 Ancheng Zheng
Ancheng Zheng Qishan Chen
Qishan Chen Li Zhang
Li Zhang- Department of Cardiology and Institute for Developmental and Regenerative Cardiovascular Medicine, Xinhua Hospital, School of Medicine, Shanghai Jiaotong University, Shanghai, China
The Hippo pathway was initially discovered in Drosophila melanogaster and mammals as a key regulator of tissue growth both in physiological and pathological states. Numerous studies depict the vital role of the Hippo pathway in cardiovascular development, heart regeneration, organ size and vascular remodeling through the regulation of YAP (yes-associated protein) translocation. Recently, an increasing number of studies have focused on the Hippo-YAP pathway in inflammation and immunology. Although the Hippo-YAP pathway has been revealed to play controversial roles in different contexts and cell types in the cardiovascular system, the mechanisms regulating tissue inflammation and the immune response remain to be clarified. In this review, we summarize findings from the past decade on the function and mechanism of the Hippo-YAP pathway in CVDs (cardiovascular diseases) such as myocardial infarction, cardiomyopathy and atherosclerosis. In particular, we emphasize the role of the Hippo-YAP pathway in regulating inflammatory cell infiltration and inflammatory cytokine activation.
Introduction
The Hippo-YAP (yes-associated protein) pathway, which was originally identified in the Drosophila genus and is highly conserved in mammals, consists of a kinase cascade as well as downstream effectors and the transcriptional coactivators (1). These core components of the Hippo pathway control transcriptional programs involved in cell growth, development, and organ size. CVDs (cardiovascular diseases) remain the leading cause of death worldwide (2–4). Numerous studies have revealed a pivotal role of the Hippo–YAP pathway and its upstream regulators in cardiac development, growth, homeostasis, disease, and regeneration using genetic models and biochemical studies (5–8).
Multiple lines of evidence continue to support that inflammation is a central process, along with multiple infiltrated immune cells and activated inflammatory cytokines, in the development of various CVDs, such as myocardial infarction (MI) (9–11), cardiomyopathy (12, 13) and atherosclerosis (14, 15). Emerging evidence shows that targeting specific inflammatory proteins or pathways can be an effective method to treat CVDs (16, 17). However, to date, there have been no clinical drugs targeting inflammation to treat CVDs; thus, more targets, pathways and their mechanisms need to be clarified.
Although the biological functions of the Hippo pathway in CVDs have been studied extensively over the last twenty years, the inflammatory regulation of this pathway, particularly in mammals, is poorly understood. Recently, the role of the Hippo-YAP pathway in regulating inflammation and the immune response has attracted considerable attention (18–25). Considering the controversial roles of the Hippo-YAP pathway in different contexts and cell types in the cardiovascular system, summarizing and clarifying the specific modulatory effect of the Hippo-YAP pathway on physiological and pathological cardiovascular system immunity and inflammation is indispensable for further clinical study. In this review, we summarize the progress on the Hippo-YAP pathway in CVDs in recent years, especially in immune and inflammatory areas.
Introduction of the Hippo-YAP pathway
Overview of the Hippo-YAP pathway
The Hippo pathway is a kinase cascade that is highly conserved from Drosophila melanogaster to mammals. Although there are still many unknowns about the Hippo-YAP pathway, abundant studies have identified that the Hippo pathway primarily functions to regulate cell proliferation and apoptosis to control organ size and tissue homeostasis during animal development and regeneration (26). The core components of the mammalian Hippo pathway include mammalian Ste20-like kinases 1/2 (MST1/2), Salvador (SAV, also called WW45), large tumor suppressor homolog 1/2 (LATS1/2), and the scaffolding proteins MOB domain kinase activator 1A/B (MOB1). MST1/2 and SAV form a complex that can phosphorylate and activate LATS1/2, which then binds to the cofactor MOB1. Then, LATS1/2 further phosphorylates the downstream transcriptional coactivators YAP and TAZ (transcriptional coactivator with a PDZ binding motif). In particular, SAV and MOB1A/B interact with MST1/2 and LATS1/2, respectively, and function as cofactors. YAP can shuttle between the cytoplasm and nucleus, which can be regulated by its phosphorylation status (Figure 1). Phosphorylation of YAP Ser127 leads to 14-3-3 binding, cytoplasmic retention (27, 28). And phosphorylation of YAP Ser381 by the Hippo pathway promotes YAP degradation through ubiquitination and reduces its nuclear localization (29). In addition, phosphorylation of YAP at Y357 increases its stability and in turn upregulates its activity (30). In the absence of Hippo pathway inhibition, YAP/TAZ accumulate in the nucleus and interact with transcription factors in which TEA domain transcription factor family members (TEADs) are major partners to regulate the transcription of downstream target genes (26, 31).
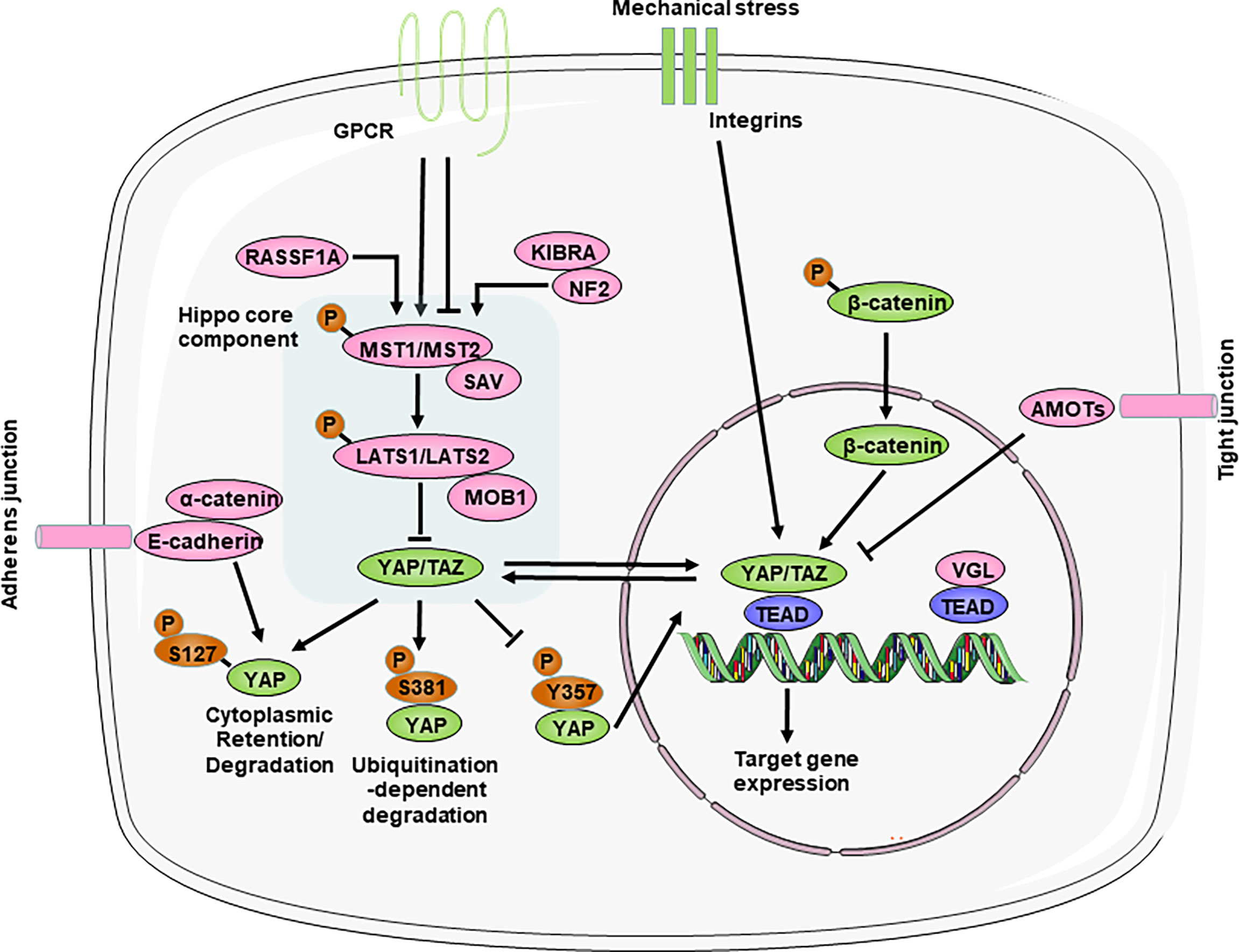
Figure 1 Overview of the Hippo-YAP pathway. The core components of the Hippo-YAP pathway contain kinases mammalian Ste20-like kinases 1/2 (MST1/2) and Large tumor suppressor homolog 1/2 (LATS1/2), their cofactors Salvador (SAV) and scaffolding proteins MOB domain kinase activator 1A/B (MOB1A/B), the transcription co-activators Yes-associated protein (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ) and the TEAD1–TEAD4 family of transcription factors. When the Hippo-YAP pathway is at the “ON” status (red), phosphorylated MST1/2 activates the phosphorylation of LATS1/2, which in turn phosphorylate and promote the degradation of the YAP and TAZ. When the Hippo-YAP pathway in at the “OFF” status (green), YAP/TAZ are dephosphorylated and accumulate in the nucleus, where they bind with TEADs to induce gene transcription. Various upstream mediators, such as adhesion proteins, mechanotransduction, and other signaling pathways, directly or indirectly to regulate the activity of YAP. For relationship among signalings, arrow represents positive regulation and transverse line represents negative regulation. AMOT, angiomotin; GPCR, G protein–coupled receptors; KIBRA, kidney and brain protein; NF2, neurofibromin 2; RASSF1A, ras association domain-containing protein 1A; VGL, vestigial like family.
Upstream regulators of the Hippo-YAP pathway
The activity of the Hippo signaling pathway is also regulated by various upstream regulators, such as cell polarity, adhesion proteins, mechanotransduction, and other signaling pathways (the Wnt/β-catenin and GPCR (G protein-coupled receptors) pathways) (Figure 1).
Importantly, the Hippo pathway, unlike other classic signal transduction pathways, does not seem to have dedicated receptors and extracellular ligands. Rather, the Hippo pathway is regulated by a network of upstream components that play roles in other processes, such as the establishment of cell–cell adhesion, including adherens junctions and tight junctions (32–37). These cell junctional complexes contain angiomotin (AMOT) family members, which have been shown to interact with the actin cytoskeleton and directly link to the Hippo pathway under different contexts (38–42); E-cadherin and its adaptor protein α-catenin, which interact with the YAP−14-3-3 protein complex (33, 43, 44); and various scaffolding components (such as neurofibromin 2 (NF2; also known as Merlin) and kidney and brain protein (KIBRA; also known as WWC1)), which regulate the activity of the core complex of the Hippo pathway (34, 45–47). Particularly, NF2 directly activate the Hippo pathway during physiological and pathological states in heart (48–50). Thus, disrupting cell–cell adhesion can have strong effects on Hippo pathway activity and lead to the activation or repression of YAP/TAZ (32).
Whether during development or adulthood, tissue homeostasis remains dependent on mechanical cues, and perturbations in extracellular matrix (ECM) stiffness or mutations affecting its perception cause pathological conditions in multiple organs, such as the heart, vessels and the liver (51). The mechanical signals exerted by ECM rigidity and cell shape also regulate the activity of Hippo pathway components (31, 52). Mechanical stress, such as when cells are grown on stiff surfaces or exposed to fluid shear stress, triggers YAP and TAZ nuclear translocation (52, 53). Moreover, integrin complexes act as upstream regulators of the Hippo pathway in response to ECM mechanical stress (54, 55). These findings identify the Hippo pathway as a sensor and mediator of mechanical cues instructed by the cellular microenvironment, but the exact mechanisms are not known and need to be further studied.
Interactions with other signaling pathways are profound regulators of Hippo activity. On the one hand, the Wnt pathway has been shown to be negatively regulated by the Hippo pathway by directly interacting with β-catenin (7, 56). On the other hand, the Hippo pathway acts as a downstream effector of the alternative Wnt signaling pathway (57, 58). In addition, these are GPCRs that are activated by lipids (lysophosphatidic acid and sphingosine-1-phosphate) or hormones (glucagon or adrenaline) and signal through F-actin to regulate YAP/TAZ (59, 60). Thus, YAP and TAZ activation–inactivation is a dynamic process involving multiple signaling pathways.
The inflammatory response in cardiovascular diseases
The development and prognosis of CVDs, including MI, cardiomyopathy and atherosclerosis, are closely related to immune and inflammatory responses. MI, including myocardial ischemia–reperfusion (I/R), and heart failure (HF), are among the leading causes of death and disability worldwide. MI is accompanied by a finely orchestrated and complex series of inflammatory events, which play critical roles in determining acute MI size and subsequent post-MI adverse LV (left ventricle) remodeling in the heart (61). First, hypoxia during ischemia impairs vascular endothelial cell (EC) integrity and barrier functions, thereby increasing vessel permeability and facilitating the infiltration of leukocytes, including neutrophils, monocytes/macrophages, and lymphocytes (62). Then, the duration of myocardial ischemia induces cellular injury and death to different constituents of the myocardium (cardiomyocytes, endothelial cells and cardiac fibroblasts (CFs)). This in turn initiates an acute proinflammatory response through the concerted action of several processes, including complement cascade activation and damage-associated molecular patterns (DAMPs), which serve as ligands for pattern recognition receptors (PRRs), such as toll-like receptors (TLRs). These factors result in the release of a cascade of proinflammatory mediators (such as cytokines, chemokines and cell adhesion molecules) and the recruitment of inflammatory cells into the ischemic region (63, 64).
Cardiomyopathy can be separated into primary (hypertrophic cardiomyopathy and dilated cardiomyopathy) and secondary (diabetic cardiomyopathy, septic cardiomyopathy) categories, which result in varied phenotypes, including myocardial dysfunction and HF (65). The immune response is closely involved in different cardiomyopathies (66–69). Damage to the myocardium, whether from a genetic or environmental cause, triggers an inflammatory response and recruits immune cells to the heart to repair the myocardium, and these cells can be identified in histopathological samples and myocardial late gadolinium enhancement by cardiac magnetic resonance imaging (69–71). A prolonged and continuous inflammatory response in turn brings about the progression of cardiomyopathy.
Vascular remodeling is a complex process that involves physical, biochemical and genetic components. It is a vital process in a wide range of CVDs, including atherosclerosis, hypertension and diabetes, and often involves the interplay of inflammatory cells (72–75). Under various stimuli, vascular ECs activate proinflammatory signaling and recruit inflammatory cells, especially macrophages, into the vessel, which is accompanied by a series of inflammatory cascade responses (76–78). Thus, targeting inflammation in vascular remodeling might be a promising therapeutic strategy.
The Hippo-YAP pathway and inflammation during myocardial infarction
Considering the complex context and diverse cell types in the heart after MI, we will discuss the role of the Hippo-YAP pathway in different cell types in the inflammatory response during MI (Figure 2).

Figure 2 Cell type-dependent Hippo-YAP pathway regulate the inflammation response after Myocardial infarction. Cardiomyocyte-specific YAP inhibits inflammatory cells infiltration, including macrophages and neutrophils, and pro-inflammatory cytokines production through interacting with TLRs (toll-like receptors), thus improve cardiac function. Epicardial-specific YAP increases Tregs (T-regulatory cells) recruitment to inhibit macrophage infiltration and pro-inflammatory cytokines production through affecting IFN-γ (interferon γ) expression. Macrophage-specific knockdown of YAP decreases the accumulation of pro-inflammatory macrophages and enhances the accumulation of anti-inflammatory macrophages through regulating the expression of Spp1 (secreted phosphoprotein 1), IL6 (interleukin 6), ARG1 (arginase 1), CD163 to improve cardiac function. Cardiac fibroblast-specific knockdown of YAP inhibits fibroinflammatory response to decrease the recruitment of inflammatory cells, the production of pro-inflammatory cytokines and improve heart function. CSF1, colony stimulating factor 1; CXCL1, C-X-C Motif Chemokine Ligand 1; CXCL12, C-X-C Motif Chemokine Ligand 12; PRRs, pattern recognition receptors.
Cardiomyocytes
Studies have shown that hypoxia during MI activates Hippo pathway kinases, which increase caspase activation and cardiomyocyte apoptosis and thereby increase the levels of phosphorylated YAP. Cardiomyocyte-specific overexpression of Hippo kinases, such as MST1 (79–81), SAV (6) and LATS2 (82), in mice results in increased cardiomyocyte apoptosis and progressive deterioration of cardiac function, whereas cardiomyocyte-specific overexpression of YAP decreases cardiomyocyte apoptosis and promotes cardiac function (83–85) (Table 1). Moreover, in large mammals such as pig, cardiomyocyte-specific knockdown of SAV reduces fibrosis and increases small blood vessels (86). Although multiple studies have depicted the role of the Hippo pathway in regulating cardiomyocyte apoptosis and cardiac function in detail, how Hippo kinases affect inflammatory cell infiltration and inflammation has yet to be defined. Dying cardiomyocytes provide the main stimulus for the postinfarction inflammatory response by releasing DAMPs in the infarcted area (64). Many studies have shown the interaction of the Hippo-YAP pathway and DAMPs in other diseases (98–101); however, to the best of our knowledge, there have been no studies on the relationship between the Hippo-YAP pathway and DAMPs during the progression of MI, which needs to be further studied.

Table 1 Hippo-YAP pathway and inflammation in myocardial infarction.
Surviving cardiomyocytes in the infarct border zone may also trigger an inflammatory response by activating PRRs, which mainly include TLRs, by binding to DAMPs to robustly produce and secrete inflammatory cytokines, which further cause the infiltration of inflammatory cells. Importantly, cardiomyocyte YAP/TEAD1 regulate the expression of TLR genes in MI (87, 88, 102). Specifically, cardiomyocyte overexpression of YAP directly suppresses the expression of TLRs, especially TLR2 and TLR4, whereas cardiomyocyte-specific Yap depletion increases the expression of TLR2 and TLR4 in vivo and in vitro (87, 88). Mechanistically, TEAD1 can directly bind genomic regions adjacent to several TLRs, especially the transcriptional start region of Tlr4 (88). In addition, TLR3-mediated regeneration and repair of the damaged neonatal myocardium occurs through glycolytic-dependent YAP activation (102). Thus, it is worthwhile to further delineate the detailed regulatory mechanisms of the Hippo-YAP pathway and TLR signaling. Cardiomyocyte-specific knockdown of Mst1 decreases proinflammatory cytokine expression, including TNF-α, IL1, and IL6, and regulates the transcriptional activity of NF-κB in the heart 4 weeks after MI (79). Similarly, using RNA sequencing on hearts with cardiomyocyte-specific knockout of Sav and control hearts, the authors showed that the downregulated genes in Sav-CKO cardiomyocytes after MI were associated with inflammation, indicating that inflammation was more effectively resolved in Sav-CKO mice (6). Chen et al. used modified mRNA (modRNA) to transiently express constitutively active human YAP in the heart, which significantly reduced cardiomyocyte death and myocardial neutrophil and macrophage infiltration after I/R (87). In conclusion, cardiomyocyte YAP decreases proinflammatory cytokine activation and macrophage or neutrophil infiltration by inhibiting TLR signaling.
Recently, a series of studies have demonstrated that the epicardium is activated following myocardial injury (103, 104). The activated adult epicardium is a source of proinflammatory signals after MI (105, 106). T-regulatory cells (Tregs), which are a subset of CD4+ T cells, have been shown to suppress the innate and adaptive immune response and limit deleterious remodeling following myocardial injury (107). Importantly, epicardial deletion of Yap/Taz exacerbates fibrosis and pericardial inflammation by decreasing the recruitment of suppressive CD4+ Tregs and decreasing the expression of genes encoding IFN-γ (interferon γ), a known Treg inducer. However, controlled local delivery of IFN-γ following MI rescued Treg infiltration into the injured myocardium in Yap/Taz mutants and decreased fibrosis (89). Future studies will be needed to identify inflammatory cytokines that are dysregulated by epicardial YAP/TAZ in the injured myocardium.
Macrophages
A growing number of studies are emerging about the role of the Hippo-YAP pathway in macrophages in various diseases, including MI (23, 108). Global deletion of Rassf1A increases macrophage accumulation and release of the proinflammatory cytokine TNFα in the heart after IR, and this pattern cannot be found in cardiomyocyte-specific Rassf1A-knockout mice relative to controls. In addition, myeloid-specific deletion of Rassf1A increases the expression of inflammatory cytokines and macrophage infiltration caused by I/R in mice, suggesting that macrophage RASSF1A plays a leading role in regulating cardiac inflammation after I/R. Cell-based studies revealed that RASSF1A negatively regulates YAP to inhibit NF-κB in macrophages and thereby attenuates inflammatory cytokine expression in macrophages (90).
Traditional views of macrophage biology defined macrophages as monocyte-derived cells that are composed of two populations: proinflammatory M1 macrophages and reparative M2 macrophages (109, 110). Early recruitment on Days 1 to 3 after MI of proinflammatory macrophages secreting high levels of proinflammatory cytokines and chemokines such as IL6 contributes to the removal of dead cells. At a later stage on Days 3 to 7 after MI, anti-inflammatory macrophage subpopulations, which are associated with Arg1 (Arginase 1), are selectively recruited and may participate in the resolution of the postinfarction inflammatory response (111). The expression of YAP and TAZ was increased in macrophages undergoing proinflammatory or reparative phenotype changes (91), and the expression of endogenous MST1 in the cardiac macrophages of wild-type mice was decreased in the first 3 days after MI (92), suggesting the potential role of the Hippo pathway in the cardiac inflammatory response after MI. Genetic deletion of Yap/Taz in macrophages impairs the proinflammatory macrophage phenotype and promotes a reparative macrophage phenotype, which is accompanied by improved post-MI ventricular remodeling and heart function after MI. In contrast, YAP activation in macrophages shows the opposite effects on macrophage polarization and cardiac function. Mechanistically, YAP/TAZ promote the proinflammatory response by directly regulating Il6 promoter activity and impair the reparative response by binding to the Arg1 promoter (91).
However, macrophages in MI are heterogeneous and are not confined to the M1 and M2 phenotypes (112, 113). Novel techniques that allow the resolution of gene expression at the single-cell level, including single-cell RNA sequencing, improve the understanding of the definitions of macrophage subsets. Combined with single-cell sequencing, Liu et al. reported three macrophage subtypes (MP1, MP2 and MP3) in MI hearts. MP1 cells highly expressed the proinflammatory genes Ccl4, Ccl2, and Spp1. In contrast, MP2 cells had high expression of anti-inflammatory genes such as Cd163, Cbr2, and Rgs10. MP3 cells have mixed properties and express anti-inflammatory (Slpi and Arg1) and proangiogenic (Cxcl2) genes (92). Although no differences were observed in the numbers of infiltrated macrophages, monocytes, or neutrophils in Mst1/2 myeloid-specific knockout hearts compared to controls, there were differences in the numbers of the three macrophage subtypes, as indicated by increased MP1 cells and decreased MP2 and MP3 cells. In particular, Mst1 mutant hearts showed increased Spp1 expression levels and reduced Cd163 and Arg1 expression in CD68+ cells, indicating that Mst1/2 knockout promoted macrophage subtype switching and impaired inflammation resolution in the mouse hearts after MI (92). Mechanistically, MST1 phosphorylates 5-lipoxygenase at its T218 residue, resulting in a reduction of leukotriene B4 production. However, in Mst1-YAP double knockout macrophage, leukotriene B4 production is similar with Mst1 knockout control, suggesting macrophage YAP and MST1 might contribute to cardiac repair post-MI independently. In summary, although YAP exhibits an anti-inflammatory phenotype in response to viral infection (21, 114), macrophage YAP exacerbates the inflammatory response and regulates macrophage subtypes in the heart after MI, resulting in impaired cardiac function.
Cardiac fibroblasts
CFs, which are one of the most abundant cells in the mammalian heart, remain quiescent and may play a role in maintaining the ECM network; however, when stimulated with DAMPs and reactive oxygen species, CFs are capable of secreting large amounts of inflammatory cytokines and chemokines and acquire a proinflammatory phenotype (111). In CFs, the phosphorylation level of YAP is reduced and thus YAP translocates into nucleus post-MI both in vivo and in vitro (93, 115). TAZ expression is increased in infarct zone after MI, but whether the increase in TAZ expression is originated from activated myofibroblasts still need to be studied (116). Furthermore, Xiao et al. conditionally deleted Lats1 and Lats2 (Lats1/2) in CFs and used single-cell sequencing to show that Lats1/2 deletion led to an ongoing CF-derived proinflammatory cascade that promoted myeloid cell influx, activation, and expansion in sham hearts and post-MI hearts (94). Another study showed that siRNA-mediated Yap knockdown in primary rat CFs revealed a specific role for Yap in controlling the expression of proinflammatory cytokines and chemokines (Tlr2, Il6, C2, C4a) (95). Fibroblast-specific deletion of Yap/Taz reduces the post-MI inflammatory response and affects the polarization and infiltration of macrophages in the infarcted hearts, further attenuating MI–induced cardiac dysfunction and fibrosis (96). In vivo, an adeno-associated virus increased the expression of YAP in CFs, increased chemokine expression (Ccl2, Ccl5) and promoted macrophage recruitment. In vitro, YAP activation in cultured CFs increased profibrotic and proinflammatory gene expression. Further study showed that YAP regulates Ccl2 expression by binding to the Ccl2 promoter (97). In summary, YAP in fibroblasts promotes inflammatory reactions and impairs heart function after MI. The differences and mechanism of the Hippo pathway in the inflammatory response and cardiac repair among different cell types after MI need to be clarified in future studies.
The Hippo-YAP pathway and the inflammatory response during heart failure and cardiomyopathy.
Heart failure affects about 40 million people worldwide (117). Molecular biology experiments showed that YAP is highly expressed and activated in hypertrophic cardiomyopathy tissue samples, as well as in TAC murine hearts, indicating that Hippo/YAP signaling is involved in the pathogenesis of cardiac hypertrophy (118) (Table 2; Figure 3). Cardiomyocyte-specific genetic modification experiments showed that YAP protected against cardiomyocyte apoptosis and protected heart function during acute pressure overload (PO) but exacerbated the progression of cardiac dysfunction during chronic PO (119–122), suggesting that the effects of YAP differ between the acute and chronic phases of PO. This discrepancy maybe caused by long-term elevation of YAP and concomitant upregulation of TEAD1 which induces cardiomyocyte dedifferentiation. In recent decades, a growing number of studies have suggested that an immune response regulated the development of cardiac hypertrophy (132, 133). Ikeda et al. used cardiomyocyte-specific knockout of WW45 (WW45-cKO) to inhibit the activity of MST1/2 and LATS1/2. Interestingly, myocardial infiltration of leukocytes, macrophages, and neutrophils was not significantly different between control and WW45-cKO mice 1 week after PO. The infiltration of macrophages and neutrophils was significantly enhanced 4 weeks after PO in WW45-cKO mice compared with control mice (122). In CFs, inhibiting Rassf1A promoted fibroblast proliferation, activation of the NF-κB pathway and selective upregulation of TNF-α expression (123), suggesting that the RASSF1A-Hippo pathway affects the inflammatory signaling pathway in fibroblasts. It is vital to further elucidate the mechanism by which the Hippo-YAP pathway regulates immune cell infiltration, including macrophage, Treg, neutrophil, and proinflammatory cytokine activation, after PO.
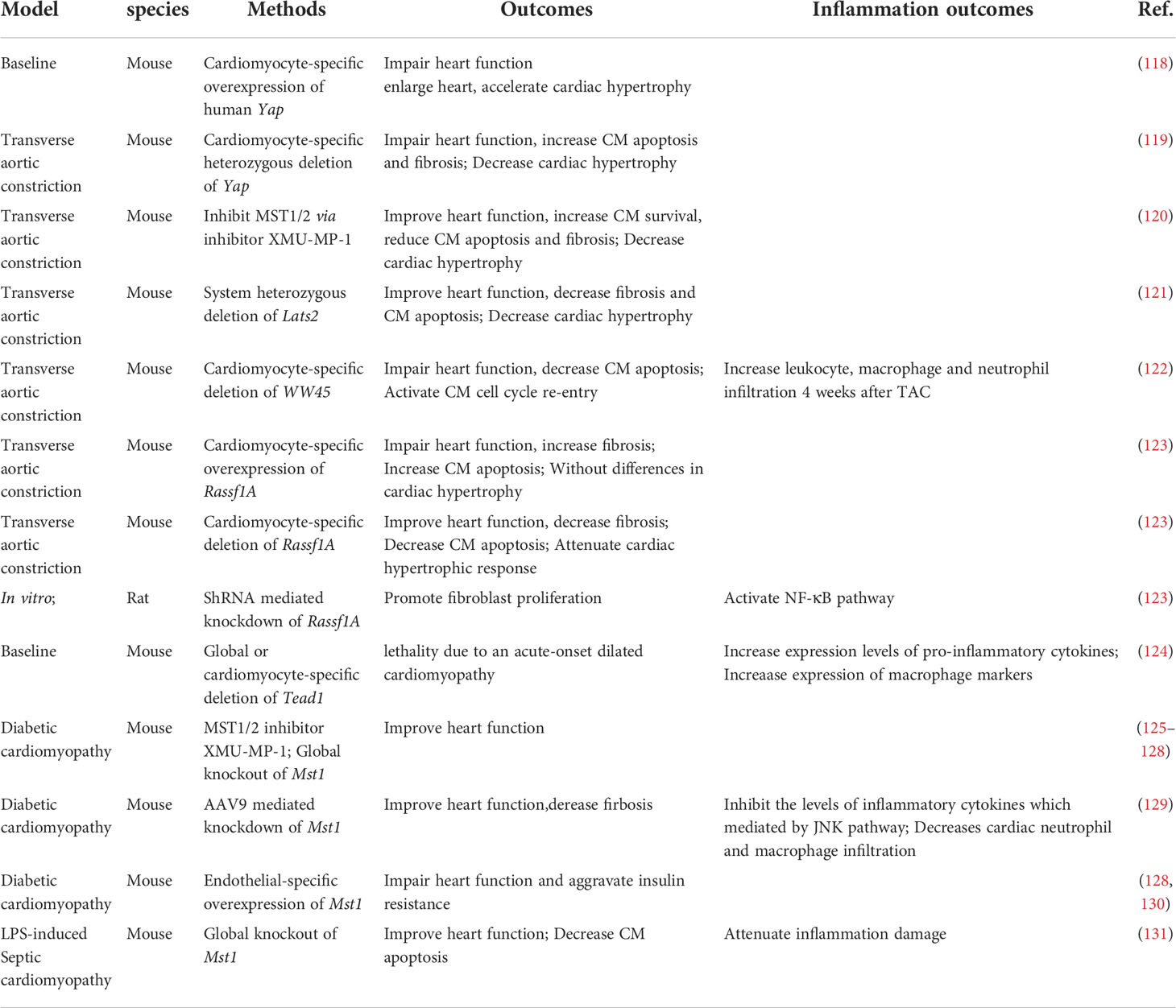
Table 2 Hippo-YAP pathway and inflammation in heart failure and cardiomyopathy.
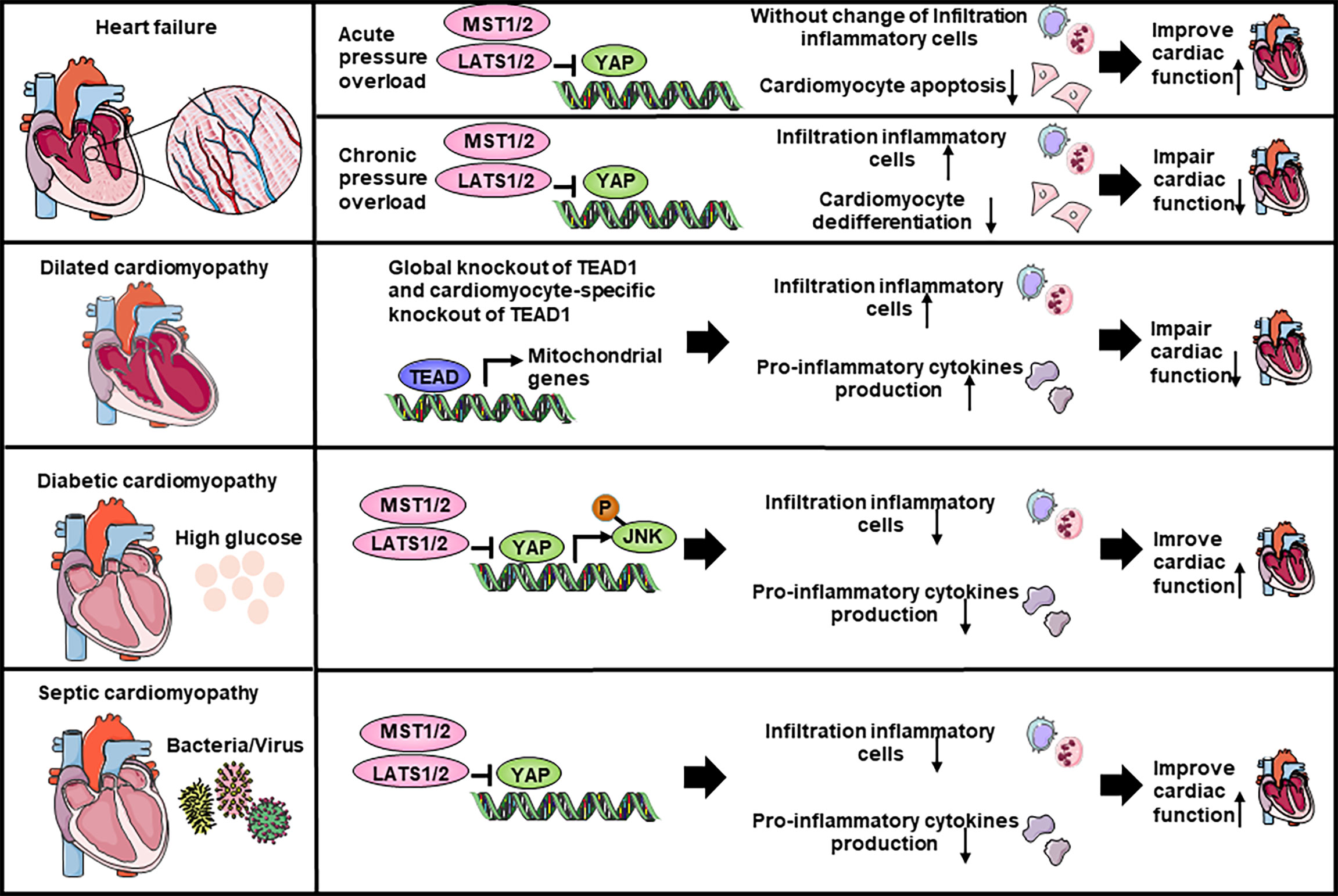
Figure 3 The Hippo-YAP pathway regulate the inflammation response in heart failure and cardiomyopathy. YAP increases the accumulation of inflammatory cells and impairs cardiac function in chronic pressure overload while hardly affects the accumulation of inflammatory cells in acute pressure overload. In dilated cardiomyopathy, global knockout of Tead1 and cardiomyocyte-specific knockout of Tead1 both can increase inflammatory cells infiltration and pro-inflammatory cytokines production to impair heart function. Both in diabetic cardiomyopathy and septic cardiomyopathy, YAP is able to inhibit inflammatory cells recruitment and pro-inflammatory cytokines production, thereby improve heart function. JNK, c-Jun N-terminal kinase.
Dilated cardiomyopathy is a nonischemic heart muscle disease with structural and functional myocardial abnormalities that can be caused by infection, autoimmunity, endocrine dysfunction, muscular dystrophy, pregnancy and gene mutation (67). Several studies have demonstrated that activation of the Hippo signaling pathway causes dilated cardiomyopathy in mice (85, 134–136). Inflammation occurs during the development of dilated cardiomyopathy, as indicated by the infiltration of inflammatory cells, such as macrophages, and the production of proinflammatory cytokines, which in turn affects the progression of dilated cardiomyopathy. Global knockout or cardiomyocyte-specific knockout of Tead1 can dramatically increase the expression levels activated macrophage markers and the proinflammatory cytokines Il6 and Ccl2 in the heart, which is accompanied by acute onset of dilated cardiomyopathy (124). However, the role of the Hippo-YAP pathway and inflammation in nonmyocytes has not been studied.
Diabetic cardiomyopathy is defined by abnormal myocardial structure and performance in the absence of other cardiac risk factors, such as coronary artery disease, hypertension, and significant valvular disease, in individuals with diabetes mellitus (137, 138). A maladaptive proinflammatory response has been implicated in the development of diabetic cardiomyopathy (137, 139–141). Inhibiting Mst1 in multiple ways, such as the pharmaceutical inhibitor XMU-MP-1 or transgenic mice, improves glucose tolerance and heart function in a diabetic mouse model (125–129), whereas the overexpression of MST1 impairs cardiac function and exacerbates insulin resistance (128, 130). Specifically, cardiomyocyte-specific Mst1 inhibition decreases cardiac neutrophil and macrophage infiltration and inhibits the levels of inflammatory cytokines mediated by the JNK (c-Jun N-terminal kinase) pathway during the development of diabetic cardiomyopathy (129). Immunostaining and immunoblot analyses showed that nuclear expression of YAP in cardiomyocytes and the level of YAP protein expression in the heart were significantly higher in HF patients with diabetes than in HF patients without diabetes (142). However, in the diabetic heart, inhibiting YAP significantly attenuated the infiltration of leukocytes, macrophages and neutrophils after PO (142), suggesting that YAP may promote HF by stimulating cardiomyocyte dedifferentiation and inflammation in diabetic hearts in the presence of high blood pressure.
Septic cardiomyopathy, which is characterized by reversible left ventricular systolic dysfunction, is increasingly recognized as a potential complication of septic shock (143). Because sepsis is an inflammatory condition, inflammatory cytokine production is seen to some extent in all affected organs, including the heart (144). LPS (lipopolysaccharide) administration in vivo can induce a model of septic cardiomyopathy (145). Global Mst1 deletion can attenuate LPS-mediated upregulation of TNFα and IL6 levels in the heart, thereby improving cardiac function (131). However, no further study has been performed on the interaction of the Hippo-YAP pathway and septic cardiomyopathy.
The Hippo-YAP pathway and the inflammatory response during vascular remodeling
Vascular remodeling refers to structural and functional alterations in the arterial wall in response to vascular injury, such as disturbed flow (DF), metabolic disorders, hypertension and exogenous pathogens. Although inflammation is a well-accepted pathological mechanism in vascular remodeling, to date, it has not been translated to specific therapies used in clinical practice (16).
Vascular endothelial cells
Vascular endothelial cells (ECs) covering the inner surface of blood vessels are constantly exposed to shear stress because of the frictional force created by the blood flow (146). Different shear forces induce distinct cellular responses. Regions of arteries with bifurcations, curvatures, or valves have weak shear stress with complex changes in direction, called disturbed flow. The endothelium of these regions have increased permeability, increased turnover and increased monocytes adhesion, thus are susceptible to developing atherosclerotic plaques. In contrast, artery regions of straight segments such as descending thoracic aorta have unidirectional shear stress that is anti-inflammatory and atheroprotective (147). Immunofluorescence staining of the normal carotid artery showed that YAP/TAZ-positive cells were abundantly present in the endothelium in the tunica intima, but few were present in the media layer. In contrast, in the atherosclerotic carotid artery and the aortic arch, YAP/TAZ staining was prominent in both the endothelium, the media layer and the intimal hyperplasia plaque, suggesting that YAP/TAZ may regulate EC functions and that the dysregulation of YAP/TAZ may contribute to the development of atherosclerosis (148) (Table 3; Figure 4). Several groups showed that pYAPSer127 was increased in HUVECs (Human umbilical vein endothelial cells) and mouse aortas exposed to USS in a time-dependent manner. Accordingly, USS suppressed the transactivation of YAP/TAZ and downregulated the expression of target genes. Conversely, DF reduced pYAPSer127 expression and increased YAP/TAZ target gene expression (54, 148–151). DF promotes inflammation through YAP/TAZ activation (148). YAP/TAZ activation induces several proinflammatory markers, such as Il6, Il8 and Ccl2 (54, 148, 149, 151), and increases monocyte adhesion to HUVECs, suggesting that endothelial YAP/TAZ activation participates in the initiation of atherosclerosis by promoting monocyte adhesion (54, 148). Furthermore, YAP/TAZ promotes endothelial activation by enhancing JNK activity. EC-specific YAP overexpression in mice significantly increased plaque formation, which was accompanied by increased expression of p-JNK and macrophage markers compared with those of control littermates. Moreover, treatment with the antiatherosclerotic drug statin inhibited EC proliferation and inflammation, which were partly mediated by the inactivation of YAP/TAZ (54, 148).
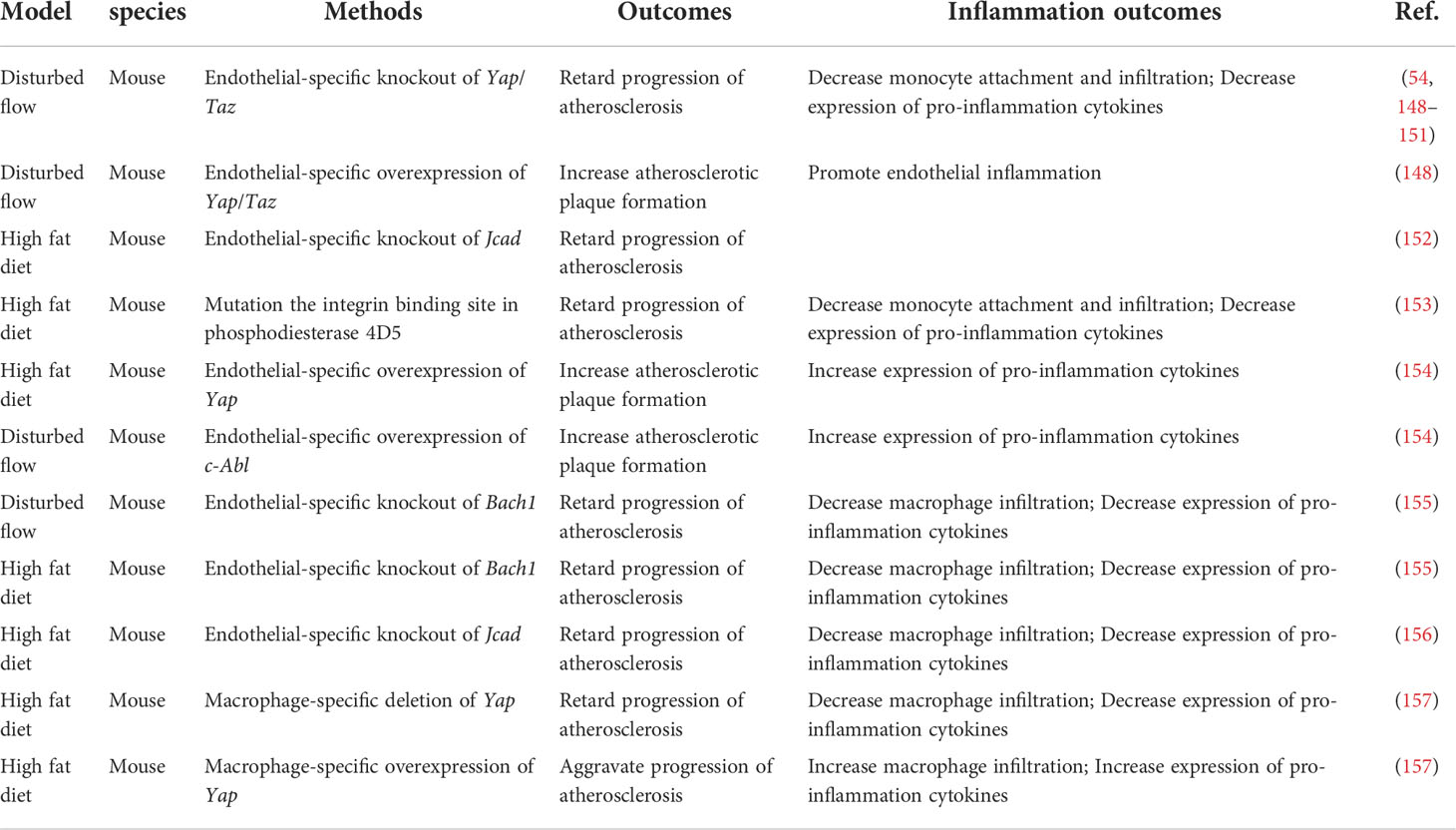
Table 3 Hippo-YAP pathway and inflammation in atherosclerosis.
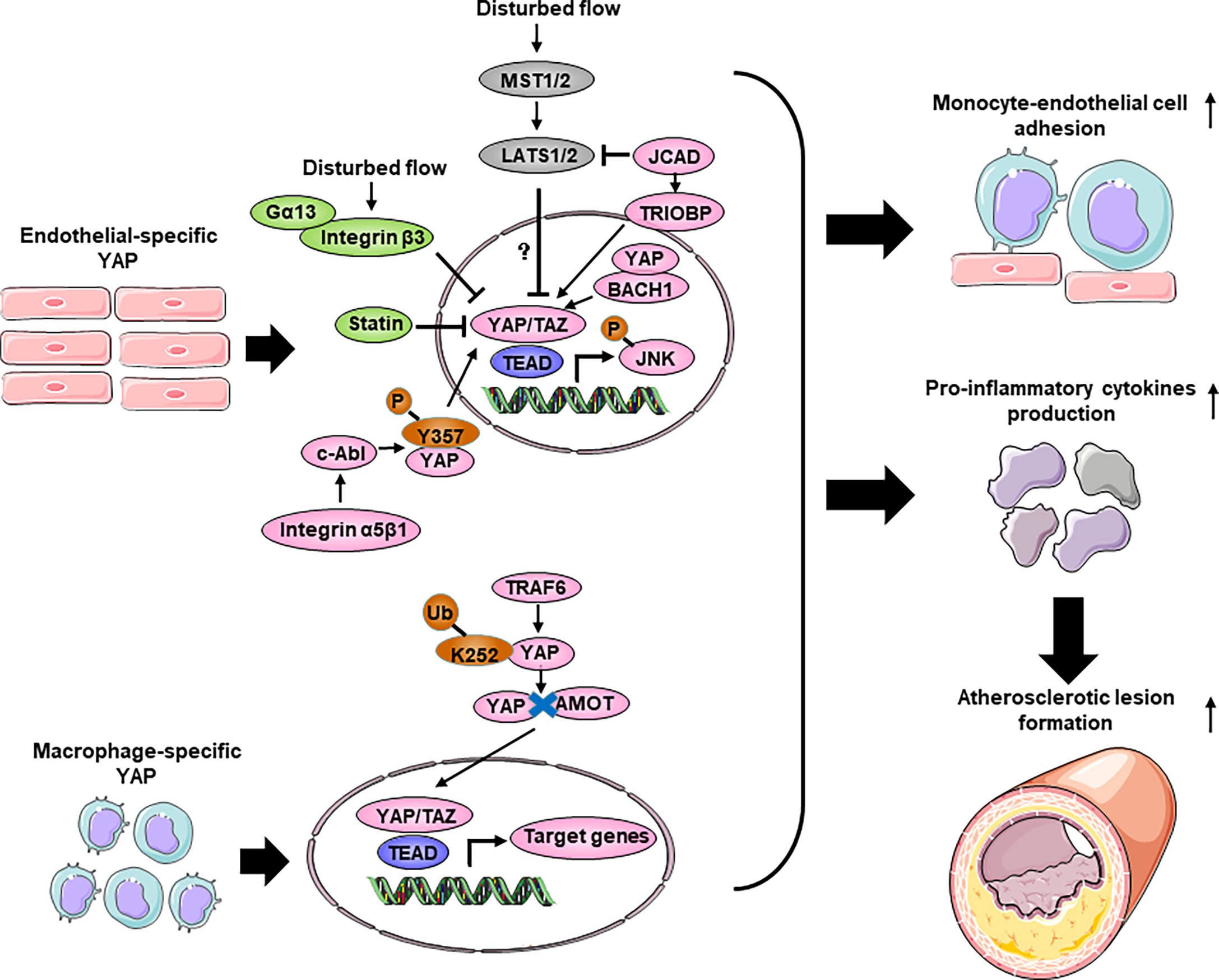
Figure 4 The role of Hippo-YAP pathway regulating inflammation response during atherosclerosis. Disturbed flow promotes YAP nuclear translocation and activates the JNK signaling without affecting the Hippo kinases, thereby accelerates monocyte-endothelial cell adhesion and pro-inflammatory cytokines production to increase the formation of atherosclerotic lesion. Several upstream regulators such as JCAD, BACH1, Integrin β3 and c-Abl affect endothelial inflammation and atherosclerosis through interacting with YAP. Moreover, statins exert anti-atherosclerosis function partly through YAP. In bone marrow derived macrophage, YAP undergoes TRAF6-dependent ubiquitination and translocate into nucleus in response to IL-1β. Macrophage-specific overexpression of YAP promotes macrophage accumulation and pro-inflammatory cytokines production, thereby aggravates atherosclerosis. BACH1, BTB domain and CNC homolog 1; Gα13, guanine nucleotide-binding protein subunit alpha 13; JCAD, junctional cadherin 5 associated; TRIOBP, TRIO and F-actin binding protein; TRAF6, TNF Receptor Associated Factor 6.
The regulation of YAP/TAZ activities, as indicated by the phosphorylation status of YAP and its cytoplasmic/nuclear localization, are controlled canonically by Hippo pathway kinases, such as LATS1/2 (5). To determine whether flow-induced YAP translocation is dependent on Hippo kinase, researchers used siRNA to knockdown Lats1. Interestingly, Lats1 knockout was not sufficient to reduce laminar shear stress-induced YAP phosphorylation in ECs (148, 150). Recent reports have shown that mechanical stimuli modulate YAP/TAZ activities via signaling pathways independent of the Hippo pathway. However, Xu et al. found that USS-mediated YAPSer127 phosphorylation was almost abolished by knockdown of Lats1/2 expression in HUVECs (149), indicating a critical role of LATS1/2 in mediating laminar flow-induced YAP phosphorylation. The differences among these studies may be attributed to the stimulation conditions, cell states and reagents. Recently, Jones et al. showed that JCAD (Junctional Cadherin 5 Associated) regulates adhesion molecule expression and monocyte-EC adhesion by interacting with LATS2, which is a kinase in the Hippo pathway, in a LATS2-dependent manner (152). However, the administration of an inhibitor of the Hippo kinase MST1/2 resulted in the attenuation of Angiotensin II-induced ascending arterial aneurysms without affecting aortic atherosclerotic plaque progression (158). Due to mouse models and the effects of exogenous inhibition, further direct investigation of the role of Hippo kinase in atherosclerosis is needed. Thus, further investigation will be required to clarify the kinases regulation of the Hippo-YAP pathway in ECs in response to different flow patterns.
In recent years, several upstream molecules have been shown to interact with YAP to regulate atherosclerotic plaque development and inflammation in ECs. The integrin family of receptors plays a critical role in cellular crosstalk with the microenvironment (159). Wang et al. demonstrated that the overexpression of integrin β3 by cytoplasmic-domain-deleted integrin (β3Δcyto) reversed DF-induced YAP phosphorylation in HUVECs, and knockdown of integrin β3 or the G-protein subunit Gα13 attenuated YAP phosphorylation and upregulated YAP/TAZ target gene expression (54). Other groups demonstrated that integrin α5β1 regulated YAP activity in response to DF (153, 154). Li et al. showed that the activation of integrin α5β1 induced tyrosine but not serine phosphorylation of YAP in ECs. Blocking integrin α5β1 with ATN161 abolished the DF-induced phosphorylation of YAP at Y357. Mechanistic studies showed that a c-Abl inhibitor attenuated integrin α5β1–induced YAP tyrosine phosphorylation (154), revealing that the integrin α5β1/c-Abl/YAP pathway may be a potential therapeutic target for early-stage atherosclerosis. BACH1 is induced and translocates into the nucleus in HUVECs and upregulates YAP expression by binding to the YAP promoter to form a complex that induces the adhesion of monocytes/macrophages (155). JCAD upregulates the activation of the YAP/TAZ pathway and the expression of downstream proatherogenic genes by interacting with the actin-binding protein TRIOBP (156). The main antiatherosclerotic drugs are statins, which are cholesterol-lowering compounds and are commonly used as first-line treatments for patients with CVDs. Intriguingly, the anti-inflammatory and anti-plaque effects of statins are now being reinterpreted as being mediated, at least in part, by their capacity to inhibit YAP and TAZ (54, 148, 160). More broadly, this finding suggests that endothelial YAP/TAZ could be specifically targeted to treat atherosclerosis.
In conclusion, YAP promotes endothelial inflammation under shear stress-induced atherosclerosis. However, the role of Hippo-YAP in endothelial inflammation is stress dependent. Diabetes-induced endothelial dysfunction and inflammation are critical factors in the mof diabetic vascular complications (161). YAP is dephosphorylated/activated by high glucose in ECs, leading to increased endothelial inflammation and monocyte attachment. Consistently, inhibiting YAP in HUVECs reduces endothelial inflammation and monocyte attachment (162, 163). Knocking down Yap/Taz in ECs using siRNA significantly reduced the TNF-α-induced expression of the leukocyte adhesion molecule VCAM1 (vascular cell adhesion protein 1) (163). Moreover, YAP aggravates the inflammatory response in the vascular endothelium under high glucose conditions. Similarly, in the aortas of Angiotensin II -induced hypertensive mice, treatment with the YAP/TAZ inhibitor verteporfin reduces macrophage infiltration and proinflammatory cytokine production (164). However, another study demonstrated that Tie2-mediated endothelial-specific knockout of Yap in mice resulted in mild endothelial inflammation and impaired microvessel permeability in lung, as evidenced by increased numbers of total cells, neutrophils, and macrophages and an increased neutrophil/macrophage ratio after LPS administration. Mechanistically, in lung ECs, exposure to LPS induces endothelial activation through TLR4 and triggers proinflammatory cytokine secretion and the expression of adhesion molecules via NF-κB signaling (165). The reasons for the differences among different groups are not fully clear. It is possible that the differences are because of the cell types and the inflammatory stimuli used.
Macrophages
During the progression of atherosclerosis, circulating monocytes enter sites of arterial hemodynamic stress by adhering to the ECs lining the lumen of susceptible arteries. Once monocytes enter the subendothelial space, they differentiate into lesional macrophages and further transform into foam cells (166). Yap/Taz knockdown inhibited the upregulation of β2 integrin and ICAM1 (intercellular adhesion molecule-1) induced by TNFα in THP1 cells and reduced their adhesion to the activated ECs without altering the plasticity of THP1 monocyte-to-macrophage differentiation (148). YAP undergoes TRAF6-dependent Lys63 chain ubiquitination in response to IL-1β in bone marrow-derived macrophages (BMDMs), which results in increased YAP nuclear localization and protein stability. In vivo, YAP overexpression in monocytes/macrophages promotes atherosclerotic lesion size and macrophage infiltration, whereas Yap deletion in monocytes/macrophages alleviates atherosclerotic plaque development. YAP significantly upregulates the production of chemokines, such as Ccl2, Ccl7, Cxcl1, Cxcl3, Cxcl5 and Cxcl12, and monocyte/macrophage migration (157).
Conclusions
Many studies have revealed the central regulatory role of the Hippo-YAP pathway in various cardiovascular physiological and pathological contexts. Hippo signaling has been shown to exert various effects according to different cell contexts and microenvironments because of its diverse interactions with a variety of signaling transduction cascades. Recently, many interesting findings have suggested that the Hippo-YAP pathway regulates inflammatory cell infiltration and inflammatory cytokine activation, which provides a deeper understanding of the Hippo-YAP pathway in the complex multicellular interaction environment. However, several issues need to be addressed in future studies. First, in cardiovascular research, most of the available studies focused on the role of Hippo-YAP in monocytes/macrophages, and little research has been done on other immune cells, such as T cells, which have been observed in other diseases (18, 167–169). Second, uncovering the classical and nonclassical regulation of the Hippo-YAP pathway in the inflammatory response is essential for controlling CVD progression. Third, future work must bring forth the data to further understand the mechanisms involved and address the cell-specific differences in the role of the Hippo-YAP pathway in CVDs. In summary, an increasing number of uncovered mechanisms of the Hippo-YAP pathway have broadened our horizon, leading to deeper thinking and the search for clinical research transitions.
Author contributions
QC and LZ conceived ideas. AZ created the figures and wrote the article. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a grant from the National Natural Science Foundation of China (81930010, 82125005).
Acknowledgments
We acknowledge the use of Servier Medical Art image bank that is used to create schematic Figures 1–4.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AMOT, angiomotin; ARG1, arginase 1; BACH1, BTB domain and CNC homolog 1; CBR2, carbonyl reductase 2; CCL, C-C Motif Chemokine Ligand; CF, cardiac fibroblast; CM, cardiomyocyte; CVDs, cardiovascular diseases; CXCL, C-X-C Motif Chemokine Ligand; DAMPs damage-associated molecular patterns; DF, disturbed flow; EC, endothelial cell; ECM, extracellular matrix; GPCR, G-protein–coupled receptors; Gα13, guanine nucleotide-binding protein subunit alpha 13; HF, heart failure; HUVECs, human umbilical vein endothelial cells; ICAM1, intercellular adhesion molecule-1; IFN-γ, interferon γ; IL, interleukin; I/R, myocardial ischemia–reperfusion; JCAD, junctional cadherin 5 associated; JNK, c-Jun N-terminal kinase; KIBRA, kidney and brain protein; LATS1/2, large tumor suppressor homolog 1/2; LPS, lipopolysaccharide; LV, left ventricle; MI, myocardial infarction; MOB1, MOB domain kinase activator 1A/B; MST1/2, mammalian Ste20-like kinases 1/2; NF2, neurofibromin 2; NF-κB, nuclear factor-κB; PRRs, pattern recognition receptors; PO, pressure overload; RASSF1A, ras association domain-containing protein 1A; RGS10, regulator of G protein signaling 10; SAV, salvador; siRNA, short interfering RNA; SLPI, secretory leukocyte peptidase inhibitor; SPP1, secreted phosphoprotein 1; TAZ, transcriptional coactivator with a PDZ binding motif; TEADs, TEA domain transcription factor family members; TLRs, toll-like receptors; TNFα, tumor necrosis factor α; TRAF6, TNF receptor associated factor 6; Tregs, T-regulatory cells; TRIOBP, TRIO and F-actin binding protein; USS, unidirectional shear stress; VCAM1, vascular cell adhesion protein 1; WW45-cKO, cardiomyocyte-specific knockout of WW45; YAP, yes-associated protein.
Glossary

References
1. Meng Z, Moroishi T, Guan KL. Mechanisms of hippo pathway regulation. Genes Dev (2016) 30(1):1–17. doi: 10.1101/gad.274027.115
2. Zhao D, Liu J, Wang M, Zhang X, Zhou M. Epidemiology of cardiovascular disease in China: Current features and implications. Nat Rev Cardiol (2019) 16(4):203–12. doi: 10.1038/s41569-018-0119-4
3. Mahmood SS, Levy D, Vasan RS, Wang TJ. The framingham heart study and the epidemiology of cardiovascular disease: a historical perspective. Lancet (2014) 383(9921):999–1008. doi: 10.1016/S0140-6736(13)61752-3
4. Leong DP, Joseph PG, McKee M, Anand SS, Teo KK, Schwalm JD, et al. Reducing the global burden of cardiovascular disease, part 2: Prevention and treatment of cardiovascular disease. Circ Res (2017) 121(6):695–710. doi: 10.1161/CIRCRESAHA.117.311849
5. Wang J, Liu S, Heallen T, Martin JF. The hippo pathway in the heart: Pivotal roles in development, disease, and regeneration. Nat Rev Cardiol (2018) 15(11):672–84. doi: 10.1038/s41569-018-0063-3
6. Leach JP, Heallen T, Zhang M, Rahmani M, Morikawa Y, Hill MC, et al. Hippo pathway deficiency reverses systolic heart failure after infarction. Nature (2017) 550(7675):260–4. doi: 10.1038/nature24045
7. Heallen T, Zhang M, Wang J, Bonilla-Claudio M, Klysik E, Johnson RL, et al. Hippo pathway inhibits wnt signaling to restrain cardiomyocyte proliferation and heart size. Science (2011) 332:458–61. doi: 10.1126/science.1199010
8. Ma S, Meng Z, Chen R, Guan KL. The hippo pathway: Biology and pathophysiology. Annu Rev Biochem (2019) 88:577–604. doi: 10.1146/annurev-biochem-013118-111829
9. Sreejit G, Abdel-Latif A, Athmanathan B, Annabathula R, Dhyani A, Noothi SK, et al. Neutrophil-derived S100A8/A9 amplify granulopoiesis after myocardial infarction. Circulation (2020) 141(13):1080–94. doi: 10.1161/CIRCULATIONAHA.119.043833
10. Takahashi M. Role of NLRP3 inflammasome in cardiac inflammation and remodeling after myocardial infarction. Biol Pharm Bulletin (2019) 42(4):518–23. doi: 10.1248/bpb.b18-00369
11. Thackeray JT, Hupe HC, Wang Y, Bankstahl JP, Berding G, Ross TL, et al. Myocardial inflammation predicts remodeling and neuroinflammation after myocardial infarction. J Am Coll Cardiol (2018) 71(3):263–75. doi: 10.1016/j.jacc.2017.11.024
12. Lubos N, van der Gaag S, Gerçek M, Kant S, Leube RE, Krusche CA. Inflammation shapes pathogenesis of murine arrhythmogenic cardiomyopathy. Basic Res Cardiol (2020) 115(4):42. doi: 10.1007/s00395-020-0803-5
13. Peterson LR, Gropler RJ. Metabolic and molecular imaging of the diabetic cardiomyopathy. Circ Res (2020) 126(11):1628–45. doi: 10.1161/CIRCRESAHA.120.315899
14. Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science (2013) 339(6116):161–6. doi: 10.1126/science.1230719
15. Silvis MJM, Demkes EJ, Fiolet ATL, Dekker M, Bosch L, van Hout GPJ, et al. Immunomodulation of the NLRP3 inflammasome in atherosclerosis, coronary artery disease, and acute myocardial infarction. J Cardiovasc Trans Res (2021) 14(1):23–34. doi: 10.1007/s12265-020-10049-w
16. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med (2017) 377(12):1119–31. doi: 10.1056/NEJMoa1707914
17. Ridker PM, MacFadyen JG, Everett BM, Libby P, Thuren T, Glynn RJ. Relationship of c-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: A secondary analysis from the CANTOS randomised controlled trial. Lancet (London England) (2018) 391(10118):319–28. doi: 10.1016/S0140-6736(17)32814-3
18. Du X, Wen J, Wang Y, Karmaus PWF, Khatamian A, Tan H, et al. Hippo/Mst signalling couples metabolic state and immune function of CD8α(+) dendritic cells. Nature (2018) 558(7708):141–5. doi: 10.1038/s41586-018-0177-0
19. Shi H, Liu C, Tan H, Li Y, Nguyen TM, Dhungana Y, et al. Hippo kinases Mst1 and Mst2 sense and amplify IL-2R-STAT5 signaling in regulatory T cells to establish stable regulatory activity. Immunity (2018) 49(5):899–914.e6. doi: 10.1016/j.immuni.2018.10.010
20. Mia MM, Singh MK. Emerging roles of the hippo signaling pathway in modulating immune response and inflammation-driven tissue repair and remodeling. FEBS J (2022) 289(14):4061–81. doi: 10.1111/febs.16449
21. Wang S, Xie F, Chu F, Zhang Z, Yang B, Dai T, et al. YAP antagonizes innate antiviral immunity and is targeted for lysosomal degradation through IKKε-mediated phosphorylation. Nat Immunol (2017) 18(7):733–43. doi: 10.1038/ni.3744
22. LaCanna R, Liccardo D, Zhang P, Tragesser L, Wang Y, Cao T, et al. Yap/Taz regulate alveolar regeneration and resolution of lung inflammation. J Clin Invest (2019) 129(5):2107–22. doi: 10.1172/JCI125014
23. Zhou X, Li W, Wang S, Zhang P, Wang Q, Xiao J, et al. YAP aggravates inflammatory bowel disease by regulating M1/M2 macrophage polarization and gut microbial homeostasis. Cell Rep (2019) 27(4):1176–89.e5. doi: 10.1016/j.celrep.2019.03.028
24. Mooring M, Fowl BH, Lum SZC, Liu Y, Yao K, Softic S, et al. Hepatocyte stress increases expression of yes-associated protein and transcriptional coactivator with PDZ-binding motif in hepatocytes to promote parenchymal inflammation and fibrosis. Hepatology (2020) 71(5):1813–30. doi: 10.1002/hep.30928
25. Liu B, Zheng Y, Yin F, Yu J, Silverman N, Pan D. Toll receptor-mediated hippo signaling controls innate immunity in drosophila. Cell (2016) 164(3):406–19. doi: 10.1016/j.cell.2015.12.029
26. Yu F-X, Zhao B, Guan K-L. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell (2015) 163(4):811–28. doi: 10.1016/j.cell.2015.10.044
27. Hao Y, Chun A, Cheung K, Rashidi B, Yang X. Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J Biol Chem (2008) 283(9):5496–509. doi: 10.1074/jbc.M709037200
28. Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, et al. Inactivation of YAP oncoprotein by the hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev (2007) 21(21):2747–61. doi: 10.1101/gad.1602907
29. Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. A coordinated phosphorylation by lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev (2010) 24(1):72–85. doi: 10.1101/gad.1843810
30. Levy D, Adamovich Y, Reuven N, Shaul Y. Yap1 phosphorylation by c-abl is a critical step in selective activation of proapoptotic genes in response to DNA damage. Mol Cell (2008) 29(3):350–61. doi: 10.1016/j.molcel.2007.12.022
31. Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, et al. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell (2013) 154(5):1047–59. doi: 10.1016/j.cell.2013.07.042
32. Moya IM, Halder G. Hippo-YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat Rev Mol Cell Biol (2019) 20(4):211–26. doi: 10.1038/s41580-018-0086-y
33. Schlegelmilch K, Mohseni M, Kirak O, Pruszak J, Rodriguez JR, Zhou D, et al. Yap1 acts downstream of α-catenin to control epidermal proliferation. Cell (2011) 144(5):782–95. doi: 10.1016/j.cell.2011.02.031
34. Yin F, Yu J, Zheng Y, Chen Q, Zhang N, Pan D. Spatial organization of hippo signaling at the plasma membrane mediated by the tumor suppressor Merlin/NF2. Cell (2013) 154(6):1342–55. doi: 10.1016/j.cell.2013.08.025
35. Chen CL, Gajewski KM, Hamaratoglu F, Bossuyt W, Sansores-Garcia L, Tao C, et al. The apical-basal cell polarity determinant crumbs regulates hippo signaling in drosophila. Proc Natl Acad Sci U S A (2010) 107(36):15810–5. doi: 10.1073/pnas.1004060107
36. Hamaratoglu F, Willecke M, Kango-Singh M, Nolo R, Hyun E, Tao C, et al. The tumour-suppressor genes NF2/Merlin and expanded act through hippo signalling to regulate cell proliferation and apoptosis. Nat Cell Biol (2006) 8(1):27–36. doi: 10.1038/ncb1339
37. Sun S, Reddy BV, Irvine KD. Localization of hippo signalling complexes and warts activation in vivo. Nat Commun (2015) 6:8402. doi: 10.1038/ncomms9402
38. Ernkvist M, Aase K, Ukomadu C, Wohlschlegel J, Blackman R, Veitonmäki N, et al. p130-angiomotin associates to actin and controls endothelial cell shape. FEBS J (2006) 273(9):2000–11. doi: 10.1111/j.1742-4658.2006.05216.x
39. Moleirinho S, Hoxha S, Mandati V, Curtale G, Troutman S, Ehmer U, et al. Regulation of localization and function of the transcriptional co-activator YAP by angiomotin. eLife (2017) 6:e23966. doi: 10.7554/eLife.23966
40. Mana-Capelli S, McCollum D. Angiomotins stimulate LATS kinase autophosphorylation and act as scaffolds that promote hippo signaling. J Biol Chem (2018) 293(47):18230–41. doi: 10.1074/jbc.RA118.004187
41. Paramasivam M, Sarkeshik A, Yates JR 3rd, Fernandes MJ, McCollum D. Angiomotin family proteins are novel activators of the LATS2 kinase tumor suppressor. Mol Biol Cell (2011) 22(19):3725–33. doi: 10.1091/mbc.e11-04-0300
42. Kim M, Kim M, Park SJ, Lee C, Lim DS. Role of angiomotin-like 2 mono-ubiquitination on YAP inhibition. EMBO Rep (2016) 17(1):64–78. doi: 10.15252/embr.201540809
43. Silvis MR, Kreger BT, Lien WH, Klezovitch O, Rudakova GM, Camargo FD, et al. α-catenin is a tumor suppressor that controls cell accumulation by regulating the localization and activity of the transcriptional coactivator Yap1. Sci Signal (2011) 4(174):ra33. doi: 10.1126/scisignal.2001823
44. Kim NG, Koh E, Chen X, Gumbiner BM. E-cadherin mediates contact inhibition of proliferation through hippo signaling-pathway components. Proc Natl Acad Sci U S A (2011) 108(29):11930–5. doi: 10.1073/pnas.1103345108
45. Harvey KF, Zhang X, Thomas DM. The hippo pathway and human cancer. Nat Rev Cancer. (2013) 13(4):246–57. doi: 10.1038/nrc3458
46. Grusche FA, Richardson HE, Harvey KF. Upstream regulation of the hippo size control pathway. Curr Biol CB (2010) 20(13):R574–82. doi: 10.1016/j.cub.2010.05.023
47. Yu J, Zheng Y, Dong J, Klusza S, Deng WM, Pan D. Kibra functions as a tumor suppressor protein that regulates hippo signaling in conjunction with merlin and expanded. Dev Cell (2010) 18(2):288–99. doi: 10.1016/j.devcel.2009.12.012
48. Guo H, Lu YW, Lin Z, Huang ZP, Liu J, Wang Y, et al. Intercalated disc protein xinβ is required for hippo-YAP signaling in the heart. Nat Commun (2020) 11(1):4666. doi: 10.1038/s41467-020-18379-8
49. Matsuda T, Zhai P, Sciarretta S, Zhang Y, Jeong JI, Ikeda S, et al. NF2 activates hippo signaling and promotes Ischemia/Reperfusion injury in the heart. Circ Res (2016) 119(5):596–606. doi: 10.1161/CIRCRESAHA.116.308586
50. Chen SN, Gurha P, Lombardi R, Ruggiero A, Willerson JT, Marian AJ. The hippo pathway is activated and is a causal mechanism for adipogenesis in arrhythmogenic cardiomyopathy. Circ Res (2014) 114(3):454–68. doi: 10.1161/CIRCRESAHA.114.302810
51. Jaalouk DE, Lammerding J. Mechanotransduction gone awry. Nat Rev Mol Cell Biol (2009) 10(1):63–73. doi: 10.1038/nrm2597
52. Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, et al. Role of YAP/TAZ in mechanotransduction. Nature (2011) 474(7350):179–83. doi: 10.1038/nature10137
53. Wada K, Itoga K, Okano T, Yonemura S, Sasaki H. Hippo pathway regulation by cell morphology and stress fibers. Development (2011) 138(18):3907–14. doi: 10.1242/dev.070987
54. Wang L, Luo JY, Li B, Tian XY, Chen LJ, Huang Y, et al. Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature (2016) 540(7634):579–82. doi: 10.1038/nature20602
55. Sansores-Garcia L, Bossuyt W, Wada K, Yonemura S, Tao C, Sasaki H, et al. Modulating f-actin organization induces organ growth by affecting the hippo pathway. EMBO J (2011) 30(12):2325–35. doi: 10.1038/emboj.2011.157
56. Azzolin L, Panciera T, Soligo S, Enzo E, Bicciato S, Dupont S, et al. YAP/TAZ incorporation in the β-catenin destruction complex orchestrates the wnt response. Cell (2014) 158(1):157–70. doi: 10.1016/j.cell.2014.06.013
57. Park HW, Kim YC, Yu B, Moroishi T, Mo JS, Plouffe SW, et al. Alternative wnt signaling activates YAP/TAZ. Cell (2015) 162(4):780–94. doi: 10.1016/j.cell.2015.07.013
58. Cai J, Maitra A, Anders RA, Taketo MM, Pan D. β-catenin destruction complex-independent regulation of hippo-YAP signaling by APC in intestinal tumorigenesis. Genes Dev (2015) 29(14):1493–506. doi: 10.1101/gad.264515.115
59. Yu FX, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH, et al. Regulation of the hippo-YAP pathway by G-protein-coupled receptor signaling. Cell (2012) 150(4):780–91. doi: 10.1016/j.cell.2012.06.037
60. Miller E, Yang J, DeRan M, Wu C, Su AI, Bonamy GM, et al. Identification of serum-derived sphingosine-1-phosphate as a small molecule regulator of YAP. Chem Biol (2012) 19(8):955–62. doi: 10.1016/j.chembiol.2012.07.005
61. Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol (2014) 11(5):255–65. doi: 10.1038/nrcardio.2014.28
62. Eltzschig HK, Eckle T. Ischemia and reperfusion–from mechanism to translation. Nat Med (2011) 17(11):1391–401. doi: 10.1038/nm.2507
63. Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res (2012) 110(1):159–73. doi: 10.1161/CIRCRESAHA.111.243162
64. Ong SB, Hernández-Reséndiz S, Crespo-Avilan GE, Mukhametshina RT, Kwek XY, Cabrera-Fuentes HA, et al. Inflammation following acute myocardial infarction: Multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol Ther (2018) 186:73–87. doi: 10.1016/j.pharmthera.2018.01.001
65. Brieler J, Breeden MA, Tucker J. Cardiomyopathy: An overview. Am Family Physician (2017) 96(10):640–6.
66. Hollenberg SM, Singer M. Pathophysiology of sepsis-induced cardiomyopathy. Nat Rev Cardiol (2021) 18(6):424–34. doi: 10.1038/s41569-020-00492-2
67. Schultheiss HP, Fairweather D, Caforio ALP, Escher F, Hershberger RE, Lipshultz SE, et al. Dilated cardiomyopathy. Nat Rev Dis Primers (2019) 5(1):32. doi: 10.1038/s41572-019-0084-1
68. Evangelista I, Nuti R, Picchioni T, Dotta F, Palazzuoli A. Molecular dysfunction and phenotypic derangement in diabetic cardiomyopathy. Int J Mol Sci (2019) 20(13):3264. doi: 10.3390/ijms20133264
69. Becker RC, Owens AP 3rd, Sadayappan S. Tissue-level inflammation and ventricular remodeling in hypertrophic cardiomyopathy. J Thromb Thrombolysis (2020) 49(2):177–83. doi: 10.1007/s11239-019-02026-1
70. Noutsias M, Rohde M, Göldner K, Block A, Blunert K, Hemaidan L, et al. Expression of functional T-cell markers and T-cell receptor vbeta repertoire in endomyocardial biopsies from patients presenting with acute myocarditis and dilated cardiomyopathy. Eur J Heart Failure (2011) 13(6):611–8. doi: 10.1093/eurjhf/hfr014
71. Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, O'Connell J, et al. Report of the 1995 world health Organization/International society and federation of cardiology task force on the definition and classification of cardiomyopathies. Circulation (1996) 93(5):841–2. doi: 10.1161/01.cir.93.5.841
72. Tuder RM. Pulmonary vascular remodeling in pulmonary hypertension. Cell Tissue Res (2017) 367(3):643–9. doi: 10.1007/s00441-016-2539-y
73. Cai Z, Gong Z, Li Z, Li L, Kong W. Vascular extracellular matrix remodeling and hypertension. Antioxidants Redox Signaling (2021) 34(10):765–83. doi: 10.1089/ars.2020.8110
74. Koenen RR, Weber C. Platelet-derived chemokines in vascular remodeling and atherosclerosis. Semin Thromb Hemostasis (2010) 36(2):163–9. doi: 10.1055/s-0030-1251500
75. Souilhol C, Serbanovic-Canic J, Fragiadaki M, Chico TJ, Ridger V, Roddie H, et al. Endothelial responses to shear stress in atherosclerosis: A novel role for developmental genes. Nat Rev Cardiol (2020) 17(1):52–63. doi: 10.1038/s41569-019-0239-5
76. Frösen J, Cebral J, Robertson AM, Aoki T. Flow-induced, inflammation-mediated arterial wall remodeling in the formation and progression of intracranial aneurysms. Neurosurg focus. (2019) 47(1):E21. doi: 10.3171/2019.5.FOCUS19234
77. Paulus WJ, Tschöpe C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol (2013) 62(4):263–71. doi: 10.1016/j.jacc.2013.02.092
78. Boniakowski AE, Kimball AS, Jacobs BN, Kunkel SL, Gallagher KA. Macrophage-mediated inflammation in normal and diabetic wound healing. J Immunol (2017) 199(1):17–24. doi: 10.4049/jimmunol.1700223
79. Odashima M, Usui S, Takagi H, Hong C, Liu J, Yokota M, et al. Inhibition of endogenous Mst1 prevents apoptosis and cardiac dysfunction without affecting cardiac hypertrophy after myocardial infarction. Circ Res (2007) 100(9):1344–52. doi: 10.1161/01.RES.0000265846.23485.7a
80. Maejima Y, Kyoi S, Zhai P, Liu T, Li H, Ivessa A, et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and bcl-2. Nat Med (2013) 19(11):1478–88. doi: 10.1038/nm.3322
81. Del Re DP, Matsuda T, Zhai P, Maejima Y, Jain MR, Liu T, et al. Mst1 promotes cardiac myocyte apoptosis through phosphorylation and inhibition of bcl-xL. Mol Cell (2014) 54(4):639–50. doi: 10.1016/j.molcel.2014.04.007
82. Chen Y, Liu C, Li J, Zhou P, Zhao X, Chen R, et al. LATS2 deletion attenuates myocardial ischemia-reperfusion injury by promoting mitochondrial biogenesis. Oxid Med Cell Longevity (2021) 2021:1058872. doi: 10.1155/2021/1058872
83. Lin Z, von Gise A, Zhou P, Gu F, Ma Q, Jiang J, et al. Cardiac-specific YAP activation improves cardiac function and survival in an experimental murine MI model. Circ Res (2014) 115(3):354–63. doi: 10.1161/CIRCRESAHA.115.303632
84. Del Re DP, Yang Y, Nakano N, Cho J, Zhai P, Yamamoto T, et al. Yes-associated protein isoform 1 (Yap1) promotes cardiomyocyte survival and growth to protect against myocardial ischemic injury. J Biol Chem (2013) 288(6):3977–88. doi: 10.1074/jbc.M112.436311
85. Xin M, Kim Y, Sutherland LB, Murakami M, Qi X, McAnally J, et al. Hippo pathway effector yap promotes cardiac regeneration. Proc Natl Acad Sci U S A. (2013) 110(34):13839–44. doi: 10.1073/pnas.1313192110
86. Liu S, Li K, Wagner Florencio L, Tang L, Heallen TR, Leach JP, et al. Gene therapy knockdown of hippo signaling induces cardiomyocyte renewal in pigs after myocardial infarction. Sci Trans Med (2021) 13(600):eabd6892. doi: 10.1126/scitranslmed.abd6892
87. Chen J, Ma Q, King JS, Sun Y, Xu B, Zhang X, et al. aYAP modRNA reduces cardiac inflammation and hypertrophy in a murine ischemia-reperfusion model. Life Sci Alliance (2020) 3(1):e201900424. doi: 10.26508/lsa.201900424
88. Gao Y, Sun Y, Ercan-Sencicek AG, King JS, Akerberg BN, Ma Q, et al. YAP/TEAD1 complex is a default repressor of cardiac toll-like receptor genes. Int J Mol Sci (2021) 22(13):6649. doi: 10.3390/ijms22136649
89. Ramjee V, Li D, Manderfield LJ, Liu F, Engleka KA, Aghajanian H, et al. Epicardial YAP/TAZ orchestrate an immunosuppressive response following myocardial infarction. J Clin Invest. (2017) 127(3):899–911. doi: 10.1172/JCI88759
90. Francisco J, Byun J, Zhang Y, Kalloo OB, Mizushima W, Oka S, et al. The tumor suppressor RASSF1A modulates inflammation and injury in the reperfused murine myocardium. J Biol Chem (2019) 294(35):13131–44. doi: 10.1074/jbc.RA119.008970
91. Mia MM, Cibi DM, Abdul Ghani SAB, Song W, Tee N, Ghosh S, et al. YAP/TAZ deficiency reprograms macrophage phenotype and improves infarct healing and cardiac function after myocardial infarction. PloS Biol (2020) 18(12):e3000941. doi: 10.1371/journal.pbio.3000941
92. Liu M, Yan M, He J, Lv H, Chen Z, Peng L, et al. Macrophage MST1/2 disruption impairs post-infarction cardiac repair via LTB4. Circ Res (2021) 129(10):909–26. doi: 10.1161/CIRCRESAHA.121.319687
93. Francisco J, Zhang Y, Jeong JI, Mizushima W, Ikeda S, Ivessa A, et al. Blockade of fibroblast YAP attenuates cardiac fibrosis and dysfunction through MRTF-a inhibition. JACC Basic to Trans Science (2020) 5(9):931–45. doi: 10.1016/j.jacbts.2020.07.009
94. Xiao Y, Hill MC, Li L, Deshmukh V, Martin TJ, Wang J, et al. Hippo pathway deletion in adult resting cardiac fibroblasts initiates a cell state transition with spontaneous and self-sustaining fibrosis. Genes Dev (2019) 33(21-22):1491–505. doi: 10.1101/gad.329763.119
95. Flinn MA, Jeffery BE, O'Meara CC, Link BA. Yap is required for scar formation but not myocyte proliferation during heart regeneration in zebrafish. Cardiovasc Res (2019) 115(3):570–7. doi: 10.1093/cvr/cvy243
96. Mia MM, Cibi DM, Binte Abdul Ghani SA, Singh A, Tee N, Sivakumar V, et al. Loss of yap/taz in cardiac fibroblasts attenuates adverse remodeling and improves cardiac function. Cardiovasc Res (2021) 118(7):1785–804. doi: 10.1093/cvr/cvab205
97. Francisco J, Zhang Y, Nakada Y, Jeong JI, Huang CY, Ivessa A, et al. AAV-mediated YAP expression in cardiac fibroblasts promotes inflammation and increases fibrosis. Sci Rep (2021) 11(1):10553. doi: 10.1038/s41598-021-89989-5
98. Yang K, Fan M, Wang X, Xu J, Wang Y, Tu F, et al. Lactate promotes macrophage HMGB1 lactylation, acetylation, and exosomal release in polymicrobial sepsis. Cell Death Differ (2022) 29(1):133–46. doi: 10.1038/s41418-021-00841-9
99. Chen R, Zhu S, Fan XG, Wang H, Lotze MT, Zeh HJ 3rd, et al. High mobility group protein B1 controls liver cancer initiation through yes-associated protein -dependent aerobic glycolysis. Hepatology (2018) 67(5):1823–41. doi: 10.1002/hep.29663
100. Rao J, Cheng F, Zhou H, Yang W, Qiu J, Yang C, et al. Nogo-b is a key mediator of hepatic ischemia and reperfusion injury. Redox Biol (2020) 37:101745. doi: 10.1016/j.redox.2020.101745
101. Li Y, Kong F, Jin C, Hu E, Shao Q, Liu J, et al. The expression of S100A8/S100A9 is inducible and regulated by the Hippo/YAP pathway in squamous cell carcinomas. BMC cancer. (2019) 19(1):597. doi: 10.1186/s12885-019-5784-0
102. Wang X, Ha T, Liu L, Hu Y, Kao R, Kalbfleisch J, et al. TLR3 mediates repair and regeneration of damaged neonatal heart through glycolysis dependent YAP1 regulated miR-152 expression. Cell Death Differ (2018) 25(5):966–82. doi: 10.1038/s41418-017-0036-9
103. Smart N, Bollini S, Dubé KN, Vieira JM, Zhou B, Davidson S, et al. De novo cardiomyocytes from within the activated adult heart after injury. Nature (2011) 474(7353):640–4. doi: 10.1038/nature10188
104. van Wijk B, Gunst QD, Moorman AF, van den Hoff MJ. Cardiac regeneration from activated epicardium. PloS One (2012) 7(9):e44692. doi: 10.1371/journal.pone.0044692
105. Huang GN, Thatcher JE, McAnally J, Kong Y, Qi X, Tan W, et al. C/EBP transcription factors mediate epicardial activation during heart development and injury. Science (2012) 338(6114):1599–603. doi: 10.1126/science.1229765
106. Spagnoli LG, Bonanno E, Mauriello A, Palmieri G, Partenzi A, Sangiorgi G, et al. Multicentric inflammation in epicardial coronary arteries of patients dying of acute myocardial infarction. J Am Coll Cardiol (2002) 40(9):1579–88. doi: 10.1016/S0735-1097(02)02376-8
107. Bansal SS, Ismahil MA, Goel M, Zhou G, Rokosh G, Hamid T, et al. Dysfunctional and proinflammatory regulatory T-lymphocytes are essential for adverse cardiac remodeling in ischemic cardiomyopathy. Circulation (2019) 139(2):206–21. doi: 10.1161/CIRCULATIONAHA.118.036065
108. Li C, Jin Y, Wei S, Sun Y, Jiang L, Zhu Q, et al. Hippo signaling controls NLR family pyrin domain containing 3 activation and governs immunoregulation of mesenchymal stem cells in mouse liver injury. Hepatology (2019) 70(5):1714–31. doi: 10.1002/hep.30700
109. Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med (2007) 204(12):3037–47. doi: 10.1084/jem.20070885
110. Peet C, Ivetic A, Bromage DI, Shah AM. Cardiac monocytes and macrophages after myocardial infarction. Cardiovasc Res (2020) 116(6):1101–12. doi: 10.1093/cvr/cvz336
111. Prabhu SD, Frangogiannis NG. The biological basis for cardiac repair after myocardial infarction: From inflammation to fibrosis. Circ Res (2016) 119(1):91–112. doi: 10.1161/CIRCRESAHA.116.303577
112. Jin K, Gao S, Yang P, Guo R, Li D, Zhang Y, et al. Single-cell RNA sequencing reveals the temporal diversity and dynamics of cardiac immunity after myocardial infarction. Small Methods (2022) 6(3):e2100752. doi: 10.1002/smtd.202100752
113. Dick SA, Macklin JA, Nejat S, Momen A, Clemente-Casares X, Althagafi MG, et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat Immunol (2019) 20(1):29–39. doi: 10.1038/s41590-018-0272-2
114. Zhang Q, Meng F, Chen S, Plouffe SW, Wu S, Liu S, et al. Hippo signalling governs cytosolic nucleic acid sensing through YAP/TAZ-mediated TBK1 blockade. Nat Cell Biol (2017) 19(4):362–74. doi: 10.1038/ncb3496
115. Sharifi-Sanjani M, Berman M, Goncharov D, Alhamaydeh M, Avolio TG, Baust J, et al. Yes-associated protein (Yap) is up-regulated in heart failure and promotes cardiac fibroblast proliferation. Int J Mol Sci (2021) 22(11):6164. doi: 10.3390/ijms22116164
116. Landry NM, Rattan SG, Filomeno KL, Meier TW, Meier SC, Foran SJ, et al. SKI activates the hippo pathway via LIMD1 to inhibit cardiac fibroblast activation. Basic Res Cardiol (2021) 116(1):25. doi: 10.1007/s00395-021-00865-9
118. Wang P, Mao B, Luo W, Wei B, Jiang W, Liu D, et al. The alteration of Hippo/YAP signaling in the development of hypertrophic cardiomyopathy. Basic Res Cardiol (2014) 109(5):435. doi: 10.1007/s00395-014-0435-8
119. Byun J, Del Re DP, Zhai P, Ikeda S, Shirakabe A, Mizushima W, et al. Yes-associated protein (YAP) mediates adaptive cardiac hypertrophy in response to pressure overload. J Biol Chem (2019) 294(10):3603–17. doi: 10.1074/jbc.RA118.006123
120. Triastuti E, Nugroho AB, Zi M, Prehar S, Kohar YS, Bui TA, et al. Pharmacological inhibition of hippo pathway, with the novel kinase inhibitor XMU-MP-1, protects the heart against adverse effects during pressure overload. Br J Pharmacol (2019) 176(20):3956–71. doi: 10.1111/bph.14795
121. Shao D, Zhai P, Hu C, Mukai R, Sciarretta S, Del Re D, et al. Lats2 promotes heart failure by stimulating p53-mediated apoptosis during pressure overload. Sci Rep (2021) 11(1):23469. doi: 10.1038/s41598-021-02846-3
122. Ikeda S, Mizushima W, Sciarretta S, Abdellatif M, Zhai P, Mukai R, et al. Hippo deficiency leads to cardiac dysfunction accompanied by cardiomyocyte dedifferentiation during pressure overload. Circ Res (2019) 124(2):292–305. doi: 10.1161/CIRCRESAHA.118.314048
123. Del Re DP, Matsuda T, Zhai P, Gao S, Clark GJ, van der Weyden L, et al. Proapoptotic Rassf1A/Mst1 signaling in cardiac fibroblasts is protective against pressure overload in mice. J Clin Invest. (2010) 120(10):3555–67. doi: 10.1172/JCI43569
124. Liu J, Wen T, Dong K, He X, Zhou H, Shen J, et al. TEAD1 protects against necroptosis in postmitotic cardiomyocytes through regulation of nuclear DNA-encoded mitochondrial genes. Cell Death Differ (2021) 28(7):2045–59. doi: 10.1038/s41418-020-00732-5
125. Faizah Z, Amanda B, Ashari FY, Triastuti E, Oxtoby R, Rahaju AS, et al. Treatment with mammalian ste-20-like kinase 1/2 (MST1/2) inhibitor XMU-MP-1 improves glucose tolerance in streptozotocin-induced diabetes mice. Molecules (2020) 25(19):4381. doi: 10.3390/molecules25194381
126. Wang S, Zhao Z, Fan Y, Zhang M, Feng X, Lin J, et al. Mst1 inhibits Sirt3 expression and contributes to diabetic cardiomyopathy through inhibiting parkin-dependent mitophagy. Biochim Biophys Acta Mol Basis Dis (2019) 1865(7):1905–14. doi: 10.1016/j.bbadis.2018.04.009
127. Lin J, Zhang L, Zhang M, Hu J, Wang T, Duan Y, et al. Mst1 inhibits CMECs autophagy and participates in the development of diabetic coronary microvascular dysfunction. Sci Rep (2016) 6:34199. doi: 10.1038/srep34199
128. Su D, Li Y, Guan L, Li Q, Shi C, Ma X, et al. Elevated MST1 leads to apoptosis via depletion of YAP1 in cardiomyocytes exposed to high glucose. Mol Med (2021) 27(1):13. doi: 10.1186/s10020-021-00267-6
129. Xiong Z, Li Y, Zhao Z, Zhang Y, Man W, Lin J, et al. Mst1 knockdown alleviates cardiac lipotoxicity and inhibits the development of diabetic cardiomyopathy in db/db mice. Biochim Biophys Acta Mol Basis Dis (2020) 1866(8):165806. doi: 10.1016/j.bbadis.2020.165806
130. Hu J, Wang S, Xiong Z, Cheng Z, Yang Z, Lin J, et al. Exosomal Mst1 transfer from cardiac microvascular endothelial cells to cardiomyocytes deteriorates diabetic cardiomyopathy. Biochim Biophys Acta Mol Basis Dis (2018) 1864(11):3639–49. doi: 10.1016/j.bbadis.2018.08.026
131. Shang X, Lin K, Zhang Y, Li M, Xu J, Chen K, et al. Mst1 deletion reduces septic cardiomyopathy via activating parkin-related mitophagy. J Cell Physiol (2020) 235(1):317–27. doi: 10.1002/jcp.28971
132. Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol (2018) 15(7):387–407. doi: 10.1038/s41569-018-0007-y
133. Shimizu I, Minamino T. Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol (2016) 97:245–62. doi: 10.1016/j.yjmcc.2016.06.001
134. Nguyen MN, Ziemann M, Kiriazis H, Su Y, Thomas Z, Lu Q, et al. Galectin-3 deficiency ameliorates fibrosis and remodeling in dilated cardiomyopathy mice with enhanced Mst1 signaling. Am J Physiol Heart Circulatory Physiol (2019) 316(1):H45–h60. doi: 10.1152/ajpheart.00609.2018
135. Yamamoto S, Yang G, Zablocki D, Liu J, Hong C, Kim SJ, et al. Activation of Mst1 causes dilated cardiomyopathy by stimulating apoptosis without compensatory ventricular myocyte hypertrophy. J Clin Invest. (2003) 111(10):1463–74. doi: 10.1172/JCI17459
136. Wu W, Ziemann M, Huynh K, She G, Pang ZD, Zhang Y, et al. Activation of hippo signaling pathway mediates mitochondria dysfunction and dilated cardiomyopathy in mice. Theranostics (2021) 11(18):8993–9008. doi: 10.7150/thno.62302
137. Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy: An update of mechanisms contributing to this clinical entity. Circ Res (2018) 122(4):624–38. doi: 10.1161/CIRCRESAHA.117.311586
138. Jia G, Whaley-Connell A, Sowers JR. Diabetic cardiomyopathy: A hyperglycaemia- and insulin-resistance-induced heart disease. Diabetologia (2018) 61(1):21–8. doi: 10.1007/s00125-017-4390-4
139. Ritchie RH, Abel ED. Basic mechanisms of diabetic heart disease. Circ Res (2020) 126(11):1501–25. doi: 10.1161/CIRCRESAHA.120.315913
140. Sharma A, Tate M, Mathew G, Vince JE, Ritchie RH, de Haan JB. Oxidative stress and NLRP3-inflammasome activity as significant drivers of diabetic cardiovascular complications: Therapeutic implications. Front Physiol (2018) 9:114. doi: 10.3389/fphys.2018.00114
141. Mariappan N, Elks CM, Sriramula S, Guggilam A, Liu Z, Borkhsenious O, et al. NF-kappaB-induced oxidative stress contributes to mitochondrial and cardiac dysfunction in type II diabetes. Cardiovasc Res (2010) 85(3):473–83. doi: 10.1093/cvr/cvp305
142. Ikeda S, Mukai R, Mizushima W, Zhai P, Oka SI, Nakamura M, et al. Yes-associated protein (YAP) facilitates pressure overload-induced dysfunction in the diabetic heart. JACC Basic to Trans science. (2019) 4(5):611–22. doi: 10.1016/j.jacbts.2019.05.006
143. Ravikumar N, Sayed MA, Poonsuph CJ, Sehgal R, Shirke MM, Harky A. Septic cardiomyopathy: From basics to management choices. Curr problems Cardiol (2021) 46(4):100767. doi: 10.1016/j.cpcardiol.2020.100767
144. Beesley SJ, Weber G, Sarge T, Nikravan S, Grissom CK, Lanspa MJ, et al. Septic cardiomyopathy. Crit Care Med (2018) 46(4):625–34. doi: 10.1097/CCM.0000000000002851
145. Saiyang X, Qingqing W, Man X, Chen L, Min Z, Yun X, et al. Activation of toll-like receptor 7 provides cardioprotection in septic cardiomyopathy-induced systolic dysfunction. Clin Trans Med (2021) 11(1):e266. doi: 10.1002/ctm2.266
146. Zhou J, Li YS, Chien S. Shear stress-initiated signaling and its regulation of endothelial function. Arteriosclerosis thrombosis Vasc Biol (2014) 34(10):2191–8. doi: 10.1161/ATVBAHA.114.303422
147. Baeyens N, Bandyopadhyay C, Coon BG, Yun S, Schwartz MA. Endothelial fluid shear stress sensing in vascular health and disease. J Clin Invest. (2016) 126(3):821–8. doi: 10.1172/JCI83083
148. Wang KC, Yeh YT, Nguyen P, Limqueco E, Lopez J, Thorossian S, et al. Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc Natl Acad Sci U S A. (2016) 113(41):11525–30. doi: 10.1073/pnas.1613121113
149. Xu S, Koroleva M, Yin M, Jin ZG. Atheroprotective laminar flow inhibits hippo pathway effector YAP in endothelial cells. Trans Res J Lab Clin Med (2016) 176:18–28.e2. doi: 10.1016/j.trsl.2016.05.003
150. Nakajima H, Yamamoto K, Agarwala S, Terai K, Fukui H, Fukuhara S, et al. Flow-dependent endothelial YAP regulation contributes to vessel maintenance. Dev Cell (2017) 40(6):523–36.e6. doi: 10.1016/j.devcel.2017.02.019
151. Yuan P, Hu Q, He X, Long Y, Song X, Wu F, et al. Laminar flow inhibits the Hippo/YAP pathway via autophagy and SIRT1-mediated deacetylation against atherosclerosis. Cell Death disease. (2020) 11(2):141. doi: 10.1038/s41419-020-2343-1
152. Jones PD, Kaiser MA, Ghaderi Najafabadi M, Koplev S, Zhao Y, Douglas G, et al. JCAD, a gene at the 10p11 coronary artery disease locus, regulates hippo signaling in endothelial cells. Arteriosclerosis thrombosis Vasc Biol (2018) 38(8):1711–22. doi: 10.1161/ATVBAHA.118.310976
153. Yun S, Hu R, Schwaemmle ME, Scherer AN, Zhuang Z, Koleske AJ, et al. Integrin α5β1 regulates PP2A complex assembly through PDE4D in atherosclerosis. J Clin Invest. (2019) 129(11):4863–74. doi: 10.1172/JCI127692
154. Li B, He J, Lv H, Liu Y, Lv X, Zhang C, et al. C-abl regulates YAPY357 phosphorylation to activate endothelial atherogenic responses to disturbed flow. J Clin Invest. (2019) 129(3):1167–79. doi: 10.1172/JCI122440
155. Jia M, Li Q, Guo J, Shi W, Zhu L, Huang Y, et al. Deletion of BACH1 attenuates atherosclerosis by reducing endothelial inflammation. Circ Res (2022) 130(7):1038–55. doi: 10.1161/CIRCRESAHA.121.319540
156. Xu S, Xu Y, Liu P, Zhang S, Liu H, Slavin S, et al. The novel coronary artery disease risk gene JCAD/KIAA1462 promotes endothelial dysfunction and atherosclerosis. Eur Heart J (2019) 40(29):2398–408. doi: 10.1093/eurheartj/ehz303
157. Liu M, Yan M, Lv H, Wang B, Lv X, Zhang H, et al. Macrophage K63-linked ubiquitination of YAP promotes its nuclear localization and exacerbates atherosclerosis. Cell Rep (2020) 32(5):107990. doi: 10.1016/j.celrep.2020.107990
158. Okuyama M, Jiang W, Yang L, Subramanian V. Mst1/2 kinases inhibitor, XMU-MP-1, attenuates angiotensin II-induced ascending aortic expansion in hypercholesterolemic mice. Circ Rep (2021) 3(5):259–66. doi: 10.1253/circrep.CR-20-0104
159. Finney AC, Stokes KY, Pattillo CB, Orr AW. Integrin signaling in atherosclerosis. Cell Mol Life Sci CMLS (2017) 74(12):2263–82. doi: 10.1007/s00018-017-2490-4
160. Panciera T, Azzolin L, Cordenonsi M, Piccolo S. Mechanobiology of YAP and TAZ in physiology and disease. Nat Rev Mol Cell Biol (2017) 18(12):758–70. doi: 10.1038/nrm.2017.87
161. Shi Y, Vanhoutte PM. Macro- and microvascular endothelial dysfunction in diabetes. J Diabetes (2017) 9(5):434–49. doi: 10.1111/1753-0407.12521
162. Ortillon J, Le Bail JC, Villard E, Léger B, Poirier B, Girardot C, et al. High glucose activates YAP signaling to promote vascular inflammation. Front Physiol (2021) 12:665994. doi: 10.3389/fphys.2021.665994
163. Choi HJ, Kim NE, Kim BM, Seo M, Heo JH. TNF-α-Induced YAP/TAZ activity mediates leukocyte-endothelial adhesion by regulating VCAM1 expression in endothelial cells. Int J Mol Sci (2018) 19(11):3428. doi: 10.3390/ijms19113428
164. Xu Q, Zhuo K, Cai R, Su X, Zhang L, Liu Y, et al. Activation of yes-associated Protein/PDZ-binding motif pathway contributes to endothelial dysfunction and vascular inflammation in AngiotensinII hypertension. Front Physiol (2021) 12:732084. doi: 10.3389/fphys.2021.732084
165. Lv Y, Kim K, Sheng Y, Cho J, Qian Z, Zhao YY, et al. YAP controls endothelial activation and vascular inflammation through TRAF6. Circ Res (2018) 123(1):43–56. doi: 10.1161/CIRCRESAHA.118.313143
166. Tabas I, Bornfeldt KE. Macrophage phenotype and function in different stages of atherosclerosis. Circ Res (2016) 118(4):653–67. doi: 10.1161/CIRCRESAHA.115.306256
167. Geng J, Yu S, Zhao H, Sun X, Li X, Wang P, et al. The transcriptional coactivator TAZ regulates reciprocal differentiation of T(H)17 cells and t(reg) cells. Nat Immunol (2017) 18(7):800–12. doi: 10.1038/ni.3748
168. Zhou D, Medoff BD, Chen L, Li L, Zhang XF, Praskova M, et al. The Nore1B/Mst1 complex restrains antigen receptor-induced proliferation of naïve T cells. Proc Natl Acad Sci U S A (2008) 105(51):20321–6. doi: 10.1073/pnas.0810773105
Keywords: Hippo-YAP pathway, inflammation, immune, myocardial infarction, atherosclerosis
Citation: Zheng A, Chen Q and Zhang L (2022) The Hippo-YAP pathway in various cardiovascular diseases: Focusing on the inflammatory response. Front. Immunol. 13:971416. doi: 10.3389/fimmu.2022.971416
Received: 17 June 2022; Accepted: 02 August 2022;
Published: 18 August 2022.
Edited by:
Guochang Hu, University of Illinois at Chicago, United StatesReviewed by:
Dominic Del Re, Rutgers, The State University of New Jersey, United StatesJixin Dong, University of Nebraska Medical Center, United States
Copyright © 2022 Zheng, Chen and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Zhang, bGkuemhhbmcudWtAZ21haWwuY29t; Qishan Chen, bGFzdHVmb0Bob3RtYWlsLmNvbQ==