
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 06 October 2022
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.974996
This article is part of the Research TopicThe Interconnection Between the Tumor Microenvironment and Immunotherapy in Brain Tumors Volume IIView all 8 articles
 Can Xu1,2†
Can Xu1,2† Menglin Xiao1,2†Xiang Li3Lei Xin1,2Jia Song2,3Qi Zhan4Changsheng Wang1,2Qisong Zhang1,2Xiaoye Yuan2,3
Menglin Xiao1,2†Xiang Li3Lei Xin1,2Jia Song2,3Qi Zhan4Changsheng Wang1,2Qisong Zhang1,2Xiaoye Yuan2,3 Yanli Tan2,3*
Yanli Tan2,3* Chuan Fang1,2*
Chuan Fang1,2*The glioma tumor microenvironment plays a crucial role in the development, occurrence, and treatment of gliomas. Glioma-associated macrophages (GAMs) are the most widely infiltrated immune cells in the tumor microenvironment (TME) and one of the major cell populations that exert immune functions. GAMs typically originate from two cell types-brain-resident microglia (BRM) and bone marrow-derived monocytes (BMDM), depending on a variety of cytokines for recruitment and activation. GAMs mainly contain two functionally and morphologically distinct activation types- classically activated M1 macrophages (antitumor/immunostimulatory) and alternatively activated M2 macrophages (protumor/immunosuppressive). GAMs have been shown to affect multiple biological functions of gliomas, including promoting tumor growth and invasion, angiogenesis, energy metabolism, and treatment resistance. Both M1 and M2 macrophages are highly plastic and can polarize or interconvert under various malignant conditions. As the relationship between GAMs and gliomas has become more apparent, GAMs have long been one of the promising targets for glioma therapy, and many studies have demonstrated the therapeutic potential of this target. Here, we review the origin and activation of GAMs in gliomas, how they regulate tumor development and response to therapies, and current glioma therapeutic strategies targeting GAMs.
Gliomas are a group of primary brain tumors of glial tissues that include astrocytomas, oligodendrogliomas, and glioblastomas (GBMs). Among them, GBM is the most commonly occurring malignant primary brain carcinoma. Even with the standard combination treatments involving surgical resection, postoperative radiotherapy, and chemotherapy, the median survival time of GBM patients is only 14.6 months. Besides, tumor recurrence and death are almost inevitable in GBM patients (1). The development of drug-resistance glioma and difficulties in designing effective treatment strategies are largely due to the high degree of heterogeneity in tumor-associated cellular and genetic signatures as well as to the preventive actions of the blood-brain barrier (BBB) (2, 3). Furthermore, the infiltration of glioma-associated macrophages (GAMs), T-regulatory cells (Tregs), and myeloid-derived suppressor cells (MDSCs) in the tumor microenvironment (TME) greatly contributes to the pathogenesis of complex malignant phenotypes and impairment of antitumor immune functions (4). In addition to the tumor cells, gliomas also contain many non-tumor infiltrates that mediate the tumor initiation, progression, and response to therapies. Most non-tumor cells in gliomas are GAMs, recruited to the glioma microenvironment under homeostatic and/or inflammatory conditions. GAMs are immunologically active and can modulate the release of various growth factors and pro-inflammatory cytokines, which generate a supportive matrix for the metastasis of tumor cells and promote the formation of an immunosuppressive TME of gliomas (5). The glioma microenvironment is characterized by high levels of immunosuppressive cytokines and significant populations of Tregs and bone marrow-derived macrophages (BMDMs) (6). In gliomas, the number of GAM infiltrates positively correlates with the glioma grading by the World Health Organization (WHO) and negatively correlates with patient survival (7, 8). Increased peritumoral BMDM infiltration has been observed in GBM patients (9). Moreover, monocytes from healthy donors have been found to acquire BMDM characteristics after treatment with a conditioned medium of a GBM cell line (10). Further explorations of GAMs and the tumor immune microenvironment are fundamental to developing novel immunotherapeutic strategies for glioma management (11).
Due to the existence of BBB, the brain has always been considered a unique immune organ (12). However, the specificity of the brain immune system is viewed as more immunologically different than an immune-specific organ (13). Gliomas originate from the primary neural stem or glial cells in the central nervous system (CNS), and the glioma microenvironment is unique in the sense that it contains a mixed population of neurons, astrocytes, resident myeloid cells, and microglia. Glioma-specific GAMs include brain-resident macroglia (BRM) or BMDM (14, 15). GAMs are the most widely infiltrated immune cells in the glioma microenvironment, mediating diverse and complex functions such as tumor metastasis, angiogenesis, treatment resistance, and development of an immunosuppressive microenvironment (Figure 1) (16). The importance of the GAM population for glioma development is reflected in the fact that GAMs account for 50% of all cells in human GBM (17, 18). There is also a marked infiltration of myeloid cells in high-grade gliomas, accounting for more than 85% of GAMs within gliomas are BMDMs, whereas BRMs predominate in the peri-tumoral areas (18, 19).
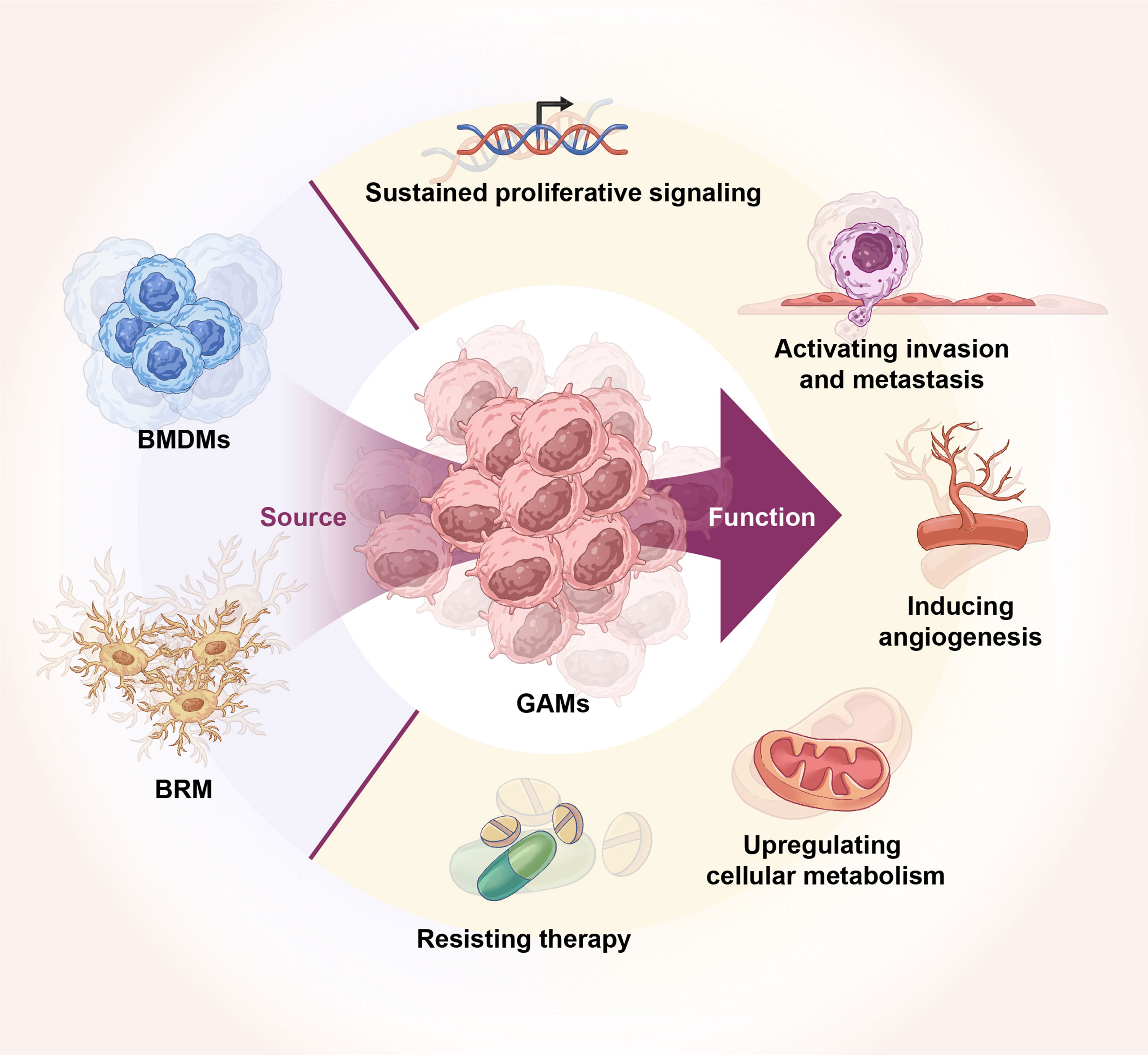
Figure 1 Origin of GAMs and their role in glioma. GAMs originate from BRM and BMDMs. GAMs regulate glioma tumor growth, invasion, angiogenesis, energy metabolism, and therapy resistance.
Although the activation states of GAMs are complex, however, it is often presented into two categories the simplicity-classically activated macrophages or M1 macrophages and alternatively activated macrophages or M2 macrophages, which exhibit antitumor/immunostimulatory and protumor/immunosuppressive effects, respectively (20). Postnatal development of macrophages occurs through the macrophage colony-stimulating factor (M-CSF) or granulocyte-macrophage colony-stimulating factor (GM-CSF)-dependent differentiation of circulating monocytes, which originate from bone marrow-derived progenitor cells (21). Previous studies have shown that the M1-type macrophages, activated by the increased exposure to stimuli like GM-CSF, interferon-gamma (IFN-γ), tumor growth factor alpha (TGF-α), and lipopolysaccharide (LPS), express surface markers such as CD68, CD80, CD86, and secretory cytokines including IL1β, IL6, IL12, IL23, CXCL9 and CXCL10 (22); while M2-type polarization is closely related to the levels of M-CSF, IL4, IL10, IL13, TGF-β and glucocorticoids, and activated M2 macrophages express surface markers like CD163, CD204, and CD206 as well as secretory cytokines including IL10, TNF-α, CCL17, CCL18, CCL22, and CCL24 (22, 23).. The primary strategy of GAM-targeted therapy for glioma aims to reduce the recruitment of BMDM and reprogram GAMs from the M2 to M1 phenotype, thereby reversing the characteristics of the anti-inflammatory tumor immune microenvironment (16). In this review, we summarize the origin, recruitment, and activation mechanisms of GAMs in gliomas, regulatory factors for the M1/M2 polarization of GAMs, and the effects of GAMs on glioma development and therapeutic outcomes, as well as the research progress on GAM-targeted treatment strategies.
Glioma-associated macrophages originate from two types of cells, namely BRM and BMDMs (24). Florent Ginhoux et al. have shown for the first time that the resident microglia in the brain originate from the extraembryonic yolk sac cells (25). Katrin Kierdorf et al. have further demonstrated that mouse microglia could be derived from the primitive c-KIT-positive erythrocyte precursors (26). In the adult brain, microglia perform multiple tasks such as functional support to neurons, phagocytosis of apoptotic cells, and immune surveillance (27, 28). Moreover, the microglia pool is maintained by their self-renewal mechanisms without the contribution of myeloid-derived progenitor cells. Therefore, microglia are the only resident immune cell population in the healthy brain (29, 30). Microglia can be discriminated from GAMs with the microglia-specific genes P2RY12 and TMEM119, higher proportion of microglia may be beneficial for patient survival in glioblastoma (31). Monocytes originate from progenitor cells in the bone marrow. During homeostasis and inflammation, circulating monocytes leave the bloodstream and migrate into tissues, where they differentiate into macrophages or dendritic cell populations depending on the presence of local growth factors, pro-inflammatory cytokines, and microbial products (32). In gliomas, the local inflammatory milieu compromises the integrity of the BBB, permitting the infiltration of inflammatory monocytes into the brain from the circulation, which then differentiates into bone marrow-derived GAMs (5) mediating the inhibition of tumor-specific immune defense mechanisms (33). However, some researchers have put forward a different point of view. Alexander Mildner et al. have used a set of bone marrow chimeric and adoptive transfer experiments to show that BRM can be derived by the differentiation of circulatory LY6CHICCR2(+) monocytes after being infiltrated into brain lesions under pathological conditions. Under the diseased conditions, microglial engraftment in the CNS is not associated with the BBB disruption but rather requires prior brain modulation (e.g., direct tissue irradiation). Experimental results have identified LY6CHICCR-2(+) monocytes as the immediate precursors of microglia in the adult brain and clarified the importance of local stimulatory factors for the microglial engraftment in the adult CNS (34).
In the past, CD45 expression was usually measured to distinguish BRM (low expression level) from BMDM-derived GAMs (high expression level). Immunofluorescence (IF) analysis of patient-derived glioma samples has shown that macrophages with high CD45 expressions are significantly more abundant than those with basal or low expressions (35). However, this notion has been challenged in recent years. New studies have indicated that although the CD45 expression level can differentiate BRM- from BMDMs-derived GAMs in mice, cell-type-specific CD45 expression profiles differ between mice and humans. Moreover, CD45 expression level could not accurately distinguish BRM- from BMDMs-derived GAMs in glioma patient samples, suggesting the need for more sensitive and specific RNA-sequencing, flow cytometry, and other comprehensive analyses to distinguish further the gene expression differences between BRM- and BMDM-derived GAMs (36).
A large-scale RNA-sequencing analysis revealed differential gene expression patterns specific to infiltrating and resident cells, suggesting that populations of GAMs from different origins may perform distinct functions (18). One study has identified the marker protein transmembrane protein 119 (TMEM119) specifically and stably expressing only in BRM-derived GAMs. Further RNA sequencing based on the TMEM119 expression profile has demonstrated unique differences in the BRM- and BMDM-derived GAM transcriptomics. The study has also reported that the gene expression pattern of BRM could be different at different developmental stages. As the microglia matures, the expression of its uniquely expressed genes (e.g., TMEM119, P2RY12, and OLFML3) increases but the cell proliferation ability decreases (37). Another study has utilized multiple genetic lineages for tracing different glioma models. Transcriptional network analysis has identified the modulator ITGA4 (CD49d) that mediates the differentiation of BRM- and BMDM-derived macrophages under a homeostatic condition. Besides, these macrophages express genes related to their specific biological functions (36). Sören Müller et al. conducted a single-cell RNA sequencing (scRNA-seq) analysis of clinical glioma specimens and identified a novel genetic signature. Two markers, CD49d and P2RY12, are detected to differentiate between BRM- and BMDM-derived GAMs under both malignant and non-malignant conditions. Compared with microglia, myeloid-derived monocytes can upregulate the expression of immunosuppressive cytokines, markers of M2 activation (like IL10 and TGF-βII), phagocytosis, and activated tricarboxylic acid (TCA) cycle (38). Notably, these gene expression and functional differences between BRM- and BMDM-derived GAMs are present in both human and mouse GAMs (36–38).
Additionally, the immune compartments within the glioma microenvironment are characterized by a level of heterogeneity and dynamicity of immune cells that cannot be recapitulated by the simplistic paradigm of M1 and M2 phenotypes. Ana et al. employed scRNA-seq and CITE-seq to map the GBM immune landscape in mouse tumors and in patients with newly diagnosed disease or recurrence. BRM- and BMDM-derived GAMs are self-renewing cell populations that compete for space and can be depleted by CSF1R blockade. Microglia-derived GAMs dominate in newly diagnosed tumors but are overtaken by monocyte-derived TAMs after recurrence, especially in hypoxic tumor environments (39). Furthermore, Natalia et al. performed scRNA-seq on microglia, monocytes, and macrophages in male and female mouse gliomas to identify distinct transcriptional programs in GAMs, the findings suggest that glioma-activated microglia Sex-specific genes (MHCII and CD74) expression in cells may be associated with morbidity and outcome in glioma patients (40). Single-cell analysis by Nourhan Abdelfattah et al. has defined the BMDM as one of nine subtypes with spatial heterogeneity, representing distinct immune states, and nominated S100A4 as a promising immunotherapy target (41).
Monocytes are divided into subpopulations based on their differences in the expression profiles of chemokine receptors (CCRs) and specific surface molecules (42, 43). In mice, monocytes can be categorized into two subtypes based on the expression of LY6C and CX3CR1 genes, such as LY6CHI and LY6CLOW (also known as CX3CR1HI) monocytes. The LY6CHI monocytes are called inflammatory monocytes, which typically express high CCR2 but low CX3CR1 levels, occupying approximately 2-5% of circulating leukocytes in healthy mice, and are rapidly recruited to sites of infection and inflammation (44). The role of CCR2 is critical in monocyte trafficking, and the deletion of this chemokine receptor significantly reduces the trafficking of LY6CHI monocytes toward the inflammation sites (45, 46). While LY6CLOW monocytes are less populated than LY6CHI monocytes and typically express high levels of CX3CR1 and low levels of CCR2 and LY6C (42, 43). In vivo microscopy studies have shown that LY6CLOW monocytes adhere and migrate along the luminal surface of endothelial cells in small blood vessels, a process called patrolling (47). In humans, monocytes are classified into three subtypes based on the differential expression of monocyte-specific antigens CD14 and CD16- classical (CD14++CD16-), non-classical (CD14+CD16++), and an intermediate (CD14++CD16+) subtypes (48). Classical monocytes, similar to those of mouse LY6CHI monocytes, highly express CCR2 and are the most prevalent monocyte subset in human blood (49). Non-classical monocytes resemble the mouse LY6CLOW monocyte population and perform patrolling functions in vivo. Although the monocyte subtypes identified in humans and mice are not identical, their process of differentiation and roles in immune defense mechanisms appear to be similar (50–52).
Chemokines are the largest subfamily of cytokines, best known for their roles in directing the movement of immune cells throughout the body (53). According to the position of the first two cysteine (C) residues in their protein sequences, chemokines are classified into four subclasses- CC, CXC, CX3C, and XC (54). Chemokine ligand 2 (CCL2) (previously known as MCP1) is the most crucial member of the CC chemokine family. CCL2 and its receptor (CCR2) are involved in regulating the monocyte/macrophage migration from the blood circulation to the brain through the vascular endothelium and are key pathogenic factors in glioma progression (55). CCL2 and CCL7 mediate the recruitment of bone-marrow-derived LY6CHICCR2+ monocytes via binding to CCR2. Furthermore, CCL2 is responsible for the recruitment of CCR4-expressing Tregs to the glioma microenvironment (56). Glial cells and macrophage-derived (especially CD163+ macrophages) CCL2 is an independent prognostic factor in GBM patients, and the CCL2-CCR2/4 axis is a potential GBM immunotherapy target (57). Loss of CCL2 or CCL7 can significantly reduce the recruitment of myeloid-derived monocytes during inflammation by approximately 40–50% (58). One possible mechanism by which CCL2 recruits BMDM to the TME could be the binding of circulating CCL2 molecules to glycosaminoglycans in gliomas to establish a concentration gradient that guides monocytes toward the site of inflammation. Another possibility could be that CCL2 and CCL7 may act in tandem to direct monocytes to lesion sites in the bone marrow, or they may act in parallel to drive the monocyte recruitment from distinct regions within the bone marrow or other tissues to the TME (59, 60).
Moreover, it has been reported that Duffy antigen receptor chemokine (DARC), which binds CCL2 and transports it to the medial lumen through the endothelium, is essential for the recruitment of monocytes from the blood to inflamed tissues (61). CCL8 and CCL12 also bind CCR2, but deletion of the genes encoding these chemokines has no detectable effect on monocyte trafficking under homeostasis conditions (56). Additionally, studies have shown that activation of the CX3CL1-CX3CR1 signaling pathway can enhance the accumulation of GAMs and promote angiogenesis during the malignant transformation (62). Activated CXCL2-CXCR2 signaling recruits and activates BRM/macrophages through the activation of extracellular signal−regulated protein kinase 1 and 2 (ERK1/2) and AKT signaling pathways, thereby promoting the GBM progression (63).
Monocyte recruitment is thought to follow the general leukocyte adhesion and transportation model, including rolling, adhesion, and migration. The migration of leukocytes, including monocytes, depends on the integrins and several other adhesion molecules (64). Monocytes of LY6CHI mice expressing selectin L (also known as CD62L), selectin P glycoprotein ligand 1 (PSGL1), integrin αLβ2 (also known as LFA1), integrin αMβ2 (also known as MAC1), platelet-endothelial cell adhesion molecule (PECAM1), and integrin α4β1 (also known as VLA4) contribute to leukocyte adhesion and migration. The patrolling of monocytes along the resting dermal vascular endothelium in LY6CLow mice is shown to be mediated by the integrin αLβ2 (47). In contrast, early recruitment of LY6CHI monocytes is not affected by αLβ2 deficiency (65). Additionally, POSTN, an extracellular matrix component produced by glioma stem cells, provides an efficient binding site for αVβ3 integrins on the cell surface of peripheral monocytes and M2-GAMs to promote the extravasation and migration in the glioma microenvironment (5). Besides, carbonic anhydrase XI (CAIX) promotes macrophage adhesion to glioma cells, cell motility, and macrophage polarization (66).
Some other proteins, amino acids, and cytokines have also been reported to be correlated with the recruitment and activation of GAMs. Studies by Quan Zhang et al. have demonstrated that overexpression of programmed cell death protein 10 (PDCD10) promotes the release of CXCL2 and activates CXCR2 and ERK1/2-mediating signaling, thereby recruiting and activating BRM/macrophages to promote the GBM progression (63). Zhuo Chen et al. have found that farnesyl diphosphate synthase (FDPS) can trigger the Wnt/β-catenin signaling pathway and promote the infiltration of GAMs by inducing the expression of CCL20 (67). The bifunctional cytokine IL33 secreted by GBM cells in humans and mouse positively correlates with the tumorigenic infiltration of GAMs. Secreted IL33 functionally regulates chemokines that co-recruit and activate circulating and resident innate immune cells. Moreover, IL34 acts through the colony-stimulating factor 1 receptor (CSF-1R) on the surface of peripheral monocytes that mediate monocyte attachment to the vascular endothelial layers (68, 69). Likewise, macrophage inhibitory factor (MIF) and intercellular adhesion molecule 1 (ICAM1) provide instructional signals to monocytes during the extravasation (70). Haitao Ge et al. have found that CD70 ablation in primary GBM cell lines can reduce the expressions of CD44 and SOX2 genes, inhibiting the tumor migration, growth, and ability to attract monocyte-derived M2 macrophages in vitro (71). In addition, kynurenine produced by GBM activates the aryl hydrocarbon receptor (AHR) in GAM cells. AHR induces the expression of CCR2 and increases the rate of recruitment of GAMs to the TME. AHR also drives the expression of KLF4 and inhibits the NF-κB-mediated inflammatory signaling in tumor associated macrophages, regulating the function of GAM cells and T-cell immunity (72).
Mian-Fu Cao et al. have found that GAM-GBM cell hybrids can exist in human GBM specimens as well as in orthotopic mice models. Following the co-culture of GBM cells with BMDM, the hybrids can undergo nuclear reprogramming with a unique gene expression profile compared to their parental cells. Moreover, glioma invasion-associated genes are enriched in hybrids with higher invasiveness. More hybrids in the invasive margin of GBM have been observed in comparison to the GBM core area, suggesting that GAM -GBM cell hybrids can enhance the invasiveness of GBM cells (73). Furthermore, GAMs can improve the invasiveness of CD133+ tumor stem cells via releasing TGF-βI, thereby increasing the production of matrix metalloproteinase 9 (MMP9). The CD133+ glioma stem-like cells (GSLCs) isolated from xenografted gliomas in mice exhibit higher invasive potential after co-culturing with GAMs (74). Continuous autocrine stimulation of macrophages by adenosine deaminase 2 or cat eye syndrome chromosome region candidate 1 gene (CECR1) enables M2-like TAMs to stimulate MAPK and c-Jun signaling in glioma cells via paracrine activation, promoting tumor proliferation and migration (75). Another study has shown that GAMs can activate the ERK1/2 phosphorylation in GBM cells by secreting CCL8, thereby inducing GBM cell invasion and stem cell-like traits. Moreover, CCR1 and CCR5 are the main receptors mediating CCL8-induced biological behaviors of gliomas. Blockade of CCL8 secreted by GAMs via neutralizing antibodies significantly reduces glioma cell invasion (76). Furthermore, GAM-secreted abundant pleiotrophin (PTN) through its receptor PTPRZ1 stimulates glioma stem cells (GSCs) and promotes GBM malignancy through the PTN-PTPRZ1 paracrine signaling (77). The RNA regulator HuR expressed in GAMs also plays a crucial role in the tumor-promoting abilities of GAMs. Suppression of HuR-induced M1-type polarization of GAMs concomitantly reduces the expression of PDL1, increases the number of infiltrating CD4 cells (including Th1 and cytotoxic effector cells), and suppresses the tumor growth in GBM mouse models (78).
GAMs promote tumor progression via regulating the angiogenesis in GBM. In GBM, CD163+ macrophages are widely distributed across tumor parenchyma vessels, especially between the proliferating microvessels (79). Perivascular GAMs in GBM are closely related to the density of microvessels and high expression of vascular endothelial growth factor A (VEGFA), heme oxygenase 1 (HO1), and thymidine phosphorylase (80). GAMs have been shown to enhance the vascular mimicry (VM) of glioma cells via upregulating the secretion of IL6 and cyclooxygenase 2 (COX2) (81, 82). Moreover, GAM-secreted IL6 promotes the angiogenesis of endothelial progenitor cells by activating the JAK-STAT signaling pathway (83). Additionally, GBM-derived C-reactive protein (CRP) induces COX2-positive GAMs to produce IL6 and IL1β, which promote endothelial cell proliferation and enhance endothelial expression of proangiogenic factors, including IL8, VEGFA, and hypoxia-inducible factor 1 alpha (HIF-1α) (84, 85). The receptor for advanced glycation end-product (RAGE) signaling in GAMs drives the GBM angiogenesis and tumor growth. Knockdown of RAGE in GBM mice models does not alter the tumor growth rate but prolongs animal survival by reducing tumor-associated inflammation (86).
Dysregulation of energy metabolism is an emerging hallmark of tumors. There is an intricate interaction network between the metabolism of tumor cells and TME. Hypoxia, acid build-up, and immune cells in the TME can regulate the metabolism of tumor cells (87, 88). M2-type macrophages promote oxidative phosphorylation (OXPHOS) and support tumor cell proliferation by releasing large amounts of VEGFA and IL10. In response to hypoxia and lactate stimulation, macrophage-produced chemokines CCL5 and CCL18 upregulate the activities of various glycolytic factors, including lactate dehydrogenase A (LDHA) and glucose-6-phosphate dehydrogenase (G6PD), promoting glycolysis in tumor cells, which leads to the accumulation of excessive lactate in the TME and suppresses tumor immune responses (89). Jian Lu et al. have found that M2-type GAM-derived IL1β, mediated by phosphatidylinositol-3-kinase-mediated protein kinase delta (PKCδ), activates phosphorylation of the glycolytic enzyme glycerol-3-phosphate dehydrogenase (GPD2) at threonine10 (GPD2 pT10) in glioma cells. GPD2 pT10 enhances its substrate affinity and increases the catalytic rate of glycolysis in glioma cells. Inhibition of PKCδ or GPD2 pT10 in glioma cells or blocking of IL1β production by macrophages reduces glioma cells’ glycolytic rate and proliferation (90). Also, Yajuan Zhang et al. have indicated that M2-type GAMs can enhance the 3-phosphoinositide-dependent protein kinase 1 (PDPK1)-mediated phosphorylation of phosphoglyceride kinase 1 (PGK1) at threonine243 by secreting IL6. This phosphorylation promotes PGK1-catalyzed glycolysis by altering substrate affinity. Inhibition of PGK1 T243 or PDPK1 phosphorylation in tumor cells or neutralization of macrophage-derived IL6 reverses the macrophage-promoted glycolysis, proliferation, and tumorigenesis (91).
The infiltration of GAMs and M2-type polarization in the glioma microenvironment lead to tumor immunosuppression and induces the resistance of GBM to chemoradiotherapies via multiple mechanisms. IL11 secreted by GAMs activates the STAT3-MYC signaling pathway, which induces the proliferation of glioma stem-like cells and confers enhanced tumorigenicity and resistance to chemotherapeutic drugs like temozolomide (TMZ) (92). Crosstalk between GBM cells and GAMs attenuates the chemotherapeutic efficacy of drugs. Studies have suggested that long non-coding (lnc) RNA TALC (lnc-TALC) is incorporated into exosomes and delivered to GAMs to promote their M2-type polarization, ultimately leading to the TMZ resistance (93). Similarly, miR-21 maturation in GAM-secreted exosomes can increase the secretion of M2-type cytokines IL6 and TGF-βI, thereby promoting the M2-type polarization of GAMs and increasing the resistance of GBM cells to the TMZ treatment (94). Furthermore, SLIT2-induced macrophage invasion and M2-type polarization in mouse GBM cells and human patient-derived GBM xenografts have exhibited enhancement of the therapeutic resistance of GBM to chemo- and immunotherapies (95). In the anti-angiogenic therapy of GBM, the recruitment and M2-type polarization of BMDMs following the administration of VEGF inhibitors constitute an immunosuppressive microenvironment, leading to treatment resistance (96). In the radiotherapy of GBM, irradiation can upregulate the expression of CSF-1R, enhancing the recruitment of BMDMs-derived GAMs, and promoting M2-type polarization, resulting in the development of radioresistance (97).
Recently, multiple studies have shown that GAMs can be activated and polarized by various modulatory factors (Table 1), including soluble molecules derived from GBM cells, thereby promoting tumor progression and metastasis. Sonic hedgehog signaling (SHH) molecule secreted by GBM cells blocks the recruitment of CD8+ T-cells to the glioma microenvironment by inhibiting CXCL9 and CXCL10 to drive M2-type polarization of GAMs (98). In contrast, the CXCL16-CXCR6 axis induces the M1-type microglia while inhibiting the polarization of the M2 phenotype upon LPS or IFN-γ stimulation (99). In addition, the Na+/H+ exchanger 1 (NHE1) protein, secreted by GBM, partially promotes the M2-type activation of GAMs by stimulating the glucose metabolism and participating in the regulation of the GBM immunosuppressive microenvironment, which is recognized as a new target for improving the efficacy of immunotherapy (124, 125). Osteopontin (OPN) is a potent macrophage-derived chemokine that maintains both the M2 genotype and phenotype of GAMs. The expression level of OPN is correlated with glioma grade and GAM infiltration. Integrin αvβ5 acts as the major receptor for OPN (100). Similarly, mucin (MUC1) and polypeptide SLIT2 have also been shown to be involved in the M2-type polarization of GAMs (95, 101). Moreover, GBM cells activate CD74 expression and induce the transformation of GAMs from M1- to M2-type polarization by secreting MIFs (70).

Table 1 Regulators of M1/M2-GAMs activation and transformation.
The GBM is highly heterogeneous at both molecular and histological levels. Not only the intratumoral heterogeneity, but GBM also exhibits a high level of intertumoral heterogeneity. Different molecular subtypes of GBM modulate different gene signatures of GAMs (102). Mesenchymal-associated glioma-associated macrophages (MA-GAMs) are the master regulators of this process, and the expression of their target genes significantly correlates with poor clinical outcomes. They are often associated with genomic aberrations in neurofibromin 1 (NF1) and phosphoinositide 3-kinases/mammalian target of rapamycin/Akt (PI3K-mTOR-AKT) pathway-related genes (103).
Cancer stem/stem-like cells are critical for cancer initiation, progression, and therapy resistance. The sc-sequencing suggests that cancer stem cells may correspond to the most malignant and proliferative glioma group of cells in gliomas, and are often the originator of other histotypes or molecularly typed glioma cells (104). Researchers have isolated a particular fraction of necrotic products spontaneously arising from glioma cells, which are morphologically and biochemically defined as autoschizis-like products (ALPs). Glioma stem cell (GSC)-derived ALPs exhibit a higher activity for the M1-GAMs polarization than those from non-GSCs (105). Furthermore, the ARS2/MAGL pathway in GSCs regulates the self-renewal and tumorigenicity of GSCs through the production of prostaglandin E2, stimulates β-catenin activation, and M2-like GAM polarization in GSCs (106), GSCs also promote the survival, and M2-like polarization of GAMs by secreting Wnt-induced signaling protein 1 (WISP1). Silencing of WISP1 has been shown to significantly disrupt the GSC maintenance and the M2-like characteristics of GAMs (107). Furthermore, GSC-secreted POSTN recruits M2-GAMs through its receptor integrin αvβ3. Knockout of POSTN in GSCs significantly reduces the GAM density, inhibits tumor growth, and increases survival in mice bearing GSCs-derived xenografts (108).
Exosomes are essential elements involved in intercellular communication and TME modulation. High expression of miR-1246 in hypoxic glioma-derived exosomes (H-GDES) significantly induces the M2-type polarization of macrophages to activate the STAT3 signaling while inhibiting the NF-κB signaling pathway, which subsequently promotes glioma proliferation, migration, and invasion both in vitro and in vivo (109). Furthermore, H-GDES containing high levels of IL6 and miR-155-3p can induce M2-like macrophage polarization via the IL6-pSTAT3-miR-155-3p-autophagy-pSTAT3 positive feedback loop, thereby promoting the glioma progression (110). Moreover, GBM-derived arginase-1+ exosomes (GBex) can reprogram M1-type GAMs to M2-type and enhance the tumor-promoting functions of macrophages (126). GBM-derived lnc-TALC gets incorporated into exosomes to be delivered to GAMs, where they promote M2-type polarization and mediate TMZ resistance (93). GAM-secreted exosomes enriched in miR-21 play a role in increasing the secretion of M2-type-related cytokines IL6 and TGF-βI, promoting M2-type polarization of GAMs, and improving the resistance of GBM cells to TMZ treatment (94).
GAMs are regulated by Tregs, MDSCs, and programmed cell death 1 ligand 1 (PD-L1). IFN-γ is the major cytokine responsible for the inhibition of M2-like polarization of GAMs. Tregs inhibit IFN-γ secreted by CD8+T-cells, preventing the sterol regulatory element-binding protein 1 (SREBP1)-mediated activation of fatty acid synthesis in immunosuppressive M2-like GAMs. Thus, Tregs indirectly but selectively maintain the metabolic homeostasis, mitochondrial integrity, and survival of M2-like GAMs (111). MDSCs have been shown to regulate GAMs’ differentiation and promote tumor proliferation by downregulating the STAT3 level (112). PD-L1 is an unfavorable prognostic marker in GBM patients. PD-L1-mediated immunosuppression can be attributed to the infiltration and M2 polarization of GAMs (113). Furthermore, the programmed cell death 1 (PD1) protein promotes the remodeling of macrophages toward M2-type polarization, and its expression on macrophages is inversely correlated with the phagocytic potency. Blockade of the PD1-PD-L1 axis increases the rate of phagocytosis in macrophages (114).
Most of the human genome is transcribed into RNA that does not code for any proteins. Any aberrant production of these non-coding RNAs (ncRNAs) is critical for the development and progression of various cancers (115). Downregulation of miR-340-5p in GBM is associated with increased tumor size, recurrence, and poor survival. MiR-340-5p directly targets POSTN, which then recruits GAMs via integrin αvβ3. The knockdown of miR-340-5p promotes GAM recruitment and M2 polarization in vitro and in vivo (116). In addition, miR-106b-5p has been reported to inhibit IRF1/IFN-β signaling and promote M2 polarization of GAMs (117). Likewise, lnc-SNHG15 promotes GBM tumorigenesis by inhibiting the maturation of tumor suppressor miR-627-5p, leading to activation of CDK66 and SOX-2, and M2 polarization of microglia (118). The exosome-associated ncRNAs, including miR-1246, miR-155-3p, miR-21, and lnc-TALC, also modulate the polarization of GAMs through the above-described mechanisms (93, 94, 109, 110).
Radiation has a significant impact on TME. The major changes that happen in the glioma microenvironment following the radiotherapy include decreased microvessel densities, increased ischemia-hypoxia lesions, and the accumulation of tumor-associated macrophages in these ischemia-hypoxic lesion sites. Irradiated hypoxic tissues exhibit different TME characteristics that favor the development of M2-type macrophage polarization under the regulation of tumor-secreted SDF-1α levels (127). BMDM-derived GAMs accumulate in irradiated glioma tissues after radiotherapy. These GAMs stimulate the restoration of blood flow in irradiated tumors, thereby promoting the recurrence of gliomas. SDF-1α/CXCR4 chemokine pathway drives the critical mechanism of the GAM accumulation. Hence, blocking this pathway to prevent the GAM accumulation in the TME enhances tumor response to radiation and protects irradiated tissues (119). Notably, radiotherapy upregulates CSF-1R expression and M2-type polarization to enhance the recruitment of BMDM to the TME, which promotes the development of tumor immunosuppression in gliomas (97, 120). A recent study using the combination of multi-tracer PET/MRI imaging to spatially visualize the regulation of TMZ on bone marrow-derived MDSCs and BMDMs has revealed that TMZ treatment can increase the expression of M2-type GAM marker genes in glioma microenvironment cells, which may, in turn, contribute to TMZ therapy resistance (121). Besides, VEGFR inhibitors and anti-PD-1 antibodies also lead to GAM recruitment and M2-type polarization in gliomas (96, 122).
Within the TME, other cellular processes, including autophagy, hypoxia, and metabolic reprogramming can also modulate the activation of GAM. Autophagy provides tumor cells with essential nutrients, nucleotides, and amino acids to promote their tumorigenic growth in the TME (123). Autophagy-dependent lysosome-secreted KRAS protein induces M2-type polarization of macrophages through the STAT3-regulated fatty acid oxidation (FAO) (128). Moreover, autophagy proteins in the bone marrow-derived glioma cells modulate LC3-associated phagocytosis (LAP) and mediate T lymphocyte-regulated immunosuppression to activate GAMs (129). M2-type polarization of GAMs correlates with angiogenic endothelial cell-macrophage and tumor cell-extracellular matrix interactions. TGF-βI and integrin αvβ3 have been shown to promote tumor-endothelial angiogenesis and M2-type polarization of GAM cells (130). Importantly, hypoxic shocks in the glioma microenvironment lead to M2-type polarization of GAMs. Both hypoxic environment and hypoxia-treated glioma cell supernatants have shown their abilities to induce M2-type polarization of GAMs. Moreover, hypoxia increases POSTN expression in glioma cells and promotes the recruitment of macrophages (131). Tumor metabolism reprogramming is crucial to the development of glioma immune tolerance. Tryptophan and adenosine metabolism have been determined to be responsible for the accumulation of Tregs and M2 macrophages, respectively, in the TME (132).
The receptor tyrosine kinase (RTK) signaling pathway regulator CSF-1R is thought to play an essential role in the recruitment and differentiation of macrophages. The CSF-1R kinase inhibitors have entered clinical trials for various cancer treatments (133). Stephanie et al. used the CSF-1R inhibitor BLZ945 to target GAMs in a mouse proneural GBM model, which revealed an improved survival and regression of established tumors in treated mice. The CSF-1R inhibitors were found to slow down the intracranial growth of patient-derived glioma xenografts, also, M2-type markers were significantly reduced in surviving mouse GAMs (134). It has also been confirmed that the CSF-1R inhibitor PLX3397 interferes with the differentiation of macrophages during carcinogenesis, thus restoring the sensitivity of glioma cells to RTK inhibitors in a preclinical combination trial (135). Additionally, PLX3397 shows promising efficacy in another preclinical glioma model (136). However, to date, CSF-1R inhibitors have demonstrated limited effectiveness in GBM clinical trials. Among 37 patients with relapsed aggressive GBM, PLX3397-treated patients showed no significant improvement in progression-free survival (PFS) during the 6-month follow-up period (137). On the other hand, targeting GAMs with CSF-1R inhibitors alone may lead to antitumor responses in GBM (138). CSF-1R inhibitors may induce resistance to therapy by promoting insulin-like growth factor 1 (IGF-1) expression and activating the PI3K pathway in GAMs (139, 140). Synergistic use of CSF-1R and IGF1R inhibitors may be a more effective glioma treatment solution (140, 141).
Additionally, GBM resistance to radiotherapy may be associated with the up-regulation of CSF-1R expression, along with enhanced recruitment of BMDM-derived GAMs. Studies have shown that both CSF-1R inhibitors BLZ945 and PLX3397 can accelerate radiotherapy efficacies by blocking the radiotherapy-induced recruitment and activation of M2-type GAMs to gliomas, thereby disrupting the tumor-promoting functions of these cells in supporting glioma proliferation and regrowth (97, 120). Inhibition of CSF-1R by BLZ-945 improves the efficacy of radiation therapy in GBM. Compared to receiving radiotherapy alone, CSF-1R inhibition prevents radiotherapy-recruited monocytes from differentiating into immunosuppressive and proangiogenic GAMs. CSF-1R inhibition may be a promising strategy to overcome the hurdles of radioresistance in GBM (97, 142).
Chemokine signaling plays a crucial role in gliomagenesis, proliferation, neovascularization, metastasis, and tumor progression (143). Chemokines promote an immunosuppressive microenvironment via recruiting BMDMs, Tregs, and MDSCs to the TME. Immunotherapy targeting chemokines constitute one of the promising strategies for glioma treatment (144). CXCR4 is one of the critical chemokines responsible for the malignant behavior of gliomas. Lentivirus-mediated knockdown of CXCR4 showed reduced proliferation, invasion, migration, and enhanced apoptosis of glioma cells. Interestingly, a better therapeutic effect was obtained after combining CXCR4 knockdown with miR-21 expression (145). A novel inhibitor of CXCR4, named peptide R, reduced the metabolic activity, proliferation, and migration capacity of U87MG cells in vitro and inhibited tumor growth in an orthotopic GBM mouse model (146). Another brain-penetrating CXCR4 antagonist, PRX177561, enhanced the efficacy of anti-angiogenic therapy in GBM after co-administration with bevacizumab or sunitinib and was considered an effective complementary strategy to anti-angiogenic treatments (147). Studies have shown that combined use of the CXCR2 inhibitor SB225002 and TMZ can reduce the TMZ-induced activation of the IL8-CXCL2-CXCR2 signaling axis, inhibit tumor angiogenesis and GAM infiltration, and enhance TMZ chemotherapy efficacy (148, 149). Furthermore, monoclonal antibodies targeting mouse and/or human CCL2 show prolonged survival in C57BL/6 mice bearing intracranial GL261 gliomas, which coincides with the reduction of GAMs and MDSCs in the TME and may enhance the therapeutic benefits of TMZ (150). In addition, the combination of chemokine- and PD1-targeted immunotherapy has shown a certain therapeutic potential. Compared with the anti-PD1 monotherapy, anti-CCR2 or anti-CXCR4 therapy combination can achieve better therapeutic efficacy in mouse glioma models (151, 152).
Anti-angiogenic therapy is one of the promising options for the treatment of gliomas. Unfortunately, despite the initial efficacy of anti-angiogenic therapies, the effective durations of these drugs are limited, and drug resistance following long-term use is almost inevitable (138). In preclinical studies, the VEGF inhibitor bevacizumab, as a single agent for GBM, only showed benefits in imaging and clinical responses but had no significant effect on the PFS (153). An essential mechanism of GBM resistance to VEGF inhibitors lies in the recruitment and M2-type polarization of BMDM-derived GAMs after the drug administration. Compared with BRM, BMDM-derived GAMs preferentially lead to treatment resistance. VEGF-targeting glioma immunotherapy needs to overcome the immunosuppressive microenvironment supported by the BMDM-derived GAMs (96). In terms of mechanism, the anti-VEGF/VEGFR therapy upregulates the expression of CXCR4, SDF-1α, and TGF-βI, leading to the recruitment of BMDMs and M2-polarization. Therefore, multi-drug regimens may be more appropriate during the anti-vascular production treatment (147). The good news is that a novel SDF-1α inhibitor, olaptesed pegol (OLA-PEG), has been shown to reduce the recruitment and activation of GAMs by anti-VEGF therapy and enhance its antitumor efficacy in GBM (154). Moreover, the dual inhibitory effect of VEGFR/ANG2 on the M1-type polarization of GAMs increases the ratio of M1 to M2 macrophages, thereby extending the survival of preclinical GBM mice (155). These findings suggest a new therapeutic strategy to suppress the recruitment of VEGFR-induced GAMs, and M2-type polarization by integrating anti-ANG2, anti-SDF-1α, and anti-CXCR4 therapies, which is expected to overcome the limitations of anti-VEGFR monotherapy in GBM patients.
Immunotherapy targeting the PD1/PD-L1 axis offers novel therapeutic options for the treatment of many cancers. However, durable antitumor responses have only been observed in a few patients, and preclinical as well as clinical studies have demonstrated that PD1/PD-L1 blockade can result in abnormalities in the TME that reduces the efficacy of anti-PD1/PD-L1 therapy (156, 157). Studies have shown that anti-PD1 monoclonal antibody treatment induces GAM polarization to the M1 phenotype, significantly inhibiting the intracranial tumor growth in GBM mice. The therapeutic effect of anti-PD1 may be regulated by the innate immune system, independent of CD8+T-cell-mediated pathways (158). What is inspiring is that multiple studies have adopted multi-drug combinations to improve the treatment efficacy of anti-PD1 therapy. The combined use of the p38MAPK inhibitor and PD-L1 antibody effectively prolonged the survival rate of TMZ-resistant GBM hosts and significantly reduced BMDM accumulation and PD-L1 abundance in BRM (159). Another study showed that treatment of GSC-derived mouse GBM tumors with nivolumab, an anti-PD1 antibody, resulted in the recruitment of intratumoral GAMs and activation of AXL, an RTK. Combining the AXL inhibitor BGB324 with nivolumab also prolonged the survival in GBM tumor-bearing mice (122). Similarly, co-administration of the CSF-1R inhibitor BLZ945 blocked the M2-type polarization of CD163+ GAMs and enhanced the function of CD154+ CD8+ T-cells and apoptosis of glioma cells, thereby enhancing the efficacy of nivolumab (160). In addition, the combined use of IL6 inhibitor and CD40 agonist reversed M2-type GAM-mediated tumor immunosuppression. These small molecule inhibitors sensitized tumors to the immune checkpoint inhibitor combination of anti-PD1 plus anti-CTLA4 antibodies and prolonged the survival of animals in two syngeneic GBM models (161). With the development of gene-editing technologies, CRISPR/Cas9-mediated PD-L1 knockout using dual single-guide RNAs (sgRNAs) and homology-directed repair (HDR) template is also a promising therapeutic option (162).
Nina Xue et al. have reported that chlorogenic acid (CGA) treatment increases the LPS/IFN-γ-induced expression of M1-type GAM markers like iNOS, MHCII, and CD11c, decreases IL4-induced expression of M2 markers Arg and CD206, suggesting that CGA may be a potential therapeutic option to inhibit glioma growth by promoting M1-type and inhibiting M2-type polarizations of GAMs (163). Sanford PC Hsu et al. could reduce the M2-type polarization of GAMs and increase phagocytic capacity and lipid droplet accumulation by combined RQ therapy with rapamycin (R) and hydroxychloroquine (Q). Whereas the RQ treatment reduced the expression levels of CD47 and SIRPα on tumor cells and macrophages in co-culture experiments. After the RQ treatment, the intratumoral ratios of M1 to M2 and CD8+ to CD4+ were significantly increased in the intracranial GL261 tumor models. Moreover, the combination of RQ and anti-PD1 therapies demonstrated synergistic efficacies (164). Jie Li et al. have shown that inhibitors targeting PI3Kγ reduce the GAM-associated IL11 secretion in the GBM microenvironment via pharmacological inhibition and enhance TMZ therapy efficacy in orthotopic GBM mice (92).
The existence of the BBB restricts the penetration of drugs from the bloodstream to the CNS. Therefore, it constitutes the main obstacle that therapeutics must overcome to enter brain tumors. Ensuring that the drug is fully penetrated the brain is essentially the decisive factor in the drug efficacy determination (165). Moreover, the application of ultrasound to increase the permeability of the BBB could improve immunotherapy efficacy (166). Multiple studies have applied liposomes and nanomaterials as drug carriers through advanced materials technology to achieve biomimetic delivery of therapeutic drugs across the BBB. Pengfei Zhao et al. have designed a bipolar-modified albumin nanoparticle, which achieved the bionic delivery of drugs to the gum tumor area through BBB. The therapeutic must reach its targeted lesion sites (M2-type GAMs) immediately following its penetration through the BBB. This drug delivery system could successfully reprogram GAMs from the M2- to M1-type polarization, thereby effectively inhibiting glioma cell proliferation (167). Feng Zhang et al. have developed a glioma-targeted infusion-based nanocarrier containing interferon regulatory factor 5 (IRF5)-encoding mRNA along with its activating kinase IKKβ that shows a reversal of the immunosuppressive microenvironment of gliomas. These drug-loaded nanoparticles can reprogram GAMs to an M1 phenotype that induces antitumor immunity and promotes tumor regression (168). The study by Xiaopeng Mo et al. has reported the co-encapsulation of simvastatin and fenretinide into D-α-tocopheryl polyethylene glycol succinate (TPGS)-TAT (a cell-penetrating peptide)-embedded lactoferrin nanoparticle system for brain-targeted biomimetic delivery via the LRP-1 receptor. The results suggest that lactoferrin nanoparticles can repolarize GAMs from the M2 phenotype to M1 by modulating the STAT6 pathway and induce reactive oxygen species (ROS)-mediated mitochondrial apoptosis by inhibiting the Ras/Raf/p-ERK pathway in glioma cells (169).
Nasha Qiu et al. have designed an IL12 delivery vector, which can embed IL12 expression plasmid to form lipid complexes to effectively transfect tumor cells and macrophages and make them IL12 production factories. This strategy has been shown to improve the M1/M2 macrophage ratio, thereby activating antitumor immune responses and remodeling the TME (170). Zening Zheng et al. have developed a brain-targeted liposome and disulfiram/copper cassette system (CDX-LIPO). CDX-LIPO activates tumor-infiltrating macrophages, dendritic cells, prime T-cells, and natural killer (NK) cells. Moreover, it can trigger tumor cell autophagy, inducing immunogenic cell death. CDX-LIPO also promotes M1-type polarization of GAMs and mTOR-mediated reprogramming of glucose metabolism in gliomas, leading to antitumor immunity and tumor regression (171).
The immunotherapy of tumor-associated macrophages has recently attracted the attention of clinicians and researchers. As a highly malignant solid tumor, gliomas have their unique pathological characteristics involving distinct TME and tumor-associated macrophage populations. Gliomas are considered immunologically “cold tumors”. There is a high degree of heterogeneity between different subtypes of glioma tumors and within the tumor itself, as well as the unique spatial structure BBBs under pathological conditions. These factors pose enormous challenges to the treatment of gliomas. As a new option for glioma treatment, immunotherapy combined with standard therapy could be a promising solution to improve the survival of glioma patients (172). In-depth exploration of the glioma tumor immune microenvironment and the macrophage recruitment and activation mechanisms are the basis for developing novel glioma immunotherapy strategies. Complex crosstalks and regulatory networks between GAMs and tumor cells contribute to the severe malignancy of gliomas and subsequent treatment resistance. Drug development targeting the critical molecules in the process of GAM recruitment and activation can reduce the accumulation of BMDM-derived GAMs, and reprogram the polarization pattern of GAMs to increase the M1/M2 macrophage ratio, ultimately reversing the tumor immunosuppressive microenvironment in gliomas (Figure 2).
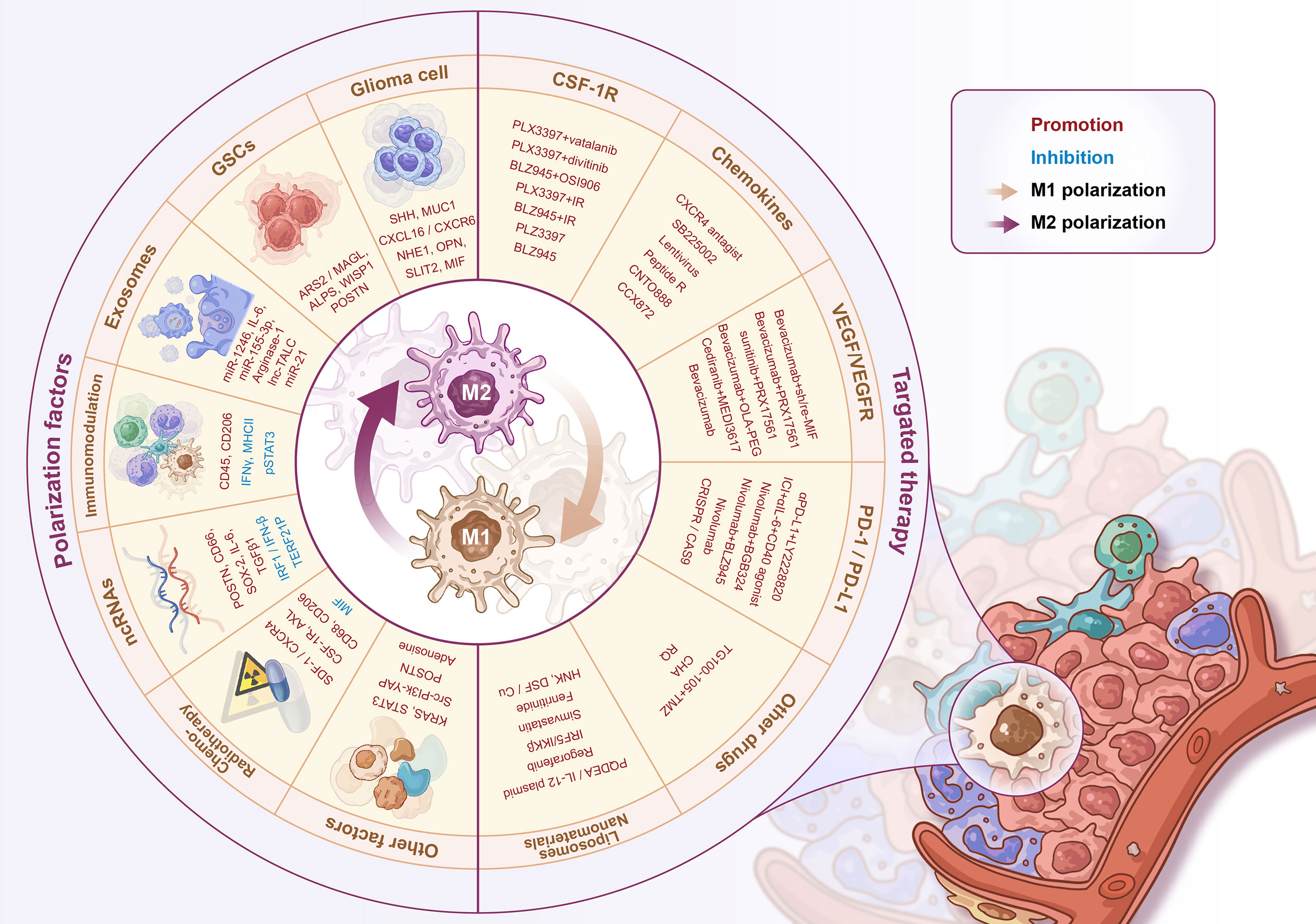
Figure 2 Regulators of M1-M2 polarization and GAMs’ targeted therapy. The regulators of GAMs polarization include a variety of soluble factors secreted by glioma cells/stem cells, exosomes, immune-related molecules, non-coding RNAs, radio-chemotherapy and some other proteins, signaling pathways, etc. The GAMs targeted therapy strategies include CSF-1R inhibitors, chemokine inhibitors, anti-angiogenic therapy, PD-1/PD-L1 inhibitors, and novel liposome/nanomaterial delivery systems for drug delivery.
Presently, various immunotherapy drugs targeting GAMs, such as CSF-1R inhibitors, PD-L1 antibodies, VEGF inhibitors, and some chemokine inhibitors have achieved a certain degree of success in the experimental stages using in vitro and in vivo xenograft models. It is worth noting that the high heterogeneity of glioma indicates that the efficacy of single-drug therapy is often limited and is prone to the development of drug resistance. The multi-drug combination therapy scheme is expected to overcome the side effects and treatment resistance brought by single-drug treatment. Preclinical trials using different compositions of a therapeutic regimen such as CSF-1R inhibitors combined with IGF1 inhibitors (140), VEGFR inhibitors combined with ANG2 inhibitors (155), CXCR2 inhibitors combined with TMZ (148, 149), and PD-L1 antibody combined with p38MAPK inhibitors, AXL inhibitors or CSF-1R inhibitors (122, 159, 160) have achieved better therapeutic effects than their monotherapies. Notably, the application of materials technology such as liposomes and nanomaterials is expected to solve the problem of drug penetration across the BBB and CNS, ensuring a selective targeting and high bioavailability of therapeutic anticancer drugs in the TME, thus providing a new option for the glioma treatment (Table 2).
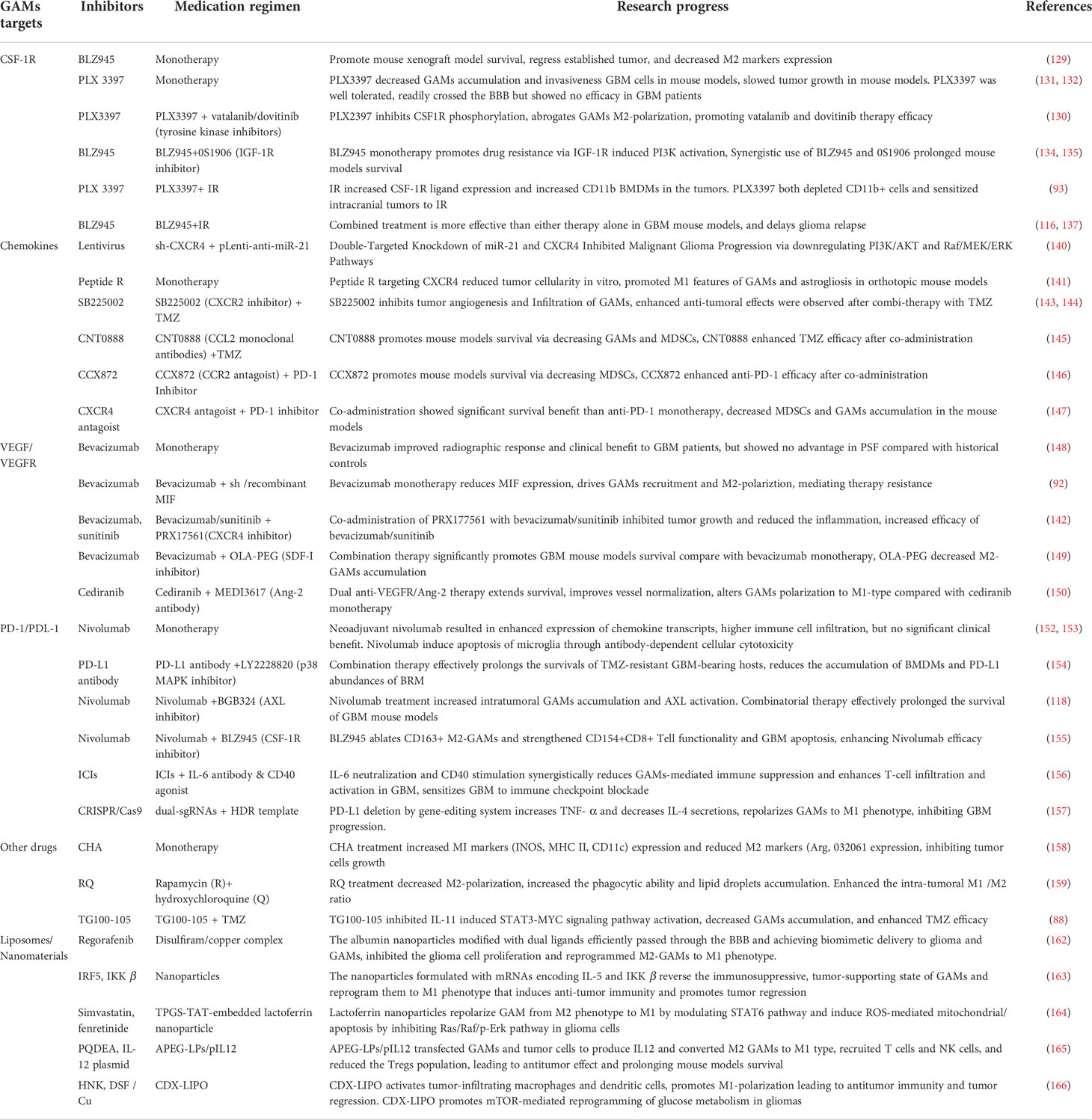
Table 2 Research progress of targeting GAMs in the treatment of GBM.
Regrettably, in these studies, liposomes and nanomaterials have been used more frequently as carriers to achieve biomimetic delivery of therapeutic drugs through the BBB. Still, the medicinal drugs loaded onto the carriers are not widely used in the targeted inhibition of key molecular switches in GAMs (such as CSF-1R, VEGF, PD-1, etc.). Further comprehensive investigations are urgently needed to achieve therapeutic breakthroughs in drug research and development through better cooperation between material scientists, immunologists, and clinicians in the future to develop effective immunotherapy or adjuvant therapy for glioma patients. Finally, with the development of single-cell sequencing technology in recent years, researchers have been able to better analyze and understand the spatial heterogeneity of glioma immune cells, where simplified M1 and M2 profiling may not be enough to fully represent the glioma microenvironment in the patients. Due to the complex immune status of macrophages, the emergence of new immune typing in the future may lead to more detailed and effective treatment strategies.
CX and MX wrote the article. XL, LX, JS, and QZ participated in literature collection and organization. CW, QZ, and XY participated in figures and tables generation. CX and YT provided critical perspectives on the article ideas. CF and YT fund the study and provide article revision. All authors contributed to the article and approved the submitted version.
This study was supported by National Natural Science Foundation of China (NSFC, No.82172660), The Hebei Provincial Government subsidized the clinical medicine outstanding talent training project (No.361007), Hebei Province Graduate Innovation Project (No.HBU2022bs012).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Geraldo LHM, Garcia C, da Fonseca ACC, Dubois LGF, de Sampaio ESTCL, Matias D, et al. Glioblastoma therapy in the age of molecular medicine. Trends Cancer. (2019) 5(1):46–65. doi: 10.1016/j.trecan.2018.11.002
2. Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell (2019) 178(4):835–49 e21. doi: 10.1016/j.cell.2019.06.024
3. Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell (2018) 33(1):152. doi: 10.1016/j.ccell.2017.12.012
4. Klemm F, Maas RR, Bowman RL, Kornete M, Soukup K, Nassiri S, et al. Interrogation of the microenvironmental landscape in brain tumors reveals disease-specific alterations of immune cells. Cell (2020) 181(7):1643–60 e17. doi: 10.1016/j.cell.2020.05.007
5. Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci (2016) 19(1):20–7. doi: 10.1038/nn.4185
6. Perng P, Lim M. Immunosuppressive mechanisms of malignant gliomas: Parallels at non-CNS sites. Front Oncol (2015) 5:153. doi: 10.3389/fonc.2015.00153
7. Zhang H, Luo YB, Wu W, Zhang L, Wang Z, Dai Z, et al. The molecular feature of macrophages in tumor immune microenvironment of glioma patients. Comput Struct Biotechnol J (2021) 19:4603–18. doi: 10.1016/j.csbj.2021.08.019
8. Sorensen MD, Dahlrot RH, Boldt HB, Hansen S, Kristensen BW. Tumour-associated microglia/macrophages predict poor prognosis in high-grade gliomas and correlate with an aggressive tumour subtype. Neuropathol Appl Neurobiol (2018) 44(2):185–206. doi: 10.1111/nan.12428
9. Fujita M, Kohanbash G, Fellows-Mayle W, Hamilton RL, Komohara Y, Decker SA, et al. COX-2 blockade suppresses gliomagenesis by inhibiting myeloid-derived suppressor cells. Cancer Res (2011) 71(7):2664–74. doi: 10.1158/0008-5472.CAN-10-3055
10. Raychaudhuri B, Rayman P, Ireland J, Ko J, Rini B, Borden EC, et al. Myeloid-derived suppressor cell accumulation and function in patients with newly diagnosed glioblastoma. Neuro Oncol (2011) 13(6):591–9. doi: 10.1093/neuonc/nor042
11. Pombo Antunes AR, Scheyltjens I, Duerinck J, Neyns B, Movahedi K, Van Ginderachter JA. Understanding the glioblastoma immune microenvironment as basis for the development of new immunotherapeutic strategies. Elife (2020) 9:e52176. doi: 10.7554/eLife.52176
12. Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol (2012) 12(9):623–35. doi: 10.1038/nri3265
13. Sampson JH, Gunn MD, Fecci PE, Ashley DM. Brain immunology and immunotherapy in brain tumours. Nat Rev Cancer. (2020) 20(1):12–25. doi: 10.1038/s41568-019-0224-7
14. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science (2010) 330(6005):841–5. doi: 10.1126/science.1194637
15. Ginhoux F, Lim S, Hoeffel G, Low D, Huber T. Origin and differentiation of microglia. Front Cell Neurosci (2013) 7:45. doi: 10.3389/fncel.2013.00045
16. Tong N, He Z, Ma Y, Wang Z, Huang Z, Cao H, et al. Tumor associated macrophages, as the dominant immune cells, are an indispensable target for immunologically cold tumor-glioma therapy? Front Cell Dev Biol (2021) 9:706286. doi: 10.3389/fcell.2021.706286
17. Quail DF, Joyce JA. The microenvironmental landscape of brain tumors. Cancer Cell (2017) 31(3):326–41. doi: 10.1016/j.ccell.2017.02.009
18. Chen Z, Feng X, Herting CJ, Garcia VA, Nie K, Pong WW, et al. Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Res (2017) 77(9):2266–78. doi: 10.1158/0008-5472.CAN-16-2310
19. Landry AP, Balas M, Alli S, Spears J, Zador Z. Distinct regional ontogeny and activation of tumor associated macrophages in human glioblastoma. Sci Rep (2020) 10(1):19542. doi: 10.1038/s41598-020-76657-3
20. Wei J, Chen P, Gupta P, Ott M, Zamler D, Kassab C, et al. Immune biology of glioma-associated macrophages and microglia: functional and therapeutic implications. Neuro Oncol (2020) 22(2):180–94. doi: 10.1093/neuonc/noz212
21. Lavin Y, Merad M. Macrophages: gatekeepers of tissue integrity. Cancer Immunol Res (2013) 1(4):201–9. doi: 10.1158/2326-6066.CIR-13-0117
22. Wu K, Lin K, Li X, Yuan X, Xu P, Ni P, et al. Redefining tumor-associated macrophage subpopulations and functions in the tumor microenvironment. Front Immunol (2020) 11:1731. doi: 10.3389/fimmu.2020.01731
23. Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: Phenotypical vs. Funct Differentiation. Front Immunol (2014) 5:514. doi: 10.3389/fimmu.2014.00514
24. Ricard C, Tchoghandjian A, Luche H, Grenot P, Figarella-Branger D, Rougon G, et al. Phenotypic dynamics of microglial and monocyte-derived cells in glioblastoma-bearing mice. Sci Rep (2016) 6:26381. doi: 10.1038/srep26381
25. Ginhoux F, Merad M. Microglia arise from extra-embryonic yolk sac primitive progenitors. Med Sci (Paris) 201127(8-9):719–24. doi: 10.1051/medsci/2011278013
26. Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, et al. Microglia emerge from erythromyeloid precursors via Pu. 1- Irf8-dependent pathways. Nat Neurosci (2013) 16(3):273–80. doi: 10.1038/nn.3318
27. Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol (2017) 35:441–68. doi: 10.1146/annurev-immunol-051116-052358
28. Salter MW, Stevens B. Microglia emerge as central players in brain disease. Nat Med (2017) 23(9):1018–27. doi: 10.1038/nm.4397
29. Crotti A, Ransohoff RM. Microglial physiology and pathophysiology: Insights from genome-wide transcriptional profiling. Immunity (2016) 44(3):505–15. doi: 10.1016/j.immuni.2016.02.013
30. Sousa C, Biber K, Michelucci A. Cellular and molecular characterization of microglia: A unique immune cell population. Front Immunol (2017) 8:198. doi: 10.3389/fimmu.2017.00198
31. Woolf Z, Swanson MEV, Smyth LC, Mee EW, Schweder P, Heppner P, et al. Single-cell image analysis reveals a protective role for microglia in glioblastoma. Neurooncol Adv (2021) 3(1):vdab031. doi: 10.1093/noajnl/vdab031
32. Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol (2011) 11(11):762–74. doi: 10.1038/nri3070
33. Peranzoni E, Zilio S, Marigo I, Dolcetti L, Zanovello P, Mandruzzato S, et al. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol (2010) 22(2):238–44. doi: 10.1016/j.coi.2010.01.021
34. Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M, et al. Microglia in the adult brain arise from ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci (2007) 10(12):1544–53. doi: 10.1038/nn2015
35. Parney IF, Waldron JS, Parsa AT. Flow cytometry and in vitro analysis of human glioma-associated macrophages. Lab Invest J Neurosurg (2009) 110(3):572–82. doi: 10.3171/2008.7.JNS08475
36. Bowman RL, Klemm F, Akkari L, Pyonteck SM, Sevenich L, Quail DF, et al. Macrophage ontogeny underlies differences in tumor-specific education in brain malignancies. Cell Rep (2016) 17(9):2445–59. doi: 10.1016/j.celrep.2016.10.052
37. Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A (2016) 113(12):E1738–46. doi: 10.1073/pnas.1525528113
38. Muller S, Kohanbash G, Liu SJ, Alvarado B, Carrera D, Bhaduri A, et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol (2017) 18(1):234. doi: 10.1186/s13059-017-1362-4
39. Pombo Antunes AR, Scheyltjens I, Lodi F, Messiaen J, Antoranz A, Duerinck J, et al. Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat Neurosci (2021) 24(4):595–610. doi: 10.1038/s41593-020-00789-y
40. Ochocka N, Segit P, Walentynowicz KA, Wojnicki K, Cyranowski S, Swatler J, et al. Single-cell RNA sequencing reveals functional heterogeneity of glioma-associated brain macrophages. Nat Commun (2021) 12(1):1151. doi: 10.1038/s41467-021-21407-w
41. Abdelfattah N, Kumar P, Wang C, Leu JS, Flynn WF, Gao R, et al. Single-cell analysis of human glioma and immune cells identifies S100A4 as an immunotherapy target. Nat Commun (2022) 13(1):767. doi: 10.1038/s41467-022-28372-y
42. Palframan RT, Jung S, Cheng G, Weninger W, Luo Y, Dorf M, et al. Inflammatory chemokine transport and presentation in HEV: a remote control mechanism for monocyte recruitment to lymph nodes in inflamed tissues. J Exp Med (2001) 194(9):1361–73. doi: 10.1084/jem.194.9.1361
43. Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity (2003) 19(1):71–82. doi: 10.1016/S1074-7613(03)00174-2
44. Serbina NV, Jia T, Hohl TM, Pamer EG. Monocyte-mediated defense against microbial pathogens. Annu Rev Immunol (2008) 26:421–52. doi: 10.1146/annurev.immunol.26.021607.090326
45. Kurihara T, Warr G, Loy J, Bravo R. Defects in macrophage recruitment and host defense in mice lacking the CCR2 chemokine receptor. J Exp Med (1997) 186(10):1757–62. doi: 10.1084/jem.186.10.1757
46. Kuziel WA, Morgan SJ, Dawson TC, Griffin S, Smithies O, Ley K, et al. Severe reduction in leukocyte adhesion and monocyte extravasation in mice deficient in CC chemokine receptor 2. Proc Natl Acad Sci U S A (1997) 94(22):12053–8. doi: 10.1073/pnas.94.22.12053
47. Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, et al. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science (2007) 317(5838):666–70. doi: 10.1126/science.1142883
48. Ziegler-Heitbrock L. The CD14+ CD16+ blood monocytes: their role in infection and inflammation. J Leukoc Biol (2007) 81(3):584–92. doi: 10.1189/jlb.0806510
49. Ziegler-Heitbrock L, Ancuta P, Crowe S, Dalod M, Grau V, Hart DN, et al. Nomenclature of monocytes and dendritic cells in blood. Blood (2010) 116(16):e74–80. doi: 10.1182/blood-2010-02-258558
50. Belge KU, Dayyani F, Horelt A, Siedlar M, Frankenberger M, Frankenberger B, et al. The proinflammatory CD14+CD16+DR++ monocytes are a major source of TNF. J Immunol (2002) 168(7):3536–42. doi: 10.4049/jimmunol.168.7.3536
51. Grage-Griebenow E, Flad HD, Ernst M. Heterogeneity of human peripheral blood monocyte subsets. J Leukoc Biol (2001) 69(1):11–20. doi: 10.1189/jlb.69.1.11
52. Ingersoll MA, Spanbroek R, Lottaz C, Gautier EL, Frankenberger M, Hoffmann R, et al. Comparison of gene expression profiles between human and mouse monocyte subsets. Blood (2010) 115(3):e10–9. doi: 10.1182/blood-2009-07-235028
53. Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol (2017) 17(9):559–72. doi: 10.1038/nri.2017.49
54. Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol (2014) 32:659–702. doi: 10.1146/annurev-immunol-032713-120145
55. Vakilian A, Khorramdelazad H, Heidari P, Sheikh Rezaei Z, Hassanshahi G. CCL2/CCR2 signaling pathway in glioblastoma multiforme. Neurochem Int (2017) 103:1–7. doi: 10.1016/j.neuint.2016.12.013
56. Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, et al. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest. (2007) 117(4):902–9. doi: 10.1172/JCI29919
57. Chang AL, Miska J, Wainwright DA, Dey M, Rivetta CV, Yu D, et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid-derived suppressor cells. Cancer Res (2016) 76(19):5671–82. doi: 10.1158/0008-5472.CAN-16-0144
58. Jia T, Serbina NV, Brandl K, Zhong MX, Leiner IM, Charo IF, et al. Additive roles for MCP-1 and MCP-3 in CCR2-mediated recruitment of inflammatory monocytes during listeria monocytogenes infection. J Immunol (2008) 180(10):6846–53. doi: 10.4049/jimmunol.180.10.6846
59. Proudfoot AE, Handel TM, Johnson Z, Lau EK, LiWang P, Clark-Lewis I, et al. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc Natl Acad Sci U S A (2003) 100(4):1885–90. doi: 10.1073/pnas.0334864100
60. Allen SJ, Crown SE, Handel TM. Chemokine: receptor structure, interactions, and antagonism. Annu Rev Immunol (2007) 25:787–820. doi: 10.1146/annurev.immunol.24.021605.090529
61. Pruenster M, Mudde L, Bombosi P, Dimitrova S, Zsak M, Middleton J, et al. The Duffy antigen receptor for chemokines transports chemokines and supports their promigratory activity. Nat Immunol (2009) 10(1):101–8. doi: 10.1038/ni.1675
62. Lee S, Latha K, Manyam G, Yang Y, Rao A, Rao G. Role of CX3CR1 signaling in malignant transformation of gliomas. Neuro Oncol (2020) 22(10):1463–73. doi: 10.1093/neuonc/noaa075
63. Zhang Q, Wang J, Yao X, Wu S, Tian W, Gan C, et al. Programmed cell death 10 mediated CXCL2-CXCR2 signaling in regulating tumor-associated Microglia/Macrophages recruitment in glioblastoma. Front Immunol (2021) 12:637053. doi: 10.3389/fimmu.2021.637053
64. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol (2007) 7(9):678–89. doi: 10.1038/nri2156
65. Henderson RB, Hobbs JA, Mathies M, Hogg N. Rapid recruitment of inflammatory monocytes is independent of neutrophil migration. Blood (2003) 102(1):328–35. doi: 10.1182/blood-2002-10-3228
66. Huang BR, Liu YS, Lai SW, Lin HJ, Shen CK, Yang LY, et al. CAIX regulates GBM motility and TAM adhesion and polarization through EGFR/STAT3 under hypoxic conditions. Int J Mol Sci (2020) 21(16):5838. doi: 10.3390/ijms21165838
67. Chen Z, Chen G, Zhao H. FDPS promotes glioma growth and macrophage recruitment by regulating CCL20 via wnt/beta-catenin signalling pathway. J Cell Mol Med (2020) 24(16):9055–66. doi: 10.1111/jcmm.15542
68. Yotsumoto F, You WK, Cejudo-Martin P, Kucharova K, Sakimura K, Stallcup WB. NG2 proteoglycan-dependent recruitment of tumor macrophages promotes pericyte-endothelial cell interactions required for brain tumor vascularization. Oncoimmunology (2015) 4(4):e1001204. doi: 10.1080/2162402X.2014.1001204
69. De Boeck A, Ahn BY, D’Mello C, Lun X, Menon SV, Alshehri MM, et al. Glioma-derived IL-33 orchestrates an inflammatory brain tumor microenvironment that accelerates glioma progression. Nat Commun (2020) 11(1):4997. doi: 10.1038/s41467-020-18569-4
70. Ghoochani A, Schwarz MA, Yakubov E, Engelhorn T, Doerfler A, Buchfelder M, et al. MIF-CD74 signaling impedes microglial M1 polarization and facilitates brain tumorigenesis. Oncogene (2016) 35(48):6246–61. doi: 10.1038/onc.2016.160
71. Ge H, Mu L, Jin L, Yang C, Chang YE, Long Y, et al. Tumor associated CD70 expression is involved in promoting tumor migration and macrophage infiltration in GBM. Int J Cancer (2017) 141(7):1434–44. doi: 10.1002/ijc.30830
72. Takenaka MC, Gabriely G, Rothhammer V, Mascanfroni ID, Wheeler MA, Chao CC, et al. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat Neurosci (2019) 22(5):729–40. doi: 10.1038/s41593-019-0370-y
73. Cao MF, Chen L, Dang WQ, Zhang XC, Zhang X, Shi Y, et al. Hybrids by tumor-associated macrophages x glioblastoma cells entail nuclear reprogramming and glioblastoma invasion. Cancer Lett (2019) 442:445–52. doi: 10.1016/j.canlet.2018.11.016
74. Ye XZ, Xu SL, Xin YH, Yu SC, Ping YF, Chen L, et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-beta1 signaling pathway. J Immunol (2012) 189(1):444–53. doi: 10.4049/jimmunol.1103248
75. Zhu C, Mustafa D, Zheng PP, van der Weiden M, Sacchetti A, Brandt M, et al. Activation of CECR1 in M2-like TAMs promotes paracrine stimulation-mediated glial tumor progression. Neuro Oncol (2017) 19(5):648–59. doi: 10.1093/neuonc/now251
76. Zhang X, Chen L, Dang WQ, Cao MF, Xiao JF, Lv SQ, et al. CCL8 secreted by tumor-associated macrophages promotes invasion and stemness of glioblastoma cells via ERK1/2 signaling. Lab Invest. (2020) 100(4):619–29. doi: 10.1038/s41374-019-0345-3
77. Shi Y, Ping YF, Zhou W, He ZC, Chen C, Bian BS, et al. Tumour-associated macrophages secrete pleiotrophin to promote PTPRZ1 signalling in glioblastoma stem cells for tumour growth. Nat Commun (2017) 8:15080. doi: 10.1038/ncomms15080
78. Wang J, Leavenworth JW, Hjelmeland AB, Smith R, Patel N, Borg B, et al. Deletion of the RNA regulator HuR in tumor-associated microglia and macrophages stimulates anti-tumor immunity and attenuates glioma growth. Glia (2019) 67(12):2424–39. doi: 10.1002/glia.23696
79. Mignogna C, Signorelli F, Vismara MF, Zeppa P, Camastra C, Barni T, et al. A reappraisal of macrophage polarization in glioblastoma: Histopathological and immunohistochemical findings and review of the literature. Pathol Res Pract (2016) 212(6):491–9. doi: 10.1016/j.prp.2016.02.020
80. Zhu C, Kros JM, Cheng C, Mustafa D. The contribution of tumor-associated macrophages in glioma neo-angiogenesis and implications for anti-angiogenic strategies. Neuro Oncol (2017) 19(11):1435–46. doi: 10.1093/neuonc/nox081
81. Zhang L, Xu Y, Sun J, Chen W, Zhao L, Ma C, et al. M2-like tumor-associated macrophages drive vasculogenic mimicry through amplification of IL-6 expression in glioma cells. Oncotarget (2017) 8(1):819–32. doi: 10.18632/oncotarget.13661
82. Rong X, Huang B, Qiu S, Li X, He L, Peng Y. Tumor-associated macrophages induce vasculogenic mimicry of glioblastoma multiforme through cyclooxygenase-2 activation. Oncotarget (2016) 7(51):83976–86. doi: 10.18632/oncotarget.6930
83. Fan Y, Ye J, Shen F, Zhu Y, Yeghiazarians Y, Zhu W, et al. Interleukin-6 stimulates circulating blood-derived endothelial progenitor cell angiogenesis in vitro. J Cereb Blood Flow Metab (2008) 28(1):90–8. doi: 10.1038/sj.jcbfm.9600509
84. Nijaguna MB, Schroder C, Patil V, Shwetha SD, Hegde AS, Chandramouli BA, et al. Definition of a serum marker panel for glioblastoma discrimination and identification of interleukin 1beta in the microglial secretome as a novel mediator of endothelial cell survival induced by c-reactive protein. J Proteomics (2015) 128:251–61. doi: 10.1016/j.jprot.2015.07.026
85. Carmi Y, Dotan S, Rider P, Kaplanov I, White MR, Baron R, et al. The role of IL-1beta in the early tumor cell-induced angiogenic response. J Immunol (2013) 190(7):3500–9. doi: 10.4049/jimmunol.1202769
86. Chen X, Zhang L, Zhang IY, Liang J, Wang H, Ouyang M, et al. RAGE expression in tumor-associated macrophages promotes angiogenesis in glioma. Cancer Res (2014) 74(24):7285–97. doi: 10.1158/0008-5472.CAN-14-1240
87. Justus CR, Sanderlin EJ, Yang LV. Molecular connections between cancer cell metabolism and the tumor microenvironment. Int J Mol Sci (2015) 16(5):11055–86. doi: 10.3390/ijms160511055
88. Libby CJ, Tran AN, Scott SE, Griguer C, Hjelmeland AB. The pro-tumorigenic effects of metabolic alterations in glioblastoma including brain tumor initiating cells. Biochim Biophys Acta Rev Cancer (2018) 1869(2):175–88. doi: 10.1016/j.bbcan.2018.01.004
89. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature (2014) 513(7519):559–63. doi: 10.1038/nature13490
90. Lu J, Xu Z, Duan H, Ji H, Zhen Z, Li B, et al. Tumor-associated macrophage interleukin-beta promotes glycerol-3-phosphate dehydrogenase activation, glycolysis and tumorigenesis in glioma cells. Cancer Sci (2020) 111(6):1979–90. doi: 10.1111/cas.14408
91. Zhang Y, Yu G, Chu H, Wang X, Xiong L, Cai G, et al. Macrophage-associated PGK1 phosphorylation promotes aerobic glycolysis and tumorigenesis. Mol Cell (2018) 71(2):201–15 e7. doi: 10.1016/j.molcel.2018.06.023
92. Li J, Kaneda MM, Ma J, Li M, Shepard RM, Patel K, et al. PI3Kgamma inhibition suppresses microglia/TAM accumulation in glioblastoma microenvironment to promote exceptional temozolomide response. Proc Natl Acad Sci U S A (2021) 118(16):e2009290118. doi: 10.1073/pnas.2009290118
93. Li Z, Meng X, Wu P, Zha C, Han B, Li L, et al. Glioblastoma cell-derived lncRNA-containing exosomes induce microglia to produce complement C5, promoting chemotherapy resistance. Cancer Immunol Res (2021) 9(12):1383–99. doi: 10.1158/2326-6066.CIR-21-0258
94. Chuang HY, Su YK, Liu HW, Chen CH, Chiu SC, Cho DY, et al. Preclinical evidence of STAT3 inhibitor pacritinib overcoming temozolomide resistance via downregulating miR-21-Enriched exosomes from M2 glioblastoma-associated macrophages. J Clin Med (2019) 8(7):959. doi: 10.3390/jcm8070959
95. Geraldo LH, Xu Y, Jacob L, Pibouin-Fragner L, Rao R, Maissa N, et al. SLIT2/ROBO signaling in tumor-associated microglia and macrophages drives glioblastoma immunosuppression and vascular dysmorphia. J Clin Invest (2021) 131(16):e141083. doi: 10.1172/JCI141083
96. Castro BA, Flanigan P, Jahangiri A, Hoffman D, Chen W, Kuang R, et al. Macrophage migration inhibitory factor downregulation: a novel mechanism of resistance to anti-angiogenic therapy. Oncogene (2017) 36(26):3749–59. doi: 10.1038/onc.2017.1
97. Stafford JH, Hirai T, Deng L, Chernikova SB, Urata K, West BL, et al. Colony stimulating factor 1 receptor inhibition delays recurrence of glioblastoma after radiation by altering myeloid cell recruitment and polarization. Neuro Oncol (2016) 18(6):797–806. doi: 10.1093/neuonc/nov272
98. Petty AJ, Li A, Wang X, Dai R, Heyman B, Hsu D, et al. Hedgehog signaling promotes tumor-associated macrophage polarization to suppress intratumoral CD8+ T cell recruitment. J Clin Invest (2019) 129(12):5151–62. doi: 10.1172/JCI128644
99. Lepore F, D’Alessandro G, Antonangeli F, Santoro A, Esposito V, Limatola C, et al. CXCL16/CXCR6 axis drives Microglia/Macrophages phenotype in physiological conditions and plays a crucial role in glioma. Front Immunol (2018) 9:2750. doi: 10.3389/fimmu.2018.02750
100. Wei J, Marisetty A, Schrand B, Gabrusiewicz K, Hashimoto Y, Ott M, et al. Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target. J Clin Invest (2019) 129(1):137–49. doi: 10.1038/s41419-018-1062-3
101. Beatson R, Tajadura-Ortega V, Achkova D, Picco G, Tsourouktsoglou TD, Klausing S, et al. The mucin MUC1 modulates the tumor immunological microenvironment through engagement of the lectin siglec-9. Nat Immunol (2016) 17(11):1273–81. doi: 10.1038/ni.3552
102. Chen Z, Hambardzumyan D. Immune microenvironment in glioblastoma subtypes. Front Immunol (2018) 9:1004. doi: 10.3389/fimmu.2018.01004
103. Sa JK, Chang N, Lee HW, Cho HJ, Ceccarelli M, Cerulo L, et al. Transcriptional regulatory networks of tumor-associated macrophages that drive malignancy in mesenchymal glioblastoma. Genome Biol (2020) 21(1):216. doi: 10.1186/s13059-020-02140-x
104. Couturier CP, Ayyadhury S, Le PU, Nadaf J, Monlong J, Riva G, et al. Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat Commun (2020) 11(1):3406. doi: 10.1038/s41467-020-17186-5
105. Tabu K, Liu W, Kosaku A, Terashima K, Murota Y, Aimaitijiang A, et al. Glioma stem cell (GSC)-derived autoschizis-like products confer GSC niche properties involving M1-like tumor-associated macrophages. Stem Cells (2020) 38(8):921–35. doi: 10.1002/stem.3193
106. Yin J, Kim SS, Choi E, Oh YT, Lin W, Kim TH, et al. ARS2/MAGL signaling in glioblastoma stem cells promotes self-renewal and M2-like polarization of tumor-associated macrophages. Nat Commun (2020) 11(1):2978. doi: 10.1038/s41467-020-16789-2
107. Tao W, Chu C, Zhou W, Huang Z, Zhai K, Fang X, et al. Dual role of WISP1 in maintaining glioma stem cells and tumor-supportive macrophages in glioblastoma. Nat Commun (2020) 11(1):3015. doi: 10.1038/s41467-020-16827-z
108. Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol (2015) 17(2):170–82. doi: 10.1038/ncb3090
109. Qian M, Wang S, Guo X, Wang J, Zhang Z, Qiu W, et al. Hypoxic glioma-derived exosomes deliver microRNA-1246 to induce M2 macrophage polarization by targeting TERF2IP via the STAT3 and NF-kappaB pathways. Oncogene (2020) 39(2):428–42. doi: 10.1038/s41388-019-0996-y
110. Xu J, Zhang J, Zhang Z, Gao Z, Qi Y, Qiu W, et al. Hypoxic glioma-derived exosomes promote M2-like macrophage polarization by enhancing autophagy induction. Cell Death Dis (2021) 12(4):373. doi: 10.1038/s41419-021-03664-1
111. Liu C, Chikina M, Deshpande R, Menk AV, Wang T, Tabib T, et al. Treg cells promote the SREBP1-dependent metabolic fitness of tumor-promoting macrophages via repression of CD8(+) T cell-derived interferon-gamma. Immunity (2019) 51(2):381–97 e6. doi: 10.1016/j.immuni.2019.06.017
112. Kumar V, Cheng P, Condamine T, Mony S, Languino LR, McCaffrey JC, et al. CD45 phosphatase inhibits STAT3 transcription factor activity in myeloid cells and promotes tumor-associated macrophage differentiation. Immunity (2016) 44(2):303–15. doi: 10.1016/j.immuni.2016.01.014
113. Zhu Z, Zhang H, Chen B, Liu X, Zhang S, Zong Z, et al. PD-L1-Mediated immunosuppression in glioblastoma is associated with the infiltration and M2-polarization of tumor-associated macrophages. Front Immunol (2020) 11:588552. doi: 10.3389/fimmu.2020.588552
114. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature (2017) 545(7655):495–9. doi: 10.1038/nature22396
115. Yan H, Bu P. Non-coding RNA in cancer. Essays Biochem (2021) 65(4):625–39. doi: 10.1016/j.immuni.2016.01.014
116. Liu Y, Li X, Zhang Y, Wang H, Rong X, Peng J, et al. An miR-340-5p-macrophage feedback loop modulates the progression and tumor microenvironment of glioblastoma multiforme. Oncogene (2019) 38(49):7399–415. doi: 10.1038/s41388-019-0952-x
117. Shi Y, Zhang B, Zhu J, Huang W, Han B, Wang Q, et al. miR-106b-5p inhibits IRF1/IFN-beta signaling to promote M2 macrophage polarization of glioblastoma. Onco Targets Ther (2020) 13:7479–92. doi: 10.2147/OTT.S238975
118. Li Z, Zhang J, Zheng H, Li C, Xiong J, Wang W, et al. Modulating lncRNA SNHG15/CDK6/miR-627 circuit by palbociclib, overcomes temozolomide resistance and reduces M2-polarization of glioma associated microglia in glioblastoma multiforme. J Exp Clin Cancer Res (2019) 38(1):380. doi: 10.1186/s13046-019-1046-x
119. Brown JM, Thomas R, Nagpal S, Recht L. Macrophage exclusion after radiation therapy (MERT): A new and effective way to increase the therapeutic ratio of radiotherapy. Radiother Oncol (2020) 144:159–64. doi: 10.1016/j.radonc.2019.11.020
120. Akkari L, Bowman RL, Tessier J, Klemm F, Handgraaf SM, de Groot M, et al. Dynamic changes in glioma macrophage populations after radiotherapy reveal CSF-1R inhibition as a strategy to overcome resistance. Sci Transl Med (2020) 12(552):7479–92. doi: 10.1126/scitranslmed.aaw7843
121. Foray C, Valtorta S, Barca C, Winkeler A, Roll W, Muther M, et al. Imaging temozolomide-induced changes in the myeloid glioma microenvironment. Theranostics (2021) 11(5):2020–33. doi: 10.7150/thno.47269
122. Sadahiro H, Kang KD, Gibson JT, Minata M, Yu H, Shi J, et al. Activation of the receptor tyrosine kinase AXL regulates the immune microenvironment in glioblastoma. Cancer Res (2018) 78(11):3002–13. doi: 10.1158/0008-5472.CAN-17-2433
123. Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature (2016) 536(7617):479–83. doi: 10.1038/nature19084
124. Guan X, Hasan MN, Begum G, Kohanbash G, Carney KE, Pigott VM, et al. Blockade of Na/H exchanger stimulates glioma tumor immunogenicity and enhances combinatorial TMZ and anti-PD-1 therapy. Cell Death Dis (2018) 9(10):1010. doi: 10.1038/s41419-018-1062-3
125. Hasan MN, Luo L, Ding D, Song S, Bhuiyan MIH, Liu R, et al. Blocking NHE1 stimulates glioma tumor immunity by restoring OXPHOS function of myeloid cells. Theranostics (2021) 11(3):1295–309. doi: 10.7150/thno.50150
126. Azambuja JH, Ludwig N, Yerneni SS, Braganhol E, Whiteside TL. Arginase-1+ exosomes from reprogrammed macrophages promote glioblastoma progression. Int J Mol Sci (2020) 21(11):3002–13. doi: 10.3390/ijms21113990
127. Chiang CS, Fu SY, Wang SC, Yu CF, Chen FH, Lin CM, et al. Irradiation promotes an m2 macrophage phenotype in tumor hypoxia. Front Oncol (2012) 2:89. doi: 10.3389/fonc.2012.00089
128. Dai E, Han L, Liu J, Xie Y, Kroemer G, Klionsky DJ, et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy (2020) 16(11):2069–83. doi: 10.1080/15548627.2020.1714209
129. Cunha LD, Yang M, Carter R, Guy C, Harris L, Crawford JC, et al. LC3-associated phagocytosis in myeloid cells promotes tumor immune tolerance. Cell (2018) 175(2):429–41 e16. doi: 10.1016/j.cell.2018.08.061
130. Cui X, Morales RT, Qian W, Wang H, Gagner JP, Dolgalev I, et al. Hacking macrophage-associated immunosuppression for regulating glioblastoma angiogenesis. Biomaterials (2018) 161:164–78. doi: 10.1016/j.biomaterials.2018.01.053
131. Guo X, Xue H, Shao Q, Wang J, Guo X, Chen X, et al. Hypoxia promotes glioma-associated macrophage infiltration via periostin and subsequent M2 polarization by upregulating TGF-beta and m-CSFR. Oncotarget (2016) 7(49):80521–42. doi: 10.18632/oncotarget.11825
132. Kesarwani P, Prabhu A, Kant S, Chinnaiyan P. Metabolic remodeling contributes towards an immune-suppressive phenotype in glioblastoma. Cancer Immunol Immunother (2019) 68(7):1107–20. doi: 10.1007/s00262-019-02347-3
133. Ramachandran SA, Jadhavar PS, Miglani SK, Singh MP, Kalane DP, Agarwal AK, et al. Design, synthesis and optimization of bis-amide derivatives as CSF1R inhibitors. Bioorg Med Chem Lett (2017) 27(10):2153–60. doi: 10.1016/j.bmcl.2017.03.064
134. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med (2013) 19(10):1264–72. doi: 10.1038/nm.3337
135. Yan D, Kowal J, Akkari L, Schuhmacher AJ, Huse JT, West BL, et al. Inhibition of colony stimulating factor-1 receptor abrogates microenvironment-mediated therapeutic resistance in gliomas. Oncogene (2017) 36(43):6049–58. doi: 10.1038/onc.2017.261
136. Coniglio SJ, Eugenin E, Dobrenis K, Stanley ER, West BL, Symons MH, et al. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol Med (2012) 18:519–27. doi: 10.2119/molmed.2011.00217
137. Butowski N, Colman H, De Groot JF, Omuro AM, Nayak L, Wen PY, et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an ivy foundation early phase clinical trials consortium phase II study. Neuro Oncol (2016) 18(4):557–64. doi: 10.1093/neuonc/nov245
138. Quail DF, Joyce JA. Molecular pathways: Deciphering mechanisms of resistance to macrophage-targeted therapies. Clin Cancer Res (2017) 23(4):876–84. doi: 10.1158/1078-0432.CCR-16-0133
139. Quail DF, Bowman RL, Akkari L, Quick ML, Schuhmacher AJ, Huse JT, et al. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science (2016) 352(6288):aad3018. doi: 10.1126/science.aad3018
140. Seoane J. The taming of the TAMs. Trends Cell Biol (2016) 26(8):562–3. doi: 10.1016/j.tcb.2016.06.007
141. Zheng Y, Bao J, Zhao Q, Zhou T, Sun X. A spatio-temporal model of macrophage-mediated drug resistance in glioma immunotherapy. Mol Cancer Ther (2018) 17(4):814–24. doi: 10.1158/1535-7163.MCT-17-0634
142. Almahariq MF, Quinn TJ, Kesarwani P, Kant S, Miller CR, Chinnaiyan P. Inhibition of colony-stimulating factor-1 receptor enhances the efficacy of radiotherapy and reduces immune suppression in glioblastoma. In Vivo (2021) 35(1):119–29. doi: 10.21873/invivo.12239
143. Groblewska M, Litman-Zawadzka A, Mroczko B. The role of selected chemokines and their receptors in the development of gliomas. Int J Mol Sci (2020) 21(10):3704. doi: 10.3390/ijms21103704
144. Takacs GP, Flores-Toro JA, Harrison JK. Modulation of the chemokine/chemokine receptor axis as a novel approach for glioma therapy. Pharmacol Ther (2021) 222:107790. doi: 10.1016/j.pharmthera.2020.107790
145. Liu F, Yang B. Double-targeted knockdown of miR-21 and CXCR4 inhibits malignant glioma progression by suppression of the PI3K/AKT and Raf/MEK/ERK pathways. BioMed Res Int (2020) 2020:7930160. doi: 10.1155/2020/7930160
146. Mercurio L, Ajmone-Cat MA, Cecchetti S, Ricci A, Bozzuto G, Molinari A, et al. Targeting CXCR4 by a selective peptide antagonist modulates tumor microenvironment and microglia reactivity in a human glioblastoma model. J Exp Clin Cancer Res (2016) 35:55. doi: 10.1186/s13046-016-0326-y
147. Gravina GL, Mancini A, Marampon F, Colapietro A, Delle Monache S, Sferra R, et al. The brain-penetrating CXCR4 antagonist, PRX177561, increases the antitumor effects of bevacizumab and sunitinib in preclinical models of human glioblastoma. J Hematol Oncol (2017) 10(1):5. doi: 10.1186/s13045-016-0377-8
148. Urbantat RM, Jelgersma C, Brandenburg S, Nieminen-Kelha M, Kremenetskaia I, Zollfrank J, et al. Tumor-associated Microglia/Macrophages as a predictor for survival in glioblastoma and temozolomide-induced changes in CXCR2 signaling with new resistance overcoming strategy by combination therapy. Int J Mol Sci (2021) 22(20):11180. doi: 10.3390/ijms222011180
149. Acker G, Zollfrank J, Jelgersma C, Nieminen-Kelha M, Kremenetskaia I, Mueller S, et al. The CXCR2/CXCL2 signalling pathway - an alternative therapeutic approach in high-grade glioma. Eur J Cancer (2020) 126:106–15. doi: 10.1016/j.ejca.2019.12.005
150. Zhu X, Fujita M, Snyder LA, Okada H. Systemic delivery of neutralizing antibody targeting CCL2 for glioma therapy. J Neurooncol (2011) 104(1):83–92. doi: 10.1007/s11060-010-0473-5
151. Flores-Toro JA, Luo D, Gopinath A, Sarkisian MR, Campbell JJ, Charo IF, et al. CCR2 inhibition reduces tumor myeloid cells and unmasks a checkpoint inhibitor effect to slow progression of resistant murine gliomas. Proc Natl Acad Sci U S A (2020) 117(2):1129–38. doi: 10.1073/pnas.1910856117
152. Wu A, Maxwell R, Xia Y, Cardarelli P, Oyasu M, Belcaid Z, et al. Combination anti-CXCR4 and anti-PD-1 immunotherapy provides survival benefit in glioblastoma through immune cell modulation of tumor microenvironment. J Neurooncol (2019) 143(2):241–9. doi: 10.1007/s11060-019-03172-5
153. Kreisl TN, Zhang W, Odia Y, Shih JH, Butman JA, Hammoud D, et al. A phase II trial of single-agent bevacizumab in patients with recurrent anaplastic glioma. Neuro Oncol (2011) 13(10):1143–50. doi: 10.1093/neuonc/nor091
154. Deng L, Stafford JH, Liu SC, Chernikova SB, Merchant M, Recht L, et al. SDF-1 blockade enhances anti-VEGF therapy of glioblastoma and can be monitored by MRI. Neoplasia (2017) 19(1):1–7. doi: 10.1016/j.neo.2016.11.010
155. Peterson TE, Kirkpatrick ND, Huang Y, Farrar CT, Marijt KA, Kloepper J, et al. Dual inhibition of ang-2 and VEGF receptors normalizes tumor vasculature and prolongs survival in glioblastoma by altering macrophages. Proc Natl Acad Sci U S A (2016) 113(16):4470–5. doi: 10.1073/pnas.1525349113
156. Hack SP, Zhu AX, Wang Y. Augmenting anticancer immunity through combined targeting of angiogenic and PD-1/PD-L1 pathways: Challenges and opportunities. Front Immunol (2020) 11:598877. doi: 10.3389/fimmu.2020.598877
157. Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, Lopez-Janeiro A, Porciuncula A, Idoate MA, et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat Med (2019) 25(3):470–6. doi: 10.1038/s41591-018-0339-5
158. Rao G, Latha K, Ott M, Sabbagh A, Marisetty A, Ling X, et al. Anti-PD-1 induces M1 polarization in the glioma microenvironment and exerts therapeutic efficacy in the absence of CD8 cytotoxic T cells. Clin Cancer Res (2020) 26(17):4699–712. doi: 10.1158/1078-0432.CCR-19-4110
159. Dang W, Xiao J, Ma Q, Miao J, Cao M, Chen L, et al. Combination of p38 MAPK inhibitor with PD-L1 antibody effectively prolongs survivals of temozolomide-resistant glioma-bearing mice via reduction of infiltrating glioma-associated macrophages and PD-L1 expression on resident glioma-associated microglia. Brain Tumor Pathol (2021) 38(3):189–200. doi: 10.1007/s10014-021-00404-3
160. Cui X, Ma C, Vasudevaraja V, Serrano J, Tong J, Peng Y, et al. Dissecting the immunosuppressive tumor microenvironments in glioblastoma-on-a-Chip for optimized PD-1 immunotherapy. Elife (2020) 9:e52253. doi: 10.7554/eLife.52253
161. Yang F, He Z, Duan H, Zhang D, Li J, Yang H, et al. Synergistic immunotherapy of glioblastoma by dual targeting of IL-6 and CD40. Nat Commun (2021) 12(1):3424. doi: 10.1038/s41467-021-23832-3
162. Fierro J Jr., DiPasquale J, Perez J, Chin B, Chokpapone Y, Tran AM, et al. Dual-sgRNA CRISPR/Cas9 knockout of PD-L1 in human U87 glioblastoma tumor cells inhibits proliferation, invasion, and tumor-associated macrophage polarization. Sci Rep (2022) 12(1):2417. doi: 10.1038/s41598-022-06430-1
163. Xue N, Zhou Q, Ji M, Jin J, Lai F, Chen J, et al. Chlorogenic acid inhibits glioblastoma growth through repolarizating macrophage from M2 to M1 phenotype. Sci Rep (2017) 7:39011. doi: 10.1038/srep39011
164. Hsu SPC, Chen YC, Chiang HC, Huang YC, Huang CC, Wang HE, et al. Rapamycin and hydroxychloroquine combination alters macrophage polarization and sensitizes glioblastoma to immune checkpoint inhibitors. J Neurooncol (2020) 146(3):417–26. doi: 10.1007/s11060-019-03360-3
165. Bajracharya R, Caruso AC, Vella LJ, Nisbet RM. Current and emerging strategies for enhancing antibody delivery to the brain. Pharmaceutics (2021) 13(12):2014. doi: 10.3390/pharmaceutics13122014
166. Sabbagh A, Beccaria K, Ling X, Marisetty A, Ott M, Caruso H, et al. Opening of the blood-brain barrier using low-intensity pulsed ultrasound enhances responses to immunotherapy in preclinical glioma models. Clin Cancer Res (2021) 27(15):4325–37. doi: 10.1158/1078-0432.CCR-20-3760
167. Zhao P, Wang Y, Kang X, Wu A, Yin W, Tang Y, et al. Dual-targeting biomimetic delivery for anti-glioma activity via remodeling the tumor microenvironment and directing macrophage-mediated immunotherapy. Chem Sci (2018) 9(10):2674–89. doi: 10.1039/C7SC04853J
168. Zhang F, Parayath NN, Ene CI, Stephan SB, Koehne AL, Coon ME, et al. Genetic programming of macrophages to perform anti-tumor functions using targeted mRNA nanocarriers. Nat Commun (2019) 10(1):3974. doi: 10.1038/s41467-019-11911-5
169. Mo X, Zheng Z, He Y, Zhong H, Kang X, Shi M, et al. Antiglioma via regulating oxidative stress and remodeling tumor-associated macrophage using lactoferrin-mediated biomimetic codelivery of simvastatin/fenretinide. J Control Release (2018) 287:12–23. doi: 10.1016/j.jconrel.2018.08.012
170. Qiu N, Wang G, Wang J, Zhou Q, Guo M, Wang Y, et al. Tumor-associated macrophage and tumor-cell dually transfecting polyplexes for efficient interleukin-12 cancer gene therapy. Adv Mater (2021) 33(8):e2100137. doi: 10.1002/adma.202006189
171. Zheng Z, Zhang J, Jiang J, He Y, Zhang W, Mo X, et al. Remodeling tumor immune microenvironment (TIME) for glioma therapy using multi-targeting liposomal codelivery. J Immunother Cancer (2020) 8(2):e000207. doi: 10.1136/jitc-2019-000207
Keywords: Glioma, glioma associated macrophages (GAMs), recruitment, polarization, therapy resistance, targeted therapy
Citation: Xu C, Xiao M, Li X, Xin L, Song J, Zhan Q, Wang C, Zhang Q, Yuan X, Tan Y and Fang C (2022) Origin, activation, and targeted therapy of glioma-associated macrophages. Front. Immunol. 13:974996. doi: 10.3389/fimmu.2022.974996
Received: 21 June 2022; Accepted: 22 September 2022;
Published: 06 October 2022.
Edited by:
Junxia Zhang, Nanjing Medical University, ChinaReviewed by:
Xiuping Zhou, Xuzhou Medical University, ChinaCopyright © 2022 Xu, Xiao, Li, Xin, Song, Zhan, Wang, Zhang, Yuan, Tan and Fang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chuan Fang, Y2h1YW5mYW5nQGhidS5lZHUuY24=; Yanli Tan, eWFubGl0YW5AaGJ1LmVkdS5jbg==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.