
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 26 January 2023
Sec. Autoimmune and Autoinflammatory Disorders
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.974078
 Margherita Giannini1,2,3Benjamin Ellezam4Valérie Leclair5Frédéric Lefebvre6,7Yves Troyanov8Marie Hudson5Jean-Luc Senécal6,7Bernard Geny1,3Océane Landon-Cardinal6,7Alain Meyer1,3,9*
Margherita Giannini1,2,3Benjamin Ellezam4Valérie Leclair5Frédéric Lefebvre6,7Yves Troyanov8Marie Hudson5Jean-Luc Senécal6,7Bernard Geny1,3Océane Landon-Cardinal6,7Alain Meyer1,3,9*Systemic sclerosis and autoimmune myositis are both associated with decreased quality of life and increased mortality. Their prognosis and management largely depend on the disease subgroups. Indeed, systemic sclerosis is a heterogeneous disease, the two predominant forms of the disease being limited and diffuse scleroderma. Autoimmune myositis is also a heterogeneous group of myopathies that classically encompass necrotizing myopathy, antisynthetase syndrome, dermatomyositis and inclusion body myositis. Recent data revealed that an additional disease subset, denominated “scleromyositis”, should be recognized within both the systemic sclerosis and the autoimmune myositis spectrum. We performed an in-depth review of the literature with the aim of better delineating scleromyositis. Our review highlights that this concept is supported by recent clinical, serological and histopathological findings that have important implications for patient management and understanding of the disease pathophysiology. As compared with other subsets of systemic sclerosis and autoimmune myositis, scleromyositis patients can present with a characteristic pattern of muscle involvement (i.e. distribution of muscle weakness) along with multisystemic involvement, and some of these extra-muscular complications are associated with poor prognosis. Several autoantibodies have been specifically associated with scleromyositis, but they are not currently integrated in diagnostic and classification criteria for systemic sclerosis and autoimmune myositis. Finally, striking vasculopathic lesions at muscle biopsy have been shown to be hallmarks of scleromyositis, providing a strong anatomopathological substratum for the concept of scleromyositis. These findings bring new insights into the pathogenesis of scleromyositis and help to diagnose this condition, in patients with subtle SSc features and/or no autoantibodies (i.e. “seronegative” scleromyositis). No guidelines are available for the management of these patients, but recent data are showing the way towards a new therapeutic approach dedicated to these patients.
Systemic sclerosis (SSc) is a rare autoimmune disease characterized by vasculopathy and fibrosis affecting multiple organs (1). Autoimmune myositis (AIM) is another rare condition characterized by myopathy with evidence of inflammation-driven muscle lesions. SSc and AIM are both associated with decreased quality of life (2, 3) and increased mortality (4, 5). However, the prognosis and care largely depend on the subtypes of these diseases, since SSc and AIM both encompass a heterogeneous group of diseases. Identification of these subgroups is fundamental because each requires different management (6). The two predominant forms of SSc are limited cutaneous (lSSc) and diffuse cutaneous scleroderma (SSc) (7). AIM is also a heterogeneous group of myopathies that classically encompasses immune-mediated necrotizing myopathy (IMNM), antisynthetase syndrome (ASS), dermatomyositis (DM) and inclusion body myositis (IBM) (8). The historical entity polymyositis (PM) is now becoming rare and even uncertain, often mistaken for more recently described patterns (6, 9, 10).
Overlap myositis (OM) has been defined as AIM with overlap clinical features (extra muscular involvement other than DM rash) and/or overlap autoantibodies (associated with other connective tissue disease than AIM) (11–13). OM has been shown to be clinically relevant since it has been reported to be the most frequent AIM subgroup and to have diagnostic, prognostic and therapeutic value (11, 12). SSc has been reported to be the most common connective tissue disease in OM patients accounting for about 40% of cases (12, 13). This AIM subgroup associating SSc and OM patients has been denominated “scleromyositis”.
Thus, historically, scleromyositis has been defined as an overlap between SSc and AIM (12, 14, 15). Yet, fulfilling the American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) classification criteria for both SSc (7) and AIM (16) is a definition for scleromyositis (17–19) that is limited by low sensitivity for the condition (20–22).
Whether scleromyositis can be recognized within both the SSc and AIM spectrum has not been reviewed.
Since of these uncertainties, an in-depth review of the literature reporting muscle involvement in SSc was performed, with the objective of better delineating scleromyositis clinically, serologically and histopathologically, and identifying implications of this diagnosis for prognosis and management.
First, all original articles in English pertaining to SSc where muscle involvement and/or SSc/AIM overlap were directly mentioned or easily calculated from the available data were collected. Second, Pubmed was searched twice in February 2022 and September 2022 using the search words “myositis” or “myopathy” or “myopathies” or “scleromyositis” or “polymyositis” or “dermatomyositis” or “antisynthetase syndrome” or “anti-synthetase syndrome” AND “scleroderma” or “systemic sclerosis” or “scleromyositis” or “anti-PM/Scl” or “anti-PMScl” or “anti-PM Scl” or “PMSCL” or “PM Scl” or “anti-PM-Scl” or “anti-PM75” or “anti-PM100” or “anti-CENPB” or “anti-CENPA” or “anti-CENP-A” or “anti-CENP-B” or “anti-CENPA/B” or “anti-centromere” or “anti–topoisomerase” or “anti-Scl70” or “anti-Scl-70” or “anti-RuvBL1/2” or “anti-RuvBL1” or “anti-RuvBL2” or “anti-ku” or “anti-RNA polymerase III” or “anti-RNA-polymerase III” or “anti-RNA pol” or “anti-POL” or “anti-RNAP III” or “anti-RNPC-3” or “anti-RNPC3” or “anti-RNP” or “anti-U1 RNP” or “anti-U1RNP” or “anti-U3 RNP” or “anti-U3RNP” or “anti-U11/U12 RNP” or “anti-U5 RNP” or “anti-U5RNP” or “anti-SMN” or “anti-fibrillarin” or “anti-Th/To”.
Reference lists of relevant articles were also manually searched to identify additional studies not captured by the search. No restrictions on publication period, type of study, nor setting, were placed on this search. Because there is currently no consensual definition of scleromyositis, all significant descriptions of association between AIM and SSc according to the authors’ opinion were included.
Two reviewers (MG and AM) independently screened titles and abstracts for inclusion. At this stage, animal and pediatric studies were excluded. Records included by at least one reviewer at the title and abstract screening stage were further included for full text review. At this stage, if consensus between the two reviewers was met, each publication was included for data extraction. All studies were included in the data synthesis irrespective of quality assessment.
Among 3263 references screened, we ultimately included 61 articles published between 1961 and 2022 reporting muscle involvement in SSc and/or SSc/AIM overlap, its characteristics and implication for prognosis and management of patients.
According to a 2013 meta-analysis, myositis is reported in 13% (95% CI 10–17) of SSc patients (23).. However, the prevalence ranges widely among surveys, from 5% to 96% (17, 23–48). This important disparity is likely due to the heterogeneity of definitions used for muscle involvement in SSc since no agreement upon diagnostic criteria of muscle involvement in SSc are currently available (49). SSc was diagnosed according to expert opinion (30–36) or ACR 1980 (50) or LeRoy (51) or ACR/EULAR 2013 (7) criteria. Muscle involvement was defined by muscle weakness and/or myalgia and/or amyotrophy and/or muscle enzymes increase and/or myopathic features at electroneuromyography (ENMG) or muscle biopsy and/or oedema at muscle magnetic resonance imaging (MRI) and/or myositis specific/associated autoantibodies or Bohan and Peter criteria (52, 53). When the authors described a myopathy associated with SSc, regardless of the criteria used, we classified these patients as possible scleromyositis. Due to such heterogeneity of definitions for myopathy in SSc, each of the above criteria for muscle involvement has been compared to items included in ACR/EULAR 2017 classification criteria, which are the most up to date criteria for AIM (16).
Conversely, among AIM patients, SSc features are also variably recognized. While the 2017 ACR/EULAR classification criteria for AIM (16) do not recognize OM as a separate entity, up to 29% of AIM cases are reported to have a concurrent SSc diagnosis (12, 18, 54).
Clinical, biological, electromyographic and radiological characteristics of muscle involvement in the whole group of SSc patients and in those possibly suffering from scleromyositis are shown in Tables 1 and 2, respectively. Although the prevalence of muscle features varied somewhat according to the composition of the SSc cohorts (relative proportions of lSSc vs dSSc) and/or the criteria selected for the definition of scleromyositis, several characteristic features of skeletal muscle involvement in SSc can be drawn and are discussed herein.
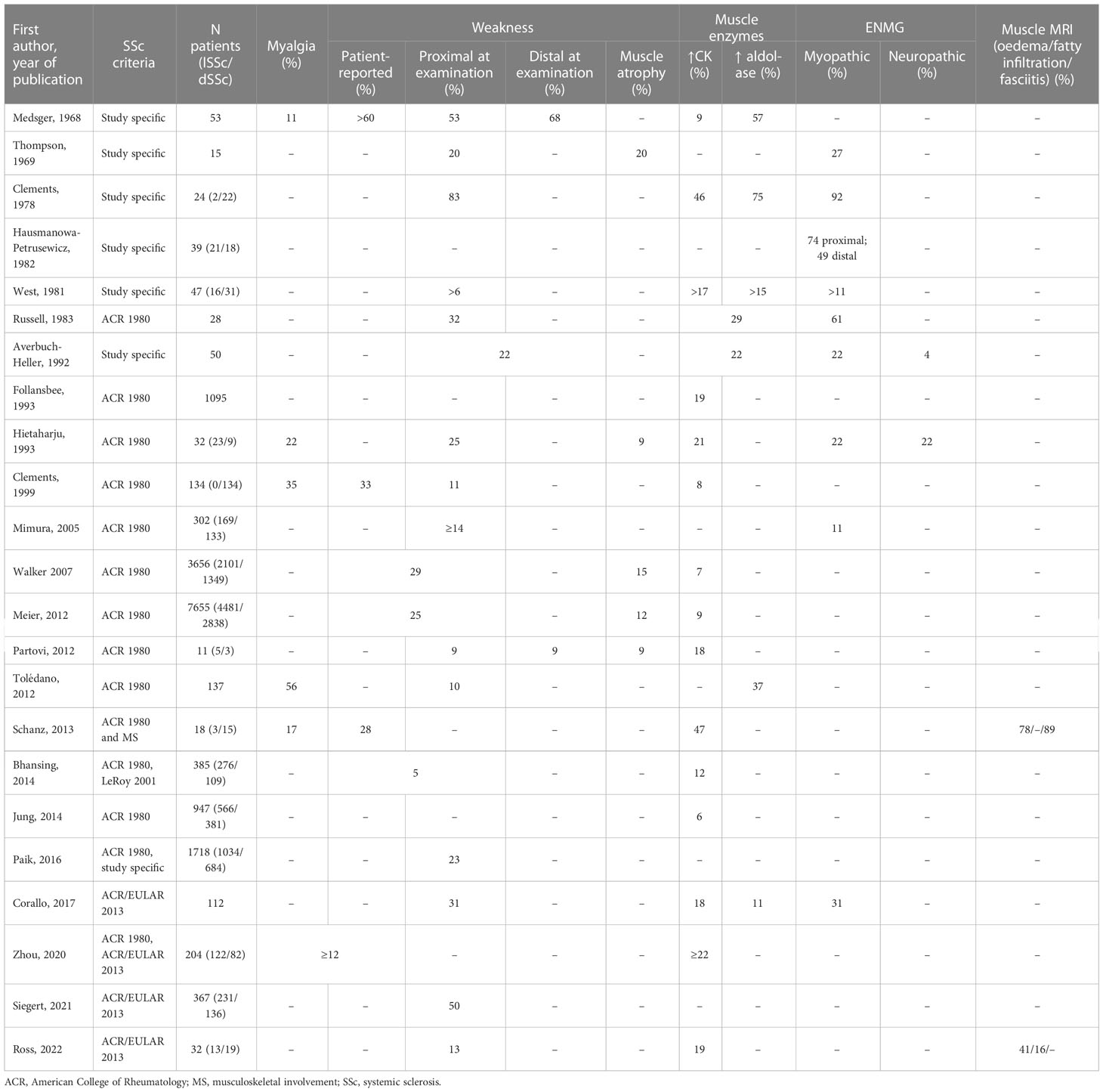
Table 1 Prevalence of muscle features in systemic sclerosis patients.
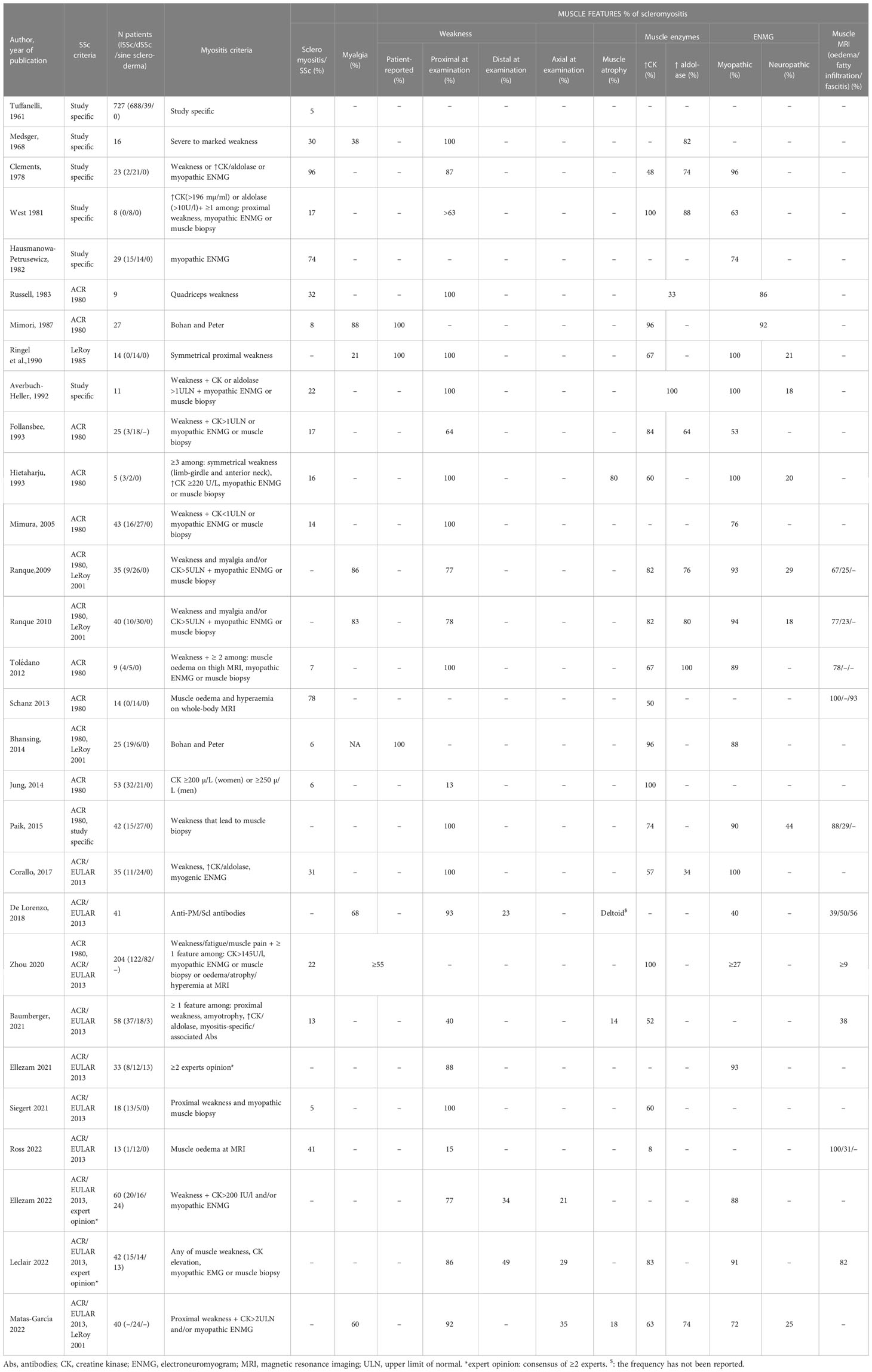
Table 2 Prevalence and characteristics of scleromyositis in systemic sclerosis patients.
Myalgias are reported by 11% to 56% of SSc patients (30, 38, 45, 46, 55, 56) compared to 21 to 88% of patients with scleromyositis (27, 28, 30, 41, 57–59). Thus, in SSc, as in other settings (16, 60, 61), myalgia lacks sensitivity and specificity for the diagnosis of myopathy. Myalgia is not included in the 2017 ACR/EULAR criteria for AIM and was selected as a criterion for scleromyositis only by a single group (27, 28).
Subjective muscle weakness was reported by 23% to 60% of SSc patients (30, 44, 46, 55) while objective muscle weakness was detected on physical examination in 9 to 83% of cases (24, 25, 30–32, 37, 38, 40, 42, 45, 47, 55, 56, 62). This may indicate that extra-muscular involvement (i.e. joint, skin thickening, interstitial lung disease [ILD], anemia and pulmonary arterial hypertension [PAH]) also contributes to the self-reported exercise limitation in some SSc patients. Muscle weakness on physical examination is a 2017 ACR/EULAR criterion for AIM whereas self-reported weakness is not (16). It was selected as a criterion for scleromyositis by most of the studies. Depending on the definition used, muscle weakness was reported in 13% to 100% of scleromyositis patients (17, 20, 21, 24–30, 32, 34, 35, 37, 38, 41–43, 45, 47, 57–59, 63, 64).
The distribution of muscle weakness was generally symmetrical and proximal both in the upper and lower limbs. Weaker upper limbs than lower limbs has been reported to be a feature of scleromyositis as compared to other myositis subgroups (57). More rarely, scleromyositis (23-49%) and SSc patients (9-68%) presented with distal weakness (20, 21, 30, 57, 62). However, skin thickening, joint contractions and arthritis may also contribute to distal muscles weakness. Finally, axial involvement leading to head drop syndrome and/or camptocormia has been reported up to one-third of scleromyositis patients (20, 21, 59, 65–68).
Muscle weakness, both self-reported and at examination, was more common in dSSc compared to lSSc (40, 44). The frequency of muscle atrophy was also greater in dSSc as compared to lSSc (40, 44), indicating a more damaging myopathy in this subgroup. Muscle weakness has been independently associated with different degrees of physical disability (56).
Serum CK level elevation is a 2017 ACR/EULAR criterion for AIM (16) and it is reported in 6% to 47% of SSc patients (17, 24, 26, 29, 30, 32, 38, 40, 44, 46, 47, 55, 62). Serum CK level elevation was selected as a criterion for scleromyositis in about half of the studies reviewed and was noted in 8% to 100% of scleromyositis patients (17, 24–29, 32, 34, 37, 41, 45–47, 58, 59, 64). Among the patients of the Canadian Scleroderma Research Group cohort, a worse survival was recorded in patients with elevated serum CK levels (29). However, in scleromyositis patients, serum CK levels > 5 times the upper limit of normal (ULN) were associated with better myositis response to corticosteroid treatment, as compared to lower serum CK levels (27). It should be noted that CK levels are also elevated in myocarditis, an important diagnostic consideration in SSc (69). Measurement of the isoenzymes (MM and MB) and serum troponins (t and i) levels are notably useful to distinguish whether the increased CK level is of skeletal and/or cardiac muscle origin (70).
Serum aldolase levels are more frequently increased than CK levels in SSc patients (11 to 75% of patients) (24, 30, 32, 45). Elevated serum level of aldolase is also a 2017 ACR/EULAR criterion for AIM (16). In a prospective cohort of SSc patients without weakness at baseline, elevated serum aldolase levels had a higher predictive value for detection of incident myopathy than elevated CK levels (45). However, unlike CK, aldolase activity is present in many tissues (71) and the specific tissue source of the increased aldolase in SSc patients remains to be determined. It was selected as a criterion for scleromyositis in only two studies (24, 37), and was increased in 34 to 100% of scleromyositis patients (21, 24, 26–28, 30, 32, 34, 45, 59).
ENMG was abnormal in 11 to 92% of SSc patients (24, 31–33, 38, 42). Although ENMG is generally very useful to explore the cause of weakness and/or elevated serum levels of muscle enzymes, it was not retained as a 2017 ACR/EULAR criterion for AIM (16). It was selected as a criterion for scleromyositis by about half of the studies reviewed and a myopathic pattern was found in 40 to 100% of scleromyositis patients (17, 20, 21, 24, 26–28, 32, 41, 42, 45, 57–59, 63, 64). Consistent with the reported clinical distribution of weakness, ENMG more frequently demonstrated myopathic patterns in proximal than in distal muscles (33). Among those myopathic patterns, spontaneous activity (positive sharp waves and/or fibrillation) has been associated with the highest CK levels (72). Interestingly, neuropathic pattern has been also reported in 18 to 29% of scleromyositis patients (27, 28, 59).
MRI allows non-invasive visualization of characteristic myositis changes, including edema, fatty replacement, atrophy, fasciitis, and subcutaneous pathology (73). However, none of these features are entirely specific to AIM (74) and therefore can lead to misdiagnosis when MRI findings are not interpreted according to the clinical context (75). Thus, muscle MRI findings were not selected as a 2017 ACR/EULAR criterion for AIM (16).
Abnormal muscle findings on whole body MRI have been found in 41 to 89% of SSc patients with musculoskeletal symptoms (muscle weakness, myalgia, arthralgia, tendon sheath discomfort and/or tendon friction rub) (46, 47). It was chosen as a criterion for scleromyositis in only three studies and 38% to 100% of scleromyositis patients had abnormal MRI muscle findings (27, 28, 37, 45, 47, 57, 64). In scleromyositis patients, MRI myositis lesions include fasciitis (thickening and/or increased signal intensity of the fascia on STIR and post-gadolinium images) and/or perifascial edema and/or muscle edema (27, 28, 46). These findings resemble those observed in DM (76). Fascial and/or muscle lesions were correlated with muscle weakness, Rodnan’s score and C-reactive protein levels (but not with serum CK levels) (46).
Myopathological analysis is an important tool to help differentiate AIM from muscular dystrophies and other non-inflammatory myopathies and to identify AIM subgroups. Myopathological lesions in AIM encompass several tissue domains that include muscle fibers, connective tissue and vasculature (77). These lesions define histopathological patterns, some of which have been linked with clinical features and outcomes. During the 119th European NeuroMuscular Centre (ENMC) international workshop, experts reached a consensus that histopathological patterns in AIM include PM, DM, IMNM, IBM and non-specific myositis (78). Thus, there is currently no estabilished histopathological criteria for scleromyositis and this entity is not recognized by the ENMC as a distinct AIM subset.
Myopathological lesions in scleromyositis patients are summarized in Table 3.
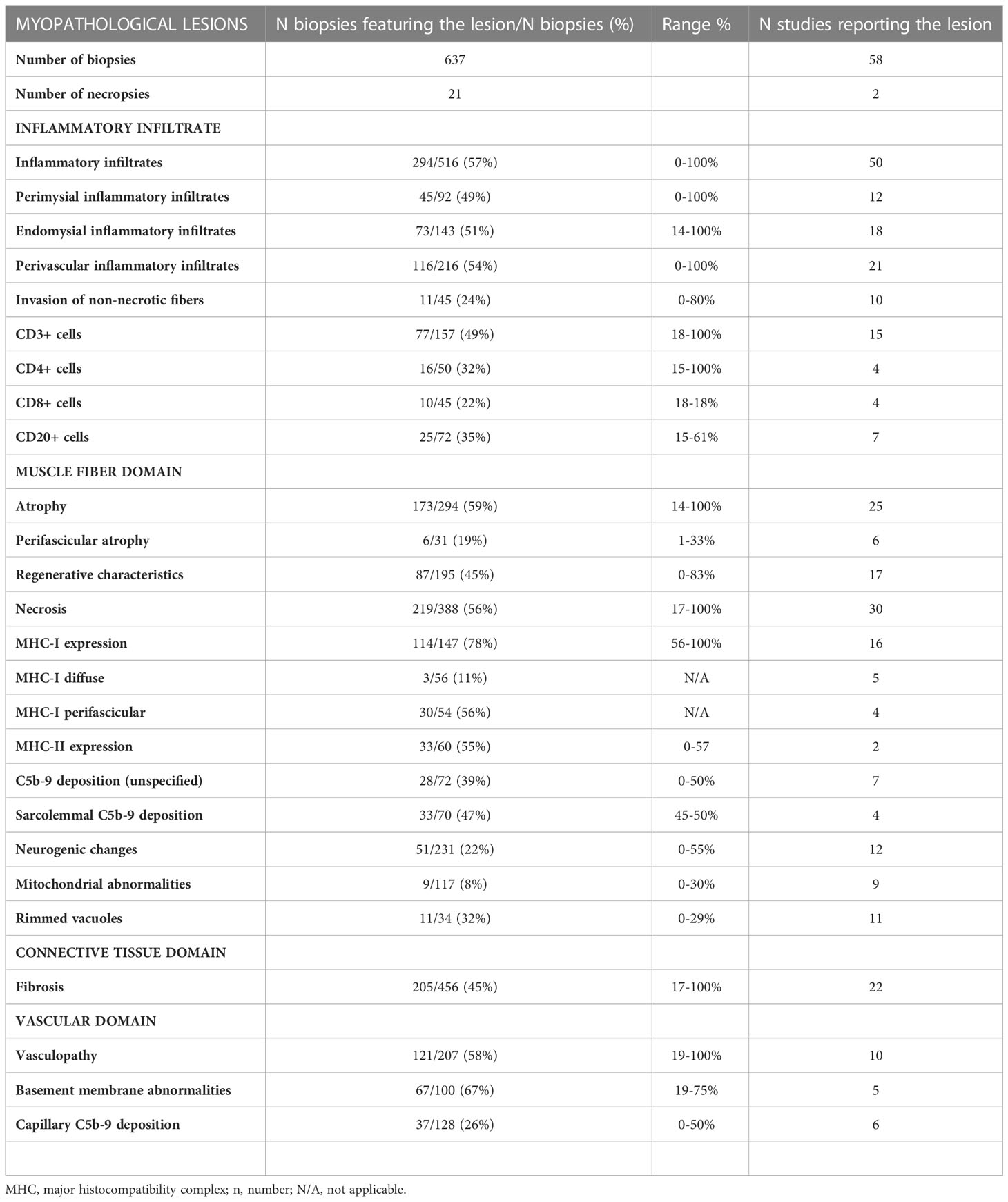
Table 3 Muscle histopathological features of scleromyositis.
In a recent scoping review (79), the most frequent myopathological lesions included sarcolemmal expression of class I major histocompatibility complex (72%), myofiber necrosis (56%), inflammatory infiltrates (57%), endomysial fibrosis (33%) and vasculopathy (33%). When these basic lesions were integrated according to the ENMC consensus, IMNM and PM were the two dominant patterns, present in up to 60% and 78% of scleromyositis patients, respectively.
Yet, several recent studies indicate that vasculopathy and fibrosis might be pathological hallmarks of scleromyositis that distinguish them from other AIM sugroups. Indeed, these two features recapitulate two cardinal pathophysiological processes of SSc (80, 81) supporting the concept of SM as an organ manifestation of SSc and a distinct subset of AIM (20).
Vasculopathy in scleromyositis has been shown in the form of capillary dropout and enlargement on light microscopy, abnormal capillary expression of VEGF or PDGFR-β on immunohistochemistry, and basement membrane reduplication with endothelial activation and/or increased numbers of ensheathed pericyte processes on electron microscopy (20, 24, 25, 63). In a recent study, capillary pathology with prominent basement membrane reduplication (≥4 layers in >50% of capillaries) was found in 65% of scleromyositis vs 0% of other AIM controls (p<0.001) (20) (Figure 1). This hallmark histopathological feature of scleromyositis provides a strong anatomopathological substratum for the thus far clinically-derived concept of scleromyositis Moreover, a predominant fibrotic pattern without inflammation or necrosis (“fibrosing myopathy”) has been described in 20-27% of scleromyositis cases and was associated with poor response to immunomodulatory drugs and increased risk of death from cardiac involvement (59, 72). On the other hand, necrosis and inflammation have been associated with good responses to those drugs (27, 59). Interestingly, “fibrosing myopathy” has generally been associated with lower increases in CK level and lack of spontaneous activity (positive sharp waves and/or fibrillation) on ENMG, while the opposite was found in scleromyositis patients with muscle inflammation and/or necrosis (27, 59, 72).
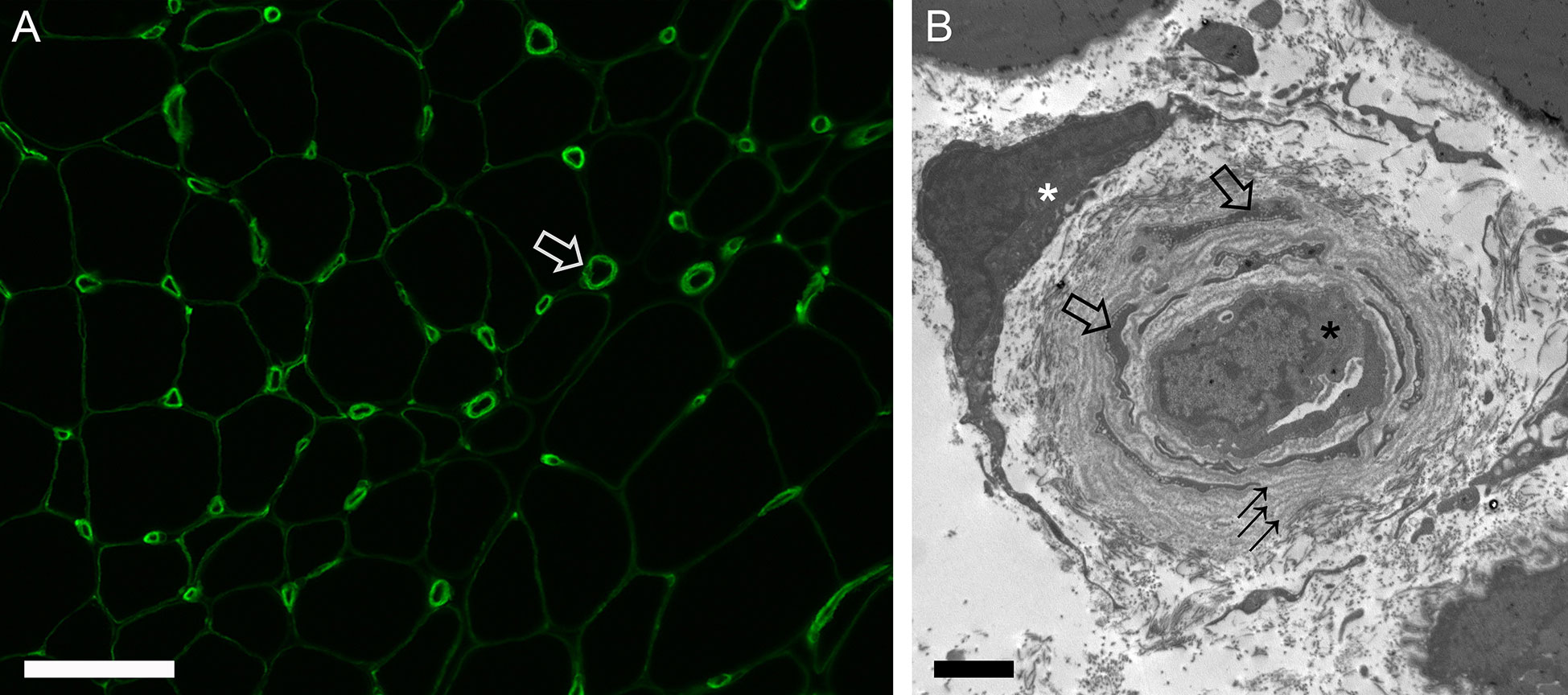
Figure 1 Capillary pathology in scleromyositis. (A) Immunofluorescence for collagen type 4 highlighting enlarged endomysial capillaries often with laminated appearance of basement membrane (open arrow). (B) Electron micrograph showing a collapsed capillary (black asterisk) with several concentric layers of reduplicated basement membrane (arrows) ensheathing many pericyte processes (open arrows). A fibroblast is also shown (white asterisk). Bars: A, 100 µm, B, 2 µm.
As compared to SSc without muscle involvement and to the other AIM subgroups, scleromyositis patients present with distinct extra-muscular complications, some of which are associated with poor prognosis.
Scleromyositis patients have been shown to have a worse survival rate than SSc patients without myositis, the most common cause of death being cardiopulmonary disease (42% to 63%) (29, 82). On the other hand, among scleromyositis patients, SSc-related complications accounted for up to half of the deaths (12). This indicates that scleromyositis is not merely an overlap between SSc and AIM, but a unique condition within both the SSc and AIM spectrum.
In the EUSTAR cohort, more than 50% of SSc-related deaths were of pulmonary causes (83). A recent meta-analysis showed that the prevalence of SSc-ILD was 56% (95% CI 49%- 63%), with high heterogeneity across studies (84). Clinically significant ILD is present in approximately 40% of patients with SSc (85), and high resolution computed tomography (HRCT) can detect interstitial abnormalities in up to 90% of the patients (86). Lung involvement negatively impacts survival (29) and quality of life (87).
Several studies have shown that scleromyositis patients more frequently present with ILD than SSc patients without muscle involvement (up to 68% vs 13%) (27–29, 42, 56, 82).
Although involvement of respiratory muscles in SSc as well as in scleromyositis has not been systematically examined, it has been described in case reports and series (21, 88–90). Moreover, in the EUSTAR cohort, patients with ILD and anti-PM/Scl (an autoantibody that is associated with scleromyositis as detailed below), vital capacity tended to improve whereas diffusion capacity of lung for carbon monoxide remained stable at one-year follow-up (91). This suggests that respiratory muscle weakness contributes to respiratory impairment in this subgroup.
In the EUSTAR cohort, 26% of SSc-related deaths were related to cardiac involvement, being the second cause of mortality in SSc patients (83). The clinical presentation may include arrhythmias (92%), congestive heart failure (68%), conduction system abnormalities (60%) and, rarely, pericardial effusion, although most patients are asymptomatic in the early stages (26, 92). Echocardiography demonstrate impaired left and right ventricle systolic dysfunction in approximately 20% of SSc patients (93). Conversely, cardiac magnetic resonance (CMR) abnormalities, such as T2-weighted changes indicating myocardial edema and late gadolinium enhancement measuring focal fibrosis, are detected in up to 96% of SSc patients even in early phases of the disease and without significant impairment of cardiac function (93–99).
Myopathy is an independent risk factor for cardiac involvement in SSc patients (26, 28, 34, 42, 100–102). Myocardial disease has been reported in up to 21% of scleromyositis patients versus 10% of SSc patients (26), although screening tools and definitions used for cardiac involvement were heterogeneous.
It should be noted that an elevation of serum CK levels can reflect either skeletal or cardiac muscle disease. Among SSc patients, those with abnormal CK levels more frequently fulfilled criteria for myocardial disease (23% vs. 9%) (e.g. atrial or ventricular arrhythmias requiring therapy, or congestive heart failure), and died of cardiac causes (9% vs 3%) (26) than those with normal CK levels. Thus, prompt recognition of heart involvement, taking advantage of CMR and cardiac biomarkers specific to the myocardium, such as cardiac troponin I (70), is recommended to adapt treatment strategy.
Scleroderma renal crisis (SRC) is the third life-threatening complication, accounting for 8% of SSc-related deaths (83). While SRC is reported in about 5% of the SSc patients (103), it occurs in up to 15% of the scleromyositis patients (27, 28, 56, 64).
In a cohort of 1718 SSc patients, those with muscle weakness more frequently developed SRC than patients without (7.1% vs 1.4%) (56). Corticosteroids (CS) are frequently used in scleromyositis (104) and are associated with SRC (103). In the EUSTAR cohort, while the risk of SRC was significantly increased in anti-PM/Scl positive patients as compared to the rest of SSc patients, multivariate analysis revealed that SRC was associated with CSbut not with anti-PM/Scl antibodies (91). This may indicate that CS use rather than anti-PM/Scl antibody itself increases the risk of SRC in SSc patients.
Gastrointestinal complications of SSc decrease the quality of life (87, 105) and cause death in about 4% of patients (83, 106).
Esophageal involvement manifests as dysphagia, acid reflux, heartburn and retrosternal pain (107). Long-term esophageal motility disorders in patients with SSc may lead to complications, such as gastroesophageal reflux, esophagitis, esophageal erosion, stricture, ulcers, diverticulum, leukoplakia, Barrett’s esophagus and adenocarcinoma (107). Although the causal relationship between gastroesophageal reflux and ILD remains unclear, early treatment of esophageal disease could reduce the severity of lung involvement (108). Manifestations related to small and large intestine dysfunctions and anorectal impairment include postprandial bloating, abdominal distension and pain, constipation and fecal incontinence, diarrhea, at times explosive, malabsorption, which can lead to severe malnutrition (109).
The frequency of these complications is similar in SSc patients with and without muscle involvement (approximately 65%) (41, 42, 44, 110), However, gastrointestinal manifestations are frequently more severe in scleromyositis patients as compared to patients without myositis (56).
Tendon friction rubs and synovitis are linked with decreased quality of life (87, 111) and disease progression in SSc (112).
Both complications are more frequently reported in scleromyositis than in SSc patients without muscle involvement (29, 56). Moreover, muscle fascial enhancement on MRI has been linked with synovitis (46).
Skin involvement negatively impacts quality of life (87), due to skin tightening, pain, pruritis and functional limitation, mainly hand disability (113). It also contributes to functional disability with positive correlation between skin severity and Health Assessment Questionnaire scores (114, 115). Moreover, survival is reduced in patients with high skin involvement score (116–118). Some evidence suggests that skin changes indicate changes in visceral disease severity (116, 117, 119). In accordance with this view, except for those with distinct autoantibody profiles as discussed below, scleromyositis patients generally have more diffuse than limited skin thickening (40-75% vs 25-60%, respectively) (27–29, 42, 44, 56, 64) and have higher modified Rodnan skin score (mRSS) compared to SSc (29, 56).
None of the 3 SSc-specific autoantibodies included in the 2013 ACR/EULAR criteria for SSc (anti-centromere, anti–topoisomerase I [anti–Scl-70], anti–RNA polymerase III) (7) have been strongly associated with scleromyositis. Indeed, anti-centromere antibodies have been consistently reported to be negatively associated with muscle involvement in SSc (18, 26, 28, 120). Anti-topoisomerase autoantibodies, when present (0% to 69% of scleromyositis patients) (17, 18, 24, 25, 27–29, 41, 42, 45–47, 56, 64, 72, 82, 121), have been associated with fibrosis on muscle biopsy (72), in accordance with the diffuse skin thickening phenotype of these patients.
In contrast, several other autoantibodies (not included in the SSc 2013 ACR/EULAR criteria), such as anti-PM/Scl, -Ku, -U1-RNP, -U3-RNP, - RuvBL1/2, -SMN, have been specifically associated with scleromyositis. This contributes to the low sensitivity of ACR/EULAR criteria to identify scleromyositis (22), and also indicates that scleromyositis may be driven by different autoimmune mechanisms among the SSc spectrum. Autoantibody expression varies widely among patients and each of the scleromyositis autoantibodies have been associated with specific clinical phenotype, response to treatment and prognosis, as summarized in Table 4.
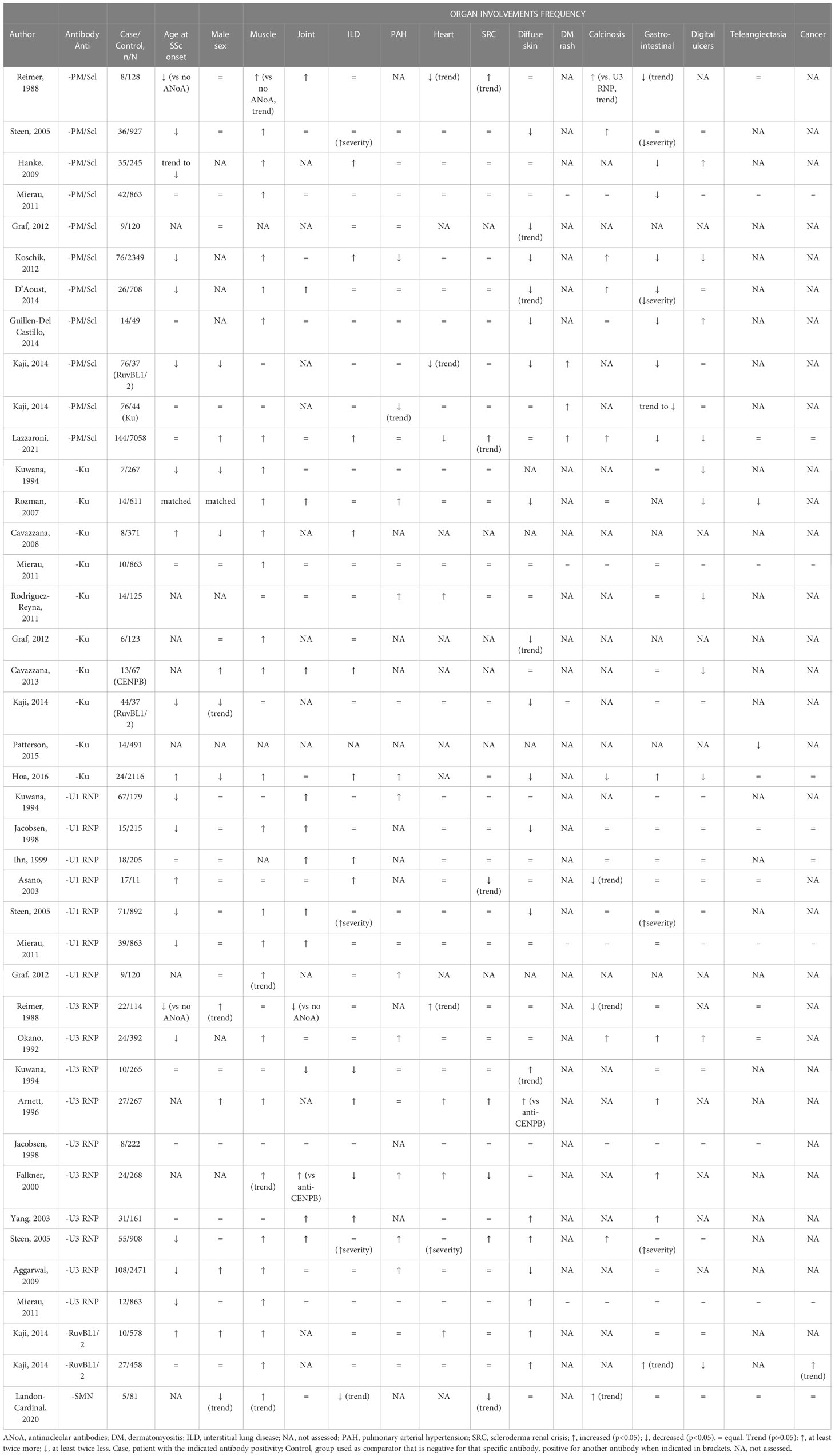
Table 4 Syndromes delineated by autoantibodies associated with scleromyositis.
Anti-PM/Scl antibodies are found in 0% to 13% of SSc patients (24, 45, 120, 122–131). A 2017 meta-analysis reported that 31% of scleromyositis patients were anti-PM/Scl positive (132). A similar prevalence was reported in independent cohorts published after 2017 (18, 37, 59).
As compared with anti-PM/Scl antibody-negative SSc patients, patients with anti-PM/Scl antibodies, in addition to having more frequent muscle involvement, were younger at disease onset and more frequently presented with ILD, arthritis, calcinosis and telangiectasias. In contrast, skin involvement was more frequently limited (sclerodactyly or puffy hands), gastrointestinal involvement was less frequent and less severe (91, 120, 122–124, 131, 133, 134), and DM rash was more frequent (91).
Despite a higher incidence of ILD, anti-PM/Scl was associated with a better response to treatment of ILD, and survival was better in this subgroup (91, 133–135).
Even though anti-PM/Scl patients may have histologic evidence of fibrosis, other histopathologic features (e.g. inflammation or necrosis) were prominent (59, 64). Moreover, in the majority of anti-PM/Scl positive patients from the Pittsburgh Scleroderma Databank (58%), inflammatory changes were found on muscle biopsy (120). In accordance with these histopathological findings, myositis had a favorable outcome when treated with CS alone or in combination with azathioprine in most cases (136).
The prevalence of anti-Ku antibodies in SSc rangs from 1 to 16% (123, 125, 126, 129–131, 137–146), and from 38% to 55% in scleromyositis patients (41, 142, 147). Several studies demonstrate the association between anti-Ku antibodies and scleromyositis (125, 130, 131, 137, 142, 148–151).
In addition, as compared with anti-Ku antibody-negative SSc patients, SSc patients with anti-Ku antibodies presented more frequently with ILD (137, 138, 140), heart involvement (69, 129), PAH (129, 130, 140), gastrointestinal involvement (140, 149) and arthritis (130, 137). They had more frequent limited skin involvement (123, 130, 137, 140) and less frequent vascular involvement (fingertip ulcers and telangiectasias) (125, 129, 130, 137, 143). Despite more frequent cardiopulmonary involvement, no difference in survival was found between subjects with and without single-specificity anti-Ku antibodies (140).
This autoantibody has been associated with a necrotizing pattern on muscle biopsy (79, 152). Patients with anti-Ku myositis have been reported to have a sustained response to CS in one study (12).
The prevalence of anti-U1RNP autoantibodies in SSc ranged from 5% to 12% (42, 45, 120, 122, 123, 129–131, 153–155). It was greater in African American SSc patients (29-32%) (126, 155) and in scleromyositis patients (10%-46%) (41, 42, 72).
In addition to muscle involvement, SSc patients with anti-U1RNP antibodies were younger at disease onset (120, 125, 131, 154) and presented more frequently with ILD (153, 156), PAH (123, 125), arthritis (125, 131, 153, 154) and limited skin involvement (120, 154) when compared with anti-U1RNP negative patients.
In accordance with the increased prevalence of ILD and PAH, the survival of patients with anti-U1RNP is reduced as compared with SSc patients with anti-centromere antibodies (123). However, in PAH-SSc patients, anti-U1RNP positivity was associated with better functional outcomes along with better 5-year and 10-year survival rates. On multivariate analysis including age at PAH diagnosis, sex, World Health Organization functional class, forced vital capacity % predicted value and hemodynamic parameters, anti-U1RNP positivity remained negatively associated with mortality in the subgroup of PAH-SSc patients (157).
Similarly to anti-Ku, this autoantibody has been associated with a necrotizing pattern on muscle biopsy (79). Patients with anti-U1RNP myositis have been reported to have a sustained response to CS in one study (12).
The prevalence of anti-U3RNP (fibrillarin) antibodies in SSc ranged from 1% to 14% (120, 125, 131, 154, 158–163) and is greater in African American patients (17-45%) (126, 155, 161).
In scleromyositis anti-U3RNP antibodies had a prevalence of 6% (72).
In addition to muscle involvement, as compared with anti-U3RNP-negative SSc patients, patients with anti-U3RNP antibodies were more frequently male (158, 159, 164) and younger at disease onset (120, 131, 158, 161, 164). They more frequently presented with diffuse cutaneous disease (125, 131, 163), digital ulcers, calcinosis (161), severe gastro-intestinal involvement (159, 161, 163, 165), myopericarditis and PAH (120, 158, 161, 165), which was the most common cause of death (158).
Some studies found a positive association with ILD (120, 159, 163) as well as with arthritis (120, 163), but others a negative association (125, 164, 165) with them.
This autoantibody has been associated with fibrosis on muscle biopsy (72). This pattern has been reported to be less responsive to immunosuppressive treatment such as CS, methotrexate or mycophenolate (72).
Anti-RuvBL1/2 are rare SSc-related antibodies (1%-2%) (149). Only 51 such patients have been described (166) and about 60% of them were diagnosed with scleromyositis (149, 166, 167). In addition to muscle involvement, these patients were characterized by older age at disease onset, male sex and diffuse skin thickening. Cardiac, gastrointestinal and peripheral vasculature involvements we also described.
Recently, a novel autoantibody targeting survival of motor neuron complex (anti-SMN) has been described in a few scleromyositis patients (167–169).
Scleromyositis patients with anti-SMN autoantibodies displayed proximal weakness, elevated serum CK levels and abnormal EMG (100%), arthritis (60%), SSc calcinosis (60%) and lSSc (80%) (167). In these patients, a nuclear dots pattern in indirect immunofluorescence (AC6/7 according to the International Consensus on Antinuclear Antibody standardized nomenclature) (170) was a useful screening test for identifying anti-SMN autoantibodies (167).
Additionally, anti-SMN autoantibodies have recently been reported in up to 59% of patients in an anti-U1RNP-positive mixed connective tissue disease cohort (171, 172). Interestingly, the presence of anti-SMN autoantibodies, especially with high titers, was associated with a higher prevalence of scleromyositis compared to patients without anti-SMN autoantibodies (171).
Almost 50% of scleromyositis patients do not have SS-associated autoantibodies (21, 28). This subgroup has been called “seronegative” and represents a that is difficult to recognize by clinicians as typically SSc skin involvement is frequently lacking (167). Cancer (20%) and deaths (15%) were common (21), highlighting the importance of correct diagnosis and adequate treatment. Furthermore, these “seronegative” patients provide the opportunity to discover new autoantibodies that could improve the accuracy of diagnosis and potentially shed light on unknown pathogenetic aspects of scleromyositis.
Randomized controlled trials of cyclophosphamide, mycophenolate mofetil, tocilizumab, rituximab, nintedanib, as well as hematopoietic stem cell transplantation (HSCT), have demonstrated efficacy (stabilization or improvement) on skin thickening and/or ILD in SSc patients (173). In contrast, the level of evidence on the effect of these treatments for muscle involvement in SSc is very low, based on uncontrolled case series and data derived from other AIM subtypes. Currently, no consensual clinical practice guidelines are available for scleromyositis management (174, 175).
High dosage CS are generally used to treat AIM patients (174). In the setting of scleromyositis, the benefits of CS should be balanced with the potential side effects as CS have been associated with an increased risk of SRC in SSc (176, 177). In a 2012 systematic review of the available literature, the rate of SRC in SSc treated with CS was approximately twice the expected rate for SSc in general (2% vs 0.94%) (177). Moreover, in the International Scleroderma Renal Crisis Survey, for every milligram of prednisone, the risk of death increased by 4% (176). Whether this is causal or the result of confounding with underlying disease severity remains unresolved (103). On the other hand, CS were effective in 78 to 100% of scleromyositis patients (12, 27, 32, 34). The EULAR experts recognized that CS are part of the therapeutic strategy in the management of musculoskeletal involvement, although the evidence regarding their efficacy in SSc is limited. They recommend that patients with SSc treated with corticosteroids should be carefully monitored with respect to the development of SRC (178). In scleromyositis, sero-pathological features may help to predict CS response of myositis. In particular, necrosis and inflammation shown on muscle biopsy were associated with a response to CS in more than 90% of cases whereas the absence of these lesions, also described as “fibrosing myopathy” (72), is associated with an improvement in only one third of cases (27). In accordance with their association with necrosis and/or inflammation on muscle biopsy (79, 152), anti-PM/SCL, -U1-RNP and -Ku autoantibodies were associated with sustained response of the myositis to CS (12). On the other hand, possibly because of their association with fibrosis on muscle biopsy (72), anti-Scl70 and anti-U3-RNP were associated with a poor response of the myositis to CS (72).
Consistent with its efficacy in SSc-ILD (179), the most frequently used immunomodulatory drug in recent series of scleromyositis patients was mycophenolate mofetil (72). However, no data have demonstrated the efficacy or the corticosteroid-sparing effect of this drug in SSc muscle involvement, nor in other AIM subgroups. Methotrexate and azathioprine were also used (27, 72, 121). Efficacy and corticosteroid-sparing effect have been only demonstrated for methotrexate in the setting of juvenile dermatomyositis (180).
Intravenous immunoglobulins (IVIg) have demonstrated efficacy in refractory DM patients (181, 182) and uncontrolled data indicated some efficacy in other subtypes of refractory myopathies (183). In a series of 52 patients with scleromyositis, 18 patients (34.6%) who received IVIg were taking lower CS doses at one year and at the end of follow-up than patients who did not receive IVIG, despite higher maximal CS dose ever, indicating a CS-sparing effect (184).
In a randomized controlled trial of refractory AIM, rituximab given early failed to demonstrate any superiority versus rituximab given 2 months. However, 83% of all the patients improved during the trial after a median interval of 20 weeks (185). Rituximab has been included as a therapeutic option in an expert consensus for several refractory AIM subgroups (183, 186). Rituximab has recently demonstrated efficacy for skin and ILD treatment in SSc (187). This treatment was shown to be effective in SSc patients with muscle involvement in case series (188).
A randomized control trial failed to demonstrate efficacy of abatacept on skin thickening in patients with SSc (189), although recent analysis by intrinsic gene expression signatures seemed to show an effect in patients in the inflammatory subset (190). Preliminary results of a recent randomized control trial indicate efficacy of abatacept in a subgroup of patients with IMNM (191). In a case series of 7 scleromyositis patients with refractory muscle involvement treated with abatacept, after a follow-up of 18 months, the myopathy tended to improve as judged on the myopathy disease activity assessed by both patients (57/100 versus 19/100) and the physicians (28/100 vs 12/100), as well as on the median serum levels of CK (456 U/L, range 166–1800 versus 192, range 109–402). Yet, the differences were not statistically significant and muscle strength was unchanged (121).
Among patients with early dSSc, autologous HSCT has been associated with increased treatment-related mortality in the first year as compared to cyclophosphamide, but with significant long-term event-free survival benefit (192, 193). Efficacy on muscle involvement was not assessed. One patient developed myositis in the HSCT group versus none in the cyclophosphamide group (192). It is currently unknown whether HSCT may be a therapeutic option in fibrosing myopathy, a condition in which other treatments have been reported to fail in two thirds of the cases (27, 72).
We reviewed in-depth studies reporting muscle involvement in SSc.
Although the definition of scleromyositis in the literature is heterogeneous, our review provided evidence that an additional disease subgroup should be recognized within both the SSc and AIM spectrum that has been termed “scleromyositis” (20, 21, 63, 167). Specifically, recent data indicate that the hallmarks of scleromyositis extend far beyond an overlap of AIM and SSc. Additionally these data include characteristic extra-muscular clinical, serological and histopathological features (17, 18, 20, 21, 24–28, 63, 72, 82, 149, 167) supporting scleromyositis as a distinct entity having important implications for prognosis and management of patients (27, 29, 72, 82, 104) (Table 5; Figure 2).

Table 5 Distinguishing clinical, serological and pathological features of scleromyositis.
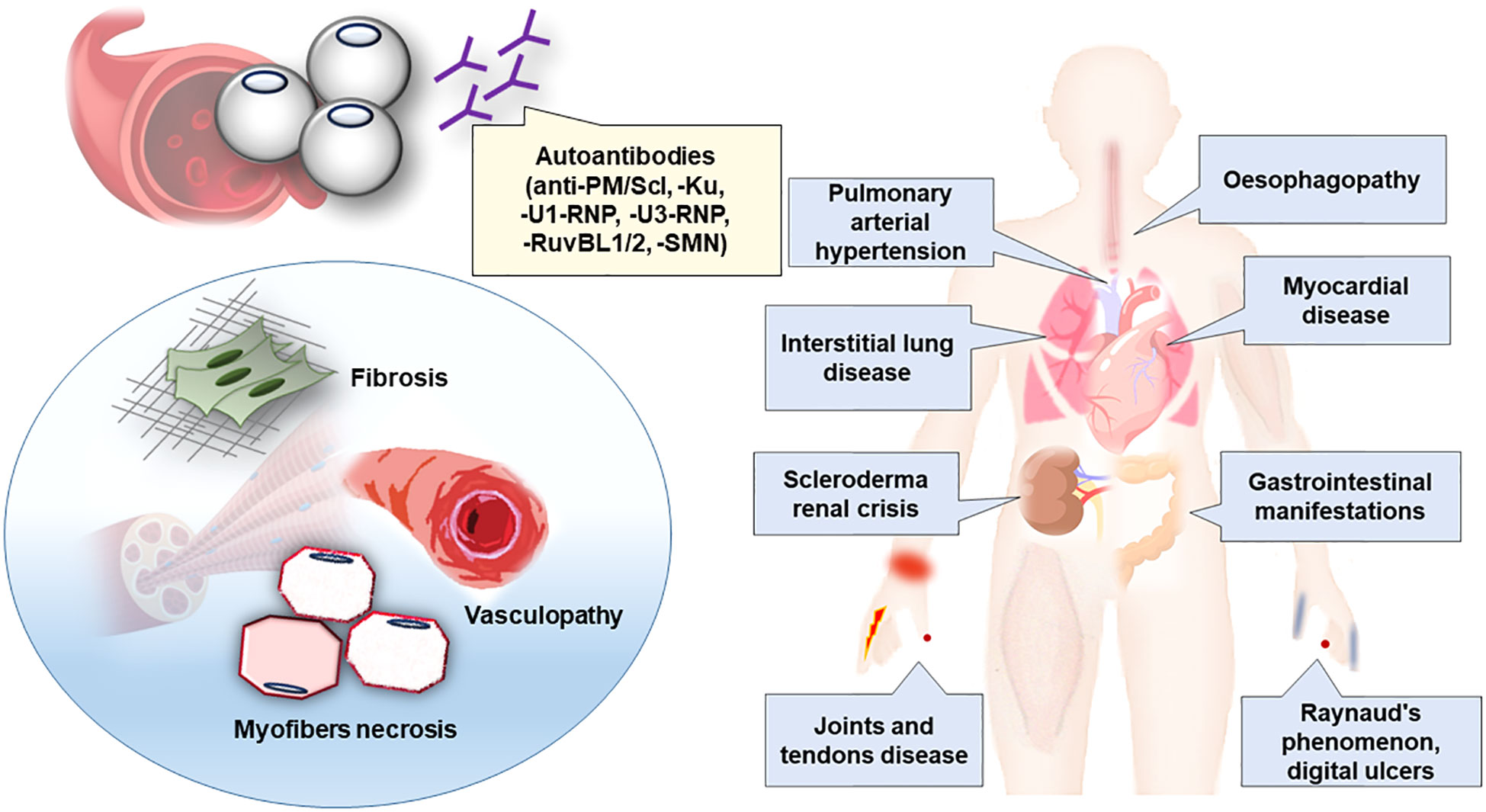
Figure 2 Clinico-sero-pathological definition of scleromyositis. Clinical phenotype, autoantibodies and main histopathological lesions associated to scleromyositis have been summarized.
Similarities and differences between scleromyositis and other AIM such as anti-SRP/-HMGCR IMNM and ASS must be emphasized in order not to confuse these conditions.
Although IMNM, as defined at the pathological level by the 2005 ENMC criteria (78), was the most common finding in muscle biopsy of scleromyositis patients, important differences were identified at the serological level by the more recent 2016 ENMC criteria (183). Muscle fiber necrosis has been associated in scleromyositis with anti-Ku, anti-PM/Scl and anti-U1-RNP autoantibodies whereas in IMNM fiber necrosis has been associated with anti-SRP and anti-HMGCR autoantibodies. Importantly, muscle biopsies of scleromyositis patients additionally display vasculopathy and fibrosis that are not features of anti-SRP and -HMGCR IMNM. Finally, extra-muscular features are frequent in scleromyositis, are linked to poor prognosis and require careful management while such features are exceptional in anti-SRP and anti-HMGCR IMNM patients. Interestingly, the two conditions are distinct even at the genetic level. Thus, anti-PM/Scl positive AIM has been linked with HLA-DQB1*02 (194). By contrast, anti-HMGCR -IMNM has been associated with HLA-DRB1*11 allele (194, 195) and no significant association with classical HLA alleles has been found for anti-SRP-IMNM.
Scleromyositis and ASS share some overlap clinical features, including ILD, arthritis, and Raynaud’s phenomenon (6, 12). Yet, the other clinical (i.e. mechanic’s hands and constitutional symptoms in ASS versus skin thickening, digital ulcers, telangiectasias, calcinosis, myocarditis and PAH in scleromyositis), serological (autoantibodies targeting aminoacyl-tRNA synthetases in ASS versus SSc-associated autoantibodies, as detailed above, in scleromyositis) and histopathological (perifascicular fibers necrosis in ASS versus necrotizing myopathy and vasculopathy in scleromyositis) (6, 20, 196) characteristics indicate that ASS and scleromyositis are two distinct entities. Consistently, at the genetic level, the HLA-B*0801 and the HLA-DRB1 03:01 alleles are associated with anti-Jo-1 ASS, while anti-PM/Scl scleromyositis is associated with HLA-DQB1 02:01 allele (194, 197).
Finally, overlaps between AIM and autoimmune diseases other than SSc (especially SLE and SjS) have also been reported (61, 198–204). Compared to scleromyositis, these associations are rarer. Yet, some clinical, histopathological, serological characteristics have been highlighted that have also implication for management and prognosis.
In patients with SLE, a biopsy proven myositis according to Bohan and Peter criteria (52, 53) has been found in only 3% of cases (201, 205). In contrast with scleromyositis patients, the frequency of ILD is lower in patients with SLE/myositis. On the other hand, the frequency of glomerulonephritis is higher (up to 39%), thus representing a hallmark life-threatening complication of this group (54, 200, 206). Anti-U1RNP are the most common antibody associated with SLE/myositis overlap syndrome (200, 201, 205, 207). Some SLE/myositis patients have anti-Ku antibodies (137, 150, 151, 200). In contrast with anti-U1RNP and anti-Ku scleromyositis patients, anti-U1RNP and anti-Ku SLE/myositis patients generally test positive for anti-dsDNA antibodies. This associations helps to predict whether lung (scleromyositis) or kidney (SLE/myositis) will drive prognosis (151, 199, 200, 206).
In patients with primary SjS, AIM is frequently suspected (10%), but rarely confirmed (1%) (61). More frequently, SjS is found in AIM (34% of patients) (198). When confirmed, AIM in SjS patients is more likely to be IBM whose diagnosis is fundamental since conventional immunomodulating agents are not effective and may even increase the risk of progression toward disability (208), while IBM-specific treatments may slowdown the progression of that disease (209). This association has been demonstrated both in patients with primary SjS (0.5% vs. 2.01/100 000 [95% CI 1.51-2.69] in the general population) (210) and in patients with AIM and secondary SjS (24% vs 6% in AIM patients without SjS) (61, 198, 211). Anti-cN1A is frequent in both SjS and IBM (212). Yet, in AIM patients, the association between anti-cN1A and SjS is independent of IBM raising caution about using anti-cN1A for the diagnosis of IBM in SjS patients (198, 211).
A systematic review was not performed because there is currently no consensual definition of scleromyositis. Thus, inclusion and exclusion criteria of studies selection were not strictly defined for the purpose of comprehensiveness. The literature research was performed without any restrictions in terms of publication period, type of study, nor setting. The key words were chosen to cover the entire scope of SSc and AIM. Our review revealed that SSc and AIM were classified using heterogeneous criteria, that probably accounts of the wide ranges in the reported prevalences.
In light of this review, Table 5 is proposed highlighting the distinguishing clinical, serological and pathological features of scleromyositis thus far.
Scleromyositis is an emerging entity in the SSc and AIM spectrum that is important for clinicians to recognize because it has many potential organ involvements (including lung, heart, gastrointestinal, skin and joint) and requires a tailored management different from AIM or SSc alone, especially regarding the risk of SRC upon CS exposure. Several autoantibodies and muscle histopathological findings are hallmarks of scleromyositis and, moreover, they assist in the prediction of extramuscular outcomes and response to treatment. An integrated clinico-sero-pathological approach is proposed (Figure 2) to recognize this novel subgroup.
MG: Data curation, Formal analysis, Investigation and Writing - original draft, Conceptualization, Methodology, Project administration, Supervision and Validation, Writing - review and editing. BE Conceptualization, Methodology, Writing - review and editing. VL: Methodology, Writing - review and editing. FL: Data curation, Formal analysis. YT: Writing - review and editing. MH: Conceptualization, Methodology, Supervision and Validation, Writing - review and editing. JL-S: Conceptualization, Methodology, Supervision and Validation, Writing - review and editing. BG: Writing - original draft, Project administration, Supervision and Validation, Writing - review and editing. OL-C: Conceptualization, Methodology, Supervision and Validation, Writing - review and editing. AM: Data curation, Formal analysis, Investigation and Writing - original draft, Conceptualization, Methodology, Project administration, Supervision and Validation, Writing - review and editing. All authors contributed to the article and approved the submitted version.
Supported by Sclérodermie Québec. J-LS holds the University of Montreal Scleroderma Research Chair.
The authors thank M.V.C. Pragnell, University of Bari, for linguistic help.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Hudson M, Fritzler MJ. Diagnostic criteria of systemic sclerosis. J Autoimmun (2014) 48–49:38–41. doi: 10.1016/j.jaut.2013.11.004
2. Hudson M, Thombs BD, Steele R, Panopalis P, Newton E, Baron M, et al. Health-related quality of life in systemic sclerosis: a systematic review. Arthritis Rheumatol (2009) 61(8):1112–20. doi: 10.1002/art.24676
3. Leclair V, Regardt M, Wojcik S, Hudson M, Canadian Inflammatory Myopathy Study (CIMS). Health-related quality of life (HRQoL) in idiopathic inflammatory myopathy: A systematic review. PloS One (2016) 11(8):e0160753. doi: 10.1371/journal.pone.0160753
4. Dobloug GC, Svensson J, Lundberg IE, Holmqvist M. Mortality in idiopathic inflammatory myopathy: Results from a Swedish nationwide population-based cohort study. Ann Rheum Dis (2018) 77(1):40–7. doi: 10.1136/annrheumdis-2017-211402
5. Pokeerbux MR, Giovannelli J, Dauchet L, Mouthon L, Agard C, Lega JC, et al. Survival and prognosis factors in systemic sclerosis: Data of a French multicenter cohort, systematic review, and meta-analysis of the literature. Arthritis Res Ther (2019) 21(1):86. doi: 10.1186/s13075-019-1867-1
6. Meyer A, Lannes B, Goetz J, Echaniz-Laguna A, Lipsker D, Arnaud L, et al. Inflammatory myopathies: A new landscape. Joint Bone Spine. (2018) 85(1):23–33. doi: 10.1016/j.jbspin.2017.03.005
7. van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, et al. Classification criteria for systemic sclerosis: An American college of rheumatology/European league against rheumatism collaborative initiative. Ann Rheum Dis (2013) 72(11):1747–55. doi: 10.1136/annrheumdis-2013-204424
8. Mariampillai K, Granger B, Amelin D, Guiguet M, Hachulla E, Maurier F, et al. Development of a new classification system for idiopathic inflammatory myopathies based on clinical manifestations and myositis-specific autoantibodies. JAMA Neurol (2018) 75(12):1528. doi: 10.1001/jamaneurol.2018.2598
9. Loarce-Martos J, Lilleker JB, Parker M, McHugh N, Chinoy H. Polymyositis: Is there anything left? a retrospective diagnostic review from a tertiary myositis centre. Rheumatology. (2021) 60(7):3398–403. doi: 10.1093/rheumatology/keaa801
10. Amato AA, Griggs RC. Unicorns, dragons, polymyositis, and other mythological beasts. Neurology. (2003) 61(3):288–9. doi: 10.1212/WNL.61.3.288
11. Cavagna L, Trallero-Araguás E, Meloni F, Cavazzana I, Rojas-Serrano J, Feist E, et al. Influence of antisynthetase antibodies specificities on antisynthetase syndrome clinical spectrum time course. J Clin Med (2019) 8(11):E2013. doi: 10.3390/jcm8112013
12. Troyanov Y, Targoff IN, Tremblay JL, Goulet JR, Raymond Y, Senécal JL. Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: Analysis of 100 French Canadian patients. Medicine. (2005) 84(4):231–49. doi: 10.1097/01.md.0000173991.74008.b0
13. Váncsa A, Gergely L, Ponyi A, Lakos G, Németh J, Szodoray P, et al. Myositis-specific and myositis-associated antibodies in overlap myositis in comparison to primary dermatopolymyositis: Relevance for clinical classification: Retrospective study of 169 patients. Joint Bone Spine. (2010) 77(2):125–30. doi: 10.1016/j.jbspin.2009.08.008
14. Wolfe JF, Adelstein E, Sharp GC. Antinuclear antibody with distinct specificity for polymyositis. J Clin Invest. (1977) 59(1):176–8. doi: 10.1172/JCI108616
15. Hausmanowa-Petrusewicz I, Kowalska-Oledzka E, Miller FW, Jarzabek-Chorzelska M, Targoff IN, Blaszczyk-Kostanecka M, et al. Clinical, serologic, and immunogenetic features in polish patients with idiopathic inflammatory myopathies. Arthritis Rheumatol (1997) 40(7):1257–66. doi: 10.1002/1529-0131(199707)40:7<1257::AID-ART10>3.0.CO;2-R
16. Lundberg IE, Tjärnlund A, Bottai M, Werth VP, Pilkington C, de Visser M, et al. European League against Rheumatism/American college of rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Ann Rheum Dis (2017) 76(12):1955–64. doi: 10.1136/annrheumdis-2017-211468
17. Bhansing KJ, Lammens M, Knaapen HKA, van Riel PLCM, van Engelen BGM, Vonk MC. Scleroderma-polymyositis overlap syndrome versus idiopathic polymyositis and systemic sclerosis: A descriptive study on clinical features and myopathology. Arthritis Res Ther (2014) 16(3):R111. doi: 10.1186/ar4562
18. Pakozdi A, Nihtyanova S, Moinzadeh P, Ong VH, Black CM, Denton CP. Clinical and serological hallmarks of systemic sclerosis overlap syndromes. J Rheumatol (2011) 38(11):2406–9. doi: 10.3899/jrheum.101248
19. Parker MJS, Oldroyd A, Roberts ME, Lilleker JB, Betteridge ZE, McHugh NJ, et al. The performance of the European league against Rheumatism/American college of rheumatology idiopathic inflammatory myopathies classification criteria in an expert-defined 10 year incident cohort. Rheumatol (Oxford). (2019) 58(3):468–75. doi: 10.1093/rheumatology/key343
20. Ellezam B, Leclair V, Troyanov Y, Bersali I, Giannini M, Hoa S, et al. Capillary pathology with prominent basement membrane reduplication is the hallmark histopathological feature of scleromyositis. Neuropathol Appl Neurobiol (2022) 48(7):e12840. doi: 10.1111/nan.12840
21. Leclair V, D’Aoust J, Gyger G, Landon-Cardinal O, Meyer A, O’Ferrall E, et al. Autoantibody profiles delineate distinct subsets of scleromyositis. Rheumatol (Oxford). (2022) 61(3):1148–57. doi: 10.1093/rheumatology/keab492
22. Meyer A, Leclair V, Landon-Cardinal O, Ellezam B, D'Aoust J, Giannini M, et al. ACR/EULAR criteria for myositis and systemic sclerosis lack sensitivity for scleromyositis [abstract]. Arthritis Rheumatol (2019) 71(suppl 10).
23. Muangchan C, Canadian Scleroderma Research Group, Baron M, Pope J. The 15% rule in scleroderma: The frequency of severe organ complications in systemic sclerosis. A systematic review. J Rheumatol (2013) 40(9):1545–56. doi: 10.3899/jrheum.121380
24. Corallo C, Cutolo M, Volpi N, Franci D, Aglianò M, Montella A, et al. Histopathological findings in systemic sclerosis-related myopathy: Fibrosis and microangiopathy with lack of cellular inflammation. Ther Adv Musculoskeletal. (2017) 9(1):3–10. doi: 10.1177/1759720X16671928
25. Siegert E, Uruha A, Goebel HH, Preuße C, Casteleyn V, Kleefeld F, et al. Systemic sclerosis-associated myositis features minimal inflammation and characteristic capillary pathology. Acta Neuropathol. (2021) 141(6):917–27. doi: 10.1007/s00401-021-02305-3
26. Follansbee WP, Zerbe TR, Medsger TA. Cardiac and skeletal muscle disease in systemic sclerosis (scleroderma): A high risk association. Am Heart J (1993) 125(1):194–203. doi: 10.1016/0002-8703(93)90075-K
27. Ranque B, Authier FJ, Le-Guern V, Pagnoux C, Berezne A, Allanore Y, et al. A descriptive and prognostic study of systemic sclerosis-associated myopathies. Ann Rheum Dis (2009) 68(9):1474–7. doi: 10.1136/ard.2008.095919
28. Ranque B, Bérezné A, Le-Guern V, Pagnoux C, Allanore Y, Launay D, et al. Myopathies related to systemic sclerosis: a case-control study of associated clinical and immunological features. Scand J Rheumatol (2010) 39(6):498–505. doi: 10.3109/03009741003774626
29. Jung M, Bonner A, Hudson M, Baron M, Pope JE, Canadian Scleroderma Research Group (CSRG). Myopathy is a poor prognostic feature in systemic sclerosis: Results from the Canadian scleroderma research group (CSRG) cohort. Scand J Rheumatol (2014) 43(3):217–20. doi: 10.3109/03009742.2013.868512
30. Medsger TA, Rodnan GP, Moossy J, Vester JW. Skeletal muscle involvement in progressive systemic sclerosis (scleroderma). Arthritis Rheumatol (1968) 11(4):554–68. doi: 10.1002/art.1780110405
31. Thompson JM, Bluestone R, Bywaters EG, Dorling J, Johnson M. Skeletal muscle involvement in systemic sclerosis. Ann Rheum Dis (1969) 28(3):281–8. doi: 10.1136/ard.28.3.281
32. Clements PJ, Furst DE, Campion DS, Bohan A, Harris R, Levy J, et al. Muscle disease in progressive systemic sclerosis: diagnostic and therapeutic considerations. Arthritis Rheumatol (1978) 21(1):62–71. doi: 10.1002/art.1780210111
33. Hausmanowa-Petrusewicz I, Jabłońska S, Błaszczyk M, Matz B. Electromyographic findings in various forms of progressive systemic sclerosis. Arthritis Rheumatol (1982) 25(1):61–5. doi: 10.1002/art.1780250110
34. West SG, Killian PJ, Lawless OJ. Association of myositis and myocarditis in progressive systemic sclerosis. Arthritis Rheumatol (1981) 24(5):662–8. doi: 10.1002/art.1780240506
35. Averbuch-Heller L, Steiner I, Abramsky O. Neurologic manifestations of progressive systemic sclerosis. Arch Neurol (1992) 49(12):1292–5. doi: 10.1001/archneur.1992.00530360094024
36. Tuffanelli DL, Winkelmann RK. Systemic scleroderma, a clinical study of 727 cases. Arch Dermatol (1961) 84:359–71. doi: 10.1001/archderm.1961.01580150005001
37. Baumberger R, Jordan S, Distler O, Baschung Pfister P, Maurer B. Diagnostic measures for patients with systemic sclerosis-associated myopathy. Clin Exp Rheumatol (2021) 39(Suppl 131(4):85–93. doi: 10.55563/clinexprheumatol/tt4ilu
38. Hietaharju A, Jääskeläinen S, Kalimo H, Hietarinta M. Peripheral neuromuscular manifestations in systemic sclerosis (scleroderma). Muscle Nerve. (1993) 16(11):1204–12. doi: 10.1002/mus.880161110
39. Mammen AL. Skeletal muscle involvement. In: Varga J, Denton CP, Wigley FM, editors. Scleroderma: From pathogenesis to comprehensive management. New York City: Springer (2011). p. 525.
40. Meier FMP, Frommer KW, Dinser R, Walker UA, Czirjak L, Denton CP, et al. Update on the profile of the EUSTAR cohort: An analysis of the EULAR scleroderma trials and research group database. Ann Rheum Dis (2012) 71(8):1355–60. doi: 10.1136/annrheumdis-2011-200742
41. Mimori T. Scleroderma-polymyositis overlap syndrome. clinical and serologic aspects. Int J Dermatol (1987) 26(7):419–25. doi: 10.1111/j.1365-4362.1987.tb00580.x
42. Mimura Y, Ihn H, Jinnin M, Asano Y, Yamane K, Tamaki K. Clinical and laboratory features of scleroderma patients developing skeletal myopathy. Clin Rheumatol (2005) 24(2):99–102. doi: 10.1007/s10067-004-0975-7
43. Russell ML, Hanna WM. Ultrastructure of muscle microvasculature in progressive systemic sclerosis: Relation to clinical weakness. J Rheumatol (1983) 10(5):741–7.
44. Walker UA, Tyndall A, Czirják L, Denton C, Farge-Bancel D, Kowal-Bielecka O, et al. Clinical risk assessment of organ manifestations in systemic sclerosis: a report from the EULAR scleroderma trials and research group database. Ann Rheum Dis (2007) 66(6):754–63. doi: 10.1136/ard.2006.062901
45. Tolédano C, Gain M, Kettaneh A, Baudin B, Johanet C, Chérin P, et al. Aldolase predicts subsequent myopathy occurrence in systemic sclerosis. Arthritis Res Ther (2012) 14(3):R152. doi: 10.1186/ar3888
46. Schanz S, Henes J, Ulmer A, Kötter I, Fierlbeck G, Claussen CD, et al. Magnetic resonance imaging findings in patients with systemic scleroderma and musculoskeletal symptoms. Eur Radiol (2013) 23(1):212–21. doi: 10.1007/s00330-012-2584-1
47. Ross L, Lindqvist A, Costello B, Hansen D, Brown Z, Day JA, et al. Using magnetic resonance imaging to map the hidden burden of muscle involvement in systemic sclerosis. Arthritis Res Ther (2022) 24(1):84. doi: 10.1186/s13075-022-02768-z
48. Zhou M, Jiang L, Nie L, Chen T, Zhang T, Sun W, et al. Myopathy is a risk factor for poor prognosis of patients with systemic sclerosis: A retrospective cohort study. Med (Baltimore). (2020) 99(33):e21734. doi: 10.1097/MD.0000000000021734
49. Paik JJ. Myopathy in scleroderma and in other connective tissue diseases. Curr Opin Rheumatol (2016) 28(6):631–5. doi: 10.1097/BOR.0000000000000336
50. Preliminary criteria for the classification of systemic sclerosis (scleroderma). subcommittee for scleroderma criteria of the American rheumatism association diagnostic and therapeutic criteria committee. Arthritis Rheum (1980) 23(5):581–90. doi: 10.1002/art.1780230510
51. LeRoy EC, Medsger TA. Criteria for the classification of early systemic sclerosis. J Rheumatol (2001) 28(7):1573–6.
52. Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med (1975) 292(7):344–7. doi: 10.1056/NEJM197502132920706
53. Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts). N Engl J Med (1975) 292(8):403–7. doi: 10.1056/NEJM197502202920807
54. Aguila LA, Lopes MRU, Pretti FZ, Sampaio-Barros PD, Carlos de Souza FH, Borba EF, et al. Clinical and laboratory features of overlap syndromes of idiopathic inflammatory myopathies associated with systemic lupus erythematosus, systemic sclerosis, or rheumatoid arthritis. Clin Rheumatol (2014) 33(8):1093–8. doi: 10.1007/s10067-014-2730-z
55. Clements PJ, Furst DE, Wong WK, Mayes M, White B, Wigley F, et al. High-dose versus low-dose d-penicillamine in early diffuse systemic sclerosis: analysis of a two-year, double-blind, randomized, controlled clinical trial. Arthritis Rheumatol (1999) 42(6):1194–203. doi: 10.1002/1529-0131(199906)42:6<1194::AID-ANR16>3.0.CO;2-7
56. Paik JJ, Wigley FM, Mejia AF, Hummers LK. Independent association of severity of muscle weakness with disability as measured by the health assessment questionnaire disability index in scleroderma. Arthritis Care Res (Hoboken). (2016) 68(11):1695–703. doi: 10.1002/acr.22870
57. De Lorenzo R, Pinal-Fernandez I, Huang W, Albayda J, Tiniakou E, Johnson C, et al. Muscular and extramuscular clinical features of patients with anti-PM/Scl autoantibodies. Neurology. (2018) 90(23):e2068–76. doi: 10.1212/WNL.0000000000005638
58. Ringel RA, Brick JE, Brick JF, Gutmann L, Riggs JE. Muscle involvement in the scleroderma syndromes. Arch Intern Med (1990) 150(12):2550–2. doi: 10.1001/archinte.1990.00390230098013
59. Matas-García A, Guillén-Del-Castillo A, Kisluk B, Selva-O’Callaghan A, Espinosa G, Prieto-González S, et al. Clinico-pathological phenotypes of systemic sclerosis associated myopathy: Analysis of a multicenter large cohort. Rheumatol (Oxford) (2022) 17:keac361. doi: 10.1093/rheumatology/keac361
60. Dieudonné Y, Allenbach Y, Benveniste O, Leonard-Louis S, Hervier B, Mariampillai K, et al. Granulomatosis-associated myositis: High prevalence of sporadic inclusion body myositis. Neurology. (2020) 94(9):e910–20. doi: 10.1212/WNL.0000000000008863
61. Felten R, Giannini M, Nespola B, Lannes B, Levy D, Seror R, et al. Refining myositis associated with primary sjögren’s syndrome: Data from the prospective cohort ASSESS. Rheumatol (Oxford). (2021) 60(2):675–81. doi: 10.1093/rheumatology/keaa257
62. Partovi S, Schulte AC, Aschwanden M, Staub D, Benz D, Imfeld S, et al. Impaired skeletal muscle microcirculation in systemic sclerosis. Arthritis Res Ther (2012) 14(5):R209. doi: 10.1186/ar4047
63. Ellezam B, Leclair V, Troyanov Y, Meyer A, Hudson M, Landon-Cardinal O. Capillary basement membrane reduplication in myositis patients with mild clinical features of systemic sclerosis supports the concept of “scleromyositis.” Acta Neuropathol. (2021) 142(2):395–7. doi: 10.1007/s00401-021-02335-x
64. Paik JJ, Wigley FM, Lloyd TE, Corse AM, Casciola-Rosen L, Shah AA, et al. Spectrum of muscle histopathologic findings in forty-two scleroderma patients with weakness: Muscle histopathologic findings in SSc patients with weakness. Arthritis Care Res (2015) 67(10):1416–25. doi: 10.1002/acr.22620
65. Fernández-Serna M, Arboleya L, Alonso S, Queiro R, Alperi M. Dropped head syndrome in a patient with scleromyositis. J Clin Rheumatol (2013) 19(1):32–4. doi: 10.1097/RHU.0b013e31827d8778
66. Garcin B, Lenglet T, Dubourg O, Mesnage V, Levy R. Dropped head syndrome as a presenting sign of scleromyositis. J Neurol Sci (2010) 292(1–2):101–3. doi: 10.1016/j.jns.2010.02.015
67. Rojana-Udomsart A, Fabian V, Hollingsworth PN, Walters SE, Zilko PJ, Mastaglia FL. Paraspinal and scapular myopathy associated with scleroderma. J Clin Neuromuscul Dis (2010) 11(4):213–22. doi: 10.1097/CND.0b013e3181c139f6
68. Rosato E, Rossi C, Salsano F. Dropped head syndrome and systemic sclerosis. Joint Bone Spine. (2009) 76(3):301–3. doi: 10.1016/j.jbspin.2008.10.013
69. Spielmann L, Séverac F, Meyer A. Response to: “Anti-Ku syndrome with elevated CK: Association with myocardial involvement in systemic sclerosis” by campochiaro et al. Ann Rheum Dis (2021) 80(7):e114. doi: 10.1136/annrheumdis-2019-216095
70. Hughes M, Lilleker JB, Herrick AL, Chinoy H. Cardiac troponin testing in idiopathic inflammatory myopathies and systemic sclerosis-spectrum disorders: Biomarkers to distinguish between primary cardiac involvement and low-grade skeletal muscle disease activity. Ann Rheum Dis (2015) 74(5):795–8. doi: 10.1136/annrheumdis-2014-206812
71. Haschek WM, Rousseaux CG, Wallig MA, Bolon B, Ochoa R. eds. Haschek and rousseaux’s handbook of toxicologic pathology, 3rd ed., Vol. III (2013). pp. 1567–665. Elsevier Inc.
72. Paik JJ, Wigley FM, Shah AA, Corse AM, Casciola-Rosen L, Hummers LK, et al. Association of fibrosing myopathy in systemic sclerosis and higher mortality. Arthritis Care Res (Hoboken). (2017) 69(11):1764–70. doi: 10.1002/acr.23291
73. Maurer B, Walker UA. Role of MRI in diagnosis and management of idiopathic inflammatory myopathies. Curr Rheumatol Rep (2015) 17(11):67. doi: 10.1007/s11926-015-0544-x
74. Marago I, Roberts M, Roncaroli F, DuPlessis D, Sewry C, Nagaraju S, et al. Limb girdle muscular dystrophy R12 (LGMD 2L, anoctaminopathy) mimicking idiopathic inflammatory myopathy: Key points to prevent misdiagnosis. Rheumatology. (2022) 61(4):1645–50. doi: 10.1093/rheumatology/keab553
75. Schulze M, Kötter I, Ernemann U, Fenchel M, Tzaribatchev N, Claussen CD, et al. MRI Findings in inflammatory muscle diseases and their noninflammatory mimics. AJR Am J Roentgenol. (2009) 192(6):1708–16. doi: 10.2214/AJR.08.1764
76. Boutry N, Hachulla E, Zanetti-Musielak C, Morel M, Demondion X, Cotten A. Imaging features of musculoskeletal involvement in systemic sclerosis. Eur Radiol (2007) 17(5):1172–80. doi: 10.1007/s00330-006-0420-1
77. Olivier PA, De Paepe B, Aronica E, Berfelo F, Colman R, Amato A, et al. Idiopathic inflammatory myopathy: Interrater variability in muscle biopsy reading. Neurology. (2019) 93(9):e889–94. doi: 10.1212/WNL.0000000000008005
78. Hoogendijk JE, Amato AA, Lecky BR, Choy EH, Lundberg IE, Rose MR, et al. 119th ENMC international workshop: Trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10–12 October 2003, naarden, the Netherlands. Neuromuscular Disord (2004) 14(5):337–45. doi: 10.1016/j.nmd.2004.02.006
79. Lefebvre F, Giannini M, Ellezam B, Leclair V, Troyanov Y, Hoa S, et al. Histopathological features of systemic sclerosis-associated myopathy: A scoping review. Autoimmun Rev (2021) 20(7):102851. doi: 10.1016/j.autrev.2021.102851
80. Fleischmajer R, Perlish JS. Capillary alterations in scleroderma. J Am Acad Dermatol (1980) 2(2):161–70. doi: 10.1016/S0190-9622(80)80396-3
81. Romano E, Rosa I, Fioretto BS, Matucci-Cerinic M, Manetti M. New insights into profibrotic myofibroblast formation in systemic sclerosis: When the vascular wall becomes the enemy. Life (Basel). (2021) 11(7):610. doi: 10.3390/life11070610
82. Bhansing KJ, van Riel PLCM, van Engelen BGM, Fransen J, Vonk MC. Patients with systemic sclerosis/polymyositis overlap have a worse survival rate than patients without it. J Rheumatol (2016) 43(10):1838–43. doi: 10.3899/jrheum.151425
83. Tyndall AJ, Bannert B, Vonk M, Airo P, Cozzi F, Carreira PE, et al. Causes and risk factors for death in systemic sclerosis: A study from the EULAR scleroderma trials and research (EUSTAR) database. Ann Rheumatic Diseases. (2010) 69(10):1809–15. doi: 10.1136/ard.2009.114264
84. Qiu M, Nian X, Pang L, Yu P, Zou S. Prevalence and risk factors of systemic sclerosis-associated interstitial lung disease in East Asia: A systematic review and meta-analysis. Int J Rheum Dis (2021) 24(12):1449–59. doi: 10.1111/1756-185X.14206
85. Steen VD, Conte C, Owens GR, Medsger TA. Severe restrictive lung disease in systemic sclerosis. Arthritis Rheumatism. (1994) 37(9):1283–9. doi: 10.1002/art.1780370903
86. Schurawitzki H, Stiglbauer R, Graninger W, Herold C, Pölzleitner D, Burghuber OC, et al. Interstitial lung disease in progressive systemic sclerosis: high-resolution CT versus radiography. Radiology. (1990) 176(3):755–9. doi: 10.1148/radiology.176.3.2389033
87. Allanore Y, Constans J, Godard D, de Pouvourville G, Bouee S, Jeanbat V, et al. Quality of life in SSc-ILD patients: Understanding the impact of the ILD and the needs of the SSc-ILD patients and their need for caregivers in France. J Scleroderma Related Disord (2022) 7(1):49–56. doi: 10.1177/23971983211013979
88. Iliffe GD, Pettigrew NM. Hypoventilatory respiratory failure in generalised scleroderma. Br Med J (Clin Res Ed) (1983) 286(6362):337–8. doi: 10.1136/bmj.286.6362.337
89. McCarthy DS, Baragar FD, Dhingra S, Sigurdson M, Sutherland JB, Rigby M, et al. The lungs in systemic sclerosis (scleroderma): a review and new information. Semin Arthritis Rheumatol (1988) 17(4):271–83. doi: 10.1016/0049-0172(88)90012-1
90. Nawata T, Shirai Y, Suzuki M, Kuwana M. Chest wall muscle atrophy as a contributory factor for forced vital capacity decline in systemic sclerosis-associated interstitial lung disease. Rheumatol (Oxford). (2021) 60(1):250–5. doi: 10.1093/rheumatology/keaa322
91. Lazzaroni MG, Marasco E, Campochiaro C, DeVries-Bouwstra J, Gonzalez-Perez MI, Rojas-Serrano J, et al. The clinical phenotype of systemic sclerosis patients with anti-PM/Scl antibodies: Results from the EUSTAR cohort. Rheumatol (Oxford). (2021) 60(11):5028–41. doi: 10.1093/rheumatology/keab152
92. Cheng CY, Baritussio A, Giordani AS, Iliceto S, Marcolongo R, Caforio ALP. Myocarditis in systemic immune-mediated diseases: Prevalence, characteristics and prognosis. A systematic review. Autoimmun Rev (2022) 21(4):103037. doi: 10.1016/j.autrev.2022.103037
93. Hachulla AL, Launay D, Gaxotte V, de Groote P, Lamblin N, Devos P, et al. Cardiac magnetic resonance imaging in systemic sclerosis: a cross-sectional observational study of 52 patients. Ann Rheum Dis (2009) 68(12):1878–84. doi: 10.1136/ard.2008.095836
94. Bezante GP, Rollando D, Sessarego M, Panico N, Setti M, Filaci G, et al. Cardiac magnetic resonance imaging detects subclinical right ventricular impairment in systemic sclerosis. J Rheumatol (2007) 34(12):2431–7.
95. Gargani L, Todiere G, Guiducci S, Bruni C, Pingitore A, De Marchi D, et al. Early detection of cardiac involvement in systemic sclerosis: The added value of magnetic resonance imaging. JACC Cardiovasc Imaging. (2019) 12(5):927–8. doi: 10.1016/j.jcmg.2018.09.025
96. Krumm P, Mueller KAL, Klingel K, Kramer U, Horger MS, Zitzelsberger T, et al. Cardiovascular magnetic resonance patterns of biopsy proven cardiac involvement in systemic sclerosis. J Cardiovasc Magn Reson (2016) 18(1):70. doi: 10.1186/s12968-016-0289-3
97. Markousis-Mavrogenis G, Bournia VK, Panopoulos S, Koutsogeorgopoulou L, Kanoupakis G, Apostolou D, et al. Cardiovascular magnetic resonance identifies high-risk systemic sclerosis patients with normal echocardiograms and provides incremental prognostic value. Diagnostics. (2019) 9(4):220. doi: 10.3390/diagnostics9040220
98. Mavrogeni SI, Schwitter J, Gargani L, Pepe A, Monti L, Allanore Y, et al. Cardiovascular magnetic resonance in systemic sclerosis: “Pearls and pitfalls.” Semin Arthritis Rheumatism. (2017) 47(1):79–85. doi: 10.1016/j.semarthrit.2017.03.020
99. Ntusi NAB, Piechnik SK, Francis JM, Ferreira VM, Rai ABS, Matthews PM, et al. Subclinical myocardial inflammation and diffuse fibrosis are common in systemic sclerosis–a clinical study using myocardial T1-mapping and extracellular volume quantification. J Cardiovasc Magn Reson (2014) 16:21. doi: 10.1186/1532-429X-16-21
100. Allanore Y, Meune C, Vonk MC, Airo P, Hachulla E, Caramaschi P, et al. Prevalence and factors associated with left ventricular dysfunction in the EULAR scleroderma trial and research group (EUSTAR) database of patients with systemic sclerosis. Ann Rheum Dis (2010) 69(1):218–21. doi: 10.1136/ard.2008.103382
101. Kerr LD, Spiera H. Myocarditis as a complication in scleroderma patients with myositis. Clin Cardiol (1993) 16(12):895–9. doi: 10.1002/clc.4960161212
102. Tager RE, Tikly M. Clinical and laboratory manifestations of systemic sclerosis (scleroderma) in black south africans. Rheumatol (Oxford). (1999) 38(5):397–400. doi: 10.1093/rheumatology/38.5.397
103. Hudson M. Scleroderma renal crisis. Curr Opin Rheumatol (2015) 27(6):549–54. doi: 10.1097/BOR.0000000000000221
104. Iudici M, van der Goes MC, Valentini G, Bijlsma JWJ. Glucocorticoids in systemic sclerosis: Weighing the benefits and risks - a systematic review. Clin Exp Rheumatol (2013) 31(2 Suppl 76):157–65.
105. Franck-Larsson K, Graf W, Rönnblom A. Lower gastrointestinal symptoms and quality of life in patients with systemic sclerosis: S population-based study. Eur J Gastroenterol Hepatol (2009) 21(2):176–82. doi: 10.1097/MEG.0b013e32831dac75
106. Sampaio-Barros PD, Bortoluzzo AB, Marangoni RG, Rocha LF, Del Rio APT, Samara AM, et al. Survival, causes of death, and prognostic factors in systemic sclerosis: analysis of 947 Brazilian patients. J Rheumatol (2012) 39(10):1971–8. doi: 10.3899/jrheum.111582
107. Li B, Yan J, Pu J, Tang J, Xu S, Wang X. Esophageal dysfunction in systemic sclerosis: An update. Rheumatol Ther (2021) 8(4):1535–49. doi: 10.1007/s40744-021-00382-0
108. Tétreault MP, Kahrilas P. GI manifestations with a focus on the esophagus: Recent progress in understanding pathogenesis. Curr Rheumatol Rep (2019) 21(8):42. doi: 10.1007/s11926-019-0841-x
109. Sakkas LI, Simopoulou T, Daoussis D, Liossis SN, Potamianos S. Intestinal involvement in systemic sclerosis: A clinical review. Dig Dis Sci (2018) 63(4):834–44. doi: 10.1007/s10620-018-4977-8
110. Hunzelmann N, Genth E, Krieg T, Lehmacher W, Melchers I, Meurer M, et al. The registry of the German network for systemic scleroderma: frequency of disease subsets and patterns of organ involvement. Rheumatol (Oxford). (2008) 47(8):1185–92. doi: 10.1093/rheumatology/ken179
111. Hyphantis TN, Tsifetaki N, Siafaka V, Voulgari PV, Pappa C, Bai M, et al. The impact of psychological functioning upon systemic sclerosis patients’ quality of life. Semin Arthritis Rheumatol (2007) 37(2):81–92. doi: 10.1016/j.semarthrit.2007.03.008
112. Avouac J, Walker UA, Hachulla E, Riemekasten G, Cuomo G, Carreira PE, et al. Joint and tendon involvement predict disease progression in systemic sclerosis: a EUSTAR prospective study. Ann Rheum Dis (2016) 75(1):103–9. doi: 10.1136/annrheumdis-2014-205295
113. Herrick AL, Assassi S, Denton CP. Skin involvement in early diffuse cutaneous systemic sclerosis: An unmet clinical need. Nat Rev Rheumatol (2022) 18(5):276–285. doi: 10.1038/s41584-022-00765-9
114. Peytrignet S, Manning J, Wragg E, Moore T, Samaranayaka M, Dinsdale G, et al. Changes in disability and their relationship with skin thickening, in diffuse and limited cutaneous systemic sclerosis: A retrospective cohort study. Scandinavian J Rheumatol (2019) 48(3):230–4. doi: 10.1080/03009742.2018.1523455
115. Zheng B, Nevskaya T, Baxter CA, Ramey DR, Pope JE, Baron M, et al. Changes in skin score in early diffuse cutaneous systemic sclerosis are associated with changes in global disease severity. Rheumatol (Oxford). (2020) 59(2):398–406. doi: 10.1093/rheumatology/kez299
116. Clements PJ, Hurwitz EL, Wong WK, Seibold JR, Mayes M, White B, et al. Skin thickness score as a predictor and correlate of outcome in systemic sclerosis: high-dose versus low-dose penicillamine trial. Arthritis Rheumatol (2000) 43(11):2445–54. doi: 10.1002/1529-0131(200011)43:11<2445::AID-ANR11>3.0.CO;2-Q
117. Ledoult E, Launay D, Béhal H, Mouthon L, Pugnet G, Lega JC, et al. Early trajectories of skin thickening are associated with severity and mortality in systemic sclerosis. Arthritis Res Ther (2020) 22(1):30. doi: 10.1186/s13075-020-2113-6
118. Shand L, Lunt M, Nihtyanova S, Hoseini M, Silman A, Black CM, et al. Relationship between change in skin score and disease outcome in diffuse cutaneous systemic sclerosis: application of a latent linear trajectory model. Arthritis Rheumatol (2007) 56(7):2422–31. doi: 10.1002/art.22721
119. Nevskaya T, Zheng B, Baxter CA, Ramey DR, Pope JE, Baron M, et al. Skin improvement is a surrogate for favourable changes in other organ systems in early diffuse cutaneous systemic sclerosis. Rheumatol (Oxford). (2020) 59(7):1715–24. doi: 10.1093/rheumatology/kez529
120. Steen VD. Autoantibodies in systemic sclerosis. Semin Arthritis Rheumatol (2005) 35(1):35–42. doi: 10.1016/j.semarthrit.2005.03.005
121. Elhai M, Meunier M, Matucci-Cerinic M, Maurer B, Riemekasten G, Leturcq T, et al. Outcomes of patients with systemic sclerosis-associated polyarthritis and myopathy treated with tocilizumab or abatacept: a EUSTAR observational study. Ann Rheum Dis (2013) 72(7):1217–20. doi: 10.1136/annrheumdis-2012-202657
122. D’Aoust J, Hudson M, Tatibouet S, Wick J, Canadian Scleroderma Research Group, Mahler M, et al. Clinical and serologic correlates of anti-PM/Scl antibodies in systemic sclerosis: A multicenter study of 763 patients. Arthritis Rheumatol (2014) 66(6):1608–15. doi: 10.1002/art.38428
123. Graf SW, Hakendorf P, Lester S, Patterson K, Walker JG, Smith MD, et al. South Australian scleroderma register: autoantibodies as predictive biomarkers of phenotype and outcome. Int J Rheum Dis (2012) 15(1):102–9. doi: 10.1111/j.1756-185X.2011.01688.x
124. Hanke K, Brückner CS, Dähnrich C, Huscher D, Komorowski L, Meyer W, et al. Antibodies against PM/Scl-75 and PM/Scl-100 are independent markers for different subsets of systemic sclerosis patients. Arthritis Res Ther (2009) 11(1):R22. doi: 10.1186/ar2614
125. Kuwana M, Kaburaki J, Okano Y, Tojo T, Homma M. Clinical and prognostic associations based on serum antinuclear antibodies in Japanese patients with systemic sclerosis. Arthritis Rheumatol (1994) 37(1):75–83. doi: 10.1002/art.1780370111
126. Kuwana M, Okano Y, Kaburaki J, Tojo T, Medsger TA. Racial differences in the distribution of systemic sclerosis-related serum antinuclear antibodies. Arthritis Rheumatol (1994) 37(6):902–6. doi: 10.1002/art.1780370619
127. Mahler M, Raijmakers R, Dähnrich C, Blüthner M, Fritzler MJ. Clinical evaluation of autoantibodies to a novel PM/Scl peptide antigen. Arthritis Res Ther (2005) 7(3):R704–713. doi: 10.1186/ar1729
128. Oddis CV, Okano Y, Rudert WA, Trucco M, Duquesnoy RJ, Medsger TA. Serum autoantibody to the nucleolar antigen PM-scl. clinical and immunogenetic associations. Arthritis Rheumatol (1992) 35(10):1211–7. doi: 10.1002/art.1780351014
129. Rodriguez-Reyna TS, Hinojosa-Azaola A, Martinez-Reyes C, Nuñez-Alvarez CA, Torrico-Lavayen R, García-Hernández JL, et al. Distinctive autoantibody profile in Mexican mestizo systemic sclerosis patients. Autoimmunity. (2011) 44(7):576–84. doi: 10.3109/08916934.2011.592886
130. Rozman B, Cucnik S, Sodin-Semrl S, Czirják L, Varjú C, Distler O, et al. Prevalence and clinical associations of anti-Ku antibodies in patients with systemic sclerosis: a European EUSTAR-initiated multi-centre case-control study. Ann Rheum Dis (2008) 67(9):1282–6. doi: 10.1136/ard.2007.073981
131. Mierau R, Moinzadeh P, Riemekasten G, Melchers I, Meurer M, Reichenberger F, et al. Frequency of disease-associated and other nuclear autoantibodies in patients of the German network for systemic scleroderma: Correlation with characteristic clinical features. Arthritis Res Ther (2011) 13(5):R172. doi: 10.1186/ar3495
132. Mahler M, Raijmakers R. Novel aspects of autoantibodies to the PM/Scl complex: Clinical, genetic and diagnostic insights. Autoimmun Rev (2007) 6(7):432–7. doi: 10.1016/j.autrev.2007.01.013
133. Guillen-Del Castillo A, Pilar Simeón-Aznar C, Fonollosa-Pla V, Alonso-Vila S, Reverte-Vinaixa MM, Muñoz X, et al. Good outcome of interstitial lung disease in patients with scleroderma associated to anti-PM/Scl antibody. Semin Arthritis Rheumatism. (2014) 44(3):331–7. doi: 10.1016/j.semarthrit.2014.07.002
134. Koschik RW, Fertig N, Lucas MR, Domsic RT, Medsger TA. Anti-PM-Scl antibody in patients with systemic sclerosis. Clin Exp Rheumatol (2012) 30(2 Suppl 71):S12–16.
135. Vandergheynst F, Ocmant A, Sordet C, Humbel RL, Goetz J, Roufosse F, et al. Anti-pm/scl antibodies in connective tissue disease: Clinical and biological assessment of 14 patients. Clin Exp Rheumatol (2006) 24(2):129–33.
136. Marguerie C, Bunn CC, Copier J, Bernstein RM, Gilroy JM, Black CM, et al. The clinical and immunogenetic features of patients with autoantibodies to the nucleolar antigen PM-scl. Medicine (1992) 71(6):327–36. doi: 10.1097/00005792-199211000-00001
137. Cavazzana I, Fredi M, Taraborelli M, Quinzanini M, Tincani A, Franceschini F. A subset of systemic sclerosis but not of systemic lupus erythematosus is defined by isolated anti-Ku autoantibodies. Clin Exp Rheumatol (2013) 31(2 Suppl 76):118–21.
138. Cavazzana I, Ceribelli A, Quinzanini M, Scarsi M, Airò P, Cattaneo R, et al. Prevalence and clinical associations of anti-Ku antibodies in systemic autoimmune diseases. Lupus. (2008) 17(8):727–32. doi: 10.1177/0961203308089442
139. Chang WSJ, Schollum J, White DHN, Solanki KK. A cross-sectional study of autoantibody profiles in the Waikato systemic sclerosis cohort, new Zealand. Clin Rheumatol (2015) 34(11):1921–7. doi: 10.1007/s10067-015-2981-3
140. Hoa S, Hudson M, Troyanov Y, Proudman S, Walker J, Stevens W, et al. Single-specificity anti-Ku antibodies in an international cohort of 2140 systemic sclerosis subjects: clinical associations. Med (Baltimore). (2016) 95(35):e4713. doi: 10.1097/MD.0000000000004713
141. Low AHL, Wong S, Thumboo J, Ng SC, Lim JY, Ng X, et al. Evaluation of a new multi-parallel line immunoassay for systemic sclerosis-associated antibodies in an Asian population. Rheumatol (Oxford). (2012) 51(8):1465–70. doi: 10.1093/rheumatology/kes055
142. Mimori T, Akizuki M, Yamagata H, Inada S, Yoshida S, Homma M. Characterization of a high molecular weight acidic nuclear protein recognized by autoantibodies in sera from patients with polymyositis-scleroderma overlap. J Clin Invest. (1981) 68(3):611–20. doi: 10.1172/JCI110295
143. Patterson KA, Roberts-Thomson PJ, Lester S, Tan JA, Hakendorf P, Rischmueller M, et al. Interpretation of an extended autoantibody profile in a well-characterized Australian systemic sclerosis (Scleroderma) cohort using principal components analysis. Arthritis Rheumatol (2015) 67(12):3234–44. doi: 10.1002/art.39316
144. Schild-Poulter C, Su A, Shih A, Kelly OP, Fritzler MJ, Goldstein R, et al. Association of autoantibodies with Ku and DNA repair proteins in connective tissue diseases. Rheumatol (Oxford). (2008) 47(2):165–71. doi: 10.1093/rheumatology/kem338
145. Villalta D, Imbastaro T, Di Giovanni S, Lauriti C, Gabini M, Turi MC, et al. Diagnostic accuracy and predictive value of extended autoantibody profile in systemic sclerosis. Autoimmun Rev (2012) 12(2):114–20. doi: 10.1016/j.autrev.2012.07.005
146. Yaneva M, Arnett FC. Antibodies against Ku protein in sera from patients with autoimmune diseases. Clin Exp Immunol (1989) 76(3):366–72.
147. Belizna C, Henrion D, Beucher A, Lavigne C, Ghaali A, Lévesque H. Anti-Ku antibodies: Clinical, genetic and diagnostic insights. Autoimmun Rev (2010) 9(10):691–4. doi: 10.1016/j.autrev.2010.05.020
148. Franceschini F, Cavazzana I, Generali D, Quinzanini M, Viardi L, Ghirardello A, et al. Anti-Ku antibodies in connective tissue diseases: Clinical and serological evaluation of 14 patients. J Rheumatol (2002) 29(7):1393–7.
149. Kaji K, Fertig N, Medsger TA, Satoh T, Hoshino K, Hamaguchi Y, et al. Autoantibodies to RuvBL1 and RuvBL2: A novel systemic sclerosis-related antibody associated with diffuse cutaneous and skeletal muscle involvement: Novel SSc-related autoantibody directed against RuvBL1/2. Arthritis Care Res (2014) 66(4):575–84. doi: 10.1002/acr.22163
150. Rigolet A, Musset L, Dubourg O, Maisonobe T, Grenier P, Charuel JL, et al. Inflammatory myopathies with anti-Ku antibodies: A prognosis dependent on associated lung disease. Med (Baltimore). (2012) 91(2):95–102. doi: 10.1097/MD.0b013e31824d9cec
151. Spielmann L, Nespola B, Séverac F, Andres E, Kessler R, Guffroy A, et al. Anti-Ku syndrome with elevated CK and anti-Ku syndrome with anti-dsDNA are two distinct entities with different outcomes. Ann Rheum Dis (2019) 78(8):1101–6. doi: 10.1136/annrheumdis-2018-214439
152. Spielmann L, Nespola B, Meyer A. Response to: “Immune-mediated necrotizing myopathies and interstitial lung disease are predominant characteristics in anti-Ku positive patients with idiopathic inflammatory myopathies” by yang et al. Ann Rheum Dis (2022) 81(3):e49. doi: 10.1136/annrheumdis-2020-217106
153. Ihn H, Yamane K, Yazawa N, Kubo M, Fujimoto M, Sato S, et al. Distribution and antigen specificity of anti-U1RNP antibodies in patients with systemic sclerosis. Clin Exp Immunol (1999) 117(2):383–7. doi: 10.1046/j.1365-2249.1999.00961.x
154. Jacobsen S, Halberg P, Ullman S, Van Venrooij WJ, Høier-Madsen M, Wiik A, et al. Clinical features and serum antinuclear antibodies in 230 Danish patients with systemic sclerosis. Br J Rheumatol (1998) 37(1):39–45. doi: 10.1093/rheumatology/37.1.39
155. Reveille JD, Fischbach M, McNearney T, Friedman AW, Aguilar MB, Lisse J, et al. Systemic sclerosis in 3 US ethnic groups: A comparison of clinical, sociodemographic, serologic, and immunogenetic determinants. Semin Arthritis Rheumatism. (2001) 30(5):332–46. doi: 10.1053/sarh.2001.20268
156. Asano Y, Ihn H, Yamane K, Kubo M, Tamaki K. The prevalence and clinical significance of anti-U1 RNA antibodies in patients with systemic sclerosis. J Invest Dermatol (2003) 120(2):204–10. doi: 10.1046/j.1523-1747.2003.12028.x
157. Sobanski V, Giovannelli J, Lynch BM, Schreiber BE, Nihtyanova SI, Harvey J, et al. Characteristics and survival of anti-U1 RNP antibody-positive patients with connective tissue disease-associated pulmonary arterial hypertension. Arthritis Rheumatol (2016) 68(2):484–93. doi: 10.1002/art.39432
158. Aggarwal R, Lucas M, Fertig N, Oddis CV, Medsger TA. Anti-U3 RNP autoantibodies in systemic sclerosis. Arthritis Rheumatol (2009) 60(4):1112–8. doi: 10.1002/art.24409
159. Arnett FC, Reveille JD, Goldstein R, Pollard KM, Leaird K, Smith EA, et al. Autoantibodies to fibrillarin in systemic sclerosis (scleroderma). an immunogenetic, serologic, and clinical analysis. Arthritis Rheumatol (1996) 39(7):1151–60. doi: 10.1002/art.1780390712
160. Hirakata M, Mimori T, Akizuki M, Craft J, Hardin JA, Homma M. Autoantibodies to small nuclear and cytoplasmic ribonucleoproteins in Japanese patients with inflammatory muscle disease. Arthritis Rheumatol (1992) 35(4):449–56. doi: 10.1002/art.1780350415
161. Okano Y, Steen VD, Medsger TA. Autoantibody to U3 nucleolar ribonucleoprotein (fibrillarin) in patients with systemic sclerosis. Arthritis Rheumatol (1992) 35(1):95–100. doi: 10.1002/art.1780350114
162. Tormey VJ, Bunn CC, Denton CP, Black CM. Anti-fibrillarin antibodies in systemic sclerosis. Rheumatol (Oxford). (2001) 40(10):1157–62. doi: 10.1093/rheumatology/40.10.1157
163. Yang JM, Hildebrandt B, Luderschmidt C, Pollard KM. Human scleroderma sera contain autoantibodies to protein components specific to the U3 small nucleolar RNP complex. Arthritis Rheumatol (2003) 48(1):210–7. doi: 10.1002/art.10729
164. Reimer G, Steen VD, Penning CA, Medsger TA, Tan EM. Correlates between autoantibodies to nucleolar antigens and clinical features in patients with systemic sclerosis (scleroderma). Arthritis Rheumatol (1988) 31(4):525–32. doi: 10.1002/art.1780310409
165. Falkner D, Wilson J, Fertig N, Clawson K, Medsger TA, Morel PA. Studies of HLA-DR and DQ alleles in systemic sclerosis patients with autoantibodies to RNA polymerases and U3-RNP (fibrillarin). J Rheumatol (2000) 27(5):1196–202.
166. Vulsteke JB, Piette Y, Bonroy C, Verschueren P, Blockmans D, Vanderschueren S, et al. Anti-RuvBL1/2 autoantibodies in patients with systemic sclerosis or idiopathic inflammatory myopathy and a nuclear speckled pattern. Ann Rheum Dis (2022) 81(5):742–4. doi: 10.1136/annrheumdis-2021-220004
167. Landon-Cardinal O, Baril-Dionne A, Hoa S, Meyer A, Leclair V, Bourré-Tessier J, et al. Recognising the spectrum of scleromyositis: HEp-2 ANA patterns allow identification of a novel clinical subset with anti-SMN autoantibodies. RMD Open (2020) 6(2):e001357. doi: 10.1136/rmdopen-2020-001357
168. Satoh M, Chan JYF, Ross SJ, Ceribelli A, Cavazzana I, Franceschini F, et al. Autoantibodies to survival of motor neuron complex in patients with polymyositis: immunoprecipitation of d, e, f, and G proteins without other components of small nuclear ribonucleoproteins. Arthritis Rheumatol (2011) 63(7):1972–8. doi: 10.1002/art.30349
169. Amlani A, Hazlewood GS, Hamilton L, Satoh M, Fritzler MJ. Autoantibodies to the survival of motor neuron complex in a patient with necrotizing autoimmune myopathy. Rheumatol (Oxford). (2018) 57(1):199–200. doi: 10.1093/rheumatology/kex392
170. Damoiseaux J, Andrade LEC, Carballo OG, Conrad K, Francescantonio PLC, Fritzler MJ, et al. Clinical relevance of HEp-2 indirect immunofluorescent patterns: the international consensus on ANA patterns (ICAP) perspective. Ann Rheum Dis (2019) 78(7):879–89. doi: 10.1136/annrheumdis-2018-214436
171. Vo C, Landon-Cardinal O, Albert A, Meyer A, Leclair V, Bourré-Tessier J, et al. Anti-SMN autoantibodies are associated with systemic sclerosis small bowel involvement in anti-U1RNP positive autoimmune myositis [abstract]. Arthritis Rheumatol (2020) 72(suppl 10).
172. El Kamouni H, Albert A, Hoa S, S. Jalaledin D, Bourre-Tessier J, Rich E, et al. Anti-SMN autoantibodies in mixed connective tissue disease are associated with a scleromyositis phenotype [abstract]. Arthritis Rheumatol (2022) 74(suppl 9).
173. Bukiri H, Volkmann ER. Current advances in the treatment of systemic sclerosis. Curr Opin Pharmacol (2022) 64:102211. doi: 10.1016/j.coph.2022.102211
174. Meyer A, Scirè CA, Talarico R, Alexander T, Amoura Z, Avcin T, et al. Idiopathic inflammatory myopathies: state of the art on clinical practice guidelines [corrected]. RMD Open (2018) 4(Suppl 1):e000784. doi: 10.1136/rmdopen-2018-000784
175. Smith V, Scirè CA, Talarico R, Airo P, Alexander T, Allanore Y, et al. Systemic sclerosis: state of the art on clinical practice guidelines. RMD Open (2018) 4(Suppl 1):e000782. doi: 10.1136/rmdopen-2018-000782
176. Hudson M, Baron M, Tatibouet S, Furst DE, Khanna D, International Scleroderma Renal Crisis Study Investigators. Exposure to ACE inhibitors prior to the onset of scleroderma renal crisis-results from the international scleroderma renal crisis survey. Semin Arthritis Rheumatol (2014) 43(5):666–72. doi: 10.1016/j.semarthrit.2013.09.008
177. Trang G, Steele R, Baron M, Hudson M. Corticosteroids and the risk of scleroderma renal crisis: A systematic review. Rheumatol Int (2012) 32(3):645–53. doi: 10.1007/s00296-010-1697-6
178. Kowal-Bielecka O, Fransen J, Avouac J, Becker M, Kulak A, Allanore Y, et al. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann Rheum Dis (2017) 76(8):1327–39. doi: 10.1136/annrheumdis-2016-209909
179. Tashkin DP, Roth MD, Clements PJ, Furst DE, Khanna D, Kleerup EC, et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): A randomised controlled, double-blind, parallel group trial. Lancet Respir Med (2016) 4(9):708–19. doi: 10.1016/S2213-2600(16)30152-7
180. Ruperto N, Pistorio A, Oliveira S, Zulian F, Cuttica R, Ravelli A, et al. Prednisone versus prednisone plus ciclosporin versus prednisone plus methotrexate in new-onset juvenile dermatomyositis: A randomised trial. Lancet (2016) 387(10019):671–8. doi: 10.1016/S0140-6736(15)01021-1
181. Dalakas MC, Illa I, Dambrosia JM, Soueidan SA, Stein DP, Otero C, et al. A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. N Engl J Med (1993) 329(27):1993–2000. doi: 10.1056/NEJM199312303292704
182. Aggarwal R, Charles-Schoeman C, Schessl J, Bata-Csörgő Z, Dimachkie MM, Griger Z, et al. Trial of intravenous immune globulin in dermatomyositis. N Engl J Med (2022) 387(14):1264–78. doi: 10.1056/NEJMoa2117912
183. Allenbach Y, Mammen AL, Benveniste O, Stenzel W, Immune-Mediated Necrotizing Myopathies Working Group. 224th ENMC international workshop:: Clinico-sero-pathological classification of immune-mediated necrotizing myopathies zandvoort, the Netherlands, 14-16 October 2016. Neuromuscul Disord (2018) 28(1):87–99. doi: 10.1016/j.nmd.2017.09.016
184. Chaigne B, Rodeia S, Benmostefa N, Bérézné A, Authier J, Cohen P, et al. Corticosteroid-sparing benefit of intravenous immunoglobulin in systemic sclerosis-associated myopathy: A comparative study in 52 patients. Autoimmun Rev (2020) 19(1):102431. doi: 10.1016/j.autrev.2019.102431
185. Oddis CV, Reed AM, Aggarwal R, Rider LG, Ascherman DP, Levesque MC, et al. Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: A randomized, placebo-phase trial. Arthritis Rheumatol (2013) 65(2):314–24. doi: 10.1002/art.37754
186. Mammen AL, Allenbach Y, Stenzel W, Benveniste O, ENMC 239th Workshop Study Group. 239th ENMC international workshop: Classification of dermatomyositis, Amsterdam, the Netherlands, 14-16 December 2018. Neuromuscul Disord (2020) 30(1):70–92. doi: 10.1016/j.nmd.2019.10.005
187. Ebata S, Yoshizaki A, Oba K, Kashiwabara K, Ueda K, Uemura Y, et al. Safety and efficacy of rituximab in systemic sclerosis (DESIRES): A double-blind, investigator-initiated, randomised, placebo-controlled trial. Lancet Rheumatol (2021) 3(7):e489–97. doi: 10.1016/S2665-9913(21)00107-7
188. Srikantharajah D, Lloyd ME, Kiely PDW. Rituximab and intravenous immunoglobulin treatment in PM/Scl antibody-associated disease: Case-based review. Rheumatol Int (2022) 42(2):359–64. doi: 10.1007/s00296-021-05075-z
189. Khanna D, Spino C, Johnson S, Chung L, Whitfield ML, Denton CP, et al. Abatacept in early diffuse cutaneous systemic sclerosis: Results of a phase II investigator-initiated, multicenter, double-blind, randomized, placebo-controlled trial. Arthritis Rheumatol (2020) 72(1):125–36. doi: 10.1002/art.41055
190. Mehta BK, Espinoza ME, Franks JM, Yuan Y, Wang Y, Wood T, et al. Machine-learning classification identifies early systemic sclerosis patients that improve with abatacept treatment by modulating CD28-pathways. JCI Insight (2022) 7(24):e155282. doi: 10.1172/jci.insight.155282
191. Aggarwal R, Lundberg IE, Song YW, Shaibani A, Werth VP, Maldonado MA. Pos0839 randomized, double-blind, placebo-controlled trial to evaluate efficacy and safety of Sc abatacept in adults with active idiopathic inflammatory. Myopathy Ann Rheumatic Dis (2022) 81:711. doi: 10.1136/annrheumdis-2022-eular.7
192. Sullivan KM, Goldmuntz EA, Keyes-Elstein L, McSweeney PA, Pinckney A, Welch B, et al. Myeloablative autologous stem-cell transplantation for severe scleroderma. N Engl J Med (2018) 378(1):35–47. doi: 10.1056/NEJMoa1703327
193. van Laar JM, Farge D, Sont JK, Naraghi K, Marjanovic Z, Larghero J, et al. Autologous hematopoietic stem cell transplantation vs intravenous pulse cyclophosphamide in diffuse cutaneous systemic sclerosis: a randomized clinical trial. JAMA. (2014) 311(24):2490–8. doi: 10.1001/jama.2014.6368
194. Rothwell S, Chinoy H, Lamb JA, Miller FW, Rider LG, Wedderburn LR, et al. Focused HLA analysis in caucasians with myositis identifies significant associations with autoantibody subgroups. Ann Rheum Dis (2019) 78(7):996–1002. doi: 10.1136/annrheumdis-2019-215046
195. Prieto-Peña D, Ocejo-Vinyals JG, Mazariegos-Cano J, Pelayo-Negro AL, Remuzgo-Martínez S, Genre F, et al. Epidemiological and genetic features of anti-3−hydroxy-3-methylglutaryl-CoA reductase necrotizing myopathy: Single-center experience and literature review. Eur J Intern Med (2022) 101:86–92. doi: 10.1016/j.ejim.2022.04.017
196. Mescam-Mancini L, Allenbach Y, Hervier B, Devilliers H, Mariampillay K, Dubourg O, et al. Anti-Jo-1 antibody-positive patients show a characteristic necrotizing perifascicular myositis. Brain (2015) 138(Pt 9):2485–92. doi: 10.1093/brain/awv192
197. Remuzgo-Martínez S, Atienza-Mateo B, Ocejo-Vinyals JG, Pulito-Cueto V, Prieto-Peña D, Genre F, et al. HLA association with the susceptibility to anti-synthetase syndrome. Joint Bone Spine. (2021) 88(3):105115. doi: 10.1016/j.jbspin.2020.105115
198. Levy D, Nespola B, Giannini M, Felten R, Severac F, Varoquier C, et al. Significance of sjögren’s syndrome and anti-cN1A antibody in myositis patients. Rheumatology (2021) 61(2):756–63. doi: 10.1093/rheumatology/keab423
199. Garton MJ, Isenberg DA. Clinical features of lupus myositis versus idiopathic myositis: A review of 30 cases. Br J Rheumatol (1997) 36(10):1067–74. doi: 10.1093/rheumatology/36.10.1067
200. Bitencourt N, Solow EB, Wright T, Bermas BL. Inflammatory myositis in systemic lupus erythematosus. Lupus. (2020) 29(7):776–81. doi: 10.1177/0961203320918021
201. Dayal NA, Isenberg DA. SLE/myositis overlap: are the manifestations of SLE different in overlap disease? Lupus (2002) 11(5):293–8. doi: 10.1191/0961203302lu186oa
202. Lim KL, Abdul-Wahab R, Lowe J, Powell RJ. Muscle biopsy abnormalities in systemic lupus erythematosus: correlation with clinical and laboratory parameters. Ann Rheum Dis (1994) 53(3):178–82. doi: 10.1136/ard.53.3.178
203. Kanellopoulos P, Baltoyiannis C, Tzioufas AG. Primary sjögren’s syndrome associated with inclusion body myositis. Rheumatol (Oxford). (2002) 41(4):440–4. doi: 10.1093/rheumatology/41.4.440
204. Colafrancesco S, Priori R, Gattamelata A, Picarelli G, Minniti A, Brancatisano F, et al. Myositis in primary sjögren’s syndrome: data from a multicentre cohort. Clin Exp Rheumatol (2015) 33(4):457–64.
205. Maazoun F, Frikha F, Snoussi M, Kaddour N, Masmoudi H, Bahloul Z. Systemic lupus erythematosusmyositis overlap syndrome: Report of 6 cases. Clin Pract (2011) 1(4):e89. doi: 10.4081/cp.2011.e89
206. Casal-Dominguez M, Pinal-Fernandez I, Corse AM, Paik J, Albayda J, Casciola-Rosen L, et al. Muscular and extramuscular features of myositis patients with anti-U1-RNP autoantibodies. Neurology. (2019) 92(13):e1416–26. doi: 10.1212/WNL.0000000000007188
207. Font J. Clusters of clinical and immunologic features in systemic lupus erythematosus: Analysis of 600 patients from a single center. Semin Arthritis Rheumatism. (2004) 33(4):217–30. doi: 10.1053/S0049-0172(03)00133-1
208. Benveniste O, Guiguet M, Freebody J, Dubourg O, Squier W, Maisonobe T, et al. Long-term observational study of sporadic inclusion body myositis. Brain (2011) 134(Pt 11):3176–84. doi: 10.1093/brain/awr213
209. Benveniste O, Hogrel JY, Belin L, Annoussamy M, Bachasson D, Rigolet A, et al. Sirolimus for treatment of patients with inclusion body myositis: A randomised, double-blind, placebo-controlled, proof-of-concept, phase 2b trial. Lancet Rheumatol (2021) 3(1):e40–8. doi: 10.1016/S2665-9913(20)30280-0
210. Meyer A, Meyer N, Schaeffer M, Gottenberg JE, Geny B, Sibilia J. Incidence and prevalence of inflammatory myopathies: A systematic review. Rheumatology. (2015) 54(1):50–63. doi: 10.1093/rheumatology/keu289
211. Giannini M, Felten R, Gottenberg JE, Geny B, Meyer A. Inclusion body myositis and sjögren’s syndrome: The association works both ways. Acta Neuropathol Commun (2022) 10(1):152. doi: 10.1186/s40478-022-01443-3
Keywords: myositis, inflammatory myopathies, dermatomyositis, antisynthetase syndrome, systemic sclerosis, scleroderma, scleromyositis, mixed connective tissue disease
Citation: Giannini M, Ellezam B, Leclair V, Lefebvre F, Troyanov Y, Hudson M, Senécal J-L, Geny B, Landon-Cardinal O and Meyer A (2023) Scleromyositis: A distinct novel entity within the systemic sclerosis and autoimmune myositis spectrum. Implications for care and pathogenesis. Front. Immunol. 13:974078. doi: 10.3389/fimmu.2022.974078
Received: 20 June 2022; Accepted: 19 December 2022;
Published: 26 January 2023.
Edited by:
Vincent Sobanski, INSERM U1286, FranceReviewed by:
Ernesto Trallero-Araguás, Vall d'Hebron University Hospital, SpainCopyright © 2023 Giannini, Ellezam, Leclair, Lefebvre, Troyanov, Hudson, Senécal, Geny, Landon-Cardinal and Meyer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alain Meyer, alain.meyer1@chru-strasbourg.fr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.