 Daiki Niizato1
Daiki Niizato1 Takeshi Isoda1*Noriko Mitsuiki1Shuya Kaneko1Dan Tomomasa1Takahiro Kamiya2
Takeshi Isoda1*Noriko Mitsuiki1Shuya Kaneko1Dan Tomomasa1Takahiro Kamiya2 Masatoshi Takagi1
Masatoshi Takagi1 Kohsuke Imai3,4Michiko Kajiwara5
Kohsuke Imai3,4Michiko Kajiwara5 Masaki Shimizu6
Masaki Shimizu6 Tomohiro Morio1
Tomohiro Morio1 Hirokazu Kanegane6
Hirokazu Kanegane6- 1Department of Pediatrics and Developmental Biology, Tokyo Medical and Dental University (TMDU), Tokyo, Japan
- 2Department of Clinical Research Center, Tokyo Medical and Dental University (TMDU), Tokyo, Japan
- 3Department of Community Pediatrics, Perinatal and Maternal Medicine, Tokyo Medical and Dental University (TMDU), Tokyo, Japan
- 4Department of Pediatrics, National Defense Medical College, Tokorozawa, Japan
- 5Center for Blood Transfusion and Cell Therapy, Tokyo Medical and Dental University Hospital, Tokyo, Japan
- 6Department of Child Health and Development, Tokyo Medical and Dental University (TMDU), Tokyo, Japan
Familial hemophagocytic lymphohistiocytosis (FHL) is a rare and fatal autosomal recessive immune disorder characterized by uncontrolled activation of T and NK cells, macrophages, and overproduction of inflammatory cytokines. Early hematopoietic cell transplantation (HCT) is required for long-term survival. Current therapy is based on the HLH-94/2004 protocol, but is insufficient to fully control disease activity. This case report describes an infant with FHL type 3 who, despite initial therapy with dexamethasone and etoposide, showed aberrant cytokine levels, including interleukin-18 (IL-18), chemokine ligand 9 (CXCL9), soluble interleukin-2 receptor (sIL-2R), and soluble tumor necrosis factor receptor type II (sTNF-RII). The Janus kinase inhibitor ruxolitinib was therefore coadministered. The patient was treated with dose-adjusted ruxolitinib guided by cytokine profiles, and was successfully prepared for HCT. The results demonstrate the effectiveness and safety of dose-adjusted ruxolitinib as a bridging therapy for FHL, and the value of monitoring cytokine levels, especially IL-18, CXCL9, sIL-2R, and sTNF-RII, as disease-activity markers for FHL.
Introduction
Familial hemophagocytic lymphohistiocytosis (FHL) is a fatal autosomal recessive immune disorder related to the dysfunction of cytotoxic, granule-mediated cell-death pathways. Prolonged activation of T and NK cells lacking the ability to kill target cells produces a high amount of cytokines, including interferon (IFN)-γ. This leads to the infiltration of activated macrophages and the production of interleukin (IL)-6, IL-18, and tumor necrosis factor (TNF)-α (1). Activation of T cells and macrophages plays a significant role in symptom development and organ damage in FHL. FHL type 3 (FHL3) is caused by a variant of the UNC13D gene encoding Munc13-4, which regulates the secretory process of cytotoxic granules of lymphocytes (2–4). In addition to the common clinical and laboratory findings, half of FHL3 patients present central nervous system (CNS) involvement and malignancy (4, 5). FHL is a fatal disease that requires early definitive diagnosis so that appropriate bridging therapy can be started until curative hematopoietic cell transplantation (HCT) is possible (4).
Immunomodulation of hemophagocytic lymphohistiocytosis (HLH) based on the pathophysiology and disease activity is warranted. However, specific cytokines that allow discrimination between FHL and secondary HLH have not been defined (6). Also, proteome analysis did not identify differential expression between primary and secondary HLH (7). Monitoring of serum cytokine profiles is effective in evaluating inflammatory status in cell therapies, infections, cancers, and auto-immune diseases (8). Time-course cytokine monitoring allows for appropriate selection and dosage of immunomodulators and is extremely important for understanding the pathogenesis of FHL. However, specific cytokines for monitoring disease activity have not been established in FHL patients.
FHL patients are predisposed to developing severe HLH even in utero or at birth, which can be fatal due to multiple organ failure, and often fail to survive until curative HCT (9–11). In the treatment strategy based on HLH-94/2004 protocols, HLH disease activity frequently recurs during steroid reduction and prolonged intervals of etoposide administration (12, 13). The HLH-2004 protocol has not improved pre-transplant patient mortality or improved survival (13), necessitating the need for novel therapeutic agents.
Ruxolitinib, a Janus kinase (JAK) inhibitor, has been used in the treatment of myelofibrosis, polycythemia vera, refractory acute and chronic graft-versus-host disease (GVHD), as well as COVID-19 acute respiratory distress syndrome and post-transplant idiopathic lung injury (14–19). Ruxolitinib is known to inhibit JAK1, JAK2, and signal transducer and activator of transcription (STAT) 3 pathways, thereby reducing downstream signaling and cytokine production, including IL-6 and IFN-γ (20–22). Reports of the use of ruxolitinib as an immunomodulator for FHL have been published (11, 23–29). However, the dose of ruxolitinib, combination of other therapies, and investigated cytokines were diversified.
In this case report, we describe an infant with FHL3 who underwent successful HCT following bridging therapy with ruxolitinib. The ruxolitinib dosage was adjusted based on monitored cytokine profiles.
Results
Case report
A 1-month-old boy was admitted to hospital with failure-to-thrive and fever. He was found to have thrombocytopenia (platelets 29 × 109/L), hyperferritinemia (ferritin 1,912 ng/mL), and elevated soluble interleukin-2 receptor (sIL-2R) (26,489 U/mL), which suggested HLH (Figures 1A, B, F), and referred to our hospital. Upon admission, physical examination revealed fever, decreased oxygenation (SpO2 93% on room air), hepatosplenomegaly, and a skin rash (Figure 1B). Complete blood count showed pancytopenia, with white blood cells 2,800/µL, neutrophils 840/µL, hemoglobin level 7.4 g/dL, and platelets 2.9 × 104/µL (Figure 1F). Bone marrow aspiration at initial diagnosis provided insufficient material with peripheral blood contamination. Except for no confirmation of hemophagocytosis in the bone marrow aspiration, seven of the eight diagnostic criteria for HLH-2004 were fulfilled, namely fever, splenomegaly, pancytopenia, hypofibrinogenemia (1.09 g/L), absent natural killer (NK) cell activity, hyperferritinemia, and an increased sIL-2R level (30). Lumbar puncture, with cytology and cytokine evaluation, and brain magnetic resonance imaging showed no CNS involvement. On day one of admission (95 days before HCT), serum cytokine levels of IL-6, IL-18, chemokine ligand 9 (CXCL9), and soluble tumor necrosis factor receptor type II (sTNF-RII) were measured using ELISA according to the manufacturer’s instructions (IL-18: MBL, Nagoya, Japan; IL-6, CXCL9, and sTNF-RII: R&D Systems, Inc., Minneapolis, MN, USA) (31). All serum cytokine levels were elevated with IL-6 6 pg/mL (normal, <3 pg/mL), IL-18 7,300 pg/mL (normal, <500 pg/mL), CXCL9 8,116 pg/mL (normal, 31-83 pg/mL), and sTNF-RII 27,008 pg/mL (normal, 829-2,262 pg/mL) (Figures 1C–E). A CD107a assay showed no degranulation of NK cells, suggesting FHL. Flow cytometry showed decreased Munc13-4 expression. Sequencing analysis of the UNC13D gene, including in the parents, revealed the c.2346-2349del (p.Arg782fs) inherited from father, and tandem duplication of exons 7-12 inherited from mother, which were previously reported as pathogenic, leading to the definitive diagnosis of FHL3 (32, 33). Further details of the genetic diagnosis are described separately (34). An HLA-matched donor could not be found in the cord blood or bone marrow bank. Induction therapy with 6 mg/m2 dexamethasone (DEX) and 33 mg/m2 etoposide weekly was started based on a modified HLH-94 protocol. DEX administration was started at a two-thirds dose (Figure 1A). The dose of etoposide was determined by body weight using the following conversion formula in accordance with the AML-05 protocol for infants (35): Dose = 100 mg body surface area × (body weight (kg)/30). The dose was further reduced to two-thirds.
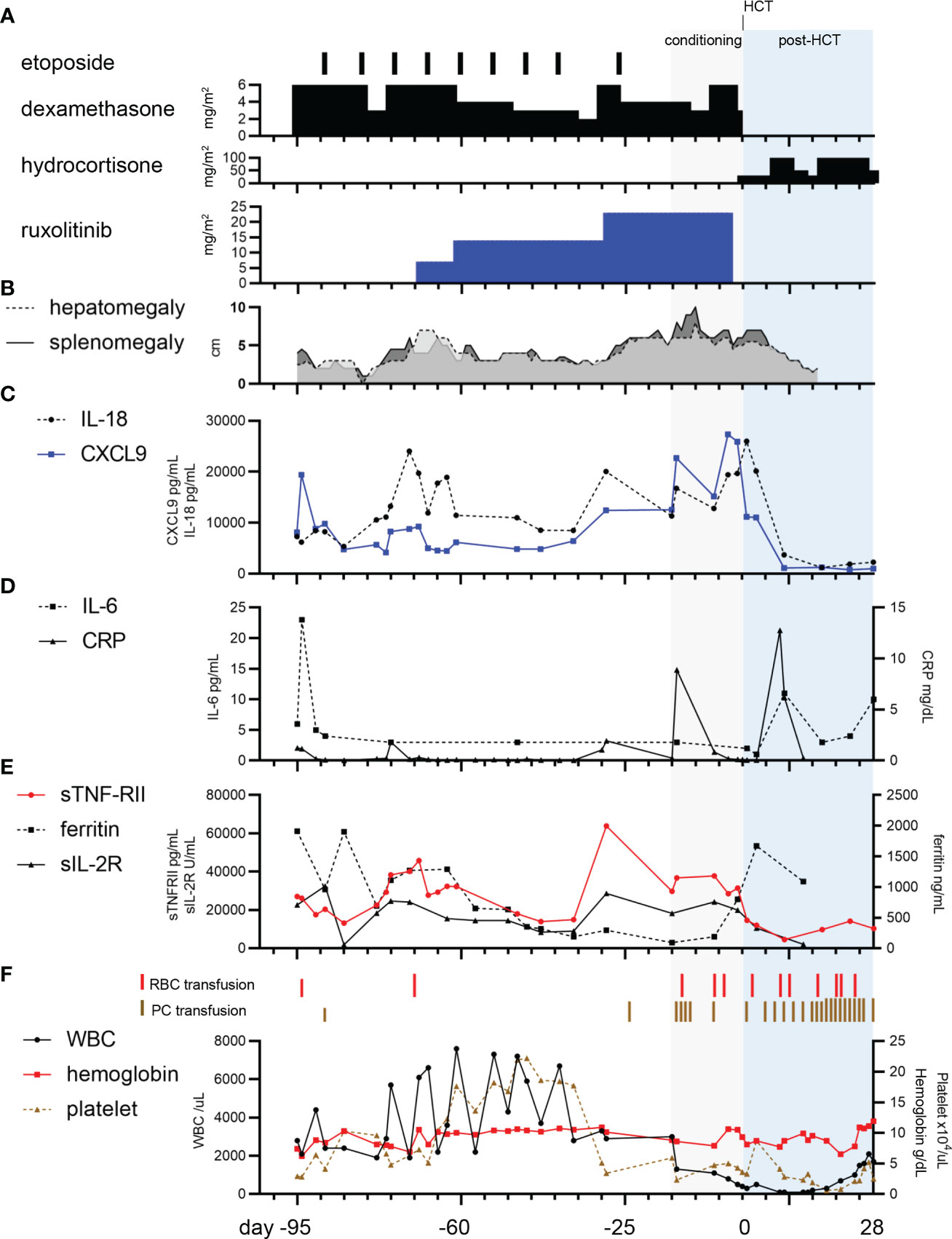
Figure 1 Clinical course for the patient. (A) Etoposide, dexamethasone, and ruxolitinib were used as bridging therapies. Hydrocortisone was given for maintenance therapy. (B) The clinical course of hepatosplenomegaly. (C) The panel shows the trajectory of chemokine ligand 9 (CXCL9) and interleukin-18 (IL-18). (D). The panel shows the course of interleukin-6 (IL-6) and C-reactive protein (CRP). (E) The panel shows the course of ferritin, soluble interleukin-2 receptor (sIL-2R), and soluble tumor necrosis factor receptor type II (sTNF-RII) (F) The panel shows the timing of red blood cell (RBC) and platelet concentrate (PC) transfusion, and complete blood count tracking.
The course of bridging therapy before conditioning for HCT
DEX was reduced from 6 to 3 mg/m2 16 days after admission, which led to elevated levels of cytokines IL-18, CXCL9, sIL-2R, and sTNF-RII, while IL-6 remained below the cut-off level (Figures 1C, D, E). Despite DEX being increased to the initial dose on day 21, worsening of recurrent fever, hepatosplenomegaly, and elevated cytokine levels, indicated that the inflammation was difficult to control with the combination of DEX and etoposide (Figures 1B, C, E). Ruxolitinib was therefore initiated at 7 mg/m2 (2.5 mg/day) on day 27. After the ruxolitinib initiation, cytokine levels for IL-18, CXCL9, sTNF-RII, and sIL-2R showed a declining trend (Figures 1C, E). Instead of reduction of DEX from 6 mg/m2 to 4 mg/m2, the dose of ruxolitinib was increased from 7 to 14 mg/m2 on day 35, which lead to a gradual decrease of cytokine levels until day 60.
To delay HCT for the young infant and reduce complications, we attempted to extend the duration of etoposide administration and reduce the dosage of systemic steroids. However, the recurrent elevation of cytokine levels required preparation for early HCT. Thus, DEX was temporarily increased to 6 mg/m2 and ruxolitinib was increased to 23 mg/m2 on day 67 (29 days before HCT). We settled the timing of HCT when he reached at least 4-month-old, which is the youngest patient with FHL using posttransplant cyclophosphamide method (36).
The course of HCT
The conditioning regimen, which included alemtuzumab 0.496 mg/kg, fludarabine 180 mg/m2, and busulfan AUC 65 mg×h/L, was started 15 days before HCT. Excellent outcomes for primary HLH have been reported with this reduced-intensity conditioning (37). Immediately after the administration of alemtuzumab, cytokine release syndrome (CRS), which includes fever, hypoxia, hepatomegaly with elevated hepatic enzymes, and rash, was observed. CRS was carefully managed with an additional dosage of the methylprednisolone, cytokine monitoring and laboratory tests. Ruxolitinib was discontinued one day before HCT because of possible negative effects on engraftment and a lack of sufficient data for continuation of ruxolitinib in GVHD prevention (38). Bone marrow transplantation from the haploidentical father (A-, B-, and C-mismatched) was performed when the patient was 4 months old. GVHD prophylaxis included posttransplant cyclophosphamide, tacrolimus, and mycophenolate mofetil (39). Cytokines IL-18, CXCL9, sIL-2R, and sTNF-RII immediately showed a profound reduction after HCT. Engraftment was achieved on day 25, complete chimerism was achieved on day 66. Although the patient developed sinusoidal obstructive syndrome (SOS), thrombotic microangiopathy (TMA) and pulmonary hypertension (PH), these complications were successfully managed. Ruxolitinib was used for TMA management after PH development during the course of haplo-HCT. Ruxolitinib was successfully used for GVHD prophylaxis instead of a calcineurin inhibitor and did not worsen the status of SOS, TMA, and PH. On day 286, the patient had remained without GVHD and recurrence of HLH, SOS, TMA, and PH after cessation of ruxolitinib and tacrolimus.
Literature search
Among 38 papers found as part of the literature review of “ruxolitinib, hemophagocytic lymphohistiocytosis, FHL, and HLH” in PubMed, we selected 12 cases with primary HLH from case reports, retrospective, and prospective studies. The dose of ruxolitinib, combination of other therapies, and monitored cytokines were varied in 13 cases including our case. Except for one case without outcome record, 10 of 12 cases treated with ruxolitinib survived without any severe adverse events (Table 1).
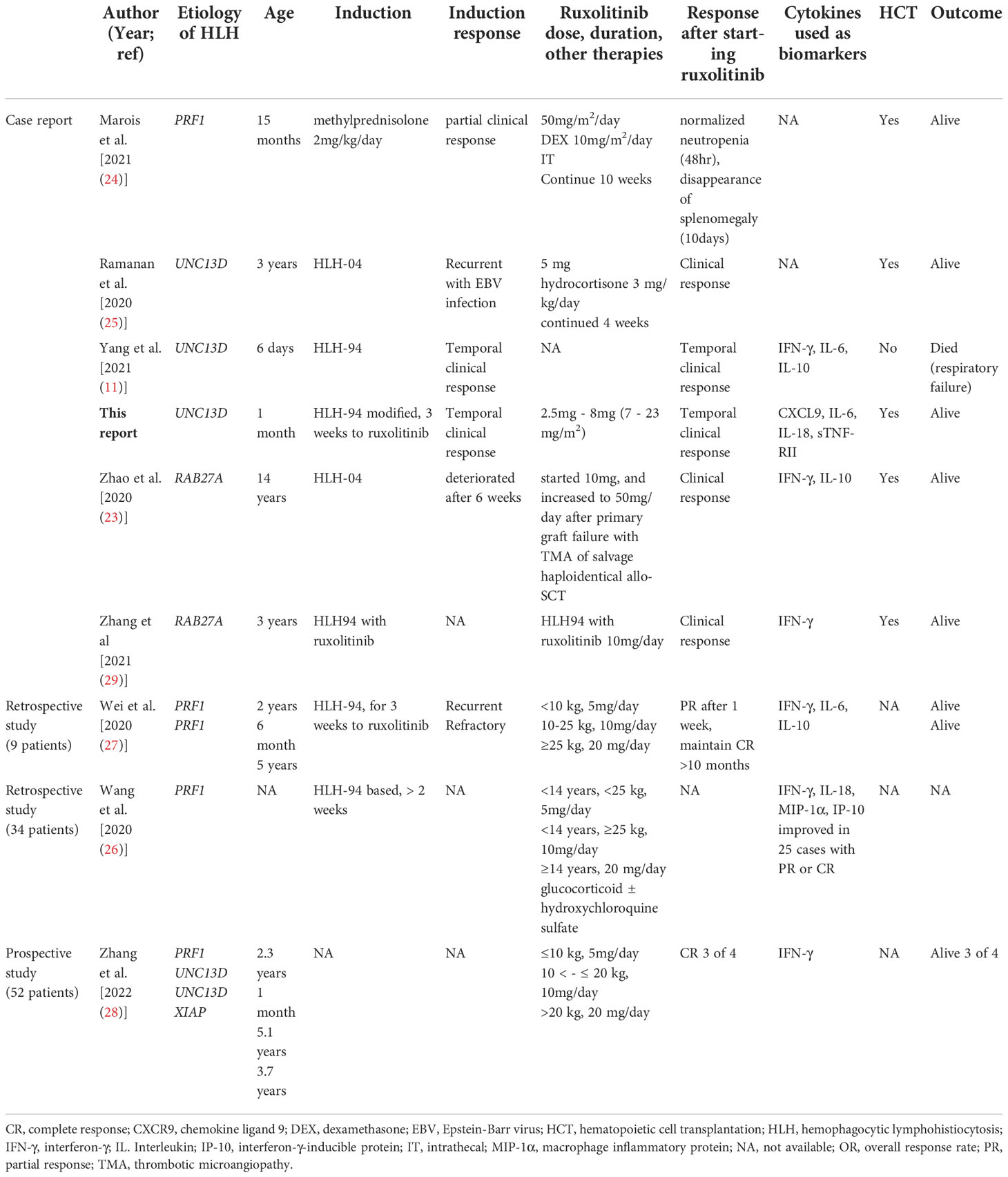
Table 1 Literature review: Ruxolitinib for FHL in case reports and prospective study.
Discussion
In this case study, the dose adjustment of ruxolitinib, guided by cytokine monitoring, was an effective bridging therapy, and minimized the required dose of etoposide and DEX. This strategy also contributed to the decision of HCT timing with maintaining performance status. However, the cytokine levels of IL-18, CXCL9, sIL-2R, and sTNF-RII remained elevated above cutoff levels, suggesting that adequate doses of ruxolitinib combined with HLH94/2004-based therapy would be necessary for the treatment of FHL.
Past reports showed that cytokine levels, including IL-6 (cutoff level, 37.25 pg/mL), at initial diagnosis predict early death in children with HLH (40). The IL-6 level in our case was consistently below this cutoff (Figure 1D). The CXCL9/IL-6 ratio is discriminately elevated in HLH compared with systemic inflammatory response syndrome/sepsis (7). Similarly, we observed that serum CXCL9 which is IFN-γ-inducible CXCR3 ligands and reflecting amplification of IFN-γ signaling wathway (41), was consistently higher than the normal range throughout the clinical course in our case (Figures 1C, D). IL-18, sTNF-RII, and sIL-2R showed a similar trend to CXCL9 (Figures 1C, E). During the disease course, the patient had three episodes of fever without any apparent sign of infection, negativity in blood cultures, and the highest CRP level was 1.9 mg/dL. Thus, we monitored cytokines, which directly reflected FHL activity in our case. Further validation is warranted to determine whether these cytokine profiles are effective to monitor disease activities of FHL.
Effective salvage therapy has not been established for refractory FHL. A retrospective study using rabbit antithymocyte globulin (ATG) showed that one-third of patients relapsed after first course of ATG, and 19 of 38 patients who received HCT after first or second-line ATG have been cured. The median time between ATG therapy and HCT was 6 weeks (42). Alemtuzumab as salvage therapy showed partial responses and response was limited to two weeks (43). Both are not suitable salvage therapies for 1-3 months old infants who are required to wait for sufficient growth before HCT. Infectious complications and cytokine release syndrome are concerns on both therapies. Thus, alternative therapies are urgently required.
Hyperactivation of effector CD8+ T cells with overproduction of IFN-γ contributes to FHL pathogenesis (44). In a mouse model of FHL2, loss of IFN-γ receptors in hepatocytes reduced liver dysfunction, suggesting that a non-hematopoietic response is also crucial in developing FHL hepatitis (45). These findings suggest that targeting IFN-γ may be a promising treatment strategy. However, a recent clinical trial using the anti-IFN-γ monoclonal antibody emapalumab has not been successful (46). Despite the combination of emapalumab and dexamethasone, only 14 out of 34 cases (41%) survived without additional therapy. In addition, only 70% of patients in the emapalumab-treated group bridged to HCT, which was inferior to the 80% in the HLH-2004 study (13, 46, 47).
Ruxolitinib inhibits the JAK/STAT pathway, which is one of the main cytokine signaling pathways in T-cell activation and the maintenance of activation (22). The mouse FHL2 model suggested that ruxolitinib inhibits HLH through IFN-γ-dependent and independent pathways (48). Ruxolitinib acts not only on T cells, but also on human macrophages and inhibits the production of both IFN-γ and IL-6 (49). Our literature review showed that ruxolitinib was used for 13 FHL patients, including our case (Table 1). In a recent single-arm study involving four FHL patients, three of them responded to ruxolitinib monotherapy. The study investigated 52 cases, including systemic autoinflammatory-associated HLH, chronic active Epstein-Barr virus infection (CAEBV), and EBV-HLH. The cytokine profiles, including sIL-2R, ferritin, and IFN-γ, of good responders normalized within two weeks (28), whereas non-responders required additional therapies. In the report, cytokine profiles of four patients with FHL were not shown (28). Cytokine profiles in our case and neonatal-onset FHL showed a non-responder pattern, suggesting that additional therapies are required for these cases (11). Except for one case without outcome information, 10 out of 12 cases were successfully managed by additional ruxolitinib administration (Table 1). In recent a case of Griscelli syndrome type 2, ruxolitinib was used with HLH-94-based therapy for initial induction, and successfully led to HCT (29). Emapalumab and ruxolitinib combination therapy for a refractory CAEBV case has been reported (50). However, the dose of ruxolitinib, combination of other therapies, and investigated laboratory markers, including cytokines, were varied. The prospective clinical trial (NCT04551131) is crucial for determining response using 50mg/m2/day of ruxolitinib with dexamethasone for FHL. Selection of inflammatory markers and determining the appropriate cutoff value will be required in prospective studies.
Concluding remark
Cytokine monitoring and trends of IL-18, CXCL9, sIL-2R, and sTNF-RII might reflect disease activity in FHL. Monitoring cytokine levels and administering ruxolitinib, combined with the HLH-94/2004 protocol or other recent target therapies, may be a potent bridging therapy for FHL patients.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by the ethics boards of the Tokyo Medical and Dental University (TMDU) (G2019-004). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
DN and TI wrote the manuscript. Literature review: DN and TI. Measurement of cytokines: SK and MS. Acquisition and interpretation of data: DN, TI, NM, SK, DT, TK, MS, and HK. Supervision of the study: MT, KI, MK, MS, and TM. Conceptualization of the study and editing the manuscript: HK. All authors contributed to the article and approved the submitted version.
Funding
This work was supported in part by JSPS KAKENHI Grant Number (21H02878) and Takeda Science Foundation to TI and by the joint research grant with Medical and Biological Laboratories Co., Ltd (MBL) to MS. KAKENHI contributed an English editing fee. KAKENHI and Takeda Science Foundation contributed open access publication fee. The Joint research grant with MBL covered the reagents for cytokine monitoring. The funder, Medical and Biological Laboratories Co., Ltd, was not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication.
Acknowledgments
We are grateful to Drs. Takayuki Miyamoto and Takahiro Yasumi for performing flow cytometry of Munc13-4. We thank the patient and his parents for participating in this study. We also thank the staff at the Department of Pediatrics, Tokyo Medical and Dental University. We thank Barbara Spathelf, PhD, from Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Fischer A, Latour S, de Saint Basile G. Genetic defects affecting lymphocyte cytotoxicity. Curr Opin Immunol (2007) 19(3):348–53. doi: 10.1016/j.coi.2007.04.006
2. Ménager MM, Ménasché G, Romao M, Knapnougel P, Ho C-H, Garfa M, et al. Secretory cytotoxic granule maturation and exocytosis require the effector protein hMunc13-4. Nat Immunol (2007) 8(3):257–67. doi: 10.1038/ni1431
3. Feldmann J, Callebaut I, Raposo G, Certain S, Bacq D, Dumont C, et al. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell (2003) 115(4):461–73. doi: 10.1016/S0092-8674(03)00855-9
4. Amirifar P, Ranjouri MR, Abolhassani H, Moeini Shad T, Almasi-Hashiani A, Azizi G, et al. Clinical, immunological and genetic findings in patients with UNC13D deficiency (FHL3): A systematic review. Pediatr Allergy Immunol (2021) 32(1):186–97. doi: 10.1111/pai.13323
5. Sieni E, Cetica V, Santoro A, Beutel K, Mastrodicasa E, Meeths M, et al. Genotype–phenotype study of familial haemophagocytic lymphohistiocytosis type 3. J Med Genet (2011) 48(5):343–52. doi: 10.1136/jmg.2010.085456
6. Wada T, Muraoka M, Yokoyama T, Toma T, Kanegane H, Yachie A. Cytokine profiles in children with primary Epstein-Barr virus infection. Pediatr Blood Cancer (2013) 60(7):E46–8. doi: 10.1002/pbc.24480
7. Lin H, Scull BP, Goldberg BR, Abhyankar HA, Eckstein OE, Zinn DJ, et al. IFN-γ signature in the plasma proteome distinguishes pediatric hemophagocytic lymphohistiocytosis from sepsis and SIRS. Blood Adv (2021) 5(17):3457–67. doi: 10.1182/bloodadvances.2021004287
8. Fajgenbaum DC, June CH. Cytokine storm. New Engl J Med (2020) 383(23):2255–73. doi: 10.1056/NEJMra2026131
9. Lipton JM, Westra S, Haverty CE, Roberts D, Harris NL. Case 28-2004. New Engl J Med (2004) 351(11):1120–30. doi: 10.1056/NEJMcpc049019
10. Hoshino A, Xi Y, Nakao T, Kato K, Fujiyama S, Koh K, et al. A cry for the development of newborn screening for familial hemophagocytic lymphohistiocytosis. J Clin Immunol (2020) 40(8):1196–8. doi: 10.1007/s10875-020-00863-x
11. Yang Y, Luo Z, Yuan T. Familial hemophagocytic lymphohistiocytosis in a neonate: Case report and literature review. Medicine (2021) 100(47):e27786. doi: 10.1097/MD.0000000000027786
12. Henter J-I, Samuelsson-Horne A, Aricò M, Egeler RM, Elinder Gr, Filipovich AH, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood (2002) 100(7):2367–73. doi: 10.1182/blood-2002-01-0172
13. Bergsten E, Horne A, Aricó M, Astigarraga I, Egeler RM, Filipovich AH, et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood (2017) 130(25):2728–38. doi: 10.1182/blood-2017-06-788349
14. Vannucchi AM, Kiladjian JJ, Griesshammer M, Masszi T, Durrant S, Passamonti F, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. New Engl J Med (2015) 372(5):426–35. doi: 10.1056/NEJMoa1409002
15. Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. New Engl J Med (2012) 366(9):799–807. doi: 10.1056/NEJMoa1110557
16. Zeiser R, von Bubnoff N, Butler J, Mohty M, Niederwieser D, Or R, et al. Ruxolitinib for glucocorticoid-refractory acute graft-versus-Host disease. New Engl J Med (2020) 382(19):1800–10. doi: 10.1056/NEJMoa1917635
17. Zeiser R, Polverelli N, Ram R, Hashmi SK, Chakraverty R, Middeke JM, et al. Ruxolitinib for glucocorticoid-refractory chronic graft-versus-Host disease. New Engl J Med (2021) 385(3):228–38. doi: 10.1056/NEJMoa2033122
18. Tomomasa D, Isoda T, Mitsuiki N, Yamashita M, Morishita A, Tomoda T, et al. Successful ruxolitinib administration for a patient with steroid-refractory idiopathic pneumonia syndrome following hematopoietic stem cell transplantation: A case report and literature review. Clin Case Rep (2021) 9(12):e05242. doi: 10.1002/ccr3.5242
19. Cao Y, Wei J, Zou L, Jiang T, Wang G, Chen L, et al. Ruxolitinib in treatment of severe coronavirus disease 2019 (COVID-19): A multicenter, single-blind, randomized controlled trial. J Allergy Clin Immunol (2020) 146(1):137–46.e3. doi: 10.1016/j.jaci.2020.05.019
20. Quintás-Cardama A, Vaddi K, Liu P, Manshouri T, Li J, Scherle PA, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood (2010) 115(15):3109–17. doi: 10.1182/blood-2009-04-214957
21. Shi JG, Chen X, McGee RF, Landman RR, Emm T, Lo Y, et al. The pharmacokinetics, pharmacodynamics, and safety of orally dosed INCB018424 phosphate in healthy volunteers. J Clin Pharmacol (2011) 51(12):1644–54. doi: 10.1177/0091270010389469
22. O'Shea John J, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity (2012) 36(4):542–50. doi: 10.1016/j.immuni.2012.03.014
23. Zhao Y, Shi J, Li X, Wang J, Sun J, Zhou J, et al. Salvage therapy with dose-escalating ruxolitinib as a bridge to allogeneic stem cell transplantation for refractory hemophagocytic lymphohistiocytosis. Bone Marrow Transplant (2020) 55(4):824–6. doi: 10.1038/s41409-019-0577-2
24. Marois L, Touzot F, Haddad E, Fernandez I, Morin MP, De Bruycker JJ, et al. Successful management of familial hemophagocytic lymphohistiocytosis by the JAK 1/2 inhibitor ruxolitinib. Pediatr Blood Cancer (2021) 68(6):e28954. doi: 10.1002/pbc.28954
25. Ramanan KM, Uppuluri R, Ravichandran N, Patel S, Swaminathan VV, Jayakumar I, et al. Successful remission induction in refractory familial hemophagocytic lymphohistiocytosis with ruxolitinib as a bridge to hematopoietic stem cell transplantation. Pediatr Blood Cancer (2020) 67(3):e28071. doi: 10.1002/pbc.28071
26. Wang J, Wang Y, Wu L, Wang X, Jin Z, Gao Z, et al. Ruxolitinib for refractory/relapsed hemophagocytic lymphohistiocytosis. Haematologica (2020) 105(5):e210–e2. doi: 10.3324/haematol.2019.222471
27. Wei A, Ma H, Li Z, Zhang L, Zhang Q, Wang D, et al. Short-term effectiveness of ruxolitinib in the treatment of recurrent or refractory hemophagocytic lymphohistiocytosis in children. Int J Hematol (2020) 112(4):568–76. doi: 10.1007/s12185-020-02936-4
28. Zhang Q, Zhao YZ, Ma HH, Wang D, Cui L, Li WJ, et al. A study of ruxolitinib-response-based stratified treatment for pediatric hemophagocytic lymphohistiocytosis. Blood (2022) 139(24):3493–504. doi: 10.1182/blood.2021014860
29. Zhang Q, Zhao YZ, Ma HH, Wang D, Zhang N, Li ZG, et al. Successful rescue of a lethal griscelli syndrome type 2 presenting with neurological involvement and hemophagocytic lymphohistiocytosis: a case report. BMC Pediatr (2021) 21(1):253. doi: 10.1186/s12887-021-02720-1
30. Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer (2007) 48(2):124–31. doi: 10.1002/pbc.21039
31. Takakura M, Shimizu M, Irabu H, Sakumura N, Inoue N, Mizuta M, et al. Comparison of serum biomarkers for the diagnosis of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. Clin Immunol (Orlando Fla) (2019) 208:108252. doi: 10.1016/j.clim.2019.108252
32. Hiejima E, Shibata H, Yasumi T, Shimodera S, Hori M, Izawa K, et al. Characterization of a large UNC13D gene duplication in a patient with familial hemophagocytic lymphohistiocytosis type 3. Clin Immunol (Orlando Fla) (2018) 191:63–6. doi: 10.1016/j.clim.2018.03.012
33. Gadoury-Levesque V, Dong L, Su R, Chen J, Zhang K, Risma KA, et al. Frequency and spectrum of disease-causing variants in 1892 patients with suspected genetic HLH disorders. Blood Adv (2020) 4(12):2578–94. doi: 10.1182/bloodadvances.2020001605
34. Tomomasa D, Hiejima E, Miyamoto T, Tanita K, Matsuoka M, Niizato D, et al. Recurrent tandem duplication of UNC13D in familial hemophagocytic lymphohistiocytosis type 3. Clin Immunol (Orlando Fla) (2022) 242:109104. doi: 10.1016/j.clim.2022.109104
35. Tomizawa D, Tawa A, Watanabe T, Saito AM, Kudo K, Taga T, et al. Appropriate dose reduction in induction therapy is essential for the treatment of infants with acute myeloid leukemia: a report from the Japanese pediatric Leukemia/Lymphoma study group. Int J Hematol (2013) 98(5):578–88. doi: 10.1007/s12185-013-1429-2
36. Medina-Valencia D, Cleves D, Beltran E, Builes N, Franco AA, Escobar-González AF, et al. Haploidentical stem cell transplant with post-transplant cyclophosphamide in pediatric hemophagocytic lymphohistiocytosis. J Clin Immunol (2021) 41(6):1172–7. doi: 10.1007/s10875-021-01009-3
37. Felber M, Steward CG, Kentouche K, Fasth A, Wynn RF, Zeilhofer U, et al. Targeted busulfan-based reduced-intensity conditioning and HLA-matched HSCT cure hemophagocytic lymphohistiocytosis. Blood Adv (2020) 4(9):1998–2010. doi: 10.1182/bloodadvances.2020001748
38. Salit RB. The role of JAK inhibitors in hematopoietic cell transplantation. Bone marrow Transplant (2022) 57(6):857–65. doi: 10.1038/s41409-022-01649-y
39. Kohli S, Rastogi N, Nivargi S, Thakkar D, Katewa S, Yadav SP. Successful haploidentical stem cell transplant with posttransplant cyclophosphamide for hemophagocytic lymphohistiocytosis. J Pediatr Hematol/Oncol (2019) 41(3):e158–e60. doi: 10.1097/MPH.0000000000001265
40. Luo Z-B, Chen Y-Y, Xu X-J, Zhao N, Tang Y-M. Prognostic factors of early death in children with hemophagocytic lymphohistiocytosis. Cytokine (2017) 97:80–5. doi: 10.1016/j.cyto.2017.03.013
41. Groom JR, Luster AD. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol (2011) 89(2):207–15. doi: 10.1038/icb.2010.158
42. Mahlaoui N, Ouacheíe-Chardin M, de Saint Basile Gv, Neven B, Picard C, Blanche Sp, et al. Immunotherapy of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins: A single-center retrospective report of 38 patients. Pediatrics (2007) 120(3):e622–e8. doi: 10.1542/peds.2006-3164
43. Marsh RA, Allen CE, McClain KL, Weinstein JL, Kanter J, Skiles J, et al. Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatr Blood Cancer (2013) 60(1):101–9. doi: 10.1002/pbc.24188
44. Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood (2004) 104(3):735–43. doi: 10.1182/blood-2003-10-3413
45. Diamond T, Burn TN, Nishiguchi MA, Minichino D, Chase J, Chu N, et al. Familial hemophagocytic lymphohistiocytosis hepatitis is mediated by IFN-γ in a predominantly hepatic-intrinsic manner. PloS One (2022) 17(6):e0269553. doi: 10.1371/journal.pone.0269553
46. Locatelli F, Jordan MB, Allen C, Cesaro S, Rizzari C, Rao A, et al. Emapalumab in children with primary hemophagocytic lymphohistiocytosis. New Engl J Med (2020) 382(19):1811–22. doi: 10.1056/NEJMoa1911326
47. Henter JI, von Bahr Greenwood T, Bergsten E. Emapalumab in primary hemophagocytic lymphohistiocytosis. New Engl J Med (2020) 383(6):596–8. doi: 10.1056/NEJMc2020754
48. Albeituni S, Verbist KC, Tedrick PE, Tillman H, Picarsic J, Bassett R, et al. Mechanisms of action of ruxolitinib in murine models of hemophagocytic lymphohistiocytosis. Blood (2019) 134(2):147–59. doi: 10.1182/blood.2019000761
49. Huarte E, Peel MT, Verbist K, Fay BL, Bassett R, Albeituni S, et al. Ruxolitinib, a JAK1/2 inhibitor, ameliorates cytokine storm in experimental models of hyperinflammation syndrome. Front Pharmacol (2021) 12:650295. doi: 10.3389/fphar.2021.650295
Keywords: cytokine, familial hemophagocytic lymphohistiocytosis (FHL), HLH, hematopoietic cell transplantation (HCT), janus kinase, ruxolitinib
Citation: Niizato D, Isoda T, Mitsuiki N, Kaneko S, Tomomasa D, Kamiya T, Takagi M, Imai K, Kajiwara M, Shimizu M, Morio T and Kanegane H (2022) Case report: Optimized ruxolitinib-based therapy in an infant with familial hemophagocytic lymphohistiocytosis type 3. Front. Immunol. 13:977463. doi: 10.3389/fimmu.2022.977463
Received: 23 June 2022; Accepted: 08 November 2022;
Published: 23 November 2022.
Edited by:
Anders Fasth, University of Gothenburg, SwedenReviewed by:
Amit Rawat, Post Graduate Institute of Medical Education and Research (PGIMER), IndiaQuan Wang, Beijing Children’s Hospital, Capital Medical University, China
Mario Abinun, Newcastle University, United Kingdom
Copyright © 2022 Niizato, Isoda, Mitsuiki, Kaneko, Tomomasa, Kamiya, Takagi, Imai, Kajiwara, Shimizu, Morio and Kanegane. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takeshi Isoda, dGlzb2RhLnBlZEB0bWQuYWMuanA=