
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 26 August 2022
Sec. Molecular Innate Immunity
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.978619
This article is part of the Research TopicIdentification, Function and Mechanisms of Interferon Induced Genes Associated with VirusesView all 12 articles
 Jia Yi1†
Jia Yi1† Jiameng Miao1†
Jiameng Miao1† Qingwei Zuo2†Felix Owusu3
Qingwei Zuo2†Felix Owusu3 Qiutong Dong1Peizhe Lin1
Qiutong Dong1Peizhe Lin1 Qilong Wang3Rui Gao4*
Qilong Wang3Rui Gao4* Xianbin Kong1*
Xianbin Kong1* Long Yang2,5*
Long Yang2,5*Coronavirus disease 2019 (COVID-19), caused by severe acute respiratory syndrome coronavirus2 (SARS-CoV-2), has spread to more than 200 countries and regions, having a huge impact on human health, hygiene, and economic activities. The epidemiological and clinical phenotypes of COVID-19 have increased since the onset of the epidemic era, and studies into its pathogenic mechanisms have played an essential role in clinical treatment, drug development, and prognosis prevention. This paper reviews the research progress on the pathogenesis of the novel coronavirus (SARS-CoV-2), focusing on the pathogenic characteristics, loci of action, and pathogenic mechanisms leading to immune response malfunction of SARS-CoV-2, as well as summarizing the pathological damage and pathological manifestations it causes. This will update researchers on the latest SARS-CoV-2 research and provide directions for future therapeutic drug development.
Since the end of December 2019, a global outbreak epidemic has emerged in several countries as an acute respiratory infection caused by a previously undiscovered strain of coronavirus (1). On December 1, 2019, the first pneumonia of unknown origin was identified in Wuhan, Hubei Province, and confirmed as a new coronavirus on January 8, 2020 (2, 3). On 12 January 2020, the World Health Organization (WHO) tentatively named this virus as “2019 new coronavirus (2019-nCoV)”, and on 11 February, the International Committee on Classification of Viruses officially called it “SARS -CoV-2”, and on the same day WHO unified pneumonia caused by SARS-CoV-2 infection as “Coronavirus disease 2019 (COVID-19)”. This pneumonia is associated with a novel strain of RNA virus from the coronavirus family. In terms of clinical manifestations, COVID-19 has a lower morbidity and mortality rate than SAS and MERS. Still, it spreads faster and more widely, and the number of infections and deaths far exceeds those of the first two viruses (4), which are highly infectious, have a long incubation period, and are prone to mutation (5). COVID-19 can lead to severe acute respiratory infections and multiple organ systems functional impairment, with 15-30% of COVID-19 patients requiring Intensive Care Unit (ICU) admission and organ function support therapy, with an overall morbidity and mortality rate of 4.3%-15% (6), with a morbidity and mortality rate of up to 61% within 28 days in critically ill patients.
Therefore, an in-depth exploration of the pathogenic mechanism of COVID-19 is crucial in achieving an accurate diagnosis, targeted therapy, vaccine development, and improved prognosis. This review provides a detailed overview of the pathogenic mechanism of the new coronavirus based on the pathogenic characteristics of the virus, the process of invasion into the human body, the dysregulation of the immune response caused, and the pathological manifestations and pathological damage of the organism, to provide clinical and scientific assistance.
The main routes of transmission of SARS-CoV-2 are currently considered to be respiratory droplets and close contact (3), with the possibility of aerosol transmission (7) and vertical transmission (8) also present. Data from a clinical study of 1145 patients suggest that the severe course of COVID-19 may be closely related to its viral load during exposure (9). Therefore, studying the pathogenic characteristics of SARS-CoV-2 and its loci of action is essential to understanding the pathogenic mechanisms of the virus.
SARS-CoV-2 is a member of the coronaviridae family, order Nestoroviridae, genus Pre-coronavirus, and is a spherical enveloped virus with a diameter of approximately 120 nm (10). Mature SARS-CoV-2 viral particles consist of a positive 5’-plus-cap and 3’-polyadenylate single-stranded RNA with a genomic sequence approximately 30,000 bases long, encoding the structural proteins nucleocapsid phosphoprotein (N), membrane glycoprotein (M), envelope (E), spines (S), and nonstructural protein (nsp) (11). Among them, it is mainly the S glycoprotein that mediates viral entry into target cells and the E and M proteins responsible for viral transcription, translation, and assembly.
The S protein consists of S1 and S2 subunits. The S1 subunit consists of the N-terminal structural domain (NTD) and the receptor binding domain (RBD), which is responsible for the direct binding of Angiotensin-converting enzyme 2 (ACE2) (12); the S2 subunit mediates the fusion of the viral envelope with the host cell membrane (13). It was shown that RBD in the SARS-CoV-2 spike-in (S) protein undergoes a specific point mutation, i.e., the asparagine is replaced by tyrosine at position 501 (N501Y) (14), and thus the N501Y S-protein binds more readily to the ACE2 receptor than the original S-protein (15). The S-protein was found to have four amino acid residues inserted at the junction of subunits S1 and S2 (PRRA) (16). This amino acid sequence can be efficiently cleaved by furin and other proteases (17), which reduce the stability of the SARS-CoV-2 S-protein, enhance viral membrane fusion and infection, and promote viral replication (18).
In addition, it has been proposed that S proteins can circulate within the Golgi and promote S protein cleavage and glycosylation, thereby infecting the plasma membrane of cells (19). Another study found that the S1/S2 cleavage site has remained constant during the human evolution of SARS-CoV-2, suggesting that it provides an adaptive advantage for the virus (14). The antiviral activity of chloroquine and its analogues are well established in the fight against SARS-CoV-2 infection (20), and clinical trials have shown that the use of some chloroquine derivatives can achieve viral reduction and improve the efficacy of the infection (21, 22). And laboratory studies have shown that its antiviral effects are attributed to multiple mechanisms, including fighting coronavirus infection by blocking the glycosylation of host receptors (23, 24), inhibiting the processing of S proteins, and suppressing the inflammatory response (25).
ACE2 is a metallocarboxypeptidase of the renin-angiotensin-aldosterone system (RAS) (26). The pathway of SARS-CoV-2 into host epithelial cells was mainly focused on ACE2 (27). Accordingly, it has been found that tetracycline and doxycycline can act as inhibitors of ACE2-peg binding (28).
ACE2 is expressed in over 150 different cell types in all major human tissues and organs, and its expression levels do not vary by age, sex, or race. Immunofluorescence data showed (29) that ACE2 is expressed at higher levels in epithelial cells of the upper respiratory tract, lung, heart, kidney, testis (30), intestine, liver, pancreas, stomach, duodenum, and rectum (31), and the higher levels of ACE2 in the cilia of the nose compared to the bronchi (32) also suggest that the nose may be the initial site of viral invasion and infection.
ACE2 decreases angiotensin II (Ang II) and is a stimulator of Nicotinamide Adenine Dinucleotide Phosphate (NADPH) oxidase (33). It is a key molecule in the body’s resistance to inflammation and oxidative damage in tissues triggered by SARS-CoV-2. After SARS-CoV-2 binds to the ACE2 receptor and begins to enter the cell and fuses with the viral particle-membrane, ACE2 expression will be downregulated (34), and the affinity of angiotensin II is significantly increased during infection, leading to the susceptibility of the virus in binding to ACE2 (35). It was shown that the affinity of SARS-CoV-2 to ACE2 receptor is about 10-20-fold higher than SARS-CoV (36). Therefore, based on the persistent downregulation of ACE2 expression, the overproduction of angiotensin II and activation of NADPH oxidase leads to enhanced oxidative stress mechanisms along with the release of inflammatory molecules (37), leading to the rapid progression of the disease.
In addition, cell surface phospholipid proteoglycans (HSPGs) interact with the S protein of SARS-CoV-2 (18), triggering a conformational change in the S protein RBD, and acting as a cofactor during viral endocytosis (33), which facilitates viral binding to its specific receptor (38). Basic studies suggest that HSPGs are modified by 3-OST isoform 3 but not 3-OST isoform 5, increasing S protein-mediated fusion between SARS-CoV-2 and cells, suggesting a role in virus transmission (39). In particular, HSPGs have been identified as adhesion receptors for SARS-CoV-2 infection in isolated human lung tissue explants from human lung epithelial cell nuclei in vitro (40).
RNA sequencing also revealed that immune cells, although not expressing ACE2, are transmembrane proteins of immunoglobulin cluster of differentiation (CD) 147, providing a pathway for the virus to enter and attack immune cells (41–43). It should be noted that one study using single-cell sequencing found that few cells in the placenta express both ACE2 and transmembrane serine proteases (TMPRSS2), thus concluding that ACE2 is not an effective route of transmission from mother to child (44). (Figure 1) Also, in combination with the replication process of new coronaviruses, the nucleoside analogue favipiravir (T-705) was found to effectively inhibit the RNA polymerase activity of RNA viruses (45). Remdesivir targets RNA-dependent RNA polymerase (RdRp) and is a nucleotide analogue. Remdesivir received emergency use authorization from the US Food and Drug Administration (FDA) and was approved as the first drug to treat patients with COVID-19 (46). The pharmacological mechanism of the drug is to interfere with the polymerization of viral RNA (47). As a broad-spectrum antiviral drug, it has significant antiviral activity against several RNA viruses such as Ebola virus, coronavirus (48), hepatitis C virus, and human immunodeficiency virus (HIV) (49). Experimental studies have shown that the drug significantly inhibited SARS-CoV-2 virus infection in Vero E6 cells (50), while reducing viral load and pulmonary pathological changes in animal models (51), and had stronger antiviral activity in combination with interferon (52). Lopinavir/ritonavir (LPV/r) are two anti-HIV protein hydrolase inhibitor (PI) drugs that act as antiviral retroviral with a pharmacological mechanism that prevents the excision of the Gag-Poll polyprotein (53), leading to the immaturity of the virus that replicates and proliferates in the organism. Clonidine is also thought to inhibit RNA virus replication by entering the infected cells during viral RNA (54).
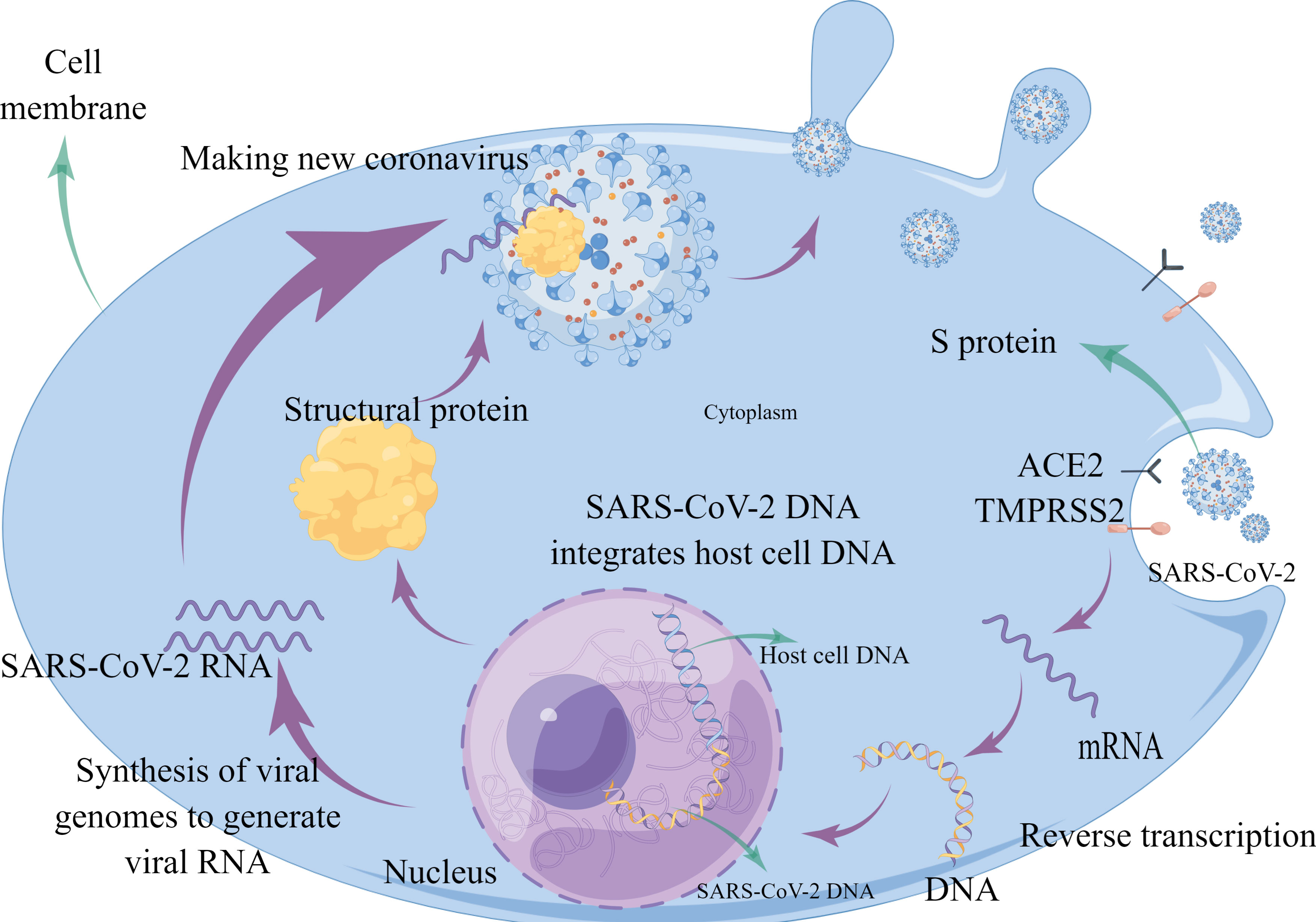
Figure 1 Schematic diagram showing SARS-CoV-2 injecting RNA into the host cell by binding to the ACE2 receptor on normal cells via the S protein. The injected RNA uses the nutrients in the host cell to replicate itself and make the structural proteins it needs. The structural proteins combine with the RNA to form a new virus.
At the same time, studies have pointed out that papain-like protease (PLpro) and main protease (Mpro/3CLpro) are two crucial proteases produced by the new coronavirus. Therefore, inhibition of PLPro and Mpro/3CLpro can effectively inhibit virus infection and replication, and is a vital target for antiviral drug development (55). The researchers screened and evaluated the applicability of a batch of FDA-approved clinical drugs targeting PLpro to SARS-CoV-2 PLpro, and found that a naphthalene-based noncovalent inhibitor GRL0617 works by occupying and blocking the PLpro substrate-binding pockets S3 and S4 exerted a potent inhibitory activity. In addition, studies have also identified inhibitors against novel coronavirus Mpro, including boceprevir, GC-376, and calpain inhibitors II and XII, which are often mimetic peptides that mimic natural peptide substrates and covalently bind to residue C145 in Mpro to exert inhibitory effects. (56). And a study selected 47 from the list of 3987 FDA-approved drugs for in vitro 3CLpro enzyme inhibitor screening test, and observed that boceprevir, ombitasvir, paritaprevir, tipranavir, and micafungin showed partial inhibition, and ivermectin blocked. The 3CLpro activity of SARS-CoV-2 was more than 85%, indicating that it has the potential to inhibit the replication of SARS-CoV-2. In addition, PF-07321332, developed by Pfizer, is the first oral coronavirus-specific major protease inhibitor approved by the U.S. FDA. The FDA has approved emergency treatment for Paxlovid (PF-07321332 and ritonavir). As a protease inhibitor, PF-07321332 binds to viral enzymes and can block the activity of proteases required for the coronavirus to replicate itself. Ritonavir, an inhibitor of a key liver enzyme called CYP3A, also increased and maintained plasma concentrations of PF-07321332 when given in combination (57).
The consensus achieved by the current study is that the entry and spread of the SARS-CoV-2 virus depend on the host ACE2 receptor and the serine protease TMPRSS2, with possible involvement of B/L7 and furin proteases (27).
Experimental studies have shown that the S protein of the SARS-CoV-2 virus binds to the receptor, acid-dependent proteolytic cleavage (58), and is assisted by the S2 subunit to mediate the fusion of the viral membrane with the cell membrane (59), leading to cytoplasmic lysis. This process is mainly mediated by certain host proteases, including furin protease, TMPRSS2, histone B, histone L, factor Xa, and elastase (60). Bertram et al. also suggested that the coronavirus protease system is transmembrane anchored, which is essential for invasion and infection (61). As previously described, after membrane fusion and protease mediation, the S1/S2 site of the S protein will insert four amino acids, providing a motif that can be recognized and cleaved by the furin protease. The virus is then cleaved by TMPRSS2, and the viral protease system forms an unlocking and fusion catalytic structure with the type II transmembrane serine protease (TTSP) family at the cell surface and mediates rapid entry into the cell and completion of ligation within the cell (61, 62), triggering irreversible and extensive conformational changes that mediate membrane fusion (63, 64).
In addition to the associated proteases, it has been proposed that coronavirus infection increases circulating exosomes containing lung-associated autoantigens as well as viral antigens and 20S proteasomes (65). It has also been shown that SARS-CoV-2 drives host cell molecular pathways to activate cellular kinases, such as casein kinase II (CK2) and p38 mitogen-activated protein kinase (MAPK), and growth factor receptor (GFR) signaling to hijack the host protein production machinery (66) for its replication, transcription, and translation purposes.
When normal, the body’s immune system can limit processes such as the entry of viruses into host cells and their replication within the host cells. The immune system has two main defense mechanisms: innate immunity and adaptive immunity. In contrast, after infection with the SARS-CoV-2 virus, the pathogen-associated molecular patterns (PAMPs) of the virus trigger specific combinations of pattern recognition receptors (PRRs) and adapter molecules, leading to an immune response adapted to the pathogen (67), resulting in abnormal immune response function and causing the associated pathological processes.
Innate immunity is the first line of defense against infection. The main cells that perform innate immunity are mast cells, NK (natural killer cells), NKT (natural killer T cells), NHC (natural helper cells), granulocytes, macrophages, and monocytes. The organism detects coronaviruses through PRRs, which trigger an innate immune response that effectively limits viral replication. And it helps to control or eliminate viral infections by releasing interferons (IFNs), while activating interferon-stimulated genes (ISGs) to exert direct antiviral effects and recruit antiviral immune effector cells to clear the virus (Table 1).

Table 1 Summary of research progress on COVID-19 innate immune response dysregulation.
Studies have shown that reduced numbers and functional failure of NK cells occur during SARS-CoV-2 infection (68, 69), and the mechanism may be closely related to the generation of reactive oxygen species (ROS) during the early stages of the immune response. In addition, Nox2 may be a key factor in the infection and development of COVID-19. It was found that the SARS-CoV-2 virus may suppress the immune response and lead to infection by activating Nox2 (NADPH oxidase 2, nicotinamide adenine dinucleotide phosphate hydrogen oxidase 2) (70). Clinical observations likewise revealed higher levels of Nox2 activation in critically ill patients with COVID-19 (37).
The main PRRs-against viruses are currently considered to be Toll-like receptors (TLRs) and RIG-I-like receptors (RLRRs), and NOD-like receptors (NLRX) (71). Among them, PRRs are present on the cytoplasmic and endosomal membranes of immune cells, and their function is to recognize foreign pathogens on the cell surface or inside. The binding of PAMPs to PRRs induces innate immune signaling (17), successively involving adaptor proteins (MYD88, TRIF, RGL-1, and MAD-5), cell membrane protein kinases (IRKs, MAPKs, and ERKs), and finally the production of transcription factors at the cell membrane (e.g., nuclear factor kappa-B, IFRs, NF-κB, and IFRs) (72). These transcription factors migrate to the nucleus and induce the expression of encoded cytokines, IFN-I, IFN-III, pro-inflammatory cytokines, and chemokines (73), which leads to a massive accumulation of neutrophils (74). Although neutrophils have an antiviral function, their secretions, cytokines, and chemokines promote the accumulation of immune cells and further produce an excessive inflammatory response (75) (Figure 2).
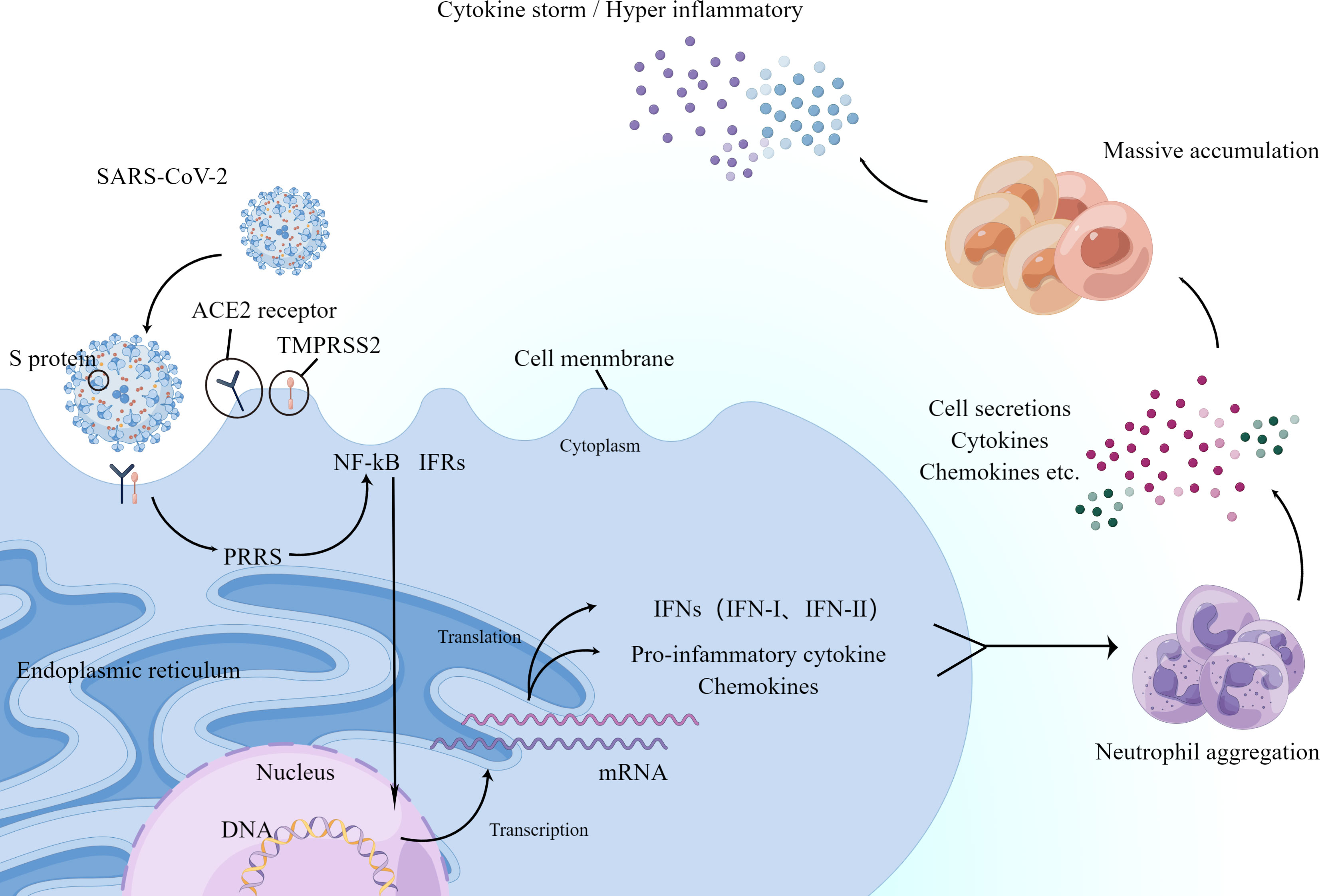
Figure 2 Schematic diagram showing the process by which the new coronavirus enters the human body and triggers an inflammatory response. ACE2 and TMPRSS2 play a decisive role in neo-coronavirus invasion. The major PRRs against viruses are present on the cytoplasmic and endosomal membranes of immune cells and recognize foreign viruses. After a series of processes, they finally produce transcription factors NF-κB and IFRs on the cell membrane. Next, they migrate to the nucleus and induce the expression of encoded cytokines and IFN-I and IFN-III, pro-inflammatory cytokines, and chemokines, which in turn accumulate large numbers of neutrophils. The secretion of neutrophils, cytokines, and chemokines promotes further accumulation of immune cells, producing an excessive inflammatory response or further triggering the cytokine storm mentioned below.
Glucocorticoids should be used for a short period of time, as appropriate, in patients with progressively worsening oxygenation indices, rapidly developing imaging, and over-activated inflammatory responses (76), and systemic corticosteroid use is effective in reducing mortality in critically ill patients with COVID-19 (77). The World Health Organization recommends using dexamethasone 6 mg daily for up to 10 days in patients with severe or critical COVID-19 (78). Calcium-release-activated calcium (CRAC) channel inhibitors block the release of pro-inflammatory cytokines, protect endothelial integrity, and may be effective in treating patients with severe COVID-19 pneumonia (79).
SARS-CoV-2 viruses possess ways to escape the natural immune system, such as modifying their own viral mRNAs, inducing mRNA translation abnormalities in host cells, and blocking interferons. It was found that nsp16 and nsp10 induce the synthesis of viral mRNAs that mimic host cell mRNAs, thereby protecting the virus from the host’s innate immune response (80). The spike protein of SARS-CoV-2 facilitates invasion of host cells and evades detection by host immune cells. It was found that the nsp1 of SARS-CoV-2 causes mRNA translation shutdown in host cells and blocks Retinoic acid-inducible gene I (RIG-I) and Immune Serum Globulin (ISG), key mediators of the innate immune response against viral infection (17). Furthermore, SARS-CoV-2 will inhibit interferon-induced and blocked IFN signaling and lead to decreased expression levels of toll-like receptor 7, TLR8, TLR2, and TLR4 receptors that recognize SARS-CoV-2 viral RNA, which will lead to immune escape as SARS-CoV-2 virus is not recognized by the host’s immune system (81).
IFNs are the first line of defense against viruses. It includes a series of antiviral IFN cytokines, classified into types I, II, and III according to their unique molecular structures, which trigger the expression of ISGs. ISGs exert various antiviral and other immunomodulatory functions by directly inhibiting viral replication (82), transcription, and translation through multiple mechanisms.
It is important to note that viruses (especially those infecting the lung) develop strategies to evade PRR detection and thus alter the host IFN response; for example, some viral proteins can inhibit PRRs in host cells (83). It was found that the immune evasion mechanism of SARS-CoV-2, in addition to the previously described, may also be related to the inhibition of IFN production and IFN signaling by viral proteins (84, 85). Experimental observations revealed that interferon regulatory factor (IRF) mediated signaling was not activated (86), suggesting that dysregulation of interferon response occurs during SARS-CoV-2 virus infection.
Studies suggest that although the organism produces ISGs, transcriptional processes regulated by the interferon regulators IRF3 or IRF7 are apparently absent in SARS-CoV-2 infection. And experiments have shown that the protease of SARS-CoV-2 can directly cleave IRF3 and lead to an attenuated production of IFN (87). Other experiments have shown that IRF7 and IRF9 are upregulated in SARS-CoV-2 infection and that severe viral load may overwhelm the IFN response and determine the outcome of the infection (86), manifesting as a dysregulated IFN response. Studies suggest that Open reading frame 6 (ORF-6) acts as an antagonist of type I interferon, promoting viral escape from the host innate immune system (11).
It was found that viral proteins or nucleic acids that trigger PRRs induce β-interferon TIR structural domain bridging proteins (TRIFs) and IRFs through TIR structural domain-containing junctions, thereby activating the interferon response (88). And triggering PRRs and interferon type I pathway leads to a further oxidative stress response. Meanwhile, the SARS-CoV-2 viral protein has an inhibitory effect on IFN-I-mediated antiviral immune responses. Its interference with interfering with the production of IFN leads to or blocks the downstream signaling pathway following the binding of IFN to ISGs (89, 90), thus antagonizing the innate immune response.
When the organism exerts a normal adaptive immune response, the SARS-CoV-2 viral antigen is recognized, processed, and presented by antigen-presenting cells (APCs), thereby activating cellular and humoral immunity. This includes the activation of CD4+ and CD8+ T cell differentiation. CD4+ T cells are activated and differentiate into Th1 and Th2 effector cells and other subpopulations (including Tfh cells, etc.) that recruit immune cells by secreting cytokines (including MIP-1s, INF γ, etc.) and chemokines, CD8+ T cells produce substances such as Perforin, CD107a, and Granzyme B, while B cell differentiation and antibody production are stimulated, which together exert adaptive immunity to destroy the virus (58) (Table 2).

Table 2 Summary of research progress on COVID-19 adaptive immune response dysregulation.
Experimental studies have shown that CD4+ T cells are significantly less responsive to various viral proteins such as S, N, and M proteins in SARS-CoV-2 infection (91). Clinical data showed a progressive decrease in peripheral blood CD4+ T and CD8+ T cells during SARS-CoV-2 infection (92). In contrast, a significant lymphocyte decrease is an important immunological marker of impending severe COVID-19 (93). Severely ill patients exhibit macrophage overreaction (also known as macrophage activation syndrome MAS) and lymphocytopenia in effective lymphocytes, including neutrophils, CD4+ T cells, and NK cells (94–96).
It was found that under normal conditions, IFN-g induces the differentiation of Th0 cells into Th1 cells. In contrast, during SARS-CoV-2 infection, lower levels of IFN-g production reduce Th1 production, leading to a further attenuation of the antiviral immune response of CD4+ T cells (97). In addition, Th2 cells normally produce IL4, IL-6, Il-8, IL-10, and IL-13, which suppress inflammatory responses, promote antibody responses, and inhibit Th1 cell-induced antiviral functions (98).
In COVID-19 patients, TNF-α and IFN-γ expression is reduced in CD4+ T cells (99); high levels of failure markers are expressed in CD8+ T cells (100); and programmed cell death protein-1 (PD-1) and T cell immunoglobulin structural domain and mucin structural domain-3 (TIM-3) expression are increased (101). T cells from patients with severe COVID-19 showed high levels of apoptosis and expression of the death receptor FAS (102), suggesting severely impaired T-cell function. It was found that in addition to the reduced number of T cells, the expression levels of T cell receptor subunits, T cell surface molecules, and their downstream signaling molecules were also severely reduced (103). In addition, SARS-CoV-2 infection also resulted in downregulation of B-cell histocompatibility complex MHC-II expression (104), which severely impaired immune function.
It should be noted that since ACE2 is not expressed in T cells, the impaired T cell response may not be due to the direct toxic effects of SARS-CoV-2 (105). It has been suggested that T-cell lymphopenia may be caused by pro-inflammatory cytokines and activation-induced cell death (69, 102).
After SARS-CoV-2 infection, cell-mediated immunity of T cells comes into play, and cytotoxic T cells recognize an attack and destroy cells containing this pathogen. Helper T cells activate the differentiation of B lymphocytes in the germinal centers of lymph nodes and other lymphoid tissues to secrete pathogen-specific antibodies (71). Studies have shown that the antibody profile against the SARS-CoV2 virus has a typical pattern of IgM and IgG production. Measurement of serological IgM and IgG titers and detecting SARS-CoV-2 nucleocapsid protein (NP) antigen by fluorescent immunochromatography showed its high specificity and relatively high sensitivity in the early stages of infection (58).
Dysregulated B-cell responses have been reported in COVID-19. Analysis of circulating B cells has shown polyclonal expansion of plasma cells and reduced memory B cells in patients with severe COVID-19 compared to patients with mild COVID-19 or healthy individuals (104, 106, 107), and synthesis of IgG and IgM antibodies also appear to be stalled or delayed (6, 108). Although studies have shown elevated anti-SARS-CoV-2 antibodies in patients with severe COVID-19 (109), their specificity and affinity appear low (110).
SARS-CoV-2 viral infection causes disseminated intravascular coagulation (DIC), septic shock (111), RAS system activation, hemodynamic changes, and cellular damage by interfering with the normal function of immune function and triggering cytokine storms and bradykinin storms, which leads to a series of pathological manifestations in the organism.
Cytokine storm, also known as cytokine release syndrome, is a potentially fatal immune disease. It is characterized by the high activation of immune cells and the overproduction of large amounts of inflammatory cytokines and chemical mediators (112). It has been proposed that cytokine storm, the excessive immune response that SARS-CoV-2 infection triggers in severe cases of COVID-19 (113), is thought to be a major cause of severe disease and death in COVID-19 patients (74). Cytokine storms begin with strong activation of cytokine-secreting cells (41), and COVID-19 cytokine storms are characterized by high expression of IL-6 and TNF-α (114). The mechanism of which may be related to SARS-CoV-2 induction of cell death and thus histone release, which triggers the secretion of pro-inflammatory molecules of the interleukin-1 (IL-1) family (115), producing such IL-6, IP-10, MIP1αβ (macrophage inflammatory protein-1αβ) and MCP1 (monocyte chemotactic protein-1), and a large number of other pro-inflammatory cytokines and chemokines (116). Among them, IL-6 is an important pleiotropic pro-inflammatory mediator and a major driver of the cytokine storm. And cytokine storm is closely associated with macrophage activation syndrome (MAS). Excessive proliferation of differentiated macrophages leads to phagocytosis and hypercytosis (117, 118), which leads to systemic inflammatory abnormalities.
In addition, CD4+ T lymphocytes rapidly differentiate into pathogenic T helper (Th)1 cells that produce IL-6 and GM-CSF (Granulocyte-macrophage Colony Stimulating Factor). GM-CSF plays an important role in mediating the cytokine storm (119). Subsequent induction of high levels of IL-6 and GM-CSF secretion by CD14+, CD16+, and monocytes (120) exacerbates the cytokine storm. Activated neutrophils can form neutrophil extracellular traps (NETs) that are involved in the pathogenesis of aseptic and nonsterile inflammation (121) and promote the development and progression of inflammation. Uncontrolled excessive inflammatory responses produce oxidative stress (imbalance between oxidants and antioxidants). Activated neutrophils and macrophages release pro-oxidant factors such as TNF-α (tumor necrosis factor-α) and release reactive oxygen species (ROS) (122–124), which in turn stimulate further cytokine production by inflammatory cells, leading to an even more intense inflammatory response (125).
In the face of the inflammatory storm generated by neo-coronavirus, it was found that timely application or combination of monoclonal antibodies can effectively reduce the rate of deterioration and mortality of neo-coronavirus pneumonia, which has broad clinical application prospects. As monoclonal antibodies against interleukin 6 receptor (IL-6), among which tocilizumab and satralizumab whose pharmacological mechanism is mainly to specifically bind to IL-6 receptor and inhibit its activation, thus inhibiting cytokine storm and reducing mortality (126), clinical studies have also confirmed that the application of this drug has significant efficacy in improving the inflammatory response in patients with COVID-19 (127). In addition, it has been shown that the administration of levilimab in patients with SARS-CoV-2 pneumonia in the absence of other signs of active infection, with or without oxygen therapy, increases the rate of sustained clinical improvement (128), that itolizumab significantly reduces the severe consequences caused by cytokine release syndrome (129), and that tocilizumab reduces the duration of hospitalization (130), the progression to mechanical ventilation (131) and the risk of transfer to the ICU (132). In addition, studies have proposed binding neutralizing antibodies to the surface of photothermal nanoparticles (NPs) to capture and inactivate novel coronaviruses. The NPs consist of a semiconductor polymer core and a biocompatible polyethylene glycol surface modified with a high-affinity neutralizing antibody. The multifunctional NP efficiently captures novel coronavirus pseudoviruses and completely blocks virus infection of host cells in vitro by surface-neutralizing antibodies. In addition to the virus capture and blocking functions, the NPs have a photothermal function to inactivate the virus by generating heat upon irradiation (133). The multifunctional nanoparticles also exhibit excellent biosafety in vitro and in vivo, and show satisfactory pulmonary delivery in mice. Most importantly, in vivo treatment with multifunctional NPs in the presence of actual novel coronaviruses was achieved, offering significantly improved therapeutic efficacy compared to soluble neutralizing antibodies and demonstrating their great potential for clinical novel coronavirus therapy. NPs are very superior to neutralizing antibodies in the treatment of actual novel coronavirus infections that occur in vivo. This versatile NP provides a flexible platform that can be easily adapted to other novel coronavirus antibodies and extended to new therapeutic proteins, and thus it promises to provide broad protection against the original novel coronavirus and its variants (134).
Clinical observations have revealed alterations in hematology associated with coagulation during COVID-19 (2, 135). In most severe cases, patients develop microvascular dysfunction such as disseminated intravascular coagulation (DIC) or infectious shock (136). Thromboembolic complications are one of the main causes of morbidity and mortality in patients with COVID-19 (137).
The cause of thrombosis is an imbalance between procoagulant and anticoagulant processes. Systemic thromboembolism, including venous thromboembolism, arterial thrombosis, and thrombotic microangiopathy, is a unique and essential feature of COVID-19. In current studies, the mechanisms of coagulation disorders may also be associated with downregulation of ACE2 activity, endothelial dysfunction (138), activation of von- Willebrand factor, activation of the complement system, neutrophil extracellular traps (139), oxidative stress injury, and high inflammatory state (140) formation. These predisposes infected individuals to the activation of Virchow’s triad, leading to arterial and venous thrombosis and vascular arrest anywhere in the body (141).
It has been studied that coagulation parameters, especially D-dimer levels, predict mortality in 2019 coronavirus disease and that patients with 2019 coronavirus disease have an increased risk of arterial and venous thrombosis. It has been suggested that anticoagulation (AC) is beneficial in these patients. That prophylactic AC with enoxaparin and apixaban is appropriate for treating hospitalized 2019 coronavirus disease patients with D-dimer levels >1µg/mL (142).
As mentioned previously, the primary site of action of the SARS-CoV-2 virus is ACE2. And viral infection may lead to a decrease in ACE2 activity, resulting in elevated angiotensin II and decreased angiotensin 1-7. Angiotensin II rapidly generates reactive oxygen species mediated by NADPH oxidase and causes oxidative stress injury (143). Angiotensin 1-7 is now considered to be an important anti-inflammatory and anti-thrombotic peptide with inhibitory effects on platelet activation (144). Therefore, these will lead to RAS dysregulation, oxidative stress injury, and coagulation disorders.
There is a strong correlation between endothelial dysfunction and thrombosis (145). Experimental studies have shown that endothelial dysfunction is a key factor in the release of the procoagulant factor fVIII (146) to generate and activate thrombus and trigger various coagulation cascades (147). This process may be associated with the endothelial expression of many prothrombotic factors and receptors. In addition, overexpression of hemagglutinin-like oxidized low-density lipoprotein receptor (LOX-1), cyclooxygenase (COX-2), and vascular endothelial growth factor (VEGF) during infection can also cause endothelial injury (148).
The underlying vascular hemophilic factor (vWF) plays a key role in COVID-19-related coagulation (149). Following endothelial injury, vWF present in the subendothelium is released, further multimerized by disulfide bonds, and exposes to the platelet-binding and collagen-binding domains (150). vWF acts as an adherent molecular glue platelet together with subendothelial collagen, activating platelet aggregation and thrombosis (151).
The complement system is capable of activating the coagulation cascade through multiple mechanisms leading to vascular thrombosis. The nucleocapsid (N) protein of SARS-CoV-2 binds to mannose-binding lectin-associated serine protease (MASP)-2, which is expressed on microvessels, leading to complement activation (121). In contrast, complement factor C3 and MAC directly activate platelets and induce platelet aggregation (152). Similarly, complement factor C5a has been shown to stimulate the expression of fibrinogen activator inhibitor 1, thereby promoting thrombosis (153).
NETs, also known as extra-neutrophil traps, contain various pro-thrombogenic molecules such as tissue factor, protein disulfide isomerase, factor XII, vWF, and fibrinogen (154). In addition, DNA released from extracellular NETs can directly activate platelets and lead to thrombosis. Circulating histones (major components of NETs) have also been found to activate Toll-like receptors on platelets and promote thrombin production (155).
Coronavirus replication can lead to lysosomal disruption, mitochondrial damage, free radical damage, disruption of membrane structure and function, destruction of mitochondria and lysosomes, cellular autolysis, and triggering ion concentration imbalance (73). Among them, reactive oxygen species (ROS) and (reactive nitrogen species) RNS may be one of the modification pathways of severe COVID-19 (156). It has been demonstrated that the downregulation of ACE2 by COVID-19 may affect the mitochondrial function of immune cells, which in turn may reduce the immune function of the host (157).
As mentioned earlier, ACE2 is an important locus for the SARS-CoV-2 virus. And one of the roles of ACE2 is to inactivate angiotensin II by converting it to angiotensin 1-7 through proteolysis, which puts ACE2 in a critical position to act as a negative regulator of the renin-angiotensin-aldosterone system (RAAS) (158) and leads to RAAS system dysfunction.
Infection with SARS-CoV-2 will cause a range of pathological injuries such as lymphopenia, and lung tissue damage (159), such as acute respiratory distress syndrome (ARDS) and respiratory failure, sepsis-induced cardiac injury and arrhythmia (58), and multi-organ failure. Enhanced granulocyte and monocyte-macrophage infiltration are common in critically patients with COVID-19. Monocytes and macrophages are involved in and exacerbate hypersensitivity reactions (160), leading to organ damage.
Studies have shown a direct relationship between apoptosis rates and the pathogenicity and severity of COVID-19 (161). COVID-19 attacks the lymphoid tissue of the body and induces apoptosis in immune cells. During SARS-CoV-2 infection, single-cell RNA sequencing showed enrichment of SARS-CoV-2 RNA in the macrophage population of bronchoalveolar lavage samples from patients phenomenon, suggesting that the virus directly infects and attacks macrophages (162) and triggers macrophage polarization toward a pro-inflammatory phenotype (163).
Several mechanisms may exist for apoptosis, decreased expression, and functional failure of immune cells. The decrease in T-cell numbers was negatively correlated with IL-6 and TNF-α levels (164), suggesting that increased inflammatory cytokines may promote T-cell failure and apoptosis. Moreover, the IL-2 signaling pathway is inhibited, negatively regulating CD8+ T cells (71) and inducing a decrease in lymphocytes. Besides, some lymphoid organs are attacked by SARS-CoV-2, which further leads to lymphocyte damage. Similarly, it has been noted that SARS-CoV-2 ORF3a induces apoptosis through the extrinsic apoptotic pathway. Caspase-8 activation/cleavage is a hallmark of the extrinsic apoptotic pathway, and SARS-CoV-2 ORF3a induces caspase-8 activation/cleavage. This process can induce epithelial apoptosis and inflammatory cytokine processing in turn, which triggers necroptotic prolapse pathway caspase-8-mediated apoptotic activation and inflammatory response, which can induce downstream immunopathogenesis in lung tissue (165).
Furthermore, elevated blood lactate levels in critically ill patients with COVID-19 inhibit lymphocyte proliferation of neutrophils with suppressive properties (e.g., granulocyte myeloid-derived suppressor cells (G-MDSCs)) (166). They may inhibit the expansion of CD4+ and CD8+ T lymphocytes (167).
Preliminary data suggest that pulmonary vascular injury and partial loss of alveolar group function are key to developing severe illness and death in patients with COVID-19 (168). Clinical data analyzed that after infection with SARS-CoV-2, most patients develop bilateral interstitial pneumonia with histology showing alveolar wall edema, protein exudates, and non-cellular focal reactive hyperplasia with vascular congestion (169), which also leads to selective death of type II pneumocytes (170). After type II pneumocyte injury, the inflammatory state will be supported by macrophage pro-inflammation (M1), cytokine release, and NF-κB support, further damaging alveolar cells in a vicious cycle (171). Loss of lung surface active gas exchange and vascular abnormalities can lead to progressive respiratory failure. Pneumonia caused by SARS-CoV-2 leads to a rapid decrease in arterial pO2 levels measured by transcutaneous saturation (136) and hypoxemia.
Some scholars have suggested that lung tissue damage may be associated with the occurrence of NETs in large numbers of neutrophils, which in turn release toxic enzymes such as elastase (172), and secrete substances such as cationic histones. These, in addition to having direct cytotoxic effects, may also enhance infection of lung cells and thus aggravate the disease (173). In addition, oxidized phospholipids in macrophages triggering cytokine production via TLR4-TRIF-TRAF6 can further aggravate lung inflammation (174).
It has been found that patients with ARDS and extrapulmonary complications have significantly elevated rates of circulating pro-inflammatory cytokines, chemokines, and systemic inflammatory markers (175), suggesting that the organism is in a state of intense inflammatory response. It has been proposed that severe lung injury in COVID-19 patients is thought to result from direct viral infection and immune hyperactivation (114). (Figure 3)
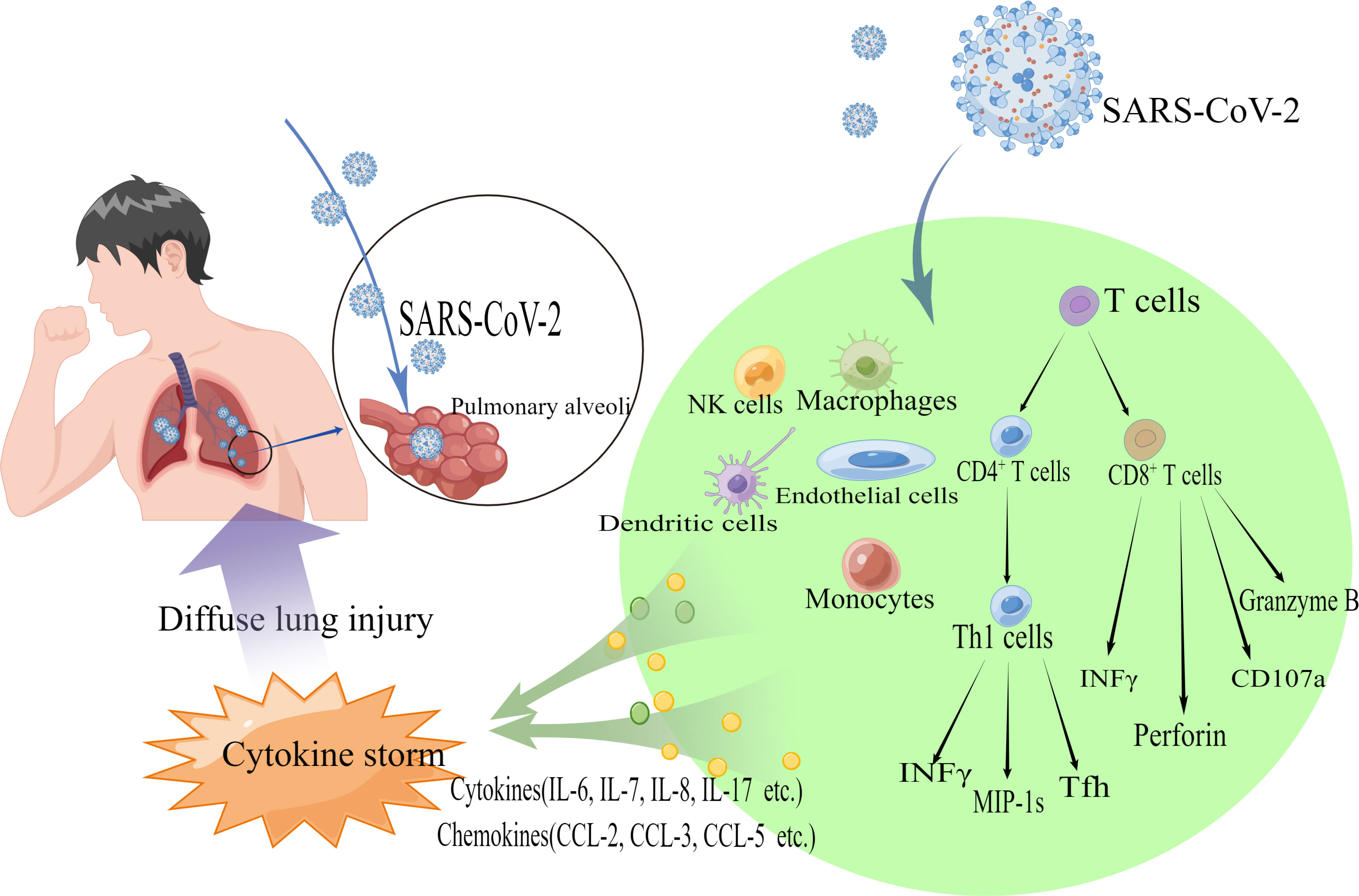
Figure 3 Schematic diagram showing the pathogenic mechanism of diffuse lung injury caused by 2019-nCoV. SARS-CoV-2 binds to ACE2 receptors on human alveolar epithelial cells via S proteins and enters the cells. NK cells, natural killer cells, macrophages, dendritic cells, monocytes, etc., release cytokines (e.g., IL-6, IL-7, IL-8, IL-17, etc.) and chemokines (e.g., CCL-2, CCL-3, CCL-5, etc.). CD8+ T cells secrete substances such as Perforin, CD107a, and Granzyme B. CD4+ T cells are activated and differentiate into Th1, Th2 effector cells, and other subpopulations (including Tfh cells, etc.), and also secrete cytokines (e.g., INFγ) and chemokines to recruit Immune cells are also secreted (e.g., INFγ) and chemokines are recruited, resulting in a cytokine storm that causes diffuse lung injury.
One study reported that phototherapy using red and near-infrared light reduced lung inflammation and pulmonary fibrosis in mice by downregulating pro-inflammatory cytokines, upregulating IL-10 secretion from fibroblasts and lung cells, and reducing collagen deposition in the lung (176). Since lung inflammation and pulmonary fibrosis are common complications in critically ill patients with novel coronavirus infections, experiments have shown that 650 nm light-emitting diode (LED) treatment may alleviate these life-threatening problems. Compared to conventional laser excitation, 650 nm LEDs have more desirable safety properties. Under its excitation, multifunctional NPs can further inactivate the virus by assisting the photothermal function. In addition, multifunctional nanoparticles have favorable properties for pulmonary delivery and retention, which can overcome the limitation of rapid clearance of antibodies in the lung (177). The unique design of multifunctional NPs not only enables antibody-mediated neutralization to capture novel coronaviruses, but also provides a strategy to mitigate the potential risk of antibody-dependent enhancement (ADE) and new, more infectious novel coronavirus variants by inactivating the virus through direct heating. Together with efficient viral inactivation capabilities, the superior therapeutic efficacy of multifunctional NPs could be further enhanced. Future research will be conducted using site-specific binding approaches, such as site-selective click chemistry (178) that can increase surface antibody binding efficiency, control antibody binding sites and orientation and purify multifunctional nanoparticles to improve their therapeutic efficacy further.
According to studies, COVID-19 has been shown over time to cause multi-system involvement of the cardiovascular system (2, 6, 179), the nervous system, the urinary system (2020), and the hematological system in addition to the respiratory and immune systems (139, 180). This is partly due to the widespread expression of ACE2 as a SARS-CoV-2 receptor in tissues and organs, and partly because SARS-CoV-2 causes a series of systemic pathological manifestations, such as cytokine storm and disorders of coagulation mechanisms (74, 181, 182).
In particular, neurological complications have become an increasingly recognized cause of morbidity and mortality in patients with COVID-19. The most common of these neurological symptoms include cerebrovascular events (183), encephalitis, Guillain-Barré syndrome, acute necrotizing encephalopathy, hemophagocytic lymphoid tissue hyperplasia, and acute ischemic cerebrovascular syndrome (184), as well as neuropsychiatric symptoms such as dizziness, sleep disturbances, cognitive deficits, delirium, hallucinations, and depression. In addition, the chronic neurological aspects of traumatic brain injury, post-stroke syndrome, long COVID-19, intractable Lyme disease, and influenza encephalopathy have close pathophysiological similarities, mainly involving positive feedback loops for TNF maintenance and activation (185), and cerebral venous sinus thrombosis (CVT) formation is also associated with infection with SARS-CoV-2 virus (139).
In addition, right ventricular (RV) dysfunction is common and correlates with poor prognosis in COVID-19 patients (186). And one experiment found that ACE2 expressed by enterocytes derived from human colon differentiation is sensitive to SARS-CoV-2 infection, revealing that IFN-c is a strong driver of epithelial cell differentiation towards the enterocyte lineage and leads to high ACE2 expression and increased susceptibility to SARS-CoV-2 (187).
In summary, the pathogenic mechanism of COVID-19 involves the combined effects of characteristic structures, genes, enzymes, and immune responses. The pathological manifestations and damage caused are manifested as a process in which the lung is the main object of damage and can cause extensive extrapulmonary partial damage as the disease progresses.
Studies on the pathogenic characteristics of neocoronaviruses and the sites of action have demonstrated that among all functional proteins of neocoronaviruses, the S protein is the main antigenic component that binds to host cell receptor proteins, promotes viral invasion of host cells, and stimulates host immune responses. ACE2 is the main receptor infection target. Serine proteases, cysteine proteases, lysosomal proteases, and other enzymes are the main proteases that activate S proteins. Therefore, the S protein can be selected as an important target for vaccine development. For clinical treatment, ACE direct injection can increase the expression of recombinant ACE2 protein and therapeutic vectors that deliver an expression of high levels of ACE2, which is used to overcome virus-induced ACE2 deficiency by increasing the expression of ACE2 protein. Alternatively, ACE inhibitors can balance ACE/ACE2 function by inhibiting the activation of S proteins through the inhibition of related enzyme activities (188). The study of viral exosomes can also be further explored to explore the possible target proteins and signaling molecules for exosomal-cell fusion, thus obtaining new ways to inhibit virus transmission.
During the immune system dysregulation caused by a new coronavirus, the virus modifies its RNA, generates an immune escape mechanism, antagonizes the immune response, and induces apoptosis of immune cells. This phenomenon suggests that viruses change by adapting to new environments, and it is speculated that a new epidemic involving coronaviruses may break out in the future. COVID-19 is highly infectious and has a long incubation period, resulting in a rampant epidemic with a complex and variable disease course, which poses a great challenge to the control of the epidemic. Up to now, no targeted vaccine or effective drug has been developed.
According to the pathogenic mechanism of neo-coronavirus, treatment is still mainly based on antiviral, anti-infective, and symptomatic supportive therapy. Many drugs have been introduced for the treatment of neo-coronavirus pneumonia; for example tetracycline and doxycycline can act as inhibitors of ACE2 binding (28); niclosamide can inhibit RNA viruses during the post-entry phase of viral RNA replication and also exhibits anti-inflammatory activity (189); chloroquine reduces the production of cytokines and damage-associated molecular patterns by interfering with the innate immune pathways of multiple immune cells, thus preventing experimental sepsis and infectious shock (190, 191). In addition, combination therapies have been introduced, such as early triple antiviral therapy with interferon beta-1b, lopinavir-ritonavir, and ribavirin, which shortens the time to viral shedding, reduces cytokine responses, relieves symptoms, and promotes the discharge of patients with mild to moderate COVID-19 (192). The discovery and study of the pathogenic mechanism of neocoronaviruses can bring the course of action of existing drugs into more explicit articulation at the cellular or molecular level to align with clinical applied medicine with more rationalized, standardized, institutionalized, and scientific research. In addition, we believe that a large number of experiments and research records can provide detailed statistics on drug efficacy, efficiency, and the number of stable cases, which can also help promote the organic combination of pathogenesis research and clinical drug action process analysis. We also suggest that a series of accurate and standardized target models may be constructed, and the criteria for clinically effective associations may be derived through model experiments. The establishment of model systems may be expected to be a reference for subsequent studies on preventing and treating other large epidemic diseases.
SASRS-CoV-2 is another highly lethal virus after SARS-CoV and MERS-CoV. COVID-19 epidemic is a major public health emergency in China and the world. The epidemic is spreading globally and the situation has been dire so far. Further experimental, as well as clinical solutions to the currently unresolved and controversial issues, are still needed. Understanding the pathogenesis of neocoronaviruses and developing therapeutic regimens based on the complex pathogenesis of the pathological damage produced by neocoronaviruses is of great importance in response to COVID-19 and possible future epidemic viruses.
Conceptualization, JY, XK, QZ and FO; writing—review and editing, JY, JM, QD, QW and PL; visualization, QZ; supervision, XK and LY; project administration, RG, LY. All authors have read and agreed to the published version of the manuscript.
This research was funded by the Scientific research project of Tianjin Education Commission, grant number 2021KJ134, Tianjin Municipal Education Commission Scientific Research Project, grant number 2019ZD11, Science and Technology Program of Tianjin, China (21ZYJDJC00070), the National Key Research and Development Program of China (2019YFC1708803), and Innovation Team and Talents Cultivation Program of National Administration of Traditional Chinese Medicine (ZYYCXTD-C-202203), Scientific and technological innovation project of China Academy of Chinese Medical Sciences (Grant No. C12021A04701 to RG)
We thank all the authors of the original work and reviewers for their time and kindness in reviewing this paper. The figures were drawn by Figdraw (https://www.figdraw.com/static/index.html#/).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Neuman BW, Kiss G, Kunding AH, Bhella D, Baksh MF, Connelly S, et al. A structural analysis of m protein in coronavirus assembly and morphology. J Struct Biol (2011) 174:11–22. doi: 10.1016/j.jsb.2010.11.021
2. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in wuhan, China. Lancet (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
3. Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A novel coronavirus from patients with pneumonia in chin. N Engl J Med (2020) 382:727–33. doi: 10.1056/NEJMoa2001017
4. Lee P, Kim DJ. Newly emerging human coronaviruses: Animal models and vaccine research for sars, mers, and covid-19. Immune Netw (2020) 20:e28. doi: 10.4110/in.2020.20.e28
5. Chan JF, Yuan S, Kok KH, To KK, Chu H, Yang J, et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet (2020) 395:514–23. doi: 10.1016/S0140-6736(20)30154-9
6. Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in wuhan, China. Jama (2020) 323:1061–9. doi: 10.1001/jama.2020.1585
7. Xia J, Tong J, Liu M, Shen Y, Guo D. Evaluation of coronavirus in tears and conjunctival secretions of patients with SARS-CoV-2 infection. J Med Virol (2020) 92:589–94. doi: 10.1002/jmv.25725
8. Xie P, Ma W, Tang H, Liu D. Severe COVID-19: A review of recent progress with a look toward the future. Front Public Health (2020) 8:189. doi: 10.3389/fpubh.2020.00189
9. Pujadas E, Chaudhry F, Mcbride R, Richter F, Zhao S, Wajnberg A, et al. SARS-CoV-2 viral load predicts COVID-19 mortality. Lancet Respir Med (2020) 8(9):e70. doi: 10.1101/2020.06.11.20128934
10. Cui J, Li F, Shi ZL. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol (2019) 17:181–92. doi: 10.1038/s41579-018-0118-9
11. Fiorino S, Zippi M, Gallo C, Sifo D, Sabbatani S, Manfredi R, et al. The rationale for a multi-step therapeutic approach based on antivirals, drugs and nutrients with immunomodulatory activity in patients with coronavirus-SARS2-induced disease of different severities. Br J Nutr (2021) 125:275–93. doi: 10.1017/S0007114520002913
12. Conceicao C, Thakur N, Human S, Kelly JT, Logan L, Bialy D, et al. The SARS-CoV-2 spike protein has a broad tropism for mammalian ACE2 proteins. PloS Biol (2020) 18:e3001016. doi: 10.1371/journal.pbio.3001016
13. Lu L, Liu X, Jin R, Guan R, Lin R, Qu Z. Potential roles of the renin-angiotensin system in the pathogenesis and treatment of COVID-19. BioMed Res Int (2020) 2020:7520746. doi: 10.1155/2020/7520746
14. Greber UF. Two years into COVID-19 - lessons in SARS-CoV-2 and a perspective from papers in FEBS letters. FEBS Lett (2021) 595:2847–53. doi: 10.1002/1873-3468.14226
15. Luan B, Wang H, Huynh T. Enhanced binding of the N501Y-mutated SARS-CoV-2 spike protein to the human ACE2 receptor: insights from molecular dynamics simulations. FEBS Lett (2021) 595:1454–61. doi: 10.1002/1873-3468.14076
16. Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS-CoV-2. Nat Med (2020) 26:450–2. doi: 10.1038/s41591-020-0820-9
17. Higashikuni Y, Liu W, Obana T, Sata M. Pathogenic basis of thromboinflammation and endothelial injury in COVID-19: Current findings and therapeutic implications. Int J Mol Sci (2021) 22(21):12081. doi: 10.3390/ijms222112081
18. De Pasquale V, Quiccione MS, Tafuri S, Avallone L, Pavone LM. Heparan sulfate proteoglycans in viral infection and treatment: A special focus on SARS-CoV-2. Int J Mol Sci (2021) 22(12):6574. doi: 10.3390/ijms22126574
19. Jennings BC, Kornfeld S, Doray B. A weak COPI binding motif in the cytoplasmic tail of SARS-CoV-2 spike glycoprotein is necessary for its cleavage, glycosylation, and localization. FEBS Lett (2021) 595:1758–67. doi: 10.1002/1873-3468.14109
20. Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res (2020) 30:269–71. doi: 10.1038/s41422-020-0282-0
21. Gao J, Tian Z, Yang X. Breakthrough: Chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. Biosci Trends (2020) 14:72–3. doi: 10.5582/bst.2020.01047
22. Gautret P, Lagier JC, Parola P, Hoang VT, Meddeb L, Mailhe M, et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: Results of an open-label non-randomized clinical trial. Int J Antimicrob Agents (2020) 56:105949. doi: 10.1016/j.ijantimicag.2020.105949
23. Vincent MJ, Bergeron E, Benjannet S, Erickson BR, Rollin PE, Ksiazek TG, et al. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol J (2005) 2:69. doi: 10.1186/1743-422X-2-69
24. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature (2020) 579:270–3. doi: 10.1038/s41586-020-2012-7
25. Guastalegname M, Vallone A. Could chloroquine/hydroxychloroquine be harmful in coronavirus disease 2019 (covid-19) treatment? Clin Infect Dis (2020) 71:888–9. doi: 10.1093/cid/ciaa321
26. Hornick MG, Olson ME, Jadhav AL. SARS-CoV-2 psychiatric sequelae: A review of neuroendocrine mechanisms and therapeutic strategies. Int J Neuropsychopharmacol (2022) 25:1–12. doi: 10.1093/ijnp/pyab069
27. Ferreira G, Blasina F, Rodríguez Rey M, Anesetti G, Sapiro R, Chavarría L, et al. Pathophysiological and molecular considerations of viral and bacterial infections during maternal-fetal and -neonatal interactions of SARS-CoV-2, zika, and mycoplasma infectious diseases. Biochim Biophys Acta Mol Basis Dis (2022) 1868:166285. doi: 10.1016/j.bbadis.2021.166285
28. Putics A, Filipowicz W, Hall J, Gorbalenya AE, Ziebuhr J. ADP-ribose-1”-monophosphatase: A conserved coronavirus enzyme that is dispensable for viral replication in tissue culture. J Virol (2005) 79:12721–31. doi: 10.1128/JVI.79.20.12721-12731.2005
29. Lei HY, Ding YH, Nie K, Dong YM, Xu JH, Yang ML, et al. Potential effects of SARS-CoV-2 on the gastrointestinal tract and liver. BioMed Pharmacother (2021) 133:111064. doi: 10.1016/j.biopha.2020.111064
30. Li MY, Li L, Zhang Y, Wang XS. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect Dis Poverty (2020) 9:45. doi: 10.1186/s40249-020-00662-x
31. Geravandi S, Mahmoudi-Aznaveh A, Azizi Z, Maedler K, Ardestani A. SARS-CoV-2 and pancreas: A potential pathological interaction? Trends Endocrinol Metab (2021) 32:842–5. doi: 10.1016/j.tem.2021.07.004
32. Hou YJ, Okuda K, Edwards CE, Martinez DR, Asakura T, Dinnon KH 3rd, et al. SARS-CoV-2 reverse genetics reveals a variable infection gradient in the respiratory tract. Cell (2020) 182:429–446.e414. doi: 10.1016/j.cell.2020.05.042
33. Soto M, Dizerega G, Rodgers KE. Countermeasure and therapeutic: A(1-7) to treat acute respiratory distress syndrome due to COVID-19 infection. J Renin Angiotensin Aldosterone Syst (2020) 21:1470320320972018. doi: 10.1177/1470320320972018
34. Zou X, Chen K, Zou J, Han P, Hao J, Han Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front Med (2020) 14:185–92. doi: 10.1007/s11684-020-0754-0
35. Kountouri A, Korakas E, Ikonomidis I, Raptis A, Tentolouris N, Dimitriadis G, et al. Type 1 diabetes mellitus in the sars-cov-2 pandemic: Oxidative stress as a major pathophysiological mechanism linked to adverse clinical outcomes. Antioxidants (Basel) (2021) 10(5):752. doi: 10.3390/antiox10050752
36. Chu H, Chan JF-W, Yuen TT-T, Shuai H, Yuan S, Wang Y, et al. Comparative tropism, replication kinetics, and cell damage profiling of SARS-CoV-2 and SARS-CoV with implications for clinical manifestations, transmissibility, and laboratory studies of COVID-19: An observational study. Lancet Microbe (2020) 1:e14–23. doi: 10.1016/S2666-5247(20)30004-5
37. Sindona C, Schepici G, Contestabile V, Bramanti P, Mazzon E. NOX2 activation in COVID-19: Possible implications for neurodegenerative diseases. Medicina (Kaunas) (2021) 57(6):604. doi: 10.3390/medicina57060604
38. Clausen TM, Sandoval DR, Spliid CB, Pihl J, Perrett HR, Painter CD, et al. SARS-CoV-2 infection depends on cellular heparan sulfate and ace2. Cell (2020) 183:1043–1057.e1015. doi: 10.1016/j.cell.2020.09.033
39. Tiwari V, Tandon R, Sankaranarayanan NV, Beer JC, Kohlmeir EK, Swanson-Mungerson M, et al. Preferential recognition and antagonism of SARS-CoV-2 spike glycoprotein binding to 3- O -sulfated heparan sulfate. bioRxiv (2020) 2020.10.08.331751. doi: 10.1101/2020.10.08.331751
40. Chu H, Hu B, Huang X, Chai Y, Zhou D, Wang Y, et al. Host and viral determinants for efficient SARS-CoV-2 infection of the human lung. Nat Commun (2021) 12:134. doi: 10.1038/s41467-020-20457-w
41. Azkur AK, Akdis M, Azkur D, Sokolowska M, Van De Veen W, Brüggen MC, et al. Immune response to SARS-CoV-2 and mechanisms of immunopathological changes in COVID-19. Allergy (2020) 75:1564–81. doi: 10.1111/all.14364
42. Hoffmann M, Kleine-Weber H, Pöhlmann S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol Cell (2020) 78:779–784.e775. doi: 10.1016/j.molcel.2020.04.022
43. Ricci D, Etna MP, Rizzo F, Sandini S, Severa M, Coccia EM. Innate immune response to SARS-CoV-2 infection: From cells to soluble mediators. Int J Mol Sci (2021) 22(13):7017. doi: 10.3390/ijms22137017
44. Pique-Regi R, Romero R, Tarca AL, Luca F, Xu Y, Alazizi A, et al. Does the human placenta express the canonical cell entry mediators for SARS-CoV-2? Elife (2020) 9:e58716. doi: 10.7554/eLife.58716
45. De Clercq E. New nucleoside analogues for the treatment of hemorrhagic fever virus infections. Chem Asian J (2019) 14:3962–8. doi: 10.1002/asia.201900841
46. Beigel JH, Tomashek KM, Dodd LE, Mehta AK, Zingman BS, Kalil AC, et al. Remdesivir for the treatment of covid-19 - final report. N Engl J Med (2020) 383:1813–26. doi: 10.1056/NEJMoa2007764
47. Lai CC, Shih TP, Ko WC, Tang HJ, Hsueh PR. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int J Antimicrob Agents (2020) 55:105924. doi: 10.1016/j.ijantimicag.2020.105924
48. Sheahan TP, Sims AC, Graham RL, Menachery VD, Gralinski LE, Case JB, et al. Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci Transl Med (2017) 9(396):eaal3653. doi: 10.1126/scitranslmed.aal3653
49. Tchesnokov EP, Feng JY, Porter DP, Götte M. Mechanism of inhibition of ebola virus rna-dependent rna polymerase by remdesivir. Viruses (2019) 11(4):326. doi: 10.3390/v11040326
50. Carothers C, Birrer K, Vo M. Acetylcysteine for the treatment of suspected remdesivir-associated acute liver failure in covid-19: A case series. pharmacotherapy. J Hum Pharmacol Drug Ther (2020) 40:1166–71. doi: 10.1002/phar.2464
51. Zhang L, Liu Y. Potential interventions for novel coronavirus in China: A systematic review. J Med Virol (2020) 92:479–90. doi: 10.1002/jmv.25707
53. Guan W, Lan W, Zhang J, Zhao S, Ou J, Wu X, et al. COVID-19: antiviral agents, antibody development and traditional chinese medicine. Virologica Sin (2020) 35:685–98. doi: 10.1007/s12250-020-00297-0
54. Rejinold NS, Piao H, Jin GW, Choi G, Choy JH. Injectable niclosamide nanohybrid as an anti-SARS-CoV-2 strategy. Colloids Surf B Biointerfaces (2021) 208:112063. doi: 10.1016/j.colsurfb.2021.112063
55. Mody V, Ho J, Wills S, Mawri A, Lawson L, Ebert M, et al. Identification of 3-chymotrypsin like protease (3CLPro) inhibitors as potential anti-SARS-CoV-2 agents. Commun Biol (2021) 4:93. doi: 10.1038/s42003-020-01577-x
56. Li J, Lin C, Zhou X, Zhong F, Zeng P, Yang Y, et al. Structural basis of the main proteases of coronavirus bound to drug candidate pf-07321332. J Virol (2022) 96:e0201321. doi: 10.1128/jvi.02013-21
57. Ma C, Sacco MD, Hurst B, Townsend JA, Hu Y, Szeto T, et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res (2020) 30:678–92. doi: 10.1038/s41422-020-0356-z
58. Devarajan A, Vaseghi M. Hydroxychloroquine can potentially interfere with immune function in COVID-19 patients: Mechanisms and insights. Redox Biol (2021) 38:101810. doi: 10.1016/j.redox.2020.101810
59. Shang J, Ye G, Shi K, Wan Y, Luo C, Aihara H, et al. Structural basis of receptor recognition by SARS-CoV-2. Nature (2020) 581:221–4. doi: 10.1038/s41586-020-2179-y
60. Rahbar Saadat Y, Hosseiniyan Khatibi SM, Zununi Vahed S, Ardalan M. Host serine proteases: A potential targeted therapy for COVID-19 and influenza. Front Mol Biosci (2021) 8:725528. doi: 10.3389/fmolb.2021.725528
61. Bertram S, Dijkman R, Habjan M, Heurich A, Gierer S, Glowacka I, et al. TMPRSS2 activates the human coronavirus 229E for cathepsin-independent host cell entry and is expressed in viral target cells in the respiratory epithelium. J Virol (2013) 87:6150–60. doi: 10.1128/JVI.03372-12
62. Glowacka I, Bertram S, Müller MA, Allen P, Soilleux E, Pfefferle S, et al. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J Virol (2011) 85:4122–34. doi: 10.1128/JVI.02232-10
63. Chen Y, Liu Q, Guo D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J Med Virol (2020) 92:418–23. doi: 10.1002/jmv.25681
64. Romano M, Ruggiero A, Squeglia F, Maga G, Berisio R. A structural view of sars-cov-2 rna replication machinery: Rna synthesis, proofreading and final capping. Cells (2020) 9(5):1267. doi: 10.20944/preprints202004.0510.v1
65. Gunasekaran M, Bansal S, Ravichandran R, Sharma M, Perincheri S, Rodriguez F, et al. Respiratory viral infection in lung transplantation induces exosomes that trigger chronic rejection. J Heart Lung Transplant (2020) 39:379–88. doi: 10.1016/j.healun.2019.12.009
66. Klann K, Bojkova D, Tascher G, Ciesek S, Münch C, Cinatl J. Growth factor receptor signaling inhibition prevents sars-cov-2 replication. Mol Cell (2020) 80:164–174.e164. doi: 10.1016/j.molcel.2020.08.006
67. Mohamed Khosroshahi L, Rokni M, Mokhtari T, Noorbakhsh F. Immunology, immunopathogenesis and immunotherapeutics of COVID-19; an overview. Int Immunopharmacol (2021) 93:107364. doi: 10.1016/j.intimp.2020.107364
68. Osman MS, Van Eeden C, Cohen Tervaert JW. Fatal COVID-19 infections: Is NK cell dysfunction a link with autoimmune HLH? Autoimmun Rev (2020) 19:102561. doi: 10.1016/j.autrev.2020.102561
69. Zheng M, Gao Y, Wang G, Song G, Liu S, Sun D, et al. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol Immunol (2020) 17:533–5. doi: 10.1038/s41423-020-0402-2
70. Violi F, Oliva A, Cangemi R, Ceccarelli G, Pignatelli P, Carnevale R, et al. Nox2 activation in covid-19. Redox Biol (2020) 36:101655. doi: 10.1016/j.redox.2020.101655
71. Kumar A, Prasoon P, Sekhawat PS, Pareek V, Faiq MA, Kumari C, et al. Pathogenesis guided therapeutic management of COVID-19: An immunological perspective. Int Rev Immunol (2021) 40:54–71. doi: 10.1080/08830185.2020.1840566
72. Vabret N, Britton GJ, Gruber C, Hegde S, Kim J, Kuksin M, et al. ). immunology of COVID-19: Current state of the science. Immunity (2020) 52:910–41. doi: 10.1016/j.immuni.2020.05.002
73. De Wit E, Van Doremalen N, Falzarano D, Munster VJ. SARS and MERS: Recent insights into emerging coronaviruses. Nat Rev Microbiol (2016) 14:523–34. doi: 10.1038/nrmicro.2016.81
74. Mehta P, Mcauley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet (2020) 395:1033–4. doi: 10.1016/S0140-6736(20)30628-0
75. Neurath MF. COVID-19 and immunomodulation in IBD. Gut (2020) 69:1335–42. doi: 10.1136/gutjnl-2020-321269
76. Lin L, Li T. Interpretation of “Guidelines for the diagnosis and treatment of novel coronavirus (2019-nCoV)) infection by the national health commission (Trial version 5)”. Natl Med J China (2020) 100:E001–1. doi: 10.3760/cma.j.cn112137-20200205-00199
77. Sterne J, Murthy S, Diaz JV, Slutsky AS, Villar J, Angus DC, et al. Association between administration of systemic corticosteroids and mortality among critically ill patients with covid-19: A meta-analysis. Jama (2020) 324:1330–41. doi: 10.1001/jama.2020.17023
78. Agarwal A, Rochwerg B, Lamontagne F, Siemieniuk RA, Agoritsas T, Askie L, et al. A living WHO guideline on drugs for covid-19. Bmj (2020) 370:m3379. doi: 10.1136/bmj.m3379
79. Bruen C, Al-Saadi M, Michelson EA, Tanios M, Mendoza-Ayala R, Miller J, et al. Auxora vs. placebo for the treatment of patients with severe COVID-19 pneumonia: A randomized-controlled clinical trial. Crit Care (2022) 26:101. doi: 10.1186/s13054-022-03964-8
80. Viswanathan T, Arya S, Chan SH, Qi S, Dai N, Misra A, et al. Structural basis of RNA cap modification by SARS-CoV-2. Nat Commun (2020) 11:3718. doi: 10.1038/s41467-020-17496-8
81. V’kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. Coronavirus biology and replication: implications for SARS-CoV-2. Nat Rev Microbiol (2021) 19:155–70. doi: 10.1038/s41579-020-00468-6
82. Kim YM, Shin EC. Type I and III interferon responses in SARS-CoV-2 infection. Exp Mol Med (2021) 53:750–60. doi: 10.1038/s12276-021-00592-0
83. Lee HC, Chathuranga K, Lee JS. Intracellular sensing of viral genomes and viral evasion. Exp Mol Med (2019) 51:1–13. doi: 10.1038/s12276-019-0299-y
84. Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science (2020) 369:718–24. doi: 10.1126/science.abc6027
85. Jiang HW, Zhang HN, Meng QF, Xie J, Li Y, Chen H, et al. SARS-CoV-2 Orf9b suppresses type I interferon responses by targeting TOM70. Cell Mol Immunol (2020) 17:998–1000. doi: 10.1038/s41423-020-0514-8
86. Hasan MZ, Islam S, Matsumoto K, Kawai T. SARS-CoV-2 infection initiates interleukin-17-enriched transcriptional response in different cells from multiple organs. Sci Rep (2021) 11:16814. doi: 10.1038/s41598-021-96110-3
87. Moustaqil M, Ollivier E, Chiu HP, Van Tol S, Rudolffi-Soto P, Stevens C, et al. SARS-CoV-2 proteases PLpro and 3CLpro cleave IRF3 and critical modulators of inflammatory pathways (NLRP12 and TAB1): Implications for disease presentation across species. Emerg Microbes Infect (2021) 10:178–95. doi: 10.1080/22221751.2020.1870414
88. Prompetchara E, Ketloy C, Palaga T. Immune responses in COVID-19 and potential vaccines: Lessons learned from SARS and MERS epidemic. Asian Pac J Allergy Immunol (2020) 38:1–9. doi: 10.12932/AP-200220-0772
89. Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann HH, Zhang Y, et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science (2020) 370(6515):eabd4585. doi: 10.1126/science.abd4585
90. Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J, et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science (2020) 370(6515):eabd4570. doi: 10.1126/science.abd4570
91. Grifoni A, Weiskopf D, Ramirez SI, Mateus J, Dan JM, Moderbacher CR, et al. Targets of T cell responses to sars-cov-2 coronavirus in humans with covid-19 disease and unexposed individuals. Cell (2020) 181:1489–1501.e1415. doi: 10.1016/j.cell.2020.05.015
92. Chen T, Wu D, Chen H, Yan W, Yang D, Chen G, et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: Retrospective study. Bmj (2020) 368:m1091. doi: 10.1136/bmj.m1091
93. Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest (2020) 130:2620–9. doi: 10.1172/JCI137244
94. Giamarellos-Bourboulis EJ, Netea MG, Rovina N, Akinosoglou K, Antoniadou A, Antonakos N, et al. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe (2020) 27:992–1000.e1003. doi: 10.1016/j.chom.2020.04.009
95. Schulte-Schrepping J, Reusch N, Paclik D, Baßler K, Schlickeiser S, Zhang B, et al. Severe COVID-19 is marked by a dysregulated myeloid cell compartment. Cell (2020) 182:1419–1440.e1423. doi: 10.1016/j.cell.2020.08.001
96. Silvin A, Chapuis N, Dunsmore G, Goubet AG, Dubuisson A, Derosa L, et al. Elevated calprotectin and abnormal myeloid cell subsets discriminate severe from mild COVID-19. Cell (2020) 182:1401–1418.e1418. doi: 10.1016/j.cell.2020.08.002
97. Han M, Ma K, Wang X, Yan W, Wang H, You J, et al. Immunological characteristics in type 2 diabetes mellitus among covid-19 patients. Front Endocrinol (Lausanne) (2021) 12:596518. doi: 10.3389/fendo.2021.596518
98. Mahlangu T, Dludla PV, Nyambuya TM, Mxinwa V, Mazibuko-Mbeje SE, Cirilli I, et al. A systematic review on the functional role of Th1/Th2 cytokines in type 2 diabetes and related metabolic complications. Cytokine (2020) 126:154892. doi: 10.1016/j.cyto.2019.154892
99. Zheng HY, Zhang M, Yang CX, Zhang N, Wang XC, Yang XP, et al. Elevated exhaustion levels and reduced functional diversity of T cells in peripheral blood may predict severe progression in COVID-19 patients. Cell Mol Immunol (2020) 17:541–3. doi: 10.1038/s41423-020-0401-3
100. Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19). Front Immunol (2020) 11:827. doi: 10.3389/fimmu.2020.00827
101. Li M, Guo W, Dong Y, Wang X, Dai D, Liu X, et al. Elevated exhaustion levels of nk and cd8(+) t cells as indicators for progression and prognosis of covid-19 disease. Front Immunol (2020) 11:580237. doi: 10.3389/fimmu.2020.580237
102. Bellesi S, Metafuni E, Hohaus S, Maiolo E, Marchionni F, D’innocenzo S, et al. Increased CD95 (Fas) and PD-1 expression in peripheral blood T lymphocytes in COVID-19 patients. Br J Haematol (2020) 191:207–11. doi: 10.1111/bjh.17034
103. Li J, Guo M, Tian X, Wang X, Yang X, Wu P, et al. Virus-host interactome and proteomic survey reveal potential virulence factors influencing sars-cov-2 pathogenesis. Med (N Y) (2021) 2:99–112.e117. doi: 10.1016/j.medj.2020.07.002
104. Wilk AJ, Rustagi A, Zhao NQ, Roque J, Martínez-Colón GJ, Mckechnie JL, et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat Med (2020) 26:1070–6. doi: 10.1038/s41591-020-0944-y
105. Wang C, Xie J, Zhao L, Fei X, Zhang H, Tan Y, et al. Alveolar macrophage dysfunction and cytokine storm in the pathogenesis of two severe COVID-19 patients. EBioMedicine (2020) 57:102833. doi: 10.1016/j.ebiom.2020.102833
106. Kuri-Cervantes L, Pampena MB, Meng W, Rosenfeld AM, Ittner CAG, Weisman AR, et al. Comprehensive mapping of immune perturbations associated with severe COVID-19. Sci Immunol (2020) 5(49):eabd7114. doi: 10.1126/sciimmunol.abd7114
107. Mathew D, Giles JR, Baxter AE, Oldridge DA, Greenplate AR, Wu JE, et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science (2020) 369(6508):eabc8511. doi: 10.1126/science.abc8511
108. Sun B, Feng Y, Mo X, Zheng P, Wang Q, Li P, et al. Kinetics of SARS-CoV-2 specific IgM and IgG responses in COVID-19 patients. Emerg Microbes Infect (2020) 9:940–8. doi: 10.1080/22221751.2020.1762515
109. Long QX, Tang XJ, Shi QL, Li Q, Deng HJ, Yuan J, et al. Clinical and immunological assessment of asymptomatic SARS-CoV-2 infections. Nat Med (2020) 26:1200–4. doi: 10.1038/s41591-020-0965-6
110. Robbiani DF, Gaebler C, Muecksch F, Lorenzi JCC, Wang Z, Cho A, et al. Convergent antibody responses to SARS-CoV-2 in convalescent individuals. Nature (2020) 584:437–42. doi: 10.1038/s41586-020-2456-9
111. Acanfora D, Acanfora C, Ciccone MM, Scicchitano P, Bortone AS, Uguccioni M, et al. The cross-talk between thrombosis and inflammatory storm in acute and long-COVID-19: Therapeutic targets and clinical cases. Viruses (2021) 13(10):1904. doi: 10.3390/v13101904
112. Teijaro JR, Walsh KB, Rice S, Rosen H, Oldstone MB. Mapping the innate signaling cascade essential for cytokine storm during influenza virus infection. Proc Natl Acad Sci USA (2014) 111:3799–804. doi: 10.1073/pnas.1400593111
113. Feng Y, Ling Y, Bai T, Xie Y, Huang J, Li J, et al. COVID-19 with different severities: A multicenter study of clinical features. Am J Respir Crit Care Med (2020) 201:1380–8. doi: 10.1164/rccm.202002-0445OC
114. Hu B, Huang S, Yin L. The cytokine storm and COVID-19. J Med Virol (2021) 93:250–6. doi: 10.1002/jmv.26232
115. Tan LY, Komarasamy TV, Rmt Balasubramaniam V. Hyperinflammatory immune response and COVID-19: A double edged sword. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.742941
116. Tsuchiya K. Inflammasome-associated cell death: Pyroptosis, apoptosis, and physiological implications. Microbiol Immunol (2020) 64:252–69. doi: 10.1111/1348-0421.12771
117. Mcgonagle D, Ramanan AV, Bridgewood C. Immune cartography of macrophage activation syndrome in the COVID-19 era. Nat Rev Rheumatol (2021) 17:145–57. doi: 10.1038/s41584-020-00571-1
118. Rodriguez-Smith JJ, Verweyen EL, Clay GM, Esteban YM, De Loizaga SR, Baker EJ, et al. Inflammatory biomarkers in COVID-19-associated multisystem inflammatory syndrome in children, Kawasaki disease, and macrophage activation syndrome: A cohort study. Lancet Rheumatol (2021) 3:e574–84. doi: 10.1016/S2665-9913(21)00139-9
119. Xu X, Han M, Li T, Sun W, Wang D, Fu B, et al. Effective treatment of severe COVID-19 patients with tocilizumab. Proc Natl Acad Sci USA (2020) 117:10970–5. doi: 10.1073/pnas.2005615117
120. Zhou Y, Fu B, Zheng X, Wang D, Zhao C, Qi Y, et al. Pathogenic T-cells and inflammatory monocytes incite inflammatory storms in severe COVID-19 patients. Natl Sci Rev (2020) 7:998–1002. doi: 10.1093/nsr/nwaa041
121. Moschonas IC, Tselepis AD. SARS-CoV-2 infection and thrombotic complications: A narrative review. J Thromb Thrombolysis (2021) 52:111–23. doi: 10.1007/s11239-020-02374-3
122. Fajgenbaum DC, June CH. Cytokine storm. N Engl J Med (2020) 383:2255–73. doi: 10.1056/NEJMra2026131
123. Zhu Z, Cai T, Fan L, Lou K, Hua X, Huang Z, et al. Clinical value of immune-inflammatory parameters to assess the severity of coronavirus disease 2019. Int J Infect Dis (2020) 95:332–9. doi: 10.1016/j.ijid.2020.04.041
124. Wieczfinska J, Kleniewska P, Pawliczak R. Oxidative stress-related mechanisms in SARS-CoV-2 infections. Oxid Med Cell Longev (2022) 2022:5589089. doi: 10.1155/2022/5589089
125. Checa J, Aran JM. Reactive oxygen species: Drivers of physiological and pathological processes. J Inflamm Res (2020) 13:1057–73. doi: 10.2147/JIR.S275595
126. Fu B, Xu X, Wei H. Why tocilizumab could be an effective treatment for severe COVID-19? J Transl Med (2020) 18:164. doi: 10.1186/s12967-020-02339-3
127. Lu CC, Chen MY, Lee WS, Chang YL. Potential therapeutic agents against COVID-19: What we know so far. J Chin Med Assoc (2020) 83:534–6. doi: 10.1097/JCMA.0000000000000318
128. Lomakin NV, Bakirov BA, Protsenko DN, Mazurov VI, Musaev GH, Moiseeva OM, et al. The efficacy and safety of levilimab in severely ill COVID-19 patients not requiring mechanical ventilation: Results of a multicenter randomized double-blind placebo-controlled phase III CORONA clinical study. Inflamm Res (2021) 70:1233–46. doi: 10.1007/s00011-021-01507-5
129. Díaz Y, Ramos-Suzarte M, Martín Y, Calderón N, Hidalgo CJ. Use of a humanized anti-CD6 monoclonal antibody (Itolizumab) in elderly patients with moderate COVID-19. Gerontology (2020) 66:1–9. doi: 10.1159/000512210
130. Cai Q, Yang M, Liu D, Chen J, Shu D, Xia J, et al. Experimental treatment with favipiravir for COVID-19: An open-label control study. Eng (Beijing) (2020) 6:1192–8. doi: 10.1016/j.eng.2020.03.007
131. Huang M, Tang T, Pang P, Li M, Ma R, Lu J, et al. Treating COVID-19 with chloroquine. J Mol Cell Biol (2020) 12:322–5. doi: 10.1093/jmcb/mjaa014
132. Stone JH, Frigault MJ, Serling-Boyd NJ, Fernandes AD, Harvey L, Foulkes AS, et al. Efficacy of tocilizumab in patients hospitalized with covid-19. N Engl J Med (2020) 383:2333–44. doi: 10.1056/NEJMoa2028836
133. Maity S, Wu WC, Tracy JB, Clarke LI, Bochinski JR. Nanoscale steady-state temperature gradients within polymer nanocomposites undergoing continuous-wave photothermal heating from gold nanorods. Nanoscale (2017) 9:11605–18. doi: 10.1039/C7NR04613H
134. Cai X, Chen M, Prominski A, Lin Y, Ankenbruck N, Rosenberg J, et al. A multifunctional neutralizing antibody-conjugated nanoparticle inhibits and inactivates SARS-CoV-2. Adv Sci (Weinh) (2022) 9:e2103240. doi: 10.1002/advs.202103240
135. Benhamou D, Keita H, Ducloy-Bouthors AS. Coagulation changes and thromboembolic risk in COVID-19 obstetric patients. Anaesth Crit Care Pain Med (2020) 39:351–3. doi: 10.1016/j.accpm.2020.05.003
136. Ryan C, Minc A, Caceres J, Balsalobre A, Dixit A, Ng BK, et al. Predicting severe outcomes in covid-19 related illness using only patient demographics, comorbidities and symptoms. Am J Emerg Med (2021) 45:378–84. doi: 10.1016/j.ajem.2020.09.017
137. Gu SX, Tyagi T, Jain K, Gu VW, Lee SH, Hwa JM, et al. Thrombocytopathy and endotheliopathy: crucial contributors to COVID-19 thromboinflammation. Nat Rev Cardiol (2021) 18:194–209. doi: 10.1038/s41569-020-00469-1
138. Roy D, Ghosh R, Dubey S, Dubey MJ, Benito-León J, Kanti Ray B. Neurological and neuropsychiatric impacts of COVID-19 pandemic. Can J Neurol Sci (2021) 48:9–24. doi: 10.1017/cjn.2020.173
139. Ghosh R, Roy D, Mandal A, Pal SK, Chandra Swaika B, Naga D, et al. Cerebral venous thrombosis in COVID-19. Diabetes Metab Syndr (2021) 15:1039–45. doi: 10.1016/j.dsx.2021.04.026
140. Ye Q, Wang B, Mao J. The pathogenesis and treatment of the `Cytokine storm’ in COVID-19. J Infect (2020) 80:607–13. doi: 10.1016/j.jinf.2020.03.037
141. Manolis AS, Manolis TA, Manolis AA, Papatheou D, Melita H. COVID-19 infection: Viral macro- and micro-vascular coagulopathy and Thromboembolism/Prophylactic and therapeutic management. J Cardiovasc Pharmacol Ther (2021) 26:12–24. doi: 10.1177/1074248420958973
142. Billett HH, Reyes-Gil M, Szymanski J, Ikemura K, Stahl LR, Lo Y, et al. Anticoagulation in covid-19: Effect of enoxaparin, heparin, and apixaban on mortality. Thromb Haemost (2020) 120:1691–9. doi: 10.1055/s-0040-1720978
143. Mehta PK, Griendling KK. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol (2007) 292:C82–97. doi: 10.1152/ajpcell.00287.2006
144. Santos R, Sampaio WO, Alzamora AC, Motta-Santos D, Alenina N, Bader M, et al. The ACE2/Angiotensin-(1-7)/MAS axis of the renin-angiotensin system: Focus on angiotensin-(1-7). Physiol Rev (2018) 98:505–53. doi: 10.1152/physrev.00023.2016
145. Yau JW, Teoh H, Verma S. Endothelial cell control of thrombosis. BMC Cardiovasc Disord (2015) 15:130. doi: 10.1186/s12872-015-0124-z
146. Do H, Healey JF, Waller EK, Lollar P. Expression of factor VIII by murine liver sinusoidal endothelial cells. J Biol Chem (1999) 274:19587–92. doi: 10.1074/jbc.274.28.19587
147. Liu S, Xu X, Kang Y, Xiao Y, Liu H. ). degradation and detoxification of azo dyes with recombinant ligninolytic enzymes from aspergillus sp. with secretory overexpression in pichia pastoris. R Soc Open Sci (2020) 7:200688. doi: 10.1098/rsos.200688
148. Watanabe T, Barker TA, Berk BC. Angiotensin ii and the endothelium: Diverse signals and effects. Hypertension (2005) 45:163–9. doi: 10.1161/01.HYP.0000153321.13792.b9
149. Rand JH, Glanville RW, Wu XX, Ross JM, Zangari M, Gordon RE, et al. The significance of subendothelial von willebrand factor. Thromb Haemost (1997) 78:445–50. doi: 10.1055/s-0038-1657567
150. Leebeek FW, Eikenboom JC. Von Willebrand’s disease. N Engl J Med (2016) 375:2067–80. doi: 10.1056/NEJMra1601561
151. Blair P, Rex S, Vitseva O, Beaulieu L, Tanriverdi K, Chakrabarti S, et al. Stimulation of toll-like receptor 2 in human platelets induces a thromboinflammatory response through activation of phosphoinositide 3-kinase. Circ Res (2009) 104:346–54. doi: 10.1161/CIRCRESAHA.108.185785
152. Subramaniam S, Jurk K, Hobohm L, Jäckel S, Saffarzadeh M, Schwierczek K, et al. Distinct contributions of complement factors to platelet activation and fibrin formation in venous thrombus development. Blood (2017) 129:2291–302. doi: 10.1182/blood-2016-11-749879
153. Wojta J, Huber K, Valent P. New aspects in thrombotic research: complement induced switch in mast cells from a profibrinolytic to a prothrombotic phenotype. Pathophysiol Haemost Thromb (2003) 33:438–41. doi: 10.1159/000083842
154. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr., et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA (2010) 107:15880–5. doi: 10.1073/pnas.1005743107
155. Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood (2011) 118:1952–61. doi: 10.1182/blood-2011-03-343061
156. Dinicolantonio JJ, Mccarty M. Thrombotic complications of COVID-19 may reflect an upregulation of endothelial tissue factor expression that is contingent on activation of endosomal NADPH oxidase. Open Heart (2020) 7(1):e001337. doi: 10.1136/openhrt-2020-001337
157. Kaundal RK, Kalvala AK, Kumar A. Neurological implications of covid-19: role of redox imbalance and mitochondrial dysfunction. Mol Neurobiol (2021) 58:4575–87. doi: 10.1007/s12035-021-02412-y
158. Ali M, Spinler SA. COVID-19 and thrombosis: From bench to bedside. Trends Cardiovasc Med (2021) 31:143–60. doi: 10.1016/j.tcm.2020.12.004
159. Barnes BJ, Adrover JM, Baxter-Stoltzfus A, Borczuk A, Cools-Lartigue J, Crawford JM, et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J Exp Med (2020) 217(6):e20200652. doi: 10.1084/jem.20200652
160. Meidaninikjeh S, Sabouni N, Marzouni HZ, Bengar S, Khalili A, Jafari R. Monocytes and macrophages in COVID-19: Friends and foes. Life Sci (2021) 269:119010. doi: 10.1016/j.lfs.2020.119010
161. Taghiloo S, Aliyali M, Abedi S, Mehravaran H, Sharifpour A, Zaboli E, et al. Apoptosis and immunophenotyping of peripheral blood lymphocytes in Iranian COVID-19 patients: Clinical and laboratory characteristics. J Med Virol (2021) 93:1589–98. doi: 10.1002/jmv.26505
162. Bost P, Giladi A, Liu Y, Bendjelal Y, Xu G, David E, et al. Host-viral infection maps reveal signatures of severe COVID-19 patients. Cell (2020) 181:1475–1488.e1412. doi: 10.1016/j.cell.2020.05.006
163. Shen B, Yi X, Sun Y, Bi X, Du J, Zhang C, et al. Proteomic and metabolomic characterization of covid-19 patient sera. Cell (2020) 182:59–72.e15. doi: 10.1016/j.cell.2020.05.032
164. Mattoo SU, Kim SJ, Ahn DG, Myoung J. Escape and over-activation of innate immune responses by sars-cov-2: two faces of a coin. Viruses (2022) 14(3):530. doi: 10.3390/v14030530
165. Li S, Zhang Y, Guan Z, Li H, Ye M, Chen X, et al. SARS-CoV-2 triggers inflammatory responses and cell death through caspase-8 activation. Signal Transduct Target Ther (2020) 5:235. doi: 10.1038/s41392-020-00334-0
166. Felsenstein S, Herbert JA, Mcnamara PS, Hedrich CM. COVID-19: Immunology and treatment options. Clin Immunol (2020) 215:108448. doi: 10.1016/j.clim.2020.108448
167. Anka AU, Tahir MI, Abubakar SD, Alsabbagh M, Zian Z, Hamedifar H, et al. Coronavirus disease 2019 (COVID-19): An overview of the immunopathology, serological diagnosis and management. Scandinavian J Immunol (2020) 93(4):e12998. doi: 10.1111/sji.12998
168. Morris G, Bortolasci CC, Puri BK, Olive L, Marx W, O’neil A, et al. The pathophysiology of SARS-CoV-2: A suggested model and therapeutic approach. Life Sci (2020) 258:118166. doi: 10.1016/j.lfs.2020.118166
169. Prete M, Favoino E, Catacchio G, Racanelli V, Perosa F. SARS-CoV-2 inflammatory syndrome. Clinical features and rationale for immunological treatment. Int J Mol Sci (2020) 21(9):3377. doi: 10.3390/ijms21093377
170. Mason RJ. Pathogenesis of COVID-19 from a cell biology perspective. Eur Respir J (2020) 55(4):2000607. doi: 10.1183/13993003.00607-2020
171. Carcaterra M, Caruso C. Alveolar epithelial cell type ii as main target of sars-cov-2 virus and covid-19 development via nf-kb pathway deregulation: A physio-pathological theory. Med Hypotheses (2021) 146:110412. doi: 10.1016/j.mehy.2020.110412
172. Tang Y, Liu J, Zhang D, Xu Z, Ji J, Wen C. Cytokine storm in covid-19: The current evidence and treatment strategies. Front Immunol (2020) 11:1708. doi: 10.3389/fimmu.2020.01708
173. Ginsburg I, Fibach E. Polycations and polyanions in SARS-CoV-2 infection. Med Hypotheses (2021) 146:110470. doi: 10.1016/j.mehy.2020.110470
174. Lin C-W, Lin K-H, Hsieh T-H, Shiu S-Y, Li J-Y. Severe acute respiratory syndrome coronavirus 3C-like protease-induced apoptosis. FEMS Immunol Med Microbiol (2006) 46:375–80. doi: 10.1111/j.1574-695X.2006.00045.x
175. Lotfi R, Kalmarzi RN, Roghani SA. A review on the immune responses against novel emerging coronavirus (SARS-CoV-2). Immunol Res (2021) 69:213–24. doi: 10.1007/s12026-021-09198-0
176. Brochetti RA, Leal MP, Rodrigues R, Da Palma RK, De Oliveira LVF, Horliana A, et al. Photobiomodulation therapy improves both inflammatory and fibrotic parameters in experimental model of lung fibrosis in mice. Lasers Med Sci (2017) 32:1825–34. doi: 10.1007/s10103-017-2281-z
177. Koussoroplis SJ, Paulissen G, Tyteca D, Goldansaz H, Todoroff J, Barilly C, et al. PEGylation of antibody fragments greatly increases their local residence time following delivery to the respiratory tract. J Control Release (2014) 187:91–100. doi: 10.1016/j.jconrel.2014.05.021
178. Welch NG, Scoble JA, Muir BW, Pigram PJ. Orientation and characterization of immobilized antibodies for improved immunoassays (Review). Biointerphases (2017) 12:02d301. doi: 10.1116/1.4978435
179. Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in wuhan, China: A descriptive study. Lancet (2020) 395:507–13. doi: 10.1016/S0140-6736(20)30211-7
180. Estrada E. Cascading from SARS-CoV-2 to parkinson’s disease through protein-protein interactions. Viruses (2021) 13(5):897. doi: 10.3390/v13050897
181. Berlin DA, Gulick RM, Martinez FJ. Severe covid-19. N Engl J Med (2020) 383:2451–60. doi: 10.1056/NEJMcp2009575
182. Jose RJ, Manuel A. COVID-19 cytokine storm: The interplay between inflammation and coagulation. Lancet Respir Med (2020) 8:e46–7. doi: 10.1016/S2213-2600(20)30216-2
183. Karadaş Ö, Öztürk B, Sonkaya AR. A prospective clinical study of detailed neurological manifestations in patients with COVID-19. Neurol Sci (2020) 41:1991–5. doi: 10.1007/s10072-020-04547-7
184. Shehata GA, Lord KC, Grudzinski MC, Elsayed M, Abdelnaby R, Elshabrawy HA. Neurological complications of covid-19: Underlying mechanisms and management. Int J Mol Sci (2021) 22(8):4081. doi: 10.3390/ijms22084081
185. Clark IA. Chronic cerebral aspects of long covid. Post-stroke syndromes and similar states share their pathogenesis and perispinal etanercept treatment logic. Pharmacol Res Perspect (2022) 10:e00926. doi: 10.1002/prp2.926
186. Gibson LE, Fenza RD, Lang M, Capriles MI, Li MD, Kalpathy-Cramer J, et al. Right ventricular strain is common in intubated covid-19 patients and does not reflect severity of respiratory illness. J Intensive Care Med (2021) 36:900–9. doi: 10.1177/08850666211006335
187. Heuberger J, Trimpert J, Vladimirova D, Goosmann C, Lin M, Schmuck R, et al. Epithelial response to IFN-γ promotes SARS-CoV-2 infection. EMBO Mol Med (2021) 13:e13191. doi: 10.15252/emmm.202013191
188. Wu Y. Compensation of ACE2 function for possible clinical management of 2019-nCoV-Induced acute lung injury. Virologica Sin (2020) 35:256–8. doi: 10.1007/s12250-020-00205-6
189. Cairns DM, Dulko D, Griffiths JK, Golan Y, Cohen T, Trinquart L, et al. Efficacy of niclosamide vs placebo in sars-cov-2 respiratory viral clearance, viral shedding, and duration of symptoms among patients with mild to moderate covid-19: A phase 2 randomized clinical trial. JAMA Netw Open (2022) 5:e2144942. doi: 10.1001/jamanetworkopen.2021.44942
190. Martinson JA, Montoya CJ, Usuga X, Ronquillo R, Landay AL, Desai SN. Chloroquine modulates HIV-1-induced plasmacytoid dendritic cell alpha interferon: implication for T-cell activation. Antimicrob Agents Chemother (2010) 54:871–81. doi: 10.1128/AAC.01246-09
191. Yang M, Cao L, Xie M, Yu Y, Kang R, Yang L, et al. Chloroquine inhibits HMGB1 inflammatory signaling and protects mice from lethal sepsis. Biochem Pharmacol (2013) 86:410–8. doi: 10.1016/j.bcp.2013.05.013
192. Hung IF, Lung KC, Tso EY, Liu R, Chung TW, Chu MY, et al. Triple combination of interferon beta-1b, lopinavir-ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: An open-label, randomised, phase 2 trial. Lancet (2020) 395:1695–704. doi: 10.1016/S0140-6736(20)31042-4
Keywords: COVID-19, SARS-CoV-2, pathogenic mechanism, virus infected, immune pathogenesis
Citation: Yi J, Miao J, Zuo Q, Owusu F, Dong Q, Lin P, Wang Q, Gao R, Kong X and Yang L (2022) COVID-19 pandemic: A multidisciplinary perspective on the pathogenesis of a novel coronavirus from infection, immunity and pathological responses. Front. Immunol. 13:978619. doi: 10.3389/fimmu.2022.978619
Received: 26 June 2022; Accepted: 04 August 2022;
Published: 26 August 2022.
Edited by:
Chang Li, Chinese Academy of Agricultural Sciences (CAAS), ChinaReviewed by:
Hualei Wang, Jilin University, ChinaCopyright © 2022 Yi, Miao, Zuo, Owusu, Dong, Lin, Wang, Gao, Kong and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rui Gao, cnVpZ2FvQDEyNi5jb20=; Xianbin Kong, a29uZ3hpYW5iaW52aXBAMTYzLmNvbQ==; Long Yang, bG9uZy55YW5nQHRqdXRjbS5lZHUuY24=
†The authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.