
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 06 October 2022
Sec. Inflammation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.980189
 Mohammad Reza Hatamnejad1‡
Mohammad Reza Hatamnejad1‡ Shaghayegh Baradaran Ghavami1*†‡
Shaghayegh Baradaran Ghavami1*†‡ Marzieh Shirvani2
Marzieh Shirvani2 Mona Asghari Ahmadabad3
Mona Asghari Ahmadabad3 Shabnam Shahrokh1*†‡
Shabnam Shahrokh1*†‡ Maryam Farmani1Ghazal Sherkat4
Maryam Farmani1Ghazal Sherkat4 Hamid Asadzadeh Aghdaei1Mohammad Reza Zali5
Hamid Asadzadeh Aghdaei1Mohammad Reza Zali5IBD, a chronic inflammatory disease, has been manifested as a growing health problem. No Crohn’s and Colitis councils have officially ratified anti-depressants as a routine regimen for IBD patients. However, some physicians empirically prescribe them to rectify functional bowel consequences such as pain and alleviate psychiatric comorbidities. On the other side, SSRIs’ prescription is accompanied by adverse effects such as sleep disturbances. Prolonged intermittent hypoxia throughout sleep disturbance such as sleep apnea provokes periodic reductions in the partial oxygen pressure gradient in the gut lumen. It promotes gut microbiota to dysbiosis, which induces intestinal inflammation. This phenomenon and evidence representing the higher amount of serotonin associated with Crohn’s disease challenged our previous knowledge. Can SSRIs worsen the IBD course? Evidence answered the question with the claim on anti-inflammatory properties (central and peripheral) of SSRIs and illuminated the other substantial elements (compared to serotonin elevation) responsible for IBD pathogenesis. However, later clinical evidence was not all in favor of the benefits of SSRIs. Hence, in this review, the molecular mechanisms and clinical evidence are scrutinized and integrated to clarify the interfering molecular mechanism justifying both supporting and disproving clinical evidence. Biphasic dose-dependent serotonin behavior accompanying SSRI shifting function when used up for the long-term can be assumed as the parameters leading to IBD patients’ adverse outcomes. Despite more research being needed to elucidate the effect of SSRI consumption in IBD patients, periodic prescriptions of SSRIs at monthly intervals can be recommended.
● Tissue-available dosage can be assumed as Serotonin’s affinity determinant in binding with receptors to represent the stimulatory or inhibitory functions.
● Complications associated with long-term SSRI use, such as dysbiosis, overwhelm its anti-inflammatory properties and stimulate the gut toward inflammation.
● Periodic prescription at monthly intervals is recommended to inhibit the biphasic dose-dependent serotonin behavior and adverse effects of long-term SSRI consumption.
One of the most prevalent manifestations of chronic inflammation is Inflammatory Bowel Disease (IBD), which principally comprises Crohn’s Disease (CD) and Ulcerative Colitis (UC). IBD affects the colon, small intestine, or both and is characterized by chronic recurrent bowel ulceration (1). The IBD pathogenesis likely involves the complex interaction between genetic, environmental, and immunological factors resulting in an upsetting and exaggerated intestinal inflammatory response to intestinal microbiota in vulnerable individuals (2, 3). A significant part of disability and malfunctioning that occurs in chronic health problems is associated with psychological disorders (4).
Psychological disorders can affect the symptomatic disease courses and increase inflammation. Anxiety and depression are known as comorbidities in IBD patients (5). They are related to the Hypothalamic-Pituitary-Adrenal (HPA) axis activation and increased circulating cortisol levels. Lengthened activation of the HPA axis, as occurs in prolonged stress or chronic inflammation, including IBD, causes chronic cortisol level elevation, leading to reduced sensitivity of glucocorticoid receptors. Reduced glucocorticoid receptor sensitivity can enhance immunological responses and augment inflammation (6, 7). On the other hand, remedies with corticosteroids can induce psychiatric symptoms (7).
The findings declare that the depression prevalence is between 15% and 25%, with possibly lower rates in UC patients than in those who suffer from CD (8, 9). Anxiety is even more rampant, with rates of nearly 30% in IBD patients (10). The rates of anxiety and depression have been higher pending periods of disease flare-up (11). Interestingly almost three-fourths of anti-depressant medications are prescribed without companioning psychiatric indications (12). Approximately 30% of IBD sufferers administrate anti-depressant medications for mental health, bowel symptoms, or both (13).
Despite the NICE guideline, which suggests taking anti-depressants for the long term when depression is accompanied by a chronic physical ailment such as IBD or cancer (14), none of the Crohn’s and Colitis councils have officially ratified anti-depressants as a routine regimen for IBD patients. However, some physicians empirically prescribe them for two aims (15). First, they recommend anti-depressants to rectify functional bowel consequences such as pain (16). A qualitative study appraising the use of anti-depressants for IBD patients demonstrated that most gastroenterology specialists (78%) had treated the patients for symptoms palliation with anti-depressants as adjunctive therapy, especially for pain (17). Second, they claim anti-depressants profitability since they can ameliorate the psychiatric comorbidities in IBD (18).
However, findings regarding the direct benefit of anti-depressants on IBD, regardless of their impact on psychiatric comorbidities, are restricted, and existing ones are controversial, especially with current investigations. Hence, in this review, the molecular mechanisms and clinical evidence are scrutinized and integrated to clarify the proper decision-making about Selective Serotonin Reuptake Inhibitor (SSRI) prescribing. This study focused on SSRIs among all the anti-depressant classes since each class’s proficiency differs. SSRIs have been the most common anti-depressants since 1974 (19) when they were introduced, so debating on this group is more necessary. Functions and pathways should be discussed to recognize better what is happening during the SSRI treatment in IBD.
The gut and brain, constructing 95% and 5% of serotonin (5-Hydroxytryptamine or 5-HT), respectively, are considered the primary sources of serotonin in the body. Serotonergic neurons generate serotonin in the central nervous system (CNS); however, this process in the gut is conducted by enterochromaffin (EC) cells and the myenteric nerve plexus (20). Considering that an enormous quantity of serotonin is attributed to the EC cells, 5-HT metabolism and homeostasis are narrated with a focus on EC cells (Figure 1).

Figure 1 Serotonin Metabolism. Tryptophan intracellular transport is carried out by an L-amino acid transporter; then, it turns to 5-hydroxytryptophan, which is facilitated by tryptophan hydroxylase-1. In the second step, aromatic L-amino acid decarboxylase converts 5-hydroxytryptophan into 5-hydroxytryptamine; eventually, serotonin is stored in vesicles by Vesicular Monoamine Transporter. With the influx of Ca2+, serotonin is exocytosed into the extracellular matrix. After affecting the targeted receptors, it is reabsorbed by the serotonin transporter and degraded by Monoamine oxidases to 5-hydroxyindoleacetic acid.
Serotonin is synthesized from the amino acid L-tryptophan (Trp) within two stages. At first, with the indole ring hydroxylation, tryptophan is converted to 5-hydroxytryptophan (5-HTP) (21). This action is mainly catalyzed by tryptophan hydroxylase (TPH), a rate-limiting enzyme identified as two isoforms (22). TPH1 originates in the pineal gland and gut, and TPH2 is located in the brain and serotonergic enteric nerves (23, 24). Within the next stage, amino acid decarboxylation is taken place by tryptophan decarboxylase (25). The final product, 5-HT, is stowed inside the vesicles by the vesicle-associated transporter (21).
By the luminal stimuli, mechano- and chemo-sensitive ion channels on the EC cells are activated, and, with the influx of Ca2+ ions, 5-HT is discharged from the basolateral and apical surfaces of EC cells (26, 27). Intraluminal-released 5-HT can affect gut microbiota’s composition and function (28), whereas serotonin in tissue-surrounding basal space acts as a pleiotropic component. 5-HT exerts diverse operations in the gastrointestinal (GI) tract through distinct receptors. Serotonin receptors can be divided into seven types (15 subtypes) from 5-HT1 to 5-HT7, which are all families of G-protein-coupled receptors. In contrast, 5-HT3 is a member of the nicotinic/Gamma-aminobutyric acid (GABAA) family of Na+/K+ channel proteins (29, 30). Except for 5-HT5 and 5-HT6 receptors, other classes can be found in the GI tissue (31). Mucosal secretion is adjusted with the effect of serotonin on epithelial 5-HTR2 and neuronal 5-HTR1P, 5-HTR3, and 5-HTR4 receptors (32). Besides, the impact of 5-HT3R and 5-HT4R agonists to reinforce muscular peristalsis has illuminated the motor regulatory role of 5-HT (26). Platelets’ serotonin transporter (SERT) can pick up released extracellular serotonin. 5-HT is a component of the clotting process (31); thus, platelets are the blood serotonin reservoirs that liberate the 5-HT to develop vasoconstriction during bleeding (33).
After all effects on the enterocytes, myenteric neurons, and leukocytes in the surrounding tissue and catching up by platelets remain accessible 5-HT should be deactivated. Serotonin is re-uptaken by the SERT, and monoamine oxidase incepts a process in which 5-HT eventually turns into 5-hydroxy indoleacetic acid (34). SERT exists on both enterocytes and presynaptic neurons; For this reason, to terminate the serotonin function, expunction can be performed within either EC cells-surrounding basal space or synaptic cleft (31, 35). Here is where exactly SSRIs act and, with the serotonin re-uptaking blockage, 5-HT consequences are continued (36).
GI upset can be a common concern of individuals with IBD and is reported with many anti-depressant medications. These side effects are dose-dependent and tend to decrease over the first weeks of treatment. As a symptom of IBD, diarrhea was reported more often with sertraline than with the rest of the SSRIs medicines (7).
Mesalazine [5-aminosalicylic acid (5-ASA)] has been used for a long time to treat IBD. It is an effective, safe, well-tolerated remedy for mild to moderate UC (37, 38). SSRI use per self has been associated with an increased risk of bleeding, particularly during the administration’s first month. The inhibition of SERT by SSRIs is supposed to be responsible for the risk of bleeding. Platelets release serotonin at the bleeding site. They do not synthesize 5-HT but acquire it from the blood and store it (39). In this way, SSRIs may also worsen the bleeding caused by ASA, the backbone of Mesalazine. The inhibition of cytochrome P450 by SSRIs has also been associated with an increased risk of drug interaction, exacerbating the bleeding (40). This risk may be increased by continuous synchronous use of other medications associated with an increased risk of bleeding (41).
Different adverse effects have been identified with SSRIs, including sleep disturbance, sexual dysfunction, and weight gain. The most troubling ones are seen during long-term SSRI medication (42). Sleep Apnea (SA), as a sleep disturbance category, is linked with many comorbidities, leading to generally augmented morbidity. The negative effect of sleep-disordered breathing and its connection with some comorbidities are supposed to be secondary to the recurrent hypoxia and sleep destruction that characterize this disorder (43).
A concept that seems to be interconnected to numerous ailments is the gut microbiome. Human microbiota refers to the broad and thorough gathering of microbes living in an organ such as the gut. Changes in gut microbiota have been presented as critical in developing some chronic diseases (44). Simultaneously, changes in the gut microbiome are related to SA, implying that the gut microbiome is an ordinary player in SA. Prolonged intermittent hypoxia (IH) throughout sleep provokes periodic reductions in the partial oxygen pressure (PO2) gradient in the gut lumen, promoting changes in the relative plenty of aerobic bacteria alongside the rise of amplified obligate and facultative anaerobes (45).
Moreover, such conversion can be achieved through the direct impact of SSRIs on the gut luminal bacterium (46, 47). SSRIs can exert antimicrobial activity and affect bacterium community diversity, especially considering they are taken for long durations (48, 49). One theory proposed for SSRIs antimicrobial relics is that they may undergo passive diffusion across the phospholipid membrane more efficiently, allowing interaction with cellular machinery (50). Also, SSRIs have been shown to inhibit microbial efflux pumps, which contribute to antimicrobial resistance and include competitive and non-competitive inhibition and clogging of the pump’s external pore (51). In addition, Diviccaro et al. have recently displayed that commencing or withdrawing paroxetine (a class of SSRI) treatment can directly alter the gut microbiota composition (52). However, for SSRIs to affect microbiota diversity, they would need to be present in the gut lumen at sufficient concentrations, especially in the microbial-enriched regions such as the ascending colon (53).
Pieces of evidence struggle to identify the possible mechanisms through which gut dysbiosis contributes to many comorbid medical circumstances such as IBD. Some bacterial species, within the normal gut microbiota composition, display mucin-degrading features and, at the same time, do the fermentation of nutritional fibers. This leads to short-chain fatty acids (SCFA) production, which are vital nutrients and energy sources for the colon epithelium (54). Gut dysbiosis reduces butyrate and acetate levels, depriving the epithelium of needed nutrients, which can cause epithelium dysfunction (55–57). In addition, IH, which represents recurrent reoxygenation cycles, induces epithelium damage (58). These perturbations cause disorders in the tight junctions among the intestinal epithelial cells (59). As one of the manifestations of alteration in gut microbiota baseline composition, toxin-producing bacteria may be increased. Endotoxins produced by these germs can efficiently translocate through the damaged gut epithelium into the systemic circulation and cause a state of systemic inflammation (Figure 3) (60). Moreover, a subsidiary duty of butyrate as the regulator of T-cells differentiation is affected by nutrition poverty, promoting inflammation (61).
With dysbiosis, the expression of genes contributing to oxidative stress mechanisms will be enhanced (62). Sulfate-reducing bacteria level elevation in UC results in hydrogen sulfate production, which can cause intestinal mucosal inflammation due to cytotoxic features (63). Other dysbiosis-mediated IBD elements are linked with Trp metabolism (Figure 3). Trp metabolism with three distinct pathways touches the immune system. Indole derivatives manufactured by the aryl hydrocarbon (Ahr) signaling pathway modulate immune balance (e.g., regulation of killer T-cells) (64). Kynurenine (Kyn) secondary production due to the Kyn pathway exhibits inflammation regulation responses (65). Serotonin, the final component of 5-HT signaling, has a crucial role in immune reactions (66), which will be explained in the subsequent section.
Abnormal regulation of 5-HT in the human gut has been implicated with various GI disorders, such as IBD (67, 68). Pieces of evidence showed that 5-HT was increased in CD (69, 70) and serum serotonin enhanced with SSRIs; therefore, our former perception of SSRIs’ advantages comes across some questions. Can SSRIs worsen the IBD course by inducing SA or providing a high amount of 5-HT? Despite these detrimental effects of SSRIs in IBD patients, for which purposes are they prescribed for these individuals?
Investigators formerly believed that serotonin-dependent mechanisms constitute a small part of IBD pathogenesis, but other substantial elements are responsible for it. Thus, an increase in 5-HT content due to SSRIs therapy cannot be mainly related to IBD. On the other hand, relieving IBD symptoms boosts the patients’ tolerance (16, 71), eliminating psychiatric comorbidities and reducing relapse rates (11, 72). The direct anti-inflammatory trace of SSRIs (73, 74) encouraged physicians to prescribe SSRIs.
The discovery of the inflammation role in the pathogenesis of depression proposed that anti-depressants may participate in inflammatory mechanisms and immune system regulation (75). A meaningful correlation between psychological stress and increased inflammation affirmed the mood disorders-inflammation association (76). A significant decrease in serum C-reactive protein concentrations four weeks after initiating SSRIs in people with major depressive disorder (77) corroborated their immune-regulatory properties. SSRIs do not significantly impact inflammatory mechanisms, but other factors modulate the complex interaction between SSRIs and inflammation (78). Although both anti-inflammatory (73, 74, 79, 80) and pro-inflammatory (75, 81–84) effects of SSRIs were reported, prescribing them seems to be based on their anti-inflammatory nature.
The anti-inflammatory effects of SSRIs can be categorized into two classes based on the mechanism’s mediators: CNS-mediated mechanisms, represented by microglial cells, and peripheral mechanisms, facilitated by tissue-habitant or circulating immune-system cells (Figure 2).
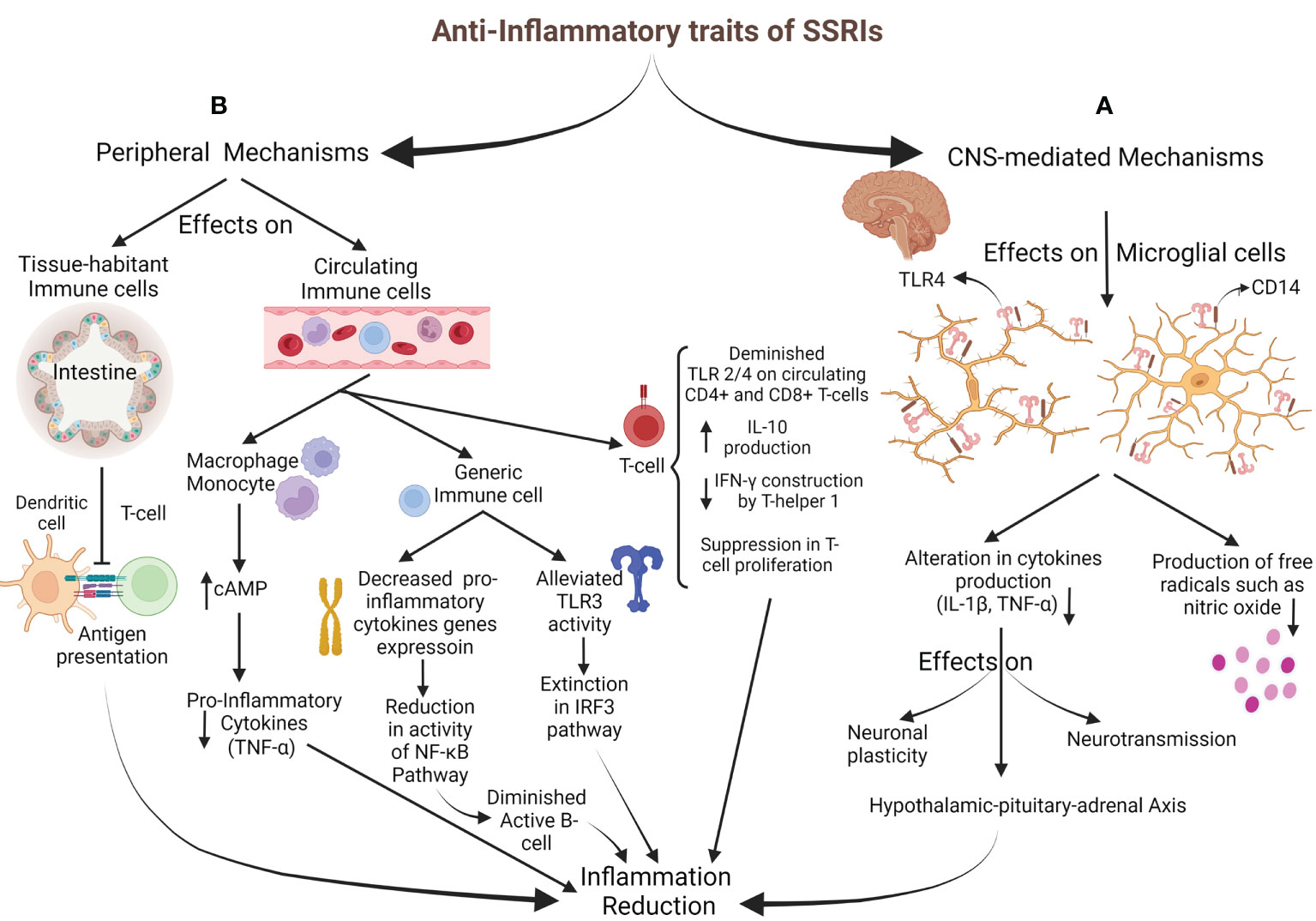
Figure 2 Anti-Inflammatory properties of SSRIs. (A) CNS-mediated mechanisms: SSRIs target microglial cells to impact through TLR4 and CD14, resulting in decreased inflammatory cytokines generation (IL-1ß and TNF-α) and reduced free radicals production. HPA-axis activation via alteration in cytokine network comes up with inflammation decrement. (B) Peripheral mechanisms: T-cell activation is inhibited by diminished antigen presentation of intestine-habitant DC. Circulating immune cells are also impressed; increasing cAMP in monocytes and macrophages brings about inflammatory cytokines drop. Generic immune cells were targeted through decreased pro-inflammatory cytokine gene expression, followed by subsidence in the NF-κB pathway activity and B-cell activation. Also, alleviating TLR3 activity with the inhibition of the IRF3 pathway leads to an inflammation drop. SSRIs directly affect T-cell by reducing TLR2/4 expression, a decline in IFN-γ generation, a rise in IL-10 production, and disturbance in their proliferation.
Microglial cells are the targets for SSRIs and Serotonin and norepinephrine reuptake inhibitors in CNS-mediated mechanisms to apply their anti-inflammatory impresses (84). They are essential in inflammatory regulation and primary inflammatory response (85, 86). The glial cells mainly express Toll-Like Receptor (TLR) 4 and CD14. TLRs mediate immune responses to exogenous and endogenous stimulations and are also vital regulators for neuroimmune reactions caused by stress and major depression. CD14 plays a co-receptor role for TLR4 when inflammatory responses release inflammatory factors like tumor necrosis factor-α (TNF-α) and interleukin (IL)-1ß (87–90). Different cytokines (such as TNF-α and IL-1ß) are released by activated glial cells to influence neurotransmission, HPA activity, neuronal plasticity, and neurogenesis. Previous studies have suggested that anti-depressant drugs act through altered cytokine networks (91). SSRIs have been shown to influence microglia’s capability to produce pro-inflammatory cytokines and free radicals, including nitric oxide (82, 92–94). In their conventional pharmacological dosage, Tynan et al. reported that SSRIs (fluoxetine, sertraline, paroxetine, fluvoxamine, and citalopram) with the impression of TLR decreased TNF-α production by microglia. However, they have established that SSRIs were moderate pro-inflammatory agents when used for lengthened periods at low doses (95).
The peripheral mechanisms mainly influence the immune system’s regulation and inflammatory cytokine production. A recent study in multiple sclerosis patients with major depression reported that the expression of TLR2 and TLR4 on circulating CD4+ and CD8+ T-cells decreased with SSRI therapy, reducing cytokine production (96). In addition, medications such as sertraline may debar the inflammation via the TLR3-IRF3 pathway (97). So, SSRI therapy in the stressful condition is associated with anti-inflammatory responses (98–100). Another suggested SSRI anti-inflammatory process is mediated by their possible effects on decreasing nuclear factor kappa-light-chain-enhancer of activated B cell (NF-κB) activity (101), which is induced by the expression of pro-inflammatory cytokine genes (14, 102). The other suggested mechanism is through the cyclic adenosine monophosphate (cAMP). Vetulani et al. declared that anti-depressants could increase intracellular cAMP (103) in microglia and macrophages, alleviating pro-inflammatory cytokines (104, 105). Another anti-inflammatory mechanism is the inhibition of interferon-gamma (IFN-γ) production by T helper 1. Also, SSRIs suppress mitogenic-stimulated T-cell proliferation and increase IL-10 (a cytokine against inflammation) production by T-cells (81). Diminished expression of inducible co-stimulatory ligand on intestinal dendritic cells (DCs) by fluoxetine down-modulates the antigens presenting from DCs to T-cells and impedes constitutes of inflammatory responses against gut microbiota (106). Stated specificities against the inflammatory processes are serotonin-independent, and the performance of SSRIs via 5-HT will be described in another section. Besides the anti-inflammatory properties of SSRIs, other components can justify their prescription.
The advantage of fundamental science is more when it becomes clear that the outcomes can be generalized to clinical science. Assumed mechanisms about the usefulness of SSRIs should experiment with under clinical conditions (Table 1).
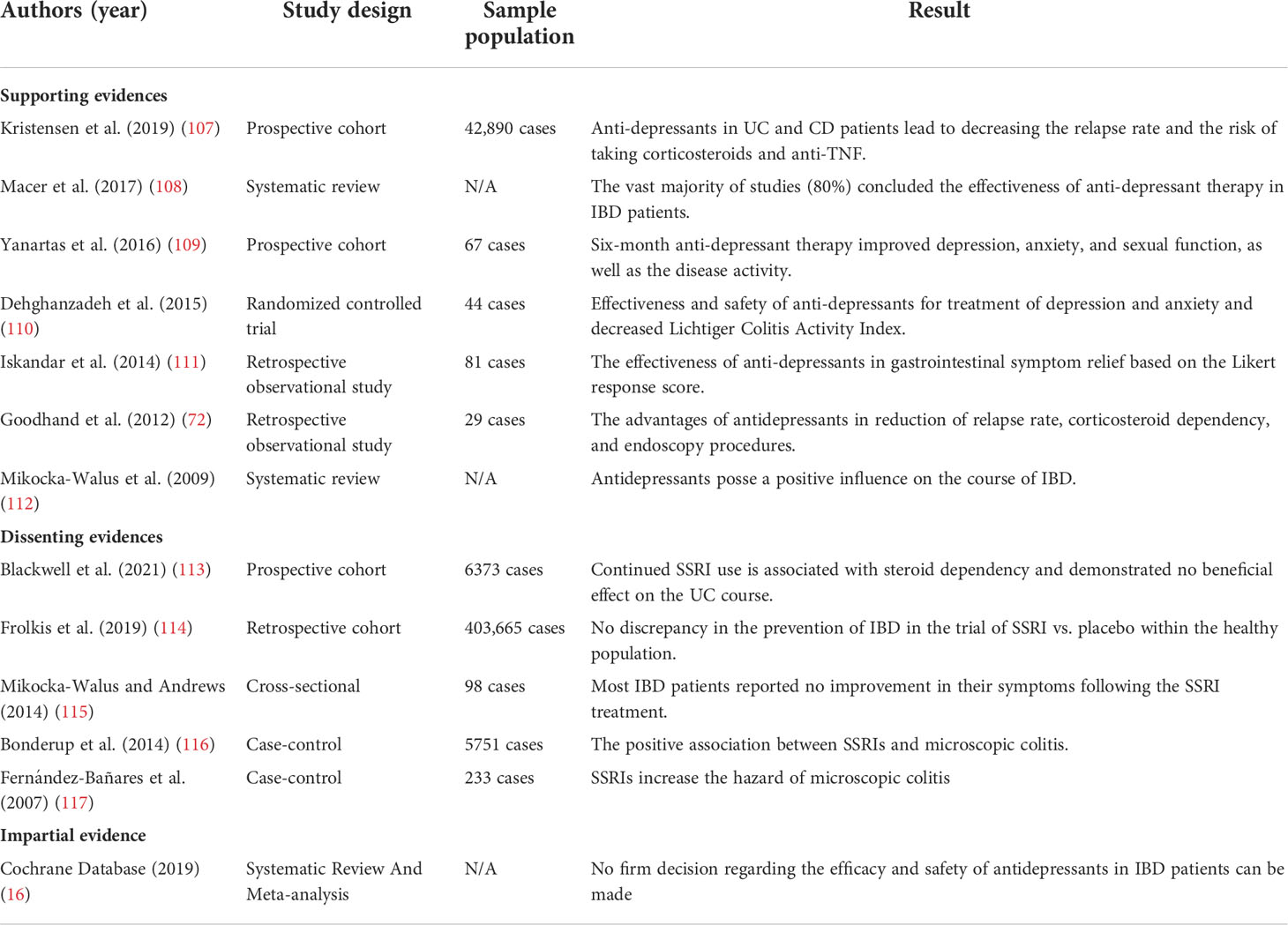
Table 1 Clinical documents of SSRI prescription.
Goodhand et al. described that prescribing anti-depressants (including SSRIs) to remedy concomitant depression in IBD patients not only ameliorates the current course of inflammation but also, by preventing inflammation relapse, diminishes the need for corticosteroid therapy and endoscopy (72). Daghaghzade et al., in a randomized, double-blind controlled clinical trial, demonstrated the significant profit of duloxetine in improving the frequency of diarrhea and severity of symptoms like pain in IBD patients (110). Also, the amending effects of SSRIs on inflammation and symptoms of IBD patients were affirmed by the CD activity index, as illustrated by Yanartas et al. (109). Another prospective cohort study conducted between 2000 and 2017 with around 44,000 subjects indicated a positive aspect of anti-depressant treatment in IBD patients, particularly those with no history of taking anti-depressants before IBD diagnosis. A better influence was found in CD patients compared to UC patients. Thus, this study showed that patients treated with anti-depressants had a significantly lower risk of receiving corticosteroids and anti-TNF medication than patients who did not take anti-depressants (107). In agreement with previous findings, Iskandar et al., in a retrospective cohort investigation, represented that the anti-depressants can appease the severity of IBD patients’ condition, more specifically in UC patients (111) (Table 1).
In a qualitative questionnaire-based online survey by Mikocka-Walus et al., most IBD patients observed no difference with taking the SSRIs, and only 25% declared an enhancement in their clinical presentations (115). Exploring the relationship between SSRIs and inflammation in the colon led to detecting a significant positive association. Fernandez et al. realized that patients with microscopic colitis (both types of lymphocytic and collagenous) possessed a higher rate of taking SSRIs than the control group, particularly in the case of sertraline (117). In a sizeable case-control study with 5,751 microscopic colitis cases based on nationwide Danish registries, Bonderup et al. figured out a positive association between SSRI exposure and microscopic colitis (116). In a retrospective cohort study conducted from 1986 to 2012 on 400,000 patients with new-onset depression, SSRIs were indicated as protective agents for UC and CD development in depressed patients. However, uncertainty was raised since they had found no discrepancy in IBD development in the trial of SSRI vs. placebo among normal individuals. SSRIs cannot provide their protective role when consumed due to any indication except depression (prevention of IBD in the healthy population) (114).
Despite documents about SSRIs’ advantages in IBD patients being controversial, comprehensive systematic reviews announced that SSRIs are beneficial for IBD courses as the conclusion (108, 112). Certainty about prescribing SSRIs for IBD management existed until 2021 when Blackwell et al. declared that continued administration of SSRIs or tricyclic antidepressants (TCA) is a red flag for IBD patients. They indicated that continuous consumption of SSRIs or TCAs is correlated with corticosteroid dependency and worse clinical outcomes in the future (113). The power of their study, comprising a large sample size (over 6,000 participants with UC) and long-term follow-up (between 2005 and 2016), challenged our previous belief about SSRIs. Also, this prospective cohort study reinforces the conclusion of a Cochrane systematic review in 2019 that declared no firm decision regarding the profits of SSRIs (16). How could these consequences be justified with all our knowledge about SSRIs? Basic science answers.
Previous evidence made the point clear that desired outcomes of prescribing SSRIs are free from their effects on 5-HT and rely on their immune-regulatory properties; nevertheless, subsequent clinical proof made us doubtful about the utility of these medications. Why should these adverse consequences appear in IBD patients after years of SSRI consumption? We hypothesized that an abundant amount of serotonin resulting from continuous consumption of SSRIs with the effect on immune system induction might overwhelm the anti-inflammatory SSRIs’ features. We need to scrutinize the exact interactions between 5-HT and the immune system for this theory (Figure 3).
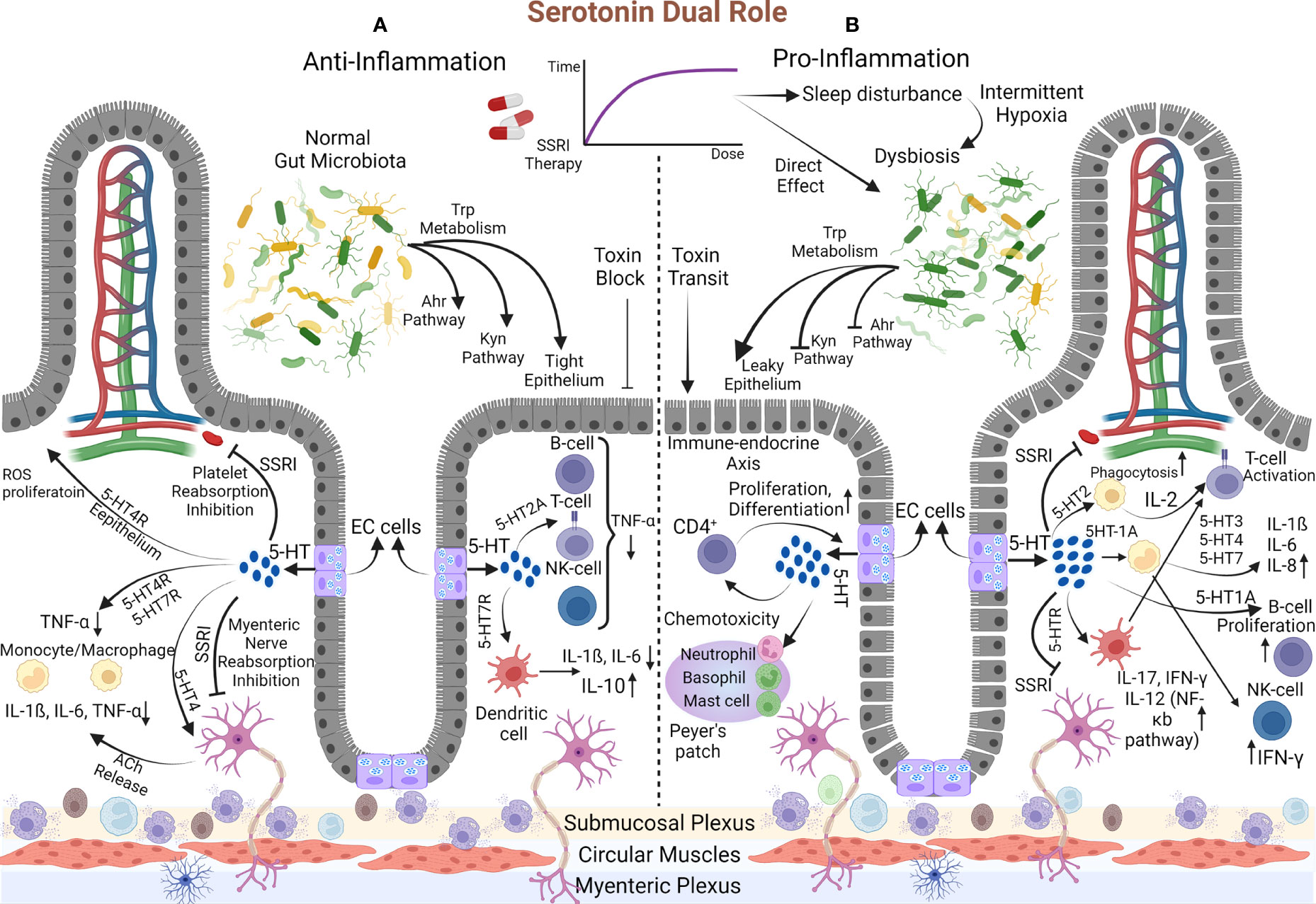
Figure 3 The dual demeanor of Serotonin. (A) Anti-Inflammatory: The gut microbiota enter the Trp into Ahr and Kyn pathways and the 5-HT synthesis process. Initially, SSRIs inhibit 5-HT reabsorption in platelets and myenteric nerves and boost mucosal serotonin content, initiating anti-inflammatory mechanisms. Proliferation and resistance to ROS arise in the epithelium via the 5-HT4 receptor. 5-HT reduces TNF-α production by monocytes and macrophages (1) directly through their 5-HT4 and 5-HT7 receptors and (2) indirectly via the 5-HT4 receptor on the myenteric nerve, which is accompanied by releasing Ach that eventually comes up with IL-1ß, IL-6, and TNF-α decrement. Similar changes in TNF-α fabrication occur via B-cell, T-cell, and NK cell 5-HT2 receptors. Additionally, 5-HT7 on the DC turns down the IL-6 and IL-1ß and elevates IL-10. (B) Pro-inflammatory: Over time, SSRI dosage augmentation affects, directly and indirectly, the gut microbiota composition, and disrupts Ahr and Kyn pathways. Pathogens’ toxins pass the leaky epithelium created by dysbiosis and stimulate the gut toward inflammation. Moreover, the inhibition of 5-HT reabsorption is amplified, resulting in much more serotonin levels. This primes pro-inflammatory mechanisms, which are known as the endocrine-immune axis. Macrophage phagocytosis is reinforced, and through the 5-HT2 receptor, they release IL-2 that activates T-cells. In addition, by acting on DC, 5-HT activates T-cells and increases IL-17, IL-12, and IFN-γ via the NF-κB pathway. Monocyte 5-HT1A receptor provocation reduces the inhibitory effect on NK-Cell to increase IFN-γ production. On the other hand, the impacts on 5-HT3, 5-HT4, and 5-HT7 monocyte receptors increase IL-6, IL-8, and INF-1ß. Also, B-Cell proliferates and activates through 5-HT1A receptor instigation. The serotonin amount can boost neutrophil, basophil, and mast cell chemo-toxicity. This process is turned into a destructive cycle since increased T-cells (CD4+ types) initiate the immune-endocrine axis (EC cells proliferation and boosting the serotonin content). Eventually, the destructive cycle exacerbates the inflammation.
Recognizing serotonin as a pivotal component in the immune system began with discovering serotonergic receptors on immune cells in 1982 (118, 119). Since then, some immune system functions have been attributed to serotonin, recognized as the endocrine-immune axis. Serotonin can enhance macrophage phagocytosis (120), increase DC, basophil, neutrophil, and mast cell chemotaxis (121), and regulate cytokine production (122, 123). Lately, it was elucidated that monocytes, macrophages, and T-cells not only can be assumed as peripheral sources of 5-HT (124, 125) but also can induce the immune-endocrine axis by affecting EC cells. Likewise, Wang et al., in a model to discover the manner of the immune-endocrine axis, compared the amount of 5-HT production between wild-type mice and severe combined immunocompromised mice in the setting of contamination with the same Trichuris muris infection. They revealed new interactions between CD4+ T cells with EC cells to enhance the generation of 5-HT in the gut via Th2-based mechanisms (126). Also, detecting the IL-13 receptor on EC cells reinforced previous findings (126).
Despite the cyclic interaction between the immune-endocrine and endocrine-immune axis, serotonin can eventually be defined as an immune system stimulatory or inhibitory modulator (Figure 3).
As a stimulatory constituent, 5-HT actuates the molecular mechanisms toward inflammation. The proliferation of T-cells and B-cells is mediated by 5-HT2 and 5-HT1A receptors, respectively (127, 128), whereas T-cells are activated via their 5-HT7 receptors (129). Treated mice by P-chlorophenyl alanine (inhibitory molecule of TPH-1) indicated that with the effect of serotonin on macrophage 5-HT2 receptors, an accessory pathway to activate CD4+ cells by releasing IL-2 is initiated (130). Serotonin binding to the 5-HT1A receptor on monocytes makes these cells less efficient in suppressing NK cells that are typically inhibited. Thus, cytotoxicity and IFN-γ production will be augmented based on NK cell activity (131, 132). Later, evidence of enhancing the NK cell proliferation and their cytosolic functions was obtained by trials of SSRI medications (133). Another study declared that serotonin directly improves NK cell function while some dopamine/serotonin antagonists suppress the CD16-mediated activity of NK cells (134). Durk et al. demonstrated that the augmentations of IL-1ß, IL-6, IL-12p40, and IL-8/CXCL8 cytokines are 5-HT3, 5-HT4, and 5-HT7 monocyte receptor-mediated (135). Serotonin by activating DCs motivates CD4+ T-cells to generate IL-17 and IFN-γ cytokines (136). Studies on disease-specific cytokine patterns have elucidated the type of CD4+ T-cells differentiation and cytokine production. Th1 and Th17, with their manufactured cytokines (IFN-γ, IL-17, and IL-22), are assumed to be related to CD pathogenicity. Meanwhile, Th2-like differentiation with increased natural killer T-cells producing IL-13 is associated with UC (137).
The inhibitory role of releasing serotonin to decrease inflammation has been portrayed in other investigations. In an asthma model, the 5-HT2A receptor on eosinophil exerts its anti-inflammatory function by preventing the recruitment to the inflammatory site (138). Suppression of IFN-γ-inducing macrophage phagocytosis through 5-HT occurs at the high concentration of IFN-γ (at the physiological dose of IFN-γ, it provides stimulatory phagocytes) (139). Inhibition of TNF-α production by peripheral blood mononuclear cells was illustrated by 5-HT2A excitation (140). As another piece of evidence, TNF-α decrement was authenticated by 5-HT4 and 5-HT7 monocyte receptors instigation (135). In an in-vitro rat model, mosapride (5-HT4 agonist) administration enhanced acetylcholine (Ach) release from myenteric neurons, resulting in macrophages/monocytes Ach receptors activation. The expression of cytokines’ (IL-1ß, IL-6, and TNF-α) mRNA decreased in addition to immune cell recruitment suppression (141).
The dual role of serotonin not only can be exerted in the GI system but also can affect other organs such as the vasculature. Vascular smooth muscle cells synthesize IL-6, possibly inducing atherogenicity of vessels in response to serotonin (142). Furthermore, downregulating the expression of pro-inflammatory genes and preventing the TNF-α-mediated inflammatory pathways are obtained by selective activation of the 5-HT2A receptor on aortic smooth muscle cells (143).
The accumulative evidence pointed to the fact that performing the stimulatory or inhibitory role of 5-HT depends on the receptor and the cell to which it attaches. The 5-HT4 receptor on epithelial cells is responsible for the proliferation, resistance to Reactive Oxygen Species (ROS)-induced apoptosis, and eventually anti-inflammatory effect (based on barrier function improvement) (144). In contrast, in colitis mice, IL-6 and IL-1ß (responsible for inflammation) excessed with the administration of intraperitoneal 5-HT and its binding to the 5-HT4 receptor on immune cells (145). In addition to cell type, the receptor is another role-determining element. Activation of 5-HT7 receptors on antigen-presenting DCs and lipopolysaccharide-stimulated macrophages, likely with the cytokine production adjustment, diminishes inflammation severity (146). In contrast, by implicating the other receptor on the same cell (DC), serotonin leads to the elevation of IL-12 (through the NF-κB pathway), IL-17, IFN-γ, and ultimately the deterioration of inflammation (136).
No literature has discussed the serotonin’s affinity determinant factors in binding to receptors to represent the stimulatory or inhibitory function. It is now obscure that with the dissemination of 5-HT, why it operates excitatory and does not exert its prohibitory action, or conversely. A theory that may justify this phenomenon is dose-dependent serotonin behavior. This theory’s origin dates back to when Kubera et al. (95) discovered a dose-dependent binary 5-HT function in cytokine production. They believed cytokine production (IL-6 and TNF-α) by macrophages and lymphocytes needs low-level serotonin, whereas a high dose of serotonin reduces these cytokines; however, in our literature review, more convincing evidence illustrated a direct correlation between serotonin dose and stimulatory action. Tolerance induced by the SSRIs’ long-term use, giving rise to consumption dose increment. A high amount of available serotonin primes its stimulatory demeanor and steers the gut toward inflammation. This process, accompanied by the SSRIs’ dose-dependent functional alteration (shifting to pro-inflammatory performance when applied for long periods (74)), exacerbates the IBD patients’ condition.
The hypothesis of inhibitory or excitatory dose-dependent serotonin role was extended, one more time, by the serotonin-gut microbiota axis in which a lower amount of mucosal 5-HT with direct and indirect (via producing the antimicrobial peptides, specifically ß-defensins) effects on gut microbiota leads to reducing pro-inflammatory cytokines, enhancing the epithelial barrier function, and eventually prohibiting gut inflammation, and vice versa (147).
The theory of SSRI intake’s role in setting up an oxidative stress response has been debated (148, 149). Therefore, SSRI-induced oxidative stress can cause cellular damage and release endogenous ligands like adenosine triphosphate. These particles have been termed damage-associated molecular patterns (DAMPs), which are recognized by the pattern recognition receptors (PRRs) on immune cells like DC cells (150, 151). The role of PRR signaling in instigating the intestinal immune cells against the microbiota and inducing inflammation has been illuminated (152). Therefore, the act of SSRIs to cause the immune response, dysbiosis, and IBD through the PRRs can fortify the previous hypothesis.
Numerous studies have claimed SSRIs’ benefits due to their anti-inflammatory properties and psychiatric comorbidities treatment. In contrast, biphasic dose-dependent serotonin behavior accompanying SSRI shifting function, when used up for the long-term, can be assumed as the reason for IBD patients’ adverse outcomes. Despite more trials and cohorts being needed to illuminate the exact effect of long-term SSRIs consumption in IBD patients, periodic prescriptions of SSRIs at monthly intervals can be recommended.
MZ, HA, and SS conceived the idea for the manuscript and, in cooperation with MH and SG, refined the latest theory. MH, MS, MA, and GS carried out the data mining and literature review. MH, MS, MA, and MF drafted the manuscript. MH, MS, and SG revised the manuscript. MH drew the figures. MZ, HA, and SS supervised the article preparation. All authors read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
IBD, Inflammatory bowel disease; CD, Crohn’s disease; UC, Ulcerative colitis; HPA, Hypothalamic-pituitary-adrenal; SSRI, Selective Serotonin Reuptake Inhibitors; 5-HT, 5-Hydroxytryptamine; CNS, central nervous system; EC, Enterochromaffin; Trp, Tryptophan; TPH, Tryptophan hydroxylase; GI: Gastrointestinal; GABA, Gamma-aminobutyric acid; SERT, Serotonin transporter; 5-ASA, 5-Aminosalicylic acid; SA, Sleep apnea; IH, Intermittent hypoxia; SCFA, Short-chain fatty acids; TLR, Toll-Like Receptor; TNF, Tumor Necrosis Factor; IL, Interleukin; NF-kB, Nuclear factor kappa-light-chain-enhancer of activated B cell; cAMP, cyclic Adenosine monophosphate; IFN-γ, Interferon-gamma; Ahr, Aryl hydrocarbon; Kyn, kynurenine; TCA, Tricyclic Antidepressant; DC, Dendritic Cell; Ach, acetylcholine; ROS, reactive oxygen species.
1. Ananthakrishnan AN. Epidemiology and risk factors for ibd. Nat Rev Gastroenterol Hepatol (2015) 12(4):205–17. doi: 10.1038/nrgastro.2015.34
2. Ko JK, Auyeung KK. Inflammatory bowel disease: Etiology, pathogenesis and current therapy. Curr Pharm design (2014) 20(7):1082–96. doi: 10.2174/13816128113199990416
3. Manichanh C, Borruel N, Casellas F, Guarner F. The gut microbiota in ibd. Nat Rev Gastroenterol Hepatol (2012) 9(10):599–608. doi: 10.1038/nrgastro.2012.152
4. Bernstein CN. Psychological stress and depression: Risk factors for ibd? Dig Dis (2016) 34(1-2):58–63. doi: 10.1159/000442929
5. Mikocka-Walus AA, Turnbull DA, Andrews JM, Moulding NT, Holtmann GJ. The effect of functional gastrointestinal disorders on psychological comorbidity and quality of life in patients with inflammatory bowel disease. Aliment Pharmacol Ther (2008) 28(4):475–83. doi: 10.1111/j.1365-2036.2008.03754.x
6. Graeff FG, Zangrossi H. The hypothalamic-Pituitary-Adrenal axis in anxiety and panic. Psychol Neurosci (2010) 3(1):3–8. doi: 10.3922/j.psns.2010.1.002
7. Graff LA, Walker JR, Bernstein CN. Depression and anxiety in inflammatory bowel disease: A review of comorbidity and management. Inflammatory bowel Dis (2009) 15(7):1105–18. doi: 10.1002/ibd.20873
8. Zhang CK, Hewett J, Hemming J, Grant T, Zhao H, Abraham C, et al. The influence of depression on quality of life in patients with inflammatory bowel disease. Inflammation Bowel Dis (2013) 19(8):1732–9. doi: 10.1097/MIB.0b013e318281f395
9. Ananthakrishnan AN, Gainer VS, Cai T, Perez RG, Cheng SC, Savova G, et al. Similar risk of depression and anxiety following surgery or hospitalization for crohn’s disease and ulcerative colitis. Am J Gastroenterol (2013) 108(4):594–601. doi: 10.1038/ajg.2012.471
10. Fuller-Thomson E, Lateef R, Sulman J. Robust association between inflammatory bowel disease and generalized anxiety disorder: Findings from a nationally representative Canadian study. Inflammation Bowel Dis (2015) 21(10):2341–8. doi: 10.1097/mib.0000000000000518
11. Bernstein CN, Singh S, Graff LA, Walker JR, Miller N, Cheang M. A prospective population-based study of triggers of symptomatic flares in ibd. Am J Gastroenterol (2010) 105(9):1994–2002. doi: 10.1038/ajg.2010.140
12. Mojtabai R, Olfson M. Proportion of antidepressants prescribed without a psychiatric diagnosis is growing. Health Aff (Millwood) (2011) 30(8):1434–42. doi: 10.1377/hlthaff.2010.1024
13. Buckley JP, Kappelman MD, Allen JK, Van Meter SA, Cook SF. The burden of comedication among patients with inflammatory bowel disease. Inflammatory Bowel Dis (2013) 19(13):2725–36. doi: 10.1097/01.MIB.0000435442.07237.a4%JInflammatoryBowelDiseases
14. National Collaborating Centre for Mental H. National institute for health and clinical excellence: Guidance. In: Depression: The treatment and management of depression in adults (Updated edition) vol. 2010. Leicester (UK: British Psychological Society Copyright © The British Psychological Society & The Royal College of Psychiatrists (2010).
15. Thorkelson G, Bielefeldt K, Szigethy E. Empirically supported use of psychiatric medications in adolescents and adults with ibd. Inflammatory bowel Dis (2016) 22(6):1509–22. doi: 10.1097/mib.0000000000000734
16. Mikocka-Walus A, Prady SL, Pollok J, Esterman AJ, Gordon AL, Knowles S, et al. Adjuvant therapy with antidepressants for the management of inflammatory bowel disease. Cochrane Database Syst Rev (2019) 4(4):Cd012680. doi: 10.1002/14651858.CD012680.pub2
17. Mikocka-Walus AA, Turnbull DA, Moulding NT, Wilson IG, Andrews JM, Holtmann GJ. “It doesn’t do any harm, but patients feel better”: A qualitative exploratory study on gastroenterologists’ perspectives on the role of antidepressants in inflammatory bowel disease. BMC Gastroenterol (2007) 7:38. doi: 10.1186/1471-230x-7-38
18. Bernstein CN, Hitchon CA, Walld R, Bolton JM, Lix LM, El-Gabalawy R, et al. The impact of psychiatric comorbidity on health care utilization in inflammatory bowel disease: A population-based study. Inflammatory Bowel Dis (2020) 27(9):1462–74. doi: 10.1093/ibd/izaa310%JInflammatoryBowelDiseases
19. Hillhouse TM, Porter JH. A brief history of the development of antidepressant drugs: From monoamines to glutamate. Exp Clin Psychopharmacol (2015) 23(1):1–21. doi: 10.1037/a0038550
20. Jonnakuty C, Gragnoli C. What do we know about serotonin? J Cell Physiol (2008) 217(2):301–6. doi: 10.1002/jcp.21533
21. Bertrand PP, Bertrand RL. Serotonin release and uptake in the gastrointestinal tract. Autonomic neuroscience: basic Clin (2010) 153(1-2):47–57. doi: 10.1016/j.autneu.2009.08.002
22. Chivite M, Leal E, Míguez JM, Cerdá-Reverter JM. Distribution of two isoforms of tryptophan hydroxylase in the brain of rainbow trout (Oncorhynchus mykiss). an in situ hybridization study. Brain Struct Funct (2021) 226(7):2265–78. doi: 10.1007/s00429-021-02322-8
23. Côté F, Thévenot E, Fligny C, Fromes Y, Darmon M, Ripoche MA, et al. Disruption of the nonneuronal Tph1 gene demonstrates the importance of peripheral serotonin in cardiac function. Proc Natl Acad Sci U.S.A. (2003) 100(23):13525–30. doi: 10.1073/pnas.2233056100
24. Coyle D, Murphy JM, Doyle B, O’Donnell AM, Gillick J, Puri P. Altered tryptophan hydroxylase 2 expression in enteric serotonergic nerves in hirschsprung’s-associated enterocolitis. World J Gastroenterol (2016) 22(19):4662–72. doi: 10.3748/wjg.v22.i19.4662
25. Park S, Kang K, Lee SW, Ahn M-J, Bae J-M, Back K. Production of serotonin by dual expression of tryptophan decarboxylase and tryptamine 5-hydroxylase in escherichia coli. Appl Microbiol Biotechnol (2011) 89(5):1387–94. doi: 10.1007/s00253-010-2994-4
26. Banskota S, Ghia JE, Khan WI. Serotonin in the gut: Blessing or a curse. Biochimie (2019) 161:56–64. doi: 10.1016/j.biochi.2018.06.008
27. Kumar A, Russell RM, Pifer R, Menezes-Garcia Z, Cuesta S, Narayanan S, et al. The serotonin neurotransmitter modulates virulence of enteric pathogens. Cell Host Microbe (2020) 28(1):41–53.e8. doi: 10.1016/j.chom.2020.05.004
28. Sun LJ, Li JN, Nie YZ. Gut hormones in microbiota-Gut-Brain cross-talk. Chin Med J (2020) 133(7):826–33. doi: 10.1097/cm9.0000000000000706
29. Qi YX, Xia RY, Wu YS, Stanley D, Huang J, Ye GY. Larvae of the small white butterfly, pieris rapae, express a novel serotonin receptor. J Neurochem (2014) 131(6):767–77. doi: 10.1111/jnc.12940
30. Goetz T, Arslan A, Wisden W, Wulff P. Gaba(a) receptors: Structure and function in the basal ganglia. Prog Brain Res (2007) 160:21–41. doi: 10.1016/s0079-6123(06)60003-4
31. Mawe GM, Hoffman JM. Serotonin signalling in the gut–functions, dysfunctions and therapeutic targets. Nat Rev Gastroenterol Hepatol (2013) 10(8):473–86. doi: 10.1038/nrgastro.2013.105
32. Fidalgo S, Ivanov DK, Wood SH. Serotonin: From top to bottom. Biogerontology (2013) 14(1):21–45. doi: 10.1007/s10522-012-9406-3
33. de Abajo FJ. Effects of selective serotonin reuptake inhibitors on platelet function: Mechanisms, clinical outcomes and implications for use in elderly patients. Drugs Aging (2011) 28(5):345–67. doi: 10.2165/11589340-000000000-00000
34. Deacon AC. The measurement of 5-hydroxyindoleacetic acid in urine. Ann Clin Biochem (1994) 31(Pt 3):215–32. doi: 10.1177/000456329403100302
35. Nagatsu T. [Molecular mechanisms of neurotransmission]. Rinsho Shinkeigaku (2000) 40(12):1185–8.
36. Apparsundaram S, Stockdale DJ, Henningsen RA, Milla ME, Martin RS. Antidepressants targeting the serotonin reuptake transporter act Via a competitive mechanism. J Pharmacol Exp Ther (2008) 327(3):982–90. doi: 10.1124/jpet.108.142315
37. Pithadia AB, Jain S. Treatment of inflammatory bowel disease (Ibd). Pharmacol Rep (2011) 63(3):629–42. doi: 10.1016/S1734-1140(11)70575-8
38. Williams C, Panaccione R, Ghosh S, Rioux K. Optimizing clinical use of mesalazine (5-aminosalicylic acid) in inflammatory bowel disease. Therap Adv Gastroenterol (2011) 4(4):237–48. doi: 10.1177/1756283x11405250
39. Halperin D, Reber G. Influence of antidepressants on hemostasis. Dialogues Clin Neurosci (2007) 9(1):47–59. doi: 10.31887/DCNS.2007.9.1/dhalperin
40. Walsky RL, Astuccio AV, Obach RS. Evaluation of 227 drugs for in vitro inhibition of cytochrome P450 2b6. J Clin Pharmacol (2006) 46(12):1426–38. doi: 10.1177/0091270006293753
41. Labos C, Dasgupta K, Nedjar H, Turecki G, Rahme E. Risk of bleeding associated with combined use of selective serotonin reuptake inhibitors and antiplatelet therapy following acute myocardial infarction. Cmaj (2011) 183(16):1835–43. doi: 10.1503/cmaj.100912
42. Ferguson JM. Ssri antidepressant medications: Adverse effects and tolerability. Prim Care Companion J Clin Psychiatry (2001) 3(1):22–7. doi: 10.4088/pcc.v03n0105
43. Mashaqi S, Gozal D. Obstructive sleep apnea and systemic hypertension: Gut dysbiosis as the mediator? J Clin Sleep Med (2019) 15(10):1517–27. doi: 10.5664/jcsm.7990
44. Kamada N, Seo S-U, Chen GY, Núñez GJ. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol (2013) 13(5):321–35. doi: 10.1038/nri3430
45. Albenberg L, Esipova TV, Judge CP, Bittinger K, Chen J, Laughlin A, et al. Correlation between intraluminal oxygen gradient and radial partitioning of intestinal microbiota. Gastroenterology (2014) 147(5):1055–63. doi: 10.1053/j.gastro.2014.07.020
46. Lyte M, Daniels KM, Schmitz-Esser S. Fluoxetine-induced alteration of murine gut microbial community structure: Evidence for a microbial endocrinology-based mechanism of action responsible for fluoxetine-induced side effects. PeerJ (2019) 7:e6199. doi: 10.7717/peerj.6199
47. Lukić I, Getselter D, Ziv O, Oron O, Reuveni E, Koren O, et al. Antidepressants affect gut microbiota and ruminococcus flavefaciens is able to abolish their effects on depressive-like behavior. Trans Psychiatry (2019) 9(1):133. doi: 10.1038/s41398-019-0466-x
48. Cussotto S, Strain CR, Fouhy F, Strain RG, Peterson VL, Clarke G, et al. Differential effects of psychotropic drugs on microbiome composition and gastrointestinal function. Psychopharmacology (2019) 236(5):1671–85. doi: 10.1007/s00213-018-5006-5
49. Ait Chait Y, Mottawea W, Tompkins TA, Hammami R. Unravelling the antimicrobial action of antidepressants on gut commensal microbes. Sci Rep (2020) 10(1):17878. doi: 10.1038/s41598-020-74934-9
50. Hancock REW, Bell A eds. Antibiotic uptake into gram-negative bacteria. Berlin, Heidelberg: Springer Berlin Heidelberg (1989).
51. Macedo D, Filho A, Soares de Sousa CN, Quevedo J, Barichello T, Júnior HVN, et al. Antidepressants, antimicrobials or both? gut microbiota dysbiosis in depression and possible implications of the antimicrobial effects of antidepressant drugs for antidepressant effectiveness. J Affect Disord (2017) 208:22–32. doi: 10.1016/j.jad.2016.09.012
52. Diviccaro S, Giatti S, Cioffi L, Falvo E, Piazza R, Caruso D, et al. Paroxetine effects in adult Male rat colon: Focus on gut steroidogenesis and microbiota. Psychoneuroendocrinology (2022) 143:105828. doi: 10.1016/j.psyneuen.2022.105828
53. Yano Jessica M, Yu K, Donaldson Gregory P, Shastri Gauri G, Ann P, Ma L, et al. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell (2015) 161(2):264–76. doi: 10.1016/j.cell.2015.02.047
54. Yuan H, Zhou J, Li N, Wu X, Huang S, Park S. Isolation and identification of mucin-degrading bacteria originated from human faeces and their potential probiotic efficacy according to host-microbiome enterotype. J Appl Microbiol (2022) 133(2):362–74. doi: 10.1111/jam.15560
55. Takaishi H, Matsuki T, Nakazawa A, Takada T, Kado S, Asahara T, et al. Imbalance in intestinal microflora constitution could be involved in the pathogenesis of inflammatory bowel disease. Int J Med Microbiol (2008) 298(5-6):463–72. doi: 10.1016/j.ijmm.2007.07.016
56. Canani RB, Di Costanzo M, Leone L, Pedata M, Meli R, Calignano A. Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World J Gastroenterol (2011) 17(12):1519. doi: 10.3748/wjg.v17.i12.1519
57. Vinolo MA, Rodrigues HG, Nachbar RT, Curi R. Regulation of inflammation by short chain fatty acids. Nutrients (2011) 3(10):858–76. doi: 10.3390/nu3100858
58. Taylor CT, Colgan SP. Hypoxia and gastrointestinal disease. J Mol Med (Berl) (2007) 85(12):1295–300. doi: 10.1007/s00109-007-0277-z
59. Moreno-Indias I, Torres M, Sanchez-Alcoholado L, Cardona F, Almendros I, Gozal D, et al. Normoxic recovery mimicking treatment of sleep apnea does not reverse intermittent hypoxia-induced bacterial dysbiosis and low-grade endotoxemia in mice. Sleep (2016) 39(10):1891–7. doi: 10.5665/sleep.6176
60. Poroyko VA, Carreras A, Khalyfa A, Khalyfa AA, Leone V, Peris E, et al. Chronic sleep disruption alters gut microbiota, induces systemic and adipose tissue inflammation and insulin resistance in mice. Sci Rep (2016) 6:35405. doi: 10.1038/srep35405
61. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature (2013) 504(7480):446–50. doi: 10.1038/nature12721
62. Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol (2012) 13(9):R79. doi: 10.1186/gb-2012-13-9-r79
63. Pitcher MC, Beatty ER, Cummings JH. The contribution of sulphate reducing bacteria and 5-aminosalicylic acid to faecal sulphide in patients with ulcerative colitis. Gut (2000) 46(1):64–72. doi: 10.1136/gut.46.1.64
64. Zhang YJ, Reddy MC, Ioerger TR, Rothchild AC, Dartois V, Schuster BM, et al. Tryptophan biosynthesis protects mycobacteria from Cd4 T-Cell-Mediated killing. Cell (2013) 155(6):1296–308. doi: 10.1016/j.cell.2013.10.045
65. Galligan JJ. Beneficial actions of microbiota-derived tryptophan metabolites. Neurogastroenterol Motil (2018) 30(2):e13283. doi: 10.1111/nmo.13283
66. Herr N, Bode C, Duerschmied D. The effects of serotonin in immune cells. Front Cardiovasc Med (2017) 4:48. doi: 10.3389/fcvm.2017.00048
67. Magro F, Vieira-Coelho MA, Fraga S, Serrão MP, Veloso FT, Ribeiro T, et al. Impaired synthesis or cellular storage of norepinephrine, dopamine, and 5-hydroxytryptamine in human inflammatory bowel disease. Dig Dis Sci (2002) 47(1):216–24. doi: 10.1023/a:1013256629600
68. Coates MD, Mahoney CR, Linden DR, Sampson JE, Chen J, Blaszyk H, et al. Molecular defects in mucosal serotonin content and decreased serotonin reuptake transporter in ulcerative colitis and irritable bowel syndrome. Gastroenterology (2004) 126(7):1657–64. doi: 10.1053/j.gastro.2004.03.013
69. Manocha M, Khan WI. Serotonin and gi disorders: An update on clinical and experimental studies. Clin Transl Gastroenterol (2012) 3(4):e13. doi: 10.1038/ctg.2012.8
70. Kidd M, Gustafsson BI, Drozdov I, Modlin IM. Il1beta- and lps-induced serotonin secretion is increased in ec cells derived from crohn’s disease. Neurogastroenterol Motil (2009) 21(4):439–50. doi: 10.1111/j.1365-2982.2008.01210.x
71. DiMatteo MR, Lepper HS, Croghan TW. Depression is a risk factor for noncompliance with medical treatment: Meta-analysis of the effects of anxiety and depression on patient adherence. Arch Intern Med (2000) 160(14):2101–7. doi: 10.1001/archinte.160.14.2101
72. Goodhand JR, Greig FI, Koodun Y, McDermott A, Wahed M, Langmead L, et al. Do antidepressants influence the disease course in inflammatory bowel disease? a retrospective case-matched observational study. Inflammation Bowel Dis (2012) 18(7):1232–9. doi: 10.1002/ibd.21846
73. Bielecka AM, Paul-Samojedny M, Obuchowicz E. Moclobemide exerts anti-inflammatory effect in lipopolysaccharide-activated primary mixed glial cell culture. Naunyn Schmiedebergs Arch Pharmacol (2010) 382(5-6):409–17. doi: 10.1007/s00210-010-0535-4
74. Tynan RJ, Weidenhofer J, Hinwood M, Cairns MJ, Day TA, Walker FR. A comparative examination of the anti-inflammatory effects of ssri and snri antidepressants on lps stimulated microglia. Brain Behavior Immun (2012) 26(3):469–79. doi: 10.1016/j.bbi.2011.12.011
75. Horowitz MA, Wertz J, Zhu D, Cattaneo A, Musaelyan K, Nikkheslat N, et al. Antidepressant compounds can be both pro-and anti-inflammatory in human hippocampal cells. Int J Neuropsychopharmacol (2015) 18(3). doi: 10.1093/ijnp/pyu076
76. Maydych V. The interplay between stress, inflammation, and emotional attention: Relevance for depression. Front Neurosci (2019) 13:384. doi: 10.3389/fnins.2019.00384
77. O’Brien SM, Scott LV, Dinan TG. Antidepressant therapy and c-reactive protein levels. Br J Psychiatry (2006) 188:449–52. doi: 10.1192/bjp.bp.105.011015
78. Kraemer HC, Frank E, Kupfer DJ. Moderators of treatment outcomes: Clinical, research, and policy importance. Jama (2006) 296(10):1286–9. doi: 10.1001/jama.296.10.1286
79. Obuchowicz E, Kowalski J, Labuzek K, Krysiak R, Pendzich J, Herman ZS. Amitriptyline and nortriptyline inhibit interleukin-1 β and tumour necrosis factor-α release by rat mixed glial and microglial cell cultures. Int J Neuropsychopharmacol (2006) 9(1):27–35. doi: 10.1017/S146114570500547X
80. Xia Z, Depierre JW, Nässberger L. Tricyclic antidepressants inhibit il-6, il-1β and tnf-α release in human blood monocytes and il-2 and interferon-Γ in T cells. Immunopharmacology (1996) 34(1):27–37. doi: 10.1016/0162-3109(96)00111-7
81. Diamond M, Kelly JP, Connor TJ. Antidepressants suppress production of the Th1 cytokine interferon-Γ, independent of monoamine transporter blockade. Eur Neuropsychopharmacol (2006) 16(7):481–90. doi: 10.1016/j.euroneuro.2005.11.011
82. Horikawa H, Kato TA, Mizoguchi Y, Monji A, Seki Y, Ohkuri T, et al. Inhibitory effects of ssris on ifn-Γ induced microglial activation through the regulation of intracellular calcium. Prog Neuropsychopharmacol Biol Psychiatry (2010) 34(7):1306–16. doi: 10.1016/j.pnpbp.2010.07.015
83. Kubera M, Maes M, Kenis G, Kim Y-K, Lasoń W. Effects of serotonin and serotonergic agonists and antagonists on the production of tumor necrosis factor α and interleukin-6. Psychiatry Res (2005) 134(3):251–8. doi: 10.1016/j.psychres.2004.01.014
84. Tynan RJ, Weidenhofer J, Hinwood M, Cairns MJ, Day TA, Walker FR. A comparative examination of the anti-inflammatory effects of ssri and snri antidepressants on lps stimulated microglia. Brain Behavior Immun (2012) 26(3):469–79. doi: 10.1016/j.bbi.2011.12.011
85. Bessis A, Béchade C, Bernard D, Roumier A. Microglial control of neuronal death and synaptic properties. Glia (2007) 55(3):233–8. doi: 10.1002/glia.20459
86. Branchi I, Alboni S, Maggi L. The role of microglia in mediating the effect of the environment in brain plasticity and behavior. Front Cell Neurosci (2014) 8:390. doi: 10.3389/fncel.2014.00390
87. Lehnardt S, Massillon L, Follett P, Jensen FE, Ratan R, Rosenberg PA, et al. Activation of innate immunity in the cns triggers neurodegeneration through a toll-like receptor 4-dependent pathway. Proc Natl Acad Sci U.S.A. (2003) 100(14):8514–9. doi: 10.1073/pnas.1432609100
88. Janova H, Böttcher C, Holtman IR, Regen T, van Rossum D, Götz A, et al. Cd14 is a key organizer of microglial responses to cns infection and injury. Glia (2016) 64(4):635–49. doi: 10.1002/glia.22955
89. Trotta T, Porro C, Calvello R, Panaro MA. Biological role of toll-like receptor-4 in the brain. J Neuroimmunol (2014) 268(1-2):1–12. doi: 10.1016/j.jneuroim.2014.01.014
90. Zhou M, Wang CM, Yang WL, Wang P. Microglial Cd14 activated by inos contributes to neuroinflammation in cerebral ischemia. Brain Res (2013) 1506:105–14. doi: 10.1016/j.brainres.2013.02.010
91. Bielecka A, Paul-Samojedny M, Obuchowicz E. Moclobemide exerts anti-inflammatory effect in lipopolysaccharide-activated primary mixed glial cell culture. Naunyn-Schmiedeberg’s Arch Pharmacol (2010) 382(5):409–17. doi: 10.1007/s00210-010-0535-4
92. Hashioka S, Klegeris A, Monji A, Kato T, Sawada M, McGeer PL, et al. Antidepressants inhibit interferon-Γ-Induced microglial production of il-6 and nitric oxide. J Exp Neurol (2007) 206(1):33–42. doi: 10.1016/j.expneurol.2007.03.022
93. Hashioka S, McGeer PL, Monji A, Kanba S. Anti-inflammatory effects of antidepressants: Possibilities for preventives against alzheimer’s disease. Cent Nervous System Agents Medicinal Chem (Formerly Curr Medicinal Chemistry-Central Nervous System Agents) (2009) 9(1):12–9. doi: 10.2174/187152409787601897
94. Hwang J, Zheng LT, Ock J, Lee MG, Kim S-H, Lee H-W, et al. Inhibition of glial inflammatory activation and neurotoxicity by tricyclic antidepressants. Neuropharmacology (2008) 55(5):826–34. doi: 10.1016/j.neuropharm.2008.06.045
95. Kubera M, Maes M, Kenis G, Kim YK, Lasoń W. Effects of serotonin and serotonergic agonists and antagonists on the production of tumor necrosis factor alpha and interleukin-6. Psychiatry Res (2005) 134(3):251–8. doi: 10.1016/j.psychres.2004.01.014
96. Sales MC, Kasahara TM, Sacramento PM, Rossi ÁD, Cafasso M, Oyamada HAA, et al. Selective serotonin reuptake inhibitor attenuates the hyperresponsiveness of Tlr2(+) and Tlr4(+) Th17/Tc17-like cells in multiple sclerosis patients with major depression. Immunology (2021) 162(3):290–305. doi: 10.1111/imm.13281
97. Zhu J, Smith K, Hsieh PN, Mburu YK, Chattopadhyay S, Sen GC, et al. High-throughput screening for Tlr3-ifn regulatory factor 3 signaling pathway modulators identifies several antipsychotic drugs as tlr inhibitors. J Immunol (2010) 184(10):5768–76. doi: 10.4049/jimmunol.0903559
98. Alboni S, Poggini S, Garofalo S, Milior G, El Hajj H, Lecours C, et al. Fluoxetine treatment affects the inflammatory response and microglial function according to the quality of the living environment. Brain Behavior Immun (2016) 58:261–71. doi: 10.1016/j.bbi.2016.07.155
99. Cheng Y, Pardo M, de Souza Armini R, Martinez A, Mouhsine H, Zagury J-F, et al. Stress-induced neuroinflammation is mediated by Gsk3-dependent Tlr4 signaling that promotes susceptibility to depression-like behavior. Brain Behavior Immun (2016) 53:207–22. doi: 10.1016/j.bbi.2015.12.012
100. Wei-Wei J, Rui-Peng L, Meng L, Shu-Yuan W, Zhang X, Xing-Xing N, et al. Antidepressant-like effect of essential oil of perilla frutescens in a chronic, unpredictable, mild stress-induced depression model mice. Chin J Nat Med (2014) 12(10):753–9. doi: 10.1016/S1875-5364(14)60115-1
101. Lim CM, Kim SW, Park JY, Kim C, Yoon SH, Lee JK. Fluoxetine affords robust neuroprotection in the postischemic brain Via its anti-inflammatory effect. J Neurosci Res (2009) 87(4):1037–45. doi: 10.1002/jnr.21899
102. Perkins ND. Integrating cell-signalling pathways with nf-κb and ikk function. Nat Rev Mol Cell Biol (2007) 8(1):49–62. doi: 10.1038/nrm2083
103. Vetulani J, Sulser F. Action of various antidepressant treatments reduces reactivity of noradrenergic cyclic amp-generating system in limbic forebrain. Nature (1975) 257(5526):495–6. doi: 10.1038/257495a0
104. Probert L, Akassoglou K, Pasparakis M, Kontogeorgos G, Kollias G. Spontaneous inflammatory demyelinating disease in transgenic mice showing central nervous system-specific expression of tumor necrosis factor alpha. Proc Natl Acad Sci (1995) 92(24):11294–8. doi: 10.1073/pnas.92.24.11294
105. Yoshikawa M, Suzumura A, Tamaru T, Takayanagi T, Sawada M. Effects of phosphodiesterase inhibitors on cytokine production by microglia. J Mult Scler (Foster City) (1999) 5(2):126–33. doi: 10.1177/135245859900500210
106. Branco-de-Almeida LS, Kajiya M, Cardoso CR, Silva MJ, Ohta K, Rosalen PL, et al. Selective serotonin reuptake inhibitors attenuate the antigen presentation from dendritic cells to effector T lymphocytes. FEMS Immunol Med Microbiol (2011) 62(3):283–94. doi: 10.1111/j.1574-695X.2011.00816.x
107. Kristensen MS, Kjærulff TM, Ersbøll AK, Green A, Hallas J, Thygesen LC. The influence of antidepressants on the disease course among patients with crohn’s disease and ulcerative colitis-a Danish nationwide register-based cohort study. Inflammation Bowel Dis (2019) 25(5):886–93. doi: 10.1093/ibd/izy367
108. Macer BJ, Prady SL, Mikocka-Walus A. Antidepressants in inflammatory bowel disease: A systematic review. Inflammation Bowel Dis (2017) 23(4):534–50. doi: 10.1097/mib.0000000000001059
109. Yanartas O, Kani HT, Bicakci E, Kilic I, Banzragch M, Acikel C, et al. The effects of psychiatric treatment on depression, anxiety, quality of life, and sexual dysfunction in patients with inflammatory bowel disease. Neuropsychiatr Dis Treat (2016) 12:673–83. doi: 10.2147/ndt.S106039
110. Daghaghzadeh H, Naji F, Afshar H, Sharbafchi MR, Feizi A, Maroufi M, et al. Efficacy of duloxetine add on in treatment of inflammatory bowel disease patients: A double-blind controlled study. J Res Med Sci (2015) 20(6):595–601. doi: 10.4103/1735-1995.165969
111. Iskandar HN, Cassell B, Kanuri N, Gyawali CP, Gutierrez A, Dassopoulos T, et al. Tricyclic antidepressants for management of residual symptoms in inflammatory bowel disease. J Clin Gastroenterol (2014) 48(5):423–9. doi: 10.1097/mcg.0000000000000049
112. Mikocka-Walus A, Clarke D, Gibson P. Can antidepressants influence the course of inflammatory bowel disease? the current state of research. Eur Gastroeneterol Hepatol Rev (2009) 5.
113. Blackwell J, Alexakis C, Saxena S, Creese H, Bottle A, Petersen I, et al. Association between antidepressant medication use and steroid dependency in patients with ulcerative colitis: A population-based study. J BMJ Open Gastroenterol (2021) 8(1):e000588. doi: 10.1136/bmjgast-2020-000588
114. Frolkis AD, Vallerand IA, Shaheen AA, Lowerison MW, Swain MG, Barnabe C, et al. Depression increases the risk of inflammatory bowel disease, which may be mitigated by the use of antidepressants in the treatment of depression. Gut (2019) 68(9):1606–12. doi: 10.1136/gutjnl-2018-317182
115. Mikocka-Walus A, Andrews JM. Attitudes towards antidepressants among people living with inflammatory bowel disease: An online Australia-wide survey. J Crohns Colitis (2014) 8(4):296–303. doi: 10.1016/j.crohns.2013.09.002
116. Bonderup OK, Fenger-Grøn M, Wigh T, Pedersen L, Nielsen GL. Drug exposure and risk of microscopic colitis: A nationwide Danish case-control study with 5751 cases. Inflammation Bowel Dis (2014) 20(10):1702–7. doi: 10.1097/mib.0000000000000143
117. Fernández-Bañares F, Esteve M, Espinós JC, Rosinach M, Forné M, Salas A, et al. Drug consumption and the risk of microscopic colitis. Am J Gastroenterol (2007) 102(2):324–30. doi: 10.1111/j.1572-0241.2006.00902.x
118. Cloëz-Tayarani I, Changeux JP. Nicotine and serotonin in immune regulation and inflammatory processes: A perspective. J Leukoc Biol (2007) 81(3):599–606. doi: 10.1189/jlb.0906544
119. Eliseeva LS, Stefanovich LE. [Specific binding of serotonin by blood leukocytes and peritoneal cells in the mouse]. Biokhimiia (1982) 47(5):810–3.
120. Nakamura K, Sato T, Ohashi A, Tsurui H, Hasegawa H. Role of a serotonin precursor in development of gut microvilli. Am J Pathol (2008) 172(2):333–44. doi: 10.2353/ajpath.2008.070358
121. Kushnir-Sukhov NM, Gilfillan AM, Coleman JW, Brown JM, Bruening S, Toth M, et al. 5-hydroxytryptamine induces mast cell adhesion and migration. J Immunol (2006) 177(9):6422–32. doi: 10.4049/jimmunol.177.9.6422
122. Idzko M, Panther E, Stratz C, Müller T, Bayer H, Zissel G, et al. The serotoninergic receptors of human dendritic cells: Identification and coupling to cytokine release. J Immunol (2004) 172(10):6011–9. doi: 10.4049/jimmunol.172.10.6011
123. Müller T, Dürk T, Blumenthal B, Grimm M, Cicko S, Panther E, et al. 5-hydroxytryptamine modulates migration, cytokine and chemokine release and T-cell priming capacity of dendritic cells in vitro and in vivo. PloS One (2009) 4(7):e6453. doi: 10.1371/journal.pone.0006453
124. O’Connell PJ, Wang X, Leon-Ponte M, Griffiths C, Pingle SC, Ahern GP. A novel form of immune signaling revealed by transmission of the inflammatory mediator serotonin between dendritic cells and T cells. Blood (2006) 107(3):1010–7. doi: 10.1182/blood-2005-07-2903
125. Finocchiaro LM, Arzt ES, Fernández-Castelo S, Criscuolo M, Finkielman S, Nahmod VE. Serotonin and melatonin synthesis in peripheral blood mononuclear cells: Stimulation by interferon-gamma as part of an immunomodulatory pathway. J Interferon Res (1988) 8(6):705–16. doi: 10.1089/jir.1988.8.705
126. Wang H, Steeds J, Motomura Y, Deng Y, Verma-Gandhu M, El-Sharkawy RT, et al. Cd4+ T cell-mediated immunological control of enterochromaffin cell hyperplasia and 5-hydroxytryptamine production in enteric infection. Gut (2007) 56(7):949–57. doi: 10.1136/gut.2006.103226
127. Nordlind K, Sundström E, Bondesson L. Inhibiting effects of serotonin antagonists on the proliferation of mercuric chloride stimulated human peripheral blood T lymphocytes. Int Arch Allergy Immunol (1992) 97(2):105–8. doi: 10.1159/000236104
128. Iken K, Chheng S, Fargin A, Goulet AC, Kouassi E. Serotonin upregulates mitogen-stimulated b lymphocyte proliferation through 5-Ht1a receptors. Cell Immunol (1995) 163(1):1–9. doi: 10.1006/cimm.1995.1092
129. León-Ponte M, Ahern GP, O’Connell PJ. Serotonin provides an accessory signal to enhance T-cell activation by signaling through the 5-Ht7 receptor. Blood (2007) 109(8):3139–46. doi: 10.1182/blood-2006-10-052787
130. Young MR, Matthews JP. Serotonin regulation of T-cell subpopulations and of macrophage accessory function. Immunology (1995) 84(1):148–52.
131. Hellstrand K, Hermodsson S. Serotonergic 5-Ht1a receptors regulate a cell contact-mediated interaction between natural killer cells and monocytes. Scand J Immunol (1993) 37(1):7–18. doi: 10.1111/j.1365-3083.1993.tb01658.x
132. Nourian M, Chaleshi V, Pishkar L, Azimzadeh P, Baradaran Ghavami S, Balaii H, et al. Evaluation of tumor necrosis factor (Tnf)-α mrna expression level and the Rs1799964 polymorphism of the tnf-α gene in peripheral mononuclear cells of patients with inflammatory bowel diseases. BioMed Rep (2017) 6(6):698–702. doi: 10.3892/br.2017.908
133. Hernandez ME, Martinez-Fong D, Perez-Tapia M, Estrada-Garcia I, Estrada-Parra S, Pavón L. Evaluation of the effect of selective serotonin-reuptake inhibitors on lymphocyte subsets in patients with a major depressive disorder. Eur Neuropsychopharmacol (2010) 20(2):88–95. doi: 10.1016/j.euroneuro.2009.11.005
134. Theorell J, Gustavsson AL, Tesi B, Sigmundsson K, Ljunggren HG, Lundbäck T, et al. Immunomodulatory activity of commonly used drugs on fc-Receptor-Mediated human natural killer cell activation. Cancer Immunol Immunother (2014) 63(6):627–41. doi: 10.1007/s00262-014-1539-6
135. Dürk T, Panther E, Müller T, Sorichter S, Ferrari D, Pizzirani C, et al. 5-hydroxytryptamine modulates cytokine and chemokine production in lps-primed human monocytes Via stimulation of different 5-htr subtypes. Int Immunol (2005) 17(5):599–606. doi: 10.1093/intimm/dxh242
136. Li N, Ghia JE, Wang H, McClemens J, Cote F, Suehiro Y, et al. Serotonin activates dendritic cell function in the context of gut inflammation. Am J Pathol (2011) 178(2):662–71. doi: 10.1016/j.ajpath.2010.10.028
137. Strober W, Fuss IJ. Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology (2011) 140(6):1756–67. doi: 10.1053/j.gastro.2011.02.016
138. Nau F Jr., Miller J, Saravia J, Ahlert T, Yu B, Happel KI, et al. Serotonin 5-Ht2 receptor activation prevents allergic asthma in a mouse model. Am J Physiol Lung Cell Mol Physiol (2015) 308(2):L191–8. doi: 10.1152/ajplung.00138.2013
139. Sternberg EM, Wedner HJ, Leung MK, Parker CW. Effect of serotonin (5-ht) and other monoamines on murine macrophages: Modulation of interferon-gamma induced phagocytosis. J Immunol (1987) 138(12):4360–5.
140. Cloëz-Tayarani I, Petit-Bertron AF, Venters HD, Cavaillon JM. Differential effect of serotonin on cytokine production in lipopolysaccharide-stimulated human peripheral blood mononuclear cells: Involvement of 5-Hydroxytryptamine2a receptors. Int Immunol (2003) 15(2):233–40. doi: 10.1093/intimm/dxg027
141. Tsuchida Y, Hatao F, Fujisawa M, Murata T, Kaminishi M, Seto Y, et al. Neuronal stimulation with 5-hydroxytryptamine 4 receptor induces anti-inflammatory actions Via α7nach receptors on muscularis macrophages associated with postoperative ileus. Gut (2011) 60(5):638–47. doi: 10.1136/gut.2010.227546
142. Ito T, Ikeda U, Shimpo M, Yamamoto K, Shimada K. Serotonin increases interleukin-6 synthesis in human vascular smooth muscle cells. Circulation (2000) 102(20):2522–7. doi: 10.1161/01.cir.102.20.2522
143. Yu B, Becnel J, Zerfaoui M, Rohatgi R, Boulares AH, Nichols CD. Serotonin 5-Hydroxytryptamine(2a) receptor activation suppresses tumor necrosis factor-Alpha-Induced inflammation with extraordinary potency. J Pharmacol Exp Ther (2008) 327(2):316–23. doi: 10.1124/jpet.108.143461
144. Spohn SN, Bianco F, Scott RB, Keenan CM, Linton AA, O’Neill CH, et al. Protective actions of epithelial 5-hydroxytryptamine 4 receptors in normal and inflamed colon. Gastroenterology (2016) 151(5):933–44.e3. doi: 10.1053/j.gastro.2016.07.032
145. Salaga M, Binienda A, Piscitelli F, Mokrowiecka A, Cygankiewicz AI, Verde R, et al. Corrigendum to “Systemic administration of serotonin exacerbates abdominal pain and colitis Via interaction with the endocannabinoid system” [Biochem. pharmacol. 161 (2019) 37-51]. Biochem Pharmacol (2019) 169:113636. doi: 10.1016/j.bcp.2019.113636
146. Guzel T, Mirowska-Guzel D. The role of serotonin neurotransmission in gastrointestinal tract and pharmacotherapy. Molecules (Basel Switzerland) (2022) 27(5). doi: 10.3390/molecules27051680
147. Kwon YH, Wang H, Denou E, Ghia JE, Rossi L, Fontes ME, et al. Modulation of gut microbiota composition by serotonin signaling influences intestinal immune response and susceptibility to colitis. Cell Mol Gastroenterol Hepatol (2019) 7(4):709–28. doi: 10.1016/j.jcmgh.2019.01.004
148. Battal D, Yalin S, Eker ED, Aktas A, Sahin NO, Cebo M, et al. Possible role of selective serotonin reuptake inhibitor sertraline on oxidative stress responses. Eur Rev Med Pharmacol Sci (2014) 18(4):477–84.
149. Ribaudo G, Bortoli M, Pavan C, Zagotto G, Orian L. Antioxidant potential of psychotropic drugs: From clinical evidence to in vitro and in vivo assessment and toward a new challenge for in silico molecular design. Antioxidants (Basel Switzerland) (2020) 9(8). doi: 10.3390/antiox9080714
150. Franklin TC, Xu C, Duman RS. Depression and sterile inflammation: Essential role of danger associated molecular patterns. Brain Behavior Immun (2018) 72:2–13. doi: 10.1016/j.bbi.2017.10.025
151. Fleshner M, Frank M, Maier SF. Danger signals and inflammasomes: Stress-evoked sterile inflammation in mood disorders. Neuropsychopharmacology (2017) 42(1):36–45. doi: 10.1038/npp.2016.125
Keywords: inflammatory bowel diseases, serotonin uptake inhibitors, pro-inflammatory, anti-inflammatory, Crohn’s disease, ulcerative colitis, antidepressant, dysbiosis
Citation: Hatamnejad MR, Baradaran Ghavami S, Shirvani M, Asghari Ahmadabad M, Shahrokh S, Farmani M, Sherkat G, Asadzadeh Aghdaei H and Zali MR (2022) Selective serotonin reuptake inhibitors and inflammatory bowel disease; Beneficial or malpractice. Front. Immunol. 13:980189. doi: 10.3389/fimmu.2022.980189
Received: 28 June 2022; Accepted: 22 September 2022;
Published: 06 October 2022.
Edited by:
Detlef Neumann, Hannover Medical School, GermanyReviewed by:
Mindy Engevik, Medical University of South Carolina, United StatesCopyright © 2022 Hatamnejad, Baradaran Ghavami, Shirvani, Asghari Ahmadabad, Shahrokh, Farmani, Sherkat, Asadzadeh Aghdaei and Zali. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shaghayegh Baradaran Ghavami, U2guYmdoYXZhbWlAeWFob28uY29t; Shabnam Shahrokh, c2hhYm5hbXNoYWhyb2toQGdtYWlsLmNvbQ==
†These authors have contributed equally to this work
‡ORCID: Mohammad Reza Hatamnejad, https://orcid.org/0000-0002-2239-5539
Shaghayegh Baradaran Ghavami, orcid.org/0000-0003-0359-8653
Shabnam Shahrokh, orcid.org/0000-0002-3420-5608
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.