113, P=0.015) and OS (HR, 1.084, 95%CI, 1.030-1.140, P=0.002). These results confirmed that the ICI-benefit score based on the ML-signature was a specific predictor for immunotherapy. These data-driven results suggested an “elbow” region between≥1 and ≥6, and the cut-point analysis demonstrated that ICI-benefit score≥2 was the break point with better survival stratification in patients treated with immunotherapy (Figure S3). Compared to ICI-non-benefit group, ICI-benefit group showed longer PFS (HR 0.47, 95%CI, 0.38-0.57, P<0.001) and OS (HR 0.61, 95%CI, 0.47-0.78, P<0.001).
The comparison of the predictive value of modified ML-signature and TMB in immunotherapy or chemotherapy arms is presented in Figure 3. In patients receiving immunotherapy (Figures 3A, B), notably, both PFS and OS were significantly greater in the ICI-benefit group vs. ICI-non-group [PFS HR: 0.47 (95%CI: 0.38-0.57, P<0.001) and OS HR: 0.61 (95%CI: 0.47-0.78, P<0.001)], compared with those of TMB-high vs. TMB-low stratification. In patients receiving chemotherapy (Figures 3C, D), the ICI-benefit group was associated with poor PFS or OS [PFS HR: 1.40 (95%CI: 1.12-1.75, P=0.002); OS HR: 1.56 (95%CI: 1.22-2.0, P<0.001)], showing a greater risk stratification compared with those of TMB-high vs. TMB-low.
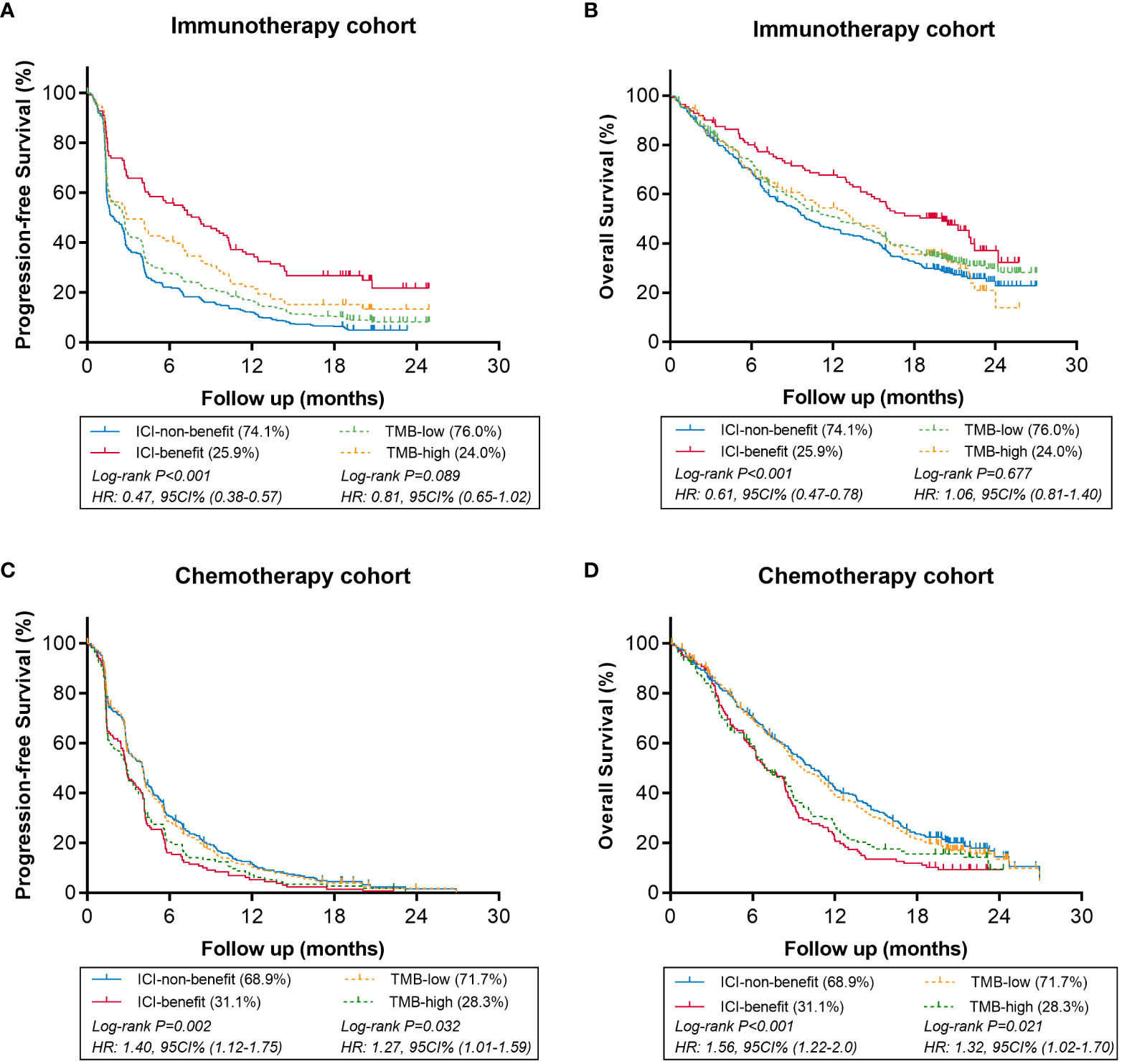
Figure 3 Comparison of survival in immunotherapy or chemotherapy arms using ML-signature (ICI-benefit vs. ICI-non-benefit) and TMB (high vs. low). (A, B) immunotherapy cohort, (C, D) chemotherapy cohort.
Efficacy comparison evaluating immunotherapy vs. chemotherapy was performed in the stratification groups based on ML-signature and TMB. When classified as benefit candidates for immunotherapy (Figures 4A, B), the PFS and OS benefits of immunotherapy vs. chemotherapy for ICI-benefit subgroup were relatively greater (HR: 0.38, 95%CI: 0.28-0.50 and HR: 0.33, 95%CI: 0.24-0.46, respectively), compared with those for TMB-high subgroup (HR: 0.63, 95%CI: 0.48-0.84; and HR: 0.58, 95%CI: 0.43-0.80, respectively). The absolute difference in median PFS and OS of immunotherapy vs. chemotherapy in the ICI-benefit subgroup was relatively greater (median PFS: 8.2 vs. 2.9 months and median OS: 20.1 vs. 6.9 months) compared with those of the TMB-high subgroup (median PFS: 2.9 vs. 2.9 months and median OS: 13.5 vs. 6.9 months).
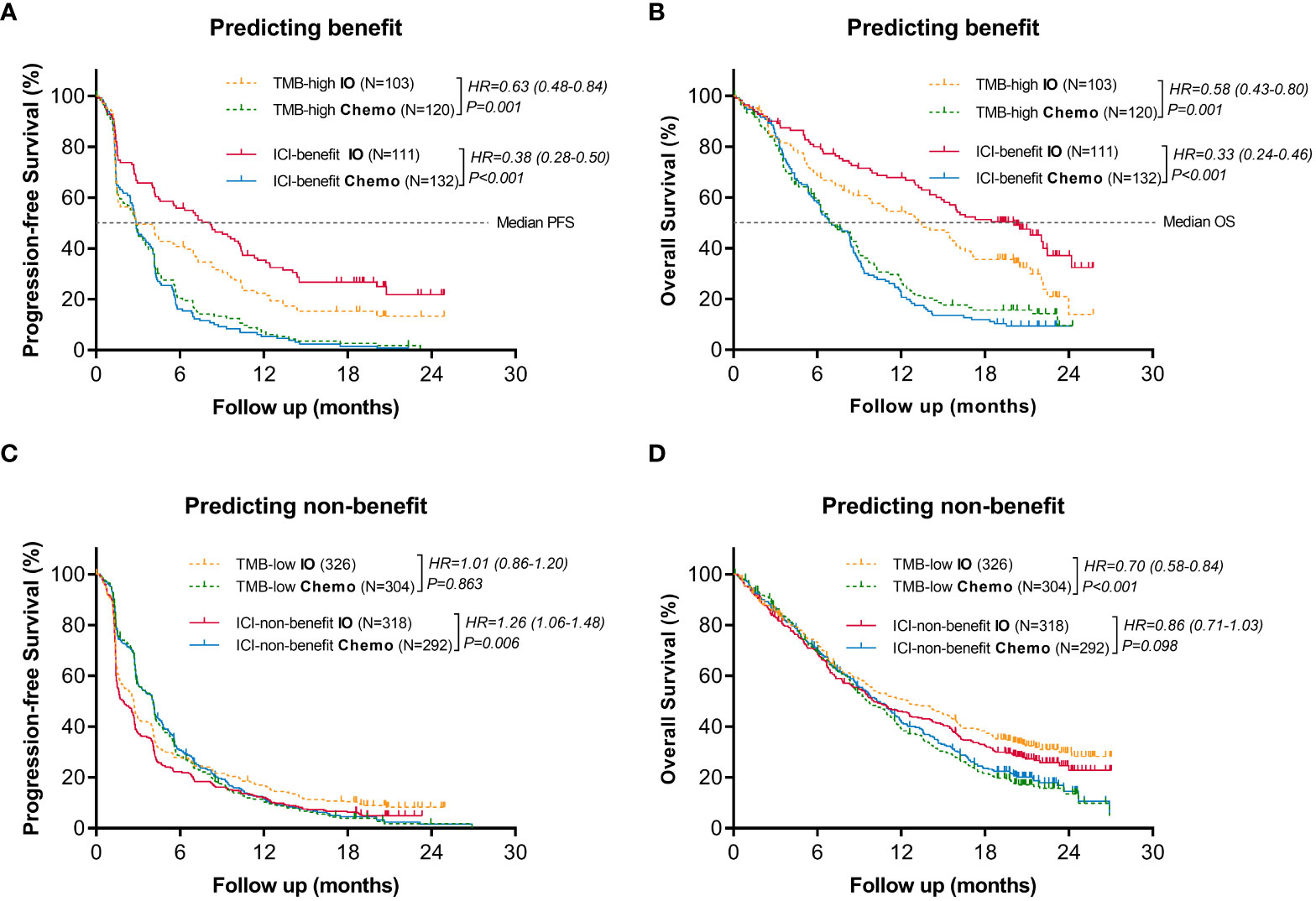
Figure 4 Comparison of the survival benefits from immunotherapy vs. chemotherapy between ML-signature and TMB stratification. (A, B) Predicting benefit cohort, (C, D) Predicting non-benefit cohort.
Additionally, when classified as non-benefit candidates for immunotherapy (Figures 4C, D), results showed that the ICI-non-benefit patients benefited more from chemotherapy than immunotherapy, suggesting that stratification based on ML-signature could help to identify those who may benefit from immunotherapy. However, TMB cannot provide sufficient treatment-efficacy stratification. Overall, these results suggested immunotherapy-benefits predictive performance of the ML-signature.
Figure 5A shows the objective response rate (ORR) in different ways of efficacy stratification. In patients classified as ICI-benefit using the ML-signature, the ORR trended towards significant benefit with immunotherapy (30.6%) vs. chemotherapy (11.4%). The absolute ORR benefit of immunotherapy vs. chemotherapy increased from 13.1% (TMB-high) to 19.2% (ICI-benefit). In the immunotherapy arm, a greater difference was observed in ORR between ICI-benefit and ICI-non-benefit subgroups (30.6% vs. 8.8%) compared with those of TMB-high and TMB-low subgroups (22.3% vs. 12.0%). Additionally, in the chemotherapy arm, the difference in ORR between ICI-benefit and ICI-non-benefit subgroups was smaller (11.4% vs. 14.4%) compared with the ORR between TMB-high and TMB-low subgroups (9.2% vs. 15.1%). In patients treated with immunotherapy, the AUC for ML-signature distinguishing between responder and non-responder patients was 0.67 (95% CI: 0.59 to 0.75), which was higher than that of TMB [0.58, 95% CI: 0.59 to 0.75)] (Figure 5B). Overall, these findings suggest a role for an immunotherapy efficacy-based ML-signature in promoting the identification of patients with better predictive benefits from immunotherapy.
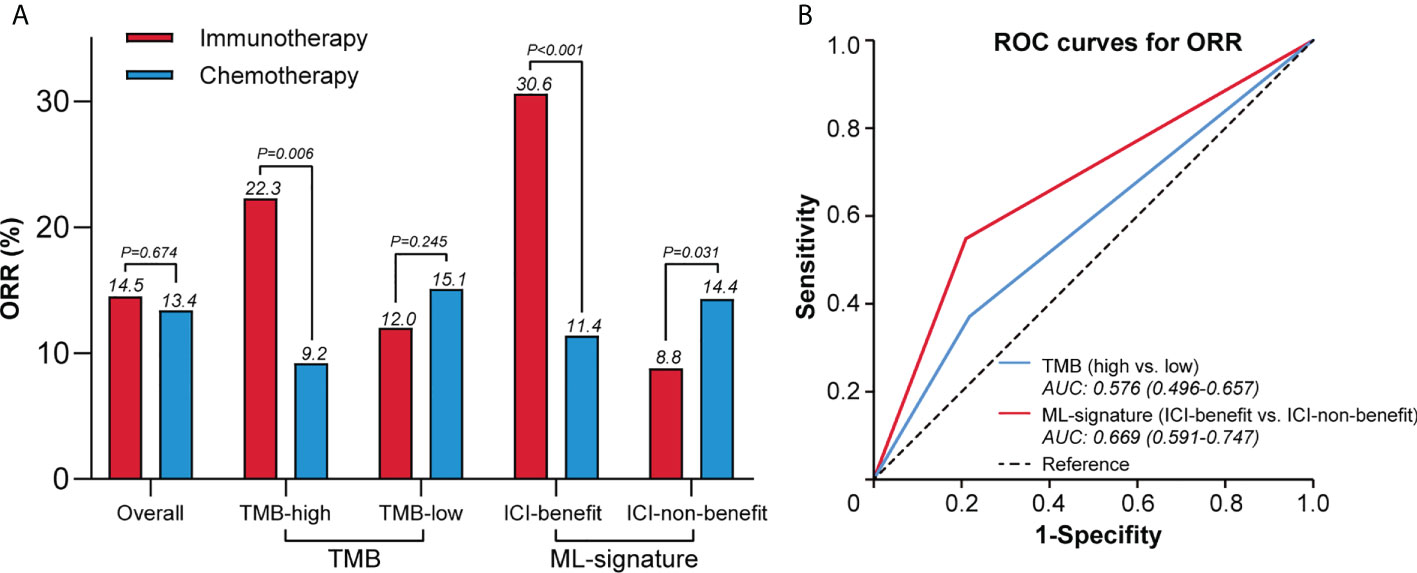
Figure 5 ML-signature associated with higher ORR of immunotherapy. (A) Difference in the ORR between ML-signature and TMB subgroups. (B) Receiver operating characteristic curves to predict ORR.
We next validate the predictive power of the ML-signature in two external cohorts of NSCLC patients treated with ICI from previously published studies (WES sequenced all patients were sequenced for gene mutations). Survival analysis of patients with different mutations in the 88-gene panel was performed. Results (Figure 6) revealed that patients classified as ICI-benefit candidates by our ML-signature had substantial survival advantages, which were remarkably similar to the PFS results obtained from the discovery cohort, thus validating the predictive power of the ML-signature independently.
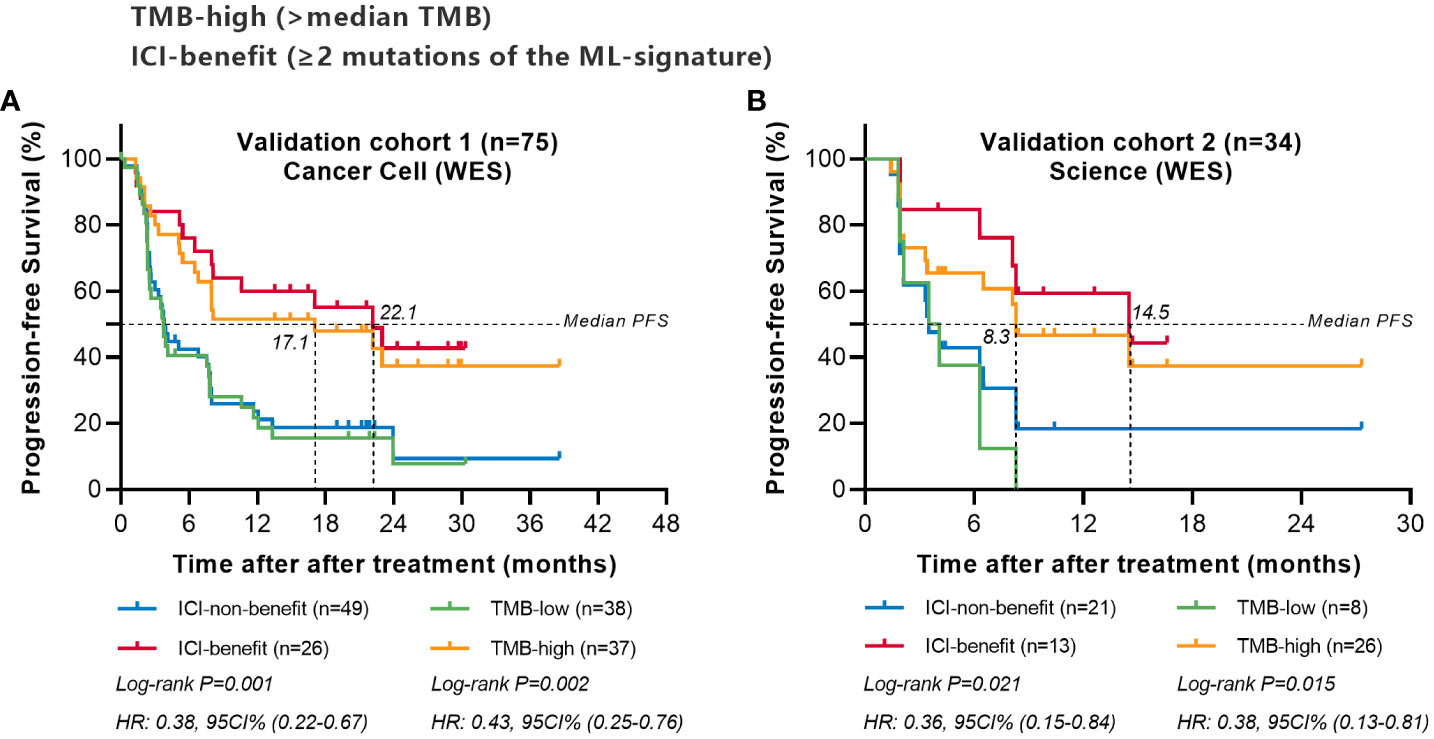
Figure 6 Comparing the predictive powers of ML-signature vs. TMB in patients treated with immunotherapy in external cohorts. (A) Cancer cell (WES) validation cohort, (B) Science (WES) validation cohort.
We also explored the predictive performance of the ML-signature in a cohort of 350 NSCLC patients treated with ICI from the MSK-IMPACT cohort (a large panel of targeted NGS sequenced all patients). There are 58 genes of the ML-signature (88 genes) that can be detected in the MSK-IMPACT panel (410 genes~468genes). Results also revealed that patients classified as ICI-benefit candidates by our ML-signature had better survival stratification, thus further validating the predictive power of the ML-signature (Figure 7).
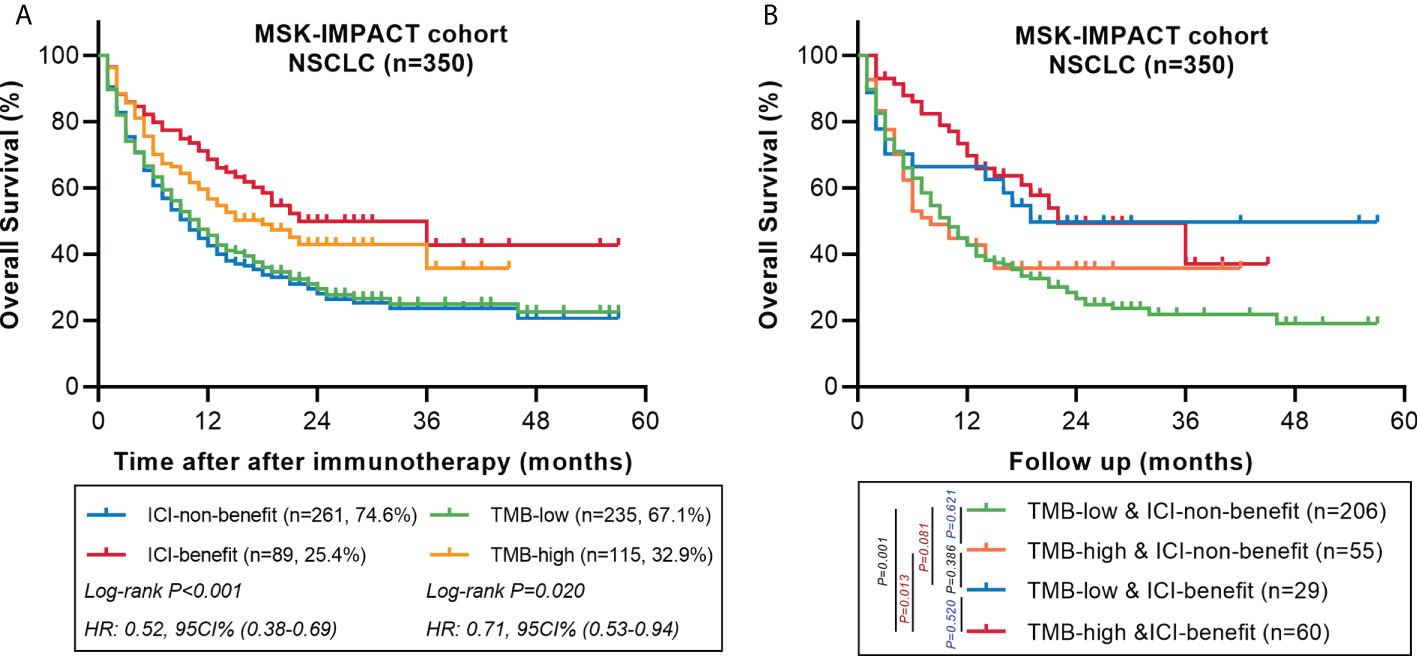
Figure 7 Comparing the predictive powers of ML-signature vs. TMB in patients treated with immunotherapy in the MSK-IMPACT cohort. (A) Overall survival of MSK-TMB patients with between ML-signature and TMB subgroups. (B) Overall survival of MSK-IMPACT patients with TMB-high and TMB-low further stratified according to groups stratified by ML-signature.
The heterogeneity of cancer patients results in various therapy efficacies and deciding whether to receive ICI is momentous (23). Previous studies reported that high expression levels of PD-L1 were related to better response to ICI for NSCLC populations (24, 25). However, gene mutation also plays a crucial role in ICI for malignant cancers (26). Prediction models involving genetic mutation information are more suitable for patients with mutations. Precedented prediction models were produced based on clinical characteristics or pathological information of tumors (27), and small sample sizes restrain their generalization performance. Therefore, we constructed a mutation signature-based prediction model using machine learning to estimate the prognosis of patients treated with ICI. We demonstrated that identifying immunotherapy efficacy-based mutations could improve prediction accuracy. Our results provided a rationale for using machine learning to develop ICI-specific mutation signatures in predicting patients suitable for immunotherapy.
In this study, we identified 88 ML-signature from almost 400 genes associated with cancer progression. These ICI-benefits related ML-signature primarily enriched in immune-related signaling pathways. Immune cells and immune cytokines in the tumor microenvironment are important during cancer development, and their biological functions change dynamically with tumor progression. In addition, these ML-signature also show decreasing transduction-related signaling pathways. Signal transduction pathways involve each stage of cancer cells, from stem cells to advanced cancer cells, including proliferation, metabolism, cell cycle, DNA repair, apoptosis, differentiation, tumor extracellular matrix remodeling, angiogenesis and metastasis (28, 29). Notably, there was no significance in metabolism, DNA repair, apoptosis or other inflammatory signaling pathways. Generally, inflammation-related signaling pathways show the transduction from pro-inflammation to anti-inflammation along with tumor evolution (30). Therefore, the complex transformation of these 88 ICI-benefits related ML-signature needs further exploration. We also observed that the distributions of more than one mutation population were similar in both immunotherapy and chemotherapy groups, which indicated that patients in the chemotherapy cohort also have potential benefits from ML-signature.
Recent evidence has shown high TMB to be associated with improved clinical outcomes from ICI in multiple cancer types (31). However, TMB as a predictive biomarker for immunotherapy remains difficult to implement because tumor heterogeneity may add to the complexity of TMB analysis and lead to misestimation of the reliability of TMB prediction for immunotherapy. In this study, we hypothesized that mutations in certain genes associated with immunotherapy efficacy rather than serving as sources of roughly TMB estimation might better predict NSCLC response to ICI therapy. With the utility of ICI-benefit score developed by machine learning, we demonstrated that ML-signature provides better efficacy prediction than TMB in patients treated with immunotherapy. More mutations represent favorable survival in the immunotherapy cohort, which is consistent with the consensus that the increasing number of mutations were processed to neo-antigens and presented by major histocompatibility complex (MHC) proteins to T lymphocytes, immune-system eliminated neo-antigens (32–34). However, cancer cells impaired the activities of T lymphocytes and achieved immune escape (35). Immunotherapies blocked immune checkpoints in a targeted manner and reduced and/or re-activated T lymphocytes (36, 37).
Our ML-signature recognized the patients most likely to benefit (longer survival time and higher objective response rate) from immunotherapy. We demonstrated that immunotherapy is not suitable for all patients. Compared with immunotherapy, ICI-non-benefit patients can gain a longer survival time from chemotherapy, which can be interpreted as immune-related adverse events (irAEs). The detailed mechanisms of irAEs are still unclear. Presented mechanisms include activated T lymphocytes attacking health issues, increasing levels of autoantibodies, and inflammatory cytokines (38, 39). Although the immune checkpoint blockade is generally regarded as a tolerable treatment (40), the long-term influence of immunotherapy should be further explored.
To further validate the value of our ML-signature, we conducted survival analyses with two external NSCLC cohorts with WES data covering the overall 88 genes of ML-signature. This result validates the consistency of the internal cohort of our main result, notably the predictive effect of the ML-signature having better survival stratification than TMB, indicating that the prediction model was steady and accurate as the predictive tool for patient selection for immunotherapy. Additionally, we used the MSK-IMPACT dataset to authenticate the ML-signature, which involved more than four hundred genes and possessed the highest degree of dissemination (41). Only 58/88 genes from our ML-signature can be detected in the MSK-IMPACT dataset; similar to the above-mentioned external validation, and these 58 genes also displayed favorable stratified performance, and ML-signature can further divide into TMB high and low subgroups. To sum up, the application of these 88 genes from our ML-signature covered large-scale commercial targeted panels.
Gene mutations have also shown a crucial role in tumor heterogeneity, and the distribution of mutations represents sub-clonal status, which may compromise the efficacy of immunotherapy. The ratio of allele frequency to maximum somatic allele frequency (AF/MSAF) has also been used to represent allele frequency heterogeneity (AFH), and AFH was recognized as the negative factor of prognosis (42). To address the problems of traditional TMB in advanced patients, including insufficient tumor tissues and substandard specimens, blood-based tumor mutational burden (bTMB) emerged for clinical requirements. Dong P et al. filtered 52 candidate genes based on the Cox proportional-hazards model and demonstrated that the 52-gene panel was superior to original TMB-H (TMB ≥10) in estimating clinical benefits for ICI therapy in NSCLC patients (43). Our teams previously reported that modified bTMB had favorable performances in estimating clinical benefits from immunotherapy (44). The special characteristics of ML-signature and modified bTMB should be compared through an additional cohort. These TMB-related studies have attempted to filter genes that predict immunotherapy benefits to calculate precise TMB, and the performance of precedent models needs further improvement. Though the performance of ML-signature does not work perfectly, it showed significant progress compared to existing models (Figure S4) and showed sound stratification capacity. In addition, the expression level of PD-L1 is also considered to be an impartial factor in the clinical decision. We also compared ML-signature and PD-L1 level, and ML-signature showed a favorable predictive performance. We observed that only high levels of PD-L1 (TC>50% or IC>10%) had predictive value, and the ROC curves plot indicated high values for ML-signature (AUC = 0.711) but low values for PD-L1 (AUC = 0.667). Other details are shown in Figure S5.
There are also some limitations in our models. As a retrospective study, datasets are incomplete inevitably and information biased. Moreover, this model excluded routine clinical information, such as patients’ characteristics, pathological data, specific therapy options and surgery status. Furthermore, the partially detected genes also suggested that our model includes some genes with low predictive value and the number of eligible genes can be further refined. The ratios of eligible genes have not been tested in real-world patients. The enrichment of genes is generally shown in immune-related signaling pathways and needs further study according to their proportion in ML-signature.
We provided an accurate ML-signature for NSCLC patients and demonstrated its feasibility with certain verified measurements. And our study supports the possibility and potential of using machine learning to screen for predictive molecular markers of immunotherapy efficacy or other treatments. In the ear of big data, further research can excavate novel biomarkers with the assistance of computing science and accelerate the process of translational medicine and precision medicine.
Publicly available datasets were analyzed in this study. This data can be found here: https://doi.org/10.1038/s41591-018-0134-3.
This study was reviewed and approved by the Research Ethics Committee of The First Affiliated Hospital of Guangzhou Medical University, IRB No.202070. Written informed consent was obtained from all participants for their participation in this study.
ZL and GL: writing. ZY: data curation. ZL, GL, LL, XW, and JS: data analysis. ZL and GL: visualization. JH, LZ, HL, and WW: supervision. All authors contributed to the article and approved the submitted version.
Guangzhou Medical University Discipline Construction Funds (Basic Medicine) (No. JCXKJS2022A11).
Author JS is employed by Tianpeng Technology Co. Ltd, Guangzhou, China.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.989275/full#supplementary-material
1. Bagchi S, Yuan R, Engleman EG. Immune checkpoint inhibitors for the treatment of cancer: Clinical impact and mechanisms of response and resistance. Annu Rev Pathol (2021) 16:223–49. doi: 10.1146/annurev-pathol-042020-042741
2. Kang JH, Bluestone JA, Young A. Predicting and preventing immune checkpoint inhibitor toxicity: Targeting cytokines. Trends Immunol (2021) 42(4):293–311. doi: 10.1016/j.it.2021.02.006
3. Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, Kashaw SK, et al. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: Mechanism, combinations, and clinical outcome. Front Pharmacol (2017) 8:561. doi: 10.3389/fphar.2017.00561
4. Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD-L1 checkpoint. Immunity (2018) 48(3):434–52. doi: 10.1016/j.immuni.2018.03.014
5. O'Donnell JS, Long GV, Scolyer RA, Teng MW, Smyth MJ, et al. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat Rev (2017) 52:71–81. doi: 10.1016/j.ctrv.2016.11.007
6. Seijo LM, Peled N, Ajona D, Boeri M, Field JK, Sozzi G, et al. Biomarkers in lung cancer screening: Achievements, promises, and challenges. J Thorac Oncol (2019) 14(3):343–57. doi: 10.1016/j.jtho.2018.11.023
7. Wu L, Qu X. Cancer biomarker detection: recent achievements and challenges. Chem Soc Rev (2015) 44(10):2963–97. doi: 10.1039/C4CS00370E
8. Timar J, Kashofer K. Molecular epidemiology and diagnostics of KRAS mutations in human cancer. Cancer Metastasis Rev (2020) 39(4):1029–38. doi: 10.1007/s10555-020-09915-5
9. Sisodiya SM. Precision medicine and therapies of the future. Epilepsia (2021) 62 Suppl 2(Suppl 2):S90–s105. doi: 10.1111/epi.16539
10. Teng MW, Ngiow SF, Ribas A, Smyth MJ. Classifying cancers based on T-cell infiltration and PD-L1. Cancer Res (2015) 75(11):2139–45. doi: 10.1158/0008-5472.CAN-15-0255
11. Bera K, Schalper KA, Rimm DL, Velcheti V, Madabhushi A. Artificial intelligence in digital pathology - new tools for diagnosis and precision oncology. Nat Rev Clin Oncol (2019) 16(11):703–15. doi: 10.1038/s41571-019-0252-y
12. Issa NT, Stathias V, Schürer S, Dakshanamurthy S. Machine and deep learning approaches for cancer drug repurposing. Semin Cancer Biol (2021) 68:132–42. doi: 10.1016/j.semcancer.2019.12.011
13. Gandara DR, Paul SM, Kowanetz M, Schleifman E, Zou W, Li Y, et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat Med (2018) 24(9):1441–8. doi: 10.1038/s41591-018-0134-3
14. Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet (London England) (2016) 387(10030):1837–46. doi: 10.1016/S0140-6736(16)00587-0
15. Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet (London England) (2017) 389(10066):255–65. doi: 10.1016/S0140-6736(16)32517-X
16. Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol (2013) 31(11):1023–31. doi: 10.1038/nbt.2696
17. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer (Oxford Engl 1990) (2009) 45(2):228–47. doi: 10.1016/j.ejca.2008.10.026
18. Livne M, Boldsen JK, Mikkelsen IK, Fiebach JB, Sobesky J, Mouridsen K. Boosted tree model reforms multimodal magnetic resonance imaging infarct prediction in acute stroke. Stroke (2018) 49(4):912–8. doi: 10.1161/STROKEAHA.117.019440
19. Zhang Z, Ho KM, Hong Y. Machine learning for the prediction of volume responsiveness in patients with oliguric acute kidney injury in critical care. Crit Care (London England) (2019) 23(1):112. doi: 10.1186/s13054-019-2411-z
20. LOWESS WSC. A program for smoothing scatterplots by robust locally weighted regression. Am Stat (1981) 35(1):54. doi: 10.2307/2683591
21. William S, Cleveland EG, Shyu WM. Local regression models, 1st Edition. New York: Routledge (1992). p. 68
22. Achim Zeileis CK, Krämer W, Hornik K. Testing and dating of structural changes in practice. Comput Stat Data Anal (2003) 44(1-2):109–23. doi: 10.1016/S0167-9473(03)00030-6
23. Prasetyanti PR, Medema JP. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol Cancer (2017) 16(1):41. doi: 10.1186/s12943-017-0600-4
24. Dantoing E, Piton N, Salaün M, Thiberville L, Guisier F. Anti-PD1/PD-L1 immunotherapy for non-small cell lung cancer with actionable oncogenic driver mutations. Int J Mol Sci (2021) 22(12):6288. doi: 10.3390/ijms22126288
25. Krieg C, Nowicka M, Guglietta S, Schindler S, Hartmann FJ, Weber LM, et al. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat Med (2018) 24(2):144–53. doi: 10.1038/nm.4466
26. Suda K, Tomizawa K, Mitsudomi T. Biological and clinical significance of KRAS mutations in lung cancer: an oncogenic driver that contrasts with EGFR mutation. Cancer metastasis Rev (2010) 29(1):49–60. doi: 10.1007/s10555-010-9209-4
27. Wang Z, Duan J, Wang G, Zhao J, Xu J, Han J, et al. Allele frequency-adjusted blood-based tumor mutational burden as a predictor of overall survival for patients with NSCLC treated with PD-(L)1 inhibitors. J Thorac Oncol (2020) 15(4):556–67. doi: 10.1016/j.jtho.2019.12.001
28. Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer's achilles' heel. Cancer Cell (2008) 13(6):472–82. doi: 10.1016/j.ccr.2008.05.005
29. Mir IH, Guha S, Behera J, Thirunavukkarasu C. Targeting molecular signal transduction pathways in hepatocellular carcinoma and its implications for cancer therapy. Cell Biol Int (2021) 45(11):2161–77. doi: 10.1002/cbin.11670
30. Coussens LM, Werb Z. Inflammation and cancer. Nature (2002) 420(6917):860–7. doi: 10.1038/nature01322
31. Ai L, Xu A, Xu J. Roles of PD-1/PD-L1 pathway: Signaling, cancer, and beyond. Adv Exp Med Biol (2020) 1248:33–59. doi: 10.1007/978-981-15-3266-5_3
32. Morad G, Helmink BA, Sharma P, Wargo JA. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell (2021) 184(21):5309–37. doi: 10.1016/j.cell.2021.09.020
33. Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harbor Perspect Biol (2010) 2(1):a001008. doi: 10.1101/cshperspect.a001008
34. Iranzo J, Martincorena I, Koonin EV. Cancer-mutation network and the number and specificity of driver mutations. Proc Natl Acad Sci USA (2018) 115(26):E6010–e9. doi: 10.1073/pnas.1803155115
35. Lei X, Lei Y, Li JK, Du WX, Li RG, Yang J, et al. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett (2020) 470:126–33. doi: 10.1016/j.canlet.2019.11.009
36. Moslehi J, Lichtman AH, Sharpe AH, Galluzzi L, Kitsis RN. Immune checkpoint inhibitor-associated myocarditis: manifestations and mechanisms. J Clin Invest (2021) 131(5):e145186. doi: 10.1172/JCI145186.
37. Kalbasi A, Ribas A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat Rev Immunol (2020) 20(1):25–39. doi: 10.1038/s41577-019-0218-4
38. Geisler AN, Phillips GS, Barrios DM, Wu J, Leung DYM, Moy AP, et al. Immune checkpoint inhibitor-related dermatologic adverse events. J Am Acad Dermatol (2020) 83(5):1255–68. doi: 10.1016/j.jaad.2020.03.132
39. Baxi S, Yang A, Gennarelli RL, Khan N, Wang Z, Boyce L, et al. Immune-related adverse events for anti-PD-1 and anti-PD-L1 drugs: systematic review and meta-analysis. BMJ (Clinical Res ed) (2018) 360:k793. doi: 10.1136/bmj.k793
40. Dolladille C, Ederhy S, Sassier M, Cautela J, Thuny F, Cohen AA, et al. Immune checkpoint inhibitor rechallenge after immune-related adverse events in patients with cancer. JAMA Oncol (2020) 6(6):865–71. doi: 10.1001/jamaoncol.2020.0726
41. Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol diagn (2015) 17(3):251–64. doi: 10.1016/j.jmoldx.2014.12.006
42. Liu Z, Xie Z, Zhao S, Ye D, Cai X, Cheng B, et al. Presence of allele frequency heterogeneity defined by ctDNA profiling predicts unfavorable overall survival of NSCLC. Trans Lung Cancer Res (2019) 8(6):1045–50. doi: 10.21037/tlcr.2019.12.10
43. Pan D, Hu AY, Antonia SJ, Li CY. A gene mutation signature predicting immunotherapy benefits in patients with NSCLC. J Thorac Oncol (2021) 16(3):419–27. doi: 10.1016/j.jtho.2020.11.021
44. Liu Z, Xie Z, Cai X, He J, Liang W. A modified algorithm adjusting both high and minor allele frequency mutation to redefine blood-based tumor mutational burden (bTMB) for optimal prediction of clinical benefits from immune checkpoint inhibitor therapy. J Thorac Oncol (2020) 15(5):e69–72. doi: 10.1016/j.jtho.2019.12.120
Keywords: non-small cell lung cancer (NSCLC), machine learning (ML), immunotherapy, gene, tumor mutational burden (TMB)
Citation: Liu Z, Lin G, Yan Z, Li L, Wu X, Shi J, He J, Zhao L, Liang H and Wang W (2022) Predictive mutation signature of immunotherapy benefits in NSCLC based on machine learning algorithms. Front. Immunol. 13:989275. doi: 10.3389/fimmu.2022.989275
Received: 08 July 2022; Accepted: 08 September 2022;
Published: 27 September 2022.
Edited by:
Massimo Guidoboni, Scientific Institute of Romagna for the Study and Treatment of Tumors, (IRCCS), ItalyReviewed by:
Dongqiang Zeng, Southern Medical University, ChinaCopyright © 2022 Liu, Lin, Yan, Li, Wu, Shi, He, Zhao, Liang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lei Zhao, emhhb2xlaUBnemhtdS5lZHUuY24=; Hengrui Liang, aGVuZ3J1aV9saWFuZ0AxNjMuY29t; Wei Wang, YmJtY3dlaUAxMjYuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.