 Lu Yu
Lu Yu Minghan Sun
Minghan Sun Qi Zhang
Qi Zhang Qiao Zhou
Qiao Zhou Yi Wang
Yi Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 08 September 2022
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.982026
With the goal of harnessing the host’s immune system to provide long-lasting remission and cures for various cancers, the advent of immunotherapy revolutionized the cancer therapy field. Among the current immunotherapeutic strategies, immune checkpoint blockades have greatly improved the overall survival rates in certain patient populations. Of note, CTLA4 and PD-1/PD-L1 are two major non-redundant immune checkpoints implicated in promoting cancer immune evasion, and ultimately lead to relapse. Antibodies or inhibitors targeting these two c+heckpoints have achieved some encouraging clinical outcomes. Further, beyond the canonical immune checkpoints, more inhibitory checkpoints have been identified. Herein, we will summarize recent progress in immune checkpoint blockade therapies, with a specific focus on key pre-clinical and clinical results of new immune checkpoint therapies for cancer. Given the crucial roles of immune checkpoint blockade in oncotherapy, drugs targeting checkpoint molecules expressed by both cancer and immune cells are in clinical trials, which will be comprehensively summarized in this review. Taken together, investigating combinatorial therapies targeting immune checkpoints expressed by cancer cells and immune cells will greatly improve immunotherapies that enhance host elimination of tumors.
The concept of immunotherapy was first introduced by McFarland Burnett and Louis Thomas in 1957, who proposed that lymphocytes were the primary actors responsible for mediating immune surveillance and tumor clearance (1–3). In the ensuing decades, immunotherapy was advanced into several major areas of study, including tumor infiltrating lymphocytes (TILs), chimeric antigen receptor (CAR)-T cells, T cell receptor (TCR)-T cells, oncolytic viruses, cytokine therapy, therapeutic cancer vaccines, and immune checkpoint blocking antibodies. The goal of these treatments was to unleash the power of the host immune systems to fight back against the cancer.
With years of research and clinical trials, immune checkpoint blocking therapies have been approved by the FDA in the United States and elsewhere. Currently, administration of immunotherapies has outweighed traditional therapeutic regimes for a variety of reasons. Firstly, immunotherapy exclusively targets immune cells and tumor cells, thus leaving other cells unscathed. Secondly, the adverse effects of immunotherapy are comparatively less severe than traditional chemotherapy and radiotherapy-treatment regimens (4). However, tumor cells are highly adaptable and can become unresponsive to immunotherapies, and therefore they continuously evolve mechanisms that evade host immunity and promote tumor persistence. Furthermore, neo-antigens, a hostile tumor microenvironment, T cell exhaustion, and other factors contribute to immune checkpoint-blockade resistance (5). Nevertheless, combinatorial use of immune checkpoint blockade with a broad spectrum of chemicals and antibodies can overcome therapy resistance, and has since produced promising clinical results (6). In this review, we comprehensively summarize the recent immunotherapy literature, with a focus on immune checkpoint blockade.
As mentioned above immune checkpoints (ICPs) could overcome the limitations of conventional cancer therapy as chemotherapy and radiotherapy. Therefore, followed by the discovery of the first immune checkpoint named CTLA4, a number of ICPs, including both stimulatory and inhibitory molecules, have been identified. Systematic assessment on the correlation between the ICPs of different cancers and the treatment responses/outcomes revealed that the ICPs varied across different cancers due to the cancer heterogeneity. Meanwhile, ICPs could be potential prognostic factors and therapeutic targets in certain types of cancer (e.g. breast cancer, lung cancer and ovarian cancer) (7). Hence, we summarized the ICPs according to their pivotal role in oncotherapy. Here is the detailed information of the major ICPs (Figure 1).
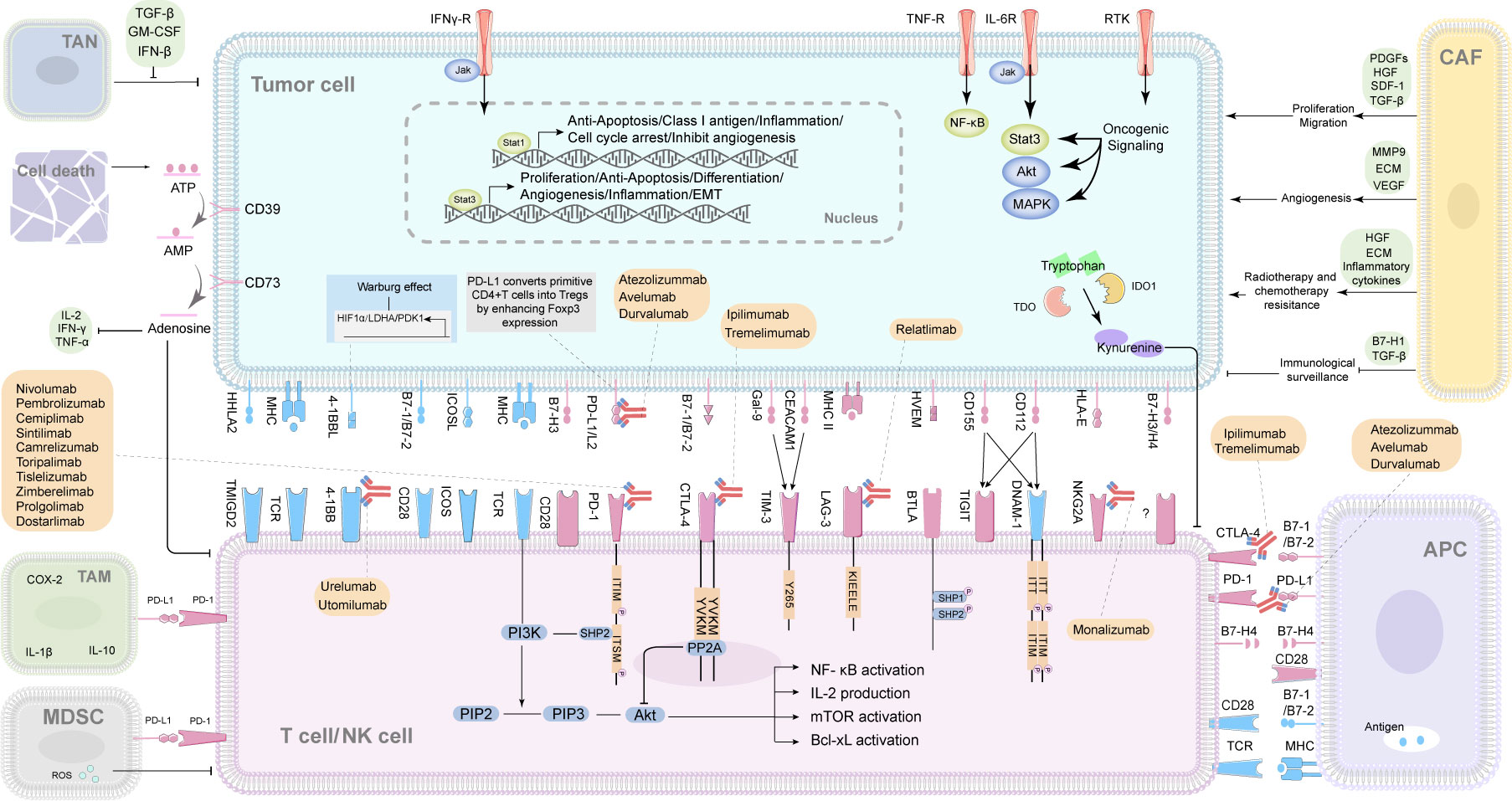
Figure 1 The interactions of immune checkpoints of cancers and immune cells, and the correlated immune checkpoint inhibitors (ICIs). TAN, tumor‐associated neutrophil; TAM, tumor-associated macrophage; CAF, cancer associated fibroblast; MDSC, myeloid-derived suppressor cell; APC, antigen-presenting cell; NK cell, natural killer cell; ATP, adenosine triphosphate; AMP, adenosine monophosphate; CD, clusters of differentiation; IL, interleukin; IFN-γ, interferon-γ; TNF-α, tumor necrosis factor; COX-2, cyclooxygenase-2; TGF-β, transforming growth factor-β; GM-CSF, granulocyte-macrophage colony stimulating factor; IFN-β, interferon-β; PDGFs, platelet-derived growth factors; HGF, hepatocyte growth factor; SDF-1, stromal cell-derived factor-1; MMP9, matrix metallopeptidase 9; ECM, extracellular matrix; VEGF, vascular endothelial growth factor; ROS, reactive oxygen species; TDO, tryptophan 2,3-dioxygenase; IDO1, indoleamine 2,3-dioxygenase1; HHLA2, The B7 family ligand HERV-H LTR–associating protein 2; TMIGD2, transmembrane and immunoglobulin domain containing 2; B7-, B7 family. B7-1(CD80), B7-2(CD86), B7-H1/PD-L1(CD274), B7-H2/ICOSL (CD275), B7-H3(CD276), B7-H4; 4-1BBL, 4-1BB ligand 4-1BB, CD137, a member of the TNF receptor superfamily, is an activation-induced T-cell costimulatory molecule; ICOSL, inducible costimulator ligand; ICOS, inducible costimulatory; MHC, major histocompatibility complex class; TCR, T cell receptor; PD-1, programmed cell death 1; PD-L1/L2, programmed cell death ligand-1/2; CTLA4, cytotoxic T-lymphocyte-associated protein 4; Gal-9, galectin-9; CEACAM1, Carcinoembryonic antigen elated cell adhesion molecule1; TIM-3, T cell immunoglobulin domain and mucin domain-3; LAG-3, Lymphocyte-activation gene 3; HVEM, Herpesvirus entry mediator; BTLA, B- and T-lymphocyte attenuator; TIGIT, T cell immunoreceptor with Ig and ITIM domains; DNAM-1, DNAX accessory molecule-1; HLA-E, major histocompatibility complex, class I, E; NKG2A, also called KLRC1, killer cell lectin like receptor C1; ITT, ITT-like motif; ITIM, immunoreceptor tyrosine-based inhibitory motif; ITSM, immunoreceptor tyrosine-based switch motif; PIP2, phosphatidylinositol-3,4-bisphosphate; PIP3, phosphatidylinositol-3,4,5-trisphosphate; Y265, a highly conserved tyrosine in the intracellular tail of Tim-3; KIEELE, conserved regions of Lag3 cytoplasmic domain, KIEELE motif; NF-κb, nuclear factor kappa-B; Bcl-xL, B-Cell Lymphoma-extra-large; SHP1/2, src-homology domain 2(SH2)-containing protein tyrosine phos-phatase-1/2.
In response to foreign pathogens, T cell activation is mediated by the T cell receptor (TCR) along with the combination of co-stimulatory positive signals such as CD28 and inducible co-stimulator (ICOS), or inhibitory signals such as cytotoxic T lymphocyte-associated antigen-4 (CTLA4, also referred to as CD152) and programmed cell death protein 1 (PD-1). The final T-cell response is determined by the balance between the positive and negative signals (8). T cell activation requires 2 signals, including: 1) the engagement of the T cell receptor (TCR) with major histocompatibility molecules (MHC) on antigen presenting cells (APCs) and 2) co-stimulation via binding of B7(CD80/CD86) on APCs to CD28 on T cells (8). As the first immune checkpoint protein discovered, CTLA4 plays a pivotal role in immune regulation (8, 9). CD28 is predominately expressed in resting T cells, while CTLA4 is absent or scarcely expressed in resting T cells. However, following T-cell activation, CTLA4 is expressed (10). Further study revealed that the intracellular localization of CTLA4 in T cells plays an essential role in its function (11). In naive T cells, CTLA4 protein is retained in the Golgi apparatus of the cell, and therefore exerts no effect on T cell activation (12). Upon engagement of TCR with MHC, CTLA4 is translocated from the cytoplasm to the plasma membrane, where it competes with CD28 for binding with CD80/CD86 by trans-endocytosis (13). CTLA4 engagement inhibits T cell activation and proliferation to maintain immune homeostasis, and protect against aberrant immune responses to self-tissues (14). In addition, CTLA4 also induces T-cell motility and inhibits the binding of T cells with antigen presenting cells such as dendritic cells (DCs) (15). CTLA4 competes with CD28 for B7 ligands for the inhibition of effector T cells, and stimulates the transcription of Foxp3 for regulatory T cells (Tregs) (16). Furthermore, by binding to its ligands B7-1 (CD80) and B7-2 (CD86), CTLA4 also reduces T cell responses by an attenuation of the induction of memory T cells (17). CTLA deficient mice show multi-organ lymphocyte infiltration and tissue destruction, implying the critical role of CTLA4 in down-regulating T cell activation and preventing autoimmunity (18). A number of investigators have identified the cytoplasmic binding partners of CTLA4 (19). The endocytosis of CTLA4 depends largely on its adaptor clathrin, where it binds to clathrin-associated adaptor complex (adaptor protein-2, AP-2) to induce CTLA4 internalization (20). When CTLA4 is internalized, it cannot deliver a negative signal. The binding domain of CTLA4 contains a YVKM motif, and when it is phosphorylated at the tyrosine site of the YVKM motif upon T cell activation, AP-2 can disengage from the binding at the same tyrosine site of CTLA4 (20, 21). With the disengagement of AP-2, CTLA4 is activated and functions as a negative regulator of T cells.
CTLA4 also plays an important role in the regulation of regulatory T cells (Tregs) functions, including Treg suppressive function, TCR hyposignaling and the induction of anergy (22). Deficient CTLA4 expression by Tregs fails to expand effector CD4+ T cells, even in the presence of interleukin-10 (IL-10) (23). Treg specific CTLA4 deficiency leads to reduced expression of CD80 and CD86 on dendritic cells (DCs), lymphocyte proliferation, T-cell mediated autoimmune diseases, and immunoglobulin E (IgE) production7 (24),. CTLA4 controls both the functional-development phase and the effector phase of Tregs (25). In addition, Tregs (which constitutively express CTLA4) regulate the infiltration of CD4+ T cell into tumors via a CTLA4/CD80-dependent manner (26).
Previous studies revealed that CTLA4 is constitutively expressed on tumor cells, and, therefore, CTLA4 blockade might stimulate tumor cell apoptosis and lead to the regression of tumors (27). Because CTLA4-inhibited immune activation relies primarily on the stimulation and activation of Tregs, the therapeutic effect of CTLA4 blockade principally acts via the regulation of Tregs (28). Administration of neutralizing monoclonal antibodies against CTLA4 are able to selectively deplete FOXP3+ Treg cells, and thereby promote tumor immunity by activating effector CD8+ T cells (29). Clinically, treatment with anti-CTLA4 monoclonal antibodies (mAbs) does not deplete Foxp3+ Tregs, but does increase CD4+ and CD8+ T cell infiltration into tumors (30).
Programmed cell death protein 1 (PD-1) was named based on its initial identification as a receptor inducing cell death in an activated T cell hybridoma (31), and is now recognized as a dominant negative regulator of T cells similarly to CTLA4 (32). Like CTLA4, PD-1 is absent from naïve and memory T cells, but is expressed upon TCR engagement (33). In contrast to CTLA4, PD-1 is expressed on the surface of activated T cells, and contains a conventional immunoreceptor tyrosine inhibitory motif (ITIM) and immunoreceptor tyrosine switch motif (ITSM). PD-1 engagement leads to the activation of the inhibitory phosphatase SHP-2, thus resulting in the inhibition of TCR mediated function, increased T cell migration, and immune evasion within the tumor microenvironment (TME) (34). Unlike the ligand of CTLA4, one of PD-1’s ligands known as programmed death- ligand 1 (PD-L1), is broadly overexpressed on tumor cells and infiltrating leukocytes (35); this allows for the induction of PD-1 mediated T cell exhaustion by tumor cells (36). However, this phenotype can be reversed via blockade of either PD-1 or PD-L1, resulting in elevated antitumor cytotoxic T cell responses and tumor regression (35). Because CTLA-4 inhibits T cells at the early stages in the lymph nodes, the CTLA4 deficient mice show severe lymphoproliferative disease, lethal lymphocytic infiltration in multiple organs and tissue destructions (37). However, because PD-1 regulates in the immune response in the peripheral tissues, the phenotypes of PD-1 and PD-L1 deficient mice are comparatively milder, as evidenced by delayed onset of inflammation and lack of severe organ destruction. These differences highlight the importance of the PD-1 pathway, specifically, in tumor immunotherapy (38, 39).
It is widely acknowledged that CTLA4 regulates T-cell proliferation in lymph nodes (40). Therefore, CTLA4 antibodies (ipilimumab and tremelimumab) can induce clinically unrestrained T-cell activation (41). And the major immune-related adverse events (irAEs) of CTLA4 blockade have been reported to be diarrhea, dermatitis, hepatitis, and endocrinopathies. However, PD-1 suppresses T cells primarily in peripheral tissues (40), and, hence, blockade of PD-1 or PD-L1 exerts less frequent and mild autoimmune adverse effects compared with CTLA4 mAb therapies (35). However, the therapeutic outcomes of PD-1 blockade on solid tumors have remained relatively poor, mainly due to the hostile TME, and the presence of immunosuppressive Tregs (42). PD-L1 has been implicated in the development and function of Tregs by enhancing the expression of Foxp3. It also converts naive CD4+ T cells to Tregs, and stimulates the immunosuppressive function of Tregs (43). The expansion of Tregs could be regulated by PD1 and PD-L1 via Notch signaling pathway (44). Additionally, in myeloid tumor cells (K562) or T cells, the PD-L1 expression was able to convert Th1 cells into Tregs (45). Furthermore, PD-1 blockade stimulates the proliferation of PD-1+ effector Treg cells, inhibiting the antitumor immunity (46, 47). Therefore, the efficacy of PD-1 blockade therapy may be predicted by the levels of PD-1 expression on effector T cells (Teff) and Tregs (48), and should be used as a biomarker to predict patient responsiveness.
T cell immunoglobulin and mucin-3 (Tim-3; also known as HAVCR2) is broadly expressed on activated T cells, FoxP3+ Treg cells, NK cells and monocytes (49). Tim-3, similarly to PD-1 and CTLA4, suppresses the immune response upon interaction with its ligand galectin-9 (49). The main functions of Tim-3 include promoting CD8+ T cell exhaustion, and inducing the expansion of myeloid-derived suppressor cells (MDSCs) and Tregs. Co-expressed with carcinoembryonic antigen cell adhesion molecule 1 (CEACAM1), the maturation and cell surface expression of Tim-3 is regulated by CEACAM1 by forming a heterodimer to mediate immune tolerance and T cell exhaustion (50). Blockade of Tim-3 accelerates the pathogenesis of autoimmune diabetes in nonobese diabetic (NOD) mice, and inhibits transplantation tolerance induced by co-stimulation blockade (51), highlighting Tim-3’s immunoregulatory role. As a T helper type 1 (Th1)–specific cell surface marker, Tim-3 binds with its ligand galectin-9, causes the hyperproliferation of Th1 cells and the release of Th1 cytokine, and thereafter induces peripheral tolerance (52, 53). Activation of the Tim-3/galectin-9 pathway is also known to increase the frequency of CD11b+Ly-6G+ myeloid cells, inhibits immune responses (54). Clinically, Tim-3 promotes T-cell exhaustion and is associated with the poor prognosis in hepatocellular carcinoma (HCC) patients (55). Tim-3 blockade also increases the pathological severity of the Th1-dependent autoimmune disease of experimental autoimmune encephalomyelitis (EAE) in mice (56).
Regarding Tregs, Tim-3+ Treg is highly effective at inhibiting T-cell proliferation (57, 58), and the blockade of Tim-3 induces a reduction in the frequency of CD4+CD25+Foxp3+ Tregs (59). Experiments using murine tumor xenograft models suggest that Tim-3 is expressed on PD-1 expressing CD8+ TILs. Functionally, Tim-3+PD-1+ TILs are severely exhausted, as evidenced by a failure to secrete interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), and interleukin-2 (IL-2) (60). Therefore, compared with the inhibition of CTLA4 pathway or PD-1/PD-L1 pathway alone, combinatorial Tim-3 and PD-1 blockade has been demonstrated to be more effective (60). For example, combinatorial therapy reverses tumor-induced T-cell exhaustion and dysfunction in patients with colorectal cancer by increasing the proliferation of tumor antigen-specific CD8+ T cells and decreasing immunosuppressive Treg populations (61). In patients with advanced melanoma, increased expression of Tim-3 and PD-1 correlates with tumor antigen-specific CD8+ T cell dysfunction (62). A recent study using triple therapy (PD-1 blockade, Tim-3 blockade and anchored granulocyte- macrophage colony- stimulating factor (GM-CSF) vaccination) demonstrated tumor regression levels higher than 50% (63). This was associated with reduced apoptosis of CD8+ TILs, decreased production of tumor-promoting cytokines, and improved cytotoxicity in bladder cancer (64) and prostate cancer (65). In another study, combinatorial Tim-3/Lag-3/PD-1 blockade significantly improved antitumor immunity in gastric cancer cell-T cell coculture models, suggesting the therapeutic potential of combinatorial therapy in gastric cancer patients (66). Tim-3 could also be used as a biomarker for cancer therapy responsiveness. In medullary thyroid cancer, a cohort study of 200 patients revealed that Tim-3, CTLA4 and PD-1/PD-L1 are promising biomarkers for tumor recurrence (67). In head and neck cancer, increased PD-1+ and Tim-3+ CD8+ TILs were inversely correlated with clinical outcome of cetuximab therapy (68). Ultimately, Tim-3 blockade has been proven in pre-clinical studies to be beneficial in improving current immune-checkpoint blockade therapies when applied in combination. These promising results provide precedence for assessing the use of Tim-3 blockade in clinical trials so as to improve clinical outcomes of cancer patients.
Discovered in 1990, lymphocyte activation gene-3 (Lag-3; also referred to as CD223), is an immune checkpoint molecule expressed by activated T cells, Tregs, NK cells and B cells (69–72). Lag-3 is structurally homologous to the CD4 co-receptor and competes with CD4 for its binding with MHC class II (73). Inhibition of Lag-3 results in the expansion of T cells, B cells, macrophages, granulocytes, and DCs (74), and increases activation of CD8+ T cells (75). With respect to Lag-3’s regulatory functions, a study by Workman et al. revealed an inability of Vβ7/8+ (T cells bearing Vβ7 and Vβ8 (Vβ7/8) T cell receptors) Lag-3-deficient T cells to expand, even when stimulated with staphylococcal enterotoxin B. Expansion of Lag-3 deficient T cells was subsequently restored by the constitutive expression of Lag-3 (76). Lag-3 is differentially expressed on induced Tregs and is required for Treg function. Upon activation, natural CD4+CD25+ Tregs express Lag-3 to suppress effector cells, whereas CD4+CD25+ Tregs from Lag-3 knockout mice exhibit reduced regulatory activity (77). Lag-3 is also a biomarker for active CD4+CD25highFoxp3+ Tregs (78). CD49b and Lag-3 are stably and selectively co-expressed on CD4+ type 1 Tregs (Tr1), which induce and maintain immune tolerance (79). Moreover, Lag-3 is expressed on CD11cl°w/B220+/PDCA-1+ plasmacytoid DCs, and regulates their homeostasis (80). Lag-3 binds with MHC II expressed within plasma membrane lipid rafts of immature human DCs, and promotes their maturation and activation (81). Due to the pivotal role of Lag-3 in immune regulation, several diseases including autoimmune diseases, cancer, chronic viral infection, and parasitic infection are correlated with aberrant Lag-3 expression (82–84). Given the immunoregulatory role of Lag-3, the combinatory therapies targeting both Lag-3 and PD-1 have gained some encouraging clinical outcomes (85–87). Lag-3 and PD-1 contribute to the rapid trafficking of the immunological synapse upon T-cell activation, and lead to a synergistic inhibitory effect on T-cell signaling (88). Dual blockade of Lag-3 and PD-1 resulted in the clearance of multiple established tumors, which were resistant to PD-1 blockade alone (89). Another study in renal cell carcinoma revealed that the inhibition of PD-1 resulted in a significant upregulation of Lag-3, and the blockade of both immune checkpoint proteins leads to increased IFN-γ secretion, emphasizing the significance and the potential of combination therapy (90). Therefore, to overcome the resistance of PD-1 blockade in resistant cancers, the synergistic blockade of Lag-3 and PD-1 may provide a promising therapy.
Discovered by genome-wide association studies (GWAS), the co-stimulatory molecule CD226 together with T cell immunoreceptor with immunoglobulin and ITIM domain (TIGIT) forms a striking pathway similarly to the CD28-CTLA4 pathway, and is correlated with multiple autoimmune diseases (91, 92). TIGIT is expressed by the activated T cells, Tregs, and NK cells (93). The ligand for TIGIT is poliovirus receptor (PVR) CD155, which also binds to CD226. Hence, in the TIGIT-CD226 axis, TIGIT can bind and disrupt CD226 homodimerization, and therefore directly inhibits T-cell activation, proliferation, and effector function by competing with CD226 for binding to CD155 (94, 95). TIGIT has been shown to suppress antitumor immunity by increasing the frequency of Tregs that express the co-inhibitory receptor Tim-3 and that induce CD8+ T-cell dysfunction (96). TIGIT+ Tregs selectively suppress the responses of proinflammatory Th1 and Th17 cells via the upregulation of fibrinogen-like protein 2 and the secretion of IL-10 (97). TIGIT represses IFN-γ secretion, promotes the nuclear translocation of forkhead box O1 (FoxO1), inhibits protein kinase B (PKB, also called Akt) function, and restores suppressor function of Tregs (98). TIGIT also plays an important role in regulating NK cell responses. In mouse models, the blockade of TIGITs interaction with its ligand PVR leads to increased NK-cell mediated cytotoxicity (99). In tumor models, TIGIT exerts its regulatory function and inhibits NK-mediated cytotoxicity on tumor cells to promote immune evasion (100). Antibody blockade of TIGIT could inhibit NK cell exhaustion and promote NK cell dependent tumor immunity (101). Clinically, dual blockade of TIGIT and PD-1 could increase the frequency of CD8+ TILs and tumor antigen specific CD8+ T cells in established melanoma patients (102). Dual PD-1 and TIGIT blockade treatment in glioblastoma multiforme (GBM) patients showed decreased effector T cell function, increased Tregs, and increased tumor infiltrating dendritic cells (TIDCs) (103). In B-cell non-Hodgkin’s lymphoma patients, the response rates (RRs) of PD-1 blockade are comparatively low, and these patients display increased CD8+ and CD4+ T effector memory cells expressing TIGIT and PD-1 with limited IFN-γ, TNF-α, and IL-2 production (104). In gastric cancer patients, TIGIT and PD-1 were found to be upregulated on infiltrating CD8+ T cells in tumor tissues, suggesting that TIGIT may serve as an emerging biomarker (105). In follicular lymphoma, increased numbers of TIGIT+ T cells are associated with poor survival, therefore TIGIT may serve as a predictive marker for therapeutic clinical outcomes (106).
The B and T lymphocyte attenuator (BTLA) is an immunoglobulin superfamily member, which downregulates T-cell activation (69). BTLA contains a glycoprotein with two immunoreceptor tyrosine-based inhibitory motifs. The ligand for BTLA is B7x, a peripheral homolog of B7. BTLA is not expressed in naïve T cells, but it is expressed on Th1 cells upon activation (107). After crosslinking BTLA with antigen receptors, BTLA attenuates IL-2 secretion, and therefore it functions as an inhibitory receptor similar to CTLA4 and PD-1 (108). Blockade of BTLA in T-cells increased T-cell proliferation, and BTLA deficient mice display increased incidence and severity of autoimmune diseases, such as EAE. A recent report suggests that PD-1 and BTLA can suppress T cell signaling via SHP-1 and SHP-2 (109). BTLA also functions as a negative regulator of B cell receptor (BCR) signaling by binding with its ligand herpesvirus entry mediator (HVEM). Engagement of BTLA4 leads to recruitment of the tyrosine phosphatase Src homology 2 domain containing phosphatase 1, and reduces the activation of BCR-signaling pathways (108). Furthermore, BTLA4 engagement inhibits the activation of B cells by targeting Syk and B cell linker proteins (110).
BTLA is also expressed on DCs and macrophages (111, 112). BTLA plays a regulatory role in peripheral tolerance, evidenced by the ability of BTLA+ DCs to induce CD8+ T cell tolerance that could alleviate the severity of type 1 diabetes (113). Regarding Tregs, BTLA governs the cell differentiation and activation of Tregs (114). BTLA deficient mice have fewer Tregs and develop autoimmune diseases such as EAE. Interestingly, adoptive transfer of myelin oligodendrocyte glycoprotein fused reovirus protein σ1-B220+CD5+ Bregs, which express elevated BTLA levels, protects against EAE development, suggesting that BTLA plays a critical role in the activation of Tregs and Bregs (115). Therapeutically, several studies have explored the application of BTLA4 blockade in improving clinical outcomes in patients with colorectal cancer, epithelial ovarian cancer, lung cancer, gastric cancer, and other cancers (116–119). BTLA blockade inhibits IL-6 and IL-10 secretion, induces CD19hi B cells through AKT and STAT3 signaling pathways, and significantly improves the therapeutic outcomes in epithelial ovarian carcinoma patients (117). Another study revealed that up-regulation of BTLA occurs independently of functional exhaustion, which is induced by high antigen load. Combinatorial blockade of PD-1, Tim-3 and BTLA could enhance the expansion, proliferation, and cytokine production of CD8+ T cells (120). Taken together, BTLA should be further explored to enhance the clinical responsiveness to immune checkpoint blockade therapies.
In addition to the immune checkpoints mentioned above, other immune checkpoint molecules have been identified including B7 family molecules (B7-H3, B7-H4 and HHLA2), indoleamine 2,3-dioxygenase 1 (IDO-1), V-domain Ig suppressor of T cell activation (VISTA), and inducible T cell co-stimulator (ICOS). B7-H3, also known as CD276, is a newly discovered immune checkpoint protein, and plays a crucial role in the adaptive immune response in human cancers (121, 122). As a transmembrane glycoprotein, B7-H3 contains single variable and several constant immunoglobulin domains. It binds with its receptor CD28 which is universally expressed on T cells, B cells, monocytes, NK cells, and APCs including DCs and myeloid-derived suppressive macrophages (123, 124). Moreover, it is also constitutively expressed on the surface of tumor cells fibroblasts, tumor endothelial cells, osteoblasts and amniotic fluid stem cells (125). Upon interaction with CD28, B7-H3 ultimately leads to immune evasion by inhibiting the activation of T cells. B7-H3 deficient mice have increased frequencies of differentiated Th1 cells associated with severe inflammation in the respiratory system and the development of autoimmune encephalomyelitis earlier than wild type littermates (126). However, B7-H3 also possesses a co-stimulatory role by inducing IFN-γ production, increasing T cell proliferation (121), and stimulating the differentiation of CD8+ T cells that drive antitumor immunity (127). CD4+CD25+Tregs inhibit DC activation via up-regulation of inhibitory B7-H3 on DCs. B7-H3 engagement also results in decreased MHC-peptide complexes and impairs T cell activation (128). The expression of B7-H3 and B7-H4 has been reported to be correlated with metastasis by stimulating the expression of immunosuppressive IL-10 and TGF-β1 (129). Studies in tumor metabolism revealed that B7-H3 increases the expression levels of hypoxia-inducible factor 1 α (HIF1α), lactate dehydrogenase A (LDHA), and pyruvate dehydrogenase kinase 1 (PDK1), and therefore stimulates the Warburg effect by increasing glucose uptake and lactate production (130). These studies provide evidence that B7-H3 can regulate aspects of both cancer persistence and immune regulation, and should be considered as a potential target in the next stage of immune checkpoint blockade therapies for cancer.
B7-H4, also referred to as B7x and B7-S1, is a highly conserved immunoglobulin superfamily and belongs to the B7 family (131). It is ubiquitously expressed by tumor tissue and immune cells, including T cells, APCs, DCs, peritoneal macrophages and B cells (131, 132). B7-H4 negatively regulates T cell activation by decreasing T cell proliferation, inhibiting the production of IL-2 and inducing cell cycle arrest of T cells (133, 134). B7-H4 regulates pro-inflammatory T-cell responses by inhibiting CD4+ T-cell proliferation and differentiation to both Th1 and Th17 cells, inducing the production of IL-10, and increasing Treg activation (135). Deficiency of B7-H4 leads to elevated Th1 and Th17 responses (136). The expression of B7-H4 was shown to be up-regulated in various cancers, including breast, pancreatic, ovarian, lung, renal cell, and endometrial cancers (137–140). The expression levels of B7-H4 have also been recognized to constitute a prognostic indicator associated with poor overall survival in pancreatic cancer patients (137).
HHLA2 (HERV-H LTR-associating 2; also referred to as B7H5 and B7H7) belongs to the B7 family and is expressed by macrophages and DCs. As a novel co-stimulatory molecule, HHLA2 binds with CD28H (also known as Transmembrane and Immunoglobulin Domain Containing 2 (TMIGD2)), stimulates T cell proliferation and increases cytokine production via induction of the Akt signaling pathway (141–143). When combined with TCR and CD28 stimulation, it plays a co-inhibitory role comparable to PD-L1 (144). In T cells and NK cells, it also binds with KIR3DL3 (killer cell immunoglobulin-like receptor, three immunoglobulin domains and long cytoplasmic tail 3) to mediate tumor immune evasion independently from PD-L1 (145). HHLA2 inhibits T-cell proliferation and cytokine production including IFN-γ, TNF-α, IL-5, IL-10, IL-13, IL-17A, and IL-22 (146). Therefore, it has been reported to be a novel prognostic predictor, similar to PD-1, in tumor immunotherapy (147, 148).
V-domain Ig-containing Suppressor of T cell Activation (VISTA; also referred to as PD-1H, DD1α, c10orf54, Gi24, Dies1 and SISP1) is a transmembrane protein containing a single N-terminal immunoglobulin (Ig) V domain (149). Phylogenetically, VISTA is similar to PD-1, CD28, and CTLA4 (150). It is the most conserved molecule among the B7 family members, sharing similar domains with CD28 and CTLA4 within its cytoplasmic tail (149). Studies on VISTAs Src homology 2 (SH2) binding motif suggest that it functions as both a ligand and receptor in controlling immune responses (149, 151). VISTA is mainly expressed on cells of the hematopoietic lineage, including macrophages, DCs, monocytes, and circulating neutrophils (152). Albeit absent in B cells, it is unanimously expressed in naïve T-cells, CD4+ memory T- cells and Tregs (150). Weak expression of VISTA is also observed in CD8+ T cells and NK cells (149). VISTA deficiency results in enhanced activation of T-cells with increased production of IFN-γ, TNF-α and IL-17, and contributes to the onset of overt autoimmunity (151, 153). Within the TME, VISTA plays a crucial role with increased expression observed on myeloid DCs, myeloid-derived suppressor cells (MDSCs) and Tregs (154). Blockade of VISTA decreases the number of MDSCs, increases activated DCs, and reduces the frequency of tumor specific Tregs (155).
The immune checkpoint molecules indoleamine 2,3-dioxygenase-1 (IDO-1) and tryptophan dioxygenase (TDO) belong to the tryptophan catabolic enzyme family. They catalyze the degradation of tryptophan (Trp) into kynurenine (Kyn) (156, 157). IDO-1 plays an immunosuppressive role in various cancers (158) by suppressing CD8+ T effector (Teff) cells and NK cells (159, 160). Further, it stimulates the proliferation of Tregs and MDSCs (161), decreases the ratio of Th17 cells versus Tregs, inhibits Th17 related cytokine production, and decreases the number of Th1 and Th22 cells (162, 163). Moreover, IDO-1 regulates the recruitment, polarization and phagocytosis capabilities of macrophages (164, 165). TDO has also been reported to be an immunosuppressive molecule capable of reducing CD8+ T cell viability, stimulating the secretion of cytokines such as IFN-γ, TNF-α, IL-10 and IL-17 by TDO reactive CD4+ T-cells, and contributes to the tumor metastasis (166–168).
Inducible Co-Stimulator (ICOS), a homodimeric protein expressed on activated T cells, is an inducible T-cell co-stimulator (169). Specifically, it shares common signaling mechanisms with CD28, and increases T cell responses by stimulating T-cell proliferation and cytokine secretion (169). By binding with its ligand (B7-H2, B7RP-1), ICOS stimulates the response of T effector cells and T cell-dependent B cells (170). Further, this interaction promotes IL-10 secretion by effector T cells even in the presence of mature DCs (171). ICOS blockade attenuated T cell proliferation and inhibited Th effector responses by decreasing Th2 cytokine secretion (172). Inhibition of ICOS attenuates cytokine productions as IL-2, IL-4, IL-5, and IFN-γ, and decreases the Th2 mediated mucosal inflammation (173). Deficiency in ICOS fails to form T follicular helper (Tfh) cells. Therefore, inhibition of ICOS decreases the immunosuppressive function of Tregs and decreases the expression of Tfh markers of the lymphoid tumor cells (174, 175). Beyond the canonical immune checkpoints of CTLA4 and PD-1/PD-L1, other immune checkpoints may, therefore, also be crucial in reducing immune responses in numerous cancers. Combination therapy with classical inhibition of CTLA4 and PD-1/PD-L1 may, thus, result in a durable clinical response in cancer therapy.
Tremendous progress has been achieved in cancer immunotherapy over the past few decades. To date, the most widely applied immunotherapeutic drugs are antibodies that block immune inhibitory receptors such as CTLA4, PD-1, and PD-L1 (Supplementary Table 1). Although the FDA has approved these drugs to treat various cancer types, a large number of immune checkpoint inhibitors (ICIs) targeting other inhibitory receptors are in clinical development. In this section, we will summarize these drugs in cancer immunotherapy.
Ipilimumab was the first FDA approved CTLA4 inhibitor to hit the market (176). In humans, a phase III clinical trial in 2010 (MDX010-20, NCT00094653) in patients with unresectable or metastatic melanoma evaluated the efficacy and safety of ipilimumab (3 mg/kg) and found it alone extended the median overall survival (OS) of patients by more than three months compared with the control group, showing that a single ICI can have potent antitumor effects (177). In 2011, ipilimumab was approved by the FDA for the treatment of late-stage (metastatic) melanoma patients (176), and 3 mg/kg administered once every 3 weeks (Q3W) for four cycles was adopted as the standard of care based on the results of several clinical trials (177–179). Besides melanoma, clinical trials using single-dose, 3 mg/kg ipilimumab have been conducted in patients with ovarian cancer (180, 181) and prostate cancer (182), but the its effect was not as effective in these cancer types compared to the success rates for melanoma patients. Thus, use of ipilimumab in combination with other treatment modalities, such as additional immunotherapies, chemotherapies and radiotherapies, are being tested in the clinic to improve success rates in multiple cancers. For example, 10 mg/kg ipilimumab plus dacarbazine (a chemotherapeutic agent) prolonged the median OS from nine months to 11 months in patients with previously untreated metastatic melanoma compared with dacarbazine plus placebo (183).
Another CTLA4 inhibitor, tremelimumab, is a fully human mAb containing an IgG2 Fc domain in order to minimize Fc domain effector functions such as complement fixation and antibody-dependent cytotoxicity (184, 185). Tremelimumab alone has displayed potential antitumor effects in advanced melanoma patients in phase I, I/II and II trials (186–188). However, in a phase III trial in patients with unresectable stage IIIc–IV melanoma who had not received prior systemic treatment, tremelimumab was found to not be superior to standard of care chemotherapy regimens (median OS 11.8 months vs 10.7 months, hazard ratio (HR) 0.96) (189). These disappointing results led to early termination of the study and has limited available data of tremelimumab in melanoma patients. Besides melanoma, tremelimumab monotherapy has been studied in several other solid tumors, including metastatic esophageal and gastric adenocarcinoma, locally advanced or metastatic non-small cell lung cancer (NSCLC), treatment-refractory colorectal cancer (CRC), etc, but no satisfactory results have been obtained (190–193).
Currently there are three anti-PD-1 and three anti-PD-L1 antibodies approved by the FDA for treatment of more than 10 cancer indications. The first PD-1 inhibitor to enter the clinic was nivolumab in 2006, and the first to receive FDA approval was pembrolizumab in 2014 for the treatment of advanced melanoma patients (194). Both of these PD-1 inhibitors contain human IgG4 Fc regions containing mutations in the stabilizing hinge region (195). Nivolumab was tested in 2010 in a phase I trial in 39 patients with advanced solid tumors that were treatment-refractory (196). These tumor types included metastatic melanoma, prostate cancer, NSCLC, renal cell carcinoma (RCC) and CRC. Different doses (0.3, 1, 3, 10 mg/kg) of nivolumab were used and antitumor activity was found in 1 CRC (complete response (CR), 3 mg/kg), 1 melanoma (partial response (PR), 10 mg/kg) and 1 RCC patient (PR, 10 mg/kg). The drug was well-tolerated and common AEs observed were reduced lymphocyte numbers, fatigue, and musculoskeletal symptoms (196). In a subsequent study, a multidose (ranging from 0.1 to 10 mg/kg) clinical trial with expanded patient numbers (N=296) was conducted in the same tumor types (197). CR or PR was observed in melanoma (28%), RCC (27%) and NSCLC (18%) patients, and phase III trials were then initiated for these indications (198–201). Considering the promising results from these trials, the FDA approved nivolumab for the treatment of refractory melanoma in 2014 and NSCLC in the following year (202). Since then, nivolumab has gained FDA approval for the treatment of multiple solid tumor types (Supplementary Table 1). Interestingly, in classical Hodgkin’s lymphoma (cHL), the surface of tumor cells also overexpresses PD-L1/PD-L2 due to genetic alterations on chromosome 9p24.1 (203, 204). In a phase I study, nivolumab showed beneficial responses in 87% (20/23) of relapsed/refractory cHL patients (205). In a phase II study, the overall objective response rate (ORR) was 69% and median PFS was 14.7 months (206). Thus nivolumab was also approved for treating cHL patients in 2016 as the first PD-1 inhibitor (207).
Pembrolizumab was produced by grafting the variable region sequences of a mouse mAb onto a homologous human IgG4-κ isotype framework containing a stabilizing S228P Fc mutation to reduce the immunogenicity of the mouse variable region (208). In a Phase I trial of patients with advanced ipilimumab-refractory melanoma, 26% of patients in the pembrolizumab group showed either CR or PR (209). Later in a Phase III trial in patients with advanced melanoma, the RR was significantly higher in pembrolizumab 10 mg/kg Q2W (33.7%) and Q3W (32.9%), compared with ipilimumab (11.9%) (210). In NSCLC patients (untreated, advanced), a Phase III trial showed the median PFS was 10.3 months in pembrolizumab, compared to 6.0 months in chemotherapy groups and the RR was 44.8% and 27.8%, respectively (211). Pembrolizumab was well tolerated, and the most common AEs reported were fatigue, pruritus, and rash. Similar to nivolumab, pembrolizumab is approved for treatment of patients with multiple cancer types (202).
Cemiplimab is another human IgG4 antibody that is approved for treating cutaneous squamous cell carcinoma (cSCC), basal cell carcinoma and certain types of NSCLC (212, 213). In a meta-analysis comparing the efficacy of cemiplimab and pembrolizumab in advanced cSCC patients, cemiplimab treatment was associated with better OS (HRs ranging from 0.21–0.52) and PFS (HRs ranging from 0.49–0.55) (214). However, this conclusion should be interpreted with caution as its results were from indirect comparison without evidence from head-to-head clinical trials. The overall number of comparator studies included in this meta-analysis was low, and some studies enrolled small numbers of patients. While data from nivolumab or pembrolizumab trials treating locally advanced or metastatic vulvar SCC has been scarce, a phase III trial (NCT03257267) investigated cemiplimab in patients with recurrent/metastatic (RM) cervical cancer, who were resistant to first-line platinum-based therapy. Results from the overall population highlighted a 31% reduction in the risk of death as well as a 25% reduction in disease progression and an ORR of 16% (n = 50) [6% (n = 19) in the chemotherapy-treated cohort]. Further, the median OS in the cemiplimab group was 12 months compared to 8.8 months with chemotherapy, and the drug was well tolerated (215). According to these data, cemiplimab was granted priority review by the FDA for patients with RM cervical cancer who have experienced disease progression while on or after chemotherapy treatment regimes.
Besides the anti-PD-1 mAbs discussed here, a number of other anti-PD-1 mAbs have been approved by China or the European Union for treatment of different cancers, such as sintilimab, camrelizumab, toripalimab, tislelizumab, zimberelimab, prolgolimab, or dostarlimab; all of which have shown promising therapeutic effects for various cancer related indications (216).
In addition to PD-1 blocking agents, antibodies targeting PD-L1 also have potent antitumor qualities. Currently, three anti-PD-L1 antibodies, atezolizumab, avelumab, and durvalumab, are approved by the FDA to treat several cancers (217). Atezolizumab is a human IgG1 that has an N297A mutation to eliminate the unwanted fragment crystallizable (Fc)-mediated functions such as antibody-dependent cytotoxicity (ADCC) by aglycosylation (218). Durvalumab also contains a modified Fc region to prevent depletion of PD-L1 expressing T cells (217). In contrast, avelumab owns a native Fc region which can bind to Fc-γ receptors on NK cells to initiate ADCC (217). Their approved indications, time and dosage are summarised in Supplementary Table 1.
Despite the success of CTLA4 or PD-1/PD-L1 inhibition, a majority of patients develop drug resistance. Thus, the identification of novel and non-redundant pathways of immune inhibition is important for advancing therapies that promote anti-tumor immunity. In a phase III clinical trial of anti-Lag-3 (relatlimab, 160 mg Q4W) combined with nivolumab (480 mg Q4W) for melanoma patients, patients receiving dual Lag-3/PD-1 blockade showed an increased PFS than those receiving nivolumab alone (median PFS 10.1 months vs 4.63 months, and PFS rates 47.7% vs 36.0% at 12 months). The combined regimen was well tolerated and total AEs were not significantly increased: 18.9% vs 9.7% grade 3/4 TRAEs and 14.6% vs 6.7% treatment discontinuations (219). Thus, this fixed-dose combination trial laid the foundation for dual inhibition apart from anti-CTLA4/anti-PD-1 pathways for clinical use and was recently FDA approved for patients with unresectable or metastatic melanoma (220).
Monalizumab is a humanized anti-NKG2A antibody that has the ability to enhance the anti-tumor activity of NK cells (221). In A20 tumor-bearing mice, combined treatment with anti-NKG2A and anti-PD-L1 mAbs rescued 75% of the mice from death compared to 40% in the anti-PD-L1 alone group (221). Although monalizumab alone was tested in patients with RM squamous cell carcinoma of the head and neck (SCCHN) in a phase II trial (222), the study did not meet its primary endpoint (no OR was observed) and it was terminated at the interim as ineffective (222). The safety profile of monalizumab was favorable, and it is now being tested in combination with durvalumab in a biomarker-based study in RM SCCHN patients. Cetuximab, an anti-EGFR mAb, promoted ADCC by binding to CD16/FcγRIII (223), and the ADCC induced by cetuximab was enhanced by monalizumab through NK cell stimulation, providing greater anti-tumor activity than cetuximab alone (221). A phase II trial of monalizumab plus cetuximab in SCCHN patients showed a 31% ORR, with the most common AEs being fatigue (17%), pyrexia (13%), and headache (10%) (221). These results led to an ongoing phase III (INTERLINK-1) trial for RM HNSCC patients who underwent prior ICI- and platinum-based chemotherapy treatments. Although monalizumab has not yet gained FDA approval, it has shown benefit in enhancing the effects of other ICIs. However, future studies are needed to carefully define the suitable tumor types and to determine which type of combination therapy provides the optimal benefits.
In several preclinical cancer models, TIM-3 blockade displayed a slight advantage in terms of efficacy, especially when combined with PD-1 blockade (60, 224). Several-in-human Phase I/II trials have been initiated for numerous TIM-3 antibodies in patients with solid tumors or lymphoma (225). Most of them have been tested in conjunction with anti-PD-1/PD-L1 mAbs. TSR-022 (TIM-3 antibody, Tesaro) plus TSR-042 (anti-PD-1 antibody) demonstrated improved antitumor immunity in NSCLC patients who failed to respond to anti-PD-1 treatment alone previously (226). A SCLC patient who received LY3321367 (a novel TIM-3 mAb) 1200 mg Q2W as a single agent also achieved PR (227). All in all, these early data suggest that blocking TIM-3 alone or together with PD-1/PD-L1 is generally safe, well tolerated, and provides a slight advantage in efficacy compared to anti-PD-1/PD-L1 therapies alone.
Despite the PD-1/PD-L1 pathway being recognized as a key checkpoint in the immune response of various cancer types, most patients do not benefit from PD-1/PD-L1 pathway blocking therapies due to primary or acquired resistance (228). The immune system can paradoxically suppress or promote cancer development and growth. Therefore, removing or blocking the factors that promote immune tolerance and boost anti-tumor immunity may facilitate better responses to anti-PD-1/PD-L1 therapy. Based on this conception, combinations of anti-PD-1/PD-L1 mAbs with chemotherapies, targeted therapies, or CTLA4 blockade have been tested clinically and some regimes become the standard of care for several cancer types including metastatic melanoma, kidney, lung, head and neck, triple negative breast (TNB), and liver cancers. Herein, we will briefly discuss the progress that has made with dual combination of ICIs and alternative therapeutic regimes (Table 1).
Although CTLA4 and PD-1 are both immune checkpoints, they can suppress the activation of T-cells in non-redundant manners, and therefore dual blockade may work synergistically to enhance anti-tumor immune responses. Neoadjuvant ipilimumab plus nivolumab in patients with macroscopic stage III melanoma showed a 2-year estimated recurrence-free survival rate of 84% in all patients, a 97% estimated survival rate in patients who achieved a pathologic response, and a 36% estimated survival rate in non-responding patients (229). In addition to melanoma, many neoadjuvant ipilimumab plus nivolumab trials have been conducted in solid tumors, and have demonstrated enhanced responses in advanced RCC (230), microsatellite high/deficient mismatch repair colorectal cancer (mCRC MSI-H/dMMR) (231), HCC (232), NSCLC [at 12 months (233) and 24 months (234)], and malignant pleural mesothelioma (MPM) (235). Based on these clinical trials, the FDA has approved the combination of ipilimumab and nivolumab for the six cancer types mentioned above.
The combination of tremelimumab and durvalumab are being tested in several phase 3 trials (236–248). Although some results were unsatisfactory, this combination significantly improved OS in patients with advanced, unresectable HCC and NSCLC patients who had a high tumor mutation burden (TMB) (248, 249), showing the importance of selecting appropriate treatment regimens for specific patient subsets.
Early in 2022, the FDA approved a fixed-dose combination of nivolumab and relatlimab for the treatment of unresectable or metastatic melanoma based on a phase II/III study (NCT03470922) (220). Data from the clinical trial showed a significantly superior PFS (median 10.2 months) in the combination group over nivolumab alone (4.6 months, p=0.0055). Combination therapy also reduced risk of death by 20% and improved OS rates versus nivolumab, although it did not reach statistical significance. The outcomes of patients with features often associated with poor prognosis such as high tumor burden, visceral metastases, increased serum LDH levels, or mucosal or acral melanoma, were improved in the dual-inhibition group compared with nivolumab alone (220). The safety profile of this combination was generally manageable and no novel or unexpected safety concerns were noted, although grade 3 or 4 TRAEs (such as fatigue, hepatitis, and adrenal insufficiency) were more frequently noted in patients receiving relatlimab-nivolumab than those receiving nivolumab (250). However, the efficacy of combined therapy in real-world patient needs to be further investigated since some populations of patients are often excluded from clinical trials, including patients with untreated or active brain metastases or with certain melanoma subtypes (e.g., uveal melanoma).
Chemotherapeutic drugs slow tumor growth primarily by inhibiting DNA replication, interfering with cellular metabolism, inducing cell cycle arrest, or inhibiting microtubule assembly (251). In addition, some cytotoxic drugs such as oxaliplatin and anthracyclines can induce cell death of immunogenic cells, thereby stimulating anti-tumor immune responses (252, 253). Based on these effects, chemotherapeutic drugs may be suitable partners for combinatorial administration with anti-PD-1/PD-L1 to achieve rapid and long-term control of cancers. A large number of clinical trials have tested the efficacy and safety of chemotherapy plus anti-PD-1/PD-L1, with FDA approved combinations summarized in Supplementary Table 1. In general, pembrolizumab combined with chemotherapy has had great clinical benefit for a wide range of cancer indications, followed by atezolizumab combined with chemotherapy.
The hypoxic tumor microenvironment (TME) leads to the upregulation of vascular endothelial growth factor (VEGF) and angiopoietin 2 (ANGPT2), which are key mediators in angiogenesis (254). By inhibiting these pro-angiogenic pathways, angiogenesis inhibitors can promote vascular normalization, restore oxygenation within the typically hypoxic TME, improve perfusion and oxygenation in tumors, and enhance the delivery of antitumor drugs (255, 256). Several tyrosine kinase inhibitors that target the VEGF-signalling axis have been approved in the United States and European Union, including sorafenib, sunitinib, pazopanib, axitinib, lenvatinib, and cabozantinib (257).
The FDA recently approved the use of nivolumab plus cabozantinib, pembrolizumab/avelumab plus axitinib, and pembrolizumab plus lenvatinib in advanced RCC; pembrolizumab plus lenvatinib in advanced endometrial carcinoma; atezolizumab plus bevacizumab (anti-VEGF mAb) in unresectable or metastatic HCC; and atezolizumab plus bevacizumab and chemotherapy in non-squamous and metastatic NSCLC based on the results of these clinical trials (258–264).
With the use of ICIs for cancer treatment rising, several side effects associated with treatment have raised concerns. Compared to treatment with standard chemotherapy or other biologics, ICIs have a different toxicity profile and most of these AEs are caused by aberrant immune responses against normal self-tissues due to impaired self-tolerance from the loss of T-cell inhibition, a phenomenon known as immune-related adverse events (irAEs) (265). Three drugs (ipilimumab, nivolumab and pembrolizumab) were reported to account for nearly 60% of reported irAEs in patients receiving immune checkpoint blockade therapies (266).
IrAEs can affect any organ system, most commonly the skin, colon and endocrine systems (267). The onset time usually ranges from a few days after treatment initiation to ≥1 year after completion of treatment, with a median time of 2–16 weeks (268–270). Most irAEs occurring soon after administration (3 weeks after initiation of therapy) of CTLA4 and PD-1 inhibitors typically involves the skin (271). Serious and often life-threatening AEs include pneumonitis and colitis (272). In general, there are fewer AEs associated with anti-PD-1/PD-L1 drugs than CTLA4 inhibitors (273), potentially due to their different mechanisms of action. CTLA4 is expressed on T cells (including Tregs), and its activation primarily suppresses the immune response in the early stages of T cell activation in lymphoid tissues. As CTLA4 blockade of Tregs would also result in the loss of immune suppression, CTLA4 inhibition would result in widespread, nonspecific activation of immune response that might explain the broad spectrum of AEs (274, 275). In contrast, PD-1 inhibitors target T cells more specifically in the tumor microenvironment and tissues, resulting in a more restricted spectrum of AEs (274, 276).
Colitis and hypophysitis often occur in patients receiving CTLA4 inhibitors, while less common AEs (pneumonitis and thyroiditis) may occur in patients using drugs targeting PD-1/PD-L1 pathway (271, 277, 278). A meta-analysis of the irAEs of individual ICIs revealed that the most common side-effects associated with ipilimumab included dermatological, gastrointestinal, and renal toxicities; and side-effects associated with nivolumab often involved endocrine toxicities. Similarly, side-effects associated with pembrolizumab included arthralgia, pneumonitis, and hepatotoxicity; and irAEs associated with atezolizumab included hypothyroidism (279). Most AEs resolved without intervention or responded to appropriate treatment (280). Severe irAEs usually require the treatment of systemic glucocorticoids or other immunosuppressive medicines, such as anti-TNF blockers.
The combination therapy such as dual PD-1/PD-L1 and CTLA4 blockade can also lead to severe irAEs such as colitis, pneumonitis, hypophysitis, and thyroiditis (267). Therefore, in these combined regimens, the dose of ipilimumab is usually reduced, which may impair the efficacy of the combination therapy (281). In a preclinical study, prophylactic TNF blockade reduced the toxicity of dual PD-1/PD-L1 and CTLA4 blocking antibodies (282). When ICIs are combined with chemotherapies, the reported irAEs were consistent with those of each drug (283, 284). In order to minimize the potential toxicity of dual PD-1/PD-L1 and CTLA4 blockade, Dovedi et al. developed a bispecific anti–PD-1/CTLA4 antibody, MEDI5752 (285, 286). The antibody could inhibit the signal transduction through PD-1 axis and preferentially block CTLA4 on activated PD-1+ T cells over PD-1- T cells, which means that CTLA4 is inhibited only in T cell populations that have already been exposed to the antigen. Therefore, the additive toxicity of this bispecific antibody is reduced compared with traditional dual blockers (285). In an ongoing in-human study in patients with advanced solid tumors, one patient with gastric cancer in whom five prior lines of chemotherapy had failed showed PR with 60% tumor reduction, and another patient with treatment-naïve renal clear cell carcinoma had PR with 68% tumor reduction. Both of the patients had manageable toxicity (285). However, more clinical studies are needed to determine the safety and efficacy of this bispecific agent or other combination therapies.
We are now in an era of innovation in cancer immunotherapy that is transforming the field of clinical oncology. Sustained durable responses from ICIs provide new hope as a treatment option for patients previously diagnosed with terminal illnesses. However, only a small percentage of tumors are responsive to these therapies and the overall response rates are low. There might be several reasons for this (1). The corresponding TMB. High TMB accompanied by elevated neoantigen expression might induce cytotoxic responses against tumor cells (287, 288). Thus tumors with low TMB and poor immunogenicity such as prostate and pancreatic cancers would be more resistant to ICIs (287) (2). Intratumor heterogeneity may lead to the selection of subclones that lack neoantigen expression and confer resistance to immune therapies (289, 290) (3). Genetic instability such as alterations in DNA-mismatch repair genes can increase immunogenicity and enhance response to ICIs (291, 292). For example, melanoma patients responded better to anti-PD-1 therapy when their tumor cells were rich in mutations in BRCA2, a gene important for homologous recombination in DNA repair (293). Mutation of B2M was also reported to be associated with initial resistance to anti PD-1 therapy in melanoma patients (294, 295) (4). Potential biomarkers may predict the response to ICIs. For example, the greater the number of tumor-infiltrating lymphocytes (TIL) in the tumor microenvironment, the better the anti-tumor effect (296); and tumors manifesting PD-L1 overexpression usually portend clinical outcomes superior to those of ICI compared with those with lower levels of ligand (297) (5). Complex interactions of immune cells and cytokines in the TME. For example, Tregs promote self-tolerance by inhibiting the function of Teff through inhibitory cytokines and direct contacts (298), and in some cancers, the infiltration of Tregs suggest an immunosuppressive environment (299). Myeloid-derived suppressor cells (MDSCs) in the TME can promote tumor growth and immune evasion (300, 301) (6). Alterations to a patient’s gut microbiome have also been associated with response or resistance to immune-checkpoint blockade, although the exact mechanism remains unknown (302) (7). The presence of tertiary lymphoid structures (TLS) might indicate improved survival in some tumor types (303, 304). Higher densities of TLS were associated with increased tumor-infiltrating CD8+ T cells density and also with an activated and cytotoxic immune signature (304–306). The existence of mature TLS was associated with improved outcomes in cancer patients treated with ICIs, regardless of their PD-L1 status and tumor-infiltrating CD8+ T-cell level after adjustment (307).
There are also other factors that could influence the anti-tumor effect of ICIs, including T-cell exhaustion (308), chromatin remodeling (309), and upregulation of alternative negative immune-checkpoint molecules (310). Resolving these issues is posited to elevate the efficacy of ICIs to a higher level. For example, personalized vaccines composed of an individual patient’s tumor neoantigens might be taken up and presented by activated APCs, leading to the activation of specific T cells that target these neoantigens, and thereby enhancing the efficacy of PD-1/PD-L1 inhibitors (311). Targeting other immunomodulatory cells or cytokines such as MDSCs, anti-TGF-β, and PI3Kγ are currently in clinical trials (311, 312). Since some results are not promising, we still need to advance our overall understanding of the molecular biology of tumors.
In addition, it is necessary to explore how to better predict patient responses to improve clinical outcomes. Although the expression of PD-L1 on tumor cells, TIL status, and the assessment of mutational burden are presently used markers (290, 313–317), they often have poor specificity and sensitivity. Accurate prediction using biomarkers therefore remains a major clinical challenge and necessitates further investigation due to the complexity of antitumor immune responses and the heterogeneity among patients and tumors.
The therapeutic landscape of oncology has been revolutionized with the advent of immunotherapies targeting immune checkpoints. A growing number of ICIs have been approved to treat different cancers. They can be used alone or in combination, or combined with other chemotherapies, vaccines and tumor immunotherapies, etc. Although these ICIs can bring survival benefits, their response rates and PFS are not high, and they also have a variety of adverse effects. A better understanding of the regulatory pathways of immune checkpoints will improve the success and efficacy of ICIs.
With in-depth investigations into immunotherapy, more novel immune checkpoints, regulators, and receptor-ligand pathways between tumor and host immune cells will continue to be discovered and further advance our understanding of the mechanisms leading to tumor immune evasion or resistance. Individualized therapeutic strategies based on a patient’s genetic background may suppress overactive T-cell responses more robustly, and thereby assist in patient selection. Further research is urgently needed to elucidate the mechanisms underlying tumor biology and immunotherapy, how one ICI affects another, and the development of novel sequential treatment options, so as to facilitate the successful use of ICIs in treating cancer patients.
YW and QZ (4th author) conceived and designed the research. LY and MS prepared the draft. QZ (3rd author) prepared the tables. QZ (3rd author) prepared the figure. YW and QZ (4th author) reviewed and edited the manuscript. All authors read and approved the final manuscript.
This study was sponsored by an grant from the National Science Foundation of China (81802504, 81872207), a grant from the Sichuan Medical Association (Q19037), grants of Sichuan Science and Technology Bureau (2019YFS0439, 2020JDJQ0067, 2021YJ0564, 2022YFH0005), and a grant from the Chengdu Science and Technology Bureau (2021-YF05-00225-SN).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.982026/full#supplementary-material
1. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol (2002) 3(11):991–8. doi: 10.1038/ni1102-991
2. Burnet FM. The concept of immunological surveillance. Prog Exp Tumor Res (1970) 13:1–27. doi: 10.1159/000386035
3. Burnet M. Cancer: a biological approach. III. viruses associated with neoplastic conditions. IV. practical applications. Br Med J (1957) 1(5023):841–7. doi: 10.1136/bmj.1.5023.841
4. Zhang Y, Zhang X, Li W, Du Y, Hu W, Zhao J. Biomarkers and risk factors for the early prediction of immune-related adverse events: a review. Hum Vaccin Immunother (2022) 18(1):2018894. doi: 10.1080/21645515.2021.2018894
5. Haddad AF, Young JS, Gill S, Aghi MK. Resistance to immune checkpoint blockade: Mechanisms, counter-acting approaches, and future directions. Semin Cancer Biol (2022), S1044–579X(22)00046-3. doi: 10.1016/j.semcancer.2022.02.019
6. Zappasodi R, Merghoub T, Wolchok JD. Emerging concepts for immune checkpoint blockade-based combination therapies. Cancer Cell (2018) 33(4):581–98. doi: 10.1016/j.ccell.2018.03.005
7. Tu L, Guan R, Yang H, Zhou Y, Hong W, Ma L, et al. Assessment of the expression of the immune checkpoint molecules PD-1, CTLA4, TIM-3 and LAG-3 across different cancers in relation to treatment response, tumor-infiltrating immune cells and survival. Int J Cancer (2020) 147(2):423–39. doi: 10.1002/ijc.32785
8. Rowshanravan B, Halliday N, Sansom DM. CTLA-4: a moving target in immunotherapy. Blood (2018) 131(1):58–67. doi: 10.1182/blood-2017-06-741033
9. McCoy KD, Le Gros G. The role of CTLA-4 in the regulation of T cell immune responses. Immunol Cell Biol (1999) 77(1):1–10. doi: 10.1046/j.1440-1711.1999.00795.x
10. Alegre ML, Noel PJ, Eisfelder BJ, Chuang E, Clark MR, Reiner SL, et al. Regulation of surface and intracellular expression of CTLA4 on mouse T cells. J Immunol (1996) 157(11):4762–70.
11. Chikuma S. CTLA-4, an essential immune-checkpoint for T-cell activation. Curr Top Microbiol Immunol (2017) 410:99–126. doi: 10.1007/82_2017_61
12. Tai X, Van Laethem F, Pobezinsky L, Guinter T, Sharrow SO, Adams A, et al. Basis of CTLA-4 function in regulatory and conventional CD4(+) T cells. Blood (2012) 119(22):5155–63. doi: 10.1182/blood-2011-11-388918
13. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science (2011) 332(6029):600–3. doi: 10.1126/science.1202947
14. Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity (1994) 1(5):405–13. doi: 10.1016/1074-7613(94)90071-x
15. Schneider H, Downey J, Smith A, Zinselmeyer BH, Rush C, Brewer JM, et al. Reversal of the TCR stop signal by CTLA-4. Science (2006) 313(5795):1972–5. doi: 10.1126/science.1131078
16. Corse E, Allison JP. Cutting edge: CTLA-4 on effector T cells inhibits in trans. J Immunol (2012) 189(3):1123–7. doi: 10.4049/jimmunol.1200695
17. Naimi A, Mohammed RN, Raji A, Chupradit S, Yumashev AV, Suksatan W, et al. Tumor immunotherapies by immune checkpoint inhibitors (ICIs); the pros and cons. Cell Commun Signal (2022) 20(1):44. doi: 10.1186/s12964-022-00854-y
18. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity (1995) 3(5):541–7. doi: 10.1016/1074-7613(95)90125-6
19. Valk E, Rudd CE, Schneider H. CTLA-4 trafficking and surface expression. Trends Immunol (2008) 29(6):272–9. doi: 10.1016/j.it.2008.02.011
20. Shiratori T, Miyatake S, Ohno H, Nakaseko C, Isono K, Bonifacino JS, et al. Tyrosine phosphorylation controls internalization of CTLA-4 by regulating its interaction with clathrin-associated adaptor complex AP-2. Immunity (1997) 6(5):583–9. doi: 10.1016/s1074-7613(00)80346-5
21. Zhang Y, Allison JP. Interaction of CTLA-4 with AP50, a clathrin-coated pit adaptor protein. Proc Natl Acad Sci U.S.A. (1997) 94(17):9273–8. doi: 10.1073/pnas.94.17.9273
22. Greenwald RJ, Boussiotis VA, Lorsbach RB, Abbas AK, Sharpe AH. CTLA-4 regulates induction of anergy in vivo. Immunity (2001) 14(2):145–55. doi: 10.1016/s1074-7613(01)00097-8
23. Sojka DK, Hughson A, Fowell DJ. CTLA-4 is required by CD4+CD25+ treg to control CD4+ T-cell lymphopenia-induced proliferation. Eur J Immunol (2009) 39(6):1544–51. doi: 10.1002/eji.200838603
24. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science (2008) 322(5899):271–5. doi: 10.1126/science.1160062
25. Tang Q, Boden EK, Henriksen KJ, Bour-Jordan H, Bi M, Bluestone JA. Distinct roles of CTLA-4 and TGF-beta in CD4+CD25+ regulatory T cell function. Eur J Immunol (2004) 34(11):2996–3005. doi: 10.1002/eji.200425143
26. Bengsch F, Knoblock DM, Liu A, McAllister F, Beatty GL. CTLA-4/CD80 pathway regulates T cell infiltration into pancreatic cancer. Cancer Immunol Immunother (2017) 66(12):1609–17. doi: 10.1007/s00262-017-2053-4
27. Contardi E, Palmisano GL, Tazzari PL, Martelli AM, Fala F, Fabbi M, et al. CTLA-4 is constitutively expressed on tumor cells and can trigger apoptosis upon ligand interaction. Int J Cancer (2005) 117(4):538–50. doi: 10.1002/ijc.21155
28. Liu Y, Zheng P. How does an anti-CTLA-4 antibody promote cancer immunity? Trends Immunol (2018) 39(12):953–6. doi: 10.1016/j.it.2018.10.009
29. Ha D, Tanaka A, Kibayashi T, Tanemura A, Sugiyama D, Wing JB, et al. Differential control of human treg and effector T cells in tumor immunity by fc-engineered anti-CTLA-4 antibody. Proc Natl Acad Sci U.S.A. (2019) 116(2):609–18. doi: 10.1073/pnas.1812186116
30. Sharma A, Subudhi SK, Blando J, Scutti J, Vence L, Wargo J, et al. Anti-CTLA-4 immunotherapy does not deplete FOXP3(+) regulatory T cells (Tregs) in human cancers. Clin Cancer Res (2019) 25(4):1233–8. doi: 10.1158/1078-0432.CCR-18-0762
31. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J (1992) 11(11):3887–95.
32. De Silva P, Aiello M, Gu-Trantien C, Migliori E, Willard-Gallo K, Solinas C. Targeting CTLA-4 in cancer: Is it the ideal companion for PD-1 blockade immunotherapy combinations? Int J Cancer (2021) 149(1):31–41. doi: 10.1002/ijc.33415
33. Mizuno R, Sugiura D, Shimizu K, Maruhashi T, Watada M, Okazaki IM, et al. PD-1 primarily targets TCR signal in the inhibition of functional T cell activation. Front Immunol (2019) 10:630. doi: 10.3389/fimmu.2019.00630
34. Honda T, Egen JG, Lammermann T, Kastenmuller W, Torabi-Parizi P, Germain RN. Tuning of antigen sensitivity by T cell receptor-dependent negative feedback controls T cell effector function in inflamed tissues. Immunity (2014) 40(2):235–47. doi: 10.1016/j.immuni.2013.11.017
35. Andrews LP, Yano H, Vignali DAA. Inhibitory receptors and ligands beyond PD-1, PD-L1 and CTLA-4: breakthroughs or backups. Nat Immunol (2019) 20(11):1425–34. doi: 10.1038/s41590-019-0512-0
36. Esch KJ, Juelsgaard R, Martinez PA, Jones DE, Petersen CA. Programmed death 1-mediated T cell exhaustion during visceral leishmaniasis impairs phagocyte function. J Immunol (2013) 191(11):5542–50. doi: 10.4049/jimmunol.1301810
37. Lo B, Abdel-Motal UM. Lessons from CTLA-4 deficiency and checkpoint inhibition. Curr Opin Immunol (2017) 49:14–9. doi: 10.1016/j.coi.2017.07.014
38. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity (1999) 11(2):141–51. doi: 10.1016/s1074-7613(00)80089-8
39. Nishimura H, Honjo T, Minato N. Facilitation of beta selection and modification of positive selection in the thymus of PD-1-deficient mice. J Exp Med (2000) 191(5):891–8. doi: 10.1084/jem.191.5.891
40. Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: Similarities, differences, and implications of their inhibition. Am J Clin Oncol (2016) 39(1):98–106. doi: 10.1097/COC.0000000000000239
41. Kahler KC, Hauschild A. Treatment and side effect management of CTLA-4 antibody therapy in metastatic melanoma. J Dtsch Dermatol Ges (2011) 9(4):277–86. doi: 10.1111/j.1610-0387.2010.07568.x
42. Aksoylar HI, Boussiotis VA. PD-1(+) treg cells: a foe in cancer immunotherapy? Nat Immunol (2020) 21(11):1311–2. doi: 10.1038/s41590-020-0801-7
43. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med (2009) 206(13):3015–29. doi: 10.1084/jem.20090847
44. Pan T, Liu Z, Yin J, Zhou T, Liu J, Qu H. Notch signaling pathway was involved in regulating programmed cell death 1 expression during sepsis-induced immunosuppression. Mediators Inflammation (2015) 2015:539841. doi: 10.1155/2015/539841
45. Amarnath S, Mangus CW, Wang JC, Wei F, He A, Kapoor V, et al. The PDL1-PD1 axis converts human TH1 cells into regulatory T cells. Sci Transl Med (2011) 3(111):111ra120. doi: 10.1126/scitranslmed.3003130
46. Kamada T, Togashi Y, Tay C, Ha D, Sasaki A, Nakamura Y, et al. PD-1(+) regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc Natl Acad Sci U.S.A. (2019) 116(20):9999–10008. doi: 10.1073/pnas.1822001116
47. Zhang Y, Liu Z, Tian M, Hu X, Wang L, Ji J, et al. The altered PD-1/PD-L1 pathway delivers the 'one-two punch' effects to promote the Treg/Th17 imbalance in pre-eclampsia. Cell Mol Immunol (2018) 15(7):710–23. doi: 10.1038/cmi.2017.70
48. Kumagai S, Togashi Y, Kamada T, Sugiyama E, Nishinakamura H, Takeuchi Y, et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat Immunol (2020) 21(11):1346–58. doi: 10.1038/s41590-020-0769-3
49. Das M, Zhu C, Kuchroo VK. Tim-3 and its role in regulating anti-tumor immunity. Immunol Rev (2017) 276(1):97–111. doi: 10.1111/imr.12520
50. Huang YH, Zhu C, Kondo Y, Anderson AC, Gandhi A, Russell A, et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature (2015) 517(7534):386–90. doi: 10.1038/nature13848
51. Sanchez-Fueyo A, Tian J, Picarella D, Domenig C, Zheng XX, Sabatos CA, et al. Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat Immunol (2003) 4(11):1093–101. doi: 10.1038/ni987
52. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol (2005) 6(12):1245–52. doi: 10.1038/ni1271
53. Sabatos CA, Chakravarti S, Cha E, Schubart A, Sanchez-Fueyo A, Zheng XX, et al. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat Immunol (2003) 4(11):1102–10. doi: 10.1038/ni988
54. Dardalhon V, Anderson AC, Karman J, Apetoh L, Chandwaskar R, Lee DH, et al. Tim-3/galectin-9 pathway: regulation of Th1 immunity through promotion of CD11b+Ly-6G+ myeloid cells. J Immunol (2010) 185(3):1383–92. doi: 10.4049/jimmunol.0903275
55. Li H, Wu K, Tao K, Chen L, Zheng Q, Lu X, et al. Tim-3/galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis b virus-associated hepatocellular carcinoma. Hepatology (2012) 56(4):1342–51. doi: 10.1002/hep.25777
56. Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature (2002) 415(6871):536–41. doi: 10.1038/415536a
57. Liu Z, McMichael EL, Shayan G, Li J, Chen K, Srivastava R, et al. Novel effector phenotype of Tim-3(+) regulatory T cells leads to enhanced suppressive function in head and neck cancer patients. Clin Cancer Res (2018) 24(18):4529–38. doi: 10.1158/1078-0432.CCR-17-1350
58. Gautron AS, Dominguez-Villar M, de Marcken M, Hafler DA. Enhanced suppressor function of TIM-3+ FoxP3+ regulatory T cells. Eur J Immunol (2014) 44(9):2703–11. doi: 10.1002/eji.201344392
59. Liu JF, Wu L, Yang LL, Deng WW, Mao L, Wu H, et al. Blockade of TIM3 relieves immunosuppression through reducing regulatory T cells in head and neck cancer. J Exp Clin Cancer Res (2018) 37(1):44. doi: 10.1186/s13046-018-0713-7
60. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med (2010) 207(10):2187–94. doi: 10.1084/jem.20100643
61. Liu J, Zhang S, Hu Y, Yang Z, Li J, Liu X, et al. Targeting PD-1 and Tim-3 pathways to reverse CD8 T-cell exhaustion and enhance ex vivo T-cell responses to autologous Dendritic/Tumor vaccines. J Immunother (2016) 39(4):171–80. doi: 10.1097/CJI.0000000000000122
62. Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med (2010) 207(10):2175–86. doi: 10.1084/jem.20100637
63. Shi X, Zhang X, Li J, Mo L, Zhao H, Zhu Y, et al. PD-1 blockade enhances the antitumor efficacy of GM-CSF surface-modified bladder cancer stem cells vaccine. Int J Cancer (2018) 142(10):2106–17. doi: 10.1002/ijc.31219
64. Zhang X, Liu G, Shi X, Shi X, Li J, Mo L, et al. Sequential administration of anti-PD-1 and anti-Tim-3 combined with an SA-GM-CSF-anchored vaccine overcomes adaptive immune resistance to reject established bladder cancer. J Cancer (2021) 12(7):2000–9. doi: 10.7150/jca.44769
65. Zhang X, Chen H, Li G, Zhou X, Shi Y, Zou F, et al. Increased Tim-3 expression on TILs during treatment with the anchored GM-CSF vaccine and anti-PD-1 antibodies is inversely correlated with response in prostate cancer. J Cancer (2020) 11(3):648–56. doi: 10.7150/jca.29705
66. Mimura K, Kua LF, Xiao JF, Asuncion BR, Nakayama Y, Syn N, et al. Combined inhibition of PD-1/PD-L1, lag-3, and Tim-3 axes augments antitumor immunity in gastric cancer-T cell coculture models. Gastric Cancer (2021) 24(3):611–23. doi: 10.1007/s10120-020-01151-8
67. Shi X, Li CW, Tan LC, Wen SS, Liao T, Zhang Y, et al. Immune Co-inhibitory receptors PD-1, CTLA-4, TIM-3, LAG-3, and TIGIT in medullary thyroid cancers: A Large cohort study. J Clin Endocrinol Metab (2021) 106(1):120–32. doi: 10.1210/clinem/dgaa701
68. Jie HB, Srivastava RM, Argiris A, Bauman JE, Kane LP, Ferris RL. Increased PD-1(+) and TIM-3(+) TILs during cetuximab therapy inversely correlate with response in head and neck cancer patients. Cancer Immunol Res (2017) 5(5):408–16. doi: 10.1158/2326-6066.CIR-16-0333
69. Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med (1990) 171(5):1393–405. doi: 10.1084/jem.171.5.1393
70. Huard B, Gaulard P, Faure F, Hercend T, Triebel F. Cellular expression and tissue distribution of the human LAG-3-encoded protein, an MHC class II ligand. Immunogenetics (1994) 39(3):213–7. doi: 10.1007/BF00241263
71. Kisielow M, Kisielow J, Capoferri-Sollami G, Karjalainen K. Expression of lymphocyte activation gene 3 (LAG-3) on b cells is induced by T cells. Eur J Immunol (2005) 35(7):2081–8. doi: 10.1002/eji.200526090
72. Solinas C, Migliori E, De Silva P, Willard-Gallo K. LAG3: The biological processes that motivate targeting this immune checkpoint molecule in human cancer. Cancers (Basel) (2019) 11(8):1213. doi: 10.3390/cancers11081213
73. Huard B, Prigent P, Tournier M, Bruniquel D, Triebel F. CD4/major histocompatibility complex class II interaction analyzed with CD4- and lymphocyte activation gene-3 (LAG-3)-Ig fusion proteins. Eur J Immunol (1995) 25(9):2718–21. doi: 10.1002/eji.1830250949
74. Workman CJ, Vignali DA. Negative regulation of T cell homeostasis by lymphocyte activation gene-3 (CD223). J Immunol (2005) 174(2):688–95. doi: 10.4049/jimmunol.174.2.688
75. Grosso JF, Kelleher CC, Harris TJ, Maris CH, Hipkiss EL, De Marzo A, et al. LAG-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. J Clin Invest (2007) 117(11):3383–92. doi: 10.1172/JCI31184
76. Workman CJ, Vignali DA. The CD4-related molecule, LAG-3 (CD223), regulates the expansion of activated T cells. Eur J Immunol (2003) 33(4):970–9. doi: 10.1002/eji.200323382
77. Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of LAG-3 in regulatory T cells. Immunity (2004) 21(4):503–13. doi: 10.1016/j.immuni.2004.08.010
78. Camisaschi C, Casati C, Rini F, Perego M, De Filippo A, Triebel F, et al. LAG-3 expression defines a subset of CD4(+)CD25(high)Foxp3(+) regulatory T cells that are expanded at tumor sites. J Immunol (2010) 184(11):6545–51. doi: 10.4049/jimmunol.0903879
79. Gagliani N, Magnani CF, Huber S, Gianolini ME, Pala M, Licona-Limon P, et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat Med (2013) 19(6):739–46. doi: 10.1038/nm.3179
80. Workman CJ, Wang Y, El Kasmi KC, Pardoll DM, Murray PJ, Drake CG, et al. LAG-3 regulates plasmacytoid dendritic cell homeostasis. J Immunol (2009) 182(4):1885–91. doi: 10.4049/jimmunol.0800185
81. Andreae S, Piras F, Burdin N, Triebel F. Maturation and activation of dendritic cells induced by lymphocyte activation gene-3 (CD223). J Immunol (2002) 168(8):3874–80. doi: 10.4049/jimmunol.168.8.3874
82. Hu S, Liu X, Li T, Li Z, Hu F. LAG3 (CD223) and autoimmunity: Emerging evidence. J Autoimmun (2020) 112:102504. doi: 10.1016/j.jaut.2020.102504
83. Long L, Zhang X, Chen F, Pan Q, Phiphatwatchara P, Zeng Y, et al. The promising immune checkpoint LAG-3: from tumor microenvironment to cancer immunotherapy. Genes Cancer (2018) 9(5-6):176–89. doi: 10.18632/genesandcancer.180
84. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: Co-inhibitory receptors with specialized functions in immune regulation. Immunity (2016) 44(5):989–1004. doi: 10.1016/j.immuni.2016.05.001
85. Burugu S, Gao D, Leung S, Chia SK, Nielsen TO. LAG-3+ tumor infiltrating lymphocytes in breast cancer: clinical correlates and association with PD-1/PD-L1+ tumors. Ann Oncol (2017) 28(12):2977–84. doi: 10.1093/annonc/mdx557
86. Matsuzaki J, Gnjatic S, Mhawech-Fauceglia P, Beck A, Miller A, Tsuji T, et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc Natl Acad Sci U.S.A. (2010) 107(17):7875–80. doi: 10.1073/pnas.1003345107
87. He Y, Yu H, Rozeboom L, Rivard CJ, Ellison K, Dziadziuszko R, et al. LAG-3 protein expression in non-small cell lung cancer and its relationship with PD-1/PD-L1 and tumor-infiltrating lymphocytes. J Thorac Oncol (2017) 12(5):814–23. doi: 10.1016/j.jtho.2017.01.019
88. Huang RY, Eppolito C, Lele S, Shrikant P, Matsuzaki J, Odunsi K. LAG3 and PD1 co-inhibitory molecules collaborate to limit CD8+ T cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget (2015) 6(29):27359–77. doi: 10.18632/oncotarget.4751
89. Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res (2012) 72(4):917–27. doi: 10.1158/0008-5472.CAN-11-1620
90. Zelba H, Bedke J, Hennenlotter J, Mostbock S, Zettl M, Zichner T, et al. PD-1 and LAG-3 dominate checkpoint receptor-mediated T-cell inhibition in renal cell carcinoma. Cancer Immunol Res (2019) 7(11):1891–9. doi: 10.1158/2326-6066.CIR-19-0146
91. Hafler JP, Maier LM, Cooper JD, Plagnol V, Hinks A, Simmonds MJ, et al. CD226 Gly307Ser association with multiple autoimmune diseases. Genes Immun (2009) 10(1):5–10. doi: 10.1038/gene.2008.82
92. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol (2009) 10(1):48–57. doi: 10.1038/ni.1674
93. Liu XG, Hou M, Liu Y. TIGIT, a novel therapeutic target for tumor immunotherapy. Immunol Invest (2017) 46(2):172–82. doi: 10.1080/08820139.2016.1237524
94. Lozano E, Dominguez-Villar M, Kuchroo V, Hafler DA. The TIGIT/CD226 axis regulates human T cell function. J Immunol (2012) 188(8):3869–75. doi: 10.4049/jimmunol.1103627
95. Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell (2014) 26(6):923–37. doi: 10.1016/j.ccell.2014.10.018
96. Kurtulus S, Sakuishi K, Ngiow SF, Joller N, Tan DJ, Teng MW, et al. TIGIT predominantly regulates the immune response via regulatory T cells. J Clin Invest (2015) 125(11):4053–62. doi: 10.1172/JCI81187
97. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity (2014) 40(4):569–81. doi: 10.1016/j.immuni.2014.02.012
98. Lucca LE, Axisa PP, Singer ER, Nolan NM, Dominguez-Villar M, Hafler DA. TIGIT signaling restores suppressor function of Th1 tregs. JCI Insight (2019) 4(3):e124427. doi: 10.1172/jci.insight.124427
99. Stanietsky N, Rovis TL, Glasner A, Seidel E, Tsukerman P, Yamin R, et al. Mouse TIGIT inhibits NK-cell cytotoxicity upon interaction with PVR. Eur J Immunol (2013) 43(8):2138–50. doi: 10.1002/eji.201243072
100. Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci U.S.A. (2009) 106(42):17858–63. doi: 10.1073/pnas.0903474106
101. Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol (2018) 19(7):723–32. doi: 10.1038/s41590-018-0132-0
102. Chauvin JM, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C, et al. TIGIT and PD-1 impair tumor antigen-specific CD8(+) T cells in melanoma patients. J Clin Invest (2015) 125(5):2046–58. doi: 10.1172/JCI80445
103. Hung AL, Maxwell R, Theodros D, Belcaid Z, Mathios D, Luksik AS, et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology (2018) 7(8):e1466769. doi: 10.1080/2162402X.2018.1466769
104. Josefsson SE, Beiske K, Blaker YN, Forsund MS, Holte H, Ostenstad B, et al. TIGIT and PD-1 mark intratumoral T cells with reduced effector function in b-cell non-Hodgkin lymphoma. Cancer Immunol Res (2019) 7(3):355–62. doi: 10.1158/2326-6066.CIR-18-0351
105. Xu D, Zhao E, Zhu C, Zhao W, Wang C, Zhang Z, et al. TIGIT and PD-1 may serve as potential prognostic biomarkers for gastric cancer. Immunobiology (2020) 225(3):151915. doi: 10.1016/j.imbio.2020.151915
106. Yang ZZ, Kim HJ, Wu H, Jalali S, Tang X, Krull JE, et al. TIGIT expression is associated with T-cell suppression and exhaustion and predicts clinical outcome and anti-PD-1 response in follicular lymphoma. Clin Cancer Res (2020) 26(19):5217–31. doi: 10.1158/1078-0432.CCR-20-0558
107. Wang XF, Chen YJ, Wang Q, Ge Y, Dai Q, Yang KF, et al. Distinct expression and inhibitory function of b and T lymphocyte attenuator on human T cells. Tissue Antigens (2007) 69(2):145–53. doi: 10.1111/j.1399-0039.2006.00710.x
108. Ning Z, Liu K, Xiong H. Roles of BTLA in immunity and immune disorders. Front Immunol (2021) 12:654960. doi: 10.3389/fimmu.2021.654960
109. Xu X, Hou B, Fulzele A, Masubuchi T, Zhao Y, Wu Z, et al. PD-1 and BTLA regulate T cell signaling differentially and only partially through SHP1 and SHP2. J Cell Biol (2020) 219(6):e201905085. doi: 10.1083/jcb.201905085
110. Vendel AC, Calemine-Fenaux J, Izrael-Tomasevic A, Chauhan V, Arnott D, Eaton DL. B and T lymphocyte attenuator regulates b cell receptor signaling by targeting syk and BLNK. J Immunol (2009) 182(3):1509–17. doi: 10.4049/jimmunol.182.3.1509
111. Simon T, Bromberg JS. BTLA(+) dendritic cells: The regulatory T cell force awakens. Immunity (2016) 45(5):956–8. doi: 10.1016/j.immuni.2016.10.030
112. Yang C, Chen Y, Guo G, Li H, Cao D, Xu H, et al. Expression of b and T lymphocyte attenuator (BTLA) in macrophages contributes to the fulminant hepatitis caused by murine hepatitis virus strain-3. Gut (2013) 62(8):1204–13. doi: 10.1136/gutjnl-2012-302239
113. Li S, Zhang M, Xiang F, Zhao J, Jiang C, Zhu J. Dendritic cells expressing BTLA induces CD8+ T cell tolerance and attenuates the severity of diabetes. Vaccine (2011) 29(44):7747–51. doi: 10.1016/j.vaccine.2011.07.125
114. Bourque J, Hawiger D. The BTLA-HVEM-CD5 immunoregulatory axis-an instructive mechanism governing pTreg cell differentiation. Front Immunol (2019) 10:1163. doi: 10.3389/fimmu.2019.01163
115. Huarte E, Jun S, Rynda-Apple A, Golden S, Jackiw L, Hoffman C, et al. Regulatory T cell dysfunction acquiesces to BTLA+ regulatory b cells subsequent to oral intervention in experimental autoimmune encephalomyelitis. J Immunol (2016) 196(12):5036–46. doi: 10.4049/jimmunol.1501973
116. Song J, Wu L. Friend or foe: Prognostic and immunotherapy roles of BTLA in colorectal cancer. Front Mol Biosci (2020) 7:148. doi: 10.3389/fmolb.2020.00148
117. Chen YL, Lin HW, Chien CL, Lai YL, Sun WZ, Chen CA, et al. BTLA blockade enhances cancer therapy by inhibiting IL-6/IL-10-induced CD19(high) b lymphocytes. J Immunother Cancer (2019) 7(1):313. doi: 10.1186/s40425-019-0744-4
118. Lan X, Li S, Gao H, Nanding A, Quan L, Yang C, et al. Increased BTLA and HVEM in gastric cancer are associated with progression and poor prognosis. Onco Targets Ther (2017) 10:919–26. doi: 10.2147/OTT.S128825
119. Li X, Xu Z, Cui G, Yu L, Zhang X. BTLA expression in stage I-III non-Small-Cell lung cancer and its correlation with PD-1/PD-L1 and clinical outcomes. Onco Targets Ther (2020) 13:215–24. doi: 10.2147/OTT.S232234
120. Fourcade J, Sun Z, Pagliano O, Guillaume P, Luescher IF, Sander C, et al. CD8(+) T cells specific for tumor antigens can be rendered dysfunctional by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and PD-1. Cancer Res (2012) 72(4):887–96. doi: 10.1158/0008-5472.CAN-11-2637
121. Chapoval AI, Ni J, Lau JS, Wilcox RA, Flies DB, Liu D, et al. B7-H3: a costimulatory molecule for T cell activation and IFN-gamma production. Nat Immunol (2001) 2(3):269–74. doi: 10.1038/85339
122. Castellanos JR, Purvis IJ, Labak CM, Guda MR, Tsung AJ, Velpula KK, et al. B7-H3 role in the immune landscape of cancer. Am J Clin Exp Immunol (2017) 6(4):66–75.
123. Sun M, Richards S, Prasad DV, Mai XM, Rudensky A, Dong C. Characterization of mouse and human B7-H3 genes. J Immunol (2002) 168(12):6294–7. doi: 10.4049/jimmunol.168.12.6294
124. Steinberger P, Majdic O, Derdak SV, Pfistershammer K, Kirchberger S, Klauser C, et al. Molecular characterization of human 4Ig-B7-H3, a member of the B7 family with four ig-like domains. J Immunol (2004) 172(4):2352–9. doi: 10.4049/jimmunol.172.4.2352
125. Picarda E, Ohaegbulam KC, Zang X. Molecular pathways: Targeting B7-H3 (CD276) for human cancer immunotherapy. Clin Cancer Res (2016) 22(14):3425–31. doi: 10.1158/1078-0432.CCR-15-2428
126. Suh WK, Gajewska BU, Okada H, Gronski MA, Bertram EM, Dawicki W, et al. The B7 family member B7-H3 preferentially down-regulates T helper type 1-mediated immune responses. Nat Immunol (2003) 4(9):899–906. doi: 10.1038/ni967
127. Hofmeyer KA, Ray A, Zang X. The contrasting role of B7-H3. Proc Natl Acad Sci U.S.A. (2008) 105(30):10277–8. doi: 10.1073/pnas.0805458105
128. Mahnke K, Ring S, Johnson TS, Schallenberg S, Schonfeld K, Storn V, et al. Induction of immunosuppressive functions of dendritic cells in vivo by CD4+CD25+ regulatory T cells: role of B7-H3 expression and antigen presentation. Eur J Immunol (2007) 37(8):2117–26. doi: 10.1002/eji.200636841
129. Han S, Wang Y, Shi X, Zong L, Liu L, Zhang J, et al. Negative roles of B7-H3 and B7-H4 in the microenvironment of cervical cancer. Exp Cell Res (2018) 371(1):222–30. doi: 10.1016/j.yexcr.2018.08.014
130. Lim S, Liu H, Madeira da Silva L, Arora R, Liu Z, Phillips JB, et al. Immunoregulatory protein B7-H3 reprograms glucose metabolism in cancer cells by ROS-mediated stabilization of HIF1alpha. Cancer Res (2016) 76(8):2231–42. doi: 10.1158/0008-5472.CAN-15-1538
131. Zang X, Loke P, Kim J, Murphy K, Waitz R, Allison JP. B7x: a widely expressed B7 family member that inhibits T cell activation. Proc Natl Acad Sci U.S.A. (2003) 100(18):10388–92. doi: 10.1073/pnas.1434299100
132. Tringler B, Zhuo S, Pilkington G, Torkko KC, Singh M, Lucia MS, et al. B7-h4 is highly expressed in ductal and lobular breast cancer. Clin Cancer Res (2005) 11(5):1842–8. doi: 10.1158/1078-0432.CCR-04-1658
133. Mao YX, Chen YJ, Ge Y, Ma HB, Yu JF, Wu HY, et al. Recombinant human B7-H4 expressed in escherichia coli inhibits T lymphocyte proliferation and IL-2 secretion in vitro. Acta Pharmacol Sin (2006) 27(6):741–6. doi: 10.1111/j.1745-7254.2006.00338.x
134. Wang X, Hao J, Metzger DL, Ao Z, Chen L, Ou D, et al. JNK, p38, and AKT activation. PloS One (2012) 7(1):e28232. doi: 10.1371/journal.pone.0028232
135. Podojil JR, Liu LN, Marshall SA, Chiang MY, Goings GE, Chen L, et al. B7-H4Ig inhibits mouse and human T-cell function and treats EAE via IL-10/Treg-dependent mechanisms. J Autoimmun (2013) 44:71–81. doi: 10.1016/j.jaut.2013.04.001
136. Wei J, Loke P, Zang X, Allison JP. Tissue-specific expression of B7x protects from CD4 T cell-mediated autoimmunity. J Exp Med (2011) 208(8):1683–94. doi: 10.1084/jem.20100639
137. Shen L, Qian Y, Wu W, Weng T, Wang FXC, Hong B, et al. B7-H4 is a prognostic biomarker for poor survival in patients with pancreatic cancer. Hum Pathol (2017) 66:79–85. doi: 10.1016/j.humpath.2017.05.023
138. Krambeck AE, Thompson RH, Dong H, Lohse CM, Park ES, Kuntz SM, et al. B7-H4 expression in renal cell carcinoma and tumor vasculature: associations with cancer progression and survival. Proc Natl Acad Sci U.S.A. (2006) 103(27):10391–6. doi: 10.1073/pnas.0600937103
139. Podojil JR, Miller SD. Potential targeting of B7-H4 for the treatment of cancer. Immunol Rev (2017) 276(1):40–51. doi: 10.1111/imr.12530
140. Smith JB, Stashwick C, Powell DJ. B7-H4 as a potential target for immunotherapy for gynecologic cancers: a closer look. Gynecol Oncol (2014) 134(1):181–9. doi: 10.1016/j.ygyno.2014.03.553
141. Zhu Y, Yao S, Iliopoulou BP, Han X, Augustine MM, Xu H, et al. B7-H5 costimulates human T cells via CD28H. Nat Commun (2013) 4:2043. doi: 10.1038/ncomms3043
142. Ni L, Dong C. New B7 family checkpoints in human cancers. Mol Cancer Ther (2017) 16(7):1203–11. doi: 10.1158/1535-7163.MCT-16-0761
143. Janakiram M, Chinai JM, Zhao A, Sparano JA, Zang X. HHLA2 and TMIGD2: new immunotherapeutic targets of the B7 and CD28 families. Oncoimmunology (2015) 4(8):e1026534. doi: 10.1080/2162402X.2015.1026534
144. Rieder SA, Wang J, White N, Qadri A, Menard C, Stephens G, et al. B7-H7 (HHLA2) inhibits T-cell activation and proliferation in the presence of TCR and CD28 signaling. Cell Mol Immunol (2021) 18(6):1503–11. doi: 10.1038/s41423-020-0361-7
145. Bhatt RS, Berjis A, Konge JC, Mahoney KM, Klee AN, Freeman SS, et al. KIR3DL3 is an inhibitory receptor for HHLA2 that mediates an alternative immunoinhibitory pathway to PD1. Cancer Immunol Res (2021) 9(2):156–69. doi: 10.1158/2326-6066.CIR-20-0315
146. Zhao R, Chinai JM, Buhl S, Scandiuzzi L, Ray A, Jeon H, et al. HHLA2 is a member of the B7 family and inhibits human CD4 and CD8 T-cell function. Proc Natl Acad Sci U.S.A. (2013) 110(24):9879–84. doi: 10.1073/pnas.1303524110
147. Qi Y, Deng G, Xu P, Zhang H, Yuan F, Geng R, et al. HHLA2 is a novel prognostic predictor and potential therapeutic target in malignant glioma. Oncol Rep (2019) 42(6):2309–22. doi: 10.3892/or.2019.7343
148. Zhou QH, Li KW, Chen X, He HX, Peng SM, Peng SR, et al. HHLA2 and PD-L1 co-expression predicts poor prognosis in patients with clear cell renal cell carcinoma. J Immunother Cancer (2020) 8(1):e000157. doi: 10.1136/jitc-2019-000157
149. Lines JL, Pantazi E, Mak J, Sempere LF, Wang L, O'Connell S, et al. VISTA is an immune checkpoint molecule for human T cells. Cancer Res (2014) 74(7):1924–32. doi: 10.1158/0008-5472.CAN-13-1504
150. Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, et al. VISTA, a novel mouse ig superfamily ligand that negatively regulates T cell responses. J Exp Med (2011) 208(3):577–92. doi: 10.1084/jem.20100619
151. Wang L, Le Mercier I, Putra J, Chen W, Liu J, Schenk AD, et al. Disruption of the immune-checkpoint VISTA gene imparts a proinflammatory phenotype with predisposition to the development of autoimmunity. Proc Natl Acad Sci U.S.A. (2014) 111(41):14846–51. doi: 10.1073/pnas.1407447111
152. ElTanbouly MA, Schaafsma E, Smits NC, Shah P, Cheng C, Burns C, et al. VISTA re-programs macrophage biology through the combined regulation of tolerance and anti-inflammatory pathways. Front Immunol (2020) 11:580187. doi: 10.3389/fimmu.2020.580187
153. Han X, Vesely MD, Yang W, Sanmamed MF, Badri T, Alawa J, et al. PD-1H (VISTA)-mediated suppression of autoimmunity in systemic and cutaneous lupus erythematosus. Sci Transl Med (2019) 11(522):eaax1159. doi: 10.1126/scitranslmed.aax1159
154. Le Mercier I, Chen W, Lines JL, Day M, Li J, Sergent P, et al. VISTA regulates the development of protective antitumor immunity. Cancer Res (2014) 74(7):1933–44. doi: 10.1158/0008-5472.CAN-13-1506
155. Xu W, Dong J, Zheng Y, Zhou J, Yuan Y, Ta HM, et al. Immune-checkpoint protein VISTA regulates antitumor immunity by controlling myeloid cell-mediated inflammation and immunosuppression. Cancer Immunol Res (2019) 7(9):1497–510. doi: 10.1158/2326-6066.CIR-18-0489
156. Zhai L, Spranger S, Binder DC, Gritsina G, Lauing KL, Giles FJ, et al. Molecular pathways: Targeting IDO1 and other tryptophan dioxygenases for cancer immunotherapy. Clin Cancer Res (2015) 21(24):5427–33. doi: 10.1158/1078-0432.CCR-15-0420
157. Amobi A, Qian F, Lugade AA, Odunsi K. Tryptophan catabolism and cancer immunotherapy targeting IDO mediated immune suppression. Adv Exp Med Biol (2017) 1036:129–44. doi: 10.1007/978-3-319-67577-0_9
158. Prendergast GC, Malachowski WJ, Mondal A, Scherle P, Muller AJ. Indoleamine 2,3-dioxygenase and its therapeutic inhibition in cancer. Int Rev Cell Mol Biol (2018) 336:175–203. doi: 10.1016/bs.ircmb.2017.07.004
159. Ufermann CM, Domrose A, Babel T, Tersteegen A, Cengiz SC, Eller SK, et al. Indoleamine 2,3-dioxygenase activity during acute toxoplasmosis and the suppressed T cell proliferation in mice. Front Cell Infect Microbiol (2019) 9:184. doi: 10.3389/fcimb.2019.00184
160. Mellor A. Indoleamine 2,3 dioxygenase and regulation of T cell immunity. Biochem Biophys Res Commun (2005) 338(1):20–4. doi: 10.1016/j.bbrc.2005.08.232
161. Munn DH, Mellor AL. IDO in the tumor microenvironment: Inflammation, counter-regulation, and tolerance. Trends Immunol (2016) 37(3):193–207. doi: 10.1016/j.it.2016.01.002
162. Yang Y, Liu K, Chen Y, Gong Y, Liang Y. Indoleamine 2,3-dioxygenase (IDO) regulates Th17/Treg immunity in experimental IgA nephropathy. Folia Biol (Praha) (2019) 65(2):101–8.
163. Azadegan-Dehkordi F, Shirzad H, Ahmadi R, Bashash D, Abdollahpour-Alitappeh M, Luzza F, et al. Increased indoleamine 2, 3-dioxygenase expression modulates Th1/Th17/Th22 and treg pathway in humans with helicobacter pylori-infected gastric mucosa. Hum Immunol (2021) 82(1):46–53. doi: 10.1016/j.humimm.2020.10.005
164. Wang XF, Wang HS, Wang H, Zhang F, Wang KF, Guo Q, et al. The role of indoleamine 2,3-dioxygenase (IDO) in immune tolerance: focus on macrophage polarization of THP-1 cells. Cell Immunol (2014) 289(1-2):42–8. doi: 10.1016/j.cellimm.2014.02.005
165. Jiang N, Zhang L, Zhao G, Lin J, Wang Q, Xu Q, et al. Indoleamine 2,3-dioxygenase regulates macrophage recruitment, polarization and phagocytosis in aspergillus fumigatus keratitis. Invest Ophthalmol Vis Sci (2020) 61(8):28. doi: 10.1167/iovs.61.8.28
166. Schramme F, Crosignani S, Frederix K, Hoffmann D, Pilotte L, Stroobant V, et al. Inhibition of tryptophan-dioxygenase activity increases the antitumor efficacy of immune checkpoint inhibitors. Cancer Immunol Res (2020) 8(1):32–45. doi: 10.1158/2326-6066.CIR-19-0041
167. Greene LI, Bruno TC, Christenson JL, D'Alessandro A, Culp-Hill R, Torkko K, et al. A role for tryptophan-2,3-dioxygenase in CD8 T-cell suppression and evidence of tryptophan catabolism in breast cancer patient plasma. Mol Cancer Res (2019) 17(1):131–9. doi: 10.1158/1541-7786.MCR-18-0362
168. Hjortso MD, Larsen SK, Kongsted P, Met O, Frosig TM, Andersen GH, et al. Tryptophan 2,3-dioxygenase (TDO)-reactive T cells differ in their functional characteristics in health and cancer. Oncoimmunology (2015) 4(1):e968480. doi: 10.4161/21624011.2014.968480
169. Hutloff A, Dittrich AM, Beier KC, Eljaschewitsch B, Kraft R, Anagnostopoulos I, et al. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature (1999) 397(6716):263–6. doi: 10.1038/16717
170. Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol (2005) 23:515–48. doi: 10.1146/annurev.immunol.23.021704.115611
171. Witsch EJ, Peiser M, Hutloff A, Buchner K, Dorner BG, Jonuleit H, et al. ICOS and CD28 reversely regulate IL-10 on re-activation of human effector T cells with mature dendritic cells. Eur J Immunol (2002) 32(9):2680–6. doi: 10.1002/1521-4141(200209)32:9<2680::AID-IMMU2680>3.0.CO;2-6
172. Gonzalo JA, Tian J, Delaney T, Corcoran J, Rottman JB, Lora J, et al. ICOS is critical for T helper cell-mediated lung mucosal inflammatory responses. Nat Immunol (2001) 2(7):597–604. doi: 10.1038/89739
173. Coyle AJ, Lehar S, Lloyd C, Tian J, Delaney T, Manning S, et al. The CD28-related molecule ICOS is required for effective T cell-dependent immune responses. Immunity (2000) 13(1):95–105. doi: 10.1016/s1074-7613(00)00011-x
174. Panneton V, Chang J, Witalis M, Li J, Suh WK. Inducible T-cell co-stimulator: Signaling mechanisms in T follicular helper cells and beyond. Immunol Rev (2019) 291(1):91–103. doi: 10.1111/imr.12771
175. Amatore F, Gorvel L, Olive D. Role of inducible Co-stimulator (ICOS) in cancer immunotherapy. Expert Opin Biol Ther (2020) 20(2):141–50. doi: 10.1080/14712598.2020.1693540
176. Specenier P. Ipilimumab in melanoma. Expert Rev Anticancer Ther (2016) 16(8):811–26. doi: 10.1080/14737140.2016.1211936
177. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363(8):711–23. doi: 10.1056/NEJMoa1003466
178. Eggermont AM, Chiarion-Sileni V, Grob JJ, Dummer R, Wolchok JD, Schmidt H, et al. Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): a randomised, double-blind, phase 3 trial. Lancet Oncol (2015) 16(5):522–30. doi: 10.1016/S1470-2045(15)70122-1
179. Ascierto PA, Del Vecchio M, Robert C, Mackiewicz A, Chiarion-Sileni V, Arance A, et al. Ipilimumab 10 mg/kg versus ipilimumab 3 mg/kg in patients with unresectable or metastatic melanoma: a randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol (2017) 18(5):611–22. doi: 10.1016/S1470-2045(17)30231-0
180. Hodi FS, Butler M, Oble DA, Seiden MV, Haluska FG, Kruse A, et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc Natl Acad Sci U.S.A. (2008) 105(8):3005–10. doi: 10.1073/pnas.0712237105
181. Hodi FS, Mihm MC, Soiffer RJ, Haluska FG, Butler M, Seiden MV, et al. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc Natl Acad Sci U.S.A. (2003) 100(8):4712–7. doi: 10.1073/pnas.0830997100
182. Small EJ, Tchekmedyian NS, Rini BI, Fong L, Lowy I, Allison JP. A pilot trial of CTLA-4 blockade with human anti-CTLA-4 in patients with hormone-refractory prostate cancer. Clin Cancer Res (2007) 13(6):1810–5. doi: 10.1158/1078-0432.CCR-06-2318
183. Robert C, Thomas L, Bondarenko I, O'Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med (2011) 364(26):2517–26. doi: 10.1056/NEJMoa1104621
184. Ribas A, Hanson DC, Noe DA, Millham R, Guyot DJ, Bernstein SH, et al. Tremelimumab (CP-675,206), a cytotoxic T lymphocyte associated antigen 4 blocking monoclonal antibody in clinical development for patients with cancer. Oncologist (2007) 12(7):873–83. doi: 10.1634/theoncologist.12-7-873
185. Tarhini AA. Tremelimumab: a review of development to date in solid tumors. Immunotherapy (2013) 5(3):215–29. doi: 10.2217/imt.13.9
186. Ribas A, Camacho LH, Lopez-Berestein G, Pavlov D, Bulanhagui CA, Millham R, et al. Antitumor activity in melanoma and anti-self responses in a phase I trial with the anti-cytotoxic T lymphocyte-associated antigen 4 monoclonal antibody CP-675,206. J Clin Oncol (2005) 23(35):8968–77. doi: 10.1200/JCO.2005.01.109
187. Camacho LH, Antonia S, Sosman J, Kirkwood JM, Gajewski TF, Redman B, et al. Phase I/II trial of tremelimumab in patients with metastatic melanoma. J Clin Oncol (2009) 27(7):1075–81. doi: 10.1200/JCO.2008.19.2435
188. Kirkwood JM, Lorigan P, Hersey P, Hauschild A, Robert C, McDermott D, et al. Phase II trial of tremelimumab (CP-675,206) in patients with advanced refractory or relapsed melanoma. Clin Cancer Res (2010) 16(3):1042–8. doi: 10.1158/1078-0432.CCR-09-2033
189. Ribas A, Hauschild A, Kefford R, Punt CJ, Haanen JB, Marmol M, et al. Open-label, randomized, comparative study of tremelimumab (CP-675,206) and chemotherapy (temozolomide [TMZ] or dacarbazine [DTIC]) in patients with advanced melanoma. J Clin Oncol (2008) 26(15_suppl):LBA9011–LBA9011. doi: 10.1200/jco.2008.26.15_suppl.lba9011
190. Ralph C, Elkord E, Burt DJ, O'Dwyer JF, Austin EB, Stern PL, et al. Modulation of lymphocyte regulation for cancer therapy: a phase II trial of tremelimumab in advanced gastric and esophageal adenocarcinoma. Clin Cancer Res (2010) 16(5):1662–72. doi: 10.1158/1078-0432.CCR-09-2870
191. Zatloukal P, Heo DS, Park K, Kang J, Butts C, Bradford D, et al. Randomized phase II clinical trial comparing tremelimumab (CP-675,206) with best supportive care (BSC) following first-line platinum-based therapy in patients (pts) with advanced non-small cell lung cancer (NSCLC). J Clin Oncol (2009) 27(15_suppl):8071–1. doi: 10.1200/jco.2009.27.15_suppl.8071
192. Melero I, Sangro B, Riezu-Boj JI, Inãarrairaegui M, Lasarte JJ, Sarobe P, et al. Prieto: Abstract 4387: Antiviral and antitumoral effects of the anti-CTLA4 agent tremelimumab in patients with hepatocellular carcinoma (HCC) and chronic hepatitis c virus (HCV) infection: Results from a phase II clinical trial. Cancer Res (2012) 72(8_Supplement):4387–7. doi: 10.1158/1538-7445.Am2012-4387
193. Chung KY, Gore I, Fong L, Venook A, Beck SB, Dorazio P, et al. Phase II study of the anti-cytotoxic T-lymphocyte-associated antigen 4 monoclonal antibody, tremelimumab, in patients with refractory metastatic colorectal cancer. J Clin Oncol (2010) 28(21):3485–90. doi: 10.1200/JCO.2010.28.3994
194. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science (2018) 359(6382):1350–5. doi: 10.1126/science.aar4060
195. Korman AJ, Garrett-Thomson SC, Lonberg N. The foundations of immune checkpoint blockade and the ipilimumab approval decennial. Nat Rev Drug Discov (2021) 21(7):509–28. doi: 10.1038/s41573-021-00345-8
196. Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol (2010) 28(19):3167–75. doi: 10.1200/JCO.2009.26.7609
197. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366(26):2443–54. doi: 10.1056/NEJMoa1200690
198. Weber JS, D'Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol (2015) 16(4):375–84. doi: 10.1016/S1470-2045(15)70076-8
199. Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med (2015) 373(19):1803–13. doi: 10.1056/NEJMoa1510665
200. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus docetaxel in advanced nonsquamous non-Small-Cell lung cancer. N Engl J Med (2015) 373(17):1627–39. doi: 10.1056/NEJMoa1507643
201. Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, et al. Nivolumab versus docetaxel in advanced squamous-cell non-Small-Cell lung cancer. N Engl J Med (2015) 373(2):123–35. doi: 10.1056/NEJMoa1504627
202. Vaddepally RK, Kharel P, Pandey R, Garje R, Chandra AB. Review of indications of FDA-approved immune checkpoint inhibitors per NCCN guidelines with the level of evidence. Cancers (Basel) (2020) 12(3):738. doi: 10.3390/cancers12030738
203. Green MR, Monti S, Rodig SJ, Juszczynski P, Currie T, O'Donnell E, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large b-cell lymphoma. Blood (2010) 116(17):3268–77. doi: 10.1182/blood-2010-05-282780
204. Roemer MG, Advani RH, Ligon AH, Natkunam Y, Redd RA, Homer H, et al. PD-L1 and PD-L2 genetic alterations define classical Hodgkin lymphoma and predict outcome. J Clin Oncol (2016) 34(23):2690–7. doi: 10.1200/JCO.2016.66.4482
205. Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory hodgkin's lymphoma. N Engl J Med (2015) 372(4):311–9. doi: 10.1056/NEJMoa1411087
206. Armand P, Engert A, Younes A, Fanale M, Santoro A, Zinzani PL, et al. Nivolumab for Relapsed/Refractory classic Hodgkin lymphoma after failure of autologous hematopoietic cell transplantation: Extended follow-up of the multicohort single-arm phase II CheckMate 205 trial. J Clin Oncol (2018) 36(14):1428–39. doi: 10.1200/JCO.2017.76.0793
207. Kasamon YL, de Claro RA, Wang Y, Shen YL, Farrell AT, Pazdur R. FDA Approval summary: Nivolumab for the treatment of relapsed or progressive classical Hodgkin lymphoma. Oncologist (2017) 22(5):585–91. doi: 10.1634/theoncologist.2017-0004
208. van Vugt MJH, Stone JA, De Greef R, Snyder ES, Lipka L, Turner DC, et al. Immunogenicity of pembrolizumab in patients with advanced tumors. J Immunother Cancer (2019) 7(1):212. doi: 10.1186/s40425-019-0663-4
209. Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet (2014) 384(9948):1109–17. doi: 10.1016/S0140-6736(14)60958-2
210. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med (2015) 372(26):2521–32. doi: 10.1056/NEJMoa1503093
211. Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Pembrolizumab versus chemotherapy for PD-L1-Positive non-Small-Cell lung cancer. N Engl J Med (2016) 375(19):1823–33. doi: 10.1056/NEJMoa1606774
212. Shalhout SZ, Emerick KS, Kaufman HL, Miller DM. Immunotherapy for non-melanoma skin cancer. Curr Oncol Rep (2021) 23(11):125. doi: 10.1007/s11912-021-01120-z
213. Akinboro O, Larkins E, Pai-Scherf LH, Mathieu LN, Ren Y, Cheng J, et al. FDA Approval summary: Pembrolizumab, atezolizumab, and cemiplimab-rwlc as single agents for first-line treatment of advanced/metastatic PD-L1 high NSCLC. Clin Cancer Res (2022) 28(11):2221–8. doi: 10.1158/1078-0432.CCR-21-3844
214. Keeping S, Xu Y, Chen CI, Cope S, Mojebi A, Kuznik A, et al. Comparative efficacy of cemiplimab versus other systemic treatments for advanced cutaneous squamous cell carcinoma. Future Oncol (2021) 17(5):611–27. doi: 10.2217/fon-2020-0823
215. Gambale E, Fancelli S, Caliman E, Petrella MC, Doni L, Pillozzi S, et al. Immune checkpoint blockade with anti-programmed cell death 1 (PD-1) monoclonal antibody (mAb) cemiplimab: ongoing and future perspectives in rare genital cancers treatment. J Immunother Cancer (2022) 10(1):e003540. doi: 10.1136/jitc-2021-003540
216. Yi M, Zheng X, Niu M, Zhu S, Ge H, Wu K. Combination strategies with PD-1/PD-L1 blockade: current advances and future directions. Mol Cancer (2022) 21(1):28. doi: 10.1186/s12943-021-01489-2
217. Akinleye A, Rasool Z. Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J Hematol Oncol (2019) 12(1):92. doi: 10.1186/s13045-019-0779-5
218. Li M, Zhao R, Chen J, Tian W, Xia C, Liu X, et al. Next generation of anti-PD-L1 atezolizumab with enhanced anti-tumor efficacy in vivo. Sci Rep (2021) 11(1):5774. doi: 10.1038/s41598-021-85329-9
219. Lipson EJ, Tawbi HA-H, Schadendorf D, Ascierto PA, Matamala L, Gutiérrez EC, et al. Relatlimab (RELA) plus nivolumab (NIVO) versus NIVO in first-line advanced melanoma: Primary phase III results from RELATIVITY-047 (CA224-047). J Clin Oncol (2021) 39(15_suppl):9503–3. doi: 10.1200/JCO.2021.39.15_suppl.9503
220. Paik J. Nivolumab plus relatlimab: First approval. Drugs (2022) 82(8):925–31. doi: 10.1007/s40265-022-01723-1
221. Andre P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell (2018) 175(7):1731–43.e13. doi: 10.1016/j.cell.2018.10.014
222. Galot R, Le Tourneau C, Saada-Bouzid E, Daste A, Even C, Debruyne P, et al. A phase II study of monalizumab in patients with recurrent/metastatic squamous cell carcinoma of the head and neck: The I1 cohort of the EORTC-HNCG-1559 UPSTREAM trial. Eur J Cancer (2021) 158:17–26. doi: 10.1016/j.ejca.2021.09.003
223. Caratelli S, Arriga R, Sconocchia T, Ottaviani A, Lanzilli G, Pastore D, et al. In vitro elimination of epidermal growth factor receptor-overexpressing cancer cells by CD32A-chimeric receptor T cells in combination with cetuximab or panitumumab. Int J Cancer (2020) 146(1):236–47. doi: 10.1002/ijc.32663
224. Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH, et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood (2011) 117(17):4501–10. doi: 10.1182/blood-2010-10-310425
225. Tian T, Li Z. Targeting Tim-3 in cancer with resistance to PD-1/PD-L1 blockade. Front Oncol (2021) 11:731175. doi: 10.3389/fonc.2021.731175
226. Berry S, Giraldo N, Nguyen P, Green B, Xu H, Ogurtsova A, et al.33rd annual meeting & pre-conference programs of the society for immunotherapy of cancer (SITC 2018). J Immunother Cancer (2018) 6(Suppl 1):115. doi: 10.1186/s40425-018-0423-x
227. Harding JJ, Patnaik A, Moreno V, Stein M, Jankowska AM, d. Mendizabal NV, et al. A phase Ia/Ib study of an anti-TIM-3 antibody (LY3321367) monotherapy or in combination with an anti-PD-L1 antibody (LY3300054): Interim safety, efficacy, and pharmacokinetic findings in advanced cancers. J Clin Oncol (2019) 37(8_suppl):12–2. doi: 10.1200/JCO.2019.37.8_suppl.12
228. Kim JM, Chen DS. Immune escape to PD-L1/PD-1 blockade: seven steps to success (or failure). Ann Oncol (2016) 27(8):1492–504. doi: 10.1093/annonc/mdw217
229. Rozeman EA, Hoefsmit EP, Reijers ILM, Saw RPM, Versluis JM, Krijgsman O, et al. Survival and biomarker analyses from the OpACIN-neo and OpACIN neoadjuvant immunotherapy trials in stage III melanoma. Nat Med (2021) 27(2):256–63. doi: 10.1038/s41591-020-01211-7
230. Motzer RJ, Rini BI, McDermott DF, Aren Frontera O, Hammers HJ, Carducci MA, et al. Nivolumab plus ipilimumab versus sunitinib in first-line treatment for advanced renal cell carcinoma: extended follow-up of efficacy and safety results from a randomised, controlled, phase 3 trial. Lancet Oncol (2019) 20(10):1370–85. doi: 10.1016/S1470-2045(19)30413-9
231. Overman MJ, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-Deficient/Microsatellite instability-high metastatic colorectal cancer. J Clin Oncol (2018) 36(8):773–9. doi: 10.1200/JCO.2017.76.9901
232. Yau T, Kang YK, Kim TY, El-Khoueiry AB, Santoro A, Sangro B, et al. Efficacy and safety of nivolumab plus ipilimumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib: The CheckMate 040 randomized clinical trial. JAMA Oncol (2020) 6(11):e204564. doi: 10.1001/jamaoncol.2020.4564
233. Hellmann MD, Paz-Ares L, Bernabe Caro R, Zurawski B, Kim SW, Carcereny Costa E, et al. Nivolumab plus ipilimumab in advanced non-Small-Cell lung cancer. N Engl J Med (2019) 381(21):2020–31. doi: 10.1056/NEJMoa1910231
234. Paz-Ares L, Ciuleanu TE, Cobo M, Schenker M, Zurawski B, Menezes J, et al. First-line nivolumab plus ipilimumab combined with two cycles of chemotherapy in patients with non-small-cell lung cancer (CheckMate 9LA): an international, randomised, open-label, phase 3 trial. Lancet Oncol (2021) 22(2):198–211. doi: 10.1016/S1470-2045(20)30641-0
235. Scherpereel A, Antonia S, Bautista Y, Grossi F, Kowalski D, Zalcman G, et al. First-line nivolumab plus ipilimumab versus chemotherapy for the treatment of unresectable malignant pleural mesothelioma: patient-reported outcomes in CheckMate 743. Lung Cancer (2022) 167:8–16. doi: 10.1016/j.lungcan.2022.03.012
236. Paz-Ares L, Dvorkin M, Chen Y, Reinmuth N, Hotta K, Trukhin D, et al. Durvalumab plus platinum-etoposide versus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): a randomised, controlled, open-label, phase 3 trial. Lancet (2019) 394(10212):1929–39. doi: 10.1016/S0140-6736(19)32222-6
237. Goldman JW, Dvorkin M, Chen Y, Reinmuth N, Hotta K, Trukhin D, et al. Durvalumab, with or without tremelimumab, plus platinum-etoposide versus platinum-etoposide alone in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): updated results from a randomised, controlled, open-label, phase 3 trial. Lancet Oncol (2021) 22(1):51–65. doi: 10.1016/S1470-2045(20)30539-8
238. Gao J, Navai N, Alhalabi O, Siefker-Radtke A, Campbell MT, Tidwell RS, et al. Neoadjuvant PD-L1 plus CTLA-4 blockade in patients with cisplatin-ineligible operable high-risk urothelial carcinoma. Nat Med (2020) 26(12):1845–51. doi: 10.1038/s41591-020-1086-y
239. Powles T, van der Heijden MS, Castellano D, Galsky MD, Loriot Y, Petrylak DP, et al. Durvalumab alone and durvalumab plus tremelimumab versus chemotherapy in previously untreated patients with unresectable, locally advanced or metastatic urothelial carcinoma (DANUBE): a randomised, open-label, multicentre, phase 3 trial. Lancet Oncol (2020) 21(12):1574–88. doi: 10.1016/S1470-2045(20)30541-6
240. Chen EX, Jonker DJ, Loree JM, Kennecke HF, Berry SR, Couture F, et al. Effect of combined immune checkpoint inhibition vs best supportive care alone in patients with advanced colorectal cancer: The Canadian cancer trials group CO.26 study. JAMA Oncol (2020) 6(6):831–8. doi: 10.1001/jamaoncol.2020.0910
241. Planchard D, Reinmuth N, Orlov S, Fischer JR, Sugawara S, Mandziuk S, et al. ARCTIC: durvalumab with or without tremelimumab as third-line or later treatment of metastatic non-small-cell lung cancer. Ann Oncol (2020) 31(5):609–18. doi: 10.1016/j.annonc.2020.02.006
242. Planchard D, Yokoi T, McCleod MJ, Fischer JR, Kim YC, Ballas M, et al. A phase III study of durvalumab (MEDI4736) with or without tremelimumab for previously treated patients with advanced NSCLC: Rationale and protocol design of the ARCTIC study. Clin Lung Cancer (2016) 17(3):232–236.e1. doi: 10.1016/j.cllc.2016.03.003
243. Kelly RJ, Lee J, Bang YJ, Almhanna K, Blum-Murphy M, Catenacci DVT, et al. Safety and efficacy of durvalumab and tremelimumab alone or in combination in patients with advanced gastric and gastroesophageal junction adenocarcinoma. Clin Cancer Res (2020) 26(4):846–54. doi: 10.1158/1078-0432.CCR-19-2443
244. Siu LL, Even C, Mesia R, Remenar E, Daste A, Delord JP, et al. Safety and efficacy of durvalumab with or without tremelimumab in patients with PD-L1-Low/Negative recurrent or metastatic HNSCC: The phase 2 CONDOR randomized clinical trial. JAMA Oncol (2019) 5(2):195–203. doi: 10.1001/jamaoncol.2018.4628
245. Kelley RK, Sangro B, Harris W, Ikeda M, Okusaka T, Kang YK, et al. Safety, efficacy, and pharmacodynamics of tremelimumab plus durvalumab for patients with unresectable hepatocellular carcinoma: Randomized expansion of a phase I/II study. J Clin Oncol (2021) 39(27):2991–3001. doi: 10.1200/JCO.20.03555
246. Necchi A, Giannatempo P, Raggi D, Mariani L, Colecchia M, Fare E, et al. An open-label randomized phase 2 study of durvalumab alone or in combination with tremelimumab in patients with advanced germ cell tumors (APACHE): Results from the first planned interim analysis. Eur Urol (2019) 75(1):201–3. doi: 10.1016/j.eururo.2018.09.010
247. Calabro L, Morra A, Giannarelli D, Amato G, D'Incecco A, Covre A, et al. Tremelimumab combined with durvalumab in patients with mesothelioma (NIBIT-MESO-1): an open-label, non-randomised, phase 2 study. Lancet Respir Med (2018) 6(6):451–60. doi: 10.1016/S2213-2600(18)30151-6
248. Rizvi NA, Cho BC, Reinmuth N, Lee KH, Luft A, Ahn MJ, et al. Durvalumab with or without tremelimumab vs standard chemotherapy in first-line treatment of metastatic non-small cell lung cancer: The MYSTIC phase 3 randomized clinical trial. JAMA Oncol (2020) 6(5):661–74. doi: 10.1001/jamaoncol.2020.0237
249. Fuerst ML. Durvalumab + tremelimumab improves survival in advanced liver cancer. Oncol Times (2022) 44(4):29. doi: 10.1097/01.Cot.0000821988.50798.78
250. Long GV, Hodi FS, Lipson EJ, Schadendorf D, Ascierto PA, Matamala L, et al. Relatlimab and nivolumab versus nivolumab in previously untreated metastatic or unresectable melanoma: Overall survival and response rates from RELATIVITY-047 (CA224-047). J Clin Oncol (2022) 40(36_suppl):360385–5. doi: 10.1200/JCO.2022.40.36_suppl.360385
251. Gotwals P, Cameron S, Cipolletta D, Cremasco V, Crystal A, Hewes B, et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat Rev Cancer (2017) 17(5):286–301. doi: 10.1038/nrc.2017.17
252. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med (2007) 13(1):54–61. doi: 10.1038/nm1523
253. Zhu H, Shan Y, Ge K, Lu J, Kong W, Jia C. Oxaliplatin induces immunogenic cell death in hepatocellular carcinoma cells and synergizes with immune checkpoint blockade therapy. Cell Oncol (Dordr) (2020) 43(6):1203–14. doi: 10.1007/s13402-020-00552-2
254. Qin S, Yi M, Jiao D, Li A, Wu K. Distinct roles of VEGFA and ANGPT2 in lung adenocarcinoma and squamous cell carcinoma. J Cancer (2020) 11(1):153–67. doi: 10.7150/jca.34693
255. Viallard C, Larrivee B. Tumor angiogenesis and vascular normalization: alternative therapeutic targets. Angiogenesis (2017) 20(4):409–26. doi: 10.1007/s10456-017-9562-9
256. Pan C, Liu H, Robins E, Song W, Liu D, Li Z, et al. Next-generation immuno-oncology agents: current momentum shifts in cancer immunotherapy. J Hematol Oncol (2020) 13(1):29. doi: 10.1186/s13045-020-00862-w
257. Hsieh JJ, Purdue MP, Signoretti S, Swanton C, Albiges L, Schmidinger M, et al. Renal cell carcinoma. Nat Rev Dis Primers (2017) 3:17009. doi: 10.1038/nrdp.2017.9
258. Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med (2018) 378(24):2288–301. doi: 10.1056/NEJMoa1716948
259. Motzer R, Alekseev B, Rha SY, Porta C, Eto M, Powles T, et al. Lenvatinib plus pembrolizumab or everolimus for advanced renal cell carcinoma. N Engl J Med (2021) 384(14):1289–300. doi: 10.1056/NEJMoa2035716
260. Choueiri TK, Powles T, Burotto M, Escudier B, Bourlon MT, Zurawski B, et al. Nivolumab plus cabozantinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med (2021) 384(9):829–41. doi: 10.1056/NEJMoa2026982
261. Motzer RJ, Robbins PB, Powles T, Albiges L, Haanen JB, Larkin J, et al. Avelumab plus axitinib versus sunitinib in advanced renal cell carcinoma: biomarker analysis of the phase 3 JAVELIN renal 101 trial. Nat Med (2020) 26(11):1733–41. doi: 10.1038/s41591-020-1044-8
262. Powles T, Plimack ER, Soulieres D, Waddell T, Stus V, Gafanov R, et al. Pembrolizumab plus axitinib versus sunitinib monotherapy as first-line treatment of advanced renal cell carcinoma (KEYNOTE-426): extended follow-up from a randomised, open-label, phase 3 trial. Lancet Oncol (2020) 21(12):1563–73. doi: 10.1016/S1470-2045(20)30436-8
263. Makker V, Rasco D, Vogelzang NJ, Brose MS, Cohn AL, Mier J, et al. Lenvatinib plus pembrolizumab in patients with advanced endometrial cancer: an interim analysis of a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol (2019) 20(5):711–8. doi: 10.1016/S1470-2045(19)30020-8
264. Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med (2020) 382(20):1894–905. doi: 10.1056/NEJMoa1915745
265. Ramos-Casals M, Brahmer JR, Callahan MK, Flores-Chavez A, Keegan N, Khamashta MA, et al. Immune-related adverse events of checkpoint inhibitors. Nat Rev Dis Primers (2020) 6(1):38. doi: 10.1038/s41572-020-0160-6
266. Ramos-Casals M, Lambotte O, Kostine M, Calabrese L, Suarez-Almazor M, Bingham C, et al. THU0628 IMMUNE-RELATED ADVERSE EVENTS INDUCED BY CANCER IMMUNOTHERAPIES. BIG DATA ANALYSIS OF 13,051 CASES (IMMUNOCANCER INTERNATIONAL REGISTRY). Ann Rheum Dis (2019) 78(Suppl 2):607–8. doi: 10.1136/annrheumdis-2019-eular.2707
267. Postow MA, Sidlow R, Hellmann MD. Immune-related adverse events associated with immune checkpoint blockade. N Engl J Med (2018) 378(2):158–68. doi: 10.1056/NEJMra1703481
268. Yoest JM. Clinical features, predictive correlates, and pathophysiology of immune-related adverse events in immune checkpoint inhibitor treatments in cancer: a short review. Immunotargets Ther (2017) 6:73–82. doi: 10.2147/ITT.S126227
269. Parakh S, Cebon J, Klein O. Delayed autoimmune toxicity occurring several months after cessation of anti-PD-1 therapy. Oncologist (2018) 23(7):849–51. doi: 10.1634/theoncologist.2017-0531
270. Kanjanapan Y, Day D, Butler MO, Wang L, Joshua AM, Hogg D, et al. Delayed immune-related adverse events in assessment for dose-limiting toxicity in early phase immunotherapy trials. Eur J Cancer (2019) 107:1–7. doi: 10.1016/j.ejca.2018.10.017
271. Weber JS, Kahler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol (2012) 30(21):2691–7. doi: 10.1200/JCO.2012.41.6750
272. Sandigursky S, Mor A. Immune-related adverse events in cancer patients treated with immune checkpoint inhibitors. Curr Rheumatol Rep (2018) 20(10):65. doi: 10.1007/s11926-018-0770-0
273. Farolfi A, Ridolfi L, Guidoboni M, Nicoletti SV, Piciucchi S, Valmorri L, et al. Ipilimumab in advanced melanoma: reports of long-lasting responses. Melanoma Res (2012) 22(3):263–70. doi: 10.1097/CMR.0b013e328353e65c
274. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi: 10.1038/nrc3239
275. Fecher LA, Agarwala SS, Hodi FS, Weber JS. Ipilimumab and its toxicities: a multidisciplinary approach. Oncologist (2013) 18(6):733–43. doi: 10.1634/theoncologist.2012-0483
276. Boutros C, Tarhini A, Routier E, Lambotte O, Ladurie FL, Carbonnel F, et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat Rev Clin Oncol (2016) 13(8):473–86. doi: 10.1038/nrclinonc.2016.58
277. Gupta A, De Felice KM, Loftus EV Jr., Khanna S. Systematic review: colitis associated with anti-CTLA-4 therapy. Aliment Pharmacol Ther (2015) 42(4):406–17. doi: 10.1111/apt.13281
278. Ribas A, Puzanov I, Dummer R, Schadendorf D, Hamid O, Robert C, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol (2015) 16(8):908–18. doi: 10.1016/S1470-2045(15)00083-2
279. Xu C, Chen YP, Du XJ, Liu JQ, Huang CL, Chen L, et al. Comparative safety of immune checkpoint inhibitors in cancer: systematic review and network meta-analysis. BMJ (2018) 363:k4226. doi: 10.1136/bmj.k4226
280. Wallis N, Bulanhagui CA, Dorazio PC, Healey DI, Marshall MA, Liang JQ, et al. Safety of tremelimumab (CP-675,206) in patients (pts) with advanced cancer. J Clin Oncol (2008) 26(15_suppl):3040–0. doi: 10.1200/jco.2008.26.15_suppl.3040
281. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med (2015) 373(1):23–34. doi: 10.1056/NEJMoa1504030
282. Perez-Ruiz E, Minute L, Otano I, Alvarez M, Ochoa MC, Belsue V, et al. Prophylactic TNF blockade uncouples efficacy and toxicity in dual CTLA-4 and PD-1 immunotherapy. Nature (2019) 569(7756):428–32. doi: 10.1038/s41586-019-1162-y
283. Langer CJ, Gadgeel SM, Borghaei H, Papadimitrakopoulou VA, Patnaik A, Powell SF, et al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: a randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol (2016) 17(11):1497–508. doi: 10.1016/S1470-2045(16)30498-3
284. Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med (2018) 379(22):2108–21. doi: 10.1056/NEJMoa1809615
285. Dovedi SJ, Elder MJ, Yang C, Sitnikova SI, Irving L, Hansen A, et al. Design and efficacy of a monovalent bispecific PD-1/CTLA4 antibody that enhances CTLA4 blockade on PD-1(+) activated T cells. Cancer Discov (2021) 11(5):1100–17. doi: 10.1158/2159-8290.CD-20-1445
286. Burton EM, Tawbi HA. Bispecific antibodies to PD-1 and CTLA4: Doubling down on T cells to decouple efficacy from toxicity. Cancer Discov (2021) 11(5):1008–10. doi: 10.1158/2159-8290.CD-21-0257
287. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science (2015) 348(6230):69–74. doi: 10.1126/science.aaa4971
288. Riaz N, Morris L, Havel JJ, Makarov V, Desrichard A, Chan TA. The role of neoantigens in response to immune checkpoint blockade. Int Immunol (2016) 28(8):411–9. doi: 10.1093/intimm/dxw019
289. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science (2011) 331(6024):1565–70. doi: 10.1126/science.1203486
290. McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science (2016) 351(6280):1463–9. doi: 10.1126/science.aaf1490
291. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med (2015) 372(26):2509–20. doi: 10.1056/NEJMoa1500596
292. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science (2017) 357(6349):409–13. doi: 10.1126/science.aan6733
293. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell (2016) 165(1):35–44. doi: 10.1016/j.cell.2016.02.065
294. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med (2016) 375(9):819–29. doi: 10.1056/NEJMoa1604958
295. Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane JP, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun (2017) 8(1):1136. doi: 10.1038/s41467-017-01062-w
296. Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, et al. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol (2019) 20(3):326–36. doi: 10.1038/s41590-019-0312-6
297. Martin AM, Nirschl TR, Nirschl CJ, Francica BJ, Kochel CM, van Bokhoven A, et al. Paucity of PD-L1 expression in prostate cancer: innate and adaptive immune resistance. Prostate Cancer Prostatic Dis (2015) 18(4):325–32. doi: 10.1038/pcan.2015.39
298. Rudensky AY. Regulatory T cells and Foxp3. Immunol Rev (2011) 241(1):260–8. doi: 10.1111/j.1600-065X.2011.01018.x
299. Chaudhary B, Elkord E. Regulatory T cells in the tumor microenvironment and cancer progression: Role and therapeutic targeting. Vaccines (Basel) (2016) 4(3):28. doi: 10.3390/vaccines4030028
300. Khaled YS, Ammori BJ, Elkord E. Myeloid-derived suppressor cells in cancer: recent progress and prospects. Immunol Cell Biol (2013) 91(8):493–502. doi: 10.1038/icb.2013.29
301. Xu J, Zhang J, Wang J. The application of traditional Chinese medicine against the tumor immune escape. J Transl Int Med (2020) 8(4):203–4. doi: 10.2478/jtim-2020-0032
302. Gopalakrishnan V, Helmink BA, Spencer CN, Reuben A, Wargo JA. The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell (2018) 33(4):570–80. doi: 10.1016/j.ccell.2018.03.015
303. Sautes-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer (2019) 19(6):307–25. doi: 10.1038/s41568-019-0144-6
304. Goc J, Germain C, Vo-Bourgais TK, Lupo A, Klein C, Knockaert S, et al. Dendritic cells in tumor-associated tertiary lymphoid structures signal a Th1 cytotoxic immune contexture and license the positive prognostic value of infiltrating CD8+ T cells. Cancer Res (2014) 74(3):705–15. doi: 10.1158/0008-5472.CAN-13-1342
305. Behr DS, Peitsch WK, Hametner C, Lasitschka F, Houben R, Schonhaar K, et al. Prognostic value of immune cell infiltration, tertiary lymphoid structures and PD-L1 expression in merkel cell carcinomas. Int J Clin Exp Pathol (2014) 7(11):7610–21.
306. Di Caro G, Bergomas F, Grizzi F, Doni A, Bianchi P, Malesci A, et al. Occurrence of tertiary lymphoid tissue is associated with T-cell infiltration and predicts better prognosis in early-stage colorectal cancers. Clin Cancer Res (2014) 20(8):2147–58. doi: 10.1158/1078-0432.CCR-13-2590
307. Vanhersecke L, Brunet M, Guegan JP, Rey C, Bougouin A, Cousin S, et al. Mature tertiary lymphoid structures predict immune checkpoint inhibitor efficacy in solid tumors independently of PD-L1 expression. Nat Cancer (2021) 2(8):794–802. doi: 10.1038/s43018-021-00232-6
309. Wang X, Haswell JR, Roberts CW. Molecular pathways: SWI/SNF (BAF) complexes are frequently mutated in cancer–mechanisms and potential therapeutic insights. Clin Cancer Res (2014) 20(1):21–7. doi: 10.1158/1078-0432.CCR-13-0280
310. Gao J, Ward JF, Pettaway CA, Shi LZ, Subudhi SK, Vence LM, et al. Sharma: VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat Med (2017) 23(5):551–5. doi: 10.1038/nm.4308
311. Liu D, Jenkins RW, Sullivan RJ. Mechanisms of resistance to immune checkpoint blockade. Am J Clin Dermatol (2019) 20(1):41–54. doi: 10.1007/s40257-018-0389-y
312. Sullivan RJ, Hong DS, Tolcher AW, Patnaik A, Shapiro G, Chmielowski B, et al. Initial results from first-in-human study of IPI-549, a tumor macrophage-targeting agent, combined with nivolumab in advanced solid tumors. J Clin Oncol (2018) 36, no. 15_suppl:3013–3. doi: 10.1200/JCO.2018.36.15_SUPPL.3013
313. Teng MW, Ngiow SF, Ribas A, Smyth MJ. Classifying cancers based on T-cell infiltration and PD-L1. Cancer Res (2015) 75(11):2139–45. doi: 10.1158/0008-5472.CAN-15-0255
314. Dong ZY, Wu SP, Liao RQ, Huang SM, Wu YL. Potential biomarker for checkpoint blockade immunotherapy and treatment strategy. Tumour Biol (2016) 37(4):4251–61. doi: 10.1007/s13277-016-4812-9
315. Wang Y, Hou K, Jin Y, Bao B, Tang S, Qi J, et al. Lung adenocarcinoma-specific three-integrin signature contributes to poor outcomes by metastasis and immune escape pathways. J Transl Int Med (2021) 9(4):249–63. doi: 10.2478/jtim-2021-0046
316. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (2015) 348(6230):124–8. doi: 10.1126/science.aaa1348
Keywords: immune checkpoints, programmed cell death protein 1 (PD-1), programmed cell death 1 ligand 1 (PD-L1), cytotoxic T lymphocyte-associated antigen-4 (CTLA4), oncotherapy, T cell immunoglobulin and mucin-3 (Tim-3), lymphocyte activation gene 3 (Lag-3)
Citation: Yu L, Sun M, Zhang Q, Zhou Q and Wang Y (2022) Harnessing the immune system by targeting immune checkpoints: Providing new hope for Oncotherapy. Front. Immunol. 13:982026. doi: 10.3389/fimmu.2022.982026
Received: 30 June 2022; Accepted: 15 August 2022;
Published: 08 September 2022.
Edited by:
Zong Sheng Guo, University at Buffalo, United StatesReviewed by:
Pushpamali De Silva, Dana–Farber Cancer Institute, United StatesCopyright © 2022 Yu, Sun, Zhang, Zhou and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiao Zhou, cWlhb3pob3VxekBvdXRsb29rLmNvbQ==; Yi Wang, d195aTIwMjJAMTYzLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.