 Steven Z. Jiang
Steven Z. Jiang Jeffrey L. To
Jeffrey L. To Michael R. Hughes
Michael R. Hughes Kelly M. McNagny
Kelly M. McNagny Hugh Kim
Hugh Kim- 1Centre for Blood Research, University of British Columbia, Vancouver, BC, Canada
- 2Department of Biochemistry and Molecular Biology, University of British Columbia, Vancouver, BC, Canada
- 3School of Biomedical Engineering, University of British Columbia, Vancouver, BC, Canada
- 4Department of Medical Genetics, University of British Columbia, Vancouver, BC, Canada
- 5Department of Oral Biological and Medical Sciences, University of British Columbia, Vancouver, BC, Canada
Rheumatoid arthritis (RA) is a debilitating autoimmune disorder characterized by chronic inflammation of the synovial tissues and progressive destruction of bone and cartilage. The inflammatory response and subsequent tissue degradation are orchestrated by complex signaling networks between immune cells and their products in the blood, vascular endothelia and the connective tissue cells residing in the joints. Platelets are recognized as immune-competent cells with an important role in chronic inflammatory diseases such as RA. Here we review the specific aspects of platelet function relevant to arthritic disease, including current knowledge of the molecular crosstalk between platelets and other innate immune cells that modulate RA pathogenesis.
Introduction: RA pathophysiology
Rheumatoid arthritis (RA) is an autoimmune disease that affects ~0.4% of the global population (1). The disease is characterized by chronic inflammation of the joints, degradation of bone and cartilage (2), and progressive pain and loss of mobility such that 37% of RA patients become disabled (3). RA decreases life expectancy and is associated with increased risk of lymphoma, cardiovascular disease, and lung cancer (4, 5). The precise etiology of RA is unknown, but genetic predisposition (6), abnormal DNA methylation patterns (7), smoking (8), infection (9), and gut microbiome dysbiosis (10) contribute to RA development. A classic hallmark of RA is the presence of the self-antigen binding anti-citrullinated protein antibodies (ACPA) that trigger chronic activation of innate immune cells. Citrullination is a protein post-translational modification where the amino acid arginine is substituted for citrulline, leading to the generation of ACPAs that initiate a pro-inflammatory response (11).
During the onset of RA, the innate immune response recruits platelets and platelet microparticles (PMPs) (12, 13) as well as leukocytes (14) to the joints. PMPs and leukocytes are a source of pro-inflammatory chemokines and cytokines (15) that signal to the resident fibroblast-like synoviocytes (FLS) lining the synovial cavity (16). FLS hyper-proliferate to form a tumor-like structure called a pannus (2, 17). Angiogenesis lays down new blood vessels that supply nutrients (18) and allow the pannus to grow until it reaches the bone and cartilage where “activated” and increasingly aggressive FLS secrete joint-destroying matrix metalloproteinases (MMP) including MMP-1, MMP-3 and MMP-13 (2, 17). Importantly, the distinct roles of platelets in mediating the innate immune response are now increasingly recognized in the context of RA.
Platelets are innate immune cells that drive RA pathogenesis
Platelets are small (2-3 μm) anucleate blood cells responsible for hemostasis (19). Resting platelets are activated by many ligands, such as collagen and thrombin, via platelet surface receptors (Figure 1A). Activated platelets change shape, aggregate and secrete granule contents that include prothrombotic mediators, cytokines, chemokines and growth factors (20), resulting in thrombus formation at the site of injury (Table 1). The process leading to activation is typically tightly regulated, allowing most platelets to circulate in their quiescent states under normal physiologic conditions. However, there is an increasing body of evidence suggesting that dysregulation of platelet activation may be a key factor in driving various chronic inflammatory diseases, including RA (101).
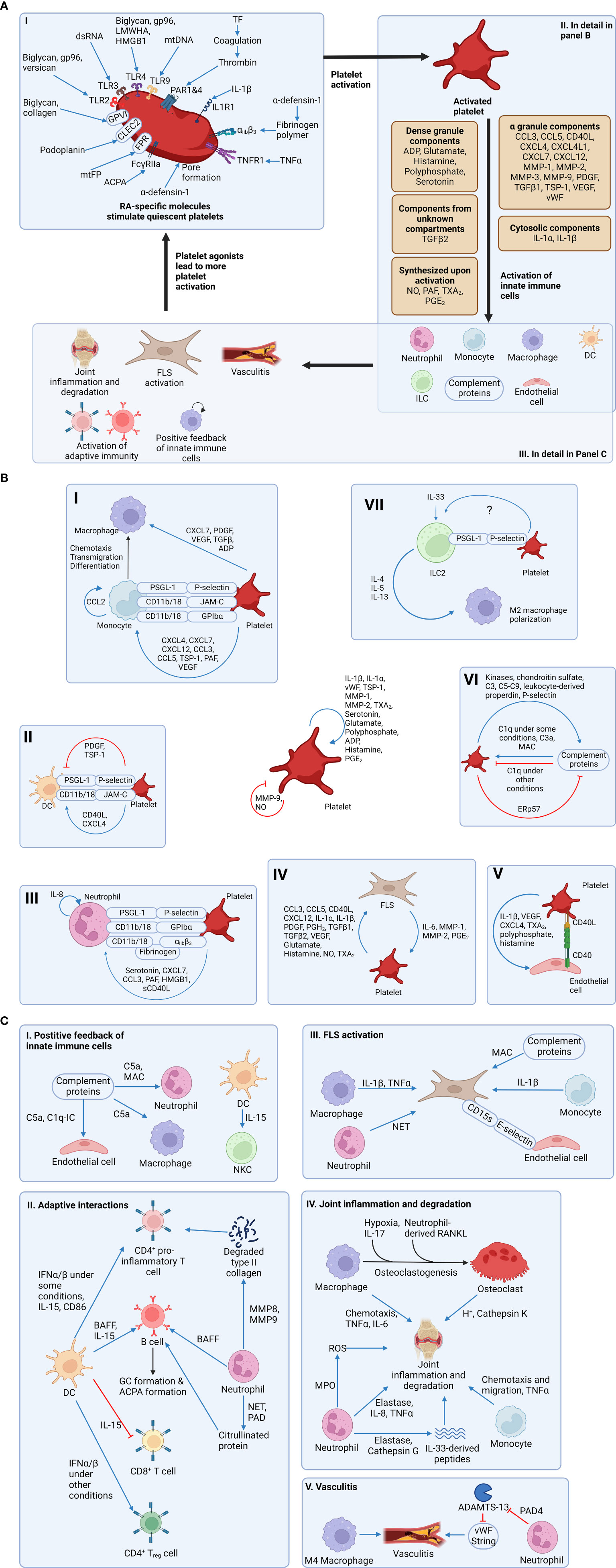
Figure 1 Platelet signaling contributes to the activation of innate immunity and the development of rheumatoid arthritis (RA). (A) Section I: RA-specific molecules activate platelets by binding to receptors on the surface of platelets. Notably, α-defensin mediates its action by forming a pore on the plasma membrane of platelets. Section II: Activated platelets secrete mediators stored in dense granules and α-granules. Notably, some mediators are freshly synthesized upon platelet activation and therefore not stored in granules. Also, IL-1 is stored in the cytosol of platelets, while TGF-β is stored in an unknown subcellular compartment. Regardless of the subcellular localization, these mediators contribute to the activation of more platelets and other cell types. Section III: Activated innate immune cells lead to more immune activation through a positive feedback loop and the generation of RA symptoms. Also, FLSs, which are activated directly and indirectly by platelets, can activate more platelets, resulting in a vicious cycle. (B) Platelets can activate many other cell types. First, platelets are capable of autocrine-activation and feedback inhibition. Section I: Platelets can support monocyte transmigration using adhesion molecules. Also, platelet-derived soluble mediators can activate monocytes. After monocytes transmigrate into subendothelial space and differentiate into macrophages, platelets can activate macrophages. Section II: Platelets support DC transmigration into the joint using adhesion molecules. Some platelet-derived soluble mediators activate DCs, while other mediators inhibit DCs. Section III: Platelets activate neutrophils using soluble mediators and support their transmigration using adhesion molecules. Section IV: Platelets and FLSs can activate each other using soluble mediators, resulting in a positive feedback loop. Section V: Activated platelets can activate endothelial cells. Section VI: Activated platelets and complement proteins can activate each other, but feedback inhibition exists in both directions. Section VII: ILC2 express PSGL-1 and adhere to platelets (likely through P-selectin) in select tissues. Platelet interaction with ILC2s potentiates their response to IL-33 and survival and/or proliferation, but it is not known if this interaction occurs in synovial tissue. Also, the mechanism of platelet-mediated enhanced ILC2 function has not been determined. ILC2s ameliorate RA pathogenesis by limiting pro-inflammatory M1 polarization and promoting M2 polarization. (C) Activated innate immune cells result in more immune activation and the development of RA symptoms. Section I: Complement proteins and DCs can activate other innate immune cells. Section II: Both neutrophils and DCs can activate proinflammatory subsets of CD4+ T cell and B cells. Notably, neutrophils activate B cells and T cells not only by releasing soluble mediators, but also by generating citrullinated proteins and degraded type II collagen, both of which serve as autoantigens. Activated proinflammatory CD4+ T cells and B cells can promote RA progression. However, DCs inhibit CD8+ T cell cytotoxicity and induce CD4+ Treg differentation, thereby inhibiting RA progression. Therefore, the role of DCs in RA is complex. Section III: Macrophages, monocytes, complement proteins, and neutrophils activate a pro-inflammatory and pro-joint degrading phenotype of FLS. Endothelial cell-derived E-selectin allows these pro-inflammatory FLS to invade distant joints. Section IV: Macrophages can differentiate into osteoclasts in RA joints. Macrophages, osteoclasts, neutrophils, and monocytes contribute to joint inflammation and degradation. Notably, neutrophil-derived elastase and cathepsin G can cleave IL-33 into three proinflammatory peptides, which are more potent. Section V: M4 macrophages contribute to vasculitis in RA. ADAMTS-13 normally cleaves vasculitis-promoting vWF strings. Neutrophil PAD4 inhibits ADAMTS-13, thereby promoting vasculitis. Figures were made using BioRender.com. αiibβ3, integrin alpha iib beta 3; ACPA, anti-citrullinated protein antibody; ADAMTS-13, a disintegrin and metalloproteinase with a thrombospondin type 1 motif member 13; ADP, adenosine diphosphate; BAFF, B cell activating factor; CCL, chemokine C-C motif ligand; CD, cluster of differentiation; CLEC2, C-type lectin-like receptor 2; CXCL, chemokine C-X-C motif ligand; CXCL4L1, non-allelic variant of CXCL4; C1q, complement component 1q; C1q-IC, a complex of C1q and immune complex (which is an antibody-antigen complex); C3, complement components 3; C5a, complement component 5a; C5-C9, complement components 5, 6, 7, 8, and 9; DC, dendritic cell; ERp57, endoplasmic reticulum protein 57; FcγRIIa, Fc-gamma receptor IIa; FLS, fibroblast-like synoviocyte (also known as synovial fibroblast); FPR, formyl peptide receptor; GP, glycoprotein; gp96, heat shock protein 96; HMGB1, high mobility group box protein 1; IFN, interferon; IL, interleukin; ILC, innate lymphoid cell; JAM-C, junctional adhesion molecule-C; LMWHA, depolymerized low molecular weight hyaluronic acid; MAC, membrane attack complex; MMP, matrix metalloproteinase; MPO, myeloperoxidase; mtDNA, extracellular mitochondrial DNA; mtFP, extracellular mitochondrial formyl peptide; M4 macrophage, a subset of macrophage induced by CXCL4; NET, neutrophil extracellular trap; NKC, natural killer cell; NO, nitric oxide; PAD, protein arginine deiminase; PAF, platelet activating factor; PAR1&4, protease activated receptor 1 and protease activated receptor 4; PDGF, platelet-derived growth factor; PGE2, prostaglandin E2; PGH2, prostaglandin H2; PSGL-1, P-selectin glycoprotein ligand 1; RA, rheumatoid arthritis; RANKL, receptor activator of nuclear factor κB ligand; ROS, reactive oxygen species; sCD40L, soluble CD40 ligand; TGF, transforming growth factor; Th1, T helper 1 cell; Th17, T helper 17 cell; TNF, tumor necrosis factor; TNFR1, tumor necrosis factor receptor 1; TSP-1, thrombospondin 1; TXA2, thromboxane A2; VEGF, vascular endothelial growth factor; vWF, von Willebrand factor.
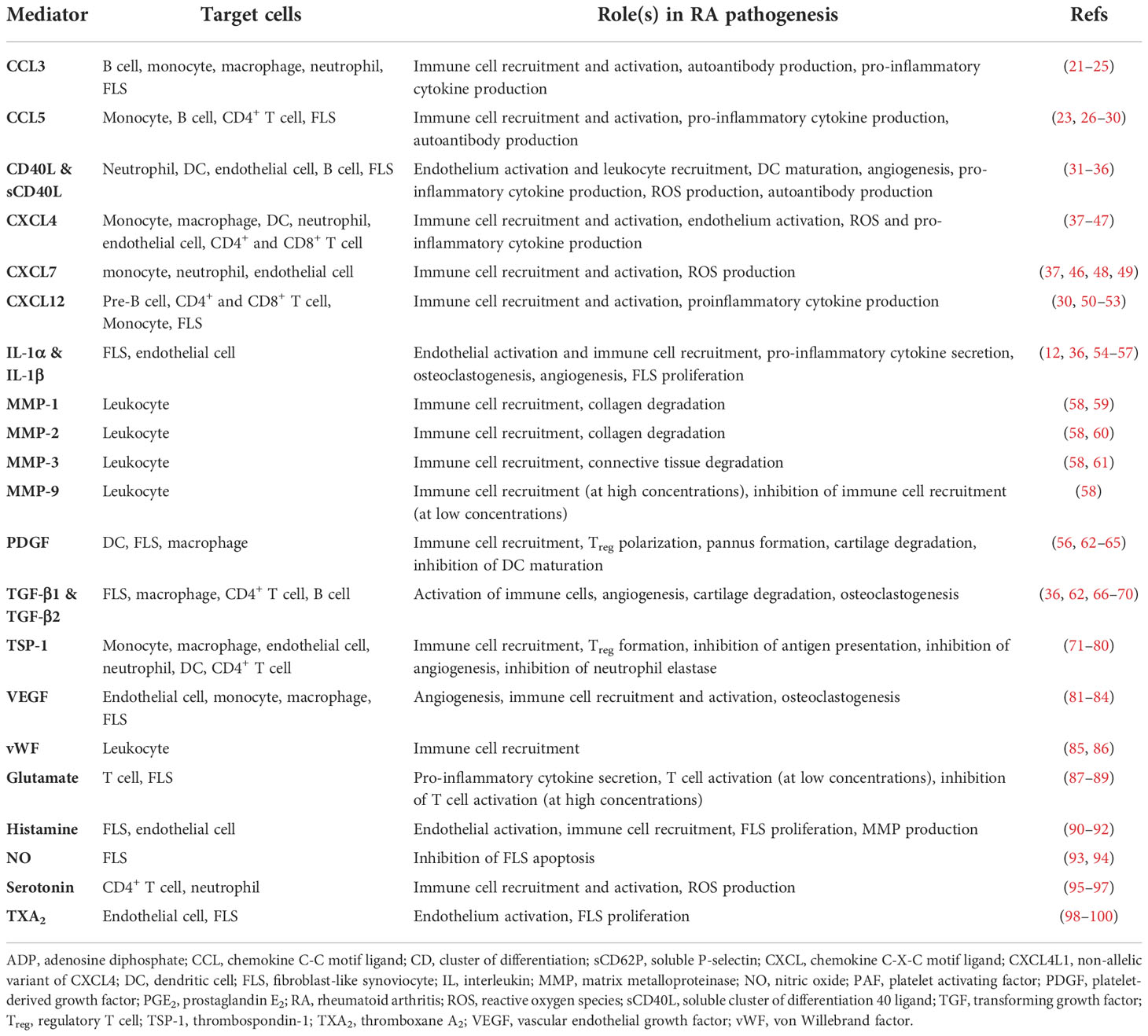
Table 1 Platelet-derived mediators with documented role(s) in RA pathogenesis.
A. Platelet activation and granule secretion
There are three types of platelet granules: alpha (α)-granules, dense granules, and lysosomes (reviewed in (102)). The first, and most abundant, are the α-granules that contain a multitude of cytokines, chemokines and growth factors (103). For example, α-granules release multiple pro-inflammatory molecules, including chemokine (C-C motif) ligand 5 (CCL5), soluble CD40 ligand (sCD40L/sCD154), and CXCL4 (aka platelet factor 4, PF4) (104). Furthermore, P-selectin (CD62P), a biomarker of α-granule secretion and platelet activation (105), is translocated from α-granules to the surface membrane upon platelet activation. Surface P-selectin promotes activation of leukocytes including neutrophils (see Platelet-neutrophil interactions section below). The second type of granule is the dense granule, so named due to its electron-dense appearance (102). The primary cargo of dense granules are nucleotides (adenosine diphosphate (ADP) and adenosine triphosphate (ATP)) and polyphosphates (102). Dense granule components primarily regulate platelet hemostasis and thrombosis (106). However, dense granules also release serotonin, which can promote inflammation by stimulating lymphocytes and neutrophils (95). The third type of granule is the lysosome, which contains tissue-degrading enzymes (such as collagenase) and acid phosphatase. As observed in other cell types, the key role of platelet lysosomes is to orchestrate catalytic breakdown of proteins (102).
B. Platelet extracellular vesicles
In addition to their granular contents, activated platelets release two types of extracellular vesicles: exosomes and microparticles (or microvesicles) (107). Firstly, exosomes are 30-100 nm vesicles contained within, and released with, larger intracellular vesicles. Secondly, microvesicles or platelet microparticles (PMPs) have a diameter of 100-1000 nm and are formed by the outward blebbing of the plasma membrane. PMPs are derived from the platelet plasma membrane and therefore contain the same markers found on platelet surfaces, including CD42 (glycoprotein Ib) and CD41/61 (integrin αIIbβ3) (107). Importantly, PMPs levels are increased in RA and may drive RA progression via release of their pro-inflammatory contents, e.g., interleukin-1 (IL-1) (12, 107).
C. Platelets are sentinels of the innate immune system
Platelet-derived cargo molecules (Table 1) include pro-inflammatory factors linked with chronic disorders including arthritis and inflammatory bowel disease (12, 108). Notably, platelets interact directly with bacteria (109), viruses (110) and complement proteins (111) and as such, are essential components of innate immunity. Platelets express toll-like receptors (TLRs) that sense pathogenic molecules (112) such as bacterial lipopolysaccharides (LPS) that induce platelet activation, aggregation, and leukocyte chemotaxis (113). Thus, platelets, as innate immune effectors, are recognized as critical mediators of RA pathogenesis (114, 115).
D. Evidence of platelet involvement in RA
Early correlational evidence suggested a role for platelets in the development and progression of RA, in that platelets were reportedly recruited to joints with their concentration in synovial fluid correlating with disease activity (13, 116, 117). Later research indicated that elevated circulating platelet count (aka thrombocytosis) was associated with more severe RA (118). Subsequently, seminal research identified PMPs (12) in the synovial fluid of RA patients. Compared to patients with non-inflammatory arthritis, RA synovial fluid contains more IL-1-rich PMPs (12). Taken together, these data indicate that platelet numbers are increased in RA, and that cytokine-laden PMPs are heavily recruited to arthritic joints.
In addition to higher platelet counts, circulating levels of platelet agonists are elevated in RA patients. For example, fibrinogen levels correlate with the number of swollen joints and overall disease activity (119). Thromboxane A2 (TXA2) is another platelet agonist whose degradation product, TXB2, is correlated with disease activity in RA (98). These data are consistent with elevated platelet activity during active RA (120). This notion is further corroborated by evidence of elevated soluble P-selectin (121) and sCD40L (122) in RA plasma. Soluble CD40L levels correlate with autoantibody levels in RA patients (123). Collectively, these data raise the possibility that platelet-derived pro-inflammatory products and cytokines play a role in propagating RA-associated joint inflammation. Animal studies provide a degree of direct evidence for the involvement of platelets in the RA pathogenesis. For example, platelet depletion prior to the induction of arthritis in mice reduces disease severity (12). Therefore, understanding the precise mechanisms by which platelets drive synovial inflammation have important implications for prospective therapies.
E. Molecules upregulated in RA promote platelet activation
In addition to driving RA pathogenesis, platelets can also be reciprocally activated by molecules that are elevated in RA plasma or joint tissue. For example, a central feature of RA is the generation of ACPA and formation of immune complexes nucleated around citrullinated proteins (11). Immune complexes bind and activate platelet Fc gamma receptor IIa (FcγRIIa) (124). Platelets are also activated by complement fragments (111). As such, complement activation during RA (125) can conceivably create a feedback loop through which platelet activity is continually elevated. Collectively, these data point to a two-way relationship between platelet function and innate responses in RA, where platelet-derived cytokines contribute to inflammation (Figure 1B) and ACPA-immune complexes (and other molecules) sustain platelet activation (Figure 1A). Moreover, RA-associated inflammatory mediators likely circulate and prime platelets systemically. These mechanisms may explain why RA patients are at higher risk for life-threatening complications such as deep vein thrombosis, myocardial infarction, and stroke (126, 127).
Platelet-driven signaling in the synovial tissues
A. Platelet growth factor signaling to FLS
Platelet α-granules store multiple forms of platelet-derived growth factor (PDGF) (128, 129), including the hetero and homo A/B dimers (PDGF-A/B) (130). PDGF-BB was recently confirmed to circulate in higher concentrations in RA patients (131). Furthermore, the invasiveness and proliferation of human FLS is increased following treatment with PDGF (62, 132). PDGF also promotes FLS production of pro-inflammatory cytokines IL-6, IL-8, as well as MMP-3 (133). In addition, platelets α-granules are an abundant source of transforming growth factor beta 1 (TGF-β1) (134). TGF-β1 stimulates the migratory behavior and invasiveness of cultured FLS obtained from RA patients (135). Interestingly, TGF-β reportedly acts synergistically along with PDGF to trigger FLS proliferation and a pro-inflammatory phenotype (132, 133). Collectively, these data provide both clinical and in vitro evidence of how synovial inflammation is likely propagated by platelet growth factors.
B. Platelet-derived cytokines and FLS
In addition to growth factors, platelet granules release several pro-inflammatory cytokines that exacerbate RA disease activity (Table 1). For example, platelets release copious quantities of CCL5 (RANTES) (26). CCL5 drives RA pathogenesis by upregulating MMPs in RA FLS (136). Activated platelets also secrete IL-1β (12, 54). IL-1β promotes synovial inflammation and RA progression by upregulating RANKL expression by FLS (55), which, in turn drives osteoclastogenesis and bone resorption (137). PMPs are another source of IL-1α (12), IL-1β (12), CCL5 (138), and sCD40L (139). Moreover, the role of PMPs in RA is supported by the observation that they increase the invasiveness and motility of cultured FLS (140, 141). Taken together, the evidence supports a role for platelet-derived cytokines and PMPs in synovial tissue degradation by promoting the release of MMPs and additional pro-inflammatory cytokines from FLS.
Interplay between platelets and other elements of the innate immune system
In addition to modulating disease by signaling to FLS, platelets interface directly with several other aspects of the innate immune system (Figure 1B). Platelets activate complement proteins and recruit leukocytes to the joint tissues. Multiple lines of evidence suggest that the pro-inflammatory leukocyte phenotype is enhanced via association with platelets. Here we detail the interactions between platelets and other aspects of the innate immune system that could explain the molecular basis underpinning the contribution of platelets to RA.
A. Platelet-neutrophil interactions
Platelets promote neutrophil chemotaxis
A feature of RA pathogenesis is the marked influx of immune cells, including neutrophils, into the synovium (14). Evidence from human (142) and murine (143) studies indicate that neutrophils are important for RA development by participating in joint tissue degradation and by releasing proinflammatory cytokines that further exacerbate RA (144, 145). Platelets can potentially recruit neutrophils into the joint by four possible mechanisms. Firstly, platelet-derived serotonin (95) and CXCL7 (146) are direct neutrophil chemoattractants; platelets stimulate FLS to secrete IL-8, CXCL2, and CXCL3 (147), all of which attract neutrophils (148); PMPs also recruit neutrophils to the joints (149). Secondly, exposed P-selectin on the platelet surface facilitates neutrophil adhesion via P-selectin glycoprotein ligand 1 (PSGL1) on the neutrophil surface (150). Thirdly, platelet-derived membrane-bound platelet activating factor (PAF) activates the neutrophil PAF receptor, leading to the activation of CD18 on the neutrophil surface (151). Neutrophil CD11b/CD18 (aka Mac-1) can then bind to glycoprotein Ibα (GPIbα) on the platelets (152). Finally, both neutrophil-derived CD11b/CD18 (153) and platelet-derived integrin αIIbβ3 (154) bind fibrinogen; CD11b/CD18 and αIIbβ3 can therefore mediate platelet-neutrophil adhesion via fibrinogen. These data provide compelling evidence of platelet-neutrophil crosstalk that can drive RA pathogenesis.
Platelets promote NET formation
Another central feature of neutrophils in the innate immune response is the release of neutrophil extracellular traps (NETs) consisting of genomic DNA, proteolytic enzymes, and reactive oxygen species (ROS) (155). NETs contribute to self-protein citrullination and correlate with ACPA levels (142) (Figure 1C). Platelets colocalize with degranulated neutrophils and NETs in coronary thrombi (156). Moreover, platelet depletion restricts NET formation in mouse models of endotoxemia (157). Activated platelets releases cytosolic stores of the alarmin high mobility group box 1 protein (HMGB1) that activates receptor for advanced glycation end products (RAGE) expressed on the surface of neutrophils, causing neutrophil autophagy and subsequent NET formation (156). These evidences implicate platelets as pivotal supporters of neutrophil-mediated inflammation and tissue degradation in RA.
B. Platelets interactions with the complement system
The complement system is an essential component of the innate immune response and is comprised of 9 serum proteins (C1-C9) activated sequentially, culminating in the formation of the membrane attack complex (MAC) that eliminates pathogens but is also implicated in autoimmune pathogenesis [reviewed in (158)]. There exist three separate pathways to complement activation: the classical pathway (elicited by binding of immunoglobulin to C1), the alternative pathway (triggered by the hydrolysis of C3 producing C3-H2O) and the lectin pathway (111).
Considerable evidence implicates complement activation as a contributing factor in RA [recently reviewed in (159)]. For example, MAC is upregulated in RA serum and synovial fluid (125), while an endogenous inhibitor of MAC, CD59, is downregulated in RA synovium (160). In addition, the role of complement proteins in neutrophil activation (161) is likely related to RA progression since neutrophils contribute to tissue degradation (144, 145) (Figure 1C).
Platelets contribute to complement activation in multiple ways. For example, a leukocyte-derived molecule termed properdin binds the platelet plasma membrane and recruits C3(H2O) thus activating the alternative pathway (162). Moreover, P-selectin on activated platelets binds C3b (163). Activated platelets also bind ficolin, which stimulates the lectin pathway (164). The evidence collectively supports a role for platelets in RA pathogenesis via the complement system.
C. Platelet interactions with monocytes/macrophages
Macrophages phagocytose invading pathogens and as such are critical elements of the innate immune response. As reviewed by Ley (165), macrophages may be polarized to a proinflammatory (M1) or an anti-inflammatory (M2) phenotype; in addition, M1 macrophages exacerbate bone degradation in RA. Accordingly, the number of macrophages in the synovium correlates with RA severity in humans (166), and depletion of monocytes attenuates the development of experimental murine arthritis (167).
Platelets and monocyte chemotaxis
As is observed with neutrophils, platelets serve to recruit monocytes. First, monocytes adhere to endothelium-bound platelets through the interaction between platelet surface P-selectin and PSGL1 expressed on the monocyte surface (168). Second, platelet-derived molecules promote adhesion between monocytes and the endothelium. Platelet-derived CCL5, CXCL4 (platelet factor 4, PF4), and CXCL7 promote monocyte chemotaxis as well as monocyte-endothelium adhesion (37). These chemokines and the anaphylatoxin C5a (generated by platelet-induced complement activation) upregulate CD11b (169), β1 integrin (170), and CD18 (β2 integrin) (170) on the monocyte surface. CD11b/CD18, which is expressed by monocytes (168), binds to platelet-derived junction adhesion molecule C (JAMC/JAM3) (171) and GPIbα (172) to facilitate firm adhesion.
Platelet-monocyte interactions
Once inside the joint, platelet-monocyte interactions contribute to joint pathology. In vitro experiments show that PAF, P-selectin, and CCL5 (RANTES) induce the secretion of CCL2, TNF-α, and IL-8 from monocytes (27, 173); these mediators can then in turn upregulate inflammation in the joint. Moreover, CXCL4 (PF4), a major platelet-derived chemokine, polarizes macrophages to an M4 subtype (distinct from M1 and M2 subtypes) that secretes pro-inflammatory TNF-α (38) and generates reactive oxygen species (ROS) (39). In addition, the differentiation of monocytes into bone-resorbing osteoclasts is accentuated by platelet-derived TGF-β1 (66). Taken together, these data illuminate multiple pathways through which platelets could promote synovial inflammation through interactions with monocytes/macrophages.
D. Platelet interactions with dendritic cells
Dendritic cells (DCs) are immune cells whose morphology is characterized by tree-like (dendritic) processes (174). Activated dendritic cells present pathogen-derived antigens to T cells, thus acting as a bridge between the innate and adaptive immune responses (175). Evidence suggests that DCs play an important role in RA (176). As is the case with neutrophils and monocytes, evidence supports a potential role for platelets in recruiting DCs. For example, in vitro data indicate that platelet-derived P-selectin and JAMC bind to PSGL1 and CD11b/CD18 expressed on DCs to promote platelet-DC adhesion (40). However, the platelets’ effects on DCs in disease are ambiguous since platelet-DC interactions can either promote or dampen inflammation. Platelet-derived sCD40L and CXCL4 (PF4) induce DC maturation, pro-inflammatory cytokine secretion from DCs, and interferon-α (IFN-α) secretion from stimulated plasmacytoid DCs (pDCs) in vitro (40, 177, 178). IFN-α causes DCs to secrete more TNF-α upon TLR4 stimulation, exacerbating RA (179). However, IFN-α can induce the polarization of both pro-inflammatory (180) and anti-inflammatory (181) T-cells, which can worsen or alleviate RA. Moreover, platelets also influence DC-mediated proliferation of T-cells (40) although it is not clear whether these T-cells adopt a pro-inflammatory (Th1/Th17) or anti-inflammatory regulatory (Treg) phenotype (Figure 1C). Further research is required to clarify the significance of DC cell function in RA pathogenesis and determine how DC function could be regulated through interactions with platelets.
E. Platelet interactions with innate lymphoid cells
Innate lymphoid cells (ILCs) include five subsets of antigen-independent effector and helper cells that mirror the phenotypes of T-cell subsets (182). This group includes natural killer (NK) cells, lymphoid tissue inducer (LTi) cells and three subtypes of innate lymphoid ‘helper-like’ cells [ILC1, ILC2 and ILC3, reviewed in (183)]. ILCs have developmental, tissue-specific, and context-dependent roles in immunity with considerable phenotypic heterogeneity that are still under investigation (183, 184). Thus, the full picture of ILC biology, particularly in the context of RA [recently reviewed in (185)], is incomplete. Generally, the ILC subsets (NK/ILC1, LTi, ILC3 cells) that promote Th1/Th17-like responses, M1-macrophage polarization or recruitment of neutrophils likely exacerbate RA (185). Conversely, ILC2s, which promote Th2/Treg-like immunity, M2-polarization, and recruitment of eosinophils likely attenuate RA severity (185).
The role of platelets in modulating ILC function is poorly understood. ILC2 cell counts are elevated in the circulation and the synovial fluid of RA patients compared to healthy controls although their presence was inversely correlated with disease severity (186). In mouse models of arthritis, ILC2 appear to diminish inflammation and bone destruction (186). Limited data are available regarding platelet interactions with ILC2 cells. Naïve ILC2 cells express PSGL-1 and adhere to platelets in murine lung tissue, presumably via P-selectin on the platelet surface (187, 188). This interaction maintains elevated ILC2 numbers in naïve lung, enhances response to IL-33, amplifies production of Th2-cytokines (IL-5 and IL-13) and, exacerbates lung inflammation in response to Alternaria (fungus) (187, 188). While documented ILC2-platelet adhesion is limited to lung tissue thus far (187), platelet-ILC2 interactions in the synovium could also conceivably attenuate RA. It is tempting to speculate that platelet-derived mediators would recruit and/or activate most ILC subsets, however, considerable further research is required to identify relevant platelet-ILC interactions in the synovium.
Perspectives
Considerable evidence now indicates that activated platelets participate in pro-inflammatory signaling in the synovial tissues, in part through activating other cellular elements of the innate immune system. However, pro-inflammatory molecules in RA also potentiate platelet activity, suggesting a reciprocal relationship between RA and platelet activation. Platelets are therefore well-positioned to occupy a central role in RA pathogenesis as it relates to the innate immune response. Indeed, the concept of platelets as a therapeutic target for RA has been explored (189) although due to the nature of many of the currently available antiplatelet drugs, it may be difficult to exploit the anti-inflammatory effects without conferring increased bleeding risks. Moreover, the sheer complexity of the biological system (Figure 1B) highlights the challenges associated with pinpointing the most clinically significant platelet-centric signaling pathways in RA. Since multiple pro-inflammatory cytokines are expressed in more than one tissue type, future research could employ experimental murine models with platelet-specific conditional knockouts of specific cytokines (and/or their cognate receptors). Such an approach could identify the exact contributions of specific platelet-derived molecules to RA pathogenesis. This information would then be applicable for identifying viable platelet-based therapeutic targets that could reduce disease severity, and mitigate the increased risk of cardiovascular complications resulting from RA.
Author contributions
SJ wrote the first draft of the manuscript. JT, MH, KM and HK designed the concept and layout of the manuscript and/or wrote sections of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a Canadian Institutes of Health Research (CIHR) Operating Grant (MY4-155400) and a Michael Smith Foundation for Health Research (MSFHR) Scholar Award (to HK).
Acknowledgments
The authors thank Angela Tether for editorial assistance. JT and SJ acknowledge support from a Graduate Student Award Program from the UBC Centre for Blood Research. SJ also acknowledges support from the UBC International Tuition Award.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Rudan I, Sidhu S, Papana A, Meng SJ, Xin-Wei Y, Wang W, et al. Prevalence of rheumatoid arthritis in low- and middle-income countries: A systematic review and analysis. J Glob Health (2015) 5(1):010409.
2. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet (2016) 388(10055):2023–38. doi: 10.1016/S0140-6736(16)30173-8
3. Sokka T, Kautiainen H, Pincus T, Verstappen SM, Aggarwal A, Alten R, et al. Work disability remains a major problem in rheumatoid arthritis in the 2000s: Data from 32 countries in the QUEST-RA study. Arthritis Res Ther (2010) 12(2):R42. doi: 10.1186/ar2951
4. Chiu YM, Lu YP, Lan JL, Chen DY, Wang JD. Lifetime risks, life expectancy, and health care expenditures for rheumatoid arthritis: A nationwide cohort followed up from 2003 to 2016. Arthritis Rheumatol (2021) 73(5):750–8. doi: 10.1002/art.41597
5. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med (2011) 365(23):2205–19. doi: 10.1056/NEJMra1004965
6. Kim K, Bang SY, Lee HS, Bae SC. Update on the genetic architecture of rheumatoid arthritis. Nat Rev Rheumatol (2017) 13(1):13–24. doi: 10.1038/nrrheum.2016.176
7. Liu Y, Aryee MJ, Padyukov L, Fallin MD, Hesselberg E, Runarsson A, et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat Biotechnol (2013) 31(2):142–7. doi: 10.1038/nbt.2487
8. Silman AJ, Newman J, MacGregor AJ. Cigarette smoking increases the risk of rheumatoid arthritis. Results from a nationwide study of disease-discordant twins. Arthritis Rheumatol (1996) 39(5):732–5.
9. Wegner N, Wait R, Sroka A, Eick S, Nguyen KA, Lundberg K, et al. Peptidylarginine deiminase from porphyromonas gingivalis citrullinates human fibrinogen and alpha-enolase: implications for autoimmunity in rheumatoid arthritis. Arthritis Rheumatol (2010) 62(9):2662–72. doi: 10.1002/art.27552
10. Scher JU, Littman DR, Abramson SB. Microbiome in inflammatory arthritis and human rheumatic diseases. Arthritis Rheumatol (2016) 68(1):35–45. doi: 10.1002/art.39259
11. Alivernini S, Galeazzi M, Peleg H, Tolusso B, Gremese E, Ferraccioli G, et al. Is ACPA positivity the main driver for rheumatoid arthritis treatment? pros and cons. Autoimmun Rev (2017) 16(11):1096–102. doi: 10.1016/j.autrev.2017.09.002
12. Boilard E, Nigrovic PA, Larabee K, Watts GF, Coblyn JS, Weinblatt ME, et al. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science. (2010) 327(5965):580–3. doi: 10.1126/science.1181928
13. Farr M, Wainwright A, Salmon M, Hollywell CA, Bacon PA. Platelets in the synovial fluid of patients with rheumatoid arthritis. Rheumatol Int (1984) 4(1):13–7. doi: 10.1007/BF00683878
14. Wright HL, Lyon M, Chapman EA, Moots RJ, Edwards SW. Rheumatoid arthritis synovial fluid neutrophils drive inflammation through production of chemokines, reactive oxygen species, and neutrophil extracellular traps. Front Immunol (2020) 11:584116. doi: 10.3389/fimmu.2020.584116
15. Liu W, Zhang Y, Zhu W, Ma C, Ruan J, Long H, et al. Sinomenine inhibits the progression of rheumatoid arthritis by regulating the secretion of inflammatory cytokines and Monocyte/Macrophage subsets. Front Immunol (2018) 9:2228. doi: 10.3389/fimmu.2018.02228
16. Bartok B, Firestein GS. Fibroblast-like synoviocytes: Key effector cells in rheumatoid arthritis. Immunol Rev (2010) 233(1):233–55. doi: 10.1111/j.0105-2896.2009.00859.x
17. Garcia-Vicuna R, Gomez-Gaviro MV, Dominguez-Luis MJ, Pec MK, Gonzalez-Alvaro I, Alvaro-Gracia JM, et al. CC and CXC chemokine receptors mediate migration, proliferation, and matrix metalloproteinase production by fibroblast-like synoviocytes from rheumatoid arthritis patients. Arthritis Rheumatol (2004) 50(12):3866–77. doi: 10.1002/art.20615
18. You S, Koh JH, Leng L, Kim WU, Bucala R. The tumor-like phenotype of rheumatoid synovium: Molecular profiling and prospects for precision medicine. Arthritis Rheumatol (2018) 70(5):637–52. doi: 10.1002/art.40406
19. Clemetson KJ. Platelets and primary haemostasis. Thromb Res (2012) 129(3):220–4. doi: 10.1016/j.thromres.2011.11.036
20. Golebiewska EM, Poole AW. Platelet secretion: From haemostasis to wound healing and beyond. Blood Rev (2015) 29(3):153–62. doi: 10.1016/j.blre.2014.10.003
21. Klinger MH, Wilhelm D, Bubel S, Sticherling M, Schroder JM, Kuhnel W. Immunocytochemical localization of the chemokines RANTES and MIP-1 alpha within human platelets and their release during storage. Int Arch Allergy Immunol (1995) 107(4):541–6. doi: 10.1159/000237097
22. Zhao X, Gu M, Xu X, Wen X, Yang G, Li L, et al. CCL3/CCR1 mediates CD14(+)CD16(-) circulating monocyte recruitment in knee osteoarthritis progression. Osteoarthr Cartilage (2020) 28(5):613–25. doi: 10.1016/j.joca.2020.01.009
23. Kimata H, Yoshida A, Ishioka C, Fujimoto M, Lindley I, Furusho K. RANTES and macrophage inflammatory protein 1 alpha selectively enhance immunoglobulin (IgE) and IgG4 production by human b cells. J Exp Med (1996) 183(5):2397–402. doi: 10.1084/jem.183.5.2397
24. Reichel CA, Rehberg M, Lerchenberger M, Berberich N, Bihari P, Khandoga AG, et al. Ccl2 and Ccl3 mediate neutrophil recruitment via induction of protein synthesis and generation of lipid mediators. Arterioscler Thromb Vasc Biol (2009) 29(11):1787–93. doi: 10.1161/ATVBAHA.109.193268
25. Zhang G, Liu HB, Zhou L, Cui XQ, Fan XH. CCL3 participates in the development of rheumatoid arthritis by activating AKT. Eur Rev Med Pharmacol Sci (2018) 22(20):6625–32.
26. von Hundelshausen P, Weber KS, Huo Y, Proudfoot AE, Nelson PJ, Ley K, et al. RANTES deposition by platelets triggers monocyte arrest on inflamed and atherosclerotic endothelium. Circulation (2001) 103(13):1772–7. doi: 10.1161/01.CIR.103.13.1772
27. Weyrich AS, Elstad MR, McEver RP, McIntyre TM, Moore KL, Morrissey JH, et al. Activated platelets signal chemokine synthesis by human monocytes. J Clin Invest (1996) 97(6):1525–34. doi: 10.1172/JCI118575
28. Machlus KR, Johnson KE, Kulenthirarajan R, Forward JA, Tippy MD, Soussou TS, et al. CCL5 derived from platelets increases megakaryocyte proplatelet formation. Blood (2016) 127(7):921–6. doi: 10.1182/blood-2015-05-644583
29. Gilat D, Hershkoviz R, Mekori YA, Vlodavsky I, Lider O. Regulation of adhesion of CD4+ T lymphocytes to intact or heparinase-treated subendothelial extracellular matrix by diffusible or anchored RANTES and MIP-1 beta. J Immunol (1994) 153(11):4899–906.
30. Nanki T, Nagasaka K, Hayashida K, Saita Y, Miyasaka N. Chemokines regulate IL-6 and IL-8 production by fibroblast-like synoviocytes from patients with rheumatoid arthritis. J Immunol (2001) 167(9):5381–5. doi: 10.4049/jimmunol.167.9.5381
31. Henn V, Slupsky JR, Grafe M, Anagnostopoulos I, Forster R, Muller-Berghaus G, et al. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature (1998) 391(6667):591–4. doi: 10.1038/35393
32. Khan SY, Kelher MR, Heal JM, Blumberg N, Boshkov LK, Phipps R, et al. Soluble CD40 ligand accumulates in stored blood components, primes neutrophils through CD40, and is a potential cofactor in the development of transfusion-related acute lung injury. Blood (2006) 108(7):2455–62. doi: 10.1182/blood-2006-04-017251
33. Czapiga M, Kirk AD, Lekstrom-Himes J. Platelets deliver costimulatory signals to antigen-presenting cells: A potential bridge between injury and immune activation. Exp Hematol (2004) 32(2):135–9. doi: 10.1016/j.exphem.2003.11.004
34. Xu H, Zhang X, Mannon RB, Kirk AD. Platelet-derived or soluble CD154 induces vascularized allograft rejection independent of cell-bound CD154. J Clin Invest (2006) 116(3):769–74. doi: 10.1172/JCI27155
35. Armitage RJ, Fanslow WC, Strockbine L, Sato TA, Clifford KN, Macduff BM, et al. Molecular and biological characterization of a murine ligand for CD40. Nature (1992) 357(6373):80–2. doi: 10.1038/357080a0
36. Cho CS, Cho ML, Min SY, Kim WU, Min DJ, Lee SS, et al. CD40 engagement on synovial fibroblast up-regulates production of vascular endothelial growth factor. J Immunol (2000) 164(10):5055–61. doi: 10.4049/jimmunol.164.10.5055
37. Baltus T, von Hundelshausen P, Mause SF, Buhre W, Rossaint R, Weber C. Differential and additive effects of platelet-derived chemokines on monocyte arrest on inflamed endothelium under flow conditions. J Leukoc Biol (2005) 78(2):435–41. doi: 10.1189/jlb.0305141
38. Scheuerer B, Ernst M, Durrbaum-Landmann I, Fleischer J, Grage-Griebenow E, Brandt E, et al. The CXC-chemokine platelet factor 4 promotes monocyte survival and induces monocyte differentiation into macrophages. Blood (2000) 95(4):1158–66. doi: 10.1182/blood.V95.4.1158.004k31_1158_1166
39. Kasper B, Brandt E, Brandau S, Petersen F. Platelet factor 4 (CXC chemokine ligand 4) differentially regulates respiratory burst, survival, and cytokine expression of human monocytes by using distinct signaling pathways. J Immunol (2007) 179(4):2584–91. doi: 10.4049/jimmunol.179.4.2584
40. Langer HF, Daub K, Braun G, Schonberger T, May AE, Schaller M, et al. Platelets recruit human dendritic cells via mac-1/JAM-C interaction and modulate dendritic cell function in vitro. Arterioscler Thromb Vasc Biol (2007) 27(6):1463–70. doi: 10.1161/ATVBAHA.107.141515
41. Briquet-Laugier V, Lavenu-Bombled C, Schmitt A, Leboeuf M, Uzan G, Dubart-Kupperschmitt A, et al. Probing platelet factor 4 alpha-granule targeting. J Thromb Haemost (2004) 2(12):2231–40. doi: 10.1111/j.1538-7836.2004.01037.x
42. von Hundelshausen P, Koenen RR, Sack M, Mause SF, Adriaens W, Proudfoot AE, et al. Heterophilic interactions of platelet factor 4 and RANTES promote monocyte arrest on endothelium. Blood (2005) 105(3):924–30. doi: 10.1182/blood-2004-06-2475
43. Woller G, Brandt E, Mittelstadt J, Rybakowski C, Petersen F. Platelet factor 4/CXCL4-stimulated human monocytes induce apoptosis in endothelial cells by the release of oxygen radicals. J Leukoc Biol (2008) 83(4):936–45. doi: 10.1189/jlb.0907592
44. Srivastava K, Cockburn IA, Swaim A, Thompson LE, Tripathi A, Fletcher CA, et al. Platelet factor 4 mediates inflammation in experimental cerebral malaria. Cell Host Microbe (2008) 4(2):179–87. doi: 10.1016/j.chom.2008.07.003
45. Aziz KA, Cawley JC, Zuzel M. via released PF4: Mechanism of priming and synergy with GM-CSF. Br J Haematol (1995) 91(4):846–53. doi: 10.1111/j.1365-2141.1995.tb05399.x
46. Schenk BI, Petersen F, Flad HD, Brandt E. Platelet-derived chemokines CXC chemokine ligand (CXCL)7, connective tissue-activating peptide III, and CXCL4 differentially affect and cross-regulate neutrophil adhesion and transendothelial migration. J Immunol (2002) 169(5):2602–10. doi: 10.4049/jimmunol.169.5.2602
47. Yu G, Rux AH, Ma P, Bdeir K, Sachais BS. Endothelial expression of e-selectin is induced by the platelet-specific chemokine platelet factor 4 through LRP in an NF-kappaB-dependent manner. Blood (2005) 105(9):3545–51. doi: 10.1182/blood-2004-07-2617
48. Zhang Y, Ma KL, Gong YX, Wang GH, Hu ZB, Liu L, et al. Platelet microparticles mediate glomerular endothelial injury in early diabetic nephropathy. J Am Soc Nephrol (2018) 29(11):2671–95. doi: 10.1681/ASN.2018040368
49. Walz A, Dewald B, von Tscharner V, Baggiolini M. Effects of the neutrophil-activating peptide NAP-2, platelet basic protein, connective tissue-activating peptide III and platelet factor 4 on human neutrophils. J Exp Med (1989) 170(5):1745–50. doi: 10.1084/jem.170.5.1745
50. Massberg S, Konrad I, Schurzinger K, Lorenz M, Schneider S, Zohlnhoefer D, et al. Platelets secrete stromal cell-derived factor 1alpha and recruit bone marrow-derived progenitor cells to arterial thrombi in vivo. J Exp Med (2006) 203(5):1221–33. doi: 10.1084/jem.20051772
51. Nagasawa T, Kikutani H, Kishimoto T. Molecular cloning and structure of a pre-b-cell growth-stimulating factor. Proc Natl Acad Sci U S A (1994) 91(6):2305–9. doi: 10.1073/pnas.91.6.2305
52. Dunussi-Joannopoulos K, Zuberek K, Runyon K, Hawley RG, Wong A, Erickson J, et al. Efficacious immunomodulatory activity of the chemokine stromal cell-derived factor 1 (SDF-1): Local secretion of SDF-1 at the tumor site serves as T-cell chemoattractant and mediates T-cell-dependent antitumor responses. Blood (2002) 100(5):1551–8. doi: 10.1182/blood.V100.5.1551.h81702001551_1551_1558
53. Chatterjee M, von Ungern-Sternberg SN, Seizer P, Schlegel F, Buttcher M, Sindhu NA, et al. Platelet-derived CXCL12 regulates monocyte function, survival, differentiation into macrophages and foam cells through differential involvement of CXCR4-CXCR7. Cell Death Dis (2015) 6:e1989. doi: 10.1038/cddis.2015.233
54. Lindemann S, Tolley ND, Dixon DA, McIntyre TM, Prescott SM, Zimmerman GA, et al. Activated platelets mediate inflammatory signaling by regulated interleukin 1beta synthesis. J Cell Biol (2001) 154(3):485–90. doi: 10.1083/jcb.200105058
55. Hashizume M, Hayakawa N, Mihara M. IL-6 trans-signalling directly induces RANKL on fibroblast-like synovial cells and is involved in RANKL induction by TNF-alpha and IL-17. Rheumatol (Oxford) (2008) 47(11):1635–40. doi: 10.1093/rheumatology/ken363
56. Bergstrom B, Carlsten H, Ekwall AH. Methotrexate inhibits effects of platelet-derived growth factor and interleukin-1beta on rheumatoid arthritis fibroblast-like synoviocytes. Arthritis Res Ther (2018) 20(1):49.
57. Bustos M, Saadi S, Platt JL. Platelet-mediated activation of endothelial cells: Implications for the pathogenesis of transplant rejection. Transplantation (2001) 72(3):509–15. doi: 10.1097/00007890-200108150-00025
58. Chung AW, Radomski A, Alonso-Escolano D, Jurasz P, Stewart MW, Malinski T, et al. Platelet-leukocyte aggregation induced by PAR agonists: regulation by nitric oxide and matrix metalloproteinases. Br J Pharmacol (2004) 143(7):845–55. doi: 10.1038/sj.bjp.0705997
59. Williams KE, Olsen DR. Matrix metalloproteinase-1 cleavage site recognition and binding in full-length human type III collagen. Matrix Biol (2009) 28(6):373–9. doi: 10.1016/j.matbio.2009.04.009
60. Rastogi A, Kim H, Twomey JD, Hsieh AH. MMP-2 mediates local degradation and remodeling of collagen by annulus fibrosus cells of the intervertebral disc. Arthritis Res Ther (2013) 15(2):R57. doi: 10.1186/ar4224
61. Okada Y, Nagase H, Harris ED Jr.A metalloproteinase from human rheumatoid synovial fibroblasts that digests connective tissue matrix components. purification and characterization. J Biol Chem (1986) 261(30):14245–55.
62. Charbonneau M, Lavoie RR, Lauzier A, Harper K, McDonald PP, Dubois CM. Platelet-derived growth factor receptor activation promotes the prodestructive invadosome-forming phenotype of synoviocytes from patients with rheumatoid arthritis. J Immunol (2016) 196(8):3264–75. doi: 10.4049/jimmunol.1500502
63. Westermark B, Wasteson A. A platelet factor stimulating human normal glial cells. Exp Cell Res (1976) 98(1):170–4. doi: 10.1016/0014-4827(76)90476-6
64. Agrawal S, Ganguly S, Hajian P, Cao JN, Agrawal A. PDGF upregulates CLEC-2 to induce T regulatory cells. Oncotarget (2015) 6(30):28621–32. doi: 10.18632/oncotarget.5765
65. Krettek A, Ostergren-Lunden G, Fager G, Rosmond C, Bondjers G, Lustig F. Expression of PDGF receptors and ligand-induced migration of partially differentiated human monocyte-derived macrophages. influence of IFN-gamma and TGF-beta. Atherosclerosis (2001) 156(2):267–75.
66. Weicht B, Maitz P, Kandler B, Fischer MB, Watzek G, Gruber R. Activated platelets positively regulate RANKL-mediated osteoclast differentiation. J Cell Biochem (2007) 102(5):1300–7. doi: 10.1002/jcb.21360
67. Assoian RK, Komoriya A, Meyers CA, Miller DM, Sporn MB. Transforming growth factor-beta in human platelets. identification of a major storage site, purification, and characterization. J Biol Chem (1983) 258(11):7155–60.
68. Cheifetz S, Weatherbee JA, Tsang ML, Anderson JK, Mole JE, Lucas R, et al. The transforming growth factor-beta system, a complex pattern of cross-reactive ligands and receptors. Cell (1987) 48(3):409–15. doi: 10.1016/0092-8674(87)90192-9
69. Takimoto T, Wakabayashi Y, Sekiya T, Inoue N, Morita R, Ichiyama K, et al. Smad2 and Smad3 are redundantly essential for the TGF-beta-mediated regulation of regulatory T plasticity and Th1 development. J Immunol (2010) 185(2):842–55. doi: 10.4049/jimmunol.0904100
70. Ehrhardt RO, Strober W, Harriman GR. Effect of transforming growth factor (TGF)-beta 1 on IgA isotype expression. TGF-beta 1 induces a small increase in sIgA+ b cells regardless of the method of b cell activation. J Immunol (1992) 148(12):3830–6.
71. Lawler JW, Slayter HS, Coligan JE. Isolation and characterization of a high molecular weight glycoprotein from human blood platelets. J Biol Chem (1978) 253(23):8609–16. doi: 10.1016/S0021-9258(17)34336-3
72. Silverstein RL, Nachman RL. Thrombospondin binds to monocytes-macrophages and mediates platelet-monocyte adhesion. J Clin Invest (1987) 79(3):867–74. doi: 10.1172/JCI112896
73. Jimenez B, Volpert OV, Crawford SE, Febbraio M, Silverstein RL, Bouck N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat Med (2000) 6(1):41–8. doi: 10.1038/71517
74. Taraboletti G, Belotti D, Borsotti P, Vergani V, Rusnati M, Presta M, et al. The 140-kilodalton antiangiogenic fragment of thrombospondin-1 binds to basic fibroblast growth factor. Cell Growth Differ (1997) 8(4):471–9.
75. Mansfield PJ, Boxer LA, Suchard SJ. Thrombospondin stimulates motility of human neutrophils. J Cell Biol (1990) 111(6 Pt 2):3077–86. doi: 10.1083/jcb.111.6.3077
76. Hogg PJ, Owensby DA, Mosher DF, Misenheimer TM, Chesterman CN. Thrombospondin is a tight-binding competitive inhibitor of neutrophil elastase. J Biol Chem (1993) 268(10):7139–46. doi: 10.1016/S0021-9258(18)53157-4
77. Grimbert P, Bouguermouh S, Baba N, Nakajima T, Allakhverdi Z, Braun D, et al. Thrombospondin/CD47 interaction: A pathway to generate regulatory T cells from human CD4+ CD25- T cells in response to inflammation. J Immunol (2006) 177(6):3534–41. doi: 10.4049/jimmunol.177.6.3534
78. Lamy L, Foussat A, Brown EJ, Bornstein P, Ticchioni M, Bernard A. Interactions between CD47 and thrombospondin reduce inflammation. J Immunol (2007) 178(9):5930–9. doi: 10.4049/jimmunol.178.9.5930
79. Doyen V, Rubio M, Braun D, Nakajima T, Abe J, Saito H, et al. Thrombospondin 1 is an autocrine negative regulator of human dendritic cell activation. J Exp Med (2003) 198(8):1277–83. doi: 10.1084/jem.20030705
80. Saban DR, Bock F, Chauhan SK, Masli S, Dana R. Thrombospondin-1 derived from APCs regulates their capacity for allosensitization. J Immunol (2010) 185(8):4691–7. doi: 10.4049/jimmunol.1001133
81. Maloney JP, Silliman CC, Ambruso DR, Wang J, Tuder RM, Voelkel NF. In vitro release of vascular endothelial growth factor during platelet aggregation. American Journal of Physiology-Heart and Circulatory Physiology (1998) 275(3):H1054–61.
82. del Rey MJ, Izquierdo E, Caja S, Usategui A, Santiago B, Galindo M, et al. Human inflammatory synovial fibroblasts induce enhanced myeloid cell recruitment and angiogenesis through a hypoxia-inducible transcription factor 1alpha/vascular endothelial growth factor-mediated pathway in immunodeficient mice. Arthritis Rheumatol (2009) 60(10):2926–34.
83. Matsumoto Y, Tanaka K, Hirata G, Hanada M, Matsuda S, Shuto T, et al. Possible involvement of the vascular endothelial growth factor-Flt-1-focal adhesion kinase pathway in chemotaxis and the cell proliferation of osteoclast precursor cells in arthritic joints. J Immunol (2002) 168(11):5824–31. doi: 10.4049/jimmunol.168.11.5824
84. Kim HR, Kim KW, Kim BM, Cho ML, Lee SH. The effect of vascular endothelial growth factor on osteoclastogenesis in rheumatoid arthritis. PLoS One (2015) 10(4):e0124909. doi: 10.1371/journal.pone.0124909
85. Maynard DM, Heijnen HF, Horne MK, White JG, Gahl WA. Proteomic analysis of platelet alpha-granules using mass spectrometry. J Thromb Haemost (2007) 5(9):1945–55. doi: 10.1111/j.1538-7836.2007.02690.x
86. Chauhan AK, Kisucka J, Brill A, Walsh MT, Scheiflinger F, Wagner DD. ADAMTS13: A new link between thrombosis and inflammation. J Exp Med (2008) 205(9):2065–74. doi: 10.1084/jem.20080130
87. Tremolizzo L, DiFrancesco JC, Rodriguez-Menendez V, Sirtori E, Longoni M, Cassetti A, et al. Human platelets express the synaptic markers VGLUT1 and 2 and release glutamate following aggregation. Neurosci Lett (2006) 404(3):262–5. doi: 10.1016/j.neulet.2006.06.015
88. Sarchielli P, Di Filippo M, Candeliere A, Chiasserini D, Mattioni A, Tenaglia S, et al. Expression of ionotropic glutamate receptor GLUR3 and effects of glutamate on MBP- and MOG-specific lymphocyte activation and chemotactic migration in multiple sclerosis patients. J Neuroimmunol (2007) 188(1-2):146–58. doi: 10.1016/j.jneuroim.2007.05.021
89. Flood S, Parri R, Williams A, Duance V, Mason D. Modulation of interleukin-6 and matrix metalloproteinase 2 expression in human fibroblast-like synoviocytes by functional ionotropic glutamate receptors. Arthritis Rheumatol (2007) 56(8):2523–34. doi: 10.1002/art.22829
90. Masini E, Di Bello MG, Raspanti S, Ndisang JF, Baronti R, Cappugi P, et al. The role of histamine in platelet aggregation by physiological and immunological stimuli. Inflammation Res (1998) 47(5):211–20. doi: 10.1007/s000110050319
91. Zenmyo M, Hiraoka K, Komiya S, Morimatsu M, Sasaguri Y. Histamine-stimulated production of matrix metalloproteinase 1 by human rheumatoid synovial fibroblasts is mediated by histamine H1-receptors. Virchows Arch (1995) 427(4):437–44. doi: 10.1007/BF00199394
92. Asako H, Kurose I, Wolf R, DeFrees S, Zheng ZL, Phillips ML, et al. Role of H1 receptors and p-selectin in histamine-induced leukocyte rolling and adhesion in postcapillary venules. J Clin Invest (1994) 93(4):1508–15. doi: 10.1172/JCI117129
93. Mehta JL, Chen LY, Kone BC, Mehta P, Turner P. Identification of constitutive and inducible forms of nitric oxide synthase in human platelets. J Lab Clin Med (1995) 125(3):370–7.
94. Migita K, Yamasaki S, Kita M, Ida H, Shibatomi K, Kawakami A, et al. Nitric oxide protects cultured rheumatoid synovial cells from fas-induced apoptosis by inhibiting caspase-3. Immunology (2001) 103(3):362–7. doi: 10.1046/j.1365-2567.2001.01252.x
95. Duerschmied D, Suidan GL, Demers M, Herr N, Carbo C, Brill A, et al. Platelet serotonin promotes the recruitment of neutrophils to sites of acute inflammation in mice. Blood (2013) 121(6):1008–15. doi: 10.1182/blood-2012-06-437392
96. Leon-Ponte M, Ahern GP, O'Connell PJ. Serotonin provides an accessory signal to enhance T-cell activation by signaling through the 5-HT7 receptor. Blood (2007) 109(8):3139–46. doi: 10.1182/blood-2006-10-052787
97. Mauler M, Herr N, Schoenichen C, Witsch T, Marchini T, Hardtner C, et al. Platelet serotonin aggravates myocardial Ischemia/Reperfusion injury via neutrophil degranulation. Circulation (2019) 139(7):918–31. doi: 10.1161/CIRCULATIONAHA.118.033942
98. Wang MJ, Huang Y, Huang RY, Chen XM, Zhou YY, Yu WL, et al. Determination of role of thromboxane A2 in rheumatoid arthritis. Discovery Med (2015) 19(102):23–32.
99. Naik MU, Patel P, Derstine R, Turaga R, Chen X, Golla K, et al. Ask1 regulates murine platelet granule secretion, thromboxane A2 generation, and thrombus formation. Blood (2017) 129(9):1197–209. doi: 10.1182/blood-2016-07-729780
100. Ishizuka T, Kawakami M, Hidaka T, Matsuki Y, Takamizawa M, Suzuki K, et al. Stimulation with thromboxane A2 (TXA2) receptor agonist enhances ICAM-1, VCAM-1 or ELAM-1 expression by human vascular endothelial cells. Clin Exp Immunol (1998) 112(3):464–70.
101. Jenne CN, Kubes P. Platelets in inflammation and infection. Platelets (2015) 26(4):286–92. doi: 10.3109/09537104.2015.1010441
102. Chen Y, Yuan Y, Li W. Sorting machineries: How platelet-dense granules differ from alpha-granules. Biosci Rep (2018) 38(5). doi: 10.1042/BSR20180458
103. Blair P, Flaumenhaft R. Platelet alpha-granules: Basic biology and clinical correlates. Blood Rev (2009) 23(4):177–89. doi: 10.1016/j.blre.2009.04.001
104. Smith CW. Release of alpha-granule contents during platelet activation. Platelets (2022) 33(4):491–502. doi: 10.1080/09537104.2021.1913576
105. Larsen E, Celi A, Gilbert GE, Furie BC, Erban JK, Bonfanti R, et al. PADGEM protein: A receptor that mediates the interaction of activated platelets with neutrophils and monocytes. Cell (1989) 59(2):305–12. doi: 10.1016/0092-8674(89)90292-4
106. Savage JS, Williams CM, Konopatskaya O, Hers I, Harper MT, Poole AW. Munc13-4 is critical for thrombosis through regulating release of ADP from platelets. J Thromb Haemost (2013) 11(4):771–5. doi: 10.1111/jth.12138
107. Boilard E, Bellio M. Platelet extracellular vesicles and the secretory interactome join forces in health and disease. Immunol Rev (2022) 312, (1):38–51. doi: 10.1111/imr.13119
108. Giannotta M, Tapete G, Emmi G, Silvestri E, Milla M. Thrombosis in inflammatory bowel diseases: what's the link? Thromb J (2015) 13:14. doi: 10.1186/s12959-015-0044-2
109. Stocker TJ, Ishikawa-Ankerhold H, Massberg S, Schulz C. Small but mighty: Platelets as central effectors of host defense. Thromb Haemost (2017) 117(4):651–61.
110. Antoniak S, Mackman N. Platelets and viruses. Platelets (2021) 32(3):325–30. doi: 10.1080/09537104.2021.1887842
111. Kim H, Conway EM. Platelets and complement cross-talk in early atherogenesis. Front Cardiovasc Med (2019) 6:131. doi: 10.3389/fcvm.2019.00131
112. Hamzeh-Cognasse H, Berthelot P, Tardy B, Pozzetto B, Bourlet T, Laradi S, et al. Platelet toll-like receptors are crucial sensors of infectious danger moieties. Platelets (2018) 29(6):533–40. doi: 10.1080/09537104.2018.1445842
113. Andonegui G, Kerfoot SM, McNagny K, Ebbert KV, Patel KD, Kubes P. Platelets express functional toll-like receptor-4. Blood (2005) 106(7):2417–23. doi: 10.1182/blood-2005-03-0916
114. Edilova MI, Akram A, Abdul-Sater AA. Innate immunity drives pathogenesis of rheumatoid arthritis. BioMed J (2021) 44(2):172–82. doi: 10.1016/j.bj.2020.06.010
115. Mott PJ, Lazarus AH. CD44 antibodies and immune thrombocytopenia in the amelioration of murine inflammatory arthritis. PLoS One (2013) 8(6):e65805. doi: 10.1371/journal.pone.0065805
116. Endresen GK. Investigation of blood platelets in synovial fluid from patients with rheumatoid arthritis. Scand J Rheumatol (1981) 10(3):204–8. doi: 10.3109/03009748109095299
117. Hutchinson RM, Davis P, Jayson MI. Thrombocytosis in rheumatoid arthritis. Ann Rheum Dis (1976) 35(2):138–42. doi: 10.1136/ard.35.2.138
118. Ertenli I, Kiraz S, Arici M, Haznedaroglu IC, Calguneri M, Celik I, et al. P-selectin as a circulating molecular marker in rheumatoid arthritis with thrombocytosis. J Rheumatol (1998) 25(6):1054–8.
119. Rooney T, Scherzer R, Shigenaga JK, Graf J, Imboden JB, Grunfeld C. Levels of plasma fibrinogen are elevated in well-controlled rheumatoid arthritis. Rheumatol (Oxford) (2011) 50(8):1458–65. doi: 10.1093/rheumatology/ker011
120. Mac Mullan PA, Peace AJ, Madigan AM, Tedesco AF, Kenny D, McCarthy GM. Platelet hyper-reactivity in active inflammatory arthritis is unique to the adenosine diphosphate pathway: A novel finding and potential therapeutic target. Rheumatology (2010) 49(2):240–5. doi: 10.1093/rheumatology/kep377
121. Sfikakis PP, Charalambopoulos D, Vaiopoulos G, Mavrikakis M. Circulating p- and l-selectin and T-lymphocyte activation and patients with autoimmune rheumatic diseases. Clin Rheumatol (1999) 18(1):28–32. doi: 10.1007/s100670050047
122. Goules A, Tzioufas AG, Manousakis MN, Kirou KA, Crow MK, Routsias JG. Elevated levels of soluble CD40 ligand (sCD40L) in serum of patients with systemic autoimmune diseases. J Autoimmun (2006) 26(3):165–71. doi: 10.1016/j.jaut.2006.02.002
123. Tamura N, Kobayashi S, Kato K, Bando H, Haruta K, Oyanagi M, et al. Soluble CD154 in rheumatoid arthritis: Elevated plasma levels in cases with vasculitis. J Rheumatol (2001) 28(12):2583–90.
124. Melki I, Allaeys I, Tessandier N, Mailhot B, Cloutier N, Campbell RA, et al. FcgammaRIIA expression accelerates nephritis and increases platelet activation in systemic lupus erythematosus. Blood (2020) 136(25):2933–45. doi: 10.1182/blood.2020004974
125. Morgan BP, Daniels RH, Williams BD. Measurement of terminal complement complexes in rheumatoid arthritis. Clin Exp Immunol (1988) 73(3):473–8.
126. Argnani L, Zanetti A, Carrara G, Silvagni E, Guerrini G, Zambon A, et al. Rheumatoid arthritis and cardiovascular risk: Retrospective matched-cohort analysis based on the RECORD study of the Italian society for rheumatology. Front Med (Lausanne) (2021) 8:745601. doi: 10.3389/fmed.2021.745601
127. Yoshimura M, Fujieda Y, Sugawara M, Kono M, Kato M, Yokota I, et al. Disease activity as a risk factor for venous thromboembolism in rheumatoid arthritis analysed using time-averaged DAS28CRP: A nested case-control study. Rheumatol Int (2022) 42, (11):1939–46. doi: 10.1007/s00296-022-05121-4
128. Rendu F, Brohard-Bohn B. The platelet release reaction: granules' constituents, secretion and functions. Platelets (2001) 12(5):261–73. doi: 10.1080/09537100120068170
129. Kaplan DR, Chao FC, Stiles CD, Antoniades HN, Scher CD. Platelet alpha granules contain a growth factor for fibroblasts. Blood (1979) 53(6):1043–52. doi: 10.1182/blood.V53.6.1043.1043
130. Colciago A, Celotti F, Casati L, Giancola R, Castano SM, Antonini G, et al. In vitro effects of PDGF isoforms (AA, BB, AB and CC) on migration and proliferation of SaOS-2 osteoblasts and on migration of human osteoblasts. Int J BioMed Sci (2009) 5(4):380–9.
131. Wang BH, Lu YH, Wu LF, Lu X, Guo W, Deng FY, et al. Evaluation of plasma cytokine protein array profile: The highlighted PDGF-BB in rheumatoid arthritis. Clin Rheumatol (2020) 39(11):3323–30. doi: 10.1007/s10067-020-05109-0
132. Shibuya H, Yoshitomi H, Murata K, Kobayashi S, Furu M, Ishikawa M, et al. TNFalpha, PDGF, and TGFbeta synergistically induce synovial lining hyperplasia via inducible PI3Kdelta. Mod Rheumatol (2015) 25(1):72–8. doi: 10.3109/14397595.2014.900847
133. Rosengren S, Corr M, Boyle DL. Platelet-derived growth factor and transforming growth factor beta synergistically potentiate inflammatory mediator synthesis by fibroblast-like synoviocytes. Arthritis Res Ther (2010) 12(2):R65. doi: 10.1186/ar2981
134. Grainger DJ, Wakefield L, Bethell HW, Farndale RW, Metcalfe JC. Release and activation of platelet latent TGF-beta in blood clots during dissolution with plasmin. Nat Med (1995) 1(9):932–7. doi: 10.1038/nm0995-932
135. Zhu D, Zhao J, Lou A, Huang Q, OuYang Q, Zhu J, et al. Transforming growth factor beta1 promotes fibroblast-like synoviocytes migration and invasion via TGF-beta1/Smad signaling in rheumatoid arthritis. Mol Cell Biochem (2019) 459(1-2):141–50. doi: 10.1007/s11010-019-03557-0
136. Agere SA, Akhtar N, Watson JM, Ahmed S. RANTES/CCL5 induces collagen degradation by activating MMP-1 and MMP-13 expression in human rheumatoid arthritis synovial fibroblasts. Front Immunol (2017) 8:1341. doi: 10.3389/fimmu.2017.01341
137. Danks L, Komatsu N, Guerrini MM, Sawa S, Armaka M, Kollias G, et al. RANKL expressed on synovial fibroblasts is primarily responsible for bone erosions during joint inflammation. Ann Rheum Dis (2016) 75(6):1187–95. doi: 10.1136/annrheumdis-2014-207137
138. Mause SF, von Hundelshausen P, Zernecke A, Koenen RR, Weber C. Platelet microparticles: a transcellular delivery system for RANTES promoting monocyte recruitment on endothelium. Arterioscler Thromb Vasc Biol (2005) 25(7):1512–8. doi: 10.1161/01.ATV.0000170133.43608.37
139. Sprague DL, Elzey BD, Crist SA, Waldschmidt TJ, Jensen RJ, Ratliff TL. Platelet-mediated modulation of adaptive immunity: Unique delivery of CD154 signal by platelet-derived membrane vesicles. Blood (2008) 111(10):5028–36. doi: 10.1182/blood-2007-06-097410
140. Wang W, Liu J, Yang B, Ma Z, Liu G, Shen W, et al. Modulation of platelet-derived microparticles to adhesion and motility of human rheumatoid arthritis fibroblast-like synoviocytes. PLoS One (2017) 12(7):e0181003. doi: 10.1371/journal.pone.0181003
141. Wang W, Deng Z, Liu G, Yang J, Zhou W, Zhang C, et al. Platelet-derived extracellular vesicles promote the migration and invasion of rheumatoid arthritis fibroblast-like synoviocytes via CXCR2 signaling. Exp Ther Med (2021) 22(4):1120. doi: 10.3892/etm.2021.10554
142. Demoruelle MK, Harrall KK, Ho L, Purmalek MM, Seto NL, Rothfuss HM, et al. Anti-citrullinated protein antibodies are associated with neutrophil extracellular traps in the sputum in relatives of rheumatoid arthritis patients. Arthritis Rheumatol (2017) 69(6):1165–75. doi: 10.1002/art.40066
143. Tanaka D, Kagari T, Doi H, Shimozato T. Essential role of neutrophils in anti-type II collagen antibody and lipopolysaccharide-induced arthritis. Immunology (2006) 119(2):195–202. doi: 10.1111/j.1365-2567.2006.02424.x
144. Assi LK, Wong SH, Ludwig A, Raza K, Gordon C, Salmon M, et al. Tumor necrosis factor alpha activates release of b lymphocyte stimulator by neutrophils infiltrating the rheumatoid joint. Arthritis Rheumatol (2007) 56(6):1776–86. doi: 10.1002/art.22697
145. Chakravarti A, Raquil MA, Tessier P, Poubelle PE. Surface RANKL of toll-like receptor 4-stimulated human neutrophils activates osteoclastic bone resorption. Blood (2009) 114(8):1633–44. doi: 10.1182/blood-2008-09-178301
146. Piccardoni P, Evangelista V, Piccoli A, de Gaetano G, Walz A, Cerletti C. Thrombin-activated human platelets release two NAP-2 variants that stimulate polymorphonuclear leukocytes. Thromb Haemost (1996) 76(5):780–5. doi: 10.1055/s-0038-1650660
147. Del Rey MJ, Fare R, Izquierdo E, Usategui A, Rodriguez-Fernandez JL, Suarez-Fueyo A, et al. Clinicopathological correlations of podoplanin (gp38) expression in rheumatoid synovium and its potential contribution to fibroblast platelet crosstalk. PLoS One (2014) 9(6):e99607.
148. Metzemaekers M, Gouwy M, Proost P. Neutrophil chemoattractant receptors in health and disease: Double-edged swords. Cell Mol Immunol (2020) 17(5):433–50. doi: 10.1038/s41423-020-0412-0
149. Duchez AC, Boudreau LH, Naika GS, Bollinger J, Belleannee C, Cloutier N, et al. Platelet microparticles are internalized in neutrophils via the concerted activity of 12-lipoxygenase and secreted phospholipase A2-IIA. Proc Natl Acad Sci U S A (2015) 112(27):E3564–73.
150. Evangelista V, Manarini S, Sideri R, Rotondo S, Martelli N, Piccoli A, et al. Platelet/polymorphonuclear leukocyte interaction: P-selectin triggers protein-tyrosine phosphorylation-dependent CD11b/CD18 adhesion: role of PSGL-1 as a signaling molecule. Blood. (1999) 93(3):876–85. doi: 10.1182/blood.V93.3.876
151. Ostrovsky L, King AJ, Bond S, Mitchell D, Lorant DE, Zimmerman GA, et al. A juxtacrine mechanism for neutrophil adhesion on platelets involves platelet-activating factor and a selectin-dependent activation process. Blood. (1998) 91(8):3028–36. doi: 10.1182/blood.V91.8.3028.3028_3028_3036
152. Simon DI, Chen Z, Xu H, Li CQ, Dong J, McIntire LV, et al. Platelet glycoprotein ibalpha is a counterreceptor for the leukocyte integrin mac-1 (CD11b/CD18). J Exp Med (2000) 192(2):193–204. doi: 10.1084/jem.192.2.193
153. Wright SD, Weitz JI, Huang AJ, Levin SM, Silverstein SC, Loike JD. Complement receptor type three (CD11b/CD18) of human polymorphonuclear leukocytes recognizes fibrinogen. Proc Natl Acad Sci U S A (1988) 85(20):7734–8. doi: 10.1073/pnas.85.20.7734
154. Durrant TN, van den Bosch MT, Hers I. Integrin alphaIIbbeta3 outside-in signaling. Blood (2017) 130(14):1607–19. doi: 10.1182/blood-2017-03-773614
155. Mutua V, Gershwin LJ. A review of neutrophil extracellular traps (NETs) in disease: Potential anti-NETs therapeutics. Clin Rev Allergy Immunol (2021) 61(2):194–211. doi: 10.1007/s12016-020-08804-7
156. Maugeri N, Campana L, Gavina M, Covino C, De Metrio M, Panciroli C, et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost (2014) 12(12):2074–88. doi: 10.1111/jth.12710
157. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med (2007) 13(4):463–9. doi: 10.1038/nm1565
158. Xie CB, Jane-Wit D, Pober JS. Complement membrane attack complex: New roles, mechanisms of action, and therapeutic targets. Am J Pathol (2020) 190(6):1138–50. doi: 10.1016/j.ajpath.2020.02.006
159. Holers VM, Banda NK. Complement in the initiation and evolution of rheumatoid arthritis. Front Immunol (2018) 9:1057. doi: 10.3389/fimmu.2018.01057
160. Konttinen YT, Ceponis A, Meri S, Vuorikoski A, Kortekangas P, Sorsa T, et al. Complement in acute and chronic arthritides: Assessment of C3c, C9, and protectin (CD59) in synovial membrane. Ann Rheum Dis (1996) 55(12):888–94. doi: 10.1136/ard.55.12.888
161. Reis ES, Chen H, Sfyroera G, Monk PN, Kohl J, Ricklin D, et al. C5a receptor-dependent cell activation by physiological concentrations of desarginated C5a: insights from a novel label-free cellular assay. J Immunol (2012) 189(10):4797–805. doi: 10.4049/jimmunol.1200834
162. Saggu G, Cortes C, Emch HN, Ramirez G, Worth RG, Ferreira VP. Identification of a novel mode of complement activation on stimulated platelets mediated by properdin and C3(H2O). J Immunol (2013) 190(12):6457–67. doi: 10.4049/jimmunol.1300610
163. Del Conde I, Cruz MA, Zhang H, Lopez JA, Afshar-Kharghan V. Platelet activation leads to activation and propagation of the complement system. J Exp Med (2005) 201(6):871–9. doi: 10.1084/jem.20041497
164. Kozarcanin H, Lood C, Munthe-Fog L, Sandholm K, Hamad OA, Bengtsson AA, et al. The lectin complement pathway serine proteases (MASPs) represent a possible crossroad between the coagulation and complement systems in thromboinflammation. J Thromb Haemost (2016) 14(3):531–45. doi: 10.1111/jth.13208
165. Ley K. M1 means kill; M2 means heal. J Immunol (2017) 199(7):2191–3. doi: 10.4049/jimmunol.1701135
166. Mulherin D, Fitzgerald O, Bresnihan B. Synovial tissue macrophage populations and articular damage in rheumatoid arthritis. Arthritis Rheumatol (1996) 39(1):115–24. doi: 10.1002/art.1780390116
167. Misharin AV, Cuda CM, Saber R, Turner JD, Gierut AK, Haines GK 3rd, et al. Nonclassical Ly6C(-) monocytes drive the development of inflammatory arthritis in mice. Cell Rep (2014) 9(2):591–604. doi: 10.1016/j.celrep.2014.09.032
168. Kuijper PH, Gallardo Torres HI, Houben LA, Lammers JW, Zwaginga JJ, Koenderman L. P-selectin and MAC-1 mediate monocyte rolling and adhesion to ECM-bound platelets under flow conditions. J Leukoc Biol (1998) 64(4):467–73. doi: 10.1002/jlb.64.4.467
169. Hamad OA, Ekdahl KN, Nilsson PH, Andersson J, Magotti P, Lambris JD, et al. Complement activation triggered by chondroitin sulfate released by thrombin receptor-activated platelets. J Thromb Haemost (2008) 6(8):1413–21. doi: 10.1111/j.1538-7836.2008.03034.x
170. Martins P, van Gils JM, Mol A, Hordijk PL, Zwaginga JJ. Platelet binding to monocytes increases the adhesive properties of monocytes by up-regulating the expression and functionality of beta1 and beta2 integrins. J Leukoc Biol (2006) 79(3):499–507. doi: 10.1189/jlb.0605318
171. Santoso S, Sachs UJ, Kroll H, Linder M, Ruf A, Preissner KT, et al. The junctional adhesion molecule 3 (JAM-3) on human platelets is a counterreceptor for the leukocyte integrin mac-1. J Exp Med (2002) 196(5):679–91. doi: 10.1084/jem.20020267
172. Wang Y, Sakuma M, Chen Z, Ustinov V, Shi C, Croce K, et al. Leukocyte engagement of platelet glycoprotein ibalpha via the integrin mac-1 is critical for the biological response to vascular injury. Circulation. (2005) 112(19):2993–3000. doi: 10.1161/CIRCULATIONAHA.105.571315
173. Weyrich AS, McIntyre TM, McEver RP, Prescott SM, Zimmerman GA. Monocyte tethering by p-selectin regulates monocyte chemotactic protein-1 and tumor necrosis factor-alpha secretion. signal integration and NF-kappa b translocation. J Clin Invest. (1995) 95(5):2297–303.
174. Gierut A, Perlman H, Pope RM. Innate immunity and rheumatoid arthritis. Rheum Dis Clin North Am (2010) 36(2):271–96. doi: 10.1016/j.rdc.2010.03.004
175. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol (2013) 31:563–604. doi: 10.1146/annurev-immunol-020711-074950
176. Victoratos P, Kollias G. Induction of autoantibody-mediated spontaneous arthritis critically depends on follicular dendritic cells. Immunity (2009) 30(1):130–42. doi: 10.1016/j.immuni.2008.10.019
177. Elzey BD, Tian J, Jensen RJ, Swanson AK, Lees JR, Lentz SR, et al. Platelet-mediated modulation of adaptive immunity. a communication link between innate and adaptive immune compartments. Immunity (2003) 19(1):9–19. doi: 10.1016/S1074-7613(03)00177-8
178. Duffau P, Seneschal J, Nicco C, Richez C, Lazaro E, Douchet I, et al. Platelet CD154 potentiates interferon-alpha secretion by plasmacytoid dendritic cells in systemic lupus erythematosus. Sci Transl Med (2010) 2(47):47ra63.
179. Roelofs MF, Wenink MH, Brentano F, Abdollahi-Roodsaz S, Oppers-Walgreen B, Barrera P, et al. Type I interferons might form the link between toll-like receptor (TLR) 3/7 and TLR4-mediated synovial inflammation in rheumatoid arthritis (RA). Ann Rheum Dis (2009) 68(9):1486–93. doi: 10.1136/ard.2007.086421
180. Rogge L, D'Ambrosio D, Biffi M, Penna G, Minetti LJ, Presky DH, et al. The role of Stat4 in species-specific regulation of Th cell development by type I IFNs. J Immunol (1998) 161(12):6567–74.
181. McRae BL, Semnani RT, Hayes MP, van Seventer GA. Type I IFNs inhibit human dendritic cell IL-12 production and Th1 cell development. J Immunol (1998) 160(9):4298–304.
182. Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells: 10 years on. Cell (2018) 174(5):1054–66. doi: 10.1016/j.cell.2018.07.017
183. Bal SM, Golebski K, Spits H. Plasticity of innate lymphoid cell subsets. Nat Rev Immunol (2020) 20(9):552–65. doi: 10.1038/s41577-020-0282-9
184. Colonna M. Innate lymphoid cells: Diversity, plasticity, and unique functions in immunity. Immunity (2018) 48(6):1104–17. doi: 10.1016/j.immuni.2018.05.013
185. Fang W, Zhang Y, Chen Z. Innate lymphoid cells in inflammatory arthritis. Arthritis Res Ther (2020) 22(1):25. doi: 10.1186/s13075-020-2115-4
186. Omata Y, Frech M, Primbs T, Lucas S, Andreev D, Scholtysek C, et al. Group 2 innate lymphoid cells attenuate inflammatory arthritis and protect from bone destruction in mice. Cell Rep (2018) 24(1):169–80. doi: 10.1016/j.celrep.2018.06.005
187. Orimo K, Tamari M, Takeda T, Kubo T, Ruckert B, Motomura K, et al. Direct platelet adhesion potentiates group 2 innate lymphoid cell functions. Allergy (2022) 77(3):843–55. doi: 10.1111/all.15057
188. Karta MR, Cavagnero K, Miller M, Badrani J, Naji L, Doherty TA, et al. Platelets attach to lung type 2 innate lymphoid cells (ILC2s) expressing p-selectin glycoprotein ligand 1 and influence ILC2 function. J Allergy Clin Immunol (2019) 144(4):1112–5.e8. doi: 10.1016/j.jaci.2019.06.001
Keywords: platelets, rheumatoid arthritis, inflammation, cell signaling, cytokines
Citation: Jiang SZ, To JL, Hughes MR, McNagny KM and Kim H (2022) Platelet signaling at the nexus of innate immunity and rheumatoid arthritis. Front. Immunol. 13:977828. doi: 10.3389/fimmu.2022.977828
Received: 24 June 2022; Accepted: 03 October 2022;
Published: 25 November 2022.
Edited by:
Zhu Chen, University of Science and Technology of China, ChinaReviewed by:
Aline Bozec, University of Erlangen Nuremberg, GermanyCopyright © 2022 Jiang, To, Hughes, McNagny and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hugh Kim, aHVnaGtpbUBkZW50aXN0cnkudWJjLmNh