 Cristian Papara1,2
Cristian Papara1,2 Christian M. Karsten3
Christian M. Karsten3 Hideyuki Ujiie4
Hideyuki Ujiie4 Enno Schmidt1,5
Enno Schmidt1,5 Leon F. Schmidt-Jiménez5
Leon F. Schmidt-Jiménez5 Adrian Baican2
Adrian Baican2 Patricia C. Freire3
Patricia C. Freire3 Kentaro Izumi4
Kentaro Izumi4 Katja Bieber5
Katja Bieber5 Matthias Peipp6
Matthias Peipp6 Admar Verschoor1,7
Admar Verschoor1,7 Ralf J. Ludwig1,5
Ralf J. Ludwig1,5 Jörg Köhl3,8
Jörg Köhl3,8 Detlef Zillikens1
Detlef Zillikens1 Christoph M. Hammers1,5*
Christoph M. Hammers1,5*- 1Department of Dermatology, University of Lübeck, Lübeck, Germany
- 2Department of Dermatology, Iuliu Hatieganu University of Medicine and Pharmacy, Cluj-Napoca, Romania
- 3Institute of Systemic Inflammation Research, University of Lübeck, Lübeck, Germany
- 4Department of Dermatology, Faculty of Medicine and Graduate School of Medicine, Hokkaido University, Sapporo, Japan
- 5Lübeck Institute of Experimental Dermatology (LIED), University of Lübeck, Lübeck, Germany
- 6Division of Antibody-Based Immunotherapy, Department of Medicine II, Christian-Albrechts-University of Kiel and University Medical Center Schleswig-Holstein, Kiel, Germany
- 7Department of Otorhinolaryngology, Klinikum rechts der Isar, Technical University Munich, Munich, Germany
- 8Division of Immunobiology, Cincinnati Children’s Hospital Medical Center and University of Cincinnati College of Medicine, Cincinnati, OH, United States
Pemphigoid diseases are autoimmune chronic inflammatory skin diseases, which are characterized by blistering of the skin and/or mucous membranes, and circulating and tissue-bound autoantibodies. The well-established pathomechanisms comprise autoantibodies targeting various structural proteins located at the dermal-epidermal junction, leading to complement factor binding and activation. Several effector cells are thus attracted and activated, which in turn inflict characteristic tissue damage and subepidermal blistering. Moreover, the detection of linear complement deposits in the skin is a diagnostic hallmark of all pemphigoid diseases. However, recent studies showed that blistering might also occur independently of complement. This review reassesses the importance of complement in pemphigoid diseases based on current research by contrasting and contextualizing data from in vitro, murine and human studies.
1 Introduction
The complement system is a complex network of more than 50 proteins, which hold crucial roles in host defense against invading microorganisms as well as in tissue homeostasis, thus representing a fundamental component of the innate immune system. There are three distinct pathways of complement activation: the classical (CP), lectin (LP), and alternative pathway (AP) (Figure 1). Under physiological circumstances, nonself recognition in an immunoglobulin (Ig)-dependent or -independent manner triggers various proteolytic cascades that result in the activation of complement component C3. All three pathways generate a C3 convertase which elicits the immediate (C3a) and downstream (C5a) release of potent proinflammatory peptides (i. e., C5a, C3a), the opsonization of susceptible pathogens (via C3b and C4b), and ultimately, the lysis of microorganisms by the assembly of the membrane attack complex (MAC; composed of C5b6789) (2, 3), thereby contributing to their efficient elimination. Several soluble and membrane-bound factors regulate complement activation on different levels to protect healthy tissue from undesired damage. C1-esterase inhibitor (C1-INH), C4b-binding protein (C4BP), carboxypeptidase N (CPN1), factor H (FH), factor I (FI), protein S, and clusterin are soluble regulators of complement activation, whereas complement receptor of the immunoglobulin superfamily (CRIg), complement receptor 1 (CR1/CD35), decay-accelerating factor (DAF/CD55), membrane-cofactor protein (MCP/CD46), and protectin (CD59) mediate complement activation and regulation on the cell membrane (4).
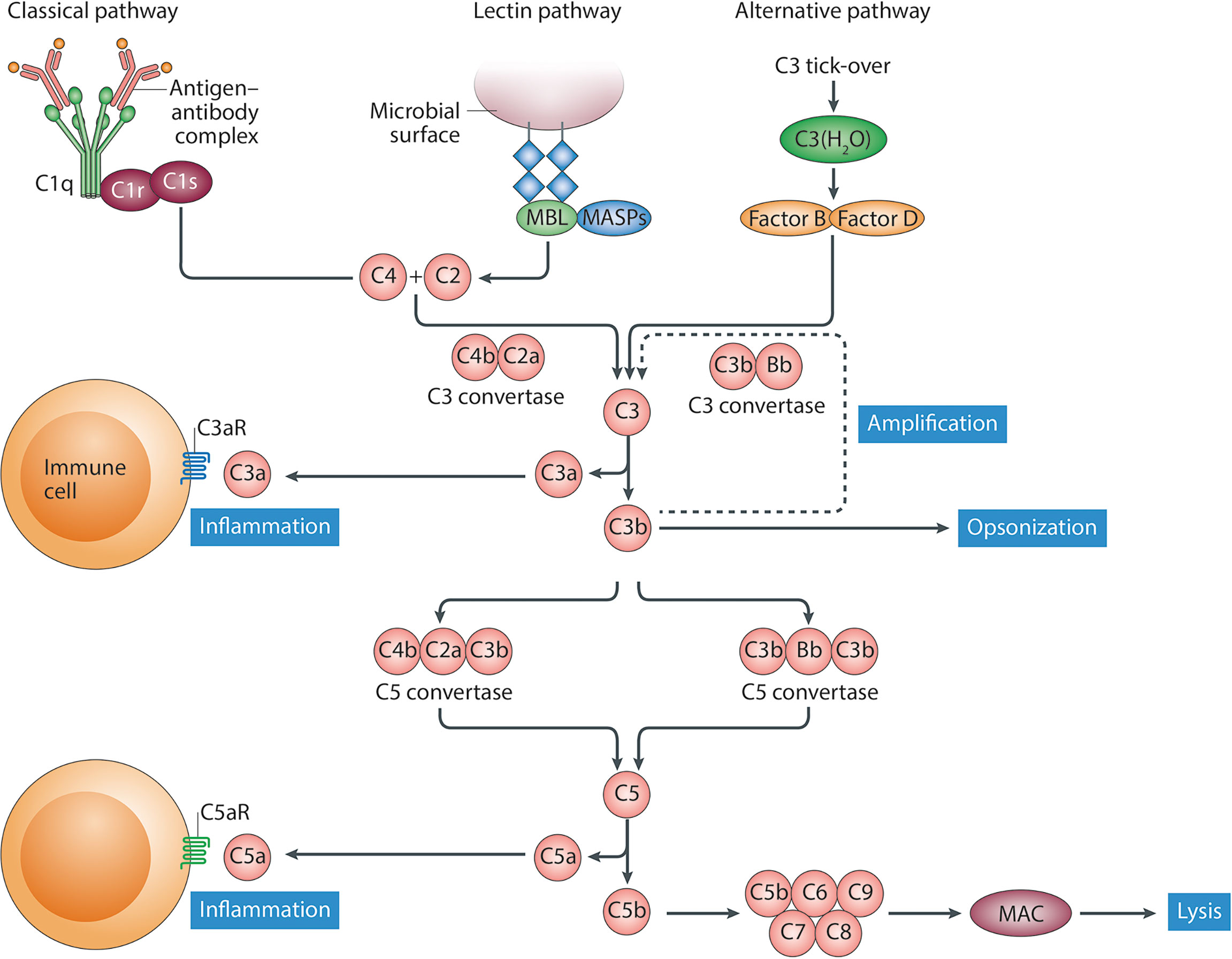
Figure 1 Schematic representation of the complement system. The classical pathway of complement activation is activated following to binding of the recognition molecule C1q to ligands such as immune complexes. The lectin pathway is activated following to binding of recognition molecules, such as mannose-binding lectin (MBL), collectins or ficolins, to their ligands, which include carbohydrate structures. Although the alternative pathway is initiated spontaneously, properdin (not shown) might also serve as a recognition molecule for directing activation of this pathway. Following activation via the initiating molecules a cascade of proteolytic activation steps leads to the formation of C3 convertases that cleave C3 into the anaphylatoxin C3a and the opsonin C3b. Next, C5 convertases generate the potent pro-inflammatory anaphylatoxins C5a and C5b, the latter of which, together with C6–C9, forms the membrane attack complex (MAC). This figure was obtained with permission from Trouw, L.A., Pickering, M.C., & Blom, A.M. Nat Rev Rheumatol. 9, 538–547 (2017), (1).
Deficiencies in complement components or impaired complement activation pathways hinder efficient host defense responses, resulting in an increased susceptibility to infections: properdin deficiency and defects in the MAC formation are associated with an increased risk of meningococcal infections, whereas C3 deficiency is linked to recurrent infections with Streptococcus pneumoniae (5–7). In contrast, C4 deficiency is associated with systemic lupus erythematosus (SLE), but also with repeated severe herpes infections (8, 9). In children, deficiencies in mannose-binding lectin (MBL) result in recurrent bacterial respiratory infections (10). Furthermore, a defective C3-dependent opsonization increases the risk of infections with Streptococcus pneumoniae and Haemophilus influenzae, while a lack of CR3 enhances the likelihood of recurrent skin infections (2, 11).
Impaired complement system activation or regulation has been observed in many dermatological diseases, such as hereditary (12) and acquired angioedema (13), cutaneous small vessel (14, 15) and hypocomplementemic urticarial vasculitis (16), SLE (17, 18), psoriasis (19, 20), acne vulgaris (21, 22) and hidradenitis suppurativa (23). Moreover, the complement system is also involved in the pathogenesis of autoimmune blistering dermatoses (AIBD), in particular the pemphigoid group, including bullous pemphigoid (BP), epidermolysis bullosa acquisita (EBA), mucous membrane pemphigoid (MMP), pemphigoid gestationis (PG), and, to a lesser degree, the pemphigus group (24–28). C3 deposits along the dermal-epidermal junction (DEJ) are observed in approximately 90% of patients with pemphigoid diseases (PDs) (29–32). Interestingly, complement activation seems to be mainly restricted to the skin (33). In fact, the detection of C3 by direct immunofluorescence (DIF) microscopy of perilesional skin is a highly valuable diagnostic marker of all PDs. In BP, the activation of complement at the DEJ, as a result of pathogenic autoantibodies binding to collagen type XVII (BP180), initiates and maintains inflammatory processes resulting in characteristic subepidermal blistering (34). Consequently, complement components of both the CP and AP have been detected at the DEJ and blister fluid. Similarly, complement-induced separation at the DEJ has also been demonstrated in EBA, MMP and PG (35, 36). In addition, complement-fixating antibodies along the DEJ represent a characteristic feature of PG, and DIF microscopy often demonstrates rather linear C3 than IgG deposition along the basement membrane (31, 37). Regarding pemphigus, DIF microscopy shows intraepidermal deposition of IgG and/or C3, and the pathogenic IgG autoantibodies belong to the IgG1 subclass, which is a notable complement activator, but also to IgG4 (38).
By contrasting and contextualizing seemingly contradictory data from in vitro-, murine- and human studies we will corroborate the central role of complement, particularly in the effector phase of PDs, with a special focus on BP, EBA and MMP as prototypic PDs.
2 Complement-dependent pathogenic pathways in PDs
2.1 Experimental lines of evidence
2.1.1 BP
BP is the most common autoimmune bullous disease that mainly affects the elderly, usually in the 7th decade of life (24). It is caused by autoantibodies targeting two hemidesmosomal proteins, BP180 (collagen XVII, BPAG2) and BP230 (BPAG1), resulting in characteristic subepidermal blister formation (39). Clinical hallmarks range from generalized, pruritic, large skin blisters to eczematous and urticarial lesions (24).
Chorzelski and Cormane were the first to demonstrate that complement binds in vivo to the basement membrane of BP patients’ skin (40), followed by the substantial work of Jordon et al., which significantly contributed to our current understanding of the role of complement activation in the pathogenesis of BP. By using DIF microscopy, Jordon and co-workers showed that BP autoantibodies are able to fix not only C3 (41–43), C1q and C4 (44), but also factor B (FB) and properdin (45, 46), thus underlining the involvement of both classical and alternative complement activation pathways. Remarkably, C3 deposition was also seen in the absence of skin-bound BP autoantibodies (47), indicating a high sensitivity of the C3 staining. Furthermore, the same group demonstrated that BP blister fluids exhibited both eosinophil and neutrophil chemotactic activity, with the latter being inhibited by the use of an anti-C5 antiserum, therefore suggesting for the first time the role of complement, namely C5a, in the effector phase of BP (48). Several studies followed that corroborated these findings, to the extent that the detection of complement deposits in the skin is now considered a highly valuable and important diagnostic hallmark of BP (49–51).
Later on, Liu et al. provided experimental evidence by transferring polyclonal rabbit antibodies against murine NC14A, a homolog of the human NC16A domain of BP180, into neonatal BALB/c mice, showing that subepidermal blistering did not occur if either (i) serum complement was depleted by cobra venom factor, (ii) mice were C5-deficient, or (iii) F(ab′)2-fragments derived from the anti-murine BP180 antibody were used (52, 53). Similar results were observed in the antibody transfer model of BP in neonatal hamsters (54). These findings suggest that complement activation is indispensable for blister formation in experimental antibody transfer models of BP. Correspondingly, Nelson et al. demonstrated, in the same model, that C4-deficient mice and wild-type (WT) mice pretreated with anti-C1q-antibody were protected from disease development, thereby underscoring the importance of the classical pathway activation in the pathogenesis of PDs (55). Interestingly, the same study showed that FB-deficient mice developed delayed and less intense subepidermal blisters, suggesting a minor role of complement activation via the alternative pathway in experimental BP.
To reduce the shortcomings of the previously mentioned antibody transfer mouse models, both Nishie et al. and Liu et al. established humanized mouse models of BP, introducing either human BP180 (56) or replacing murine BP180NC14A with the homologous human BP180NC16A epitope cluster region (57). The CP of the complement system appeared to be pivotal also in these humanized mouse models: F(ab′)2-fragments derived from pathogenic antibodies did not induce blisters, complement C3 depletion with cobra venom factor protected from disease development, and transfer of humanized IgG1 against BP180NC16A that were previously mutated at the C1q binding site resulting in reduced C1q binding, induced less blisters compared to the unmutated humanized antigen-specific IgG1 (57, 58).
Another crucial pathway underlining the importance of complement activation in BP is the interaction between C5 and its receptors, C5a receptor 1 (C5aR1) and C5a receptor 2 (C5aR2) (Figure 2A). By the injection of anti-BP180 IgG in adult mice, Karsten et al. recently demonstrated that C5 knockout mice had an up to 50% reduction in disease activity when compared to corresponding WT-mice (59). Unlike previous findings, this intriguing result could be explained by the use of exclusively murine models, as well as by the opposing roles of the two receptors: C5aR1-deficient mice were protected from disease development, whereas C5aR2-deficient were rather prone to develop inflammatory skin lesions (59). In addition, the pharmacological inhibition of C5aR1 by the PMX53 peptide mitigated disease development, but only when applied in a preventive setting (59).
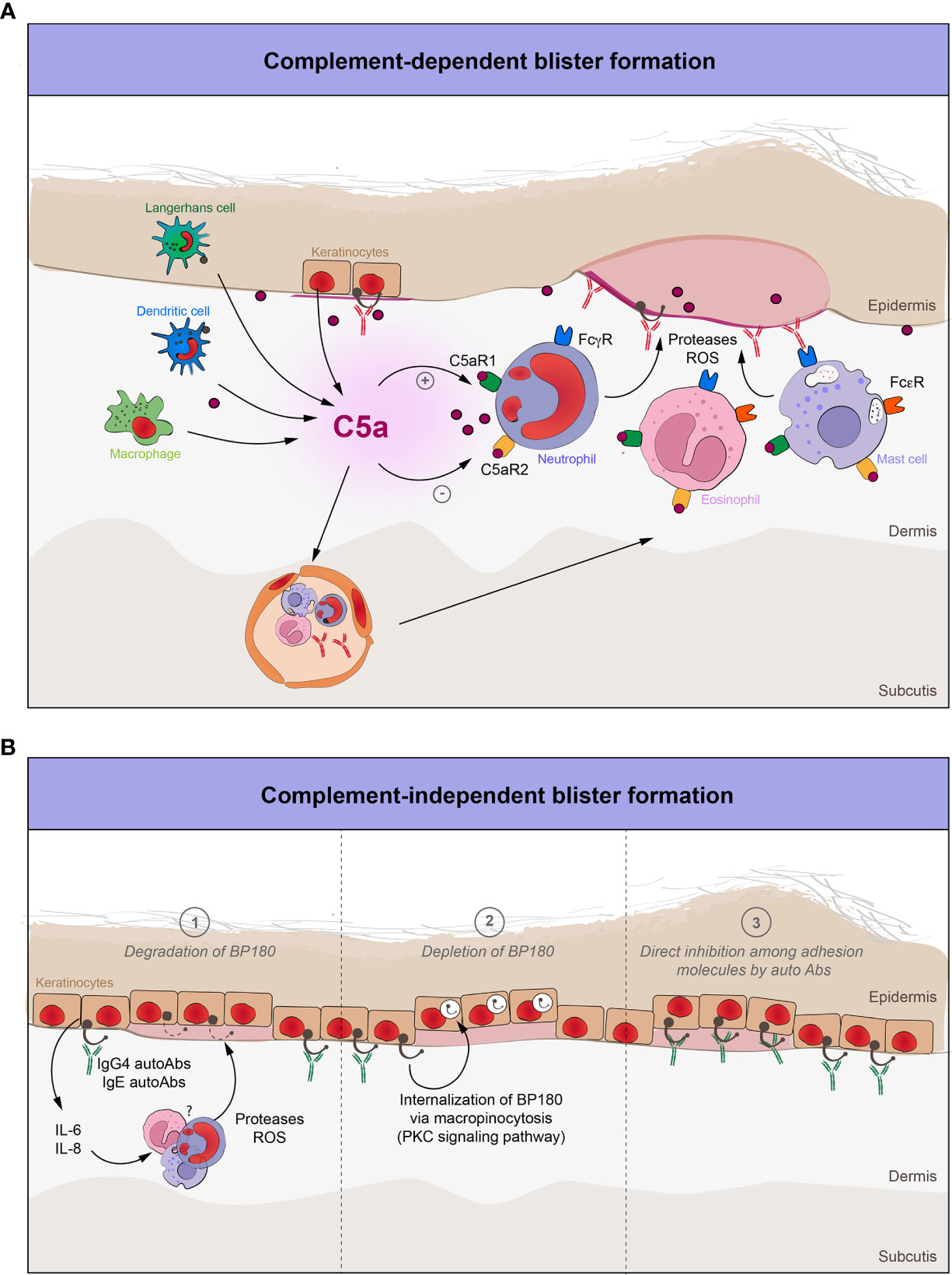
Figure 2 The role of complement in blister formation in pemphigoid diseases. (A) Complement-dependent pathways in pemphigoid diseases. Complement-fixing autoantibodies target various structural antigens of the basement membrane. The complement system is activated, thus leading to the recruitment and activation of various effector cells. C5a, which is the most potent proinflammatory peptide of the complement cascade, will interact with these cells through their C5a receptors (C5aR1, C5aR2), enabling them to release several proteases and ROS, thereby inflicting tissue damage and characteristic subepidermal blistering. (B) Complement-independent pathways in pemphigoid diseases. Non-complement fixing autoantibodies (IgG4) and IgE against BP180 bind to the dermal-epidermal junction and (1) degrade BP180 by increasing the expression of IL-6 and IL-8, which in turn will attract and activate neutrophils, as a major source of various proteases and ROS; (2) decrease the BP180 hemidesmosomal content by internalization of the IgG-BP180 immune complex via macropinocytosis; (3) directly inhibit the adhesion molecules of the dermal-epidermal junction.
These findings emphasize the role of complement activation in the early effector phase of the disease when neutrophils, eosinophils and mast cells are attracted by C5a to the DEJ to inflict characteristic tissue damage and subsequent blister formation (60–63). Remarkably, the depletion of neutrophils protected against BP in WT-mice, whereas the use of C5a and interleukin (IL)-8 in C5-deficient mice made them susceptible for disease development (64).
In fact, complement-dependent neutrophil infiltration in BP is contingent on prior mast cell degranulation (65), which is also induced by the activation of complement: upon the interaction between C5a and C5aR1, mast cells degranulate and release various pro-inflammatory cytokines that in turn attract neutrophils and eosinophils, as well as proteases, including the mouse mast cell protease-4 (mMCP-4) (63). The latter activates the key neutrophil protease, matrix metalloproteinase-9, both cleaving BP180 and thus, leading to blister formation (66, 67). Given this, mice deficient in mast cells, mMCP-4, or C5aR1 failed to develop BP (63, 65, 66).
In addition to neutrophils and mast cells, eosinophils appear to be involved in, at least, IgE-mediated pathology in BP, which may occur independently of neutrophils and relies on prior mast cell degranulation (61, 68). In this regard, eosinophils from most BP patients do highly express the high-affinity IgE receptor (FcϵRI) (69), and, as previously shown in a humanized FcϵRI mouse model of BP, human anti-BP180 IgE autoantibodies recruit and activate eosinophils via FcϵRI, resulting in eosinophil degranulation and subsequent blister formation (61). Of note, anaphylatoxins C5a and C3a can however directly induce degranulation of eosinophils via selective, receptor-mediated processes (70, 71), potentially connecting complement and eosinophils in human disease.
Taken together, all these data suggest that infiltration and activation of specific effector cells responsible for the characteristic BP skin pathology depend, at least indirectly, on the activated complement cascade.
2.1.2 EBA
EBA is a rare autoimmune blistering disease, which is characterized by autoantibodies against type VII collagen (COL7) (72). Clinically, it can present with various phenotypes, with the classical/mechano-bullous and the non-classical/non-mechano-bullous variant being the most common forms.
Our current understanding of the role of complement in the pathogenesis of EBA is mainly relying on the use of experimental murine models which, by design, mimic human disease clinically, immunologically and histologically. EBA can be induced in mice by either transfer of human anti-COL7 IgG or rabbit anti-mouse-COL7 IgG, or by immunization of mice with an immunodominant fragment of the murine COL7 antigen (73).
Sitaru et al. showed in the active EBA model, that diseased mice presented with significantly higher C3 skin deposition compared to non-diseased animals (36). Furthermore, the complement-fixing IgG2a and IgG2b autoantibody subclasses were considerably elevated in the diseased group (36), whereas F(ab′)2-fragments of pathogenic rabbit anti-COL7 IgG did not induce blistering in the passive EBA mouse model (74). Similar results were obtained for F(ab′)2-fragments generated from affinity-purified anti-COL7 antibodies from EBA patients’ sera (75), thus underlining a key role of the IgG-fragment crystallizable region (Fc), likely mediating inflammation via C1q interaction and CP complement activation in these EBA models.
Experimental evidence also illustrates the involvement of the AP in passive EBA. Mihai et al. demonstrated that FB-deficient mice as well as WT-mice treated with an anti-FB antibody developed a delayed and significantly less severe blistering phenotype when compared to controls (76, 77). In contrast, C1q-, C6- and MBL-deficient mice did develop the disease, implying that the MAC formation and the LP may be dispensable for blister formation. The rather unexpected finding that C1q-deficient mice were not fully protected from disease could be explained by either the presence of non-canonical complement activation or by different Fc N-glycosylation autoantibody patterns associated with proinflammatory responses and high neutrophil activating potential and ROS release (78–80). Moreover, the interaction between T cells and antigen-presenting cells was shown to unexpectedly induce the secretion of C3 or C5, as well as the formation of their corresponding convertases, thereby generating activated complement fragments and yielding characteristic effector responses without depending on CP or AP (i.e., non-canonical complement activation) (81). Correspondingly, macrophage-generated C4 restored the altered humoral response against tumor antigens in C4-deficient mice (82). Interestingly, it was previously shown that C5a can also be generated in the absence of C3-dependent convertases (83). In addition, human keratinocytes are able to express and produce various complement proteins, including C3 (84).
Similarly to murine BP models, the C5-C5aR1 axis is engaged in downstream tissue damage and subsequent blister formation in EBA: C5-deficient mice were partially or fully protected from developing EBA in the antibody transfer model (74, 85), while the application of an anti-murine-C5 monoclonal antibody significantly reduced the blistering phenotype in this system (77). Moreover, C5aR1-deficient mice did not develop disease, and the pharmacological blockade of this receptor also led to a notable improvement of the blistering phenotype (77, 86). In contrast to the protective role of C5aR2 in experimental BP, C5aR2-deficient mice developed an attenuated disease phenotype in the antibody transfer mouse model of EBA, with C5aR2 being essential for neutrophil activation and recruitment by regulating Fcγ receptor (FcγR) expression levels in this model (59, 87). In line with these findings, Sezin et al. demonstrated that dual inhibition of both C5 and leukotriene B4 (LTB4) with coversin (i.e., nomacopan) suppressed disease in passive EBA much more efficiently than the inhibitor of LTB4 alone (88).
2.1.3 MMP
MMP is a group of pemphigoid diseases with predominant mucous membrane involvement and a significant tendency towards scarring (89). Autoantibodies target various antigens, mainly BP180 and laminin 332. The mechanisms of blistering in MMP were also studied in different animal models (73). In contrast to BP and EBA, no immunization-induced mouse models have been yet described, but are in the pipeline (89). Since initial antibody transfer mouse models developed in the 90s were not completely replicating human disease, Heppe et al. recently introduced a novel model by transferring rabbit IgG against the middle and the C-terminal part of the murine laminin α3 chain, that reproduced disease clinically and immunopathologically (90–92).
Current evidence on the role of complement in experimental MMP models is scarce and contradictory at this point of time. Lazarova et al. showed that C5-deficient mice were not protected from disease development (90). Moreover, F(ab′)2-fragments from pathogenic rabbit anti-laminin 332 IgG elicited blistering in mice (91). Conversely, a recent study by Heppe et al., demonstrated that C5aR1- and FcγR-deficient mice developed little or no disease (92). The use of different mouse models for MMP could explain these discrepancies: the first study group induced MMP by passively transferring rabbit anti-laminin 332 IgG into neonatal mice, whereas the latter injected adult mice with rabbit anti-laminin 332 IgG against 2 immunodominant regions of laminin 332. Moreover, the latter mouse model was shown to fully replicate human disease, and dapsone, which is the first-choice in the therapeutic armamentarium of MMP, has proven effective in this passive MMP mouse model, thus further underlining its utility for the study of MMP (92, 93). Nevertheless, more experimental studies are needed to clarify the impact of complement in the pathogenesis of MMP.
2.2 Clinical lines of evidence
The detection of linear C3 deposits at the DEJ by DIF microscopy studies is routinely used to diagnose PDs and considered as the “gold standard” (31, 32, 94, 95).
In a large cohort study, Romeijn et al. demonstrated that over 80% of patients with BP showed C3c deposition along the DEJ in their (peri)lesional cutaneous biopsies (30). Moreover, this finding also significantly correlated with both clinical and serological disease activity (96). Similar results were also obtained by Ständer et al., who have shown that BP patients with C3 deposition had higher levels of seropositivity and autoantibodies (97).
Other complement components and activation factors, including C1q, C3, C3c, C3d, C4, C4d, C5, C5b-9, FB, FH, and properdin were also detected in the skin and blister fluid of BP patients, thus pointing towards the involvement of both CP and AP in the pathogenesis of human BP in vivo (42, 98). Interestingly, the detection of the two complement split products C3d and C4d at the DEJ by immunoperoxidase staining in formalin-fixed paraffin-embedded tissue proved as an effective novel diagnostic option for BP (99–103).
The pathogenic role of autoantibodies against BP180 is intimately intertwined with the activation of complement. Analysis of the IgG subclass distribution revealed that BP patients present a predominance of IgG1 antibodies in the skin (38). In addition, serum levels of IgG1 autoantibodies against the NC16A domain correlated with disease severity in these patients (104). Since IgG1 strongly binds complement via the C1q binding site of the Fc region, this adds further evidence to the decisive role of the CP in BP. Correspondingly, Sitaru et al. demonstrated that F(ab’)2 fragments of pathogenic antibodies against BP180-NC16A, which lack the Fc tail required for complement activation, did not induce dermal-epidermal separation in cryosections of human skin (105). Furthermore, Chiorean et al. recently demonstrated that the functional complement activation capacity of autoantibodies ex vivo in BP correlates with disease severity and autoantibody levels (96).
An IgA autoimmune response to BP180 and BP230 was also demonstrated in PDs, especially in MMP (106). It was shown to induce dermal epidermal separation via FcαRI-mediated neutrophil activation (107). However, there is contradictory data regarding IgA’s potential to activate complement (108, 109). With regard to PDs, neither IgA1 nor IgA2 were able to induce complement deposition at the DEJ in cryosections of human skin. However, they still could amplify the complement activation pathways via AP activation, which is a major source of the proinflammatory C5a (110, 111). This might explain the stronger ability of both IgA1 and IgA2 to activate neutrophils, as well as to induce blistering without the CP activation.
3 Complement-independent pathogenic pathways in PDs
3.1 Experimental lines of evidence
First evidence pointing towards direct, complement-independent blistering in PDs was presented by Kitajima et al.: Here, binding of anti-BP180 IgG to the lateral-apical cell surface of basal cells led to internalization of the BP180-IgG immune complex in both cultured keratinocytes and biopsy specimens from BP patients (112, 113). Furthermore, Iwata et al. corroborated this finding by demonstrating that pathogenic autoantibodies from BP patients not only significantly depleted the hemidesmosomal BP180 content from cultured keratinocytes, but also reduced their adhesive strength to the basement membrane, as determined in a standardized detachment assay using vibration (114). Interestingly, the α6 and β4 integrin levels of hemidesmosomes were not altered, thus underlining the putative specificity of the anti-BP180 IgG-mediated effect. The mechanism behind the internalization of the whole immune complex is attributed to the macropinocytosis pathway via calcium-dependent phosphorylation of the intracellular domain of BP180 by the protein kinase C (115, 116) (Figure 2B).
In line, in a neonatal BP180-humanized mouse model, Natsuga et al. were able to show that transfer of rabbit and human F(ab′)2-fragments against the immunodominant human NC16A domain of BP180 induced dermal-epidermal separation by mechanical stress, and also reduced the expression of BP180 in mouse skin as shown by immunoblotting (117). However, not all mice injected with F(ab′)2-fragments against NC16A showed skin detachment, pointing towards several synergistic pathways including Fc-mediated complement activation, potentially contributing to autoantibody-induced tissue pathology in BP (118).
Further in vivo data revealed that BP180 internalization and degradation via the ubiquitin/proteasome pathway is sufficient to induce blister formation in a C3-deficient BP180-humanized mouse model (119). Mice injected with a recombinant human IgG4 monoclonal antibody against the human NC16A domain of BP180 developed blisters, even though the IgG4 subclass does not fix complement, and also a proteasome inhibitor was added simultaneously. These results imply that BP180 internalization with subsequent hemidesmosomal weakening, followed by BP180 degradation via proteasome pathway may suffice for blister formation. Surprisingly, IgG4 antibodies from BP patients were found to induce dermal-epidermal separation in human cryosections, although with a much lower potential compared to IgG1 antibodies (120). Conversely, Zuo et al. suggested a protective role for IgG4 anti-NC16A antibodies in BP. He showed that the transfer of human IgG4 anti-NC16A to humanized BP180 mice inhibited human IgG1 and IgG3 induced complement activation with subsequent neutrophil infiltration, preventing both clinical and histological blistering in a dose-dependent manner (121). Correspondingly, IgG4 mitigates allergic diseases by inhibiting the activity of IgE (122). Based on these findings, it is tempting to speculate that IgG4 anti-NC16A might abrogate complement-dependent blister formation in BP. More studies are needed to fully clarify the role of IgG4 antibodies in PDs (38, 123).
Previously cited studies were nonetheless performed in neonatal mice, making data interpretation difficult at times. It was shown that neonatal mice do not entirely reproduce the clinical disease, since the majority of patients with PDs are adults and elderly, and lesions develop only with the application of friction in neonatal mice (24, 124). Moreover, there are known immunological differences between species, as well as in neutrophil function and skin physiology between neonatal and adult mice, and between murine and human skin, respectively (124, 125).
3.2 Clinical lines of evidence
Dainichi et al. reported two unusual BP cases with no complement deposition at the DEJ (126). Both patients had predominant IgG4 antibody involvement, which has restricted complement activating abilities. In line, approximately 20% of BP patients did not show C3 deposition at the DEJ in a large cohort of patients, and of these, the majority had prevalent IgG4 antibodies (30). Moreover, BP patients presenting with the non-blistering phenotype showed also less C3 deposition and previous studies hinted towards a IgG4 predominance in C3-negative cases (30, 127). In addition, BP-IgG4 antibodies induced subepidermal split formation ex vivo (120). Therefore, Dainichi et al. proposed a new entity in PDs, the so called “C3-negative BP” or “IgG4-dominant BP” (128). Given that the detection of C3 at the basement membrane is highly more sensitive than IgG for the diagnosis of BP by DIF microscopy, C3-negative DIF may require additional diagnostic methods or reconsidering the initial diagnosis (39). On the other hand, Boch et al. recently described 3 PD patients with weak or no C3 deposition, but with exclusive IgM reactivity at the cutaneous basement membrane (129). Interestingly, these patients also manifested with pruritic erythematous lesions without macroscopic or microscopic blistering. Since IgM is usually a strong inducer of complement activation, these findings further suggest that the activation of complement at the DEJ is indeed required for blister formation in PDs (108).
Both IgG1 and IgG4 against the NC16A domain of BP180 antibodies prevail in BP, and their serum levels significantly correlate with disease severity and poor prognosis (38, 104, 123). Interestingly, the IgG4 subclass was the first to be detected as well as the most prevalent in the prodromal, papular and urticarial BP variant (130). Since almost half of the IgG4 positive BP patients had also other IgG subclasses, and that IgG4 prevented IgG1 and IgG3 induced complement activation and final blister formation in mice, one might easily assume that IgG4 antibodies may play an essential role in disease induction, especially in the non-blistering phase, exerting also potential inhibitory effects as shown by Zuo et al. (121, 130). Conversely, the antibody switch to complement-fixing IgG subclasses will promote characteristic blister formation in BP (38, 107). Despite the fact that IgG4 antibodies do not fix complement, all patients with the non-blistering phenotype and IgG4 predominance still presented C3 and/or C5-9 deposition, suggesting early complement activation even with less IgG deposition at the DEJ and without blister formation (130). These findings underscore the utility of complement detection in the diagnosis of PDs, even in earlier disease stages. Recent data suggests that IgG4 might unexpectedly activate complement through both the CP and LP (131).
The report of a C4-deficient BP patient further questioned the role of complement as a prerequisite for blister development (132). Interestingly, this patient still showed linear C3 deposition at the basement membrane, implying non-canonical complement activation pathways. In contrast, C4-deficient neonatal mice were protected from BP development (55, 132). However, the administration of IL-8, which is an important polymorphonuclear neutrophil chemoattractant, restored disease susceptibility of these C4-deficient mice (55, 64). In line with this, cultured NHEKs with BP-IgG or -IgE anti-NC16A antibodies directly induced the expression of IL-6 and IL-8, whereas control IgG or BP180-deficient keratinocytes did not (133, 134). These findings suggest that the binding of BP180 IgG may induce blister formation without complement activation, rather by attracting neutrophils, which release various proteases and ROS that inflict characteristic tissue damage. Accordingly, neutrophils were shown to be indispensable for dermal-epidermal separation in both ex vivo murine cryosections and in vivo passive mouse models of PDs (64, 105, 134–136). In this sense, complement may at least maintain the inflammatory response in PD patients in a positive feedback loop pattern (39, 59, 117). On the other hand, it was shown that proinflammatory cytokines (i.e., IL-6) may actually activate complement (137, 138). More studies are needed to evaluate these complement-dependent and -independent pathways concomitantly, since one does not rule out the other, and most PD patients do feature complement fixation at the DEJ.
Notwithstanding the critical role of neutrophils in mice, eosinophil-predominant inflammatory infiltrates in the papillary dermis and eosinophilic spongiosis represent the main histological hallmarks of BP in humans (39). Furthermore, the majority of BP patients show also elevated levels of eosinophils and IgE antibodies in their sera, skin and blister fluid, respectively (139, 140). In addition, about 70% have specific IgE autoantibodies against both the NC16A and non-NC16A domains of BP180 (139, 141). Eosinophils are considered the liaison between IgE autoantibodies and skin blistering in BP. Lin et al. showed in a humanized IgE receptor mouse model of BP that IgE-mediated blistering relies on eosinophils, since eosinophil-deficient mice were protected from IgE-induced blister formation (61). Moreover, the degree of eosinophil infiltration correlated with disease severity. It seems that eosinophils are most prevalent in early urticarial lesions and that they degranulate at the basement membrane before blister formation (142). Given the fact that IgE antibodies are indicative of type I hypersensitivity responses, together with eosinophils they may be the driver of the prodromal, non-blistering phase of BP, which could not be unraveled by the clearly established IgG deposition-induced complement activation and immune effector cells recruitment paradigm. Even though IgE antibodies do not activate complement, both in vitro and in vivo data demonstrated that specific anti-BP180 IgE antibodies were still able to induce dermal-epidermal separation (62, 134, 143). However, many BP patients have both specific IgG and IgE antibodies in their sera, with the latter predominantly found on eosinophils and mast cells, and very rarely, in a discontinuous pattern along the basement membrane (144–146). Since eosinophils express the high-affinity IgE receptor FcϵRI, this might explain how anti-BP180 IgE antibodies can activate them (69). On the other hand, in vitro studies showed that full-length Ig as well as F(ab’)2 fragments from BP-IgG and -IgE are able to decrease the hemidesmosomes number by cytokine secretion, thereby contributing to blister formation in a FcR-independent manner (134). Moreover, BP-IgE antibodies seem to target the same NC16A domain as pathogenic IgG antibodies (146). Interestingly, Messingham et al. demonstrated by using a cryosection model of BP that eosinophil localization along the DEJ is dependent on IgG and complement deposition rather than on IgE (147). No subepidermal split was however observed in this study. To induce blister formation eosinophils required IL-5 mediated activation, also prompted by IgG deposition and complement fixation (148). Considering that most BP patients have both IgG and IgE antibodies, and that each of them may not fully explain all pathologies observed in blister formation, a potential way to integrate all the above-mentioned findings is the following: to induce blistering, specific anti-BP180 IgE autoantibodies require the presence of eosinophils, which are recruited and activated mainly as a result of IgG deposition and subsequent complement activation. Given these results, future studies should sequentially connect and integrate these distinct pathways rather than dissect and presume the existence of solely one in the intricate pathophysiology of blister formation in PDs.
4 Complement – useful or dispensable?
In the 90s numerous studies showed that complement is indispensable for blister formation in ex vivo cryosections and in vivo experimental mouse models of PDs (52, 53, 64, 90, 95, 149). Furthermore, the detection of C3 along the basement membrane by DIF was used since then as an important diagnostic hallmark of PDs (24, 30, 32). However, Iwata et al. questioned the necessity of complement by demonstrating that BP autoantibodies were able to deplete BP180 content in cultured keratinocytes, which in turn led to an increase in cells’ detachment implicating loss of function (114). Further research showed potential complement-independent pathways also in experimental mouse models (117, 119). However, these studies were performed in neonatal and complement knock-out mice, with the former exhibiting many limitations when compared to adult mice, and the latter being not physiologic, since patients with PDs with an additional complement component deficiency are rather an exception than the rule. Notwithstanding that Natsuga et al. demonstrated that BP antigen specific human or rabbit derived F(ab′)2-fragments were still able to induce disease, this reflects merely a theoretical potential of antibodies than a suggestion of the dispensability of complement, considering that full, complement-activating Ig molecules are prevalent in human sera and tissue, whereas F(ab’)2 fragments are artificial (117, 150).
Even though these data may refute the necessity of complement in PDs, they rather provide new insights into PD pathogenesis, suggesting that complement-dependent and -independent mechanisms may indeed coexist in patients at the same time. Even if complement is not absolutely indispensable to induce blister formation, we propose that it is essential in the amplification of characteristic inflammation and tissue damage, thus contributing to disease severity. Therefore, eliminating complement activation in PDs may significantly ameliorate disease, as data from in vitro and in vivo studies suggest (151, 152).
In view of this, different complement-targeting therapies have been specifically developed for PDs. Among these are sutimlimab and nomacopan. In a phase I trial, sutimlimab, a humanized monoclonal IgG4 antibody directed against the C1s subunit of human complement component C1, was shown to partially or completely abrogate C3 deposition along the DEJ in BP patients (153). Nomacopan (formerly known as coversin) is a bifunctional inhibitor of both C5 and leukotriene B4 (88, 154). EBA mice treated with coversin were almost completely protected from disease development mainly due to the C5-inhibitory effect of this compound (88). More recently, Sadik et al. demonstrated in a phase IIa clinical trial that nomacopan successfully reduced the clinical disease severity in BP patients, without any serious adverse event (152). Furthermore, several other complement-targeting treatments have been developed that so far have not been evaluated in preclinical and clinical settings (151, 154).
Piecing these data together, we conclude from the evidence published so far that complement remains an important and, in most cases, an indispensable hallmark of both human and experimental models of PDs and that complement-targeting therapies are effective and safe treatment strategies for these patients.
Author contributions
All authors contributed to the writing and review of this manuscript. All authors approved the final version of this manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by DFG funding for the Collaborative Research Center (CRC) 1526 – Pathomechanisms of Antibody-mediated Autoimmunity (PANTAU) – Insights from Pemphigoid Diseases (Grant No.: 454193335), the CRU 303 P7, the Research Training Group “Autoimmune Pre-Disease” (GRK 2633), the Schleswig-Holstein Excellence-Chair Program from the State of Schleswig Holstein, the Excellence Cluster EXC 2167 Precision Medicine in Chronic Inflammation (TI-4), and the Sinergia Unravel principles of self-organization in injured tissue (CRSII5_202301/1) from the Swiss National Science Foundation.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Trouw LA, Pickering MC, Blom AM. The complement system as a potential therapeutic target in rheumatic disease. Nat Rev Rheumatol (2017) 13(9):538–47. doi: 10.1038/nrrheum.2017.125
2. Walport MJ. Complement. first of two parts. New Engl J Med (2001) 344(14):1058–66. doi: 10.1056/NEJM200104053441406
3. Walport MJ. Complement. second of two parts. New Engl J Med (2001) 344(15):1140–4. doi: 10.1056/NEJM200104123441506
4. Meri S, Jarva H. Complement regulation. Vox Sanguinis (1998) 74(S2):291–302. doi: 10.1111/j.1423-0410.1998.tb05434.x
5. Bathum L, Hansen H, Teisner B, Koch C, Garred P, Rasmussen K, et al. Association between combined properdin and mannose-binding lectin deficiency and infection with neisseria meningitidis. Mol Immunol (2006) 43(5):473–9. doi: 10.1016/j.molimm.2005.02.017
6. Schneider MC, Exley RM, Ram S, Sim RB, Tang CM. Interactions between neisseria meningitidis and the complement system. Trends Microbiol (2007) 15(5):233–40. doi: 10.1016/j.tim.2007.03.005
7. Linton SM, Morgan BP. Properdin deficiency and meningococcal disease–identifying those most at risk. Clin Exp Immunol (2001) 118(2):189–91. doi: 10.1046/j.1365-2249.1999.01057.x
8. Kallio-Laine K, Seppänen M, Aittoniemi J, Kautiainen H, Seppälä I, Valtonen V, et al. HLA-DRB1*01 allele and low plasma immunoglobulin G1 concentration may predispose to herpes-associated recurrent lymphocytic meningitis. Hum Immunol (2010) 71(2):179–81. doi: 10.1016/j.humimm.2009.10.014
9. Yang Y, Lhotta K, Chung EK, Eder P, Neumair F, Yu CY. Complete complement components C4A and C4B deficiencies in human kidney diseases and systemic lupus erythematosus. J Immunol (2004) 173(4):2803–14. doi: 10.4049/jimmunol.173.4.2803
10. Koch A. Acute respiratory tract infections and mannose-binding lectin insufficiency during early childhood. JAMA (2001) 285(10):1316. doi: 10.1001/jama.285.10.1316
11. Mold C. Role of complement in host defense against bacterial infection. Microbes Infect (1999) 1(8):633–8. doi: 10.1016/S1286-4579(99)80063-X
12. Frank MM. Complement disorders and hereditary angioedema. J Allergy Clin Immunol (2010) 125(2):S262–71. doi: 10.1016/j.jaci.2009.10.063
13. Cicardi M, Zanichelli A. Acquired angioedema. Allergy Asthma Clin Immunol (2010) 6(1):14. doi: 10.1186/1710-1492-6-14
14. Dauchel H, Joly P, Delpech A, Thomine E, Sauger E, Loet X, et al. Local and systemic activation of the whole complement cascade in human leukocytoclastic cutaneous vasculitis; C3d,g and terminal complement complex as sensitive markers. Clin Exp Immunol (2008) 92(2):274–83. doi: 10.1111/j.1365-2249.1993.tb03392.x
15. Fischetti F, Tedesco F. Cross-talk between the complement system and endothelial cells in physiologic conditions and in vascular diseases. Autoimmunity (2006) 39(5):417–28. doi: 10.1080/08916930600739712
16. Jara LJ, Navarro C, Medina G, Vera-Lastra O, Saavedra MA. Hypocomplementemic urticarial vasculitis syndrome. Curr Rheumatol Rep (2009) 11(6):410–5. doi: 10.1007/s11926-009-0060-y
17. Truedsson L, Bengtsson AA, Sturfelt G. Complement deficiencies and systemic lupus erythematosus. Autoimmunity (2007) 40(8):560–6. doi: 10.1080/08916930701510673
18. Weinstein A, Alexander RV, Zack DJ. A review of complement activation in SLE. Curr Rheumatol Rep (2021) 23(3):16. doi: 10.1007/s11926-021-00984-1
19. Pasch M, Bos JD, Asghar SS. Activation of complement in psoriasis. Clin Exp Dermatol (1998) 23(4):189–90. doi: 10.1046/j.1365-2230.1998.00360.x
20. Giacomassi C, Buang N, Ling GS, Crawford G, Cook HT, Scott D, et al. Complement C3 exacerbates imiquimod-induced skin inflammation and psoriasiform dermatitis. J Invest Dermatol (2017) 137(3):760–3. doi: 10.1016/j.jid.2016.11.011
21. Scott DG, Cunliffe WJ, Gowland G. Activation of complement-a mechanism for the inflammation in acne. Br J Dermatol (2006) 101(3):315–20. doi: 10.1111/j.1365-2133.1979.tb05625.x
22. Leeming JP, Ingham E, Cunliffe WJ. The microbial content and complement C3 cleaving capacity of comedones in acne vulgaris. Acta Derm Venereol (1988) 68(6):468–73.
23. Kanni T, Zenker O, Habel M, Riedemann N, Giamarellos-Bourboulis EJ. Complement activation in hidradenitis suppurativa: a new pathway of pathogenesis? Br J Dermatol (2018) 179(2):413–9. doi: 10.1111/bjd.16428
24. Schmidt E, Zillikens D. Pemphigoid diseases. Lancet (2013) 381(9863):320–32. doi: 10.1016/S0140-6736(12)61140-4
25. Schmidt E, Kasperkiewicz M, Joly P. Pemphigus. Lancet (2019) 394(10201):882–94. doi: 10.1016/S0140-6736(19)31778-7
26. Ujiie H, Rosmarin D, Schön MP, Ständer S, Boch K, Metz M, et al. Unmet medical needs in chronic, non-communicable inflammatory skin diseases. Front Med (2022) 9. doi: 10.3389/fmed.2022.875492
27. Hashimoto T, Nishikawa T, Kurihara S, Hatano H. Complement-fixing pemphigus antibodies. Arch Dermatol (1978) 114(8):1191–2. doi: 10.1001/archderm.1978.01640200045011
28. Hashimoto T, Sugiura M, Kurihara S, Nishikawa T, Hatano H. Experimental acantholysis by complement-fixing intercellular antibodies. Arch Dermatol Res (1982) 273(1–2):129–35. doi: 10.1007/BF00509037
29. Arbache ST, Nogueira TG, Delgado L, Miyamoto D, Aoki V. Immunofluorescence testing in the diagnosis of autoimmune blistering diseases: overview of 10-year experience. Anais Brasileiros Dermatol (2014) 89(6):885–9. doi: 10.1590/abd1806-4841.20143221
30. Romeijn TR, Jonkman MF, Knoppers C, Pas HH, Diercks GFH. Complement in bullous pemphigoid: results from a large observational study. Br J Dermatol (2017) 176(2):517–9. doi: 10.1111/bjd.14822
31. Intong LRA, Murrell DF. Pemphigoid gestationis: Pathogenesis and clinical features. Dermatol Clinics (2011) 29(3):447–52. doi: 10.1016/j.det.2011.03.002
32. van Beek N, Zillikens D, Schmidt E. Bullous autoimmune dermatoses. Deutsches Ärzteblatt Int (2021) 118(24):413–20. doi: 10.3238/arztebl.m2021.0136
33. Kasprick A, Holtsche MM, Rose EL, Hussain S, Schmidt E, Petersen F, et al. The anti-C1s antibody TNT003 prevents complement activation in the skin induced by bullous pemphigoid autoantibodies. J Invest Dermatol (2018) 138(2):458–61. doi: 10.1016/j.jid.2017.08.030
34. Genovese G, di Zenzo G, Cozzani E, Berti E, Cugno M, Marzano AV. New insights into the pathogenesis of bullous pemphigoid: 2019 update. Front Immunol (2019) 10:1506/full(1506). doi: 10.3389/fimmu.2019.01506/full
35. Lessey E, Li N, Diaz L, Liu Z. Complement and cutaneous autoimmune blistering diseases. Immunol Res (2008) 41(3):223–32. doi: 10.1007/s12026-008-8028-y
36. Sitaru C, Chiriac MT, Mihai S, Büning J, Gebert A, Ishiko A, et al. Induction of complement-fixing autoantibodies against type VII collagen results in subepidermal blistering in mice. J Immunol (2006) 177(5):3461–8. doi: 10.4049/jimmunol.177.5.3461
37. Chimanovitch I, Schmidt E, Döpp R, Bröcker EB, Zillikens D, Messer G, et al. IgG1 and IgG3 are the major immunoglobulin subclasses targeting epitopes within the NC16A domain of BP180 in pemphigoid gestationis. J Invest Dermatol (1999) 113(1):140–2. doi: 10.1046/j.1523-1747.1999.00622.x
38. Sitaru C, Mihai S, Zillikens D. The relevance of the IgG subclass of autoantibodies for blister induction in autoimmune bullous skin diseases. Arch Dermatol Res (2007) 299(1):1–8. doi: 10.1007/s00403-007-0734-0
39. Hammers CM, Stanley JR. Mechanisms of disease: Pemphigus and bullous pemphigoid. Annu Rev Pathol: Mech Dis (2016) 11(1):175–97. doi: 10.1146/annurev-pathol-012615-044313
40. Chorzelski TP, Cormane RH. The presence of complement “Bound” in vivo in the skin of patients with pemphigoid. Dermatology (1968) 137(3):134–8. doi: 10.1159/000254042
41. Jordon RE, Sams WM, Beutner EH. Complement immunofluorescent staining in bullous pemphigoid. J Lab Clin Med (1969) 74(4):548–56.
42. Jordon RE, Day NK, Sams WM, Good RA. The complement system in bullous pemphigoid. i. complement and component levels in sera and blister fluids. J Clin Invest (1973) 52(5):1207–14. doi: 10.1172/JCI107288
43. Jordon RE, McDuffie FC. Serum and blister fluid anticomplementary activity in pemphigus and bullous pemphigoid. sucrose density gradient studies. Proc Soc Exp Biol Med (1976) 151(3):594–8. doi: 10.3181/00379727-151-39267
44. Jordon RE, Nordby JM, Milstein H. The complement system in bullous pemphigoid. III. fixation of C1q and C4 by pemphigoid antibody. J Lab Clin Med (1975) 86(5):733–40. doi: 10.1016/0090-1229(75)90017-3
45. Provost TT, Tomasi TB. Evidence for complement activation via the alternate pathway in skin diseases, i. herpes gestationis, systemic lupus erythematosus, and bullous pemphigoid. J Clin Invest (1973) 52(7):1779–87. doi: 10.1172/JCI107359
46. Jordon RE. Complement activation in bullous skin diseases. J Invest Dermatol (1975) 65(1):162–9. doi: 10.1111/1523-1747.ep12598113
47. Millns JL, Meurer M, Jordon RE. The complement system in bullous pemphigoid. VI. C3 fixing activity in the absence of detectable antibody. Clin Immunol Immunopathol (1979) 13(4):475–83. doi: 10.1016/0090-1229(79)90090-4
48. Diaz-Perez JL, Jordon RE. The complement system in bullous pemphigoid. IV. chemotactic activity in blister fluid. Clin Immunol Immunopathol (1976) 5(3):360–70. doi: 10.1016/0090-1229(76)90045-3
49. Jordon RE, Bushkell LL. THE COMPLEMENT SYSTEM IN PEMPHIGUS, BULLOUS PEMPHIGOID AND HERPES GESTATIONIS. Int J Dermatol (1979) 18(4):271–81. doi: 10.1111/j.1365-4362.1979.tb01927.x
50. Yamada H, Hashimoto T, Nishikawa T. IgG subclasses of intercellular and basement membrane zone antibodies: The relationship to the capability of complement fixation. J Invest Dermatol (1989) 92(4):585–7. doi: 10.1111/1523-1747.ep12709613
51. Schmidt-Ullrich B, Rule A, Schaumburg-Lever G, Leblanc C. Ultrastructural localization of in vivo-bound complement in bullous pemphigoid. J Invest Dermatol (1975) 65(2):217–9. doi: 10.1111/1523-1747.ep12598218
52. Liu Z, Diaz LA, Troy JL, Taylor AF, Emery DJ, Fairley JA, et al. A passive transfer model of the organ-specific autoimmune disease, bullous pemphigoid, using antibodies generated against the hemidesmosomal antigen, BP180. J Clin Invest (1993) 92(5):2480–8. doi: 10.1172/JCI116856
53. Liu Z, Giudice GJ, Swartz SJ, Fairley JA, Till GO, Troy JL, et al. The role of complement in experimental bullous pemphigoid. J Clin Invest (1995) 95(4):1539–44. doi: 10.1172/JCI117826
54. Yamamoto K, Inoue N, Masuda R, Fujimori A, Saito T, Imajoh-Ohmi S, et al. Cloning of hamster type XVII collagen cDNA, and pathogenesis of anti-type XVII collagen antibody and complement in hamster bullous pemphigoid. J Invest Dermatol (2002) 118(3):485–92. doi: 10.1046/j.0022-202x.2001.01683.x
55. Nelson KC, Zhao M, Schroeder PR, Li N, Wetsel RA, Diaz LA, et al. Role of different pathways of the complement cascade in experimental bullous pemphigoid. J Clin Invest (2006) 116(11):2892–900. doi: 10.1172/JCI17891
56. Nishie W, Sawamura D, Goto M, Ito K, Shibaki A, McMillan JR, et al. Humanization of autoantigen. Nat Med (2007) 13(3):378–83. doi: 10.1038/nm1496
57. Liu Z, Sui W, Zhao M, Li Z, Li N, Thresher R, et al. Subepidermal blistering induced by human autoantibodies to BP180 requires innate immune players in a humanized bullous pemphigoid mouse model. J Autoimmun (2008) 31(4):331–8. doi: 10.1016/j.jaut.2008.08.009
58. Li Q, Ujiie H, Shibaki A, Wang G, Moriuchi R, Qiao HJ, et al. Human IgG1 monoclonal antibody against human collagen 17 noncollagenous 16A domain induces blisters via complement activation in experimental bullous pemphigoid model. J Immunol (2010) 185(12):7746–55. doi: 10.4049/jimmunol.1000667
59. Karsten CM, Beckmann T, Holtsche MM, Tillmann J, Tofern S, Schulze FS, et al. Tissue destruction in bullous pemphigoid can be complement independent and may be mitigated by C5aR2. Front Immunol (2018) 9:488. doi: 10.3389/fimmu.2018.00488
60. Sezin T, Krajewski M, Wutkowski A, Mousavi S, Chakievska L, Bieber K, et al. The leukotriene B4 and its receptor BLT1 act as critical drivers of neutrophil recruitment in murine bullous pemphigoid-like epidermolysis bullosa acquisita. J Invest Dermatol (2017) 137(5):1104–13. doi: 10.1016/j.jid.2016.12.021
61. Lin L, Hwang BJ, Culton DA, Li N, Burette S, Koller BH, et al. Eosinophils mediate tissue injury in the autoimmune skin disease bullous pemphigoid. J Invest Dermatol (2018) 138(5):1032–43. doi: 10.1016/j.jid.2017.11.031
62. Zone JJ, Taylor T, Hull C, Schmidt L, Meyer L. IgE basement membrane zone antibodies induce eosinophil infiltration and histological blisters in engrafted human skin on SCID mice. J Invest Dermatol (2007) 127(5):1167–74. doi: 10.1038/sj.jid.5700681
63. Heimbach L, Li Z, Berkowitz P, Zhao M, Li N, Rubenstein DS, et al. The C5a receptor on mast cells is critical for the autoimmune skin-blistering disease bullous pemphigoid. J Biol Chem (2011) 286(17):15003–9. doi: 10.1074/jbc.M111.221036
64. Liu Z, Giudice GJ, Zhou X, Swartz SJ, Troy JL, Fairley JA, et al. A major role for neutrophils in experimental bullous pemphigoid. J Clin Invest (1997) 100(5):1256–63. doi: 10.1172/JCI119639
65. Chen R, Ning G, Zhao ML, Fleming MG, Diaz LA, Werb Z, et al. Mast cells play a key role in neutrophil recruitment in experimental bullous pemphigoid. J Clin Invest (2001) 108(8):1151–8. doi: 10.1172/JCI11494
66. Lin L, Bankaitis E, Heimbach L, Li N, Abrink M, Pejler G, et al. Dual targets for mouse mast cell protease-4 in mediating tissue damage in experimental bullous pemphigoid. J Biol Chem (2011) 286(43):37358–67. doi: 10.1074/jbc.M111.272401
67. Tchougounova E, Lundequist A, Fajardo I, Winberg JO, Åbrink M, Pejler G. A key role for mast cell chymase in the activation of pro-matrix metalloprotease-9 and pro-matrix metalloprotease-2. J Biol Chem (2005) 280(10):9291–6. doi: 10.1074/jbc.M410396200
68. Dvorak AM, Mihm MC, Osage JE, Kwan TH, Austen KF, Wintroub BU. Bullous pemphigoid, an ultrastructural study of the inflammatory response: eosinophil, basophil and mast cell granule changes in multiple biopsies from one patient. J Invest Dermatol (1982) 78(2):91–101. doi: 10.1111/1523-1747.ep12505711
69. Messingham KN, Holahan HM, Frydman AS, Fullenkamp C, Srikantha R, Fairley JA. Human eosinophils express the high affinity IgE receptor, FcϵRI, in bullous pemphigoid. PloS One (2014) 9(9):e107725. doi: 10.1371/journal.pone.0107725
70. Zeck-Kapp G, Kroegel C, Riede UN, Kapp A. Mechanisms of human eosinophil activation by complement protein C5a and platelet-activating factor: similar functional responses are accompanied by different morphologic alterations. Allergy (1995) 50(1):34–47. doi: 10.1111/j.1398-9995.1995.tb02481.x
71. Takafuji S, Tadokoro K, lto K, Dahinden CA. Degranulation from human eosinophils stimulated with C3a and C5a. Int Arch Allergy Immunol (1994) 104(1):27–9. doi: 10.1159/000236743
72. Koga H, Prost-Squarcioni C, Iwata H, Jonkman MF, Ludwig RJ, Bieber K. Epidermolysis bullosa acquisita: The 2019 update. Front Med (Lausanne) (2018) 5:362. doi: 10.3389/fmed.2018.00362
73. Bieber K, Koga H, Nishie W. In vitro and in vivo models to investigate the pathomechanisms and novel treatments for pemphigoid diseases. Exp Dermatol (2017) 26(12):1163–70. doi: 10.1111/exd.13415
74. Sitaru C, Mihai S, Otto C, Chiriac MT, Hausser I, Dotterweich B, et al. Induction of dermal-epidermal separation in mice by passive transfer of antibodies specific to type VII collagen. J Clin Invest (2005) 115(4):870–8. doi: 10.1172/JCI200521386
75. Woodley DT, Ram R, Doostan A, Bandyopadhyay P, Huang Y, Remington J, et al. Induction of epidermolysis bullosa acquisita in mice by passive transfer of autoantibodies from patients. J Invest Dermatol (2006) 126(6):1323–30. doi: 10.1038/sj.jid.5700254
76. Mihai S, Chiriac MT, Takahashi K, Thurman JM, Holers VM, Zillikens D, et al. The alternative pathway of complement activation is critical for blister induction in experimental epidermolysis bullosa acquisita. J Immunol (2007) 178(10):6514–21. doi: 10.4049/jimmunol.178.10.6514
77. Mihai S, Hirose M, Wang Y, Thurman JM, Holers VM, Morgan BP, et al. Specific inhibition of complement activation significantly ameliorates autoimmune blistering disease in mice. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.00535
78. Peschke B, Keller CW, Weber P, Quast I, Lünemann JD. Fc-galactosylation of human immunoglobulin gamma isotypes improves C1q binding and enhances complement-dependent cytotoxicity. Front Immunol (2017) 8. doi: 10.3389/fimmu.2017.00646
79. Clauder AK, Kordowski A, Bartsch YC, Köhl G, Lilienthal GM, Almeida LN, et al. IgG Fc N-glycosylation translates MHCII haplotype into autoimmune skin disease. J Invest Dermatol (2021) 141(2):285–94. doi: 10.1016/j.jid.2020.06.022
80. Thielens NM, Tedesco F, Bohlson SS, Gaboriaud C, Tenner AJ. C1q: A fresh look upon an old molecule. Mol Immunol (2017) 89:73–83. doi: 10.1016/j.molimm.2017.05.025
81. Kolev M, le Friec G, Kemper C. Complement — tapping into new sites and effector systems. Nat Rev Immunol (2014) 14(12):811–20. doi: 10.1038/nri3761
82. Gadjeva M, Verschoor A, Brockman MA, Jezak H, Shen LM, Knipe DM, et al. Macrophage-derived complement component C4 can restore humoral immunity in C4-deficient mice. J Immunol (2002) 169(10):5489–95. doi: 10.4049/jimmunol.169.10.5489
83. Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, et al. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med (2006) 12(6):682–7. doi: 10.1038/nm1419
84. Jiang Y, Tsoi LC, Billi AC, Ward NL, Harms PW, Zeng C, et al. Cytokinocytes: the diverse contribution of keratinocytes to immune responses in skin. JCI Insight (2020) 5(20):e142067. doi: 10.1172/jci.insight.142067
85. Iwata H, Witte M, Samavedam UKSRL, Gupta Y, Shimizu A, Ishiko A, et al. Radiosensitive hematopoietic cells determine the extent of skin inflammation in experimental epidermolysis bullosa acquisita. J Immunol (2015) 195(5):1945–54. doi: 10.4049/jimmunol.1501003
86. Karsten CM, Pandey MK, Figge J, Kilchenstein R, Taylor PR, Rosas M, et al. Anti-inflammatory activity of IgG1 mediated by Fc galactosylation and association of FcγRIIB and dectin-1. Nat Med (2012) 18(9):1401–6. doi: 10.1038/nm.2862
87. Seiler DL, Kleingarn M, Kähler KH, Gruner C, Schanzenbacher J, Ehlers-Jeske E, et al. C5aR2 deficiency ameliorates inflammation in murine epidermolysis bullosa acquisita by regulating FcγRIIb expression on neutrophils. J Invest Dermatol (2022) S0022-202X(22)00001-X. doi: 10.1016/j.jid.2021.12.029
88. Sezin T, Murthy S, Attah C, Seutter M, Holtsche MM, Hammers CM, et al. Dual inhibition of complement factor 5 and leukotriene B4 synergistically suppresses murine pemphigoid disease. JCI Insight (2019) 4(15):e128239. doi: 10.1172/jci.insight.128239
89. Du G, Patzelt S, van Beek N, Schmidt E. Mucous membrane pemphigoid. Autoimmun Rev (2022) 21(4):103036. doi: 10.1016/j.autrev.2022.103036
90. Lazarova Z, Yee C, Darling T, Briggaman RA, Yancey KB. Passive transfer of anti-laminin 5 antibodies induces subepidermal blisters in neonatal mice. J Clin Invest (1996) 98(7):1509–18. doi: 10.1172/JCI118942
91. Lazarova Z, Hsu R, Briggaman RA, Yancey KB. Fab fragments directed against laminin 5 induce subepidermal blisters in neonatal mice. Clin Immunol (2000) 95(1):26–32. doi: 10.1006/clim.2000.4845
92. Heppe EN, Tofern S, Schulze FS, Ishiko A, Shimizu A, Sina C, et al. Experimental laminin 332 mucous membrane pemphigoid critically involves C5aR1 and reflects clinical and immunopathological characteristics of the human disease. J Invest Dermatol (2017) 137(8):1709–18. doi: 10.1016/j.jid.2017.03.037
93. Murthy S, Schilf P, Patzelt S, Thieme M, Becker M, Kröger L, et al. Dapsone suppresses disease in preclinical murine models of pemphigoid diseases. J Invest Dermatol (2021) 141(11):2587–2595.e2. doi: 10.1016/j.jid.2021.04.009
94. Rogers RS, Perry HO, Bean SF, Jordon RE. IMMUNOPATHOLOGY OF CICATRICIAL PEMPHIGOID: STUDIES OF COMPLEMENT DEPOSITION. J Invest Dermatol (1977) 68(1):39–43. doi: 10.1111/1523-1747.ep12485162
95. Mooney E, Falk RJ, Gammon WR. Studies on complement deposits in epidermolysis bullosa acquisita and bullous pemphigoid. Arch Dermatol (1992) 128(1):58–60. doi: 10.1001/archderm.1992.01680110068008
96. Chiorean RM, Baican A, Mustafa MB, Lischka A, Leucuta DC, Feldrihan V, et al. Complement-activating capacity of autoantibodies correlates with disease activity in bullous pemphigoid patients. Front Immunol (2018) 9:2687. doi: 10.3389/fimmu.2018.02687/full
97. Ständer S, Holtsche MM, Schmidt E, Hammers CM, Zillikens D, Ludwig RJ, et al. Presence of cutaneous complement deposition distinguishes between immunological and histological features of bullous pemphigoid–insights from a retrospective cohort study. J Clin Med (2020) 9(12):3928. doi: 10.3390/jcm9123928
98. Jordon RE, Kawana S, Fritz KA. Immunopathologic mechanisms in pemphigus and bullous pemphigoid. J Invest Dermatol (1985) 85(1):S72–8. doi: 10.1111/1523-1747.ep12275497
99. Pfaltz K, Mertz K, Rose C, Scheidegger P, Pfaltz M, Kempf W. C3d immunohistochemistry on formalin-fixed tissue is a valuable tool in the diagnosis of bullous pemphigoid of the skin. J Cutaneous Pathol (2010) 37(6):654–8. doi: 10.1111/j.1600-0560.2009.01450.x
100. Magro CM, Dyrsen ME. The use of C3d and C4d immunohistochemistry on formalin-fixed tissue as a diagnostic adjunct in the assessment of inflammatory skin disease. J Am Acad Dermatol (2008) 59(5):822–33. doi: 10.1016/j.jaad.2008.06.022
101. Chandler W, Zone J, Florell S. C4d immunohistochemical stain is a sensitive method to confirm immunoreactant deposition in formalin-fixed paraffin-embedded tissue in bullous pemphigoid. J Cutaneous Pathol (2009) 36(6):655–9. doi: 10.1111/j.1600-0560.2008.01129.x
102. Villani AP, Chouvet B, Kanitakis J. Application of C4d immunohistochemistry on routinely processed tissue sections for the diagnosis of autoimmune bullous dermatoses. Am J Dermatopathol (2016) 38(3):186–8. doi: 10.1097/DAD.0000000000000333
103. Kassaby SS, Hicks A, Leicht S, Youngberg GA. Bullous pemphigoid: Use of C4d immunofluorescent staining in a case with repeated negative conventional direct immunofluorescence studies. Am J Dermatopathol (2017) 39(12):932–4. doi: 10.1097/DAD.0000000000000943
104. Zhou XP, Liu B, Xu Q, Yang Y, He CX, Zuo YG, et al. Serum levels of immunoglobulins G1 and G4 targeting the non-collagenous 16A domain of BP180 reflect bullous pemphigoid activity and predict bad prognosis. J Dermatol (2016) 43(2):141–8. doi: 10.1111/1346-8138.13051
105. Sitaru C, Schmidt E, Petermann S, Munteanu LS, Bröcker EB, Zillikens D. Autoantibodies to bullous pemphigoid antigen 180 induce dermal–epidermal separation in cryosections of human skin. J Invest Dermatol (2002) 118(4):664–71. doi: 10.1046/j.1523-1747.2002.01720.x
106. Horváth B, Niedermeier A, Podstawa E, Müller R, Hunzelmann N, Kárpáti S, et al. IgA autoantibodies in the pemphigoids and linear IgA bullous dermatosis. Exp Dermatol (2010) 19(7):648–53. doi: 10.1111/j.1600-0625.2010.01080.x
107. Sitaru C, Zillikens D. Mechanisms of blister induction by autoantibodies. Exp Dermatol (2005) 14(12):861–75. doi: 10.1111/j.1600-0625.2005.00367.x
108. Daha NA, Banda NK, Roos A, Beurskens FJ, Bakker JM, Daha MR, et al. Complement activation by (auto-) antibodies. Mol Immunol (2011) 48(14):1656–65. doi: 10.1016/j.molimm.2011.04.024
109. Maillard N, Wyatt RJ, Julian BA, Kiryluk K, Gharavi A, Fremeaux-Bacchi V, et al. Current understanding of the role of complement in IgA nephropathy. J Am Soc Nephrol (2015) 26(7):1503–12. doi: 10.1681/ASN.2014101000
110. Banda NK, Levitt B, Wood AK, Takahashi K, Stahl GL, Holers VM, et al. Complement activation pathways in murine immune complex-induced arthritis and in C3a and C5a generation in vitro. Clin Exp Immunol (2009) 159(1):100–8. doi: 10.1111/j.1365-2249.2009.04035.x
111. Recke A, Trog LM, Pas HH, Vorobyev A, Abadpour A, Jonkman MF, et al. Recombinant human IgA1 and IgA2 autoantibodies to type VII collagen induce subepidermal blistering ex vivo. J Immunol (2014) 193(4):1600–8. doi: 10.4049/jimmunol.1400160
112. Kitajima Y, Hirako Y, Owaribe K, Mori S, Yaoita H. Antibody-binding to the 180-kD bullous pemphigoid antigens at the lateral cell surface causes their internalization and inhibits their assembly at the basal cell surface in cultured keratinocytes. J Dermatol (1994) 21(11):838–46. doi: 10.1111/j.1346-8138.1994.tb03299.x
113. Kitajima Y, Nojiri M, Yamada T, Hirako Y, Owaribe K. Internalization of the 180 kDa bullous pemphigoid antigen as immune complexes in basal keratinocytes: an important early event in blister formation in bullous pemphigoid. Br J Dermatol (1998) 138(1):71–6. doi: 10.1046/j.1365-2133.1998.02028.x
114. Iwata H, Kamio N, Aoyama Y, Yamamoto Y, Hirako Y, Owaribe K, et al. IgG from patients with bullous pemphigoid depletes cultured keratinocytes of the 180-kDa bullous pemphigoid antigen (Type XVII collagen) and weakens cell attachment. J Invest Dermatol (2009) 129(4):919–26. doi: 10.1038/jid.2008.305
115. Hiroyasu S, Ozawa T, Kobayashi H, Ishii M, Aoyama Y, Kitajima Y, et al. Bullous pemphigoid IgG induces BP180 internalization via a macropinocytic pathway. Am J Pathol (2013) 182(3):828–40. doi: 10.1016/j.ajpath.2012.11.029
116. Iwata H, Kamaguchi M, Ujiie H, Nishimura M, Izumi K, Natsuga K, et al. Macropinocytosis of type XVII collagen induced by bullous pemphigoid IgG is regulated via protein kinase c. Lab Invest (2016) 96(12):1301–10. doi: 10.1038/labinvest.2016.108
117. Natsuga K, Nishie W, Shinkuma S, Ujiie H, Nishimura M, Sawamura D, et al. Antibodies to pathogenic epitopes on type XVII collagen cause skin fragility in a complement-dependent and -independent manner. J Immunol (2012) 188(11):5792–9. doi: 10.4049/jimmunol.1003402
118. Ludwig RJ, Vanhoorelbeke K, Leypoldt F, Kaya Z, Bieber K, McLachlan SM, et al. Mechanisms of autoantibody-induced pathology. Front Immunol (2017) 8:603. doi: 10.3389/fimmu.2017.00603
119. Ujiie H, Sasaoka T, Izumi K, Nishie W, Shinkuma S, Natsuga K, et al. Bullous pemphigoid autoantibodies directly induce blister formation without complement activation. J Immunol (2014) 193(9):4415–28. doi: 10.4049/jimmunol.1400095
120. Mihai S, Chiriac MT, Herrero-González JE, Goodall M, Jefferis R, Savage COS, et al. IgG4 autoantibodies induce dermal–epidermal separation. J Cell Mol Med (2007) 11(5):1117–28. doi: 10.1111/j.1582-4934.2007.00081.x
121. Zuo Y, Evangelista F, Culton D, Guilabert A, Lin L, Li N, et al. IgG4 autoantibodies are inhibitory in the autoimmune disease bullous pemphigoid. J Autoimmun (2016) 73:111–9. doi: 10.1016/j.jaut.2016.06.019
122. James LK, Till SJ. Potential mechanisms for IgG4 inhibition of immediate hypersensitivity reactions. Curr Allergy Asthma Rep (2016) 16(3):23. doi: 10.1007/s11882-016-0600-2
123. Khil’chenko S, Boch K, van Beek N, Vorobyev A, Zillikens D, Schmidt E, et al. Alterations of total serum immunoglobulin concentrations in pemphigus and pemphigoid: Selected IgG2 deficiency in bullous pemphigoid. Front Med (2020) 7:472. doi: 10.3389/fmed.2020.00472
124. Bieber K, Sun S, Ishii N, Kasperkiewicz M, Schmidt E, Hirose M, et al. Animal models for autoimmune bullous dermatoses. Exp Dermatol (2010) 19(1):2–11. doi: 10.1111/j.1600-0625.2009.00948.x
125. Liechty KW, Schibler KR, Ohls RK, Perkins SL, Christensen RD. The failure of newborn mice infected with Escherichia coli to accelerate neutrophil production correlates with their failure to increase transcripts for granulocyte colony-stimulating factor and interleukin-6. Neonatology (1993) 64(5):331–40. doi: 10.1159/000244007
126. Dainichi T, Nishie W, Yamagami Y, Sonobe H, Ujiie H, Kaku Y, et al. Bullous pemphigoid suggestive of complement-independent blister formation with anti- BP 180 IgG4 autoantibodies. Br J Dermatol (2016) 175(1):187–90. doi: 10.1111/bjd.14411
127. Bird P, Friedmann PS, Ling N, Bird AG, Thompson RA. Subclass distribution of IgG autoantibodies in bullous pemphigoid. J Invest Dermatol (1986) 86(1):21–5. doi: 10.1111/1523-1747.ep12283737
128. Dainichi T, Chow Z, Kabashima K. IgG4, complement, and the mechanisms of blister formation in pemphigus and bullous pemphigoid. J Dermatol Sci (2017) 88(3):265–70. doi: 10.1016/j.jdermsci.2017.07.012
129. Boch K, Hammers CM, Goletz S, Kamaguchi M, Ludwig RJ, Schneider SW, et al. Immunoglobulin m pemphigoid. J Am Acad Dermatol (2021) 85(6):1486–92. doi: 10.1016/j.jaad.2021.01.017
130. Lamb PM, Patton T, Deng JS. The predominance of IgG4 in prodromal bullous pemphigoid. Int J Dermatol (2008) 47(2):150–3. doi: 10.1111/j.1365-4632.2008.03361.x
131. Kawa S. The immunobiology of immunoglobulin G4 and complement activation pathways in IgG4-related disease. Curr Top Microbiol Immunol (2017) 401:61–73. doi: 10.1007/82_2016_39
132. Shiraishi S, Iio T, Shirakata Y, Sayama K, Nishimukai H, Miki Y. Bullous pemphigoid in a patient with a C4 deficiency. Br J Dermatol (1991) 124(3):296–8. doi: 10.1111/j.1365-2133.1991.tb00579.x
133. Schmidt E, Reimer S, Jainta S, Bröcker EB, Zillikens D, Kruse N, et al. Autoantibodies to BP180 associated with bullous pemphigoid release interleukin-6 and interleukin-8 from cultured human keratinocytes. J Invest Dermatol (2000) 115(5):842–8. doi: 10.1046/j.1523-1747.2000.00141.x
134. Messingham KN, Srikantha R, DeGueme AM, Fairley JA. FcR-independent effects of IgE and IgG autoantibodies in bullous pemphigoid. J Immunol (2011) 187(1):553–60. doi: 10.4049/jimmunol.1001753
135. Shimanovich I, Mihai S, Oostingh GJ, Ilenchuk TT, Bröcker EB, Opdenakker G, et al. Granulocyte-derived elastase and gelatinase b are required for dermal–epidermal separation induced by autoantibodies from patients with epidermolysis bullosa acquisita and bullous pemphigoid. J Pathol (2004) 204(5):519–27. doi: 10.1002/path.1674
136. Sitaru C, Kromminga A, Hashimoto T, Bröcker EB, Zillikens D. Autoantibodies to type VII collagen mediate Fcγ-dependent neutrophil activation and induce dermal-epidermal separation in cryosections of human skin. Am J Pathol (2002) 161(1):301–11. doi: 10.1016/S0002-9440(10)64182-X
137. Katz Y, Revel M, Strunk RC. Interleukin 6 stimulates synthesis of complement proteins factor b and C3 in human skin fibroblasts. Eur J Immunol (1989) 19(6):983–8. doi: 10.1002/eji.1830190605
138. Hyc A, Osiecka-Iwan A, Strzelczyk P, Moskalewski S. Effect of IL-1beta, TNF-alpha and IL-4 on complement regulatory protein mRNA expression in human articular chondrocytes. Int J Mol Med (2003) 11(1):91–4. doi: 10.3892/ijmm.11.1.91
139. Messingham KN, Crowe TP, Fairley JA. The intersection of IgE autoantibodies and eosinophilia in the pathogenesis of bullous pemphigoid. Front Immunol (2019) 10:2331/full. doi: 10.3389/fimmu.2019.02331/full
140. Amber KT, Valdebran M, Kridin K, Grando SA. The role of eosinophils in bullous pemphigoid: A developing model of eosinophil pathogenicity in mucocutaneous disease. Front Med (2018) 5:201. doi: 10.3389/fmed.2018.00201
141. Fairley JA, Fu CL, Giudice GJ. Mapping the binding sites of anti-BP180 immunoglobulin E autoantibodies in bullous pemphigoid. J Invest Dermatol (2005) 125(3):467–72. doi: 10.1111/j.0022-202X.2005.23853.x
142. Borrego L, Maynard B, Peterson EA, George T, Iglesias L, Peters MS, et al. Deposition of eosinophil granule proteins precedes blister formation in bullous pemphigoid. comparison with neutrophil and mast cell granule proteins. Am J Pathol (1996) 148(3):897–909.
143. Fairley JA, Burnett CT, Fu CL, Larson DL, Fleming MG, Giudice GJ. A pathogenic role for IgE in autoimmunity: Bullous pemphigoid IgE reproduces the early phase of lesion development in human skin grafted to nu/nu mice. J Invest Dermatol (2007) 127(11):2605–11. doi: 10.1038/sj.jid.5700958
144. Pomponi D, di Zenzo G, Zennaro D, Calabresi V, Eming R, Zuzzi S, et al. Detection of IgG and IgE reactivity to BP180 using the ISAC ® microarray system. Br J Dermatol (2013) 168(6):1205–14. doi: 10.1111/bjd.12161
145. Hashimoto T, Ohzono A, Teye K, Numata S, Hiroyasu S, Tsuruta D, et al. Detection of IgE autoantibodies to BP180 and BP230 and their relationship to clinical features in bullous pemphigoid. Br J Dermatol (2017) 177(1):141–51. doi: 10.1111/bjd.15114
146. Freire PC, Muñoz CH, Stingl G. IgE autoreactivity in bullous pemphigoid: eosinophils and mast cells as major targets of pathogenic immune reactants. Br J Dermatol (2017) 177(6):1644–53. doi: 10.1111/bjd.15924
147. Messingham KN, Wang JW, Holahan HM, Srikantha R, Aust SC, Fairley JA. Eosinophil localization to the basement membrane zone is autoantibody- and complement-dependent in a human cryosection model of bullous pemphigoid. Exp Dermatol (2016) 25(1):50–5. doi: 10.1111/exd.12883
148. de Graauw E, Sitaru C, Horn M, Borradori L, Yousefi S, Simon HU, et al. Evidence for a role of eosinophils in blister formation in bullous pemphigoid. Allergy (2017) 72(7):1105–13. doi: 10.1111/all.13131
149. Zillikens D, Rose PA, Balding SD, Liu Z, Olague-Marchan M, Diaz LA, et al. Tight clustering of extracellular BP180 epitopes recognized by bullous pemphigoid autoantibodies. J Invest Dermatol (1997) 109(4):573–9. doi: 10.1111/1523-1747.ep12337492
150. Engelberts PJ, Voorhorst M, Schuurman J, van Meerten T, Bakker JM, Vink T, et al. Type I CD20 antibodies recruit the B cell receptor for complement-dependent lysis of malignant b cells. J Immunol (2016) 197(12):4829–37. doi: 10.4049/jimmunol.1600811
151. Gutjahr A, Heck F, Emtenani S, Hammers A -K., Hundt JE, Muck P, et al. Bullous pemphigoid autoantibody-mediated complement fixation is abolished by the low-molecular-weight heparin tinzaparin sodium. Br J Dermatol (2019) 181(3):593–4. doi: 10.1111/bjd.18156
152. Sadik CD, Rashid H, Hammers CM, Diercks GFH, Weidinger A, Beissert S, et al. Evaluation of nomacopan for treatment of bullous pemphigoid. JAMA Dermatol (2022) 158(6):641–9. doi: 10.1001/jamadermatol.2022.1156
153. Freire PC, Muñoz CH, Derhaschnig U, Schoergenhofer C, Firbas C, Parry GC, et al. Specific inhibition of the classical complement pathway prevents C3 deposition along the dermal-epidermal junction in bullous pemphigoid. J Invest Dermatol (2019) 139(12):2417–24. doi: 10.1016/j.jid.2019.04.025
Keywords: complement, relevance, pemphigoid, bullous pemphigoid, BP, EBA, MMP, pathophysiology
Citation: Papara C, Karsten CM, Ujiie H, Schmidt E, Schmidt-Jiménez LF, Baican A, Freire PC, Izumi K, Bieber K, Peipp M, Verschoor A, Ludwig RJ, Köhl J, Zillikens D and Hammers CM (2022) The relevance of complement in pemphigoid diseases: A critical appraisal. Front. Immunol. 13:973702. doi: 10.3389/fimmu.2022.973702
Received: 20 June 2022; Accepted: 26 July 2022;
Published: 16 August 2022.
Edited by:
Takashi Hashimoto, Osaka City University, JapanReviewed by:
Jong Hoon Kim, Yonsei University, South KoreaZhi Liu, University of North Carolina at Chapel Hill, United States
Ana Maria Abreu-Velez, Georgia Dermatopathology Associates (GDA), United States
Copyright © 2022 Papara, Karsten, Ujiie, Schmidt, Schmidt-Jiménez, Baican, Freire, Izumi, Bieber, Peipp, Verschoor, Ludwig, Köhl, Zillikens and Hammers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christoph M. Hammers, Y2hyaXN0b3BoLmhhbW1lcnNAdWtzaC5kZQ==