 Yoko Yoshida
Yoko Yoshida Hiroshi Nishi
Hiroshi Nishi- Division of Nephrology and Endocrinology, The University of Tokyo Graduate School of Medicine, Tokyo, Japan
The complement system is part of the innate immune system. The crucial step in activating the complement system is the generation and regulation of C3 convertase complexes, which are needed to generate opsonins that promote phagocytosis, to generate C3a that regulates inflammation, and to initiate the lytic terminal pathway through the generation and activity of C5 convertases. A growing body of evidence has highlighted the interplay between the complement system, coagulation system, platelets, neutrophils, and endothelial cells. The kidneys are highly susceptible to complement-mediated injury in several genetic, infectious, and autoimmune diseases. Atypical hemolytic uremic syndrome (aHUS) and lupus nephritis (LN) are both characterized by thrombosis in the glomerular capillaries of the kidneys. In aHUS, congenital or acquired defects in complement regulators may trigger platelet aggregation and activation, resulting in the formation of platelet-rich thrombi in the kidneys. Because glomerular vasculopathy is usually noted with immunoglobulin and complement accumulation in LN, complement-mediated activation of tissue factors could partly explain the autoimmune mechanism of thrombosis. Thus, kidney glomerular capillary thrombosis is mediated by complement dysregulation and may also be associated with complement overactivation. Further investigation is required to clarify the interaction between these vascular components and develop specific therapeutic approaches.
Introduction
Complement system
The complement system is essential for the innate immune system to eliminate invading pathogens. The crucial step in activating the complement system is the generation and regulation of C3 convertase. All complement mechanisms in immune defense, namely opsonization, phagocytosis, inflammation, and target cell lysis, rely on the enzymatic step that generates C3 convertase. The complement system is activated by three different pathways: the classical (CP), lectin (LP), and alternative (AP) pathways (Figure 1). Although each of these routes has a different activation mechanism, they all produce C3 convertase, which cleaves C3 into C3a and C3b to activate the terminal complement pathway and generate C5b-9 (1).
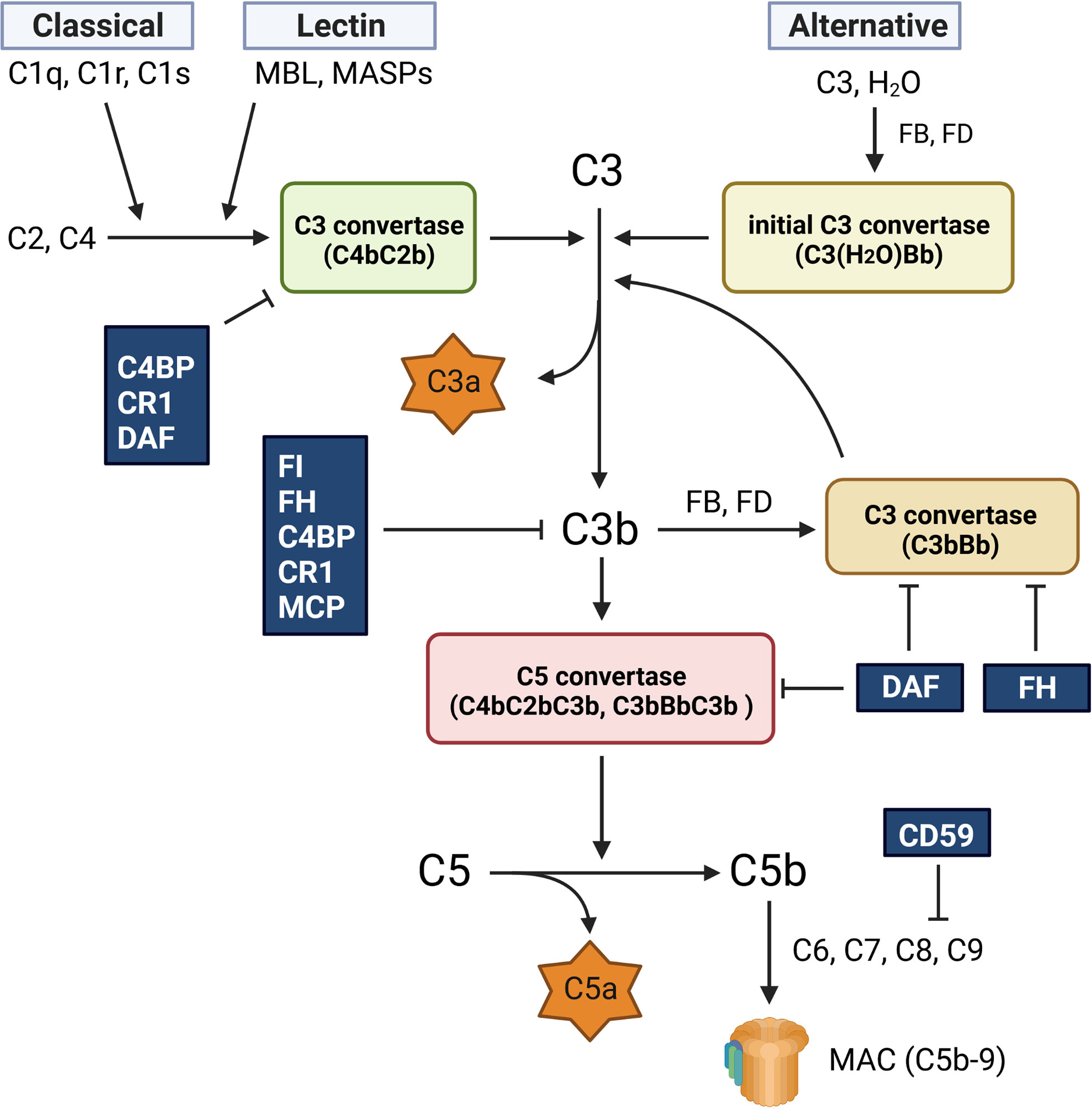
Figure 1 Complement activation pathway. The complement system is activated via three different pathways: classical (CP), lectin (LP), and alternative (AP). The CP is activated by the binding of C1q to antigen-bound antibodies. This reaction activates C1s and C1r, leading to the formation of the CP C3 convertase (C4bC2b). The LP generates the same C3 convertase as the CP, but its activation is caused by mannose-binding lectin (MBL) and MBL-associated serine proteases (MASPs). The AP is spontaneously activated via hydrolysis of C3 (C3 (H2O)), which generates the initial C3 convertase (C3(H2O)Bb). All three activation routes merge at the cleavage of C3 and lead to the formation of the C5 convertases (C4bC2bC3b and C3bBbC3b), which cleave C5 into C5a and C5b. The C5b fragment forms the membrane attack complex (C5b-9, MAC) by binding to C6, C7, C8, and C9. C9 polymerization is required for C5b-9 generation. C5b-9 creates pores in the membrane and lyses the target cells. The fragment of C3b is opsonin, and C3a and C5a are also known as anaphylatoxins and chemotactic factors, respectively.
The CP is activated by C1q recognition of antigen-bound antibodies, which eventually induces C1s to produce the CP C3 convertase (C4bC2b) through the cleavage of C2 and C4. In the LP, the binding of mannose-binding lectin (MBL) to microbial carbohydrates triggers the generation of C3 convertase (C4bC2b) via the MBL-associated serine proteases 1 and 2 (MASP-1 and MASP-2). In contrast, the AP is constitutively activated at low levels by the hydrolysis of C3 (C3(H2O)). C3(H2O) rapidly reacts with complement factors B (FB) and D, resulting in the formation of an initial fluid-phase C3 convertase (C3(H2O)Bb). This initial convertase can cleave C3 and generate C3b, which is generally inactivated by various complement regulatory proteins. However, in the presence of pathogens, C3b binds to the target surface and induces the formation of C3 convertase (C3bBb). All C3 convertases (CP/LP C4bC2b and AP C3bBb) can attach to the C3b fragment and form C5 convertases (CP/LP C4bC2bC3b and AP C3bBbC3b), which cleave C5 into C5a and C5b. The C5b molecule sequentially binds to C6, C7, and C8 to form C5b-8. Finally, C5b-8 binds to C9, which polymerizes and forms a transmembrane ring, leading to the formation of C5b-9, which is also known as the membrane attack complex (MAC).
Although cell lysis by C5b-9 is effective against gram-negative bacteria, the complement system also acts against gram-positive bacteria by promoting opsonization and neutrophil phagocytosis. The C3b and C4b fragments are opsonins, and opsonized pathogens are recognized by complement receptor type 1 (CR1) on neutrophils, which then phagocytose them. In addition to opsonization, complement C3b enhances antibody generation by B cells, and another important role of complement is the generation of two anaphylatoxins, C3a and C5a. These peptides support inflammation and activate cells expressing anaphylatoxin receptors (1).
Negative regulators of the complement system and their expression in the kidneys
Various complement-regulatory proteins tightly control complement activation to protect autologous tissues from complement attack (2–4). The plasma protein C1 inhibitor (C1-INH) prevents the initiation of the CP and LP by binding to and inactivating C1r, C1s, and MASPs (5). During C3 convertase formation, various complement regulators function as cofactors for factor I (FI), a serine protease that inactivates C3b and C4b. Some regulators also exhibit decay acceleration activity, which decreases the stability of C3 convertases by accelerating the dissociation of Bb from C3bBb and/or C2b from C4bC2b (1, 2, 4).
Factor H (FH) and C4b-binding protein (C4BP) are fluid-phase proteins associated with FI-mediated C3b or C4b cleavage, as well as the decay acceleration activity of the AP or CP C3 convertase (1, 2, 4). Cell membrane inhibitors, such as CR1 and membrane cofactor protein (MCP), also function as cofactors for the inactivation of C3b and C4b via FI. CR1 also shows decay acceleration activity with respect to the CP C3 convertase, but MCP does not. Decay accelerating factor (DAF or CD55) and CD59 are glycosylphosphatidylinositol (GPI)-anchored membrane inhibitors of the complement system. As its name implies, DAF accelerates the decay of C3/C5 convertases, and CD59 inhibits C5b-9 formation by binding to C8 and C9. CR1-related gene/-protein y (Crry) is a rodent-specific membrane regulator with both cofactor and decay-accelerating activities, and is similar to human MCP and DAF (6). Animal experiments using Crry-neutralizing antibodies have revealed that Crry is a critical complement regulator in rodent kidneys (2).
In the context of renal physiology, MCP, CR1, DAF, and CD59 are expressed in the glomeruli and protect them from complement attacks. The expression levels of these regulatory proteins can be altered by complement attack and in various glomerular disorders (2). MCP, DAF, and CD59 are ubiquitously expressed in all resident glomerular cells, although DAF is barely detectable in the glomerular cells of normal kidneys (2). The expression of MCP and DAF, but not that of CD59, is concentrated in the juxtaglomerular apparatus (3). In contrast, the expression of CR1 is mainly restricted to podocytes (2).
Although FH is a fluid phase complement regulatory protein, it is also present on the cell surface. The C-terminal region of FH can bind to glycosaminoglycans and sialic acid on the cell surface. After binding to the cell surface, it can trap deposited C3b to induce FI-mediated C3b inactivation (4). Clinically, FH dysfunction is highly associated with renal impairment caused by atypical hemolytic uremic syndrome (aHUS) and C3 glomerulopathy, suggesting that FH-mediated complement regulation plays an essential role in kidney health.
Roles of the complement system and inflammation in thrombosis
The complement system, coagulation-fibrinolytic system, platelets, and leukocytes all form a close network and interact with each other (7). Therefore, dysregulation of any component can lead to multiple diseases with different pathological conditions and clinical manifestations (8) (Figure 2).
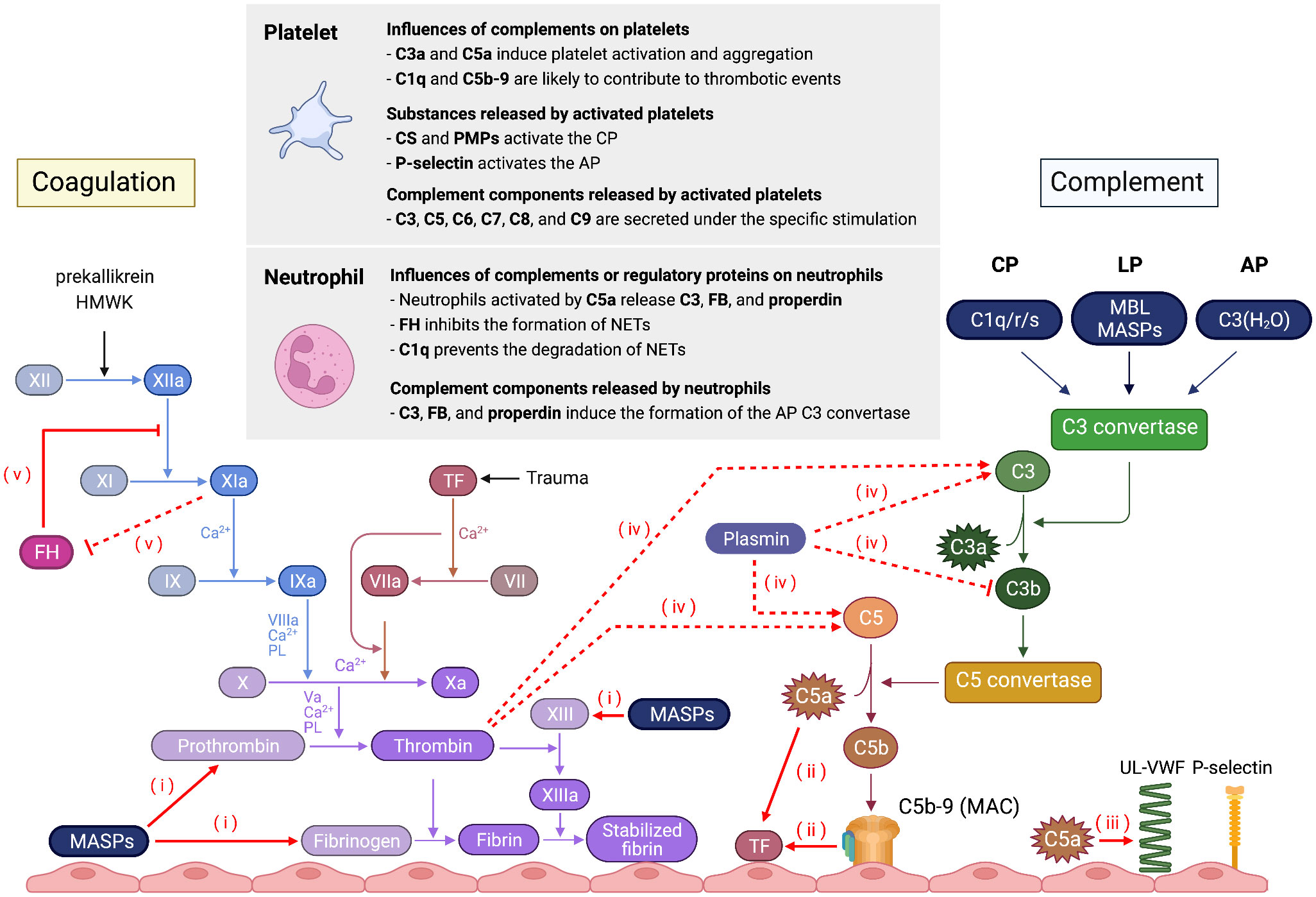
Figure 2 Schematic overview of the interaction between the complement system and the coagulation system, platelets, and neutrophils. (i) MASPs can cleave coagulation factors, such as prothrombin, fibrinogen, and factor XIII. (ii) C5a and C5b-9 enhance blood thrombogenicity through upregulation of TF in endothelial cells and neutrophils. (iii) C5a induces UL-VWF secretion and P-selectin expression. Conversely, coagulation factors activate the complement cascade. (iv) Thrombin and plasmin can generate C3a and C5a, whereas plasminogen enhances FI-mediated C3b cleavage in the presence of FH and plasmin degrades C3b. (v) Factor XIa decreases the cofactor and decay acceleration activity of FH and the ability binding of FH to bind to human endothelial cells. Conversely, FH inhibits FXIa activation.
Complement and coagulation systems
Both the complement and coagulation systems share common cascade pathways that involve proteolysis and enable an inflammatory response as a way of defending the host (9–11).
The complement system modulates the coagulation cascade in several ways. Both C5- and C3-deficient mice have longer tail bleeding time and reduced susceptibility to thrombosis, indicating that the complement system plays an essential role in the coagulation process (12). MASP-1 and MASP-2 cleave coagulation factors such as prothrombin, fibrinogen, factor XIII, and thrombin-activatable fibrinolysis inhibitors in vitro (13, 14). C5a and C5b-9 induce tissue factor (TF) expression, which initiates the extrinsic coagulation pathway in both endothelial cells and neutrophils (9, 15). C5a also induces the secretion of ultra large von Willebrand factor multimers and P-selectin and increases neutrophil adhesion to cultured endothelial cells (16). These suggest that C5a is an important inflammatory mediator between neutrophils and endothelial cells during the acute inflammatory response.
Coagulation factors can also activate the complement cascade at different levels. Thrombin cleaves C3 and C5 into C3a and C5a in vitro, which amplifies the activation of the complement system and the induction of chemotaxis and neutrophil activation (17, 18). Plasmin also cleaves C3 and C5 (17). Plasminogen, on the other hand, enhances FI-mediated cleavage of C3b in the presence of FH, and plasmin degrades C3b (19). Factor XIa (FXIa) and FH modulate each other. FXIa cleaves FH, which decreases the cofactor and decay acceleration activity of FH and the ability of FH to bind to human endothelial cells. Conversely, FH inhibits FXIa activation via either thrombin or factor XIIa (20).
Complement system and platelets
Various complement components specifically activate platelets (10, 11). Thrombin-mediated platelet aggregation and release are enhanced by several complement components, such as C3, C5, C6, C7, C8, and C9 (21). Most of these proteins are stored in platelets and are secreted following activation (22–24). The binding of C1q to the C1q receptor (gC1qR/p33) expressed on platelets activates glycoprotein (GP) IIb-IIIa fibrinogen binding sites and P-selectin expression, which contributes to the thrombotic events associated with complement activation (25). The binding of C5b-9 induces a change in the membrane potential of platelets, thereby exposing the binding sites to factor Va and serving as a basis for the proteolytic generation of thrombin (26). In addition, anaphylatoxins C3a and C5a induce platelet activation and aggregation (27).
Platelets also release various molecules that activate or modulate the complement system (10, 11). Chondroitin sulfate released by thrombin-activated platelets induces fluid-phase activation of the CP in a C1q-dependent manner (28). Platelet microparticles also support the activation of the CP, whereas they induce the expression of C1-INH from the α-granules of platelets (29). Intriguingly, P-selectin expressed in activated platelets acts as a receptor for C3b, thereby initiating the activation of the AP (30). Experimental animal data have shown that C3, but not C5, is not redundant in platelet activation (12).
Furthermore, platelets have a complement regulatory system that limits complement activation on their surfaces. Platelets express MCP, CD55, and CD59 and carry C1-INH and FH (31, 32) in their α granules. In addition, activated platelets and neutrophils can remove C5b-9 in the form of microparticles (33). The absence or impairment of such regulatory proteins is associated with platelet dysfunction, alterations in platelet activation, or thrombocytopenia (8).
Complement system, leukocytes, and the endothelium
Neutrophils accumulate at inflamed sites with the help of the chemoattractant C5a and phagocytose bacteria with surface deposits of C3b or iC3b via CR1 and CR3 (34, 35). Neutrophil extracellular traps (NETs) are also considered part of the human innate immune system. These form when neutrophils respond to bacteria or immune complexes by ejecting nuclear chromatin and digestive enzymes to kill pathogens (36) and can sometimes evoke autoimmune tissue injury (37, 38).
Properdin is a positive complement regulator that stabilizes C3 convertase in the AP (39). Intriguingly, activated neutrophils (including those activated by C5a) release C3, FB, and properdin (40), and induce the formation of AP C3 convertase, leading to C5a generation. C5a activates additional neutrophils, which secrete key components of the AP. C3- and C3a receptor (C3aR)-deficient knockout mice fail to form NETs (41, 42), suggesting that the complement system also affects the formation of NETs. Furthermore, C3b opsonization promoted the release of NETs (43). In addition, neutrophils stimulated with phorbol myristate acetate (PMA) secrete properdin and deposit it on NETs and certain bacteria to induce the formation of C5b–9 (40).
FH, a negative regulator of the AP, binds to neutrophils, inhibits the formation of PMA-stimulated NETs (44), and reduces the inflammatory response (45). Conversely, binding of C1q prevents the degradation of NETs by directly inhibiting DNAse-I by C1q (46). NETs also exert thrombogenic activity through their expression of functionally active TF (47), which is disrupted by complement C3 inhibition (48).
Role of the complement system in glomerular capillary thrombosis in kidney disorders
Thrombotic microangiopathy
Thrombotic microangiopathy (TMA) is a pathological condition caused by the formation of microvascular thrombi that leads to thrombocytopenia, microangiopathic hemolytic anemia, and end-organ damage (49). TMA is caused by various hereditary or acquired factors, and is classified into four main categories: thrombotic thrombocytopenic purpura (TTP), hemolytic uremic syndrome (HUS) caused by Shiga toxin-producing Escherichia coli (STEC), atypical HUS (aHUS), and secondary TMA.
TTP is caused by a severe deficiency of ADAMTS13 (a disintegrin-like metalloprotease with thrombospondin type 1 motif, member 13) resulting from genetic or acquired defects (50). STEC-induced HUS is predominantly found in children and is diagnosed based on the direct detection of Shiga toxins in feces and the presence of anti-lipopolysaccharide immunoglobulin M antibodies (51).
aHUS is a complement-mediated TMA caused by the overactivation of the AP as a result of inherited and/or acquired complement abnormalities (52, 53). aHUS mainly targets the kidneys, and 50-70% of patients develop end-stage kidney disease (ESKD) unless they receive early and appropriate treatment (52). The efficacy and safety of the complement-inhibiting drug, eculizumab (54, 55) and ravulizumab (56, 57), have been described in many reports. Genetic variants of several complement regulators (FH, FI, and MCP) and activators (C3 and FB) have been identified in up to 50% of patients with aHUS (58). Thus, complementary diagnostic approaches have been developed to address the limitations of comprehensive gene analysis (59, 60). In addition, the acquisition of inhibitory autoantibodies against FH can cause aHUS (52, 61). In both genetic and acquired defects, impaired complement control on self-cell surfaces via the formation of C5b-9 leads to endothelial tissue damage and the generation of thrombi in the microvasculature (62).
Thrombocytopenia is a typical feature of TMA in which platelet thrombi are found in the capillaries and arterioles (62, 63). As mentioned above, various complement regulators normally protect platelets from complement attack; thus, defects in these regulators in aHUS may cause platelet activation and the formation of platelet-rich microvascular thrombi. Although there are few reports regarding the coagulation profile of aHUS (64), genetic variants of some coagulation-related proteins are associated with the pathogenesis of aHUS (65–67). Because the activations of complement and coagulation systems synergistically amplify each other as discussed earlier, this vicious cycle may be associated with the formation of microvascular thrombi in aHUS.
Patients with FH variants have a significantly increased risk of ESKD than those with variants in CD46 (MCP) (68). FH variants associated with aHUS are primarily located at the C-terminus, and inhibitory autoantibodies also target the C-terminal region of FH (68–70). These genetic and acquired defects inhibit FH binding to the cell surface, resulting in C5b-9 formation in endothelial cells and platelets (62, 71). Although it is still unclear why complement damage seems to be restricted to the kidneys, heparan sulfate expressed in the kidneys appears critical for FH binding on cell surfaces (72).
In addition to TTP, STEC-induced HUS, and aHUS, a variety of pathological conditions such as autoimmune disease (73, 74), drug use (75), infection (76, 77), malignancy (78), malignant hypertension (79–81), pregnancy (82, 83), and transplantation (84, 85) can trigger secondary TMA. Although TTP and STEC-HUS have well-established diagnostic tests, the differentiation between aHUS and secondary TMA remains controversial because pathogenic variants in complement-related genes are only identified in about half of the patients with aHUS. In addition, complement abnormalities have been found in some secondary TMA (86).
Complement abnormalities may play a role in secondary TMA caused by malignant hypertension (79–81) and pregnancy (82). Rare genetic variants in complement-related genes have been identified in approximately 30-70% of patients with malignant hypertension-associated TMA (79–81). In these cases, ex vivo analysis showed that patient sera induced massive C5b-9 formation in microvasculature endothelial cells, suggesting that these variants induced excessive complement activation. In addition, patients with over-deposition of C5b-9 in vascular endothelial cells had a higher incidence of ESKD, and the administration of anti-C5 antibodies improved renal function in these cases.
Lupus nephritis
Systemic lupus erythematosus (SLE) is a autoimmune disease that primarily affects young women. SLE affects the kidneys in approximately 50% of such patients as lupus nephritis (LN) (87). A variety of abnormal immune responses, such as defects in the clearance of immune complexes and apoptotic cells, nucleic acid-sensing abilities, lymphocyte signaling, and interferon-production have a central role in the pathogenesis of this disease (88, 89). Because immune complexes are deposited in tissues, and the deposition of C1q in tissues is relatively characteristic of SLE, it is plausible that these immune complexes activate the complement system and decrease complement levels (C3, C4, and CH50) in the peripheral blood. Activation of the complement system may cause inflammatory injury to tissues, as C3 or C4 fragments bind to complement receptors on B lymphocytes to enhance antibody generation (90, 91). However, this cannot explain why the deficiency of complement components such as C1q, C1s, C1r, C2, and C4 causes lupus-like symptoms (92). Deficiencies in this component activity may disturb the efficient disposal of dying and dead cells (93) or the normal tolerance mechanisms of lymphocytes (94), with both leading to the generation of autoantibodies.
The International Society of Nephrology/Renal Pathology Society meeting report mentioned the importance to have a standardized approach and terminology to distinguish ordinary arterial or arteriolar sclerosis from lupus-related lesions such as vasculopathy associated with immune complex deposition, vasculitis, and TMA (95). Indeed, within the glomerular capillaries and small arterioles in LN kidney specimens, microvascular thrombosis is conventionally recognized as one of the most common histopathological findings (96, 97). This characteristic finding has been variously described as “lupus vasculopathy (LV)” (97, 98), “immunoglobulin microvascular cast” (96), and “glomerular thrombosis” (99). LV is documented predominantly in diffuse proliferative LN, but the underlying etiology and its prognostic value remain undetermined (96, 97, 100). LV is characterized by the accumulation of immunoglobulins and complements in the vascular wall (98). This results in luminal narrowing and suggests the presence of immune-mediated vascular injury (98). Notably, unlike lupus vasculitis, LV does not involve the deposition of inflammatory cells in the vascular wall (98).
In lupus-prone MRL/lpr mice, treatment with aspirin and dexamethasone partially attenuates glomerular thrombosis (101). In rodents with nephrotoxic nephritis that mimics the downstream effector phase of LN (102), Fcγ receptors (103, 104) and components of the complement system (105, 106) have been implicated in the pathogenesis of tissue injury, including glomerular thrombosis. These experimental findings in animals suggest that inflammation may promote kidney glomerular thrombosis in LN.
Antiphospholipid syndrome (APS) frequently occurs in SLE and is characterized by vascular thrombosis, repeated miscarriages, and the presence of antiphospholipid antibodies (APLA) (107). Although patients with APS show a prolonged activated partial thromboplastin time, APLA exert prothrombotic effects because they inhibit β2-glycosylphosphatidylinisotol, which is an inhibitory regulator of phospholipid-dependent coagulation, protein C activation, thrombomodulin, and heparan sulfate on vascular endothelial cells (108, 109). Intriguingly, C4 deficiency attenuates fetal loss in mice with APS (110). Moreover, APLA stimulate TF activation in myelomonocytic cells, and mice deficient in C3, unlike mice deficient in C5, are protected from in vivo thrombus formation induced by cofactor-independent APLA, suggesting that C3 is required for TF activation and APLA-induced thrombosis (111). Because complement mediates TF enrichment in NETs (47, 48), neutrophils may be another player in thrombin formation in this context.
Primary glomerulonephritis
Glomerulonephritis refers to a group of kidney diseases affecting the glomeruli due to the damage mediated by immunological dysregulation. Hypocomplementemia is a significant feature of kidney glomerular diseases such as post-infectious glomerulonephritis, immune complex-mediated membranoproliferative glomerulonephritis, C3 glomerulopathy, dense deposit diseases, and IgG4-related kidney disease (112, 113). In addition, the majority of patients with IgA nephropathy (114) or membranous nephropathy (115) have local C3 deposits in the glomeruli. Other complement elements, such as the FH-related proteins 1 and 5 and lectin pathway products, also accumulate (114–116). These findings suggest that systemic or local activation of the complement system may cause kidney tissue damage.
However, thrombosis is rarely observed in these patients unless they develop heavy proteinuria (117), which evokes a hypercoagulable state due to the urinary loss of coagulation regulatory proteins, including antithrombin and protein S, which counterbalances the increase in the synthesis rate of hemostatic proteins in the liver (118). Further studies are needed to determine the association of the development of thrombosis with complement-mediated kidney injury.
Conclusion
The complement system has long been recognized as a central mediator of innate immune defenses that eliminate invading pathogens. Accumulating evidence has revealed how the complement system can modulate the function of the coagulation system, platelets, and neutrophils, and contribute to thrombosis. Further research on the link between the complement system and kidney disorders may deepen our understanding of complement-dependent mechanisms that promote glomerular capillary thrombosis and severely impair glomerular filtration. Such insights may provide novel therapeutic options for patients and clinicians.
Author contributions
YY drafted the original manuscript and HN revised it. Both authors approved the submitted version of the manuscript.
Funding
This work was supported by the Japan Society for the Promotion of Science Grant-in-Aid for Scientific Research 21K08272 (to YY) and the SENSHIN Medical Research Foundation (to HN).
Acknowledgments
The figures were created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Merle NS, Church SE, Fremeaux-Bacchi V. Roumenina, LT: Complement system part I - molecular mechanisms of activation and regulation. Front Immunol (2015) 6:262. doi: 10.3389/fimmu.2015.00262
2. Nangaku M. Complement regulatory proteins in glomerular diseases. Kidney Int (1998) 54:1419–28. doi: 10.1046/j.1523-1755.1998.00130.x
3. Thurman JM, Renner B. Dynamic control of the complement system by modulated expression of regulatory proteins. Lab Invest (2011) 91:4–11. doi: 10.1038/labinvest.2010.173
4. Lesher AM, Song WC. Review: Complement and its regulatory proteins in kidney diseases. Nephrol (Carlton) (2010) 15:663–75. doi: 10.1111/j.1440-1797.2010.01373.x
5. Mollnes TE, Kirschfink M. Strategies of therapeutic complement inhibition. Mol Immunol (2006) 43:107–21. doi: 10.1016/j.molimm.2005.06.014
6. Naik A, Sharma S, Quigg RJ. Complement regulation in renal disease models. Semin Nephrol (2013) 33:575–85. doi: 10.1016/j.semnephrol.2013.08.008
7. Borkowska S, Suszynska M, Mierzejewska K, Ismail A, Budkowska M, Salata D, et al. : Novel evidence that crosstalk between the complement, coagulation and fibrinolysis proteolytic cascades is involved in mobilization of hematopoietic stem/progenitor cells (HSPCs). Leukemia (2014) 28:2148–54. doi: 10.1038/leu.2014.115
8. Verschoor A, Langer HF. Crosstalk between platelets and the complement system in immune protection and disease. Thromb Haemost (2013) 110:910–9. doi: 10.1160/TH13-02-0102
9. Markiewski MM, Nilsson B, Ekdahl KN, Mollnes TE, Lambris JD. Complement and coagulation: Strangers or partners in crime? Trends Immunol (2007) 28:184–92. doi: 10.1016/j.it.2007.02.006
10. Nording H, Langer HF. Complement links platelets to innate immunity. Semin Immunol (2018) 37:43–52. doi: 10.1016/j.smim.2018.01.003
11. Rawish E, Sauter M, Sauter R, Nording H, Langer HF. Complement, inflammation and thrombosis. Br J Pharmacol (2021) 178:2892–904. doi: 10.1111/bph.15476
12. Subramaniam S, Jurk K, Hobohm L, Jackel S, Saffarzadeh M, Schwierczek K, et al. Distinct contributions of complement factors to platelet activation and fibrin formation in venous thrombus development. Blood (2017) 129:2291–302. doi: 10.1182/blood-2016-11-749879
13. Hess K, Ajjan R, Phoenix F, Dobo J, Gal P, Schroeder V. Effects of MASP-1 of the complement system on activation of coagulation factors and plasma clot formation. PloS One (2012) 7:e35690. doi: 10.1371/journal.pone.0035690
14. Krarup A, Wallis R, Presanis JS, Gal P, Sim RB. Simultaneous activation of complement and coagulation by MBL-associated serine protease 2. PloS One (2007) 2:e623. doi: 10.1371/journal.pone.0000623
15. Ikeda K, Nagasawa K, Horiuchi T, Tsuru T, Nishizaka H, Niho Y. C5a induces tissue factor activity on endothelial cells. Thromb Haemost (1997) 77:394–8. doi: 10.1055/s-0038-1655974
16. Foreman KE, Vaporciyan AA, Bonish BK, Jones ML, Johnson KJ, Glovsky MM, et al. C5a-induced expression of p-selectin in endothelial cells. J Clin Invest (1994) 94:1147–55. doi: 10.1172/JCI117430
17. Amara U, Flierl MA, Rittirsch D, Klos A, Chen H, Acker B, et al. Molecular intercommunication between the complement and coagulation systems. J Immunol (2010) 185:5628–36. doi: 10.4049/jimmunol.0903678
18. Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, et al. Generation of C5a in the absence of C3: A new complement activation pathway. Nat Med (2006) 12:682–7. doi: 10.1038/nm1419
19. Barthel D, Schindler S, Zipfel PF. Plasminogen is a complement inhibitor. J Biol Chem (2012) 287:18831–42. doi: 10.1074/jbc.M111.323287
20. Puy C, Pang J, Reitsma SE, Lorentz CU, Tucker EI, Gailani D, et al. Cross-talk between the complement pathway and the contact activation system of coagulation: Activated factor XI neutralizes complement factor h. J Immunol (2021) 206:1784–92. doi: 10.4049/jimmunol.2000398
21. Polley MJ, Nachman RL. Human complement in thrombin-mediated platelet function: uptake of the C5b-9 complex. J Exp Med (1979) 150:633–45. doi: 10.1084/jem.150.3.633
22. Tedesco F, Densen P, Villa MA, Presani G, Roncelli L, Rosso di san Secondo VE. Functional C8 associated with human platelets. Clin Exp Immunol (1986) 66:472–80.
23. Jenkinson ML, Bliss MR, Brain AT, Scott DL. Rheumatoid arthritis and senile dementia of the alzheimer’s type. Br J Rheumatol (1989) 28:86–8. doi: 10.1093/rheumatology/28.1.86-b
24. Martel C, Cointe S, Maurice P, Matar S, Ghitescu M, Theroux P, et al. Requirements for membrane attack complex formation and anaphylatoxins binding to collagen-activated platelets. PloS One (2011) 6:e18812. doi: 10.1371/journal.pone.0018812
25. Peerschke EI, Reid KB, Ghebrehiwet B. Platelet activation by C1q results in the induction of alpha IIb/beta 3 integrins (GPIIb-IIIa) and the expression of p-selectin and procoagulant activity. J Exp Med (1993) 178:579–87. doi: 10.1084/jem.178.2.579
26. Wiedmer T, Sims PJ. Effect of complement proteins C5b-9 on blood platelets. evidence for reversible depolarization of membrane potential. J Biol Chem (1985) 260:8014–9.
27. Polley MJ, Nachman RL. Human platelet activation by C3a and C3a des-arg. J Exp Med (1983) 158:603–15. doi: 10.1084/jem.158.2.603
28. Hamad OA, Ekdahl KN, Nilsson PH, Andersson J, Magotti P, Lambris JD, et al. Complement activation triggered by chondroitin sulfate released by thrombin receptor-activated platelets. J Thromb Haemost (2008) 6:1413–21. doi: 10.1111/j.1538-7836.2008.03034.x
29. Yin W, Ghebrehiwet B, Peerschke EI. Expression of complement components and inhibitors on platelet microparticles. Platelets (2008) 19:225–33. doi: 10.1080/09537100701777311
30. Del Conde I, Cruz MA, Zhang H, Lopez JA, Afshar-Kharghan V. Platelet activation leads to activation and propagation of the complement system. J Exp Med (2005) 201:871–9. doi: 10.1084/jem.20041497
31. Schmaier AH, Smith PM, Colman RW. Platelet C1- inhibitor. A secreted alpha-granule protein. J Clin Invest (1985) 75:242–50. doi: 10.1172/JCI111680
32. Devine DV, Rosse WF. Regulation of the activity of platelet-bound C3 convertase of the alternative pathway of complement by platelet factor h. Proc Natl Acad Sci USA (1987) 84:5873–7. doi: 10.1073/pnas.84.16.5873
33. Morgan BP. The membrane attack complex as an inflammatory trigger. Immunobiology (2016) 221:747–51. doi: 10.1016/j.imbio.2015.04.006
34. Ehrengruber MU, Geiser T, Deranleau DA. Activation of human neutrophils by C3a and C5A. comparison of the effects on shape changes, chemotaxis, secretion, and respiratory burst. FEBS Lett (1994) 346:181–4. doi: 10.1016/0014-5793(94)00463-3
35. van Kessel KP, Bestebroer J, van Strijp JA. Neutrophil-mediated phagocytosis of staphylococcus aureus. Front Immunol (2014) 5:467. doi: 10.3389/fimmu.2014.00467
36. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303:1532–5. doi: 10.1126/science.1092385
37. Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol (2011) 187:538–52. doi: 10.4049/jimmunol.1100450
38. Chen K, Nishi H, Travers R, Tsuboi N, Martinod K, Wagner DD, et al. Endocytosis of soluble immune complexes leads to their clearance by FcgammaRIIIB but induces neutrophil extracellular traps via FcgammaRIIA in vivo. Blood (2012) 120:4421–31. doi: 10.1182/blood-2011-12-401133
39. Kimura Y, Miwa T, Zhou L, Song WC. Activator-specific requirement of properdin in the initiation and amplification of the alternative pathway complement. Blood (2008) 111:732–40. doi: 10.1182/blood-2007-05-089821
40. Yuen J, Pluthero FG, Douda DN, Riedl M, Cherry A, Ulanova M, et al. NETosing neutrophils activate complement both on their own NETs and bacteria via alternative and non-alternative pathways. Front Immunol (2016) 7:137. doi: 10.3389/fimmu.2016.00137
41. Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med (2012) 18:1386–93. doi: 10.1038/nm.2847
42. Guglietta S, Chiavelli A, Zagato E, Krieg C, Gandini S, Ravenda PS, et al. Coagulation induced by C3aR-dependent NETosis drives protumorigenic neutrophils during small intestinal tumorigenesis. Nat Commun (2016) 7:11037. doi: 10.1038/ncomms11037
43. Palmer LJ, Damgaard C, Holmstrup P, Nielsen CH. Influence of complement on neutrophil extracellular trap release induced by bacteria. J Periodontal Res (2016) 51:70–6. doi: 10.1111/jre.12284
44. Schneider AE, Sandor N, Karpati E, Jozsi M. Complement factor h modulates the activation of human neutrophil granulocytes and the generation of neutrophil extracellular traps. Mol Immunol (2016) 72:37–48. doi: 10.1016/j.molimm.2016.02.011
45. Halder LD, Abdelfatah MA, Jo EA, Jacobsen ID, Westermann M, Beyersdorf N, et al. Factor h binds to extracellular DNA traps released from human blood monocytes in response to candida albicans. Front Immunol (2016) 7:671. doi: 10.3389/fimmu.2016.00671
46. Leffler J, Martin M, Gullstrand B, Tyden H, Lood C, Truedsson L, et al. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J Immunol (2012) 188:3522–31. doi: 10.4049/jimmunol.1102404
47. Stakos D, Skendros P, Konstantinides S, Ritis K. Traps n’ clots: NET-mediated thrombosis and related diseases. Thromb Haemost (2020) 120:373–83. doi: 10.1055/s-0039-3402731
48. Skendros P, Mitsios A, Chrysanthopoulou A, Mastellos DC, Metallidis S, Rafailidis P, et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J Clin Invest (2020) 130:6151–7. doi: 10.1172/JCI141374
49. George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med (2014) 371:654–66. doi: 10.1056/NEJMra1312353
50. Joly BS, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura. Blood (2017) 129:2836–46. doi: 10.1182/blood-2016-10-709857
51. Trachtman H, Austin C, Lewinski M, Stahl RA. Renal and neurological involvement in typical shiga toxin-associated HUS. Nat Rev Nephrol (2012) 8:658–69. doi: 10.1038/nrneph.2012.196
52. Kavanagh D, Goodship TH, Richards A. Atypical hemolytic uremic syndrome. Semin Nephrol (2013) 33:508–30. doi: 10.1016/j.semnephrol.2013.08.003
53. Yoshida Y, Kato H, Ikeda Y, Nangaku M. Pathogenesis of atypical hemolytic uremic syndrome. J Atheroscler Thromb (2019) 26:99–110. doi: 10.5551/jat.RV17026
54. Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med (2013) 368:2169–81. doi: 10.1056/NEJMoa1208981
55. Licht C, Greenbaum LA, Muus P, Babu S, Bedrosian CL, Cohen DJ, et al. Efficacy and safety of eculizumab in atypical hemolytic uremic syndrome from 2-year extensions of phase 2 studies. Kidney Int (2015) 87:1061–73. doi: 10.1038/ki.2014.423
56. Rondeau E, Scully M, Ariceta G, Barbour T, Cataland S, Heyne N, et al. The long-acting C5 inhibitor, ravulizumab, is effective and safe in adult patients with atypical hemolytic uremic syndrome naive to complement inhibitor treatment. Kidney Int (2020) 97:1287–96. doi: 10.1016/j.kint.2020.01.035
57. Ariceta G, Dixon BP, Kim SH, Kapur G, Mauch T, Ortiz S, et al. The long-acting C5 inhibitor, ravulizumab, is effective and safe in pediatric patients with atypical hemolytic uremic syndrome naive to complement inhibitor treatment. Kidney Int (2021) 100:225–37. doi: 10.1016/j.kint.2020.10.046
58. Noris M, Bresin E, Mele C, Remuzzi G. Genetic atypical hemolytic-uremic syndrome. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews. Seattle (WA): University of Washington, Seattle (2021).
59. Sanchez-Corral P, Gonzalez-Rubio C, Rodriguez de Cordoba S, Lopez-Trascasa M. Functional analysis in serum from atypical hemolytic uremic syndrome patients reveals impaired protection of host cells associated with mutations in factor h. Mol Immunol (2004) 41:81–4. doi: 10.1016/j.molimm.2004.01.003
60. Yoshida Y, Miyata T, Matsumoto M, Shirotani-Ikejima H, Uchida Y, Ohyama Y, et al. A novel quantitative hemolytic assay coupled with restriction fragment length polymorphisms analysis enabled early diagnosis of atypical hemolytic uremic syndrome and identified unique predisposing mutations in Japan. PloS One (2015) 10:e0124655. doi: 10.1371/journal.pone.0124655
61. Jozsi M, Licht C, Strobel S, Zipfel SL, Richter H, Heinen S, et al. Factor h autoantibodies in atypical hemolytic uremic syndrome correlate with CFHR1/CFHR3 deficiency. Blood (2008) 111:1512–4. doi: 10.1182/blood-2007-09-109876
63. Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med (2009) 361:1676–87. doi: 10.1056/NEJMra0902814
64. Sakurai S, Kato H, Yoshida Y, Sugawara Y, Fujisawa M, Yasumoto A, et al. Profiles of coagulation and fibrinolysis activation-associated molecular markers of atypical hemolytic uremic syndrome in the acute phase. J Atheroscler Thromb (2020) 27:353–62. doi: 10.5551/jat.49494
65. Delvaeye M, Noris M, De Vriese A, Esmon CT, Esmon NL, Ferrell G, et al. Conway, EM: Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N Engl J Med (2009) 361:345–57. doi: 10.1056/NEJMoa0810739
66. Lemaire M, Fremeaux-Bacchi V, Schaefer F, Choi M, Tang WH, Le Quintrec M, et al. Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat Genet (2013) 45:531–6. doi: 10.1038/ng.2590
67. Bu F, Maga T, Meyer NC, Wang K, Thomas CP, Nester CM, et al. Comprehensive genetic analysis of complement and coagulation genes in atypical hemolytic uremic syndrome. J Am Soc Nephrol (2014) 25:55–64. doi: 10.1681/ASN.2013050453
68. Fremeaux-Bacchi V, Fakhouri F, Garnier A, Bienaime F, Dragon-Durey MA, Ngo S, et al. Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol (2013) 8:554–62. doi: 10.2215/CJN.04760512
69. Jozsi M, Strobel S, Dahse HM, Liu WS, Hoyer PF, Oppermann M, et al. Anti factor h autoantibodies block c-terminal recognition function of factor h in hemolytic uremic syndrome. Blood (2007) 110:1516–8. doi: 10.1182/blood-2007-02-071472
70. Noris M, Caprioli J, Bresin E, Mossali C, Pianetti G, Gamba S, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol (2010) 5:1844–59. doi: 10.2215/CJN.02210310
71. Lehtinen MJ, Rops AL, Isenman DE, van der Vlag J, Jokiranta TS. Mutations of factor h impair regulation of surface-bound C3b by three mechanisms in atypical hemolytic uremic syndrome. J Biol Chem (2009) 284:15650–8. doi: 10.1074/jbc.M900814200
72. Loeven MA, Rops AL, Berden JH, Daha MR, Rabelink TJ, van der Vlag J. The role of heparan sulfate as determining pathogenic factor in complement factor h-associated diseases. Mol Immunol (2015) 63:203–8. doi: 10.1016/j.molimm.2014.08.005
73. Kwok SK, Ju JH, Cho CS, Kim HY, Park SH. Thrombotic thrombocytopenic purpura in systemic lupus erythematosus: Risk factors and clinical outcome: a single centre study. Lupus (2009) 18:16–21. doi: 10.1177/0961203308094360
74. Yamashita H, Kamei R, Kaneko H. Classifications of scleroderma renal crisis and reconsideration of its pathophysiology. Rheumatol (Oxford) (2019) 58:2099–106. doi: 10.1093/rheumatology/kez435
75. Al-Nouri ZL, Reese JA, Terrell DR, Vesely SK, George JN. Drug-induced thrombotic microangiopathy: A systematic review of published reports. Blood (2015) 125:616–8. doi: 10.1182/blood-2014-11-611335
76. Bitzan M, Zieg J. Influenza-associated thrombotic microangiopathies. Pediatr Nephrol (2018) 33:2009–25. doi: 10.1007/s00467-017-3783-4
77. Tiwari NR, Phatak S, Sharma VR, Agarwal SK. COVID-19 and thrombotic microangiopathies. Thromb Res (2021) 202:191–8. doi: 10.1016/j.thromres.2021.04.012
78. Thomas MR, Scully M. How I treat microangiopathic hemolytic anemia in patients with cancer. Blood (2021) 137:1310–7. doi: 10.1182/blood.2019003810
79. Timmermans S, Abdul-Hamid MA, Vanderlocht J, Damoiseaux J, Reutelingsperger CP, van Paassen P, et al. Patients with hypertension-associated thrombotic microangiopathy may present with complement abnormalities. Kidney Int (2017) 91:1420–5. doi: 10.1016/j.kint.2016.12.009
80. Timmermans S, Abdul-Hamid MA, Potjewijd J, Theunissen R, Damoiseaux J, Reutelingsperger CP, et al. C5b9 formation on endothelial cells reflects complement defects among patients with renal thrombotic microangiopathy and severe hypertension. J Am Soc Nephrol (2018) 29:2234–43. doi: 10.1681/ASN.2018020184
81. Timmermans S, Werion A, Damoiseaux J, Morelle J, Reutelingsperger CP, van Paassen P. Diagnostic and risk factors for complement defects in hypertensive emergency and thrombotic microangiopathy. Hypertension (2020) 75:422–30. doi: 10.1161/HYPERTENSIONAHA.119.13714
82. Fakhouri F, Roumenina L, Provot F, Sallee M, Caillard S, Couzi L, et al. Pregnancy-associated hemolytic uremic syndrome revisited in the era of complement gene mutations. J Am Soc Nephrol (2010) 21:859–67. doi: 10.1681/ASN.2009070706
83. Yoshida Y, Matsumoto M, Yagi H, Isonishi A, Sakai K, Hayakawa M, et al. Severe reduction of free-form ADAMTS13, unbound to von willebrand factor, in plasma of patients with HELLP syndrome. Blood Adv (2017) 1:1628–31. doi: 10.1182/bloodadvances.2017006767
84. Nishi H, Hanafusa N, Kondo Y, Nangaku M, Sugawara Y, Makuuchi M, et al. Clinical outcome of thrombotic microangiopathy after living-donor liver transplantation treated with plasma exchange therapy. Clin J Am Soc Nephrol (2006) 1:811–9. doi: 10.2215/CJN.01781105
85. Schoettler M, Chonat S, Williams K, Lehmann L. Emerging therapeutic and preventive approaches to transplant-associated thrombotic microangiopathy. Curr Opin Hematol (2021) 28:408–16. doi: 10.1097/MOH.0000000000000687
86. Palma LMP, Sridharan M, Sethi S. : Complement in secondary thrombotic microangiopathy. Kidney Int Rep (2021) 6:11–23. doi: 10.1016/j.ekir.2020.10.009
87. Almaani S, Meara A. Rovin, BH: Update on lupus nephritis. Clin J Am Soc Nephrol (2017) 12:825–35. doi: 10.2215/CJN.05780616
88. Tsokos GC, Lo MS, Costa Reis P. Sullivan, KE: New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol (2016) 12:716–30. doi: 10.1038/nrrheum.2016.186
89. Nishi H, Mayadas TN. Neutrophils in lupus nephritis. Curr Opin Rheumatol (2019) 31:193–200. doi: 10.1097/BOR.0000000000000577
90. Villiers MB, Villiers CL, Laharie AM, Marche PN. Amplification of the antibody response by C3b complexed to antigen through an ester link. J Immunol (1999) 162:3647–52.
91. Rossbacher J, Shlomchik MJ. The b cell receptor itself can activate complement to provide the complement receptor 1/2 ligand required to enhance b cell immune responses in vivo. J Exp Med (2003) 198:591–602. doi: 10.1084/jem.20022042
92. Macedo AC, Isaac L. Systemic lupus erythematosus and deficiencies of early components of the complement classical pathway. Front Immunol (2016) 7:55. doi: 10.3389/fimmu.2016.00055
93. Korb LC, Ahearn JM. C1q binds directly and specifically to surface blebs of apoptotic human keratinocytes: Complement deficiency and systemic lupus erythematosus revisited. J Immunol (1997) 158:4525–8.
94. Mitchell DA, Pickering MC, Warren J, Fossati-Jimack L, Cortes-Hernandez J, Cook HT, et al. C1q deficiency and autoimmunity: The effects of genetic background on disease expression. J Immunol (2002) 168:2538–43. doi: 10.4049/jimmunol.168.5.2538
95. Bajema IM, Wilhelmus S, Alpers CE, Bruijn JA, Colvin RB, Cook HT, et al. Revision of the international society of Nephrology/Renal pathology society classification for lupus nephritis: Clarification of definitions, and modified national institutes of health activity and chronicity indices. Kidney Int (2018) 93:789–96. doi: 10.1016/j.kint.2017.11.023
96. Descombes E, Droz D, Drouet L, Grunfeld JP, Lesavre P. Renal vascular lesions in lupus nephritis. Med (Baltimore) (1997) 76:355–68. doi: 10.1097/00005792-199709000-00003
97. Barber C, Herzenberg A, Aghdassi E, Su J, Lou W, Qian G, et al. Evaluation of clinical outcomes and renal vascular pathology among patients with lupus. Clin J Am Soc Nephrol (2012) 7:757–64. doi: 10.2215/CJN.02870311
98. Gonzalez-Suarez ML, Waheed AA, Andrews DM, Ascherman DP, Zeng X, Nayer A. Lupus vasculopathy: Diagnostic, pathogenetic and therapeutic considerations. Lupus (2014) 23:421–7. doi: 10.1177/0961203313520340
99. Miranda JM, Garcia-Torres R, Jara LJ, Medina F, Cervera H, Fraga A. Renal biopsy in systemic lupus erythematosus: significance of glomerular thrombosis. analysis of 108 cases. Lupus (1994) 3:25–9. doi: 10.1177/096120339400300106
100. Wu LH, Yu F, Tan Y, Qu Z, Chen MH, Wang SX, et al. : Inclusion of renal vascular lesions in the 2003 ISN/RPS system for classifying lupus nephritis improves renal outcome predictions. Kidney Int (2013) 83:715–23. doi: 10.1038/ki.2012.409
101. Gonzalo-Gil E, Garcia-Herrero C, Toldos O, Usategui A, Criado G, Perez-Yague S, et al. Microthrombotic renal vascular lesions are associated to increased renal inflammatory infiltration in murine lupus nephritis. Front Immunol (2018) 9:1948. doi: 10.3389/fimmu.2018.01948
102. Du Y, Fu Y, Mohan C. Experimental anti-GBM nephritis as an analytical tool for studying spontaneous lupus nephritis. Arch Immunol Ther Exp (Warsz) (2008) 56:31–40. doi: 10.1007/s00005-008-0007-4
103. Park SY, Ueda S, Ohno H, Hamano Y, Tanaka M, Shiratori T, et al. Resistance of fc receptor- deficient mice to fatal glomerulonephritis. J Clin Invest (1998) 102:1229–38. doi: 10.1172/JCI3256
104. Nishi H, Furuhashi K, Cullere X, Saggu G, Miller MJ, Chen Y, et al. Neutrophil FcgammaRIIA promotes IgG-mediated glomerular neutrophil capture via Abl/Src kinases. J Clin Invest (2017) 127:3810–26. doi: 10.1172/JCI94039
105. Sogabe H, Nangaku M, Ishibashi Y, Wada T, Fujita T, Sun X, et al. Increased susceptibility of decay-accelerating factor deficient mice to anti-glomerular basement membrane glomerulonephritis. J Immunol (2001) 167:2791–7. doi: 10.4049/jimmunol.167.5.2791
106. Robson MG, Cook HT, Botto M, Taylor PR, Busso N, Salvi R, et al. Accelerated nephrotoxic nephritis is exacerbated in C1q-deficient mice. J Immunol (2001) 166:6820–8. doi: 10.4049/jimmunol.166.11.6820
107. Afeltra A, Vadacca M, Conti L, Galluzzo S, Mitterhofer AP, Ferri GM, et al. Thrombosis in systemic lupus erythematosus: Congenital and acquired risk factors. Arthritis Rheum (2005) 53:452–9. doi: 10.1002/art.21172
108. Atsumi T, Khamashta MA, Amengual O, Donohoe S, Mackie I, Ichikawa K, et al. Binding of anticardiolipin antibodies to protein c via beta2-glycoprotein I (beta2-GPI): A possible mechanism in the inhibitory effect of antiphospholipid antibodies on the protein c system. Clin Exp Immunol (1998) 112:325–33. doi: 10.1046/j.1365-2249.1998.00582.x
109. Sang Y, Roest M, de Laat B, de Groot PG, Huskens D. Interplay between platelets and coagulation. Blood Rev (2021) 46:100733. doi: 10.1016/j.blre.2020.100733
110. Girardi G, Berman J, Redecha P, Spruce L, Thurman JM, Kraus D, et al. Complement C5a receptors and neutrophils mediate fetal injury in the antiphospholipid syndrome. J Clin Invest (2003) 112:1644–54. doi: 10.1172/JCI200318817
111. Muller-Calleja N, Ritter S, Hollerbach A, Falter T, Lackner KJ, Ruf W. Complement C5 but not C3 is expendable for tissue factor activation by cofactor-independent antiphospholipid antibodies. Blood Adv (2018) 2:979–86. doi: 10.1182/bloodadvances.2018017095
112. Hebert LA, Cosio FG, Neff JC. Diagnostic significance of hypocomplementemia. Kidney Int (1991) 39:811–21. doi: 10.1038/ki.1991.102
113. Couser WG, Johnson RJ. The etiology of glomerulonephritis: Roles of infection and autoimmunity. Kidney Int (2014) 86:905–14. doi: 10.1038/ki.2014.49
114. Tortajada A, Gutierrez E, Pickering MC, Praga Terente M, Medjeral-Thomas N. The role of complement in IgA nephropathy. Mol Immunol (2019) 114:123–32. doi: 10.1016/j.molimm.2019.07.017
115. Ma H, Sandor DG, Beck LH Jr. The role of complement in membranous nephropathy. Semin Nephrol (2013) 33:531–42. doi: 10.1016/j.semnephrol.2013.08.004
116. Medjeral-Thomas NR, Cook HT, Pickering MC. Complement activation in IgA nephropathy. Semin Immunopathol (2021) 43:679–90. doi: 10.1007/s00281-021-00882-9
117. Kerlin BA, Ayoob R, Smoyer WE. Epidemiology and pathophysiology of nephrotic syndrome-associated thromboembolic disease. Clin J Am Soc Nephrol (2012) 7:513–20. doi: 10.2215/CJN.10131011
Keywords: complement, kidney, platelet, neutrophil, atypical hemolytic uremic syndrome, thrombosis, lupus nephritis, coagulation
Citation: Yoshida Y and Nishi H (2022) The role of the complement system in kidney glomerular capillary thrombosis. Front. Immunol. 13:981375. doi: 10.3389/fimmu.2022.981375
Received: 29 June 2022; Accepted: 25 August 2022;
Published: 14 September 2022.
Edited by:
Mihály Józsi, Eötvös Loránd University, HungaryReviewed by:
Bradley Patton Dixon, University of Colorado Anschutz Medical Campus, United StatesPilar Sánchez-Corral, University Hospital La Paz Research Institute (IdiPAZ), Spain
Copyright © 2022 Yoshida and Nishi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yoko Yoshida, eW9rb3kxMjJAZ21haWwuY29t