 Yerly Useche
Yerly Useche Ana Rosa Pérez
Ana Rosa Pérez Juliana de Meis1,4,5
Juliana de Meis1,4,5 Adriana Bonomo
Adriana Bonomo Wilson Savino
Wilson Savino
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 10 November 2022
Sec. Parasite Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.975106
The involvement of the central nervous system (CNS) during human acute and chronic Chagas disease (CD) has been largely reported. Meningoencephalitis is a frequent finding during the acute infection, while during chronic phase the CNS involvement is often accompanied by behavioral and cognitive impairments. In the same vein, several studies have shown that rodents infected with Trypanosoma cruzi (T. cruzi) display behavior abnormalities, accompanied by brain inflammation, in situ production of pro-inflammatory cytokines and parasitism in diverse cerebral areas, with involvement of microglia, macrophages, astrocytes, and neurons. However, the mechanisms used by the parasite to reach the brain remain now largely unknown. Herein we discuss the evidence unravelling the CNS involvement and complexity of neuroimmune interactions that take place in acute and chronic CD. Also, we provide some clues to hypothesize brain infections routes in human and experimental acute CD following oral infection by T. cruzi, an infection route that became a major CD related public health issue in Brazil.
Chagas disease (CD) is caused by the protozoan parasite Trypanosoma cruzi (T. cruzi) and affects approximately 6–8 million people, being endemic in 21 countries from the south of the United States to southern Argentina and Chile. Migration of infected people has spread the disease in non-endemic regions in Europe, North America, Asia, and Oceania (1). In human CD, around 20-30% of chronically infected patients develop cardiac and/or gastrointestinal damage, with cardiovascular disease-induced early mortality and loss of productivity. These data illustrate to what extent CD remains a public health issue, also providing an economic argument for the need of urgent efforts towards controlling CD (2, 3).
Transmission of CD was classically described as being dependent on the insect vector bite. Nowadays, infection through the oral route had gained attention, while transmission through an insect bite, placenta, transfusions, and transplants have been better controlled (4). In the period between 1968-2000, more than 50% of CD acute cases in the Brazilian Amazon region were attributable to micro-epidemics due to orally transmitted infection (5). Moreover, oral infection outbreaks have been reported in several South American countries, being associated with T. cruzi presence in food or beverage consumed by the habitants of endemic regions (6). A recent epidemiological study of acute CD indicates a total of 5,184 cases recorded with an annual incidence rate of 0.16 per 100,000 habitants/year. This study reported an increased frequency of oral transmission in acute cases in the North Brazilian region, from 493 cases in the first period from 2005-2009 to 1770 cases in the second period 2010-2018, thus increasing almost 30% in the north of Brazil (7).
The involvement of the CNS during human acute and chronic phases of CD has been reported. Meningoencephalitis has been described more frequently in children during the acute disease, while in the chronic phase neuritis is related to sensory impairment reported in up to 10% of patients. Dementia, confusion, chronic encephalopathy, and sensitive and motor deficits are less frequent. Moreover, experimental infections with several parasite strains demonstrated behavioral alterations (8–10). In line with these evidences, HIV and transplant patients, both immunesuppressed, often show parasite reactivation with meningoencephalitis and brain abscesses (11). In this regard, experimental model of oral acute infection cleary showed the presence of parasite in brain regions a few days after infection (12).
Herein we will focus on the neuroimmune interactions in acute and chronic CD, mostly studied following vector infection route. Moreover, we provide some clues concerning posible CNS changes in orally transmitted T. cruzi in experimental infections.
The involvement of the CNS during T. cruzi infection in both acute and chronic phases has been reported decades ago. Encephalitis may occur during the acute phase, accompanied by elevated levels of albumin, leukocytes, and trypomastigotes in the cerebral spinal fluid -CSF- (13). Also, acutely infected patients manifest difficulty in mental concentration, cephalea, muscular disturbances (myoclonus, bradykinesia, dyspraxia), weakness, and speech disturbances (14). Headaches, seizures, lethargy, or mood changes are probably due to meningoencephalitis, exhibited by 5-10% of T. cruzi acutely infected patients (11). This condition more frequently affects children under 2 years of age and is almost always fatal when coexists with myocarditis and cardiac insufficiency (15). Of note, the acute congenital phase may be associated with seizures and meningoencephalitis in a small proportion of children (16).
Chronically infected patients with dementia, confusion, as well as sensorial and motor deficits occur, have been reported. Additionally, motor deficits associated with neuritis result in altered tendon reflexes and sensory impairment (11). Decreased orientation, attention, and cognitive performance (i.e. lower Mini-Mental State Exam scores) were reported in chronic CD patients compared with healthy controls. Moreover, lower scores of the Weschler Memory Scale and the WAIS global test of Intelligence were associated with chronic CD. The cognitive dysfunction is compatible with signs characteristic of White Matter Disease, with impairment in non-verbal reasoning, information processing, problem-solving and learning (17).
It is noteworthy that CNS is the most frequent site of T. cruzi reactivation in immunosuppressed HIV-infected patients (18). Also, the reactivation in this situation is accompanied by brain tissue damage related to increased IL-17 expression (19), as well as low peripheral blood CD4 T-cell counts (11). Of note, meningoencephalitis usually coincides with abundant trypomastigotes in the CSF (20).
We should also point out that proinflammatory cytokines, as well as pathogen associated molecular patterns (LPS and poli:IC) induce depressive behavior (21). Accordingly, during CNS infection by T. cruzi, macrophages, microglia and astrocytes can release TNF-α, IL-1β, and nitric oxide -NO- (22, 23), which seems to be related to neurological alterations observed in CD. Also, glutamate release is a common result of CNS infection, being induced by TNF-α and LPS (24). These data should be placed in the context that glutamate is the most prevalent excitatory neurotransmitter in the CNS and plays a role in basic brain capacities, being altered in several psychiatric and neurological disorders (25).
Experimental evidence showed CNS alterations and parasite presence in the brain after T. cruzi infection, despite the entry routes (Table 1). Frequently, CNS infection occurs during the acute infections around the peak of parasitemia, although parasite load and number of inflammatory foci are more conspicuos in the basal ganglia when parasitemia is no longer detectable (32). Interestingly, amastigote nests appear in the neuropil of a cerebellar gray matter nucleus, in the absence of a detectable inflammatory process (28). Actually, diverse brain regions can be parasitized by T. cruzi. Concurrent to hypothalamic and pituitary detection of T. cruzi after infection, several alterations and injury in the hypothalamic–pituitary–adrenal (HPA) axis were reported, caused either directly by parasite invasion or indirectly as a consequence of local and/or systemic inflammation in response to infection (33–38). Brain regions frequently infected by T. cruzi, beside the ones described above, are the meninges, cerebral and cerebellar cortices (28, 29), which leads to meningoencephalitis. Other less frequently infected regions are the hippocampus (27), as well as areas without BBB, such as the choroid plexus (29).
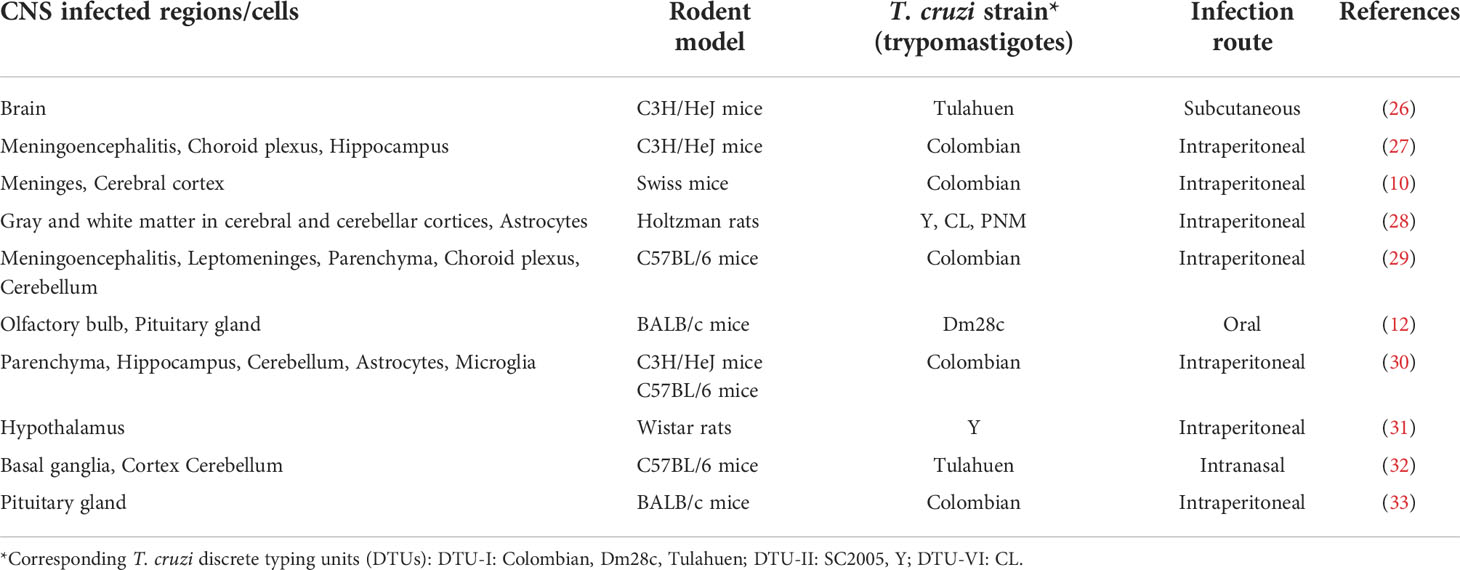
Table 1 Experimental T. cruzi infection models with CNS involvement.
Histological studies of T. cruzi-infected rodents showed injured CNS regions with neuronal loss (31), glial nodules (due to astrocyte proliferation), edema, and enlargement of perivascular spaces (27, 31), as well as the appearance of perivascular and intraparenchymal mononuclear cellular infiltrates (10, 18, 27–29, 31).
Experimental infections with Colombian and H4 T. cruzi strains also demonstrated behavioral abnormalities, such as sleep and memory deficits, anxiety, and depression (8–10, 30). In the same vein, depressive behavior, probably associated with oxidative stress and enhanced pro-inflammatory cytokines has been recorded in chronic CD patients [see review by (39)]. Colombian strain-infected mice showed significant increased immobility and signs of depression both during the acute and chronic infections. Interestingly, TNF-α inhibitors (i.e. pentoxifylline or anti-TNF), as well as the antidepressant fluoxetine, ameliorate depressive-like alterations in this model (30), suggesting a role for systemically TNF-α release in T. cruzi-induced depression.
Chronic depressive-like behavior appears to be parasite strain-dependent, since depression was triggered by the Colombian but not by the Y strain. Probably the depressive-like behavior was driven by the increased indoleamine 2,3-dioxygenase (IDO) expression in the CNS in acute and chronic T. cruzi infection (30). IDO is a tryptophan-catabolizing enzyme, induced by IFN-γ and TNF-α in infected tissues, including the brain. In addition to depression, chronically infected C57BL/6 mice with the Colombian strain exhibit anxiety and alterations of motor coordination, not associated with sickness behavior signs such as temperature variations or weight loss (9). Other behavioral distubances in chronically T. cruzi-infected mice may occur whithout neuroinflammation, including deficits in spatial habituation, novel object recognition, and aversive memory recall. Of note, T. cruzi persistence and increased lipid peroxidation in the CNS hippocampus and cortex were associated with these cognitive alterations (40). All these data are summarized in Figure 1. Interestingly, in the brain of patients suffering from depression, tryptophan depletion by IDO affects serotonin synthesis and low serotonin levels ocurr, being a possible mechanism related to depression (41).
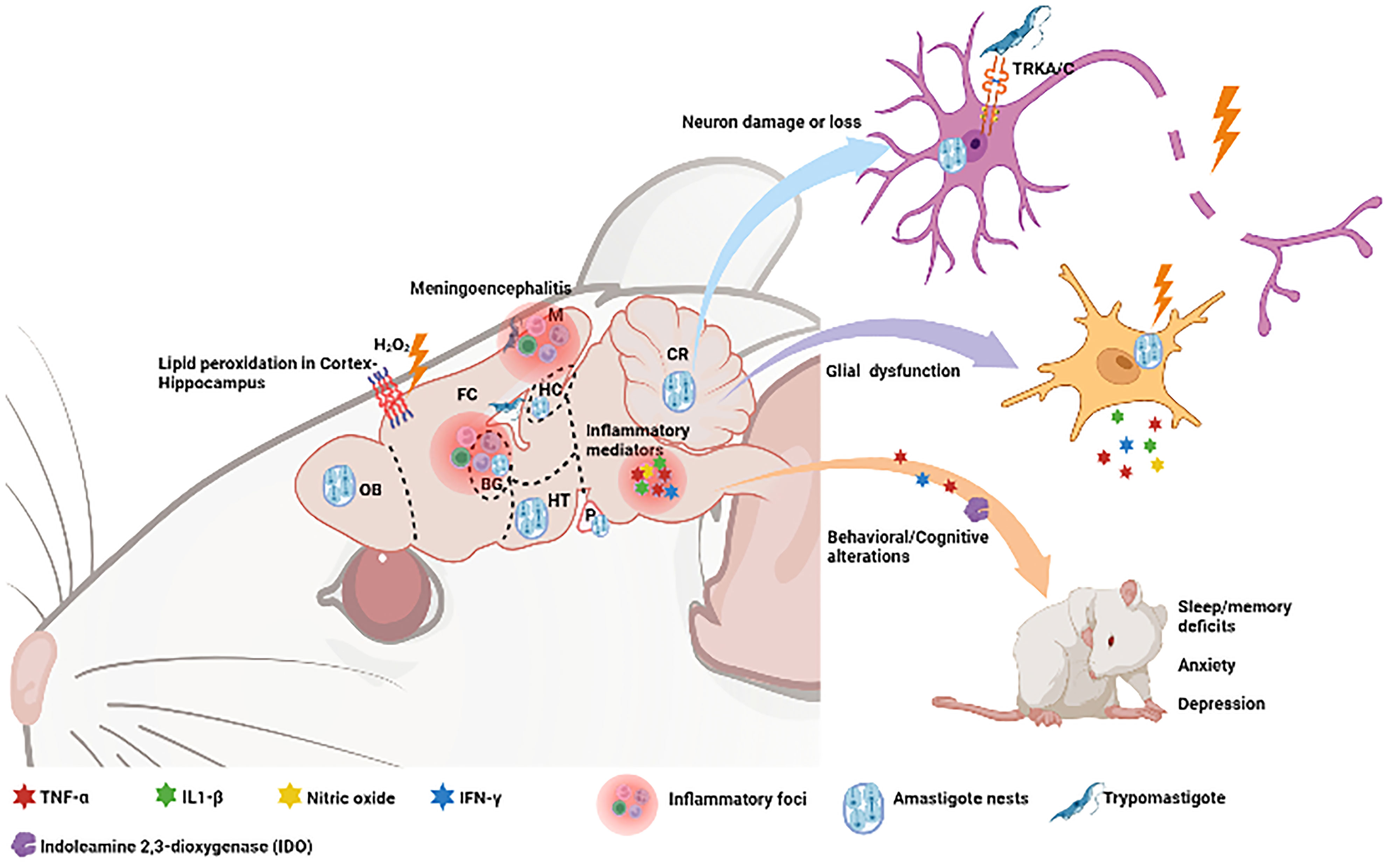
Figure 1 Consequences of experimental T. cruzi infection upon the Central Nervous System and respective involved regions. The presence of the parasite in these regions can cause lipid peroxidation as well as neuroinflammation with release of inflammatory mediators, glial dysfunction and neural damage. Secretion of proinflammatory cytokine triggers behavioural and congnitive changes with deficits in sleep and memory, as well as symptoms of anxiety and depression. Olfactory Bulb (OB), Frontal Cortex (FC), Meninges (M), Basal Ganglia (BG), Hippocampus (HC), Hypothalamus (HT), Cerebellum (CR). Neurotrophin receptors (TRKA/C). Created with BioRender.com.
As mentioned above, several studies have shown that T. cruzi does infect the CNS (12, 26–33). Yet, the mechanisms used by the parasite to reach brain tissues remain largely unknown.
Successful spread from a point of entry to the CNS, either by crossing or disrupting the blood brain barrier (BBB) or the cerebrospinal fluid barrier (BCSFB), involves evasion from both peripheral and intra-CNS immune responses. The BBB and BCSFB are selectively permeable to macromolecules and hydrophilic molecules, due to tight junctions, particularly prominent in the BBB (42). Moreover, leptomeningeal vessels as well as CNS regions bearing BCSFB (i.e. choroid plexus and brain circumventricular organs) are more permeable than vessels in the parenchyma (43). Additionally, the immune response in the meninges is stronger than that seen in the brain parenchyma (42). Also, circulating immune cells, including monocytes, T cells, and macrophages, increase their passage through the brain endothelial cells (BEC) when activated; a process mediated by astrocyte-released cytokines and chemokines, which induce the expression of BEC adhesion molecules in response to infection (44).
In T. cruzi infected newborn rats, parasites were detected at the peak of parasitemia inside glial cells, near neuronal somas, capillaries, and venules in the cerebral and cerebellar cortices and the thalamic subcortical nuclei (28). This parasite localization suggests a possible invasion through the BBB vessels, as for example, by the BBB endothelial junction’s impairment mediated by bradykinin, as occurs in cerebral malaria (45). It is known that parasite-derived cruzipain cleaves plasma kininogens into bradykinin, which activates the endothelial bradykinin-B2 receptors and protein-G immune pathway (46). As shown in vitro, it is conceivable that T. cruzi migrates through a paracellular way across BECs without monolayer disruption, mediated by bradykinin and CCL2 gradient (47).
Increment in plasma TNF-α levels or CNS Tnf mRNA expression during murine T. cruzi acute infection has been reported. For instance, orally T. cruzi infected mice present higher parasitemia, mortality rates, and TNF-α serum levels (48, 49). Interestingly, Trypanosoma brucei (T. brucei), which is another trypanosomatid parasite, crosses the disrupted BCSFB mediated by TNF-α overexpression in the choroid plexus and circumventricular organs during infection in rodents (50). Actually, T. brucei requires the TLR-MyD88 signal mediation to penetrate the brain and generate microglial activation (51).
An increase in metalloproteinases (MMPs) is another inductor factor of BBB dysfunction, as reported in T. brucei CNS infection in mice (52). MMPs cleave specific collagen types in the basement membranes and degrade endothelial tight junctions, improving the parasite invasion through the BBB (53). Although the activation of the TLR-MyD88 pathway has not been reported in CNS infection by T. cruzi, it is a plausible scenario.
Several areas with BCSFB, as the choroid plexus and hippocampus, presented mild to intense inflammatory infiltrates during experimental acute and chronic T. cruzi infection (27, 29). Moreover, amastigote nests were seen in the region between the cerebellum and the spinal cord (cerebellar neuropil) (28), and below the pia mater in the ependymal cells that form the glia limitans at the brain surfaces (10). These findings indicate that T. cruzi can cross the endothelium of the leptomeningeal venules in the subarachnoid space, traverse the pia mater till the glia limitans, and gain access to the brain parenchyma. The infection of the glia limitans may explain some reports of cerebellar T. cruzi infection (28, 32) as there is a glia-cerebellum neural connection.
The neurovascular unit (NVU) comprises BBB-endothelial cells, pericytes, astrocytes, glia, neurons and the extracellular matrix (ECM); all being involved in regulating cerebral blood flow and BBB function. Therefore, any dysregulation of NVU components should alter brain homeostasis, leading to disease (54). In this way, experimental studies have shown that T. cruzi infects microglia, macrophages, astrocytes, and neurons (30, 55, 56). Furthermore, in CD patients, the parasite was found in microglia, endothelial cells, and macrophages (18). Also, histological and in vitro evidence of T. cruzi parasitism revealed high frequency of parasite nests without surrounding membrane in non-glial cells and parasites in astrocyte processes among Purkinje cerebellar cell bodies (28, 49).
Interestingly, astrocytes seem incapable to control T. cruzi by the secretion of IL-1β and NO, turning them more permissive to parasite replication than microglial cells (57). In addition, astrocytic processes surrounding the CNS vessels may facilitate parasite invasion, especially in the gray matter where capillary density is higher than in the white matter (28). Some biological processes have been described during CNS T. cruzi infection, which could promote parasite replication and tissue damage through the ligation between the parasite-derived trans-sialidase (TS) and the neurotrophin receptors TrkA and TrkC (55, 58), which are commonly upregulated during CNS injury, infections, or degenerative diseases. These interactions facilitate parasite adherence and efficient invasion of neuronal, epithelial, and phagocytic cells in vitro and in vivo (59); also activating MAP kinase (MAPK) and phosphatidylinositol 3-kinase/Akt signaling pathways, as seen in infected PC12 cell line, an established model of neuronal differentiation (60).
Lastly, systemic or in situ pro-inflammatory cytokine release can promote T. cruzi invasion. In vitro and in vivo experiments showed that IFN-γ-expressing glial cells as well as TNF-α levels correlate with astrocyte invasion (49, 56). Also, parasite infection drives astrocytes to a pro-inflammatory profile with enhanced IL-6 and TNF-α production and TNFR1 expression, potentially favoring TNF signaling (49). This suggests a self-sustaining inflammatory loop creating a favorable environment for parasitic replication.
The oral infection in humans is characterized by more severe manifestations than those associated with vectorial transmission: prolonged fever, acute myocarditis, heart failure, and meningoencephalitis (61). At least in Brazil, acute human CD occurs after oral infection, with a potential higher inoculum in patients infected by the oral route, as compared to those infected by the vectorial route (61). Importantly, the presence of T. cruzi in various tissues located in the anatomical pathway between the oral mucosa and the CNS has been reported (12, 32, 62). Therefore, in principle, after invading the oral mucosa, parasites might migrate to the blood and/or lymphatic fluids from the submucosa, through the palate and tissues of the nasal cavity. These data unravel the existence of multiple interactions between the T. cruzi and the diverse anatomical regions of the oral mucosa (12).
We found that some soft regions of the oral cavity and underlying tissues are invaded by T. cruzi in orally infected mice, including the cheek muscle, salivary glands, and submandibular lymph nodes (12). Hypothetically, the lymph node invasion occurs by drainage of infected tissues, implying a route of infection towards more distant tissues such as the heart, liver and spleen following the drainage route.
We also found that the naso-maxillary region, nasal cavities, and subjacent tissues are invaded by T. cruzi in orally infected mice. T. cruzi DNA was also observed in brain tissues after oral infection (12), suggesting that the parasite could reach the brain via invasion of the olfactory nerve and later the olfactory bulb. Corroborating this idea, intranasally T. cruzi infected mice showed infection of several brain regions (32), indicating that the oral cavity and the adjacent nasal compartment invasion represent a putative anatomical route for parasite spreading to the brain.
Active T. cruzi penetration into the host cell involves first a step of adhesion for later penetration of cells. ECM proteins could be relevant players in early oral mucosal infection. In humans, the major basement membrane components of palatal mucosa are laminin, type IV collagen, and fibronectin (63). Similarly, T. cruzi can take advantage of ECM to invade CNS tissues. For instance, fibronectin is present in the meninges and the choroid plexus (27), and together with laminin, composes the blood brain barrier (BBB) related basal lamina surrounding the cerebral endothelial cells (53). Actually, several T. cruzi target molecules are ECM components of the oral mucosa and the CNS. T. cruzi-derived peptidases, like the Tc80 oligopeptidase B, hydrolyses human type-I and type-IV collagens, as well as fibronectin, which is important for the parasite transit through the ECM (64). Similarly, Tc85 is capable of binding host cell derived molecules, such as fibronectin, laminin and heparin (64). Also, parasite penetrin has affinity for ECM elements such as heparin, heparan sulfate proteoglicans (HSPGs), and collagen, promoting fibroblast adhesion and penetration (65), suggesting a role of T. cruzi penetrin and neuronal tissue alterations.
Other molecular group includes the parasite-derived TS family. Inactive forms function as parasite adhesins, binding to sialic acid and β-galactose residues, whereas active TSs transfer sialic acid from host glycoconjugates to acceptor molecules on the parasite surface, regulating the process of cell adhesion, migration, and invasion (66). Strikingly, the expression of active TSs during the trypomastigote life cycle stage seems to favor the epithelial cell invasion (67). Considering that epithelial cells from the oral cavity are enriched in sialic acid, it is conceivable that T. cruzi takes advance of sialoglycoproteins, invading the cavity and evade host defense. Interestingly, TSs have also neuraminidase activity and can interact with neurotrophin receptors TrkA and TrkC, promoting both invasion and survival of neurons and glial cells (58, 59).
Whichever route is used to spread from the oral cavity, parasites can ultimately disseminate, crossing the endothelium of both blood and lymphatic vessels, the oral epithelium at different location and the blood-brain barrier (BBB) to invade the brain, as schematically depicted in Figure 2.
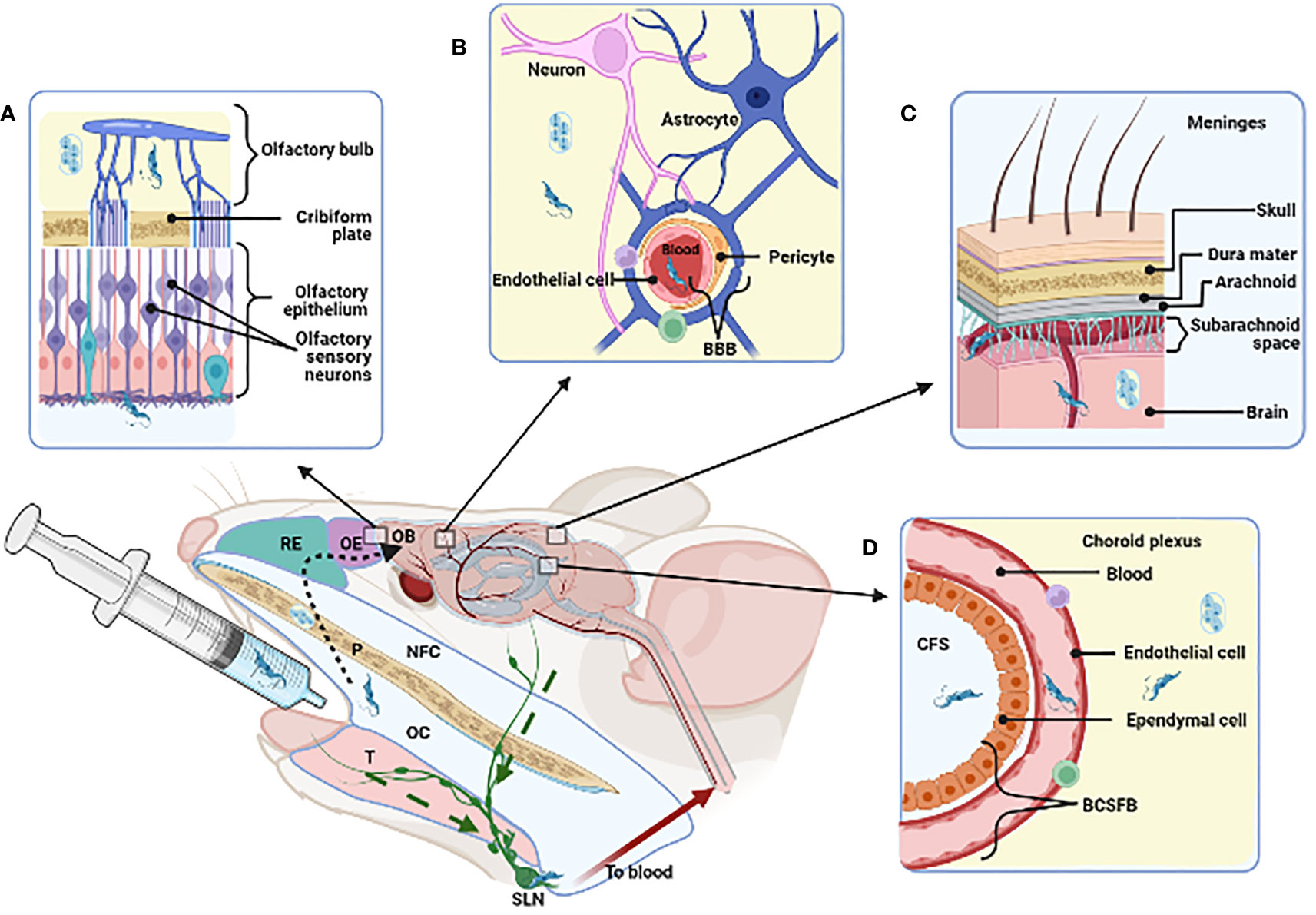
Figure 2 Strategies that T. cruzi may use for reaching and invading the Central Nervous System and possible dissemination following oral cavity (OC) infection. The figure stands for murine experimental oral infection. Accordingly, after delivering the parasites into de oral cavity, they may take the following pathway(s): 1) olfactory way (A, dotted arrow), traversing the palate (P), nasopharyngeal cavity (NFC), respiratory and olfactory epithelia (RE, OE) reaching the olfactory bulb (OB); 2) disrupture of the blood brain barrier (BBB) in the parenchyma (B) or in the meninges (C); 3) disrupture of the cerebrospinal fluid barrier (BCSFB) in the choroid plexus (D); 4) invasion of the submandibular lymph node (SLN) by drainage of infected tissues (green dashed arrow). Tongue (T). Created with BioRender.com.
The data summarized herein clearly show the CNS commitment in acute and chronic CD. Different cell types in the brain can be infected by T. cruzi, including neurons, astrocytes and microglia. Also, various regions of the CNS can be affected, such as choroid plexus, hippocampus, meninges, olfactory bulb, pituitary gland, among others. The cellular mechanisms involved in T. cruzi infection in the central nervours tissues has also been largely documented, comprising both parasite-derived and host-derived moieties; among others ECM ligands such as fibronectin, laminin and glycosaminoglycans (summarized in Table 2).
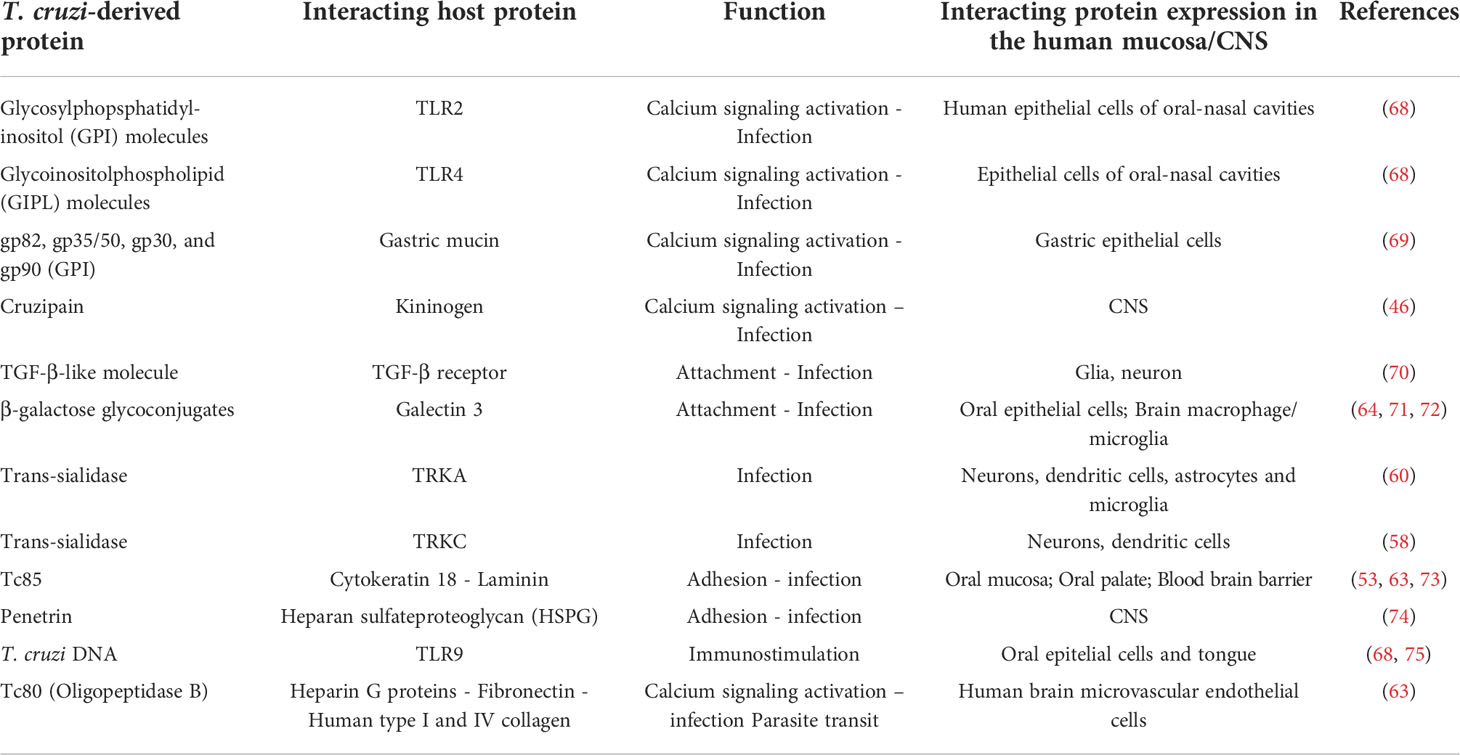
Table 2 Molecular interactions between T. cruzi and host molecules.
The possibility of T. cruzi CNS invasion after oral ingestion raises a series of questions that still need to be answered. Which ligands and/or mechanisms does the parasite use to cross the mucosa and reach the brain tissue? Can the immune system adequately respond to parasite infection in the mucosa and brain? Are there differences regarding CNS commitment after oral infection compared to those caused by reactivation due to immunosuppression? Which brain areas and functions are mostly affected after infection? Which are the systemic consequences due to CNS infection by the oral route? Addressing these issues will provide new clues that may allow earlier diagnosis and treatment for oral Chagas Disease.
JM and YU contributed to the conception and design of the manuscript. YU wrote the first draft and AP, AB and WS wrote sections of the manuscript and made a substantial intellectual contribution to the work. All authors approved it for publication.
This work was partially funded by Fiocruz CNPq, CAPEs, FAPERJ (Brazil), PIP CONICET 0715 (Argentina), and Mercosur Fund for structural convergenge (FOCEM, Mercosur). AB and WS are recipients with grants from CNPq, whereas YU received a Post-doctoral Felowship from FAPERJ (Grant E-26/202.139/2020), Brazil. This work was conducted in the frameworks of the National Institute o Science and Technology on Neuroimmunomodulatiom (CNPq), and the Rio de Janeiro Network on Neuroinflammation (FAPERJ).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. World Health Organization. Chagas disease (American trypanosomiasis) (2022). Available at: https://www.who.int/news-room/fact-sheets/detail/chagas-disease-(american-trypanosomiasis) (Accessed May 1, 2022).
2. Lee BY, Bacon KM, Bottazzi ME, Hotez PJ. Global economic burden of chagas disease: A computational simulation model. Lancet Infect Dis (2013) 13:342–8. doi: 10.1016/S1473-3099(13)70002-1
3. Mendes FSNS, Perez-Molina JA, Angheben A, Meymandi SK, Sosa-Estani S, Molina I. Critical analysis of chagas disease treatment in different countries. Memorias do Instituto Oswaldo Cruz (2021) 116:1–5. doi: 10.1590/0074-02760210034
4. de Noya BA, González ON. An ecological overview on the factors that drives to Trypanosoma cruzi oral transmission. Acta Tropica (2015) 151:94–102. doi: 10.1016/j.actatropica.2015.06.004
5. Coura JR, Junqueira ACV, Fernandes O, Valente SAS, Miles MA. Emerging Chagas disease in Amazonian Brazil. Trends Parasitol (2002) 18:171–6. doi: 10.1016/S1471-4922(01)02200-0
6. Toso M, Vial U, Galanti N. Oral transmission of Chagas’ disease. Rev Med Chil. (2011) 139:258–66. doi: 10.4067/S0034-98872011000200017
7. Santos EF, Silva Â.AO, Leony LM, Freitas NEM, Daltro RT, Regis-Silva CG, et al. Acute chagas disease in brazil from 2001 to 2018: A nationwide spatiotemporal analysis. PloS Negl Trop Dis (2020) 14:1–16. doi: 10.1371/journal.pntd.0008445
8. Arankowsky-Sandoval G, Mut-Martín M, Solís-Rodríguez F, Góngora-Alfaro JL, Barrera-Pérez M. Sleep and memory deficits in the rat produced by experimental infection with Trypanosoma cruzi. Neurosci Lett (2001) 306:65–8. doi: 10.1016/S0304-3940(01)01866-3
9. Vilar-Pereira G, de Souza Ruivo LA, Lannes-Vieira J. Behavioural alterations are independent of sickness behaviour in chronic experimental chagas disease. Memorias do Instituto Oswaldo Cruz (2015) 110:1042–50. doi: 10.1590/0074-02760150300
10. Baldissera MD, Souza CF, Carmo GM, Monteiro SG, Mendes RE, Stefani LM, et al. Relation between acetylcholinesterase and na+, k+-ATPase activities with impaired memory of mice experimentally infected by Trypanosoma cruzi. Microbial Pathogenesis (2017) 111:75–80. doi: 10.1016/j.micpath.2017.08.019
11. Córdova E, Maiolo E, Corti M, Orduña T. Neurological manifestations of Chagas’ disease. Neurological Res (2010) 32:238–44. doi: 10.1179/016164110X12644252260637
12. Silva-dos-Santos D, Barreto-de-Albuquerque J, Guerra B, Moreira OC, Berbert LR, Ramos MT, et al. Unraveling chagas disease transmission through the oral route: Gateways to Trypanosoma cruzi infection and target tissues. PloS Negl Trop Dis (2017) 11:1–26. doi: 10.1371/journal.pntd.0005507
13. Hoff R, Teixeira R, Carvalho J, Mott K. T. cruzi in the cerebrospinal fluid during the acute stage of chagas’ disease. N Engl J Med (1978) 298:604–6. doi: 10.1056/NEJM197803162981106
14. Jorg M, Zalazar I. Formas encefalopáticas de enfermedad de chagas crónica observadas en Argentina. Mem Inst Oswaldo Cruz (1981) 76:353–60. doi: 10.1590/S0074-02761981000400003
15. Coura JR. Chagas’ disease: What is known and what is needed – a background article. Mem Inst Oswaldo Cruz. (2007) 102 (Suppl 1):113–22. doi: 10.1590/S0074-02762007007500001
16. Zaidenberg M. La enfermedad de chagas congenita en la provincia de salta, Argentina, años 1980-1997. Rev Soc Bras Med Trop (1999) 32:689–95. doi: 10.1590/s0037-86821999000600012
17. Mangone CA, Sica RE, Pereyra S, Genovese O, Segura E, Riarte A, et al. Cognitive impairment in human chronic chagas’ disease. Arq Neuropsiquiatr (1994) 52:200–3. doi: 10.1590/s0004-282x1994000200008
18. Pittella JE. Central nervous system involvement in Chagas’ disease. an updating. Rev Inst Med Trop Sao Paulo (1993) 35:111–6. doi: 10.1590/S0036-46651993000200001
19. Gattoni CM, Aleixo IF, de Araujo MF, Teixeira V, de PA, Rocha Rodrigues DB, et al. Chagas disease reactivation in HIV-coinfected patients: Histopathological aspects. Immunobiology (2015) 220:656–62. doi: 10.1016/j.imbio.2014.11.013
20. Simioli F, Sánchez-Cunto M, Velázquez E, Lloveras S, Orduna T. Chagas disease in the central nervous system in patient infected with HIV: Diagnostic and therapeutic difficulties. Rev Chil Infectol (2017) 34:62–6. doi: 10.4067/S0716-10182017000100009
21. Gibb J, Hayley S, Poulter MO, Anisman H. Effects of stressors and immune activating agents on peripheral and central cytokines in mouse strains that differ in stressor responsivity. Brain Behavior Immun (2011) 25:468–82. doi: 10.1016/j.bbi.2010.11.008
22. de Almeida-Leite CM, Silva ICC, da Cunha Galvão LM, Arantes RME. Sympathetic glial cells and macrophages develop different responses to Trypanosoma cruzi infection or lipopolysaccharide stimulation. Memorias do Instituto Oswaldo Cruz (2014) 109:459–65. doi: 10.1590/0074-0276130492
23. Borghi SM, Fattori V, Carvalho TT, Tatakihara VLH, Zaninelli TH, Pinho-Ribeiro FA, et al. Experimental Trypanosoma cruzi infection induces pain in mice dependent on early spinal cord glial cells and NFκB activation and cytokine production. Front Immunol (2021) 11:539086. doi: 10.3389/fimmu.2020.539086
24. Yawata I, Takeuchi H, Doi Y, Liang J, Mizuno T, Suzumura A. Macrophage-induced neurotoxicity is mediated by glutamate and attenuated by glutaminase inhibitors and gap junction inhibitors. Life Sci (2008) 82:1111–6. doi: 10.1016/j.lfs.2008.03.010
25. Miladinovic T, Nashed MG, Singh G. Overview of glutamatergic dysregulation in central pathologies. Biomolecules (2015) 5:3112–41. doi: 10.3390/biom5043112
26. Buckner FS, Wilson AJ, Van Voorhis WC. Detection of live Trypanosoma cruzi in tissues of infected mice by using histochemical stain for-galactosidase. Infect Immun (1999) 67(1):403–9. doi: 10.1128/IAI.67.1.403-409.1999
27. Silva A, Roffê E, Marino AP, dos Santos PVA, Quirico-Santos T, Paiva CN, et al. Chagas’ disease encephalitis: Intense CD8+ lymphocytic infiltrate is restricted to the acute phase, but is not related to the presence of Trypanosoma cruzi antigens. Clin Immunol (1999) 92:56–66. doi: 10.1006/clim.1999.4716
28. Da-Mata JR, Camargos MR, Chiari E, Machado CR. Trypanosoma cruzi infection and the rat central nervous system: Proliferation of parasites in astrocytes and the brain reaction to parasitism. Brain Res Bull (2000) 53:153–62. doi: 10.1016/S0361-9230(00)00326-9
29. Roffê E, Silva AA, Marino APMP, dos Santos PVA, Lannes-Vieira J. Essential role of VLA-4/VCAM-1 pathway in the establishment of CD8 + T-cell-mediated Trypanosoma cruzi-elicited meningoencephalitis. J Neuroimmunol (2003) 142:17–30. doi: 10.1016/S0165-5728(03)00254-6
30. Vilar-Pereira G, Silva AA, Pereira IR, Silva RR, Moreira OC, de Almeida LR, et al. Trypanosoma cruzi-induced depressive-like behavior is independent of meningoencephalitis but responsive to parasiticide and TNF-targeted therapeutic interventions. Brain Behavior Immun (2012) 26:1136–49. doi: 10.1016/j.bbi.2012.07.009
31. Jardim E. Moléstia de chagas aguda experimental: Parasitismo do hipotálamo. Arq Neuropsiquiatr (1971) 29:190–7. doi: 10.1590/S0004-282X1971000200007
32. Caradonna K, Pereira-Perrin M. Preferential brain homing following intranasal administration of Trypanosoma cruzi. Infection Immun (2009) 77:1349–56. doi: 10.1128/IAI.01434-08
33. Corrêa-De-Santana E, Paez-Pereda M, Theodoropoulou M, Kenji Nihei O, Gruebler Y, Bozza M, et al. Hypothalamus-pituitary-adrenal axis during Trypanosoma cruzi acute infection in mice. J Neuroimmunol (2006) 173:12–22. doi: 10.1016/j.jneuroim.2005.08.015
34. Pérez AR, Bottasso O, Savino W. The impact of infectious diseases upon neuroendocrine circuits. NeuroImmunoModulation (2009) 16:96–105. doi: 10.1159/000180264
35. Roggero E, Pérez AR, Bottasso OA, Besedovsky HO, del Rey A. Neuroendocrine-immunology of experimental chagas’ disease. Ann N Y Acad Sci (2009) 1153:264–71. doi: 10.1111/j.1749-6632.2008.03982.x
36. Roggero E, del Rey A, Wildmann J, Besedovsky H. Glucocorticoids and sympathetic neurotransmitters modulate the acute immune response to Trypanosoma cruzi. Ann N Y Acad Sci (2019) 1437:83–93. doi: 10.1111/nyas.13946
37. Morrot A, Villar SR, González FB, Pérez AR. Evasion and immuno-endocrine regulation in parasite infection: Two sides of the same coin in chagas disease? Front Microbiol (2016) 7:704. doi: 10.3389/fmicb.2016.00704
38. González FB, Villar SR, Pacini MF, Bottasso OA, Pérez AR. Immune-neuroendocrine and metabolic disorders in human and experimental T. cruzi infection: New clues for understanding chagas disease pathology. Biochim Biophys Acta - Mol Basis Dis (2020) 1866:1–14. doi: 10.1016/j.bbadis.2019.165642
39. Duarte-Silva E, Maes M, Macedo D, Savino W, Peixoto CA. Shared neuroimmune and oxidative pathways underpinning chagas disease and major depressive disorder. Trans Psychiatry (2020) 10:1–10. doi: 10.1038/s41398-020-01105-9
40. Vilar-Pereira G, Barrios LC, da Silva AA, MartinsBatista A, Pereira IR, Moreira OC, et al. Memory impairment in chronic experimental chagas disease: Benznidazole therapy reversed cognitive deficit in association with reduction of parasite load and oxidative stress in the nervous tissue. PloS One (2021) 16:1–21. doi: 10.1371/journal.pone.0244710
41. Svensson JE, Svanborg C, Plavén-Sigray P, Kaldo V, Halldin C, Schain M, et al. Serotonin transporter availability increases in patients recovering from a depressive episode. Trans Psychiatry (2021) 11:1–10. doi: 10.1038/s41398-021-01376-w
42. Adalid-Peralta L, Sáenz B, Fragoso G, Cárdenas G. Understanding host-parasite relationship: The immune central nervous system microenvironment and its effect on brain infections. Parasitology (2018) 145:988–99. doi: 10.1017/S0031182017002189
43. Kristensson K. Microbes’ roadmap to neurons. Nat Rev Neurosci (2011) 12:345–57. doi: 10.1038/nrn3029
44. Hudson LC, Bragg DC, Tompkins MB, Meeker RB. Astrocytes and microglia differentially regulate trafficking of lymphocyte subsets across brain endothelial cells. Brain Res (2005) 1058:148–60. doi: 10.1016/j.brainres.2005.07.071
45. Silva LS, Pinheiro AS, Teixeira DE, Silva-Aguiar RP, Peruchetti DB, Scharfstein J, et al. Kinins released by erythrocytic stages of Plasmodium falciparum enhance adhesion of infected erythrocytes to endothelial cells and increase blood brain barrier permeability via activation of bradykinin receptors. Front Med (2019) 6:75. doi: 10.3389/fmed.2019.00075
46. Scharfstein J, Schmitz V, Morandi V, Capella MMA, Paula A, Lima CA, et al. Host cell invasion by Trypanosoma cruzi is potentiated by activation of bradykinin b 2 receptors. New York, NY, USA:Rockefeller University Press (2000). Available at: http://www.jem.org/cgi/content/full/192/9/1289.
47. Coates BM, Sullivan DP, Makanji MY, Du NY, Olson CL, Muller WA, et al. Endothelial transmigration by Trypanosoma cruzi. PloS One (2013) 8:1–10. doi: 10.1371/journal.pone.0081187
48. Barreto-de-Albuquerque J, Silva-dos-Santos D, Pérez AR, Berbert LR, de Santana-van-Vliet E, Farias-de-Oliveira DA, et al. Trypanosoma cruzi infection through the oral route promotes a severe infection in mice: New disease form from an old infection? PloS Negl Trop Dis (2015) 9:1–21. doi: 10.1371/journal.pntd.0003849
49. Silva AA, Silva RR, Gibaldi D, Mariante RM, dos Santos JB, Pereira IR, et al. Priming astrocytes with TNF enhances their susceptibility to Trypanosoma cruzi infection and creates a self-sustaining inflammatory milieu. J Neuroinflamm (2017) 14:1–15. doi: 10.1186/s12974-017-0952-0
50. Quan N, Mhlanga JD, Whiteside MB, Mccoy AN, Kristensson K, Herkenham M. Chronic overexpression of proinflammatory cytokines and histopathology in the brains of rats infected with Trypanosoma brucei. J Comp Neurol (1999) 414(1):114–30. doi: 10.1002/(SICI)1096-9861(19991108)414:1<114::AID-CNE9>3.0.CO;2-G
51. Amin DN, Vodnala SK, Masocha W, Sun B, Kristensson K, Rottenberg ME. Distinct toll-like receptor signals regulate cerebral parasite load and interferon α/β and tumor necrosis factor α-dependent T-cell infiltration in the brains of Trypanosoma brucei-infected mice. J Infect Dis (2012) 205:320–32. doi: 10.1093/infdis/jir734
52. Masocha W, Rottenberg ME. Minocycline impedes African trypanosome invasion of the brain in a murine model. Antimicrobial Agents Chemother (2006) 50:1798–804. doi: 10.1128/AAC.50.5.1798-1804.2006
53. Geurts N, Opdenakker G, van den Steen PE. Matrix metalloproteinases as therapeutic targets in protozoan parasitic infections. Pharmacol Ther (2012) 133:257–79. doi: 10.1016/j.pharmthera.2011.11.008
54. Grab DJ, Chakravorty SJ, van der Heyde H, Stins MF. How can microbial interactions with the blood-brain barrier modulate astroglial and neuronal function? Cell Microbiol (2011) 13:1470–8. doi: 10.1111/j.1462-5822.2011.01661.x
55. Chuenkova MV, PereiraPerrin M. Chagas’ disease parasite promotes neuron survival and differentiation through TrkA nerve growth factor receptor. J Comp Neurol. 91:385–94. doi: 10.1111/j.1471-4159.2004.02724.x
56. Silva RR, Mariante RM, Silva AA, dos Santos ALB, Roffê E, Santiago H, et al. Interferon-gamma promotes infection of astrocytes by Trypanosoma cruzi. PloS One (2015) 10:1–23. doi: 10.1371/journal.pone.0118600
57. Pacheco AL, Vicentini G, Matteucci KC, Ribeiro RR, Weinlich R, Bortoluci KR. The impairment in the NLRP3-induced NO secretion renders astrocytes highly permissive to T. cruzi replication. J Leukocyte Biol (2019) 106:201–7. doi: 10.1002/JLB.4AB1118-416RR
58. Weinkauf C, PereiraPerrin M. Trypanosoma cruzi promotes neuronal and glial cell survival through the neurotrophic receptor TrkC. Infection Immun (2009) 77:1368–75. doi: 10.1128/IAI.01450-08
59. de Melo-Jorge M, PereiraPerrin M. The chagas’ disease parasite Trypanosoma cruzi exploits nerve growth factor receptor TrkA to infect mammalian hosts. Cell Host Microbe (2007) 1:251–61. doi: 10.1016/j.chom.2007.05.006
60. Chuenkova MV, Pereira MA. The T. cruzi trans-sialidase induces PC12 cell differentiation via MAPK/ERK pathway. NeuroReport (2001) 12:3715–8. doi: 10.1097/00001756-200112040-00022
61. Filigheddu MT, Górgolas M, Ramos JM. Orally-transmitted chagas disease. Medicina Clínica (English Edition) (2017) 148:125–31. doi: 10.1016/j.medcle.2017.02.007
62. Giddings OK, Eickhoff CS, Smith TJ, Bryant LA, Hoft DF. Anatomical route of invasion and protective mucosal immunity in Trypanosoma cruzi conjunctival infection. Infection Immun (2006) 74:5549–60. doi: 10.1128/IAI.00319-06
63. Poulopoulos A, Belazi M, Epivatianos A, Velegraki A, Antoniades D. The role of Candida in inflammatory papillary hyperplasia of the palate. J Oral Rehabil (2007) 34:685–92. doi: 10.1111/j.1365-2842.2007.01758.x
64. De-Souza W, De-Carvalho TMU, Barrias ES. Review on Trypanosoma cruzi: Host cell interaction. Int J Cell Biol (2010) 2010:1–18. doi: 10.1155/2010/295394
65. Ortega-Barria E, Pereira MEA. A novel T. cruzi heparin-binding protein promotes fibroblast adhesion and penetration of engineered bacteria and trypanosomes into mammalian cells. Cell (1991) 67:411–21. doi: 10.1016/0092-8674(91)90192-2
66. Schenkman S, Eichinger D, Pereira MEA, Nussenzweig V. Structural and functional properties of Trypanosoma cruzi trans-sialidase. Annu Rev Microbiol (1994) 48:499–523. doi: 10.1146/annurev.mi.48.100194.002435
67. Butler CE, de Carvalho TMU, Grisard EC, Field RA, Tyler KM. Trans-sialidase stimulates eat me response from epithelial cells. Traffic (2013) 14:853–69. doi: 10.1111/tra.12078
68. McClure R, Massari P. TLR-dependent human mucosal epithelial cell responses to microbial pathogens. Front Immunol (2014) 5. doi: 10.3389/fimmu.2014.00386
69. Yoshida N. Molecular basis of mammalian cell invasion by Trypanosoma cruzi. Anais da Academia Bras Cienc (2006) 78:87–111. doi: 10.1590/s0001-37652006000100010
70. Dobolyi A, Vincze C, Pál G, Lovas G. The neuroprotective functions of transforming growth factor beta proteins. Int Mol Sci. (2012) 13(7):8219–58. doi: 10.3390/ijms13078219
71. Quenum Zangbede FO, Chauhan A, Sharma J, Mishra BB. Galectin-3 in M2 macrophages plays a protective role in resolution of neuropathology in brain parasitic infection by regulating neutrophil turnover. J Neurosci (2018) 38 (30):6737–50. doi: 10.1523/JNEUROSCI.3575-17.2018
72. Yohannes E, Ghosh SK, Jiang B, McCormick TS, Weinberg A, Hill E, et al. Proteomic signatures of human oral epithelial cells in HIV-infected subjects. PloS One (2011) 6 (11):e27816. doi: 10.1371/journal.pone.0027816
73. Kellokoski J, Syrjänen S, Tosi P, Cintorino M, Leoncini P, Syrjänen K. Cytokeratin pattern in normal and HPV infected oral mucosa in women with genital HPV infections. J Oral Pathol Med (1991) 20(1):26–31. doi: 10.1111/j.1600-0714.1991.tb00883.x
74. O'Callaghan P, Zhang X, Li JP. Heparan sulfate proteoglycans as relays of neuroinflammation. J Histochem Cytochem (2018) 66(4):305–19. doi: 10.1369/0022155417742147
Keywords: Chagas disease, central nervous system, neuroimmune interactions, encephalitis, brain inflammation, oral Trypanosoma cruzi transmission
Citation: Useche Y, Pérez AR, de Meis J, Bonomo A and Savino W (2022) Central nervous system commitment in Chagas disease. Front. Immunol. 13:975106. doi: 10.3389/fimmu.2022.975106
Received: 22 June 2022; Accepted: 21 October 2022;
Published: 10 November 2022.
Edited by:
José Roberto Mineo, Federal University of Uberlandia, BrazilReviewed by:
Phileno Pinge-Filho, State University of Londrina, BrazilCopyright © 2022 Useche, Pérez, de Meis, Bonomo and Savino. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yerly Useche, eWVybHkudXNlY2hlc0BnbWFpbC5jb20=; Wilson Savino, d2lsc29uLnNhdmlub0BmaW9jcnV6LmJy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.