
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 09 December 2022
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.983181
This article is part of the Research Topic Transcriptional Control in Normal and Malignant B-Lymphocytes View all 8 articles
 Sara Ovejero1,2
Sara Ovejero1,2 Elena Viziteu1,2
Elena Viziteu1,2 Laure Dutrieux1,2
Laure Dutrieux1,2 Julie Devin1,2
Julie Devin1,2 Yea-Lih Lin2Elina Alaterre1,2Michel Jourdan1,2
Yea-Lih Lin2Elina Alaterre1,2Michel Jourdan1,2 Jihane Basbous2Guilhem Requirand1Nicolas Robert1Hugues de Boussac3
Jihane Basbous2Guilhem Requirand1Nicolas Robert1Hugues de Boussac3 Anja Seckinger4Dirk Hose4
Anja Seckinger4Dirk Hose4 Laure Vincent5Charles Herbaux2,5
Laure Vincent5Charles Herbaux2,5 Angelos Constantinou2
Angelos Constantinou2 Philippe Pasero2
Philippe Pasero2 Jérôme Moreaux1,2,6*
Jérôme Moreaux1,2,6*Multiple myeloma (MM) is a hematologic cancer characterized by accumulation of malignant plasma cells in the bone marrow. To date, no definitive cure exists for MM and resistance to current treatments is one of the major challenges of this disease. The DNA helicase BLM, whose depletion or mutation causes the cancer-prone Bloom’s syndrome (BS), is a central factor of DNA damage repair by homologous recombination (HR) and genomic stability maintenance. Using independent cohorts of MM patients, we identified that high expression of BLM is associated with a poor outcome with a significant enrichment in replication stress signature. We provide evidence that chemical inhibition of BLM by the small molecule ML216 in HMCLs (human myeloma cell lines) leads to cell cycle arrest and increases apoptosis, likely by accumulation of DNA damage. BLM inhibition synergizes with the alkylating agent melphalan to efficiently inhibit growth and promote cell death in HMCLs. Moreover, ML216 treatment re-sensitizes melphalan-resistant cell lines to this conventional therapeutic agent. Altogether, these data suggest that inhibition of BLM in combination with DNA damaging agents could be of therapeutic interest in the treatment of MM, especially in those patients with high BLM expression and/or resistance to melphalan.
Multiple myeloma (MM) is the second most common hematologic cancer after non-Hodgkin lymphoma. It mainly affects patients over 70 years of age and to date there is no definitive treatment. Treatments of choice include a combination of immunomodulatory drugs, proteasome inhibitors, DNA damaging agents, and monoclonal antibodies among others, together with autologous stem cell transplantation in transplant-eligible patients [reviewed in (1)]. Although these treatments can extend the life expectancy of the patients, eventually almost all of them develop resistance to chemotherapy and relapse. Therefore, MM remains a non-curable disease with significant morbidity and a median survival of 10 years for patients eligible to high dose melphalan (2–5). These facts point at an urgent need of better and targeted therapeutic approaches for MM patients to reduce the morbidity and overcome the resistance to current treatments.
At the cellular level, MM is characterized by the accumulation of malignant plasma cells, called multiple myeloma cells (MMCs), in the bone marrow (BM). These MMCs present high somatic hypermutation of immunoglobulin genes, characteristic aberrant chromosomal translocations (3, 6, 7), and a strong dependence on BM microenvironment, which provides survival signals and mediates drug resistance (8–10). Recent advances in treatment with the approval of several novel agents and their combinations have significantly improved patient outcome (11). However, patients invariably relapse after multiple lines of treatment, with shortened intervals between relapses, and finally become resistant to all treatments, resulting in loss of clinical control over the disease. MM is a genetically and clinically heterogeneous disease. Genome sequencing studies have revealed considerable heterogeneity and genomic instability, a complex mutational landscape and a branching pattern of clonal evolution (12). Epigenetics has also been shown to play a role in the disease progression and resistance to treatments, and deregulation of epigenetic factors, notably of those associated with DNA methylation, are related to bad prognosis in MM patients (13–15). In addition, intraclonal heterogeneity adds more complexity to MM pathophysiology and is most likely crucial for the progression of the disease and the relapse after treatment (16–19). In this context, genetic and epigenetic-wide screens constitute attractive strategies to understand the onset and development of MM as well as to identify new candidates to overcome drug resistance.
In our effort to identify new therapeutic targets in MM, we found that the BLM gene, which encodes the Bloom’s syndrome (BS) protein BLM, an ATP-dependent 3’-5’ DNA helicase (20–22), is associated with a poor prognostic value in MM patients. Moreover, we have recently reported that BLM has a role in the regulation of cell proliferation and survival during human normal B to plasma cell (PC) differentiation (23). BLM belongs to the highly evolutionary conserved RECQ family of DNA helicases; four of the five human genes of this family, BLM, WRN, RECQL4 and RECQL1, are associated with inheritable premature-aging and cancer-prone diseases (22, 24–26). The mutations of BLM disrupting its ATPase or helicase activity cause BS (27), a rare autosomal recessive genetic disorder characterized by developmental problems, growth retardation, immunodeficiency, sunlight sensitivity, fertility defects, and cancer predisposition associated with genomic and chromosomic instability (28–30). Soon after BS was first described, it was reported that lymphocytes from BS patients present a large increase in sister chromatid exchanges, which to date remains one of the cellular hallmarks of the disease (29, 31, 32). This cellular phenotype is due to the role of BLM in preventing sister chromatid and homolog chromosome exchanges during homologous recombination (HR) (33). Indeed, BS patients present a high sensitivity to DNA damaging agents commonly used in chemotherapy because the loss of BLM activity causes deficient DNA repair (34, 35). In particular, BLM is necessary for normal replication completion and is involved in several steps of the HR process. First, BLM is recruited to DNA double strand breaks (DSBs) in a manner dependent on the presence of NBS1, MRE11 and ATM. ATM activity is essential only for the early recruitment of BLM, whereas polyubiquitination of BLM and its subsequent interaction with NBS1 are required for its retention at DSBs (35). Then, BLM together with the endonuclease DNA2 is involved in 5’-end DNA resection during the initiation step of DSBs repair by HR (21, 36, 37). Later, once DNA synthesis has been completed across the DNA break, BLM resolves the Holliday junctions to restore the separated DNA duplexes (38–40). However, BLM can also have anti-recombinogenic activity by disrupting the D-loops formed during the strand invasion step of HR, and other studies have underlined the dual role of BLM through its pro- and anti-recombinogenic activities to balance HR and non-homologous end joining (NHEJ) in order to preserve genome stability (34, 41–45). BLM also maintains genome stability at stalled replication forks by promoting fork regression and restart (46–48), resolves mitotic chromosome bridges (49–51), and participates in telomere maintenance by resolving G-quadruplexes that can interfere with telomere replication (52–54).
As most cancers, MM and other hematological malignancies are also characterized by genomic instability that may arise from defective DNA replication and repair pathways (55–57). It has been highlighted that a subgroup of MM patients displays high chromosomal instability and replication stress which correlate with poor outcome (58–60). Along this line, we previously reported the importance of RECQ1 helicase in the survival to replication stress and drug resistance of MM cells (61, 62). BLM helicase, which belongs to the same family as RECQ1, is crucial in the maintenance of chromosomal stability and has been clearly associated with cancer development in BS patients. Here, we report that BLM expression is deregulated in several MM patient cohorts and that its overexpression is associated with poor prognosis. A novel BLM inhibitor, ML216, is a small molecule that inhibits the catalytic activity of BLM more than other RECQ family helicases (63, 64). ML216 has been used to characterize BLM function in HR (35), but its potential as an anti-cancer therapy has barely been addressed and to date no available studies have evaluated it in the context of MM.
Here, we characterized the importance of BLM for MM pathophysiology and resistance to treatments as well as the use of ML216 alone and in combination with current MM chemotherapies. Using a unique collection of human MM cell lines (HMCLs) that recapitulate the heterogeneity and complexity of MM patients (65, 66), we found that different HMCLs display different sensitivity to BLM inhibition by ML216. Characterizing the impact of BLM inhibition on MM plasma cell survival, we found that ML216 induces DNA damage and apoptosis. Moreover, co-treatment of HMCLs with ML216 and melphalan, a common anti-myeloma drug, has a synergistic effect leading to increased MMC death. Our results suggest that BLM inhibition in combination with melphalan could be of therapeutic interest in the treatment of MM.
Patients’ MMCs were purified using anti-CD138 MACS microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany) and their gene expression profile (GEP) obtained using Affymetrix U133 plus 2.0 microarrays as described (Array Express public database [E-MTAB-372]) (67). Publicly available cohorts of newly-diagnosed MM patients treated with high dose melphalan and autologous hematopoietic stem cell transplantation (UAMS-TT2 and TT3 (GSE24080), and Hovon (GSE19784) cohorts) were also used. Gene expression data were normalized with the MAS5 algorithm and analyses processed with GenomicScape (http://www.genomicscape.com) (68). Gene Set Expression Analysis (GSEA) was used to identify genes and pathways differentially expressed between populations. Difference in overall survival between groups of patients was assayed with a log-rank test and survival curves plotted using the Kaplan–Meier method (Maxstat R package) (69).
XG1, XG2, XG7, XG12, XG19 and XG21 HMCLs are IL-6 dependent cell lines obtained as previously described (65). Upon removal of IL-6, these cell lines progressively apoptose within 10 to 14 days and are routinely maintained in RPMI 1640 GlutaMAX medium (61870044, Gibco) supplemented with 10% fetal calf serum (CVFSVF00 01, Eurobio) and with IL-6 (2 ng/ml) (65). AMO-1, LP1 and OPM2 were purchased from DSMZ (Braunsweig, Germany) and RPMI8226 from ATCC (Rockville, MD, USA). These cell lines were grown in RPMI 1640 GlutaMAX medium (61870044, Gibco) supplemented with 10% fetal calf serum (CVFSVF00 01, Eurobio). Melphalan-resistant XG2 and XG7 cell lines were derived from XG2 and XG7 parental cell lines after sequential in vitro treatment and selection (70). HMCLs were authenticated according to their short tandem repeat profiling and their gene expression profiling using Affymetrix U133 plus 2.0 microarrays deposited in the ArrayExpress public database under accession numbers E-TABM-937 and E-TABM-1088. Whole exome sequencing analysis was performed on XG2 and XG7 melphalan-resistant cell lines and the corresponding parental cell lines as previously reported (66). The WES library preparation was done with 1000 ng of input DNA. Sequences of exome were enriched using SureSelectxt kit and SureSelectxt All Exons v5 library (Agilent Technologies, Santa Clara, California, USA). Paired-end exome sequencing was performed on the enriched exome sequences using the illumina NextSeq500 sequencing instrument (Helixio, Clermont-Ferrand, France), generating 75 bp paired-end reads with 100X average coverage per sample.
Drugs used in this study: ML216 (SML0661, Sigma) and melphalan (Y0001457, European Pharmacopoeia Reference Standard).
XG2 cells were transduced with control or BLM miRNA lentiviral particles. BLM miRNAs (Invitrogen, Carlsbad, USA) were cloned in the pLenti4-EZ-mIR plasmid (Invitrogen) as described (61). This plasmid contains the shRNA sequence and also the GFP gene under the control of Tet operators. Cells were selected with 12.5 μg/ml of zeocin for 2 weeks. When selection was completed, cells were maintained in the presence of 6.25 μg/ml zeocin to keep the selection pressure. Before every experiment, zeocin was removed from the medium by extensive washing and cells were plated in zeocin-free fresh medium. BLM depletion was validated by western blot using anti-BLM (ab476, Abcam).
Bone marrow samples from untreated MM patients (n = 7) were obtained at the University Hospital of Montpellier after patients’ written informed consent in accordance with the Declaration of Helsinki and agreement of the Montpellier University Hospital Centre for Biological Resources (DC-2008-417). Bone marrow mononuclear cells are cultured with IL-6 (2ng/ml) (61, 71) seeded at 5x105cells/mL in RPMI 1640 medium, 5% FCS, 2ng/mL IL-6, and cultured with or without ML216 (3 μM, 6 μM or 10 μM) for 4 days as described (61, 71). In each culture group, viability and cell count were assayed and MM cell cytotoxicity was assessed by flow cytometry (61, 71). MM plasma cells (CD138+) were detected using anti-CD138-phycoerythrin monoclonal antibody (Immunotech, Marseille, France) and all CD138- cells were analyzed as non-myeloma cells.
For IC50 determination, HMCLs were seeded at 10000 cells/well and cultured for 4 days in 96-well flat-bottom plates in presence of ML216 at concentrations ranging from 0.78 μM to 100 μM. Cell proliferation was evaluated using CellTiter-Glo (CTG) Luminiscent Assay (G7573, Promega) according to manufacturer’s protocol and luminescence was measured using a Centro LB 960 luminometer (Berthold Technologies, Bad Wildbad, Germany). IC50 for each HMCL was calculated using non-linear regression analysis in GraphPrism software.
For evaluation of ML216 and melphalan synergy, increasing concentrations of each single drug were combined with all concentrations of the other drug so all possible combinations were evaluated. Cell growth was evaluated with CTG reagent as described above. For each combination, the percentage of expected growing cells in the case of effect independence was calculated with Bliss equation using R package “SynergyFinder”.
Cells were treated with the indicated concentrations of ML216 and melphalan for 48 and 96h. Cells were collected, counted and 105 cells per condition were processed with the Annexin V kit (556421, BD Biosciences) according to manufacturer’s instructions. Apoptotic cells (AnnexinV+) were quantified by flow cytometry.
For cell cycle analysis, cells were cultured and treated as described above. To mark replicating cells, culture medium was supplemented with 10 μg/ml BrdU (bromodeoxyuridine) during the last hour of each treatment and samples were processed with the APC BrdU flow kit (552598, BD Biosciences) according to manufacturer’s instructions. Cells cycle phases were analyzed by flow cytometry. BrdU+ cells were assigned to S-phase and BrdU- cells were classified as G0/G1 or G2/M phases based on their DNA content.
All flow cytometry acquisitions were done on a Fortessa flow cytometer (BD biosciences) and quantifications were done with Kaluza software.
For comparison of BLM protein levels in HMCLs, cells were lysed with RIPA buffer (sc-24948, Santa Cruz) supplemented with halt protease and phosphatase inhibitor cocktail (78442, Thermo Scientific). Cell extracts were quantified by BCA assay (23225, Thermo Scientific), absorbance at 570 nm was measured using a spectrophotometer (Tecan) and a linear regression was performed to determine protein concentration in each sample. Typically, 20 μg of protein extract were loaded per sample in 8% polyacrylamide gels.
For analysis of apoptotic and DDR pathways, 1 million cells were directly lysed in 300 μl of Laemmli buffer (1x), vortexed, boiled at 95°C for 5 minutes and 20 μl were loaded per sample in 8%, 10% and 14% polyacrylamide gels.
Antibodies used in this study are: anti-BLM (ab476, Abcam), anti-WRN (4666, Cell Signaling), anti-RECQ1 (ab22830, Abcam), anti-RECQL5 (sc-515050, Santa Cruz), anti-Tubulin (2144S, Cell Signaling), anti-pSer15_p53 (9284S, Cell Signaling), anti-p53 (9282S, Cell Signaling), anti-p21 (2946S, Cell Signaling), anti-p27 (3688S, Cell Signaling), anti-γH2AX (05-636, Millipore), anti-PARP (9532S, Cell Signaling), anti-pThr68_Chk2 (2197S, Cell Signaling), anti-Chk2 (2662, Cell Signaling), anti-pSer345_Chk1 (2348S, Cell Signaling), anti-Chk1 (2345S, Cell Signaling), anti-Caspase 3 (9662S, Cell Signaling), anti-Caspase 8 (9746, Cell Signaling), anti-Caspase 9 (9502S, Cell Signaling).
Cells were deposited on poly-lysine coated slides (J2800AMNZ, Thermo Scientific) using a Cytospin 4 centrifuge (Thermo Scientific) at 600 rpm for 10 minutes. Soluble cell fraction was pre-extracted by incubation with cold cytoskeleton buffer (CSK: 10 mM PIPES, pH 7, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 0.7% Triton X-100) (2 x 3 minutes), fixed with 4% PFA-PBS, and saturated with 3% BSA-PBS for 1h at RT. Anti-BLM antibodies (ab476, Abcam; sc-365753, Santa Cruz) were diluted at 1:200 and anti-nucleolin (ab22758, Abcam) was diluted at 1:1000 in saturation buffer and incubated on slides in a humid chamber for 90 minutes. Slides were washed 3 x 5 minutes with PBS-0.01% Tween, incubated protected from light in a humid chamber with secondary antibody (A11008, Invitrogen) 1:500 for 45 minutes at RT. Washed again 3 x 5 minutes with PBS-0.01% Tween, incubated with DAPI (20 μg/ml) in H2O for 5 minutes and washed 3 times with H2O. Slides were air dried and mounted with Prolong Gold (P36930, Invitrogen) and let to dry overnight. Image acquisition was performed with a ZEISS Axio Imager Z1 Apotome microscope and analysis was done with Omero server.
We previously reported that BLM gene had a bad prognostic value in the Heidelberg-Montpellier (HM) MM cohort of patients (58) and that BLM expression was significantly upregulated in MM according to bioinformatics analysis of one publicly available cohort of MM patients with gene expression dataset (62). Therefore, we further studied BLM in the pathophysiology of MM and validate our previous observations with several independent cohorts of patients. No significant difference in BLM expression between normal bone marrow plasma cells (BMPCs; n=5; median: 1133; range: 863-1328) and MMCs from patients (n=206; median: 935; range: 113-5206) was found (Figure 1A). Furthermore, although we did not observe a statistical difference with BMPCs, BLM mRNA levels appeared heterogeneous in MMCs ranging from 236 to 3079 in Affymetrix signal (Figure 1A, MMCs outliers marked by *). However, BLM expression was significantly higher in HMCLs (n=42; median: 1848; range: 246-10901) when compared to both normal BMPCs and primary MMCs (P = 0.0001) (Figure 1A and Supplementary Figure 1A), suggesting an increase in BLM levels with the progression of the disease. Primary MMCs of untreated patients can be classified into seven molecular groups associated with different patient survival (72). According to this classification, BLM expression was significantly higher in the poor prognosis “proliferation” subgroup (PR; P < 0.5) and “low bone disease” subgroup (LB; P < 0.01) (Figure 1B). Finally, we investigated the BLM expression in a cohort of 18 patients with paired samples at diagnosis and relapse, and identified a significant higher expression of BLM at relapse (P=0.04) (Figure 1C).
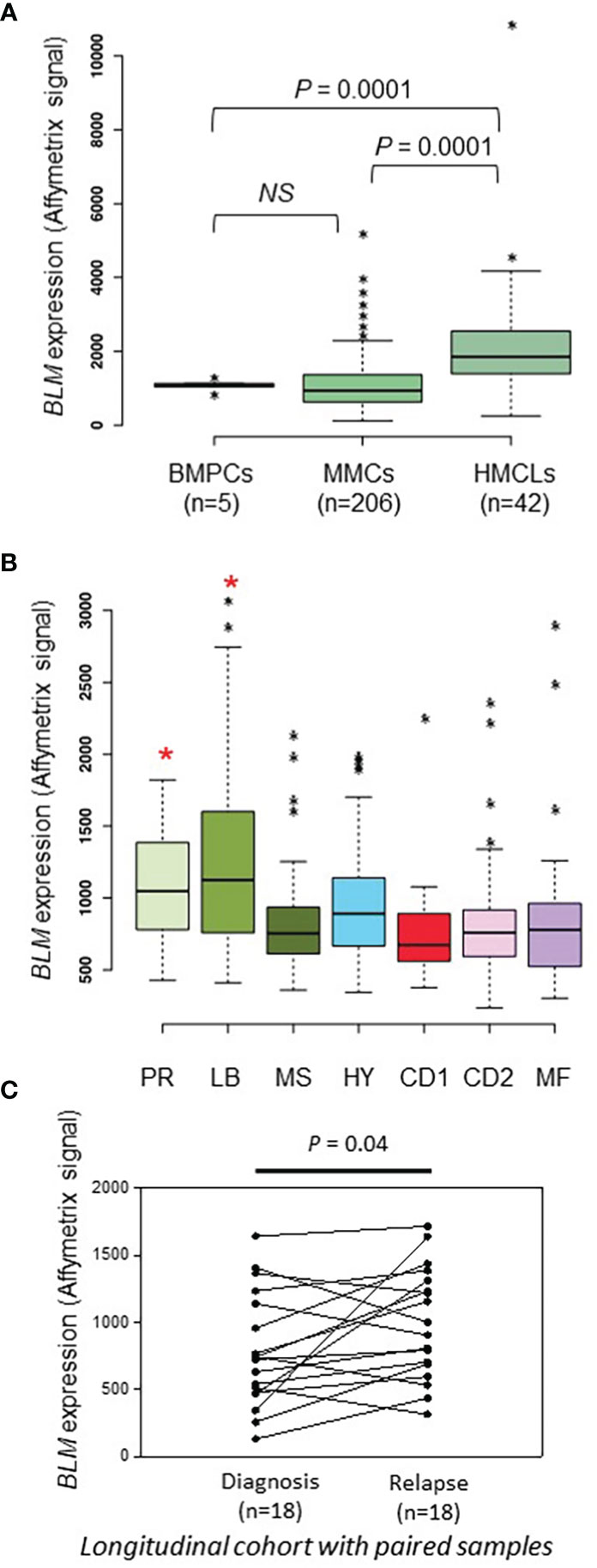
Figure 1 BLM expression in MM. (A) BLM expression analysis by Affimetrix microarrays from normal BMPCs from 5 healthy donors (median: 1133; range: 863-1328), MMCs from 206 MM patients from the HM cohort (median: 935; range: 113-5206), and 42 HMCLs (median: 1848; range: 246-10901). * marks outliers. Statistical analysis used a Student’s t-test of paired samples. (B) Gene expression profiling of MMCs of the patients of UAMS-TT2 cohort were used. Patients (n = 250) were classified in the 7 molecular groups of MM. PR: cell cycle and proliferation, LB: low bone disease, MSET: MMSET overexpression, HY: hyperdiploid signature, CDNN1: Cyclin D1 overexpression, CDNN2: Cyclin D2 overexpression, MAF: overexpression of MAF and MAFB genes. Small asterisks mark outliers in each group. Big red asterisks indicate that BLM expression is significantly higher in the group compared to all the patients of the cohort (P < 0.05) (Student’s t-test). (C) BLM expression is significantly higher at relapse compared to diagnosis in a longitudinal cohort of 18 paired patient’s samples (paired Student’s t-test). * p-value < 0.05.
Using Maxstat R package (69), we determined that high BLM expression in MMCs could predict shorter overall survival (OS) in four independent cohorts of previously untreated patients that were homogenously treated with high melphalan dose (HDT) followed by autologous stem cell transplantation (ASCT), a standard-of-care therapy for newly diagnosed MM (73) (HM cohort (n = 206): P = 0.003; UAMS-TT2 cohort (n = 250): P = 0.0002; TT3 cohort (n = 158): P = 0.0008; Hovon cohort (n = 282): P = 0.04) (Figures 2A–C). A high BLM expression could also predict for shorter event free survival (EFS) in the HM and TT2 cohorts (Figures 2A, B, right panels). In a COX multivariate analysis [including ISS, B2M level, t (4, 14), del17p, Gep70 score (74), IFM score (75), Growth Proliferation Index (GPI) (67) and RS (76)], BLM expression, B2M level, t (4, 14) and RS remain independent prognostic factors (Supplementary Table S1). In addition, Gene Set Enrichment Analysis (GSEA) showed that patients with high BLM expression level and high-risk present a significant enrichment of proliferation-associated genes (Reactome cell cycle and mitotic: P = 0.01; reactome G1/S transition: P = 0.01) (Supplementary Figure S1B). However, no significant correlation between BLM expression and MMC plasma cell labeling index (77) was found (Supplementary Figure S1C). Furthermore, no significant difference was identified comparing GPI subgroups (Supplementary Figure S2A). BLM expression was significantly higher in high-risk MM patients defined by RS score (76) (Supplementary Figure S2A). (High BLM expression was also associated with a poor outcome in a cohort of MM patients at relapse treated by anti-CD38 MoAb (78) (Figure 2D) at diagnosis non eligible to HDT and ASCT (n=63) (Figure 2E). These data indicate that high BLM expression is associated with poor prognosis in MM patients and correlates with increased expression of cell cycle progression genes, even though cells with high BLM expression do not show increased proliferation.
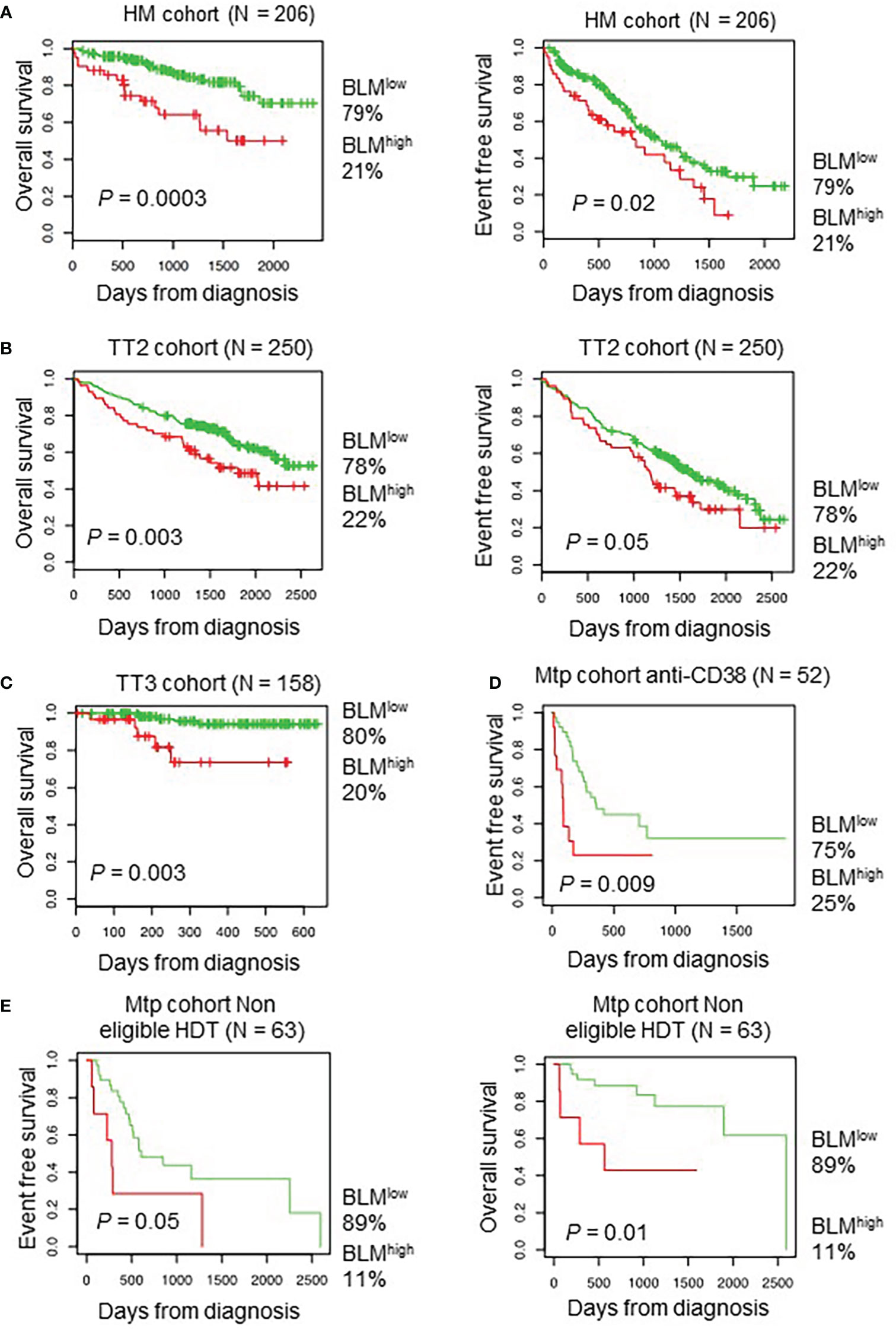
Figure 2 High BLM expression is associated with a poor outcome in MM. Correlation between BLM expression and overall survival was analyzed using Maxstat R package in independent cohorts of MM patients: including patients at diagnosis treated by HDT and ASCT (A, left panel) HM cohort; (B, left panel) UAMS-TT2 cohort; (C) TT3 cohort; a cohort of patients at relapse treated by Anti-CD38 antibody (Daratumumab) (D) Mtp cohort anti-CD38 and a cohort of patients at diagnosis non eligible to HDT and ASCT (E) Mtp cohort non eligible HDT. (A, B, right panels) High expression of BLM is associated with shorter event free survival in the HM, TT2 and Mtp cohort non eligible HDT cohorts.
ML216 is a small molecule that inhibits BLM DNA unwinding activity by blocking its nucleic acid binding site. This inhibitor has been shown to be selective for BLM over other members of the RECQ family in vivo (63, 64). Hence, the response to ML216 was tested in a panel of ten HMCLs representative in part of the molecular heterogeneity of MM (65). Treatment with this drug inhibited cell proliferation in a dose-dependent manner with a median IC50 of 2.78 µM (range: 1.2-16.9 µM) (Figure 3A). RNA-seq and western blot analysis showed marked differences in BLM expression among HMCLs (Figures 3B, C). However, there was not any correlation between BLM gene expression or BLM protein levels and HMCLs sensitivity to ML216. Importantly, BLM was not mutated in any of these HMCLs according to our sequencing data [data not shown published in (66)]. The differences in the protein levels of the RECQ helicases WRN, RECQ1 and RECQL5, and the basal level of DDR activation were also not correlated with the sensitivity to ML216 (Figure 3C), thus ruling out that the effect of the BLM inhibitor on cell proliferation was due to a non-specific effect on another helicase or a higher level of basal DNA damage specific to certain cell lines. Additionally, we did not find any correlation between the sensitivity to the BLM inhibitor and the MM molecular subgroups (65), nor the recurrent mutations reported in MM, nor mutations in genes involved in the DDR (66, 79) (Figure 3D). However, our sequencing data identified in each cell line mutations in numerous genes involved in transcription (66). Therefore, it is possible that deregulation of transcription may have an indirect impact in the DDR and other molecular pathways, making some cells more vulnerable to BLM inhibition than others. In addition, BLM is involved in other cellular processes other than DNA repair, such as replication, telomeric maintenance or ribosomal DNA regulation among others (80), which suggests that differences in the regulation of these processes may also account for the differential sensitivity of the cell lines to ML216.
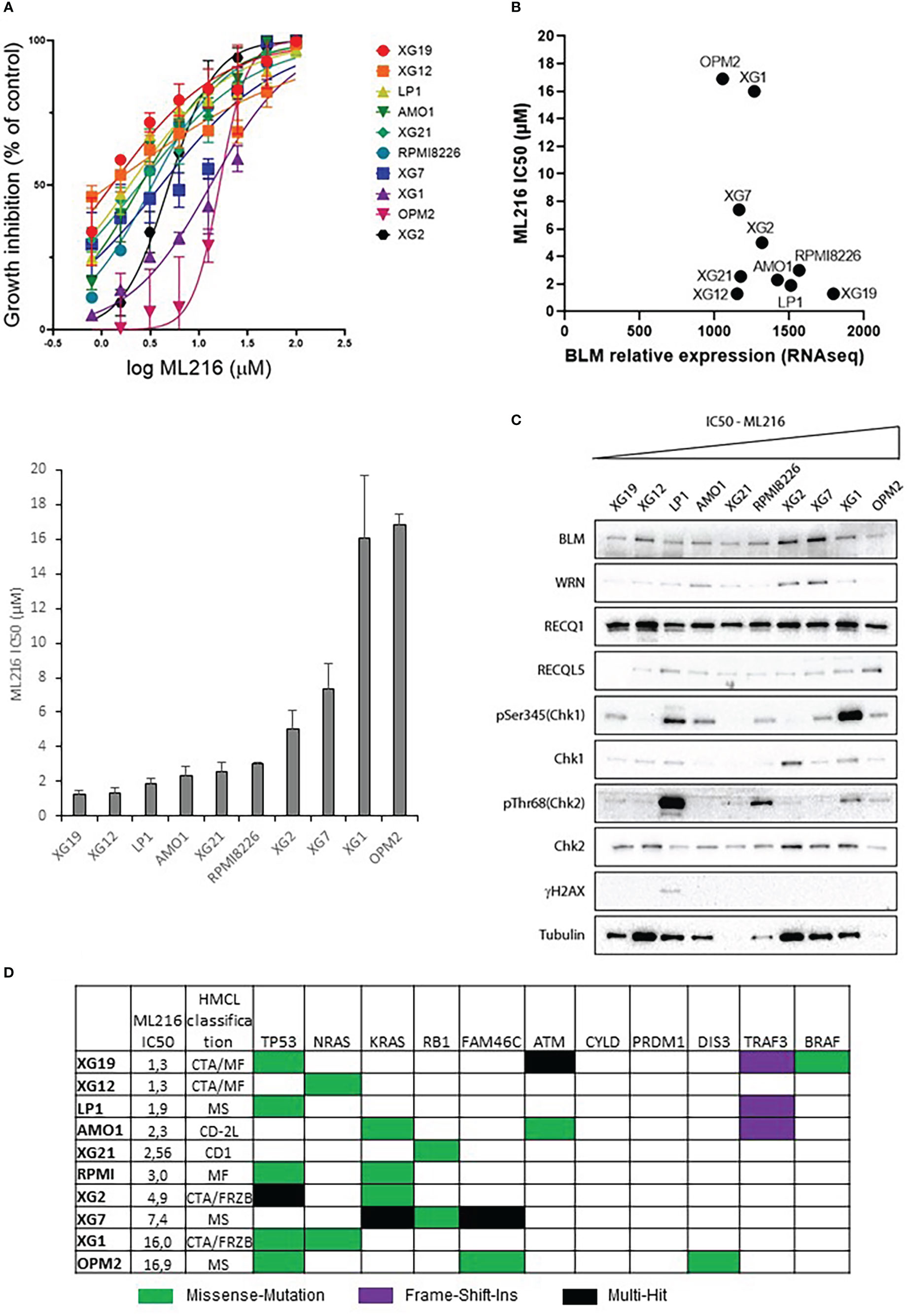
Figure 3 Effect of ML216 in HMCLs. (A) 10 HMCLs were treated with increasing doses of ML216 (0.78 – 100 μM). At day 4, cell viability was assessed using CellTiter-Glo Luminiscent Cell Viability Assay. IC50 for each cell line was calculated using GraphPrism software. Upper panel shows the non-linear regression curve of growth inhibition for all HMCLs. Low panel shows the IC50 value for each HMCL. Data are based on at least 3 independent experiments. (B) BLM expression and sensitivity to ML216 (IC50) do not correlate after applying a Spearman’s test. (C) Western blot analysis of the protein level of the RECQ helicases and basal DDR markers in 10 HMCLs. Cell lines are ranked from left to right in order of increasing IC50 for ML216. Note that in most cell lines protein levels do not correlate with BLM expression levels determined by RNAseq in Figure 3B. (D) Mutational status of MM frequently mutated genes in the 10 HMCLs used for the other experiments in this figure. “HMCL classification” refers to the MM molecular group classification. PR (cell cycle and proliferation), LB (low bone disease), MSET (aberrant expression of FGFR3 and MMSET genes), HY (hyperdiploid signature), CDNN1 (overexpression of Cyclin D1), CDNN2 (overexpression of Cyclin D2), and MAF (overexpression of MAF and MAFB genes) (72). Data extracted from (66).
ML216 has been described to exclusively inhibit BLM unwinding activity without any further impact on its biological regulation (63, 64). To confirm that this is the case in MM cells, we analyzed BLM protein levels upon ML216 treatment in the sensitive cell lines XG2 and XG19. The levels of BLM and the RECQ helicases WRN, RECQ1 and RECQL5 did not significantly change after 48h and 96h in presence of ML216 (Supplementary Figure S3A). BLM is a nuclear protein recruited to chromatin in response to DNA damage. It can form foci and micro-speckles (81) and also localizes to the nucleolus (82). To determine the subcellular localization of BLM in HMCLs, XG19 and XG2 cells were treated with ML216 for 48h, soluble proteins were pre-extracted by cytoskeleton (CSK) buffer prior to fixation and immunofluorescence detection. Chromatin bound BLM localized to the nucleolus and nuclear foci in basal conditions in XG19 and XG2 cells and no significant change was observed upon ML216 treatment (Figure S3B). Intriguingly, in XG2 cells ML216 treatment induce a partial relocalisation of nucleolin in the chromatin, and a conformational change of the nucleoli, which appeared more elongated (Figure S3B, right), although BLM remained localized in them. Together, these data indicate that the chemical inhibition by ML216 does not affect BLM levels or localization in MM cells, and that the observed cytotoxicity is likely due to the inhibition of BLM helicase activity.
In order to study the effect of BLM inhibition on MM cell survival, one ML216-resistant (XG1; IC50 = 13.2 µM) and two ML216-sensitive (XG19 IC50 = 1.2 µM; XG2 IC50 = 4.9 µM) HMCLs were chosen. ML216 induced marked apoptosis at all doses in the sensitive XG19 and XG2 cell lines already at 48 hours of treatment, whereas this effect was only mild on the resistant XG1 cell line even at 96 hours (Figure 4A). To confirm that BLM is also required for the survival of primary MMCs from patients, BM samples containing malignant MMCs with their BM environment and recombinant IL-6 were cultured with ML216 as described (13, 70, 83). ML216 treatment significantly reduced the median number of viable myeloma cells by more than 50% (P < 0.001; N = 7) compared to untreated control (Figure 4B). Of interest, the normal BM non-myeloma cells were less affected by ML216 treatment at low and mild doses (Figure 4C). We therefore conclude that viability of MM cell lines and primary MM cells depends on high BLM expression levels.
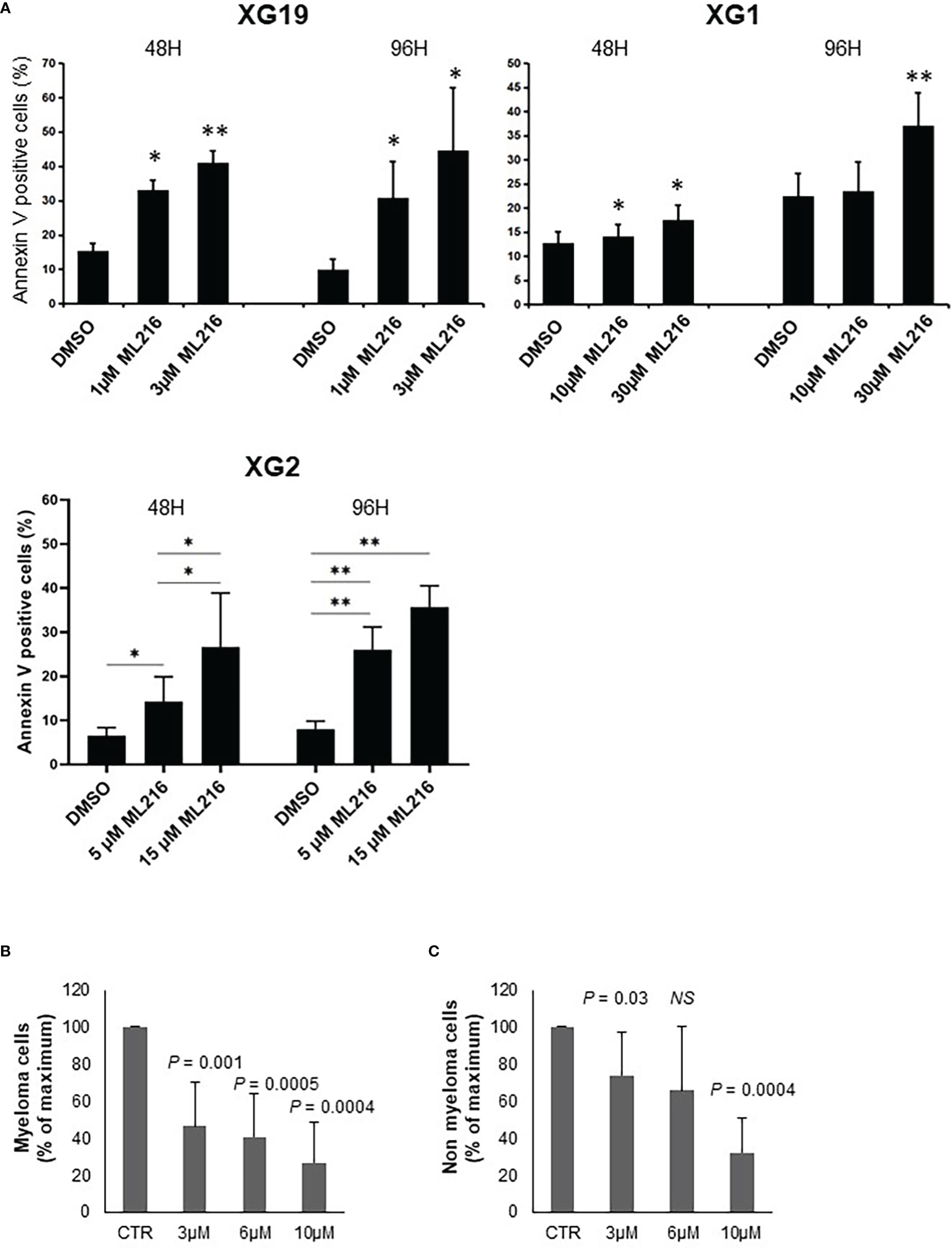
Figure 4 ML216 selectively induces apoptosis in MMCs. (A) XG19, XG2 and XG1 cell lines were treated for 48 and 96 hours with the indicated concentrations of ML216. Apoptotic cells were detected as Annexin V+ cells by flow cytometry. Results are the average of 3 independent experiments. * indicates a significant increase in apoptosis compared to DMSO controls using a Student’s t-test. (B, C) BM cells extracted from 7 MM patients were cultured with recombinant IL-6 and 3, 6 or 10 μM ML216 for 96 hours. Cytotoxicity was assessed by flow cytometry. (B) MM plasma cells (CD138+) were detected using an anti-CD138 antibody and (C) all CD138- cells were analyzed as non-myeloma cells. Number of cells in each condition was normalized with respect to the control. P-values indicate the significance of the observed differences after applying a Wilcoxon test for pairs. NS, not significant. * p-value < 0.05, ** p-value < 0.01.
BLM is a DNA helicase that unwinds DNA resulting from HR-mediated processes occurring during DNA replication and repair, such as D-loops (34, 41–45) or Holliday junctions (38–40). Therefore, a higher sensitivity of replicative or post-replicative cells to ML216 treatment was predicted. To address this possibility, we investigated the cell-cycle distribution of HMCLs treated with ML216 by flow cytometry. In the case of the XG1 cell line, ML216 induced a decrease in the percentage of cells in S-phase concomitant with an increase in G0/G1, indicative of a cell cycle arrest at the G1/S transition (Figure 5), and correlated also with more Annexin V positive cells (Figure 4A). In the XG19 sensitive cell line, ML216 treatment caused a decrease in the S-phase population (Figure 5) that correlated with an increase in the apoptotic population (Figure 4A) and no change was observed in the other phases of the cell cycle. In the XG2 cell line, ML216 treatment induced an accumulation in S-phase correlated to a decrease in the G2/M cell population (Figure 4A).Together, these data indicate that ML216 prevents entry and progression through S-phase in myeloma cells, causing an alteration of the cell cycle distribution. When cell cycle arrest due to ML216 treatment is sustained over time, apoptosis is triggered in all of the cell lines, suggesting that permanent inhibition of BLM causes DNA damage to a level that is lethal for myeloma cells.
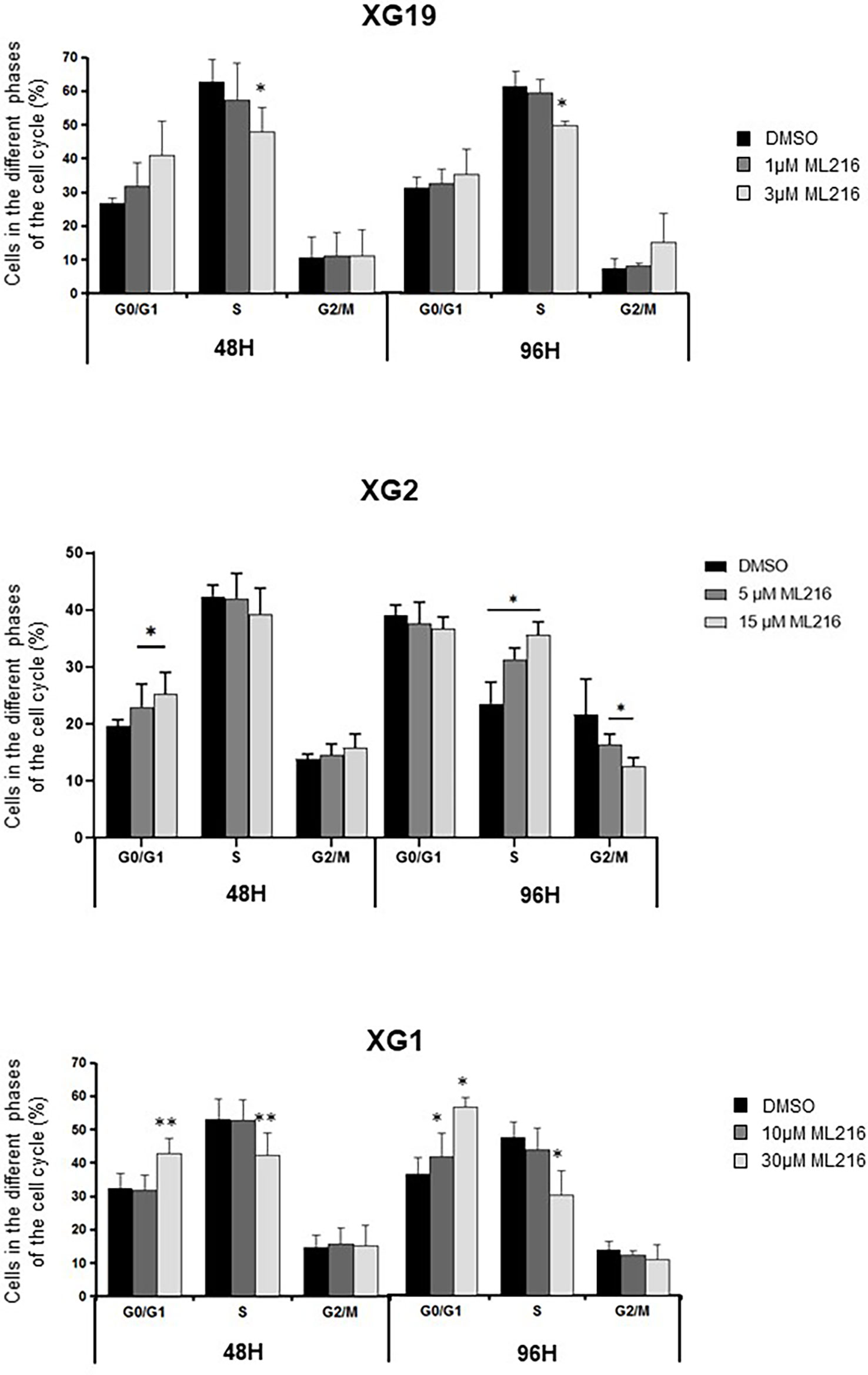
Figure 5 ML216 effect on cell cycle distribution in HMCLs. XG19, XG2 and XG1 cell lines were treated for 48 and 96 hours with the indicated concentrations of ML216. BrdU (10 μg/ml) was added during the last 1.5 hours of treatment. Cells were fixed and processed to detect BrdU incorporation and total DNA (see Materials and Methods for more details). BrdU+ cells were assigned to S-phase. BrdU- cells were assigned to G0/G1 or G2/M phases based on their DNA content. * indicates a significant difference compared to DMSO treated (control) cells after applying a Student’s t-test for pairs. Results are the mean of 3 independent experiments. ** p-value < 0.01.
Various chemotherapeutic drugs are currently used to treat MM patients, and their rational and combinatorial use has proven to be a good treatment strategy to improve MM patient survival [reviewed in (1)]. Since another RECQ helicase was associated with resistance to genotoxic agents in MM (61), we next investigated whether ML216 could synergize with conventional MM treatment including melphalan, lenalidomide, and bortezomib. No synergy of ML216 with bortezomib (Supplementary Figure S4A), whereas a synergy between ML216 and lenalidomide was observed in XG2 and to a lesser extent in XG19 (Supplementary Figure S4B). However, ML216 treatment of XG2 did not induce any change in the protein levels of Myc and IRF4 (84) (Supplementary Figure S4C), and the molecular mechanism of this potential synergy remains to be explored. Interestingly, treatment with ML216 mildly potentiated the cytotoxic effect of melphalan in the XG1 and XG2 cell lines (Figure 6), whereas it had no major effect in the XG19 cell line (Figure 6). Since melphalan generates DNA damage and BLM is involved in DNA double-strand breaks (DSBs) resolution, we hypothesized that co-treatment with both drugs may increase DNA damage above a threshold that myeloma cells cannot cope with, leading to cell death. Indeed, combination of ML216 and melphalan induced a significant increase in apoptosis (Figure 7A), and affected cell cycle distribution inducing a decrease in the fraction of S-phase cells and an increase in G0/G1 cells (Figure 7B). As already mentioned, BLM promotes the HR-mediated repair of DNA DSBs (21, 36, 37). DSBs are marked by the presence of γH2AX, resulting from the phosphorylation of the H2AX histone variant on Ser139 by the checkpoint apical kinases ATM and ATR (85–87). Then, MDC1 (mediator of DNA damage checkpoint protein 1) binds to γH2AX and together orchestrate the recruitment of downstream DNA repair factors such as BRCA1, 53BP1, the MRN complex or RAD51 among others (88–90). Interestingly, the effect on cell cycle and cell death of ML216 and melphalan co-treatment (Figures 7A, B) correlated with an increase in γH2AX positive cells (Figures 7C, D), indicative of higher DNA damage levels in the presence of both drugs.
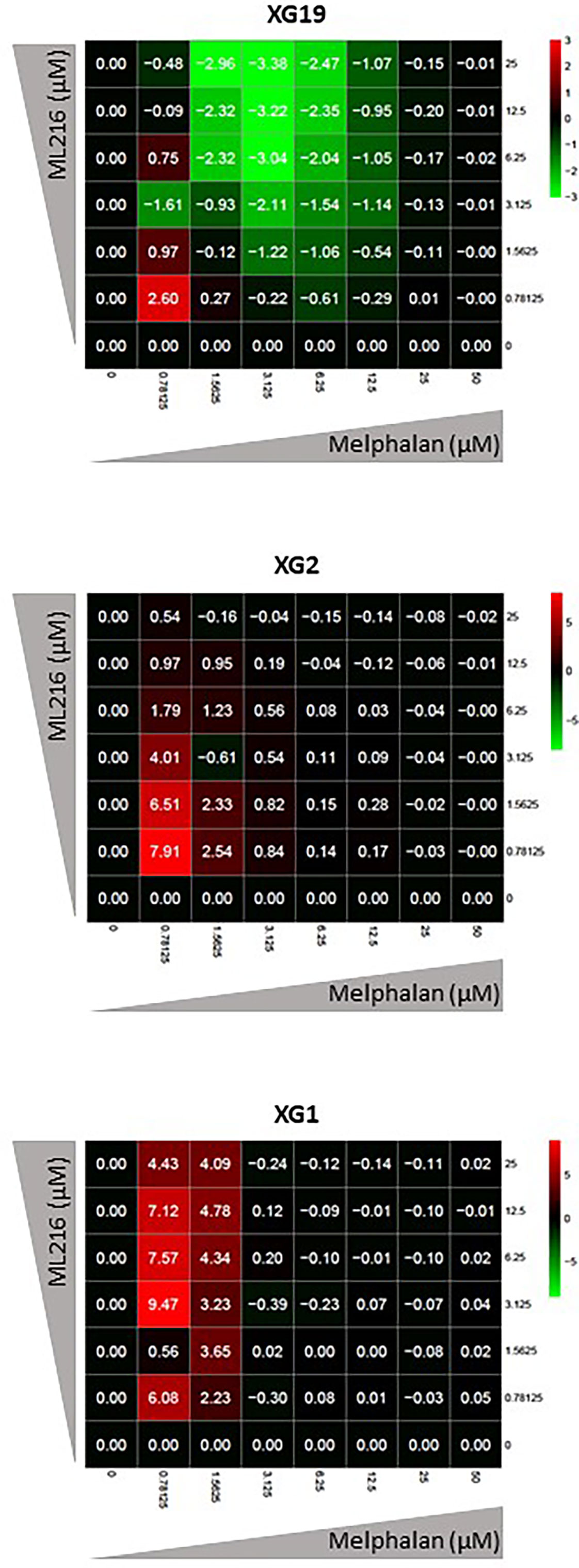
Figure 6 ML216 treatment synergizes with melphalan to inhibit HMCLs proliferation. Dose-response matrixes to measure synergy of ML216 and Melphalan co-treatment. Synergy scores are shown using a continuous pseudo-color scale ranging from bright-green (=antagonism) to bright-red (=synergism). XG19, XG2 and XG1 were treated with increasing concentrations of ML216 (0.78125 – 25 μM), and of the alkylating agent melphalan (0.78125 – 50 μM), for 4 days. Cell viability was assessed using the CellTiter-Glo Luminiscent Cell Viability Assay and was normalized to untreated conditions. Matrixes show the average of 3-4 independent experiments.
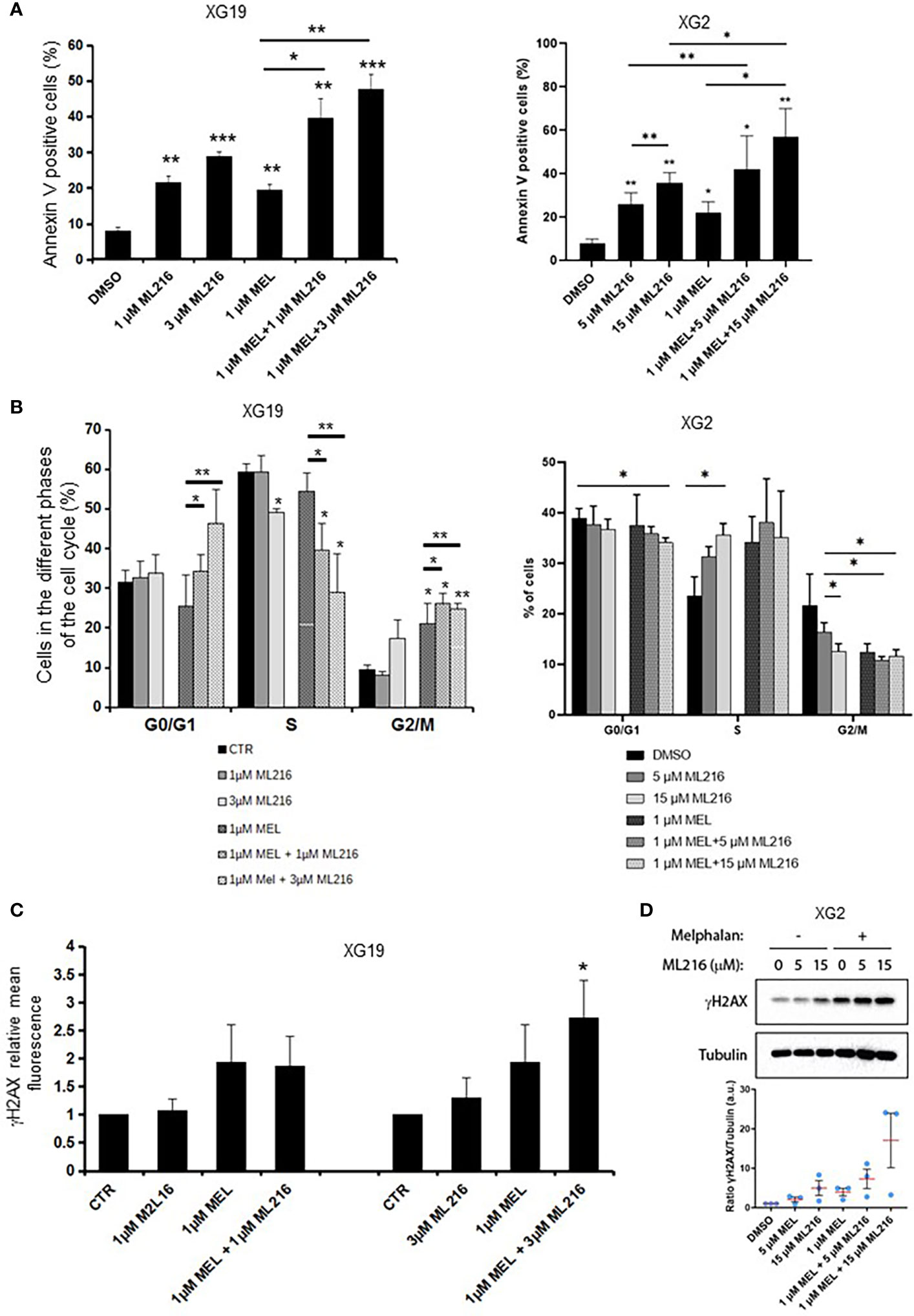
Figure 7 Effect of combination of ML216 and melphalan. (A) XG19 and XG2 cells were treated with ML216 and melphalan as indicated for 96 hours. Apoptotic cells were detected as Annexin V+ cells by flow cytometry. Results are the average of 3 independent experiments. * indicates a significant increase in apoptosis compared to DMSO controls using a Student’s t-test. (B) XG19 and XG2 cells were treated for 96 hours with the indicated concentrations of ML216 and melphalan. BrdU (10 μg/ml) was added during the last 1.5 hours of treatment. Cells were fixed and processed to detect BrdU incorporation and total DNA (see Materials and Methods for more details). BrdU+ cells were assigned to S-phase. BrdU- cells were assigned to G0/G1 or G2/M phases based on their DNA content. Asterisks indicate a significant difference compared to DMSO treated (control) cells after applying a Student’s t-test: * p-value < 0.05, ** p-value < 0.01. Results are the mean of 3 independent experiments. (C) XG19 cells treated as in (B) were processed to quantify γH2AX intensity by flow cytometry (see Materials and Methods for more details). * indicate a significant difference compared to DMSO treated (control) cells after applying a Student’s t-test for pairs: p-value < 0.05. (D) XG2 cells were treated with the indicated doses of ML216 and 1 μM melphalan for 48 hours. Cells were harvested and γH2AX levels were analyzed by western blot. One representative experiment out of three is shown. The graph shows the quantification of the γH2AX signal (a.u.: arbitrary units) with respect to tubulin, normalized to the control condition (DMSO) in 3 independent experiments. *** p-value < 0.001.
In order to further investigate the molecular mechanism at the origin of this synergism, we next analyzed the activation of apoptosis pathways in HMCLs treated with ML216 and melphalan by western blot. BLM inhibition in combination with melphalan induced PARP cleavage, a marker of cell death (91), without significant changes in cell cycle regulators or DDR factors, such as the phosphorylation of p53, Chk1 and Chk2, and p21 levels (Figure S4A). In all conditions, Caspases 3/8/9 presented already a low degree of cleavage, which was slightly increased in the case of Caspase 3 in XG19 and Caspases 3/9 in XG2 by melphalan and ML216 combination (Figure S4B). Thus, our data suggest that combination of BLM inhibition with melphalan induces cell death (Figure 7), likely through DNA damage accumulation above the cell’s tolerance level.
In order to confirm the specificity of our results with ML216, we transduced the XG2 cell line with lentivirus containing specific miRNA to knock-down BLM expression (KD BLM). We validated the depletion of BLM protein by western blot, and confirmed that BLM downregulation did not significantly affect the level of expression of the other RECQ helicases (Figure 8A). XG2 control and KD BLM cell lines were treated with 1 μM melphalan for 4 days, and the DDR and caspases activation were analyzed by western blot. In agreement with our results with BLM inhibition in the XG2 parental cell line (Figure 7 and Supplementary Figure S5), the treatment with melphalan induced DDR activation (Figure 8B) and more PARP and caspases cleavage (Figure 8C) in KD BLM cells than in control cells. Moreover, apoptosis level was higher in KD BLM in response to melphalan (Figure 8D). The treatment with the alkylating agent also induced an accumulation of cells in S-phase both in control and KD BLM cells, with small but significant differences also in the repartition of G0/G1 and G2/M populations (Figure 8E). These results validate the specificity of the phenotypes obtained with the chemical inhibition of BLM with ML216 in combination with melphalan, and strengthen the notion that BLM activity and levels are important for the response to DNA damage agents in myeloma cells.
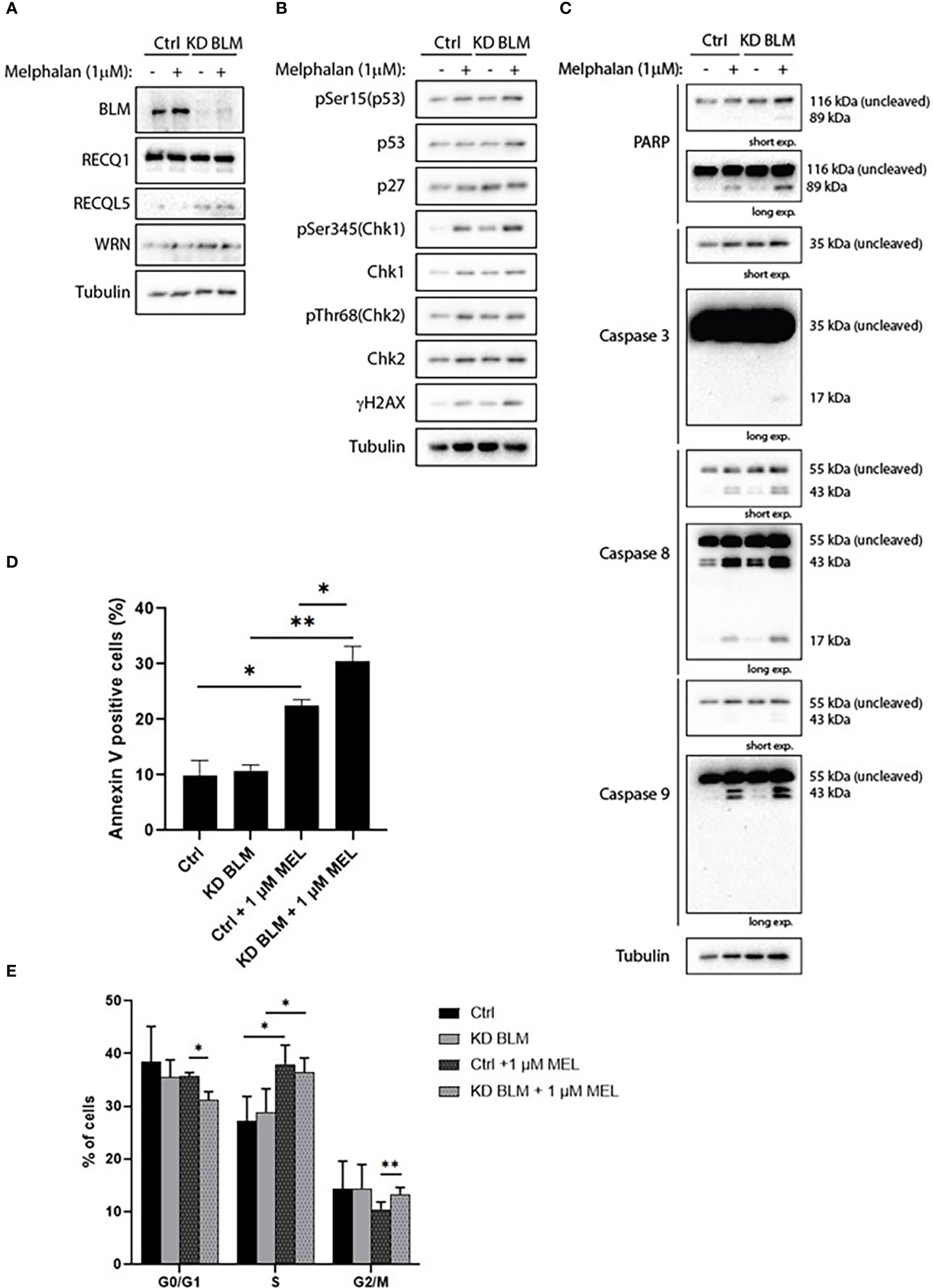
Figure 8 BLM depletion sensitizes MM cells to melphalan. XG2 cells were transduced with control lentiviruses (Ctrl) or miRNA against BLM to stably knock-down its expression (KD BLM). Both cell lines were treated with 1 μM melphalan for 96 hours and samples were collected to analyze (A) BLM depletion and protein levels of the other RECQ helicases; (B) cell cycle and DDR markers by western blot; (C) PARP and caspases cleavage as apoptosis markers by western blot; (D) apoptotic cells (Annexin V+) by flow cytometry; (E) cell cycle distribution using BrdU incorporation and DAPI staining as in Figures 5 and 7. Asterisks indicate a significant difference after applying a Student’s t-test for pairs. * p-value < 0.05, ** p-value < 0.01. All experiments in this figure were repeated 3 times independently.
Finally, based on our previous observation of a synergy between ML216 and melphalan (Figure 6), we hypothesized that the inhibition of BLM could increase the sensitivity of cells to melphalan, notably in melphalan-resistant MM cells, which would be of therapeutic interest. We explored this possibility by comparing the sensitivity of XG2 and XG7 cell lines to their melphalan-resistant counterparts (XG2 MR and XG7 MR) (70). Interestingly, ML216 synergized with melphalan to inhibit cell growth specifically in XG2 and XG7 melphalan-resistant cell lines (Figures 9A, B). These data indicate that BLM inhibition could represent a new therapeutic option to overcome resistance to melphalan in MM cells (Figure 9C). A mutational signature (named SBS-MM1) linked to exposure to high-dose melphalan in MM patients and related to relapse has been described (92–94). These mutations mostly occur in the late-replicating and non-coding parts of the genome (92). In addition, another work reported that TP53 mutation or loss are linked to melphalan resistance and that inactivation of DDR genes such as ATM, FANCA, RAD54B, and BRCC3, enhances the response to the treatment (95), which argues in favor of the combination of BLM inhibition with melphalan to increase melphalan effect. We compared the mutational burden of both XG2 (TP53 mutated) and XG7 (TP53 wild type) melphalan-resistant cell lines to their parental counterparts and identified 16 mutated genes (Supplementary Table S2) common to both melphalan-resistant cell lines. Four of those genes, TBP (TATA-binding protein) (96), TRRAP (Transformation/transcription domain-associated protein) (97), CTBP2 (C-terminal-binding protein 2) and CTDSP2 (Carboxy-terminal domain RNA polymerase II polypeptide A small phosphatase 2), play a role in transcription regulation, either directly or indirectly, which suggests that resistance to melphalan may be mediated not only by mutation of genes involved in DDR, but also by altered regulation of this and other pathways due to changes in transcription. In addition, TRRAP and CTBP2 also play roles in DNA repair. On the one hand, TRRAP, a member of the PIKK family as ATM, has been involved in the regulation of DDR by mediating DSB repair (98) and we have recently reported that its mutation is associated with high-risk MM (99). On the other hand, CTBP2 has roles in diminishing cell cycle arrest and BRCA-mediated DDR, and in apoptosis regulation (100). Thus, in future studies, it would be interesting to further analyze the implications of TRRAP and CTBP2 mutations in the development of resistance to melphalan in MMCs.
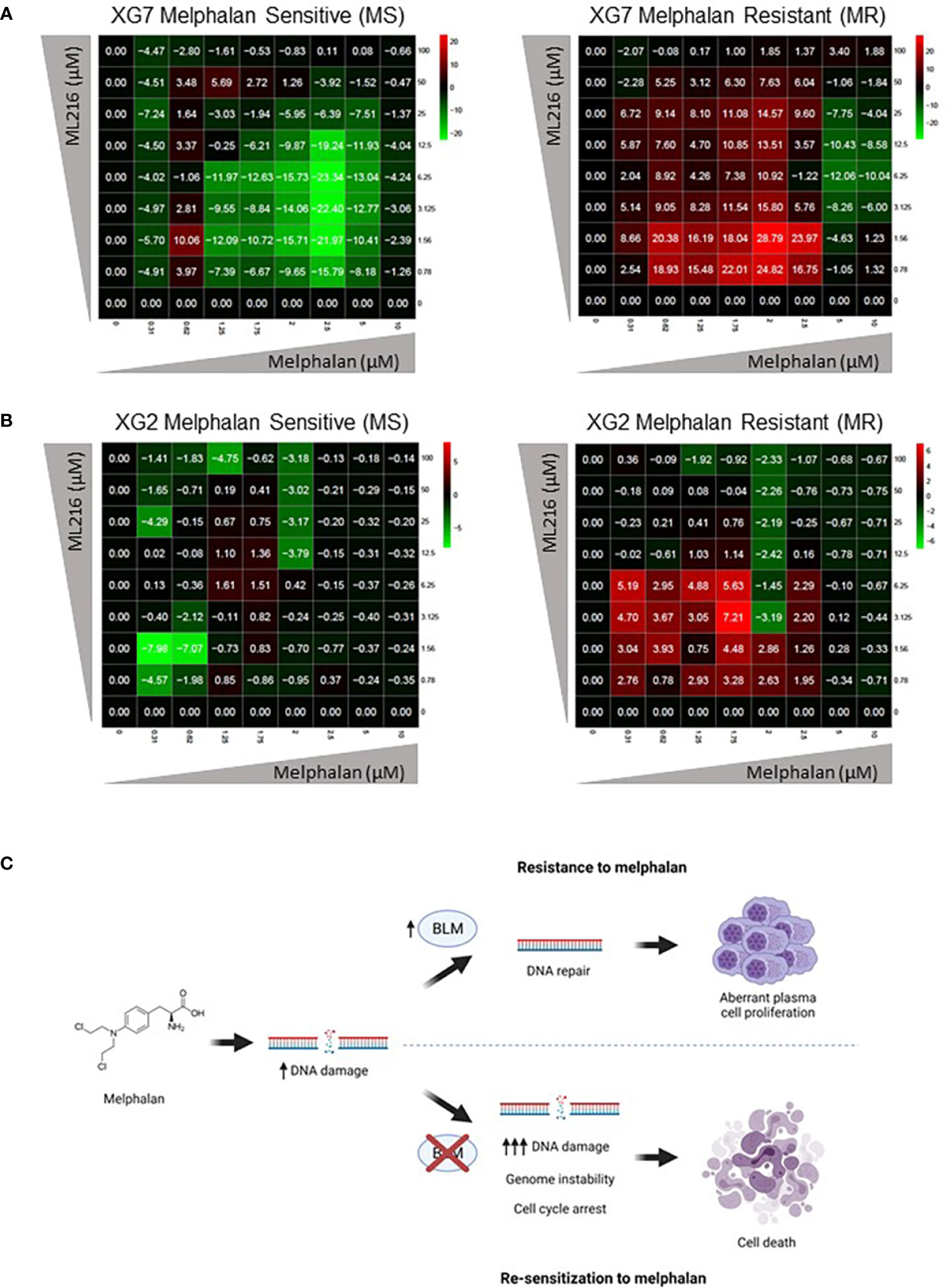
Figure 9 ML216 re-sensitizes Melphalan-resistant MM cells to Melphalan. Dose-response matrixes to measure synergy of ML216 and Melphalan co-treatment. Synergy scores are shown using a continuous pseudo-color scale ranging from dark-green (=antagonism) to dark-red (=synergism). (A) XG7 parental cells and XG7 MR (Melphalan Resistant) (Melphalan IC50: 0.625 μM and 7.5 μM respectively) were treated for 4 days with the indicated doses of ML216 and melphalan. Cell viability was assessed using CellTiter-Glo Luminiscent Cell Viability Assay and was normalized with respect to untreated conditions. Matrixes show the mean of 3-4 independent experiments. (B) Same as in (A) but comparing XG2 parental and XG2 MR (Melphalan Resistant) (Melphalan IC50: 0.625 μM and 2 μM respectively). Matrixes show the mean of 3 independent experiments. (C) Treatment of MM cells with melphalan produces DNA damage. Cells that overexpress BLM can cope better with the drug-induced DNA damage and therefore survive to the treatment, showing a resistant phenotype. On the contrary, BLM inhibition in combination with melphalan increases DNA damage to levels that tumoral plasma cells cannot efficiently repair, leading to cell cycle arrest and eventually to cell death, overcoming melphalan-resistance. Figure was created with BioRender.com.
During the past decades, the development of new therapies has significantly prolonged the survival of MM patients. However, resistance to chemotherapy and relapse remain frequent causes of death (101). The DNA alkylating agent melphalan is one of the main anti-myeloma treatments, alone or in combination with other drugs. Resistance to DNA damaging agents like melphalan could be caused by deregulation or mutation of DDR pathways or increased antioxidant defenses among others (58, 60, 61, 102–106). We have previously demonstrated that RECQ1, a DNA helicase important for the response to replication stress, has a role in cell survival to replication problems and is related to drug resistance in MM cells (61). Another helicase from the RECQ family, namely BLM, is a DDR factor necessary for correct HR, whose mutations are associated with the cancer-prone Bloom’s syndrome (27). In this study, we present evidence that the DNA helicase BLM is also associated with MM cell survival and resistance to DNA damaging chemotherapy. We found that BLM expression increases along the progression of the disease and that BLM is differentially expressed among MM patients, with high BLM expression associated with a bad prognosis (Figures 1, 2).
It has been proposed that BLM can act both as a tumor suppressor and as a proto-oncogene. On the one hand, BLM loss or mutation leads to increased genetic instability and BS development, which points to a role as a tumor suppressor. On the other hand, increased BLM expression has been associated with multiple types of cancers, suggesting a proto-oncogenic function [reviewed in (107)]. For example, a recent study reported a correlation between BLM overexpression and poor overall survival in lung and gastric cancer patients (108). Similarly, our data showed that high BLM expression correlates with worse overall and event free survival in MM patients (Figure 2), confirming a proto-oncogenic role in MM as well. High levels of BLM were also found in hematological malignancies such as myeloid leukemia, lymphoma and myeloma (62, 109). Furthermore, transcriptomics analysis of fibroblasts from BS patients identified cell proliferation and survival genes, as well as immunological pathways, as the topmost deregulated in this disease (110, 111). Similarly, our GSEA results showed an increase in the expression of proliferation-associated genes in MM patients with high level of BLM expression and bad prognosis (Figure S1).
We took advantage of model MM cell lines derived from patients to further characterize the role of BLM in MM. A panel of HMCLs showed different responses to the BLM inhibitor ML216, with IC50 concentrations ranging from 1.3 μM for the most sensitive cells to 16.9 μM for the most resistant one. Intriguingly, sensitivity to ML216 did not correlate with the levels of expression of any tested RECQ helicase (BLM, RECQ1, RECQL5, and WRN), the cell lines’ basal DDR activation, their MM molecular subgroup or mutations in several oncogenes (Figure 3). Loss of a particular DDR pathway in cancer cells can make cells more dependent on other pathways. Thus, one possibility is that deregulation of other molecular factors, likely involved in DDR and/or cell cycle regulation, could account for the differential sensitivity to BLM inhibition. However, BLM has other cellular functions other than DNA repair (reviewed in [80)], that could also be responsible for the differences in the response to ML216. For example, in physiological conditions, BLM is required for fork progression and stability, resolution of R-loops or ultrafine anaphase bridges, and acts during replication stress by unwinding unusual DNA structures. BLM also regulates the correct replication of telomeric regions and BLM-deficient cells show a slowdown of replication forks and an increase in G4 at telomeres (52). In addition, BLM has been reported to facilitate pre-mRNA synthesis by direct binding with RNA Pol I and DNA topoisomerase I (82, 112, 113). Thus, inhibiting BLM would also affect ribosome biogenesis, which is known to be of particular importance for fast-proliferating cells such as tumor cells (114). Finally, it has been suggested that BLM would regulate transcription of a set of genes via its interaction to their G4 motifs (110). Therefore, given the plethora of molecular mechanisms in which BLM is involved in the cells, it is likely that the different sensitivity to ML216 inhibitor shown by our panel of MM cell lines would depend not only on each cell line tolerance to DNA damage, but also to alterations in the other BLM-regulated processes.
We demonstrated that continuous BLM inhibition induces cell cycle arrest and eventually leads to apoptosis in HMCLs. Importantly, this toxicity seems specific to myeloma cells, since ML216 poorly affected non-myeloma primary cells from patients (Figure 4B, C). This is likely due to the role of BLM as a safeguard of genomic stability, making rapidly dividing tumoral cells more dependent on its activity to cope with DNA damage and replication-transcription conflicts. This notion provides a strong rationale to combine BLM inhibition with DNA damaging agents, in order to overload the cell with DNA damage while impairing its repair. Indeed, our data show that ML216 potentiated the effect of melphalan to kill the 3 tested HMCLs (Figure 6). Double treatment induced PARP and caspases cleavage, concomitant with a strong cell cycle arrest and increased cell death, supporting the idea of causing a DNA damage overload to kill tumor cells (Figure 9C and Supplementary Figure S5). A recent work has reported a synergy between ML216 and the PARP inhibitor olaparib with irradiation to kill NSCLC (non-small cell lung cancer) cells by inhibiting HR and promoting NHEJ (115). Therefore, the interest of the combination of ML216 with chemotherapies currently in clinical use deserves further investigation.
Resistance to drugs remains a major concern in the therapeutic management of MM. We have previously reported that BLM expression correlates with sensitivity to the immunomodulatory agent lenalidomide, a standard-of-care drug in MM (23). Interestingly, we have found a synergy between ML216 and lenalidomide in HMCLs (Supplementary Figure S3B). However, the mechanism of this synergy remains to be explored, since no significant effect on IMiD targets including Myc and IRF4 levels was observed upon treatment with ML216 (Supplementary Figure S4C). Melphalan is another standard-of-care drug used both in patients eligible and non-eligible for autologous hematopoietic stem cell transplantation, alone or in combination with other chemotherapeutic agents, respectively (56, 116, 117). However, patients often acquire mutations in DDR pathways that result in resistance (58, 60). By using melphalan-resistant cell models, we showed that inhibition of BLM may be a good strategy to overcome such resistance (Figure 9A, B). Similarly, we previously reported that sublethal concentrations of the inhibitors of Chk1 (AZD7762) and Cdc7-Dbf4 (XL413), both kinases involved in DNA damage and replication stress response, overcome resistance to melphalan in HMCLs (70). Thus, the combination of DDR inhibitors and DNA damaging agents should be further explored as a therapeutic option for MM patients with resistance to DNA damage chemotherapies.
Targeting DDR factors to enhance sensitivity to melphalan in MM has already been proposed. In particular, treatment with bortezomib reduced the levels of Fanconi Anemia (FA) factors BRCA1/2 and FANCD2, and proved effective to induce DNA damage and cell death in combination with melphalan (103). FA is an autosomal recessive disorder characterized by genetic instability that leads to developmental abnormalities, BM failure and elevated risk of developing certain types of cancer like acute myeloid leukemia and squamous cell carcinomas (118, 119). BS and FA disease present some overlapping phenotypes, like predisposition to cancer and immunodeficiency, and are both characterized by genetic instability. Although their genetic origins are different, it has been proposed that the BS complex (BLM, RMI1, RMI2 and Topoisomerase III-a) and the FA pathway are related by protein interactions, forming the BRAFT multiprotein complex (BLM, RPA, FA and Topoisomerase III-α), which has various DNA-processing activities (120, 121). In addition, BLM interacts with the helicase FANCJ (122), the DNA translocase FANCM interacts with FANCF, RMI1 and Topoisomerase III-a, linking both complexes (123). Moreover, BLM has been shown to promote the activation of the FA pathway through FANCD2 in response to cross-linking agents (124). Thus, it is not surprising that BLM inhibition also increases the toxicity of melphalan.
The notion to combine DNA damaging agents with drugs that target DDR pathways is gaining attention as a potential way to increase anticancer therapy effectiveness. For instance, inhibitors of RAD51 and WRN sensitize cancer cells to DNA damaging agents (125, 126). Thus, development of new inhibitors that target other DDR factors to be used in combination with current chemotherapeutic drugs becomes an appealing option to treat cancer more efficiently and confront drug resistance. BLM, a critical proven factor for genomic stability maintenance, is therefore an interesting candidate for combination therapies. Indeed, several experimental and computational studies have proposed RECQ helicases as potential targets in a range of different cancers (127–130). However, till recently only the small molecule ML216 had been developed to target BLM, which is still poorly characterized in treating cancer cells. Of note, isaindigotone and quinazolinone derivatives have been recently reported as potential new BLM inhibitors. Both inhibit proliferation and trigger apoptosis and DDR activation in the human colon cancer cell line HCT116 (131, 132). Also recently, a screen for antiproliferative drugs in breast cancer identified HJNO, a tetrandrine derivative which inhibits BLM DNA binding, unwinding and ATPase activities, diminishing breast cancer cells proliferation (133). Another study has analyzed derivatives of ML216 that seem highly specific allosteric inhibitors of BLM, that could be used to cause highly cytotoxic BLM-DNA complexes to kill cancer cells (134). In the next years, further work to develop and characterize new BLM inhibitors is warranted.
In MM, most patients develop resistance to the existing therapies, including melphalan. Our data suggest that BLM expression can be a good biomarker for MM and that combination of BLM inhibitors with DNA damaging drugs could be of therapeutic interest to treat MM patients who have developed resistance to melphalan. It is important to keep in mind that chemotherapy drugs used to target tumor cells are also toxic to other types of healthy cells, leading to toxicity and, ultimately, the development of secondary cancers in many patients later in life. Several mechanisms associated with MMC resistance to genotoxic treatments have been described, underlining the myeloma endemic heterogeneous landscape (135). These findings provide several therapeutic strategies to overcome drug resistance and limit mutagenic effects of genotoxic agents in MM (135). Exploiting synthetic lethality between DNA repair inhibitors and DNA-damaging agents would allow lower concentrations of the latter to be used, limiting undesirable side effects. The clinical manifestations of patients with Bloom’s syndrome indicate that BLM activity is crucial for the maintenance of genetic stability at the organismal level. However, the toxicity associated with BLM inhibition in the context of therapeutic treatment, i.e. inhibition that is not sustained over time for years, is not likely to have such a dramatic impact on the fitness of cancer patients compared to patients with BS. Yet, BLM inhibitor’s toxicity needs to be carefully addressed using in vivo models to assess the benefits and risks of its use in cancer treatment.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ebi.ac.uk/arrayexpress/, E-MTAB-37, https://www.ebi.ac.uk/arrayexpress/, E-TABM-937, https://www.ebi.ac.uk/arrayexpress/, E-TABM-1088.
SO performed the research and wrote the paper. EV, LD and JD participated in the research and in the writing of the paper. MJ, GR, NR and HB participated in the research. EA participated in biocomputational analyses. AS, DH, LV and CH participated in clinical data analysis and participated in the writing of the paper. Y-LL, JB, AC and PP participated in the research and in the writing of the paper. JM supervised the research and the writing of the paper. All authors contributed to the article and approved the submitted version.
The JM research group was supported by grants from INCA PLBIO18-362PIT-MM and PLBIO19 FATidique, ANR (TIE-Skip; 2017-CE15-0024-01), ANR-18-CE15-0010-01 PLASMADIFF-3D, SIRIC Montpellier Cancer (INCa_Inserm_D-GOS_12553), Carnot FINDMED, Institut Carnot CALYM, Labex EpiGenMed, FFRMG (AAP-FFRMG-2021), INSERM PSCI 2020 Smooth-MM, Labex Epigenmed and Institut Universitaire de France. Work in PP’s lab is supported by grants from INCA, the Ligue Nationale Contre le Cancer (équipe labellisée), and the Fondation MSDAvenir. LD is supported by a grant from Ligue Nationale Contre le Cancer LNCC.
The authors thank the imaging facility MRI, member of the national infrastructure France-Bioimaging supported by the French National Research Agency (ANR-10-INBS-04, Investments for the future).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.983181/full#supplementary-material
1. Pinto V, Bergantim R, Caires HR, Seca H, Guimarães JE, Vasconcelos MH. Multiple myeloma: Available therapies and causes of drug resistance. Cancers (2020) 12:407. doi: 10.3390/cancers12020407
2. Gay F, Mina R. Redefining the treatment paradigm for multiple myeloma. Lancet Oncol (2019) 20:743–4. doi: 10.1016/S1470-2045(19)30295-5
3. Röllig C, Knop S, Bornhäuser M. Multiple myeloma. Lancet (2015) 385:2197–208. doi: 10.1016/S0140-6736(14)60493-1
4. Kumar SK, Dispenzieri A, Lacy MQ, Gertz MA, Buadi FK, Pandey S, et al. Continued improvement in survival in multiple myeloma: Changes in early mortality and outcomes in older patients. Leukemia (2014) 28:1122–8. doi: 10.1038/leu.2013.313
5. Tacchetti P, Pantani L, Patriarca F, Petrucci MT, Zamagni E, Dozza L, et al. Bortezomib, thalidomide, and dexamethasone followed by double autologous haematopoietic stem-cell transplantation for newly diagnosed multiple myeloma (GIMEMA-MMY-3006): Long-term follow-up analysis of a randomised phase 3, open-label study. Lancet Haematol (2020) 7:e861–73. doi: 10.1016/S2352-3026(20)30323-9
6. González D, Balanzategui A, García-Sanz R, Gutiérrez N, Seabra C, van Dongen JJM, et al. Incomplete DJH rearrangements of the IgH gene are frequent in multiple myeloma patients: Immunobiological characteristics and clinical implications. Leukemia (2003) 17:1398–403. doi: 10.1038/sj.leu.2402964
7. Kuehl WM, Bergsagel PL. Molecular pathogenesis of multiple myeloma and its premalignant precursor. J Clin Invest (2012) 122:3456–63. doi: 10.1172/JCI61188
8. Hazlehurst LA, Damiano JS, Buyuksal I, Pledger WJ, Dalton WS. Adhesion to fibronectin via β1 integrins regulates p27kip1 levels and contributes to cell adhesion mediated drug resistance (CAM-DR). Oncogene (2000) 19:4319–27. doi: 10.1038/sj.onc.1203782
9. Chauhan D, Uchiyama H, Akbarali Y, Urashima M, Yamamoto K, Libermann TA, et al. Multiple myeloma cell adhesion-induced interleukin-6 expression in bone marrow stromal cells involves activation of NF-kappa b. Blood (1996) 87:1104–12. doi: 10.1182/blood.V87.3.1104.bloodjournal8731104
10. Gupta VA, Matulis SM, Conage-Pough JE, Nooka AK, Kaufman JL, Lonial S, et al. Bone marrow microenvironment–derived signals induce mcl-1 dependence in multiple myeloma. Blood (2017) 129:1969–79. doi: 10.1182/blood-2016-10-745059
11. Bal S, Giri S, Godby KN, Costa LJ. New regimens and directions in the management of newly diagnosed multiple myeloma. Am J Hematol (2021) 96:367–78. doi: 10.1002/ajh.26080
12. Manier S, Salem KZ, Park J, Landau DA, Getz G, Ghobrial IM. Genomic complexity of multiple myeloma and its clinical implications. Nat Rev Clin Oncol (2017) 14:100–13. doi: 10.1038/nrclinonc.2016.122
13. Herviou L, Kassambara A, Boireau S, Robert N, Requirand G, Müller-Tidow C, et al. PRC2 targeting is a therapeutic strategy for EZ score defined high-risk multiple myeloma patients and overcome resistance to IMiDs. Clin Epigenet (2018) 10:121. doi: 10.1186/s13148-018-0554-4
14. Bruyer A, Maes K, Herviou L, Kassambara A, Seckinger A, Cartron G, et al. DNMTi/HDACi combined epigenetic targeted treatment induces reprogramming of myeloma cells in the direction of normal plasma cells. Br J Cancer (2018) 118:1062–73. doi: 10.1038/s41416-018-0025-x
15. Alzrigat M, Párraga AA, Jernberg-Wiklund H. Epigenetics in multiple myeloma: From mechanisms to therapy. Semin Cancer Biol (2018) 51:101–15. doi: 10.1016/j.semcancer.2017.09.007
16. Cremer FW, Kartal M, Hose D, Bila J, Buck I, Bellos F, et al. High incidence and intraclonal heterogeneity of chromosome 11 aberrations in patients with newly diagnosed multiple myeloma detected by multiprobe interphase FISH. Cancer Genet Cytogenet (2005) 161:116–24. doi: 10.1016/j.cancergencyto.2005.02.015
17. Egan JB, Shi C-X, Tembe W, Christoforides A, Kurdoglu A, Sinari S, et al. Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, evolution, and clonal tides. Blood (2012) 120:1060–6. doi: 10.1182/blood-2012-01-405977
18. Lopez-Corral L, Gutierrez NC, Vidriales MB, Mateos MV, Rasillo A, Garcia-Sanz R, et al. The progression from MGUS to smoldering myeloma and eventually to multiple myeloma involves a clonal expansion of genetically abnormal plasma cells. Clin Cancer Res (2011) 17:1692–700. doi: 10.1158/1078-0432.CCR-10-1066
19. Gonsalves WI, Rajkumar SV, Gupta V, Morice WG, Timm MM, Singh PP, et al. Quantification of clonal circulating plasma cells in newly diagnosed multiple myeloma: Implications for redefining high-risk myeloma. Leukemia (2014) 28:2060–5. doi: 10.1038/leu.2014.98
20. Karow JK, Chakraverty RK, Hickson ID. The bloom’s syndrome gene product is a 3′-5′ DNA helicase. J Biol Chem (1997) 272:30611–4. doi: 10.1074/jbc.272.49.30611
21. Nimonkar AV, Genschel J, Kinoshita E, Polaczek P, Campbell JL, Wyman C, et al. BLM–DNA2–RPA–MRN and EXO1–BLM–RPA–MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev (2011) 25:350–62. doi: 10.1101/gad.2003811
22. Croteau DL, Popuri V, Opresko PL, Bohr VA. Human RecQ helicases in DNA repair, recombination, and replication. Annu Rev Biochem (2014) 83:519–52. doi: 10.1146/annurev-biochem-060713-035428
23. Kassambara A, Herviou L, Ovejero S, Jourdan M, Thibaut C, Vikova V, et al. RNA-Sequencing data-driven dissection of human plasma cell differentiation reveals new potential transcription regulators. Leukemia (2021) 35:1451–62. doi: 10.1038/s41375-021-01234-0
24. Bernstein KA, Gangloff S, Rothstein R. The RecQ DNA helicases in DNA repair. Annu Rev Genet (2010) 44:393–417. doi: 10.1146/annurev-genet-102209-163602
25. Lu H, Davis AJ. Human RecQ helicases in DNA double-strand break repair. Front Cell Dev Biol (2021) 9:640755. doi: 10.3389/fcell.2021.640755
26. Abu-Libdeh B, Jhujh SS, Dhar S, Sommers JA, Datta A, Longo GM, et al. RECON syndrome is a genome instability disorder caused by mutations in the DNA helicase RECQL1. J Clin Invest (2022) 132:e147301. doi: 10.1172/JCI147301
27. Bahr A, De Graeve F, Kedinger C, Chatton B. Point mutations causing bloom’s syndrome abolish ATPase and DNA helicase activities of the BLM protein. Oncogene (1998) 17:2565–71. doi: 10.1038/sj.onc.1202389
28. Bloom D. Congenital telangiectatic erythema resembling lupus erythematosus in dwarfs; probably a syndrome entity. AMA Am J Dis Child (1954) 88:754–8. doi: 10.1001/archpedi.1954.02050100756008
29. German J, Archibald R, Bloom D. Chromosomal breakage in a rare and probably genetically determined syndrome of man. Science (1965) 148:506–7. doi: 10.1126/science.148.3669.506
30. German J. Bloom’s syndrome. i. genetical and clinical observations in the first twenty-seven patients. Am J Hum Genet (1969) 21:196–227.
31. German J, Crippa LP, Bloom D. Bloom’s syndrome. III. analysis of the chromosome aberration characteristic of this disorder. Chromosoma (1974) 48:361–6. doi: 10.1007/BF00290993
32. Chaganti RSK, Schonberg S, German J. A manyfold increase in sister chromatid exchanges in bloom’s syndrome lymphocytes. Proc Natl Acad Sci USA (1974) 71:4508–12. doi: 10.1073/pnas.71.11.4508
33. Wu L, Hickson ID. The bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature (2003) 426:870–4. doi: 10.1038/nature02253
34. Dhar S, Brosh RM. BLM’s balancing act and the involvement of FANCJ in DNA repair. Cell Cycle (2018) 17:2207–20. doi: 10.1080/15384101.2018.1520567
35. Tripathi V, Agarwal H, Priya S, Batra H, Modi P, Pandey M, et al. MRN complex-dependent recruitment of ubiquitylated BLM helicase to DSBs negatively regulates DNA repair pathways. Nat Commun (2018) 9:1016. doi: 10.1038/s41467-018-03393-8
36. Gravel S, Chapman JR, Magill C, Jackson SP. DNA Helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev (2008) 22:2767–72. doi: 10.1101/gad.503108
37. Nimonkar AV, Özsoy AZ, Genschel J, Modrich P, Kowalczykowski SC. Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proc Natl Acad Sci USA (2008) 105:16906–11. doi: 10.1073/pnas.0809380105
38. Karow JK, Constantinou A, Li J-L, West SC, Hickson ID. The bloom’s syndrome gene product promotes branch migration of holliday junctions. Proc Natl Acad Sci USA (2000) 97:6504–8. doi: 10.1073/pnas.100448097
39. Bizard AH, Hickson ID. The dissolution of double holliday junctions. Cold Spring Harbor Perspect Biol (2014) 6:a016477–a016477. doi: 10.1101/cshperspect.a016477
40. Kowalczykowski SC. An overview of the molecular mechanisms of recombinational DNA repair. Cold Spring Harb Perspect Biol (2015) 7:a016410. doi: 10.1101/cshperspect.a016410
41. Bugreev DV, Yu X, Egelman EH, Mazin AV. Novel pro- and anti-recombination activities of the bloom’s syndrome helicase. Genes Dev (2007) 21:3085–94. doi: 10.1101/gad.1609007
42. Bachrati CZ. Mobile d-loops are a preferred substrate for the bloom’s syndrome helicase. Nucleic Acids Res (2006) 34:2269–79. doi: 10.1093/nar/gkl258
43. van Brabant AJ, Ye T, Sanz M, German JL, Ellis NA, Holloman WK. Binding and melting of d-loops by the bloom syndrome helicase. Biochemistry (2000) 39:14617–25. doi: 10.1021/bi0018640
44. Patel DS, Misenko SM, Her J, Bunting SF. BLM helicase regulates DNA repair by counteracting RAD51 loading at DNA double-strand break sites. J Cell Biol (2017) 216:3521–34. doi: 10.1083/jcb.201703144
45. Tripathi V, Nagarjuna T, Sengupta S. BLM helicase–dependent and –independent roles of 53BP1 during replication stress–mediated homologous recombination. J Cell Biol (2007) 178:9–14. doi: 10.1083/jcb.200610051
46. Ralf C, Hickson ID, Wu L. The bloom’s syndrome helicase can promote the regression of a model replication fork. J Biol Chem (2006) 281:22839–46. doi: 10.1074/jbc.M604268200
47. Davies SL, North PS, Hickson ID. Role for BLM in replication-fork restart and suppression of origin firing after replicative stress. Nat Struct Mol Biol (2007) 14:677–9. doi: 10.1038/nsmb1267
48. Sengupta S, Robles AI, Linke SP, Sinogeeva NI, Zhang R, Pedeux R, et al. Functional interaction between BLM helicase and 53BP1 in a Chk1-mediated pathway during s-phase arrest. J Cell Biol (2004) 166:801–13. doi: 10.1083/jcb.200405128
49. Rouzeau S, Cordelières FP, Buhagiar-Labarchède G, Hurbain I, Onclercq-Delic R, Gemble S, et al. Bloom’s syndrome and PICH helicases cooperate with topoisomerase IIα in centromere disjunction before anaphase. PloS One (2012) 7:e33905. doi: 10.1371/journal.pone.0033905
50. Ke Y, Huh J-W, Warrington R, Li B, Wu N, Leng M, et al. PICH and BLM limit histone association with anaphase centromeric DNA threads and promote their resolution: PICH and BLM limit histone association. EMBO J (2011) 30:3309–21. doi: 10.1038/emboj.2011.226
51. Chan K-L, North PS, Hickson ID. BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J (2007) 26:3397–409. doi: 10.1038/sj.emboj.7601777
52. Drosopoulos WC, Kosiyatrakul ST, Schildkraut CL. BLM helicase facilitates telomere replication during leading strand synthesis of telomeres. J Cell Biol (2015) 210:191–208. doi: 10.1083/jcb.201410061
53. Lansdorp P, van Wietmarschen N. Helicases FANCJ, RTEL1 and BLM act on guanine quadruplex DNA in vivo. Genes (2019) 10:870. doi: 10.3390/genes10110870
54. Tan J, Wang X, Phoon L, Yang H, Lan L. Resolution of ROS-induced G-quadruplexes and r-loops at transcriptionally active sites is dependent on BLM helicase. FEBS Lett (2020) 594:1359–67. doi: 10.1002/1873-3468.13738
55. Hoeijmakers JHJ. Genome maintenance mechanisms for preventing cancer. Nature (2001) 411:366–74. doi: 10.1038/35077232
56. Deans AJ, West SC. DNA Interstrand crosslink repair and cancer. Nat Rev Cancer (2011) 11:467–80. doi: 10.1038/nrc3088
57. Economopoulou P, Pappa V, Papageorgiou S, Dervenoulas J, Economopoulos T. Abnormalities of DNA repair mechanisms in common hematological malignancies. Leuk Lymph (2011) 52:567–82. doi: 10.3109/10428194.2010.551155
58. Kassambara A, Gourzones-Dmitriev C, Sahota S, Rème T, Moreaux J, Goldschmidt H, et al. A DNA repair pathway score predicts survival in human multiple myeloma: the potential for therapeutic strategy. Oncotarget (2014) 5:2487–98. doi: 10.18632/oncotarget.1740
59. Cottini F, Hideshima T, Suzuki R, Tai Y-T, Bianchini G, Richardson PG, et al. Synthetic lethal approaches exploiting DNA damage in aggressive myeloma. Cancer Discovery (2015) 5:972–87. doi: 10.1158/2159-8290.CD-14-0943
60. Gourzones-Dmitriev C, Kassambara A, Sahota S, Rème T, Moreaux J, Bourquard P, et al. DNA Repair pathways in human multiple myeloma: Role in oncogenesis and potential targets for treatment. Cell Cycle (2013) 12:2760–73. doi: 10.4161/cc.25951
61. Viziteu E, Klein B, Basbous J, Lin Y-L, Hirtz C, Gourzones C, et al. RECQ1 helicase is involved in replication stress survival and drug resistance in multiple myeloma. Leukemia (2017) 31:2104–13. doi: 10.1038/leu.2017.54
62. Viziteu E, Kassambara A, Pasero P, Klein B, Moreaux J. RECQ helicases are deregulated in hematological malignancies in association with a prognostic value. biomark Res (2016) 4:3. doi: 10.1186/s40364-016-0057-4
63. Rosenthal AS, Dexheimer TS, Gileadi O, Nguyen GH, Chu WK, Hickson ID, et al. Synthesis and SAR studies of 5-(pyridin-4-yl)-1,3,4-thiadiazol-2-amine derivatives as potent inhibitors of bloom helicase. Bioorg Med Chem Lett (2013) 23:5660–6. doi: 10.1016/j.bmcl.2013.08.025
64. Nguyen GH, Dexheimer TS, Rosenthal AS, Chu WK, Singh DK, Mosedale G, et al. A small molecule inhibitor of the BLM helicase modulates chromosome stability in human cells. Chem Biol (2013) 20:55–62. doi: 10.1016/j.chembiol.2012.10.016
65. Moreaux J, Klein B, Bataille R, Descamps G, Maiga S, Hose D, et al. A high-risk signature for patients with multiple myeloma established from the molecular classification of human myeloma cell lines. Haematologica (2011) 96:574–82. doi: 10.3324/haematol.2010.033456
66. Vikova V, Jourdan M, Robert N, Requirand G, Boireau S, Bruyer A, et al. Comprehensive characterization of the mutational landscape in multiple myeloma cell lines reveals potential drivers and pathways associated with tumor progression and drug resistance. Theranostics (2019) 9:540–53. doi: 10.7150/thno.28374
67. Hose D, Reme T, Hielscher T, Moreaux J, Messner T, Seckinger A, et al. Proliferation is a central independent prognostic factor and target for personalized and risk-adapted treatment in multiple myeloma. Haematologica (2011) 96:87–95. doi: 10.3324/haematol.2010.030296
68. Kassambara A, Rème T, Jourdan M, Fest T, Hose D, Tarte K, et al. GenomicScape: An easy-to-Use web tool for gene expression data analysis. application to investigate the molecular events in the differentiation of b cells into plasma cells. PloS Comput Biol (2015) 11:e1004077. doi: 10.1371/journal.pcbi.1004077
69. Kassambara A, Hose D, Moreaux J, Walker BA, Protopopov A, Reme T, et al. Genes with a spike expression are clustered in chromosome (sub)bands and spike (sub)bands have a powerful prognostic value in patients with multiple myeloma. Haematologica (2012) 97:622–30. doi: 10.3324/haematol.2011.046821
70. de Boussac H, Bruyer A, Jourdan M, Maes A, Robert N, Gourzones C, et al. Kinome expression profiling to target new therapeutic avenues in multiple myeloma. Haematologica (2020) 105:784–95. doi: 10.3324/haematol.2018.208306
71. Moreaux J, Legouffe E, Jourdan E, Quittet P, Rème T, Lugagne C, et al. BAFF and APRIL protect myeloma cells from apoptosis induced by interleukin 6 deprivation and dexamethasone. Blood (2004) 103:3148–57. doi: 10.1182/blood-2003-06-1984
72. Zhan F, Huang Y, Colla S, Stewart JP, Hanamura I, Gupta S, et al. The molecular classification of multiple myeloma. Blood (2006) 108:2020–8. doi: 10.1182/blood-2005-11-013458
73. Engelhardt M, Terpos E, Kleber M, Gay F, Wasch R, Morgan G, et al. European Myeloma network recommendations on the evaluation and treatment of newly diagnosed patients with multiple myeloma. Haematologica (2014) 99:232–42. doi: 10.3324/haematol.2013.099358
74. Shaughnessy JD, Zhan F, Burington BE, Huang Y, Colla S, Hanamura I, et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood (2007) 109:2276–84. doi: 10.1182/blood-2006-07-038430
75. Decaux O, Lodé L, Magrangeas F, Charbonnel C, Gouraud W, Jézéquel P, et al. Prediction of survival in multiple myeloma based on gene expression profiles reveals cell cycle and chromosomal instability signatures in high-risk patients and hyperdiploid signatures in low-risk patients: A study of the intergroupe francophone du myélome. JCO (2008) 26:4798–805. doi: 10.1200/JCO.2007.13.8545
76. Rème T, Hose D, Theillet C, Klein B. Modeling risk stratification in human cancer. Bioinformatics (2013) 29:1149–57. doi: 10.1093/bioinformatics/btt124
77. Requirand G, Robert N, Boireau S, Vincent L, Seckinger A, Bouhya S, et al. BrdU incorporation in multiparameter flow cytometry: A new cell cycle assessment approach in multiple myeloma. Cytometry (2019) 96:209–14. doi: 10.1002/cyto.b.21730
78. Pochard C, Herbaux C, Gabellier L, Fornero L, Robert N, Requirand G, et al. Integrative approach to find predictive markers of response to daratumumab in multiple myeloma. European Hematology Association(EHA): Library (2021). Available at: https://library.ehaweb.org/eha/2021/eha2021-virtual-congress/325708/camille.pochard.integrative.approach.to.find.predictive.markers.of.response.to.html?f=menu%3D6%2Abrowseby%3D8%2Asortby%3D2%2Amedia%3D3%2Ace_id%3D2035%2Alabel%3D21989%2Aot_id%3D25571%2Amarker%3D1286%2Afeatured%3D17286.
79. Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D, et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell (2014) 25:91–101. doi: 10.1016/j.ccr.2013.12.015
80. Ababou M. Bloom syndrome and the underlying causes of genetic instability. Mol Genet Metab (2021) 133:35–48. doi: 10.1016/j.ymgme.2021.03.003
81. Ellis NA, Proytcheva M, Sanz MM, Ye T-Z, German J. Transfection of BLM into cultured bloom syndrome cells reduces the sister-chromatid exchange rate toward normal. Am J Hum Genet (1999) 65:1368–74. doi: 10.1086/302616
82. Tangeman L, McIlhatton M, Grierson P, Groden J, Acharya S. Regulation of BLM nucleolar localization. Genes (2016) 7:69. doi: 10.3390/genes7090069
83. Herviou L, Ovejero S, Izard F, Karmous-Gadacha O, Gourzones C, Bellanger C, et al. Targeting the methyltransferase SETD8 impairs tumor cell survival and overcomes drug resistance independently of p53 status in multiple myeloma. Clin Epigenet (2021) 13:174. doi: 10.1186/s13148-021-01160-z
84. Lopez-Girona A, Heintel D, Zhang L-H, Mendy D, Gaidarova S, Brady H, et al. Lenalidomide downregulates the cell survival factor, interferon regulatory factor-4, providing a potential mechanistic link for predicting response: Lenalidomide downregulates IRF4. Br J Haematol (2011) 154:325–36. doi: 10.1111/j.1365-2141.2011.08689.x
85. Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA Double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem (1998) 273:5858–68. doi: 10.1074/jbc.273.10.5858
86. Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol (2000) 10:886–95. doi: 10.1016/S0960-9822(00)00610-2
87. Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem (2001) 276:47759–62. doi: 10.1074/jbc.C100569200
88. Furuta T, Takemura H, Liao Z-Y, Aune GJ, Redon C, Sedelnikova OA, et al. Phosphorylation of histone H2AX and activation of Mre11, Rad50, and Nbs1 in response to replication-dependent DNA double-strand breaks induced by mammalian DNA topoisomerase I cleavage complexes. J Biol Chem (2003) 278:20303–12. doi: 10.1074/jbc.M300198200
89. Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell (2005) 123:1213–26. doi: 10.1016/j.cell.2005.09.038
90. Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, et al. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell (2007) 131:887–900. doi: 10.1016/j.cell.2007.09.040
91. Kaufmann SH, Desnoyers S, Ottaviano Y, Davidson NE, Poirier GG. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: An early marker of chemotherapy-induced apoptosis. Cancer Res (1993) 53:3976–85.
92. Rustad EH, Yellapantula V, Leongamornlert D, Bolli N, Ledergor G, Nadeu F, et al. Timing the initiation of multiple myeloma. Nat Commun (2020) 11:1917. doi: 10.1038/s41467-020-15740-9
93. Giesen N, Paramasivam N, Toprak UH, Huebschmann D, Xu J, Uhrig S, et al. Comprehensive genomic analysis of refractory multiple myeloma reveals a complex mutational landscape associated with drug resistance and novel therapeutic vulnerabilities. Haematologica 2022 107(8):1891–901. doi: 10.3324/haematol.2021.279360
94. Landau HJ, Yellapantula V, Diamond BT, Rustad EH, Maclachlan KH, Gundem G, et al. Accelerated single cell seeding in relapsed multiple myeloma. Nat Commun (2020) 11:3617. doi: 10.1038/s41467-020-17459-z
95. Bohl SR, Schmalbrock LK, Bauhuf I, Meyer T, Dolnik A, Szyska M, et al. Comprehensive CRISPR-Cas9 screens identify genetic determinants of drug responsiveness in multiple myeloma. Blood Adv (2021) 5:2391–402. doi: 10.1182/bloodadvances.2020003541
96. Mishal R, Luna-Arias JP. Role of the TATA-box binding protein (TBP) and associated family members in transcription regulation. Gene (2022) 833:146581. doi: 10.1016/j.gene.2022.146581
97. Murr R, Vaissière T, Sawan C, Shukla V, Herceg Z. Orchestration of chromatin-based processes: Mind the TRRAP. Oncogene (2007) 26:5358–72. doi: 10.1038/sj.onc.1210605
98. Murr R, Loizou JI, Yang Y-G, Cuenin C, Li H, Wang Z-Q, et al. Histone acetylation by trrap–Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat Cell Biol (2006) 8:91–9. doi: 10.1038/ncb1343
99. Alaterre E, Vikova V, Kassambara A, Bruyer A, Robert N, Requirand G, et al. RNA-Sequencing-Based transcriptomic score with prognostic and theranostic values in multiple myeloma. JPM (2021) 11:988. doi: 10.3390/jpm11100988
100. Stankiewicz TR, Gray JJ, Winter AN, Linseman DA. C-terminal binding proteins: Central players in development and disease. Biomol Concepts (2014) 5:489–511. doi: 10.1515/bmc-2014-0027
101. Robak P, Drozdz I, Szemraj J, Robak T. Drug resistance in multiple myeloma. Cancer Treat Rev (2018) 70:199–208. doi: 10.1016/j.ctrv.2018.09.001
102. Gourzones C, Bellanger C, Lamure S, Gadacha O, De Paco E, Vincent L, et al. Antioxidant defenses confer resistance to high dose melphalan in multiple myeloma cells. Cancers (2019) 11:439. doi: 10.3390/cancers11040439
103. Yarde DN, Oliveira V, Mathews L, Wang X, Villagra A, Boulware D, et al. Targeting the fanconi Anemia/BRCA pathway circumvents drug resistance in multiple myeloma. Cancer Res (2009) 69:9367–75. doi: 10.1158/0008-5472.CAN-09-2616
104. Xiong T, Wei H, Chen X, Xiao H. PJ34, a poly(ADP-ribose) polymerase (PARP) inhibitor, reverses melphalan-resistance and inhibits repair of DNA double-strand breaks by targeting the FA/BRCA pathway in multidrug resistant multiple myeloma cell line RPMI8226/R. Int J Oncol (2015) 46:223–32. doi: 10.3892/ijo.2014.2726
105. Patel PR, Senyuk V, Sweiss K, Calip GS, Pan D, Rodriguez N, et al. PARP inhibition synergizes with melphalan but does not reverse resistance completely. Biol Blood Marrow Transplant (2020) 26:1273–9. doi: 10.1016/j.bbmt.2020.03.008
106. Alagpulinsa DA, Yaccoby S, Ayyadevara S, Shmookler Reis RJ. A peptide nucleic acid targeting nuclear RAD51 sensitizes multiple myeloma cells to melphalan treatment. Cancer Biol Ther (2015) 16:976–86. doi: 10.1080/15384047.2015.1040951
107. Kaur E, Agrawal R, Sengupta S. Functions of BLM helicase in cells: Is it acting like a double-edged sword? Front Genet (2021) 12:634789. doi: 10.3389/fgene.2021.634789
108. Alzahrani FA, Ahmed F, Sharma M, Rehan M, Mahfuz M, Baeshen MN, et al. Investigating the pathogenic SNPs in BLM helicase and their biological consequences by computational approach. Sci Rep (2020) 10:12377. doi: 10.1038/s41598-020-69033-8
109. Turley H, Wu L, Canamero M, Gatter KC, Hickson ID. The distribution and expression of the bloom’s syndrome gene product in normal and neoplastic human cells. Br J Cancer (2001) 85:261–5. doi: 10.1054/bjoc.2001.1874
110. Nguyen GH, Tang W, Robles AI, Beyer RP, Gray LT, Welsh JA, et al. Regulation of gene expression by the BLM helicase correlates with the presence of G-quadruplex DNA motifs. Proc Natl Acad Sci USA (2014) 111:9905–10. doi: 10.1073/pnas.1404807111
111. Montenegro MM, Quaio CR, Palmeira P, Gasparini Y, Rangel-Santos A, Damasceno J, et al. Gene expression profile suggesting immunological dysregulation in two Brazilian bloom’s syndrome cases. Mol Genet Genom Med (2020) 8(4):e1133. doi: 10.1002/mgg3.1133
112. Grierson PM, Lillard K, Behbehani GK, Combs KA, Bhattacharyya S, Acharya S, et al. BLM helicase facilitates RNA polymerase I-mediated ribosomal RNA transcription. Hum Mol Genet (2012) 21:1172–83. doi: 10.1093/hmg/ddr545
113. Grierson PM, Acharya S, Groden J. Collaborating functions of BLM and DNA topoisomerase I in regulating human rDNA transcription. Mutat Res/Fundam Mol Mech Mutagen (2013) 743–744:89–96. doi: 10.1016/j.mrfmmm.2012.12.002
114. Derenzini M, Montanaro L, Trerè D. Ribosome biogenesis and cancer. Acta Histochem (2017) 119:190–7. doi: 10.1016/j.acthis.2017.01.009
115. Kong Y, Xu C, Sun X, Sun H, Zhao X, He N, et al. BLM helicase inhibition synergizes with PARP inhibition to improve the radiosensitivity of olaparib resistant non-small cell lung cancer cells by inhibiting homologous recombination repair. Cancer Biol Med (2022) 19:1150–71. doi: 10.20892/j.issn.2095-3941.2021.0178
116. Hashimoto S, Anai H, Hanada K. Mechanisms of interstrand DNA crosslink repair and human disorders. Genes Environ (2016) 38:9. doi: 10.1186/s41021-016-0037-9
117. Esma F, Salvini M, Troia R, Boccadoro M, Larocca A, Pautasso C. Melphalan hydrochloride for the treatment of multiple myeloma. Expert Opin Pharmacother (2017) 18:1127–36. doi: 10.1080/14656566.2017.1349102
118. Kee Y, D’Andrea AD. Molecular pathogenesis and clinical management of fanconi anemia. J Clin Invest (2012) 122:3799–806. doi: 10.1172/JCI58321
119. Kottemann MC, Smogorzewska A. Fanconi anaemia and the repair of Watson and crick DNA crosslinks. Nature (2013) 493:356–63. doi: 10.1038/nature11863
120. Meetei AR, Sechi S, Wallisch M, Yang D, Young MK, Joenje H, et al. A multiprotein nuclear complex connects fanconi anemia and bloom syndrome. Mol Cell Biol (2003) 23:3417–26. doi: 10.1128/MCB.23.10.3417-3426.2003
121. Wang W. Emergence of a DNA-damage response network consisting of fanconi anaemia and BRCA proteins. Nat Rev Genet (2007) 8:735–48. doi: 10.1038/nrg2159
122. Suhasini AN, Brosh RM. Fanconi anemia and bloom’s syndrome crosstalk through FANCJ–BLM helicase interaction. Trends Genet (2012) 28:7–13. doi: 10.1016/j.tig.2011.09.003
123. Deans AJ, West SC. FANCM connects the genome instability disorders bloom’s syndrome and fanconi anemia. Mol Cell (2009) 36:943–53. doi: 10.1016/j.molcel.2009.12.006
124. Panneerselvam J, Wang H, Zhang J, Che R, Yu H, Fei P. BLM promotes the activation of fanconi anemia signaling pathway. Oncotarget (2016) 7:32351–61. doi: 10.18632/oncotarget.8707
125. Budke B, Logan HL, Kalin JH, Zelivianskaia AS, Cameron McGuire W, Miller LL, et al. RI-1: a chemical inhibitor of RAD51 that disrupts homologous recombination in human cells. Nucleic Acids Res (2012) 40:7347–57. doi: 10.1093/nar/gks353
126. Aggarwal M, Sommers JA, Shoemaker RH, Brosh RM. Inhibition of helicase activity by a small molecule impairs Werner syndrome helicase (WRN) function in the cellular response to DNA damage or replication stress. Proc Natl Acad Sci USA (2011) 108:1525–30. doi: 10.1073/pnas.1006423108
127. Chan EM, Shibue T, McFarland JM, Gaeta B, Ghandi M, Dumont N, et al. WRN helicase is a synthetic lethal target in microsatellite unstable cancers. Nature (2019) 568:551–6. doi: 10.1038/s41586-019-1102-x
128. Datta A, Dhar S, Awate S, Brosh RM. Synthetic lethal interactions of RECQ helicases. Trends Cancer (2021) 7:146–61. doi: 10.1016/j.trecan.2020.09.001
129. Kategaya L, Perumal SK, Hager JH, Belmont LD. Werner Syndrome helicase is required for the survival of cancer cells with microsatellite instability. iScience (2019) 13:488–97. doi: 10.1016/j.isci.2019.02.006
130. Pearl LH, Schierz AC, Ward SE, Al-Lazikani B, Pearl FMG. Therapeutic opportunities within the DNA damage response. Nat Rev Cancer (2015) 15:166–80. doi: 10.1038/nrc3891
131. Yin Q-K, Wang C-X, Wang Y-Q, Guo Q-L, Zhang Z-L, Ou T-M, et al. Discovery of isaindigotone derivatives as novel bloom’s syndrome protein (BLM) helicase inhibitors that disrupt the BLM/DNA interactions and regulate the homologous recombination repair. J Med Chem (2019) 62:3147–62. doi: 10.1021/acs.jmedchem.9b00083
132. Wang C-X, Zhang Z-L, Yin Q-K, Tu J-L, Wang J-E, Xu Y-H, et al. Design, synthesis, and evaluation of new quinazolinone derivatives that inhibit bloom syndrome protein (BLM) helicase, trigger DNA damage at the telomere region, and synergize with PARP inhibitors. J Med Chem (2020) 63:9752–72. doi: 10.1021/acs.jmedchem.0c00917
133. Zhang W, Yang S, Liu J, Bao L, Lu H, Li H, et al. Screening antiproliferative drug for breast cancer from bisbenzylisoquinoline alkaloid tetrandrine and fangchinoline derivatives by targeting BLM helicase. BMC Cancer (2019) 19:1009. doi: 10.1186/s12885-019-6146-7
134. Chen X, Ali YI, Fisher CE, Arribas-Bosacoma R, Rajasekaran MB, Williams G, et al. Uncovering an allosteric mode of action for a selective inhibitor of human bloom syndrome protein. eLife (2021) 10:e65339. doi: 10.7554/eLife.65339
Keywords: multiple myeloma, BLM, DNA damage, replication stress, drug resistance
Citation: Ovejero S, Viziteu E, Dutrieux L, Devin J, Lin Y-L, Alaterre E, Jourdan M, Basbous J, Requirand G, Robert N, de Boussac H, Seckinger A, Hose D, Vincent L, Herbaux C, Constantinou A, Pasero P and Moreaux J (2022) The BLM helicase is a new therapeutic target in multiple myeloma involved in replication stress survival and drug resistance. Front. Immunol. 13:983181. doi: 10.3389/fimmu.2022.983181
Received: 30 June 2022; Accepted: 24 November 2022;
Published: 09 December 2022.
Edited by:
Jo Caers, University of Liège, BelgiumReviewed by:
Richard L. Frock, Stanford University, United StatesCopyright © 2022 Ovejero, Viziteu, Dutrieux, Devin, Lin, Alaterre, Jourdan, Basbous, Requirand, Robert, de Boussac, Seckinger, Hose, Vincent, Herbaux, Constantinou, Pasero and Moreaux. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jérôme Moreaux, amVyb21lLm1vcmVhdXhAaWdoLmNucnMuZnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.