 Jingfei Yao1
Jingfei Yao1 Yifu Qiu
Yifu Qiu- 1Institute of Molecular Medicine, Beijing Key Laboratory of Cardiometabolic Molecular Medicine, College of Future Technology, Peking University, Beijing, China
- 2Peking-Tsinghua Center for Life Sciences, Peking University, Beijing, China
Adipose tissue macrophage (ATM) has been appreciated for its critical contribution to obesity-associated metabolic diseases in recent years. Here, we discuss the regulation of ATM on both metabolic homeostatsis and dysfunction. In particular, the macrophage polarization and recruitment as well as the crosstalk between ATM and adipocyte in thermogenesis, obesity, insulin resistance and adipose tissue fibrosis have been reviewed. A better understanding of how ATM regulates adipose tissue remodeling may provide novel therapeutic strategies against obesity and associated metabolic diseases.
Introduction
Obesity is an accumulation of adipose tissue resulting from an energy imbalance, which has been linked to numerous comorbid conditions including type 2 diabetes mellitus, nonalcoholic fatty liver disease (NAFLD), atherosclerosis, cancers as well as COVID-19 (1, 2). Adipose tissues, which include brown adipose tissue (BAT) and white adipose tissue (WAT), play critical roles in the maintenance of energy homeostasis. WAT store energy when nutrition is abundant, while BAT dissipate energy for heat production through a mitochondrial uncoupled respiration.
Besides adipocytes, many types of immune cells reside in both BAT and WAT to control adipose tissue homeostasis (3). Among these immune cells, macrophage is the most abundant population, constituting 5%-10% cell numbers of the adipose tissue in the lean state and increasing to 50% or more in the condition of extreme obesity both in humans and in mice (4). Macrophage is derived from embryo or adult bone marrow-derived circulating monocytes, which are essential in the maintenance of tissue homeostasis and play a vital role in different pathologies. Macrophage is a heterogeneous population of immune cells, such as Kupffer cell in liver, alveolar macrophage in lung, microglia in brain among many others. They play tissue-specific functions in homeostatic and immune-related responses shaped by different local microenvironment (5, 6).
Adipose tissue is an energy reservoir which contains lots of lipids and acts as an important endocrine organ by secreting numerous factors. These lipids and factors generate a specific microenvironment that distinguishes adipose tissue from others and distinguishes adipose tissue macrophage (ATM) from macrophage in other tissues. There are two types of activated macrophage in adipose tissue, named M1 macrophage and M2 macrophage. In the adipose tissue of lean mice, most macrophages are M2 activated, which produce anti-inflammatory cytokines including interleukin-10 (IL-10) and TGF-β, contributing to resolution of inflammation and tissue homeostasis. But in obese mice, the adipose tissue recruits many M1 macrophages, which generate proinflammatory cytokines, causing adipose tissue inflammation and metabolic dysfunction (7, 8).
Here, we summarize the latest progresses of the metabolic implications of ATMs. We describe the polarization and recruitment of adipose tissue macrophages, and discuss their functions both in health and metabolic diseases, including thermogenesis, obesity, insulin resistance as well as adipose tissue fibrosis.
Macrophage polarization
M1/M2 polarization of macrophage is a process by which macrophages produce distinct functional phenotypes driven by microenvironmental stimuli in specific conditions. There are two types of macrophage polarization in adipose tissue, M1 and M2 macrophages. M1 macrophages are generated when stimulated with lipopolysaccharide (LPS) or Th1 proinflammatory cytokines such as IFN-γ. Meanwhile, M2 macrophages are induced by Th2 cytokines such as IL-4 and IL-13. M1 macrophages are usually characterized by enhanced phagocytic activity and increased secretion of proinflammatory cytokines (9). Phenotypically, M1 macrophages show enhanced expression of main histocompatibility complex class II (MHC-II), CD68, CD80 and CD86 both in mice and humans (10). These characteristics are mainly promoted by IFN-γ-mediated Janus kinase-signal transducer and activator of transcription (JAK-STAT) signaling or directly by pathogen associated molecular patterns (PAMPs) such as LPS. Thus, M1 macrophages, along with other innate immune cells, provide the first line of defense to fight against infectious pathogens and promotes Th1 immune response. Several pathways have been discoveried to regulate M1 activation. Transcription factor interferon regulatory factor 5 (IRF5) has been reported as a key player in the polarization of both human and mouse macrophages towards a proinflammatory M1-like phenotype by controlling expression of M1 markers, as well as Th1 and Th17 responses (11). STAT1, which is activated by LPS/TLR4 pathway, plays a critical role in M1 polarization (12, 13). Suppressor of cytokine signaling 3 (SOCS3) activates nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway to produce NO, which promotes expression of M1 markers and inhibits IL-10 expression (14). M2 macrophages have been initially identified during helminth infection, which promotes a Th2-polarized response. They are usually characterized by the expression of M2 markers including arginase 1 (ARG1), chitinase 3-like 3 (also known as YM1), FIZZ1 and CD206. Depending on the contexts and the expression of phenotypic markers, M2 macrophages can be subtyped into M2a, M2b, M2c and M2d ones (15, 16). M2a macrophages play a role in the Th2 response during parasite infections. They are typically induced by stimulation of IL-4 and IL-13, which are produced by eosinophils. M2a macrophages are characterized by high surface expression of CD206, ARG1, YM1, FIZZ1 and TGF-β, and they can promote fibrosis and wound healing. M2b macrophages show immune-regulated and anti-inflammatory effects which induced by IL-1 and TLR agonists such as LPS, expressing high levels of TNF superfamily, C-C motif chemokine ligand 1 (CCL1) and IL-10.M2c macrophages are induced in the presence of IL-10, TGF-β and glucocorticoids. They are usually considered as deactivated or anti-inflammatory macrophages, and involved in phagocytosis of apoptotic cells. M2c macrophages secret large amounts of IL-10 and TGF-β, and express multiple markers including CD163, CD206, RAGE and other scavenger receptors. M2d macrophages, also known as tumor-associated macrophages (TAMs), are induced by the TLR antagonists, and they release IL-10, TGF-βand vascular endothelial growth factors (VEGF) to contribute to tumor angiogenesis (17–21) (Figure 1). Transcription factors such as Krueppel-like factor 4 (KLF4), STAT6 and peroxisome proliferator-activated receptor-γ (PPARγ) are all involved in the polarization of M2 macrophages (22–24). Besides, recent studies identified PI3K/AKT signaling as another critical mediator in mouse M2 macrophage polarization, which is independent of the well-established JAK1/STAT6 pathway (25). IL-4 stimulates the phosphorylation of IRS-2 that leads to the recruitment and activation of PI3K/AKT pathway (26). Interestingly, different AKT isoforms seem to play different roles in macrophage polarization, with Akt1 isoform deficiency leading to an M1 activation while Akt2 isoform ablation causing an M2 phenotype (27).
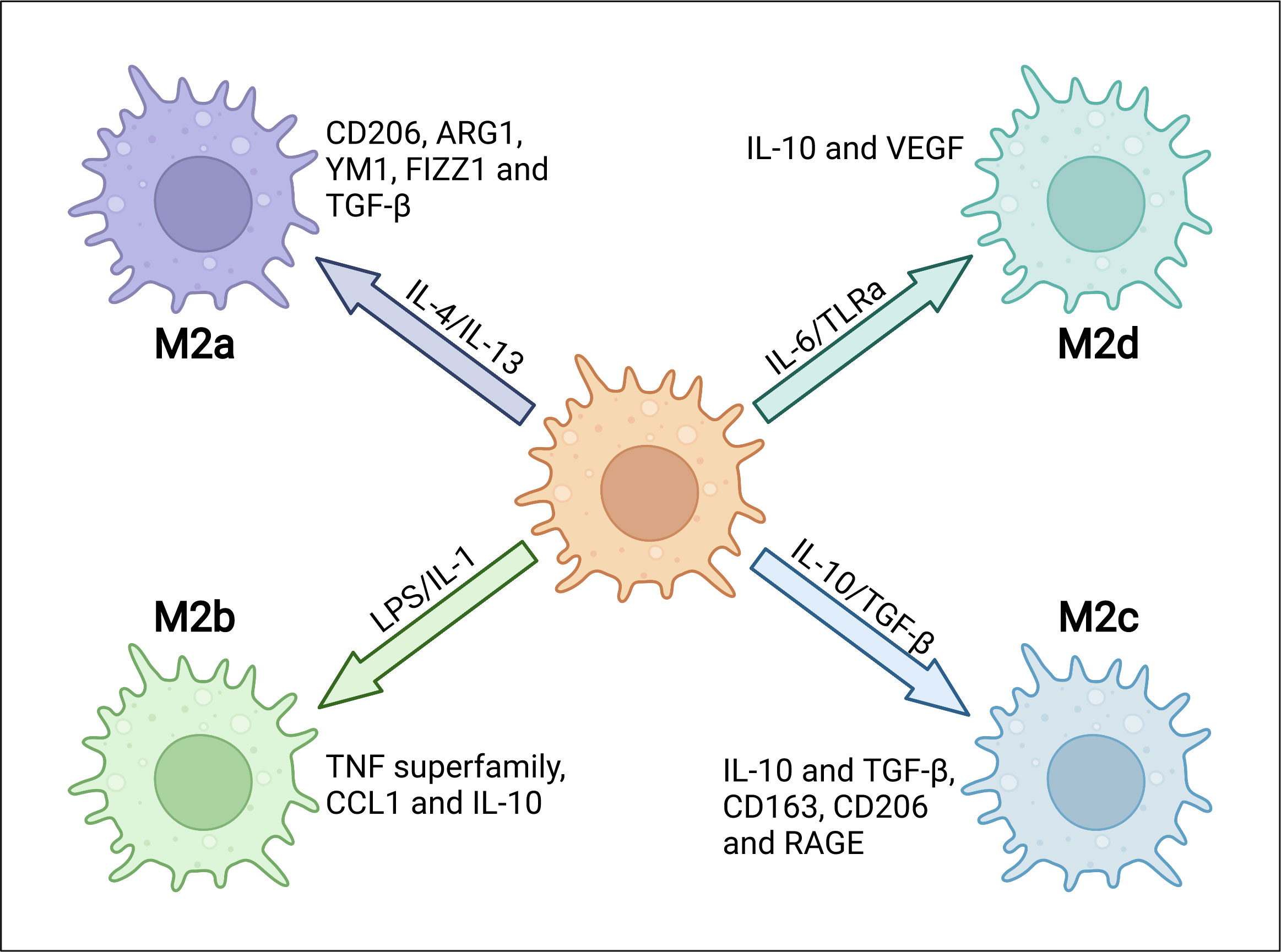
Figure 1 The heterogeneity and characterizations of M2 macrophages. M2 macrophages can be subgrouped into M2a, M2b, M2c and M2d depending on different microenvironmental stimuli. Specific stimuli include, but are not limited to, IL-4 or IL-13 for M2a, LPS or IL-1 receptor ligands for M2b, IL-10 or TGF-β for M2c and IL-6 or Toll-like receptor agonists (TLRa) for M2d. Different subtypes express distinct markers, including intracellular proteins and secreted cytokines.
In obese adipose tissue, ATMs tend to polarize to M1 macrophages, which are mainly regulated by adipocytes. Adipocytes exert effects on ATM phenotypes via a variety of mechanisms. Obese adipocytes secrete many proinflammatory cytokines, including monocyte chemoattractant protein 1 (MCP-1/CCL2), which recruit macrophages and induce their polarization to proinflammatory M1 type. Besides cytokines, ATM polarization is affected by lipids and glucose which are much more abundant in the obese condition. Macrophages treated by very low-density lipoproteins (VLDLs) and short chain fatty acids increase a secretion of proinflammatory cytokines (28). Free fatty acids (FFAs), which is increased in the serum of obese animals, can induce TLR4 signaling activation in murine ATMs and polarize ATM to M1 (29–31). High levels of glucose directly promotes macrophage M1 activation via the Rho-associated protein kinase (ROCK)/c-Jun amino-terminal kinase (JNK) and ROCK/extracellular signal-regulated kinase (ERK) pathways (32, 33). Besides, miR-155, which is secreted by adipocyte-derived microvesicles (ADM), regulates M1 macrophage polarization (34). Moreover, DPP4, a dipeptidyl protease expressed and released by hapatocytes, can activate ERK1/2 and NF-κB signaling to induce MCP-1 and IL-6 expression in ATMs that promotes adipose tissue inflammation (35).
Lean adipocytes normally secrete adiponectin that stimulates M2 ATM polarization (36). PPAR-δ, a nuclear hormone receptor, plays an important role in the activation of M2 macrophage, and alleviates diet-induced insulin resistance (37). PPAR-δ is induced by cellular lipids when apoptotic cells are engulfed by macrophages and further regulates the clearance of these apoptotic cells (38). Helminth infection significantly promotes Th2 responses and M2 macrophage polarization, which alleviate obesity. Adoptive transfer of M2 macrophages treated by helminth to recipient mice significantly improve high-fat diet (HFD)-induced obesity (39). Furthermore, PPAR-γ has been reported to polarize human monocytes to M2 macrophages in vitro (40), while deletion of PPAR-γ in myeloid cells inhibits M2 macrophage activation and accelerates diet-induced obesity and insulin resistance in mice (41).
A number of regulators govern the polarization of M1 or M2 macrophage and the switch between M1 and M2 activation. TLR4 is the key component in LPS-mediated M1 polarization, and TLR4 deficiency inhibits HFD-induced recruitment of proinflammatory M1 macrophages and induces M2 macrophage polarization (42). 11β-HSD1, a reductase reactivating glucocorticoids, was reported to promote the switch from M2 to M1 macrophages in human obesity (43). In addition, inositol-requiring enzyme 1α (IRE1α) wasalso reported to be a key factor controlling ATM polarization and energy balance in mice. Deficiency of IRE1α promotes M2 macrophage polarization, and transcriptomic profiling revealed that expression of IRF4 and KLF4 can be inhibited by IRE1α, both of which are critical players controlling M2 polarization (44). MicroRNA has rencently been implicated in the regulation of macrophage M1/M2 polarization as well as insulin resistance. miR-495 promotes M1 macrophage activation by targeting and inhibiting the expression of Fto (45). However, another study reported that Fto silencing significantly suppressed both M1 and M2 polarization, through inhibiting the expression of STAT1 and of STAT6 and PPAR-γ respectively (46). FTO gene has been considered as the strongest genetic effector in human polygenic obesity, in which IRX3 may participate by mediating this effect. Recently, using cell-specific knockout mouse models, we demonstrated that macrophage IRX3 regulates body weight through acting as a transcriptional factor to control the expression of proinflammatory cytokines (47). Mechanistically, we found that IRX3 promotes M1 but not M2 gene expression when it is phosphorylated and activated by JNK1/2 in macrophages (47).
ATM recruitment
Healthy adipose tissue predominantly contains anti-inflammatory M2 macrophages that originate from yolk sac, with a little contribution of circulating monocytes (48). An extreme increase in adipocyte size is accompanied by an inadequate supply of oxygen due to expanding adipose tissue, causing an increased frequency of adipocyte death and following macrophage recruitment (49, 50). Over 90% of macrophages recruited to adipose tissue are arranged around dead adipocytes, which form a structure called “crown-like structure (CLS)”, both in obese animals and humans (49, 51). These recruited macrophages exert their phagocytotic function to clear dead adipocytes. Deficiency of mannose-binding lectin (MBL), which can bind apoptotic cells and promote engulfment by phagocytes, inhibits the clearance of apoptotic cells in adipose tissue (52). Meanwhile, macrophages in CLS store and buffer excess lipids released from dead adipocytes, which are named lipid-laden macrophages (53). The number of CLS is highly positively correlated with adipose tissue inflammation and metabolic disorders of obese subjects (54, 55). Proinflammatory adipokines including MCP-1 and TNF, as well as saturated fatty acids secreted by obese adipocytes, can recruit and activate ATMs (56). Activated macrophages release proinflammatory chemokines including MCP-1 to recruit more monocytes from blood into adipose tissues by binding to its receptor C-C chemokine receptor type 2 (CCR2) (37). After infiltrating into the adipose tissue, monocytes differentiate to macrophages and interact with adipocytes in a paracrine manner, further increasing the secretion of proinflammatory cytokines (57). This interaction between adipocytes and macrophages establishes a vicious spiral in obese adipose tissue and persistently recruits more and more macrophages from circulation (55). Besides, obesity promotes the expression of chemokine receptors in adipose tissues from both mice and humans, which further enhance the vicious spiral (58, 59).
Many cytokines and their receptors participate in the recruitment of monocytes/macrophages. As previously mentioned, MCP-1-CCR2 is reported as the most important cascade in macrophage recruitment. MCP-1 in adipocytes promotes ATM recruitment and insulin resistance in mice (57), while HFD-induced macrophage accumulation in adipose tissue was extensively reduced in MCP-1 KO mice (60), which indicates a critical role of MCP-1 in the trafficking of macrophages. In addition to adipose tissue, the MCP-1-CCR2 circuit plays an important role in recruiting monocyte in many other tissues, such as in liver, heart and lung (61–64). Moreover, MCP-1 has been reported to induce local proliferation of macrophages, which is another important mechanism underlying obesity-elicited macrophage accumulation (65). Besides MCP-1, CCR2 can also be activated by other ligands, including CCL7 (MCP-2) (66), CCL8 (MCP-3) (67), CCL13 (MCP-4) (68), and CCL12 (MCP-5) (69), many of which are expressed in obese adipose tissue and affect monocyte/macrophage recruitment (58). On the other hand, CCR5 expression is highly upregulated in obesity and FACS analysis further illustrated that WAT from obese mice have significant accumulation of CCR5 positive macrophages. Consistently, CCR5 deficient improves obesity-induced insulin resistance in mice (70). CCL3 and CCL5 have been reported as ligands of CCR5 (71). Inhibition of CCL3 reduces macrophage infiltration and activation by downregulating CCR5 (72). CCL5 recruits macrophages mainly by promoting cell adhesion and transmigration of monocyte vascular endothelial cells (73). CX3CL1-CX3CR1 axis also precipitates in macrophage infiltration and inflammation in both atherosclerosis and rheumatologic disorders (74, 75). It has been suggested that adipocytes express CX3CL1 that can activate the CX3CR1 signaling in macrophages (76).Cx3cr1-deficient mice fed HFD displayed significantly declined monocytes and produced less proinflammatory cytokines in the WAT (77). However, another study reported that Cx3cr1-dificient mice showed a reduction of M2-polarized macrophage migration, and exacerbated adipose tissue inflammation, insulin resistance and hepatic steatosis when fed HFD (78). Serum amyloid A (SAA) promotes monocyte recruitment by inducing the expression of the adhesion antigens CD11b, intracellular adhesion molecule-1 (ICAM-1) and vascular adhesion molecule-1(VCAM-1) through a NF-κB-dependent signaling (79, 80). Myeloid cell-specific ablation of GPR105, which is activated by UDP and UDP-linked glucose, prevents macrophage recruitment to liver or adipose tissue in mice fed HFD (81). C-X-C motif chemokine ligand 14 (CXCL14), which is required for the activation of dendritic cells, is another chemoattractant participates in the recruitment of macrophages into adipose tissue and insulin resistance, although its receptor has not yet been identified (82, 83). Using knockout mouse model, CXCL14 has recently been reported to be produced by brown adipocytes upon thermogenic activation and promotes the recruitment and activation of M2 macrophages in BAT (84).
Collectively, there are several steps in the recruitment of ATMs. Initially, Obesity-induced adipocyte death and adipose tissue inflammation promote a secretion of CCL2 and other chemokines, which bind to their receptors on monocytes circulating in the blood. Then, activated monocytes adhere to endothelial cells of blood vessel via upregulated adhesion molecules including ICAM-1, VCAM-1 and integrin. After integrin-dependent lateral migration, monocytes transmigrate from blood vessel to target adipose tissue. Eventually, recruited monocytes differentiate into proinflammatory macrophages in response to local microenvironmental stimuli (Figure 2) (85).
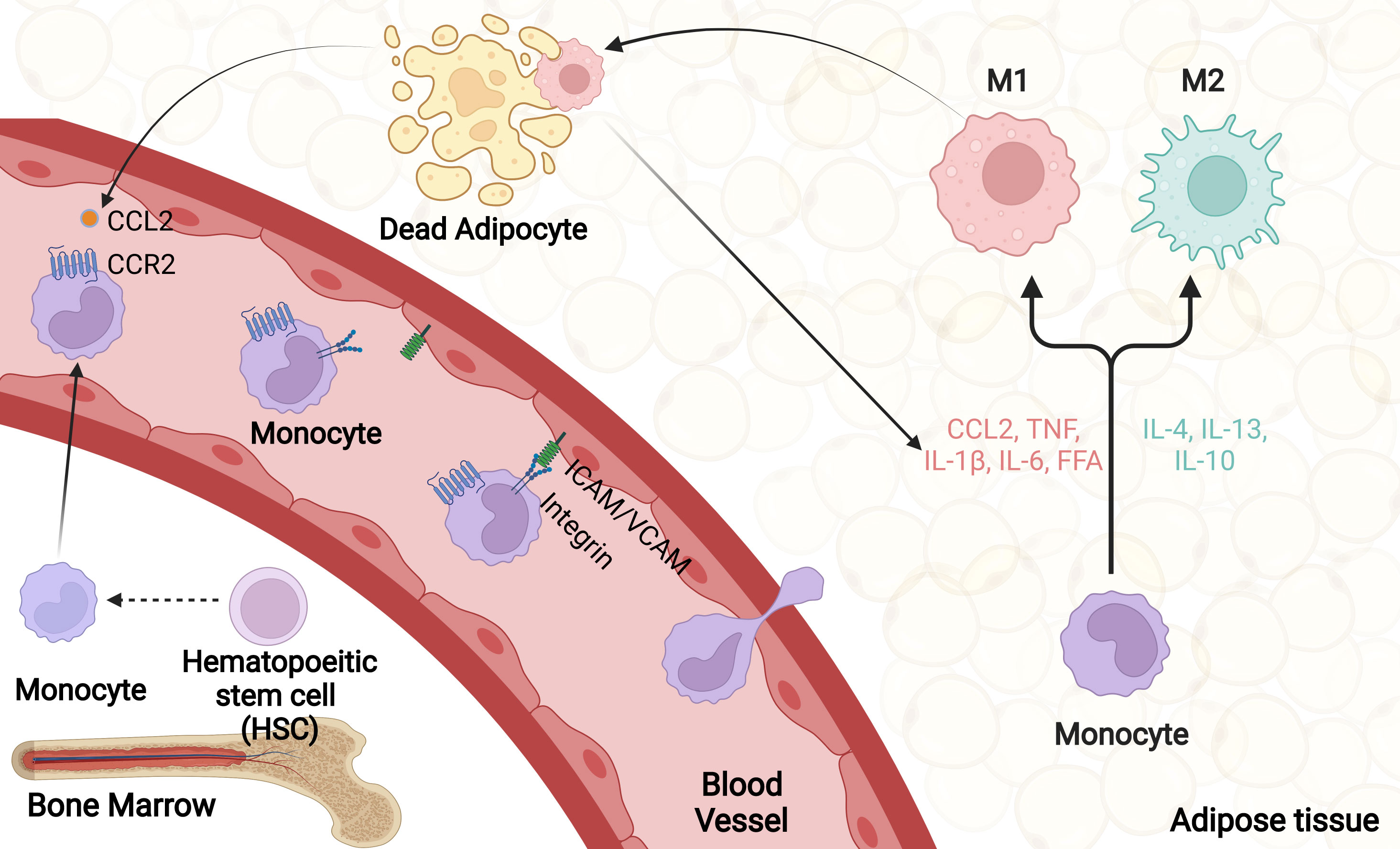
Figure 2 ATM recruitment. Obesity promotes adipocyte death as well as adipose tissue inflammation, which firstly trigger a large number of chemokines secretion, including CCL2. Upon activation, monocytes and vascular endothelial cells produce various cellular adhesion molecules, mainly integrin, ICAM-1 and VCAM-1. Through rolling and adhesion process, monocytes bind to adhesion molecules on vascular endothelial cells and transmigrate from blood vessel to target adipose tissue. Eventually, recruited monocytes differentiate into proinflammatory macrophages in response to local microenvironmental stimuli.
ATM in adaptive thermogenesis and lipolysis
Brown adipocyte and beige adipocyte, which highly express mitochondrial uncoupling protein 1 (UCP1), are responsible for adaptive thermogenesis and protect against metabolic diseases in mice and humans. FFAs, which are produced as a result of lipolysis, serve both as direct activators of UCP1 and fuel sources for thermogenesis (86). Thermogenesis and lipolysis of adipose tissues and browning of WAT are dynamic processes, in which both M1 and M2 macrophages play critical roles.
M2 macrophages in adaptive thermogenesis and lipolysis
M2 macrophage has been demonstrated as an activator to promote fat thermogenesis and lipolysis through different mechanisms. In 2011, cold-induced BAT thermogenesis and WAT lipolysis were first linked to macrophage M2 recruitment and activation (87). Using myeloid cell-specific ILR4α-deficient and IL4 administration mouse models, M2 macrophage has been revealed to be required and sufficient for BAT and beige fat thermogenesis and lipolysis (87, 88). Mechanistically, cold exposure induces the expression of tyrosine hydroxylase (TH) and resultant catecholamine production in M2 macrophages to sustain thermogenesis and lipolysis, although whether M2 macrophages expresse a significant amount of TH is under debate (87–90). Another study reported a similar recruitment and pro-thermogenic effects of M2 macrophage in cold-induced browning of subcutaneous white adipose tissue (scWAT) in mice, further supporting a critical role of M2 macrophage in adaptive thermogenesis (88, 90). Moreover, CD44+ M2 macrophage is recruited by CL316.243 (CL)-mediated adipocyte death to produce high level of 9-hydroxyoctadecadienoic acid (9-HODE) and 13-HODE, two known PPARγ ligands, which promote differentiation of platelet-derived growth factor receptor alpha (PDGFRα+) progenitors to beige adipocytes (91). Besides, macrophage-derived osteopontin (OPN) triggers a recruitment of PDGFRα+ progenitors, which contribute to beige adipogenesis (92). In addition to the above paracrine manner, M2 macrophage has also been reported to promote beige adipogenesis in a direct-contact manner both in humans and mice (93). More recently, it has been reported that brown adipocyte ejectes damaged mitochondria via extracellular vesicles, whose removal by M2 macrophage ensures optimal BAT thermogenesis in mice (94). Started from the M2 macrophage, multiple studies have explored the importance of other anti-inflammatory cytokines in the regulation of adipose thermogenesis and lipolysis.IL-4, IL-13 and IL-33, as key members in type 2 cytokines, all have been demonstrated to promote thermogenesis and lipolysis (88, 95). Different from other type 2 cytokines, ablation of IL-10 elicits thermogenesis and browning of scWAT and protects against diet-induced obesity. ATAC-seq, ChIP-seq, and RNA-seq analyses revealed that IL-10 affects chromatin structure and CCAAT/enhancer binding protein-β (C/EBPβ) and activating transcription factor 2 (ATF2) occupancy at the promoters of thermogenic genes (96).
M1 macrophages in adaptive thermogenesis and lipolysis
In contrast to M2 macrophage, M1 macrophage and its secreted proinflammatory cytokines usually exert negative effects on thermogenesis and lipolysis. Prolonged treatment with TNF cytokine inhibits the sensitivity of adipocytes to β-adrenergic stimulation, and thus inhibits Ucp1 gene expression, thermogenesis and lipolysis (97, 98). TNF-activated inhibitor of nuclear factor kappa-B kinase subunit epsilon (IKKϵ) and TANK-binding kinase 1 (TBK1) desensitize lipolytic signaling by phosphorylating and activating phosphodiesterase 3B (PDE3B), which decreases cAMP levels (98, 99). Overexpression of IKKϵ blunts β-adrenoreceptor-stimulated Ucp1 expression in adipocytes, while an inhibitor of IKKϵ and TBK1 restores catecholamine sensitivity and reversed the effects of HFD feeding on thermogenesis and weight gain (98, 100). IL-1β inhibits β-adrenoreceptor-stimulated Ucp1 expression which was significantly abrogated by the inhibition of ERK (101, 102). Genetic ablation of Jnk1, a major intracellular mediator of inflammatory signaling, enhances Ucp1 expression and thermogenesis in adipose tissues (103). Through singly or in combination treatments of beige adipocytes in vitro with different proinflammatory cytokines, we found that TNF and IL-1β moderately inhibited adrenergic signaling separately, while a mixture of four cytokines (IL-1α, IL-1β, IL-6 and TNF) achieved a dramatic inhibition of thermogenesis and lipolysis (47). Besides proinflammatory cytokines, adipocytes express TLRs and key components of their downstream signaling pathway. LPS or palmitic acid-stimulated TLR4 signaling abolished cAMP-induced upregulation of Ucp1 and thermogenesis through activating NF-κB and MAPK pathways (104, 105). Consistently, TLR3 and TLR4, upstream of interferon regulatory factor 3 (IRF3) signalling, induce insulin resistance and thermogenesis in adipocytes (106). IRF3-deficient mice exhibit systemic inflammation and enhanced browning of scWAT when fed HFD (107). Inflammation-imposed inhibition of beige adipogenesis and thermogenesis are also mediated by a direct adhesion of inflammatory macrophages to adipocytes. Specifically, α4 integrin-mediated adhesion of inflammatory M1 macrophages to VCAM-1, which is expressed by adipocytes, inhibits thermogenesis in an ERK-dependent manner. Genetic or pharmacologic inhibition of α4 integrin resulted in an increase of beige adipogenesis and UCP1 expression of the scWAT (93).
ATM also regulates adipocyte thermogenesis and lipolysis indirectly through other cells like sympathetic nerves. Fasting and cold exposure increase the release of catecholamines from sympathetic nerves, which bind to adipocyte adrenergic receptors and activate cAMP-PKA signaling to trigger lipolysis and thermogenesis. Yochai Wolf et al. reported a homeostatic role of macrophages in the control of brown adipose tissue innervation in mice (108). They found that BAT resident CX3CR1+ macrophages inhibit sympathetic innervation and decrease the local level of catecholamine. Mecp2 deficiency in CX3CR1+ macrophages decreased thermogenesis and led to spontaneous obesity (108). Two recent reports identified a subtype of macrophages in human and mouse WAT that take up and degrade norepinephrine (NE), then inhibit adipocyte lipolysis and thermogenesis. These macrophages are termed either sympathetic neuron associated macrophages (SAMs) or nerve-associated macrophages (NAMs). Different from CX3CR1+ macrophage that inhibits sympathetic neuronal innervation in BAT, SAMs/NAMs function in eWAT and scWAT (109, 110). Mechanistically, NE-degrading macrophages are activated via NLR family pyrin domain containing 3 (NLRP3) inflammasome system in aged WAT. NLRP3 activation upregulates the expression of growth differentiation factor-3 (GDF3) and GDF3-dependent expression of monoamine oxidase A (MAOA) that degrade NE. Macrophages that lack NRLP3 or GDF3 decreased adipose NE removal and increased lipolysis upon aging. The MAOA inhibitor treatment of aged mice restored fasting-induced lipolysis and increased expression of UCP1 (110). Moreover, activin receptor-like kinase 7 (ALK7) signaling, which is activated by GDF3, contributes to diet-induced catecholamine resistance in adipose tissue. Fat-specific Alk7 knock-out enhances adipose β-adrenergic signaling, lipolysis and thermogenesis, resulting in reduced fat mass and resistance to HFD-induced obesity (111). SAMs in mouse WAT can specifically express the NE transporter SLC6A2. Genetic ablation of Slc6a2 in SAMs increases thermogenesis and weight loss in obese mice (109). CX3CR1+ macrophages produce IL-27 to activate p38 MAPK-PGC-1α pathway in adipocytes and promote thermogenesis of BAT and scWAT (112). However, it is unknown whether these SAMs/NAMs can be categorized as M1 or M2.
ATM in insulin resistance and diabetes
Type 2 diabetes is associated with obesity and occurs as a consequence of insulin resistance, which emerges when three major insulin-sensitive tissues (skeletal muscle, liver, and adipose tissue) can not respond well to insulin and can not effectively take up glucose from blood. Insulin is a peptide hormone secreted by pancreatic β cells, which plays a crucial role in carbohydrate metabolism, lipid anabolic regulation, cell growth and proliferation (113). Blood glucose induces β-cells to produce and secrete insulin, which stimulates glucose uptake in different types of cells, including adipocyte, muscle cell, liver cell and others, thereby decreasing blood glucose level. The effects of insulin on whole-body metabolism result from its binding to insulin receptor (IR), leading to autophosphorylation of specific tyrosine residues of IR and subsequently phosphorylation of proteins known as insulin receptor substrates (IRS). PI3K, a key component of IRS downstream, mediates insulin signaling mainly by activating PKB/AKT and PKC signaling pathways (114).
TNF
Adipose tissue macrophage modulates insulin action through different mechanisms, with M1 macrophage promoting insulin resistance while M2 macrophage enhancing insulin sensitivity (115, 116). The major difference between M1 and M2 is the expression of proinflammatory cytokines. In 1990s, the first study reported the inflammatory origin of obesity and diabetes. They found that adipose tissue from different obese rodents and humans has an enhanced secretion of proinflammatory cytokines, mainly TNF, which was linked to insulin resistance (117, 118). TNF‐deficient obese mice are protected from obesity‐induced insulin resistance in muscle and adipose tissues (119). The important role of TNF is further evidenced by TNF neutralization, which improves an increased peripheral glucose uptake and insulin sensitivity in obese mice (117). These studies showed that blocking a single cytokine can restore insulin sensitivity, and macrophage was further identified as the major cell source of TNF and other proinflammatory molecules in obesity (4). Binding of TNF to its receptors results in the activation of JNK and causes phosphorylation of IRS1 at serine 307, which impairs IR-mediated tyrosine phosphorylation of IRS1 and downstream signaling (120).
IL-1β
IL-1β is another important proinflammatory cytokine that is produced by ATM (121). IL-1β exerts its biological effects by binding directly to IL-1Rα and activates the IKK/NF-κB pathway (122). It was reported that adipose tissue appears to be a major source of IL-1R antagonist production, which prompted an interest in the role of IL-1β in obesity-induced diabetes and insulin resistance (123). IL-1β, released by ATM, alters insulin sensitivity of adipose tissue by inhibiting insulin signaling, so it decreases insulin-stimulated glucose uptake and lipogenesis in both murine and human adipocytes (124, 125). In vitro studies revealed that IL-1β treatment of adipocytes disturbs insulin signaling via downregulation of IRS1 expression, leading to a reduction of translocation of insulin-stimulated glucose transporter type 4 (GLUT-4), an essential process for glucose uptake (124, 125). Consistently, insulin resistance of human adipocytes imposed by macrophage-derived conditioned medium can be reversed by neutralizing IL-1β (124).
IL-6
Many studies have established a positive correlation between IL‐6 and insulin resistance (126). Adipose tissue‐derived IL‐6 enters circulation and exerts systemic regulation on insulin action. Up to 35% of systemic IL‐6 originates from adipose tissue under basal condition, secreted by both adipocytes and macrophages (4, 127). IL-6 has been described to impair insulin signaling, primarily through inhibiting insulin-stimulated tyrosine phosphorylation of IRS in adipose tissue (128). Modest increase of basal glucose transporter GLUT1 was observed in 3T3‐L1 adipocytes when incubated with IL‐6 (129), while the expression of Glut4 and Irs1 genes was inhibited by chronic IL‐6 treatment (128, 130). Besides, IL‐6 induces the expression of SOCS3, which is a negative regulator of insulin signaling in adipocytes (131).
NF-κB
ATM-released proinflammatory cytokines activate different signaling pathways in adipocytes to modulate insulin action. NF-κB is a master inflammatory transcriptional factor involved in a variety of physiological and pathological processes such as inflammation and innate and adaptive immune responses. The activation of NF-κB signaling can increase the expression of several proinflammatory genes, which exacerbate insulin resistance progression (132, 133). IKKβ specific deficiency in adipocytes completely prevents FFA-induced IL-6 and TNF expression, and improves glucose tolerance and insulin sensitivity (134, 135). Mechanistically, IKKβ activation promotes IRS1 serine phosphorylation through activation of the TSC1/TSC2/mTORC1/S6 kinase-1 pathway, which impairs IR-mediated tyrosine phosphorylation of IRS1 (136). In addition, activation of the IKKβ/NF-κB pathway increases the expression of protein-tyrosine phosphatase 1B (PTP1B), a tyrosine phosphatase that catalyzes dephosphorylation of tyrosine residues of IRS1, further inhibiting insulin signaling in adipose tissue (137).
JNK
JNK might be the most investigated stress kinase in obesity-related insulin resistance. The activity of JNK in increased upon exposure to inflammatory stimuli which include cytokines, FFAs, and then phosphorylates transcription factor activator protein-1 (AP-1) (120). Like IKKβ, JNK inhibits insulin signaling through an inhibitory serine-threonine phosphorylation of IRS1, thereby decreases PI3K/AKT signaling (138, 139). It should be noticed that JNK seems more implicated in the direct regulation of IRS serine phosphorylation than IKKβ (140). JNK activity could be induced in adipose tissue of obese mice compared to lean mice. Adipose tissue-specific JNK1-deficient mice are protected against the development of insulin resistance under HFD feeding. Interestingly, this protective effect is not systemic as JNK1 deficiency in adipocytes only restore liver but not muscle insulin sensitivity (141). Besides JNK1, JNK2 isoform is also involved in insulin resistance but to a lesser extent (142). Moreover, using a myeloid cell-specific JNK1/2 double knock-out mouse model, another study demonstrated that macrophage JNK1/2 are required for the establishment of obesity-induced adipose tisse inflammation and insulin resistance through promoting macrophage M1 activation (143).
ERK1/2
ERK1/2 is activated in adipose tissue of obese mice or human (144). Multiple cellular studies have reported that activated ERK1/2 in diabetes induces IRS1 serine phosphorylation, which inhibits IRS1 tyrosine phosphorylation. In addition this serine phosphorylation decreases the interaction between IRS1 and PI3K and inhibits the association between IRS1 and insulin receptor, further diminishing the metabolic effects of insulin (140). ERK1-deficient mice are protected against diet-induced obesity and insulin resistance by inhibiting adipogenesis and promoting energy expenditure (145). Besides, ERK activation promotes insulin resistance indirectly, mainlythrough a stimulation of adipocyte lipolysis and FFA release, and mice deficient in the signaling adapter p62 (an ERK inhibitor) show similar phenotypes (Figure 3) (146, 147).
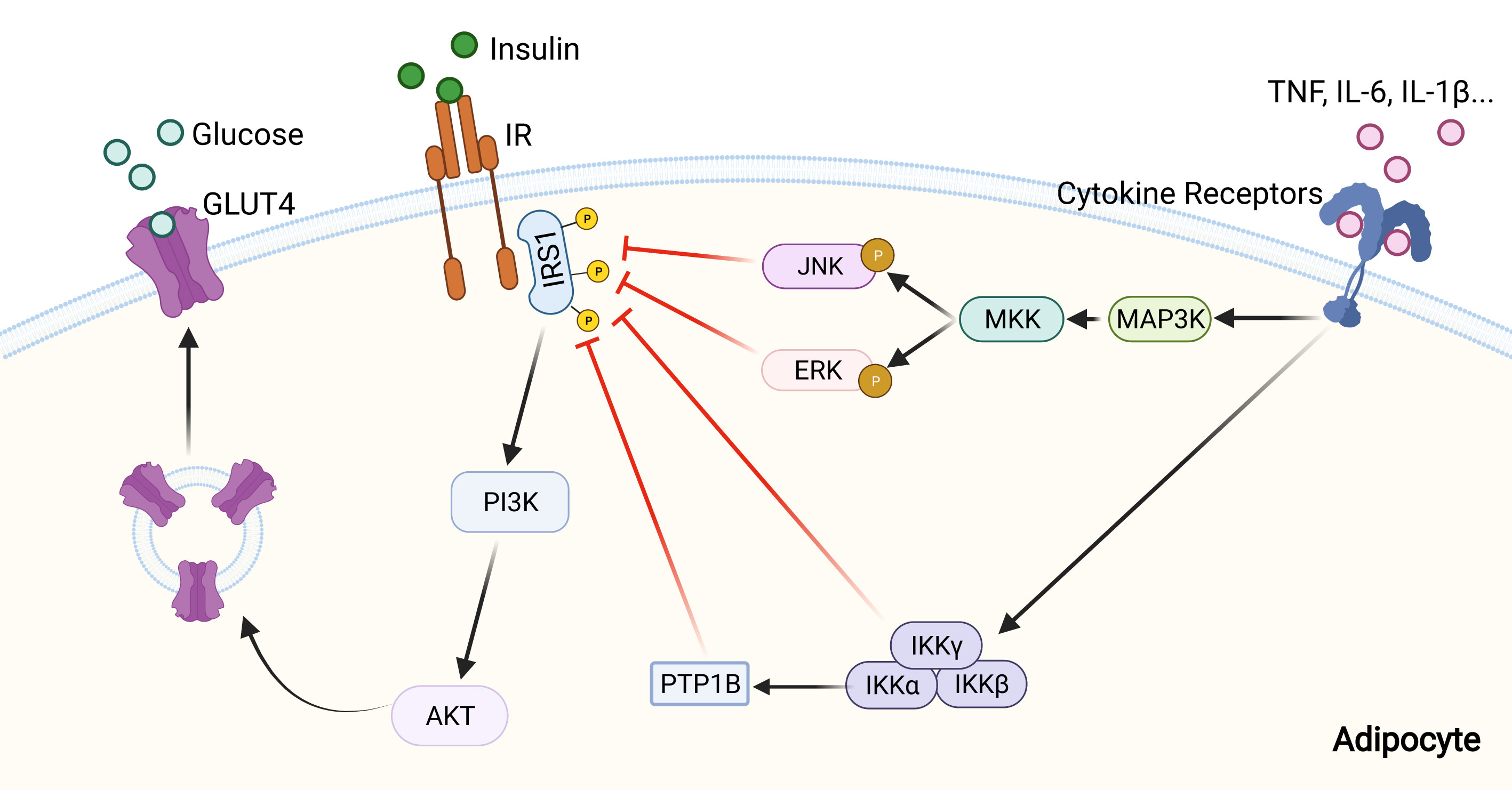
Figure 3 Inhibition of insulin signaling pathway by inflammatory signaling. Pro-inflammatory cytokines, including IL-1β, IL-6, and TNF, can activate inflammatory signaling pathways through their receptors. Then activated MAPK and NF-κB signaling pathways inhibit insulin signaling by altering the phosphorylation status of IRS-1, and further lead to a reduction of glucose uptake by adipocyte.
The regulation of insulin sensitivity by ATMs is also mediated by miRNAs, which are contained and transferred by ATM-derived exosome (148). Treatment with obese ATM-derived exosome leads to insulin resistance, whereas lean ATM-derived exosome increases insulin sensitivity in obese mice. Mechanistically, miR-155 is among the differentially expressed miRNAs in obese ATM-derived exosome, and it causes systemic insulin resistance and glucose intolerance (148).
In addition to M1 and M2 macrophages, other macrophage populations that regulate insulin resistance have been reported recently, including neuropilin-1 (NRP1)+ macrophages and TREM2+ lipid-associated macrophage (LAM) (149, 150). NRP1+ macrophages accumulate in adipose tissue and protect against obesity and metabolic syndrome. Conditional deletion of NRP1 in macrophages compromised lipid uptake and led to insulin resistance (149). Using single-cell RNA sequencing, one study identified a subset of TREM2+ lipid-associated macrophages (LAMs) that prominently arise under obesity condition both in humans and mice. TREM2 deficient mice showed inhibited recruitment of macrophages to CLS and led to adipocyte hypertrophy as well as insulin resistance (150). However, bone marrow transplantation experiments by another group argued that hematopoietic-expressed TREM2 is dispensable for obesity-induced metabolic dysfunction, including insulin resistance (151).
ATM in fibrosis
Adipocytes are surrounded by a network of extracellular matrix (ECM) proteins that not only serve as a structural support and protection but also regulate adipose tissue homeostasis by responding to different signals (152). In other words, ECM ensures adipose tissue to expand and maintain in a healthy manner. However, the development of obesity promotes an excessive accumulation of ECM proteins and elicits adipose tissue fibrosis, which reduces tissue plasticity and results in adipocyte dysfunction such as insulin resistance (153, 154). Thus, fibrosis is considered as a hallmark of metabolically dysfunctional adipose tissue.
During the development of obesity, rapid adipose tissue expansion and adipocyte enlargement cause adipose tissue hypoxia and lead to an activation of hypoxia-inducible factor 1-alpha (HIF1-α), which induces the production of ECM proteins (155, 156). Hypoxia promotes expression of many proinflammatory genes through HIF1-α induction. Low-grade inflammation contributed by hypoxia in obesity further deteriorates adipose tissue fibrosis (157).
In addition, adipocyte hypertrophy and adipose tissue hypoxia are tightly associated with increased infiltration of macrophages, which promote local ECM accumulation (154, 158). Proinflammatory M1 macrophages make up significant proportion in fibrotic adipose tissue. Besides proinflammatory cytokines, ATM produces many other cytokines, including TGF-β1 and PDGF, which directly activate fibroblasts and increase ECM accumulation (159). As a vicious spiral, macrophages promote fibrogenesis by releasing chemokines that attract fibroblasts and more proinflammatory cells (160). Besides, macrophage inducible C-type (Mincle), which is induced in macrophage by TLR4 activation, regulate ECM prodaction and degredation, as well as fibroblast proliferation (161, 162). Additionally, saturated fatty acids (SFA)-mediated inflammation is potentiated by TLR4 activation and contributes to WAT fibrosis by fueling local inflammation (31). Besides their role in promoting fibrogenesis, macrophages participate in ECM clearance through collagen uptake and degradation. Collagen phagocytosis by macrophages depends on mannose receptor 1 (MRC1) and urokinase plasminogen activator receptor-associated protein (uPARAP/endo180) (163). In adipose tissue, it has been widely accepted that there is no single signaling or single cell type responsible for ECM production. Adipose tissue collagens are contributed by both preadipocytes and macrophages, and the fibrosis is coordinated through intimate crosstalk between macrophages and preadipocytes under different physiological and pathological conditions (164). In vitro studies suggest that preadipocytes in contact with proinflammatory macrophages can produce ECM proteins, including collagen I and fibronectin (164). Meanwhile, macrophage is found to be the master regulator of fibrosis through producing TGF-β1 and PDGF, which have been proved to directly activate fibroblasts and control ECM dynamics by regulating the balance between various matrix metalloproteinases (MMPs) and their inhibitors (159).
ATM and adipokines
Adipose tissue secretes many kinds of hormones, called adipokines, which exert their biological functions in an autocrine, paracrine, and/or systemic manner and influence many physiological/pathological processes, such as thermogenesis, insulin resistance and fibrosis (165). The most well-known adipokines are adiponectin and leptin, both of which can exhibit either proinflammatory or anti-inflammatory property, thereby contributing to adipose tissue functions (165).
Adiponectin
Adiponectin, a 30-kDa adipokine exclusively secreted from adipocytes, exists in cells and plasma (166, 167). As the most aboundent peptide secreted by adipocytes, adiponectin shows protective activity in multiple diseases such as inflammation, obesity and insulin resistance (166, 168–170). Many evidences proved that adiponectin acts as anti-inflammatory factor by regulating the polarization of adipose tissue macrophages (36, 104). Recombinant adiponectin treatment results in an increased expression of M2 markers and a decreased expression of M1 markers in adipose tissue, while macrophages from adiponectin knock-out mice display increased M1 markers (36, 171). Interestingly, adiponectin has also been reported as a proinflammatory factor to increase TNF-α and IL-6 secretion directly. The authors further suggested that the anti-inflammatory property of adiponectin may be due to its desensitized effects on cells for further proinflammatory response, although the specific molecular mechanism is still unknown (172, 173). Additionally, adiponectin has been reported to induce adipose tissue M2 macrophage proliferation both in vivo and in vitro, further promoting cold-induced adipose tissue thermogenesis (174). Mechanistically, adiponectin is recruited to the cell surface of M2 macrophages via T-cadherin and promotes cell proliferation by activation of AKT signaling (174). Besides, several intracellular signaling pathways have been reported to mediate adiponectin action in regulating macrophages. Adiponectin suppresses M1 macrophage proliferation via inhibiting NF-κB signaling (175). A mutual antagonistic action was observed between adiponection and TNF/IL-6 expression. LPS-induced TNF/IL-6 is suppressed by adiponectin, and TNF/IL-6 conversely inhibit adiponectin expression (176, 177). Besides, both oxidative stress and ROS release inhibit adiponectin expression in obesity, therefore forming a vicious circle that lowers adiponectin level while increases proinflammatory cytokines and oxidative stress in obese adipose tissue (56).
Leptin
Leptin, another pivotal adipokine, exerts its function through modulating immunity and inflammation (178). Leptin is a 16-kDa peptide hormone secreted mainly from adipose tissue, and the most evident function of leptin is its control of energy balance by inhibiting appetite through hypothalamus (179, 180). However, leptin receptor (LEPR) is ubiquitously expressed on the surface of many cells like immune cells, suggesting pleiotropic actions of leptin (181, 182). High levels of thymocyte apoptosis and reduced thymic cellularity were observed in in obese mice with mutation in leptin (ob/ob mice) or LEPR (db/db mice), which were reversed by peripheral administration of recombinant leptin, revealing an important role of lepin in immunity (183). Consistently, ob/ob mice show impaired cellular and humoral immue activities, and they are protected against inflammation in different models (184–186). Besides acting on adaptive immunity, leptin regulates innate immune cells such as macrophages, to promote inflammation. Macrophages generated from ob/ob or db/db mice showed a decrease of phagocytosis and inflammatory cytokine production, whereas exogenous leptin administration upregulated both of them (178, 181). Several in vitro or ex vivo studies in wild-type mice also support that leptin acts as a proinflammatory factor in immune cells. They showed that exogenous leptin administration upregulated both phagocytosis and production of proinflammatory cytokines (4, 187–189). Leptin stimulates production of proinflammatory cytokines through activation of JAK2-STAT3 pathway in macropahges (189, 190). Moreover, leptin-deficient mast cells polarize macrophages from M1 to M2 and thus protects mice from obesity (191). Leptin has proinflammatory properties, and the expression of leptin in adipose tissue as well as circulating leptin are promoted by administration of proinflammatory stimuli (192, 193). Thus, it appears that proinflammatory cytokines and leptin form a vicious circle that promotes the development of chronic inflammation and obesity.
Conclusions and perspective
Macrophage is the most abundant cell population and believed to play a dominant role in the homeostasis of adipose tissue and whole-body energy metabolism, whose dysregulation significantly contributes to metabolic diseases (Figure 4). Noticebaly, many other immune cells exist in adipose tissue as well, including both innate and adaptive immune cells like ILC2s, eosinophils, invariant natural killer T (iNKT) and T lymphocytes. They play critically important roles in the maintenance of energy homeostasis and contribute to the metabolic dysfunction, directly or indirectly through crosstalk with macrophages (194). ILC2s recruit and activate eosinophils through IL-5, and then eosinophil-secreted IL-4 and IL-13 promote macrophage M2 activation. Besides, ILC2s directly secret IL-13, which induces a physiological expansion and differentiation of beige adipocyte precursors, further promoting adipose tissue thermogenesis (95). iNKT cells are a type of innate immune cell, and their activation induces a production of fibroblast growth factor 21 (FGF21) that promotes thermogenic browning of WAT (195). Infiltration of CD8+ T lymphocytes is an early event during the development of obesity and contribute to macrophage accumulation. Adoptive transfer of CD8+ T cells into CD8-deficient obese mice induces M1 macrophage infiltration and promotes systemic insulin resistance (196).
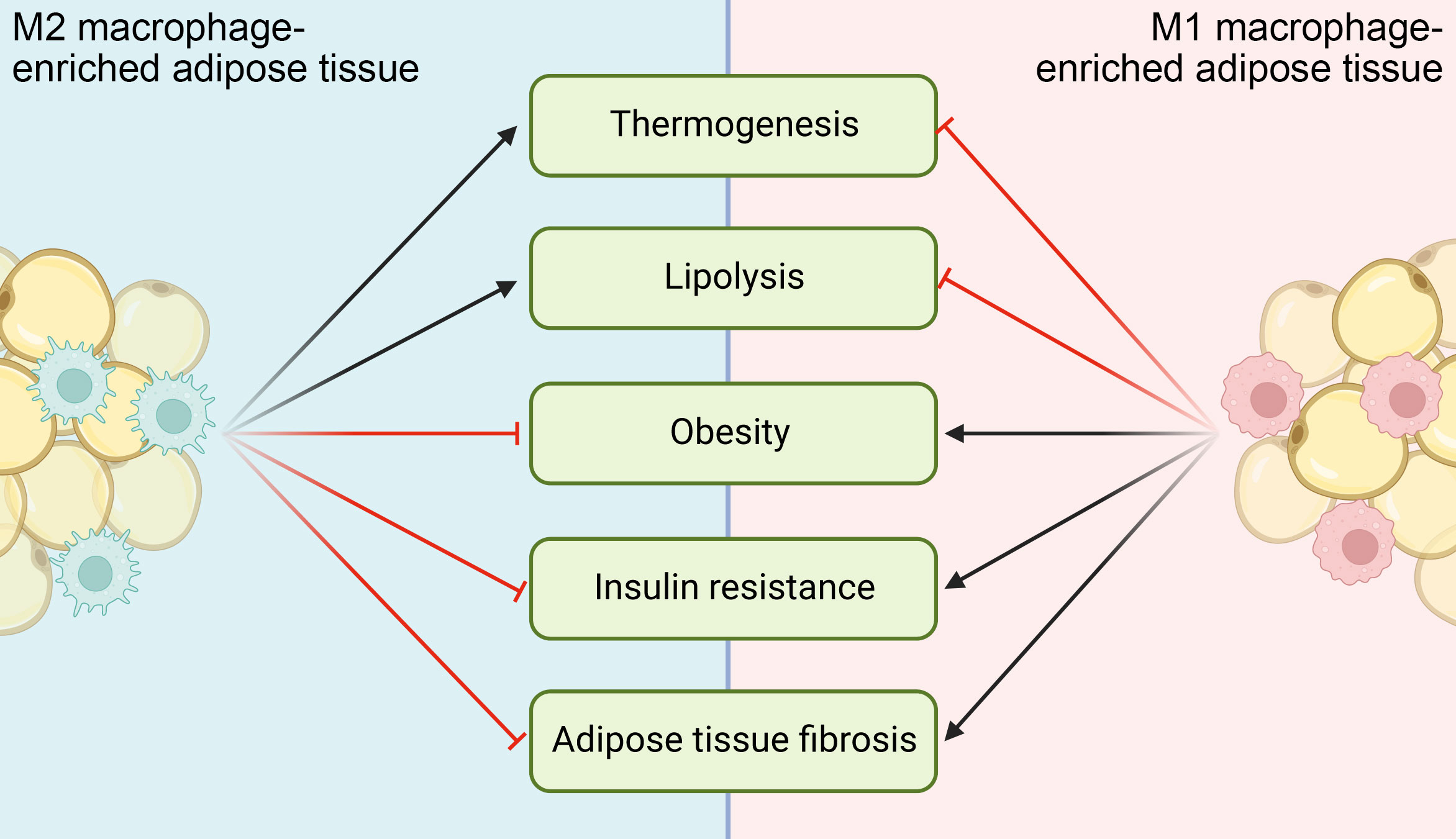
Figure 4 Functional implications of ATMs in different metabolic diseases. M1 ATMs inhibit adipocyte lipolysis and thermogenesis, while M2 ATMs do the opposites. When M1 activation of ATMs chronically exceeds M2 activation, metabolic diseases like obesity, insulin resistance and adipose tissue fibrosis ensue and/or deteriorate.
One goal of the mouse ATM investigation is to exploit its specific characteristics and functions to treat human metabolic diseases. Even though ATMs from human and mouse models are highly similar in both gene expression pattern and function, considerable differences exist (197, 198). Mouse models have their own limitations and can not replicate the properties of humans in many aspects, thus the validation of findings from mouse models in humans is critical for their translation. But, there are still many challenges for mouse-to-human validation because of the technical limitations, including limited tools to safely manipulate ATM in humans.
The great progress of ATM studies generates many further questions that need to be addressed in the future. Firstly, it is difficult to manipulate ATM specifically. Several genetically modified animal models are used to investigate tissue-specific macrophages, including Lyz2-Cre, Cx3cr1-Cre, CD11b-Cre and F4/80-Cre, whereas all of them are ubiquitously expressed in macrophages from different tissues but not specifically in ATM (199). Besides, some of them are expressed in other types of cells. For example, the widely used Lyz2-Cre is expressed in other myelomonocytic cells, including most granulocytes, few CD11c+ dendritic cells (DCs), and a small percentage of non-hematopoietic cells (200). Thus, more specific markers and animal models need to be identified and established, which will greatly facilitate our understanding on ATM in health and metabolic diseases. Secondly, macrophage constitutes a plastic and heterogeneous cell populations modulated by and interacted with their microenvironment in different adipose tissues. Distinct fat pads in different locations show different molecular, cellular and anatomical features (201). Accordingly, the physiological characteristics of ATM in these adipose tissues may be quite diverse, which have not been well investigated yet. Thirdly, ATM was oversimplified to be divided into two groups, M1 and M2 macrophages. However, this M1/M2 classification has been questioned as a result of the identifications of many distinct ATM subtypes, including CD9+ macrophage (202), TREM2+ LAM (150) and SAM (109). Although single cell RNA-sequencing provides an objective view on the identity and function of ATM (150), more unbiased approaches and new technologies should be used to identify and characterize all the different ATM populations and their regulations on health and metabolic diseases.
Author contributions
JY wrote the manuscript and DW and YQ edited it. All authors approved the submitted version. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from National Key R&D Program of China (2018YFA0800702 and 2021YFA0804801), National Natural Science Foundation of China (31671227 and 91642113) and the Thousand Young Talents Program of the Chinese government (to YQ).
Acknowledgments
Figures were created using BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Popkin BM, Du S, Green WD, Beck MA, Algaith T, Herbst CH, et al. Individuals with obesity and covid-19: A global perspective on the epidemiology and biological relationships. Obes Rev (2020) 21(11):e13128. doi: 10.1111/obr.13128
2. Muller TD, Bluher M, Tschop MH, DiMarchi RD. Anti-obesity drug discovery: Advances and challenges. Nat Rev Drug Discovery (2022) 21(3):201–23. doi: 10.1038/s41573-021-00337-8
3. Kane H, Lynch L. Innate immune control of adipose tissue homeostasis. Trends Immunol (2019) 40(9):857–72. doi: 10.1016/j.it.2019.07.006
4. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest (2003) 112(12):1796–808. doi: 10.1172/JCI19246
5. Okabe Y, Medzhitov R. Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell (2014) 157(4):832–44. doi: 10.1016/j.cell.2014.04.016
6. Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell (2014) 159(6):1312–26. doi: 10.1016/j.cell.2014.11.018
7. Wang Y, Smith W, Hao D, He B, Kong L. M1 and M2 macrophage polarization and potentially therapeutic naturally occurring compounds. Int Immunopharmacol (2019) 70:459–66. doi: 10.1016/j.intimp.2019.02.050
8. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol (2010) 72:219–46. doi: 10.1146/annurev-physiol-021909-135846
9. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity (2014) 41(1):14–20. doi: 10.1016/j.immuni.2014.06.008
10. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat Immunol (2010) 11(10):889–96. doi: 10.1038/ni.1937
11. Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, Sahgal N, et al. Irf5 promotes inflammatory macrophage polarization and Th1-Th17 responses. Nat Immunol (2011) 12(3):231–8. doi: 10.1038/ni.1990
12. Shuai K, Stark GR, Kerr IM, Darnell JE Jr. A single phosphotyrosine residue of Stat91 required for gene activation by interferon-gamma. Science (1993) 261(5129):1744–6. doi: 10.1126/science.7690989
13. Toshchakov V, Jones BW, Perera PY, Thomas K, Cody MJ, Zhang S, et al. Tlr4, but not Tlr2, mediates ifn-Beta-Induced Stat1alpha/Beta-dependent gene expression in macrophages. Nat Immunol (2002) 3(4):392–8. doi: 10.1038/ni774
14. Arnold CE, Whyte CS, Gordon P, Barker RN, Rees AJ, Wilson HM. A critical role for suppressor of cytokine signalling 3 in promoting M1 macrophage activation and function in vitro and in vivo. Immunology (2014) 141(1):96–110. doi: 10.1111/imm.12173
15. Colin S, Chinetti-Gbaguidi G, Staels B. Macrophage phenotypes in atherosclerosis. Immunol Rev (2014) 262(1):153–66. doi: 10.1111/imr.12218
16. Roszer T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediators Inflammation (2015) 2015:816460. doi: 10.1155/2015/816460
17. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity (2016) 44(3):450–62. doi: 10.1016/j.immuni.2016.02.015
18. Wang LX, Zhang SX, Wu HJ, Rong XL, Guo J. M2b macrophage polarization and its roles in diseases. J Leukoc Biol (2019) 106(2):345–58. doi: 10.1002/JLB.3RU1018-378RR
19. Chistiakov DA, Bobryshev YV, Nikiforov NG, Elizova NV, Sobenin IA, Orekhov AN. Macrophage phenotypic plasticity in atherosclerosis: The associated features and the peculiarities of the expression of inflammatory genes. Int J Cardiol (2015) 184:436–45. doi: 10.1016/j.ijcard.2015.03.055
20. Ambarus CA, Santegoets KC, van Bon L, Wenink MH, Tak PP, Radstake TR, et al. Soluble immune complexes shift the tlr-induced cytokine production of distinct polarized human macrophage subsets towards il-10. PLoS One (2012) 7(4):e35994. doi: 10.1371/journal.pone.0035994
21. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol (2004) 25(12):677–86. doi: 10.1016/j.it.2004.09.015
22. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140(6):805–20. doi: 10.1016/j.cell.2010.01.022
23. Charo IF. Macrophage polarization and insulin resistance: Ppargamma in control. Cell Metab (2007) 6(2):96–8. doi: 10.1016/j.cmet.2007.07.006
24. Liao X, Sharma N, Kapadia F, Zhou G, Lu Y, Hong H, et al. Kruppel-like factor 4 regulates macrophage polarization. J Clin Invest (2011) 121(7):2736–49. doi: 10.1172/JCI45444
25. Ruckerl D, Jenkins SJ, Laqtom NN, Gallagher IJ, Sutherland TE, Duncan S, et al. Induction of il-4r alpha-dependent micrornas identifies Pi3k/Akt signaling as essential for il-4-Driven murine macrophage proliferation in vivo. Blood (2012) 120(11):2307–16. doi: 10.1182/blood-2012-02-408252
26. Kubota T, Inoue M, Kubota N, Takamoto I, Mineyama T, Iwayama K, et al. Downregulation of macrophage Irs2 by hyperinsulinemia impairs il-4-Indeuced M2a-subtype macrophage activation in obesity. Nat Commun (2018) 9(1): 1–15. doi: 10.1038/s41467-018-07358-9
27. Arranz A, Doxaki C, Vergadi E, de la Torre YM, Vaporidi K, Lagoudaki ED, et al. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. P Natl Acad Sci U S A (2012) 109(24):9517–22. doi: 10.1073/pnas.1119038109
28. Shin KC, Hwang I, Choe SS, Park J, Ji Y, Kim JI, et al. Macrophage vldlr mediates obesity-induced insulin resistance with adipose tissue inflammation. Nat Commun (2017) 8(1):1087. doi: 10.1038/s41467-017-01232-w
29. Lee JY, Sohn KH, Rhee SH, Hwang D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through toll-like receptor 4. J Biol Chem (2001) 276(20):16683–9. doi: 10.1074/jbc.M011695200
30. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. Tlr4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest (2006) 116(11):3015–25. doi: 10.1172/JCI28898
31. Lancaster GI, Langley KG, Berglund NA, Kammoun HL, Reibe S, Estevez E, et al. Evidence that Tlr4 is not a receptor for saturated fatty acids but mediates lipid-induced inflammation by reprogramming macrophage metabolism. Cell Metab (2018) 27(5):1096–110.e5. doi: 10.1016/j.cmet.2018.03.014
32. Cheng CI, Chen PH, Lin YC, Kao YH. High glucose activates Raw264.7 macrophages through rhoa kinase-mediated signaling pathway. Cell Signal (2015) 27(2):283–92. doi: 10.1016/j.cellsig.2014.11.012
33. Torres-Castro I, Arroyo-Camarena UD, Martinez-Reyes CP, Gomez-Arauz AY, Duenas-Andrade Y, Hernandez-Ruiz J, et al. Human monocytes and macrophages undergo M1-type inflammatory polarization in response to high levels of glucose. Immunol Lett (2016) 176:81–9. doi: 10.1016/j.imlet.2016.06.001
34. Zhang Y, Mei H, Chang X, Chen F, Zhu Y, Han X. Adipocyte-derived microvesicles from obese mice induce M1 macrophage phenotype through secreted mir-155. J Mol Cell Biol (2016) 8(6):505–17. doi: 10.1093/jmcb/mjw040
35. Ghorpade DS, Ozcan L, Zheng Z, Nicoloro SM, Shen Y, Chen E, et al. Hepatocyte-secreted Dpp4 in obesity promotes adipose inflammation and insulin resistance. Nature (2018) 555(7698):673–7. doi: 10.1038/nature26138
36. Ohashi K, Parker JL, Ouchi N, Higuchi A, Vita JA, Gokce N, et al. Adiponectin promotes macrophage polarization toward an anti-inflammatory phenotype. J Biol Chem (2010) 285(9):6153–60. doi: 10.1074/jbc.M109.088708
37. Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, Vats D, Morel CR, Goforth MH, et al. Alternative M2 activation of kupffer cells by ppardelta ameliorates obesity-induced insulin resistance. Cell Metab (2008) 7(6):496–507. doi: 10.1016/j.cmet.2008.04.003
38. Mukundan L, Odegaard JI, Morel CR, Heredia JE, Mwangi JW, Ricardo-Gonzalez RR, et al. Ppar-delta senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat Med (2009) 15(11):1266–72. doi: 10.1038/nm.2048
39. Su CW, Chen CY, Li Y, Long SR, Massey W, Kumar DV, et al. Helminth infection protects against high fat diet-induced obesity Via induction of alternatively activated macrophages. Sci Rep (2018) 8(1):4607. doi: 10.1038/s41598-018-22920-7
40. Bouhlel MA, Derudas B, Rigamonti E, Dievart R, Brozek J, Haulon S, et al. Ppargamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab (2007) 6(2):137–43. doi: 10.1016/j.cmet.2007.06.010
41. Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. Macrophage-specific ppargamma controls alternative activation and improves insulin resistance. Nature (2007) 447(7148):1116–20. doi: 10.1038/nature05894
42. Griffin C, Eter L, Lanzetta N, Abrishami S, Varghese M, McKernan K, et al. Tlr4, trif, and Myd88 are essential for myelopoiesis and Cd11c(+) adipose tissue macrophage production in obese mice. J Biol Chem (2018) 293(23):8775–86. doi: 10.1074/jbc.RA117.001526
43. Nakajima S, Koh V, Kua LF, So J, Davide L, Lim KS, et al. Accumulation of Cd11c+Cd163+ adipose tissue macrophages through upregulation of intracellular 11beta-Hsd1 in human obesity. J Immunol (2016) 197(9):3735–45. doi: 10.4049/jimmunol.1600895
44. Shan B, Wang X, Wu Y, Xu C, Xia Z, Dai J, et al. The metabolic er stress sensor Ire1alpha suppresses alternative activation of macrophages and impairs energy expenditure in obesity. Nat Immunol (2017) 18(5):519–29. doi: 10.1038/ni.3709
45. Hu F, Tong J, Deng B, Zheng J, Lu C. Mir-495 regulates macrophage M1/M2 polarization and insulin resistance in high-fat diet-fed mice via targeting fto. Pflugers Arch (2019) 471(11-12):1529–37. doi: 10.1007/s00424-019-02316-w
46. Gu X, Zhang Y, Li D, Cai H, Cai L, Xu Q. N6-methyladenosine demethylase fto promotes M1 and M2 macrophage activation. Cell Signal (2020) 69:109553. doi: 10.1016/j.cellsig.2020.109553
47. Yao J, Wu D, Zhang C, Yan T, Zhao Y, Shen H, et al. Macrophage Irx3 promotes diet-induced obesity and metabolic inflammation. Nat Immunol (2021) 22(10):1268–79. doi: 10.1038/s41590-021-01023-y
48. Daemen S, Schilling JD. The interplay between tissue niche and macrophage cellular metabolism in obesity. Front Immunol (2019) 10:3133. doi: 10.3389/fimmu.2019.03133
49. Cinti S, Mitchell G, Barbatelli G, Murano I, Ceresi E, Faloia E, et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res (2005) 46(11):2347–55. doi: 10.1194/jlr.M500294-JLR200
50. Patel PS, Buras ED, Balasubramanyam A. The role of the immune system in obesity and insulin resistance. J Obes (2013) 2013:616193. doi: 10.1155/2013/616193
51. Murano I, Barbatelli G, Parisani V, Latini C, Muzzonigro G, Castellucci M, et al. Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. J Lipid Res (2008) 49(7):1562–8. doi: 10.1194/jlr.M800019-JLR200
52. Stienstra R, Dijk W, van Beek L, Jansen H, Heemskerk M, Houtkooper RH, et al. Mannose-binding lectin is required for the effective clearance of apoptotic cells by adipose tissue macrophages during obesity. Diabetes (2014) 63(12):4143–53. doi: 10.2337/db14-0256
53. Xu XY, Grijalva A, Skowronski A, van Eijk M, Serlie MJ, Ferrante AW. Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab (2013) 18(6):816–30. doi: 10.1016/j.cmet.2013.11.001
54. Apovian CM, Bigornia S, Mott M, Meyers MR, Ulloor J, Gagua M, et al. Adipose macrophage infiltration is associated with insulin resistance and vascular endothelial dysfunction in obese subjects. Arterioscl Throm Vas (2008) 28(9):1654–9. doi: 10.1161/ATVBAHA.108.170316
55. Bremer AA, Devaraj S, Afify A, Jialal I. Adipose tissue dysregulation in patients with metabolic syndrome. J Clin Endocr Metab (2011) 96(11):E1782–E8. doi: 10.1210/jc.2011-1577
56. Suganami T, Nishida J, Ogawa Y. A paracrine loop between adipocytes and macrophages aggravates inflammatory changes: Role of free fatty acids and tumor necrosis factor alpha. Arterioscler Thromb Vasc Biol (2005) 25(10):2062–8. doi: 10.1161/01.ATV.0000183883.72263.13
57. Kamei N, Tobe K, Suzuki R, Ohsugi M, Watanabe T, Kubota N, et al. Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J Biol Chem (2006) 281(36):26602–14. doi: 10.1074/jbc.M601284200
58. Huber J, Kiefer FW, Zeyda M, Ludvik B, Silberhumer GR, Prager G, et al. Cc chemokine and cc chemokine receptor profiles in visceral and subcutaneous adipose tissue are altered in human obesity. J Clin Endocrinol Metab (2008) 93(8):3215–21. doi: 10.1210/jc.2007-2630
59. Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest (2003) 112(12):1821–30. doi: 10.1172/JCI19451
60. Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, et al. Mcp-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest (2006) 116(6):1494–505. doi: 10.1172/JCI26498
61. Obstfeld AE, Sugaru E, Thearle M, Francisco AM, Gayet C, Ginsberg HN, et al. C-c chemokine receptor 2 (Ccr2) regulates the hepatic recruitment of myeloid cells that promote obesity-induced hepatic steatosis. Diabetes (2010) 59(4):916–25. doi: 10.2337/db09-1403
62. Dewald O, Zymek P, Winkelmann K, Koerting A, Ren GF, Abou-Khamis T, et al. Ccl2/Monocyte chemoattractant protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res (2005) 96(8):881–9. doi: 10.1161/01.RES.0000163017.13772.3a
63. Vozzelli MA, Mason SN, Whorton MH, Auten RL. Antimacrophage chemokine treatment prevents neutrophil and macrophage influx in hyperoxia-exposed newborn rat lung. Am J Physiol-Lung C (2004) 286(3):L488–L93. doi: 10.1152/ajplung.00414.2002
64. Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (Mcp-1): An overview. J Interf Cytok Res (2009) 29(6):313–26. doi: 10.1089/jir.2008.0027
65. Amano SU, Cohen JL, Vangala P, Tencerova M, Nicoloro SM, Yawe JC, et al. Local proliferation of macrophages contributes to obesity-associated adipose tissue inflammation. Cell Metab (2014) 19(1):162–71. doi: 10.1016/j.cmet.2013.11.017
66. Gong X, Gong W, Kuhns DB, Ben-Baruch A, Howard OM, Wang JM. Monocyte chemotactic protein-2 (Mcp-2) uses Ccr1 and Ccr2b as its functional receptors. J Biol Chem (1997) 272(18):11682–5. doi: 10.1074/jbc.272.18.11682
67. Combadiere C, Ahuja SK, Van Damme J, Tiffany HL, Gao JL, Murphy PM. Monocyte chemoattractant protein-3 is a functional ligand for cc chemokine receptors 1 and 2b. J Biol Chem (1995) 270(50):29671–5. doi: 10.1074/jbc.270.50.29671
68. Berkhout TA, Sarau HM, Moores K, White JR, Elshourbagy N, Appelbaum E, et al. Cloning, in vitro expression, and functional characterization of a novel human cc chemokine of the monocyte chemotactic protein (Mcp) family (Mcp-4) that binds and signals through the cc chemokine receptor 2b. J Biol Chem (1997) 272(26):16404–13. doi: 10.1074/jbc.272.26.16404
69. Sarafi MN, Garcia-Zepeda EA, MacLean JA, Charo IF, Luster AD. Murine monocyte chemoattractant protein (Mcp)-5: A novel cc chemokine that is a structural and functional homologue of human mcp-1. J Exp Med (1997) 185(1):99–109. doi: 10.1084/jem.185.1.99
70. Kitade H, Sawamoto K, Nagashimada M, Inoue H, Yamamoto Y, Sai Y, et al. Ccr5 plays a critical role in obesity-induced adipose tissue inflammation and insulin resistance by regulating both macrophage recruitment and M1/M2 status. Diabetes (2012) 61(7):1680–90. doi: 10.2337/db11-1506
71. Zhang Z, Wang Q, Yao J, Zhou X, Zhao J, Zhang X, et al. Chemokine receptor 5, a double-edged sword in metabolic syndrome and cardiovascular disease. Front Pharmacol (2020) 11:146. doi: 10.3389/fphar.2020.00146
72. Noh HJ, Kim CS, Kang JH, Park JY, Choe SY, Hong SM, et al. Quercetin suppresses mip-1alpha-Induced adipose inflammation by downregulating its receptors Ccr1/Ccr5 and inhibiting inflammatory signaling. J Med Food (2014) 17(5):550–7. doi: 10.1089/jmf.2013.2912
73. Keophiphath M, Rouault C, Divoux A, Clement K, Lacasa D. Ccl5 promotes macrophage recruitment and survival in human adipose tissue. Arterioscler Thromb Vasc Biol (2010) 30(1):39–45. doi: 10.1161/ATVBAHA.109.197442
74. D'Haese JG, Demir IE, Friess H, Ceyhan GO. Fractalkine/Cx3cr1: Why a single chemokine-receptor duo bears a major and unique therapeutic potential. Expert Opin Ther Targets (2010) 14(2):207–19. doi: 10.1517/14728220903540265
75. Lesnik P, Haskell CA, Charo IF. Decreased atherosclerosis in Cx3cr1-/- mice reveals a role for fractalkine in atherogenesis. J Clin Invest (2003) 111(3):333–40. doi: 10.1172/JCI15555
76. Digby JE, McNeill E, Dyar OJ, Lam V, Greaves DR, Choudhury RP. Anti-inflammatory effects of nicotinic acid in adipocytes demonstrated by suppression of fractalkine, rantes, and mcp-1 and upregulation of adiponectin. Atherosclerosis (2010) 209(1):89–95. doi: 10.1016/j.atherosclerosis.2009.08.045
77. Polyak A, Ferenczi S, Denes A, Winkler Z, Kriszt R, Pinter-Kubler B, et al. The Fractalkine/Cx3cr1 system is implicated in the development of metabolic visceral adipose tissue inflammation in obesity. Brain Behav Immun (2014) 38:25–35. doi: 10.1016/j.bbi.2014.01.010
78. Nagashimada M, Sawamoto K, Ni Y, Kitade H, Nagata N, Xu L, et al. Cx3cl1-Cx3cr1 signaling deficiency exacerbates obesity-induced inflammation and insulin resistance in Male mice. Endocrinology (2021) 162(6): 1–20. doi: 10.1210/endocr/bqab064
79. Badolato R, Wang JM, Murphy WJ, Lloyd AR, Michiel DF, Bausserman LL, et al. Serum amyloid a is a chemoattractant: Induction of migration, adhesion, and tissue infiltration of monocytes and polymorphonuclear leukocytes. J Exp Med (1994) 180(1):203–9. doi: 10.1084/jem.180.1.203
80. Mullan RH, Bresnihan B, Golden-Mason L, Markham T, O'Hara R, FitzGerald O, et al. Acute-phase serum amyloid a stimulation of angiogenesis, leukocyte recruitment, and matrix degradation in rheumatoid arthritis through an nf-Kappab-Dependent signal transduction pathway. Arthritis Rheum (2006) 54(1):105–14. doi: 10.1002/art.21518
81. Xu J, Morinaga H, Oh D, Li P, Chen A, Talukdar S, et al. Gpr105 ablation prevents inflammation and improves insulin sensitivity in mice with diet-induced obesity. J Immunol (2012) 189(4):1992–9. doi: 10.4049/jimmunol.1103207
82. Hromas R, Broxmeyer HE, Kim C, Nakshatri H, Christopherson K 2nd, Azam M, et al. Cloning of brak, a novel divergent cxc chemokine preferentially expressed in normal versus malignant cells. Biochem Biophys Res Commun (1999) 255(3):703–6. doi: 10.1006/bbrc.1999.0257
83. Nara N, Nakayama Y, Okamoto S, Tamura H, Kiyono M, Muraoka M, et al. Disruption of cxc motif chemokine ligand-14 in mice ameliorates obesity-induced insulin resistance. J Biol Chem (2007) 282(42):30794–803. doi: 10.1074/jbc.M700412200
84. Cereijo R, Gavalda-Navarro A, Cairo M, Quesada-Lopez T, Villarroya J, Moron-Ros S, et al. Cxcl14, a brown adipokine that mediates brown-Fat-to-Macrophage communication in thermogenic adaptation. Cell Metab (2018) 28(5):750–63.e6. doi: 10.1016/j.cmet.2018.07.015
85. Dalmas E, Clement K, Guerre-Millo M. Defining macrophage phenotype and function in adipose tissue. Trends Immunol (2011) 32(7):307–14. doi: 10.1016/j.it.2011.04.008
86. Cannon B, Nedergaard J. Brown adipose tissue: Function and physiological significance. Physiol Rev (2004) 84(1):277–359. doi: 10.1152/physrev.00015.2003
87. Nguyen KD, Qiu Y, Cui X, Goh YP, Mwangi J, David T, et al. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature (2011) 480(7375):104–8. doi: 10.1038/nature10653
88. Qiu Y, Nguyen KD, Odegaard JI, Cui X, Tian X, Locksley RM, et al. Eosinophils and type 2 cytokine signaling in macrophages orchestrate development of functional beige fat. Cell (2014) 157(6):1292–308. doi: 10.1016/j.cell.2014.03.066
89. Fischer K, Ruiz HH, Jhun K, Finan B, Oberlin DJ, van der Heide V, et al. Alternatively activated macrophages do not synthesize catecholamines or contribute to adipose tissue adaptive thermogenesis. Nat Med (2017) 23(5):623–30. doi: 10.1038/nm.4316
90. Spadaro O, Camell CD, Bosurgi L, Nguyen KY, Youm YH, Rothlin CV, et al. Igf1 shapes macrophage activation in response to immunometabolic challenge. Cell Rep (2017) 19(2):225–34. doi: 10.1016/j.celrep.2017.03.046
91. Lee YH, Kim SN, Kwon HJ, Maddipati KR, Granneman JG. Adipogenic role of alternatively activated macrophages in beta-adrenergic remodeling of white adipose tissue. Am J Physiol Regul Integr Comp Physiol (2016) 310(1):R55–65. doi: 10.1152/ajpregu.00355.2015
92. Lee YH, Petkova AP, Granneman JG. Identification of an adipogenic niche for adipose tissue remodeling and restoration. Cell Metab (2013) 18(3):355–67. doi: 10.1016/j.cmet.2013.08.003
93. Chung KJ, Chatzigeorgiou A, Economopoulou M, Garcia-Martin R, Alexaki VI, Mitroulis I, et al. A self-sustained loop of inflammation-driven inhibition of beige adipogenesis in obesity. Nat Immunol (2017) 18(6):654–64. doi: 10.1038/ni.3728
94. Rosina M, Ceci V, Turchi R, Chuan L, Borcherding N, Sciarretta F, et al. Ejection of damaged mitochondria and their removal by macrophages ensure efficient thermogenesis in brown adipose tissue. Cell Metab (2022) 34(4):533–48.e12. doi: 10.1016/j.cmet.2022.02.016
95. Lee MW, Odegaard JI, Mukundan L, Qiu Y, Molofsky AB, Nussbaum JC, et al. Activated type 2 innate lymphoid cells regulate beige fat biogenesis. Cell (2015) 160(1-2):74–87. doi: 10.1016/j.cell.2014.12.011
96. Rajbhandari P, Thomas BJ, Feng AC, Hong C, Wang J, Vergnes L, et al. Il-10 signaling remodels adipose chromatin architecture to limit thermogenesis and energy expenditure. Cell (2018) 172(1-2):218–33.e17. doi: 10.1016/j.cell.2017.11.019
97. Sakamoto T, Nitta T, Maruno K, Yeh YS, Kuwata H, Tomita K, et al. Macrophage infiltration into obese adipose tissues suppresses the induction of Ucp1 level in mice. Am J Physiol Endocrinol Metab (2016) 310(8):E676–E87. doi: 10.1152/ajpendo.00028.2015
98. Mowers J, Uhm M, Reilly SM, Simon J, Leto D, Chiang SH, et al. Inflammation produces catecholamine resistance in obesity Via activation of Pde3b by the protein kinases ikkepsilon and Tbk1. Elife (2013) 2:e01119. doi: 10.7554/eLife.01119
99. Chiang SH, Bazuine M, Lumeng CN, Geletka LM, Mowers J, White NM, et al. The protein kinase ikkepsilon regulates energy balance in obese mice. Cell (2009) 138(5):961–75. doi: 10.1016/j.cell.2009.06.046
100. Reilly SM, Chiang SH, Decker SJ, Chang L, Uhm M, Larsen MJ, et al. An inhibitor of the protein kinases Tbk1 and ikk-varepsilon improves obesity-related metabolic dysfunctions in mice. Nat Med (2013) 19(3):313–21. doi: 10.1038/nm.3082
101. Goto T, Naknukool S, Yoshitake R, Hanafusa Y, Tokiwa S, Li Y, et al. Proinflammatory cytokine interleukin-1beta suppresses cold-induced thermogenesis in adipocytes. Cytokine (2016) 77:107–14. doi: 10.1016/j.cyto.2015.11.001
102. Sakamoto T, Takahashi N, Sawaragi Y, Naknukool S, Yu R, Goto T, et al. Inflammation induced by raw macrophages suppresses Ucp1 mrna induction Via erk activation in 10t1/2 adipocytes. Am J Physiol Cell Physiol (2013) 304(8):C729–38. doi: 10.1152/ajpcell.00312.2012
103. Diaz-Delfin J, Hondares E, Iglesias R, Giralt M, Caelles C, Villarroya F. Tnf-alpha represses beta-klotho expression and impairs Fgf21 action in adipose cells: Involvement of Jnk1 in the Fgf21 pathway. Endocrinology (2012) 153(9):4238–45. doi: 10.1210/en.2012-1193
104. Okla M, Wang W, Kang I, Pashaj A, Carr T, Chung S. Activation of toll-like receptor 4 (Tlr4) attenuates adaptive thermogenesis Via endoplasmic reticulum stress. J Biol Chem (2015) 290(44):26476–90. doi: 10.1074/jbc.M115.677724
105. Bae J, Ricciardi CJ, Esposito D, Komarnytsky S, Hu P, Curry BJ, et al. Activation of pattern recognition receptors in brown adipocytes induces inflammation and suppresses uncoupling protein 1 expression and mitochondrial respiration. Am J Physiol Cell Physiol (2014) 306(10):C918–30. doi: 10.1152/ajpcell.00249.2013
106. Doyle S, Vaidya S, O'Connell R, Dadgostar H, Dempsey P, Wu T, et al. Irf3 mediates a Tlr3/Tlr4-specific antiviral gene program. Immunity (2002) 17(3):251–63. doi: 10.1016/s1074-7613(02)00390-4
107. Kumari M, Wang X, Lantier L, Lyubetskaya A, Eguchi J, Kang S, et al. Irf3 promotes adipose inflammation and insulin resistance and represses browning. J Clin Invest (2016) 126(8):2839–54. doi: 10.1172/JCI86080
108. Wolf Y, Boura-Halfon S, Cortese N, Haimon Z, Sar Shalom H, Kuperman Y, et al. Brown-Adipose-Tissue macrophages control tissue innervation and homeostatic energy expenditure. Nat Immunol (2017) 18(6):665–74. doi: 10.1038/ni.3746
109. Pirzgalska RM, Seixas E, Seidman JS, Link VM, Sanchez NM, Mahu I, et al. Sympathetic neuron-associated macrophages contribute to obesity by importing and metabolizing norepinephrine. Nat Med (2017) 23(11):1309–18. doi: 10.1038/nm.4422
110. Camell CD, Sander J, Spadaro O, Lee A, Nguyen KY, Wing A, et al. Inflammasome-driven catecholamine catabolism in macrophages blunts lipolysis during ageing. Nature (2017) 550(7674):119–23. doi: 10.1038/nature24022
111. Guo T, Marmol P, Moliner A, Bjornholm M, Zhang C, Shokat KM, et al. Adipocyte Alk7 links nutrient overload to catecholamine resistance in obesity. Elife (2014) 3:e03245. doi: 10.7554/eLife.03245
112. Wang Q, Li D, Cao G, Shi Q, Zhu J, Zhang M, et al. Il-27 signalling promotes adipocyte thermogenesis and energy expenditure. Nature (2021) 600(7888):314–8. doi: 10.1038/s41586-021-04127-5
113. Fu Z, Gilbert ER, Liu D. Regulation of insulin synthesis and secretion and pancreatic beta-cell dysfunction in diabetes. Curr Diabetes Rev (2013) 9(1):25–53. doi: 10.2174/1573399811309010025
114. Ramalingam L, Oh E, Thurmond DC. Novel roles for insulin receptor (Ir) in adipocytes and skeletal muscle cells Via new and unexpected substrates. Cell Mol Life Sci (2013) 70(16):2815–34. doi: 10.1007/s00018-012-1176-1
115. Lumeng CN, DelProposto JB, Westcott DJ, Saltiel AR. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes (2008) 57(12):3239–46. doi: 10.2337/db08-0872
116. Patsouris D, Li PP, Thapar D, Chapman J, Olefsky JM, Neels JG. Ablation of Cd11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab (2008) 8(4):301–9. doi: 10.1016/j.cmet.2008.08.015
117. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science (1993) 259(5091):87–91. doi: 10.1126/science.7678183
118. Uysal KT, Wiesbrock SM, Hotamisligil GS. Functional analysis of tumor necrosis factor (Tnf) receptors in tnf-Alpha-Mediated insulin resistance in genetic obesity. Endocrinology (1998) 139(12):4832–8. doi: 10.1210/endo.139.12.6337
119. Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking tnf-alpha function. Nature (1997) 389(6651):610–4. doi: 10.1038/39335
120. Aguirre V, Uchida T, Yenush L, Davis R, White MF. The c-jun Nh(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J Biol Chem (2000) 275(12):9047–54. doi: 10.1074/jbc.275.12.9047
121. Bing C. Is interleukin-1beta a culprit in macrophage-adipocyte crosstalk in obesity? Adipocyte (2015) 4(2):149–52. doi: 10.4161/21623945.2014.979661
122. Scholz CC, Cavadas MA, Tambuwala MM, Hams E, Rodriguez J, von Kriegsheim A, et al. Regulation of il-1beta-Induced nf-kappab by hydroxylases links key hypoxic and inflammatory signaling pathways. Proc Natl Acad Sci U.S.A. (2013) 110(46):18490–5. doi: 10.1073/pnas.1309718110
123. Juge-Aubry CE, Somm E, Giusti V, Pernin A, Chicheportiche R, Verdumo C, et al. Adipose tissue is a major source of interleukin-1 receptor antagonist: Upregulation in obesity and inflammation. Diabetes (2003) 52(5):1104–10. doi: 10.2337/diabetes.52.5.1104
124. Lagathu C, Yvan-Charvet L, Bastard JP, Maachi M, Quignard-Boulange A, Capeau J, et al. Long-term treatment with interleukin-1beta induces insulin resistance in murine and human adipocytes. Diabetologia (2006) 49(9):2162–73. doi: 10.1007/s00125-006-0335-z
125. Jager J, Gremeaux T, Cormont M, Le Marchand-Brustel Y, Tanti JF. Interleukin-1beta-Induced insulin resistance in adipocytes through down-regulation of insulin receptor substrate-1 expression. Endocrinology (2007) 148(1):241–51. doi: 10.1210/en.2006-0692
126. Feve B, Bastard JP. The role of interleukins in insulin resistance and type 2 diabetes mellitus. Nat Rev Endocrinol (2009) 5(6):305–11. doi: 10.1038/nrendo.2009.62
127. Fried SK, Bunkin DA, Greenberg AS. Omental and subcutaneous adipose tissues of obese subjects release interleukin-6: Depot difference and regulation by glucocorticoid. J Clin Endocrinol Metab (1998) 83(3):847–50. doi: 10.1210/jcem.83.3.4660
128. Lagathu C, Bastard JP, Auclair M, Maachi M, Capeau J, Caron M. Chronic interleukin-6 (Il-6) treatment increased il-6 secretion and induced insulin resistance in adipocyte: Prevention by rosiglitazone. Biochem Biophys Res Commun (2003) 311(2):372–9. doi: 10.1016/j.bbrc.2003.10.013
129. Stouthard JM, Oude Elferink RP, Sauerwein HP. Interleukin-6 enhances glucose transport in 3t3-L1 adipocytes. Biochem Biophys Res Commun (1996) 220(2):241–5. doi: 10.1006/bbrc.1996.0389
130. Rotter V, Nagaev I, Smith U. Interleukin-6 (Il-6) induces insulin resistance in 3t3-L1 adipocytes and is, like il-8 and tumor necrosis factor-alpha, overexpressed in human fat cells from insulin-resistant subjects. J Biol Chem (2003) 278(46):45777–84. doi: 10.1074/jbc.M301977200
131. Fasshauer M, Kralisch S, Klier M, Lossner U, Bluher M, Klein J, et al. Insulin resistance-inducing cytokines differentially regulate socs mrna expression Via growth factor- and Jak/Stat-signaling pathways in 3t3-L1 adipocytes. J Endocrinol (2004) 181(1):129–38. doi: 10.1677/joe.0.1810129
132. Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest (2006) 116(7):1793–801. doi: 10.1172/JCI29069
133. Panahi G, Pasalar P, Zare M, Rizzuto R, Meshkani R. High glucose induces inflammatory responses in Hepg2 cells Via the oxidative stress-mediated activation of nf-kappab, and mapk pathways in Hepg2 cells. Arch Physiol Biochem (2018) 124(5):468–74. doi: 10.1080/13813455.2018.1427764
134. Jiao P, Ma J, Feng B, Zhang H, Diehl JA, Chin YE, et al. Ffa-induced adipocyte inflammation and insulin resistance: Involvement of er stress and ikkbeta pathways. Obes (Silver Spring) (2011) 19(3):483–91. doi: 10.1038/oby.2010.200
135. Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, et al. Ikk-beta links inflammation to obesity-induced insulin resistance. Nat Med (2005) 11(2):191–8. doi: 10.1038/nm1185
136. Lee DF, Kuo HP, Chen CT, Wei Y, Chou CK, Hung JY, et al. Ikkbeta suppression of Tsc1 function links the mtor pathway with insulin resistance. Int J Mol Med (2008) 22(5):633–8. doi: 10.3892/ijmm_00000065
137. Zabolotny JM, Kim YB, Welsh LA, Kershaw EE, Neel BG, Kahn BB. Protein-tyrosine phosphatase 1b expression is induced by inflammation in vivo. J Biol Chem (2008) 283(21):14230–41. doi: 10.1074/jbc.M800061200
138. Tanti JF, Gremeaux T, van Obberghen E, Le Marchand-Brustel Y. Serine/Threonine phosphorylation of insulin receptor substrate 1 modulates insulin receptor signaling. J Biol Chem (1994) 269(8):6051–7. doi: 10.1016/S0021-9258(17)37568-3
139. Gual P, Le Marchand-Brustel Y, Tanti JF. Positive and negative regulation of insulin signaling through irs-1 phosphorylation. Biochimie (2005) 87(1):99–109. doi: 10.1016/j.biochi.2004.10.019
140. Tanti JF, Jager J. Cellular mechanisms of insulin resistance: Role of stress-regulated serine kinases and insulin receptor substrates (Irs) serine phosphorylation. Curr Opin Pharmacol (2009) 9(6):753–62. doi: 10.1016/j.coph.2009.07.004
141. Sabio G, Das M, Mora A, Zhang Z, Jun JY, Ko HJ, et al. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science (2008) 322(5907):1539–43. doi: 10.1126/science.1160794
142. Tuncman G, Hirosumi J, Solinas G, Chang L, Karin M, Hotamisligil GS. Functional in vivo interactions between Jnk1 and Jnk2 isoforms in obesity and insulin resistance. Proc Natl Acad Sci U S A (2006) 103(28):10741–6. doi: 10.1073/pnas.0603509103
143. Han MS, Jung DY, Morel C, Lakhani SA, Kim JK, Flavell RA, et al. Jnk expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science (2013) 339(6116):218–22. doi: 10.1126/science.1227568
144. Bost F, Aouadi M, Caron L, Binetruy B. The role of mapks in adipocyte differentiation and obesity. Biochimie (2005) 87(1):51–6. doi: 10.1016/j.biochi.2004.10.018
145. Bost F, Aouadi M, Caron L, Even P, Belmonte N, Prot M, et al. The extracellular signal-regulated kinase isoform Erk1 is specifically required for in vitro and in vivo adipogenesis. Diabetes (2005) 54(2):402–11. doi: 10.2337/diabetes.54.2.402
146. Souza SC, Palmer HJ, Kang YH, Yamamoto MT, Muliro KV, Paulson KE, et al. Tnf-alpha induction of lipolysis is mediated through activation of the extracellular signal related kinase pathway in 3t3-L1 adipocytes. J Cell Biochem (2003) 89(6):1077–86. doi: 10.1002/jcb.10565
147. Rodriguez A, Duran A, Selloum M, Champy MF, Diez-Guerra FJ, Flores JM, et al. Mature-onset obesity and insulin resistance in mice deficient in the signaling adapter P62. Cell Metab (2006) 3(3):211–22. doi: 10.1016/j.cmet.2006.01.011
148. Ying W, Riopel M, Bandyopadhyay G, Dong Y, Birmingham A, Seo JB, et al. Adipose tissue macrophage-derived exosomal mirnas can modulate in vivo and in vitro insulin sensitivity. Cell (2017) 171(2):372–84.e12. doi: 10.1016/j.cell.2017.08.035
149. Wilson AM, Shao Z, Grenier V, Mawambo G, Daudelin JF, Dejda A, et al. Neuropilin-1 expression in adipose tissue macrophages protects against obesity and metabolic syndrome. Sci Immunol (2018) 3(21): eaan4626. doi: 10.1126/sciimmunol.aan4626
150. Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, et al. Lipid-associated macrophages control metabolic homeostasis in a Trem2-dependent manner. Cell (2019) 178(3):686–98.e14. doi: 10.1016/j.cell.2019.05.054
151. Sharif O, Brunner JS, Korosec A, Martins R, Jais A, Snijder B, et al. Beneficial metabolic effects of Trem2 in obesity are uncoupled from its expression on macrophages. Diabetes (2021) 70(9):2042–57. doi: 10.2337/db20-0572
152. Khan T, Muise ES, Iyengar P, Wang ZV, Chandalia M, Abate N, et al. Metabolic dysregulation and adipose tissue fibrosis: Role of collagen vi. Mol Cell Biol (2009) 29(6):1575–91. doi: 10.1128/MCB.01300-08
153. Mariman EC, Wang P. Adipocyte extracellular matrix composition, dynamics and role in obesity. Cell Mol Life Sci (2010) 67(8):1277–92. doi: 10.1007/s00018-010-0263-4
154. Sun K, Tordjman J, Clement K, Scherer PE. Fibrosis and adipose tissue dysfunction. Cell Metab (2013) 18(4):470–7. doi: 10.1016/j.cmet.2013.06.016
155. Halberg N, Khan T, Trujillo ME, Wernstedt-Asterholm I, Attie AD, Sherwani S, et al. Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol Cell Biol (2009) 29(16):4467–83. doi: 10.1128/MCB.00192-09
156. Engin A. Adipose tissue hypoxia in obesity and its impact on preadipocytes and macrophages: Hypoxia hypothesis. Adv Exp Med Biol (2017) 960:305–26. doi: 10.1007/978-3-319-48382-5_13
157. Henegar C, Tordjman J, Achard V, Lacasa D, Cremer I, Guerre-Millo M, et al. Adipose tissue transcriptomic signature highlights the pathological relevance of extracellular matrix in human obesity. Genome Biol (2008) 9(1):R14. doi: 10.1186/gb-2008-9-1-r14
158. Sun K, Kusminski CM, Scherer PE. Adipose tissue remodeling and obesity. J Clin Invest (2011) 121(6):2094–101. doi: 10.1172/JCI45887
159. Song E, Ouyang N, Horbelt M, Antus B, Wang M, Exton MS. Influence of alternatively and classically activated macrophages on fibrogenic activities of human fibroblasts. Cell Immunol (2000) 204(1):19–28. doi: 10.1006/cimm.2000.1687
160. Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol (2008) 214(2):199–210. doi: 10.1002/path.2277
161. Vila IK, Badin PM, Marques MA, Monbrun L, Lefort C, Mir L, et al. Immune cell toll-like receptor 4 mediates the development of obesity- and endotoxemia-associated adipose tissue fibrosis. Cell Rep (2014) 7(4):1116–29. doi: 10.1016/j.celrep.2014.03.062
162. Tanaka M, Ikeda K, Suganami T, Komiya C, Ochi K, Shirakawa I, et al. Macrophage-inducible c-type lectin underlies obesity-induced adipose tissue fibrosis. Nat Commun (2014) 5:4982. doi: 10.1038/ncomms5982
163. Madsen DH, Leonard D, Masedunskas A, Moyer A, Jurgensen HJ, Peters DE, et al. M2-like macrophages are responsible for collagen degradation through a mannose receptor-mediated pathway. J Cell Biol (2013) 202(6):951–66. doi: 10.1083/jcb.201301081
164. Keophiphath M, Achard V, Henegar C, Rouault C, Clement K, Lacasa D. Macrophage-secreted factors promote a profibrotic phenotype in human preadipocytes. Mol Endocrinol (2009) 23(1):11–24. doi: 10.1210/me.2008-0183
165. Waki H, Tontonoz P. Endocrine functions of adipose tissue. Annu Rev Pathol (2007) 2:31–56. doi: 10.1146/annurev.pathol.2.010506.091859
166. Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF. A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem (1995) 270(45):26746–9. doi: 10.1074/jbc.270.45.26746
167. Hu E, Liang P, Spiegelman BM. Adipoq is a novel adipose-specific gene dysregulated in obesity. J Biol Chem (1996) 271(18):10697–703. doi: 10.1074/jbc.271.18.10697
168. Yokota T, Oritani K, Takahashi I, Ishikawa J, Matsuyama A, Ouchi N, et al. Adiponectin, a new member of the family of soluble defense collagens, negatively regulates the growth of myelomonocytic progenitors and the functions of macrophages. Blood (2000) 96(5):1723–32. doi: 10.1182/blood.V96.5.1723
169. Gil-Campos M, Canete RR, Gil A. Adiponectin, the missing link in insulin resistance and obesity. Clin Nutr (2004) 23(5):963–74. doi: 10.1016/j.clnu.2004.04.010
170. Haluzik M, Parizkova J, Haluzik MM. Adiponectin and its role in the obesity-induced insulin resistance and related complications. Physiol Res (2004) 53(2):123–9.
171. Wolf AM, Wolf D, Rumpold H, Enrich B, Tilg H. Adiponectin induces the anti-inflammatory cytokines il-10 and il-1ra in human leukocytes. Biochem Biophys Res Commun (2004) 323(2):630–5. doi: 10.1016/j.bbrc.2004.08.145
172. Tsatsanis C, Zacharioudaki V, Androulidaki A, Dermitzaki E, Charalampopoulos I, Minas V, et al. Adiponectin induces tnf-alpha and il-6 in macrophages and promotes tolerance to itself and other pro-inflammatory stimuli. Biochem Biophys Res Commun (2005) 335(4):1254–63. doi: 10.1016/j.bbrc.2005.07.197
173. Park PH, McMullen MR, Huang H, Thakur V, Nagy LE. Short-term treatment of Raw264.7 macrophages with adiponectin increases tumor necrosis factor-alpha (Tnf-alpha) expression Via Erk1/2 activation and egr-1 expression: Role of tnf-alpha in adiponectin-stimulated interleukin-10 production. J Biol Chem (2007) 282(30):21695–703. doi: 10.1074/jbc.M701419200
174. Hui X, Gu P, Zhang J, Nie T, Pan Y, Wu D, et al. Adiponectin enhances cold-induced browning of subcutaneous adipose tissue Via promoting M2 macrophage proliferation. Cell Metab (2015) 22(2):279–90. doi: 10.1016/j.cmet.2015.06.004
175. Ajuwon KM, Spurlock ME. Adiponectin inhibits lps-induced nf-kappab activation and il-6 production and increases Ppargamma2 expression in adipocytes. Am J Physiol Regul Integr Comp Physiol (2005) 288(5):R1220–5. doi: 10.1152/ajpregu.00397.2004
176. Park PH, Huang H, McMullen MR, Mandal P, Sun L, Nagy LE. Suppression of lipopolysaccharide-stimulated tumor necrosis factor-alpha production by adiponectin is mediated by transcriptional and post-transcriptional mechanisms. J Biol Chem (2008) 283(40):26850–8. doi: 10.1074/jbc.M802787200
177. Fasshauer M, Kralisch S, Klier M, Lossner U, Bluher M, Klein J, et al. Adiponectin gene expression and secretion is inhibited by interleukin-6 in 3t3-L1 adipocytes. Biochem Biophys Res Commun (2003) 301(4):1045–50. doi: 10.1016/s0006-291x(03)00090-1
178. Loffreda S, Yang SQ, Lin HZ, Karp CL, Brengman ML, Wang DJ, et al. Leptin regulates proinflammatory immune responses. FASEB J (1998) 12(1):57–65. doi: 10.1096/fsb2fasebj.12.1.57
179. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature (1994) 372(6505):425–32. doi: 10.1038/372425a0
180. Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature (1998) 395(6704):763–70. doi: 10.1038/27376
181. Procaccini C, Jirillo E, Matarese G. Leptin as an immunomodulator. Mol Aspects Med (2012) 33(1):35–45. doi: 10.1016/j.mam.2011.10.012
182. Fernandez-Riejos P, Najib S, Santos-Alvarez J, Martin-Romero C, Perez-Perez A, Gonzalez-Yanes C, et al. Role of leptin in the activation of immune cells. Mediators Inflammation (2010) 2010:568343. doi: 10.1155/2010/568343
183. Howard JK, Lord GM, Matarese G, Vendetti S, Ghatei MA, Ritter MA, et al. Leptin protects mice from starvation-induced lymphoid atrophy and increases thymic cellularity in Ob/Ob mice. J Clin Invest (1999) 104(8):1051–9. doi: 10.1172/JCI6762
184. Faggioni R, Feingold KR, Grunfeld C. Leptin regulation of the immune response and the immunodeficiency of malnutrition. FASEB J (2001) 15(14):2565–71. doi: 10.1096/fj.01-0431rev
185. Siegmund B, Lear-Kaul KC, Faggioni R, Fantuzzi G. Leptin deficiency, not obesity, protects mice from con a-induced hepatitis. Eur J Immunol (2002) 32(2):552–60. doi: 10.1002/1521-4141(200202)32:2<552::AID-IMMU552>3.0.CO;2-H
186. Matarese G, Di Giacomo A, Sanna V, Lord GM, Howard JK, Di Tuoro A, et al. Requirement for leptin in the induction and progression of autoimmune encephalomyelitis. J Immunol (2001) 166(10):5909–16. doi: 10.4049/jimmunol.166.10.5909
187. Zarkesh-Esfahani H, Pockley G, Metcalfe RA, Bidlingmaier M, Wu ZD, Ajami A, et al. High-dose leptin activates human leukocytes Via receptor expression on monocytes. J Immunol (2001) 167(8):4593–9. doi: 10.4049/jimmunol.167.8.4593
188. Mancuso P, Curtis JL, Freeman CM, Peters-Golden M, Weinberg JB, Myers MG. Ablation of the leptin receptor in myeloid cells impairs pulmonary clearance of streptococcus pneumoniae and alveolar macrophage bactericidal function. Am J Physiol-Lung C (2018) 315(1):L78–86. doi: 10.1152/ajplung.00447.2017
189. Kiguchi N, Maeda T, Kobayashi Y, Fukazawa Y, Kishioka S. Leptin enhances cc-chemokine ligand expression in cultured murine macrophage. Biochem Bioph Res Co (2009) 384(3):311–5. doi: 10.1016/j.bbrc.2009.04.121
190. Conde J, Scotece M, Gomez R, Gomez-Reino JJ, Lago F, Gualillo O. At The crossroad between immunity and metabolism: Focus on leptin. Expert Rev Clin Immunol (2010) 6(5):801–8. doi: 10.1586/eci.10.48
191. Zhou Y, Yu X, Chen H, Sjoberg S, Roux J, Zhang L, et al. Leptin deficiency shifts mast cells toward anti-inflammatory actions and protects mice from obesity and diabetes by polarizing M2 macrophages. Cell Metab (2015) 22(6):1045–58. doi: 10.1016/j.cmet.2015.09.013
192. Landman RE, Puder JJ, Xiao E, Freda PU, Ferin M, Wardlaw SL. Endotoxin stimulates leptin in the human and nonhuman primate. J Clin Endocrinol Metab (2003) 88(3):1285–91. doi: 10.1210/jc.2002-021393
193. Faggioni R, Fantuzzi G, Fuller J, Dinarello CA, Feingold KR, Grunfeld C. Il-1 beta mediates leptin induction during inflammation. Am J Physiol (1998) 274(1):R204–8. doi: 10.1152/ajpregu.1998.274.1.R204
194. Villarroya F, Cereijo R, Villarroya J, Gavalda-Navarro A, Giralt M. Toward an understanding of how immune cells control brown and beige adipobiology. Cell Metab (2018) 27(5):954–61. doi: 10.1016/j.cmet.2018.04.006
195. Lynch L, Hogan AE, Duquette D, Lester C, Banks A, LeClair K, et al. Inkt cells induce Fgf21 for thermogenesis and are required for maximal weight loss in Glp1 therapy. Cell Metab (2016) 24(3):510–9. doi: 10.1016/j.cmet.2016.08.003
196. Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, et al. Cd8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med (2009) 15(8):914–20. doi: 10.1038/nm.1964
197. Arguello RJ, Combes AJ, Char R, Gigan JP, Baaziz AI, Bousiquot E, et al. Scenith: A flow cytometry-based method to functionally profile energy metabolism with single-cell resolution. Cell Metab (2020) 32(6):1063–75.e7. doi: 10.1016/j.cmet.2020.11.007
198. Artyomov MN, Van den Bossche J. Immunometabolism in the single-cell era. Cell Metab (2020) 32(5):710–25. doi: 10.1016/j.cmet.2020.09.013
199. Shi J, Hua L, Harmer D, Li P, Ren G. Cre driver mice targeting macrophages. Methods Mol Biol (2018) 1784:263–75. doi: 10.1007/978-1-4939-7837-3_24
200. Faust N, Varas F, Kelly LM, Heck S, Graf T. Insertion of enhanced green fluorescent protein into the lysozyme gene creates mice with green fluorescent granulocytes and macrophages. Blood (2000) 96(2):719–26. doi: 10.1182/blood.V96.2.719
201. Zhang F, Hao G, Shao M, Nham K, An Y, Wang Q, et al. An adipose tissue atlas: An image-guided identification of human-like bat and beige depots in rodents. Cell Metab (2018) 27(1):252–62.e3. doi: 10.1016/j.cmet.2017.12.004
Keywords: adipose tissue macrophage, obesity, adaptive thermogenesis, insulin resistance, fibrosis, adipokines
Citation: Yao J, Wu D and Qiu Y (2022) Adipose tissue macrophage in obesity-associated metabolic diseases. Front. Immunol. 13:977485. doi: 10.3389/fimmu.2022.977485
Received: 24 June 2022; Accepted: 18 August 2022;
Published: 02 September 2022.
Edited by:
Lisardo Bosca, University of Madrid, SpainReviewed by:
Yuxiang Sun, Texas A&M University, United StatesJingbo Pang, University of Illinois at Chicago, United States
Copyright © 2022 Yao, Wu and Qiu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yifu Qiu, eWlmdS5xaXVAcGt1LmVkdS5jbg==