 Ana V. Marin
Ana V. Marin Anaïs Jiménez-Reinoso
Anaïs Jiménez-Reinoso Marina S. Mazariegos1
Marina S. Mazariegos1 José R. Regueiro
José R. Regueiro- 1Department of Immunology, Ophthalmology and Ear, Nose and Throat (ENT), Complutense University School of Medicine and 12 de Octubre Health Research Institute (imas12), Madrid, Spain
- 2Pediatric Nephrology, Hospital Universitari i Politècnic la Fe, Valencia, Spain
Schimke immuno-osseous dysplasia (SIOD) caused by mutations in SMARCAL1 is an ultra-rare disease characterized by specific facial features, skeletal dysplasia, and steroid-resistant nephrotic syndrome, which often leads to kidney failure and requires transplantation. Cellular (T-cell) deficiency, lymphopenia, and infections have been frequently reported, but whether they are due to T-cell-intrinsic defects in T-cell receptor (TCR) signaling associated with SMARCAL1 deficiency or to T-cell-extrinsic effects such as the impaired proliferation of hematopoietic precursors or T-cell-specific immunosuppression after renal transplantation remains unknown. We have explored the effects of SMARCAL1 deficiency on T-cell receptor signaling in primary and immortalized T cells from a 9-year-old SIOD patient under immunosuppression treatment when compared to healthy donors. Immortalized T cells recapitulated the SMARCAL1 deficiency of the patient, as judged by their impaired response to gamma irradiation. The results indicated that TCR-mediated signaling was normal in SIOD-derived immortalized T cells but strongly impaired in the primary T cells of the patient, although rescued with TCR-independent stimuli such as PMA + ionomycin, suggesting that SIOD-associated T-cell signaling is not intrinsically defective but rather the result of the impaired proliferation of hematopoietic precursors or of T-cell-specific immunosuppression. The lack of early thymic emigrants in our patients may support the former hypothesis.
Introduction
Schimke immuno-osseous dysplasia (SIOD) is a multisystem disorder that is inherited in an autosomal recessive pattern and characterized by the combination of specific facial features, skeletal dysplasia, steroid-resistant nephrotic syndrome, and defective cellular immunity with episodic severe lymphopenia. Less than 100 cases have been reported and its prevalence is 1 in 1–3 million new births, so Orphanet considers it an ultra-rare disease (OMIM: 242900). Early-onset affected patients show severe symptoms and die at around 10 years of age due to strokes, severe opportunistic infections, bone marrow failure, kidney failure, cardiovascular issues, and other complications (1).
SIOD is the consequence of biallelic mutations in SMARCAL1 (SWI/SNF-related Matrix-associated Actin-dependent Regulator of Chromatin, subfamily A-Like 1), also known as HARP or HepA-Related Protein. SMARCAL1 is a member of the Sucrose Non-Fermenting 2 (SNF2) family of ATP-dependent chromatin remodeling enzymes, highly conserved in evolution. It is expressed in the nucleus and has a role in the maintenance of genome stability and reactivation of stalled DNA replication forks (2). The N-terminal domain of SMARCAL1 contains a replication protein A (RPA)-binding region followed by two tandem HARP domains. SMARCAL1 catalyzes replication fork remodeling when it binds to RPA to maintain genome stability activity (3) and has annealing ATP-dependent helicase activity relying on the HARP domains (4). The C-terminal domains of SMARCAL1 contain the SNF2 ATPase region, composed of a region shared by the SNF2 family, the Helicase ATP-binding region, and a Helicase C-terminal region, both necessary to bind and hydrolyze ATP and transduce the resulting energy for chromatin remodeling (5). SMARCAL1 mutations accumulate in the Helicase ATP-binding region, but mutations along the entire gene have been reported (6). While missense changes seem to be more frequent in severe SIOD cases, frameshift and truncation mutations predominate in patients with mild phenotypes (7).
SIOD patients suffer from severe renal pathology (8) and eventually defective immunity that may cause recurrent infections (9). Although lymphopenia is a frequent finding in SIOD patients (10), the mechanism remains unknown, but some authors suggest that it may be due to either T-cell intrinsic or hematopoietic stem cell defects (11). Before kidney transplantation, patients receive immunosuppressive therapy, which can delay, but not fully prevent, kidney failure (12), probably due to the important roles of SMARCAL1 in kidney development (8). Under these circumstances, it is not easy to determine if lymphopenia is T-cell-intrinsic or secondary to hematopoietic stem cell defects or immunosuppression, and whether the latter should be maintained after kidney transplantation, considering the risk of associated life-threatening infectious events. While SMARCAL1 biology has been addressed in renal tissue pathology because of its relevance to the main life-threatening symptoms (8), it has not been studied deeply in T lymphocytes, because defective immunity is not present in all patients. Low responses to T-cell mitogens and defective IL-7 receptor expression and thus signaling have been reported in T cells from SIOD patients (13), but impaired T-cell development could explain such findings. Early membrane-proximal signaling has not been specifically studied in SIOD patients, likely because SMARCAL1 is a nuclear DNA helicase, so mechanistically it is unlikely to be involved in cytoplasmic events. Deficiencies of enzymes that participate in DNA damage and repair often cause T- and B-cell lymphopenia and thus opportunistic infections (14), but TCR signaling events remain unaffected (15).
Here we report a new SIOD patient with a homozygous c1920_1921insG frameshift mutation in SMARCAL1 that generates a stop codon and likely leads to the synthesis of a truncated protein without the helicase domain. The patient received immunosuppressive treatment due to kidney transplantation and showed lymphopenia. We generated an immortalized T-cell line that allowed us to study if T-cell signaling defects in SIOD were T-cell-intrinsic or -extrinsic (i.e., induced by immunosuppression or lymphopenia). The results indicated that T-cell receptor (TCR)-mediated signaling was normal in SIOD-derived immortalized T cells, suggesting that the T-cell immunodeficiency is not due to intrinsic early TCR signaling defects, but rather to extrinsic defects caused by immunosuppression or lymphopenia.
Materials and methods
Case report
The patient was the third child of healthy consanguineous parents, with two healthy untested siblings. No prenatal pathology was detected during pregnancy (APGAR test 9/10). Born by urgent cesarean section due to oligohydramnios at week 33 of gestation with low weight (1,200 g; height, 38 cm). He was admitted to Neonatology for 30 days due to an intrauterine growth delay. At 3 years of age, he was diagnosed with spondyloepiphyseal dysplasia and short stature. Also, subclinical hypothyroidism and empty sella syndrome with growth hormone deficit and a short lower segment. At 5 years of age, he presented with cortico-resistant nephrotic syndrome with rapid evolution to secondary chronic kidney disease with hypertension that progressed to kidney failure at 8 years of age. The renal biopsy determined focal segmental glomerulosclerosis. Genetic testing identified a homozygous mutation (c.1921dup) in SMARCAL1, a diagnostic of SIOD (Figure 1A). This mutation was previously reported in a compound heterozygote Spanish SIOD patient (16). The insertion causes a frameshift and a missense sequence of around 50 amino acids within the region that codifies for the Helicase ATP-dependent region (Mutation Taster prediction), and the loss of the Helicase C-terminal region (Figure 1B).
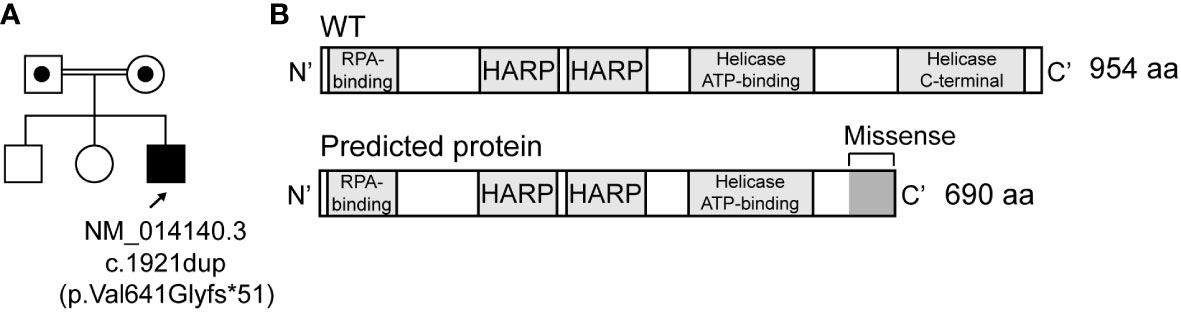
Figure 1 Homozygous mutations most likely cause a frameshift in SMARCAL1. (A) Pedigree for studied individuals, including the germline mutation (NM_014140.3, c.1921dup). Circles and squares indicate females and males, respectively. (B) Wild type and predicted SMARCAL1 proteins.
He received a cadaveric donor kidney transplantation at the age of 9 and responded well to low immunosuppressants (1.4 mg/kg prednisone, 0.4 mg/kg tacrolimus, and everolimus 2–3 ng/ml) and other symptomatic treatments. Three months post-transplantation he presented with normal graft function, serum creatinine of 0.37 mg/dl, and asymptomatic bacteriuria in relation to the JJ catheter in ureteral implant, uncomplicated non-specific pneumonia, and transitory BK viruria. He did not have opportunistic infections and his HLA antibodies were negative. He only had a partial focal seizure at 9 years old that was resolved with treatment 18 months post-transplantation. A basic immunological test determined an inverted CD4/CD8 ratio, CD3 and CD19 lymphopenia, decreased proliferation of T lymphocytes, and decreased oxidation capacity of neutrophils. At age 11, he was admitted due to an acute respiratory process with hypoxemia. A lung CT scan showed normal perfusion ventilation. He maintained clinical hypoxemia. Secondarily, he developed moderate pulmonary hypertension, left ventricular hypertrophy, and mild ventricular dysfunction. Two months later, he was readmitted due to severe neutropenia, so everolimus was suspended. The second lung CT scan showed a diffuse ground glass pattern compatible with cellular bronchiolitis. Laboratory tests for Aspergillus, pneumocystis, respiratory viruses, and mycoplasma were negative. He died at 13 years of age due to a cardiorespiratory arrest with a functioning graft.
PBMC isolation and immunophenotyping
Peripheral blood mononuclear cells (PBMC) were isolated by centrifugation on a Ficoll-Hypaque (GE Healthcare, Little Chalfont, United Kingdom) gradient. Blood samples were obtained after written informed consent from all participants or their tutors, according to the local ethics policy guidelines and the Declaration of Helsinki. Multiparametric flow cytometry was performed with mAbs against CD3 (UCHT-1), CD4 (13B8.2), CD19 (J3-119), CD45RA (ALB11), CD45RO (UCHL-1) and CD16 (3G8) from Beckman Coulter (Brea, Calif, USA); αβTCR (IP26) from Invitrogen/Thermo Fischer Scientific (Waltham, Mass, USA); γδTCR (11F2), IgD (IA6-2), CD31 (WM59), CD27 (M-T271), CD56 (B159), and CD8 (RPA-T8) from BD Biosciences (San Jose, Calif). Data were acquired with a FACSCalibur flow cytometer (BD Biosciences) at the Center for Cytometry and Fluorescence Microscopy of Complutense University of Madrid (Spain) and analyzed with FlowJo software (TreeStar, Ashland, Ore, USA).
Generation of immortalized T-cell lines
Human T-lymphotropic virus type 1 (HTLV-I) cell lines were generated as described (17). Briefly, PBMC were stimulated with 10 μg/ml phytohemagglutinin-L (PHA-L) from Phaseolus vulgaris (only at day 0) from Sigma-Aldrich (St. Louis, Mo) for 24 h and co-cultured with gamma-irradiated (150 Gy) HTLV-1-producing cell line (MT2) at 1:2 ratio in RPMI 1640 medium from Lonza (Basel, Switzerland), supplemented with 100 IU/ml recombinant interleukin-2 (rIL-2) (provided by Craig W. Reynolds, Frederick Cancer Research and Development Center, NCI, NIH, Frederick, Maryland, USA), 10% fetal bovine serum (FBS) and 1% L-glutamine and antibiotic–antimycotic from Life Technologies (Carlsbad, Calif, USA). Cells were maintained at 1–2 × 106 cells/ml and culture medium supplemented with 100 IU/ml rhIL-2 was refreshed twice per week.
Genomics
Genomic DNA for mutation determination was extracted from 3 million immortalized T cells using QIAamp® DNA Mini (Qiagen, Düsseldorf, Germany) and resuspended in H2O. A total of 100 ng were used for PCR amplification using Easy™ Oligo primers purchased from Sigma Aldrich (Burlington, MA, USA), designed as indicated in Supplementary Table S1, and TAQ PCR MASTER KIT (1,000, Cat. No. 201443) from Qiagen. PCR products were Sanger sequenced in the Genomics Unit of the Complutense University of Madrid (Spain) on a 3730xl DNA Analyzer and studied by Chromas software.
To analyze SMARCAL1 expression, total RNA was isolated using the RNeasy Mini Kit (250, Cat. No. 7410) from Qiagen and resuspended in H2O. A total of 2 µg was used for reverse transcription using the High-Capacity cDNA Reverse Transcription Kit from Applied Biosystems™ (Waltham, MA, USA). Different amounts of cDNA were used for PCR amplifications using Easy™ Oligo primers purchased from Sigma Aldrich (Supplementary Table S1) and TAQ PCR MASTER KIT (1,000, Cat. No. 201443) from Qiagen. PCR products were loaded onto a 2% agarose (Tris-acetate-EDTA buffer) gel with a 1/30,000 dilution of SYBR® Green nucleic acid gel stain from Invitrogen (Waltham, MA, USA) and visualized with the GelDoc Go Gel Imaging System from BIO-RAD (Hercules, CA, USA).
T-cell function assays
TCR signaling was studied using standard protocols (18). Briefly, 0.2 × 106 PBMC or immortalized T cells were plated in RPMI-1640 supplemented with 10% FBS in flat-bottom 96-well plates coated with 10 μg/ml anti-CD3 mAb (UCHT-1 from BD Biosciences) and stimulated for 24 h. T-cell activation was analyzed by flow cytometry with anti-CD69 mouse anti-human antibody (L-78 from BD Biosciences). Alternatively, 0.2 × 106 PBMC were plated in RPMI-1640 supplemented with 10% FBS in flat-bottom 96-well plates coated with 1 μg/ml anti-CD3 mAb (OKT-3; eBioscience, Waltham, Mass, USA), or with 5 μg/ml PHA-L, or with 10 ng/ml phorbol 12-myristate 13-acetate (PMA) and 1 mM ionomycin or 10 ng/ml superantigens (Staphylococcal Enterotoxin B or E; Toxin Technology, Inc, Sarasota, Fla) and stimulated for 5 days, and the percentage of blastic cells within the lymphocyte SSC-H vs FSC-H gate was quantified by flow cytometry.
DNA repair assay
Immortalized T-cell lines were harvested in an RPMI-1640 medium and treated with 10 Gy gamma irradiation (using the Irradiator Gammacell 1000 of the Central Radioactive Facility of Complutense University of Madrid, Spain) as described (19). DNA repair was evaluated at different time points, the phosphorylation of γH2AX was evaluated by intracellular flow cytometry using Alexa Fluor® 647 Mouse anti-H2AX antibody (pS139 clone, BD Biosciences) and eBioscience™ Foxp3/Transcription Factor Staining Buffer Set and protocol (BD Biosciences). Data were acquired with a FACSCalibur flow cytometer and analyzed with FlowJo software.
Standard biosecurity and institutional safety procedures have been adhered to, following European and Universidad Complutense guidelines.
Results
Primary T cells
The patient showed selective severe T-cell lymphopenia at 9 years of age (Table 1).
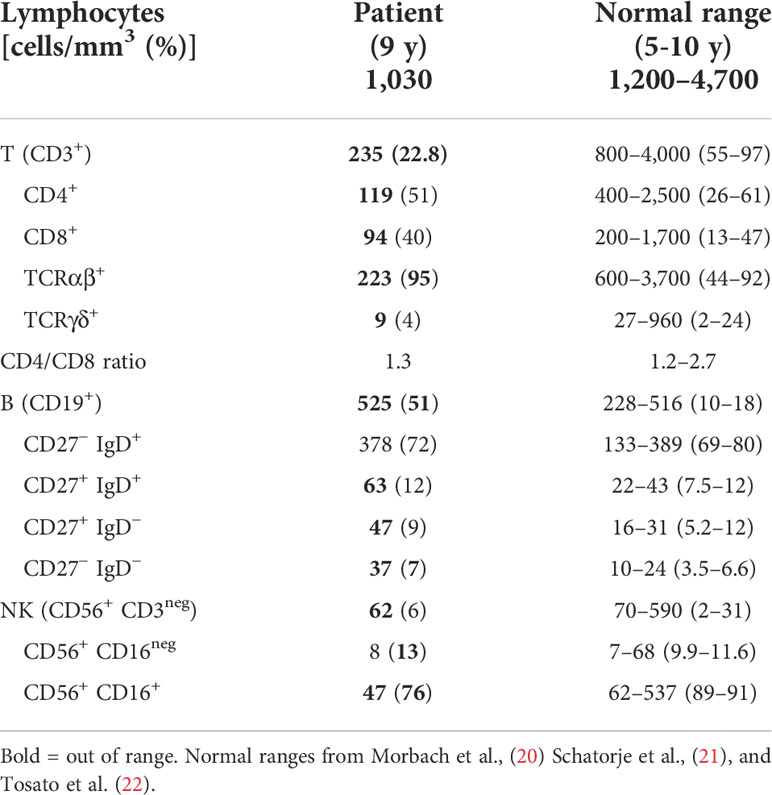
Table 1 Immunological phenotype.
Accordingly, thymus output measured as recent thymic emigrants was strongly reduced and most peripheral T cells were CD45RO+ (memory) in the patient (Figure 2). The evolution of the leucocyte and lymphocyte cell numbers indicated lymphopenia and a decrease after immunosuppression (see Figure S1 in Supplementary Material).
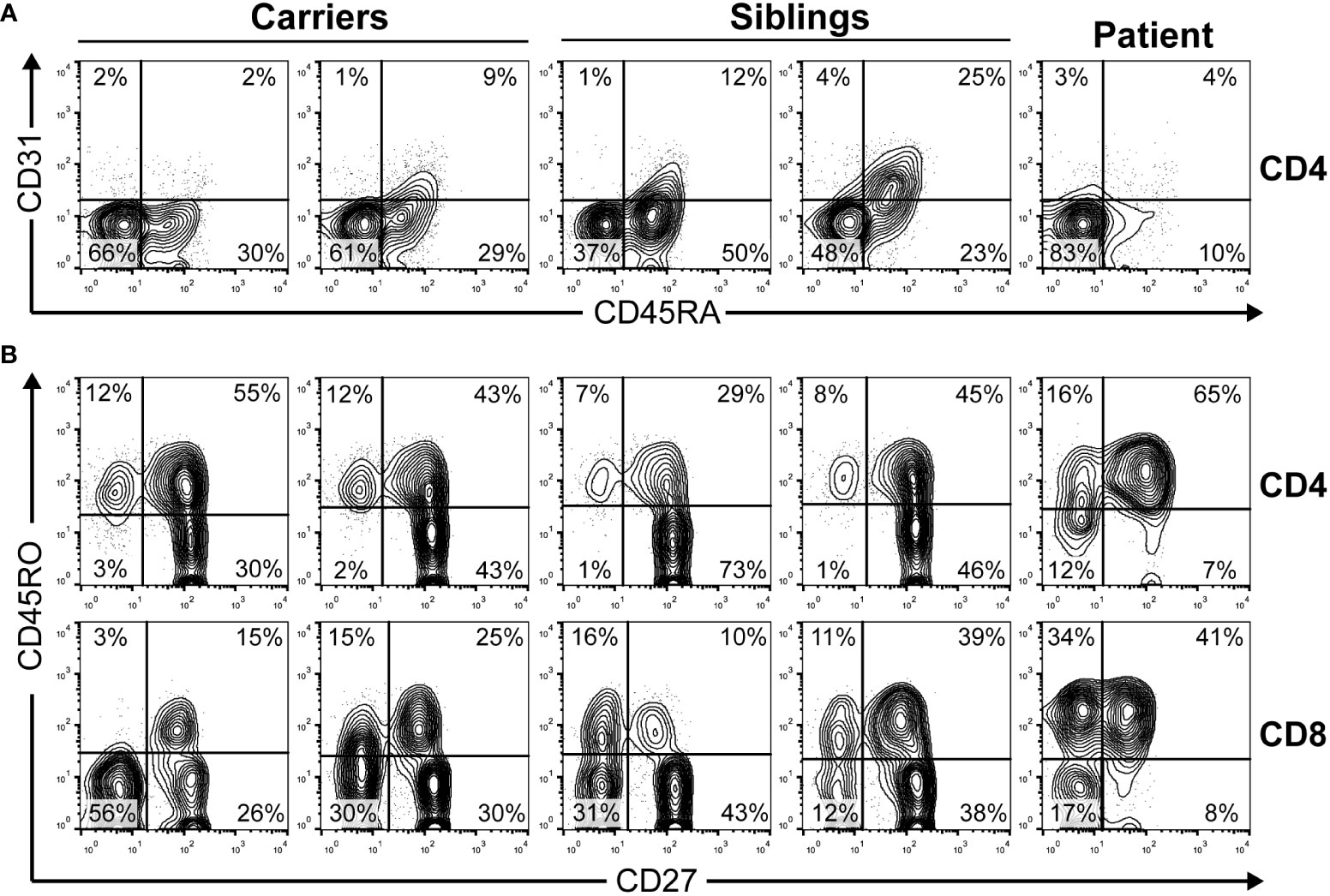
Figure 2 The peripheral blood phenotype of T cells was determined by extracellular flow cytometry in the indicated family members. (A) Recent thymic emigrants CD4 T cells defined as CD45RA+CD31+ lymphocytes as reported (23). (B) T-cell maturation stages defined in CD4 and CD8 bright T-cells as naive (CD45RO−CD27+), central memory (CD45RO+CD27+), effector memory (CD45RO+CD27−), or effector (CD45RO−CD27−). Carriers, 39–40 years old (yo); siblings, 12–15 yo; patient, 9 yo.
Immortalized T cells
To understand the relevance of SMARCAL1 in TCR signaling ex vivo, excluding immunosuppression effects, HTLV-I-immortalized T-cell lines were generated from all family members and an unrelated healthy donor. All cell lines showed similar cell growth kinetics (see Figure S2 in Supplementary Material). SMARCAL1 Sanger sequencing of all immortalized T-cell lines confirmed the expected segregation of the mutation (compare Figure 1A and Figure 3A). To estimate the effect of the homozygous mutation on SMARCAL1 transcription, the sequence containing the mutation was amplified by PCR using different amounts of cDNA from immortalized, healthy, and patient T cells. CD3E was used as a T cell-specific internal positive control. The results showed a strong reduction of SMARCAL1 PCR product in the patient compared to a healthy donor, relative to CD3E (Figure 3B).
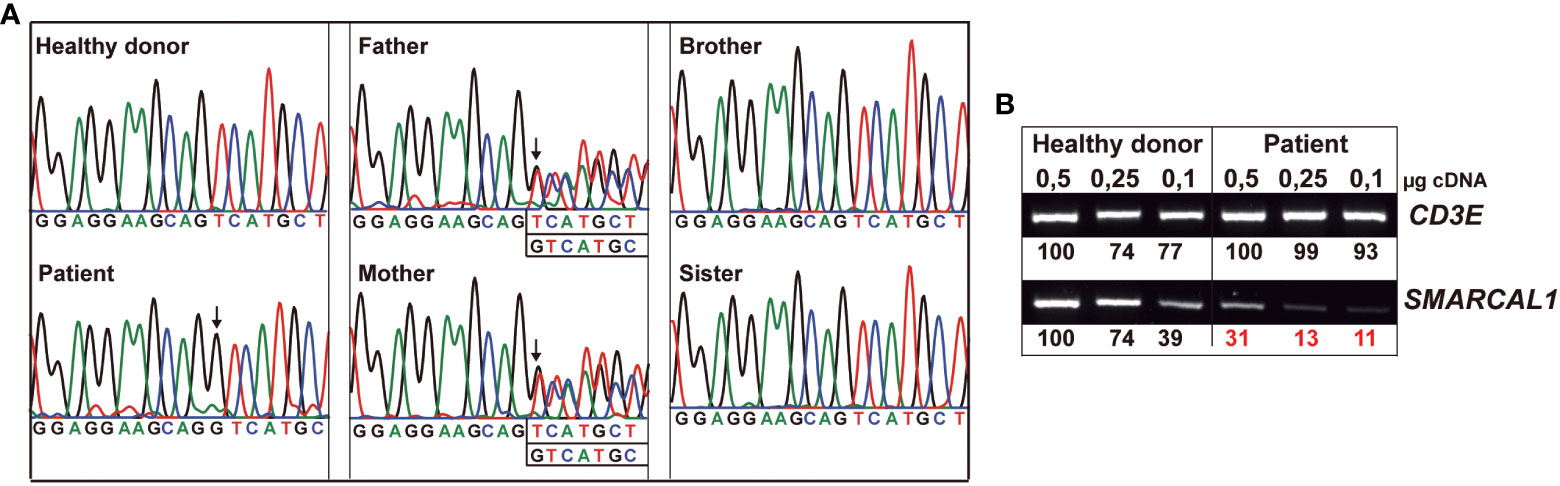
Figure 3 Heterozygous and homozygous SMARCAL1 c.1921dup mutations in immortalized T cells. (A) Genomic DNA SMARCAL1 Sanger sequencing of immortalized T-cell lines from the indicated donors. (B) Different amounts of T-cell line cDNA from healthy donors and patients were amplified by PCR using primers for CD3E as an internal control and SMARCAL1, as indicated in Table S1 (see Supplementary Material). The numbers represent the intensities of SMARCAL1 vs CD3E PCR product bands.
DNA repair kinetics can be studied by treating cells with insults that induce DNA breaks and recruit the phosphorylated histone γH2AX (p-γH2AX) (24). As SMARCAL1 is a component of the DNA damage response, epithelial SMARCAL1-deficient cells are hypersensitive to replication stress agents like hydroxyurea (HU) (25, 26), and increased DNA damage markers but not survival have been described in Smarcal1 KO mouse thymocytes after gamma radiation (11). Therefore, we studied DNA damage response sensitivity in vitro in the immortalized T-cell lines. To that end, cells were treated with 10 Gy gamma irradiation and p-γH2AX levels were measured at different time points. At early time points, gamma irradiation induced significantly lower p-γH2AX levels in immortalized patient T cells compared to healthy donors (Figure 4).
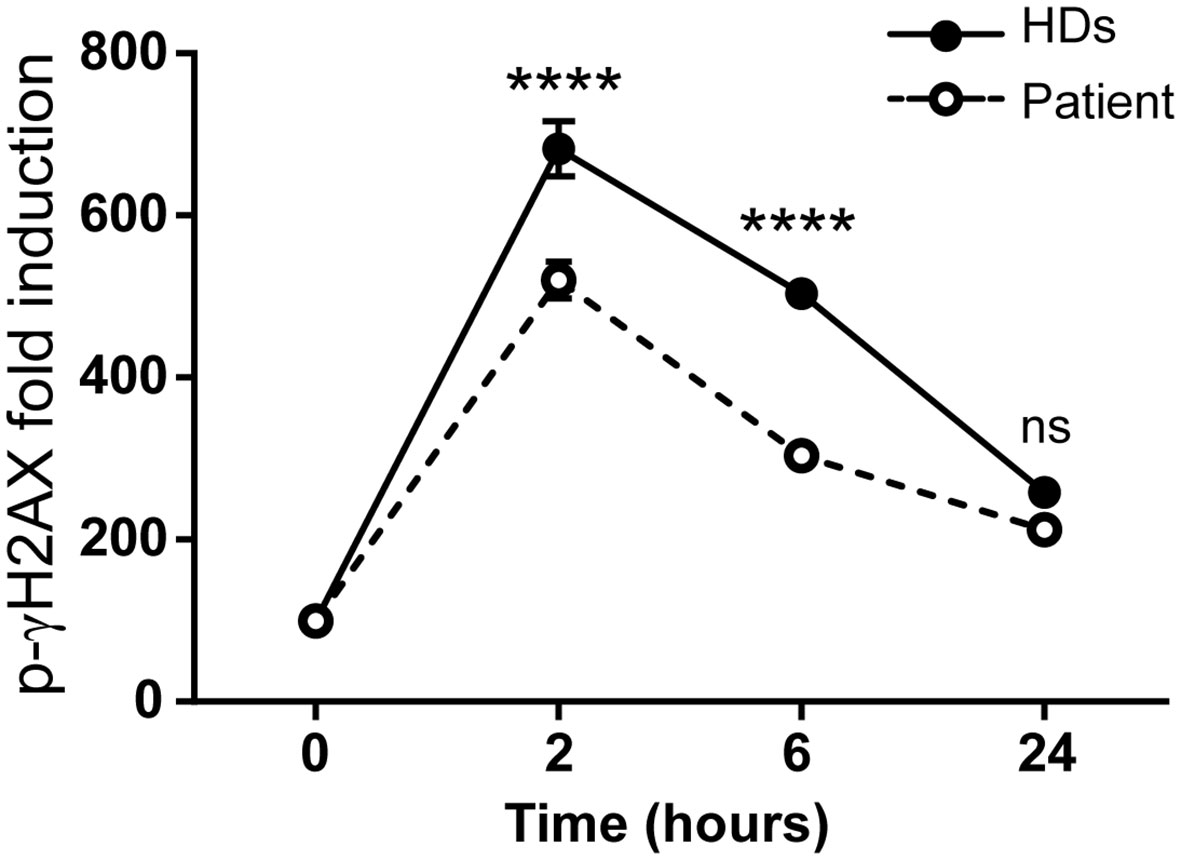
Figure 4 The SMARCAL1 c.1921dup mutation affects gamma irradiation DNA repair in T-cell lines. Immortalized T-cell line DNA repair after gamma irradiation treatment was evaluated by intracellular p-γH2AX levels by flow cytometry at different time points and the geometric mean fluorescence intensity (geo-MFI) relative to non-treated cells was calculated. HDs = 3; n = 6; ****p<0.0001; ns, not significant.
Taken together, these results validated the immortalized T-cell lines for the SMARCAL1 mutation, gene expression, and functional defects.
Primary vs immortalized T cells
Next, TCR function was studied in primary and immortalized T cells. Early (CD69 induction) and late (proliferation against different stimuli) events were studied after TCR-dependent (anti-CD3, PHA) or independent (PMA and ionomycin or ION) stimuli. CD69 induction (Figure 4, top) and T-cell proliferation (see Figure S3 in Supplementary Material) after anti-CD3 or PHA stimulation were strongly impaired in primary T cells from the patient compared to carriers and siblings. The defect was TCR-dependent, as PMA+ION induced normal responses in the primary T cells of the patient. In sharp contrast, immortalized T cells analyzed in parallel for CD69 induction showed completely normal responses to TCR engagement (Figure 4, bottom). These results indicate that SMARCAL1 does not have an intrinsic role in TCR signaling.
Discussion
We have shown that a lethal homozygous SMARCAL1 mutation causing severe kidney disease and lymphopenia in a boy did not impair TCR signaling in a T-cell model derived from the patient. The immortalized T cells were shown to carry the same mutations originally detected in the patient (Figure 3), which impaired SMARCAL1 gene expression (Figure 3B) as well as the response of the cells to gamma irradiation (Figure 4), but not TCR-mediated signaling (Figure 5). Impaired response to gamma-irradiation has also been reported in other cell systems (shRNA and knockdown chicken cell lines), where a reduction or loss of SMARCAL1 increased radiosensitivity (27, 28).
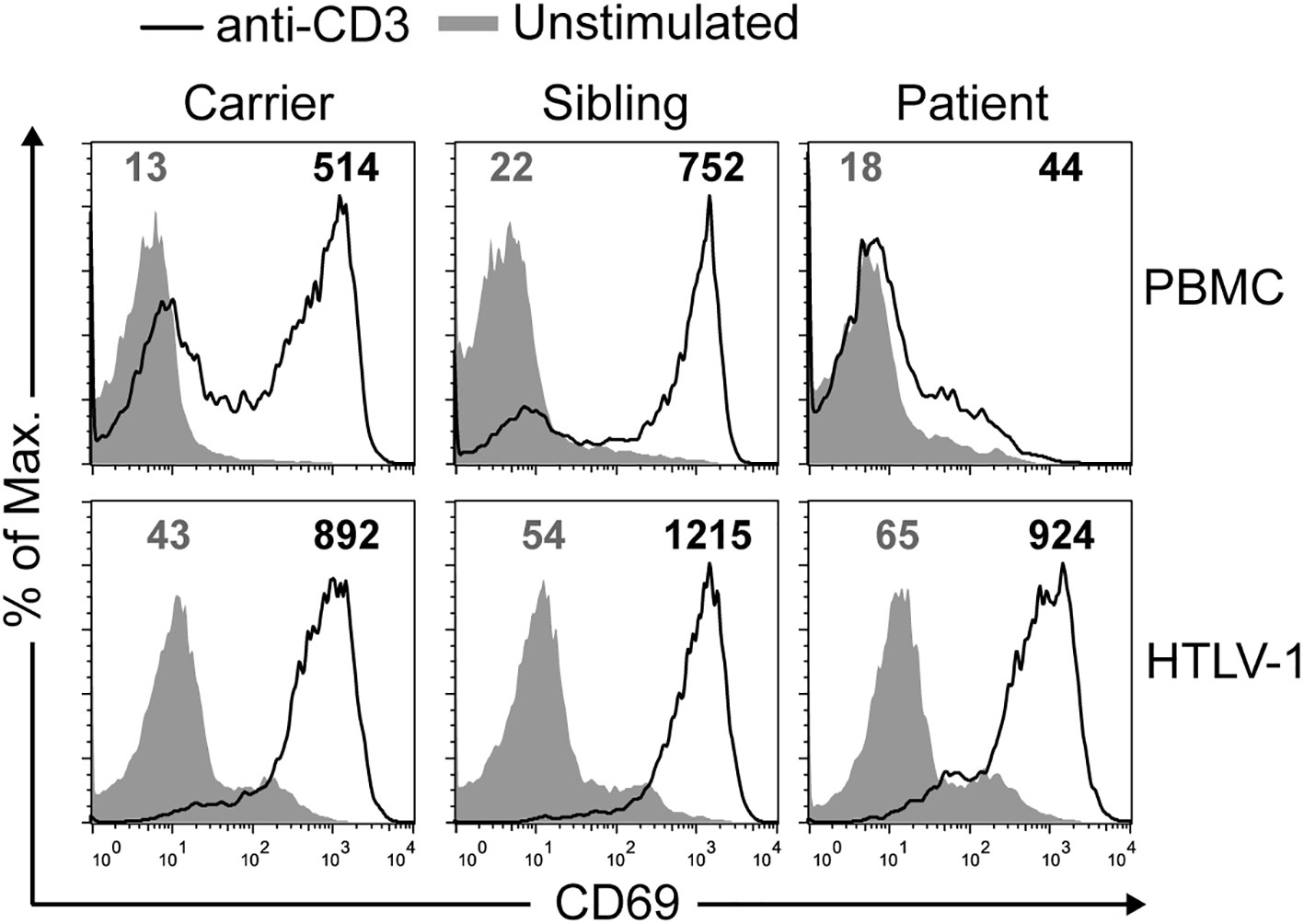
Figure 5 Normal signaling in immortalized but not primary SIOD T cells. CD69 mean fluorescence intensity (MFI) was quantified in non-stimulated cells (gray lines) and after stimulation (black lines) with anti-CD3 antibody for 24 h by flow cytometry on primary (top) or immortalized (bottom) T cells. This experiment was repeated twice with similar results.
Similar results were found with immortalized T cells derived from ataxia telangiectasia patients (15). Despite a long-term belief in the field that the affected molecule (ATM, a nuclear kinase activated by double-strand breaks or oxidative stress, which phosphorylates several targets including SMARCAL1) was involved in TCR or BCR signaling and thus caused lymphopenia and immunodeficiency, we showed that immortalized T cells derived from patients carried the causing mutations and thus preserved the sensitivity to ionizing radiation of ataxia telangiectasia cells, but did not show any intrinsic immune functional defects when stimulated through their TCR.
As TCR-mediated signaling is SMARCAL1-independent, how can the observed immune defects observed in SIOD patients be connected to SMARCAL1 function? This includes frequent lymphopenia and less frequent immunodeficiency (i.e., susceptibility to infection; see Table 2). Our hypothesis is that SMARCAL1 impairs nuclear functions required for early T-cell development checkpoints that require strong cellular expansion. Such a developmental defect would cause lymphopenia and, in some cases, also immunodeficiency. This hypothesis has been proved right by others for a different DNA helicase termed BLM involved in Bloom’s syndrome in humans, which is associated with mild lymphopenia and an increased number of infections (54). T-lineage-specific mouse BLM ablation was shown to decrease thymocyte numbers due to a developmental block at the T-cell β-selection checkpoint (55). The hypothesis has not been directly addressed in SIOD patients, but some results in Smarcal1-deficient mice strongly point to a hematopoietic progenitor cell defect (11). Our hypothesis could be tested by studying SIOD T-cell differentiation using CD34+ lymphoid precursors in artificial thymic organoids as reported elsewhere for intrinsic (such as reticular dysgenesis) vs extrinsic (such as DiGeorge syndrome) T-cell lymphopenia (56). Experiments are underway to test this hypothesis. The fact that early thymic emigrants in peripheral blood are essentially absent in our patient (Figure 2A) and other studied patients (13) lends support to the hypothesis of an SMARCAL1-dependent expansion defect in T-cell development.
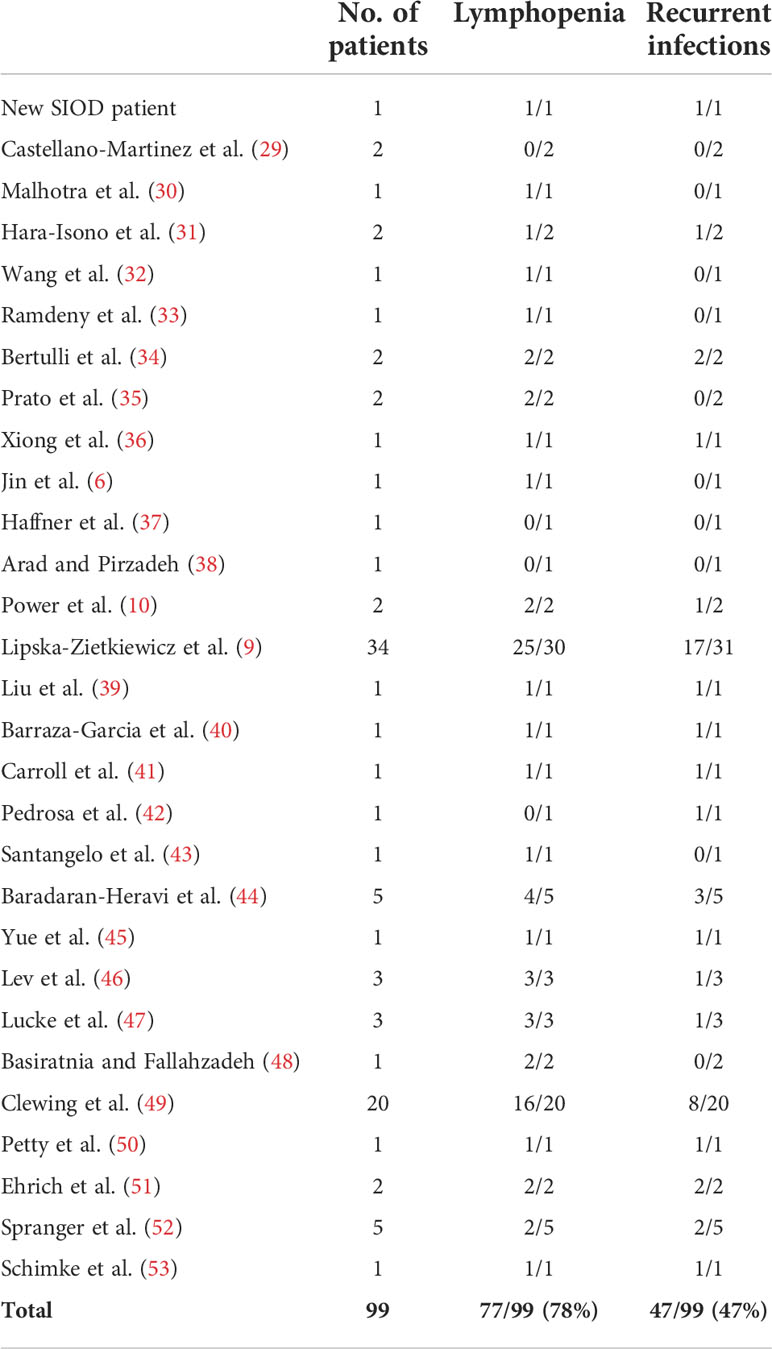
Table 2 Immunological features of SIOD patients.
It may be argued that our findings refer to a single patient, but numerous single patient studies have revealed crucial pathways underlying physiological and pathological processes by establishing a causal relationship between the candidate genotype and the clinical phenotype via a relevant cellular phenotype, as we show here (57). Also, the literature has not addressed the role of SMARCAL1 in TCR-mediated signaling in patient-derived immortalized T cells, as we have done here for the first time. An obvious caveat to using a single patient is, naturally, that different mutations may differently affect T-cell development since disease severity has been shown to be inversely proportionate to overall SMARCAL1 activity (7). Thus, the spectrum of the disease may vary from mild phenotypes, as observed in patients with frameshift and truncation mutations (43), to very severe phenotypes in patients with homozygous missense mutations, as described in our work and others' (7, 58).
Although HTLV-1 transformation could affect T-cell function by itself, the use of HTLV-1 T-cell lines as in vitro models of human T lymphocytes has been validated in the past (59) and we have extensively demonstrated that the immortalization process does not affect the functional status of proximal TCR (15, 60–65) and other (18, 66) signaling pathways. We do not rule out that SMARCAL1 has a role in T-cell proliferation, however, and this would fit better with the reported role for that nuclear protein, although we favour the hypothesis of an SMARCAL1-dependent expansion defect in early T-cell development.
The immortalized T cells carrying the lethal SMARCAL1 mutation characterized here may help in understanding the distinct biological role of SMARCAL1 in different cell types and developing rational therapies for the T-cell immunological dysfunction of SIOD patients with severe mutations. However, Table 2 clearly shows that there is a great variability in terms of T-cell lymphopenia, which is present in most SIOD patients (78%), and T-cell immunodeficiency as estimated by susceptibility to infections (present only in 47% of the patients). Thus, immunosuppression regimens must necessarily be personalized according to the immune profile of the patient.
Conclusions
T-cell receptor early signaling in Schimke immuno-osseous dysplasia is SMARCAL1-independent, as demonstrated by the fact that transformed T cells carrying lethal SMARCAL1 mutations showed impaired SMARCAL1 gene expression and impaired response to gamma irradiation but not impaired T-cell receptor signaling for CD69 induction.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
This study was reviewed and approved by CEIm Hospital Clínico San Carlos. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
JRR and ER-O conceived the experimental study. AVM, AJ-R, and MSM designed the experiments and analyzed the data. JRR and AVM wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the Ministerio de Economía y Competitividad (MINECO PID2021-125501OB-I00 and RTI2018-095673-B-I00), the Comunidad Autónoma de Madrid (CAM B2017/BMD3673), and the Asociación Española Contra el Cáncer (AECC PROYE20084REGU). AVM was supported by the Complutense/Harvard University (CT46/15); AJ-R by the MINECO (grant no. BES-2012-055054).
Acknowledgments
We thank Alejandro Briones for manuscript revision, Carmen Alonso and Julia Fernandez-Boraita for technical help, and the patient, his family, the Asociación Española de Displasias Óseas Minoritarias (www.facebook.com/asociacionaedom/), and clinicians for their collaboration.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.979722/full#supplementary-material
References
1. Lou S, Lamfers P, McGuire N, Boerkoel CF. Longevity in schimke immuno-osseous dysplasia. J Med Genet (2002) 39(12):922–5. doi: 10.1136/jmg.39.12.922
2. Lugli N, Sotiriou SK, Halazonetis TD. The role of SMARCAL1 in replication fork stability and telomere maintenance. DNA Repair (Amst) (2017) 56:129–34. doi: 10.1016/j.dnarep.2017.06.015
3. Bhat KP, Betous R, Cortez D. High-affinity DNA-binding domains of replication protein a (RPA) direct SMARCAL1-dependent replication fork remodeling. J Biol Chem (2015) 290(7):4110–7. doi: 10.1074/jbc.M114.627083
4. Ghosal G, Yuan J, Chen J. The HARP domain dictates the annealing helicase activity of HARP/SMARCAL1. EMBO Rep (2011) 12(6):574–80. doi: 10.1038/embor.2011.74
5. Ryan DP, Owen-Hughes T. Snf2-family proteins: Chromatin remodellers for any occasion. Curr Opin Chem Biol (2011) 15(5):649–56. doi: 10.1016/j.cbpa.2011.07.022
6. Jin J, Wu K, Liu Z, Chen X, Jiang S, Wang Z, et al. Whole exome sequencing identified a novel biallelic SMARCAL1 mutation in the extremely rare disease SIOD. Front Genet (2019) 10:565. doi: 10.3389/fgene.2019.00565
7. Elizondo LI, Cho KS, Zhang W, Yan J, Huang C, Huang Y, et al. Schimke immuno-osseous dysplasia: SMARCAL1 loss-of-function and phenotypic correlation. J Med Genet (2009) 46(1):49–59. doi: 10.1136/jmg.2008.060095
8. Sarin S, Javidan A, Boivin F, Alexopoulou I, Lukic D, Svajger B, et al. Insights into the renal pathogenesis in schimke immuno-osseous dysplasia: A renal histological characterization and expression analysis. J Histochem Cytochem (2015) 63(1):32–44. doi: 10.1369/0022155414558335
9. Lipska-Zietkiewicz BS, Gellermann J, Boyer O, Gribouval O, Zietkiewicz S, Kari JA, et al. Low renal but high extrarenal phenotype variability in schimke immuno-osseous dysplasia. PloS One (2017) 12(8):e0180926. doi: 10.1371/journal.pone.0180926
10. Power BD, Walsh KP, Awan A, Waldron M, O’Grady MJ. Klippel-feil syndrome as a novel feature of schimke immunoosseous dysplasia. Am J Med Genet A (2019) 179(5):862–3. doi: 10.1002/ajmg.a.61087
11. Puccetti MV, Fischer MA, Arrate MP, Boyd KL, Duszynski RJ, Bétous R, et al. Defective replication stress response inhibits lymphomagenesis and impairs lymphocyte reconstitution. Oncogene (2017) 36(18):2553–64. doi: 10.1038/onc.2016.408
12. Zivicnjak M, Franke D, Zenker M, Hoyer J, Lucke T, Pape L, et al. SMARCAL1 mutations: A cause of prepubertal idiopathic steroid-resistant nephrotic syndrome. Pediatr Res (2009) 65(5):564–8. doi: 10.1203/PDR.0b013e3181998a74
13. Sanyal M, Morimoto M, Baradaran-Heravi A, Choi K, Kambham N, Jensen K, et al. Lack of IL7Rα expression in T cells is a hallmark of T-cell immunodeficiency in schimke immuno-osseous dysplasia (SIOD). Clin Immunol (2015) 161(2):355–65. doi: 10.1016/j.clim.2015.10.005
14. Gullickson P, Xu YW, Niedernhofer LJ, Thompson EL, Yousefzadeh MJ. The role of DNA repair in immunological diversity: From molecular mechanisms to clinical ramifications. Front Immunol (2022) 13:834889. doi: 10.3389/fimmu.2022.834889
15. Rivero-Carmena M, Porras O, Pelaez B, Pacheco-Castro A, Gatti RA, Regueiro JR. Membrane and transmembrane signaling in herpesvirus saimiri-transformed human CD4(+) and CD8(+) T lymphocytes is ATM-independent. Int Immunol (2000) 12(6):927–35. doi: 10.1093/intimm/12.6.927
16. Boerkoel CF, Takashima H, John J, Yan J, Stankiewicz P, Rosenbarker L, et al. Mutant chromatin remodeling protein SMARCAL1 causes schimke immuno-osseous dysplasia. Nat Genet (2002) 30(2):215–20. doi: 10.1038/ng821
17. Nutman TB. Generation of HTLV-I-transformed T cell lines. Curr Protoc Immunol (2002) 7. doi: 10.1002/0471142735.im0720s47
18. Jiménez-Reinoso A, Marin AV, Subias M, López-Lera A, Román-Ortiz E, Payne K, et al. Human plasma C3 is essential for the development of memory b, but not T, lymphocytes. J Allergy Clin Immunol (2018) 141(3):1151–1154.e1114. doi: 10.1016/j.jaci.2017.09.037
19. Recio MJ, Dominguez-Pinilla N, Perrig MS, Rodriguez Vigil-Iturrate C, Salmón-Rodriguez N, Martinez Faci C, et al. Extreme phenotypes with identical mutations: Two patients with same non-sense NHEJ1 homozygous mutation. Front Immunol (2018) 9:2959. doi: 10.3389/fimmu.2018.02959
20. Morbach H, Eichhorn EM, Liese JG, Girschick HJ. Reference values for b cell subpopulations from infancy to adulthood. Clin Exp Immunol (2010) 162(2):271–9. doi: 10.1111/j.1365-2249.2010.04206.x
21. Schatorje EJ, Gemen EF, Driessen GJ, Leuvenink J, van Hout RW, de Vries E. Paediatric reference values for the peripheral T cell compartment. Scand J Immunol (2012) 75(4):436–44. doi: 10.1111/j.1365-3083.2012.02671.x
22. Tosato F, Bucciol G, Pantano G, Putti MC, Sanzari MC, Basso G, et al. Lymphocytes subsets reference values in childhood. Cytometry A (2015) 87(1):81–5. doi: 10.1002/cyto.a.22520
23. Ravkov E, Slev P, Heikal N. Thymic output: Assessment of CD4(+) recent thymic emigrants and T-cell receptor excision circles in infants. Cytometry B Clin Cytom (2017) 92(4):249–57. doi: 10.1002/cyto.b.21341
24. Mah LJ, El-Osta A, Karagiannis TC. Gammah2ax: A sensitive molecular marker of DNA damage and repair. Leukemia (2010) 24(4):679–86. doi: 10.1038/leu.2010.6
25. Bansbach CE, Bétous R, Lovejoy CA, Glick GG, Cortez D. The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev (2009) 23(20):2405–14. doi: 10.1101/gad.1839909
26. Postow L, Woo EM, Chait BT, Funabiki H. Identification of SMARCAL1 as a component of the DNA damage response. J Biol Chem (2009) 284(51):35951–61. doi: 10.1074/jbc.M109.048330
27. Ciccia A, Bredemeyer AL, Sowa ME, Terret ME, Jallepalli PV, Harper JW, et al. The SIOD disorder protein SMARCAL1 is an RPA-interacting protein involved in replication fork restart. Genes Dev (2009) 23(20):2415–25. doi: 10.1101/gad.1832309
28. Keka IS, Mohiuddin, Maede Y, Rahman MM, Sakuma T, Honma M, et al. Smarcal1 promotes double-strand-break repair by nonhomologous end-joining. Nucleic Acids Res (2015) 43(13):6359–72. doi: 10.1016/j.clim.2009.08.017
29. Castellano-Martinez A, Acuñas-Soto S, de la Varga Martínez R, Rodriguez-Gonzalez M, Mora-Lopez F, Iriarte-Gahete M, et al. Different phenotypes of schimke immuno-osseous dysplasia (SIOD) in two sisters with the same mutation in the SMARCAL1 gene: Case reports. Endocr Metab Immune Disord Drug Targets (2022) 22(8):888–894. doi: 10.2174/1871530322666220223154028
30. Malhotra R, Sharma M, Dwivedi A, Kalra S. A case of schimke immunoosseous dysplasia caused by Large deletion of SMARCAL1 gene. Indian J Endocrinol Metab (2021) 25(4):358–60. doi: 10.4103/ijem.ijem_148_21
31. Hara-Isono K, Matsubara K, Hamada R, Shimada S, Yamaguchi T, Wakui K, et al. A patient with silver-Russell syndrome with multilocus imprinting disturbance, and schimke immuno-osseous dysplasia unmasked by uniparental isodisomy of chromosome 2. J Hum Genet (2021) 66(11):1121–6. doi: 10.1038/s10038-021-00937-7
32. Wang L, Li J, Wu G, Kong X. A novel compound heterozygous variant in SMARCAL1 leading to mild schimke immune-osseous dysplasia identified using whole-exome sequencing. J Int Med Res (2021) 49(4):3000605211010644. doi: 10.1177/03000605211010644
33. Ramdeny S, Chaudhary A, Worth A, Ghorashian S, Slatter M, Lum SH, et al. Activity of blinatumomab in lymphoblastic leukemia with impaired T-cell immunity due to congenital immunodeficiency. Blood Adv (2021) 5(8):2153–5. doi: 10.1182/bloodadvances.2021004284
34. Bertulli C, Marzollo A, Doria M, Di Cesare S, La Scola C, Mencarelli F, et al. Expanding phenotype of schimke immuno-osseous dysplasia: Congenital anomalies of the kidneys and of the urinary tract and alteration of NK cells. Int J Mol Sci (2020) 21(22):8604. doi: 10.3390/ijms21228604
35. Prato G, De Grandis E, Mancardi MM, Cordani R, Giacomini T, Pisciotta L, et al. Schimke immuno-osseous dysplasia, two new cases with peculiar EEG pattern. Brain Dev (2020) 42(5):408–13. doi: 10.1016/j.braindev.2020.01.008
36. Xiong S, Shuai L, Li X, Dang X, Wu X, He Q. Podocytic infolding in schimke immuno-osseous dysplasia with novel SMARCAL1 mutations: A case report. BMC Nephrol (2020) 21(1):170. doi: 10.1186/s12882-020-01809-6
37. Haffner DN, Rollins NK, Dowling MM. Reversible cerebral vasoconstriction syndrome: A novel mechanism for neurological complications in schimke immuno-osseous dysplasia. Pediatr Neurol (2019) 92:67–70. doi: 10.1016/j.pediatrneurol.2018.10.022
38. Arad B, Pirzadeh Z. A case report of schimke immuno-osseous dysplasia: A rare autosomal recessive disorder. Int J Pediatr (2018) 6(2):7151–5. doi: 10.22038/ijp.2017.26522.2282
39. Liu S, Zhang M, Ni M, Zhu P, Xia X. A novel compound heterozygous mutation of the SMARCAL1 gene leading to mild schimke immune-osseous dysplasia: A case report. BMC Pediatr (2017) 17(1):217. doi: 10.1186/s12887-017-0968-8
40. Barraza-Garcia J, Rivera-Pedroza CI, Belinchon A, Fernandez-Camblor C, Valenciano-Fuente B, Lapunzina P, et al. A novel SMARCAL1 missense mutation that affects splicing in a severely affected schimke immunoosseous dysplasia patient. Eur J Med Genet (2016) 59(8):363–6. doi: 10.1016/j.ejmg.2016.06.002
41. Carroll C, Hunley TE, Guo Y, Cortez D. A novel splice site mutation in SMARCAL1 results in aberrant exon definition in a child with schimke immunoosseous dysplasia. Am J Med Genet A (2015) 167a(10):2260–4. doi: 10.1002/ajmg.a.37146
42. Pedrosa AK, Torres LF, Silva AC, Dantas AB, Zuntini KL, Aguiar LC. Rare case of nephrotic syndrome: Schimke syndrome. J Bras Nefrol (2016) 38(3):370–3. doi: 10.5935/0101-2800.20160057
43. Santangelo L, Gigante M, Netti GS, Diella S, Puteo F, Carbone V, et al. A novel SMARCAL1 mutation associated with a mild phenotype of schimke immuno-osseous dysplasia (SIOD). BMC Nephrol (2014) 15:41. doi: 10.1186/1471-2369-15-41
44. Baradaran-Heravi A, Lange J, Asakura Y, Cochat P, Massella L, Boerkoel CF. Bone marrow transplantation in schimke immuno-osseous dysplasia. Am J Med Genet A (2013) 161a(10):2609–13. doi: 10.1002/ajmg.a.36111
45. Yue Z, Xiong S, Sun L, Huang W, Mo Y, Huang L, et al. Novel compound mutations of SMARCAL1 associated with severe schimke immuno-osseous dysplasia in a Chinese patient. Nephrol Dial Transplant (2010) 25(5):1697–702. doi: 10.1093/ndt/gfq071
46. Lev A, Amariglio N, Levy Y, Spirer Z, Anikster Y, Rechavi G. Molecular Assessment of Thymic Capacities in Patients with Schimke Immuno-Osseous Dysplasia. Clin Immunol (2009) 133(3):375–81. doi: 10.1016/j.clim.2009.08.017
47. Lucke T, Clewing JM, Boerkoel CF, Hartmann H, Das AM, Knauth M, et al. Cerebellar atrophy in schimke-immuno-osseous dysplasia. Am J Med Genet A (2007) 143a(17):2040–5. doi: 10.1002/ajmg.a.31878
48. Basiratnia M, Fallahzadeh MH. Schimke immuno-osseous dysplasia. Saudi Med J (2007) 28(3):457–60.
49. Clewing JM, Fryssira H, Goodman D, Smithson SF, Sloan EA, Lou S, et al. Schimke immunoosseous dysplasia: Suggestions of genetic diversity. Hum Mutat (2007) 28(3):273–83. doi: 10.1002/humu.20432
50. Petty EM, Yanik GA, Hutchinson RJ, Alter BP, Schmalstieg FC, Levine JE, et al. Successful bone marrow transplantation in a patient with schimke immuno-osseous dysplasia. J Pediatr (2000) 137(6):882–6. doi: 10.1067/mpd.2000.109147
51. Ehrich JH, Burchert W, Schirg E, Krull F, Offner G, Hoyer PF, et al. Steroid resistant nephrotic syndrome associated with spondyloepiphyseal dysplasia, transient ischemic attacks and lymphopenia. Clin Nephrol (1995) 43(2):89–95.
52. Spranger J, Hinkel GK, Stöss H, Thoenes W, Wargowski D, Zepp F. Schimke immuno-osseous dysplasia: A newly recognized multisystem disease. J Pediatr (1991) 119(1 Pt 1):64–72. doi: 10.1016/s0022-3476(05)81040-6
53. Schimke RN, Horton WA, King CR. Chondroitin-6-Sulphaturia, Defective Cellular Immunity, and Nephrotic Syndrome. Lancet (2071) 2(7733):1088–9. doi: 10.1016/s0140-6736(71)90400-4
54. Cunniff C, Bassetti JA, Ellis NA. Bloom’s syndrome: Clinical spectrum, molecular pathogenesis, and cancer predisposition. Mol Syndromol (2017) 8(1):4–23. doi: 10.1159/000452082
55. Babbe H, Chester N, Leder P, Reizis B. The bloom’s syndrome helicase is critical for development and function of the alphabeta T-cell lineage. Mol Cell Biol (2007) 27(5):1947–59. doi: 10.1128/mcb.01402-06
56. Bosticardo M, Pala F, Calzoni E, Delmonte OM, Dobbs K, Gardner CL, et al. Artificial thymic organoids represent a reliable tool to study T-cell differentiation in patients with severe T-cell lymphopenia. Blood Adv (2020) 4(12):2611–6. doi: 10.1182/bloodadvances.2020001730
57. Casanova JL, Conley ME, Seligman SJ, Abel L, Notarangelo LD. Guidelines for genetic studies in single patients: Lessons from primary immunodeficiencies. J Exp Med (2014) 211(11):2137–49. doi: 10.1084/jem.20140520
58. Baradaran-Heravi A, Cho KS, Tolhuis B, Sanyal M, Morozova O, Morimoto M, et al. Penetrance of biallelic SMARCAL1 mutations is associated with environmental and genetic disturbances of gene expression. Hum Mol Genet (2012) 21(11):2572–87. doi: 10.1093/hmg/dds083
59. Moro H, Iwai K, Mori N, Watanabe M, Fukushi M, Oie M, et al. Interleukin-2-dependent but not independent T-cell lines infected with human T-cell leukemia virus type 1 selectively express CD45RO, a marker for persistent infection in vivo. Virus Genes (2001) 23(3):263–71. doi: 10.1023/a:1012565105098
60. Alvarez-Zapata D, de Miguel Olalla S, Fontán G, Ferreira A, García-Rodríguez MC, Madero L, et al. Phenotypical and functional characterization of herpesvirus saimiri-immortalized human major histocompatibility complex class II-deficient T lymphocytes. Tissue Antigens (1998) 51(3):250–7. doi: 10.1111/j.1399-0039.1998.tb03099.x
61. Zapata DA, Pacheco-Castro A, Torres PS, Ramiro AR, San José E, Alarcón B, et al. Conformational and biochemical differences in the TCR.CD3 complex of CD8(+) versus CD4(+) mature lymphocytes revealed in the absence of CD3gamma. J Biol Chem (1999) 274(49):35119–28. doi: 10.1074/jbc.274.49.35119
62. Torres PS, Zapata DA, Pacheco-Castro A, Rodríguez-Fernández JL, Cabañas C, Regueiro JR. Contribution of CD3 gamma to TCR regulation and signaling in human mature T lymphocytes. Int Immunol (2002) 14(11):1357–67. doi: 10.1093/intimm/dxf095
63. Torres PS, Alcover A, Zapata DA, Arnaud J, Pacheco A, Martín-Fernández JM, et al. TCR dynamics in human mature T lymphocytes lacking CD3 gamma. J Immunol (2003) 170(12):5947–55. doi: 10.4049/jimmunol.170.12.5947
64. Martín-Fernández JM, Cabanillas JA, Rivero-Carmena M, Lacasa E, Pardo J, Anel A, et al. Herpesvirus saimiri-transformed CD8+ T cells as a tool to study chediak-higashi syndrome cytolytic lymphocytes. J Leukoc Biol (2005) 77(5):661–8. doi: 10.1189/jlb.0904500
65. Reiné J, Busto EM, Muñoz-Ruiz M, Rossi NE, Rodríguez-Fernández JL, Martínez-Naves E, et al. CD3γ-independent pathways in TCR-mediated signaling in mature T and iNKT lymphocytes. Cell Immunol (2011) 271(1):62–6. doi: 10.1016/j.cellimm.2011.06.009
66. Bravo García-Morato M, Aracil Santos FJ, Briones AC, Blázquez Moreno A, Del Pozo Maté Á., Domínguez-Soto Á., et al. New human combined immunodeficiency caused by interferon regulatory factor 4 (IRF4) deficiency inherited by uniparental isodisomy. J Allergy Clin Immunol (2018) 141(5):1924–1927.e1918. doi: 10.1016/j.jaci.2017.12.995
Keywords: Schimke, SIOD, SMARCAL1, immunosuppression, T-cell, lymphopenia, kidney failure
Citation: Marin AV, Jiménez-Reinoso A, Mazariegos MS, Román-Ortiz E and Regueiro JR (2022) T-cell receptor signaling in Schimke immuno-osseous dysplasia is SMARCAL1-independent. Front. Immunol. 13:979722. doi: 10.3389/fimmu.2022.979722
Received: 27 June 2022; Accepted: 22 September 2022;
Published: 18 October 2022.
Edited by:
Martin Perez-Andres, University of Salamanca, SpainReviewed by:
Rohini Muthuswami, Jawaharlal Nehru University, IndiaCatarina Gregório Martins, New University of Lisbon, Portugal
Copyright © 2022 Marin, Jiménez-Reinoso, Mazariegos, Román-Ortiz and Regueiro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: José R. Regueiro, cmVndWVpcm9AdWNtLmVz