 Aldo Spanjaard1†
Aldo Spanjaard1† Maria Stratigopoulou2†Daniël de Groot1‡Muhammad Aslam1‡
Maria Stratigopoulou2†Daniël de Groot1‡Muhammad Aslam1‡ Paul C. M. van den Berk1‡Chantal Stappenbelt1§Matilda Ayidah1§
Paul C. M. van den Berk1‡Chantal Stappenbelt1§Matilda Ayidah1§ Joyce J. I. Catsman1
Joyce J. I. Catsman1 Iris N. Pardieck3Maaike Kreft1
Iris N. Pardieck3Maaike Kreft1 Ramon Arens3
Ramon Arens3 Jeroen E. J. Guikema2*‖
Jeroen E. J. Guikema2*‖ Heinz Jacobs1*‖
Heinz Jacobs1*‖- 1Division of Tumor Biology and Immunology, Netherlands Cancer Institute, Amsterdam, Netherlands
- 2Department of Pathology, Amsterdam University Medical Centers, Location Academic Medical Center (AMC), Lymphoma and Myeloma center Amsterdam (LYMMCARE), Amsterdam, Netherlands
- 3Department of Immunology, Leiden University Medical Center, Leiden, Netherlands
The development and differentiation of B cells is intimately linked to cell proliferation and the generation of diverse immunoglobulin gene (Ig) repertoires. The ubiquitin E3 ligase HUWE1 controls proliferation, DNA damage responses, and DNA repair, including the base excision repair (BER) pathway. These processes are of crucial importance for B-cell development in the bone marrow, and the germinal center (GC) response, which results in the clonal expansion and differentiation of B cells expressing high affinity immunoglobulins. Here, we re-examined the role of HUWE1 in B-cell proliferation and Ig gene diversification, focusing on its involvement in somatic hypermutation (SHM) and class switch recombination (CSR). B-cell-specific deletion of Huwe1 resulted in impaired development, differentiation and maturation of B cells in the bone marrow and peripheral lymphoid organs. HUWE1 deficiency diminished SHM and CSR by impairing B-cell proliferation and AID expression upon activation in vitro and in vivo, and was unrelated to the HUWE1-dependent regulation of the BER pathway. Interestingly, we found that HUWE1-deficient B cells showed increased mRNA expression of Myc target genes upon in vitro activation despite diminished proliferation. Our results confirm that the E3 ligase HUWE1 is an important contributor in coordinating the rapid transition of antigen naïve, resting B cells into antigen-activated B cells and regulates mutagenic processes in B cells by controlling AID expression and the post-transcriptional output of Myc target genes.
1 Introduction
The ability of B cells to protect the host from pathogens relies on their ability to terminally differentiate into plasma cells that secrete high-affinity antibodies in response to foreign antigen. During an immune reaction, B cells bind to and are activated by foreign antigen, resulting in the formation of a transient histological structure that include follicular dendritic cells and T follicular helper cells. Within the GC, B cells rapidly proliferate, and are licensed to undergo somatic hypermutation (SHM) to increase affinity of antibodies for target antigen (1, 2). Class switch recombination (CSR), which alters the effector function of antigen takes place in pre-GC B cells (3).
During SHM, mutations are introduced in the variable (V) regions of the immunoglobulin genes (Ig), which are instigated by activation-induced cytidine deaminase (AID). AID binds single-stranded DNA and processively deaminates cytosine (C) bases to uracils (U). Upon replication and if left unrepaired, the resulting U:G mismatches give rise to C-to-T and G-to-A transitions. Alternatively, the Us may be removed by the base excision repair (BER) pathway through the action of uracil glycosylase (UNG), yielding apurinic/apyrimidinic (AP) sites. AP sites can be bypassed by translesion synthesis (TLS) polymerases, generating C-to-T transitions, as well as C-to-A and C-to-G transversions. Conversely, the mutagenic fate of AP sites is counteracted by the AP endonucleases 1 (APE1) and APE2 that cleave the DNA phosphodiester backbone at the AP site, yielding a 5’ deoxyribose-phosphate (dRP) moiety and a 3’ hydroxyl group. The BER protein DNA polymerase beta (POLB) removes the 5’ dRP through its lyase activity and inserts C opposite G, thereby faithfully repairing the AID-instigated lesion. Lastly, the AID-instigated U:G mismatch may be recognized by the mismatch repair (MMR) components MSH2 and MSH6, which in concert with PMS2 and MLH1 recruit exonuclease 1 (EXO1). Subsequently, EXO1 excises a patch of DNA containing the U base(s) triggering fill-in synthesis by DNA polymerase eta (POLH), where POLH is recruited by PCNA-ubiquitination (4, 5). POLH is particularly error-prone when encountering A or T bases. This non-canonical MMR (ncMMR) activity results in A/T mutations in the V genes. Importantly, EXO1 activity requires a DNA nick as entry point to initiate excision. It was shown that PMS2 contains endonuclease activity that nicks 5’ from the U:G mismatch (6). Alternatively, the nick may be provided by the concerted action of UNG and APE1/2, suggesting that BER and ncMMR not only compete for the same U:G mismatch but also may cooperate to generate mutations (7).
During CSR the concerted action of AID, UNG, and AP endonucleases introduces staggered DNA double strand breaks (DSBs) at switch (S) regions located upstream of the constant regions within the Igh loci, enabling the deletion of gene fragments located between two active S regions in antibody Igh constant region (Ch), thereby exchanging Cμ (coding for IgM) for one of the downstream Ch regions. DSBs are generated when AP sites on opposite strands are sufficiently close and are cleaved by APE1/2. Alternatively, U:Gs are recognized as G mismatches by the MMR to generate DSBs by triggering DNA patch excision on both strands (8). As a result, the clonotypic class switched antibody of a B-cell is equipped with an alternate Ch with distinct effector functions and tissue distribution. Interestingly, the generation of DNA nicks required for CSR may be counteracted by conventional BER involving POLB, which faithfully repairs AID-instigated U lesions (9, 10).
Given these insights, it is apparent that both SHM and CSR involve the complex interplay between the BER and the MMR pathways, where the proximal part of the BER pathway (UNG and APE1/2) contributes to mutagenesis during SHM and to DNA nicks during CSR, whereas the distal BER component POLB counteracts mutagenesis and CSR. In agreement, we have recently shown that loss of POLB stimulates in vitro A/T mutagenesis by triggering EXO1 patch excision and ncMMR activity (10). Furthermore, we showed that human GC B cells are characterized by low protein expression of POLB, whereas B-cell lines that are used to study SHM and CSR in vitro display ample POLB protein expression, which may explain the relative paucity of A/T mutations in these cell lines.
The contribution of the BER and MMR pathways towards SHM and CSR has been extensively studied, however, the regulation of its components in a physiological context has received little attention. Previously, it was shown that the E3 ligase HUWE1 (also known as Mule or ARF-BP1) is an important modulator of the BER pathway, regulating the steady-state levels of POLB by ubiquitination (11). However, this was contested by Fang et al showing that lysine-to-arginine mutations of the HUWE1 target residues on POLB did not affect proteasome-mediated degradation (12). HUWE1 was shown to regulate additional DNA repair proteins by monoubiquitination, such as H2A.X histone variant (H2AX) and histone variant 2B (13, 14), breast cancer type 1 susceptibility protein (BRCA1) (15, 16), DNA polymerase lambda (POLL) (11, 17). In addition, HUWE1 was shown to be involved in DNA replication by interacting with proliferating cell nuclear antigen (PCNA) (13) and ubiquitination of cell division cycle 6 (CDC6) (18). Importantly, the proliferation-associated MYC proto-oncogene was shown to be a HUWE1 substrate. HUWE1-mediated lysine 63-linked (K63) polyubiquitination of MYC was required for recruitment of the transcriptional coactivator p300, thereby switching a repressive state of MYC into a transactivation state (19). MYC was shown to promote transcriptional elongation, which requires the interaction with polymerase II-associated factor 1 (PAF1c). Interestingly, the interaction between MYC and PAF1c is driven by HUWE1-mediated ubiquitination, showing that the transcriptional output of MYC is in part controlled by HUWE1 (14).
Previously, using B-cell-specific conditional Huwe1-deficient mice it was demonstrated that HUWE1 plays an important role in B-cell homeostasis (15, 20, 21) by preventing activation of the DNA damage response (DDR) involving the ataxia telangiectasia mutated (ATM) kinase and p53. It was shown that CSR in vitro and in vivo was impaired in Huwe1-deficient B cells. However, the GC response, the transcriptional consequences of Huwe1 deletion in B cells and the effects on SHM were not reported (15). This prompted us to revisit the role of HUWE1 in B-cell activation and Ig diversification in vivo. In agreement with previous studies, we observed that HUWE1 was a crucial regulator of B-cell activation (15, 21). Huwe1;Mb1-cre mice harboured reduced B-cell numbers in the spleen and bone marrow. Stimulation of Huwe1;Mb1cre B cells failed to induce proliferation, despite increased upregulation of Myc target genes. In line with reduced proliferation, SHM, CSR, and GC formation were reduced in Huwe1;Mb1cre B cells. However, in contrast to previous reports we did not observe obvious effect of Huwe1 deletion on POLB protein expression in vivo. Transcriptome analyses revealed that HUWE1 significantly impacted on the transcriptional output of Myc in resting and activated mature B cells. Our data highlight a critical contribution of Huwe1 to B-cell homeostasis and Ig diversification mechanisms including CSR and SHM, by regulating proliferation and acting independently of the regulation of BER.
2 Materials and methods
2.1 Mice
Huwe1-floxed mice have been described elsewhere and were kindly provided by professor Wei Gu (Columbia University, Herbert Irving Comprehensive Cancer Center, New York, NY) (22). To obtain B-cell-specific deletions of Huwe1, Mb1-Cre homozygous male mice were crossed with Huwe1-FL/wildtype (WT) female mice. As HUWE1 is X-linked, this crossing generated male Mb1-Cre mice that were either Huwe1-WT or Huwe1-Floxed (Huwe1FL/yMb1cre, indicated as Huwe1;Mb1cre in this study). The Mb1-Cre mouse model was chosen as it achieves efficient gene deletion in the B-cell lineage. Moreover, it allowed the recapitulation of previous findings on the role of Huwe1 in B-cell development and B-cell activation (15). For this study, 7 to 15-week-old mice were used. All animal experiments were approved by the Animal Ethics Committee of the Netherlands Cancer Institute (NKI) and the Dutch Experiments on Animals Act and the Council of Europe.
The genotyping primers to detect the WT, floxed, and deleted version of the Huwe1 allele were 5’-CTAATCACAGGAAGCGGTTACAAG-3’, 5’-CTCCTATAGCAAAGTAAAAGTATAG-3’, 5’-GTTATTAGGTGGATCCGTACGAT-3’, and 5’-TTAGCTTGTTCTGCAGGTGGCGAC-3’. Primers for Mb1cre genotyping were 5’-CTGCGGGTAGAAGGGGGTC-3’ and 5’-CCTTGC
GAGGTCAGGGAGCC-3’. To assess Huwe1 deletion in sorted B cells the following primers were used: 5’-GTGCAAGCTGAACAACAGGAA-3’ and 5’-AAGGAGAATGTGGATGCTGG-3’, which only yield a PCR product upon successful LoxP gene deletion. PCR settings were: 95°C (3min), 75°C (5min), 72°C (1.5min), followed by 40 cycles of 94°C (30sec), 58°C (45 sec), 72°C (45sec), and a final extension at 72°C.
2.2 Flow cytometry and FACS
To examine early B-cell development, bone marrow (BM) was flushed from the femurs using a 21-gauge syringe with cold PBEA buffer (1× PBS 0.5% BSA, 2 mM EDTA, and 0.02% sodium azide). Erylysis buffer (15.5mM NH4Cl, 1mM KHCO3, 0.01mM EDTA, pH 7.4) was added for 3min on ice to lyse red blood cells. Identical procedures were performed to obtain B cells from the spleen, except that here, spleens were first filtered through 70 µm strainers. Samples were measured using LSR Fortessa (BD Biosciences, Franklin Lakes, NJ) flow cytometers, and sorted using LSR Fortessa Aria2 cell sorter. Data was analyzed with FlowJo software v10.7.1 (Tree Star Inc., Ashland, OR).
2.3 Class switch recombination and proliferation
Spleens were isolated from 11-15 weeks-old mice. Single cell suspensions were generated and erylysis buffer was added to remove erythrocytes. Naïve splenic B cells were enriched by performing CD43 depletion using biotinylated anti-CD43 antibody (1:100, BD Pharmingen, San Diego, CA) and sorted from the spleen by flow cytometry-coupled sorting. 5 x 106 B cells were labeled for 10 minutes at 37°C at a final concentration of 0,05 μM carboxyfluorescein succinimidyl ester (CFSE, Molecular probes, Eugene, OR) in Iscove’s Modified Dulbecco’s Medium (IMDM) containing 8% fetal bovine serum (FCS). For both the CSR and CFSE assays, the cells were seeded at 1 x 105 cells per well. To trigger proliferation and CSR, the isolated B cells were stimulated with lipopolysaccharide (Escherichia coli, LPS, 5 μg/mL, Sigma-Aldrich, St. Louis, MO) and mouse recombinant interleukin-4 (IL-4) (10 ng/mL, Peprotech, London, UK), IL-4 and anti-CD40 (clone HM40-3, 1 μg/mL, BD Biosciences), and LPS combined with IL-4 and αIgD-dextran (1:5000, Fina Biosolutions LLC, Rockville, MD) to induce class switching. Cells were cultured for four days, at which the cells were harvested daily to measure proliferation. Additionally, after 72h and 96h, the percentage of cells that had class switched to IgG1 was measured.
2.4 Immunization and phenotyping the spleen
To analyze B-cell responses against infection, 7 – 11 weeks-old WT and Huwe1;Mb1cre mice were immunized by an intraperitoneal injection of 2 x 107 lymphocytic choriomeningitis virus particles (LCMV-Armstrong, kindly gifted by Dr. Ramon Arens, Leiden). Two weeks after immunization, the spleens were isolated and single cell suspensions were made as previously described to investigate B-cell development (23). Splenocytes were labeled using the following markers for plasma cell identification (CD19, B220, IgD, IgM, Sca1, CD138, CD3e and Ter119), and for splenic mature B-cell subsets (CD19, B220, IgD, IgM, CD93, CD23and CD21/35) and GC staining (CD19, CD95, GL7 and IgM). Serum was prepared from blood to determine levels of LCMV-specific IgG and IgM by enzyme-linked immunosorbent assay (ELISA): In short, Nunc-Immuno Maxisorp plates (ThermoFisher Scientific, Waltham, MA) were coated overnight at 4°C with virus particles in bicarbonate buffer. Plates were subsequently incubated for 1h with blocking buffer (PBS/5% milk powder). Serum samples from mice were diluted in PBS/1% milk powder and incubated for 1h at 37°C. Next, Horseradish peroxidase (HRP)-conjugated IgG and IgM antibodies (Southern Biotech, Birmingham, AL) were diluted 1:4,000 in PBS/1% milk powder and incubated 1 h at 37°C. Plates were developed with 3,3’,5,5’-tetramethylbenzidine (TMB) substrate (Sigma Aldrich), and the color reaction was stopped by the addition of 1 M H2SO4. Optical density was read at 450 nm (OD450) using a Microplate reader (Model 680, Bio-Rad, Hercules, CA). Pre-immune serum samples were used as controls.
2.5 Somatic hypermutation analysis
To perform SHM analysis, splenic CD19+CD3-IgD-IgK+ memory B cells from unimmunized mice were obtained by FACS and the JH4 intron region was analyzed for mutations by DNA sequencing. This is a well-established and convenient method to analyze SHM in post-germinal center B cells (4). Briefly, memory B cells were sorted into tubes containing Tris-HCl and Proteinase K (ProtK). To lyse the cells, tubes were incubated for 1hr at 55°C, and ProtK was inactivated for 5 minutes at 85°C. Tubes were stored at -20°C until further use. DNA was purified by ethanol precipitation. The JH4 intron region was amplified by PCR using the VHB1-8 framework three primer forward primer (5’-CAGCCTGACATCTGAGGACTC-3’) and reverse primer (5’-CTCCACCAGACCTCTCTAGACA-3’). 2% E-gel with SYBR safe (Invitrogen) were used to extract the JH4 amplified product, corresponding to a fragment of around 650bp in length and sequenced by Illumina MiSeq Nano using paired-end sequencing. For mutation frequency calculations, identical mutated sequences were considered clonal and were counted once. Statistical analysis to determine the mutational spectra was performed in R to determine the mutational frequencies.
2.6 Protein extraction and Western blot
Spleens were isolated from 11-15 weeks-old unimmunized mice. Single cell suspensions were generated and erylysis buffer (hybridoma, red cell lysis buffer, Sigma-Aldrich) was added to remove erythrocytes. Naïve splenic B cells were enriched by performing magnetic-activated cell sorting (MACS) with anti-CD43 (Ly-48) MicroBeads, (miltenyi-Biotec, Bergisch Gladbach, Germany) according to the manufacturer’s protocol. The cells were seeded in in 24-well-plates (0.5 x 106 cells per well) and activated with lipopolysaccharide (Escherichia coli, LPS, 50 μg/mL, Sigma-Aldrich) and IL-4 (20 ng/mL, ProspecBio, Hamada, Israel), αIgD-dextran (10ng/ml, Fina Biosolution) and recombinant human BAFF (100ng/ml, ProspecBio) for three days. After three days of activation the cells were harvested and washed with ice-cold PBS and lysed in ice-cold lysis buffer (10 mM Tris-HCl pH 8, 140 mM of NaCl, 1% Nonidet P-40, 0.1% sodium deoxycholate, 0.1% SDS, 1 mM of EDTA) supplemented with protease and phosphatase inhibitors (EDTA-free protease mixture inhibitor; Roche Diagnostics, Almere, the Netherlands). The protein pellets were homogenized passing through a 25G needle, and protein concentrations were measured using the bicinchoninic acid assay (BioRad). For each sample, 10 μg of total protein lysate and cell equivalents were used for protein separation in Precise 4–12% gradient Bis-Tris gels (Invitrogen, Waltham, MA). Subsequently, separated protein lysates were transferred onto polyvinylidene difluoride membranes (Immobilon-P; EMD Millipore, Burlington, MA); membranes were blocked in 5% BSA (BSA Fraction V; Roche Life Sciences, Almere, The Netherlands) in tris-buffered saline with 0.1% Tween-20 (TBS-T) for 1 h. Primary antibodies were incubated overnight at 4°C. After a series of thorough washes with TBS-T, membranes were incubated with secondary antibodies (goat anti-rabbit HRP or rabbit anti-mouse HRP; DAKO, Agilent Technologies, Heverlee, Belgium) for 2 h at room temperature. Antibody binding (protein expression) was visualized using Amersham ECL Prime Western blotting detection reagent (GE Healthcare Bio-Sciences AB, Uppsala, Sweden).
The following antibodies were used in this study: MSH6 (BD, 610919), MSH2 (clone FE11, Millipore), β-Actin antibody (clone AC-15, Sigma-Aldrich), DNA Polymerase beta antibody (ab26343, Abcam, Cambridge, UK), c-Myc (051203, Epitomics), p21 (SX118, Santa Cruz), AID [kind gift from Prof. Dr. Hans-Martin Jäck, University of Erlangen (24)].
2.7 Immunohistochemistry
Tissue sections were deparaffinized with xylene and hydrated through an alcohol rinse series followed by 15 minutes incubation in methanol + 0.3% H2O2. After heat-inducible epitope retrieval in citrate buffer (pH=6) in a pressure cooker for 20 minutes, slides were treated with Super Block (AAA125, ScyTek Laboratories, West Logan, UT) for 15 minutes at room temperature. Then, the tissue sections were incubated with the primary antibodies (PNA, Vector laboratories, B-1075; POLB, Abcam, ab26343, POLB antibody specificity was confirmed by immunohistochemistry on paraffin-embedded CH12-F3 Polb-knockdown mouse lymphoma cells that were described previously (10); Ki-67, ThermoFisher scientific, SP6). Antibody–antigen detection was performed with goat anti-rabbit poly horseradish peroxidase (HRP) (Bright Vision poly-HRP-anti-rabbit IgG, Immunologic, VWR, Duiven, The Netherlands). The end products were visualized with 3,3’-diaminobenzidine (DAB) (Bright DAB substrate kit, Immunologic). For double immunohistochemistry stainings, similar to single staining, tissue sections were deparaffinized. After heat induced-epitope retrieval and blocking with Super Block, the cells were stained for c-Myc (051203, Epitomics) overnight at 4°C. After several washing, slides were incubated with secondary goat-anti-rabbit-poly-alkaline phosphatase (AP) (Bright Vision poly-AP-anti-rabbit IgG, Immunologic) for 30 minutes at room temperature, followed by adding VECTOR® Red AP substrate, which resulted in a red precipitate. The slides were then boiled in citrate buffer (pH=6) again to remove the bound antibodies, follow by blocking with Super Block and restained with rat anti-p21(HUGO291, Abcam) overnight at 4°C. After several washing steps, slides were incubated with secondary rabbit-anti-rat antibodies (DAKO) for 30 minutes at room temperature and subsequently with goat anti-rabbit-poly-AP (Bright Vision poly AP-anti-rabbit IgG, Immunologic) for 30 minutes at room temperature followed by adding VECTOR® Blue AP substrate, resulting in blue precipitate. The slides were covered with VECTAMOUNT® permanent mounting medium (H-5000, Vector laboratories, Burlingame, CA).
2.8 Statistics
Statistical analyses were performed using Graphpad V7.0. Statistical significance was calculated using an unpaired student t-test with Welch correction to account for unequal standard deviations. For the somatic hypermutation analysis, Fisher’s exact chi-square test was performed.
3 Results
3.1 Huwe1 controls B-cell development in the bone marrow and spleen
To re-examine the effects of HUWE1 deficiency in murine B cells, we crossed male homozygous Mb1cre mice with female Huwe1fl/wt mice from a strain (22) that is different than the mouse strain for which B-cell specific defects were reported previously (15). Resulting male offspring featured either a WT or floxed version of Huwe1 (indicated as Huwe1/Mb1cre), which could be detected via PCR. We could also identify deletions of Huwe1 via PCR in sorted splenic B cells (Supplementary Figures 1A-E). Comprehensive analysis of B-cell development in the bone marrow of Huwe1;Mb1cre mice confirmed a decrease of B-lineage cells in the bone marrow (Figures 1A, B). While the cellularity of the bone marrow was not altered in Huwe1;Mb1cre mice (Figure 1B), B cells were reduced in number (Figure 1B). Pro-B cells were increased in numbers, in contrast to what has previously been reported (15) and pre-B cells were significantly reduced in Huwe1;Mb1cre mice (Figure 1B), suggestive of a pro- to pre-B cell differentiation block. We did not observe significant differences in the number of immature B cells (Figure 1B). While in WT mice, we could clearly distinguish immature from mature B cells on the basis of B220 staining, this discrimination was less apparent in Huwe1;Mb1cre mice due to reduced expression of B220 (Figure 1A). We therefore gated on IgD-positive B220-positive mature B cells and found that their numbers were significantly decreased in Huwe1;Mb1cre mice (Figures 1A, B).
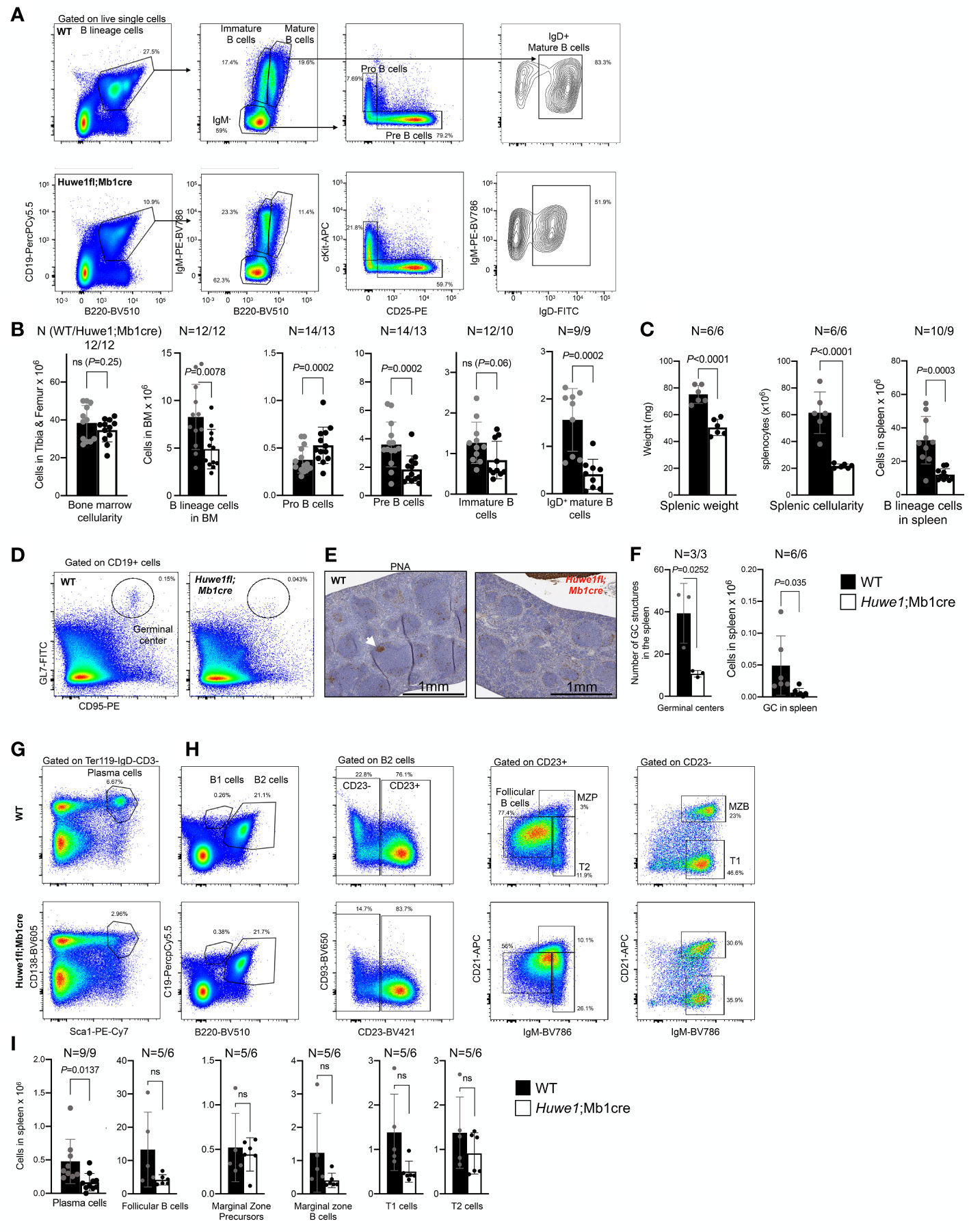
Figure 1 Impaired B-cell development in the bone marrow of Huwe1;Mb1cre mice. (A) Representative flow cytometry gating strategy to assess B-cell development in the bone marrow of wildtype (WT) mice (top) and Huwe1;Mb1cre (bottom) mice. Left panels show the B-cell percentages in the nucleated cell compartment, middle panels show the IgM-negative, mature, and immature B cells, and the right panels depict the pro-B cells and pre-B cells. (B) Absolute numbers of cell populations indicated in (A) in the bone marrow. Mean ± SD is shown. N for WT (left) and Huwe1;Mb1cre is depicted above the graph. Black bars depict WT mice, white bars depict Huwe1;Mb1cre mice. (C) Splenic weight, cellularity, and B lineage cells (CD19+ B220+) in the spleen. (D) Flow cytometry gating strategy of spontaneous germinal centers in the spleen. Percentages of GL7+ CD95+ GC B cells are indicated. (E) Peanut agglutinin (PNA) immunohistochemistry staining in the spleen. Magnification is 20X. (F) Germinal centers in the spleen as identified by counting PNA+ GCs in IHC slides (left), and by flow cytometry staining for GL7 and CD95 (right). (G, H) Flow cytometry gating strategy for plasma cells in the spleen and splenic follicular B cells, marginal zone progenitors (MZP) and transitional 1 (T1) and 2 (T2) B-cell subsets (H). (I) Absolute numbers of subsets as indicated in (G, H), bar graphs depict means ± SD. P-values were calculated using an unpaired t-test with Welch correction. ns, not significant.
The effects of Huwe1-deficiency were even more pronounced in the spleen. We noted a significant decrease in splenic weight, splenic cellularity, and splenic B-cell numbers in absence of Huwe1 (Figures 1D–I). Moreover, fewer plasma cells and spontaneous GCs were observed in the spleens from unimmunized mice (Figures 1D–F), as shown by both flow cytometry and by immunohistochemical staining with the peanut agglutinin (PNA) (Figures 1D–F). Consistent with the reduction of GC B cells as defined by flow cytometry, we observed less and far smaller spontaneous GC structures in the spleens of unimmunized animals (Figure 1F). These results are in line with prior observations of Huwe1;Mb1cre mice, and firmly establish the role of Huwe1 in B-cell development in the bone marrow and spleen (15, 20, 21). However, while we observed a decrease of multiple types of B-cell subsets, such as B1a, B2, T1, T2, cells etc., except for plasma cells, none of these subsets was significantly decreased, suggesting that differentiation of B cells to differentiated subsets is not affected significantly by the absence of Huwe1.
3.2 Reduced but not abolished immune responses in Huwe1-deficient B cells
To assess whether GC responses were also affected in immunized Huwe1;Mb1cre mice we examined immune responses to lymphocytic choriomeningitis (LCMV)-virus, which elicit robust and relevant T-cell dependent B-cell responses that includes GC reaction, memory B-cell and long-lived plasma cell formation. At day 14 post LCMV-infection, spleens and serum samples were collected for analysis. Immunohistochemistry revealed reduced levels of B cells and GCs in the spleen after LCMV infection, which was confirmed using flow cytometry (Figures 2A, B; see (23) for extended gating strategy). In accordance, splenic plasma cells were decreased in Huwe1;Mb1cre mice (Figure 2B). In parallel, LCMV-specific IgM and IgG serum titres were determined. We noted a significant decrease in serum levels of LCMV-specific IgG and IgM antibodies (Figure 2C), the decrease being most prominent for IgM titers. The LCMV-specific IgG response was approximately 10-fold reduced in the Huwe1;Mb1cre mice, but not ablated, suggesting that a GC-independent humoral response, perhaps involving the differentiation of marginal zone (MZ) B cells, may support the LCMV-specific IgG in Huwe1;Mb1cre mice. Importantly, loss of HUWE1 did not significantly affect differentiation of splenic B cells, although there was a trend that B-cell levels were reduced (Figure 2B). However, these results suggest that while development of the B lineage cells relies on HUWE1, once B-cell identity has been determined, much of the downstream differentiation is not significantly affected, with the exception for GC B cells and plasma cells.
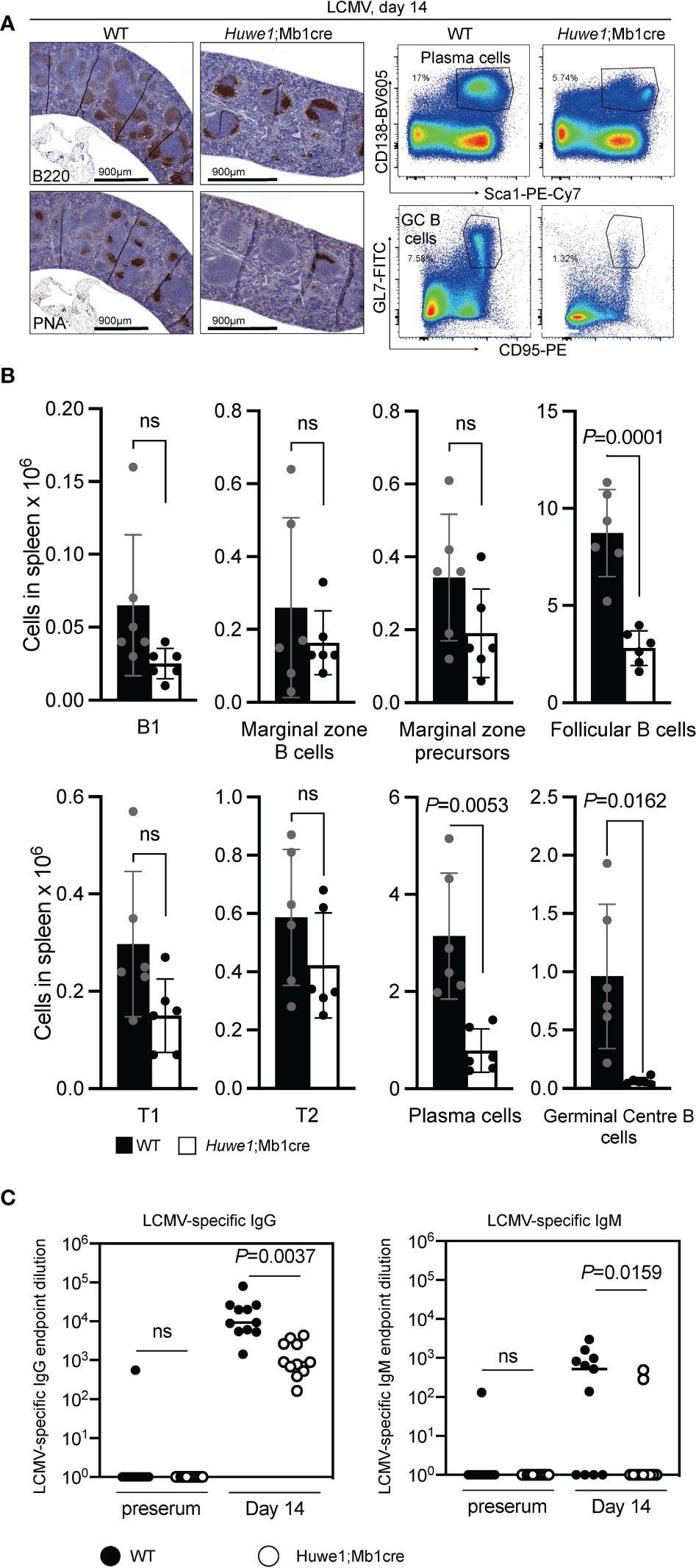
Figure 2 Huwe1-deficiency reduces the immune response to lymphocytic choriomeningitis virus. (LCMV). Wildtype (WT) and Huwe1;Mb1cre mice were infected with LCMV and analyzed 14 days post-infection. (A) Left panels show immunohistochemistry for B220 (total B cells) and peanut-agglutinin (PNA) (GCs). Right panels show flow cytometry gating strategy for plasma cells (CD138+ Sca1+) and GC B cells (GL7+ CD95+) in spleens from LCMV-infected mice. Percentage of gated cells are indicated. (B) Absolute numbers of indicated B-cell subsets from LCMV-infected spleens. Means ± SD are depicted. Black bars represent WT mice, white bars represent Huwe1;Mb1cre mice (N=6 for both groups). Flow cytometry gating strategy for B-cell subsets was similar as in Figure 1. (C) LCMV-specific IgG and IgM serum titres 14 days post-infection. Serum isolated before infection (preserum) was used as control. Numbers indicate the endpoint dilution of the sera. N=12 for both groups. P-values were calculated using an unpaired t-test with Welch correction. ns, not significant.
3.3 HUWE1 is essential for in vitro B-cell proliferation and CSR and affects SHM in vivo
The reduced B-cell development in bone marrow and spleen, and the blunted GC responses in absence of Huwe1 could result from defects in proliferation, differentiation, or increased apoptotic signalling. To determine the effects of Huwe1 deficiency on the proliferative capacity of B cells we activated resting splenic B cells for three days with various B-cell stimulation cocktails that induce CSR to IgG1, as CSR is is intimately linked to B-cell proliferation (25). Proliferation of Huwe1;Mb1cre B cells was decreased compared to WT B cells after stimulation with LPS and IL-4, IL-4 and CD40, and LPS and IL-4 and anti IgD-dextran as shown by CFSE dilution analysis (Figure 3A). Concomitantly, IgG1 CSR was severely impaired in all stimulations tested (Figures 3B, C). Since CSR requires cell division, and as Huwe1;Mb1cre B cells featured less cell division after stimulations, we analyzed the level of CSR in equally divided B cells (Figure 3C). Of interest, even in B cells that underwent an equal number of cell divisions, Huwe1-deficiency impaired CSR, indicating that Huwe1 controls CSR in a proliferation-independent manner.
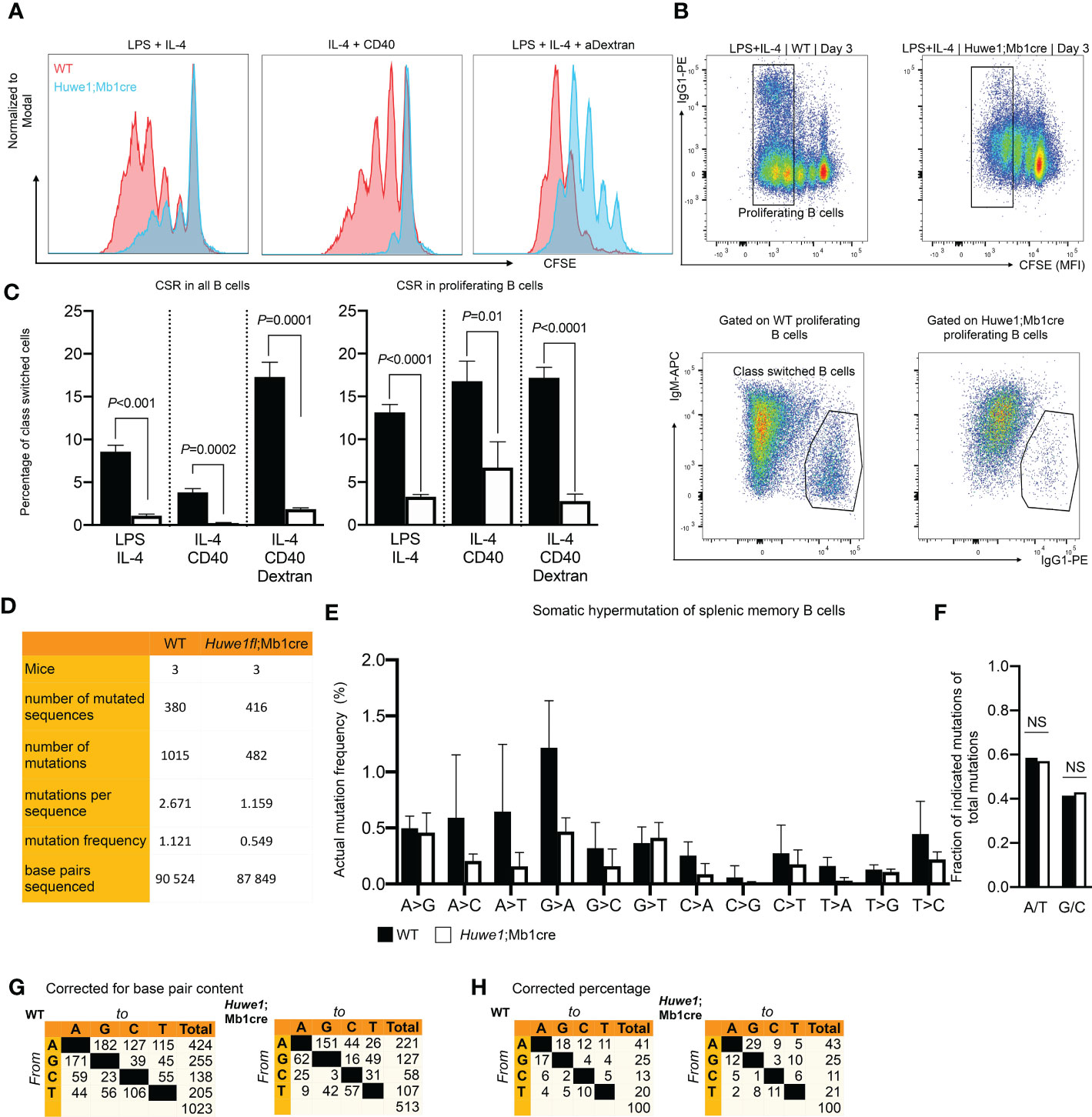
Figure 3 Reduced proliferation, class switch recombination (CSR), and somatic hypermutation. (SHM) in Huwe1;Mb1cre mice. (A Proliferation of activated B cells as measured by carboxyfluoroscein succinimidyl ester (CFSE) labeling. Resting splenic B-cells were activated in vitro for 3 days as indicated and analyzed by flow cytometry. CFSE overlay histograms are shown. Splenic B cells from wildtype (WT) mice are depicted as red histograms, B-cells from Huwe1;Mb1cre are shown as blue histograms. One of three representative experiments is shown. (B) Splenic B-cells activated for 3 days with lipopolysaccharide (LPS) and interleukin-4 (IL-4) to induce IgG1 CSR. Activated B cells were analyzed by flow cytometry. CSR was determined by staining for IgG1. (C) Percentages of IgM- IgG1+ cells in all activated B cells (left panel, no gate applied on CFSE channel) and in proliferating B cells (right panel: gating strategy shown in B, designated as ‘proliferating B cells’). Bar graphs depict means ± SD, one representative of three independent experiments is shown. (D) SHM was determined in splenic memory B cells (CD19+ CD3- IgD- IgK+) isolated from unimmunized mice. SHM was assessed by next-generation sequencing of JH4 intron amplicons. Table indicates numbers of mice, sequences and mutations analyzed. (E) Frequencies of mutations, defined as number of specific template base pairs to be sequenced to find the indicated mutation in WT and Huwe1;Mb1cre B-cells. Means ± SD of three mice are depicted. (F) Bar graph depicting frequencies of mutations at A and T bases (A/T), or G and C bases (G/C) (no significant differences as determined by Fisher’s exact chi-square test). (G-H) Cross tables showing the mutation spectrum, indicated as total number of mutations (G) or the percentage of total mutations (H). P-values were calculated using an unpaired t-test with Welch correction. ns, not significant.
To assess whether the defective GC response also affected SHM we isolated Ig-Kappa-positive, IgD-negative splenic memory B cells from unimmunized mice, and used JH4-intron next generation sequencing to determine SHM. To circumvent any potential bias due to fewer GCs found in Huwe1;Mb1cre mice we chose to analyze SHM in positively selected post-GC (memory) B cells. We noted a strong decrease in the mutation load of SHM affecting nearly the entire spectrum of mutations, indicating that absence of HUWE1 does not affect the generations specific classes of mutations (transversions and transitions at specific bases, Figures 3D–G). In line with this notion, there was no significant difference (Fisher’s exact chi-square, p=0.6) in the frequency of mutations at A:T and C:G bases (Figure 3F). These results argue against any direct role of HUWE1 in SHM with regards to regulating DNA repair proteins implicated in the SHM mutation spectrum (7), but rather argue that lower mutation loads of HUWE1-deficient B cells is the result of reduced proliferation and/or lower AID expression.
3.4 HUWE1 regulates the transcriptional output of MYC in B cells
To further investigate these effects of Huwe1 deficiency we examined the transcriptomes of WT and Huwe1;Mb1cre B cells by RNA-seq. We isolated resting B cells from the spleens of unimmunized mice, and either directly isolated RNA, or stimulated B cells for three days with LPS and IL-4 and anti-IgD-dextran. Importantly, upon stimulation, we noted decreased expression of Huwe1 mRNA in Huwe1;Mb1cre cells, indicating efficient deletion of Huwe1. Moreover, stimulated Huwe1;Mb1cre B cells featured reduced mRNA expression of Aicda, encoding AID, which partly explains the impaired CSR in stimulated Huwe1;Mb1cre B cells (Supplementary Figure 2A). Gene set enrichment analysis (GSEA) revealed that in unstimulated B cells, HUWE1-deficiency resulted in lower expression of MYC target genes and p53 pathway genes, in line with the reported role of HUWE1 on Myc (20, 21). In stimulated B cells, however, we noted significantly increased mRNA expression of the Myc target genes, as well as DNA repair genes, while p53 pathway genes remained downregulated upon stimulation in Huwe1-deficient B cells compared to WT B cells (Figure 4A). Strikingly, the genes that contributed most significantly to the MYC gene set enrichment (‘leading-edge genes’) in unstimulated WT B cells largely overlapped with the leading-edge genes responsible for MYC gene set enrichment in stimulated Huwe1-deficient B cells (Supplementary Figures 3, 4), suggesting that HUWE1 maintains the promoters of this set of genes in a poised state in unstimulated B cells and is involved in the post-transcriptional control of these genes in stimulated B cells.
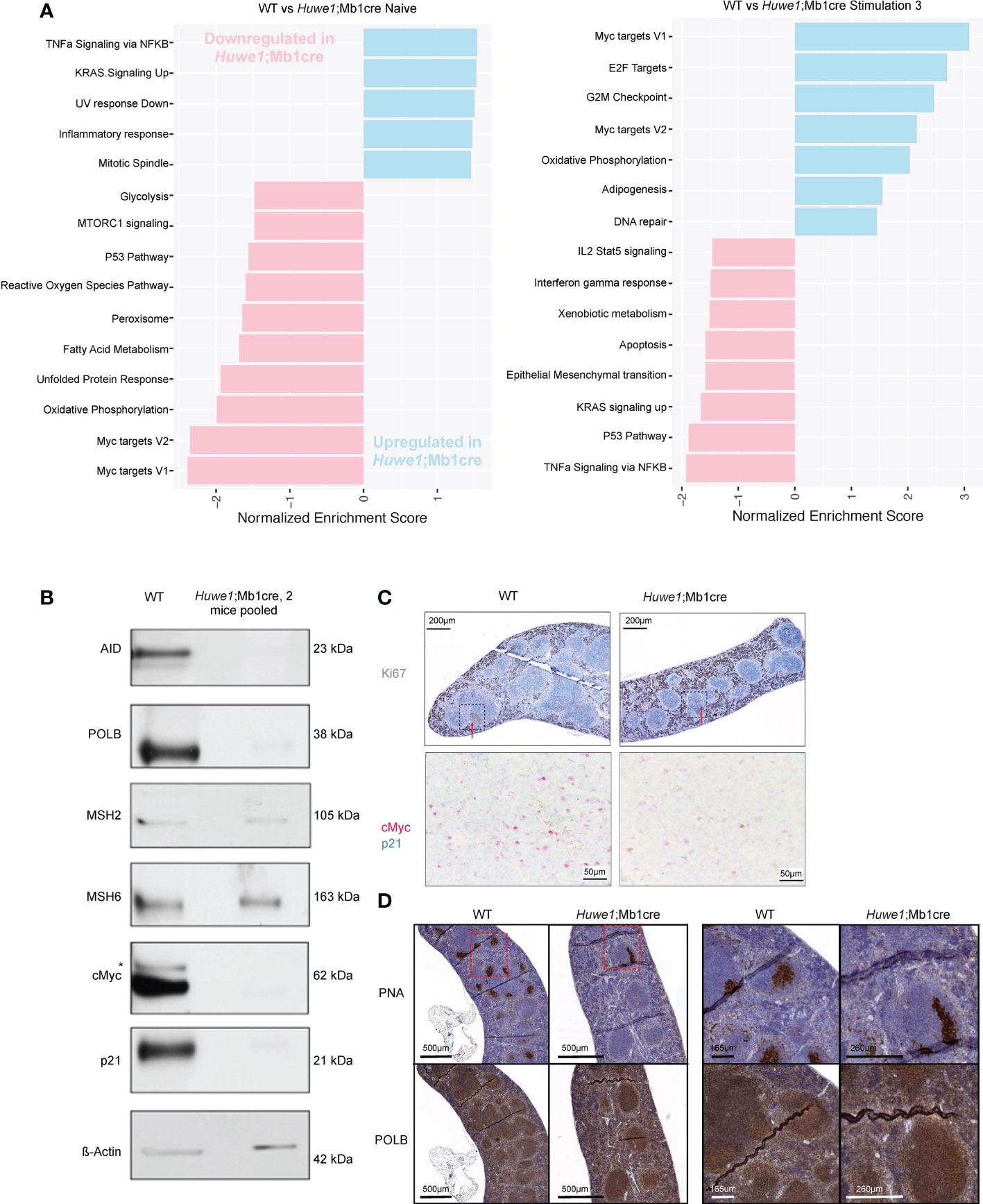
Figure 4 RNA-sequencing gene expression profiling and protein expression analysis of HUWE1-deficient B cells. (A) Fast gene set enrichment analysis (FGSEA) of naive and stimulated B cells. Plotted is the normalized enrichment score (NES) of genes down and upregulated in Huwe1;Mb1cre (pink and blue, respectively.) For each condition, RNA-sequencing was performed on B cells from three independent mice. (B) Immunoblotting analysis of protein expression in activated splenic B cells stimulated with LPS+IL4+anti-IgD-dextran (‘Stimulation type 3’). For each sample, 10 μg of total protein were used for protein separation. To obtain sufficient material, B cells from 2 Huwe1;Mb1cre mice were pooled. Aspecific band in the cMyc immunoblot is indicated with an asterisk. β-actin is used as a loading control. (C) Immunohistochemistry (IHC) staining for Ki67 (visualized by 3,3’-diaminobenzidine substrate, brown color) to identify proliferating cells and germinal centers in formalin-fixed paraffin-embedded spleen sections from LCMV-infected wildtype (WT) and Huwe1;Mb1cre mice 14 days post-infection. The red arrows indicate the GC structures. The area surrounded by the box with the black dashed line corresponds to the area shown in the bottom panel of the figure studying by IHC the cells that express cMyc (pink) and p21 (blue) in splenic GCs from WT and Huwe1;Mb1cre unimmunized mice. (D) IHC staining for peanut agglutinin (PNA) to identify GCs, and DNA polymerase beta (POLB) in the spleens from LCMV-infected mice 14 days post-infection. Panels on the right are magnifications of the indicated boxes with the red dashed lines.
3.5 HUWE1-deficiency does not result in POLB stabilization in activated B cells
In addition, we analyzed the expression of relevant proteins by comparative immunoblotting of lysates obtained from LPS and IL-4 and anti-IgD-dextran-stimulated WT and Huwe1;Mb1cre B cells. In agreement with the transcriptome data, activated Huwe1;Mb1cre splenic B cells showed reduced protein expression levels of AID (Figure 4B). Opposite of what we expected, the expression of POLB was decreased, indicating that loss of HUWE1 did not result in POLB protein stabilization (Figure 4B), as was previously suggested (11). Strikingly, Polb mRNA expression was not affected in activated Huwe1;Mb1cre B cells, suggesting a post-translational regulation of POLB that did not involve HUWE1-dependent ubiquitination and proteasomal degradation. Of interest, other proteins associated with (error-prone) processing of AID-instigated lesions, such as the mismatch repair proteins MSH2, MSH6 were not affected by HUWE1 deficiency, which showed that lower expression of DNA repair proteins is not a general feature of activated Huwe1;Mb1cre B cells. While the mRNA transcription of Myc target genes was significantly enriched, the Myc protein level was reduced in activated Huwe1;Mb1cre cells compared to WT cells, as was the level of cell cycle regulator p21, which is a direct transcriptional target of p53 (Figure 4B). We studied the expression of Myc and p21 also in situ and we confirmed their downregulation in the splenic GCs of LCMV-infected Huwe1;Mb1cre mice at day 14 compared to WT mice (Figure 4C). POLB was expressed in splenic GCs from LCMV-immunized mice, in stark contrast to continual GC in human tonsils, which we reported previously to be characterized by low POLB expression (10). Specificity of the POLB antibody used for immunohistochemistry was confirmed by immunoblotting and immunohistochemistry on paraffin-embedded CH12/F3 Polb knockdown mouse GC-derived lymphoma cells (Supplementary Figures 5A, B). Although POLB protein expression was decreased in in vitro activated Huwe1;Mb1cre B cells, expression in GCs in situ was not affected (Figure 4D).
Our results show that HUWE1 crucially regulates B-cell proliferation, which impinges on B-cell progenitor expansion in the bone marrow, splenic B-cell numbers, CSR and SHM. Our data are not consistent with a role for HUWE1 in the regulation of POLB or other DNA repair proteins that may affect CSR and SHM. However, HUWE1 appears to participate in the control of MYC-target genes. Our finding that the mRNA expression of many DNA replication-associated genes was consistently upregulated in activated Huwe1/Mb1cre B cells, although these cells were severely hampered with regards to proliferation, suggests that HUWE1 may play a crucial role in the post-transcriptional/post-translational regulation of the products of these genes.
4 Discussion
In this study we aimed to delineate the relevance of the E3 ligase HUWE1, also known as Mule or ARF-BP1 in B-cell-dependent immunity. Huwe1 is essential for life, as a bi-allelic inactivation was found to be embryonic lethal (15, 22). However, consistent with a previous study (15), B-lineage cells are able to tolerate partially the loss of Huwe1 at distinct stages of B-cell development. An ablation of both Huwe1 alleles early in the B-cell lineage caused a developmental block of B-cell progenitors at the transition from pro- to pre-B cells. Yet, immature and mature B cells do develop and are present in the peripheral blood and in the spleen, albeit at reduced numbers, indicating that development is hampered but not prohibited by the loss of Huwe1 expression. These insights argue against a critical role of HUWE1 in B-cell development and differentiation, but rather highlights an important contribution of the regulation of cell proliferation.
Given the relevance of HUWE1 in controlling central processes like cell proliferation and DNA-repair (11, 13, 14), we wanted to determine to what extent this affects the intimate link between GC B-cell proliferation and secondary Ig gene diversification, two important hallmarks of the GC reaction. In line with a previous report on the role of HUWE1 in B-cell functioning, proliferation and differentiation of B cells were hampered in the absence of HUWE1 (15). Moreover, detailed ex vivo, as well as in vitro CSR and SHM analyses combined with transcriptomics identify HUWE1 as an important driver of B-cell activation, and in extension, the GC reaction. In agreement with the failure of Huwe1-deficient B cells to upregulate AID expression at the transcriptional and translational level, activated B cells were grossly impaired in their CSR activity in vitro, as well as their SHM capacity in vivo. The failure to acquire normal AID expression levels in the absence of Huwe1 explained the global reduction of the mutation load in somatically mutated Ig genes and the decreased CSR activity. We hypothesize that reduced proliferation resulted in lower AID expression, which is linked to cell division. We observed fewer GCs in Huwe1;Mb1cre mice, and consequently, fewer output cells (memory B cells and plasma cells). SHM was generally lower in positively selected post-GC memory B cells without affecting the mutation spectrum, consistent with lower AID levels during the GC reaction in Huwe1;Mb1cre mice.
Interestingly, it was shown that Myc overexpression was able to complement the CSR defect in Huwe1-deficient B cells (21). However, Myc has not been reported to directly drive AID transcription in GC B cells. This suggests that the effect of Huwe1 deficiency on AID expression indirectly involved Myc, most likely by diminished Myc output resulting in hampered proliferation, thereby limiting AID expression.
In the hematopoietic stem cell (HSC) compartment, Huwe1 controls lineage differentiation, stress responses, and stem cell gene expression (20). The strong reduction of B cells in Huwe1;Mb1cre mice suggested HUWE1-dependent deregulation of pathways crucial for B-cell development and lineage choice. Analyses of the transcriptomes of WT and Huwe1-deficient B cells showed however, that very few pathways were significantly differentially expressed. The most significantly affected genes belonged to the MYC_v1/MYC_v2 gene sets, whose expression was downregulated in naïve (unstimulated) Huwe1-deficient B cells compared to WT B cells, as expected. Counterintuitively, most of these genes were found to be upregulated in activated (LPS+IL-4+anti-IgD-dextran stimulated) Huwe1-deficient B cells compared to activated WT B cells. Many of these genes are important for cell-cycle progression, such as CDK4, MCM2, MCM6, PCNA and POLD2, yet the Huwe1-deficient B cells showed severely hampered in vitro proliferation upon activation. Moreover, MYC protein expression was clearly lower in activated Huwe1;Mb1cre B cells, arguing that even in the absence of MYC, gene expression of MYC target genes can occur, although this clearly does not promote efficient replication. Various studies suggest that HUWE1 critically regulates MYC family members in a context-dependent manner (20, 21). For instance, in HSCs it was demonstrated that N-myc was upregulated upon loss of Huwe1 leading to increased proliferation and stem cell exhaustion, which was reversed by Mycn depletion (20). Whereas in B cells it was shown that the homeostatic defects caused by loss of Huwe1 could be mitigated by overexpression of C-myc (21). Based on this we speculate that in the absence of HUWE1, proliferation of B cells may be initiated but cannot proceed normally due to the absence of functional MYC. This may be accompanied by the inappropriate mRNA expression of DNA replication-associated genes due to defective cell-cycle progression and cell-cycle regulated transcription. Alternatively, but not mutually exclusive, HUWE1 may be involved in the post-transcriptional/post-translational regulation of these genes, by which increased mRNA expression is not effectuated in enhanced function of the gene products. Also, our results suggest that the increased expression of MYC target genes is not necessarily related to increased MYC protein expression, indicating that other transcription factors may crucially regulate the mRNA expression of MYC target genes in the absence of MYC protein. To further elucidate this mechanism additional molecular investigations are required.
A previous study in human cell lines identified DNA Polymerase Beta (POLB), which exerts a critical function in short patch BER, as a target of the E3 ligase HUWE1 (11). However, in contrast to an expected stabilization of POLB, POLB production was reduced in activated murine Huwe1-deficient B cells, perhaps due to differential proliferation. Moreover, in stark contrast to human GCs, levels of POLB were not found to be reduced in murine GCs compared to the surrounding tissue. Hence, in the murine setting, the lack of Huwe1 could not be associated with a stabilization of POLB, neither in vitro nor in situ. The decreased level of POLB in activated B cells could moreover be explained by the decreased levels of p53 since there is evidence of p53 implication in the BER and most specifically POLB in vivo (26). At the same time, members of the MMR machinery, specifically the mismatch recognition complex MSH2/6 were found unaffected in vitro. These insights are consistent with the notion that despite an overall reduction in the SHM load, the spectrum of SHM was found remarkably stable between both conditions, having no impact on A:T mutations versus G:C mutations.
Our findings identify HUWE1 as a critical regulator of B-cell mediated immunity. In addition to proliferation, HUWE1 regulates critical pathways associated with B-cell activation, SHM, and CSR. Our study demonstrates the complexity of Huwe-1 in controlling cell proliferation. To distinguish from direct and indirect effects, future studies will have to identify direct and indirect targets of HUWE1 and define their spatio-temporal relation during the cell-cycle and define the targets steering B-cell development and differentiation.
Data availability statement
RNA sequencing data has been deposited to the NCBI GEO database and can be accessed under accession number GSE221351. The somatic hypermutation NGS dataset has been deposited at the NCBI SRA database and can be accessed under accession number PRJNA913839.
Ethics statement
All animal experiments were approved by the Animal Ethics Committee of the Netherlands Cancer Institute (NKI) and the Dutch Experiments on Animals Act and the Council of Europe.
Author contributions
AS, PB, MuA, JG and HJ designed the study. AS, MS, JG and HJ wrote the manuscript. AS, MS, CS, MA, JC MK, and IP performed wet-lab experiments. DG and MuA analyzed the transcriptome data and performed the somatic hypermutation analysis. All authors contributed to the article and approved the submitted version.
Funding
This work was supported and made possible by two grants from the Dutch cancer foundation; KWF NKI-2017– 10032 and NKI2017-10796 to HJ, and by the COSMIC project that has received funding from the EU Horizon 2020 and the innovation program under the Marie Skłodowska-Curie grant agreement No. 765158. to JG.
Acknowledgments
We thank the biotechnical staff of the NKI-AVL for support, especially Msc. Martijn van Baalen, Frank van Diepen, Anita Pfauth, and Dr. Debajit Bowmick for their expertise and help with flow-cytometry experiments and cell sorting. We moreover like to thank Tijmen van Dam and Nike Claessen for wet-lab insights and expertise, and Ji-Ying Song from the animal pathology facility to examine the mouse organs. We thank Dr. Ron van Kerkhoven, Marja Nieuwland, and Roel Kluin for their help with the RNA sequencing and sequencing for the somatic hypermutation analysis.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.986863/full#supplementary-material
References
1. Mesin L, Ersching J, Victora GD. Germinal center b cell dynamics. Immunity (2016) 45:471–82. doi: 10.1016/j.immuni.2016.09.001
2. De Silva NS, Klein U. Dynamics of b cells in germinal centres. Nat Rev Immunol (2015) 15:137–48. doi: 10.1038/nri3804
3. Roco JA, Mesin L, Binder SC, Nefzger C, Gonzalez-Figueroa P, Canete PF, et al. Class-switch recombination occurs infrequently in germinal centers. Immunity (2019) 51:337–350.e7. doi: 10.1016/j.immuni.2019.07.001
4. Langerak P, Nygren AOH, Krijger PHL, van den Berk PCM, Jacobs H. A/T mutagenesis in hypermutated immunoglobulin genes strongly depends on PCNAK164 modification. J Exp Med (2007) 204:1989–98. doi: 10.1084/jem.20070902
5. Krijger PHL, Van Den Berk PCM, Wit N, Langerak P, Jansen JG, Reynaud CA, et al. PCNA ubiquitination-independent activation of polymerase η during somatic hypermutation and DNA damage tolerance. DNA Repair (Amst) (2011) 10:1051–9. doi: 10.1016/j.dnarep.2011.08.005
6. Van Oers JMM, Roa S, Werling U, Liu Y, Genschel J, Hou H, et al. PMS2 endonuclease activity has distinct biological functions and is essential for genome maintenance. Proc Natl Acad Sci U.S.A. (2010) 107:13384–9. doi: 10.1073/pnas.1008589107
7. Pilzecker B, Jacobs H. Mutating for good: DNA damage responses during somatic hypermutation. Front Immunol (2019) 10:438. doi: 10.3389/fimmu.2019.00438
8. Schrader CE, Guikema JEJ, Linehan EK, Selsing E, Stavnezer J. Activation-induced cytidine deaminase-dependent DNA breaks in class switch recombination occur during G 1 phase of the cell cycle and depend upon mismatch repair. J Immunol (2007) 179:6064–71. doi: 10.4049/jimmunol.179.9.6064
9. Wu X, Stavnezer J. DNA Polymerase β is able to repair breaks in switch regions and plays an inhibitory role during immunoglobulin class switch recombination. J Exp Med (2007) 204:1677–89. doi: 10.1084/jem.20070756
10. Bahjat M, Stratigopoulou M, Pilzecker B, Van Dam TP, Mobach S, Bende RJ, et al. DNA Polymerase β prevents AID-instigated mutagenic non-canonical mismatch DNA repair. BioRxiv (2020). doi: 10.1101/2020.01.30.926964
11. Parsons JL, Tait PS, Finch D, Dianova II, Edelmann MJ, Khoronenkova SV, et al. Ubiquitin ligase ARF-BP1/Mule modulates base excision repair. EMBO J (2009) 28:3207–15. doi: 10.1038/emboj.2009.243
12. Fang Q, Inanc B, Schamus S, Wang XH, Wei L, Brown AR, et al. HSP90 regulates DNA repair via the interaction between XRCC1 and DNA polymerase β. Nat Commun (2014) 5. doi: 10.1038/ncomms6513
13. Choe KN, Nicolae CM, Constantin D, Imamura Kawasawa Y, Delgado-Diaz MR, De S, et al. HUWE 1 interacts with PCNA to alleviate replication stress. EMBO Rep (2016) 17:874–86. doi: 10.15252/embr.201541685
14. Endres T, Solvie D, Heidelberger JB, Andrioletti V, Baluapuri A, Ade CP, et al. Ubiquitylation of MYC couples transcription elongation with double-strand break repair at active promoters. Mol Cell (2021) 81:830–844.e13. doi: 10.1016/j.molcel.2020.12.035
15. Hao Z, Duncan GS, Su YW, Li WY, Silvester J, Hong C, et al. The E3 ubiquitin ligase mule acts through the ATM-p53 axis to maintain b lymphocyte homeostasis. J Exp Med (2012) 209:173–86. doi: 10.1084/jem.20111363
16. Wang X, Lu G, Li L, Yi J, Yan K, Wang Y, et al. HUWE1 interacts with BRCA1 and promotes its degradation in the ubiquitin-proteasome pathway. Biochem Biophys Res Commun (2014) 444:290–5. doi: 10.1016/j.bbrc.2013.12.053
17. Markkanen E, van Loon B, Ferrari E, Hübscher U. Ubiquitylation of DNA polymerase λ. FEBS Lett (2011) 585:2826–30. doi: 10.1016/j.febslet.2011.03.069
18. Hall JR, Kow E, Nevis KR, Lu CK, Luce KS, Zhong Q, et al. Cdc6 stability is regulated by the Huwe1 ubiquitin ligase after DNA damage. Mol Biol Cell (2007) 18:3340–50. doi: 10.1091/mbc.e07-02-0173
19. Adhikary S, Marinoni F, Hock A, Hulleman E, Popov N, Beier R, et al. The ubiquitin ligase HectH9 regulates transcriptional activation by myc and is essential for tumor cell proliferation. Cell (2005) 123:409–21. doi: 10.1016/j.cell.2005.08.016
20. King B, Boccalatte F, Moran-Crusio K, Wolf E, Wang J, Kayembe C, et al. The ubiquitin ligase Huwe1 regulates the maintenance and lymphoid commitment of hematopoietic stem cells. Nat Immunol (2016) 17:1312–21. doi: 10.1038/ni.3559
21. Qi C-F, Zhang R, Sun J, Li Z, Shin D-M, Wang H, et al. Homeostatic defects in b cells deficient in the E3 ubiquitin ligase ARF-BP1 are restored by enhanced expression of MYC. Leuk Res (2013) 37:1680–9. doi: 10.1016/j.leukres.2013.09.009
22. Kon N, Zhong J, Qiang L, Accili D, Gu W. Inactivation of arf-bp1 induces p53 activation and diabetic phenotypes in mice. J Biol Chem (2012) 287:5102–11. doi: 10.1074/jbc.M111.322867
23. Aslam MA, Alemdehy MF, Kwesi-Maliepaard EM, Muhaimin FI, Caganova M, Pardieck IN, et al. Histone methyltransferase DOT1L controls state-specific identity during b cell differentiation. EMBO Rep (2021) 22:1–21. doi: 10.15252/embr.202051184
24. Metzner M, Schuh W, Roth E, Jäck HM, Wabl M. Two forms of activation-induced cytidine deaminase differing in their ability to bind agarose. PloS One (2010) 5:1–6. doi: 10.1371/journal.pone.0008883
25. Rush JS, Liu M, Odegard VH, Unniraman S, Schatz DG. Expression of activation-induced cytidine deaminase is regulated by cell division, providing a mechanistic basis for division-linked class switch recombination. Proc Natl Acad Sci U.S.A. (2005) 102:13242–7. doi: 10.1073/pnas.0502779102
Keywords: HUWE1, B-cell, immune responses, homeostasis, development, somatic hypermutation, class switch recombination
Citation: Spanjaard A, Stratigopoulou M, de Groot D, Aslam M, van den Berk PCM, Stappenbelt C, Ayidah M, Catsman JJI, Pardieck IN, Kreft M, Arens R, Guikema JEJ and Jacobs H (2023) Huwe1 supports B-cell development, B-cell-dependent immunity, somatic hypermutation and class switch recombination by regulating proliferation. Front. Immunol. 13:986863. doi: 10.3389/fimmu.2022.986863
Received: 05 July 2022; Accepted: 12 December 2022;
Published: 09 January 2023.
Edited by:
Yu Nee Lee, Sheba Medical Center, IsraelReviewed by:
Javier Marcelo Di Noia, Montreal Clinical Research Institute (IRCM), CanadaBerit Jungnickel, Friedrich Schiller University Jena, Germany
Copyright © 2023 Spanjaard, Stratigopoulou, de Groot, Aslam, van den Berk, Stappenbelt, Ayidah, Catsman, Pardieck, Kreft, Arens, Guikema and Jacobs. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Heinz Jacobs, aC5qYWNvYnNAbmtpLm5s; Jeroen E. J. Guikema, ai5lLmd1aWtlbWFAYW1zdGVyZGFtdW1jLm5s
†These authors contributed equally to this work and share first authorship
‡These authors contributed equally to this work and share second authorship
§These authors contributed equally to this work and share third authorship
‖These authors contribute equally to this work and share senior authorship