
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 13 September 2022
Sec. B Cell Biology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.972723
This article is part of the Research Topic Unraveling Mechanisms and Implications of Anti-RBC Antibodies in Transfusion Medicine View all 7 articles
 Annie Qiu1
Annie Qiu1 Anabel Miller1
Anabel Miller1 Flavia Dei Zotti1
Flavia Dei Zotti1 Manjula Santhanakrishnan2
Manjula Santhanakrishnan2 Jeanne E. Hendrickson2Maria Tredicine3
Jeanne E. Hendrickson2Maria Tredicine3 Sean R. Stowell4Chance John Luckey5
Sean R. Stowell4Chance John Luckey5 James C. Zimring6,7†
James C. Zimring6,7† Krystalyn E. Hudson1*†
Krystalyn E. Hudson1*†Passive immunization with anti-D can prevent maternal alloimmunization to RhD thereby preventing hemolytic disease of the fetus and newborn. Unexpectedly, anti-D fails in some cases and some monoclonal anti-D preparations paradoxically enhances alloimmunization. The underlying mechanisms modulating humoral alloimmunization by anti-D are unknown. We previously reported that IgG antibody subclasses differentially regulate alloimmunity in response to red blood cell (RBC) transfusions in a mouse model; in particular, IgG2c significantly enhanced RBC alloantibody responses. Initial mechanistic studies revealed that IgG2c:RBC immune complexes were preferentially consumed by the splenic dendritic cell (DC) subsets that play a role in RBC alloimmunization. The deletion of activating Fc-gamma receptors (FcγRs) (i.e., FcγRI, FcγRIII, and FcγRIV) on DCs abrogated IgG2c-mediated enhanced alloimmunization. Because DCs express high levels of FcγRIV, which has high affinity for the IgG2c subclass, we hypothesized that FcγRIV was required for enhanced alloimmunization. To test this hypothesis, knockout mice and blocking antibodies were used to manipulate FcγR expression. The data presented herein demonstrate that FcγRIV, but not FcγRI or FcγRIII, is required for IgG2c-mediated enhancement of RBC alloantibody production. Additionally, FcγRI is alone sufficient for IgG2c-mediated RBC clearance but not for increased alloimmunization, demonstrating that RBC clearance can occur without inducing alloimmunization. Together, these data, combined with prior observations, support the hypothesis that passive immunization with an RBC-specific IgG2c antibody increases RBC alloantibody production through FcγRIV ligation on splenic conventional DCs (cDCs). This raises the question of whether standardizing antibody subclasses in immunoprophylaxis preparations is desirable and suggests which subclasses may be optimal for generating monoclonal anti-D therapeutics.
Prevention of maternal alloimmunization to RhD by passive immunization with polyclonal anti-D (i.e., immunoprophylaxis) dramatically decreased rates of hemolytic disease of the fetus and newborn. Nonetheless, some anti-D preparations, under certain conditions, paradoxically enhance alloimmunization (1, 2). The underlying mechanisms by which anti-D modulates humoral alloimmunization remain unknown, but different hypotheses involve antigen modulation, increased RhD+ red blood cell (RBC) clearance, preferential ligation of inhibitory Fc receptors on B cells, and/or steric hindrance/antigen masking (3–5). Although polyclonal anti-D preparations offer numerous advantages (e.g., polyvalent specificity), several limitations apply, including the number of human volunteers required, highly variable time-to-response conversion to produce anti-D alloantibodies, and batch-to-batch variations (5, 6). Thus, there is a considerable need to develop a monoclonal antibody approach, which would provide a cost-effective, scalable option that offers batch-to-batch consistency. Although many monoclonal anti-D antibodies have been tested, some enhanced RBC alloantibody responses whereas others suppressed (7–9). Additionally, there was discordance between RhD+ RBC clearance and immunoprophylaxis among the anti-D monoclonal antibodies evaluated (8), challenging the explanation that anti-D works by clearing RhD+ RBCs. Recently, two new monoclonal anti-D antibodies demonstrated efficacy in preventing RhD alloimmunization in pregnant women (10, 11), although additional studies with a larger number of participants and re-exposure to RhD+ RBCs are required to fully evaluate the efficacy of these new therapeutics.
Human polyclonal anti-D consists of four IgG subclasses: IgG1, IgG2, IgG3, and IgG4, each with distinct effector functions, including complement fixation and ligation of specific Fc-gamma (γ) receptors (FcγRs) (12). To gain insight into how different subclasses modulate immune responses, we previously reported the generation of monoclonal antibodies specific for model RBC alloantigens in mice, and the generation of panels of IgG switch variants for each murine IgG subclass: IgG1, IgG2a/c, IgG2b, and IgG3 (13, 14). Across these models, passive immunization with the RBC-specific IgG2c subclass consistently enhanced RBC alloimmunization rates and alloantibody levels (13–15). In initial mechanistic studies, IgG2c:RBC immune complexes were preferentially consumed by splenic CD11c+CD11b+ and CD11c+CD8+ conventional dendritic cells (cDCs), which are known to be important in immune responses to RBCs (13, 16, 17). In addition, the IgG2c-mediated enhancement of alloimmunization depended on specific FcγR expression; that is, deleting the common γ chain required for expression of all activating FcγRs (i.e., FcγRI, FcγRIII, FcγRIV) on splenic cDCs abrogated alloantibody production (13). By cellular surface plasmon resonance (cSPR), FcγRI and FcγRIV have been shown to have strong affinity for IgG2c (14). Based on prior observations demonstrating the involvement of FcγRIV in immune responses mediated by IgG2a (i.e., a genetic variant of IgG2c) (18), in initiating autoimmune responses (19, 20), and in CD4+ T cell priming by cDCs (21), we hypothesized that FcγRIV is required for IgG2c-mediated enhanced alloimmunization to RBC antigens.
Herein, we report that FcγRIV is required for enhanced RBC alloimmunization following passive immunization with IgG2c; that is, inhibiting FcγRIV abrogated RBC alloantibody production. FcγRIII was not required and FcγRI was not sufficient for enhancing alloimmune responses. Unexpectedly, FcγRI was not sufficient for increased alloimmunization but was alone sufficient for IgG2c-mediated RBC clearance from the circulation, providing further evidence that RBC clearance need not be directly linked to alloantibody production. Together, these data provide additional information into which FcγRs are responsible for that IgG2c-mediated enhancement of RBC alloimmunization.
B6 (C57BL/6NCrl, stock #027) were purchased from Charles River and FcγRIII-/+ (B6.129P2-Fcgr3tm1Jsv/J; stock #003171) were purchased from The Jackson Laboratory; these mice were generated on a 129/B6 mixed background and backcrossed to C57BL/6 for 7 generations. Thus, to control for genetic background differences, FcγRIII-/+ mice were bred to generate FcγRIII+/+ mice for control recipients. For experiments, FcγRIII-/+ mice were bred to generate FcγRIII-/- mice. Mice expressing an RBC-specific triple fusion protein consisting of hen egg lysozyme, ovalbumin, and the human blood group molecule Duffy (HOD) were generated, as previously described (22). All mice were maintained in a pathogen-free environment on standard rodent chow and water in a light and temperature-controlled environment. Unless otherwise stated, mice were 8-24 weeks old and both male and female animals were used. All protocols used were approved by the Columbia University Irving Medical Campus Institutional Animal Care and Use Committee (IACUC).
Anti-HOD mAb IgG2c antibody (1 μg/mouse, which recognizes an epitope common to Duffy (Fya and Fyb) blood group molecule contained within the HOD antigen) was passively infused into recipients 2 hours before an RBC transfusion, as previously described (13). Transfusions consisted of 50μL of leukoreduced CellTrace-CFSE (ThermoFisher) labeled HOD packed RBCs (pRBCs) and 50μL of leukoreduced CellTrace-Far Red (ThermoFisher) labeled B6 pRBCs diluted to a 20% hematocrit with PBS. In some experiments, recipients were infused with 200μg of the purified monoclonal anti-mouse CD16.2 (FcγRIV) blocking antibody (Biolegend, Clone 9e9) or Armenian hamster IgG isotype control with anti-glutathione S-transferase specificity (BioXCell #BE0260, Clone PIP) 30 minutes prior to passive immunization. To evaluate the specificity of the FcγRIV blocking antibody, spleens were collected and stained with antibodies to delineate leukocyte subsets and Fc receptor expression, as previously described (13). Post-transfusion survival of allogeneic RBCs, and subsequent alloantibody production, were performed, as previously described (13).
Sera was collected from experimental mice, diluted 1:100 and added to HOD target RBCs or B6 control target RBCs in FACS buffer (phosphate-buffered saline + 0.2mg/mL bovine serum albumin + 0.9mg/mL ethylenediaminetetraacetic acid). RBCs and sera were incubated for 20 minutes at room temperature, washed three times with FACS buffer and then stained with 1:100 goat anti-mouse immunoglobulins (IgM + IgG + IgA) conjugated to APC as secondary detection reagent. To determine the isotype and subclass of alloantibodies, we used directly-conjugated antibodies against IgM, IgG1, IgG2b, IgG2c, and IgG3. Samples were incubated at 4°C for 30 minutes and then washed three times with FACS buffer prior to analysis with a flow cytometer. To calculate the adjusted MFI, the sera incubated with B6 RBCs (negative control/background signal) was subtracted from the signal from sera incubated with HOD RBCs. The only difference between HOD and B6 RBCs is the expression of the HOD antigen, which is confirmed with PUMA6 and goat anti-mouse immunoglobulin-APC staining.
HOD and B6 whole blood was collected into 14% CPDA-1 from donor animals by retro-orbital exsanguination and leukoreduced with an Acrodisc PSF syringe filter (Pall Life Sciences). For CellTrace-CFSE labeling, 10mL of CellTrace-CFSE (5 μ M) was added for every 1mL of pRBCs. For CellTrace-Far Red, 2mL of CellTrace-Far Red (5 μ M) was added for every 1mL of pRBCs. pRBCs were added directly into either working solutions of CellTrace-CFSE or CellTrace-Far Red, swirled to mix, and incubated at 37°C in the dark for 20 minutes, inverting after 10 minutes. RBCs were washed twice with PBS and resuspended at 20% hematocrit in PBS. Labelled HOD and B6 RBCs were then mixed at a 1:1 ratio. To determine pre-transfusion ratio, an aliquot of mixed RBCs was analyzed by flow cytometry.
Spleens from mice were collected into complete RPMI, collagenase digested, and filtered, as previously described (13, 23). Single cells were washed with FACS buffer, RBCs were lysed, and the remaining cells were stained with antibodies to delineate leukocyte subsets (Supplemental Table 1). To enable detection of FcγRs, cells were stained with antibodies against FcγRI, FcγRII, FcγRIII, and FcγRIV; to prevent potential steric hindrance, cells were stained for an individual FcγR to evaluate expression levels. Antibodies specific for Thy1.2, Ter119, PDCA1, CD115, Ly6G, CD11c, F4/80, and CD8 were purchased from ThermoFisher those recognizing CD19, CD11b, FcγRI, FcγRII, and FcγRIV were purchased from BioLegend. An antibody to FcγRIII was purchased from Bio-Rad. All staining was performed in FACS buffer, and cells were interrogated on an Attune NxT flow cytometer (ThermoFisher), and data analyzed with FlowJo software.
Statistical analysis was performed with a repeated measures 2-way ANOVA or a one-way ANOVA with Tukey’s multiple comparisons test; p<0.05 was considered significant. Analyses were performed using Prism, version X (GraphPad Software, Inc.).
Passive immunization with anti-HOD mAb IgG2c was previously shown to enhance alloantibody production following transfusion with RBCs expressing the HOD alloantigen (13). This enhancement required expression of activating FcγRs on CD11c+CD8+ and CD11c+CD11b+ cDC subsets (13), although which individual FcγR(s) were required remained unknown. To elucidate specific FcγRs, knockout mice and blocking antibodies were used. FcγRIV activity was inhibited by infusion of an anti-CD16.2 antibody (clone 9e9; herein referred to as “9e9”). Nonspecific effects were controlled for using either an IgG isotype control or PBS. 9e9 treatment inhibited detection of FcγRIV on all splenic antigen presenting cell subsets evaluated, effectively reducing the staining signal to background levels (Figure 1A). Unexpectedly, 9e9 infusion also led to decreased expression of FcγRIII on red pulp macrophages, inflammatory monocytes, resident monocytes, and neutrophils (Figure 1B) and decreased expression of inhibitory FcγRIIb on inflammatory monocytes (Figure 1C). No significant differences were observed with FcγRI expression (Figure 1D). Absolute counts of each cell subset revealed 9e9 infusion blocked FcγRIV but did not promote cell death (Supplemental Figure 1). In contrast, levels of each individual FcγR were similar between the IgG isotype control-treated and PBS-treated animals.
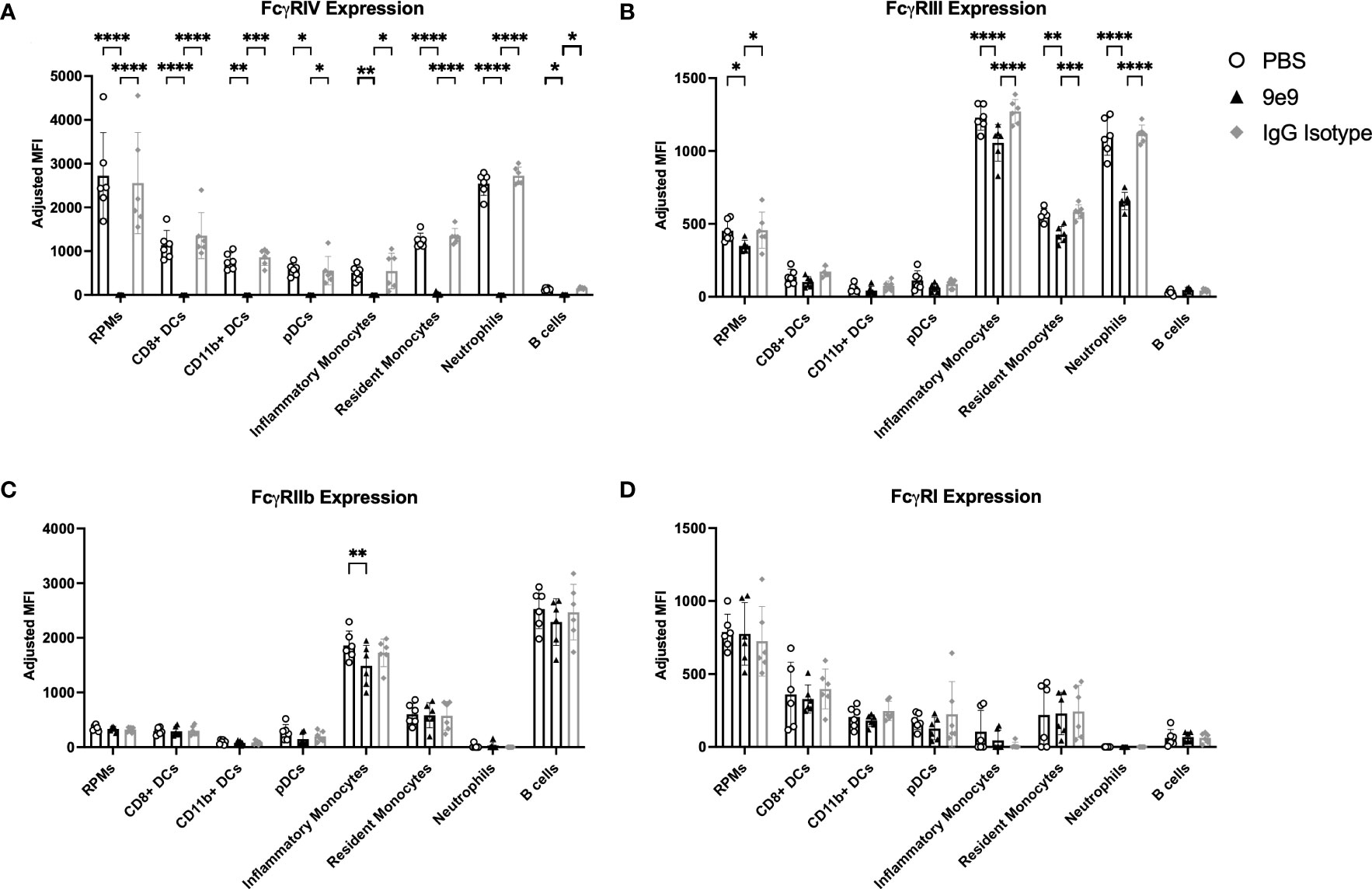
Figure 1 Anti-FcγRIV (clone 9e9) inhibits FcγRIV. B6 recipient mice received an infusion of 200μg of anti-FcγRIV clone 9e9, IgG isotype control, or PBS. Spleens were collected 30 minutes later and processed into single cell suspensions. Splenocytes were stained with antibodies to delineate cell subsets and expression of (A) FcγRIV, (B) FcγRIII, (C) FcγRIIb, or (D) FcγRI. The mean fluorescence intensity (MFI) for each individual FcγR was calculated; a fluorescence minus one (FMO) was used as a negative control. To determine the adjusted MFI, the MFI of the FMO was subtracted from the MFI of the stain containing the antibody for the FcγR. The following phenotypes were used to delineate cell subsets: red pulp macrophages (RPMs): CD11c−/loCD11b−/loF4/80+; CD8+ DCs: CD11chiCD11b−CD8+; CD11b+ DCs: CD11chiCD11b+CD8−; plasmacytoid DCs (pDCs): PDCA1+CD11cintLy6Chi; inflammatory monocytes: CD11c−/loCD11b+Ly6G−Ly6ChiCD115+; resident monocytes: CD11c−/loCD11b+Ly6G−Ly6CloCD115−; neutrophils: CD11c−/loCD11b+Ly6G+ and a high side scatter. Data shown are an aggregate of 2 independent experiments with 3 mice per group. Statistical analysis was performed with a one-way ANOVA with Tukey’s multiple comparisons test; ****p<0.0001, ***p<0.001, **p<0.05, *p<0.05.
To assess whether FcγRIV inhibition affected RBC alloimmunization, recipient B6 mice were infused with 9e9, an IgG isotype control antibody, or PBS 30 minutes before passive immunization with anti-HOD mAb IgG2c or PBS control. Each recipient was then transfused 2 hours later with a 1:1 mix of fluorescently-labeled allogeneic HOD and syngeneic B6 RBCs (Supplemental Figure 2). Consistent with prior observations (13), B6 mice passively immunized with anti-HOD mAb IgG2c had significantly higher anti-HOD alloantibody production throughout the 21-day time course, as compared to (PBS + PBS) control mice (Figure 2A). In contrast, 9e9 pre-treatment significantly reduced anti-HOD alloantibodies over the 21-day time course, as compared to (PBS + anti-HOD mAb IgG2c) treated animals. No significant differences were observed between the (PBS + PBS) and the (9e9 + anti-HOD mAb IgG2c) treated groups, demonstrating that inhibiting FcγRIV abrogated enhanced RBC alloantibody production. Additionally, levels of anti-HOD alloantibody production were similar between anti-HOD mAb IgG2c groups pre-treated with PBS or the IgG isotype control, providing evidence that reduction in alloantibody production was specific to 9e9 recognizing its epitope, and not due to nonspecific antibody effects. Analysis of IgM and IgG subclasses showed similar a similar trend as total immunoglobulins (Supplemental Figure 3).
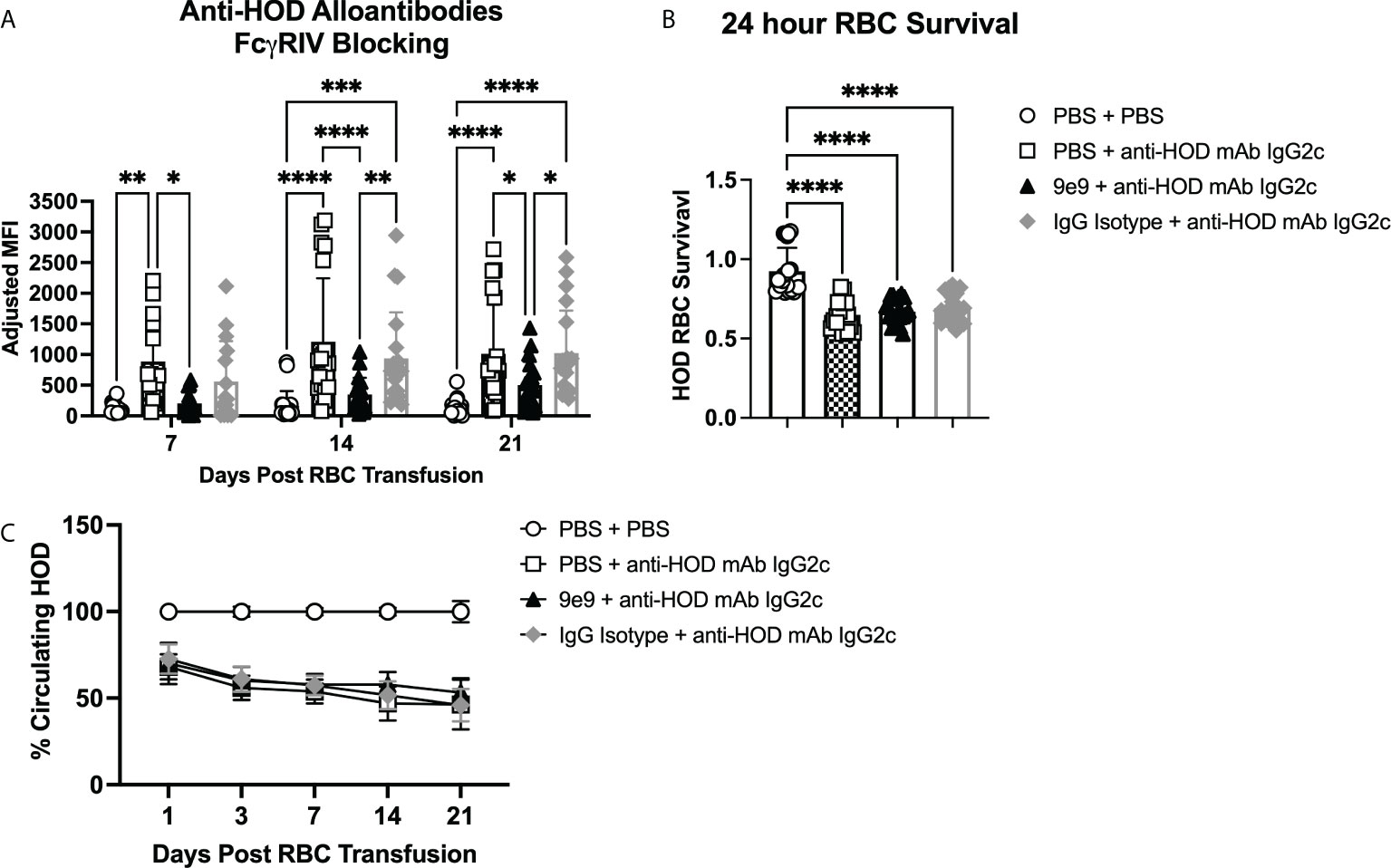
Figure 2 RBC alloantibody production requires FcγRIV. B6 recipient mice were passively immunized with 1μg of anti-HOD IgG2c followed by an RBC transfusion 2 hours later. Some animals received an infusion of 1μg of 9e9, an anti-FcγRIV blocking antibody, or IgG isotype control 30 minutes prior to passive immunization. Sera was collected weekly and (A) analyzed for anti-HOD alloantibodies by flow crossmatch. To assess RBC clearance, survival of HOD RBCs was determined at (B) 24 hours and (C) over a 3-week time course. Data shown are cumulative of 4 independent experiments with 4-5 mice per group. Statistical analysis was performed with a repeated measures 2-way ANOVA or a one-way ANOVA with Tukey’s multiple comparisons test, ****p<0.0001, ***p<0.001, **p<0.05, *p<0.05, comparisons without significance are not shown.
We previously reported that passive immunization with IgG2c induces significant allogeneic RBC clearance, which is mitigated by decreased expression of activating FcγRI, FcγRIII, and FcγRIV (13). To test whether inhibiting FcγRIV affected HOD RBC clearance, post-transfusion survival was measured over a the 3-week time course. Consistent with prior observations (13), HOD RBC survival at 24-hours was significantly reduced in (PBS + anti-HOD mAb IgG2c) treated animals compared to (PBS + PBS) controls with no anti-HOD mAb IgG2c (Figure 2B). However, no differences in clearance were observed between groups that received 9e9 or an IgG isotype control prior to anti-HOD IgG2c mAb. To evaluate potential long-term differences between experimental groups, HOD RBC survival was normalized to the (PBS + PBS) treated control group. No significant differences in HOD RBC survival were noted between groups passively immunized with anti-HOD IgG2c mAb (Figure 2C). Together, these data demonstrate that, although FcγRIV is required for IgG2c-mediated alloantibody production, but it is not required for allogeneic RBC clearance.
To test the roles of FcγRIII and FcγRI in IgG2c-mediated enhanced alloantibody production, knockout mice and 9e9 were used together. Passive immunization with anti-HOD IgG2c mAb followed by RBC transfusion into FcγRIII-/- animals induced significantly increased anti-HOD alloantibody levels at all time points, as compared to (PBS + PBS) treated control animals (Figure 3A). Because FcγRIII-/- mice were generated on a 129/B6 mixed background, FcγRIII+/+ replete mice were used as controls to isolate the contribution of FcγRIII in alloimmune responses. These data demonstrate that FcγRIII is not required for IgG2c-mediated enhanced RBC alloimmunization. To test whether FcγRI alone was sufficient for enhancing alloantibodies with IgG2c, FcγRIII-/- mice were pre-treated with 9e9 or IgG isotype control. Pre-treating FcγRIII-/- mice with 9e9 (thereby eliminating both FcγRIII and FcγRIV, and leaving FcγRI signaling intact) significantly reduced anti-HOD alloantibodies to levels comparable to (PBS + PBS) treated control mice (Figure 3A, black triangles). RBC alloantibodies were not reduced in the IgG isotype control group (Figure 3A, gray diamonds). Thus, FcγRI is not sufficient for increasing alloantibody production upon passive immunization with anti-HOD IgG2c mAb. To exclude background genetics as a potential confounder, experiments were performed in FcγRIII+/+ mice, in parallel, and similar trends were observed as in FcγRIII-/- mice (Figure 3B).
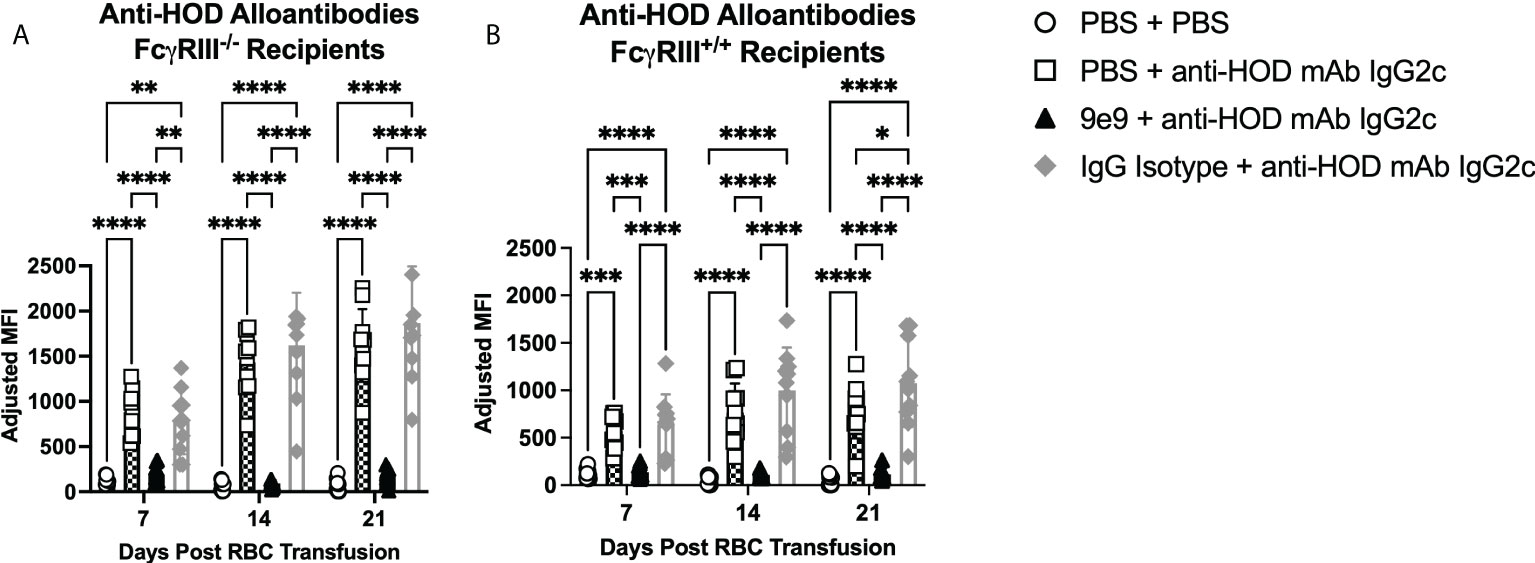
Figure 3 Neither FcγRI nor FcγRIII are sufficient for enhanced alloantibodies with IgG2c. Recipient mice were passively immunized with 1μg of anti-HOD IgG2c followed by an RBC transfusion 2 hours later. Some animals received an infusion of 1μg of 9e9 or IgG isotype control 30 minutes prior to passive immunization. Sera was collected from (A) FcγRIII-/- and (B) FcγRIII+/+ recipient mice weekly and analyzed for anti-HOD alloantibodies by flow crossmatch. Data shown are cumulative of 2 independent experiments with 5 mice per group. Statistical analysis was performed with a repeated measures 2-way ANOVA with Tukey’s multiple comparisons test; ****p<0.0001, ***p<0.001, **p<0.05, *p<0.05, comparisons without significance are not shown.
Passive immunization with anti-HOD IgG2c mAb leads to rapid HOD RBC clearance (Figures 2B, C). Similar levels and kinetics of clearance were observed upon blocking FcγRIV with 9e9 (Figure 2B), suggesting clearance is mediated by FcγRI, FcγRIII, or both. To evaluate FcγRIII-mediated clearance of allogeneic HOD RBCs by anti-HOD IgG2c mAb, 24-hour survival was determined in FcγRIII-/- and FcγRIII+/+ mice. HOD RBC clearance by IgG2c was equivalent in FcγRIII+/+ and FcγRIII-/- mice (Figure 4A). Thus, FcγRIII is not required for IgG2c-mediated clearance of HOD RBCs. To evaluate FcγRI function alone, Fcγ RIV was inhibited in FcγRIII-/- animals; this approach leaves only FcγRI signaling intact. No significant change was observed in HOD RBC clearance between 9e9 or IgG isotype control treated FcγRIII-/- animals (Figure 4B), demonstrating that, FcγRI expression is alone sufficient for HOD RBC clearance by anti-HOD IgG2c mAb; similar results were observed in FcγRIII+/+ control mice (Figure 4C). No significant differences in HOD RBC survival were noted between groups passively immunized with anti-HOD IgG2c mAb in FcγRIII+/+ or FcγRIII-/- mice (Supplemental Figure 4). Together, these data demonstrate that FcγRI is sufficient for IgG2c-mediated RBC clearance, and that FcγRIII is not required.
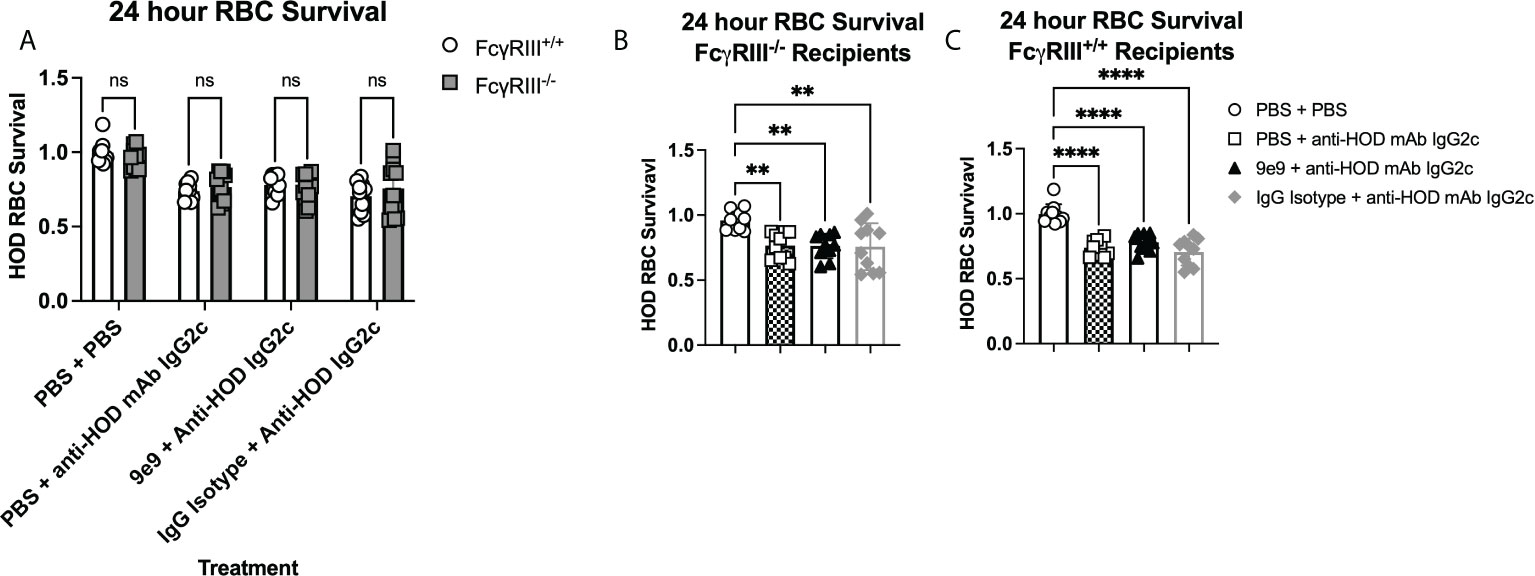
Figure 4 FcγRI is sufficient to mediate RBC clearance with IgG2c. Recipient mice were passively immunized with 1μg of anti-HOD IgG2c followed by an RBC transfusion 2 hours later. Some animals received an infusion of 1μg of 9e9 or IgG isotype control 30 minutes prior to passive immunization. To assess RBC clearance, 24 hour survival of HOD RBCs was determined in (A) all treatment groups and recipients, or between treatment groups within (B) FcyRIII-/- , or (C) FcyRIII+/+ recipient mice. Data shown are cumulative of 2 independent experiments with 5 mice per group. Statistical analysis was performed with a one-way ANOVA with Tukey’s multiple comparisons test; ****p<0.0001, **p<0.05, ns, not significant.
A murine model of RBC alloimmunization was used to test the hypothesis that FcγRIV is required for enhanced alloantibody production upon passive immunization with IgG2c. Inhibiting FcγRIV with 9e9, an antibody that blocks its binding site, reduced RBC alloantibody production to background levels. Passive immunization with anti-HOD IgG2c mAb into FcγRIII-/- mice, which still express FcγRI and FcγRIV, enhanced RBC alloantibody levels, demonstrating that FcγRIII was not required. Likewise, inhibiting FcγRIV in FcγRIII-/- animals, but leaving FcγRI signaling intact, failed to induce RBC alloantibodies, indicating that FcγRI was not sufficient for enhanced alloantibody production. In parallel, FcγRI was shown to be sufficient for anti-HOD IgG2c mAb-mediated RBC clearance, demonstrating that the mechanisms of IgG2c-mediated clearance and alloantibody production involve distinct FcγRs. These data, together with our prior publication (13), provide evidence that enhanced alloantibody production following passive immunization with antigen-specific IgG2c and transfusion of the corresponding antigen-expressing RBCs, requires Fcγ RIV expression on splenic cDCs.
The reliance on FcγRIV for IgG2a-mediated effector functions has been demonstrated in several models including B cell depletion, phagocytosis of platelets, and autoimmune responses such as autoimmune hemolytic anemia, rheumatoid arthritis, and nephritis (18, 24–27). Mechanistically, ligation of FcγRIV promotes cellular activation (e.g., CD86 expression) and increases antigen presentation (28). We, and others, have shown that immune complexes consisting of antigen:IgG2a (or IgG2c) are preferentially taken up by splenic cDC subsets, which promotes increased T cell activation and proliferation (13, 21). Because blocking FcγRIV can ameliorate autoimmune and alloimmune IgG2a/c-mediated pathology, FcγRIV is an attractive therapeutic target.
Treatment with the anti-FcγRIV 9e9 antibody significantly reduced detectable FcγRIV on the surface of all splenic antigen presenting cell subsets that were analyzed. Unexpectedly, there was also reduced expression of FcγRIII and FcγRII (but not FcγRI) on different subsets. Thus, one must consider that some of the effects of 9e9 may be due to altering levels of FcγRIII and/or FcγRII. However, we show herein that FcγRIII-/- mice have normal induction of alloimmunization by IgG2c. Also, we have shown previously that FcγRII is not sufficient for alloimmunization, as common γ chain knockout mice have not IgG2c-mediated enhancement of RBC alloimmunization (13). Thus, we reject the interpretation that decreased FcγRII and FcγRIII contributed to blockade of alloimmunization by 9e9.
Interestingly, passive immunization with IgG2c anti-HOD mAb induced higher levels of enhancement in FcγRIII-/- than in FcγRIII+/+ mice. The reason for this difference is unclear, as FcγRIII+/+ is not typically considered inhibitory; however, it could be the result of decreased competition with FcγRIV for the common γ chain required for FcγR signaling or contributions of the background genetics (29), which may influence immune responses.
Blocking FcγRIV with 9e9 significantly reduced HOD RBC alloantibody levels following passive immunization with anti-HOD IgG2c mAb. However, throughout a 3-week time course, alloantibody levels did gradually increase. This may be attributed to the half-life of the 9e9 antibody, as only one infusion was given. Repeating these studies in FcγRI and FcγRIV knockout mice would address the limitations of using a blocking antibody; however, FcγR deficiency modulates expression of other FcγRs (18, 30). Finally, it is worth noting that passive immunization with anti-HOD IgG2c mAb in 9e9 treated FcγRIII-/- animals resulted in much lower levels of RBC alloantibodies, as compared to wild-type B6 recipients. Thus, although FcγRIII was not required for enhancing RBC alloantibody levels, it may still contribute to alloimmune responses; in particular, inhibiting both FcγRIII and FcγRIV simultaneously is required to prevent all such RBC alloantibody production (Figure 3A).
Polyclonal antibodies, such as human anti-D, contain a heterogenous mixture of IgG subclasses, consisting of multiple epitope specificities that may work in an additive, synergistic, or even an inhibitory, manner. Because the therapeutic anti-D used for immunoprophylaxis against Rh disease is derived by pooling material from multiple human volunteers, the efficacy of anti-D could depend on numerous variables, including donor-specific and antibody-specific characteristics. For example, a recent analysis of 23 such human monoclonal antibodies revealed that glycosylation profiles influenced biological activity (31), and IgG glycosylation can vary from human to human. Moreover, studies in mice highlight additional complexities that contribute to antibody-mediated immune responses, including affinity, the RBC antigen target and/or epitope, and RBC antigen copy number (13–15, 32–34). In addition to modulating immune enhancement or suppression, subclass can also dictate which effector pathways are employed (e.g., complement, FcRs, etc.) (35, 36). Thus, additional mechanistic investigations to elucidate how polyclonal anti-D prevents RhD alloimmunization will instruct the future development of human monoclonal anti-D antibodies, such that subclass, affinity, glycosylation, and epitope recognition can be fine-tuned to exert the desired function(s). Of note, human FcγRIIIA is the human ortholog to mouse FcγRIV (25, 37). Human IgG1 and IgG3, the main subclasses evaluated for monoclonal anti-D preparations, have high affinities for FcγRIIIA; however, this affinity is influenced by FcγRIIIA polymorphisms and the concentration of the antibody:antigen immune complexes (38). Thus, in addition to donor-specific and antibody-specific characteristics, recipient-specific characteristics will also need to be considered. In addition to the generalizable advantages of monoclonal antibody therapy (e.g., cost-effective, scalable, high specificity, lot-to-lot consistency), the substantial advances in antibody technology and increased understanding of immune function, now allow antibodies to be engineered to have increased affinity for the neonatal Fc receptor (FcRn), thereby facilitating an extended half-life to make passive immunization more durable. Moreover, antibodies can be engineered to inhibit antibody:FcRn interactions at the maternal-fetal interface, which is especially important for preventing hemolytic disease of the fetus and newborn mediated by alloantibodies to non-D RBC antigens (39, 40).
There are several limitations to the current studies. Although there is a clear reduction in alloantibody production upon blocking FcγRIV with 9e9, these studies would be strengthened by validating these findings in FcγRIV deficient animals or with another blocking antibody with a different binding site for FcγRIV. Additionally, passive immunization with anti-HOD IgG2c mAb followed by HOD transfusion leads to endogenous production of both IgM and IgG antibodies (13). Thus, while Fc-dependent FcγRIV ligation and signaling is blocked and signaling by IgG2c subclass antibodies is blocked, these antibodies may signal through other FcRs (e.g., FcmR) and modulate alloimmune responses (41). Finally, although the current analyses were limited to splenic antigen presenting cells, it is also likely that immune complex clearance also occurs in the liver.
The data presented herein demonstrate that FcγRIV, but neither FcγRIII nor FcγRI, is required for IgG2c-mediated enhanced alloimmunization to RBCs. These results, combined with our prior findings (13), support a model whereby passive immunization with an RBC-specific IgG2c antibody increases RBC alloantibody production to the corresponding antigen through FcγRIV ligation on splenic cDCs. To apply these mechanistic insights human anti-D, the future design of monoclonal antibodies should be tailored and optimized to leverage distinct subclass effector functions (12), Fc receptor expression patterns (41), and signaling (42).
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by Columbia University Irving Medical Campus Institutional Animal Care and Use Committee (IACUC).
AQ and KH designed the studies and experiments. AQ, AM, FD, MS, and MT set up experiments, collected and processed samples, and performed data analysis. All authors participated in data interpretation, revised the manuscript, and approved of the submitted version.
These studies were supported in part by the National Institutes of Health R01HL135248 (KH) and P01HL132819 (JZ) by National Heart, Lung, and Blood Institute.
The authors thank Drs. Steven L. Spitalnik and Eldad Hod for critical review of the manuscript.
Although unrelated to the contents of this manuscript, KH has a sponsored research agreement with Alpine Immune Sciences. JZ is a consultant for Rubius Therapeutics and is the founder and CSO of Svalinn Therapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.972723/full#supplementary-material
1. Bowman JM. Controversies in Rh prophylaxis. Who needs Rh Immune globulin when should it be given? Am J Obstet Gynecol (1985) 151:289–94. doi: 10.1016/0002-9378(85)90288-1
2. Contreras M, Mollison PL. Rh Immunization facilitated by passively-administered anti-Rh? Br J Haematol (1983) 53:153–9. doi: 10.1111/j.1365-2141.1983.tb01997.x
3. Sullivan HC, Arthur CM, Thompson L, Patel SR, Stowell SR, Hendrickson JE, et al. Anti-RhD reduces levels of detectable RhD antigen following anti-RhD infusion. Transfusion (2018) 58:542–4. doi: 10.1111/trf.14452
4. Crow AR, Lazarus AH. The mechanisms of action of intravenous immunoglobulin and polyclonal anti-d immunoglobulin in the amelioration of immune thrombocytopenic purpura: What do we really know? Transfusion Med Rev (2008) 22:103–16. doi: 10.1016/j.tmrv.2007.12.001
5. Brinc D, Lazarus AH. Mechanisms of anti-d action in the prevention of hemolytic disease of the fetus and newborn. Hematology (2009) 2009:185–91. doi: 10.1182/asheducation-2009.1.185
6. Urbaniak SJ, Robertson AE. A successful program of immunizing Rh-negative male volunteers for anti-d production using frozen/thawed blood. Transfusion (1981) 21:64–9. doi: 10.1046/j.1537-2995.1981.21181127486.x
7. Béliard R. Monoclonal anti-d antibodies to prevent alloimmunization: lessons from clinical trials. Transfusion Clinique Biologique (2006) 13:58–64. doi: 10.1016/j.tracli.2006.03.013
8. Kumpel BM. Efficacy of RhD monoclonal antibodies in clinical trials as replacement therapy for prophylactic anti-d immunoglobulin: More questions than answers. Vox Sang (2007) 93:99–111. doi: 10.1111/j.1423-0410.2007.00945.x
9. Kumpel BM. Lessons learnt from many years of experience using anti-d in humans for prevention of RhD immunization and haemolytic disease of the fetus and newborn. Clin Exp Immunol (2008) 154:1–5. doi: 10.1111/j.1365-2249.2008.03735.x
10. Mayekar RV, Paradkar GV, Bhosale AA, Sachan R, Beeram S, Anand AR, et al. Recombinant anti-d for prevention of maternal-foetal Rh(D) alloimmunization: A randomized multi-centre clinical trial. Obstet Gynecol Sci (2020) 63:315–22. doi: 10.5468/ogs.2020.63.3.315
11. Chauhan AR, Nandanwar YS, Ramaiah A, Yelikar KA, Rashmi MD, Sachan R, et al. A multicenter, randomized, open-label trial comparing the efficacy and safety of monoclonal anti-Rh (D) immunoglobulin with polyclonal anti-Rh (D) immunoglobulin for the prevention of maternal Rh-isoimmunization. J Obstet Gynaecol India (2019) 69:420–5. doi: 10.1007/s13224-019-01234-2
12. Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol (2014) 5:520–. doi: 10.3389/fimmu.2014.00520
13. Gruber DR, Richards AL, Howie HL, Hay AM, Lebedev JN, Wang X, et al. Passively transferred IgG enhances humoral immunity to a red blood cell alloantigen in mice. Blood Adv (2020) 4:1526–37. doi: 10.1182/bloodadvances.2019001299
14. Shinde P, Howie HL, Stegmann TC, Hay AM, Waterman HR, Szittner Z, et al. IgG subclass determines suppression versus enhancement of humoral alloimmunity to kell RBC antigens in mice. Front Immunol (2020) 11:1516. doi: 10.3389/fimmu.2020.01516
15. Jash A, Usaneerungrueng C, Howie H, Qiu A, Luckey JC, Zimring JC, et al. Antibodies to low-copy number RBC alloantigen convert a tolerogenic stimulus to an immunogenic stimulus in mice. Front Immunol (2021) 12:629608. doi: 10.3389/fimmu.2021.629608
16. Richards AL, Qiu A, Zotti FD, Sheldon K, Usaneerungrueng C, Gruber DR, et al. Autoantigen presentation by splenic dendritic cells is required for RBC-specific autoimmunity. Transfusion (2021) 61:225–35. doi: 10.1111/trf.16191
17. Calabro S, Gallman A, Gowthaman U, Liu D, Chen P, Liu J, et al. Bridging channel dendritic cells induce immunity to transfused red blood cells. J Exp Med (2016) 213:887–96. doi: 10.1084/jem.20151720
18. Nimmerjahn F, Lux A, Albert H, Woigk M, Lehmann C, Dudziak D, et al. FcyRIV deletion reveals its central role for IgG2a and IgG2b activity in vivo. Proc Natl Acad Sci USA (2010) 107:19396–401. doi: 10.1073/pnas.1014515107
19. Hobday P, Auger J, Schuneman G, Haasken S, Verbeek J, Binstadt B. Fcγ receptor III and fcγ receptor IV on macrophages drive autoimmune valvular carditis in mice. Arthritis Rheumatol (2014) 66:852–62. doi: 10.1002/art.38311
20. Seeling M, Hillenhoff U, David Jean P, Schett G, Tuckermann J, Lux A, et al. Inflammatory monocytes and fcγ receptor IV on osteoclasts are critical for bone destruction during inflammatory arthritis in mice. Proc Natl Acad Sci (2013) 110:10729–34. doi: 10.1073/pnas.1301001110
21. Lehmann CHK, Baranska A, Heidkamp GF, Heger L, Neubert K, Luhr JJ, et al. DC Subset–specific induction of T cell responses upon antigen uptake via fcγ receptors in vivo. J Exp Med (2017) 214:1509–28. doi: 10.1084/jem.20160951
22. Desmarets M, Cadwell CM, Peterson KR, Neades R, Zimring JC. Minor histocompatibility antigens on transfused leukoreduced units of red blood cells induce bone marrow transplant rejection in a mouse model. Blood (2009) 114:2315–22. doi: 10.1182/blood-2009-04-214387
23. Richards AL, Hendrickson JE, Zimring JC, Hudson KE. Erythrophagocytosis by plasmacytoid dendritic cells and monocytes is enhanced during inflammation. Transfusion (2016) 56:905–16. doi: 10.1111/trf.13497
24. Hamaguchi Y, Xiu Y, Komura K, Nimmerjahn F, Tedder TF. Antibody isotype-specific engagement of fcγ receptors regulates b lymphocyte depletion during CD20 immunotherapy. J Exp Med (2006) 203:743–53. doi: 10.1084/jem.20052283
25. Nimmerjahn F, Bruhns P, Horiuchi K, Ravetch JV. FcyRIV: A novel FcR with distinct IgG subclass specificity. Immunity (2005) 23:41–51. doi: 10.1016/j.immuni.2005.05.010
26. Syed SN, Konrad S, Wiege K, Nieswandt B, Nimmerjahn F, Schmidt RE, et al. Both FcγRIV and FcγRIII are essential receptors mediating type II and type III autoimmune responses via FcRγ-LAT-dependent generation of C5a. Eur J Immunol (2009) 39:3343–56. doi: 10.1002/eji.200939884
27. Baudino L, Nimmerjahn F, Azeredo da Silveira S, Martinez-Soria E, Saito T, Carroll M, et al. Differential contribution of three activating IgG fc receptors (FcγRI, FcγRIII, and FcγRIV) to IgG2a- and IgG2b-induced autoimmune hemolytic anemia in mice. J Immunol (2008) 180:1948. doi: 10.4049/jimmunol.180.3.1948
28. Junker F, Gordon J, Qureshi O. Fc gamma receptors and their role in antigen uptake, presentation, and T cell activation. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.01393
29. Hay AM, Howie HL, Gorham JD, D’Alessandro A, Spitalnik SL, Hudson KE, et al. Mouse background genetics in biomedical research: The devil’s in the details. Transfusion (2021) 61:3017–25. doi: 10.1111/trf.16628
30. Kerntke C, Nimmerjahn F, Biburger M. There is (Scientific) strength in numbers: A comprehensive quantitation of fc gamma receptor numbers on human and murine peripheral blood leukocytes. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.00118
31. Kumpel BM, Saldova R, Koeleman CAM, Abrahams JL, Ederveen AH, Armour KL, et al. Anti-d monoclonal antibodies from 23 human and rodent cell lines display diverse IgG fc-glycosylation profiles that determine their clinical efficacy. Sci Rep (2020) 10:1464. doi: 10.1038/s41598-019-57393-9
32. Yu H, Stowell SR, Bernardo L, Hendrickson JE, Zimring JC, Amash A, et al. Antibody-mediated immune suppression of erythrocyte alloimmunization can occur independently from red cell clearance or epitope masking in a murine model. J Immunol (2014) 193:2902. doi: 10.4049/jimmunol.1302287
33. Cruz-Leal Y, Marjoram D, Lazarus AH. Erythrocyte saturation with IgG is required for inducing antibody-mediated immune suppression and impacts both erythrocyte clearance and antigen-modulation mechanisms. J Immunol (2018) 200(4):ji1700874. doi: 10.4049/jimmunol.1700874
34. Bernardo L, Amash A, Marjoram D, Lazarus AH. Antibody-mediated immune suppression is improved when blends of anti-RBC monoclonal antibodies are used in mice. Blood (2016) 128:1076–80. doi: 10.1182/blood-2016-01-692178
35. Mener A, Patel SR, Arthur CM, Stowell SR. Antibody-mediated immunosuppression can result from RBC antigen loss independent of fcγ receptors in mice. Transfusion (2019) 59:371–84. doi: 10.1111/trf.14939
36. Escamilla-Rivera V, Santhanakrishnan M, Liu J, Gibb DR, Forsmo JE, Foxman EF, et al. Complement plays a critical role in inflammation-induced immunoprophylaxis failure in mice. Front Immunol (2021) 12:704072–. doi: 10.3389/fimmu.2021.704072
37. Bournazos S, Ravetch JV. Fcγ receptor pathways during active and passive immunization. Immunol Rev (2015) 268:88–103. doi: 10.1111/imr.12343
38. Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, et al. Specificity and affinity of human fcγ receptors and their polymorphic variants for human IgG subclasses. Blood (2009) 113:3716–25. doi: 10.1182/blood-2008-09-179754
39. Wyckoff SL, Hudson KE. Targeting the neonatal fc receptor (FcRn) to treat autoimmune diseases and maternal-fetal immune cytopenias. Transfusion (2021) 61:1350–4. doi: 10.1111/trf.16341
40. Ohto H, Denomme GA, Ito S, Ishida A, Nollet KE, Yasuda H. Three non-classical mechanisms for anemic disease of the fetus and newborn, based on maternal anti-kell, anti-Ge3, anti-m, and anti-jr(a) cases. Transfus Apher Sci (2020) 59:102949. doi: 10.1016/j.transci.2020.102949
41. Bruhns P, Jönsson F. Mouse and human FcR effector functions. Immunol Rev (2015) 268:25–51. doi: 10.1111/imr.12350
Keywords: Fc receptor, antibody, red blood cell, alloantibody, antibody mediated enhancement
Citation: Qiu A, Miller A, Zotti FD, Santhanakrishnan M, Hendrickson JE, Tredicine M, Stowell SR, Luckey CJ, Zimring JC and Hudson KE (2022) FcγRIV is required for IgG2c mediated enhancement of RBC alloimmunization. Front. Immunol. 13:972723. doi: 10.3389/fimmu.2022.972723
Received: 18 June 2022; Accepted: 18 August 2022;
Published: 13 September 2022.
Edited by:
David Nemazee, The Scripps Research Institute, United StatesReviewed by:
Randall S. Davis, University of Alabama at Birmingham, United StatesCopyright © 2022 Qiu, Miller, Zotti, Santhanakrishnan, Hendrickson, Tredicine, Stowell, Luckey, Zimring and Hudson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Krystalyn E. Hudson, a2VoMjE5N0BjdW1jLmNvbHVtYmlhLmVkdQ==
†These authors share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.