 Yu-Jyun Li
Yu-Jyun Li Chi-Yuan Chen
Chi-Yuan Chen Jeng-How Yang
Jeng-How Yang Ya-Fang Chiu
Ya-Fang Chiu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 13 September 2022
Sec. Microbial Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.982264
This article is part of the Research Topic Role of Lipid Rafts in Anti-Microbial Immune Response Volume II View all 6 articles
Influenza A virus (IAV) is widely disseminated across different species and can cause recurrent epidemics and severe pandemics in humans. During infection, IAV attaches to receptors that are predominantly located in cell membrane regions known as lipid rafts, which are highly enriched in cholesterol and sphingolipids. Following IAV entry into the host cell, uncoating, transcription, and replication of the viral genome occur, after which newly synthesized viral proteins and genomes are delivered to lipid rafts for assembly prior to viral budding from the cell. Moreover, during budding, IAV acquires an envelope with embedded cholesterol from the host cell membrane, and it is known that decreased cholesterol levels on IAV virions reduce infectivity. Statins are commonly used to inhibit cholesterol synthesis for preventing cardiovascular diseases, and several studies have investigated whether such inhibition can block IAV infection and propagation, as well as modulate the host immune response to IAV. Taken together, current research suggests that there may be a role for statins in countering IAV infections and modulating the host immune response to prevent or mitigate cytokine storms, and further investigation into this is warranted.
Influenza A virus (IAV) is a member of the Orthomyxoviridae family that causes seasonal outbreaks of respiratory infections in humans and animals. Influenza infections can induce life-threatening conditions such as pneumonia, particularly in the elderly (1, 2). Although influenza vaccines are available, even under the best conditions, when circulating viruses match the viral strains used to make vaccines, vaccination only reduces the risks of illness by 40% to 60% (3). This is because IAV mutates constantly, as the RNA-dependent RNA polymerase (RdRP) used in IAV viral RNA (vRNA) replication lacks proofreading activity (4, 5). Furthermore, genome reassortment across different species also generates IAV mutants that may be able to evade immune recognition and cause severe disease in hosts (6, 7). This constant changing of the virus poses a serious challenge to influenza vaccination prevention strategies (8). As for treatment after IAV infection, most mild to moderate infections are treated with a combination of over-the-counter drugs that can include antipyretics, analgesics, decongestants, antihistamines, and antitussives, which provide relief from symptoms but do not attack IAV or interfere with its propagation in any way (9). However, for treatment of severe cases, or for treatment or prophylaxis following confirmed exposure in groups at high risk of postinfection complications, antiviral medications that target the IAV life cycle can be used (10). These antivirals are summarized in Table 1 and can be divided into five main classes: M2 protein inhibitors (amantadine and rimantadine), which disrupt the acidification of endosomes by blocking the M2 ion channel, thereby preventing the release of viral genomes to the cytoplasm (16); neuraminidase (NA) inhibitors (oseltamivir, zanamivir, laninamivir, and peramivir), which block the release of viral progeny by inhibiting NA activity (17); acidic endonuclease inhibitors (baloxavir marboxil), which inhibit the endonuclease activity required for viral gene transcription in the IAV RNA-dependent RNA polymerase (RdRP) complex (18); RNA polymerase inhibitors (favipiravir), which inhibit RdRP activity (19); and membrane fusion inhibitors (umifenovir), which block IAV from binding to and fusing with host cell membranes (15). However, new therapeutic approaches are constantly needed, as the high frequency of IAV mutation and reassortment drives resistance to treatment over time (20). One potential target is cholesterol (21, 22), which is abundantly present in areas known as lipid rafts on host cell membranes. Lipid rafts serve as hubs for cross talk and coordination of many regulatory and signaling events (23), and studies have shown that lipid rafts play important roles in the IAV life cycle; moreover, lipid rafts also serve as platforms for host immune responses. Interestingly, recent studies have shown that statins, a class of cholesterol-synthesis inhibitors, can act through cholesterol-dependent or -independent mechanisms to disrupt several stages of the IAV life cycle, as well as mediate host immune responses against IAV, and this may serve as a novel therapeutic approach to influenza treatment (21, 24, 25). In this review, the potential opportunities for statins at different stages of the IAV life cycle and host immune response are discussed, and current research is summarized to provide a comprehensive overview of the evidence surrounding the use of statins against IAV. Further research and examination of this topic is warranted, as statins do not target specific components of IAV, and mutational changes are less likely to confer resistance to treatment. This may mark an important paradigm shift in the decades-long arms race between humans and IAV.

Table 1 Antiviral drugs approved for use against influenza A viruses.
Statins, including atorvastatin, fluvastatin, lovastatin, pitavastatin, pravastatin, rosuvastatin, and simvastatin (26–31), are a class of drugs that block cholesterol synthesis through the inhibition of hydroxyl methylglutaryl-coenzyme A (HMG-CoA) reductase (26–31). Statins are now widely used in the primary and secondary prevention of cardiovascular disease (32). In addition, as the proliferation of many viruses requires cholesterol and cholesterol-rich lipid rafts on host cell membranes, the role of statins in countering viral infections has been examined in several studies (26, 28, 30, 31, 33). For instance, lovastatin has been shown to inhibit human immunodeficiency virus (HIV) entry to host cells by blocking the interaction between the cellular lymphocyte function-associated antigen-1 (LFA-1) receptor and intercellular adhesion molecule-1 (ICAM-1), which is present on the envelope of HIV viral particles. The inhibition by lovastatin decreases the propagation of HIV by 50% (28). Statins have also been reported to reduce the risk of severe COVID-19 by 70% (30). These findings suggest that statins may be useful in countering viral infections.
IAV is an enveloped virus that contains a genome consisting of eight single-stranded, negative-sense RNA segments (34). These segments encode at least 10 viral proteins, including hemagglutinin (HA); NA (35); the RdRP subunits PB1, PB2, and PA; nucleocapsid protein (NP); matrix proteins (M1 and M2); and non-structural proteins (NS1 and NS2) (1, 2, 36). The vRNA segments wrap around NPs and are bound by RdRP to form ribonucleoproteins (vRNPs), which are packaged within the virion. During infection, IAV is internalized by endocytosis through interactions between HA on the viral envelope with sialylated host receptors on the plasma membrane (37, 38). Following internalization of the virus, the low pH of the endosome environment activates M2 pH-gated proton channels on the viral envelope to acidify the viral interior, which in turn causes dissociation of the M1 matrix protein from the viral nucleoproteins (37, 38). The structure of HA is also altered inside the acidified endosomes, which leads to the fusion of the viral envelope with the endosomal membrane, followed by the release of vRNPs to the cytoplasm (37, 38).
After their release from virions, vRNPs are translocated to the nucleus, where their attached RdRP facilitate vRNA transcription and replication (36). IAV mRNA is then exported to the cytoplasm and translated into viral proteins (39). The vRNA is also used as a template for the synthesis of positive-stranded complementary RNAs (cRNAs), which are then used by RdRP as templates for vRNA replication (2, 40). The newly replicated vRNA is then packaged with NP and RdRP to form vRNPs, which are then exported to the cytoplasm through M1 and Rab-dependent recycling endosomes (37, 41, 42). HA and NA are also transported to lipid rafts, microdomains enriched with cholesterol, sphingomyelin, glycolipids, glycoproteins, and receptors on the host cell membrane, where they accumulate and facilitate viral particle assembly (Figure 1) (43–47). In the final stage, progeny viruses are assembled and released outside host cells by budding, and NA subsequently cleaves off sialic acid from the cellular receptor to prevent viral aggregation at the cell surface (37, 48, 49). Lipid rafts play an important role in the budding process, and the released virions are also coated in an envelope containing embedded cholesterol from the host cell membrane (43, 44). Given the prominent role of cholesterol and lipid rafts in the IAV life cycle, previous research has sought to identify and exploit opportunities where statins could make an impact on IAV infection and propagation.
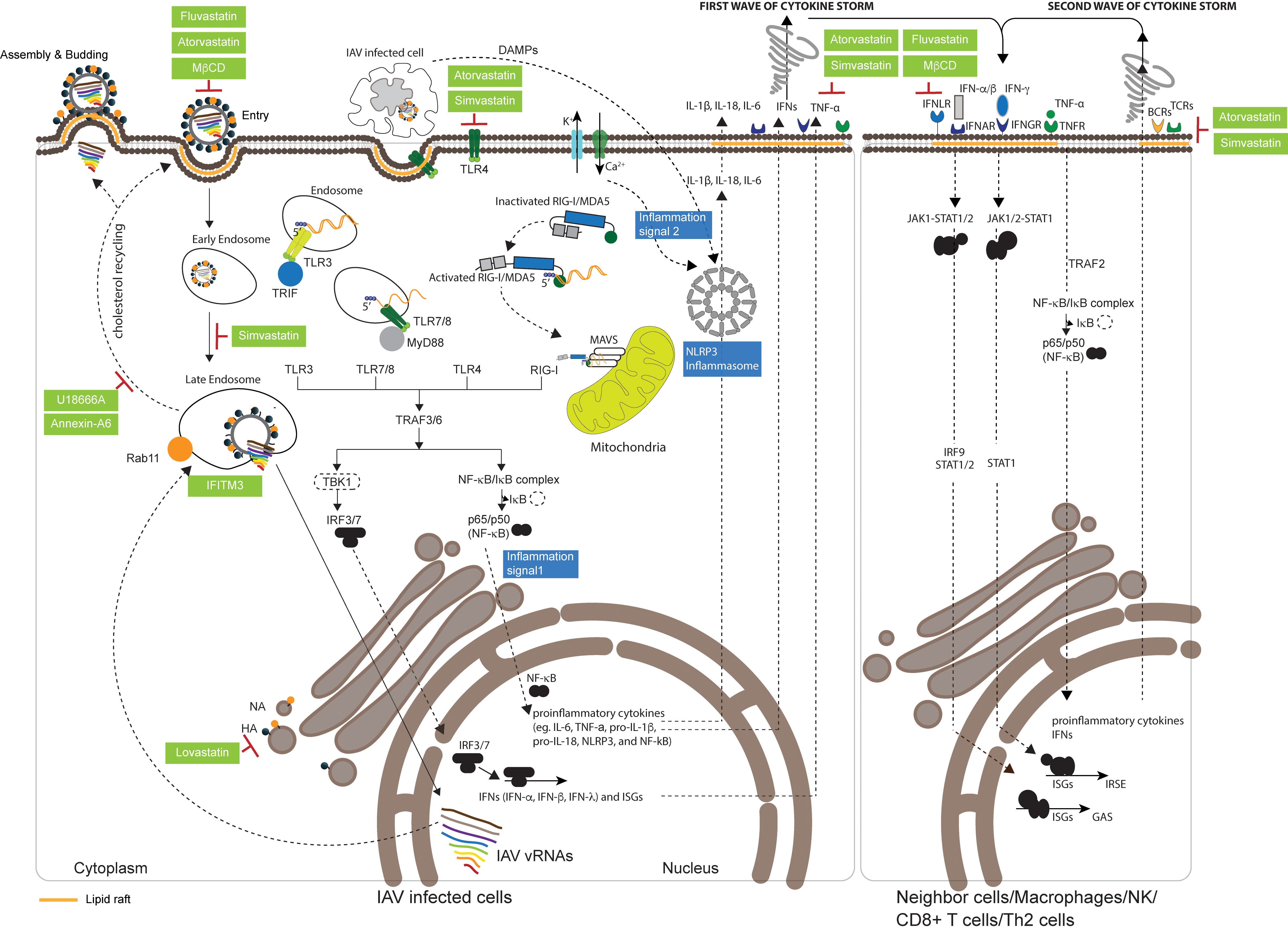
Figure 1 Effect of statins on the IAV life cycle and the host immune response. Influenza A virus (IAV) binds to sialylated host receptors at lipid rafts on the plasma membrane to initiate endocytosis for cellular entry. After internalization, acidification within the endosome causes the structural alteration of hemagglutinin (HA), which leads to the fusion of the viral envelope with the endosomal membrane and the release of viral genomes to the cytosol. Viral genomes are then transported to the nucleus for transcription and replication of viral RNA (vRNA). Several cholesterol biosynthesis regulators or inhibitors are known to be involved in the infection process: Methyl-β-cyclodextrin (MβCD) prevents IAV attachment; simvastatin reduces the efficiency of viral genome transport to the nucleus; fluvastatin and atorvastatin reduce IAV infectivity; and interferon-induced transmembrane protein 3 (IFITM3) inhibits the release of viral genomes from the endosome to the cytosol. Incoming single-stranded vRNA, double-stranded RNA intermediates that are formed during vRNA transcription and replication, and newly synthesized vRNA are recognized by immunosensors that can subsequently activate innate immunity, including toll-like receptor 3 (TLR3) and TLR7/TLR8 within the endosome, and retinoic acid-inducible gene I (RIG-I)/melanoma differentiation-associated gene 5 (MDA5), which interact with MyD88, TRIF, and mitochondrial antiviral signaling protein (MAVS) in the mitochondria. All TLRs and intracellular RIG-I initiating signals activate TANK-binding kinase 1 (TBK1) and the nuclear factor (NF)-kB/IkB complex, leading to the translocation of interferon regulatory factor-3 (IRF-3)/IRF-7 and NF-kB from the cytosol to the nucleus to induce the expression of interferons (IFNs), interferon-stimulated genes (ISGs), and proinflammatory cytokines. Following the maturation of proinflammatory cytokines induced by the nucleotide-binding oligomerization domain (NOD)-like receptor family pyrin domain (PYD)-containing 3 (NLRP3) inflammasome, inflammatory cytokines, tumor necrosis factors (TNFs), and IFNs are secreted extracellularly as the first wave of a cytokine storm. These secreted defense molecules are recognized by interferon-α receptors (IFNARs), interferon-γ receptors (IFNGRs), and TNF receptors (TNFRs) on neighboring cells, macrophages, natural killer (NK) cells, CD8+ T cells, or Th2 cells, which act to amplify innate immune signaling against IAV infection. Damage-associated molecular patterns (DAMPs) released from damaged or dying cells, including macrophages that take up the aggregated viral PB1-F2 protein, are recognized by TLR4. In addition, IAV M2 embedded in the trans-Golgi network (TGN) changes proton flux, which then activates NLRP3 complexes to induce the second wave of a cytokine storm. Atorvastatin, simvastatin, fluvastatin, and MβCD are capable of depleting or preventing the expression of IFNAR, IFNGR, and TNFR at lipid rafts, resulting in the stimulation of innate responses. T-cell receptors (TCRs) or B-cell receptors (BCRs), respectively presented at lipid rafts on T cells or B cells, are depleted by atorvastatin and simvastatin. IAV exploits the cholesterol recycling process to deliver newly synthesized vRNAs to lipid rafts on the plasma membrane for viral assembly through PB2–Rab11 interactions. U18666A and Annexin-A6, which retain and accumulate cholesterol in late endosomes to reduce the amount of cholesterol at lipid rafts, can decrease IAV production. Lovastatin, which depletes the cholesterol component on plasma membranes, prevents the trafficking of HA to the plasma membrane and alters the composition of cholesterol incorporated into viral particles.
Many viruses are known to enter host cells through endocytosis and hijack endosomes for viral trafficking (50). It is known that IAV infection is mainly mediated by the binding of HA on the virion to clusters of sialyated glycoproteins or glycolipids on the cell surface (51–53). Although several glycoproteins, including epidermal growth factor receptor (EGFR), liver/lymph node-specific intracellular adhesion molecule-2 grabbing non-integrin (L-SIGN), and dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN), have been shown to facilitate IAV attachment on cell surface for entry, whether these molecules are specific receptors for IAV uptake remain to be elucidated (54, 55). For example, a recent study has shown that IAV does not directly bind to EGFR, but the binding of IAV with multivalent sialic acid clusters can trigger EGFR activation (56). However, the glycoprotein carcinoembryonic antigen-related cell adhesion molecule 6 (CD66c or CEACAM6) has recently been identified as a receptor for IAV infection (57), and further research will undoubtedly identify more such interacting receptors in the future. After binding to cellular receptors, IAV is taken up by cells via either clathrin- or caveolin-dependent endocytosis, which usually occurs at lipid rafts (Figure 1) (50, 58). Lipid rafts serve as a platform for the cross talk and coordination of many regulatory proteins and signaling molecules, and cholesterol plays a key role in lipid raft structure and function. Several studies have shown that depletion of cholesterol from lipid rafts or viral envelopes with methyl-β-cyclodextrin (MβCD) prevents IAV attachment and reduces IAV infectivity; however, IAV infectivity is restored after exogenous cholesterol supplementation (Figure 1, Table 2) (55, 59–61). Moreover, sphingomyelin is known to coexist and interact with cholesterol on the plasma membrane (73), and disruption of sphingomyelin on lipid rafts with sphingomyelinases (SMase) also prevents IAV attachment and viral internalization (74, 75).

Table 2 Effects of molecules that deplete cholesterol, prevent cholesterol biosynthesis, or inhibit cholesterol trafficking, and ISGs on IAV infection.
In addition, cholesterol is known to modulate IAV trafficking, and depletion of cholesterol disrupts IAV transport after cellular entry (60, 62, 64, 76). Mehrbod et al. (62) showed that simvastatin prevents RhoA prenylation, which is key to arrangement of the actin cytoskeleton for endosome trafficking, and results in inefficient transport of viral genomes into the nucleus for replication (Figure 1). Fluvastatin and atorvastatin treatment also reduces vRNA replication and viral protein synthesis (Figure 1) (63, 64). These findings show that cholesterol is critical to IAV binding and entry to host cells, and preliminary studies show that statins may be able to disrupt this process and prevent IAV proliferation (63, 64).
Following their glycosylation in the trans-Golgi network (TGN), HA and NA are transported to and accumulate at lipid rafts (Figure 1) (77, 78). An electron microscopy study showed that the cytoplasmic tails of HA and NA interact with the glycosphingolipid GM1 at lipid rafts, forming a structure called the budozone, where IAV budding occurs (44) (Figure 1). Substituting the amino acids in HA that are required for interaction with GM1 not only changes the amounts of HA on the viral particles but also reduces the number of IAV particles budding through lipid rafts (43).
Cholesterol is an essential component of lipid rafts (60), and the availability of cholesterol in lipid rafts has been shown to be critical to IAV replication. It has been shown that IAV production is significantly reduced after treatment with a cholesterol transport inhibitor, U18666A (Figure 1), and treatment of IAV-infected cells with Annexin A6, which causes retention and accumulation of cholesterol in the late endosomes to reduce cholesterol levels at lipid rafts, similarly reduces IAV production (Figure 1; Table 2) (65–67). During budding, cholesterol is also incorporated into the IAV envelope, constituting up to 44% of lipids, or 12% of the total mass of an IAV virion (60). Previous studies have shown that using lovastatin to reduce intracellular cholesterol and deplete lipid rafts not only influences the trafficking of HA to the plasma membrane but also alters the composition of cholesterol incorporated into the viral particles (Figure 1) (21, 25, 47).
Cholesterol in the endosomal membrane is acquired from the plasma membrane during endocytosis (79), and this endosomal cholesterol is recycled back to the plasma membrane by Rab11 (Figure 1) (80). IAV appears to exploit this cholesterol-recycling process to deliver vRNPs to lipid rafts on the plasma membrane for viral assembly, as PB2, a component of vRNP, has been shown to interact with the Rab11-cholesterol complex (Figure 1; Table 2) (68, 69). Furthermore, knockdown in the expression of Rab11 by shRNA, or expression of a mutant Rab11 that decreases the efficiency of cholesterol recycling back to the plasma membrane, was shown to reduce the amount of vRNPs conveyed to lipid rafts (Figure 1; Table 2) (68, 69). This indicates the importance of the cholesterol-recycling process to IAV maturation.
Many immunosensors, including toll-like receptors (TLRs) and c-type lectin receptors (CLRs), which are known as pattern recognition receptors (PRRs; 81), are localized at lipid rafts, where they detect viral infection and trigger defense machinery for viral clearance (Figure 1) (82, 83). In addition to PRRs on the plasma membrane, there are intracellular PRRs, such as RIG-I-like receptors (RLRs), which detect intracellular pathogens (84). After IAV infection, host cells utilize both types of PRRs to recognize either viral proteins or genomes and activate the immune response accordingly to defend against invasion (85, 86). Considering that the depletion of lipid rafts will not only decrease IAV production but also may affect immune responses against IAV infection (64, 87, 88), it is important to consider the role of statins in this context as well. Therefore, in this section, the main types of PRRs involved in IAV infection will be presented, and the impact of statins on PRRs and PRR-mediated immune responses will be discussed.
At least 10 different types of TLRs have been identified to date, all of which are capable of recognizing either extracellular or intracellular pathogen‐associated molecular patterns to trigger the secretion of proinflammatory cytokines and interferons (89–92). TLR1, TLR2, TLR4, TLR5, and TLR6 are present in lipid rafts on the cell surface, and their signaling is triggered by extracellular microorganisms and ligands (93–95). TLR3, TLR7, TLR8, and TLR9 are present on the cytosolic organelles and engage with either foreign ligands that are imported across plasma membrane barriers, or newly synthesized pathogenic components (85, 96, 97). However, some TLRs are found to shuttle between the plasma membrane and the cytosolic organelles to recognize incoming pathogens (95). For instance, TLR3 and TLR9 are transported to the cell surface to recognize extracellular pathogens, while cytosolic forms of TLR2 and TLR4 are found in the cytoplasm, TGN, endoplasmic reticulum (ER), or nucleolus, where they are known to engage the HA of measles virus, the glycoprotein B (gB) of herpes simplex virus (HSV), the F protein of respiratory syncytial virus (RSV), or the envelope protein of mouse mammary tumor virus (MMTV) (97–99).
After internalization of IAV through endocytosis, vRNPs are released to the cytosol following the acidification of endosomes and fusion of the viral envelope with endosomal membranes (100). The IAV genome is recognized by several TLRs, including TLR3, TLR7, and TLR8 (Table 3) (85, 96, 108), among which endosomal TLR7 and TLR8 serve as the first immunosensors for incoming vRNPs (96). TLR7 recruits MyD88 to induce proinflammatory cytokines and chemokines such as tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), and IL-1β and can further promote type I interferon release, dendritic cell (DC) maturation, and antiviral immunity (Figure 1) (109, 110). In addition, TLR3 senses IAV double-stranded RNA (dsRNA) intermediates formed during IAV transcription and vRNA replication within the endosome, and binding of the dsRNA to TLR3 triggers the expression of IFN-β and proinflammatory cytokines (Figure 1) (85). As TLR3-defective mice produce significantly few cytokines after IAV infection, TLR3 is believed to play a critical role in IAV clearance (111). In contrast to TLR7 and TLR3, TLR4 is present on the cell surface and detects IAV infection by recognizing S100A9, which is a damage-associated molecular pattern (DAMP) secreted by macrophages after IAV infection (Figure 1) (108). Preliminary evidence shows that depletion of TLR4 from lipid rafts by atorvastatin and simvastatin may reduce IAV clearance (Figure 1; Table 3) (101, 102), although further research is needed to better understand the extent of such effects.
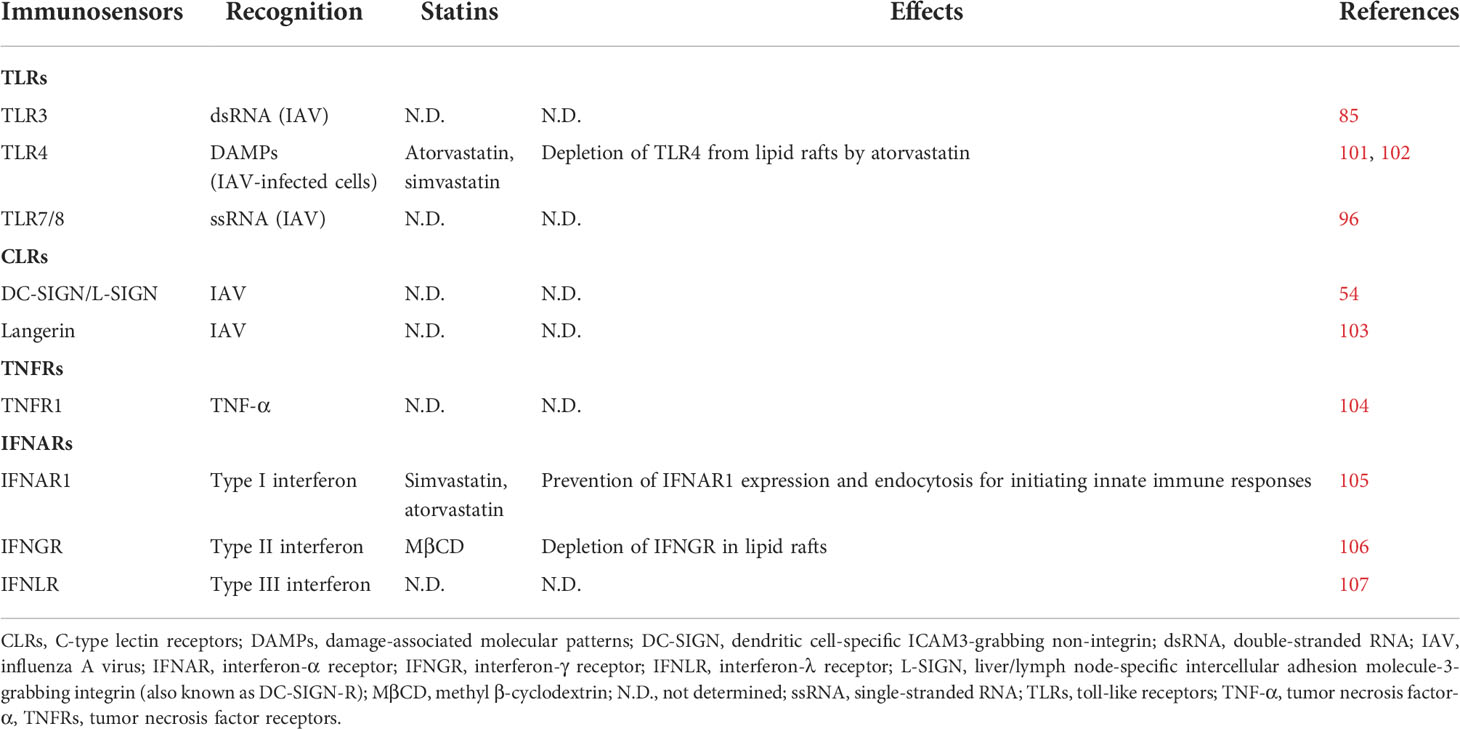
Table 3 Immunosensors and receptors embedded in lipid rafts for IAV recognition.
Another group of immunosensors in lipid rafts that are known to influence IAV infection are CLRs, which are transmembrane glycoproteins expressed by monocytes, macrophages, DCs, and Langerhans cells (LCs; 112–114). CLRs are recruited to lipid rafts and function as PRRs against the glycans of glycoproteins on pathogens (115). CLRs have been found to be important for IAV infection (54, 103), and several studies have shown that CLRs such as DC-SIGN, L-SIGN (also known as DC-SIGN-R), and langerin can facilitate IAV infection (Table 3) (54, 103). Londrigan et al. (54) showed that IAV is recognized and internalized through DC-SIGN and L-SIGN on Lec2 Chinese hamster ovary cells, which lack sialic acid-modified glycoproteins that are usually recognized by IAV and mediate infection. Moreover, Ng et al. (103) showed that the internalization of IAV is mediated through the binding of HA to langerin, suggesting that CLRs can function as receptors for IAV infection. The effect of statins on CLRs remains unclear, and further research is needed to ascertain if statins can interact with CLRs and disrupt their facilitating effect on IAV infection.
In addition to PRRs that are embedded on the cell surface, many intracellular soluble PRRs such as NOD-like receptors (NLRs) recognize IAV to trigger innate immunity (81, 116, 117). At the early stage of IAV infection, proteins encoded by interferon-stimulated genes (ISGs), such as serine/threonine kinase protein kinase R (PKR), trigger the formation of stress granules when newly synthesized uncapped vRNA and retinoic acid-inducible gene-I (RIG-I) are recruited (118). Following the recognition of uncapped vRNA, RIG-I is activated and subsequently induces the polymerization of mitochondrial antiviral signaling protein (MAVS) on the outer mitochondrial membrane (Figure 1) (119). As MAVS preferentially oligomerizes at the sites of mitochondria with high cholesterol content (120), this oligomerization may be modulated by cholesterol as well. RIG-I-MAVS signaling leads to either the activation of nuclear factor-κB (NF-κB) through TNF receptor-associated factor (TRAF)-3, TRAF-6, and receptor-interacting protein 1 (RIP-1) or the phosphorylation and activation of interferon regulator factor (IRF)-3 and IRF-7 via TRAF3 (121–124). Activated IRF-3/IRF-7 and NF-κB subsequently translocate to the nucleus to activate the expression of IFNs, ISGs, and proinflammatory genes, including those encoding IL-1β and IL-18 (Figure 1) (117, 123, 125, 126). Following secretion, IFNs bind to the receptors on the cell surface of IAV-infected cells or their neighbor cells to activate the JAK-STAT pathway, which induces the expression of ISGs to act against IAV infection (Figure 1) (127, 128). Similar to RIG-I, melanoma differentiation-associated gene 5 (MDA5) is also activated by IAV dsRNA intermediates and then recruited to the outer mitochondrial membrane to trigger the IRF-3/IRF-7 and NF-κB signaling pathway, which in turn promotes the expression of IFN, ISGs, and proinflammatory genes (Figure 1) (129–133).
Similarly, two major NOD-like receptor (NLR) molecules, nucleotide-binding oligomerization domain 2 (NOD2) and NOD-, leucine-rich repeat (LRR)-, and pyrin domain-containing protein 3 (NLRP3), serve as PRRs upon IAV infection (134, 135). NOD2 recognizes IAV single-stranded RNA (ssRNA) and triggers the activation and translocation of IRF-3/IRF-7 and MAPK, by respectively recruiting the adaptor proteins, MAVS and RIPK2, to induce IFN-α/β and proinflammatory cytokine production (135); NLRP3 is also a critical component of the inflammasome, which induces the secretion of IL-1β and IL-18 and triggers a cytokine storm upon IAV infection (Figure 1) (134, 136).
Recognition of incoming or newly synthesized IAV genomes by either transmembrane or intracellular PRRs activates innate immunity and initiates the expression of first-wave IFNs, ISGs, chemokines, and proinflammatory cytokines, which stimulate immune cell infiltration to activate the expression of second-wave cytokines for IAV clearance. However, innate immunity may cause uncontrolled and excessive release of inflammatory cytokines to result in acute respiratory distress syndrome (ARDS), also known as a cytokine storm (Figure 1) (137) (Please also see the section, “IAV-induced inflammation and cytokine storms”). Lipophilic statins have been reported to exert a number of pleiotropic effects on the NLRP3 complex, acting to reduce inflammatory activity (138). This may have an effect in preventing or mitigating cytokine storms, which are a major cause of morbidity and mortality in severe influenza infections (139).
Immunosensors stimulated by infection trigger signaling to activate the expression of IFNs, TNFs, cytokines, and ISGs (140–143). These defense molecules then interact with immunoreceptors, including TNF-α receptors (TNFRs) and interferon receptors (IFNRs), to induce immunosignaling cascades against pathogens (127, 144).
TNF-α is a proinflammatory cytokine that is upregulated after IAV infection (145) and is secreted from infected cells to trigger warning signals in neighboring cells via binding to TNFR1 or TNFR2 on cell surfaces (Figure 1; Table 3) (146). The secreted TNF-α also attracts immune cells and stimulates infiltration (147). In addition, release of TNF-α from vesicles through fusion with the plasma membrane is mediated by the N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex, which is enriched at lipid rafts (148). MβCD treatment to deplete cholesterol at lipid rafts reduced the secretion of TNF-α, and Legler et al. (104) further showed that TNFR1 translocates to lipid rafts, where it associates with the serine/threonine kinase RIP, TRADD, and TRAF2 as a signaling complex. Depletion of lipid rafts by MβCD abrogates TNF-α-mediated NF-κB activation, suggesting that TNFR1 assembly at lipid rafts is essential for NF-κB activation during IAV infection. Embedded TNFR2 in the lipid rafts of CD8+ T cells is essential for interaction with TNF-α and induction of the immune response against IAV infection; however, excessive TNFR2 expression can lead to cytokine storms that may cause severe and lethal lung injury (149).
Three types of interferon receptors, IFN-α receptors (IFNARs), IFN-γ receptors (IFNGRs), and INF-λ receptors (INFLRs), can recognize their respective IFNs when these are released from immune cells or pathogen-infected cells (Figure 1; Table 3) (150–152). IFN-α/β is expressed in immune cells, including macrophages, alveolar cells, DCs, and inflammatory monocytes, while IFN-γ is expressed by NK cells and cytotoxic T cells (CTLs) (153–155). This recognition process triggers immunopathology during a cytokine storm. IFNLRs are present on mucosal epithelial cells and recognize IFN-λ, which is typically released from myeloid cells, epithelial cells, and DCs (Figure 1) (107). Reduction of cholesterol with MβCD is known to disrupt localization and assembly of IFNGR at lipid rafts (106), while depletion of cholesterol by simvastatin and atorvastatin prevents IFNAR1 expression and endocytosis (Figure 1; Table 3) (105).
During IAV infection, IFNs are produced and released from IAV-infected cells after recognizing IAV ssRNA or dsRNA (85, 109), and when these IFNs bind with their respective receptors, both IFNARs and IFNGRs are recruited to lipid rafts and internalized through endocytosis (Figure 1) (156). This interaction induces the recruitment and phosphorylation of the JAK-STAT and tyrosine kinase 2 (TYK2) pathways (150, 157). Following the recruitment and autophosphorylation of STAT1/2 (158), phosphorylated STAT1/2 forms a transcription factor complex with IRF-9, termed IFN-stimulated gene factor 3 (ISGF3; 159), which translocates from the cytosol to the nucleus and binds to IFN-stimulated response elements (ISREs) in the ISG promoters to initiate the transcription of genes against viral infection (160, 161). Among ISGs activated by viruses, IFN-induced transmembrane protein 3 (IFITM3) has been reported to restrict the replication of dengue virus, West Nile virus, coronavirus, and IAV (72, 162). In the early stages of IAV infection, IFITM3 is upregulated after activation by IFN signaling (Figure 1) (Table 2). IFITM3 prevents the transport of cholesterol from ER to late endosomes, thus affecting fusion with the IAV envelope, and also blocks the formation of fusion pores to disrupt the release of vRNPs (Figure 1; Table 2) (65, 70–72). Several recent studies have shown that statins can inhibit IFN signaling and activity (87, 105), likely through the inhibition of IRF-3 and JAK/STAT signaling in macrophages (105), and the impact of this on the host antiviral response is worthy of further investigation. Interestingly, a study of gammaherpesvirus infection showed that type I interferon counters the antiviral effects of statins derived through the reduction of cholesterol, and therefore the reported inhibition of IFN activity by statins may be expected to enhance their cholesterol-dependent antiviral activity (163).
Although innate immune responses are known to limit IAV replication and transmission (86, 109, 145, 164), IAV clearance requires substantial activation, clonal expansion, recruitment, and acquisition of effector immune cells at the respiratory tract, as part of the adaptive immune response (165–167). However, activated adaptive immunity can lead to excessive inflammatory responses that are prone to induce cytokine storms and cause severe or fatal lung injury (168, 169). Therefore, a well-controlled adaptive immune response is essential to avoid triggering cytokine storms (170, 171).
After IAV infection, B-cell receptors (BCRs) on mature B cells interact with antigens presented on antigen-presenting cells (APCs) and then translocate to lipid rafts, where they recruit co-stimulatory factors to trigger downstream activating signals (Figure 1) (172–174). Antigens recognized by BCR are then internalized through BCR-mediated endocytosis and processed within major histocompatibility complex class II (MHCII)-containing lysosomes; ultimately, the processed antigens are presented on the cell surface (172–174). The presented antigens on the MHCII of B cells are recognized by T-cell receptors (TCRs) of CD4+ T cells, leading to expression of the surface protein CD40L, as well as the cytokines IL-4 and IL-21, to enable activation of B cells via the interaction with CD40 and cytokine receptors on B cells (175, 176). In this way, B cells present antigens to stimulate CD4+ T cells, and this in turn enables the activation of B cells and the synthesis of antigen-specific antibodies (177, 178). In the case of IAV infection, the assistance from CD4+ T cells enables B cells to mature as plasmablasts (PBs) at germinal centers (GCs), and the matured B cells then produce antibodies targeting the surface HA or NA on IAV virions, thereby preventing IAV infection or egress (179–181). Mature B cells also produce anti-M2 antibodies to prevent IAV production (182). Moreover, secreted antibodies against IAV can also serve to mediate antibody-dependent cell-mediated cytotoxicity (ADCC) through NK cells, macrophages, γδ T cells, and leukocytes (183, 184).
In addition to antibodies generated by B cells, T cells can also play a critical role in preventing IAV infection (165). T cells differentiate in the thymus into CD4+ T and CD8+ cells (185), which subsequently differentiate further into cytotoxic T cells (CTLs) after recognizing IAV-associated antigens presented on major histocompatibility complex class I (MHCI) molecules on DCs (185). Activated CTLs recognize IAV-infected cells, and in response, they produce cytokines (TNF-α/β, IFN-γ, and IL-2) and cytotoxic granules containing granzymes and perforin, which induce the formation of pores on CTL-targeted cells and restrict IAV replication (186). CTLs also induce apoptosis of IAV-infected cells by delivering granzymes through perforin-mediated pores and secreting cytokines such as TNF, Fas ligand (FasL), and TNF-related apoptosis-inducing ligand (TRAIL) to recruit death receptors (187, 188); however, CTL infiltration in respiratory tracts often causes excessive production of proinflammatory cytokines by respiratory cells or immune cells recruited to the airways, and severe injury of lung tissues may follow as a result (168, 189). IFN-γ and TNF-α secreted by CD8+ T cells are known to enhance the release of lung epithelial chemokines, which promote inflammatory cell infiltration, lung pathogenic injury, and apoptosis of IAV-infected or non-infected lung epithelial cells, thereby raising the risk of severe cytokine storms (168, 189, 190). It has been shown that treatment with anti-IFN-γ significantly reduced lung pathology, inflammatory cell infiltration, and mortality of mouse models infected by IAV, indicating that IFN-γ is a key molecule involved in the development of a cytokine storm. Statins are known to inhibit the production of several inflammatory cytokines (87, 102, 105) and may serve to modulate the cytotoxic activity of CD8+ T cells during IAV clearance. Unlike CD8+ T cells, CD4+ T cells are activated by antigens presented on MHCII molecules, followed by the binding of CD40L to CD40 on APCs such as DCs (191). Activated CD4+ T cells facilitate B-cell activation and antibody production (192) and can also differentiate into various subtypes, depending on the co-stimulatory cytokines received from the microenvironment (193–196).
Similar to BCRs, TCRs are located at non-raft regions during the resting state but are translocated to lipid rafts following T-cell activation (Figure 1) (197, 198). After TCRs recognize antigens presented on MHCs present on APCs, TCRs interact with the CD4- or CD8-lymphocyte-specific protein tyrosine kinase complex (199) to initiate the T-cell activation signaling cascade (200, 201). Failure of the Src-family kinase Lck to localize to lipid rafts (202), depletion of lipid rafts by atorvastatin (203), or reduction of intracellular cholesterol by simvastatin and atorvastatin (204) can abort the activation of adaptive immune responses. During the activation of T and B cells, several co-stimulatory factors, including CD40, CD83, and CD86, also localize to lipid rafts and are required for T-cell and B-cell activation (205, 206). Shimabukuro-Vornhagen et al. (204) showed that simvastatin and atorvastatin inhibit B-cell activation and proliferation by downregulating the expression of CD40 and other co-stimulatory factors such as CD80 and CD86, and MHCII, in a dose-dependent manner. Additionally, statins can reduce the expression of CD40, CD83, and CD86, as well as the secretion of IL-6, IL-8, IL-12, and TNF-α by DC, resulting in the inhibition of DC-induced T-cell proliferation and activation (207). These studies show that lipid rafts serve as a platform to regulate adaptive immune responses (173, 197, 206, 208), and the modulation of lipid rafts with statins may represent a promising approach to manage the adaptive immune response to IAV infection.
Inflammation is an innate immune response that protects cells from IAV infection (134, 209). Inflammasome formation is tightly regulated by two sequential signals, a priming signal and an activating signal (210–214). Inflammasome components (e.g., NLRP3) and proinflammatory cytokines are upregulated by NF-κB signaling via TLRs, RLRs, TNFR1, and IL-1 receptors, which serve as a priming signal (Figure 1) (210, 215, 216). DAMPs released from damaged or dying cells infected by IAV are then sensed by NLRP3, and this triggers the activating signal (Figure 1) (108, 134). A variety of molecules can serve as DAMPs, including IAV ssRNAs that stimulate the release of IL-1β, IAV M2 embedded in the TGN that can alter proton flux, and aggregated PB1-F2 derived from dying infected cells that are taken up by macrophages and transported to lysosomes (136, 217). Activated NLRP3 recruits an adaptor, ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain (CARD), also known as PYCARD), along with caspase 1, to activate inflammatory caspases and promote the maturation of IL-1β and IL-18 (Figure 1); this process also stimulates pyroptosis, which is a rapid, inflammatory form of lytic programmed cell death induced after infections (217–221).
Following internalization of IAV by host cells, the virus is recognized by TLRs, CLRs, or RLRs, which initiate the innate immune response within infected cells to release cytokines (81). These cytokines are in turn recognized by NK cells, CD8+ T cells, Th2 cells, macrophages, and neutrophils, resulting in the stimulation and production of a second wave of cytokines, which ultimately enhance the release of lung epithelial chemokines (168, 222–225) (Please also see the section, “Lipid rafts serve as a platform for the host adaptive immune response against IAV”). The chemokines aggravate apoptosis of lung cells but can also increase inflammatory cell infiltration to promote IAV clearance (154, 226). However, when the production of proinflammatory cytokines, including TNF-α, IFN-α/β, IL-6, and IL-1β, becomes excessive and spirals out of control, or if anti-inflammatory factors fail to curb the growing inflammatory response, a cytokine storm develops (Figure 1). The condition often causes severe or fatal lung injury (168, 169, 227, 228). Therefore, cytokine dysregulation is regarded as a major pathophysiological mechanism in IAV infection, with potentially fatal consequences, as seen in the 1997 H5N1 Hong Kong avian influenza epidemic and the 1918 influenza pandemic (229, 230). Inhibition of the excessive inflammatory responses driven by a cytokine storm is considered to be an effective approach in preventing fatal IAV infections (231, 232). Several therapies, including the TNF-α inhibitor etanercept, the sphingosine analog AAL-R, and tyrosine kinase inhibitors such as ponatinib, have been assessed for efficacy in blocking IAV-induced cytokine storms (233–237). Statins have also been shown to inhibit the production of inflammatory cytokines, including TNF-α, IFN-γ, and IL-6 or IL-8 (62, 64), and these effects may have utility in preventing cytokine storms and fatal IAV infections (Figure 1) (234).
Statins have been shown to prevent IAV propagation and transmission in cell culture and animal studies (62, 63, 209), suggesting that there may be a role for statins in the treatment of IAV infection. Several studies show benefits of statin use during IAV infection, including a 40% reduction in the risks of pneumonia death caused by IAV infection (238), reduction of fatal IAV infection cases in hospitalized IAV-infected inpatients during the 2007–2008 epidemic season (239), and reduced risk of developing influenza-associated pneumonia in patients regularly taking statins to prevent cardiovascular disease (240). This was also noted in a study by Brassard et al. (241), who analyzed the records of approximately 10,000 patients in the UK Clinical Practice Research Datalink and found that regular use of statins significantly lowered hospitalization and mortality rates during IAV infection. Recently, a meta-analysis on statin efficacy in IAV infection also found that the use of statins significantly reduced influenza prevalence among both flu-vaccinated and unvaccinated subjects and was associated with significantly reduced mortality after IAV infection, including both 30- and 90-day mortality after diagnosis of infection (242). This suggests that statins can indeed have a positive clinical impact on both preventing IAV infection and mitigating the severity of disease after infection.
However, many other studies have failed to confirm that statins provide substantial protection from IAV infection. In a retrospective cohort study that examined patients from administrative healthcare databases in Ontario from a 10-year period (1996 to 2006), statins were found to provide slight protective effects against IAV-induced pneumonia hospitalization, 30-day pneumonia mortality, and all-cause mortality among approximately 2 million people aged 65 and older (243). A single-center retrospective study investigating statin uses and outcome in hospitalized patients during the 2009 influenza pandemic found that the use of statins lowered the number of cases with severe or lethal lung injury, but the benefits of statin treatment on the reduction of fatal infections was not statistically significant (244). Izurieta et al. (245) analyzed about 1,400,000 patients prescribed with statin treatment but did not find any benefits regarding IAV infection. Cutrell et al. (246) also did not find any positive correlation between statin usage and the reduction of acute illness caused by IAV infection. Similarly, Brett et al. (247) found that statins had no effect on the reduction of severe illness caused by IAV infection.
In light of the complex factors involved in these observational clinical studies, Izurieta et al. (245) studied whether statins could be used against IAV infection in mice under well-controlled etiology and pathology conditions. However, the study showed that statins provided only marginal inhibitory effects on protection from IAV infection. In a similar mouse model system, Belser et al. (248) showed that simvastatin reduced levels of IFN-γ, IL-10, and TNF-α, all cytokines known to be involved in lung infiltration, but the survival rate of the mice did not increase after infection. In a similar study, Radigan et al. (249) found that rosuvastatin did not increase the survival of infected experimental animals. The lack of increase in survival rates following treatment might be due to the use of high titers of IAV in these studies, which caused rapid death in the animal models studied and may not have allowed sufficient exploration of the benefits of statins against IAV infection.
The inconsistency of clinical benefits for statin use in the treatment of severe influenza infection may be multifactorial, being partly due to the variation in timing and duration of statin administration between participants, and partly due to the differences in pathogenic mechanism(s) of pneumonia induced by viral, bacterial, or other pathogens, course of acute respiratory infection, vaccination against IAV infection, and other risk factors, such as chronic or cardiovascular diseases (241). Moreover, statins may also affect innate and adaptive immunity (102, 203), and thus any antiviral benefit from statin treatment may be offset by these effects on the innate immune response. Therefore, comprehensive observational studies on individuals who do not regularly use cardioprotective statins or immunomodulatory agents should be conducted, to provide better information on the therapeutic potential of statins. Interestingly, Karlsson et al. (250) found that simvastatin reduced symptoms of IAV-induced pneumococcal pneumonia in obese mice, suggesting that lipid metabolic status may influence the protective capability of statins in IAV-associated respiratory diseases.
From another perspective, the triggering of cytokine storms can be influenced by age, gender, and pregnancy (251). The risk of developing secondary bacterial infections after IAV infection ranges from 2% to 65% (252) and is closely associated with obesity, as excess lipids in obese individuals can increase the number of leukocytes and monocytes in the blood, upregulate activating interactions between B cells and T cells, and raise the number of Th1 and Th17 cells (253). These changes often lead to chronic cell infiltration and inflammation, which can heighten the risk of cytokine storms during IAV infection (254). A phase 2 clinical trial (ClinicalTrials.gov Identifier: NCT02056340) conducted from October 2013 to June 2018 at Beth Israel Deaconess Medical Center showed that atorvastatin treatment of inpatients diagnosed with IAV infection but without statin pretreatment or liver- or cardiovascular-associated diseases significantly reduced levels of the inflammatory cytokine IL-6, which may help to prevent the occurrence of cytokine storms (30). These promising findings offer hope regarding the use of statins to prevent excess mortality in IAV pandemics.
There is no solid clinical evidence to support the benefits of treating severe influenza illness with statins as yet, but data from some observational cohorts suggest that statin therapy is associated with a reduction in poor outcomes and mortality. The efficacy of statins in influenza management should be examined in larger double-blind, placebo-controlled, and randomized trials for hospitalized statin-naïve patients with IAV infection, and the metabolic status of patients should be taken into account as a key variable in future studies. Clinicians should also be mindful of the effects on immunity when weighing the benefits and risks of prescribing statins to patients.
The original contributions presented in the study are included in the article/supplementary materials. Further inquiries can be directed to the corresponding author/s.
Conception and design: All authors. Literature analysis and interpretation: All authors. Writing the manuscript: All authors, and Y-JL and C-YC contributed equally to this work. Final approval: All authors. All authors contributed to the article and approved the submitted version.
This work was financially supported by the Ministry of Science and Technology (MOST), Taiwan (MOST 109-2320-B-182-028-MY3); the Chang Gung Medical Research Program (CMRPD1K0321 and CMRPD1K0322); Chang Gung Memorial Hospital, Linkou (BMRPF14); and the Research Center for Emerging Viral Infections from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) and MOST in Taiwan (MOST 110-2634-F-182-001, MOST 109-2327-B-182-002).
We thank Dr. Chih-Ho Lai for his insights and suggestions.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Ferhadian D, Contrant M, Printz-Schweigert A, Smyth RP, Paillart J-C, Marquet R. Structural and functional motifs in influenza virus RNAs. Front Microbiol (2018) 9:559. doi: 10.3389/fmicb.2018.00559
2. Matsuoka Y, Matsumae H, Katoh M, Eisfeld AJ, Neumann G, Hase T, et al. A comprehensive map of the influenza a virus replication cycle. BMC Syst Biol (2013) 7:1–18. doi: 10.1186/1752-0509-7-97
3. Jefferson T, Rivetti D, Rivetti A, Rudin M, Di Pietrantonj C, Demicheli V. Efficacy and effectiveness of influenza vaccines in elderly people: A systematic review. Lancet (2005) 366:1165–74. doi: 10.1016/S0140-6736(05)67339-4
4. Drake JW. Rates of spontaneous mutation among RNA viruses. Proc Natl Acad Sci U. S. A. (1993) 90:4171–5.
5. Penhoet E, Miller H, Doyle M, Blatti S. RNA-Dependent RNA polymerase activity in influenza virions. Proc Natl Acad Sci U. S. A. (1971) 68:1369–71. doi: 10.1073/pnas.68.6.1369
6. Pleschka S. Overview of influenza viruses. Curr Top Microbiol Immunol (2013) 370:1–20. doi: 10.1007/82_2012_272
7. Zambon MC. Epidemiology and pathogenesis of influenza. J Antimicrob Chemother (1999) 44 Suppl B:3–9. doi: 10.1093/jac/44.suppl_2.3
8. Carrat F, Flahault A. Influenza vaccine: The challenge of antigenic drift. Vaccine (2007) 25:6852–62. doi: 10.1016/j.vaccine.2007.07.027
9. George BO, Etzel JV, Ambizas EM. The common cold: A review of OTC options. U. S. Pharm (2018) 43:6–10. Available at: https://www.uspharmacist.com/article/the-common-cold-a-review-of-otc-options
10. Norikoshi Y, Ikeda T, Sasahara K, Hamada M, Torigoe E, Nagae M, et al. A comparison of the frequency of prescription and pharmacy revisits between baloxavir marboxil and neuraminidase inhibitors in influenza-infected pediatric patients during the 2019–2020 influenza season. Biol Pharm Bull (2020) 43:1960–5. doi: 10.1248/bpb.b20-00543
11. Hayden FG, Gwaltney JJM, Van de Castle RL, Adams KF, Giordani B. Comparative toxicity of amantadine hydrochloride and rimantadine hydrochloride in healthy adults. Antimicrob Agents Chemother (1981) 19:226–33. doi: 10.1128/AAC.19.2.226
12. Toovey S, Prinssen EP, Rayner CR, Thakrar BT, Dutkowski R, Koerner A, et al. Post-marketing assessment of neuropsychiatric adverse events in influenza patients treated with oseltamivir: An updated review. Adv Ther (2012) 29:826–48. doi: 10.1007/s12325-012-0050-8
13. Freund B, Gravenstein S, Elliott M, Miller I. Zanamivir: A review of clinical safety. Drug Saf. (1999) 21:267–81. doi: 10.2165/00002018-199921040-00003
14. Ergur FO, Yildiz M, Sener MU, Kavurgaci S, Ozturk A. Adverse effects associated with favipiravir in patients with COVID-19 pneumonia: A retrospective study. Sao Paulo Med J (2022) 140:372–7. doi: 10.1590/1516-3180.2021.0489.r1.13082021
15. Haviernik J, Štefánik M, Fojtíková M, Kali S, Tordo N, Rudolf I, et al. Arbidol (Umifenovir): A broad-spectrum antiviral drug that inhibits medically important arthropod-borne flaviviruses. Viruses (2018) 10:184. doi: 10.3390/v10040184
16. Beigel J, Bray M. Current and future antiviral therapy of severe seasonal and avian influenza. Antiviral Res (2008) 78:91–102. doi: 10.1016/j.antiviral.2008.01.003
17. De Clercq E. Antiviral agents active against influenza a viruses. Nat Rev Drug Discovery (2006) 5:1015–25. doi: 10.1038/nrd2175
18. Demicheli V, Jefferson T, Ferroni E, Rivetti A, Di Pietrantonj C. Vaccines for preventing influenza in healthy adults. Cochrane Database Syst Rev. (2018) 2:CD001269. doi: 10.1002/14651858.CD001269.pub6
19. Liu J-W, Lin S-H, Wang L-C, Chiu H-Y, Lee J-A. Comparison of antiviral agents for seasonal influenza outcomes in healthy adults and children: A systematic review and network meta-analysis. JAMA Netw Open (2021) 4:e2119151. doi: 10.1001/jamanetworkopen.2021.19151
20. Fedson DS. Confronting an influenza pandemic with inexpensive generic agents: Can it be done? Lancet Infect Dis (2008) 8:571–6. doi: 10.1016/S1473-3099(08)70070-7
21. Bajimaya S, Frankl T, Hayashi T, Takimoto T. Cholesterol is required for stability and infectivity of influenza a and respiratory syncytial viruses. Virology (2017) 510:234–41. doi: 10.1016/j.virol.2017.07.024
22. Veit M, Thaa B. Association of influenza virus proteins with membrane rafts. Adv Virol (2011) 2011:370606. doi: 10.1155/2011/370606
23. Silvius JR. Role of cholesterol in lipid raft formation: lessons from lipid model systems. Biochim Biophys Acta-Biomembranes (2003) 1610:174–83. doi: 10.1016/S0005-2736(03)00016-6
24. Fedson DS. Pandemic influenza: A potential role for statins in treatment and prophylaxis. Clin Infect Dis (2006) 43:199–205. doi: 10.1086/505116
25. Keller P, Simons K. Cholesterol is required for surface transport of influenza virus hemagglutinin. J Cell Biol (1998) 140:1357–67. doi: 10.1083/jcb.140.6.1357
26. Armitage J. The safety of statins in clinical practice. Lancet (2007) 370:1781–90. doi: 10.1016/S0140-6736(07)60716-8
27. Cohen DE, Armstrong EJ. Pharmacology of cholesterol and lipoprotein metabolism. In: Golan DE, Tashjian AH, Amstrong EJ, Galanter JM, Armstrong AW, Arnaout RA, Rose HS, editors. Principles of pharmacology: The pathophysiologic basis of drug therapy, 2nd edition. Philadelphia, PA: Lippincott Williams & Wilkins (2007). p. 417–38.
28. Giguere J-F, Tremblay MJ. Statin compounds reduce human immunodeficiency virus type 1 replication by preventing the interaction between virion-associated host intercellular adhesion molecule 1 and its natural cell surface ligand LFA-1. J Virol (2004) 78:12062–5. doi: 10.1128/JVI.78.21.12062-12065.2004
29. Rosenson RS. Statins: Actions, side effects, and administration (2012). Available at: https://www.uptodate.com/contents/statins-actions-side-effects-and-administration (Accessed June 26, 2022).
30. Tan WY, Young BE, Lye DC, Chew DE, Dalan R. Statin use is associated with lower disease severity in COVID-19 infection. Sci Rep (2020) 10:17458. doi: 10.1038/s41598-020-74492-0
31. Yang Y-H, Chen W-C, Tsan Y-T, Chen M-J, Shih W-T, Tsai Y-H, et al. Statin use and the risk of cirrhosis development in patients with hepatitis c virus infection. J hepatology. J Hepatol (2015) 63:1111–7. doi: 10.1016/j.jhep.2015.07.006
32. Zhou Q, Liao JK. Statins and cardiovascular diseases: From cholesterol lowering to pleiotropy. Curr Pharm Des (2009) 15:467–78. doi: 10.2174/138161209787315684
33. Tsan Y-T, Lee C-H, Wang J-D, Chen P-C. Statins and the risk of hepatocellular carcinoma in patients with hepatitis b virus infection. J Clin Oncol (2012) 30:623–30. doi: 10.1200/JCO.2011.36.0917
34. McGeoch D, Fellner P, Newton C. Influenza virus genome consists of eight distinct RNA species. Proc Natl Acad Sci U. S. A. (1976) 73:3045–9. doi: 10.1073/pnas.73.9.3045
36. Eisfeld AJ, Neumann G, Kawaoka Y. At The centre: Influenza a virus ribonucleoproteins. Nat Rev Microbiol (2015) 13:28–41. doi: 10.1038/nrmicro3367
37. Shaw ML, Palese P. Orthomyxoviridae: The viruses and their replication. In: Knipe DM, Howley PM, editors. Fields virology, 6th edition. Philadelphia, PA: Lippincott Williams & Wilkins (2013). p. 1691–740.
38. Wright PF, Neumann G, Kawaoka Y. Orthomyxoviruses. In: Knipe DM, Howley PM, editors. Fields virology, 6th edition. Philadelphia, PA: Lippincott Williams & Wilkins (2013). p. 1186–243.
39. Fodor E. The RNA polymerase of influenza a virus: Mechanisms of viral transcription and replication. Acta Virol (2013) 57:113–22. doi: 10.4149/av_2013_02_113
40. Chiu Y-F, Huang Y-W, Chen C-Y, Chen Y-C, Gong Y-N, Kuo R-L, et al. Visualizing influenza a virus vRNA replication. Front Microbiol (2022) 13:812711. doi: 10.3389/fmicb.2022.812711
41. Chase GP, Rameix-Welti MA, Zvirbliene A, Zvirblis G, Gotz V, Wolff T, et al. Influenza virus ribonucleoprotein complexes gain preferential access to cellular export machinery through chromatin targeting. PloS Pathog (2011) 7:e1002187. doi: 10.1371/journal.ppat.1002187
42. Neumann G, Hughes MT, Kawaoka Y. Influenza a virus NS2 protein mediates vRNP nuclear export through NES-independent interaction with hCRM1. EMBO J (2000) 19:6751–8. doi: 10.1093/emboj/19.24.6751
43. Bruce EA, Abbink TE, Wise HM, Rollason R, Galao RP, Banting G, et al. Release of filamentous and spherical influenza a virus is not restricted by tetherin. J Gen Virol (2012) 93:963–9. doi: 10.1099/vir.0.038778-0
44. Chazal N, Gerlier D. Virus entry, assembly, budding, and membrane rafts. microbiol. Mol Biol Rev (2003) 67:226–37. doi: 10.1128/MMBR.67.2.226-237.2003
45. Lorent JH, Levental I. Structural determinants of protein partitioning into ordered membrane domains and lipid rafts. Chem Phys Lipids (2015) 192:23–32. doi: 10.1016/j.chemphyslip.2015.07.022
46. Pike LJ. Rafts defined: A report on the keystone symposium on lipid rafts and cell function. J Lipid Res (2006) 47:1597–8. doi: 10.1194/jlr.E600002-JLR200
47. Sato R, Okura T, Kawahara M, Takizawa N, Momose F, Morikawa Y. Apical trafficking pathways of influenza a virus HA and NA via Rab17- and Rab23-positive compartments. Front Microbiol (2019) 10:1857. doi: 10.3389/fmicb.2019.01857
48. Air GM, Laver WG. The neuraminidase of influenza virus. Proteins (1989) 6:341–56. doi: 10.1002/prot.340060402
49. Bucher D, Palese P. The biologically active proteins of influenza virus: neuraminidase. In: Kilbourne ED, editor. The influenza viruses and influenza. New York, NY: Academic Press (1975). p. 83–123.
50. Lakadamyali M, Rust MJ, Zhuang X. Endocytosis of influenza viruses. Microbes Infect (2004) 6:929–36. doi: 10.1016/j.micinf.2004.05.002
51. Edinger TO, Pohl MO, Stertz S. Entry of influenza a virus: Host factors and antiviral targets. J Gen Virol (2014) 95:263–77. doi: 10.1099/vir.0.059477-0
52. Gambaryan AS, Matrosovich TY, Boravleva EY, Lomakina NF, Yamnikova SS, Tuzikov AB, et al. Receptor-binding properties of influenza viruses isolated from gulls. Virology (2018) 522:37–45. doi: 10.1016/j.virol.2018.07.004
53. Russell RJ, Kerry PS, Stevens DJ, Steinhauer DA, Martin SR, Gamblin SJ, et al. Structure of influenza hemagglutinin in complex with an inhibitor of membrane fusion. Proc Natl Acad Sci U. S. A. (2008) 105:17736–41. doi: 10.1073/pnas.0807142105
54. Londrigan SL, Turville SG, Tate MD, Deng YM, Brooks AG, Reading PC. N-linked glycosylation facilitates sialic acid-independent attachment and entry of influenza a viruses into cells expressing DC-SIGN or l-SIGN. J Virol (2011) 85:2990–3000. doi: 10.1128/JVI.01705-10
55. Eierhoff T, Hrincius ER, Rescher U, Ludwig S, Ehrhardt C. The epidermal growth factor receptor (EGFR) promotes uptake of influenza a viruses (IAV) into host cells. PloS Pathog (2010) 6:e1001099. doi: 10.1371/journal.ppat.1001099
56. Sieben C, Sezgin E, Eggeling C, Manley S. Influenza a viruses use multivalent sialic acid clusters for cell binding and receptor activation. PloS Pathog (2020) 16:e1008656. doi: 10.1371/journal.ppat.1008656
57. Rahman SK, Ansari MA, Gaur P, Ahmad I, Chakravarty C, Verma DK, et al. The immunomodulatory CEA cell adhesion molecule 6 (CEACAM6/CD66c) is a protein receptor for the influenza a virus. Viruses (2021) 13:726. doi: 10.3390/v13050726
58. Nunes-Correia I, Eulálio A, Nir S, Lima MC. Caveolae as an additional route for influenza virus endocytosis in MDCK cells. Cell Mol Biol Lett (2004) 9:47–60.
59. Mahammad S, Parmryd I. Cholesterol depletion using methyl-β-cyclodextrin. Methods Mol Biol (2015) 1232:91–102. doi: 10.1007/978-1-4939-1752-5_8
60. Sun X, Whittaker GR. Role for influenza virus envelope cholesterol in virus entry and infection. J Virol (2003) 77:12543–51. doi: 10.1128/JVI.77.23.12543-12551.2003
61. Verma DK, Gupta D, Lal SK. Host lipid rafts play a major role in binding and endocytosis of influenza a virus. Viruses (2018) 10:650. doi: 10.3390/v10110650
62. Mehrbod P, Hair-Bejo M, Tengku Ibrahim TA, Omar AR, El Zowalaty M, Ajdari Z, et al. Simvastatin modulates cellular components in influenza a virus-infected cells. Int J Mol Med (2014) 34:61–73. doi: 10.3892/ijmm.2014.1761
63. Episcopio D, Aminov S, Benjamin S, Germain G, Datan E, Landazuri J, et al. Atorvastatin restricts the ability of influenza virus to generate lipid droplets and severely suppresses the replication of the virus. FASEB J (2019) 33:9516–25. doi: 10.1096/fj.201900428RR
64. Peng J, Zhang D, Ma Y, Wang G, Guo Z, Lu J. Protective effect of fluvastatin on influenza virus infection. Mol Med Rep (2014) 9:2221–6. doi: 10.3892/mmr.2014.2076
65. Kühnl A, Musiol A, Heitzig N, Johnson DE, Ehrhardt C, Grewal T, et al. Late endosomal/lysosomal cholesterol accumulation is a host cell-protective mechanism inhibiting endosomal escape of influenza a virus. MBio (2018) 9:e01345–18. doi: 10.1128/mBio.01345-18
66. Ma H, Kien F, Manière M, Zhang Y, Lagarde N, Tse KS, et al. Human annexin A6 interacts with influenza a virus protein M2 and negatively modulates infection. J Virol (2012) 86:1789–801. doi: 10.1128/JVI.06003-11
67. Musiol A, Gran S, Ehrhardt C, Ludwig S, Grewal T, Gerke V, et al. Annexin A6-balanced late endosomal cholesterol controls influenza a replication and propagation. mBio (2013) 4:e00608–13. doi: 10.1128/mBio.00608-13
68. Amorim MJ, Bruce EA, Read EK, Foeglein A, Mahen R, Stuart AD, et al. A Rab11-and microtubule-dependent mechanism for cytoplasmic transport of influenza a virus viral RNA. J Virol (2011) 85:4143–56. doi: 10.1128/JVI.02606-10
69. Momose F, Sekimoto T, Ohkura T, Jo S, Kawaguchi A, Nagata K, et al. Apical transport of influenza a virus ribonucleoprotein requires Rab11-positive recycling endosome. PloS One (2011) 6:e21123. doi: 10.1371/journal.pone.0021123
70. Amini-Bavil-Olyaee S, Choi YJ, Lee JH, Shi M, Huang IC, Farzan M, et al. The antiviral effector IFITM3 disrupts intracellular cholesterol homeostasis to block viral entry. Cell Host Microbe (2013) 13:452–64. doi: 10.1016/j.chom.2013.03.006
71. Desai TM, Marin M, Chin CR, Savidis G, Brass AL, Melikyan GB. IFITM3 restricts influenza a virus entry by blocking the formation of fusion pores following virus-endosome hemifusion. PloS Pathog (2014) 10:e1004048. doi: 10.1371/journal.ppat.1004048
72. Kummer S, Avinoam O, Kräusslich H-G. IFITM3 clusters on virus containing endosomes and lysosomes early in the influenza a infection of human airway epithelial cells. Viruses (2019) 11:548. doi: 10.3390/v11060548
73. García-Arribas AB, Alonso A, Goñi FM. Cholesterol interactions with ceramide and sphingomyelin. Chem Phys Lipids (2016) 199:26–34. doi: 10.1016/j.chemphyslip.2016.04.002
74. Audi A, Soudani N, Dbaibo G, Zaraket H. Depletion of host and viral sphingomyelin impairs influenza virus infection. Front Microbiol (2020) 11:612. doi: 10.3389/fmicb.2020.00612
75. Chen Y, Xu SC, Duan RD. Mevalonate inhibits acid sphingomyelinase activity, increases sphingomyelin levels and inhibits cell proliferation of HepG2 and caco-2 cells. Lipids Health Dis (2015) 14:130. doi: 10.1186/s12944-015-0137-8
76. Lu H, Talbot S. Down regulation of hmgcr in response to influenza a infection is independent of the IFN response in human cells (2019). Available at: https://www.biorxiv.org/content/10.1101/650465v1 (Accessed June 26, 2022).
77. Ohkura T, Momose F, Ichikawa R, Takeuchi K, Morikawa Y. Influenza a virus hemagglutinin and neuraminidase mutually accelerate their apical targeting through clustering of lipid rafts. J Virol (2014) 88:10039–55. doi: 10.1128/JVI.00586-14
78. Rodriguez-Boulan E, Paskiet KT, Salas PJ, Bard E. Intracellular transport of influenza virus hemagglutinin to the apical surface of madin-Darby canine kidney cells. . J Cell Biol (1984) 98:308–19. doi: 10.1083/jcb.98.1.308
79. Chen H, Yang J, Low PS, Cheng J-X. Cholesterol level regulates endosome motility via rab proteins. Biophys J (2008) 94:1508–20. doi: 10.1529/biophysj.106.099366
80. Holtta-Vuori M, Tanhuanpaa K, Mobius W, Somerharju P, Ikonen E. Modulation of cellular cholesterol transport and homeostasis by Rab11. Mol Biol Cell (2002) 13:3107–22. doi: 10.1091/mbc.e02-01-0025
81. Iwasaki A, Pillai PS. Innate immunity to influenza virus infection. Nat Rev Immunol (2014) 14:315–28. doi: 10.1038/nri3665
82. Cambi A, de Lange F, van Maarseveen NM, Nijhuis M, Joosten B, van Dijk EM, et al. Microdomains of the c-type lectin DC-SIGN are portals for virus entry into dendritic cells. J Cell Biol (2004) 164:145–55. doi: 10.1083/jcb.200306112
83. Tatematsu M, Nishikawa F, Seya T, Matsumoto M. Toll-like receptor 3 recognizes incomplete stem structures in single-stranded viral RNA. Nat Commun (2013) 4:1833. doi: 10.1038/ncomms2857
84. Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol (2005) 175:2851–8. doi: 10.4049/jimmunol.175.5.2851
85. Guillot L, Le Goffic R, Bloch S, Escriou N, Akira S, Chignard M, et al. Involvement of toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza a virus. J Biol Chem (2005) 280:5571–80. doi: 10.1074/jbc.M410592200
86. Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature (2006) 441:101–5. doi: 10.1038/nature04734
87. Koike A, Tsujinaka K, Fujimori K. Statins attenuate antiviral IFN-beta and ISG expression via inhibition of IRF3 and JAK/STAT signaling in poly(I:C)-treated hyperlipidemic mice and macrophages. FEBS J (2021) 288:4249–66. doi: 10.1111/febs.15712
88. Omer SB, Phadke VK, Bednarczyk RA, Chamberlain AT, Brosseau JL, Orenstein WA. Impact of statins on influenza vaccine effectiveness against medically attended acute respiratory illness. J Infect Dis (2016) 213:1216–23. doi: 10.1093/infdis/jiv457
89. Chuang T, Ulevitch RJ. Identification of hTLR10: A novel human toll-like receptor preferentially expressed in immune cells. Biochim Biophys Acta (2001) 1518:157–61. doi: 10.1016/S0167-4781(00)00289-X
90. Chuang TH, Ulevitch RJ. Cloning and characterization of a sub-family of human toll-like receptors: HTLR7, hTLR8 and hTLR9. Eur Cytokine Netw (2000) 11:372–8.
91. Rock FL, Hardiman G, Timans JC, Kastelein RA, Bazan JF. A family of human receptors structurally related to drosophila toll. proc. natl. acad. Sci U. S. A. (1998) 95:588–93. doi: 10.1073/pnas.95.2.588
92. Takeuchi O, Kawai T, Sanjo H, Copeland NG, Gilbert DJ, Jenkins NA, et al. TLR6: A novel member of an expanding toll-like receptor family. Gene (1999) 231:59–65. doi: 10.1016/S0378-1119(99)00098-0
93. Huh JW, Shibata T, Hwang M, Kwon EH, Jang MS, Fukui R, et al. UNC93B1 is essential for the plasma membrane localization and signaling of toll-like receptor 5. Proc Natl Acad Sci U. S. A. (2014) 111:7072–7. doi: 10.1073/pnas.1322838111
94. Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM couples endocytosis of toll-like receptor 4 to the induction of interferon-beta. Nat Immunol (2008) 9:361–8. doi: 10.1038/ni1569
95. Triantafilou M, Gamper FG, Haston RM, Mouratis MA, Morath S, Hartung T, et al. Membrane sorting of toll-like receptor (TLR)-2/6 and TLR2/1 heterodimers at the cell surface determines heterotypic associations with CD36 and intracellular targeting. J Biol Chem (2006) 281:31002–11. doi: 10.1074/jbc.M602794200
96. de Marcken M, Dhaliwal K, Danielsen AC, Gautron AS, Dominguez-Villar M. TLR7 and TLR8 activate distinct pathways in monocytes during RNA virus infection. Sci Signal (2019) 12:eaaw1347. doi: 10.1126/scisignal.aaw1347
97. Sorensen LN, Reinert LS, Malmgaard L, Bartholdy C, Thomsen AR, Paludan SR. TLR2 and TLR9 synergistically control herpes simplex virus infection in the brain. J Immunol (2008) 181:8604–12. doi: 10.4049/jimmunol.181.12.8604
98. Hahm B, Cho JH, Oldstone MB. Measles virus-dendritic cell interaction via SLAM inhibits innate immunity: Selective signaling through TLR4 but not other TLRs mediates suppression of IL-12 synthesis. Virology (2007) 358:251–7. doi: 10.1016/j.virol.2006.10.004
99. Uematsu S, Akira S. Toll-like receptors (TLRs) and their ligands. Handb. Exp Pharmacol (2008) 183:1–20. doi: 10.1007/978-3-540-72167-3_1
100. Liu SL, Zhang ZL, Tian ZQ, Zhao HS, Liu H, Sun EZ, et al. Effectively and efficiently dissecting the infection of influenza virus by quantum-dot-based single-particle tracking. ACS Nano (2012) 6:141–50. doi: 10.1021/nn2031353
101. Chansrichavala P, Chantharaksri U, Sritara P, Ngaosuwankul N, Chaiyaroj SC. Atorvastatin affects TLR4 clustering via lipid raft modulation. Int Immunopharmacol. (2010) 10:892–9. doi: 10.1016/j.intimp.2010.04.027
102. Methe H, Kim JO, Kofler S, Nabauer M, Weis M. Statins decrease toll-like receptor 4 expression and downstream signaling in human CD14+ monocytes. arterioscler. Thromb Vasc Biol (2005) 25:1439–45. doi: 10.1161/01.ATV.0000168410.44722.86
103. Ng WC, Londrigan SL, Nasr N, Cunningham AL, Turville S, Brooks AG, et al. The c-type lectin langerin functions as a receptor for attachment and infectious entry of influenza a virus. J Virol (2016) 90:206–21. doi: 10.1128/JVI.01447-15
104. Legler DF, Micheau O, Doucey MA, Tschopp J, Bron C. Recruitment of TNF receptor 1 to lipid rafts is essential for TNFalpha-mediated NF-kappaB activation. Immunity (2003) 18:655–64. doi: 10.1016/S1074-7613(03)00092-X
105. Tenesaca S, Vasquez M, Alvarez M, Otano I, Fernandez-Sendin M, Di Trani CA, et al. Statins act as transient type I interferon inhibitors to enable the antitumor activity of modified vaccinia Ankara viral vectors. J Immunother. Cancer. (2021) 9:e001587. doi: 10.1136/jitc-2020-001587
106. Sen S, Roy K, Mukherjee S, Mukhopadhyay R, Roy S. Restoration of IFNgammaR subunit assembly, IFNgamma signaling and parasite clearance in leishmania donovani infected macrophages: Role of membrane cholesterol. PloS Pathog (2011) 7:e1002229. doi: 10.1371/journal.ppat.1002229
107. Peterson ST, Kennedy EA, Brigleb PH, Taylor GM, Urbanek K, Bricker TL, et al. Disruption of type III interferon (IFN) genes Ifnl2 and Ifnl3 recapitulates loss of the type III IFN receptor in the mucosal antiviral response. J Virol (2019) 93:e01073–19. doi: 10.1128/JVI.01073-19
108. Tsai S-Y, Segovia JA, Chang T-H, Morris IR, Berton MT, Tessier PA, et al. DAMP molecule S100A9 acts as a molecular pattern to enhance inflammation during influenza a virus infection: Role of DDX21-TRIF-TLR4-MyD88 pathway. PloS Pathog (2014) 10:e1003848. doi: 10.1371/journal.ppat.1003848
109. Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science (2004) 303:1529–31. doi: 10.1126/science.1093616
110. Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, et al. Recognition of single-stranded RNA viruses by toll-like receptor 7. Proc Natl Acad Sci U. S. A. (2004) 101:5598–603. doi: 10.1073/pnas.0400937101
111. Le Goffic R, Balloy V, Lagranderie M, Alexopoulou L, Escriou N, Flavell R, et al. Detrimental contribution of the toll-like receptor (TLR)3 to influenza a virus-induced acute pneumonia. PloS Pathog (2006) 2:e53. doi: 10.1371/journal.ppat.0020053
112. Puig-Kroüger A, Serrano-Gómez D, Caparrós E, Domiínguez-Soto A, Relloso M, Colmenares MA, et al. Regulated expression of the pathogen receptor dendritic cell-specific intercellular adhesion molecule 3 (ICAM-3)-grabbing nonintegrin in THP-1 human leukemic cells, monocytes, and macrophages. J Biol Chem (2004) 279:25680–8. doi: 10.1074/jbc.M311516200
113. Soilleux EJ, Morris LS, Leslie G, Chehimi J, Luo Q, Levroney E, et al. Constitutive and induced expression of DC-SIGN on dendritic cell and macrophage subpopulations in situ and in vitro. J Leukoc. Biol (2002) 71:445–57.
114. Stoitzner P, Romani N. Langerin, the “Catcher in the rye”: An important receptor for pathogens on langerhans cells. Eur J Immunol (2011) 41:2526–9. doi: 10.1002/eji.201141934
115. Cambi A, Koopman M, Figdor CG. How c-type lectins detect pathogens. Cell Microbiol (2005) 7:481–8. doi: 10.1111/j.1462-5822.2005.00506.x
116. Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, et al. RIG-i-mediated antiviral responses to single-stranded RNA bearing 5'-phosphates. Science (2006) 314:997–1001. doi: 10.1126/science.1132998
117. Thomas PG, Dash P, Aldridge JJR, Ellebedy AH, Reynolds C, Funk AJ, et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza a virus via the regulation of caspase-1. Immunity (2009) 30:566–75. doi: 10.1016/j.immuni.2009.02.006
118. Onomoto K, Jogi M, Yoo J-S, Narita R, Morimoto S, Takemura A, et al. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PloS One (2012) 7:e43031. doi: 10.1371/journal.pone.0043031
119. Kowalinski E, Lunardi T, McCarthy AA, Louber J, Brunel J, Grigorov B, et al. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell (2011) 147:423–35. doi: 10.1016/j.cell.2011.09.039
120. Horner SM, Liu HM, Park HS, Briley J, Gale M. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis c virus. Proc Natl Acad Sci U. S. A. (2011) 108:14590–5. doi: 10.1073/pnas.1110133108
121. Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, et al. IPS-1, an adaptor triggering RIG-i-and Mda5-mediated type I interferon induction. Nat Immunol (2005) 6:981–8. doi: 10.1038/ni1243
122. Saha SK, Pietras EM, He JQ, Kang JR, Liu SY, Oganesyan G, et al. Regulation of antiviral responses by a direct and specific interaction between TRAF3 and cardif. EMBO J (2006) 25:3257–63. doi: 10.1038/sj.emboj.7601220
123. Seth RB, Sun L, Ea C-K, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF3. Cell (2005) 122:669–82. doi: 10.1016/j.cell.2005.08.012
124. Xu L-G, Wang Y-Y, Han K-J, Li L-Y, Zhai Z, Shu H-B. VISA is an adapter protein required for virus-triggered IFN-β signaling. Mol Cell (2005) 19:727–40. doi: 10.1016/j.molcel.2005.08.014
125. Kawai T, Akira S. Antiviral signaling through pattern recognition receptors. J Biochem (2007) 141:137–45. doi: 10.1093/jb/mvm032
126. Schroder K, Tschopp J. The inflammasomes. Cell (2010) 140:821–32. doi: 10.1016/j.cell.2010.01.040
127. Darnell JE Jr., Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science (1994) 264:1415–21. doi: 10.1126/science.8197455
128. Levy D, Lew D, Decker T, Kessler D, Darnell JJ. Synergistic interaction between interferon-alpha and interferon-gamma through induced synthesis of one subunit of the transcription factor ISGF3. EMBO J (1990) 9:1105–11. doi: 10.1002/j.1460-2075.1990.tb08216.x
129. Benitez AA, Panis M, Xue J, Varble A, Shim JV, Frick AL, et al. In vivo RNAi screening identifies MDA5 as a significant contributor to the cellular defense against influenza a virus. Cell Rep (2015) 11:1714–26. doi: 10.1016/j.celrep.2015.05.032
130. Broquet AH, Hirata Y, McAllister CS, Kagnoff MF. RIG-I/MDA5/MAVS are required to signal a protective IFN response in rotavirus-infected intestinal epithelium. J Immunol (2011) 186:1618–26. doi: 10.4049/jimmunol.1002862
131. Lee N-R, Kim H-I, Choi M-S, Yi C-M, Inn K-S. Regulation of MDA5-MAVS antiviral signaling axis by TRIM25 through TRAF6-mediated NF-κB activation. Mol Cells (2015) 38:759. doi: 10.14348/molcells.2015.0047
132. Sirén J, Imaizumi T, Sarkar D, Pietilä T, Noah DL, Lin R, et al. Retinoic acid inducible gene-I and mda-5 are involved in influenza a virus-induced expression of antiviral cytokines. Microbe Infect (2006) 8:2013–20. doi: 10.1016/j.micinf.2006.02.028
133. Wang JP, Cerny A, Asher DR, Kurt-Jones EA, Bronson RT, Finberg RW. MDA5 and MAVS mediate type I interferon responses to coxsackie b virus. J Virol (2010) 84:254–60. doi: 10.1128/JVI.00631-09
134. Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza a virus through recognition of viral RNA. Immunity (2009) 30:556–65. doi: 10.1016/j.immuni.2009.02.005
135. Sabbah A, Chang TH, Harnack R, Frohlich V, Tominaga K, Dube PH, et al. Activation of innate immune antiviral responses by Nod2. Nat Immunol (2009) 10:1073–80. doi: 10.1038/ni.1782
136. Pandey KP, Zhou Y. Influenza a virus infection activates NLRP3 inflammasome through trans-golgi network dispersion. Viruses (2022) 14:88. doi: 10.3390/v14010088
137. Yokota S. [Influenza-associated encephalopathy–pathophysiology and disease mechanisms]. Nihon Rinsho (2003) 61:1953–8.
138. Koushki K, Shahbaz SK, Mashayekhi K, Sadeghi M, Zayeri ZD, Taba MY, et al. Anti-inflammatory action of statins in cardiovascular disease: The role of inflammasome and toll-like receptor pathways. Clin Rev Allergy Immunol (2021) 60:175–99. doi: 10.1007/s12016-020-08791-9
139. Gu Y, Zuo X, Zhang S, Ouyang Z, Jiang S, Wang F, et al. The mechanism behind influenza virus cytokine storm. Viruses (2021) 13:1362. doi: 10.3390/v13071362
140. Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci U. S. A. (1975) 72:3666–70. doi: 10.1073/pnas.72.9.3666
141. Isaacs A, Lindenmann J. Virus interference. I. Proc R Soc Lond B Biol Sci (1957) 147:258–67. doi: 10.1098/rspb.1957.0048
142. Isaacs A, Lindenmann J, Valentine RC. Virus interference. II. some properties of interferon. Proc R Soc Lond B Biol Sci (1957) 147:268–73. doi: 10.1098/rspb.1957.0049
143. Larner AC, Jonak G, Cheng YS, Korant B, Knight E, Darnell JE Jr. Transcriptional induction of two genes in human cells by beta interferon. Proc Natl Acad Sci U. S. A. (1984) 81:6733–7. doi: 10.1073/pnas.81.21.6733
144. Aggarwal BB, Eessalu TE, Hass PE. Characterization of receptors for human tumour necrosis factor and their regulation by gamma-interferon. Nature (1985) 318:665–7. doi: 10.1038/318665a0
145. Seo SH, Webster RG. Tumor necrosis factor alpha exerts powerful anti-influenza virus effects in lung epithelial cells. J Virol (2002) 76:1071–6. doi: 10.1128/JVI.76.3.1071-1076.2002
146. McDermott MF, Aksentijevich I, Galon J, McDermott EM, Ogunkolade BW, Centola M, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell (1999) 97:133–44. doi: 10.1016/S0092-8674(00)80721-7
147. Hennessy E, Griffin É.W., Cunningham C. Astrocytes are primed by chronic neurodegeneration to produce exaggerated chemokine and cell infiltration responses to acute stimulation with the cytokines IL-1β and TNF-α. J Neurosci (2015) 35:8411–22. doi: 10.1523/JNEUROSCI.2745-14.2015
148. Kay JG, Murray RZ, Pagan JK, Stow JL. Cytokine secretion via cholesterol-rich lipid raft-associated SNAREs at the phagocytic cup. J Biol Chem (2006) 281:11949–54. doi: 10.1074/jbc.M600857200
149. DeBerge MP, Ely KH, Wright PF, Thorp EB, Enelow RI. Shedding of TNF receptor 2 by effector CD8+ T cells by ADAM17 is important for regulating TNF-α availability during influenza infection. J Leukoc. Biol (2015) 98:423–34. doi: 10.1189/jlb.3A0914-432RR
150. Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, et al. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol (2003) 4:69–77. doi: 10.1038/ni875
151. Valente G, Ozmen L, Novelli F, Geuna M, Palestro G, Forni G, et al. Distribution of interferon-gamma receptor in human tissues. Eur J Immunol (1992) 22:2403–12. doi: 10.1002/eji.1830220933
152. Vanden Broecke C, Pfeffer LM. Characterization of interferon-alpha binding sites on human cell lines. J Interferon Res (1988) 8:803–11. doi: 10.1089/jir.1988.8.803
153. Abramovich C, Shulman L, Ratovitski E, Harroch S, Tovey M, Eid P, et al. Differential tyrosine phosphorylation of the IFNAR chain of the type I interferon receptor and of an associated surface protein in response to IFN-alpha and IFN-beta. EMBO J (1994) 13:5871–7. doi: 10.1002/j.1460-2075.1994.tb06932.x
154. Guo XJ, Thomas PG. New fronts emerge in the influenza cytokine storm. Semin Immunopathol (2017) 39:541–50. doi: 10.1007/s00281-017-0636-y
156. Takaoka A, Mitani Y, Suemori H, Sato M, Yokochi T, Noguchi S, et al. Cross talk between interferon-gamma and -alpha/beta signaling components in caveolar membrane domains. Science (2000) 288:2357–60. doi: 10.1126/science.288.5475.2357
157. Domanski P, Witte M, Kellum M, Rubinstein M, Hackett R, Pitha P, et al. Cloning and expression of a long form of the beta subunit of the interferon alpha beta receptor that is required for signaling. J Biol Chem (1995) 270:21606–11. doi: 10.1074/jbc.270.37.21606
158. Pilz A, Ramsauer K, Heidari H, Leitges M, Kovarik P, Decker T. Phosphorylation of the Stat1 transactivating domain is required for the response to type I interferons. EMBO Rep (2003) 4:368–73. doi: 10.1038/sj.embor.embor802
159. Qureshi SA, Salditt-Georgieff M, Darnell JE Jr. Tyrosine-phosphorylated Stat1 and Stat2 plus a 48-kDa protein all contact DNA in forming interferon-stimulated-gene factor 3. Proc Natl Acad Sci U. S. A. (1995) 92:3829–33. doi: 10.1073/pnas.92.9.3829
160. Tolomeo M, Cavalli A, Cascio A. STAT1 and its crucial role in the control of viral infections. Int J Mol Sci (2022) 23:4095. doi: 10.3390/ijms23084095
161. Zhao L, Xia M, Wang K, Lai C, Fan H, Gu H, et al. A long non-coding RNA IVRPIE promotes host antiviral immune responses through regulating interferon β1 and ISG expression. Front Microbiol (2020) 11:260. doi: 10.3389/fmicb.2020.00260
162. Huang I-C, Bailey CC, Weyer JL, Radoshitzky SR, Becker MM, Chiang JJ, et al. Distinct patterns of IFITM-mediated restriction of filoviruses, SARS coronavirus, and influenza a virus. PloS Pathog (2011) 7:e1001258. doi: 10.1371/journal.ppat.1001258
163. Lange PT, Darrah EJ, Vonderhaar EP, Mboko WP, Rekow MM, Patel SB, et al. Type I interferon counteracts antiviral effects of statins in the context of gammaherpesvirus infection. J Virol (2016) 90:3342–54. doi: 10.1128/JVI.02277-15
164. Wang X, Hinson ER, Cresswell P. The interferon-inducible protein viperin inhibits influenza virus release by perturbing lipid rafts. Cell Host Microbe (2007) 2:96–105. doi: 10.1016/j.chom.2007.06.009
165. Doherty PC, Topham DJ, Tripp RA, Cardin RD, Brooks JW, Stevenson PG. Effector CD4+ and CD8+ T-cell mechanisms in the control of respiratory virus infections. Immunol Rev (1997) 159:105–17. doi: 10.1111/j.1600-065X.1997.tb01010.x
166. Koyama S, Ishii KJ, Kumar H, Tanimoto T, Coban C, Uematsu S, et al. Differential role of TLR- and RLR-signaling in the immune responses to influenza a virus infection and vaccination. J Immunol (2007) 179:4711–20. doi: 10.4049/jimmunol.179.7.4711
167. Wrammert J, Koutsonanos D, Li GM, Edupuganti S, Sui J, Morrissey M, et al. Broadly cross-reactive antibodies dominate the human b cell response against 2009 pandemic H1N1 influenza virus infection. J Exp Med (2011) 208:181–93. doi: 10.1084/jem.20101352
168. Ramana CV, DeBerge MP, Kumar A, Alia CS, Durbin JE, Enelow RI. Inflammatory impact of IFN-gamma in CD8+ T cell-mediated lung injury is mediated by both Stat1-dependent and -independent pathways. am. j. physiol. lung cell. Mol Physiol (2015) 308:L650–657. doi: 10.1152/ajplung.00360.2014
169. Wiley JA, Cerwenka A, Harkema JR, Dutton RW, Harmsen AG. Production of interferon-gamma by influenza hemagglutinin-specific CD8 effector T cells influences the development of pulmonary immunopathology. Am J Pathol (2001) 158:119–30. doi: 10.1016/S0002-9440(10)63950-8
170. Dutta A, Miaw SC, Yu JS, Chen TC, Lin CY, Lin YC, et al. Altered T-bet dominance in IFN-gamma-decoupled CD4+ T cells with attenuated cytokine storm and preserved memory in influenza. J Immunol (2013) 190:4205–14. doi: 10.4049/jimmunol.1202434
171. Gogishvili T, Langenhorst D, Luhder F, Elias F, Elflein K, Dennehy KM, et al. Rapid regulatory T-cell response prevents cytokine storm in CD28 superagonist treated mice. PloS One (2009) 4:e4643. doi: 10.1371/journal.pone.0004643
172. Braun J, Hochman PS, Unanue ER. Ligand-induced association of surface immunoglobulin with the detergent-insoluble cytoskeletal matrix of the b lymphocyte. J Immunol (1982) 128:1198–204.
173. Cheng PC, Dykstra ML, Mitchell RN, Pierce SK. A role for lipid rafts in b cell antigen receptor signaling and antigen targeting. J Exp Med (1999) 190:1549–60. doi: 10.1084/jem.190.11.1549
174. Sun Y, Li X, Wang T, Li W. Core fucosylation regulates the function of pre-BCR, BCR and IgG in humoral immunity. Front Immunol (2022) 13:844427. doi: 10.3389/fimmu.2022.844427
175. Ding BB, Bi E, Chen H, Yu JJ, Ye BH. IL-21 and CD40L synergistically promote plasma cell differentiation through upregulation of blimp-1 in human b cells. J Immunol (2013) 190:1827–36. doi: 10.4049/jimmunol.1201678
176. Rush JS, Hodgkin PD. B cells activated via CD40 and IL-4 undergo a division burst but require continued stimulation to maintain division, survival and differentiation. Eur J Immunol (2001) 31:1150–9. doi: 10.1002/1521-4141(200104)31:4<1150::AID-IMMU1150>3.0.CO;2-V
177. Okada T, Miller MJ, Parker I, Krummel MF, Neighbors M, Hartley SB, et al. Antigen-engaged b cells undergo chemotaxis toward the T zone and form motile conjugates with helper T cells. PloS Biol (2005) 3:e150. doi: 10.1371/journal.pbio.0030150
178. Warren WD, Berton MT. Induction of germ-line gamma 1 and epsilon ig gene expression in murine b cells. IL-4 and the CD40 ligand-CD40 interaction provide distinct but synergistic signals. J Immunol (1995) 155:5637–46.
179. Ekiert DC, Kashyap AK, Steel J, Rubrum A, Bhabha G, Khayat R, et al. Cross-neutralization of influenza a viruses mediated by a single antibody loop. Nature (2012) 489:526–32. doi: 10.1038/nature11414
180. Monto AS, Petrie JG, Cross RT, Johnson E, Liu M, Zhong W, et al. Antibody to influenza virus neuraminidase: An independent correlate of protection. J Infect Dis (2015) 212:1191–9. doi: 10.1093/infdis/jiv195
181. Ng S, Nachbagauer R, Balmaseda A, Stadlbauer D, Ojeda S, Patel M, et al. Novel correlates of protection against pandemic H1N1 influenza a virus infection. Nat Med (2019) 25:962–7. doi: 10.1038/s41591-019-0463-x
182. El Bakkouri K, Descamps F, De Filette M, Smet A, Festjens E, Birkett A, et al. Universal vaccine based on ectodomain of matrix protein 2 of influenza a: Fc receptors and alveolar macrophages mediate protection. J Immunol (2011) 186:1022–31. doi: 10.4049/jimmunol.0902147
183. Bangaru S, Zhang H, Gilchuk IM, Voss TG, Irving RP, Gilchuk P, et al. A multifunctional human monoclonal neutralizing antibody that targets a unique conserved epitope on influenza HA. Nat Commun (2018) 9:2669. doi: 10.1038/s41467-018-04704-9
184. DiLillo DJ, Palese P, Wilson PC, Ravetch JV. Broadly neutralizing anti-influenza antibodies require fc receptor engagement for in vivo protection. J Clin Invest. (2016) 126:605–10. doi: 10.1172/JCI84428
185. Heer AK, Harris NL, Kopf M, Marsland BJ. CD4+ and CD8+ T cells exhibit differential requirements for CCR7-mediated antigen transport during influenza infection. . J Immunol (2008) 181:6984–94. doi: 10.4049/jimmunol.181.10.6984
186. Hamada H, Bassity E, Flies A, Strutt TM, Garcia-Hernandez Mde L, McKinstry KK, et al. Multiple redundant effector mechanisms of CD8+ T cells protect against influenza infection. J Immunol (2013) 190:296–306. doi: 10.4049/jimmunol.1200571
187. Brincks EL, Katewa A, Kucaba TA, Griffith TS, Legge KL. CD8 T cells utilize TRAIL to control influenza virus infection. J Immunol (2008) 181:4918–25. doi: 10.4049/jimmunol.181.7.4918
188. Topham DJ, Tripp RA, Doherty PC. CD8+ T cells clear influenza virus by perforin or fas-dependent processes. J Immunol (1997) 159:5197–200.
189. DeBerge MP, Ely KH, Cheng G-S, Enelow RI. ADAM17-mediated processing of TNF-α expressed by antiviral effector CD8+ T cells is required for severe T-cell-mediated lung injury. PloS One (2013) 8:e79340. doi: 10.1371/journal.pone.0079340
190. Liu B, Zhang X, Deng W, Liu J, Li H, Wen M, et al. Severe influenza A(H1N1)pdm09 infection induces thymic atrophy through activating innate CD8(+)CD44(hi) T cells by upregulating IFN-gamma. Cell Death Dis (2014) 5:e1440. doi: 10.1038/cddis.2014.323
191. Elgueta R, Benson MJ, De Vries VC, Wasiuk A, Guo Y, Noelle RJ. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev (2009) 229:152–72. doi: 10.1111/j.1600-065X.2009.00782.x
192. Janeway JCA, Travers P, Walport M, Shlomchik MJ. B-cell activation by armed helper T cells, in: Immunobiology: The immune system in health and disease (2001). New York, NY: Garland Science. Available at: https://www.ncbi.nlm.nih.gov/books/NBK27142/ (Accessed June 26, 2022).
193. Bao K, Reinhardt RL. The differential expression of IL-4 and IL-13 and its impact on type-2 immunity. Cytokine (2015) 75:25–37. doi: 10.1016/j.cyto.2015.05.008
194. Deng N, Weaver JM, Mosmann TR. Cytokine diversity in the Th1-dominated human anti-influenza response caused by variable cytokine expression by Th1 cells, and a minor population of uncommitted IL-2+ IFNγ-thpp cells. PloS One (2014) 9:e95986. doi: 10.1371/journal.pone.0095986
195. Janeway JCA, Travers P, Walport M, Shlomchik MJ. Macrophage activation by armed CD4 TH1 cells, in: Immunobiology: The immune system in health and disease (2001). New York, NY: Garland Science. Available at: https://www.ncbi.nlm.nih.gov/books/NBK27153/ (Accessed June 26, 2022).
196. Martinez GJ, Nurieva RI, Yang XO, Dong C. Regulation and function of proinflammatory TH17 cells. Ann N Y. Acad Sci (2008) 1143:188–211. doi: 10.1196/annals.1443.021
197. Dinic J, Riehl A, Adler J, Parmryd I. The T cell receptor resides in ordered plasma membrane nanodomains that aggregate upon patching of the receptor. Sci Rep (2015) 5:10082. doi: 10.1038/srep10082
198. Xavier R, Brennan T, Li Q, McCormack C, Seed B. Membrane compartmentation is required for efficient T cell activation. Immunity (1998) 8:723–32. doi: 10.1016/S1074-7613(00)80577-4
199. Trbojević-Akmačić I, Abdel-Mohsen M, Falck D, Rapp E. Immunoglobulin glycosylation analysis: State-of-the-Art methods and applications in immunology. Front Immunol (2022) 13:2022.923393. doi: 10.3389/fimmu.2022.923393
200. Beck-García K, Beck-García E, Bohler S, Zorzin C, Sezgin E, Levental I, et al. Nanoclusters of the resting T cell antigen receptor (TCR) localize to non-raft domains. Biochim Biophys Acta-Mol Cell Res (2015) 1853:802–9. doi: 10.1016/j.bbamcr.2014.12.017
201. Varshney P, Yadav V, Saini N. Lipid rafts in immune signalling: Current progress and future perspective. Immunology (2016) 149:13–24. doi: 10.1111/imm.12617
202. Dykstra M, Cherukuri A, Sohn HW, Tzeng S-J, Pierce SK. Location is everything: lipid rafts and immune cell signaling. Annu Rev Immunol (2003) 21:457–81. doi: 10.1146/annurev.immunol.21.120601.141021
203. Blank N, Schiller M, Krienke S, Busse F, Schatz B, Ho AD, et al. Atorvastatin inhibits T cell activation through 3-hydroxy-3-methylglutaryl coenzyme a reductase without decreasing cholesterol synthesis. J Immunol (2007) 179:3613–21. doi: 10.4049/jimmunol.179.6.3613
204. Shimabukuro-Vornhagen A, Zoghi S, Liebig TM, Wennhold K, Chemitz J, Draube A, et al. Inhibition of protein geranylgeranylation specifically interferes with CD40-dependent b cell activation, resulting in a reduced capacity to induce T cell immunity. J Immunol (2014) 193:5294–305. doi: 10.4049/jimmunol.1203436
205. Meyer zum Bueschenfelde CO, Unternaehrer J, Mellman I, Bottomly K. Regulated recruitment of MHC class II and costimulatory molecules to lipid rafts in dendritic cells. J Immunol (2004) 173:6119–24. doi: 10.4049/jimmunol.173.10.6119
206. Pham LV, Tamayo AT, Yoshimura LC, Lo P, Terry N, Reid PS, et al. A CD40 signalosome anchored in lipid rafts leads to constitutive activation of NF-kappaB and autonomous cell growth in b cell lymphomas. Immunity (2002) 16:37–50. doi: 10.1016/S1074-7613(01)00258-8
207. Yilmaz A, Reiss C, Weng A, Cicha I, Stumpf C, Steinkasserer A, et al. Differential effects of statins on relevant functions of human monocyte-derived dendritic cells. J Leukoc. Biol (2006) 79:529–38. doi: 10.1189/jlb.0205064
208. Kabouridis PS, Janzen J, Magee AL, Ley SC. Cholesterol depletion disrupts lipid rafts and modulates the activity of multiple signaling pathways in T lymphocytes. Eur J Immunol (2000) 30:954–63. doi: 10.1002/1521-4141(200003)30:3<954::AID-IMMU954>3.0.CO;2-Y
209. Ramos I, Fernandez-Sesma A. Modulating the innate immune response to influenza a virus: Potential therapeutic use of anti-inflammatory drugs. Front Immunol (2015) 6:361. doi: 10.3389/fimmu.2015.00361
210. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol (2009) 183:787–91. doi: 10.4049/jimmunol.0901363
211. Choudhury SM, Ma X, Abdullah SW, Zheng H. Activation and inhibition of the NLRP3 inflammasome by RNA viruses. J Inflammation Res (2021) 14:1145. doi: 10.2147/JIR.S295706
212. Hamarsheh SA, Zeiser R. NLRP3 inflammasome activation in cancer: A double-edged sword. Front Immunol (2020) 11:1444. doi: 10.3389/fimmu.2020.01444
213. Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature (2006) 440:228–32. doi: 10.1038/nature04515
214. Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity (2013) 38:1142–53. doi: 10.1016/j.immuni.2013.05.016
215. Franchi L, Eigenbrod T, Núñez G. Cutting edge: TNF-α mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J Immunol (2009) 183:792–6. doi: 10.4049/jimmunol.0900173
216. Zhao C, Zhao W. NLRP3 inflammasome–a key player in antiviral responses. Front Immunol (2020) 11:211. doi: 10.3389/fimmu.2020.00211
217. McAuley JL, Tate MD, MacKenzie-Kludas CJ, Pinar A, Zeng W, Stutz A, et al. Activation of the NLRP3 inflammasome by IAV virulence protein PB1-F2 contributes to severe pathophysiology and disease. PloS Pathog (2013) 9:e1003392. doi: 10.1371/journal.ppat.1003392
218. He W-T, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin d is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res (2015) 25:1285–98. doi: 10.1038/cr.2015.139
219. Malik A, Kanneganti T-D. Inflammasome activation and assembly at a glance. J Cell Sci (2017) 130:3955–63. doi: 10.1242/jcs.207365
220. Pothlichet J, Meunier I, Davis BK, Ting JP, Skamene E, von Messling V, et al. Type I IFN triggers RIG-I/TLR3/NLRP3-dependent inflammasome activation in influenza a virus infected cells. PloS Pathog (2013) 9:e1003256. doi: 10.1371/journal.ppat.1003256
221. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526:660–5. doi: 10.1038/nature15514
222. Bersinger NA, Günthert AR, McKinnon B, Johann S, Mueller MD. Dose–response effect of interleukin (IL)-1β, tumour necrosis factor (TNF)-α, and interferon-γ on the in vitro production of epithelial neutrophil activating peptide-78 (ENA-78), IL-8, and IL-6 by human endometrial stromal cells. Arch Gynecol. Obstet. (2011) 283:1291–6. doi: 10.1007/s00404-010-1520-3
223. Hoshino T, Okamoto M, Sakazaki Y, Kato S, Young HA, Aizawa H. Role of proinflammatory cytokines IL-18 and IL-1β in bleomycin-induced lung injury in humans and mice. Am J Respir Cell Mol (2009) 41:661–70. doi: 10.1165/rcmb.2008-0182OC
224. Nussbaum JC, Van Dyken SJ, Von Moltke J, Cheng LE, Mohapatra A, Molofsky AB, et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature (2013) 502:245–8. doi: 10.1038/nature12526
225. Weiss ID, Wald O, Wald H, Beider K, Abraham M, Galun E, et al. IFN-γ treatment at early stages of influenza virus infection protects mice from death in a NK cell-dependent manner. J Interferon Cytokine Res (2010) 30:439–49. doi: 10.1089/jir.2009.0084
226. Laghlali G, Lawlor KE, Tate MD. Die another way: Interplay between influenza a virus, inflammation and cell death. Viruses (2020) 12:401. doi: 10.3390/v12040401
227. Bhaskar S, Sinha A, Banach M, Mittoo S, Weissert R, Kass JS, et al. Cytokine storm in COVID-19–immunopathological mechanisms, clinical considerations, and therapeutic approaches: The REPROGRAM consortium position paper. Front Immunol (2020) 11:1648. doi: 10.3389/fimmu.2020.01648
228. Ragab D, Salah Eldin H, Taeimah M, Khattab R, Salem R. The COVID-19 cytokine storm; what we know so far. Front Immunol (2020) 11:1446. doi: 10.3389/fimmu.2020.01446
229. Seo SH, Hoffmann E, Webster RG. Lethal H5N1 influenza viruses escape host anti-viral cytokine responses. Nat Med (2002) 8:950–4. doi: 10.1038/nm757
230. Tumpey TM, Garcia-Sastre A, Taubenberger JK, Palese P, Swayne DE, Pantin-Jackwood MJ, et al. Pathogenicity of influenza viruses with genes from the 1918 pandemic virus: Functional roles of alveolar macrophages and neutrophils in limiting virus replication and mortality in mice. J Virol (2005) 79:14933–44. doi: 10.1128/JVI.79.23.14933-14944.2005
231. Hussell T, Pennycook A, Openshaw PJ. Inhibition of tumor necrosis factor reduces the severity of virus-specific lung immunopathology. Eur J Immunol (2001) 31:2566–73. doi: 10.1002/1521-4141(200109)31:9<2566::AID-IMMU2566>3.0.CO;2-L
232. Tate MD, Ong JDH, Dowling JK, McAuley JL, Robertson AB, Latz E, et al. Reassessing the role of the NLRP3 inflammasome during pathogenic influenza a virus infection via temporal inhibition. Sci Rep (2016) 6:27912. doi: 10.1038/srep27912
233. Chen S, Liu G, Chen J, Hu A, Zhang L, Sun W, et al. Ponatinib protects mice from lethal influenza infection by suppressing cytokine storm. Front Immunol (2019) 10:1393. doi: 10.3389/fimmu.2019.01393
234. Davidson S. Treating influenza infection, from now and into the future. Front Immunol (2018) 9:1946. doi: 10.3389/fimmu.2018.01946
235. Marsolais D, Rosen H. Chemical modulators of sphingosine-1-phosphate receptors as barrier-oriented therapeutic molecules. Nat Rev Drug Discovery (2009) 8:297–307. doi: 10.1038/nrd2356
236. Shi X, Zhou W, Huang H, Zhu H, Zhou P, Zhu H, et al. Inhibition of the inflammatory cytokine tumor necrosis factor-alpha with etanercept provides protection against lethal H1N1 influenza infection in mice. Crit Care (2013) 17:R301. doi: 10.1186/cc13171
237. Walsh KB, Teijaro JR, Wilker PR, Jatzek A, Fremgen DM, Das SC, et al. Suppression of cytokine storm with a sphingosine analog provides protection against pathogenic influenza virus. Proc Natl Acad Sci U. S. A. (2011) 108:12018–23.
238. Frost FJ, Petersen H, Tollestrup K, Skipper B. Influenza and COPD mortality protection as pleiotropic, dose-dependent effects of statins. Chest (2007) 131:1006–12. doi: 10.1378/chest.06-1997
239. Vandermeer ML, Thomas AR, Kamimoto L, Reingold A, Gershman K, Meek J, et al. Association between use of statins and mortality among patients hospitalized with laboratory-confirmed influenza virus infections: A multistate study. J Infect Dis (2012) 205:13–9. doi: 10.1093/infdis/jir695
240. Mehrbod P, Omar AR, Hair-Bejo M, Haghani A, Ideris A. Mechanisms of action and efficacy of statins against influenza. BioMed Res Int (2014) 2014:872370. doi: 10.1155/2014/872370
241. Brassard P, Wu JW, Ernst P, Dell'Aniello S, Smiechowski B, Suissa S. The effect of statins on influenza-like illness morbidity and mortality. pharmacoepidemiol. Drug Saf. (2017) 26:63–70.
242. Vahedian-Azimi A, Mannarino MR, Shojaie S, Rahimibashar F, Esmaeili Gouvarchin Galeh H, Banach M, et al. Effect of statins on prevalence and mortality of influenza virus infection: A systematic review and meta-analysis. Arch Med Sci (2022) 18(6). doi: 10.5114/aoms/149633
243. Kwong JC, Li P, Redelmeier DA. Influenza morbidity and mortality in elderly patients receiving statins: A cohort study. PloS One (2009) 4:e8087. doi: 10.1371/journal.pone.0008087
244. Atamna A, Babitch T, Bracha M, Sorek N, Haim B-Z, Elis A, et al. Statins and outcomes of hospitalized patients with laboratory-confirmed 2017–2018 influenza. Eur J Clin Microbiol (2019) 38:2341–8. doi: 10.1007/s10096-019-03684-y
245. Izurieta HS, Chillarige Y, Kelman JA, Forshee R, Qiang Y, Wernecke M, et al. Statin use and risks of influenza-related outcomes among older adults receiving standard-dose or high-dose influenza vaccines through Medicare during 2010–2015. Clin Infect Dis (2018) 67:378–87. doi: 10.1093/cid/ciy100
246. Cutrell JB, Drechsler H, Bedimo R, Alvarez CA, Mansi IA. Statin use and medically attended acute respiratory illness among influenza vaccine recipients. Vaccine (2019) 37:6707–13. doi: 10.1016/j.vaccine.2019.09.024
247. Brett SJ, Myles P, Lim WS, Enstone JE, Bannister B, Semple MG, et al. Pre-admission statin use and in-hospital severity of 2009 pandemic influenza A(H1N1) disease. PloS One (2011) 6:e18120. doi: 10.1371/journal.pone.0018120
248. Belser JA, Szretter KJ, Katz JM, Tumpey TM. Simvastatin and oseltamivir combination therapy does not improve the effectiveness of oseltamivir alone following highly pathogenic avian H5N1 influenza virus infection in mice. Virology (2013) 439:42–6. doi: 10.1016/j.virol.2013.01.017
249. Radigan KA, Urich D, Misharin AV, Chiarella SE, Soberanes S, Gonzalez A, et al. The effect of rosuvastatin in a murine model of influenza a infection. PloS One (2012) 7:e35788. doi: 10.1371/journal.pone.0035788
250. Karlsson EA, Schultz-Cherry S, Rosch JW. Protective capacity of statins during pneumonia is dependent on etiological agent and obesity. Front Cell Infect Microbiol (2018) 8:41. doi: 10.3389/fcimb.2018.00041
251. Chang DH, Bednarczyk RA, Becker ER, Hockenberry JM, Weiss PS, Orenstein WA, et al. Trends in U.S. hospitalizations and inpatient deaths from pneumonia and influenza 1996-2011. Vaccine (2016) 34:486–94. doi: 10.1016/j.vaccine.2015.12.003
252. Klein EY, Monteforte B, Gupta A, Jiang W, May L, Hsieh YH, et al. The frequency of influenza and bacterial coinfection: A systematic review and meta-analysis. Influenza Other Respir Viruses (2016) 10:394–403. doi: 10.1111/irv.12398
253. Kintscher U, Hartge M, Hess K, Foryst-Ludwig A, Clemenz M, Wabitsch M, et al. T-Lymphocyte infiltration in visceral adipose tissue: A primary event in adipose tissue inflammation and the development of obesity-mediated insulin resistance. Arterioscler Thromb Vasc Biol (2008) 28:1304–10. doi: 10.1161/ATVBAHA.108.165100
Keywords: influenza A virus, statins, inflammatory cytokines, lipid rafts, cholesterol
Citation: Li Y-J, Chen C-Y, Yang J-H and Chiu Y-F (2022) Modulating cholesterol-rich lipid rafts to disrupt influenza A virus infection. Front. Immunol. 13:982264. doi: 10.3389/fimmu.2022.982264
Received: 30 June 2022; Accepted: 15 August 2022;
Published: 13 September 2022.
Edited by:
Kazuhisa Iwabuchi, Juntendo University, JapanReviewed by:
Jose M. Reyes-Ruiz, Mexican Social Security Institute (IMSS), MexicoCopyright © 2022 Li, Chen, Yang and Chiu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ya-Fang Chiu, eWZjaGl1QG1haWwuY2d1LmVkdS50dw==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.