 Liguang Fang
Liguang Fang Kunjing Liu1†
Kunjing Liu1† Cun Liu
Cun Liu Xiaomin Wang
Xiaomin Wang Wenzhe Ma
Wenzhe Ma Wenhua Xu
Wenhua Xu Jibiao Wu
Jibiao Wu Changgang Sun
Changgang Sun
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 02 September 2022
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.979116
This article is part of the Research Topic Understanding Regulation of T cell’s Cytotoxic Function to Optimize Immunotherapy for Cancer Patients View all 9 articles
The development and response to treatment of tumor are modulated by inflammation, and chronic inflammation promotes tumor progression and therapy resistance. This article summarizes the dynamic evolution of inflammation from acute to chronic in the process of tumor development, and its effect on T cells from activation to the promotion of exhaustion. We review the mechanisms by which inflammatory cells and inflammatory cytokines regulate T cell exhaustion and methods for targeting chronic inflammation to improve the efficacy of immunotherapy. It is great significance to refer to the specific state of inflammation and T cells at different stages of tumor development for accurate clinical decision-making of immunotherapy and improving the efficiency of tumor immunotherapy.
There seems to be a role for chronic inflammation in modulating all phases of malignant disease, including morbidity and mortality (1). There is substantial evidence that chronic inflammation caused by persistent infections, autoimmune reactions, or exposure to toxic chemicals increases the risk of tumor (2). However, not all chronic inflammation leads to tumors, the tissue where chronic inflammation occurs also affects cancer development. Inflammation of the gut or liver can greatly increase the risk of tumor, while inflammation of the joints or muscles rarely affects the development of tumor (3).
Tumor-associated inflammation underwent a transition from acute inflammation to chronic inflammation. Acute inflammation is a protective response elicited by injury and infection, being positive for immune activation. While, chronic inflammation supports immunosuppression rather than immune activation, thereby resulting in the growth and progression of tumors (4). Acute inflammation makes a difference in tissue regeneration and plays an important immunostimulatory function (5). However, due to the persistence of damage, a repair can not be completed in time, and inflammation is transformed from acute inflammation to chronic inflammation. Chronic inflammation can lead to genetic mutations and epigenetic changes in normal tissues that drive malignant transformation (6).
Largely due to the exhaustion of T cells, chronic inflammation inhibits immunity, leading to tumor progression. T cell exhaustion is one of the mechanisms that tumor cells are able to escape from immune control (7).
As monoclonal antibodies are increasingly used in tumor medicine, more options are available across a wide range of oncological indications. Some patients, nevertheless, may not benefit from an inhibitor of immune checkpoint receptors. Even patients who benefit do not last. It appears that tumor-induced inflammation is an important driver of malignant progression in many solid malignancies. A chronic inflammatory response is a significant contributing factor to a majority of solid and hematopoietic tumors (3). Immune-related adverse events (irAEs) are also significant factors that limit the use of immune checkpoint blockers besides drug resistance in patients who are suitable for immunotherapy (8). T-cell exhaustion caused by chronic inflammation is a negative feedback regulator in response to infection and tissue damage. Immune-related adverse reactions occur partly because immunotherapy disrupts this regulatory mechanism (9). Immune checkpoint blockers partially reversed T cell exhaustion, breaking this negative feedback mechanism, increasing the immunotherapeutic efficiency. It also unties the reins of immune cells and increases the damage of immune cells to normal tissues, resulting in serious immune-related events. Therefore, it is greatly significant to deeply learn the complex interaction between the chronic inflammation microenvironment and immunity to improve the effect of immunotherapy and reduce related adverse events.
In addition to immunotherapy, traditional radiotherapy and chemotherapy remain clinically mainstream. However, radio-chemotherapy induced dying cells are of great concern, a variety of cell death patterns may occur, as well as the release of complex factors, and promote the occurrence of chronic inflammation in the tumor microenvironment, thus orchestrating repopulation cascades of tumor (10). Dead cells enhance the survival of residual viable tumor cells, boost proliferation, and hasten tumor cell metastasis (11). Therefore, further study of tumor-related chronic inflammation and finding appropriate intervention targets is not only necessary to improve the effect of immunotherapy, but also necessary to improve the long-term therapeutic effect of radiotherapy and chemotherapy.
For tumor cells, the immune cells that play the main effect are CD8+ T cells, but due to the chronic inflammatory continuous stimulation, the killing function of T cells will gradually decline, presenting a state of exhaustion. T cell exhaustion is the body’s self-protective mechanism to prevent excessive immunity from damaging normal tissues in the tumor microenvironment. T cell exhaustion is a compromise of the autoimmune system for the repair of long-standing viral, bacterial infections, or chronic tissue damage machine-processed. In these settings, perhaps (partial) T cell exhaustion strikes the balance between maintaining limited infection control capacity and moderating immuno-pathology. For a long time, exhaustion is thought to represent a series of cellular dysfunctions. Exhausted cells do make a significant contribution to infection control, although they can not clear it all (12).
The loss of function in exhausted CD8+ T cells occurs hierarchically, with some properties being lost before others (13). Usually, the first thing that gets lost in exhausted T cells is their ability to produce IL-2 and to proliferate. Other properties are lost during the intermediate stages of cell exhaustion, including the ability to produce tumor necrosis factor. When cells become severely exhausted, they can no longer produce large quantities of interferon-gamma (IFN-γ) or beta-chemokines or to degranulate. When T cells are completely exhausted, they become exhausted T cells with total disappearance of their effector functions (14). Exhaustion is often consistent with the expression of inhibitory surface receptors, including PD1, CD160, 2B4, LAG-3, and CTLA-4 (7). Poor effector function and persistent expression of inhibitory receptors are key features of T cell exhaustion (15).
In the inflammatory microenvironment, T cell failure is regulated by inflammatory factors (IL-10/IL-35/IL-7/IL-21/TGF-β, etc.) (16) inflammatory cells (Treg, MDSCS, macrophages, neutrophils, mesenchymal cells, etc.), and related nutrients (glucose, amino acids, fatty acids, etc.) (12). The specific mechanism of action will be discussed in the following article.
In tissues, inflammation occurs earlier than in tumors. Local inflammation is largely caused by infection or the stimulation of environmental factors, and systemic inflammation is mostly related to obesity and metabolic diseases. Both local and systemic inflammation interact through the circulatory system. The tumor and related radiotherapy and chemotherapy can promote both local inflammation and systemic inflammation. During tumor progression, inflammation undergoes a transition from acute to chronic, and its effect on T cells also switches from activation to promotion of exhaustion. It is very essential to understand how inflammation transforms in different stages of tumor growth to improve the effectiveness of immunotherapy.
Pathogen microbial infection and local stimuli are the main causes of chronic local inflammation, such as Helicobacter pylori infection, Hepatitis virus infection pancreatitis, colitis, esophagitis, cholangitis, etc. (17). Environmental and chemical human carcinogens induce pro-tumor inflammation, including UV, aflatoxins, nitrosamines, and tobacco (18, 19). Local chronic inflammation activates the NF-κB pathway, which suppresses apoptosis with malignant potential, leading to a malignant transformation in the tissue (20). The activation of NF-κB plays a vital role in the control of the communication between tumor cells and inflammatory cells (21). The microenvironment of a tumor and normal tissues are always filled with activation signals. NF-κB pathway activation occurs when p53 is dysfunctional, issuing in increased expression of inflammatory genes (22). NF-κB activity can be induced by many factors, such as TNFα, IL-1β, LPS, ionizing radiation, ROS, etc. (23). The ROS produced by neutrophils and macrophages can not only activate the NF-κB pathway, but also cause DNA disruption, induce gene mutations, and increase susceptibility to tumors (24). In addition, in the chronic inflammatory microenvironment, the formation of tumor progression is conducive to the immune suppression microenvironment, this inhibitory microenvironment will not only lead to immune escape but also a screening of tissues, only to adapt to this inhibitory environment of malignant cells that can survive, this selection process becomes immune editing (25). In conclusion, chronic inflammation plays an important role in the malignant change of cells, and later immune escape.
Systemic inflammation can evolve from local inflammation or result from systemic metabolic diseases. The local immune response consists of cytokines derived from a tumor and inflammatory proteins derived from the host, as well as infiltrating immune cells, acting in the local tumor micro-environment (26). On the other hand, systemic inflammation is also associated with small molecules in local inflammation. The difference is that these mediators flow in the systemic circulation and lead to paraneoplastic syndromes in cancer patients. It is evident that mediators in the systematic inflammatory microenvironment and the local microenvironment communicate extensively (26).
As a result of adipose expansion and chronic obesity, an inflammatory program is activated, permanently skewing the immune system in favor of inflammation (27). There is ample evidence that obesity is closely associated with colon tumors, oesophageal tumors (adenocarcinoma), renal tumor (renal cell carcinoma), breast tumor (postmenopausal), and endometrial tumor (28).
Adipose tissue necrosis causes macrophage infiltration, while the metabolism of adipose tissue itself, produces chemokines and resident factors, causing the indwelling of macrophages. Invasive macrophages produce inflammatory factors to further shape the inflammatory microenvironment. In addition to TAMs, MDSCs and DCs are also recruited by adipose tissue, both of which can promote chronic inflammation in the context of obesity (29, 30). In adipose tissue, the number or proportion of Treg cells decreases will further promote the development of chronic inflammation (31). The adipose tissue is directly resulted from the nutrient excess, which also leads to reactive oxygen species (ROS) production sourced from mitochondrial (32). ROS stimulates chronic inflammation by activating the upstream kinases I-κB and JNK to provoke proinflammatory transcription factors, such as AP-1 and NF-κB (33). Inflammation in adipose tissue may also be influenced by commensal flora metabolism, through bacterial products (34). The inflammation caused by adipose tissue is chronic, and metabolic. It does not disappear unless the adipose tissue disappears.
According to the current study, the gut microbiota and its metabolites can trigger inflammation in obese and diabetic individuals. The chronic inflammatory response further increases insulin resistance, and the two promote each other, forming a bad cycle (35).
Chronic inflammation plays an important role in the occurrence, development, distant metastasis, and recurrence of most tumors (Figure 1). That is, the occurrence of certain tumors and inflammation are not closely related, but the treatment of inflammation to intervene, can still achieve positive efficacy (36, 37).
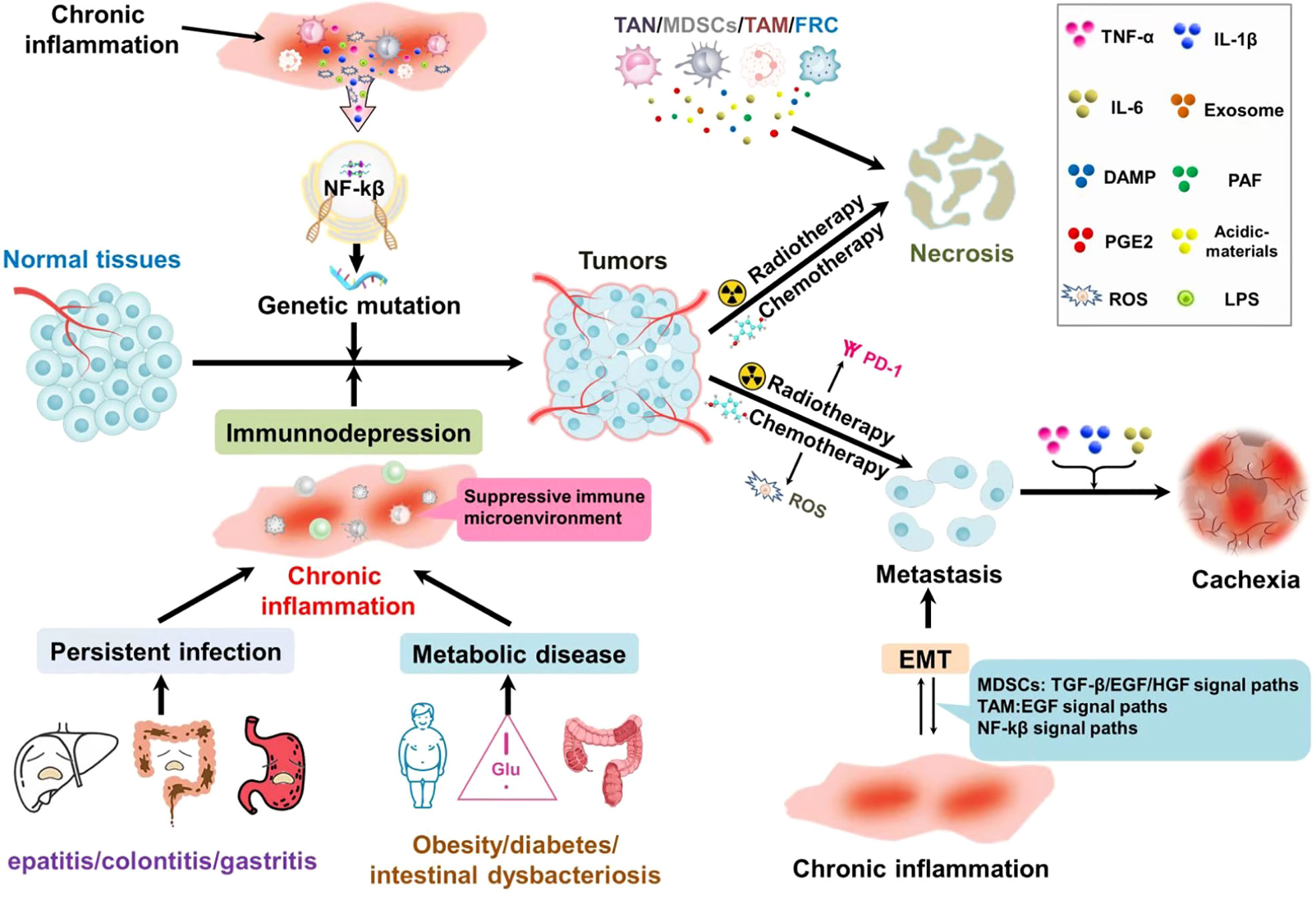
Figure 1 Chronic inflammation of different sources accompanies the whole process of tumor development: in precancerous lesions, chronic local irritation, and chronic inflammation caused by systemic metabolic diseases not only activate the NF-κB pathway, but also induce gene mutations that lead to tissue malignant transformation. At the same time, it can inhibit the immune monitoring and make the tumor cells escape. Tumor treatment could result in the necrosis of tumor cells. Necrosis of the tumor cells chemotactic inflammatory cell aggregation, suppression of immunity; At the same time, a large number of cytokines are released to generate factor storms leading to the aggregation and recurrence of tumor cells. Chronic inflammation and epithelial-mesenchymal transformation promote each other and form a vicious cycle, which is conducive to distant metastasis of tumors. Inflammatory factors are closely related to the production of tumor cachexia.
Acute inflammation is caused by the recognition of pathogen-associated molecular patterns (PAMPs), or damage-associated molecular patterns (DAMPs), which activate pro-inflammatory cytokines and chemokines, thereby enhancing immune responses (38). Granulocytes constitute the majority of infiltrating inflammatory cells in acute inflammation (39). Acute inflammatory responses typically stimulate dendritic cell (DC) maturation and antigen presentation, favoring the activation of CD8+ T cells. During the tumor, acute inflammation mostly occurs in the early stage of the tumor (40), which is caused by tumor-specific antigens or tumor-associated antigens (41). Acute inflammation can also be led by tumor growth and invasion to destroy normal tissue (42), tumor-specific chemotherapy and low-dose, radiation (43).
The simultaneous occurrence of tissue destruction and repair is a hallmark of chronic inflammation. The main infiltrating immune cells at sites of chronic inflammation are macrophages and lymphocytes (44). Chronic inflammation leads to the presence of high amount of immunosuppressive cells (TAM2,Treg cells, MDSCs etc.) and cytokines in the tumor microenvironment (45), which induces T cell exhaustion and produces an immunosuppressive microenvironment. Before the occurrence of tumors, chronic inflammation already exists and produces immunosuppression, which is a risk factor for tumorigenesis. In the process of the tumor, most solid tumors develop chronic inflammation that promotes tumor progression (45). Tumor cells and stromal cells release chemokines, recruit macrophages and neutrophils (46), tumor growth and invasion can damage normal tissues and release damage-associated molecular patterns that activate granulocytes (42). Tumor-specific metabolic patterns result in an acidic and hypoxia TME, leading to neovascularization and recruitment of macrophages (47).
Chemoradiation-induced death of tumor cells releases complex factors, thereby coordinating the cascade of tumor regeneration. Dead cells enhance the survival of residual live tumor cells and promote metastasis. In addition, dead cells can also contribute to chemoradiation-induced side effects (11, 48). Cells dying from chemoradiotherapy also secrete platelet activation factor (PAF)-like phospholipids, which further exacerbate immunosuppression by binding to the PAF receptor (49, 50). Tumor-related therapy can also lead to drug resistance in patients (51).
Chemotherapy can trigger inflammation through ROS and damage-related proteins (DAMP) (52). Chemotherapy or immunotherapy damages the normal tissue, thereby inducing the migration of macrophages, and cytotoxic lymphocytes to damaged and non-damaged sites, causing chronic inflammation of the tissue (53). Tumor treatment will also promote the production of ROS (54). Reactive oxygen species result in mitochondrial DNA (mtDNA) and nuclear DNA damage. DAMP from the mitochondria enters the cellular cytoplasm, activates intercellular spatial receptors, and further triggers the recruitment of immune effector cells (55). Computer modeling study shows that increased tumor cell death from chemoradiotherapy leads to short-term tumor shrinkage, but ultimately can accelerate tumor growth and metastasis in the long-term (56).
Antitumor immunity can be stimulated by low-dose radiation therapy (57). Radiotherapy can contribute to the expression of proinflammatory chemicals, cytokines, and NK cells, stimulate the formation of inflammation and be conducive to activating the immune response (58). High-dose radiotherapy acts directly on sphingal membrane-bound sphingolipid, decomposes sphingolipid, and generates ceramide by sphingingholipase. This process results in the accumulation of ceramides in damaged tissues. Angiogenesis is suppressed by ceramides, which damage endothelial cells (59). Immune cells are less likely to be attracted to damaged/inflammatory regions through blood flow areas when angiogenic inhibition is present. It is a failure to recruit immune cells (macrophages, T cells, and endocytic) resulting in PD-L1 accumulation and immunosuppression.
EMT is important for tumor metastasis. The EMT program not only enables tumor cells to exhibit enhanced invasiveness, stem cell-like characteristics, and resistance to apoptosis, but also stimulates tumor cells to produce pro-inflammatory factors. At the same time, inflammation further induces EMT. Therefore, in an alliance of metastatic growth, these phenomena may support each other (60). In the tumor microenvironment, inflammatory cells can effectively promote EMT. Macrophages induce EMT by the production of EGF (61). Bone marrow-derived suppressive cells infiltrate the primary tumor and induce EMT in tumor cells through the TGF-β, EGF, and HGF signaling pathways, leading to metastasis and the spread of tumor cells (62). In addition to inflammatory cells, many inflammatory factors also play important roles in EMT, such as TGF-β、IL-6、CCL-20/CCL-8 et al. (63–65). In conclusion, chronic inflammation not only suppresses the anti-tumor immunity but also promotes distant tumor metastasis by promoting EMT.
Tumor cachexia, also known as wasting syndrome, is closely associated with chronic inflammation. Cachexia is characterized by severe atrophy of skeletal muscle and gradual weight loss due to partial adipose tissue. Epidemiological data suggest that circulating CRP levels are associated with cachexia levels and mortality risk in patients. Inflammatory cytokines such as IL-6, IL-1β, and TNF-α are boosted in the TME in patients with systemic or cachexia (66). Pro-inflammatory factors have direct causal relationships with cachexia. TNF-α, tumor proteolytic inducible factors and lipid mobilization factors are directly relevant to the breakdown of muscle and adipose tissue (67). Other pro-inflammatory cytokines, including IL-1 and IL-6 induced a strong reduction in transporters involved in bile formation and bile acid secretion, further aggravating the development of cachexia (68).
Different aspects of inflammation seem to play a role in all stages of malignant disease. Although acute inflammation can stimulate immunity, tumor-associated inflammation suppresses immunity and promotes tumor progression (69). The suppression of chronic inflammation on immunity is mainly reflected in the induction of T cell exhaustion, so this section discusses in detail how chronic inflammation regulates T cell exhaustion (Figure 2). It is concluded that the signals regulating T cell exhaustion are mainly divided into the following three categories. Sustained antigenic stimulation (signal 1) from viruses or tumor cells is a central factor in T cell exhaustion (70). Proinflammatory cytokines and inhibitory cytokines (signal 2) produced by virus-infected cells, immunoregulatory cells, and tumors are important players in regulating T cell exhaustion in chronic inflammation. Inhibitory receptors on the surface of T cells (signal 3) provide a negative costimulatory signal, which blocks the activation of T cell effectors and renders T cells unable to mount a robust immune response (71).
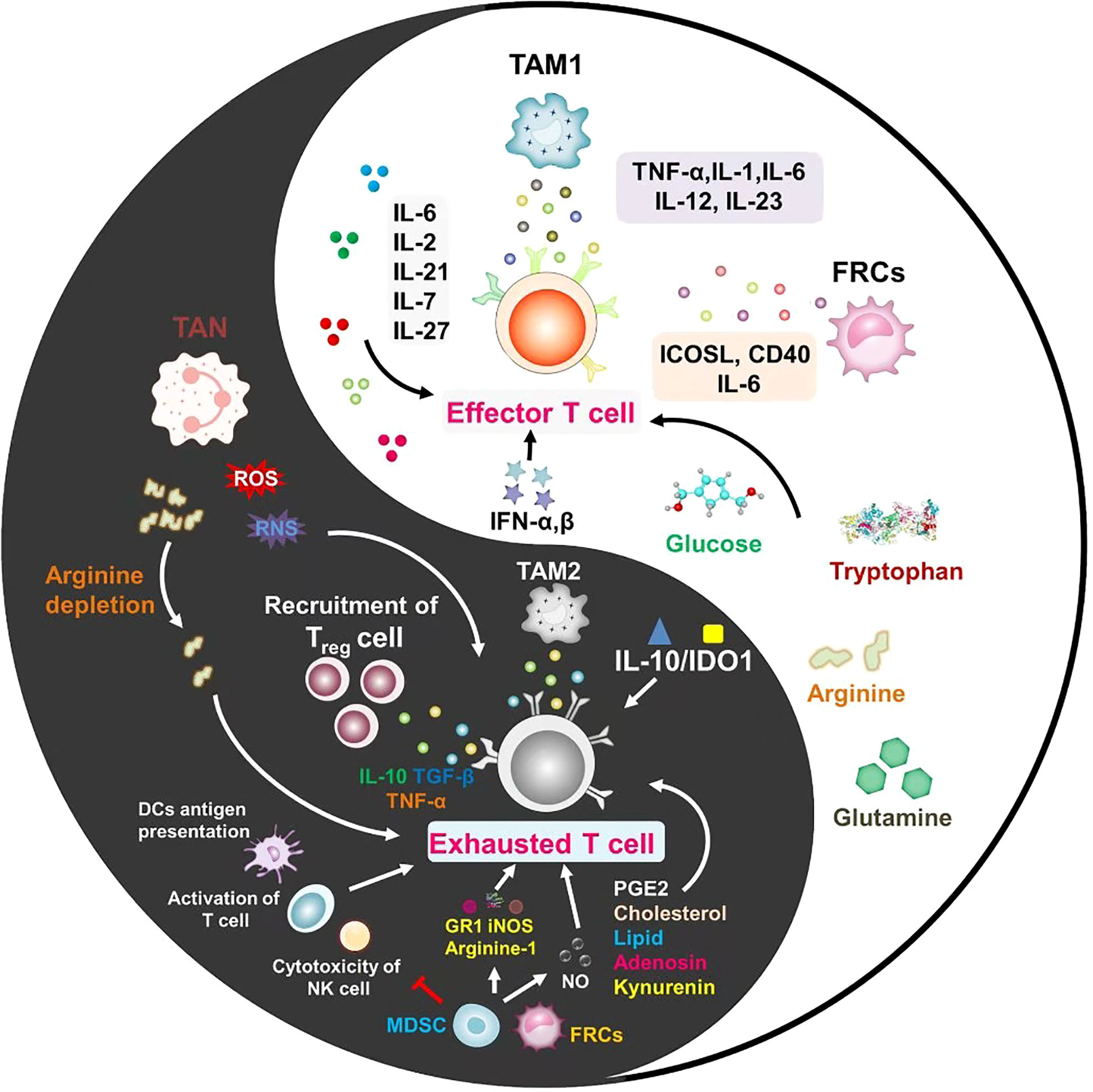
Figure 2 Every coin has two sides, and inflammation is no exception. In the tumor microenvironment, the role of inflammatory cells and inflammatory factors on T cells is not static. The different mechanisms between inflammatory factors for promoting T cell activation or exhaustion are important issues to be considered in future studies.
It has been conducted for the research on immunosuppressive receptors such as PD-1, TIM-3, LAG3, CTLA-4 and BTLA (72). However, there are still few studies on the mechanisms regulating receptor and ligand expression in tumor-associated chronic inflammation. The expression of inhibitory receptors and their ligands plays a critical role in T cell exhaustion (73).
It is one of the hallmarks of T cell exhaustion that the expression of inhibitory receptors on the surface of T cells. The nuclear factor of activated T cells (NFAT) controls CD8+ T cell exhaustion by binding to the promoter of exhaustion-related genes like Pdcd1 (PD-1) and Havcr2 (TIM3) (74). STAT3/STAT4 can induce the expression of PDCD1 by acting on the transcription factor NFAT (75). This may partly explain the specific mechanism how chronic inflammation induces T cell exhaustion (76). Nuclear receptor 4A (Nr4a) and Thymocyte selection-associated high mobility group box protein (Tox) transcription factors promote T cell exhaustion by acting on NFAT. Tox2 transcription factor and Nr4a transcription factor may be promising targets for future immunotherapy (77). T-bet directly inhibits the transcription of PD-1-encoding genes, resulting in decreased expression of other inhibitory receptors. Sustained antigenic stimulation leads to T-bet downregulation, leading to more severe exhaustion of CD8+ T cells (78). The progression of T cell exhaustion is associated with additional repressor genes and transcriptional pathways, such as Forkhead box transcription factor O1 (Foxo1) (79), B lymphocyte-induced maturation protein 1 (Blimp-1) (80), Basic leucine zipper transcription factor ATF-like -interferon-regulatory factor (BATF-IRF) interaction (79).
Inhibitory ligands on the surface of immune cells and tumor cells are also important factors affecting T cell exhaustion. Hyperactivated ALK signaling pathway promotes PD-L1 expression via STAT3 which is activated by NPM-ALK gene fusion (80). Other pathways that are associated with chronic inflammation such as the NF-kB pathway, PTEN/PI3K pathway and MAPK pathway are all involved in the regulation of PD-L1 expression (81–84).
In conclusion, soluble microkinis have an important role to play in regulating immunity (Table 1), in inflammatory microenvironment and even in a sense, the composition of different types of cytokines determines the quality of T cell pathogen-specific response. Inflammation-related factors can not only induce the expression of inhibitory receptors on the T cell surface, but also activate oncogenic pathways and alter the expression of inhibitory ligands (Table 1). Notch signaling promotes T cell exhaustion by affecting NFAT transcription factors (103). Notch signaling is involved in both malignancies and chronic inflammatory diseases (104, 105). IL-6 can promote T cell exhaustion through IL-6/STAT3/PD-1 transcription regulation (106). Other pro-inflammatory cytokines in TME promote the expression of PD-1 on the surface of T cells, leading to the exhaustion of T cells such as Il-10,TGF-β (106–109). Inflammatory factors (IFN-a, IFN-b, TNF-α, TGF-β) can also induce PD-L1 expression and attenuate immune cell activation (110).
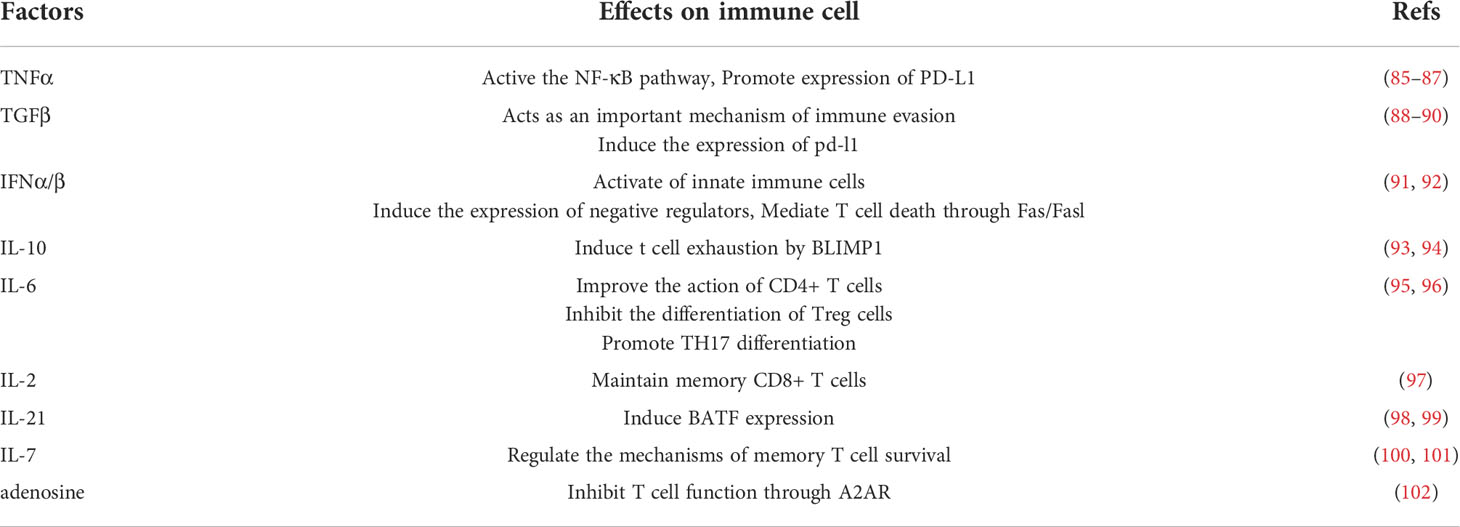
Table 1 Factors in chronic inflammation.
Inflammation-related cells can promote T cell exhaustion, possibly through cell-surface immunosuppressive ligands or by self-secreting inhibitory inflammatory factors (Table 2). Immunosuppressive ligands on the surface of tumor cells and macrophages (127), bind to receptors on the T cell surface, inhibiting the killing function of T cells and promoting the exhaustion of T cells (128). TAMs and MDSCs were associated with the expression of PD-1 on the T cell surface (129, 130). Treg cell-derived IL-10 and IL-35 promote BLIMP1-dependent depletion of CD8+ TILs, thereby limiting effective antitumor immunity (94).
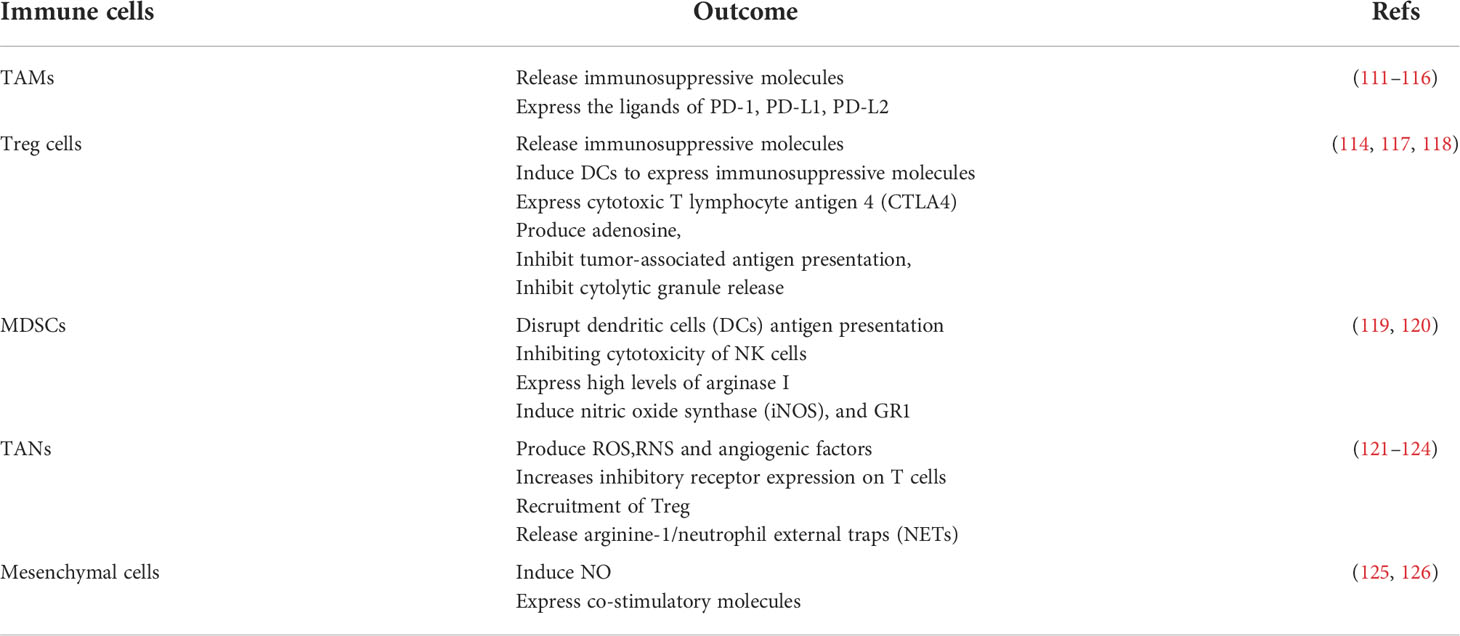
Table 2 Immune cells in chronic inflammation.
When the inhibitory receptor on the surface of T cells binds to the corresponding ligand, it can block the binding of other stimulatory ligands and inhibit the activation of T cells (131). Inhibitory receptors can induce a decrease or disappearance of the expression of intracellular receptors that positively regulate immunity or costimulatory factors (132). In addition, inhibitory receptors induce the expression of certain inhibitory genes that promote T cell exhaustion (133).
As tumor treatment enters the immune era, the application of immune checkpoint blockers has become more and more extensive. T-cell exhaustion is a negative feedback mechanism in the body, and immune checkpoint blockers directly disrupt this negative feedback regulatory state, sometimes resulting in immune cells destroying normal tissues in the body. This phenomenon is known as immune-related adverse reactions, which can sometimes be more deadly than tumors. Therefore, it is greatly important for the safe application of immunotherapy to keep the balance between the state of T cells and the body’s protective mechanism.
Moreover, immune checkpoint blockers can partially reverse T cell exhaustion, but sometimes the treatment is not durable. One of the reasons for this is the continuous stimulation of the chronic inflammatory environment in the tumor (131).
For the T cell exhaustion induced by chronic inflammation, researchers have developed many treatments that are combined with immune checkpoint blockers to improve treatment responsiveness and persistence of efficacy. The first is the simultaneous use of multiple immune checkpoint blockers (134). Since T cell failure is not only manifested by elevated PD-1 expression, other immune checkpoints also make a difference. The simultaneous application of multiple immune checkpoints blockers can not only directly reverse the T cell exhaustion, but also increase the glucose uptake and utilization ability of T cells, thus conducive to the activation of T cells (135).
While applying immunotherapy, the combination with traditional radiotherapy can also increase the effect of immunotherapy. Radiation therapy can increase the infiltration of T cells while causing acute inflammation can enhance cellular immunity (136). Certain chemotherapeutic drugs or radiation can induce immunogenic death of tumor cells, followed by the release of tumor antigens which are recognized by antigen-presenting cells and facilitate the activation of T cells (137).
Both local and systemic chronic inflammation increases the incidence of malignancies and is closely associated with a poor prognosis (27). Anti-inflammatory treatment (Table 3) and the treatment for metabolic diseases are beneficial to prevent tumors; the combined application of immune checkpoint blockers is expected to enhance therapeutic efficiency of immune checkpoint blockers and delay tumor progression (143).

Table 3 Anti-inflammatory treatment are beneficial to prevent tumors.
In addition to systemic metabolism, the lack of amino acids, glucose, cholesterol, and the accumulation of lactate and lipid can also affect T cell exhaustion, and targeting these metabolic targets for treatment will also strongly promote T cell activation (144). Immune checkpoints inhibit T cell function by inhibiting glycolysis in immune cells. So immune checkpoint blockers can restore glucose uptake by immune cells (145). Blocking the tumor metabolism of glutamine not only brings about reduced hypoxia, acidosis, and nutrient consumption in the tumor microenvironment but also promotes T cell activation and extends the life span of T cells (146). Targeted arginase (ARG) restores arginine levels, leading to tumor regression and improved T cell function (147). The combination of the ARG1 (arginase 1) targeted vaccine with anti-PD-1 also leads to increased T cell infiltration and promotes T cell activation (148). Adenosine-producing enzyme CD73 is inhibited by mAb19 and adenosine-mediated T cell exhaustion is suppressed in vitro by mAb19 (149).
Strategies that target fatty acid synthases or fatty acid oxidation to improve lipid abundance and thereby improve immune efficacy are beneficial in mouse tumor models (150). The accumulation of lactic acid can inhibit immunity, and one way to target lactic acid is to inhibit lactic acid transporters to reduce lactate acid accumulation (151).
Cytokines in the inflammatory microenvironment have different directions on T cell exhaustion. Increasing factors that antagonize T cell exhaustion or blocking factors that promote T cell exhaustion, combined with immune checkpoint blockers, has been actively explored in anti-tumor therapy. During chronic viral infection, increasing the IL-2 or IL-7 content in the inflammatory microenvironment can improve the virus-specific CD8+ T cell response and accelerate viral clearance (152, 153). Blocking the interleukin-10 receptor increases the effect of CD8+ T cells and accelerates the rate of virus clearance (154). The blockade of TGF-β, TNF-α, also showed meaningful results in reversing T cell exhaustion (155, 156).
In addition to targeting their secreted inflammatory factors, direct intervention in the polarization and recruitment of inflammatory cells is also a promising therapeutic modality. Reduction of Treg cells or block of Treg cell differentiation may allow CD8+ T cells to increase their immune surveillance of tumor cells (157). Targeting MDSCs with monoclonal antibodies restored the function of TILs (119). Colony-stimulating factor 1 (CSF-1) as a signal for macrophage recruitment and aggregation, using a small molecule PLX3397 that effectively inhibits the activity of the CSF-1R tyrosine kinase, can effectively reduce macrophage recruitment and improve tumor recurrence (158). In addition to blocking recruitment, M2 macrophages can be reeducated. It has been shown that blocking NF-κB activation in TAMs can convert TAMs from a tumor-promoting M2 phenotype to an m1-similar cytotoxic phenotype, thereby alleviating immunosuppression and enhancing the tumor control effect (159). Neutrophils are able to promote angiogenesis and promote distant tumor metastasis. Targeted granulineolytic colony-stimulating factors (G-CSF) can inhibit the recruitment of neutrophils and enhance the efficacy of antiangiogenic agents (160). The combination of therapy targeting the inflammatory microenvironment with immune checkpoint blockers is expected to further improve the efficiency of immunotherapy in clinical application.
Cancer development and its response to therapy are modulated by chronic inflammation, and chronic inflammation promotes tumor progression and therapy resistance. We discuss in detail how chronic inflammation induces T cell exhaustion in tumors and the prospects of targeting chronic inflammation in combination with immune checkpoint blockers for tumor therapy.
In addition, this article summarizes the dynamic evolution of inflammation and the results of different effects on the body’s immunity during tumor development. Discuss the positive role of inflammation and T cell exhaustion in maintaining health from a new perspective. The research in this paper has important reference significance for making the accurate clinical decision in different stages of tumor development, by combining with the specific state of inflammation and T cells.
In the treatment of reversing T cell exhaustion, in addition to directly using immune checkpoint blockers to act on CD8+ T cells, targeting related helper cells can also effectively promote T cell activation. At present, there is further research on the exhaustion of CD4+ T cells, which is beneficial to stimulating stronger anti-tumor immunity (161). Fibroblasts retain an effective inflammatory environment in chronic inflammation, resulting in immunosuppression in malignant tumors (162). Future studies could further focus on the link between associated helper cells and T cell exhaustion, providing more targets for reversing T cell exhaustion.
Inflammation factors in the tumor microenvironment play an dual-directional and dynamic role in the regulation of T cell exhaustion (163). IL-2 has a dual role in regulating tumor-associated immunity (164). IL-2 can promote the activation and proliferation of CD8+ T cells in the early stage of tumor, but it can switch its function to induce the exhaustion of CD8+ T cells in the late stage of tumor. This change may be related to the increase of IL-2 content (165). Oncostatin (OSM), as a member of the Il-6 family, also has two-sided effects on immunity. In the early stages of tumors, OSM can activate immunity and inhibit tumor progression (166). However, in the late stage of the tumor, it will inhibit the immune system and promote the progression of the tumor (167). The mechanism of this effect may be related to the activation of JAK-STAT signaling pathway (168). It may also be due to the accumulation of the concentration (169), which has transformed its effect. The tumor microenvironment is a small ecosystem (170), and the interaction of tumor cells with immune cells and cytokines cannot be viewed in isolation (171).
Bioinformatics and single-cell sequencing technologies are playing an increasingly important role in tumor research. Under the background of the era of precision therapy, it is very necessary to apply relevant analysis techniques to perform accurate subgroup analysis and marker analysis for T cell exhaustion. Therefore, for the degree of T cell exhaustion and the evaluation of the effect of immune checkpoint blockers therapy, it is necessary to further study for related markers to clarify the indication of the application of immune checkpoint and the timing of stopping the immune checkpoint inhibitors, so as to improve the anti-tumor efficacy and reduce the damage of excessive immunity to the body (146). With the application of single-cell sequencing, recent studies have defined the heterogeneity of T lymphocyte populations at the genetic level (150). This approach may be used to monitor the effects of different immunotherapies on specific subsets of TIL and specific diseases. This paves the way for the use of blockers at specific immune checkpoints in the future.
The gut microbiota directly or indirectly affects the differentiation and function of immune cells, and the gut microbiota is a promising target for the treatment of inflammation-related cancers. Little is known about the mechanisms by which gut microbiota regulates T cell exhaustion. It is believed that the intervention of intestinal flora can further improve the effect of immunotherapy.
There are many studies on the mechanism of inhibitory receptors on T cells, but little is known about how the expression of inhibitory receptors is regulated in the tumor microenvironment. This also indirectly leads to less research on the specific regulatory mechanism of inflammatory mediators and inflammatory cells intervening in T cell exhaustion, only focusing on the correlation between inflammatory factors or cells and the expression of inhibitory receptors. There are some studies on the regulation of T-bet and NFAT on the expression of immunosuppressive receptors. While relative to the complex inhibitory receptors, the current research is still insufficient. Moreover, the current mechanism research is still far from the clinical application of targeted inhibitory receptor combined immunotherapy. Therefore, future research can focus more on the mechanism of the tumor microenvironment regulating the expression of immunosuppressive receptors on the basis of modern technologies such as bioinformatics and single-cell sequencing, combined with existing experimental techniques.
LF and CS conceived and designed this review. LF and KL wrote the paper. CL, XW, WM, WX, and JW provided direction and guidance throughout the preparation of this manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (81973677; 82174222) and Shandong Province Natural Science Foundation (ZR2021LZY015, ZR202103030292).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer QW declared a shared affiliation with the author WM at the time of review.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
TILs, Tumor-infiltrating T lymphocytes; TAM, Tumor-associated macrophages; TAN, Tumor-associated neutrophils; MDSCs, Myeloid-derived suppressor cells; DC, Dendritic cells; Treg, T regulatory cell; FRC, Fibroblast reticulate cells; TRCs, T region reticulate cells; CTLA4,T lymphocyte antigen 4; PD-L1, Programmed death ligand-1; PD-1, Programmed death-1; DAMPs, Damage-related proteins; PAMPs, Pathogen-associated molecular patterns; TGFβ, Transforming growth factor-beta; TNF-α, Tumor necrosis factor-alpha; PGE2, Prostaglandin E2; ROS, Reactive oxygen species; RNS, Reactive nitrogen; LPS, Lipopolysaccharides; EMT, Epithelial-to-mesenchymal transition; EGF, Epidermal growth factor; NETs, Neutrophil external traps; IDO1, Indindamine-2,3-dioxygenase; iNOS, Inducible Nitric oxide synthase; TLR5, Toll-like receptor 5; PAF, Platelet activation factor; TME, Tumor microenvironment; TSP1, thrombospondin-1; ARG, Arginase; CSF-1, Colony-stimulating factor 1; GPCRs, G protein-coupled receptors; TDAG8, T cell death-associated gene 8; OGR1,Ovarian tumor G-protein-coupled receptor 1; mtDNA, Mitochondrial DNA; TCA cycle, Tricarboxylic acid cycle; T2D, Type 2 diabetes; TOX, thymocyte selection-associated high mobility group box protein; Nr4a, Nuclear receptor 4A; Foxo1, Forkhead box transcription factor O1; Blimp-1, B lymphocyte-induced maturation protein 1; BATF, The basic leucine zipper transcription factor activating transcription factor-like; IRF, interferon-regulatory factor; OSM, Oncostatin.
1. Trinchieri G. Cancer and inflammation: An old intuition with rapidly evolving new concepts. Annu Rev Immunol (2012) 30:677–706. doi: 10.1146/annurev-immunol-020711-075008
2. Plummer M, de Martel C, Vignat J, Ferlay J, Bray F, Franceschi S. Global burden of cancers attributable to infections in 2012: A synthetic analysis. Lancet Global Health (2016) 4(9):e609–16. doi: 10.1016/s2214-109x(16)30143-7
3. Taniguchi K, Karin M. Nf-κb, inflammation, immunity and cancer: Coming of age. Nat Rev Immunol (2018) 18(5):309–24. doi: 10.1038/nri.2017.142
4. Shalapour S, Karin M. Pas de deux: Control of anti-tumor immunity by cancer-associated inflammation. Immunity (2019) 51(1):15–26. doi: 10.1016/j.immuni.2019.06.021
5. Karin M, Clevers H. Reparative inflammation takes charge of tissue regeneration. Nature (2016) 529(7586):307–15. doi: 10.1038/nature17039
6. Balkwill F, Charles K, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell (2005) 7(3):211–7. doi: 10.1016/j.ccr.2005.02.013
7. Wang J, Xu Y, Huang Z, Lu X. T Cell exhaustion in cancer: Mechanisms and clinical implications. J Cell Biochem (2018) 119(6):4279–86. doi: 10.1002/jcb.26645
8. Champiat S, Lambotte O, Barreau E, Belkhir R, Berdelou A, Carbonnel F, et al. Management of immune checkpoint blockade dysimmune toxicities: A collaborative position paper. Ann Oncol Off J Eur Soc Med Oncol (2016) 27(4):559–74. doi: 10.1093/annonc/mdv623
9. Cornberg M, Kenney L, Chen A, Waggoner S, Kim S, Dienes H, et al. Clonal exhaustion as a mechanism to protect against severe immunopathology and death from an overwhelming Cd8 T cell response. Front Immunol (2013) 4:475. doi: 10.3389/fimmu.2013.00475
10. Karagiannis G, Condeelis J, Oktay M. Chemotherapy-induced metastasis: Molecular mechanisms, clinical manifestations, therapeutic interventions. Cancer Res (2019) 79(18):4567–76. doi: 10.1158/0008-5472.Can-19-1147
11. Jiang M, Gu D, Dai J, Huang Q, Tian L. Dark side of cytotoxic therapy: Chemoradiation-induced cell death and tumor repopulation. Trends Cancer (2020) 6(5):419–31. doi: 10.1016/j.trecan.2020.01.018
12. Speiser D, Ho P, Verdeil G. Regulatory circuits of T cell function in cancer. Nat Rev Immunol (2016) 16(10):599–611. doi: 10.1038/nri.2016.80
14. Wherry E, Blattman J, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters Cd8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol (2003) 77(8):4911–27. doi: 10.1128/jvi.77.8.4911-4927.2003
15. Thommen D, Schumacher T. T Cell dysfunction in cancer. Cancer Cell (2018) 33(4):547–62. doi: 10.1016/j.ccell.2018.03.012
16. Wherry E, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol (2015) 15(8):486–99. doi: 10.1038/nri3862
17. Fishbein A, Hammock B, Serhan C, Panigrahy D. Carcinogenesis: Failure of resolution of inflammation? Pharmacol Ther (2021) 218:107670. doi: 10.1016/j.pharmthera.2020.107670
18. Kadariya Y, Menges C, Talarchek J, Cai K, Klein-Szanto A, Pietrofesa R, et al. Inflammation-related Il1β/Il1r signaling promotes the development of asbestos-induced malignant mesothelioma. Cancer Prev Res (Philadelphia Pa) (2016) 9(5):406–14. doi: 10.1158/1940-6207.Capr-15-0347
19. Takahashi H, Ogata H, Nishigaki R, Broide DH, Karin M. Tobacco smoke promotes lung tumorigenesis by triggering ikkbeta- and Jnk1-dependent inflammation. Cancer Cell (2010) 17(1):89–97. doi: 10.1016/j.ccr.2009.12.008
20. Perkins ND. Nf-kappab: Tumor promoter or suppressor? Trends Cell Biol (2004) 14(2):64–9. doi: 10.1016/j.tcb.2003.12.004
21. Fan Y, Mao R, Yang J. Nf-κb and Stat3 signaling pathways collaboratively link inflammation to cancer. Protein Cell (2013) 4(3):176–85. doi: 10.1007/s13238-013-2084-3
22. Andriani G, Almeida V, Faggioli F, Mauro M, Tsai W, Santambrogio L, et al. Whole chromosome instability induces senescence and promotes sasp. Sci Rep (2016) 6:35218. doi: 10.1038/srep35218
23. Lin Y, Bai L, Chen W, Xu S. The nf-kappab activation pathways, emerging molecular targets for cancer prevention and therapy. Expert Opin Ther Targets (2010) 14(1):45–55. doi: 10.1517/14728220903431069
24. Qin H, Wilson C, Lee S, Zhao X, Benveniste E. Lps induces Cd40 gene expression through the activation of nf-kappab and stat-1alpha in macrophages and microglia. Blood (2005) 106(9):3114–22. doi: 10.1182/blood-2005-02-0759
25. Galon J, Bruni D. Tumor immunology and tumor evolution: Intertwined histories. Immunity (2020) 52(1):55–81. doi: 10.1016/j.immuni.2019.12.018
26. Diakos CI, Charles KA, McMillan DC, Clarke SJ. Cancer-related inflammation and treatment effectiveness. Lancet Oncol (2014) 15(11):e493–503. doi: 10.1016/S1470-2045(14)70263-3
27. Saltiel A, Olefsky J. Inflammatory mechanisms linking obesity and metabolic disease. J Clin Invest (2017) 127(1):1–4. doi: 10.1172/jci92035
28. Lauby-Secretan B, Scoccianti C, Loomis D, Grosse Y, Bianchini F, Straif K. Body fatness and cancer–viewpoint of the iarc working group. N Engl J Med (2016) 375(8):794–8. doi: 10.1056/NEJMsr1606602
29. Turbitt WJ, Collins SD, Meng H, Rogers CJ. Increased adiposity enhances the accumulation of mdscs in the tumor microenvironment and adipose tissue of pancreatic tumor-bearing mice and in immune organs of tumor-free hosts. Nutrients (2019) 11(12):3012. doi: 10.3390/nu11123012
30. Ivanov S, Merlin J, Lee MKS, Murphy AJ, Guinamard RR. Biology and function of adipose tissue macrophages, dendritic cells and b cells. Atherosclerosis (2018) 271:102–10. doi: 10.1016/j.atherosclerosis.2018.01.018
31. Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med (2009) 15(8):930–9. doi: 10.1038/nm.2002
32. Wellen K, Thompson C. Cellular metabolic stress: Considering how cells respond to nutrient excess. Mol Cell (2010) 40(2):323–32. doi: 10.1016/j.molcel.2010.10.004
33. Naik E, Dixit VM. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J Exp Med (2011) 208(3):417–20. doi: 10.1084/jem.20110367
34. Le Roy T, Moens de Hase E, Van Hul M, Paquot A, Pelicaen R, Regnier M, et al. Dysosmobacter welbionis is a newly isolated human commensal bacterium preventing diet-induced obesity and metabolic disorders in mice. Gut (2022) 71(3):534–43. doi: 10.1136/gutjnl-2020-323778
35. Scheithauer T, Rampanelli E, Nieuwdorp M, Vallance B, Verchere C, van Raalte D, et al. Gut microbiota as a trigger for metabolic inflammation in obesity and type 2 diabetes. Front Immunol (2020) 11:571731. doi: 10.3389/fimmu.2020.571731
36. Greten F, Grivennikov S. Inflammation and cancer: Triggers, mechanisms, and consequences. Immunity (2019) 51(1):27–41. doi: 10.1016/j.immuni.2019.06.025
37. Huang Q, Li F, Liu X, Li W, Shi W, Liu F, et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat Med (2011) 17(7):860–6. doi: 10.1038/nm.2385
38. Medzhitov R. Origin and physiological roles of inflammation. Nature (2008) 454(7203):428–35. doi: 10.1038/nature07201
39. Ariel A, Serhan CN. Resolvins and protectins in the termination program of acute inflammation. Trends Immunol (2007) 28(4):176–83. doi: 10.1016/j.it.2007.02.007
40. Shalapour S, Karin M. Immunity, inflammation, and cancer: An eternal fight between good and evil. J Clin Invest (2015) 125(9):3347–55. doi: 10.1172/JCI80007
41. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science (2015) 348(6230):69–74. doi: 10.1126/science.aaa4971
42. Hernandez C, Huebener P, Schwabe RF. Damage-associated molecular patterns in cancer: A double-edged sword. Oncogene (2016) 35(46):5931–41. doi: 10.1038/onc.2016.104
43. Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H, et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity (2013) 38(4):729–41. doi: 10.1016/j.immuni.2013.03.003
44. Ploeger HE, Takken T, de Greef MH, Timmons BW. The effects of acute and chronic exercise on inflammatory markers in children and adults with a chronic inflammatory disease: A systematic review. Exerc Immunol Rev (2009) 15:6–41. doi: 10.1080/17461390903038470
45. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature (2008) 454(7203):436–44. doi: 10.1038/nature07205
46. Bonavita E, Galdiero MR, Jaillon S, Mantovani A. Phagocytes as corrupted policemen in cancer-related inflammation. Adv Cancer Res (2015) 128:141–71. doi: 10.1016/bs.acr.2015.04.013
47. Fan C, Zhang S, Gong Z, Li X, Xiang B, Deng H, et al. Emerging role of metabolic reprogramming in tumor immune evasion and immunotherapy. Sci China Life Sci (2021) 64(4):534–47. doi: 10.1007/s11427-019-1735-4
48. Soares M, Feres G, Salluh J. Systemic inflammatory response syndrome and multiple organ dysfunction in patients with acute tumor lysis syndrome. Clinics (Sao Paulo Brazil) (2009) 64(5):479–81. doi: 10.1590/s1807-59322009000500016
49. REVESZ L. Effect of tumour cells killed by X-rays upon the growth of admixed viable cells. Nature (1956) 178(4547):1391–2. doi: 10.1038/1781391a0
50. Rofstad E, Mathiesen B, Kindem K, Galappathi K. Acidic extracellular ph promotes experimental metastasis of human melanoma cells in athymic nude mice. Cancer Res (2006) 66(13):6699–707. doi: 10.1158/0008-5472.Can-06-0983
51. Obenauf A, Zou Y, Ji A, Vanharanta S, Shu W, Shi H, et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature (2015) 520(7547):368–72. doi: 10.1038/nature14336
52. Boopathi E, Thangavel C. Dark side of cancer therapy: Cancer treatment-induced cardiopulmonary inflammation, fibrosis, and immune modulation. Int J Mol Sci (2021) 22(18):10126. doi: 10.3390/ijms221810126
53. Wirsdörfer F, Jendrossek V. The role of lymphocytes in radiotherapy-induced adverse late effects in the lung. Front Immunol (2016) 7:591. doi: 10.3389/fimmu.2016.00591
54. Terasaki Y, Ohsawa I, Terasaki M, Takahashi M, Kunugi S, Dedong K, et al. Hydrogen therapy attenuates irradiation-induced lung damage by reducing oxidative stress. Am J Physiol Lung Cell Mol Physiol (2011) 301(4):L415–26. doi: 10.1152/ajplung.00008.2011
55. Liu X, Chen Z. The pathophysiological role of mitochondrial oxidative stress in lung diseases. J Trans Med (2017) 15(1):207. doi: 10.1186/s12967-017-1306-5
56. Enderling H, Anderson A, Chaplain M, Beheshti A, Hlatky L, Hahnfeldt P. Paradoxical dependencies of tumor dormancy and progression on basic cell kinetics. Cancer Res (2009) 69(22):8814–21. doi: 10.1158/0008-5472.Can-09-2115
57. Shigematsu A, Adachi Y, Koike-Kiriyama N, Suzuki Y, Iwasaki M, Koike Y, et al. Effects of low-dose irradiation on enhancement of immunity by dendritic cells. J Radiat Res (2007) 48(1):51–5. doi: 10.1269/jrr.06048
58. Chen J, Liu X, Zeng Z, Li J, Luo Y, Sun W, et al. Immunomodulation of nk cells by ionizing radiation. Front Oncol (2020) 10:874. doi: 10.3389/fonc.2020.00874
59. Bodas M, Pehote G, Silverberg D, Gulbins E, Vij N. Autophagy augmentation alleviates cigarette smoke-induced cftr-dysfunction, ceramide-accumulation and copd-emphysema pathogenesis. Free Radical Biol Med (2019) 131:81–97. doi: 10.1016/j.freeradbiomed.2018.11.023
60. Suarez-Carmona M, Lesage J, Cataldo D, Gilles C. Emt and inflammation: Inseparable actors of cancer progression. Mol Oncol (2017) 11(7):805–23. doi: 10.1002/1878-0261.12095
61. Ravi J, Elbaz M, Wani N, Nasser M, Ganju R. Cannabinoid receptor-2 agonist inhibits macrophage induced emt in non-small cell lung cancer by downregulation of egfr pathway. Mol carcinogenesis (2016) 55(12):2063–76. doi: 10.1002/mc.22451
62. Toh B, Wang X, Keeble J, Sim W, Khoo K, Wong W, et al. Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS Biol (2011) 9(9):e1001162. doi: 10.1371/journal.pbio.1001162
63. Pickup M, Owens P, Moses H. Tgf-β, bone morphogenetic protein, and activin signaling and the tumor microenvironment. Cold Spring Harbor Perspect Biol (2017) 9(5):a022285. doi: 10.1101/cshperspect.a022285
64. Kim MS, Jeong J, Seo J, Kim HS, Kim SJ, Jin W. Dysregulated Jak2 expression by trkc promotes metastasis potential, and emt program of metastatic breast cancer. Sci Rep (2016) 6:33899. doi: 10.1038/srep33899
65. Cheng XS, Li YF, Tan J, Sun B, Xiao YC, Fang XB, et al. Ccl20 and Cxcl8 synergize to promote progression and poor survival outcome in patients with colorectal cancer by collaborative induction of the epithelial-mesenchymal transition. Cancer Lett (2014) 348(1-2):77–87. doi: 10.1016/j.canlet.2014.03.008
66. Baazim H, Antonio-Herrera L, Bergthaler A. The interplay of immunology and cachexia in infection and cancer. Nat Rev Immunol (2021) 22(5):309–21. doi: 10.1038/s41577-021-00624-w
67. Moreli J, Corrêa-Silva S, Damasceno D, Sinzato Y, Lorenzon-Ojea A, Borbely A, et al. Changes in the tnf-Alpha/Il-10 ratio in hyperglycemia-associated pregnancies. Diabetes Res Clin Pract (2015) 107(3):362–9. doi: 10.1016/j.diabres.2015.01.005
68. Thibaut M, Sboarina M, Roumain M, Pötgens S, Neyrinck A, Destrée F, et al. Inflammation-induced cholestasis in cancer cachexia. J cachexia sarcopenia Muscle (2021) 12(1):70–90. doi: 10.1002/jcsm.12652
69. Balkwill F, Mantovani A. Inflammation and cancer: Back to virchow? Lancet (2001) 357(9255):539–45. doi: 10.1016/S0140-6736(00)04046-0
70. Utzschneider DT, Alfei F, Roelli P, Barras D, Chennupati V, Darbre S, et al. High antigen levels induce an exhausted phenotype in a chronic infection without impairing T cell expansion and survival. J Exp Med (2016) 213(9):1819–34. doi: 10.1084/jem.20150598
71. McLane LM, Abdel-Hakeem MS, Wherry EJ. Cd8 T cell exhaustion during Chronic viral infection and cancer. Annu Rev Immunol (2019) 37:457–95. doi: 10.1146/annurev-immunol-041015-055318
72. Shin DS, Ribas A. The evolution of checkpoint blockade as a cancer therapy: What's here, what's next? Curr Opin Immunol (2015) 33:23–35. doi: 10.1016/j.coi.2015.01.006
73. Borkner L, Kaiser A, van de Kasteele W, Andreesen R, Mackensen A, Haanen JB, et al. Rna interference targeting programmed death receptor-1 improves immune functions of tumor-specific T cells. Cancer Immunol Immunother (2010) 59(8):1173–83. doi: 10.1007/s00262-010-0842-0
74. Martinez G, Pereira R, Äijö T, Kim E, Marangoni F, Pipkin M, et al. The transcription factor nfat promotes exhaustion of activated Cd8+ T cells. Immunity (2015) 42(2):265–78. doi: 10.1016/j.immuni.2015.01.006
75. Agnellini P, Wolint P, Rehr M, Cahenzli J, Karrer U, Oxenius A. Impaired nfat nuclear translocation results in split exhaustion of virus-specific Cd8+ T cell functions during chronic viral infection. Proc Natl Acad Sci USA (2007) 104(11):4565–70. doi: 10.1073/pnas.0610335104
76. Austin JW, Lu P, Majumder P, Ahmed R, Boss JM. Stat3, Stat4, Nfatc1, and ctcf regulate pd-1 through multiple novel regulatory regions in murine T cells. J Immunol (2014) 192(10):4876–86. doi: 10.4049/jimmunol.1302750
77. Seo H, Chen J, González-Avalos E, Samaniego-Castruita D, Das A, Wang Y, et al. Tox and Tox2 transcription factors cooperate with Nr4a transcription factors to impose Cd8 T cell exhaustion. Proc Natl Acad Sci USA (2019) 116(25):12410–5. doi: 10.1073/pnas.1905675116
78. Kao C, Oestreich KJ, Paley MA, Crawford A, Angelosanto JM, Ali MA, et al. Transcription factor T-bet represses expression of the inhibitory receptor pd-1 and sustains virus-specific Cd8+ T cell responses during chronic infection. Nat Immunol (2011) 12(7):663–71. doi: 10.1038/ni.2046
79. Seo H, González-Avalos E, Zhang W, Ramchandani P, Yang C, Lio C, et al. Batf and Irf4 cooperate to counter exhaustion in tumor-infiltrating car T cells. Nat Immunol (2021) 22(8):983–95. doi: 10.1038/s41590-021-00964-8
80. Marzec M, Zhang Q, Goradia A, Raghunath PN, Liu X, Paessler M, et al. Oncogenic kinase Npm/Alk induces through Stat3 expression of immunosuppressive protein Cd274 (Pd-L1, B7-H1). Proc Natl Acad Sci USA (2008) 105(52):20852–7. doi: 10.1073/pnas.0810958105
81. Yang Z, Wang Y, Liu S, Deng W, Lomeli SH, Moriceau G, et al. Enhancing pd-L1 degradation by itch during mapk inhibitor therapy suppresses acquired resistance. Cancer Discov (2022) 12(8):1942–59. doi: 10.1158/2159-8290.CD-21-1463
82. Cedres S, Ponce-Aix S, Pardo-Aranda N, Navarro-Mendivil A, Martinez-Marti A, Zugazagoitia J, et al. Analysis of expression of Pten/Pi3k pathway and programmed cell death ligand 1 (Pd-L1) in malignant pleural mesothelioma (Mpm). Lung Cancer (2016) 96:1–6. doi: 10.1016/j.lungcan.2016.03.001
83. Li T, Zhang C, Zhao G, Zhang X, Hao M, Hassan S, et al. Igfbp2 regulates pd-L1 expression by activating the egfr-Stat3 signaling pathway in malignant melanoma. Cancer Lett (2020) 477:19–30. doi: 10.1016/j.canlet.2020.02.036
84. Antonangeli F, Natalini A, Garassino MC, Sica A, Santoni A, Di Rosa F. Regulation of pd-L1 expression by nf-kappab in cancer. Front Immunol (2020) 11:584626. doi: 10.3389/fimmu.2020.584626
85. Wang X, Yang L, Huang F, Zhang Q, Liu S, Ma L, et al. Inflammatory cytokines il-17 and tnf-α up-regulate pd-L1 expression in human prostate and colon cancer cells. Immunol Lett (2017) 184:7–14. doi: 10.1016/j.imlet.2017.02.006
86. Bertrand F, Montfort A, Marcheteau E, Imbert C, Gilhodes J, Filleron T, et al. Tnfα blockade overcomes resistance to anti-Pd-1 in experimental melanoma. Nat Commun (2017) 8(1):2256. doi: 10.1038/s41467-017-02358-7
87. Li Z, Zhang C, Du JX, Zhao J, Shi MT, Jin MW, et al. Adipocytes promote tumor progression and induce pd-L1 expression via tnf-Alpha/Il-6 signaling. Cancer Cell Int (2020) 20:179. doi: 10.1186/s12935-020-01269-w
88. Sia D, Jiao Y, Martinez-Quetglas I, Kuchuk O, Villacorta-Martin C, Castro de Moura M, et al. Identification of an immune-specific class of hepatocellular carcinoma, based on molecular features. Gastroenterology (2017) 153(3):812–26. doi: 10.1053/j.gastro.2017.06.007
89. Batlle E, Massague J. Transforming growth factor-beta signaling in immunity and cancer. Immunity (2019) 50(4):924–40. doi: 10.1016/j.immuni.2019.03.024
90. Park B, Freeman Z, Ghasemzadeh A, Chattergoon M, Rutebemberwa A, Steigner J, et al. Tgfβ1-mediated Smad3 enhances pd-1 expression on antigen-specific T cells in cancer. Cancer Discov (2016) 6(12):1366–81. doi: 10.1158/2159-8290.Cd-15-1347
91. McNab F, Mayer-Barber K, Sher A, Wack A, O'Garra A. Type I interferons in infectious disease. Nat Rev Immunol (2015) 15(2):87–103. doi: 10.1038/nri3787
92. Papatriantafyllou M. Regulatory T cells: Distilling regulatory T cell inducers. Nat Rev Immunol (2013) 13(8):546. doi: 10.1038/nri3506
93. Brooks D, Trifilo M, Edelmann K, Teyton L, McGavern D, Oldstone M. Interleukin-10 determines viral clearance or persistence in vivo. Nat Med (2006) 12(11):1301–9. doi: 10.1038/nm1492
94. Sawant DV, Yano H, Chikina M, Zhang Q, Liao M, Liu C, et al. Adaptive plasticity of il-10(+) and il-35(+) treg cells cooperatively promotes tumor T cell exhaustion. Nat Immunol (2019) 20(6):724–35. doi: 10.1038/s41590-019-0346-9
95. Harker J, Lewis G, Mack L, Zuniga E. Late interleukin-6 escalates T follicular helper cell responses and controls a chronic viral infection. Sci (New York NY) (2011) 334(6057):825–9. doi: 10.1126/science.1208421
96. Tanaka T, Narazaki M, Kishimoto T. Interleukin (Il-6) immunotherapy. Cold Spring Harbor Perspect Biol (2018) 10(8):a028456. doi: 10.1101/cshperspect.a028456
97. Liu Y, Zhou N, Zhou L, Wang J, Zhou Y, Zhang T, et al. Il-2 regulates tumor-reactive Cd8 T cell exhaustion by activating the aryl hydrocarbon receptor. Nat Immunol (2021) 22(3):358–69. doi: 10.1038/s41590-020-00850-9
98. Xin G, Schauder D, Lainez B, Weinstein J, Dai Z, Chen Y, et al. A critical role of il-21-Induced batf in sustaining Cd8-T-Cell-Mediated chronic viral control. Cell Rep (2015) 13(6):1118–24. doi: 10.1016/j.celrep.2015.09.069
99. Loschinski R, Böttcher M, Stoll A, Bruns H, Mackensen A, Mougiakakos DJO. Il-21 modulates memory and exhaustion phenotype of T-cells in a fatty acid oxidation-dependent manner. Oncotarget (2018) 9(17):13125–38. doi: 10.18632/oncotarget.24442
100. Cui G, Staron MM, Gray SM, Ho PC, Amezquita RA, Wu J, et al. Il-7-Induced glycerol transport and tag synthesis promotes memory Cd8+ T cell longevity. Cell (2015) 161(4):750–61. doi: 10.1016/j.cell.2015.03.021
101. He C, Zhou Y, Li Z, Farooq M, Ajmal I, Zhang H, et al. Co-Expression of il-7 improves Nkg2d-based car T cell therapy on prostate cancer by enhancing the expansion and inhibiting the apoptosis and exhaustion. Cancers (Basel) (2020) 12(7):1969. doi: 10.3390/cancers12071969
102. Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, et al. Expression of ectonucleotidase Cd39 by Foxp3+ treg cells: Hydrolysis of extracellular atp and immune suppression. Blood (2007) 110(4):1225–32. doi: 10.1182/blood-2006-12-064527
103. Mathieu M, Cotta-Grand N, Daudelin JF, Thebault P, Labrecque N. Notch signaling regulates pd-1 expression during Cd8(+) T-cell activation. Immunol Cell Biol (2013) 91(1):82–8. doi: 10.1038/icb.2012.53
104. Meurette O, Mehlen P. Notch signaling in the tumor microenvironment. Cancer Cell (2018) 34(4):536–48. doi: 10.1016/j.ccell.2018.07.009
105. Christopoulos PF, Gjolberg TT, Kruger S, Haraldsen G, Andersen JT, Sundlisaeter E. Targeting the notch signaling pathway in chronic inflammatory diseases. Front Immunol (2021) 12:668207. doi: 10.3389/fimmu.2021.668207
106. Kuo IY, Yang YE, Yang PS, Tsai YJ, Tzeng HT, Cheng HC, et al. Converged Rab37/Il-6 trafficking and Stat3/Pd-1 transcription axes elicit an immunosuppressive lung tumor microenvironment. Theranostics (2021) 11(14):7029–44. doi: 10.7150/thno.60040
107. Li C, Zuo W. Il-10 in vitro could enhance ifngamma expression and suppress pd-1 expression in Cd8 T cells from esophageal cancer patients. Exp Cell Res (2019) 379(2):159–65. doi: 10.1016/j.yexcr.2019.03.038
108. Jimbu L, Mesaros O, Neaga A, Nanut AM, Tomuleasa C, Dima D, et al. The potential advantage of targeting both pd-L1/Pd-L2/Pd-1 and il-10-Il-10r pathways in acute myeloid leukemia. Pharm (Basel) (2021) 14(11):1105. doi: 10.3390/ph14111105
109. Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. Tgfbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature (2018) 554(7693):538–43. doi: 10.1038/nature25492
110. Cha JH, Chan LC, Li CW, Hsu JL, Hung MC. Mechanisms controlling pd-L1 expression in cancer. Mol Cell (2019) 76(3):359–70. doi: 10.1016/j.molcel.2019.09.030
111. Ruffell B, Coussens L. Macrophages and therapeutic resistance in cancer. Cancer Cell (2015) 27(4):462–72. doi: 10.1016/j.ccell.2015.02.015
112. Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho C, Pryer N, et al. Macrophage il-10 blocks Cd8+ T cell-dependent responses to chemotherapy by suppressing il-12 expression in intratumoral dendritic cells. Cancer Cell (2014) 26(5):623–37. doi: 10.1016/j.ccell.2014.09.006
113. Wang L, Rubinstein R, Lines J, Wasiuk A, Ahonen C, Guo Y, et al. Vista, a novel mouse ig superfamily ligand that negatively regulates T cell responses. J Exp Med (2011) 208(3):577–92. doi: 10.1084/jem.20100619
114. Ju X, Zhang H, Zhou Z, Chen M, Wang Q. Tumor-associated macrophages induce pd-L1 expression in gastric cancer cells through il-6 and tnf-a signaling. Exp Cell Res (2020) 396(2):112315. doi: 10.1016/j.yexcr.2020.112315
115. Shan T, Chen S, Chen X, Wu T, Yang Y, Li S, et al. M2−Tam subsets altered by lactic acid promote T−Cell apoptosis through the Pd−L1/Pd−1 pathway. Oncol Rep (2020) 44(5):1885–94. doi: 10.3892/or.2020.7767
116. Ju X, Zhang H, Zhou Z, Chen M, Wang Q. Tumor-associated macrophages induce pd-L1 expression in gastric cancer cells through il-6 and tnf-a signaling. Exp Cell Res (2020) 396(2):112315. doi: 10.1016/j.yexcr.2020.112315
117. Facciabene A, Peng X, Hagemann I, Balint K, Barchetti A, Wang L, et al. Tumour hypoxia promotes tolerance and angiogenesis via Ccl28 and T(Reg) cells. Nature (2011) 475(7355):226–30. doi: 10.1038/nature10169
118. Deaglio S, Dwyer K, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by Cd39 and Cd73 expressed on regulatory T cells mediates immune suppression. J Exp Med (2007) 204(6):1257–65. doi: 10.1084/jem.20062512
119. Gabrilovich D, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9(3):162–74. doi: 10.1038/nri2506
120. Deryugina E, Zajac E, Juncker-Jensen A, Kupriyanova T, Welter L, Quigley J. Tissue-infiltrating neutrophils constitute the major in vivo source of angiogenesis-inducing mmp-9 in the tumor microenvironment. Neoplasia (New York NY) (2014) 16(10):771–88. doi: 10.1016/j.neo.2014.08.013
121. Koyama S, Akbay E, Li Y, Aref A, Skoulidis F, Herter-Sprie G, et al. Stk11/Lkb1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T-cell activity in the lung tumor microenvironment. Cancer Res (2016) 76(5):999–1008. doi: 10.1158/0008-5472.Can-15-1439
122. Coffelt S, Kersten K, Doornebal C, Weiden J, Vrijland K, Hau C, et al. Il-17-Producing Γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature (2015) 522(7556):345–8. doi: 10.1038/nature14282
123. Xiong S, Dong L, Cheng L. Neutrophils in cancer carcinogenesis and metastasis. J Hematol Oncol (2021) 14(1):173. doi: 10.1186/s13045-021-01187-y
124. Cools-Lartigue J, Spicer J, McDonald B, Gowing S, Chow S, Giannias B, et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest (2013) 123(8):3446–58. doi: 10.1172/jci67484
125. Perez-Shibayama C, Gil-Cruz C, Ludewig B. Fibroblastic reticular cells at the nexus of innate and adaptive immune responses. Immunol Rev (2019) 289(1):31–41. doi: 10.1111/imr.12748
126. Saucillo D, Gerriets V, Sheng J, Rathmell J, Maciver N. Leptin metabolically licenses T cells for activation to link nutrition and immunity. J Immunol (Baltimore Md 1950) (2014) 192(1):136–44. doi: 10.4049/jimmunol.1301158
127. Fang W, Zhou T, Shi H, Yao M, Zhang D, Qian H, et al. Progranulin induces immune escape in breast cancer via up-regulating pd-L1 expression on tumor-associated macrophages (Tams) and promoting Cd8(+) T cell exclusion. J Exp Clin Cancer Res (2021) 40(1):4. doi: 10.1186/s13046-020-01786-6
128. Deng J, Le Mercier I, Kuta A, Noelle RJ. A new vista on combination therapy for negative checkpoint regulator blockade. J Immunother Cancer (2016) 4:86. doi: 10.1186/s40425-016-0190-5
129. Loeuillard E, Yang J, Buckarma E, Wang J, Liu Y, Conboy C, et al. Targeting tumor-associated macrophages and granulocytic myeloid-derived suppressor cells augments pd-1 blockade in cholangiocarcinoma. J Clin Invest (2020) 130(10):5380–96. doi: 10.1172/JCI137110
130. Holtzhausen A, Harris W, Ubil E, Hunter DM, Zhao J, Zhang Y, et al. Tam Family receptor kinase inhibition reverses mdsc-mediated suppression and augments anti-Pd-1 therapy in melanoma. Cancer Immunol Res (2019) 7(10):1672–86. doi: 10.1158/2326-6066.CIR-19-0008
131. Chen D, Mellman I. Oncology meets immunology: The cancer-immunity cycle. Immunity (2013) 39(1):1–10. doi: 10.1016/j.immuni.2013.07.012
132. Xu X, Hou B, Fulzele A, Masubuchi T, Zhao Y, Wu Z, et al. Pd-1 and btla regulate T cell signaling differentially and only partially through Shp1 and Shp2. J Cell Biol (2020) 219(6):e201905085. doi: 10.1083/jcb.201905085
133. Quigley M, Pereyra F, Nilsson B, Porichis F, Fonseca C, Eichbaum Q, et al. Transcriptional analysis of hiv-specific Cd8+ T cells shows that pd-1 inhibits T cell function by upregulating batf. Nat Med (2010) 16(10):1147–51. doi: 10.1038/nm.2232
134. Sakuishi K, Apetoh L, Sullivan J, Blazar B, Kuchroo V, Anderson A. Targeting Tim-3 and pd-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med (2010) 207(10):2187–94. doi: 10.1084/jem.20100643
135. Chang C, Qiu J, O'Sullivan D, Buck M, Noguchi T, Curtis J, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell (2015) 162(6):1229–41. doi: 10.1016/j.cell.2015.08.016
136. Gameiro S, Jammeh M, Wattenberg M, Tsang K, Ferrone S, Hodge J. Radiation-induced immunogenic modulation of tumor enhances antigen processing and calreticulin exposure, resulting in enhanced T-cell killing. Oncotarget (2014) 5(2):403–16. doi: 10.18632/oncotarget.1719
137. Motz G, Coukos G. Deciphering and reversing tumor immune suppression. Immunity (2013) 39(1):61–73. doi: 10.1016/j.immuni.2013.07.005
138. Bosetti C, Santucci C, Gallus S, Martinetti M, La Vecchia C. Aspirin and the risk of colorectal and other digestive tract cancers: An updated meta-analysis through 2019. Ann Oncol Off J Eur Soc Med Oncol (2020) 31(5):558–68. doi: 10.1016/j.annonc.2020.02.012
139. Liu F, Wu Q, Han W, Laster K, Hu Y, Ma F, et al. Targeting integrin αvβ3 with indomethacin inhibits patient-derived xenograft tumour growth and recurrence in oesophageal squamous cell carcinoma. Clin Trans Med (2021) 11(10):e548. doi: 10.1002/ctm2.548
140. Xia W, Qi X, Li M, Wu Y, Sun L, Fan X, et al. Metformin promotes anticancer activity of nk cells in a P38 mapk dependent manner. Oncoimmunology (2021) 10(1):1995999. doi: 10.1080/2162402x.2021.1995999
141. Kwon M, Nam G, Jung H, Kim S, Kim S, Choi Y, et al. Statin in combination with cisplatin makes favorable tumor-immune microenvironment for immunotherapy of head and neck squamous cell carcinoma. Cancer Lett (2021) 522:198–210. doi: 10.1016/j.canlet.2021.09.029
142. Soldati L, Di Renzo L, Jirillo E, Ascierto P, Marincola F, De Lorenzo A. The influence of diet on anti-cancer immune responsiveness. J Trans Med (2018) 16(1):75. doi: 10.1186/s12967-018-1448-0
143. Lee BY, Hogg EKJ, Below CR, Kononov A, Blanco-Gomez A, Heider F, et al. Heterocellular osm-osmr signalling reprograms fibroblasts to promote pancreatic cancer growth and metastasis. Nat Commun (2021) 12(1):7336. doi: 10.1038/s41467-021-27607-8
144. Chapman N, Boothby M, Chi H. Metabolic coordination of T cell quiescence and activation. Nat Rev Immunol (2020) 20(1):55–70. doi: 10.1038/s41577-019-0203-y
145. Qorraj M, Bruns H, Böttcher M, Weigand L, Saul D, Mackensen A, et al. The pd-1/Pd-L1 axis contributes to immune metabolic dysfunctions of monocytes in chronic lymphocytic leukemia. Leukemia (2017) 31(2):470–8. doi: 10.1038/leu.2016.214
146. Leone R, Zhao L, Englert J, Sun I, Oh M, Sun I, et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Sci (New York NY) (2019) 366(6468):1013–21. doi: 10.1126/science.aav2588
147. Miret J, Kirschmeier P, Koyama S, Zhu M, Li Y, Naito Y, et al. Suppression of myeloid cell arginase activity leads to therapeutic response in a nsclc mouse model by activating anti-tumor immunity. J immunotherapy Cancer (2019) 7(1):32. doi: 10.1186/s40425-019-0504-5
148. Aaboe Jørgensen M, Ugel S, Hübbe M, Carretta M, Perez-Penco M, Weis-Banke S, et al. Arginase 1-based immune modulatory vaccines induce anti-cancer immunity and synergize with anti-Pd-1 checkpoint blockade. Cancer Immunol Res (2021) 9(11):1316–26. doi: 10.1158/2326-6066.Cir-21-0280
149. Wurm M, Schaaf O, Reutner K, Ganesan R, Mostbock S, Pelster C, et al. A novel antagonistic Cd73 antibody for inhibition of the immunosuppressive adenosine pathway. Mol Cancer Ther (2021) 20(11):2250–61. doi: 10.1158/1535-7163.MCT-21-0107
150. Herber D, Cao W, Nefedova Y, Novitskiy S, Nagaraj S, Tyurin V, et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med (2010) 16(8):880–6. doi: 10.1038/nm.2172
151. de la Cruz-López K, Castro-Muñoz L, Reyes-Hernández D, García-Carrancá A, Manzo-Merino J. Lactate in the regulation of tumor microenvironment and therapeutic approaches. Front Oncol (2019) 9:1143. doi: 10.3389/fonc.2019.01143
152. Blattman J, Grayson J, Wherry E, Kaech S, Smith K, Ahmed R. Therapeutic use of il-2 to enhance antiviral T-cell responses in vivo. Nat Med (2003) 9(5):540–7. doi: 10.1038/nm866
153. Pellegrini M, Calzascia T, Toe J, Preston S, Lin A, Elford A, et al. Il-7 engages multiple mechanisms to overcome chronic viral infection and limit organ pathology. Cell (2011) 144(4):601–13. doi: 10.1016/j.cell.2011.01.011
154. Ejrnaes M, Filippi C, Martinic M, Ling E, Togher L, Crotty S, et al. Resolution of a chronic viral infection after interleukin-10 receptor blockade. J Exp Med (2006) 203(11):2461–72. doi: 10.1084/jem.20061462
155. Totzke J, Gurbani D, Raphemot R, Hughes P, Bodoor K, Carlson D, et al. Takinib, a selective Tak1 inhibitor, broadens the therapeutic efficacy of tnf-α inhibition for cancer and autoimmune disease. Cell Chem Biol (2017) 24(8):1029–39.e7. doi: 10.1016/j.chembiol.2017.07.011
156. Rodon J, Carducci M, Sepulveda-Sánchez J, Azaro A, Calvo E, Seoane J, et al. First-in-Human dose study of the novel transforming growth factor-β receptor I kinase inhibitor Ly2157299 monohydrate in patients with advanced cancer and glioma. Clin Cancer Res an Off J Am Assoc Cancer Res (2015) 21(3):553–60. doi: 10.1158/1078-0432.Ccr-14-1380
157. Kurebayashi Y, Olkowski C, Lane K, Vasalatiy O, Xu B, Okada R, et al. Rapid depletion of intratumoral regulatory T cells induces synchronized Cd8 T- and nk-cell activation and ifnγ-dependent tumor vessel regression. Cancer Res (2021) 81(11):3092–104. doi: 10.1158/0008-5472.Can-20-2673
158. Stafford J, Hirai T, Deng L, Chernikova S, Urata K, West B, et al. Colony stimulating factor 1 receptor inhibition delays recurrence of glioblastoma after radiation by altering myeloid cell recruitment and polarization. Neuro-oncology (2016) 18(6):797–806. doi: 10.1093/neuonc/nov272
159. Hagemann T, Lawrence T, McNeish I, Charles K, Kulbe H, Thompson R, et al. "Re-educating" tumor-associated macrophages by targeting nf-kappab. J Exp Med (2008) 205(6):1261–8. doi: 10.1084/jem.20080108
160. Phan V, Wu X, Cheng J, Sheng R, Chung A, Zhuang G, et al. Oncogenic ras pathway activation promotes resistance to anti-vegf therapy through G-Csf-Induced neutrophil recruitment. Proc Natl Acad Sci USA (2013) 110(15):6079–84. doi: 10.1073/pnas.1303302110
161. Miggelbrink AM, Jackson JD, Lorrey SJ, Srinivasan ES, Waibl-Polania J, Wilkinson DS, et al. Cd4 T-cell exhaustion: Does it exist and what are its roles in cancer? Clin Cancer Res (2021) 27(21):5742–52. doi: 10.1158/1078-0432.CCR-21-0206
162. Davidson S, Coles M, Thomas T, Kollias G, Ludewig B, Turley S, et al. Fibroblasts as immune regulators in infection, inflammation and cancer. Nat Rev Immunol (2021) 21(11):704–17. doi: 10.1038/s41577-021-00540-z
163. Rodriguez-Vita J, Lawrence T. The resolution of inflammation and cancer. Cytokine Growth Factor Rev (2010) 21(1):61–5. doi: 10.1016/j.cytogfr.2009.11.006
164. Majidpoor J, Mortezaee K. Interleukin-2 therapy of cancer-clinical perspectives. Int Immunopharmacol (2021) 98:107836. doi: 10.1016/j.intimp.2021.107836
165. Liu Y, Zhou N, Zhou L, Wang J, Zhou Y, Zhang T, et al. Il-2 regulates tumor-reactive Cd8(+) T cell exhaustion by activating the aryl hydrocarbon receptor. Nat Immunol (2021) 22(3):358–69. doi: 10.1038/s41590-020-00850-9
166. Jones SA, Jenkins BJ. Recent insights into targeting the il-6 cytokine family in inflammatory diseases and cancer. Nat Rev Immunol (2018) 18(12):773–89. doi: 10.1038/s41577-018-0066-7
167. Abe H, Takeda N, Isagawa T, Semba H, Nishimura S, Morioka MS, et al. Macrophage hypoxia signaling regulates cardiac fibrosis via oncostatin m. Nat Commun (2019) 10(1):2824. doi: 10.1038/s41467-019-10859-w
168. Johnson DE, O'Keefe RA, Grandis JR. Targeting the il-6/Jak/Stat3 signalling axis in cancer. Nat Rev Clin Oncol (2018) 15(4):234–48. doi: 10.1038/nrclinonc.2018.8
169. Shi J, Xu X, Du J, Cui H, Luo Q. Expression of oncostatin m in early gastric cancer and precancerous lesions. Gastroenterol Res Pract (2019) 2019:3616140. doi: 10.1155/2019/3616140
170. Korolev KS, Xavier JB, Gore J. Turning ecology and evolution against cancer. Nat Rev Cancer (2014) 14(5):371–80. doi: 10.1038/nrc3712
Keywords: T cell exhaustion, chronic inflammation, tumor immune escape, immunotherapy, tumor microenvironment
Citation: Fang L, Liu K, Liu C, Wang X, Ma W, Xu W, Wu J and Sun C (2022) Tumor accomplice: T cell exhaustion induced by chronic inflammation. Front. Immunol. 13:979116. doi: 10.3389/fimmu.2022.979116
Received: 27 June 2022; Accepted: 22 August 2022;
Published: 02 September 2022.
Edited by:
Bin Xu, Renmin Hospital of Wuhan University, ChinaReviewed by:
Hongli Du, South China University of Technology, ChinaCopyright © 2022 Fang, Liu, Liu, Wang, Ma, Xu, Wu and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Changgang Sun, c2NnZG9jdG9yQDEyNi5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.