 Anna Krutyhołowa1
Anna Krutyhołowa1 Karolina Strzelec2Agata Dziedzic2
Karolina Strzelec2Agata Dziedzic2 Grzegorz P. Bereta1Katarzyna Łazarz-Bartyzel3Jan Potempa1,4*†
Grzegorz P. Bereta1Katarzyna Łazarz-Bartyzel3Jan Potempa1,4*† Katarzyna Gawron2*†
Katarzyna Gawron2*†- 1Department of Microbiology, Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University, Krakow, Poland
- 2Department of Molecular Biology and Genetics, Faculty of Medical Sciences in Katowice, Medical University of Silesia, Katowice, Poland
- 3Department of Periodontology and Oral Medicine, Faculty of Medicine, Medical College, Jagiellonian University, Krakow, Poland
- 4Department of Oral Immunology and Infectious Diseases, School of Dentistry, University of Louisville, Louisville, KY, United States
Observations from numerous clinical, epidemiological and serological studies link periodontitis with severity and progression of rheumatoid arthritis. The strong association is observed despite totally different aetiology of these two diseases, periodontitis being driven by dysbiotic microbial flora on the tooth surface below the gum line, while rheumatoid arthritis being the autoimmune disease powered by anti-citrullinated protein antibodies (ACPAs). Here we discuss genetic and environmental risk factors underlying development of both diseases with special emphasis on bacteria implicated in pathogenicity of periodontitis. Individual periodontal pathogens and their virulence factors are argued as potentially contributing to putative causative link between periodontal infection and initiation of a chain of events leading to breakdown of immunotolerance and development of ACPAs. In this respect peptidylarginine deiminase, an enzyme unique among prokaryotes for Porphyromonas gingivalis, is elaborated as a potential mechanistic link between this major periodontal pathogen and initiation of rheumatoid arthritis development.
Introduction
Periodontal diseases affect the gingiva, the supporting connective tissue and the alveolar bone. They are one of the most common inflammatory disorders, affecting nearly 30% of the population worldwide (1). Specifically, two diseases can be distinguished: the first is gingivitis, which is inflammation of the gingiva and is limited to the soft-tissue compartment of the gingival epithelium and connective tissue; the second is periodontitis (PD), defined as inflammation of the tooth-supportive tissues, which results in attachment loss and bone destruction. Many years of research have identified a number of microbial aetiologies for periodontal diseases. Socransky and Haffajee (2) summarized the hypotheses regarding possible causes and postulated that periodontal diseases are triggered by infection. The oral cavity is colonized by many different microorganisms. More than 700 species of oral bacteria have been identified in biofilms (dental plaque) (3). Extensive analysis of dental plaques has resulted in a detailed description of polymicrobial communities associated with general health or periodontal disease (2, 4–10). These studies also re-classified PD as a microbial shift disease in which a Gram-positive microbiota shifts to a mostly Gram-negative microbiota (11). However, the exact mechanisms underlying changes in microbial composition remain unclear. Recent metagenomic and mechanistic studies suggest that PD is not caused by the presence of a few specific periodontal pathogens; rather, it results from polymicrobial synergy and dysbiosis (12–16). Thus, the concept of polymicrobial synergy and dysbiosis was proposed (17). Many studies report an association between PD and three periodontal pathogens, namely, Porphyromonas gingivalis (P. gingivalis), Tannerella forsythia (T. forsythia) and Treponema denticola (T. denticola) (designated as “red complex” bacteria). Recent whole genome DNA probe studies of the oral microbiota identified new bacterial species that correlate with PD, namely, Aggregatibacter actinomycetemcomitans (A. actinomycetemcomitans), Filifactor alocis (F. alocis) and other species belonging to the genera Peptostreptococcaceae, Desulfobulbus and Synergistetes (12, 18–20). The presence of oral polymicrobial communities cannot stay unnoticed by the immune system. Innate defence mechanisms regulate the composition of the microbiome and help to maintain periodontal health. For example, healthy periodontal tissue is characterized by a high number of neutrophils transiting through the junctional epithelium and expression of numerous host innate mediators, such as defensins (BD1, BD2 and BD3), cytokines (IL-8) and lipopolysaccharide-binding protein (LBP) (21–23). Moreover, even though cytokines associated with tissue damage, i.e., interleukin 1β (IL-1β), tumour necrosis factor α (TNFα) and prostaglandin E2 (PGE2) are present in gingival crevicular fluid (GCF) from clinically healthy sites, these levels are lower than those in fluid from diseased sites (24, 25). Innate immune mechanisms make a marked contribution to bone resorption in PD. This process is controlled by the ratio of RANKL (receptor activator of nuclear factor-κB ligand), which induces differentiation of osteoclast precursors, to osteoprotegerin (OPG), which is a soluble receptor for RANKL (26, 27). RANKL expression is regulated by proinflammatory cytokines, such as TNFα and IL-1β, thus an increase in the concentration of these cytokines in healthy tissue can lead to bone loss (28, 29). Both, commensal and pathogenic bacteria activate innate immune responses via Toll-like receptors, which are responsible for recognition of bacterial components. This suggests that a microbiome of appropriate composition ensures precise equilibrium between native immune responses within healthy tissues. Dysbiosis or a microbiome shift may disturb the balance between expression of inflammatory and anti-inflammatory mediators and lead to destruction of bone and tooth-supporting tissues. Hence, PD results from the combined influence of a dysbiotic microbiome that forms dental plaques and host inflammatory responses that destroy the periodontium. Many studies demonstrate a central role for P. gingivalis in pathogenesis of PD, particularly since this bacterium can cause bone loss after implantation into the oral cavity of animals (30–32). However, recent studies show that the pathogen itself does not cause PD in mice lacking commensal bacteria (15). This suggests, that the actual role of P. gingivalis is to manipulate host responses and convert a symbiotic community into a dysbiotic one, resulting in destructive inflammation. It should be noted, however, that species, such as T. forsythia, T. denticola and A. actinomycetemcomitans can modulate host immune responses, suggesting that they also impact on PD (33–35).
Rheumatoid arthritis (RA) is a systemic autoimmune disease that affects 0.5–1% of the population worldwide (36). It is characterized by chronic inflammation of synovial joints and bone erosion, which together result in joint destruction, disability, susceptibility to other pathological conditions and shorter life expectancy. The aetiology of the disease remains unknown, but many studies suggest involvement of both, genetic and environmental factors; indeed, genetic influences contribute to approximately 60% of RA cases (37). Importantly, an infectious agent in a susceptible host may be a trigger factor for this disease (38). Rheumatoid factor (RF), an antibody specific for the Fc fragment of IgG, is an established diagnostic marker for RA. Thus, IgG antibodies were considered major autoantigens in RA. However, further studies identified RF in other autoimmune and infectious diseases, as well as in 5% of the healthy population; in these cases, it may be a response to polyclonal B cell activation (39). More recent studies focused on the role of citrullinated proteins and peptides as possible autoantigens in RA. Citrullination, also known as deimination, is a post-translational enzymatic modification that converts a positively charged arginine residue into a neutral citrulline residue (Figure 1). This reaction is catalyzed by calcium ion-dependent enzymes called peptidylarginine deiminases (PADs) (40). The human genome encodes five members of the PAD family (PAD1, PAD2, PAD3, PAD4 and PAD6), which differ with respect to tissue distribution, cellular sub-localisation and substrate specificity (41). Many studies confirmed that citrullination is required for physiological processes, such as citrullination of keratin and filaggrin during keratinocyte differentiation or for citrullination of myelin basic protein, which ensures electrical insulation of myelin sheets (42–44). There are also strong suggestions that citrullination plays a role in transcriptional regulation and chromatin decondensation during neutrophil trap formation (45). However, extensive or inadequate deimination can lead to pathological conditions. Replacement of arginine, which often plays a central role in the structural and functional integrity of proteins, may alter protein folding, thereby exposing new epitopes to the immune system. PAD2 and PAD4 seem to play a crucial role in RA due to their presence in the rheumatoid synovial membrane, synovial fluid cells and synovial fluid (46–49). To date, four well established citrullinated autoantigens have been identified, i.e., collagen type II, fibrinogen, vimentin and α-enolase (50–53). All of these proteins can be found in the joints and all can form immune complexes with antibodies, which then mediate further inflammatory reactions.
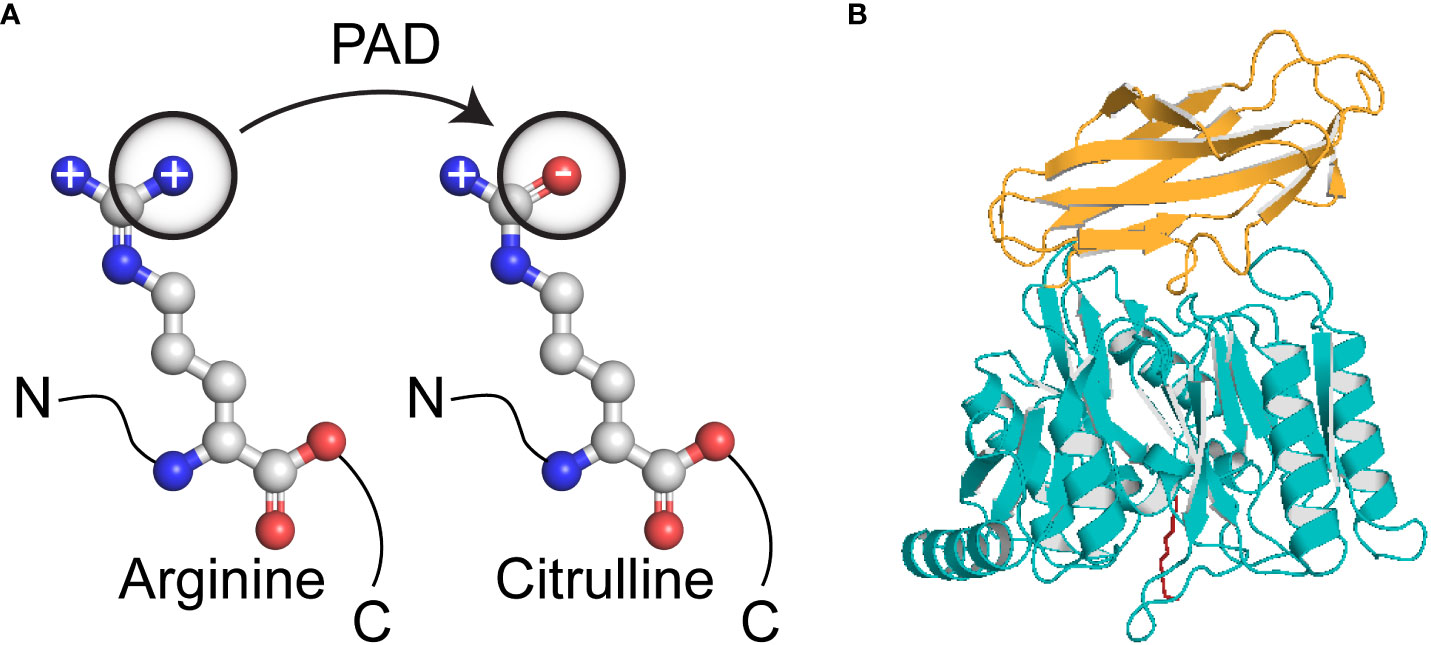
Figure 1 Conversion of arginine to citrulline. (A) Biochemical reaction resulting in loss of positive charge on a side chain; (B) The crystallised structure of Porphyromonas gingivalis peptidylarginine deiminase comprises two domains; deiminase domain aa490-360 (yellow) and Ig-like fold aa361-461 (blue). The substrate in the active site was marked as red.
Epidemiologic correlation between PD and RA
A scientific concept claiming that dental sepsis can cause systemic inflammation, including arthritis, arose in the 19th century and was developed further in the 20th century. Despite the fact, that in 1952 the American Medical Association pronounced that tooth extraction was not based on scientific evidence, and that the practice should not be considered as a treatment approach to reduce the severity or symptoms of RA, the method was used continuously up until the 1970s, when the first effective RA drugs, e.g., penicillamine appeared (54–56).
Recently, links and associations between PD and RA have been investigated intensively and many studies show a correlation between these two diseases (57–60). The earliest studies investigated the prevalence of RA in patients with PD. Mercado et al. (61) showed that individuals with moderate to severe PD are more susceptible to RA. They examined 1412 individuals attending a dental clinic and divided them into two groups, i.e., those with PD and a control group comprising individuals attending the dental clinic for general treatment. The prevalence of RA in the PD group was significantly higher than that in the control group. Moreover, patients with RA were more likely to have a moderate to severe form of PD than patients without RA. However, it should be kept in mind that this study relied on self-reported RA and used non-validated parameters to classify PD.
Subsequent studies tried to dissect whether PD occurs more often in patients with established RA. Most studies used criteria for PD defined by the American Association of Periodontology and criteria for RA defined by the American College of Rheumatology (62). To evaluate the association between PD and RA, several factors were analysed, namely, genetic factors, proinflammatory factors and the presence of different oral bacterial DNA species in periodontal pocket samples, serum and synovial fluids from patients with RA (63). PD severity was identified as the third most potent predictor of RA, immediately after female gender and smoking (57). Occurrence of PD correlated strongly with RA, hence it was proposed that both diseases are driven by a common molecular mechanism.
PD and RA are characterized by chronic inflammation, and TNFα is considered a major proinflammatory mediator. The main source of TNFα in RA are joint macrophages (64). A study by Nilsson et al. (65) shows that plasma levels of TNFα are related to the degree of systemic inflammation and may affect PD development in patients with RA. Patients suffering from RA that have higher levels of circulating TNFα show worse clinical parameters with respect to the periodontium, i.e., enhanced bleeding on probing, lower clinical attachment level and greater probing pocket depth. However, the association between TNFα and PD is not exclusive to RA. Increased levels of TNFα are also associated with severity of PD in patients with diabetes type 2 (66). One problem with studies evaluating levels of proinflammatory mediators in PD and RA is treatment of RA symptoms during the study. In such cases, lack of higher levels of proinflammatory mediators, such as TNFα and/or C-reactive protein in patients suffering from both PD and RA is likely due to pharmacologic treatment of RA (65).
“Risk factors” that link PD and RA
PD and RA are similar in several ways. Both diseases are characterized by chronic inflammation, which leads to tissue destruction. The aetiology of both diseases is multifactorial and includes genetic and environmental components (Figure 2). Although the hypothesis that bacteria are the main cause of PD is well established, there is no direct proof that RA has a microbial origin. By contrast, an evidence exists that autoimmunity plays a role in PD (67). Some studies report detection of antibodies specific for host components, such as collagen and DNA, as well as aggregation of antibodies and increased lymphotoxicity toward oral epithelial cells and fibroblasts (67).
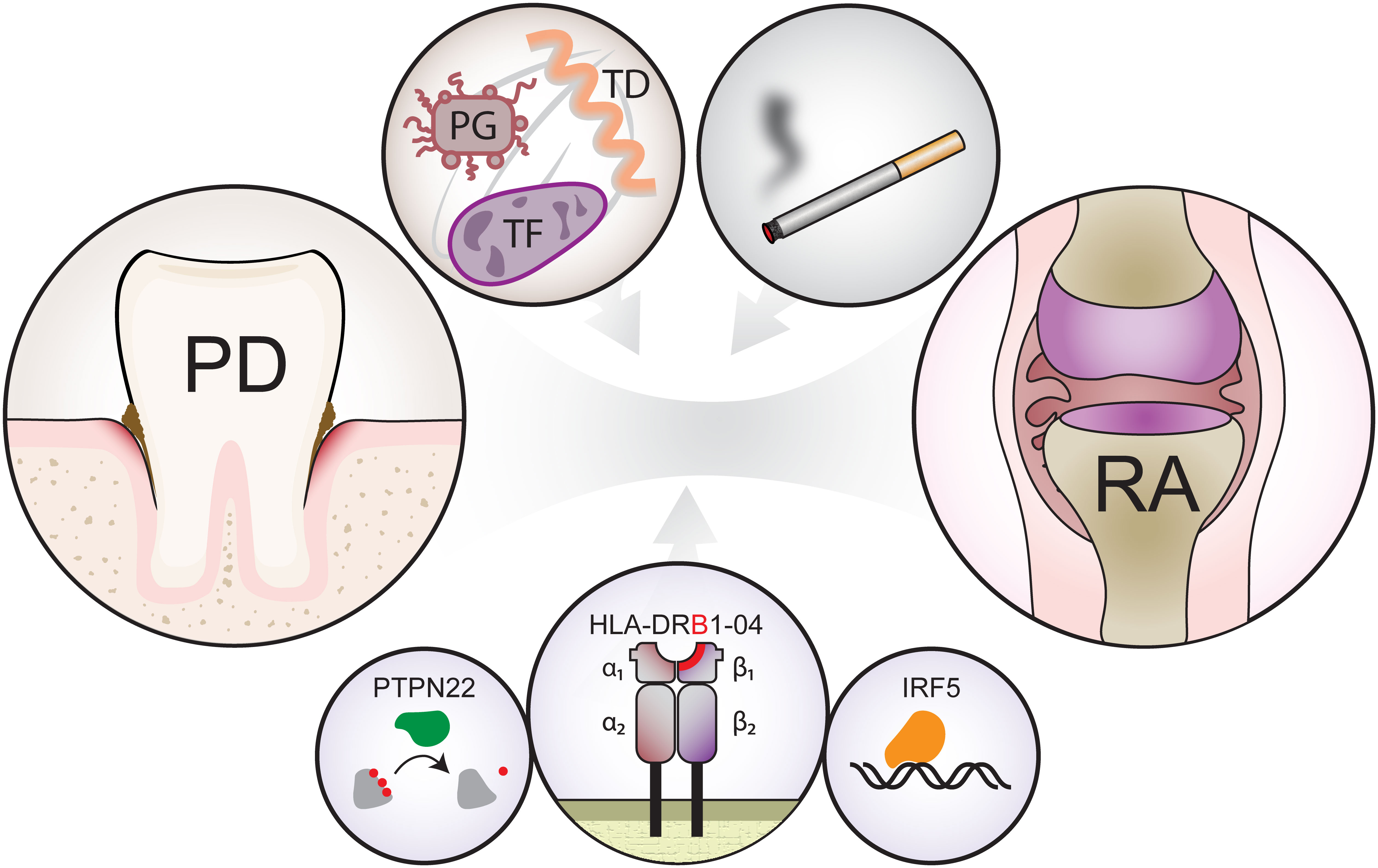
Figure 2 Shared risk factors that contribute to development of periodontal disease (left circle, PD) and rheumatoid arthritis (right circle, RA). The top left circle shows bacteria forming the “red complex”, namely, Porphyromonas gingivalis (red, PG), Tannerella forsythia (violet, TF) and Treponema denticola (orange, TD) and the top right circle shows an environmental factor (smoking). Bottom three circles show the main genetic risk factors, namely shared epitopes within the β-chain of human leukocyte antigen (HLA), tyrosine phosphatase (PTPN22) and Interferon Regulatory Factor 5 (IRF5).
The main genetic risk factor for autoimmune diseases is related to major histocompatibility complex class II (MHC II) molecules, certain alleles of which may strongly favour presentation of citrullinated epitopes and promote generation of autoimmune antibodies. The most significant alleles responsible for RA susceptibility are present on HLA-DR4, -DR1 and -DR10. These genetic variants mainly affect six amino acids, namely, EQKRAA, situated in the peptide-binding cleft of the MHC II molecule. These alleles, collectively called “shared epitopes”, are important for RA development (68, 69). Shared epitopes are associated mainly with the presence of antibodies against citrullinated proteins rather than with the disease itself (70). In particular, HLA-DR4 is associated with rapidly progressive PD; in patients with PD, the frequency of shared epitopes is higher than that in controls (71, 72).
In addition, genetic variants of tyrosine phosphatase (PTPN22), which may be involved in regulating T cell and B cell activation may contribute to a connection between PD and RA (73, 74). Another study suggests, that PD and RA share interferon regulatory factor 5 (IRF5) and PR domain zinc finger protein 1 (PRDM1) as susceptibility factors (75). Also, epigenetic alterations within the X chromosome may, at least partially, explain the higher prevalence of RA in women with PD (76).
Smoking is considered an environmental risk factor for both PD and RA. Epidemiological studies of large cohorts show significant association between smoking and RA (77). Indeed, smoking stimulates peptide citrullination by PAD2 and PAD4 (78). This finding strongly supports the concept of pathological importance of anti-citrullinated protein antibodies (ACPAs) in development of ACPA-positive RA, however, smoking-induced citrullination fails to explain ACPA-negative RA. Smoking worsens both PD and RA by promoting growth of bacteria. A study of patients with PD where smokers were distinguished from non-smokers showed, that smoking was associated with increased load of T. forsythia, Peptostreptococcus micros (P. micros), Fusobacterium nucleatum (F. nucleatum) and Campylobacter rectus (C. rectus) in subgingival dental plaque (79).
Another factor that may affect bacterial growth is infection with Epstein-Barr virus (EBV-1) and cytomegalovirus. These viruses promote colonization of the gingiva by P. gingivalis, A. actinomycetemcomitans, T. forsythia, Prevotella intermedia (P. intermedia), Prevotella nigrescens (P. nigrescens) or T. denticola (80). PCR-based studies of gingival tissue detected EBV in 71–89% and cytomegalovirus in 65–78% of patients with severe PD, but in only 6% of healthy control subjects (81).
Impact of periodontal pathogens on pathogenesis of PD and RA
Periodontal bacteria play a crucial role in progression of PD and may be a factor linking this disease with RA. This thesis is supported by several studies showing, that serum and synovial fluid from RA patients contain higher levels of antibodies against periodontal pathogens than serum from control subjects. The detected antibodies were specific for P. gingivalis, P. intermedia, Prevotella melaninogenica (P. melaninogenica) and T. forsythia (82, 83). In addition to antibodies, DNA from periodontal bacteria was detected in serum and synovial fluids from RA patients. Martinez-Martinez et al. (84) detected periodontal bacterial DNA in 100% of synovial fluid samples and in 83.5% of serum samples from RA patients. The most common bacterial DNAs in synovial fluid were derived from P. intermedia (73.6%) and P. gingivalis (42.1%). However, synovial fluid samples from RA patients plated on agar did not show bacterial growth, and bacterial DNA was not detected in leukocytes. A similar result was reported by Moen et al. (85), who reported that the most common DNAs in synovial fluid and serum were derived from P. intermedia, P. gingivalis and T. denticola. Interestingly, there was no significant difference between the amount of bacterial DNA in subgingival dental plaques, serum and synovial fluid in cases with A. actinomycetemcomitans and P. gingivalis. By contrast, P. intermedia, T. forsythia, P. nigrescens and T. denticola were significantly more abundant in subgingival dental plaques than in serum and synovial fluid. It should be noted, that species detected in synovial fluid and serum were also present in subgingival dental plaques. Detection of bacterial DNA in synovial fluid suggests transport of DNA from periodontal tissue to the joints of RA patients. The mechanism underlying such transport is unclear, however, several hypotheses have been proposed: (i) transport of free DNA via the bloodstream, (ii) transport via viable non-immune cells and (iii) intracellular capture and transport by phagocytes and other immune cells (86). Many studies were conducted to elucidate the mechanism(s) underlying transport of bacterial DNA. These studies included testing lymphocytes isolated from blood for the presence of bacterial DNA and inoculation of synovial fluid onto different growth media under both, aerobic and anaerobic conditions, however, neither approach showed a positive result, suggesting, that bacterial DNA is transported in the blood as free DNA (84).
Another interesting study in patients with RA showed that oral and gut microbiota are different from those in healthy control subjects (87). The results revealed, that P. gingivalis was more common in control saliva and in control dental plaques, and that there was no association between this species, peptidylarginine deiminase levels and RA. These results are in agreement with those of other studies confirming a lack of correlation between P. gingivalis and RA (88, 89). By contrast, anaerobic species, such as Lactobacillus salivarius, Atopobium spp. and Cryptobacterium curtum (C. curtum) were enriched in both salivary and dental samples from subjects with RA, whereas aerobes, such as Neisseria spp. and Rothia aeria were enriched in control samples. Appropriate treatment of RA patients resulted in partial resolution of these alterations (87). A study conducted on periodontally healthy subjects with and without RA showed, that Gram-negative anaerobes were significantly more abundant in RA (90). Such a dysbiotic state could be an indication for further development of PD. P. gingivalis and A. actinomycetemcomitans were not dominant components of the microbiome, and there was no significant difference in their abundance between the groups. Therefore, it is likely, that other bacteria play a role in initiating RA, but P. gingivalis and A. actinomycetemcomitans contribute at later stages to maintain the disease. Interestingly, this study identified C. curtum in the periodontal microbiome of RA patients (100-fold greater abundance in RA, with 39-fold greater odds of detection) (90). The fact, that C. curtum is more abundant in the oral and gut microbiota of those with early RA does not imply an aetiopathogenic role for C. curtum, however, this species may be an interesting candidate for future studies. Moreover, PD is significantly more common in RA patients compared to healthy controls: the incidence is higher in people in the earliest stages of the disease, which is probably related to the role of P. gingivalis in inducing citrullination and the development of new antigens.
P. gingivalis
P. gingivalis is an obligate, anaerobic, non-motile, asaccharolytic Gram-negative bacterium, that forms black-pigmented colonies on blood agar plates. It is found in 85.75% of subgingival plaque samples from patients with chronic PD (91). As an obligate anaerobe, P. gingivalis occupies dental pockets and is considered a secondary colonizer of dental plaques; secondary colonizers adhere to primary colonizers, such as Streptococcus gordonii and P. intermedia. There are invasive and non-invasive strains of P. gingivalis. This classification is based on their ability to form abscesses in a mouse model (91). In vitro and in vivo studies show that invasive strains are more pathogenic than non-invasive strains (92, 93). P. gingivalis harbours a wide variety of virulence factors, including fimbriae, capsules, lipopolysaccharide (LPS), lipoteichoic acids, haemagglutinins, gingipains, outer membrane proteins, outer membrane vesicles and peptidylarginine deiminase (PPAD) (94, 95). Expression of virulence factors is often regulated by changes in the external environment (96–98). Active virulence factors are responsible for manipulation of host immune responses, induction of host responses via cytokine production and inhibition of host protective mechanisms via citrullination and protease degradation, all of which lead to rapid destruction of periodontal tissues and bone resorption (95).
P. gingivalis has a unique ability to express a special PPAD, which is considered to be one of the virulence factors harboured by this periodontal pathogen (99). Biochemical studies show, that PPAD deiminates preferentially the C-terminal arginine of peptides and proteins, and in contrast to human PADs, it is able to citrullinate L-arginine in a calcium-independent manner (100). P. gingivalis strains lacking PPAD or harbouring an inactive form of this enzyme are less adherent and invade human gingival fibroblasts less efficiently than the wild-type strain (101). Moreover, mutant strains show an impaired ability to stimulate PGE2-dependent signalling pathways, and these properties are restored by addition of purified enzyme to the cell cultures. Also, PPAD is an important factor that alters host immune responses. For example, citrullination of LL-37 and components of the complement system, e.g., the C5a anaphylatoxin results in loss of function (102, 103). Moreover, in both, RA and PD, local inflammation of the gingival mucosa or the synovial membrane of the joint is exacerbated by the influx of inflammatory cells from the circulation. The conditions in the gingival pockets in combination with the biofilm, which includes P. gingivalis, increase the production of ACPA antibodies due to the high activity of PPAD. The above factors contribute to the stimulation of subsequent inflammatory processes.
PPAD is not the only virulence factor of P. gingivalis that has a possible role in initiating RA onset. Some of the most important virulence factors harboured by P. gingivalis are lysine/arginine-specific cysteine proteases, called gingipains. They play a major role in both, bacterial development and infection. They are responsible for maturation of fimbriae, which enable bacteria to attach to and invade host cells, such as human gingival fibroblasts and epithelial cells (104, 105). The major component of long fimbriae is the FimA protein. A recent study showed, that both, collagen-induced arthritis and PD in mice infected with P. gingivalis pre-incubated with an anti-FimA antibody were markedly less severe, suggesting that disrupting functional fimbriae with an anti-FimA antibody may ameliorate RA (106). As proteolytic enzymes, gingipains degrade host extracellular matrix proteins, such as laminin, fibronectin and collagen, as well as complement system components. Arginine-specific gingipain cooperates with PPAD, and the cleavage of the substrate leaves arginine as a C-terminal residue, which can then be modified by PPAD (107). Similar to P. gingivalis, other oral bacteria express extracellular proteases, which may reveal new antigens via proteolytic cleavage. The blood of individuals with pre-RA and established RA contains higher levels of anti-RgpB than that of healthy controls. Unlike ACPA, the level of which increases over time, the level of anti-RgpB decreases over time (108). In contrast, a study of a Southern European cohort revealed no association between pre-RA and anti-RgpB (109). However, the study did not evaluate periodontal status and anti-P. gingivalis antibody levels.
Moreover, some reports indicate that in RA patients DNA of P. gingivalis can be detected in the synovial fluid, and antibodies in serum against this bacterium. Additionally, the immunologic response against P. gingivalis is also present in people genetically predisposed to RA development in whom serum antibodies against citrullinated proteins and RF are detected. Importantly, there was no evidence of an immune response in these patients against other associated periopathogens with the infectious etiology of chronic periodontitis. P. gingivalis, as the only finely system, produces PPAD and the citrullination of proteins, which may be important in the development of an immune response against citrullinated proteins in RA patients, thus constituting a significant risk factor for the development of RA.
Most recently it was shown, that repeated oral infections with P. gingivalis directly caused development of seropositive arthritis accompanied by systemic inflammation and bone destruction in Lewis rats. Conversely, infection with P. intermedia resulted only in mild gingivitis but no bone erosion (110). The observed bone erosion strongly resembled pathological manifestation of RA in an adjuvant-induced arthritis model in rats (111). Moreover, the bone destruction pattern was consistent with results from human RA, which also showed an increase of IgM, anti-IgG and anti-CCP2 levels and their correlation with anti-P. gingivalis antibodies (83, 110).
T. denticola
T. denticola is a common oral bacterium closely associated with both, PD and aetiology of implant-related periarthritis (112). This Gram-negative, obligatory anaerobic bacterium inhabits subgingival plaques. Numerous studies allowed identification of T. denticola virulence factors, which include leucine-rich repeat proteins, metabolic end-products, biofilm creation, toxin-antitoxin systems, dentilisin, lipoproteins, trypsin-like protease activity and sheath proteins (113–118). Unlike P. gingivalis and T. forsythia, T. denticola is motile and able to respond chemotactically to environmental changes (33). Several studies suggest, that the presence of P. gingivalis is required for colonization by T. denticola, however, interactions between “red complex” bacteria are unclear (119, 120). Mouse subcutaneous abscess models of disease have been used to investigate T. denticola virulence factors, as well as those of other species from that genus (121, 122). These studies showed that mono-infection with Treponemes may cause localised lesions, but only co-infection with P. gingivalis and T. denticola leads to increased tissue damage as compared with that after mono-infection by P. gingivalis (123). Although the relevance of these abscess models has been questioned, and more adequate models have been developed, more recent studies confirm these results. For instance, inoculation with the “red bacterial complex” at a 1:1:1 ratio resulted in greater bone resorption than mono-inoculations with the same total number of bacterial cells (124). At the same time, co-infection with P. gingivalis and T. denticola causes the same level of damage as a 40-fold higher number of P. gingivalis alone (125). These studies underline the importance of synergistic interactions of periodontal pathogens in development and severity of periodontal disease and probably with PD-associated diseases, such as RA.
T. forsythia
T. forsythia is a Gram-negative, anaerobic bacterium and a member of the “red complex”. A growing body of evidence implicates T. forsythia in the pathogenesis of PD, however, the species is relatively understudied due to its demanding growth requirements and difficulties with genetic manipulations (126, 127). A number of studies demonstrated, that T. forsythia is the first colonizer of a dental plaque, and that it may be a necessary precursor species for P. gingivalis and T. denticola colonization. Moreover, T. forsythia is more likely to cause PD in overweight women than in women of normal weight (128). Recent studies show overgrowth of T. forsythia in overweight and obese individuals when compared with those of a normal weight (129). A study conducted on patients with RA showed that T. forsythia was associated with highly active RA and high salivary ammonium levels, which may be a result of PPAD activity (130). There are several putative virulence factors expressed by T. forsythia, i.e., KLIKK proteases and PrtH protease, sialidases, a leucine-rich repeat cell surface-associated and secreted protein (BspA), a hemagglutinin, components of the bacterial S-layer and methylglyoxal production (131). To fully understand the T. forsythia virulence mechanisms that contribute to pathogenesis of PD and RA, future studies should be mostly focused on interaction between its virulence factors, other bacteria and host responses.
A. actinomycetemcomitans
A. actinomycetemcomitans is a Gram-negative bacterium associated with aggressive PD, chronic PD and several non-oral infections (132). It is found in the oral cavity of more than one-third of the population (133, 134). This species harbours a wide variety of virulence factors, including adhesins, fimbriae, exotoxins and endotoxins, all of which vary among individual strains (serotypes). Certain serotypes are more prevalent in individuals suffering from aggressive PD (132). Its ability to produce leukotoxin-A (LtxA) is considered to be the most important virulence factor. This toxin kills white blood cells, which results in untamed bacterial growth and potent stimulation of the host immune response (135). LtxA from A. actinomycetemcomitans belongs to the RTX (Repeats-in-Toxin) family of bacterial proteins and is a pore-forming toxin. Its expression is regulated by both, environmental and genetic factors, however, the exact expression trigger is unknown. Macrophages are the cells that are most sensitive to LtxA-induced cell lysis, which leads to the release of high amounts of IL-1β, a strong proinflammatory mediator (136, 137). It was shown, that LtxA induces hypercitrullination in neutrophils, and leukotoxic A. actinomycetemcomitans strains were found associated with both, PD and RA. Despite association between exposure to A. actinomycetemcomitans and ACPA in RA, the presence of A. actinomycetemcomitans cannot be attributed solely to RA (138). In a recent case study reported by Mukherjee et al. (139), a 59-year-old man with ACPA-positive RA was diagnosed with highly leukotoxic strain of A. actinomycetemcomitans endocarditis. After treatment with a ceftriaxone, an antibiotic from the beta-lactam group, previously elevated levels of ACPA and CRP normalized, joint symptoms resolved and did not reappear for one year after the treatment.
C. curtum
There are many other bacteria in the oral microbiota, and these have been suggested as possible factors that contribute to development of RA. Recently, C. curtum was identified as a predominant component of the oral microbiota of patients with RA (90). This species is an oral pathogen, however, its translocation can cause several infections, including pelvic abscesses, gynaecologic infections and wounds (140). It is a Gram-positive, anaerobic, asaccharolytic rod, which has an ability to produce citrulline along with ornithine and ammonia due to arginine degradation (141). C. curtum can be found in the oral and gut microbiota of patients with early RA (87).
F. nucleatum
F. nucleatum is a periodontal bacterium that facilitates and promotes growth of other periodontal pathogens, thus it contributes indirectly to RA development. This early colonizing anaerobic bacterium belonging to the Bacteroidaceae family is a dominant species in the periodontium (142). F. nucleatum interacts with both, Gram-positive and Gram-negative bacteria present in dental biofilms (143, 144). An increased number of F. nucleatum are found at sites of PD, however, it is not responsible directly for periodontal tissue destruction associated with PD (145). Several studies show that co-infection with F. nucleatum increases attachment and invasion of human gingival epithelial cells by P. gingivalis and A. actinomycetemcomitans (146, 147). These phenomena are inhibited by galactose. Another example of a synergistic interaction between F. nucleatum and P. gingivalis is that co-infection with these two bacterial species increases alveolar bone loss to a greater extent than infection by either bacterium alone (14). However, interactions between bacteria are not always synergistic. Recent study has demonstrated, that collagen-induced arthritis in mice inoculated with a mix of P. gingivalis, F. nucleatum and A. actinomycetemcomitans showed less alveolar bone loss compared to mice inoculated with P. gingivalis alone. At the same time, F. nucleatum and A. actinomycetemcomitans alone accelerated the onset and progression of RA (148). Moreover, F. nucleatum reacts to host immune responses in PD. Unlike other Gram-negative bacteria, F. nucleatum is susceptible to host β-defensins, particularly β-defensin 3 (hβD-3) and LL-37 (145). Therefore, F. nucleatum is thought to downregulate expression of hβD-1 and LL-37, nevertheless, hβD-2 and hβD-3 are upregulated after epithelial exposure to this bacterium (145). Genes upregulated by F. nucleatum include those encoding host antimicrobial peptides and proteins, as well as chemokines (IL-8, CXCL1, 3, 5 and 10), which attract neutrophils, monocytes, lymphocytes and macrophages, and CSF2 and -3, which stimulate neutrophil development (145). Additionally, F. nucleatum secretes serine proteases, which enhance its pathogenicity. Like proteases from other periodontal pathogens, such as P. gingivalis and T. denticola, they are capable of degrading extracellular matrix proteins, such as fibrinogen, fibronectin and collagen type I and IV (145, 149, 150). Moreover, the 65 kDa protease cleaves the α-chains of immunoglobulin A, but not immunoglobulin G, which makes it particularly suited to the oral environment. However, it should be noted that, in contrast to the high proteolytic activity of other periodontal bacteria, the activity of this protease is relatively low. To reach the same activity as the phenylalanine-specific protease of T. denticola, the purified protease from F. nucleatum requires nearly ten times longer incubation. This can be explained by the fact, that F. nucleatum can be found together with microorganisms showing strong proteolytic activity; therefore, it does not itself require a highly active protease (145).
F. alocis
F. alocis is a very recently discovered Gram-positive anaerobic rod, which may play a significant role in development of PD (20). It has several unique characteristics responsible for mediating its pathogenic activity. F. alocis is often found in patients with PD, and in contrast to other periodontal pathogens, the species is absent from healthy subjects (151). This suggests, that F. alocis plays an important role in development of inflammation and is associated with several oral infections, for example, endodontic infections and in peri-implantitis (152, 153). F. alocis interacts with other microbial species present in dental biofilms, and these interactions enhance its invasive capacity, which may result in chronic inflammation. It has been shown, that this bacterium accumulates preferentially at sites rich in F. nucleatum, but cannot colonize niches occupied by Gram-positive streptococci, e.g., Streptococcus gordonii. Interactions with A. actinomycetemcomitans are strain-dependent, but some strains of A. actinomycetemcomitans facilitate accumulation of F. alocis (154). Moreover, F. alocis induces release of host proinflammatory cytokines resulting in apoptosis of gingival epithelial cells and most importantly, it has an ability to adhere to and invade epithelial cells, which is enhanced in the presence of P. gingivalis, although the exact mechanism underlying their interaction remains unclear (155, 156). Co-culture of P. gingivalis and F. alocis results in a significant increase in biofilm formation. It is driven by the fact, that F. alocis shows very high resistance to oxidative stress. Therefore, its presence increases the survival of P. gingivalis under oxidative stress conditions (156). F. alocis also exhibits increased activity of non-gingipain-type proteases (its gingipain-type activity is low), that primarily recognize arginine and lysine at a cleavage site. In silico studies of the F. alocis genome revealed the presence of multiple genes involved in degrading arginine to ornithine and citrulline (20). Due to the presence of specific deiminases, F. alocis decomposes a C-terminal arginine to ornithine and ammonia, which counteracts acidic conditions generated by carbohydrate metabolism in biofilms (157).
P. intermedia
P. intermedia is a Gram-negative, obligate anaerobic bacterium associated with PD (158). It is found in both, healthy and inflamed periodontal tissue in PD patients. This species shows high degradative enzyme activity, which also contributes to progression of PD (159). P. intermedia expresses nucleases that degrade neutrophil extracellular traps, leading to release of endogenous PADs (160, 161). Although P. intermedia’s DNA along with antibodies against this pathogen were detected in synovial fluid from patients with RA, infection did not exacerbate collagen-induced arthritis (82, 84, 162). Moreover, a study on Lewis rat model has demonstrated, that P. intermedia causes only mild gingivitis; moreover, bone erosion was not observed and rats did not develop systemic inflammation. However, antibodies against P. intermedia were detected in rats serum after 1 month of exposure and their concentration increased after 4, and 8 months, respectively (110). A recent study identified a new citrullinated peptide of cytokeratin 13 (CK13-1) in GCF against which RA patients mounted an antibody response. It should be noted, that anti-cCK13-1 antibodies, together with anti-cTNC5 (tenascin C) antibodies are associated with the presence of P. intermedia.
Brief summary of modulatory effects exerted by periodontal pathogens on host immune response is presented in Figure 3.
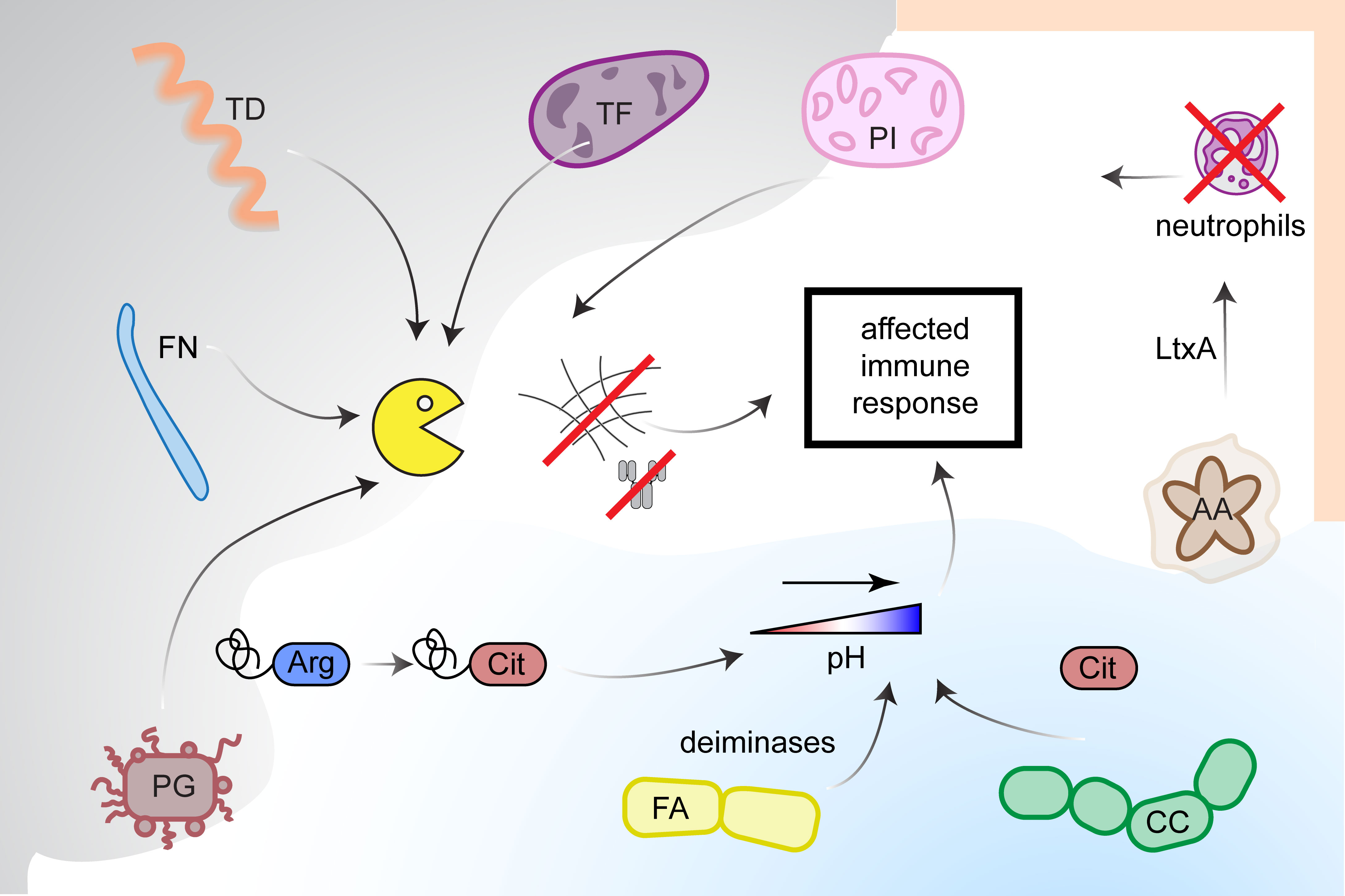
Figure 3 Modulation of immune response by periodontal pathogens. Host immune response can be affected in several ways. Proteases (yellow icon) secreted by Fusobacterium nucleatum (FN), Prevotella intermedia (PI), Treponema denticola (TD), Tannerella forsythia (TF) and Porphyromonas gingivalis (PG) cleave proteins of extracellular matrix, what leads to tissue degradation. Moreover, PG secretes peptidylarginine deiminase (PPAD), which citrullinates C-terminal arginines of proteins and peptides creating new epitopes recognized by anti-citrullinated proteins antibodies (ACPA). Activity of PPAD together with deiminases from Filifactor alocis (FA) and Cryptobacterium curtum (CC) contributes to the increase of pH in oral cavity. Leukotoxin A from Aggregatibacter actinomycetemcomitans (AA) affects white blood cells (mostly macrophages) inducing their lysis and stimulation of NETs release from neutrophils, which in turn are degradated by nucleases expressed by PI. Degradation of NETs leads to endogenous PADs release.
Molecular mechanisms linking PD and RA
Rheumatoid factors
Rheumatoid factors (RFs) are antibodies specific for the Fc region of IgG, and comprise immunoglobulins of any type, however, predominantly belong to the IgM class (163). RFs were first discovered in RA patients over 70 years ago, and since then, together with ACPA, they have been used as diagnostic and predictive markers for RA. Although RFs are associated mainly with RA, they are also present in those with other autoimmune and inflammatory diseases, as well as in healthy individuals (164). Moreover, RFs are detected in the gingiva, subgingival plaques and serum of patients with PD, and in seropositive patients cross-react with bacterial epitopes (165–167). Gingipains, an important virulence factor of P. gingivalis, are responsible for epitope exposure in the RF-Fc region. The Fc regions of IgG molecules contain sequences of lysine and arginine, thus gingipains play a role in IgG3 CH2 and CH3 domains processing and have a key function in RFs production (168). It should be also mentioned, that IgG Fc regions in RA patients harbour significantly fewer galactose residues than those in age-matched healthy controls, thus lack of terminal galactose residues may play a role in more severe disease progression (169). P. melaninogenica is a saccharolytic bacterium, that metabolises galactose molecules, then it binds to the Fc region of IgG antibodies and metabolises galactose residues (170). The resulting alteration in the composition of sugar moieties affects the activity of antibodies associated with autoimmune diseases.
Antigens and autoantibodies
Recently, autoantigens involved in RA disease pathology have been well studied and characterized. Interests are focused mainly on citrullinated proteins as true autoantigens in RA. It is assumed, that inflammation and proinflammatory mediators activate PADs in calcium-rich environments via cleavage and activation of human proteinase-activated receptor-2 (PAR-2), which contributes greatly to accumulation of citrullinated proteins (171, 172). At present, there are four well established citrullinated autoantigens, i.e., collagen type II, fibrinogen, vimentin and α-enolase. However, Wegner et al. (39) argue, that identification of all citrullinated proteins is necessary before we can fully explore pathogenic mechanisms. Serum ACPAs are present in 70% of patients with RA (173, 174). They are associated with RA progression and can be detected 10 years before onset of clinical symptoms, and also correlate with progression of PD (175). Based on these findings, there is a hypothesis, that citrullination associated with PD in genetically susceptible subjects can lead to localised oral immune responses, which may result in systemic ACPA responses followed by synovial inflammation and onset of RA (176). However, Konig et al. (177) argue, that the central role of ACPA is to break tolerance during RA development and they propose, that loss of tolerance is a consequence of the presence of antibodies against native proteins, which precede generation of ACPA.
Another post-translational modification that can affect protein structure and function is carbamylation. This involves non-enzymatic binding of cyanate to the primary amine of lysine, which forms a carbamyl group to generate homocitrulline. Carbamylated proteins induce production of autoantibodies (anti-CarP) (178). Neutrophils and their associated myeloperoxidases (MPOs) contribute to carbamylation by promoting conversion of thiocyanate to cyanate (179). Anti-CarP antibodies can predict RA development independently of anti-CCP2 (citrullinated cyclic peptide 2) antibodies. Their presence can be detected in both, ACPA-positive and ACPA-negative pre-RA patients, and in those with established RA. Detection of anti-CarP in ACPA-negative patients may indicate a more severe disease course (180). Carbamylated proteins have been detected in inflamed gingival tissue, along with elevated levels of MPO, however, there is no significant correlation between PD and anti-CarP (181, 182). Moreover, there is no association between anti-CarP and RA risk factors, either genetic or environmental, e.g., smoking (183).
Patients with established RA show elevated levels of antibodies against proteins modified with malondialdehyde-acetaldehyde adducts (MAAs), which are also associated with ACPA and RFs (184). MAAs result from modification of lysine residues in proteins, which is mediated by highly reactive malondialdehyde and acetaldehyde molecules formed by ROS during lipid peroxidation. ROS are generated mainly under conditions of oxidative stress, however, they can also be released by neutrophils (185). There are indications, that P. gingivalis may contribute to production of anti-MAA antibodies in mice (186).
Citrullination
PPAD contributes to RA development by generating citrullinated antigens. Evidence suggests, that human fibrinogen and α-enolase (targeted by ACPA in RA) are substrates for PPAD, and that antibodies against citrullinated α-enolase from P. gingivalis cross-react with human anti-α-enolase antibodies (187). It is suggested, that PPAD can autocitrullinate its own arginine residues, however this was observed only in a single case of PPAD expressed by E. coli (188). Anti-PPAD antibodies detected in RA are not directed against citrullinated PPAD (189). There is no consensus concerning the role of PPAD and anti-PPAD antibodies in RA. According to the study by Quirke et al. (188), anti-PPAD antibody levels in patients with RA are higher than those in controls. However, another study showed no correlation between these antibodies and ACPA levels or severity of RA, where the ACPA levels decreased in patients with both, RA and PD (89). The discrepancy between these studies may be due to methodological differences. Recently, another study showed a correlation between anti-PPAD IgG and anti-CCP IgG in RA patients treated with disease-modifying anti-rheumatic drugs (DMARDs). Subjects with lower anti-PPAD levels showed better responses to DMARDs treatment than those with higher levels (190). These results may promote anti-PPAD antibody levels as a tool for predicting responses to therapy. Autoantibody profiles may influence treatment responses. Indeed, patients with a broad baseline of autoantibodies respond better during early stages of treatment, however, they are less likely to achieve initial drug-free remission (191). A broad baseline of autoantibodies may be due to more active humoral immune responses to several different antigens and to switching of antibody isotypes. This process can be targeted efficiently by medications (191). The hypothesis of PPAD as a possible link between PD and RA is supported by studies using animal models. Mice infected with a P. gingivalis strain expressing PPAD developed collagen-induced arthritis more rapidly than controls, and the course of disease was more severe. Moreover, the levels of citrullinated proteins at disease sites were higher (162). Although the link between PD and RA is well established, the particular role of P. gingivalis and its PPAD is less clear. PD is a polymicrobial disease, therefore, interaction between P. gingivalis and other oral bacteria should be taken into account. For example, A. actinomycetemcomitans produce pore-forming LtxA, which induces hypercitrullination in neutrophils. Deregulation of neutrophil-citrullinating enzymes by LtxA may result in neutrophil trap formation and subsequent release of hypercitrullinated proteins and peptides. Moreover, the presence of LtxA-producing A. actinomycetemcomitans was confirmed in RA patients, and correlated with ACPA and RF (138). In addition, a recent study postulates, that A. actinomycetemcomitans is able to induce autoimmune responses associated with RA in genetically predisposed individuals (139). However, it should be noted, that there is a negative association between P. gingivalis and A. actinomycetemcomitans in subgingival samples (192). In addition, P. gingivalis inhibits growth and attachment of A. actinomycetemcomitans within co-cultured biofilms (193, 194). Furthermore, the presence of P. gingivalis, P. intermedia and P. nigrescens inhibits activity of LtxA (195).
Heat shock proteins (HSPs)
HSPs interact reversibly with non-folded abnormal proteins and peptides, thereby protecting the cell from proteotoxic stress. They play a role in innate and acquired immune responses and are associated with RA pathogenesis. Bacterial HSPs were found at high levels in serum samples from patients with RA, and the 70 kDa HSP of P. melanogenica and P. intermedia was found in serum samples from patients with PD (196, 197). Interestingly, antibodies specific for HSP70 and HSP40 of A. actinomycetemcomitans are present in the synovium of patients with RA (196). Expression of HSP70 is triggered by stress factors, such as heat, trauma, endotoxins and anti-inflammatory drugs, but its expression in synovium is mainly triggered by proinflammatory cytokines, such as TNFα, IL-1 and IL-6 (198). Therefore, these results may indicate a role of HSPs produced by A. actinomycetemcomitans, but not other oral bacteria in patients with RA.
Immune cells
Neutrophils play an important role in both, PD and RA, as they are responsible for deregulated immune responses that result in tissue damage. Moreover, they contribute to production of autoantibodies. Neutrophils in patients with PD, RA and other inflammatory diseases display an activated phenotype and high levels of NETs (neutrophil extracellular traps) release. Oral bacteria promote neutrophil-mediated production of autoantibodies. Several factors are involved in this process, i.e., LtxA from A. actinomycetemcomitans, degranulation of neutrophils promoted by F. alocis, and release of NETs triggered by P. gingivalis, A. actinomycetemcomitans and F. nucleatum (199–201).
The common feature of inflamed tissue in PD and RA is infiltration by high numbers of other immune cell types, including dendrocytes, macrophages and B and T lymphocytes. Joint and blood samples from patients suffering from RA harbour unusual populations of CD4+CD28- T cells (202). These cells are similar to NK T cells and have the ability to produce large amounts of IFN γ (interferon γ), which is a marker of activated T cells (known as TH1 helper cells) and contain intracellular perforin and granzyme B, providing them with the ability to lyse target cells. Their outgrowth into large clonal populations may be partially attributed to a defect in down-regulating Bcl-2 when deprived of T cell growth factors. In the absence of the CD28 molecule, these unusual CD4 T cells use alternate costimulatory pathways, while the secretion of pro-inflammatory factors stimulates the development of RA (202). TH1 cells play a role in promoting autoimmune diseases (203). Moreover, IFN γ increases expression of MHC class II and stimulates production of proinflammatory mediators by macrophages (204). Th17 cells are another T cell subset found at sites of chronic inflammation in PD and in the synovium of RA patients (205). These cells secrete IL-17, which plays an important role in RA pathogenesis. In brief, IL-17 promotes production of proinflammatory cytokines by macrophages and fibroblasts, facilitates infiltration of joints by immune cells, induces expression of matrix metalloproteinases, and also contributes to bone resorption (206). It has been shown that P. gingivalis antigens induce expression of IL-17 (207).
Plasma cells and B cells are the most common cell types found in periodontal lesions comprising about 50% and 18% of all immune cells, respectively (208). P. gingivalis promotes B cell hyper-reactivity by stimulating dendritic cells (209). In addition, B cell survival and maturation into plasma cells is enhanced in gingival epithelium. The presence of B cells in periodontal tissue is crucial for maintaining periodontal health. Antibodies secreted by plasma cells control bacterial growth via neutralisation, opsonisation and complement activation. However, hyper-reactivity of B cells may be harmful due to their ability to present antigens effectively to T cells, which leads to osteoclastogenesis and increased bone resorption (210).
Conclusions
We summarized herein epidemiological studies that establish a correlation between PD and RA on multiple levels. While many studies show higher prevalence and severity of PD in patients with RA, others demonstrate that patients with PD are more prone to developing RA. However, since correlation does not imply causation, the precise mechanisms connecting these two diseases remain unclear.
Both, PD and RA share the same risk factors, including HLA-DRB1-04 as a genetic factor, smoking and infection with EBV and cytomegalovirus. Moreover, both conditions are characterized by chronic inflammation, a crucial role played by B cells and plasma cells, and tissue destruction evidenced by alveolar bone resorption and joint erosion. These common features may suggest a similar underlying mechanisms of both diseases.
Furthermore, we discussed in this review the associations among bacteria responsible for the onset, development and progression of PD and RA. We focused mainly on oral pathogens, which are designated as “red complex” bacteria, and are established the aetiological agents of PD. We also attempted to cover the role of other, less commonly described bacterial species present in the oral microbiome of patients with PD and/or RA. We analyzed the main virulence factors of these microorganisms, their mechanisms of action and their effects on host immune responses. We strived to provide examples of cross-talk between bacterial pathogens and to indicate potential overlap of pathogenic mechanisms that may lead to synergistic effects. In general, the evidence presented herein supports a dominant paradigm involving a microbiome shift that results in PD and, possibly, RA. Hopefully, future in depth investigations of the oral microbiome and the molecular mechanisms utilized by oral bacteria will pave the way for novel treatments and diagnostic tools for PD and RA, so far, two incurable diseases.
Author contributions
AK, conceptualization, data acquisition and analysis, and writing-original draft preparation; KS and AD, literature search, data analysis and visualization, writing-editing; GPB, data acquisition and analysis, writing-reviewing, and editing; KŁ-B: design, data analysis, writing-reviewing and editing; JP, conceptualization and design, writing-editing, and supervision; KG, conceptualization and design, writing-original draft preparation, and supervision; All authors contributed to the article and approved the submitted version.
Acknowledgments
We thank the National Science Centre of Poland (UMO-2018/29/B/NZ2/01930 to KG, UMO-2018/30/A/NZ5/00650 to JP) and NIH/NIDCR (DE 022597 to JP) for providing funding to many of the experiments reviewed in this publication.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Dye BA. Global periodontal disease epidemiology. Periodontol 2000 (2012) 58(1):10–25. doi: 10.1111/j.1600-0757.2011.00413.x
2. Socransky SS, Haffajee AD. Evidence of bacterial etiology: a historical perspective. Periodontol 2000 (1994) 5:7–25. doi: 10.1111/j.1600-0757.1994.tb00016.x
3. Dewhirst FE, Chen T, Izard J, Paster BJ, Tanner ACR, Yu W-H, et al. The human oral microbiome. J Bacteriol (2010) 192(19):5002–17. doi: 10.1128/JB.00542-10
4. Kroes I, Lepp PW, Relman DA. Bacterial diversity within the human subgingival crevice. Proc Natl Acad Sci U.S.A. (1999) 96(25):14547–52. doi: 10.1073/pnas.96.25.14547
5. Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL. Microbial complexes in subgingival plaque. J Clin Periodontol (1998) 25(2):134–44. doi: 10.1111/j.1600-051x.1998.tb02419.x
6. Ximénez-Fyvie LA, Haffajee AD, Socransky SS. Comparison of the microbiota of supra- and subgingival plaque in health and periodontitis. J Clin Periodontol (2000) 27(9):648–57. doi: 10.1034/j.1600-051x.2000.027009648.x
7. Lin X, Wu J, Xie H. Porphyromonas gingivalis minor fimbriae are required for cell-cell interactions. Infect Immun (2006) 74(10):6011–5. doi: 10.1128/IAI.00797-06
8. Tanner A, Kent R, Maiden MF, Taubman MA. Clinical, microbiological and immunological profile of healthy, gingivitis and putative active periodontal subjects. J Periodontal Res (1996) 31(3):195–204. doi: 10.1111/j.1600-0765.1996.tb00484.x
9. Darveau RP, Tanner A, Page RC. The microbial challenge in periodontitis. Periodontol 2000 (1997) 14:12–32. doi: 10.1111/j.1600-0757.1997.tb00190.x
10. Huyghe A, Francois P, Charbonnier Y, Tangomo-Bento M, Bonetti E-J, Paster BJ, et al. Novel microarray design strategy to study complex bacterial communities. Appl Environ Microbiol (2008) 74(6):1876–85. doi: 10.1128/AEM.01722-07
11. Marsh PD. Microbial ecology of dental plaque and its significance in health and disease. Adv Dent Res (1994) 8(2):263–71. doi: 10.1177/08959374940080022001
12. Griffen AL, Beall CJ, Campbell JH, Firestone ND, Kumar PS, Yang ZK, et al. Distinct and complex bacterial profiles in human periodontitis and health revealed by 16S pyrosequencing. ISME J (2012) 6(6):1176–85. doi: 10.1038/ismej.2011.191
13. Ramsey MM, Rumbaugh KP, Whiteley M. Metabolite cross-feeding enhances virulence in a model polymicrobial infection. PloS Pathog (2011) 7(3):e1002012. doi: 10.1371/journal.ppat.1002012
14. Settem RP, El-Hassan AT, Honma K, Stafford GP, Sharma A. Fusobacterium nucleatum and tannerella forsythia induce synergistic alveolar bone loss in a mouse periodontitis model. Infect Immun (2012) 80(7):2436–43. doi: 10.1128/IAI.06276-11
15. Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, et al. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe (2011) 10(5):497–506. doi: 10.1016/j.chom.2011.10.006
16. Orth RK-H, O’Brien-Simpson NM, Dashper SG, Reynolds EC. Synergistic virulence of porphyromonas gingivalis and treponema denticola in a murine periodontitis model. Mol Oral Microbiol (2011) 26(4):229–40. doi: 10.1111/j.2041-1014.2011.00612.x
17. Hajishengallis G, Lamont RJ. Beyond the red complex and into more complexity: the polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Mol Oral Microbiol (2012) 27(6):409–19. doi: 10.1111/j.2041-1014.2012.00663.x
18. Abusleme L, Dupuy AK, Dutzan N, Silva N, Burleson JA, Strausbaugh LD, et al. The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. ISME J (2013) 7(5):1016–25. doi: 10.1038/ismej.2012.174
19. Curtis MA, Zenobia C, Darveau RP. The relationship of the oral microbiotia to periodontal health and disease. Cell Host Microbe (2011) 10(4):302–6. doi: 10.1016/j.chom.2011.09.008
20. Aruni AW, Mishra A, Dou Y, Chioma O, Hamilton BN, Fletcher HM. Filifactor alocis-a new emerging periodontal pathogen. Microbes Infect (2015) 17(7):517–30. doi: 10.1016/j.micinf.2015.03.011
21. Ren L, Jin L, Leung WK. Local expression of lipopolysaccharide-binding protein in human gingival tissues. J Periodontal Res (2004) 39(4):242–8. doi: 10.1111/j.1600-0765.2004.00732.x
22. Lu Q, Samaranayake LP, Darveau RP, Jin L. Expression of human β-defensin-3 in gingival epithelia. J Periodontal Res (2005) 40(6):474–81. doi: 10.1111/j.1600-0765.2005.00827.x
23. Tonetti MS, Imboden MA, Gerber L, Lang NP, Laissue J, Mueller C. Localized expression of mRNA for phagocyte-specific chemotactic cytokines in human periodontal infections. Infect Immun (1994) 62(9):4005–14. doi: 10.1128/iai.62.9.4005-4014.1994
24. Kamma J, Mombelli A, Tsinidou K, Vasdekis V, Giannopoulou C. Cytokines in gingival crevicular fluid of adolescents and young adults. Oral Microbiol Immunol (2009) 24(1):7–10. doi: 10.1111/j.1399-302X.2008.00466.x
25. Górska R, Gregorek H, Kowalski J, Laskus-Perendyk A, Syczewska M, Madaliński K. Relationship between clinical parameters and cytokine profiles in inflamed gingival tissue and serum samples from patients with chronic periodontitis. J Clin Periodontol (2003) 30(12):1046–52. doi: 10.1046/j.0303-6979.2003.00425.x
26. Cochran DL. Inflammation and bone loss in periodontal disease. J Periodontol (2008) 79(8 Suppl):1569–76. doi: 10.1902/jop.2008.080233
27. Nagasawa T, Kiji M, Yashiro R, Hormdee D, Lu H, Kunze M, et al. Roles of receptor activator of nuclear factor-kappaB ligand (RANKL) and osteoprotegerin in periodontal health and disease. Periodontol 2000 (2007) 43:65–84. doi: 10.1111/j.1600-0757.2006.00185.x
28. Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL. TNF-α induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest (2000) 106(12):1481–8. doi: 10.1172/JCI11176
29. Wei S, Kitaura H, Zhou P, Ross FP, Teitelbaum SL. IL-1 mediates TNF-induced osteoclastogenesis. J Clin Invest (2005) 115(2):282–90. doi: 10.1172/JCI23394
30. Baker PJ, Evans RT, Roopenian DC. Oral infection with porphyromonas gingivalis and induced alveolar bone loss in immunocompetent and severe combined immunodeficient mice. Arch Oral Biol (1994) 39(12):1035–40. doi: 10.1016/0003-9969(94)90055-8
31. Meka A, Bakthavatchalu V, Sathishkumar S, Lopez MC, Verma RK, Wallet SM, et al. Porphyromonas gingivalis infection-induced tissue and bone transcriptional profiles. Mol Oral Microbiol (2010) 25(1):61–74. doi: 10.1111/j.2041-1014.2009.00555.x
32. Zhang W, Ju J, Rigney T, Tribble G. Porphyromonas gingivalis infection increases osteoclastic bone resorption and osteoblastic bone formation in a periodontitis mouse model. BMC Oral Health (2014) 14:89. doi: 10.1186/1472-6831-14-89
33. Dashper SG, Seers CA, Tan KH, Reynolds EC. Virulence factors of the oral spirochete treponema denticola. J Dent Res (2011) 90(6):691–703. doi: 10.1177/0022034510385242
34. Sharma A. Virulence mechanisms of tannerella forsythia. Periodontol 2000 (2010) 54(1):106–16. doi: 10.1111/j.1600-0757.2009.00332.x
35. Åberg CH, Kelk P, Johansson A. Aggregatibacter actinomycetemcomitans: virulence of its leukotoxin and association with aggressive periodontitis. Virulence (2015) 6(3):188–95. doi: 10.4161/21505594.2014.982428
36. Silman AJ, Pearson JE. Epidemiology and genetics of rheumatoid arthritis. Arthritis Res (2002) 4 Suppl 3(Suppl 3):S265–72. doi: 10.1186/ar578
37. MacGregor AJ, Snieder H, Rigby AS, Koskenvuo M, Kaprio J, Aho K, et al. Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum (2000) 43(1):30–7. doi: 10.1002/1529-0131(200001)43:1<30::AID-ANR5>3.0.CO;2-B
38. Carty SM, Snowden N, Silman AJ. Should infection still be considered as the most likely triggering factor for rheumatoid arthritis? Ann Rheum Dis (2004) 63 Suppl 2(Suppl 2):ii46–9. doi: 10.1136/ard.2004.028241
39. Wegner N, Lundberg K, Kinloch A, Fisher B, Malmström V, Feldmann M, et al. Autoimmunity to specific citrullinated proteins gives the first clues to the etiology of rheumatoid arthritis. Immunol Rev (2010) 233(1):34–54. doi: 10.1111/j.0105-2896.2009.00850.x
40. Nakayama-Hamada M, Suzuki A, Kubota K, Takazawa T, Ohsaka M, Kawaida R, et al. Comparison of enzymatic properties between hPADI2 and hPADI4. Biochem Biophys Res Commun (2005) 327(1):192–200. doi: 10.1016/j.bbrc.2004.11.152
41. Vossenaar ER, Zendman AJW, van Venrooij WJ, Pruijn GJM. PAD, a growing family of citrullinating enzymes: genes, features and involvement in disease. BioEssays (2003) 25(11):1106–18. doi: 10.1002/bies.10357
42. György B, Tóth E, Tarcsa E, Falus A, Buzás EI. Citrullination: a posttranslational modification in health and disease. Int J Biochem Cell Biol (2006) 38(10):1662–77. doi: 10.1016/j.biocel.2006.03.008
43. Tarcsa E, Marekov LN, Mei G, Melino G, Lee SC, Steinert PM. Protein unfolding by peptidylarginine deiminase. substrate specificity and structural relationships of the natural substrates trichohyalin and filaggrin. J Biol Chem (1996) 271(48):30709–16. doi: 10.1074/jbc.271.48.30709
44. Pritzker LB, Joshi S, Gowan JJ, Harauz G, Moscarello MA. Deimination of myelin basic protein. 1. effect of deimination of arginyl residues of myelin basic protein on its structure and susceptibility to digestion by cathepsin d. Biochemistry (2000) 39(18):5374–81. doi: 10.1021/bi9925569
45. Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol (2009) 184(2):205–13. doi: 10.1083/jcb.200806072
46. Foulquier C, Sebbag M, Clavel C, Chapuy-Regaud S, Badine RA, Méchin M-C, et al. Peptidyl arginine deiminase type 2 (PAD-2) and PAD-4 but not PAD-1, PAD-3, and PAD-6 are expressed in rheumatoid arthritis synovium in close association with tissue inflammation. Arthritis Rheum (2007) 56(11):3541–53. doi: 10.1002/art.22983
47. Chang X, Yamada R, Suzuki A, Sawada T, Yoshino S, Tokuhiro S, et al. Localization of peptidylarginine deiminase 4 (PADI4) and citrullinated protein in synovial tissue of rheumatoid arthritis. Rheumatol (Oxford) (2005) 44(1):40–50. doi: 10.1093/rheumatology/keh414
48. Vossenaar ER, Radstake TRD, van der Heijden A, van Mansum MAM, Dieteren C, de Rooij D-J, et al. Expression and activity of citrullinating peptidylarginine deiminase enzymes in monocytes and macrophages. Ann Rheum Dis (2004) 63(4):373–81. doi: 10.1136/ard.2003.012211
49. Kinloch A, Lundberg K, Wait R, Wegner N, Lim NH, Zendman AJW, et al. Synovial fluid is a site of citrullination of autoantigens in inflammatory arthritis. Arthritis Rheum (2008) 58(8):2287–95. doi: 10.1002/art.23618
50. Masson-Bessière C, Sebbag M, Girbal-Neuhauser E, Nogueira L, Vincent C, Senshu T, et al. The major synovial targets of the rheumatoid arthritis-specific antifilaggrin autoantibodies are deiminated forms of the alpha- and beta-chains of fibrin. J Immunol (2001) 166(6):4177–84. doi: 10.4049/jimmunol.166.6.4177
51. Vossenaar ER, Després N, Lapointe E, van der Heijden A, Lora M, Senshu T, et al. Rheumatoid arthritis specific anti-sa antibodies target citrullinated vimentin. Arthritis Res Ther (2004) 6(2):R142–50. doi: 10.1186/ar1149
52. Burkhardt H, Sehnert B, Bockermann R, Engström A, Kalden JR, Holmdahl R. Humoral immune response to citrullinated collagen type II determinants in early rheumatoid arthritis. Eur J Immunol (2005) 35(5):1643–52. doi: 10.1002/eji.200526000
53. Kinloch A, Tatzer V, Wait R, Peston D, Lundberg K, Donatien P, et al. Identification of citrullinated alpha-enolase as a candidate autoantigen in rheumatoid arthritis. Arthritis Res Ther (2005) 7(6):R1421–9. doi: 10.1186/ar1845
54. Easlick KA. An evaluation of the effect of dental foci of infection on health. J Am Dent Assoc (1951) 42(6):615–97. doi: 10.14219/jada.archive.1951.0112
55. Jaffe IA. Penicillamine in rheumatoid arthritis: clinical pharmacology and biochemical properties. Scand J Rheumatol Suppl (1979) 28:58–64. doi: 10.3109/03009747909108238
56. Lau CS, Chia F, Dans L, Harrison A, Hsieh TY, Jain R, et al. 2018 update of the APLAR recommendations for treatment of rheumatoid arthritis. Int J Rheum Dis (2019) 22(3):357–75. doi: 10.1111/1756-185X.13513
57. Dissick A, Redman RS, Jones M, Rangan BV, Reimold A, Griffiths GR, et al. Association of periodontitis with rheumatoid arthritis: a pilot study. J Periodontol (2010) 81(2):223–30. doi: 10.1902/jop.2009.090309
58. de Pablo P, Dietrich T, McAlindon TE. Association of periodontal disease and tooth loss with rheumatoid arthritis in the US population. J Rheumatol (2008) 35(1):70–6.
59. Mirrielees J, Crofford LJ, Lin Y, Kryscio RJ, Dawson 3DR, Ebersole JL, et al. Rheumatoid arthritis and salivary biomarkers of periodontal disease. J Clin Periodontol (2010) 37(12):1068–74. doi: 10.1111/j.1600-051X.2010.01625.x
60. Trombone AP, Claudino M, Colavite P, de Assis GF, Avila-Campos MJ, Silva JS, et al. Periodontitis and arthritis interaction in mice involves a shared hyper-inflammatory genotype and functional immunological interferences. Genes Immun (2010) 11(6):479–89. doi: 10.1038/gene.2010.13
61. Mercado F, Marshall RI, Klestov AC, Bartold PM. Is there a relationship between rheumatoid arthritis and periodontal disease? J Clin Periodontol (2000) 27(4):267–72. doi: 10.1034/j.1600-051x.2000.027004267.x
62. Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American rheumatism association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum (1988) 31(3):315–24. doi: 10.1002/art.1780310302
63. Luan Y-Z, Chen BS, Ma KS-K. Sequencing of the 16S ribosomal DNA gene and virulence of the oral microbiome in patients with rheumatoid arthritis: comment on the article by kroese et al. Arthritis Rheumatol (2022) 74(7):1296–97. doi: 10.1002/art.42106
64. Choy EHS, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis. N Engl J Med (2001) 344(12):907–16. doi: 10.1056/NEJM200103223441207
65. Nilsson M, Kopp S. Gingivitis and periodontitis are related to repeated high levels of circulating tumor necrosis factor-alpha in patients with rheumatoid arthritis. J Periodontol (2008) 79(9):1689–96. doi: 10.1902/jop.2008.070599
66. Engebretson S, Chertog R, Nichols A, Hey-Hadavi J, Celenti R, Grbic J. Plasma levels of tumour necrosis factor-alpha in patients with chronic periodontitis and type 2 diabetes. J Clin Periodontol (2007) 34(1):18–24. doi: 10.1111/j.1600-051X.2006.01017.x
67. Kaur G, Mohindra K, Singla S. Autoimmunity-basics and link with periodontal disease. Autoimmun Rev (2017) 16(1):64–71. doi: 10.1016/j.autrev.2016.09.013
68. Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis. an approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum (1987) 30(11):1205–13. doi: 10.1002/art.1780301102
69. Weyand CM, Goronzy JJ. Association of MHC and rheumatoid arthritis. HLA polymorphisms in phenotypic variants of rheumatoid arthritis. Arthritis Res (2000) 2(3):212–6. doi: 10.1186/ar90
70. McHugh NJ, Owen P, Cox B, Dunphy J, Welsh K. MHC class II, tumour necrosis factor alpha, and lymphotoxin alpha gene haplotype associations with serological subsets of systemic lupus erythematosus. Ann Rheum Dis (2006) 65(4):488–94. doi: 10.1136/ard.2005.039842
71. Katz J, Goultschin J, Benoliel R, Brautbar C. Human leukocyte antigen (HLA) DR4. positive association with rapidly progressing periodontitis. J Periodontol (1987) 58(9):607–10. doi: 10.1902/jop.1987.58.9.607
72. Bonfil JJ, Dillier FL, Mercier P, Reviron D, Foti B, Sambuc R, et al. A "case control" study on the rôle of HLA DR4 in severe periodontitis and rapidly progressive periodontitis. identification of types and subtypes using molecular biology (PCR.SSO). J Clin Periodontol (1999) 26(2):77–84. doi: 10.1034/j.1600-051x.1999.260203.x
73. Begovich AB, Carlton VEH, Honigberg LA, Schrodi SJ, Chokkalingam AP, Alexander HC, et al. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet (2004) 75(2):330–37. doi: 10.1086/422827
74. Kallberg H, Padyukov L, Plenge RM, Ronnelid J, Gregersen PK, van der Helm-van Mil AHM, et al. Gene-gene and gene-environment interactions involving HLA-DRB1, PTPN22, and smoking in two subsets of rheumatoid arthritis. Am J Hum Genet (2007) 80(5):867–75. doi: 10.1086/516736
75. Schaefer AS, Jochens A, Dommisch H, Graetz C, Jockel-Schneider Y, Harks I, et al. A large candidate-gene association study suggests genetic variants at IRF5 and PRDM1 to be associated with aggressive periodontitis. J Clin Periodontol (2014) 41(12):1122–31. doi: 10.1111/jcpe.12314
76. Viatte S, Plant D, Raychaudhuri S. Genetics and epigenetics of rheumatoid arthritis. Nat Rev Rheumatol (2013) 9(3):141–53. doi: 10.1038/nrrheum.2012.237
77. Di Giuseppe D, Discacciati A, Orsini N, Wolk A. Cigarette smoking and risk of rheumatoid arthritis: a dose-response meta-analysis. Arthritis Res Ther (2014) 16(2):R61. doi: 10.1186/ar4498
78. Lugli EB, Correia RESM, Fischer R, Lundberg K, Bracke KR, Montgomery AB, et al. Expression of citrulline and homocitrulline residues in the lungs of non-smokers and smokers: implications for autoimmunity in rheumatoid arthritis. Arthritis Res Ther (2015) 17(1):9. doi: 10.1186/s13075-015-0520-x
79. van Winkelhoff AJ, Bosch-Tijhof CJ, Winkel EG, van der Reijden WA. Smoking affects the subgingival microflora in periodontitis. J Periodontol (2001) 72(5):666–71. doi: 10.1902/jop.2001.72.5.666
80. Contreras A, Umeda M, Chen C, Bakker I, Morrison JL, Slots J. Relationship between herpesviruses and adult periodontitis and periodontopathic bacteria. J Periodontol (1999) 70(5):478–84. doi: 10.1902/jop.1999.70.5.478
81. Slots J, Kamma JJ, Sugar C. The herpesvirus-porphyromonas gingivalis-periodontitis axis. J Periodontal Res (2003) 38(3):318–23. doi: 10.1034/j.1600-0765.2003.00659.x
82. Ogrendik M, Kokino S, Ozdemir F, Bird PS, Hamlet S. Serum antibodies to oral anaerobic bacteria in patients with rheumatoid arthritis. Med Gen Med (2005) 7(2):2.
83. Mikuls TR, Payne JB, Reinhardt RA, Thiele GM, Maziarz E, Cannella AC, et al. Antibody responses to porphyromonas gingivalis (P. gingivalis) in subjects with rheumatoid arthritis and periodontitis. Int Immunopharmacol (2009) 9(1):38–42. doi: 10.1016/j.intimp.2008.09.008
84. Martinez-Martinez RE, Abud-Mendoza C, Patiño-Marin N, Rizo-Rodríguez JC, Little JW, Loyola-Rodríguez JP. Detection of periodontal bacterial DNA in serum and synovial fluid in refractory rheumatoid arthritis patients. J Clin Periodontol (2009) 36(12):1004–10. doi: 10.1111/j.1600-051X.2009.01496.x
85. Moen K, Brun JG, Valen M, Skartveit L, Ribs Eribe EK, Olsen I, et al. Synovial inflammation in active rheumatoid arthritis and psoriatic arthritis facilitates trapping of a variety of oral bacterial DNAs. Clin Exp Rheumatol (2006) 24(6):656–63.
86. Loyola-Rodriguez JP, Martinez-Martinez RE, Abud-Mendoza C, Patiño-Marin N, Seymour GJ. Rheumatoid arthritis and the role of oral bacteria. J Oral Microbiol (2010) 2. doi: 10.3402/jom.v2i0.5784
87. Zhang X, Zhang D, Jia H, Feng Q, Wang D, Liang D, et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat Med (2015) 21(8):895–905. doi: 10.1038/nm.3914
88. Scher JU, Ubeda C, Equinda M, Khanin R, Buischi Y, Viale A, et al. Periodontal disease and the oral microbiota in new-onset rheumatoid arthritis. Arthritis Rheum (2012) 64(10):3083–94. doi: 10.1002/art.34539
89. Konig MF, Paracha AS, Moni M, Bingham 3CO, Andrade F. Defining the role of porphyromonas gingivalis peptidylarginine deiminase (PPAD) in rheumatoid arthritis through the study of PPAD biology. Ann Rheum Dis (2015) 74(11):2054–61. doi: 10.1136/annrheumdis-2014-205385
90. Lopez-Oliva I, Paropkari AD, Saraswat S, Serban S, Yonel Z, Sharma P, et al. Dysbiotic subgingival microbial communities in periodontally healthy patients with rheumatoid arthritis. Arthritis Rheumatol (2018) 70(7):1008–13. doi: 10.1002/art.40485
91. How KY, Song KP, Chan KG. Porphyromonas gingivalis: An overview of periodontopathic pathogen below the gum line. Front Microbiol (2016) 7:53 (. doi: 10.3389/fmicb.2016.00053(
92. Dorn BR, Burks JN, Seifert KN, Progulske-Fox A. Invasion of endothelial and epithelial cells by strains of Porphyromonas gingivalis. FEMS Microbiol Lett (2000) 187(2):139–44. doi: 10.1111/j.1574-6968.2000.tb09150.x
93. Baek KJ, Ji S, Kim YC, Choi Y. Association of the invasion ability of Porphyromonas gingivalis with the severity of periodontitis. Virulence (2015) 6(3):274–81. doi: 10.1080/21505594.2014.1000764
94. Holt SC, Kesavalu L, Walker S, Genco CA. Virulence factors of porphyromonas gingivalis. Periodontol 2000 (1999) 20:168–238. doi: 10.1111/j.1600-0757.1999.tb00162.x
95. Hajishengallis G, Lamont RJ. Breaking bad: manipulation of the host response by porphyromonas gingivalis. Eur J Immunol (2014) 44(2):328–38. doi: 10.1002/eji.201344202
96. Curtis MA, Percival RS, Devine D, Darveau RP, Coats SR, Rangarajan M, et al. Temperature-dependent modulation of Porphyromonas gingivalis lipid a structure and interaction with the innate host defenses. Infect Immun (2011) 79(3):1187–93. doi: 10.1128/IAI.00900-10
97. Roy F, Vanterpool E, Fletcher HM. HtrA in porphyromonas gingivalis can regulate growth and gingipain activity under stressful environmental conditions. Microbiology (2006) 152(Pt 11):3391–98. doi: 10.1099/mic.0.29147-0
98. Xie H, Cai S, Lamont RJ. Environmental regulation of fimbrial gene expression in porphyromonas gingivalis. Infect Immun (1997) 65(6):2265–71. doi: 10.1128/iai.65.6.2265-2271.1997
99. Gawron K, Montgomery A, Łazarz-Bartyzel K, Bereta G, Chomyszyn-Gajewska M, Venables P, et al. Porphyromonas gingivalis peptidyl arginine deiminase: a unique bacterial PAD with implications for periodontal disease and rheumatoid arthritis. In: Nicholas AP, Bhattacharya SK, Thompson PR, editors. Protein deimination in human health and disease, 2nd edition. Springer International Publishing (2017). p. 99–135. doi: 10.1007/978-3-319-58244-3_7
100. Goulas T, Mizgalska D, Garcia-Ferrer I, Kantyka T, Guevara T, Szmigielski B, et al. Structure and mechanism of a bacterial host-protein citrullinating virulence factor, porphyromonas gingivalis peptidylarginine deiminase. Sci Rep (2015) 5:11969. doi: 10.1038/srep11969
101. Gawron K, Bereta G, Nowakowska Z, Lazarz-Bartyzel K, Lazarz M, Szmigielski B, et al. Peptidylarginine deiminase from Porphyromonas gingivalis contributes to infection of gingival fibroblasts and induction of prostaglandin e 2-signaling pathway. Mol Oral Microbiol (2014) 29(6):321–32. doi: 10.1111/omi.12081
102. Koziel J, Bryzek D, Sroka A, Maresz K, Glowczyk I, Bielecka E, et al. Citrullination alters immunomodulatory function of LL-37 essential for prevention of endotoxin-induced sepsis. J Immunol (2014) 192(11):5363–72. doi: 10.4049/jimmunol.1303062
103. Bielecka E, Scavenius C, Kantyka T, Jusko M, Mizgalska D, Szmigielski B, et al. Peptidyl arginine deiminase from porphyromonas gingivalis abolishes anaphylatoxin C5a activity. J Biol Chem (2014) 289(47):32481–7. doi: 10.1074/jbc.C114.617142
104. Lee JY, Miller DP, Wu L, Casella CR, Hasegawa Y, Lamont RJ. Maturation of the Mfa1 fimbriae in the oral pathogen porphyromonas gingivalis. Front Cell Infect Microbiol (2018) 8:137. doi: 10.3389/fcimb.2018.00137
105. Weinberg A, Belton CM, Park Y, Lamont RJ. Role of fimbriae in porphyromonas gingivalis invasion of gingival epithelial cells. Infect Immun (1997) 65(1):313–6. doi: 10.1128/iai.65.1.313-316.1997
106. Jeong SH, Nam Y, Jung H, Kim J, Rim YA, Park N, et al. Interrupting oral infection of porphyromonas gingivalis with anti-FimA antibody attenuates bacterial dissemination to the arthritic joint and improves experimental arthritis. Exp Mol Med (2018) 50(3):e460. doi: 10.1038/emm.2017.301
107. Wegner N, Wait R, Sroka A, Eick S, Nguyen K-A, Lundberg K, et al. Peptidylarginine deiminase from porphyromonas gingivalis citrullinates human fibrinogen and α-enolase: implications for autoimmunity in rheumatoid arthritis. Arthritis Rheum (2010) 62(9):2662–72. doi: 10.1002/art.27552
108. Johansson L, Sherina N, Kharlamova N, Potempa B, Larsson B, Israelsson L, et al. Concentration of antibodies against porphyromonas gingivalis is increased before the onset of symptoms of rheumatoid arthritis. Arthritis Res Ther (2016) 18(1):201. doi: 10.1186/s13075-016-1100-4
109. Fisher BA, Cartwright AJ, Quirke A-M, de Pablo P, Romaguera D, Panico S, et al. Smoking, porphyromonas gingivalis and the immune response to citrullinated autoantigens before the clinical onset of rheumatoid arthritis in a southern European nested case–control study. BMC Musculoskelet Disord (2015) 16:331. doi: 10.1186/s12891-015-0792-y
110. Courbon G, Rinaudo-Gaujous M, Blasco-Baque V, Auger I, Caire R, Mijola L, et al. Porphyromonas gingivalis experimentally induces periodontis and an anti-CCP2-associated arthritis in the rat. Ann Rheum Dis (2019) 78(5):594–9. doi: 10.1136/annrheumdis-2018-213697
111. Courbon G, Lamarque R, Gerbaix M, Caire R, Linossier M-T, Laroche N, et al. Early sclerostin expression explains bone formation inhibition before arthritis onset in the rat adjuvant-induced arthritis model. Sci Rep (2018) 8(1):3492. doi: 10.1038/s41598-018-21886-w
112. Zhou X, Li Y. Subgingival microbes. In: Atlas of oral microbiology: From healthy microflora to disease (2015). p. 67–93. doi: 10.1016/B978-0-12-802234-4.00004-5
113. Magalhães MAO, Sun CX, Glogauer M, Ellen RP. The major outer sheath protein of treponema denticola selectively inhibits Rac1 activation in murine neutrophils. Cell Microbiol (2008) 10(2):344–54. doi: 10.1111/j.1462-5822.2007.01045.x
114. Jobin M-C, Virdee I, McCulloch CA, Ellen RP. Activation of MAPK in fibroblasts by treponema denticola major outer sheath protein. Biochem Biophys Res Commun (2007) 356(1):213–8. doi: 10.1016/j.bbrc.2007.02.111
115. Miyamoto M, Ishihara K, Okuda K. The treponema denticola surface protease dentilisin degrades interleukin-1 beta (IL-1 beta), IL-6, and tumor necrosis factor alpha. Infect Immun (2006) 74(4):2462–7. doi: 10.1128/IAI.74.4.2462-2467.2006
116. Mitchell HL, Dashper SG, Catmull DV, Paolini RA, Cleal SM, Slakeski N, et al. Treponema denticola biofilm-induced expression of a bacteriophage, toxin-antitoxin systems and transposases. Microbiology (2010) 156(Pt 3):774–88. doi: 10.1099/mic.0.033654-0
117. Chu L, Lai Y, Xu X, Eddy S, Yang S, Song L, et al. A 52-kDa leucyl aminopeptidase from Treponema denticola is a cysteinylglycinase that mediates the second step of glutathione metabolism. J Biol Chem (2008) 283(28):19351–8. doi: 10.1074/jbc.M801034200
118. Ikegami A, Honma K, Sharma A, Kuramitsu HK. Multiple functions of the leucine-rich repeat protein LrrA of treponema denticola. Infect Immun (2004) 72(8):4619–27. doi: 10.1128/IAI.72.8.4619-4627.2004
119. Simonson LG, McMahon KT, Childers DW, Morton HE. Bacterial synergy of treponema denticola and porphyromonas gingivalis in a multinational population. Oral Microbiol Immunol (1992) 7(2):111–2. doi: 10.1111/j.1399-302x.1992.tb00519.x
120. Sela MN. Role of treponema denticola in periodontal diseases. Crit Rev Oral Biol Med (2001) 12(5):399–413. doi: 10.1177/10454411010120050301
121. Gemmell E, Bird PS, Carter CL, Drysdale KE, Seymour GJ. Effect of fusobacterium nucleatum on the T and b cell responses to porphyromonas gingivalis in a mouse model. Clin Exp Immunol (2002) 128(2):238–44. doi: 10.1046/j.1365-2249.2002.01852.x
122. Kesavalu L, Walker SG, Holt SC, Crawley RR, Ebersole JL. Virulence characteristics of oral treponemes in a murine model. Infect Immun (1997) 65(12):5096–102. doi: 10.1128/iai.65.12.5096-5102.1997
123. Kesavalu L, Holt SC, Ebersole JL. Virulence of a polymicrobic complex, treponema denticola and porphyromonas gingivalis, in a murine model. Oral Microbiol Immunol (1998) 13(6):373–7. doi: 10.1111/j.1399-302x.1998.tb00694.x
124. Kesavalu L, Sathishkumar S, Bakthavatchalu V, Matthews C, Dawson D, Steffen M, et al. Rat model of polymicrobial infection, immunity, and alveolar bone resorption in periodontal disease. Infect Immun (2007) 75(4):1704–12. doi: 10.1128/IAI.00733-06
125. O’Brien-Simpson NM, Pathirana RD, Paolini RA, Chen Y-Y, Veith PD, Tam V, et al. An immune response directed to proteinase and adhesin functional epitopes protects against porphyromonas gingivalis-induced periodontal bone loss. J Immunol (2005) 175(6):3980–9. doi: 10.4049/jimmunol.175.6.3980
126. Sakakibara J, Nagano K, Murakami Y, Higuchi N, Nakamura H, Shimozato K, et al. Loss of adherence ability to human gingival epithelial cells in s-layer protein-deficient mutants of tannerella forsythensis. Microbiology (2007) 153(Pt 3):866–76. doi: 10.1099/mic.0.29275-0
127. Honma K, Kuramitsu HK, Genco RJ, Sharma A. Development of a gene inactivation system for bacteroides forsythus: construction and characterization of a BspA mutant. Infect Immun (2001) 69(7):4686–90. doi: 10.1128/IAI.69.7.4686-4690.2001
128. Brennan RM, Genco RJ, Wilding GE, Hovey KM, Trevisan M, Wactawski-Wende J. Bacterial species in subgingival plaque and oral bone loss in postmenopausal women. J Periodontol (2007) 78(6):1051–61. doi: 10.1902/jop.2007.060436
129. Haffajee AD, Socransky SS. Relation of body mass index, periodontitis and Tannerella forsythia. J Clin Periodontol (2009) 36(2):89–99. doi: 10.1111/j.1600-051X.2008.01356.x
130. Martínez-Rivera J-I, Xibillé-Friedmann DX, González-Christen J, de la Garza-Ramos MA, Carrillo-Vázquez SM, Montiel-Hernández JL. Salivary ammonia levels and tannerella forsythia are associated with rheumatoid arthritis: A cross sectional study. Clin Exp Dent Res (2017) 3(3):107–14. doi: 10.1002/cre2.68
131. Sharma A. Virulence mechanisms of tannerella forsythia. Periodontol 2000 (2010) 54(1):106–16. doi: 10.1111/j.1600-0757.2009.00332.x
132. Henderson B, Ward JM, Ready D. Aggregatibacter (Actinobacillus) actinomycetemcomitans: a triple a* periodontopathogen? Periodontol 2000 (2010) 54(1):78–105. doi: 10.1111/j.1600-0757.2009.00331.x
133. Leys EJ, Griffen AL, Strong SJ, Fuerst PA. Detection and strain identification of actinobacillus actinomycetemcomitans by nested PCR. J Clin Microbiol (1994) 32(5):1288–94. doi: 10.1128/jcm.32.5.1288-1294.1994
134. Lamell CW, Griffen AL, McClellan DL, Leys EJ. Acquisition and colonization stability of actinobacillus actinomycetemcomitans and porphyromonas gingivalis in children. J Clin Microbiol (2000) 38(3):1196–9. doi: 10.1128/JCM.38.3.1196-1199.2000
135. Johansson A. Aggregatibacter actinomycetemcomitans leukotoxin: a powerful tool with capacity to cause imbalance in the host inflammatory response. Toxins (Basel) (2011) 3(3):242–59. doi: 10.3390/toxins3030242
136. Kelk P, Johansson A, Claesson R, Hänström L, Kalfas S. Caspase 1 involvement in human monocyte lysis induced by actinobacillus actinomycetemcomitans leukotoxin. Infect Immun (2003) 71(8):4448–55. doi: 10.1128/IAI.71.8.4448-4455.2003
137. Kelk P, Claesson R, Hänström L, Lerner UH, Kalfas S, Johansson A. Abundant secretion of bioactive interleukin-1beta by human macrophages induced by actinobacillus actinomycetemcomitans leukotoxin. Infect Immun (2005) 73(1):453–8. doi: 10.1128/IAI.73.1.453-458.2005
138. Konig MF, Abusleme L, Reinholdt J, Palmer RJ, Teles RP, Sampson K, et al. Aggregatibacter actinomycetemcomitans-induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci Transl Med (2016) 8(369):369ra176. doi: 10.1126/scitranslmed.aaj1921
139. Mukherjee A, Jantsch V, Khan R, Hartung W, Fischer R, Jantsch J, et al. Rheumatoid arthritis-associated autoimmunity due to aggregatibacter actinomycetemcomitans and its resolution with antibiotic therapy. Front Immunol (2018) 9:2352. doi: 10.3389/fimmu.2018.02352
140. Brook I, Frazier EH. Significant recovery of nonsporulating anaerobic rods from clinical specimens. Clin Infect Dis (1993) 16(4):476–80. doi: 10.1093/clind/16.4.476
141. Uematsu H, Sato N, Djais A, Hoshino E. Degradation of arginine by slackia exigua ATCC 700122 and cryptobacterium curtum ATCC 700683. Oral Microbiol Immunol (2006) 21(6):381–4. doi: 10.1111/j.1399-302X.2006.00307.x
142. Dzink JL, Socransky SS, Haffajee AD. The predominant cultivable microbiota of active and inactive lesions of destructive periodontal diseases. J Clin Periodontol (1988) 15(5):316–23. doi: 10.1111/j.1600-051x.1988.tb01590.x
143. Kolenbrander PE. Oral microbial communities: biofilms, interactions, and genetic systems. Annu Rev Microbiol (2000) 54:413–37. doi: 10.1146/annurev.micro.54.1.413
144. Kolenbrander PE, Andersen RN, Blehert DS, England PG, Foster JS, Palmer RJ Jr. Communication among oral bacteria. Microbiol Mol Biol Rev (2002) 66(3):486–505. doi: 10.1128/MMBR.66.3.486-505.2002
145. Signat B, Roques C, Poulet P, Duffaut D. Fusobacterium nucleatum in periodontal health and disease. Curr Issues Mol Biol (2011) 13(2):25–36.
146. Saito A, Inagaki S, Kimizuka R, Okuda K, Hosaka Y, Nakagawa T, et al. Fusobacterium nucleatum enhances invasion of human gingival epithelial and aortic endothelial cells by Porphyromonas gingivalis. FEMS Immunol Med Microbiol (2008) 54(3):349–55. doi: 10.1111/j.1574-695X.2008.00481.x
147. Li Y, Guo H, Wang X, Lu Y, Yang C, Yang P. Coinfection with fusobacterium nucleatum can enhance the attachment and invasion of porphyromonas gingivalis or aggregatibacter actinomycetemcomitans to human gingival epithelial cells. Arch Oral Biol (2015) 60(9):1387–93. doi: 10.1016/j.archoralbio.2015.06.017
148. Ebbers M, Lübcke PM, Volzke J, Kriebel K, Hieke C, Engelmann R, et al. Interplay between p. gingivalis, f. nucleatum and a. actinomycetemcomitans in murine alveolar bone loss, arthritis onset and progression. Sci Rep (2018) 8(1):15129. doi: 10.1038/s41598-018-33129-z
149. Doron L, Coppenhagen-Glazer S, Ibrahim Y, Eini A, Naor R, Rosen G, et al. Identification and characterization of fusolisin, the fusobacterium nucleatum autotransporter serine protease. PloS One (2014) 9(10):e111329. doi: 10.1371/journal.pone.0111329
150. Bachrach G, Rosen G, Bellalou M, Naor R, Sela MN. Identification of a fusobacterium nucleatum 65 kDa serine protease. Oral Microbiol Immunol (2004) 19(3):155–9. doi: 10.1111/j.0902-0055.2004.00132.x
151. Kumar PS, Leys EJ, Bryk JM, Martinez FJ, Moeschberger ML, Griffen AL. Changes in periodontal health status are associated with bacterial community shifts as assessed by quantitative 16S cloning and sequencing. J Clin Microbiol (2006) 44(10):3665–73. doi: 10.1128/JCM.00317-06
152. Gomes BPFA, Jacinto RC, Pinheiro ET, Sousa ELR, Zaia AA, Ferraz CCR, et al. Molecular analysis of filifactor alocis, tannerella forsythia, and treponema denticola associated with primary endodontic infections and failed endodontic treatment. J Endod (2006) 32(10):937–40. doi: 10.1016/j.joen.2006.05.003
153. Tamura N, Ochi M, Miyakawa H, Nakazawa F. Analysis of bacterial flora associated with peri-implantitis using obligate anaerobic culture technique and 16S rDNA gene sequence. Int J Oral Maxillofac Implants (2013) 28(6):1521–9. doi: 10.11607/jomi.2570
154. Wang Q, Wright CJ, Dingming H, Uriarte SM, Lamont RJ. Oral community interactions of filifactor alocis in vitro. PloS One (2013) 8(10):e76271. doi: 10.1371/journal.pone.0076271
155. Moffatt CE, Whitmore SE, Griffen AL, Leys EJ, Lamont RJ. Filifactor alocis interactions with gingival epithelial cells. Mol Oral Microbiol (2011) 26(6):365–73. doi: 10.1111/j.2041-1014.2011.00624.x
156. Aruni AW, Roy F, Fletcher HM. Filifactor alocis has virulence attributes that can enhance its persistence under oxidative stress conditions and mediate invasion of epithelial cells by porphyromonas gingivalis. Infect Immun (2011) 79(10):3872–86. doi: 10.1128/IAI.05631-11
157. Uematsu H, Sato N, Hossain MZ, Ikeda T, Hoshino E. Degradation of arginine and other amino acids by butyrate-producing asaccharolytic anaerobic gram-positive rods in periodontal pockets. Arch Oral Biol (2003) 48(6):423–9. doi: 10.1016/s0003-9969(03)00031-1
158. Stingu CS, Jentsch H, Eick S, Schaumann R, Knöfler G, Rodloff A. Microbial profile of patients with periodontitis compared with healthy subjects. Quintessence Int (2012) 43(2):e23–31.
159. Maeda N, Okamoto M, Kondo K, Ishikawa H, Osada R, Tsurumoto A, et al. Incidence of prevotella intermedia and prevotella nigrescens in periodontal health and disease. Microbiol Immunol (1998) 42(9):583–9. doi: 10.1111/j.1348-0421.1998.tb02328.x
160. Doke M, Fukamachi H, Morisaki H, Arimoto T, Kataoka H, Kuwata H. Nucleases from Prevotella intermedia can degrade neutrophil extracellular traps. Mol Oral Microbiol (2017) 32(4):288–300. doi: 10.1111/omi.12171
161. Spengler J, Lugonja B, Ytterberg AJ, Zubarev RA, Creese AJ, Pearson MJ, et al. Release of active peptidyl arginine deiminases by neutrophils can explain production of extracellular citrullinated autoantigens in rheumatoid arthritis synovial fluid. Arthritis Rheumatol (2015) 67(12):3135–45. doi: 10.1002/art.39313
162. Maresz KJ, Hellvard A, Sroka A, Adamowicz K, Bielecka E, Koziel J, et al. Porphyromonas gingivalis facilitates the development and progression of destructive arthritis through its unique bacterial peptidylarginine deiminase (PAD). PloS Pathog (2013) 9(9):e1003627. doi: 10.1371/journal.ppat.1003627
163. Gioud-Paquet M, Auvinet M, Raffin T, Girard P, Bouvier M, Lejeune E, et al. IgM rheumatoid factor (RF), IgA RF, IgE RF, and IgG RF detected by ELISA in rheumatoid arthritis. Ann Rheum Dis (1987) 46(1):65–71. doi: 10.1136/ard.46.1.65
164. Tasliyurt T, Kisacik B, Uzun Kaya S, Yildirim B, Pehlivan Y, Kutluturk F, et al. The frequency of antibodies against cyclic citrullinated peptides and rheumatoid factor in healthy population: a field study of rheumatoid arthritis from northern Turkey. Rheumatol Int (2013) 33(4):939–42. doi: 10.1007/s00296-012-2458-5
165. Gargiulo AV Jr., Robinson J, Toto PD, Gargiulo AW. Identification of rheumatoid factor in periodontal disease. J Periodontol (1982) 53(9):568–77. doi: 10.1902/jop.1982.53.9.568
166. Thé J, Ebersole JL. Rheumatoid factor (RF) distribution in periodontal disease. J Clin Immunol (1991) 11(3):132–42. doi: 10.1007/BF00918681
167. Thé J, Ebersole JL. Rheumatoid factor from periodontitis patients cross-reacts with epitopes on oral bacteria. Oral Dis (1996) 2(4):253–62. doi: 10.1111/j.1601-0825.1996.tb00235.x
168. Bonagura VR, Artandi SE, Davidson A, Randen I, Agostino N, Thompson K, et al. Mapping studies reveal unique epitopes on IgG recognized by rheumatoid arthritis-derived monoclonal rheumatoid factors. J Immunol (1993) 151(7):3840–52.
169. Lacki JK, Porawska W, Mackiewicz U, Mackiewicz S, Müller W. Changes in agalactosyl IgG levels correlate with radiological progression in early rheumatoid arthritis. Ann Med (1996) 28(3):265–9. doi: 10.3109/07853899609033129
170. Haraldsson G, Meurman JH, Könönen E, Holbrook WP. Properties of hemagglutination by prevotella melaninogenica. Anaerobe (2005) 11(5):285–9. doi: 10.1016/j.anaerobe.2005.02.004
171. Belton CM, Goodwin PC, Fatherazi S, Schubert MM, Lamont RJ, Izutsu KT. Calcium oscillations in gingival epithelial cells infected with porphyromonas gingivalis. Microbes Infect (2004) 6(5):440–7. doi: 10.1016/j.micinf.2004.01.007
172. Izutsu KT, Belton CM, Chan A, Fatherazi S, Kanter JP, Park Y, et al. Involvement of calcium in interactions between gingival epithelial cells and porphyromonas gingivalis. FEMS Microbiol Lett (1996) 144(2-3):145–50. doi: 10.1111/j.1574-6968.1996.tb08521.x
173. Suzuki K, Sawada T, Murakami A, Matsui T, Tohma S, Nakazono K, et al. High diagnostic performance of ELISA detection of antibodies to citrullinated antigens in rheumatoid arthritis. Scand J Rheumatol (2003) 32(4):197–204. doi: 10.1080/03009740310003677
174. Schellekens GA, Visser H, de Jong BA, van den Hoogen FH, Hazes JM, Breedveld FC, et al. The diagnostic properties of rheumatoid arthritis antibodies recognizing a cyclic citrullinated peptide. Arthritis Rheum (2000) 43(1):155–63. doi: 10.1002/1529-0131(200001)43:1<155::AID-ANR20>3.0.CO;2-3
175. Terao C, Asai K, Hashimoto M, Yamazaki T, Ohmura K, Yamaguchi A, et al. Significant association of periodontal disease with anti-citrullinated peptide antibody in a Japanese healthy population – the nagahama study. J Autoimmun (2015) 59:85–90. doi: 10.1016/j.jaut.2015.03.002
176. Ma KS-K, Chiang C-H, Chen Y-W, Wang L-T. Correspondence to 'Bacterial citrullinated epitopes generated by Porphyromonas gingivalis infection-a missing link for ACPA production'. Ann Rheum Dis (2021), annrheumdis–2020-219255. doi: 10.1136/annrheumdis-2020-219255
177. Konig MF, Giles JT, Nigrovic PA, Andrade F. Antibodies to native and citrullinated RA33 (hnRNP A2/B1) challenge citrullination as the inciting principle underlying loss of tolerance in rheumatoid arthritis. Ann Rheum Dis (2016) 75(11):2022–28. doi: 10.1136/annrheumdis-2015-208529
178. Klareskog L, Amara K, Malmström V. Adaptive immunity in rheumatoid arthritis: anticitrulline and other antibodies in the pathogenesis of rheumatoid arthritis. Curr Opin Rheumatol (2014) 26(1):72–9. doi: 10.1097/BOR.0000000000000016
179. Wang Z, Nicholls SJ, Rodriguez ER, Kummu O, Hörkkö S, Barnard J, et al. Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nat Med (2007) 13(10):1176–84. doi: 10.1038/nm1637
180. Shi J, Knevel R, Suwannalai P, van der Linden MP, Janssen GMC, van Veelen PA, et al. Autoantibodies recognizing carbamylated proteins are present in sera of patients with rheumatoid arthritis and predict joint damage. Proc Natl Acad Sci USA (2011) 108(42):17372–7. doi: 10.1073/pnas.1114465108
181. Leppilahti JM, Hernández-Ríos PA, Gamonal JA, Tervahartiala T, Brignardello-Petersen R, Mantyla P, et al. Matrix metalloproteinases and myeloperoxidase in gingival crevicular fluid provide site-specific diagnostic value for chronic periodontitis. J Clin Periodontol (2014) 41(4):348–56. doi: 10.1111/jcpe.12223
182. Bright R, Proudman SM, Rosenstein ED, Bartold PM. Is there a link between carbamylation and citrullination in periodontal disease and rheumatoid arthritis? Med Hypotheses (2015) 84(6):570–6. doi: 10.1016/j.mehy.2015.03.006
183. Jiang X, Trouw LA, van Wesemael TJ, Shi J, Bengtsson C, Källberg H, et al. Anti-CarP antibodies in two large cohorts of patients with rheumatoid arthritis and their relationship to genetic risk factors, cigarette smoking and other autoantibodies. Ann Rheum Dis (2014) 73(10):1761–8. doi: 10.1136/annrheumdis-2013-205109
184. Thiele GM, Duryee MJ, Anderson DR, Klassen LW, Mohring SM, Young KA, et al. Malondialdehyde-acetaldehyde adducts and anti-malondialdehyde-acetaldehyde antibodies in rheumatoid arthritis. Arthritis Rheumatol (2015) 67(3):645–55. doi: 10.1002/art.38969
185. Darrah E, Andrade F. Editorial: citrullination, and carbamylation, and malondialdehyde-acetaldehyde! oh my! entering the forest of autoantigen modifications in rheumatoid arthritis. Arthritis Rheumatol (2015) 67(3):604–8. doi: 10.1002/art.38970
186. Cheng Z, Meade J, Mankia K, Emery P, Devine DA. Periodontal disease and periodontal bacteria as triggers for rheumatoid arthritis. Best Pract Res Clin Rheumatol (2017) 31(1):19–30. doi: 10.1016/j.berh.2017.08.001
187. Lundberg K, Kinloch A, Fisher BA, Wegner N, Wait R, Charles P, et al. Antibodies to citrullinated α-enolase peptide 1 are specific for rheumatoid arthritis and cross-react with bacterial enolase. Arthritis Rheum (2008) 58(10):3009–19. doi: 10.1002/art.23936
188. Quirke A-M, Lugli EB, Wegner N, Hamilton BC, Charles P, Chowdhury M, et al. Heightened immune response to autocitrullinated porphyromonas gingivalis peptidylarginine deiminase: a potential mechanism for breaching immunologic tolerance in rheumatoid arthritis. Ann Rheum Dis (2014) 73(1):263–9. doi: 10.1136/annrheumdis-2012-202726
189. Konig MF, Bingham 3CO, Andrade F. PPAD is not targeted as a citrullinated protein in rheumatoid arthritis, but remains a candidate for inducing autoimmunity. Ann Rheum Dis (2015) 74(1):e8. doi: 10.1136/annrheumdis-2014-206681
190. Shimada A, Kobayashi T, Ito S, Okada M, Murasawa A, Nakazono K, et al. Expression of anti-porphyromonas gingivalis peptidylarginine deiminase immunoglobulin G and peptidylarginine deiminase-4 in patients with rheumatoid arthritis and periodontitis. J Periodontal Res (2016) 51(1):103–11. doi: 10.1111/jre.12288
191. de Moel EC, Derksen VFAM, Stoeken G, Trouw LA, Bang H, Goekoop RJ, et al. Baseline autoantibody profile in rheumatoid arthritis is associated with early treatment response but not long-term outcomes. Arthritis Res Ther (2018) 20(1):33. doi: 10.1186/s13075-018-1520-4
192. Chen C, Wang T, Chen W. Occurrence of aggregatibacter actinomycetemcomitans serotypes in subgingival plaque from united states subjects. Mol Oral Microbiol (2010) 25(3):207–14. doi: 10.1111/j.2041-1014.2010.00567.x
193. Takasaki K, Fujise O, Miura M, Hamachi T, Maeda K. Porphyromonas gingivalis displays a competitive advantage over Aggregatibacter actinomycetemcomitans in co-cultured biofilm. J Periodontal Res (2013) 48(3):286–92. doi: 10.1111/jre.12006
194. Haraguchi A, Miura M, Fujise O, Hamachi T, Nishimura F. Porphyromonas gingivalis gingipain is involved in the detachment and aggregation of Aggregatibacter actinomycetemcomitans biofilm. Mol Oral Microbiol (2014) 29(3):131–43. doi: 10.1111/omi.12051
195. Johansson A, Hänström L, Kalfas S. Inhibition of actinobacillus actinomycetemcomitans leukotoxicity by bacteria from the subgingival flora. Oral Microbiol Immunol (2000) 15(4):218–25. doi: 10.1034/j.1399-302x.2000.150402.x
196. Yoshida A, Nakano Y, Yamashita Y, Oho T, Ito H, Kondo M, et al. Immunodominant region of actinobacillus actinomycetemcomitans 40-kilodalton heat shock protein in patients with rheumatoid arthritis. J Dent Res (2001) 80(1):346–50. doi: 10.1177/00220345010800010901
197. Ando T, Kato T, Ishihara K, Ogiuchi H, Okuda K. Heat shock proteins in the human periodontal disease process. Microbiol Immunol (1995) 39(5):321–7. doi: 10.1111/j.1348-0421.1995.tb02208.x
198. Schett G, Redlich K, Xu Q, Bizan P, Gröger M, Tohidast-Akrad M, et al. Enhanced expression of heat shock protein 70 (hsp70) and heat shock factor 1 (HSF1) activation in rheumatoid arthritis synovial tissue. differential regulation of hsp70 expression and hsf1 activation in synovial fibroblasts by proinflammatory cytokines, shear stress, and antiinflammatory drugs. J Clin Invest (1998) 102(2):302–11. doi: 10.1172/JCI2465
199. Konig MF, Andrade F. A critical reappraisal of neutrophil extracellular traps and NETosis mimics based on differential requirements for protein citrullination. Front Immunol (2016) 7:461. doi: 10.3389/fimmu.2016.00461
200. Armstrong CL, Miralda I, Neff AC, Tian S, Vashishta A, Perez L, et al. Filifactor alocis promotes neutrophil degranulation and chemotactic activity. Infect Immun (2016) 84(12):3423–33. doi: 10.1128/IAI.00496-16
201. White PC, Chicca IJ, Cooper PR, Milward MR, Chapple ILC. Neutrophil extracellular traps in periodontitis: A web of intrigue. J Dent Res (2016) 95(1):26–34. doi: 10.1177/0022034515609097
202. Warrington KJ, Takemura S, Goronzy JJ, Weyand CM. CD4+,CD28- T cells in rheumatoid arthritis patients combine features of the innate and adaptive immune systems. Arthritis Rheum (2001) 44(1):13–20. doi: 10.1002/1529-0131(200101)44:1<13::AID-ANR3>3.0.CO;2-6
203. Dolhain RJ, van der Heiden AN, ter Haar NT, Breedveld FC, Miltenburg AM. Shift toward T lymphocytes with a T helper 1 cytokine-secretion profile in the joints of patients with rheumatoid arthritis. Arthritis Rheum (1996) 39(12):1961–9. doi: 10.1002/art.1780391204
204. Firestein GS. Immunologic mechanisms in the pathogenesis of rheumatoid arthritis. J Clin Rheumatol (2005) 11(3 Suppl):S39–44. doi: 10.1097/01.rhu.0000166673.34461.33
205. Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. N Engl J Med (2009) 361(9):888–98. doi: 10.1056/NEJMra0707449
206. Stamp LK, James MJ, Cleland LG. Interleukin-17: the missing link between T-cell accumulation and effector cell actions in rheumatoid arthritis? Immunol Cell Biol (2004) 82(1):1–9. doi: 10.1111/j.1440-1711.2004.01212.x
207. Oda T, Yoshie H, Yamazaki K. Porphyromonas gingivalis antigen preferentially stimulates T cells to express IL-17 but not receptor activator of NF-kappaB ligand in vitro. Oral Microbiol Immunol (2003) 18(1):30–6. doi: 10.1034/j.1399-302x.2003.180105.x
208. Berglundh T, Donati M. Aspects of adaptive host response in periodontitis. J Clin Periodontol (2005) 32 Suppl 6:87–107. doi: 10.1111/j.1600-051X.2005.00820.x
209. Cutler CW, Jotwani R, Palucka KA, Davoust J, Bell D, Banchereau J. Evidence and a novel hypothesis for the role of dendritic cells and porphyromonas gingivalis in adult periodontitis. J Periodontal Res (1999) 34(7):406–12. doi: 10.1111/j.1600-0765.1999.tb02274.x
Keywords: periodontitis, rheumatoid arthritis, oral microbiome, citrullination, ACPA, autoimmune disease, infection
Citation: Krutyhołowa A, Strzelec K, Dziedzic A, Bereta GP, Łazarz-Bartyzel K, Potempa J and Gawron K (2022) Host and bacterial factors linking periodontitis and rheumatoid arthritis. Front. Immunol. 13:980805. doi: 10.3389/fimmu.2022.980805
Received: 28 June 2022; Accepted: 27 July 2022;
Published: 25 August 2022.
Edited by:
James Cheng-Chung Wei, Chung Shan Medical University Hospital, TaiwanReviewed by:
Kevin Sheng-Kai Ma, University of Pennsylvania, United StatesAnders Johansson, Umeå University, Sweden
Copyright © 2022 Krutyhołowa, Strzelec, Dziedzic, Bereta, Łazarz-Bartyzel, Potempa and Gawron. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katarzyna Gawron, a2dhd3JvbkBzdW0uZWR1LnBs; Jan Potempa, amFuLnBvdGVtcGFAdWouZWR1LnBs
†ORCID: Katarzyna Gawron, orcid.org/0000-0003-3430-6270
Jan Potempa, orcid.org/0000-0002-3600-7461