 Bouchra Ghazi
Bouchra Ghazi Adil El Ghanmi2
Adil El Ghanmi2 Amina Ghouzlani
Amina Ghouzlani Abdallah Badou
Abdallah Badou
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 15 November 2022
Sec. Vaccines and Molecular Therapeutics
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.978195
This article is part of the Research Topic Global Excellence in Molecular Immunology and Therapeutics: Africa 2021 View all 8 articles
Chimeric antigen receptor (CAR) T-cells represent a new genetically engineered cell-based immunotherapy tool against cancer. The use of CAR T-cells has revolutionized the therapeutic approach for hematological malignancies. Unfortunately, there is a long way to go before this treatment can be developed for solid tumors, including colorectal cancer. CAR T-cell therapy for colorectal cancer is still in its early stages, and clinical data are scarce. Major limitations of this therapy include high toxicity, relapses, and an impermeable tumor microenvironment for CAR T-cell therapy in colorectal cancer. In this review, we summarize current knowledge, highlight challenges, and discuss perspectives regarding CAR T-cell therapy in colorectal cancer.
State-of-the-art in cancer immunotherapy consists of approaches that combine cellular engineering with synthetic biology to generate a powerful immune weapon. Current strategies include therapeutic tumor-associated antigen (TAA)-specific monoclonal antibodies, immune checkpoint inhibitors (ICIs) (1–4), bispecific antibodies, and four generations of chimeric antigen receptor (CAR) T-cells for targeting multiple tumor antigens. All these strategies are widely used in clinical practice for treating hematologic and solid tumors.
Colorectal cancer (CRC) currently ranks third in cancer incidence and continues to be the leading cause of cancer-related death. In 2020, there were 1,931,590 new cases of CRC worldwide and 935,173 deaths, accounting for 10.0% and 9.4% of the total number of cancer cases and deaths, respectively (5, 6). In CRC, the most effective therapeutic strategy for increasing patient survival involves surgical resection alone or in combination with chemotherapy and radiotherapy. However, in advanced stages of CRC, representing approximately 25% of cases, therapeutic strategies based on surgery are not useful and are associated with higher risk of poor outcomes (7, 8).
Current pharmacological approaches for optimizing CRC management include non-specific drugs, such as fluoropyrimidines, irinotecan, and oxiplatin, as well as targeted drugs, such as bevacizumab, angiogenesis inhibitors, cetuximab, epidermal growth factor receptor (EGFR) inhibitors, regorafenib, and multikinase inhibitors (7, 9, 10).
In recent years, combinational regimes and targeted drugs have moved rapidly from the bench to the bedside, and many others are currently under investigation in clinical trials to improve the outcomes of patients with CRC, especially those with metastatic disease (5, 11, 12). However, despite these efforts, the 5-year overall survival (OS) rate of patients with CRC ranges from 90 to 10%, highlighting the urgent need for better therapeutic strategies (7, 10, 13, 14).
In the latest few decades, immunotherapy has been proven to aid in the treatment of many solid tumors, notably melanoma and non-small cell lung cancer (15, 16). Immune checkpoint blockade (ICB), such as blocking of programmed death-1 (PD-1), has emerged as a successful approach that provides long-lasting benefits for patients with cancer and significantly improves prognosis (17). Currently, this immunotherapeutic approach is used as the first-line treatment for patients with advanced solid tumors (18–21). However, in CRC, ICB targeting PD-1 or PD-ligand 1 (PD-L1) has shown no clinical benefit. In 2017, pembrolizumab, an anti-PD-1, was approved by the Food and Drug Administration (FDA) as a second-line treatment for patients with mCRC associated with microsatellite instability (MSI-H) (22). In 2020, the treatment paradigm was revolutionized by the FDA approval of pembrolizumab, which was used as the first-line treatment for patients with unresectable or metastatic MSI-H CRC, with no prior systemic treatment for advanced disease; the approval was based on data from the Keynote-177 study (5, 23). However, only a few patients with deficient DNA mismatch repair (dMMR)/MSH-H respond to immunotherapy, and approximately 50% of them rapidly develop primary immune resistance (15–17).
In view of the sustainable clinical responses in the treatment of hematological indications, CAR T-cell therapy against solid tumors has shown poor efficacy and did not achieve the desired outcome (24–28). These unsuccessful outcomes are mainly due to two major obstacles associated with poor response or relapse during CAR T-cell therapy. The first major issue is the lack of suitable target antigens for CAR T-cells in solid tumors; in most individuals, target antigens are present in healthy tissues as well, leading to on-target, off-tumor toxicities (29). The second dilemma is the poor accessibility of target antigens from CAR T-cells, leading to inefficient activation, expansion, and function. Other obstacles are the heterogeneous pattern of tumor antigens expressed by tumor cells, escape mechanisms for evading single tumor antigen-specific CAR T-cells, immunosuppressive tumor microenvironment (TME) (30), CAR T cells non-responsiveness or exhaustion, insufficient infiltration of CAR T-cells into the TME, and metabolic privation. These are implicated in the poor efficacy of CAR T-cell therapy, leading to negative results in clinical trials (31–33).
In this review, we aim to provide state-of-the-art knowledge in preclinical and clinical studies of CAR T-cell therapies for CRC treatment, the most important success-limiting challenges in the application of CAR T-cells and ultimately future directions needed to render this therapeutic tool useful for CRC to achieve efficient clinical translation.
Modified T-cell therapies, including CAR T-cell therapy, have already emerged in the realm of hematologic cancers. CAR T-cell strategy is a personalized immunotherapy based on genetically modified allogeneic or autologous synthetic CAR-expressing T-cells (34). The CAR molecule is composed of extracellular binding moieties, often an antibody-derived single-chain fragment variable or tumor-specific antigen (TSA)-sensing element. Atransmembrane anchor fused to signaling domains from the T-cell-receptor zeta chain complex and costimulatory molecules, such as CD28 and 4-1BB is also found. Specific and direct recognition of tumor antigens through the extracellular domain drives CAR T-cell activation, which results in cancer cell death (35). CARs are most commonly transduced into a patient’s T-cells using lentiviral or gammaretroviral vectors. After the manufacture of CAR T-cells ex vivo, the patient receives lymphodepleting chemotherapy, if needed, followed by CAR T-cell infusion (Figure 1).
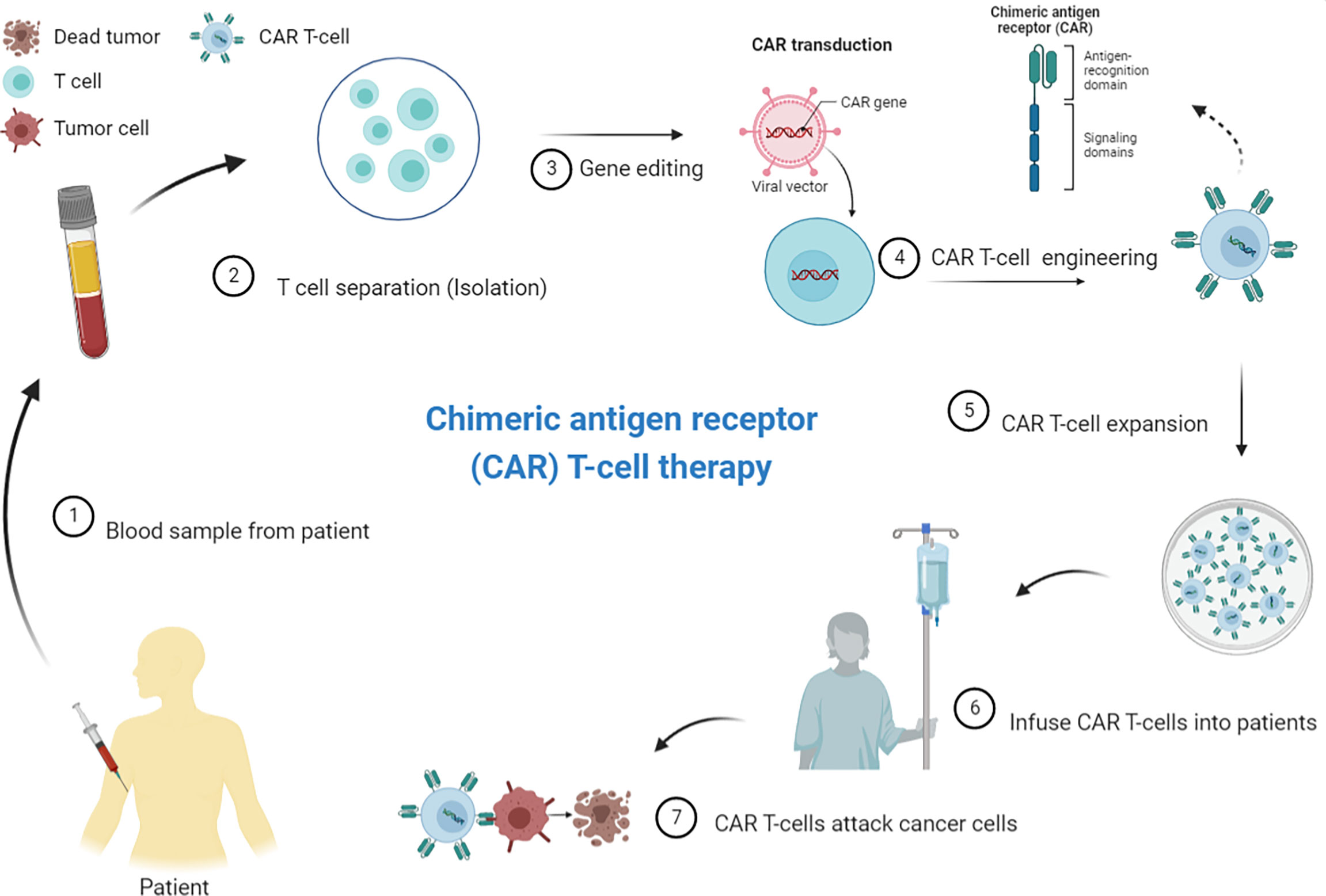
Figure 1 Principles of chimeric antigen receptor (CAR) T-cell therapy/CAR T-cell manufacturing process. CAR T-cell production includes normal T-cell extraction from the patient’s peripheral blood, followed by enrichment and CAR vector delivery, using viral or non-viral vector systems, into the T-cells in vitro. The CAR T-cells is subjected to expansion before being re-injected into the patient’s bloodstream. Patients usually receive a lymphodepletion before CAR T-cell administration to improve the expansion of adoptively transferred T-cells. The CAR T-cells proliferate and attack tumor cells that carry the specific antigen against which the CARs are directed (Source: Author’s personal collection, 2021). Created with BioRender.com.
CAR T-cell receptors are designed to (5) deliver strong activation, proliferation, and survival signals via a single binding event (6); recognize tumor antigens in a manner independent of the major histocompatibility complex, bypassing the downregulation of the major histocompatibility complex by certain tumors; and (7) exhibit high affinity even at low antigen density (36–38). The evolution of CAR T-cell strategies from the original design, ectodomain antibody single-chain fragment variable fragment engineered to the T-cell receptor (TCR) ζ-chain, has resulted in several CAR generations. Hence, CAR design has evolved from conventional structures to more sophisticated moieties, enabling combinatorial antigen selection with different signaling abilities (37). Thus, this development supports the production of armored CAR T-cells with substantial enhancement of antitumor efficacy and adequate control of toxicity (39, 40).
The growing demand for CAR T-cells in cancer therapy has been accompanied by an effort to ameliorate a few undesirable aspects of CAR T-cells.
CAR T-cells can amplify or attenuate the immune response due to well-thought out the intracellular domains design, by generating signals of varying strengths, durations, and intensities. An expected complication of this therapy is that excessive CAR T-cell activation drives a cytokine storm, leading to “on-target toxicity.” Another potential issue is “off-target toxicity” to normal cells when the current targets do not allow distinction between normal and cancer cells (37). On-target and off-target toxicities of CAR T-cells need to be monitored and measures taken quickly to abrogate them. In contrast, CAR T-cell exhaustion induced by sustained signaling of chimeric receptors contributes to poor persistence and limits the durability of CAR T-cell immunotherapy.
T-cell exhaustion is a dysfunctional state of differentiated T-cells that differs from anergy and senescence. The exhausted state is a complex phenotype associated with progressive loss of effector functions and poor memory T-cell response (41). This exhausted state is associated with increased expression of multiple inhibitory receptors, including PD1 (42). Another concern about conventional CAR T-cells is fixed antigen specificity, which means that only one TAA can be targeted at a time (37). This limitation harbors the risk of tumor-variant appearance and limits the eficacy of CAR T-cell therapy.
CAR T-cells do not follow the classical T-cell–target cell immunological synapse topology. The epitope location, structural dimensions of the target molecule, and extracellular spacer length of CAR allows for the specificity of CAR T-cell dimensional interactions (43, 44). Steric constraints due to the location of the target epitope may require structural modeling of the CAR for adapting/modulating the extracellular spacer length and promoting synapse formation between effector T-cells and tumor cells (45, 46). Other aspects, including the density and accessibility of target molecules to tumor cells, need to be considered (46). In the context of immunological synapse dynamics, tumor cells and their microenvironment underpin specificity and complexity of immune responses through secretion of immunosuppressive soluble factors, such as indoleamine-2,3-dioxygenase-1 (IDO1), PD-L1, and IL-10 (47–50), and recruitment of immunosuppressive cells, such as tumor-associated macrophages and myeloid-derived suppressor cells (51). This immunosuppressive microenvironment limits immune cell homing and infiltration and inhibits effector T-cell activity (52).
The metabolic landscape created by cancer cells in the TME dampens the ability of T-cells to respond (53). The energetic state of the TME and catabolites generated by tumor metabolism play a critical role in T-cell biology, affecting T-cell infiltration, survival, and function (54). The production of immunosuppressive enzymes, such as IDO, tryptophan-2,3-dioxygenase, nitric oxide synthase, and arginase-1, impairs the activation and effector function of T-cells by causing tryptophan and L-arginine deficiency. These aspects of the TME impact CAR T-cell functions and lead to significant clinical implications (55).
According to data from clinical trials on CAR T-cells in solid tumors, immunotherapy outcomes are associated with multiple parameters that reflect cross-talk between the TME and the immune system, including tumor mutational burden (TMB), immunologically hot tumors with inflammatory microenvironments containing high levels of infiltrating cytotoxic T-cells, immune escape mechanisms, timing of administration, and delivery systems of immunotherapeutic agents (56).
These considerations indicate that the following strategies are key for the generation of potent CAR T-cells with limited side effects: improving CAR T-cell chimeric receptor flexibility, engineering an appropriate extracellular domain for antigen recognition and binding ability, and improving intracellular signaling capacities. Further understanding of TME variables will help in the design of therapeutic strategies that would enhance the clinical effectiveness of CAR T-cells, especially in solid tumors (Figure 2).
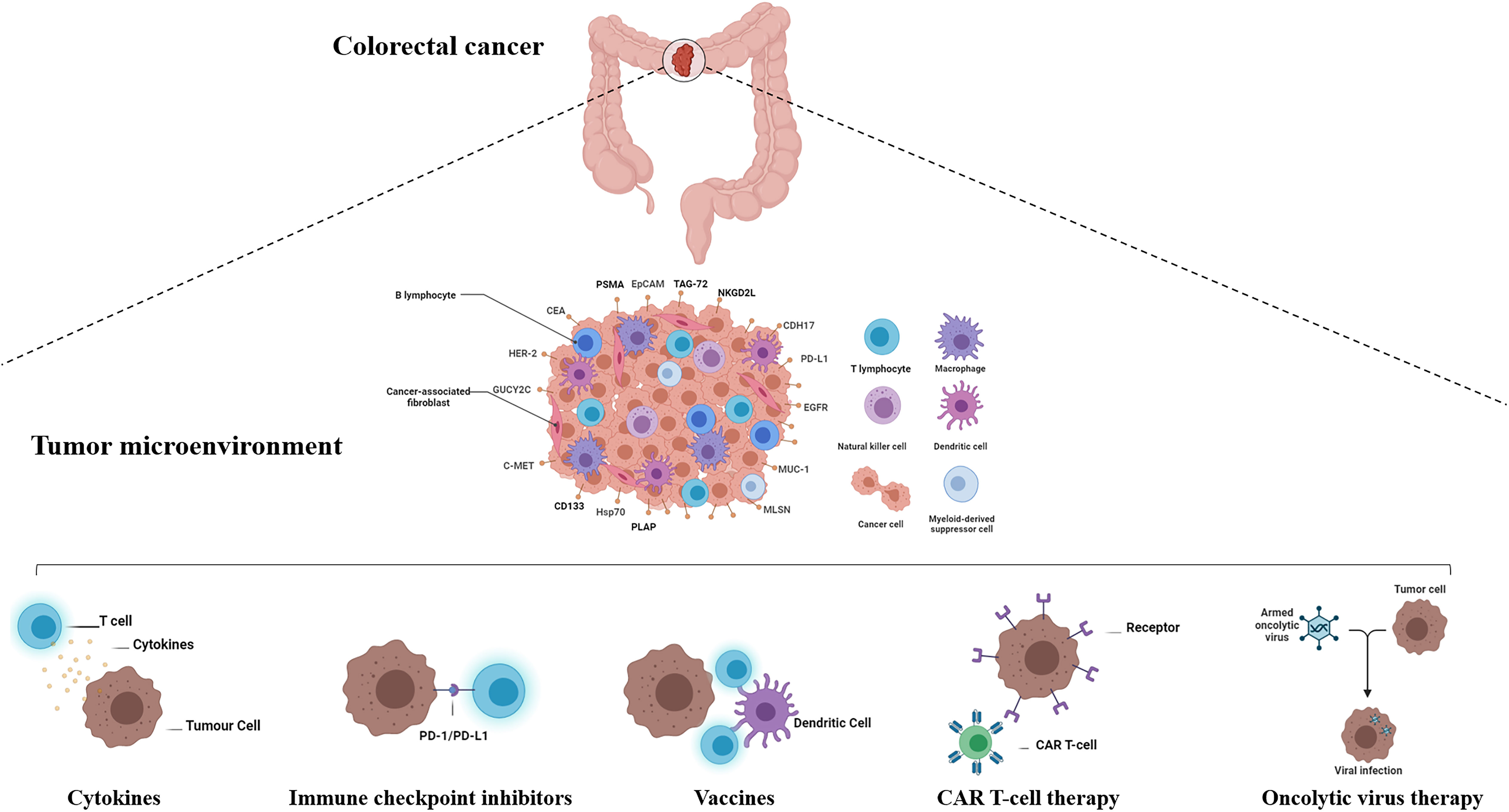
Figure 2 Active and passive immunotherapy strategies in colorectal cancer. The immunotherapy treatments approved in recent years has widened the arsenal against colorectal cancer. Active immunotherapy includes cancer vaccines, checkpoint inhibitors, and oncolytic viruses. Cancer vaccine immunotherapy approaches are based on immunizing patients with cancer against their autologous cancers using either autologous tumor cell-derived vaccines, dendritic cell vaccines, peptide vaccines, DNA vaccines, or viral vector vaccines. Passive immunotherapy includes the use of immune checkpoint blockade, cytokines, and adoptive cell transfer of ex vivo educated immune cells (Source: Author’s personal collection, 2021). Created with BioRender.com.
CRC is one of the most common malignancies and the fourth leading cause of cancer-related death worldwide (57). According to estimates, the CRC burden is expected to increase by 60% to more than 2.2 million new cases and 1.1 million cancer deaths by 2030 (58). First-line therapeutic approaches for patients with CRC, including surgery and chemotherapy, have long been associated with poor prognosis (59). Even if the response rate to chemotherapy can reach up to 50%, the treatment of far-advanced or recurrent diseases remains modest (60). A better understanding of the pathways contributing to tumor evolution and proliferation has enabled the development of effective and target-selective therapies. Targeted therapies are now being used in daily clinical practice for metastatic CRC with significantly improved survival outcomes (61). Following success with EGFR-targeted therapy (cetuximab) and angiogenesis inhibition (bevacizumab), immunotherapy is now taking center stage and is widely considered one of the most promising potential treatments (61–63). Three CAR T-cell therapies have been approved by the FDA and the European Medicines Agency; all of them target CD19-positive hematological malignancies (axicabtagene ciloleucel, tisagenlecleucel lecleucel, and brexucabtagene autoleucel).
Despite CAR T-cells being promising for hematological malignancies, their therapeutic efficacy in solid tumors, including CRC, remains unproven. Several groups have focused on pre-clinical studies involving CAR T-cell biology, aiming to develop safe therapeutic strategies and validate their use in CRC.
Several CAR T-cell approaches have been developed to target different tumor antigens. TSAs, also called neoantigens, are of high value as therapeutic targets because of their low on-target off-tumor effects. TSAs are dysfunctional peptides derived from the expression of non-synonymous somatic mutations and typically result from tumor-specific mutations; thus, they are only expressed in tumor cells with a high immunogenic potential (64). However, due to the low frequency of TSA mutations among patients with CRC, the investigated CAR T-cells are directed against TAAs, which are expressed in normal cells but overexpressed in tumor cells (64, 65). The different antigens targeted by CAR T-cell therapies in CRC and those reported in preclinical studies are summarized in Table 1.
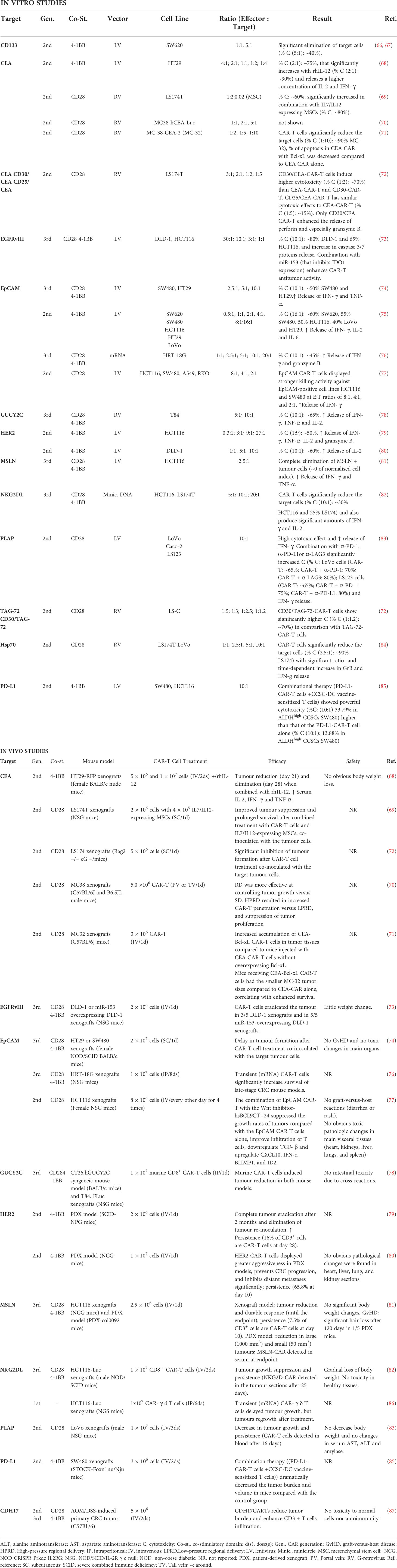
Table 1 Antigens targeted by CAR-T cell therapies in CRC and reported in preclinical studies.
One of the first preclinical studies, published by Ang et al., focused on the cytotoxic effects of epithelial cell adhesion molecule (EpCAM or CD326)-directed CAR T-cells. EpCAM is a transmembrane glycoprotein expressed on the surface of normal epithelial cells (88). EpCAM is one of the first cancer stem cell antigens to be reported. Its overexpression is associated with increased cell proliferation, migration, invasion, and tumor metastasis (88, 89). In CRC, EpCAM is overexpressed in more than 90% of all cancer cells and is associated with poor prognosis (89, 90). In the study conducted by Ang et al., EpCAM-targeting CAR T-cells were developed using a humanized monoclonal antibody, and their ability to treat CRC-xenografted mice was assessed (76). This in vivo immunodeficient mouse model displayed a phenotype similar to late-stage metastatic cancer in humans, including extensive peritoneal metastasis and ascites production (76). Interestingly, repeated injections of EpCAM-CAR T-cells suppressed peritoneal disease progression in tumor-bearing xenografted mice.
Along with EpCAM, carcinoembryonic antigen (CEA) is another studied target of anti-CRC CAR T-cells. CEA is a glycoprotein belonging to the immunoglobulin family and is overexpressed in many types of human cancers, including colon, pancreatic, gastric, lung, and ovarian cancers. CEA is overexpressed in 98.8% of CRC tissue samples and is one of the most important diagnostic and prognostic tumor markers (91, 92). Thus, CEA-targeted therapies hold promise for generating novel therapeutic strategies for CRC treatment. CAR T-cells targeting CEA have demonstrated excellent antitumor activity both in vitro and in vivo, which was significantly increased when combined with interleukins, such as IL-12 (68). Schlimper et al. generated cytokine-induced killer cells from blood lymphocytes of patients with CRC and transformed them into CEA-targeting cells by engineering a new CAR. Such cytokine-induced killer cells showed enhanced selectivity towards autologous CEA+ colon carcinoma cells and increased secretion of proinflammatory cytokines, such as IFN-γ and cytolysis (57, 93). Owing to the potent effects of IL-12 on T-cell functions, Chi et al. designed CEA-specific CAR T-cells and used them in combination with recombinant human IL‐12 to improve CRC tumor responses (68). The authors showed that CAR T-cells used simultaneously with recombinant human IL‐12 significantly enhanced antitumor activity in a xenografted tumor model. These data highlight the importance of adding cytokines to increase CAR T-cell activity in solid tumors.
To improve efficacy against peritoneal metastases, intraperitoneal (IP) delivery of CAR T-cells have been assessed. Compared with systemic intravenous delivery, regional IP infusion of anti-CEA CAR T-cells effectively protects against CEA+ peritoneal tumors and metastasis, enhances the effector memory response and is associated with increased IFNγ production (70). Therefore, it has been suggested that regional delivery improves CAR T-cell penetration deep inside solid tumors without increasing toxicity or overcoming barriers associated with systemic delivery. Furthermore, intratumoral accumulation of myeloid-derived suppressor cells and regulatory T-cells (Tregs) was detected in IP tumors, and the removal of these cells enhanced intra-tumoral T-cell cytotoxicity (94). Combination of IP CAR T-cell delivery and suppressor cell targeting appears to be a viable strategy for circumventing the immunosuppressive microenvironment in solid tumors.
In a xenograft model induced by co-inoculation of CAR T-cells and tumor cells, Huang et al. reported complete eradication of the tumor using EGFRvIII-CAR T-cells in combination with miR-153. These results indicate that miR-153 inhibits IDO1 expression in CRC cells and enhances CAR T-cell therapy. Thus, combination of IDO1 inhibitors and CAR T-cells can prospectively serve as an attractive therapeutic strategy for treating CRC and solid tumors (73).
To generate an adaptable gene-based vector that confers immune-cell specificity to various cancer types, Tamada et al. genetically engineered cells to express a CAR that binds a fluorescein isothiocyanate (FITC) molecule (anti-FITC CAR T-cells). The results indicated an improvement in the ability of the CAR T-cells to treat EGFR-positive CRC tumor-bearing mice (95). This versatile model confers T-cell specificity to a tag, which is easily conjugated with various clinically used tumor-reactive antibodies such as cetuximab, rendering the process economical. The results presented by the authors showed that specific interactions between FITC-labeled cetuximab and anti-FITC CAR T-cells in an immunocompromised mouse model, delayed colon cancer growth (57, 95).
A pivotal challenge in solid tumor CAR T-cell therapy is the potential toxicity of on-target/off-tumor effects owing to shared antigens in normal tissues. Placental alkaline phosphatase (PLAP) is a TSA that is not expressed in normal tissues but is overexpressed in CRC, rendering it an attractive target for CAR T-cell therapy (96). PLAP has been detected in more than 20% of colorectal adenocarcinomas. To assess the therapeutic potential of PLAP, a second-generation humanized PLAP-CAR T-cell construct was used in a xenograft model of CRC, which showed significantly decreased tumor growth in vivo (83). Humanized PLAP-CAR T-cell activity was further amplified by the addition of ICB therapy (anti-PD-1 or LAG-3 antibodies). This suggests that combining CAR T-cell therapy with ICB may be an effective therapeutic approach for CRC. Hence, the synergistic effect of the combination can be achieved through two steps: infiltration of immunogenically silent tumors by CAR T-cells followed by ICB therapy, which reverses CAR T-cell inhibition and restores functional persistence.
Similarly, Dezfouli et al. reported that the stress-inducible membrane form of heat shock protein 70 (mHsp70) was an ideal CAR T-cell target for CRC treatment, due to its sufficient affinity, specificity, density, and valence, while being undetectable in normal cells and tissues (97–99). The antitumor activity of anti-Hsp70 CAR T-cells was demonstrated by increased granzyme B degranulation and IFN‐g secretion, associated with enhanced cytotoxicity against CRC cells expressing mHsp70. Furthermore, the greater in vitro persistence of anti-Hsp70 CAR T-cells compared to that of normal T-cells promises long-term persistence in a clinical context (84). Despite the promising in vitro results, which support the applicability and specificity of the anti-Hsp70 CAR T-cell approach, and the reduced likelihood of off-target effects, further studies are needed for exploring the safety and efficacy of this approach in relevant in vivo models.
Daly et al. used genetically modified T-cells to target the CRC-associated antigen EGP40. The CAR construct contained two EGP40-specific monoclonal antibodies fused in line with the Fc receptor gamma-signaling chain (57, 100). Human redirected T-cells specifically recognize and lyse EGP40-expressing colon carcinoma cells (100).
A major drawback in the development of CAR T-cell therapy is the toxicity to vital organs (78). Guanylyl cyclase C (GUCY2C) is a membrane-bound receptor that is expressed in >95% of CRC metastases (101, 102). The GUCY2C receptor expressed in intestinal epithelial cells triggers signaling pathways that regulate epithelial cell proliferation, differentiation, and apoptosis (103). In a syngeneic murine CRC model with lung metastases, Magee et al. reported that GUCY2C-targeting CAR T-cells provided long-term protection in the absence of intestinal toxicity (104). CAR T-cells exhibited T-cell activation, cytokine production, and enhanced cytolytic activity in an antigen-dependent manner. In a human xenograft model, GUCY2C-targeting CAR T-cells eliminated human CRC metastatic cells, resulting in survival benefits (105).
Natural killer group 2 member D (NKG2D) is an activating immunoreceptor associated with tumor immunosurveillance owing its ability to recognize tumor cells and initiate an antitumor immune response. In humans, it is expressed on NK cells, invariant natural killer T-cells, CD8+ T-cells, γ δ T-cells, and certain autoreactive or immunosuppressive CD4+ T-cells (106). Ligands for NKG2D are expressed in response to external signals, such as stress or pathogens, and during neoplastic cell transformation; however, they are typically absent in healthy tissues, rendering it a promising CAR candidate (106, 107). Soluble NKG2D ligands impair NKG2D-dependent functions, resulting in an impaired antitumor response. Thus, they are associated with poor clinical prognosis and metastasis (108). High expression of NKG2DL in human colorectal carcinomas and its correlation with improved disease-free survival rationalized the use of NKG2D as a potential immunotherapy target (109). Deng et al. reported the role of third-generation NKG2D CAR T-cells and investigated their cytotoxicity against human CRC cells in a xenograft model (82). In this model, NKG2D CAR T-cells showed significantly high killing activity, inhibited tumor growth, reduced tumor size, and produced longer overall survival with no severe toxicity to vital organs. Moreover, tumor sections from treated mice showed lymphocyte infiltration (82). However, regarding the genetic modification strategy, the authors showed that electroporation of T-cells in the presence of a NKG2D-CAR minicircle DNA vector reduced cell viability and CAR expression in a time-dependent manner (82). This strategy, based on CAR transient expression, produces less toxic effects but requires re-inoculation for achieving stable antitumor activity (76).
There is growing evidence that CRC is derived from cancer stem cells, which are slow-cycling, self-renewing, and pluripotent (110–112). This small fraction of the total cell population can undergo epithelial–mesenchymal transition, a key process for metastasis formation and therapeutic drug resistance (113, 114). Double cortin-like kinase 1 (DCLK1), a microtubule-associated protein, maintains stemness in CRC cells, and it is a putative tumor stem cell (TSC) marker in CRC mouse models (115–118). Sureban et al. described DCLK1-targeting CAR T-cells as an efficient therapeutic approach for eradicating CRC stem cells (110). DCLK1-targeting CAR T-cells have been shown to display higher cytotoxicity in vitro and exhibit antitumor activity in in vivo xenograft mouse models in vivo. Intravenous injections of DCLK1-directed CAR T-cells have been shown to reduce tumor size in tumor-bearing mice, strongly suggesting that DCLK1-based CAR T-cells inhibit CRC tumor growth in vivo without obvious cytotoxicity to normal tissues (110).
The availability of clinically relevant CRC models is essential for CAR T-cell testing and molecular analysis. In this regard, patient-derived xenograft mouse models of CRC have been generated and used in preclinical studies of anti-CRC CAR T-cells. These models reflect the clinical and molecular heterogeneity of patients, and they have been used in two preclinical studies on anti-CRC CAR T-cells (79, 81, 119). One study selected highly expressed mesothelin (MSLN/MESO) as a target for CAR T-cells, and MSLN-CAR T-cells showed significant antitumor efficacy (81). MSLN expressed at low levels in normal tissues acts as a tumor differentiation antigen.MSLN is a glycosylphosphatidylinositol-anchored protein that was first described as a membrane protein expressed in normal and neoplastic mesothelial cells. In subsequent studies, a broader expression pattern has been demonstrated mainly in cancer, with overexpression observed in multiple solid tumor types, including CRC (120). A significant expression of MSLN is significantly expressed in CRC, with up to 60% of cases showing positivity (120–122). In in vitro and iIn vivo models, MSLN overexpression activates the downstream PI3K/AkT, NF-κB, and MAPK/ERK pathways, which promote cancer cell proliferation, migration, and metastasis and hinder apoptosis (120–128). Although the biological significance of MSLN expression in CRC remains poorly understood, it is certain that MSLN plays a positive role in CRC cell proliferation. Furthermore, MSLN has shown potential as a prognostic biomarker and potential target for CRC treatment.
In another study, a patient-derived xenograft mouse model implanted with CRC xenograft has been used for assessing the antitumor capacity of human epidermal growth factor receptor 2 (HER2)-CAR T-cells (79). HER2 is an oncoprotein that is overexpressed in a small but relevant proportion of patients with CRC, especially in anti-EGFR-resistant and RAS wild-type tumors (79, 129). HER2-targeted CAR T-cells have been engineered and their in vivo efficacy and safety was tested in a patient-derived xenograft model in vivo (79, 80). Significant regression of CRC xenografts has been observed, with protection from relapses upon tumor rechallenge in this mouse model. This enhanced efficacy results in a significant survival benefit (79). Consequently, HER2 has become a promising target for CAR T-cell therapy in CRC. Nevertheless, as clinical trials are full of uncertainty, one of the first clinical trials testing third-generation HER2 CAR T-cellsin patients with metastatic cancers (NCT00924287) was terminated after the first patient died of severe acute respiratory failure as a result of the treatment.
Recently, Feng and his coworkers used CDH17 as a target for CAR T-cells. In humans, CDH17 is a CA2+-dependent adhesion switch mainly expressed in the intestinal system (130, 131). CDH17 is upregulated in gastrointestinal cancers and is used as a marker for adenocarcinomas of the gastrointestinal system (132, 133). The results demonstrated that CDH17-CAR T-cells eradicated tumors in a xenografted NSG mouse model without damaging healthy normal tissues expressing CDH17 as it is sequestered and hidden between normal cells (87). This study is the first preclinical investigation of CDH17 as a target for CAR T-cell development, and it uncovers a new class of TAAs that are accessible to CAR T-cells in tumors but are masked in normal cells, protecting the latter from CAR T-cell attack. Hence, in solid tumors, re-investigation of previously identified TAAs is of great interest.
As the clinical safety and effectiveness of CAR T-cell therapy are closely associated with the intrinsic quality and fitness of CAR T-cells, comprehensive preclinical investigations of CAR T-cell biological products are crucial. Such investigations are usually performed in different in vitro and in vivo models that can prove to be powerful and versatile tools for disease modeling and therapeutic experimentation and generate reliable data that indicate the clinical therapeutic potency of CAR T-cell therapies.
Owing to the promising results obtained in preclinical studies, many CAR T-cell therapies developed against CRC are being evaluated for antitumor efficacy and safety in clinical trials.
CAR T-cell therapy has shown great success in hematological diseases, and progress in the clinical evaluation of CAR T-cells has provided evidence for the promising application of CAR T-cells to solid tumors. Currently, CAR T-cell therapy is transitioning from experimental trials to mainstream clinical practice. However, CAR T-cell therapy for solid tumors shows limited efficacy and poor therapeutic response due, in part, to restricted trafficking, antigen escape and/or lack of antigen expression, and inefficient tumor infiltration.
Several CAR T-cell therapies against various CRC antigens have entered clinical trials. In Table 2, we summarize the CAR T-cell clinical studies for CRC, reported on the clinicaltrials.gov website.
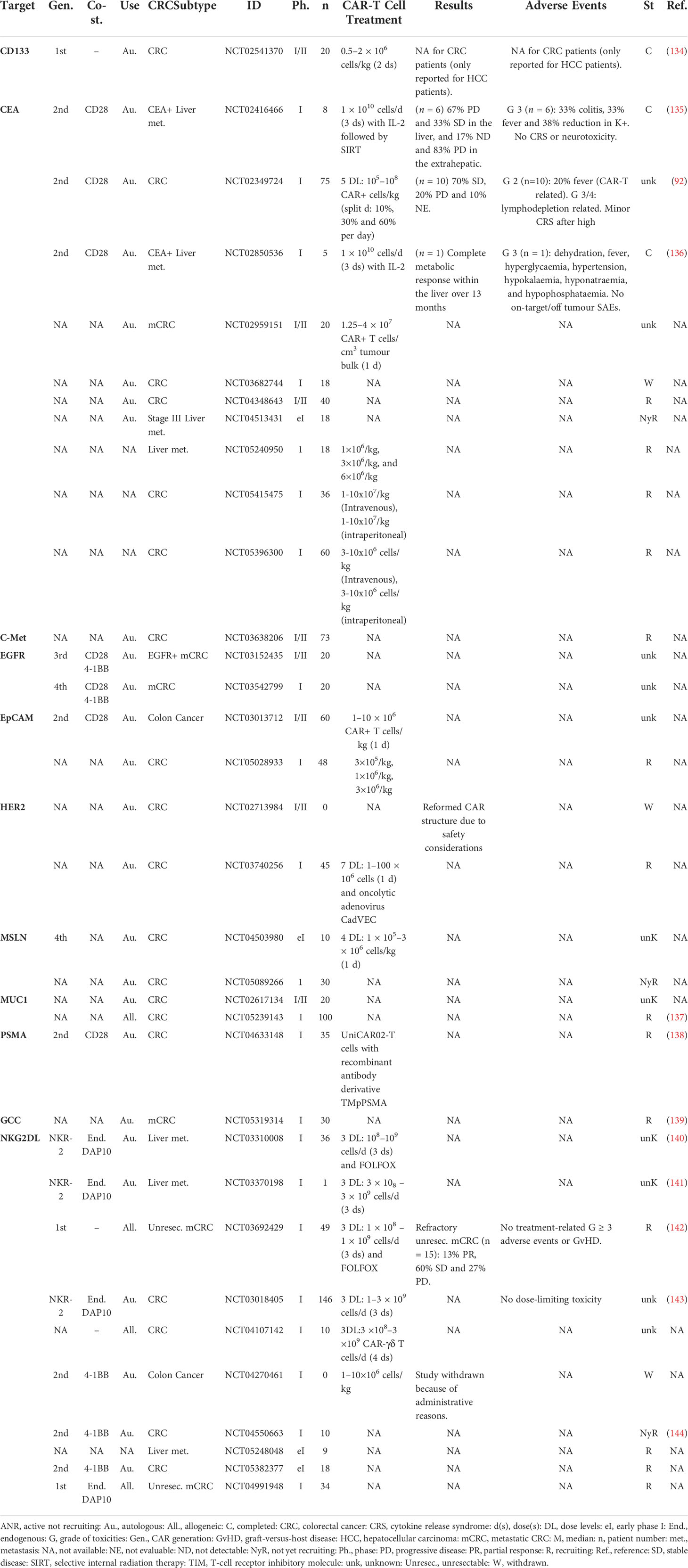
Table 2 Clinical trials of CAR-T cells in patients with colorectal cancer.
In their first attempts to apply CAR T-cell therapy to CRC, several groups have conducted clinical trials to assess the anticancer activity of CAR T-cells directed against HER2, tumor-associated glycoprotein 72 (TAG-72), CEA, or CEACAM5 (33, 92, 145, 146). One of the first clinical trials to evaluate CAR T-cell therapies for CRC targeted HER2 in a patient with colon cancer that was metastatic to the lungs and liver and refractory to multiple standard treatments. HER2-specific CAR T-cells were developed on the basis of the widely used humanized monoclonal antibody trastuzumab. Unfortunately, the patient who received the HER2-specific CAR T-cell infusion experienced respiratory distress with a cytokine storm and died of CAR T-cell-related toxicity (33).
No significant therapeutic benefits were observed with CAR T-cells, which were directed against the other three targets (TAG-72, CEA, or CEACAM5) (92, 145, 146). In another study, Hege et al. reported the results of one of the first CAR T-cell trials for metastatic CRC. The study was based on TAG-72-targeting CART72 cells (145). Two phase 1 trials were conducted using systemic (C9701) or regional hepatic artery infusion (C9702) CART72 delivery routes. Despite its safety profile, CART72 exhibited limited persistence in the blood because of an anti-CAR immune response and incomplete penetration into the tumor masses (145).
A phase I trial was conducted in CEA-positive patients with CRC presenting metastases for evaluating the safety and efficacy of CEA-targeting CAR T-cells (92). The endpoint showed good tolerability of CEA CAR T-cells even at high doses, persistence of CAR T-cells in peripheral blood, and eficacy in most treated patients (92). No significant beneficial clinical response was observed upon CEACAM5-specific CAR T-cell use with dose escalation in a phase I study (146). Respiratory toxicity combined with the lack of demonstrable clinical efficacy resulted in the premature closure of the trial.
Several trials are being conducted for assessing the safety and efficacy of CAR T-cells in CRC treatment. More than 50% of ongoing CAR T-cell clinical trials are in East Asia. A phase I, open-label clinical trial is underway for testing the safety and efficacy of EGFR CAR T-cells in the treatment of metastatic CRC (147, 148). To enhance EGFR CAR T-cell cytotoxicity, persistence, and to overcome a hostile TME in CRC, fourth-generation EGFR-IL12 CAR T-cells, also known as T-cells redirected for universal cytokines (TRUCKs), are engineered to secrete IL-12, a proinflammatory cytokine EGFR-IL12 TRUCKs express IL-12 upon activation and deliver it to enhance local antitumor immune responses, such as activating NK cells and repolarizing macrophages. The maximum tolerated doses and safety of EGFR-IL12 CAR T-cells are being currently assessed in patients with metastatic CRC (147, 148).
The other fourth-generation CAR T-cell therapy tested in CRC is αPD-1-MESO CAR T-cells, which exhibit inducible production and secretion of anti-PD-1 nanobodies (NCT04503980). A Chinese trial is underway to test an αPD-1-MESO CAR T-cell therapy in MSLN-positive advanced solid tumors (colorectal and ovarian cancer). During CAR T-cell production, patients receive a premedication regimen (cyclophosphamide) to deplete lymphocytes. The estimated completion date of the study is June 2022.
Ongoing trials are investigating the use of CEA-specific CAR T-cells in CEA-positive CRC. The objective is to confirm efficacy and safety and to set the appropriate doses and infusion plan (149–153). Another goal of these studies is to establish adverse events, most notably the cytokine release syndrome. Administration protocols include systemic and hepatic transarterial administration, vascular intervention, or intraperitoneal infusion, and the results are awaited (149–153). A new combinatorial strategy using HER2-specific CAR T-cells and an oncolytic adenovirus (CAdVEC) is under clinical investigation (154). Oncolytic adenoviruses replicate and spread specifically inside tumors, enhancing their cytotoxicity, improving tumor penetration, and reversing immune suppression. CAdVEC is a modified oncolytic adenovirus that contains various immunostimulatory molecules (155). Phase 1 trials are currently investigating the efficiency and safety of combining HER2 CAR T-cells with oncolysis (154). The intratumoral administration of CAdVEC is thought to promote an anti-tumorigenic proinflammatory TME that triggers recruitment and local expansion of the infused HER2 CAR T-cells. A phase I study of MUC1+ relapsed or refractory solid tumors was conducted for determining the safety and efficacy of MUC1 CAR T-cell therapy (156). In the phase I/II multi-target gene-modified CAR T-cell/TCR T-cell trial, c-MET was chosen as a target for CRC. Overexpression of c-MET in patients with CRC predicts poor survival (157). The safety and efficacy of CAR T-cells directed against EpCAM and CD133 are being explored in two clinical trials (158, 159). Preliminary data with CD133 CAR T-cells are encouraging, and they further indicate that CD133 can be an interesting target for relapsed or refractory CRC. NKG2D receptors bind to stress-induced ligands that are upregulated in infected and transformed, stressed cells. NKR-2, a second-generation autologous CAR T-cell line, uses the NKG2D complex to deliver primary and costimulatory signals. In contrast to classical target-specific CAR T-cells, NKR-2 recognizes various ligands on tumor cells and exhibits the potential to target several tumors (160). In addition to direct recognition of tumor cells, NKR2 is expected to target NKG2D-positive non-tumor cells within the stroma, tumor blood vessels, and immunosuppressive TME, which disrupts the support mechanisms essential for tumor cell survival and growth (161). Two clinical trials are currently assessing the safety and efficiency of NKR-2 CAR T-cells in patients with CRC, and the results are eagerly awaited. The first trial investigated the hepatic transarterial administration of NKR-2 CAR T-cells in patients with CRC presenting unresectable liver metastases, while the second investigated the safety and efficiency of NKR-2 cells combined with chemotherapy in resectable CRC liver metastases (162, 163).
CYAD-101 is an allogeneic CAR T-cell therapy engineered to co-express NKG2D-based CAR T-cells and the novel inhibitory peptide TCR inhibitory molecule (TIM). The expression of TIM prevents graft-versus-host disease by reducing the TCR signaling complex (164). In an open-label phase I trial, CYAD-101 treatment was administered simultaneously with FOLFOX chemotherapy in patients with unresectable metastatic CRC for assessing its safety and tolerability, clinical activity, and cell kinetics (165).
To date, although no particularly encouraging results have been reported for CAR T-cell therapy in CRC, several promising preclinical studies have shown success. Several major hurdles must be overcome to achieve the expected effect and eficacy in clinical trials. In the application of CAR T-cell therapies for CRC, the physical barrier and immunosuppressive microenvironment are the main hurdles in the achievement of desirable efficacy. Elegant strategies are currently under development for improving CAR T-cell function in solid tumors by prolonging their persistence, trafficking, tumor inflltration, and tumor elimination. For precise tumor targeting, potential solutions can be used, such as targeting T-cells with tandem CARs, universal CARs, affinity tuning, and regulation of CAR expression levels (166). In addition, preliminary data have provided promising evidence for combination therapies of CAR T-cells and other immunotherapies, chemotherapy, or radiotherapy (167).
The efforts of CAR T-cell research teams have yielded outstanding results for the treatment of leukemia (168–174). However, clinical evaluation of this new immunological weapon for fighting solid cancers is surrounded by disappointment. Past experiences, notably with ICIs, have highlighted significant contribution of multiple factors that significantly contribute to the strengths and weaknesses of immunotherapies and forecast their clinical outcome in solid cancer. These aspects must be considered when identifying the therapeutic strategies for solid tumor treatment.
Nevertheless, it must be considered that even for hematological malignancies with complete remission rates as high as 90%, patients remain at risk of relapse because of the poor persistence of CAR T-cells in vivo. Limited CAR T-cell persistence is closely related to the immunological properties of the cellular infusion product.
In the last decade, cancer treatment has undergone a major revolution owing to therapies that target the TME. Deep characterization of the immune response and TME, a continuously evolving entity, along with the molecular and phenotypic analysis of tumor cells, is an attractive strategy for cancer treatment. Using accumulated data from recent immunotherapy studies, notably ICI, to design more effective CAR T cells, can be of a great interest.
The complex spatiotemporal changes in the TME complicate cancer treatment by driving various responses to immunotherapies. The landscape of tumor-infiltrating immune cells defines prognosis and treatment efficacy (175, 176), supporting the idea that treatment strategy designs need to be guided by mapping the composition and functional state of immune cell infiltrates (177). Schematically, the TME can be broadly classified into cold (non-T-cell inflamed) or hot (T-cell inflamed), on the basis of accumulation of proinflammatory cytokines and T-cell infiltration (178). Commonly, hot tumors, characterized by T-cell infiltration and immune activation signatures, exhibit better response to ICB treatment (179). Meanwhile, cold tumors lack such infiltrates because of several factors: poor tumor antigenicity, impaired antigen presentation, T-cell activation failure, and/or lack of T-cell homing into the tumor bed. A limited number of T-cells surround cold tumors and localize to the tumor periphery with poor T-cell activation and infiltration (180, 181). Thus, the high-inflammatory state of the TME is associated with high TMB and increased tumor-related antigenicity, resulting in T-cell tumor-infiltration, amplification, and tumor necrosis induction (178, 182). Remarkably, CRC tumors with MSI and a higher mutational burden respond well to ICI-based immunotherapy (183–186). Primary lesions, such as metastatic colon cancer, are highly enriched in immune infiltrating cells with increased mutational load, and a high immunoscore is associated with a significantly lower number of metastases (187).
Several research teams are focused on converting cold tumors into hot tumors to render them more receptive to immunotherapies, leading to an objective antitumor response. The second goal was to prevent hot tumors from cooling. In this way, creating a beneficial TME goes through vascular bed remodeling to enhance the circulation of CAR T-cells and activate the immune response at the tumor side. Innate immune sensing and signaling, dysregulated oncologic pathways, altered cellular metabolism, and epigenetic changes in the TME must be integrated as tightly as possible into the global antitumor strategy. Immunogenic cell death induced by chemo-, radio-, and targeted therapies plays a key role in stimulating altered antitumor immune responses, potentially converting non-inflamed cold tumors into hot tumors (188–192). Hence, the future of antitumor combination therapies lies in well-designed sequential strategies that successfully prepare patients for immunotherapeutic regimens, including CAR T-cell adoptive cell transfer (193, 194). Tumor immune microenvironment characterization can help identify good responders to immunotherapies, such as CAR T-cell therapy.
Currently, 28 ongoing clinical trials are evaluating TMB as a stratification biomarker for predicting immunotherapy responses (195–197). The frequency of tumor mutations varies widely across, and within cancer subtypes (198). Greater mutational load, notably nonsynonymous mutations, increases the possibility of neoantigen generation and expression by tumor cells, allowing the conversion of immune tolerance to antitumor immune responses. Consistent with this hypothesis, TMB has been shown to predict the clinical benefit of anti-PD-1/PD-L1 immunotherapy (199, 200). Patients in the immune desert cluster with a low T-cell infiltration microenvironment respond less favorably to ICI therapies (201–203). Moreover, several recent preclinical and clinical reports have clearly highlighted the major role of neoantigen-specific T-cell activity in recognizing and eliminating cancer cells (204–213). Several studies have reported a highly significant association between neoantigen burden and clinical benefits (183, 214–216). Therefore, TMB has emerged as a powerful determinant in response to immunotherapies.
Based on current data and trends, TMB appears to be a robust predictive biomarker of the response to immune checkpoint immunotherapy. Generally, two types of cancer tend to harbor high TMB: (i) tumors induced by carcinogenic and mutagenic substances and (ii) tumors caused by germline mutations in DNA repair and replication factors. Thus, TMB promises to be useful for MSI-H CRCs, marked by frequent mutations in dMMR and DNA replication genes (195, 196). The model combining TMB with other immunotherapy biomarkers provides a promising method with greater predictive power to identify immunotherapy responders and predict outcomes.
In dMMR and MSI-H CRC tumors, neoplastic foci present lymphoid tertiary structures deeply infiltrated with activated CD8+ T-cells (217–219). Consistent with this observation, dMMR cancers are sensitive to checkpoint inhibitors, leading to pembrolizumab approval for MSI-H or dMMR cancers (22, 220, 221). In colon cancer, the potent local immune response associated with high mutational load allows neoantigens to be targeted by T and B lymphocytes with an appreciable clinical antitumor reactivity under ICI treatment (183, 222–224).
The complex TME plays an essential role in the clinical response of solid tumors to CAR T-cell therapy. Thus, the inhibition of proliferation and/or lack of persistence can limit effective antitumor immune response and lead to the poor treatment response in CRC. The immunosuppressive microenvironment plays a decisive role in inhibiting CAR T-cell persistence in the tumor milieu. On the one hand, immunosuppressive cells and cytokines hamper the function of CAR T-cells by blocking them from tumor sites or inducing apoptosis (36). On the other hand, the immunosuppressive microenvironment lacking the cytokines necessary for T-cell persistence can foster the apoptotic pathway.
Once at the tumor site, CAR T-cells can undergo activation-induced cell death upon functional activation and proliferation/expansion. B-cell lymphoma-extra-large (Bcl-xL), a critical regulator for T-cell survival, has been reported to contribute to CAR T-cell persistence and antitumor activity (225–227). Interestingly, the inclusion of exogenous Bcl-xL gene into a second-generation anti-CEA CAR retroviral construct promoted CAR T-cell persistence both in vitro and in vivo, with enhanced antitumor potency and sustained survival in a CRC mouse model. The novel CAR T-cell platform which is based on the exogenous expression of persistent genes, can help overcome the lack of persistence of CAR T-cells in solid tumors.
Exploring better combination therapies is another approach to improve CAR T-cell efficacy and decrease their shortcomings for solid-tumor treatment. Emerging novel combined CAR T-cell therapies and immunotherapy or small-molecule drugs have shown promise in preclinical studies, and they can overcome the limitations of CAR T-cells targeting solid tumors. The combination of EpCAM CAR T-cells with the Wnt pathway inhibitor hsBCL9CT-24 has shown a synergistic effect against EpCAM-positive colon cells in vitro and in vivo. The combined strategies modulate the TME to induce the inflltration of T-cells, effector function and prevent the exhaustion of CAR T-cells (77). Induction of antibody-dependent cell cytotoxicity (ADCC) via the Fc-gamma receptor is another multi-target CAR T-cell approach that enables ADCC to CAR T-cells. Hence, by endowing CAR T-cells with ADCC activity via the Fc-gamma receptor IIIa (CD16), they exhibit sustained proliferation and cytotoxicity to antibody-targeted cancer cells (228). In CRC, CD16-CAR T-cells combined with cetuximab decreased the viability of KRAS-mutated HCT116 CRC cells in vitro, decreased tumor growth in a SCID mouse model, and increased disease-free survival (229). Recently, dendritic cell (DC) vaccines have been studied in combination with immunotherapy strategies, such as those targeting PD-1/PD-L1. Regarding cancer stem cell resistance to conventional therapies, the synergistic killing effects of PD-L1-CAR T-cells and CRC stem cell (CCSC)-dendritic cell vaccine-sensitized T-cells in CCSC have been investigated. A study has shown that PD-L1 is highly expressed in CCSCs and that PD-L1-CAR T-cells specifically recognize the PD-L1 molecule on CCSCs (85). In this study, combined therapy markedly relieved the tumor burden in mice compared to monotherapy with PD-L1-CAR T-cells or CCSC-DC vaccine. This showed that there was moderate tumor remission both in vitro and in vivo.
Finally, combination therapies are being developed for improving immune response, minimizing on-target off-tumor toxicities, and transforming immunologically “cold” to “hot” tumors (230). A combination of CAR NK and CAR T-cells can harness the advantages of both CAR NK and CAR T-cells, allowing for both rapid and persistent killing while potentially minimizing the toxicities associated with CAR-T cells (228, 231–233). The development of these therapies is poised to be the next frontier in immunotherapy as it will result in more robust immune attacks and effective clinical outcomes.
Data continues to accumulate supporting CAR T-cells as a promising immunological form of cancer immunotherapy. In hematological malignancies, this concept has considerably improved patient care. However, CAR T-cell therapy also has substantial limitations, including the requirement of certified laboratories and clinical centers with specifically trained personnel to manage the side effects. The use of CAR T-cells in solid tumors faces additional challenges, including inefficient T-cell trafficking and a hostile TME. Overcoming these limitations would enable the achievement of the full potential benefit of this novel approach.
CRC is one of the few cancers in which immunotherapy has shown limited promise. CAR T-cell therapies could be very beneficial in CRC, as suggested by preclinical and early phase clinical studies. However, several questions remain unanswered (1): What is the appropriate target for CAR T-cells? (2) what is the right therapeutic strategy with regard to novel checkpoint inhibitors and monoclonal antibodies: a combination of immunotherapies (ICB, virotherapy, IDO inhibitor, or CAR T-cells) or immunotherapies administered in sequence? The efficacy and safety of cancer treatment appear to depend on the drug combination, timing, and sequence of administration. The treatment schedule design should be driven by changes in the tumor-immune microenvironment. Ongoing clinical trials can answer these questions and provide insights into the spectrum of patients who can benefit from this treatment.
BG and AEG conceived and drafted the manuscript. AG designed the figures. BG and SK designed the tables. AB made substantial contributions to manuscript modifications. All authors contributed to the article and approved the submitted version.
This work was supported by the Moroccan Ministry of Higher Education, Research and innovation through a “PPR1” project and by the Moroccan Ministry of Higher Education, Research and innovation and the Digital Development Agency "ADD” through an “Al-khawarizmi” project.
We would like to thank Konala Priyanka Reddy at London North West Healthcare NHS Trust Harrow on the Hill, United Kingdom who helped us to improve the linguistic quality of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.

1. Ghouzlani, Kandoussi S, Tall M, Reddy KP, Rafii S, Badou A. Immune checkpoint inhibitors in human glioma microenvironment. Front. Immunol. (2021) 12:679425. doi: 10.3389/fimmu.2021.679425
2. Ghouzlani A., Kandoussi S, Rafii S, Lakhdar A, Badou A. High expression levels of Foxp3 and VISTA in advanced human gliomas and impact on patient’s prognosis. Arch Clin. Biomed. Res. (2020) 4(6):691–703. doi: 10.26502/acbr
3. Ghouzlani A., Rafii S, Karkouri M, Lakhdar A, Badou A. The promising IgSF11 immune checkpoint is highly expressed in advanced human gliomas and associates topoor prognosis. Front. Oncol. (2021) 10: 608609. doi: 10. 3389/fonc.2020.608609.
4. Ghouzlani A., Lakhdar A., Rafii S., Karkouri M, Badou A. The immune checkpoint VISTA exhibits high expression levels in human gliomas and associates with a poor prognosis. Sci Rep (2021) 11: 21504. doi: 10.1038/s41598-021-00835-0
5. Weng J, Li S, Zhu Z, Liu Q, Zhang R, Yang Y, et al. Exploring immunotherapy in colorectal cancer. J Hematol Oncol déc (2022) 15(1):95. doi: 10.1186/s13045-022-01294-4
6. Siegel RL, Miller KD, Jemal A. Cancer statistics. CA A Cancer J Clin janv (2019) 69(1):7−34. doi: 10.3322/caac.21551
7. Aparicio C, Belver M, Enríquez L, Espeso F, Núñez L, Sánchez A, et al. Cell therapy for colorectal cancer: The promise of chimeric antigen receptor (CAR)-T cells. IJMS. 29 oct (2021) 22(21):11781. doi: 10.3390/ijms222111781
8. Rawla P, Sunkara T, Barsouk A. Epidemiology of colorectal cancer: incidence, mortality, survival, and risk factors. Prz Gastroenterol (2019) 14(2):89−103. doi: 10.5114/pg.2018.81072
9. Wolpin BM, Mayer RJ. Systemic treatment of colorectal cancer. Gastroenterology. mai (2008) 134(5):1296–310.e1. doi: 10.1053/j.gastro.2008.02.098
10. El Bali M, Bakkach J, Bennani Mechita M. Colorectal cancer: From genetic landscape to targeted therapy. J Oncol (2021) 2021:1−17. doi: 10.1155/2021/9918116
11. Benson AB, Venook AP, Al-Hawary MM, Arain MA, Chen YJ, Ciombor KK, et al. Colon cancer, version 2.2021, NCCN clinical practice guidelines in oncology. J Natl Compr Cancer Network (2021) 19(3):329−59. doi: 10.6004/jnccn.2021.0012
12. Ajani JA, D’Amico TA, Bentrem DJ, Chao J, Cooke D, Corvera C, et al. Gastric cancer, version 2.2022, NCCN clinical practice guidelines in oncology. J Natl Compr Cancer Network (2022) 20(2):167−92. doi: 10.6004/jnccn.2022.0008
13. Van Cutsem E, Cervantes A, Adam R, Sobrero A, Van Krieken JH, Aderka D, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol (2016) 27(8):1386−422. doi: 10.1093/annonc/mdw235
14. Chibaudel B, Tournigand C, Bonnetain F, Richa H, Benetkiewicz M, André T, et al. Therapeutic strategy in unresectable metastatic colorectal cancer: an updated review. Ther Adv Med Oncol (2015) 7(3):153−69. doi: 10.1177/1758834015572343
15. Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WEE, Poddubskaya E, et al. Nivolumab versus docetaxel in advanced squamous-cell non–Small-Cell lung cancer. N Engl J Med (2015) 373(2):123−35. doi: 10.1056/NEJMoa1504627
16. Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med (2011) 364(26):2517−26. doi: 10.1056/NEJMoa1104621
17. Wolchok JD, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, et al. CheckMate 067: 6.5-year outcomes in patients (pts) with advanced melanoma. JCO (2021) 39(15_suppl):9506−9506. doi: 10.1200/JCO.2021.39.15_suppl.9506
18. Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Durvalumab after chemoradiotherapy in stage III non–Small-Cell lung cancer. N Engl J Med (2017) 377(20):1919−29. doi: 10.1056/NEJMoa1709937
19. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. science. Science (2017) 357(6349):409−13. doi: 10.1126/science.aan6733
20. Peng L, Liang WH, Mu DG, Xu S, Hong SD, Stebbing J, et al. First-line treatment options for PD-L1–negative non-small cell lung cancer: A Bayesian network meta-analysis. Front Oncol 23 juin (2021) 11:657545. doi: 10.3389/fonc.2021.657545
21. Insa A, Martín-Martorell P, Di Liello R, Fasano M, Martini G, Napolitano S, et al. Which treatment after first line therapy in NSCLC patients without genetic alterations in the era of immunotherapy? critical reviews in Oncology/Hematology. janv (2022) 169:103538. doi: 10.1016/j.critrevonc.2021.103538
22. Marcus L, Lemery SJ, Keegan P, Pazdur R. FDA Approval summary: Pembrolizumab for the treatment of microsatellite instability-high solid tumors. Clin Cancer Res (2019) 25(13):3753−8. doi: 10.1158/1078-0432.CCR-18-4070
23. Casak SJ, Marcus L, Fashoyin-Aje L, Mushti SL, Cheng J, Shen YL, et al. FDA Approval summary: Pembrolizumab for the first-line treatment of patients with MSI-H/dMMR advanced unresectable or metastatic colorectal carcinoma. Clin Cancer Res (2021) 27(17):4680−4. doi: 10.1158/1078-0432.CCR-21-0557
24. Safarzadeh Kozani P, Safarzadeh Kozani P, Ahmadi Najafabadi M, Yousefi F, Mirarefin SMJ, Rahbarizadeh F. Recent advances in solid tumor CAR-T cell therapy: Driving tumor cells from hero to zero? front immunol. 11 mai (2022) 13:795164. doi: 10.3389/fimmu.2022.795164
25. Safarzadeh Kozani P, Safarzadeh Kozani P, Rahbarizadeh F. Optimizing the clinical impact of CAR-T cell therapy in b-cell acute lymphoblastic leukemia: Looking back while moving forward. Front Immunol 28 oct (2021) 12:765097. doi: 10.3389/fimmu.2021.765097
26. O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med (2017) 9(399):eaaa0984. doi: 10.1126/scitranslmed.aaa0984
27. Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human epidermal growth factor receptor 2 (HER2) –specific chimeric antigen receptor–modified T cells for the immunotherapy of HER2-positive sarcoma. JCO (2015) 33(15):1688−96. doi: 10.1200/JCO.2014.58.0225
28. Yu WL, Hua ZC. Chimeric antigen receptor T-cell (CAR T) therapy for hematologic and solid malignancies: Efficacy and safety–a systematic review with meta-analysis. Cancers. 7 janv (2019) 11(1):47. doi: 10.3390/cancers11010047
29. Ruella M, Barrett DM, Kenderian SS, Shestova O, Hofmann TJ, Perazzelli J, et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J Clin Invest (2016) 126(10):3814−26. doi: 10.1172/JCI87366
30. Chraa D, Naim A, Olive D, Badou A. T lymphocyte subsets in cancer immunity: Friends or foes. J Leukocyte Biol (2018) 105(2):243–255. doi: 10.1002/JLB.MR0318-097R
31. Anderson KG, Stromnes IM, Greenberg PD. Obstacles posed by the tumor microenvironment to T cell activity: A case for synergistic therapies. Cancer Cell (2017) 31(3):311−25. doi: 10.1016/j.ccell.2017.02.008
32. Choi BD, Yu X, Castano AP, Bouffard AA, Schmidts A, Larson RC, et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat Biotechnol (2019) 37(9):1049−58. doi: 10.1038/s41587-019-0192-1
33. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther (2010) 18(4):843−51. doi: 10.1038/mt.2010.24
34. Brandt LJB, Barnkob MB, Michaels YS, Heiselberg J, Barington T. Emerging approaches for regulation and control of CAR T cells: A mini review. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.00326
35. June CH, Sadelain M. Chimeric antigen receptor therapy. New Engl J Med (2018) 379(1):64−73. doi: 10.1056/NEJMra1706169
36. Martinez M, Moon EK. CAR T cells for solid tumors: New strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.00128
37. Sacchetti B, Botticelli A, Pierelli L, Nuti M, Alimandi M. CAR-T with license to kill solid tumors in search of a winning strategy. Int J Mol Sci 17 avr (2019) 20(8):1903. doi: 10.3390/ijms20081903
38. Park S, Shevlin E, Vedvyas Y, Zaman M, Park S, Hsu YMS, et al. Micromolar affinity CAR T cells to ICAM-1 achieves rapid tumor elimination while avoiding systemic toxicity. Sci Rep (2017) 7(1):14366. doi: 10.1038/s41598-017-14749-3
39. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-t-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci (1989) 86(24):10024−8. doi: 10.1073/pnas.86.24.10024
40. Heyman B, Yang Y. Chimeric antigen receptor T cell therapy for solid tumors: Current status, obstacles and future strategies. Cancers. 6 févr (2019) 11(2):191. doi: 10.3390/cancers11020191
41. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol (2015) 15(8):486−99. doi: 10.1038/nri3862
42. Chen DS, Mellman I. Elements of cancer immunity and the cancer–immune set point. Nature. (2017) 541(7637):321−30. doi: 10.1038/nature21349
43. James JR, Vale RD. Biophysical mechanism of T-cell receptor triggering in a reconstituted system. Nature. (2012) 487(7405):64−9. doi: 10.1038/nature11220
44. Benmebarek MR, Karches C, Cadilha B, Lesch S, Endres S, Kobold S. Killing mechanisms of chimeric antigen receptor (CAR) T cells. Int J Mol Sci 14 mars (2019) 20(6):1283. doi: 10.3390/ijms20061283
45. James SE, Greenberg PD, Jensen MC, Lin Y, Wang J, Till BG, et al. Antigen sensitivity of CD22-specific chimeric TCR is modulated by target epitope distance from the cell membrane. J Immunol (2008) 180(10):7028−38. doi: 10.4049/jimmunol.180.10.7028
46. Riddell SR, Jensen MC, June CH. Chimeric antigen receptor–modified T cells: Clinical translation in stem cell transplantation and beyond. Biol Blood Marrow Transplantation. (2013) 19(1):S2−5. doi: 10.1016/j.bbmt.2012.10.021
47. Zhou K, Guo S, Li F, Sun Q, Liang G. Exosomal PD-L1: New insights into tumor immune escape mechanisms and therapeutic strategies. Front Cell Dev Biol (2020) 8. doi: 10.3389/fcell.2020.569219
48. Cha JH, Chan LC, Li CW, Hsu JL, Hung MC. Mechanisms controlling PD-L1 expression in cancer. Mol Cell (2019) 76(3):359−70. doi: 10.1016/j.molcel.2019.09.030
49. Mannino MH, Zhu Z, Xiao H, Bai Q, Wakefield MR, Fang Y. The paradoxical role of IL-10 in immunity and cancer. Cancer Letters. (2015) 367(2):103−7. doi: 10.1016/j.canlet.2015.07.009
50. Meireson A, Devos M, Brochez L. IDO expression in cancer: Different compartment, different functionality? Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.531491
51. Labani-Motlagh A, Ashja-Mahdavi M, Loskog A. The tumor microenvironment: A milieu hindering and obstructing antitumor immune responses. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.00940
52. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med (2018) 24(5):541−50. doi: 10.1038/s41591-018-0014-x
53. Rivadeneira DB, Delgoffe GM. Antitumor T-cell reconditioning: Improving metabolic fitness for optimal cancer immunotherapy. Clin Cancer Res (2018) 24(11):2473−81. doi: 10.1158/1078-0432.CCR-17-0894
54. Le Bourgeois T, Strauss L, Aksoylar HI, Daneshmandi S, Seth P, Patsoukis N, et al. Targeting T cell metabolism for improvement of cancer immunotherapy. Front Oncol (2018) 8. doi: 10.3389/fonc.2018.00237
55. Mussai F, Egan S, Hunter S, Webber H, Fisher J, Wheat R, et al. Neuroblastoma arginase activity creates an immunosuppressive microenvironment that impairs autologous and engineered immunity. Cancer Res (2015) 75(15):3043−53. doi: 10.1158/0008-5472.CAN-14-3443
56. Wagner J, Wickman E, DeRenzo C, Gottschalk S. CAR T cell therapy for solid tumors: Bright future or dark reality?, Mol Ther. 28 (11):2320–39. doi: 10.1016/j.ymthe.2020.09.015
57. Zhang Q, Zhang H, Ding J, Liu H, Li H, Li H, et al. Combination therapy with EpCAM-CAR-NK-92 cells and regorafenib against human colorectal cancer models. J Immunol Res (2018) 2018:1−11. doi: 10.1155/2018/4263520
58. Arnold M, Sierra MS, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global patterns and trends in colorectal cancer incidence and mortality. Gut. avr (2017) 66(4):683−91. doi: 10.1136/gutjnl-2015-310912
59. Xie YH, Chen YX, Fang JY. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduction Targeted Ther (2020) 5(1):22. doi: 10.1038/s41392-020-0116-z
60. Hu T, Li Z, Gao CY, Cho CH. Mechanisms of drug resistance in colon cancer and its therapeutic strategies. World J Gastroenterology. (2016) 22(30):6876. doi: 10.3748/wjg.v22.i30.6876
61. Piawah S, Venook AP. Targeted therapy for colorectal cancer metastases: A review of current methods of molecularly targeted therapy and the use of tumor biomarkers in the treatment of metastatic colorectal cancer. Cancer. (2019) 125(23):4139−47. doi: 10.1002/cncr.32163
62. Price TJ, Tang M, Gibbs P, Haller DG, Peeters M, Arnold D, et al. Targeted therapy for metastatic colorectal cancer. Expert Rev Anticancer Ther (2018) 18(10):991−1006. doi: 10.1080/14737140.2018.1502664
63. Caiazza F, Elliott L, Fennelly D, Sheahan K, Doherty GA, Ryan EJ. Targeting EGFR in metastatic colorectal cancer beyond the limitations of KRAS status: alternative biomarkers and therapeutic strategies. Biomarkers Med (2015) 9(4):363−75. doi: 10.2217/bmm.15.5
64. Rus Bakarurraini NAA, Ab Mutalib NS, Jamal R, Abu N. The landscape of tumor-specific antigens in colorectal cancer. Vaccines 10 juill (2020) 8(3):371. doi: 10.3390/vaccines8030371
65. Feng M, Zhao Z, Yang M, Ji J, Zhu D. T-Cell-based immunotherapy in colorectal cancer. Cancer Letters. (2021) 498:201−9. doi: 10.1016/j.canlet.2020.10.040
66. Wang Y, Chen M, Wu Z, Tong C, Dai H, Guo Y, et al. CD133-directed CAR T cells for advanced metastasis malignancies: A phase I trial. OncoImmunology. (2018) 7(7):e1440169. doi: 10.1080/2162402X.2018.1440169
67. Feng Kc, Guo Yl, Liu Y, Dai Hr, Wang Y, Lv Hy, et al. Cocktail treatment with EGFR-specific and CD133-specific chimeric antigen receptor-modified T cells in a patient with advanced cholangiocarcinoma. J Hematol Oncol (2017) 10(1):4. doi: 10.1186/s13045-016-0378-7
68. Chi X, Yang P, Zhang E, Gu J, Xu H, Li M, et al. Significantly increased anti-tumor activity of carcinoembryonic antigen-specific chimeric antigen receptor T cells in combination with recombinant human IL-12. Cancer Med (2019) 8(10):4753−65. doi: 10.1002/cam4.2361
69. Hombach AA, Geumann U, Günther C, Hermann FG, Abken H. IL7-IL12 engineered mesenchymal stem cells (MSCs) improve a CAR T cell attack against colorectal cancer cells. Cells. (2020) 9(4):E873. doi: 10.3390/cells9040873
70. Chai LF, Hardaway JC, Heatherton KR, O’Connell KP, LaPorte JP, Guha P, et al. Antigen receptor T cells (CAR-T) effectively control tumor growth in a colorectal liver metastasis model. J Surg Res (2022) 272:37−50. doi: 10.1016/j.jss.2021.11.001
71. Fan J, Das JK, Xiong X, Chen H, Song J. Development of CAR-T cell persistence in adoptive immunotherapy of solid tumors. Front Oncol (2021) 10:574860. doi: 10.3389/fonc.2020.574860
72. Hombach AA, Rappl G, Abken H. Blocking CD30 on T cells by a dual specific CAR for CD30 and colon cancer antigens improves the CAR T cell response against CD30– tumors. Mol Ther (2019) 27(10):1825−35. doi: 10.1016/j.ymthe.2019.06.007
73. Huang Q, Xia J, Wang L, Wang X, Ma X, Deng Q, et al. miR-153 suppresses IDO1 expression and enhances CAR T cell immunotherapy. J Hematol Oncol déc (2018) 11(1):58. doi: 10.1186/s13045-018-0600-x
74. Zhang BL, Li D, Gong YL, Huang Y, Qin DY, Jiang L, et al. Preclinical evaluation of chimeric antigen receptor–modified T cells specific to epithelial cell adhesion molecule for treating colorectal cancer. Hum Gene Ther (2019) 30(4):402−12. doi: 10.1089/hum.2018.229
75. Zhou Y, Wen P, Li M, Li Y, Li X. Construction of chimeric antigen receptor−modified T cells targeting EpCAM and assessment of their anti−tumor effect on cancer cells. Mol Med Rep (2019). 20(3):2355–64. doi: 10.3892/mmr.2019.10460
76. Ang WX, Li Z, Chi Z, Du SH, Chen C, Tay JCK, et al. Intraperitoneal immunotherapy with T cells stably and transiently expressing anti-EpCAM CAR in xenograft models of peritoneal carcinomatosis. Oncotarget. (2017) 8(8):13545−59. doi: 10.18632/oncotarget.14592
77. Li W, Zhou Y, Wu Z, Shi Y, Tian E, Zhu Y, et al. Targeting wnt signaling in the tumor immune microenvironment to enhancing EpCAM CAR T-cell therapy. Front Pharmacol (2021) 12:724306. doi: 10.3389/fphar.2021.724306
78. Magee MS, Abraham TS, Baybutt TR, Flickinger JC, Ridge NA, Marszalowicz GP, et al. Human GUCY2C-targeted chimeric antigen receptor (CAR)-expressing T cells eliminate colorectal cancer metastases. Cancer Immunol Res (2018) 6(5):509−16. doi: 10.1158/2326-6066.CIR-16-0362
79. Teng R, Zhao J, Zhao Y, Gao J, Li H, Zhou S, et al. Chimeric antigen receptor–modified T cells repressed solid tumors and their relapse in an established patient-derived colon carcinoma xenograft model. J Immunother (2019) 42(2):33−42. doi: 10.1097/CJI.0000000000000251
80. Xu J, Meng Q, Sun H, Zhang X, Yun J, Li B, et al. HER2-specific chimeric antigen receptor-T cells for targeted therapy of metastatic colorectal cancer. Cell Death Dis (2021) 12(12):1109. doi: 10.1038/s41419-021-04100-0
81. Zhang Q, Liu G, Liu J, Yang M, Fu J, Liu G, et al. The antitumor capacity of mesothelin-CAR-T cells in targeting solid tumors in mice. Mol Ther - Oncolytics. (2021) 20:556−68. doi: 10.1016/j.omto.2021.02.013
82. Deng X, Gao F, Li N, Li Q, Zhou Y, Yang T, et al. Antitumor activity of NKG2D CAR-T cells against human colorectal cancer cells in vitro and in vivo. Am J Cancer Res (2019) 9(5):945−58.
83. Golubovskaya V. PLAP -CAR T nbsp cells mediate high specific cytotoxicity against colon cancer cells. Front Biosci (2020) 25(9):1765−86. doi: 10.2741/4877
84. Bashiri Dezfouli A, Yazdi M, Benmebarek MR, Schwab M, Michaelides S, Miccichè A, et al. CAR T cells targeting membrane-bound Hsp70 on tumor cells mimic Hsp70-primed NK cells. Front Immunol 1 juin (2022) 13:883694. doi: 10.3389/fimmu.2022.883694
85. Liu L, Liu Y, Xia Y, Wang G, Zhang X, Zhang H, et al. Synergistic killing effects of PD-L1-CAR T cells and colorectal cancer stem cell-dendritic cell vaccine-sensitized T cells in ALDH1-positive colorectal cancer stem cells. J Cancer. (2021) 12(22):6629−39. doi: 10.7150/jca.62123
86. Ang WX, Ng YY, Xiao L, Chen C, Li Z, Chi Z, et al. Electroporation of NKG2D RNA CAR improves Vγ9Vδ2 T cell responses against human solid tumor xenografts. Mol Ther - Oncolytics. (2020) 17:421−30. doi: 10.1016/j.omto.2020.04.013
87. Feng Z, He X, Zhang X, Wu Y, Xing B, Knowles A, et al. Potent suppression of neuroendocrine tumors and gastrointestinal cancers by CDH17CAR T cells without toxicity to normal tissues. Nat Cancer. (2022) 3(5):581−94. doi: 10.1038/s43018-022-00344-7
88. Liang KH, Tso HC, Hung SH, Kuan II, Lai JK, Ke FY, et al. Extracellular domain of EpCAM enhances tumor progression through EGFR signaling in colon cancer cells. Cancer Letters. (2018) 433:165−75. doi: 10.1016/j.canlet.2018.06.040
89. Eyvazi S, Farajnia S, Dastmalchi S, Kanipour F, Zarredar H, Bandehpour M. Antibody based EpCAM targeted therapy of cancer, review and update. CCDT. (2018) 18(9):857−68. doi: 10.2174/1568009618666180102102311
90. Gires O, Pan M, Schinke H, Canis M, Baeuerle PA. Expression and function of epithelial cell adhesion molecule EpCAM: where are we after 40 years? Cancer Metastasis Rev (2020) 39(3):969−87. doi: 10.1007/s10555-020-09898-3
91. Han ZW, Lyv ZW, Cui B, Wang YY, Cheng JT, Zhang Y, et al. The old CEACAMs find their new role in tumor immunotherapy. Invest New Drugs (2020) 38(6):1888−98. doi: 10.1007/s10637-020-00955-w
92. Zhang C, Wang Z, Yang Z, Wang M, Li S, Li Y, et al. Phase I escalating-dose trial of CAR-T therapy targeting CEA + metastatic colorectal cancers. Mol Ther (2017) 25(5):1248−58. doi: 10.1016/j.ymthe.2017.03.010
93. Schlimper C, Hombach AA, Abken H, Schmidt-Wolf IGH. Improved activation toward primary colorectal cancer cells by antigen-specific targeting autologous cytokine-induced killer cells. Clin Dev Immunol (2012) 2012:1−8. doi: 10.1155/2012/238924
94. Law AMK, Valdes-Mora F, Gallego-Ortega D. Myeloid-derived suppressor cells as a therapeutic target for cancer. Cells 27 févr (2020) 9(3):561. doi: 10.3390/cells9030561
95. Tamada K, Geng D, Sakoda Y, Bansal N, Srivastava R, Li Z, et al. Redirecting gene-modified T cells toward various cancer types using tagged antibodies. Clin Cancer Res (2012) 18(23):6436−45. doi: 10.1158/1078-0432.CCR-12-1449
96. Reiswich V, Gorbokon N, Luebke AM, Burandt E, Menz A, Kluth M, et al. Pattern of placental alkaline phosphatase ( PLAP ) expression in human tumors: a tissue microarray study on 12,381 tumors. J Pathol Clin Res (2021) 7(6):577−89. doi: 10.1002/cjp2.237
97. Multhoff G, Botzler C, Wiesnet M, Müller E, Meier T, Wilmanns W, et al. A stress-inducible 72-kDa heat-shock protein (HSP72) is expressed on the surface of human tumor cells, but not on normal cells. Int J Cancer. (1995) 61(2):272−9. doi: 10.1002/ijc.2910610222
98. Pfister K, Radons J, Busch R, Tidball JG, Pfeifer M, Freitag L, et al. Patient survival by Hsp70 membrane phenotype: Association with different routes of metastasis. Cancer. (2007) 110(4):926−35. doi: 10.1002/cncr.22864
99. Soleimani A, Zahiri E, Ehtiati S, Norouzi M, Rahmani F, Fiuji H, et al. Therapeutic potency of heat-shock protein-70 in the pathogenesis of colorectal cancer: current status and perspectives. Biochem Cell Biol (2019) 97(2):85−90. doi: 10.1139/bcb-2018-0177
100. Daly T, Royal RE, Kershaw MH, Treisman J, Wang G, Li W, et al. Recognition of human colon cancer by T cells transduced with a chimeric receptor gene. Cancer Gene Ther (2000) 7(2):284−91. doi: 10.1038/sj.cgt.7700121
101. Birbe R, Palazzo JP, Walters R, Weinberg D, Schulz S, Waldman SA. Guanylyl cyclase c is a marker of intestinal metaplasia, dysplasia, and adenocarcinoma of the gastrointestinal tract. Hum Pathology. (2005) 36(2):170−9. doi: 10.1016/j.humpath.2004.12.002
102. Carrithers SL, Barber MT, Biswas S, Parkinson SJ, Park PK, Goldstein SD, et al. Guanylyl cyclase c is a selective marker for metastatic colorectal tumors in human extraintestinal tissues. Proc Natl Acad Sci (1996) 93(25):14827−32. doi: 10.1073/pnas.93.25.14827
103. Lisby AN, Flickinger JC, Bashir B, Weindorfer M, Shelukar S, Crutcher M, et al. GUCY2C as a biomarker to target precision therapies for patients with colorectal cancer. Expert Rev Precis Med Drug Dev (2021) 6(2):117−29. doi: 10.1080/23808993.2021.1876518
104. Magee MS, Kraft CL, Abraham TS, Baybutt TR, Marszalowicz GP, Li P, et al. GUCY2C-directed CAR-T cells oppose colorectal cancer metastases without autoimmunity. OncoImmunology. 2 oct (2016) 5(10):e1227897. doi: 10.1080/2162402X.2016.1227897
105. Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DAN, Feldman SA, et al. T Cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther (2011) 19(3):620−6. doi: 10.1038/mt.2010.272
106. Zingoni A, Molfetta R, Fionda C, Soriani A, Paolini R, Cippitelli M, et al. NKG2D and its ligands: “One for all, all for one”. Front Immunol 12 mars (2018) 9:476. doi: 10.3389/fimmu.2018.00476
107. Lanier LL. NKG2D receptor and its ligands in host defense. Cancer Immunol Res (2015) 3(6):575−82. doi: 10.1158/2326-6066.CIR-15-0098
108. Dhar P, Wu JD. NKG2D and its ligands in cancer. Curr Opin Immunol (2018) 51:55−61. doi: 10.1016/j.coi.2018.02.004
109. Curio S, Jonsson G, Marinović S. A summary of current NKG2D-based CAR clinical trials. Immunotherapy Advances. (2021) 1(1):ltab018. doi: 10.1093/immadv/ltab018
110. Sureban SM, Berahovich R, Zhou H, Xu S, Wu L, Ding K, et al. DCLK1 monoclonal antibody-based CAR-T cells as a novel treatment strategy against human colorectal cancers. Cancers. 23 déc (2019) 12(1):54. doi: 10.3390/cancers12010054
111. Fan F, Samuel S, Evans KW, Lu J, Xia L, Zhou Y, et al. Overexpression of snail induces epithelial-mesenchymal transition and a cancer stem cell-like phenotype in human colorectal cancer cells. Cancer Med (2012) 1(1):5−16. doi: 10.1002/cam4.4
112. Wang Y, Liu Y, Lu J, Zhang P, Wang Y, Xu Y, et al. Rapamycin inhibits FBXW7 loss-induced epithelial–mesenchymal transition and cancer stem cell-like characteristics in colorectal cancer cells. Biochem Biophys Res Commun (2013) 434(2):352−6. doi: 10.1016/j.bbrc.2013.03.077
113. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial–mesenchymal transition. Nat Rev Mol Cell Biol (2014) 15(3):178−96. doi: 10.1038/nrm3758
114. Brabletz T. EMT and MET in metastasis: Where are the cancer stem cells? Cancer Cell (2012) 22(6):699−701. doi: 10.1016/j.ccr.2012.11.009
115. Sureban SM, May R, Weygant N, Qu D, Chandrakesan P, Bannerman-Menson E, et al. XMD8-92 inhibits pancreatic tumor xenograft growth via a DCLK1-dependent mechanism. Cancer Letters. (2014) 351(1):151−61. doi: 10.1016/j.canlet.2014.05.011
116. Nakanishi Y, Seno H, Fukuoka A, Ueo T, Yamaga Y, Maruno T, et al. Dclk1 distinguishes between tumor and normal stem cells in the intestine. Nat Genet (2013) 45(1):98−103. doi: 10.1038/ng.2481
117. Weygant N, Ge Y, Qu D, Kaddis JS, Berry WL, May R, et al. Survival of patients with gastrointestinal cancers can be predicted by a surrogate microRNA signature for cancer stem–like cells marked by DCLK1 kinase. Cancer Res (2016) 76(14):4090−9. doi: 10.1158/0008-5472.CAN-16-0029
118. Dai T, Hu Y, Lv F, Ozawa T, Sun X, Huang J, et al. Analysis of the clinical significance of DCLK1+ colorectal cancer using novel monoclonal antibodies against DCLK1. OncoTargets Ther (2018) Volume 11:5047−57. doi: 10.2147/OTT.S169928
119. Bürtin F, Mullins CS, Linnebacher M. Mouse models of colorectal cancer: Past, present and future perspectives. WJG. (2020) 26(13):1394−426. doi: 10.3748/wjg.v26.i13.1394
120. Weidemann S, Gagelmann P, Gorbokon N, Lennartz M, Menz A, Luebke AM, et al. Mesothelin expression in human tumors: A tissue microarray study on 12,679 tumors. Biomedicines. (2021) 9(4):397. doi: 10.3390/biomedicines9040397
121. Bharadwaj U, Marin-Muller C, Li M, Chen C, Yao Q. Mesothelin confers pancreatic cancer cell resistance to TNF-α-induced apoptosis through Akt/PI3K/NF-κB activation and IL-6/Mcl-1 overexpression. Mol Cancer. (2011) 10(1):106. doi: 10.1186/1476-4598-10-106
122. Chang MC, Chen CA, Chen PJ, Chiang YC, Chen YL, Mao TL, et al. Mesothelin enhances invasion of ovarian cancer by inducing MMP-7 through MAPK/ERK and JNK pathways. Biochem J (2012) 442(2):293−302. doi: 10.1042/BJ20110282
123. Chang MC, Chen CA, Hsieh CY, Lee CN, Su YN, Hu YH, et al. Mesothelin inhibits paclitaxel-induced apoptosis through the PI3K pathway. Biochem J (2009) 424(3):449−58. doi: 10.1042/BJ20082196
124. He X, Wang L, Riedel H, Wang K, Yang Y, Dinu CZ, et al. Mesothelin promotes epithelial-to-mesenchymal transition and tumorigenicity of human lung cancer and mesothelioma cells. Mol Cancer. (2017) 16(1):63. doi: 10.1186/s12943-017-0633-8
125. Servais EL, Colovos C, Rodriguez L, Bograd AJ, Nitadori Ji, Sima C, et al. Mesothelin overexpression promotes mesothelioma cell invasion and MMP-9 secretion in an orthotopic mouse model and in epithelioid pleural mesothelioma patients. Clin Cancer Res (2012) 18(9):2478−89. doi: 10.1158/1078-0432.CCR-11-2614
126. Uehara N, Matsuoka Y, Tsubura A. Mesothelin promotes anchorage-independent growth and prevents anoikis via extracellular signal-regulated kinase signaling pathway in human breast cancer cells. Mol Cancer Res (2008) 6(2):186−93. doi: 10.1158/1541-7786.MCR-07-0254
127. Wang Y, Wang L, Li D, Wang H, Chen Q. Mesothelin promotes invasion and metastasis in breast cancer cells. J Int Med Res (2012) 40(6):2109−16. doi: 10.1177/030006051204000608
128. Kaur S, Kumar S, Momi N, Sasson AR, Batra SK. Mucins in pancreatic cancer and its microenvironment. Nat Rev Gastroenterol Hepatol (2013) 10(10):607−20. doi: 10.1038/nrgastro.2013.120
129. La Salvia A, Lopez-Gomez V, Garcia-Carbonero R. HER2-targeted therapy: an emerging strategy in advanced colorectal cancer. Expert Opin Investigational Drugs (2019) 28(1):29−38. doi: 10.1080/13543784.2019.1555583
130. Gessner R, Tauber R. Intestinal cell adhesion molecules: Liver-intestine cadherin. Ann New York Acad Sci (2006) 915(1):136−43. doi: 10.1111/j.1749-6632.2000.tb05236.x
131. Wendeler MW, Drenckhahn D, Geßner R, Baumgartner W. Intestinal LI-cadherin acts as a Ca2+-dependent adhesion switch. J Mol Biol (2007) 370(2):220−30. doi: 10.1016/j.jmb.2007.04.062
133. Liu LX, Lee NP, Chan VW, Xue W, Zender L, Zhang C, et al. Targeting cadherin-17 inactivates wnt signaling and inhibits tumor growth in liver carcinoma. Hepatology. (2009) 50(5):1453−63. doi: 10.1002/hep.23143
134. Dai H, Tong C, Shi D, Chen M, Guo Y, Chen D, et al. Efficacy and biomarker analysis of CD133-directed CAR T cells in advanced hepatocellular carcinoma: a single-arm, open-label, phase II trial. OncoImmunology. (2020) 9(1):1846926. doi: 10.1080/2162402X.2020.1846926
135. Katz SC, Hardaway J, Prince E, Guha P, Cunetta M, Moody A, et al. HITM-SIR: phase ib trial of intraarterial chimeric antigen receptor T-cell therapy and selective internal radiation therapy for CEA+ liver metastases. Cancer Gene Ther (2020) 27(5):341−55. doi: 10.1038/s41417-019-0104-z
136. Katz SC, Moody AE, Guha P, Hardaway JC, Prince E, LaPorte J, et al. HITM-SURE: Hepatic immunotherapy for metastases phase ib anti-CEA CAR-T study utilizing pressure enabled drug delivery. J Immunother Cancer. (2020) 8(2):e001097. doi: 10.1136/jitc-2020-001097
137. Zhang Y, Kozlowska A, Fritz J, Zhao Y, Torre CPL, Cranert S, et al. 123 p-MUC1C-ALLO1: A fully allogeneic stem cell memory T cell (TSCM) CAR-T therapy with broad potential in solid tumor. J Immunother Cancer. (2021) 9(Suppl 2):A132−A132. doi: 10.1136/jitc-2021-SITC2021.123
138. Definition of autologous universal CAR-expressing T lymphocytes UniCAR02-T-NCI drug dictionary-national cancer institute. Available at: https://www.cancer.gov/publications/dictionaries/cancer-drug/def/autologous-universal-car-expressingt-lymphocytes-unicar02-t (Accessed on 28 August 2022).
139. Cui J, Chen N, Pu C, Zhao L, Li N, Wang C, et al. A phase 1 dose-escalation study of GCC19 CART a novel coupled CAR therapy for subjects with metastatic colorectal cancer. JCO. (2022) 40(16_suppl):3582−3582. doi: 10.1200/JCO.2022.40.16_suppl.3582
140. Lonez C, Hendlisz A, Shaza L, Aftimos P, Awada A, Machiels JPH, et al. Abstract CT123: A phase I study assessing the safety and clinical activity of multiple doses of a NKG2D-based CAR-T therapy, CYAD-01, administered concurrently with the neoadjuvant FOLFOX treatment in patients with potentially resectable liver metastases from colorectal cancer. Cancer Res (2018) 78(13_Supplement):CT123−CT123. doi: 10.1158/1538-7445.AM2018-CT123
141. Braun N, Hendlisz A, Shaza L, Vouche M, Donckier V, Aftimos P, et al. Abstract CT134: A phase I study assessing the safety and clinical activity of multiple hepatic transarterial administrations of a NKG2D-based CAR-T therapy CYAD-01, in patients with unresectable liver metastases from colorectal cancer. Cancer Res (2018) 78(13_Supplement):CT134−CT134. doi: 10.1158/1538-7445.AM2018-CT134
142. Prenen H, Dekervel J, Hendlisz A, Anguille S, Awada A, Cerf E, et al. Updated data from alloSHRINK phase I first-in-human study evaluating CYAD-101, an innovative non-gene edited allogeneic CAR-T in mCRC. JCO. (2021) 39(3_suppl):74−74. doi: 10.1200/JCO.2021.39.3_suppl.74
143. Sallman DA, Brayer JB, Poire X, Kerre T, Lewalle P, Wang ES, et al. Abstract CT129: The THINK clinical trial: Preliminary evidence of clinical activity of NKG2D chimeric antigen receptor T cell therapy (CYAD-01) in acute myeloid leukemia. Cancer Res (2018) 78(13_Supplement):CT129−CT129. doi: 10.1158/1538-7445.AM2018-CT129
144. Definition of autologous NKG2D CAR T cells KD-025-NCI drug dictionary-national cancer institute. Available at: https://www.cancer.gov/publications/dictionaries/cancer-drug/def/autologous-nkg2d-car-t-cells-kd-025 (Accessed on 28 August 2022).
145. Hege KM, Bergsland EK, Fisher GA, Nemunaitis JJ, Warren RS, McArthur JG, et al. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J ImmunoTherapy Cancer (2017) 5(1):22. doi: 10.1186/s40425-017-0222-9
146. Thistlethwaite FC, Gilham DE, Guest RD, Rothwell DG, Pillai M, Burt DJ, et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunology Immunother (2017) 66(11):1425−36. doi: 10.1007/s00262-017-2034-7
147. EGFR-IL12-CART cells for patients with metastatic colorectal cancer (EGFRCART). Available at: https://clinicaltrials.gov/ct2/show/NCT03542799 (Accessed on 20 June 2020).
148. EGFR CART cells for patients with metastatic colorectal cancer. Available at: https://clinicaltrials.gov/ct2/show/NCT03152435 (Accessed on 20 June 2020).
149. A study of chimeric antigen receptor T cells combined with interventional therapy in advanced liver malignancy. Available at: https://clinicaltrials.gov/ct2/show/NCT02959151 (Accessed on 20 June 2020).
150. A clinical research of CAR T cells targeting CEA positive cancer. Available at: https://clinicaltrials.gov/ct2/show/NCT02349724 (Accessed on 20 June 2020).
151. CAR-T intraperitoneal infusions for CEA-expressing adenocarcinoma peritoneal metastases or malignant ascites (IPC). Available at: https://clinicaltrials.gov/ct2/show/NCT03682744 (Accessed on 20 June 2020).
152. CAR-T hepatic artery infusions or pancreatic venous infusions for CEA-expressing liver metastases or pancreas cancer (HITM-SURE). Available at: https://clinicaltrials.gov/ct2/show/NCT02850536 (Accessed on 20 June 2020).
153. CAR-T hepatic artery infusions and sir-spheres for liver metastases (HITM-SIR). Available at: https://clinicaltrials.gov/ct2/show/NCT02416466 (Accessed on 20 June 2020).
154. Binary oncolytic adenovirus in combination with HER2-specific autologous CAR VST, advanced HER2 positive solid tumors (VISTA). Available at: https://clinicaltrials.gov/ct2/show/NCT03740256 (Accessed on 20 June 2020).
155. Rosewell Shaw A, Suzuki M. Recent advances in oncolytic adenovirus therapies for cancer. Curr Opin Virology. (2016) 21:9−15. doi: 10.1016/j.coviro.2016.06.009.
156. CAR-T cell immunotherapy in MUC1 positive solid tumor. Available at: https://clinicaltrials.gov/ct2/show/NCT02617134 (Accessed on 20 June 2020).
157. Autologous CAR-T/TCR-T cell immunotherapy for malignancies. Available at: https://clinicaltrials.gov/ct2/show/record/NCT03638206 (Accessed on 20 June 2020).
158. Treatment of relapsed and/or chemotherapy refractory advanced malignancies by CART133. Available at: https://clinicaltrials.gov/ct2/show/record/NCT02541370?cond=Colorectal+Cancer&intr=chimeric+antigen+receptor+t-cell (Accessed on 20 June 2020).
159. A clinical research of CAR T cells targeting EpCAM positive cancer (CARTEPC). Available at: https://clinicaltrials.gov/ct2/show/NCT03013712 (Accessed on 20 June 2020).
160. Lonez C, Verma B, Hendlisz A, Aftimos P, Awada A, Van Den Neste E, et al. Study protocol for THINK: a multinational open-label phase I study to assess the safety and clinical activity of multiple administrations of NKR-2 in patients with different metastatic tumour types. BMJ Open (2017) 7(11):e017075. doi: 10.1136/bmjopen-2017-017075
161. Murad JM, Graber DJ, Sentman CL. Advances in the use of natural receptor- or ligand-based chimeric antigen receptors (CARs) in haematologic malignancies. Best Pract Res Clin Haematology. (2018) 31(2):176−83. doi: 10.1016/j.beha.2018.03.003
162. Hepatic transarterial administrations of NKR-2 in patients with unresectable liver metastases from colorectal cancer (LINK). Available at: https://clinicaltrials.gov/ct2/show/NCT03370198 (Accessed on 20 June 2020).
163. Dose escalation and dose expansion phase I study to assess the safety and clinical activity of multiple doses of NKR-2 administered concurrently with FOLFOX in colorectal cancer with potentially resectable liver metastases (SHRINK). Available at: https://clinicaltrials.gov/ct2/show/NCT03310008 (Accessed on 20 June 2020).
164. Bailey SR, Maus MV. Gene editing for immune cell therapies. Nat Biotechnol (2019) 37(12):1425−34. doi: 10.1038/s41587-019-0137-8
165. alloSHRINK - standard cHemotherapy regimen and immunotherapy with allogeneic NKG2D-based CYAD-101 chimeric antigen receptor T-cells (alloSHRINK). Available at: https://clinicaltrials.gov/ct2/show/NCT03692429 (Accessed on 20 June 2020).
166. Ponterio E, De Maria R, Haas TL. Identification of targets to redirect CAR T cells in glioblastoma and colorectal cancer: An arduous venture. Front Immunol (2020) 11:565631. doi: 10.3389/fimmu.2020.565631
167. Wang D, Zhang H, Xiang T, Wang G. Clinical application of adaptive immune therapy in MSS colorectal cancer patients. Front Immunol (2021) 12:762341. doi: 10.3389/fimmu.2021.762341
168. Zhao L, Cao YJ. Engineered T cell therapy for cancer in the clinic. Front Immunol (2019) 10:2250. doi: 10.3389/fimmu.2019.02250
169. Coscia M, Bruno B, Neelapu S. Editorial: CAR T-cell therapies in hematologic tumors. Front Oncol (2020) 10:588134. doi: 10.3389/fonc.2020.588134
170. Greenbaum U, Mahadeo KM, Kebriaei P, Shpall EJ, Saini NY. Chimeric antigen receptor T-cells in b-acute lymphoblastic leukemia: State of the art and future directions. Front Oncol (2020) 10:1594. doi: 10.3389/fonc.2020.01594
171. Vitale C, Strati P. CAR T-cell therapy for b-cell non-Hodgkin lymphoma and chronic lymphocytic leukemia: Clinical trials and real-world experiences. Front Oncol (2020) 10:849. doi: 10.3389/fonc.2020.00849
172. Rodríguez-Lobato LG, Ganzetti M, Fernández de Larrea C, Hudecek M, Einsele H, Danhof S. CAR T-cells in multiple myeloma: State of the art and future directions. Front Oncol (2020) 10:1243. doi: 10.3389/fonc.2020.01243
173. Mardiana S, Gill S. CAR T cells for acute myeloid leukemia: State of the art and future directions. Front Oncol (2020) 10:697. doi: 10.3389/fonc.2020.00697
174. Sievers S, Watson G, Johncy S, Adkins S. Recognizing and grading CAR T-cell toxicities: An advanced practitioner perspective. Front Oncol (2020) 10:885. doi: 10.3389/fonc.2020.00885
175. Giraldo NA, Becht E, Remark R, Damotte D, Sautès-Fridman C, Fridman WH. The immune contexture of primary and metastatic human tumours. Curr Opin Immunol (2014) 27:8−15. doi: 10.1016/j.coi.2014.01.001
176. Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. (2012) 12(4):298−306. doi: 10.1038/nrc3245
177. Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. (2013) 39(4):782−95. doi: 10.1016/j.immuni.2013.10.003
178. Gajewski TF. The next hurdle in cancer immunotherapy: Overcoming the non–T-Cell–Inflamed tumor microenvironment. Semin Oncol (2015) 42(4):663−71. doi: 10.1053/j.seminoncol.2015.05.011
179. Zemek RM, De Jong E, Chin WL, Schuster IS, Fear VS, Casey TH, et al. Sensitization to immune checkpoint blockade through activation of a STAT1/NK axis in the tumor microenvironment. Sci Trans Med (2019) 11(501):eaav7816. doi: 10.1126/scitranslmed.aav7816
180. Chow MT, Ozga AJ, Servis RL, Frederick DT, Lo JA, Fisher DE, et al. Intratumoral activity of the CXCR3 chemokine system is required for the efficacy of anti-PD-1 therapy. Immunity. (2019) 50(6):1498–512.e5. doi: 10.1016/j.immuni.2019.04.010
181. Garris CS, Arlauckas SP, Kohler RH, Trefny MP, Garren S, Piot C, et al. Successful anti-PD-1 cancer immunotherapy requires T cell-dendritic cell crosstalk involving the cytokines IFN-γ and IL-12. Immunity. (2018) 49(6):1148–61.e7. doi: 10.1016/j.immuni.2018.09.024
182. Hegde PS, Karanikas V, Evers S. The where, the when, and the how of immune monitoring for cancer immunotherapies in the era of checkpoint inhibition. Clin Cancer Res (2016) 22(8):1865−74. doi: 10.1158/1078-0432.CCR-15-1507
183. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science. (2015) 348(6230):124−8. doi: 10.1126/science.aaa1348
184. Passardi A, Canale M, Valgiusti M, Ulivi P. Immune checkpoints as a target for colorectal cancer treatment. Int J Mol Sci (2017) 18(6):1324. doi: 10.3390/ijms18061324
185. Leal AD, Paludo J, Finnes HD, Grothey A. Response to pembrolizumab in patients with mismatch repair deficient (dMMR) colorectal cancer (CRC). J Clin Oncol (2017) 35(15_suppl):3558−3558. doi: 10.1200/JCO.2017.35.15_suppl.3558
186. Tran E, Robbins PF, Rosenberg SA. « final common pathway » of human cancer immunotherapy: targeting random somatic mutations. Nat Immunol (2017) 18(3):255−62. doi: 10.1038/ni.3682
187. Van den Eynde M, Mlecnik B, Bindea G, Fredriksen T, Church SE, Lafontaine L, et al. The link between the multiverse of immune microenvironments in metastases and the survival of colorectal cancer patients. Cancer Cell (2018) 34(6):1012–26.e3. doi: 10.1016/j.ccell.2018.11.003
188. Duan Q, Zhang H, Zheng J, Zhang L. Turning cold into hot: Firing up the tumor microenvironment. Trends Cancer. (2020) 6(7):605−18. doi: 10.1016/j.trecan.2020.02.022
189. Dewan MZ, Galloway AE, Kawashima N, Dewyngaert JK, Babb JS, Formenti SC, et al. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res (2009) 15(17):5379−88. doi: 10.1158/1078-0432.CCR-09-0265
190. Teng F, Kong L, Meng X, Yang J, Yu J. Radiotherapy combined with immune checkpoint blockade immunotherapy: Achievements and challenges. Cancer Letters. (2015) 365(1):23−9. doi: 10.1016/j.canlet.2015.05.012
191. Ngiow SF, McArthur GA, Smyth MJ. Radiotherapy complements immune checkpoint blockade. Cancer Cell (2015) 27(4):437−8. doi: 10.1016/j.ccell.2015.03.015
192. Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discovery. (2019) 18(3):197−218. doi: 10.1038/s41573-018-0007-y
193. Zhang J, Endres S, Kobold S. Enhancing tumor T cell infiltration to enable cancer immunotherapy. Immunotherapy. (2019) 11(3):201−13. doi: 10.2217/imt-2018-0111
194. Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell (2015) 28(6):690−714. doi: 10.1016/j.ccell.2015.10.012
195. Galuppini F, Dal Pozzo CA, Deckert J, Loupakis F, Fassan M, Baffa R. Tumor mutation burden: from comprehensive mutational screening to the clinic. Cancer Cell Int (2019) 19:209. doi: 10.1186/s12935-019-0929-4
196. Fancello L, Gandini S, Pelicci PG, Mazzarella L. Tumor mutational burden quantification from targeted gene panels: major advancements and challenges. J ImmunoTherapy Cancer (2019) 7(1):183. doi: 10.1186/s40425-019-0647-4
197. Chan TA, Yarchoan M, Jaffee E, Swanton C, Quezada SA, Stenzinger A, et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol (2019) 30(1):44−56. doi: 10.1093/annonc/mdy495
198. Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. New Engl J Med (2017) 377(25):2500−1. doi: 10.1056/NEJMc1713444
199. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non–Small-Cell lung cancer. New Engl J Med (2015) 372(21):2018−28. doi: 10.1056/NEJMoa1501824
200. Iwai Y, Hamanishi J, Chamoto K, Honjo T. Cancer immunotherapies targeting the PD-1 signaling pathway. J Biomed Sci (2017) 24(1): 26. doi: 10.1186/s12929-017-0329-9
201. Boumber Y. Tumor mutational burden (TMB) as a biomarker of response to immunotherapy in small cell lung cancer. J Thorac Disease. (2018) 10(8):4689−93. doi: 10.21037/jtd.2018.07.120
202. Goto Y. Tumor mutation burden: Is it ready for the clinic? J Clin Oncol (2018) 36(30):2978−9. doi: 10.1200/JCO.2018.79.3398
203. Johnson DB, Frampton GM, Rioth MJ, Yusko E, Xu Y, Guo X, et al. Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol Res (2016) 4(11):959−67. doi: 10.1158/2326-6066.CIR-16-0143
204. Castle JC, Kreiter S, Diekmann J, Lower M, van de Roemer N, de Graaf J, et al. Exploiting the mutanome for tumor vaccination. Cancer Res (2012) 72(5):1081−91. doi: 10.1158/0008-5472.CAN-11-3722
205. Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. (2012) 482(7385):400−4. doi: 10.1038/nature10755
206. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. (2014) 515(7528):577−81. doi: 10.1038/nature13988
207. Schumacher T, Bunse L, Pusch S, Sahm F, Wiestler B, Quandt J, et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature. (2014) 512(7514):324−7. doi: 10.1038/nature13387
208. Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. (2014) 515(7528):572−6. doi: 10.1038/nature14001
209. Duan F, Duitama J, Al Seesi S, Ayres CM, Corcelli SA, Pawashe AP, et al. Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules to predict anticancer immunogenicity. J Exp Med (2014) 211(11):2231−48. doi: 10.1084/jem.20141308
210. Linnemann C, van Buuren MM, Bies L, Verdegaal EME, Schotte R, Calis JJA, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med (2015) 21(1):81−5. doi: 10.1038/nm.3773
211. Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med (2013) 19(6):747−52. doi: 10.1038/nm.3161
212. Rajasagi M, Shukla SA, Fritsch EF, Keskin DB, DeLuca D, Carmona E, et al. Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia. Blood. (2014) 124(3):453−62. doi: 10.1182/blood-2014-04-567933
213. van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol (2013) 31(32):e439−42. doi: 10.1200/JCO.2012.47.7521
214. Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. (2015) 350(6257):207−11. doi: 10.1126/science.aad0095
215. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. New Engl J Med (2014) 371(23):2189−99. doi: 10.1056/NEJMoa1406498
216. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. (2015) 160(1−2):48−61. doi: 10.1016/j.cell.2014.12.033
217. Guidoboni M, Gafà R, Viel A, Doglioni C, Russo A, Santini A, et al. Microsatellite instability and high content of activated cytotoxic lymphocytes identify colon cancer patients with a favorable prognosis. Am J Pathology. (2001) 159(1):297−304. doi: 10.1016/S0002-9440(10)61695-1
218. Buckowitz A, Knaebel HP, Benner A, Bläker H, Gebert J, Kienle P, et al. Microsatellite instability in colorectal cancer is associated with local lymphocyte infiltration and low frequency of distant metastases. Br J Cancer. (2005) 92(9):1746−53. doi: 10.1038/sj.bjc.6602534
219. Dolcetti R, Viel A, Doglioni C, Russo A, Guidoboni M, Capozzi E, et al. High prevalence of activated intraepithelial cytotoxic T lymphocytes and increased neoplastic cell apoptosis in colorectal carcinomas with microsatellite instability. Am J Pathology. (1999) 154(6):1805−13. doi: 10.1016/S0002-9440(10)65436-3
220. Boyiadzis MM, Kirkwood JM, Marshall JL, Pritchard CC, Azad NS, Gulley JL. Significance and implications of FDA approval of pembrolizumab for biomarker-defined disease. J immunotherapy cancer. (2018) 6(1):35. doi: 10.1186/s40425-018-0342-x
221. Romero D. New first-line therapy for dMMR/MSI-h CRC. Nat Rev Clin Oncol (2021) 18(2):63. doi: 10.1038/s41571-020-00464-y
222. Saeterdal I, Bjorheim J, Lislerud K, Gjertsen MK, Bukholm IK, Olsen OC, et al. Frameshift-mutation-derived peptides as tumor-specific antigens in inherited and spontaneous colorectal cancer. Proc Natl Acad Sci (2001) 98(23):13255−60. doi: 10.1073/pnas.231326898
223. Duval A, Raphael M, Brennetot C, Poirel H, Buhard O, Aubry A, et al. The mutator pathway is a feature of immunodeficiency-related lymphomas. Proc Natl Acad Sci (2004) 101(14):5002−7. doi: 10.1073/pnas.0400945101
224. Radvanyi LG, Bernatchez C, Zhang M, Fox PS, Miller P, Chacon J, et al. Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin Cancer Res (2012) 18(24):6758−70. doi: 10.1158/1078-0432.CCR-12-1177
225. Zhao B, Song A, Haque R, Lei F, Weiler L, Xiong X, et al. Cooperation between molecular targets of costimulation in promoting T cell persistence and tumor regression. J Immunol (2009) 182(11):6744−52. doi: 10.4049/jimmunol.0804387
226. Horton BL, Williams JB, Cabanov A, Spranger S, Gajewski TF. Intratumoral CD8+ T-cell apoptosis is a major component of T-cell dysfunction and impedes antitumor immunity. Cancer Immunol Res (2018) 6(1):14−24. doi: 10.1158/2326-6066.CIR-17-0249
227. Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell–mediated tumor eradication. Mol Ther (2010) 18(2):413−20. doi: 10.1038/mt.2009.210
228. Nguyen A, Johanning G, Shi Y. Emerging novel combined CAR-T cell therapies. Cancers. (2022) 14(6):1403. doi: 10.3390/cancers14061403
229. Arriga R, Caratelli S, Lanzilli G, Ottaviani A, Cenciarelli C, Sconocchia T, et al. CD16-158-valine chimeric receptor T cells overcome the resistance of KRAS-mutated colorectal carcinoma cells to cetuximab. Int J Cancer. (2020) 146(9):2531−8. doi: 10.1002/ijc.32618
230. Li H, Yang C, Cheng H, Huang S, Zheng Y. CAR-T cells for colorectal cancer: Target-selection and strategies for improved activity and safety. J Cancer. (2021) 12(6):1804−14. doi: 10.7150/jca.50509
231. Pan K, Farrukh H, Chittepu VCSR, Xu H, Pan Cx, Zhu Z. CAR race to cancer immunotherapy: from CAR T, CAR NK to CAR macrophage therapy. J Exp Clin Cancer Res (2022) 41(1):119. doi: 10.1186/s13046-022-02327-z
232. Lu H, Zhao X, Li Z, Hu Y, Wang H. From CAR-T cells to CAR-NK cells: A developing immunotherapy method for hematological malignancies. Front Oncol (2021) 11:720501. doi: 10.3389/fonc.2021.720501
233. Li G, Wu X, Chan IH, Trager JB. Abstract 4235: A combination of CAR-NK and CAR-T cells results in rapid and persistent anti-tumor efficacy while reducing CAR-T cell mediated cytokine release and T-cell proliferation. Cancer Res (2020) 80(16_Supplement):4235−4235. doi: 10.1158/1538-7445.AM2020-4235
Keywords: colorectal cancer, CAR T cells, immunotherapies, tumor microenvironement, tumor mutational burden, clinical data
Citation: Ghazi B, El Ghanmi A, Kandoussi S, Ghouzlani A and Badou A (2022) CAR T-cells for colorectal cancer immunotherapy: Ready to go? Front. Immunol. 13:978195. doi: 10.3389/fimmu.2022.978195
Received: 25 June 2022; Accepted: 14 October 2022;
Published: 15 November 2022.
Edited by:
Lynn Morris, National Institute of Communicable Diseases (NICD), South AfricaReviewed by:
Thanh Huong Phung, Hanoi University of Pharmacy, VietnamCopyright © 2022 Ghazi, El Ghanmi, Kandoussi, Ghouzlani and Badou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Abdallah Badou, YWJkYWxsYWguYmFkb3VAdW5pdmgyYy5tYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.