 Zhen Tan
Zhen Tan Li Wang
Li Wang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Immunol. , 13 September 2022
Sec. Autoimmune and Autoinflammatory Disorders
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.967304
This article is part of the Research Topic Genetic and Environmental Factors in Rheumatic Diseases View all 14 articles
Primary Sjögren’s syndrome (pSS) is a systemic autoimmune disease characterized by exocrine gland dysfunction and inflammation. Patients often have dry mouth and dry eye symptoms, which seriously affect their lives. Improving dry mouth and eye symptoms has become a common demand from patients. For this reason, researchers have conducted many studies on external secretory glands. In this paper, we summarize recent studies on the salivary glands of pSS patients from the perspective of the immune microenvironment. These studies showed that hypoxia, senescence, and chronic inflammation are the essential characteristics of the salivary gland immune microenvironment. In the SG of pSS, genes related to lymphocyte chemotaxis, antigen presentation, and lymphocyte activation are upregulated. Interferon (IFN)-related genes, DNA methylation, sRNA downregulation, and mitochondrial-related differentially expressed genes are also involved in forming the immune microenvironment of pSS, while multiple signaling pathways are involved in regulation. We further elucidated the regulation of the salivary gland immune microenvironment in pSS and relevant, targeted treatments.
Primary Sjögren’s syndrome (pSS) is a systemic autoimmune disease that occurs mostly in middle-aged women and is characterized by impaired glandular function and the appearance of autoantibodies caused by infiltrating exocrine glands with lymphocytes, with an estimated prevalence of 0.3-3/1000 in the general population (1, 2). PSS is a heterogeneous disease; approximately 5% to 35% of the population has dry eye, approximately 20% of patients have dry mouth, and up to 34% of patients have parotid gland swelling (3–5). In addition to glandular involvement, arthritis occurs in approximately 50% of patients, lung involvement occurs in 9-12% of patients, skin lesions occur in 10% of patients, kidneys involvement occur in 5% of patients, and sensory neuropathy occurs in 10-25% of patients (6–8). In blood samples, 40%-75% of pSS patients have anti-RO/SSA, and 23%-52% have LA/SSB antibodies (9).
The etiology and pathogenesis of SS are still unclear, and it is generally believed that genetic susceptibility related to environmental factors is an important cause of the occurrence of Sjogren’s syndrome (10). Currently, genome-wide association studies on pSS have been completed, among which HLA genes have the strongest association signal (11). Epigenetic mechanisms such as DNA methylation, histone modification, and noncoding RNA play a role in the pathogenesis of pSS by regulating gene expression and may form a dynamic link between the genome and phenotypic expression. Bacteria and viruses are essential components of environmental factors. Bacteria can cause autoimmune diseases through various mechanisms, such as pathogen persistence, epitope spread, molecular mimicry, epigenetic changes, and Toll-like receptor activation. Type I IFN is a critical immune mediator involved in viral defense and immune response activation, which suggests the important role of viral infection in the pathogenesis of the disease. A recent analysis of the gene expression of SGECs showed that the IFN signaling pathway and genes involved in the immune response (HLA-DRA, IL-7, and B-cell activator receptor) in pSS were upregulated (12). Other studies found dysregulation of the IFN signaling pathway in SG and peripheral blood of some patients with SS (13), especially the upregulation of type I IFN-induced genes. Various factors, such as infection and hypoxia, induce the activation of SG epithelial cells, leading to lymphocyte infiltration (especially CD4+ T cells) and the release of inflammatory factors. New cell populations, such as follicular T cells, TH17 cells, dendritic (IFN-producing) cells, and B lymphocytes, gradually develop into B lymphocyte-dominated ectopic germinal centers (GCs) with autoantibody production.
The occurrence and development of pSS is a complex process involving many kinds of cells. The salivary gland, as the most commonly affected organ, has attracted increasing attention. Early SG lesions in pSS are rarely reported, which may be related to the delayed diagnosis of pSS. However, many studies have attempted to find targeted therapies for pSS by intervening in the inflammatory process of pSS. Among them, the improvement of dry mouth symptoms caused by impaired salivary gland function is an important goal of treatment. Autoantibodies in the salivary glands induce abnormal immune responses, which together with a large number of infiltrating inflammatory cells destroy normal salivary gland cells, atrophy of salivary gland cells and disappearance of salivary duct. As time goes on, the normal secretory function of salivary glands can be affected, resulting in dry mouth (14). Salivary gland cells, inflammatory cells, inflammatory mediators, autoantibodies and cytokines produced by each of them or by each other constitute the unique immune microenvironment of salivary gland cells. Changes in the immune microenvironment may cause changes in glandular function. Therefore, obtaining more salivary gland tissue samples, analyzing the potential differences between peripheral blood and salivary gland tissue, and further understanding the composition and regulation of the salivary gland immune microenvironment can help us find more targeted treatments for pSS xerostomia. In addition, the salivary gland immune microenvironment of pSS, a type of autoimmune epithelitis, can provide a model for the study of other autoimmune epithelitis (celiac disease, primary biliary cirrhosis, etc.).
The salivary gland microenvironment directly affects salivary gland secretion function and is very important for the occurrence and development of pSS. It generally has three characteristics: hypoxia, chronic inflammation, and senescence.
Hypoxia is a state of reduced available oxygen caused by reduced blood flow, anemia, metabolic changes, and inflammation (15, 16). Hypoxia has been shown to accelerate cell apoptosis in the renal epithelium (17) while downregulating Cl- ion secretion in the intestinal epithelium, resulting in decreased epithelial fluid transport activity and destruction of tight connections between epithelial cells (18). Hypoxia can also lead to macrophage polarization, regulatory T-cell aggregation, and inhibitory T-cell maturation, leading to immune tolerance and tissue damage. A recent study of minor salivary glands in pSS patients found that hypoxia and IFN-related genes were closely associated with the expression of interleukin (IL)-21 signaling genes, which were significantly increased in pSS patients (19), suggesting a correlation between hypoxia and pSS morbidity. Hypoxia-inducible factor 1α (HIF1α), a transcription factor, is a major regulator of oxygen homeostasis and can be regarded as a hypoxia marker. HIF1α is also a key player in integrating the T-cell receptor (TCR) and cytokine receptor-mediated signals of CD4+ helper T cells (20). In addition, HIF1α enhances Th17 development through direct transcriptional activation of RAR-related orphan receptor gamma t (RORt) (21). This subpopulation is highly increased in salivary gland tissue of patients with SS and a mouse model of SS (22). Longyun Ye et al. cultured mouse submandibular glands (SMGs) in vitro and showed that hypoxia (5% O2) induced HIF-1α, glucose transporter 1 and VEGF expression, while BAY 87-2243-mediated HIF-1α inhibited salivary gland development (23). There are also differing viewpoints. Recent studies have shown that HIF1α expressed in epithelial cells protects against hypoxia-induced tight junction integrity loss and epithelial secretory function loss. The genotype and the allele of the HIF1A Pro582Ser polymorphism were associated with a reduced risk of pSS, suggesting that HIF1α activity may be involved in the development of pSS disease (24). Such different results may be related to different degrees of hypoxia. Antonela Romina Terrizzi et al. performed a comparative analysis of adult Wistar rats exposed to persistent or intermittent hypoxia over 90 days (25). The results suggested that salivary secretion decreased and prostaglandin E2 (PGE2) content increased in animals exposed to hypoxia. The persistent hypoxia group showed higher HIF-1α staining. This suggests that PGE2 plays a negative role during gland adaptation to hypoxia.
Chronic inflammation is another feature of salivary gland involvement in pSS patients, mainly manifested by periductal lymphocytic infiltration. The lesions are mainly T and B lymphocytes, with a few monocytes, including macrophages, myeloid cells, plasmoid dendritic cells, and follicular dendritic cells (FDC) (26). The corresponding plasma cells attack normal tissues and organs, including salivary glands, and cause tissue damage when they produce autoantibodies. It has been reported that in a mouse salivary gland inflammation model, the degree of salivary gland inflammation is related to the titer of antinuclear antibodies (27). Some authors have described the presence of anti-Ro/SSA and anti-La/SSB autoantibodies in the saliva of pSS patients but no circulating antibodies in serum, suggesting that the salivary glands of pSS patients can specifically produce these autoantibodies (28). In addition to anti-Ro/SSA and anti-La/SSB autoantibodies, anti-salivary gland protein 1, anti-carboxylase 6, and anti-parotid secreted protein autoantibodies have also been reported, which recognize salivary gland- and lacrimal gland-specific antigens (29). Moreover, IL-2, IL-6, IL-21, BCL6, Foxp3, and other cytokines and transcription factors were detected in the salivary glands, which further proved the persistence of inflammation (30–33).
Senescence is a permanent state of cell cycle arrest, with the upregulation of antiapoptotic pathways (34). Using organoid culture techniques, some researchers found that salivary gland progenitor cells (SGPCs) in pSS patients showed insufficient self-renewal ability and differential ability compared with the control group. The telomeres of pSS SGPCs were shorter than normal, suggesting the existence of an aging phenotype (35). In addition, p16+ expression increased in the basal striated duct cell (BSD) progenitor cell niche and the whole parotid epithelium, a marker of aging (36). Senescent cells express and secrete proinflammatory cytokines (i.e., senescence-related secretion phenotypes, SASP) that play a role in spreading senescence and promoting tissue inflammation (37).
In conclusion, pSS SGPCs tend to be senescent, and SASPs maintain the senescent SG microenvironment. The salivary function of pSS patients could not recover after the improvement of salivary gland inflammation, which proved the existence of senescence from another aspect. Mie Kurosawa et al. found an accumulation of senescence-associated T cells (SA-TS) in salivary glands of PSs model mice, which were involved in the pathogenesis of SS-associated sialadenitis through upregulation of the epithelial chemokine CXCL13 (38), and they may become another target for pSS treatment.
First, let us review the SGs. The salivary gland is the general name for the exocrine gland opening in the mouth through the duct because its secretions are discharged into the mouth and mixed into saliva. There are three pairs of SGs: parotid, sublingual and submandibular, and the largest pair is the parotid gland. SGs are composed of repeatedly branching ducts and terminal acinus forming the gland parenchyma. The acinar is divided into serous, mucinous, and mixed acinar, producing and secreting watery or mucus-rich saliva from serous and mucinous acinar cells. The secreted saliva passes through intercalated ducts into striate tubes of basal cells and lumen cells and finally into the mouth through larger excretory ducts. Luminal striated duct cells, basal striated duct cells, intercalated ducts, acinar cells, and myoepithelial cells constitute the salivary gland’s epithelial cells (SGECs). Salivary progenitor cells reside in striatal canals and proliferate and differentiate to maintain gland homeostasis.
SGECs are not only a critical immune target of pSS but also play an essential immune function in the pathogenesis of pSS, mediating the initiation and persistence of inflammation and autoimmune response. Human pSS SG epithelial cells show increased proapoptotic molecules (such as Fas and Bax) and decreased antiapoptotic molecules (Bcl-2) compared with healthy individuals (39–41). Endoplasmic reticulum stress leads to autophagy and apoptosis, which may lead to redistribution of Ro/SSA and La/SSB autoantigens, initially on the cell surface and eventually in apoptotic blisters (42). These autoantigens are upregulated in pSS SGECs and regulated by the TLR/IFN TYPE I signaling pathway (43). SGECs are not only important sources of pSS autoantigens Ro/SSA and La/SSB but also express MHC class I and II and T-cell costimulatory molecules (CD80/CD86), enabling them to function as autoantigen presenting cells (44–46). SGECs have been shown to express not only virus-associated toll-like receptors (TLRs) (3 and 7) but also bacterial infection-associated TLRs (1, 2, 4) (47). SGECs bind to multiple pathogen-associated molecular patterns (PAMPs) through the expression of TLRs 1, 2, 3, 4, and 7. In addition to the B-cell activator CD40, SGECs also express CXCL10, CXCL12, CXCL13, IFNα, IFNβ, IFNλ, TNFα, and other receptors. They also produce a variety of cytokines/chemokines under various stimulus conditions, including IL-18, IL-21, IL-1, IL-6, TNFα, B-cell activating factor (BAFF), CXCL-10, CXCL-12, CXCL-13, and CCL-21 (47–59). IL-6 and the costimulatory molecule ICOSL contribute to follicle-assisted T-cell induction, which is critical for B-cell activation and differentiation (60). IFNλ stimulation of SG epithelial cells also induces the expression of BAFF and CXCL10, suggesting that type III IFN plays a role in developing SG pathology in pSS (61).
Abundant evidence suggests that SGECs can drive the activation, differentiation, and survival of B cells through direct interaction and cytokine production and promote the pathogenesis of SS (12, 62, 63). In vitro, culture results showed that SGECs from pSS patients promoted the differentiation of B cells into mature B-cell phenotypes and improved the survival rate (12, 63). SGECs may also indirectly induce B-cell differentiation. SGECs have been reported to promote T follicular helper cell differentiation and IL-21 production (60), which may further enhance B-cell hyperactivity in salivary glands in pSS patients. In the salivary glands of pSS patients, high levels of CXCL12 were detected in ductal epithelial cells (64), and CXCL12 expression and IL-6 were associated with high focusing scores and high levels of CD138+ plasma cell infiltration (51). Riviere et al. showed the presence of IL-7/IFNγ amplification loops involving SGEC and T cells in primary SS (65). They stimulated primary cultures of SGECs from control and primary SS patients with poly (I-C), interferon α, or interferon γ. SG explants were cultured with an anti-IL-7 receptor (IL-7R) antagonist antibody (OSE-127) for 4 days, and transcriptome analysis was performed using the NanoString platform. The results suggested that the expression of IL7R was decreased in T cells. Il-7 is secreted by SGECs stimulated by poly (I-C), IFNα, or IFNγ. IL-7 stimulation increases T-cell activation and IFNγ secretion. Transcriptome analysis of SG explants showed a correlation between IL7 and IFN expression, and explants cultured with anti-IL-7R antibodies showed reduced IFN-stimulated gene expression. These results indicate the presence of IL-7/IFNγ amplification loops involving SGEC and T cells in primary SS. Il-7 is secreted by the SGEC stimulated by type I or TYPE II IFN, which in turn activates T cells that secrete type II IFN.
The stromal component of the salivary glands is composed of mesenchymal stromal cells (MSCs), which provide tissue-homeostatic properties, including regeneration, repair, and immune regulation. It has been shown that human bone marrow mesenchymal stem cells (hMSCs) cocultured with purified immune cell subpopulations alter the cytokine secretion profile of dendritic cells (DC), primary and effector T cells (Th1 and Th2), and natural killer cells (NK) to induce a more anti-inflammatory or tolerant phenotype (66). This suggests that MSCs may reduce inflammation by acting as immunomodulators and promoting tissue regeneration. In recent studies, IFNγ stimulated cultured resident MSG-derived MSCs (MSG-Mscs) isolated from the small salivary glands of pSS patients, and the protein levels of indoleamine 2,3-dioxygenase (IDO), programmed death ligand 1 (PD-L1), and intercellular adhesion marker 1 (ICAM-1) increased. These results suggest that MSG-Mscs have normal immunomodulatory functions in small salivary glands. In addition, MSG-Mscs inhibited T-cell proliferation in a dose-dependent manner and were not associated with 17-β-estradiol exposure (67). In addition, follicular dendritic cells (FDCs) are stromal cells located in primary follicles and germinal centers (GCs) of secondary and tertiary lymphoid organs and have the unique ability to retain natural antigens in B-cell follicles for several months (68).
With the progression of epithelial cell activation and disease, new cells appear in the salivary gland, such as follicle T cells and Th17 cells, dendritic cells (producing IFN), macrophages (MFs), natural killer cells, and B lymphocytes (T cells are usually found in mild lesions, and B cells and MFs dominate in the most severe lesions). Furthermore, they gradually develop into ectopic germinal centers (69, 70).
There are many different types of B cells in salivary gland tissue. FcRL4+B cells were found in or near the ductal epithelium of the inflammatory salivary gland tissue of pSS. FcRL4 is closely related to lymphoma and is expressed in almost all MUCOSAL-associated lymphoid tissue (MALT) B-cell lymphomas associated with pSS, especially in the parotid gland. RNA sequencing of FcRL4+ B cells isolated from parotid cell suspensions from pSS patients showed that FcRL4+ B cells were not enriched in the blood of pSS patients compared with non-SS-sicCA patients, but these cells generally displayed a proinflammatory phenotype. Genes encoding CD11c (ITGAX), T-BET (TBX21), TACI (TNFRSF13B), Src tyrosine kinase and NFκB pathway-related genes were significantly upregulated in glandular FcRL4+B cells compared with FCRL4-B cells. Therefore, FcRL4+B cells in pSS exhibit many characteristics of chronic activation and proinflammatory B cells (71). Some researchers found that through immunohistochemistry and mRNA analysis, the expression level of FcRL4 mRNA in parotid MALT lymphoma was increased compared with the parotid tissue of pSS patients without lymphoma, which may explain why MALT lymphoma in pSS patients preferentially occurred in this specific site (15). In addition, MZB cells were detected in saliva and lacrimal glands in both patients with salivary gland disease and mouse models. The C57BL/6. Nod-aec1aec2 mouse model, as well as several SS gene knockout mouse models, showed that B lymphocytes, especially peripheral zone B (MZB) cells, are necessary for the development of clinical manifestations and pathogenesis, although destruction of lacrimal and salivary gland cells involves a typical T-cell-mediated autoimmune response. Peck et al., through in vitro temporal global RNA transcriptomic analysis, showed that MZB cells from C57BL/6. Nod-aec1aec2 mice were recruited by the upregulated Cxcl13 chemokine to the exocrine gland, where they recognized complement-modified antigens through their sphingosin-1-phosphate and B-cell receptors (72). BAFF transgenic (TG) mice developed autoimmune diseases characterized by autoantibody production, leading to salivary gland destruction (salivary adenitis), which was associated with enlargement of the B-cell compartment in the marginal region (MZ) and abnormal presence of MZ-like B cells in blood and inflamed salivary glands (15). In the IL14aTG mouse model, elimination of MZB from mice by B-cell-specific deletion of RBP-J resulted in complete elimination of all SS disease manifestations (73). Daridon and others, through classification and reverse transcription polymerase chain reaction analysis of salivary gland specimens in the presence of B cells and its polyclonal form in 18 patients, found that pSS patients with heterotopic salivary glands in GC sample structure transition - 2 B-cell amplification, locally produce autoantibodies, which may help and influence subsequent epithelial damage (74). Immunoglobulin rearrangement in single parotid B cells isolated from the parotid gland was analyzed by fluorescence-activated cell sorting, and the results showed that compared with peripheral blood, most parotid B cells in pSS showed the mutant status and phenotype of memory B cells, which accumulated in the salivary glands of pSS patients (75). Hansen et al. analyzed chemokine receptor expression in CD27- naive and CD27+ memory B cells from primary SS patients and healthy controls using flow cytometry, single-cell reverse transcription polymerase chain reaction (RT−PCR), and migration assays. The results showed that CD27+ memory B cells overexpressed the chemokine receptors CXCR4 and CXCR5, which may promote the infiltration of memory B cells into inflammatory glands through the chemokine receptors CXCL12 and CXCL13 from epithelial cells (76). Similar to transition B cells, CD27+ memory B cells seem to promote the formation of ectopic GC-like structures in the exocrine glands of pSS patients (77). Skarstein et al. found that in pSS patients, the increase in CD138+ plasma cells and CD20+ B cells is associated with fat infiltration and focal infiltration, suggesting that they are actively involved in promoting inflammation (78). Szyszko et al. performed single, double, and triple immunohistochemical and immunofluorescence staining of small salivary gland tissues from pSS, chronic inflammatory, and normal subjects, suggesting that plasma cells were located near CXCL12- and IL-6-expressing cells. A salivary gland environment with a high focus score provided a critical factor for plasma cell survival (51). In addition, Cui et al. developed an enzyme-linked immunosorbent assay for autoantibodies with good quantitative ability and found that the expression levels of saliva anti-Cofilin-1, anti-α enolase and anti-RGI2 in parotid gland tissues of pSS/MALT patients were significantly higher than those of healthy controls (79). These results suggest the promoting role of plasma cells in MALT lymphoma.
In the salivary glands, T cells mainly assist B cells. Th1 cells are believed to play a major role in pSS and are the most relevant CD4+ cell population infiltrating inflammatory SGs (80). Th2 cytokine levels are closely associated with SG lymphocyte infiltration (30). Th17 cells also play a key role in pSS. Th17 cells in SGs can develop into Th17.1 cells and produce IL-17 and IFN-γ, which are involved in the pathogenesis of pSS (81). It has been reported that Tfh cells selectively accumulate in the SGs of pSS patients (82, 83). Tfh cells appear as a unique subpopulation of CD4+ T helper cells that promote the development and activation of B cells. Tfh cells express CXCR5, which migrates and localizes in B-cell follicles and induces the expression of T-cell costimulatory (ICOS) molecules, coinhibitory programmed cell death protein-1 (PD-1), and the transcription factor Bcl6 (84). Tfh cells release large amounts of IL-21, a key cytokine that activates the molecular mechanism of somatic excessive mutation and analog switching of B cells (85, 86). Another subpopulation of CD4+ T cells, follicular regulatory T cells (Tfr), also express CXCR5 but have the typical inhibitory function of regulatory T cells, negatively regulating GC responses to prevent abnormal GC responses (87). In addition, pSS patients showed a high degree of infiltration of pathogenic T peripheral helper cells (Tph) in SGs, which lacked typical Tfh markers such as CXCR5 and Bcl6 but could assist homologous B cells through IL-21 and CD40-L (82, 88). By studying the peripheral blood of pSS patients, Dupre et al. found that Tfh and Tph were amplified in the peripheral blood of patients and correlated with disease activity and B-cell marker (RF and anti-SSB) levels (89). Pontarini et al. performed transcriptome (microarray and quantitative PCR) analysis, FACS T-cell immunophenotyping and intracellular cytokine detection, polychromatic immunofluorescence microscopy and in situ hybridization. It was found that damaged CD4+CD45RO+ICOS+PD1+ cells selectively infiltrated ELS+ tissues in SG and amplified abnormally in parotid malt-L. In ELS+SG and MALT-L parotid glands, the traditional CXCR5+CD4+PD1+ICOS+ FOXP3-TFH cell population and the uniquely enlarged CXCR5-CD4+PD1HIICOS+ Foxp3-TPH cell population showed frequent IL-21/interferon dual production. The results highlight Tfh and Tph cells and IL-21 and ICOS costimulation pathways as key pathogenic factors in SS immunopathology (90). Cytotoxic T lymphocytes (CTLs) can specifically recognize and lyse their targets. Recently, antigen-specific cytotoxicity expressed by cytotoxic T cells in vitro and in vivo has been shown to be Fas based (91). Kong et al. used immunohistochemical staining and reverse transcription polymerase chain reaction in situ to detect the expression of Fas and Fas ligand (FasL) in salivary gland biopsy materials and evaluated the DNA fragments in apoptotic cells by enzymatic incorporation of labeled nucleotide (digoxin -dUTP). The results showed that the acinar epithelial cells of SS were Fas+ and FasL+, and the cells died by apoptosis. Fas+ and Bcl-2+ were the dominant infiltrating lymphocytes in SS, and FasL was expressed in a few lymphocytes. In situ detection of apoptosis showed minimal cell death of lymphocytes, especially in dense periductal lesions. These results suggest that the Fas pathway may be an important mechanism of SS gland destruction (39).
Dendritic cells (DCs) can be divided into antigen presenting myeloid DCs (MDCs), which are effector cells, and plasma cell DCs (PDCs), which mainly produce type I interferon. Among them, plasmacytoid dendritic cells (PDCs) produce type I interferon (IFN) and contribute to the pathogenesis of various autoimmune diseases. PDC and type I IFN activity are elevated in the salivary glands of SS patients. Zhou et al. applied pdC-consuming anti-BST2/CD317 antibodies to female NOD mice aged 4 to 7 weeks at the early stage of SS and assessed the pathology of SS at 10 weeks of age. The results suggested that PDC treatment inhibited the development of inflammation and secretory dysfunction of SMG and significantly reduced the number of type I interferon mRNA, total white blood cells, T lymphocytes and B lymphocytes in SMG. This suggests a role for PDC in the pathogenesis of pSS (92). In patients with pSS, immature myeloid dendritic cells (DCS) are reduced in the blood, and mature myeloid dendritic cells accumulate in the salivary glands. As the duration of pSS syndrome increases, the reduction in myeloid dendritic cells in the blood spontaneously recovers. Myeloid DCs may play an important role in the pathogenesis of pSS by initiating the helper T-cell immune response (93).
Fibroblasts are an extremely heterogeneous population of cells with a spindle shape, oval nuclei, and the ability to adhere to collagen fibers. In addition to synthesizing and reshaping the structure and function of the extracellular matrix, they also have the ability to secrete cytokines, chemokines and growth factors. Furthermore, immune fibroblasts affect the homeostasis of immune cells, and are one of the important stromal cells constituting the tertiary lymphoid structures (TLS) (94). Nayar et al. showed that immune fibroblast activation and expansion were observed during TLS formation in wild-type (WT) mice that induced salivary gland inflammation. In Dm2 mice, the loss of PDPN+/FAP+ fibroblasts disrupted the establishment of TLS and impaired the establishment of local pathology. Meanwhile, in salivary glands of pSS patients, the phenotype and proliferation of TLS immune fibroblasts are regulated by IL-13 and IL-22 (95). In addition, Korsunsky et al. performed single-cell RNA sequencing of fibroblasts and found that CXCL10+CCL19+ immune-interacting and SPARC+COL3A1+ vascular-interacting fibroblasts were expanded in a variety of inflammatory tissues (salivary glands of pSS, synovium of RA, colon of ulcerative colitis, etc.) (96). In ulcerative colitis, fibroblasts are the main source of IL-6, and many cytokines and inflammatory mediators have been found to significantly induce IL-6 expression in fibroblasts, including TNF, IL-17, IL-1, LPS, and IFN (97).
SS is essentially a kind of epithelial inflammation, and the integrity, structure, and function of epithelial cells largely depend on the homeostasis of the extracellular matrix (ECM). The ECM is a network of many components, including fibrin, glycosaminoglycan, growth factor, protease, and inhibitors. An increasing number of studies have shown that changes in the morphology and function of acini and ducts, accompanied by the degradation and remodeling of ECM, are critical events in salivary gland changes in pSS patients. ECM not only supports glandular cells, but its components are also important components of damage-related molecular patterns (DAMPs). DAMPs are potential endogenous inflammatory sources that drive autoimmunity by activating pattern recognition receptors (98). When the glandular tissue of pSS patients is damaged by internal and external environmental factors, the ECM releases soluble DAMPs (Biglycan, decorin, etc.) under the action of matrix metalloproteinases (MMPs). Soluble DAMPs activate homologous receptors that mediate inflammation (e.g., MyD88-dependent TLRs), leading to aseptic inflammation and enhancing pathogen-mediated inflammation (99–101). The salivary mucins MUC1, MUC7, and MUC5B secreted into the intercellular space have been reported to activate proinflammatory molecules and Toll-like receptors, which are also involved in inflammatory responses (99). Aberrant decorin levels (DCN) induce damage to human salivary gland epithelial cells and the polarization of macrophages (102). The increased level of DCN in the parotid gland of pSS patients was positively correlated with several chemokines (CXCL13, CXCL9, and CCL20), IL-1ß, and caspase3 but negatively correlated with the proliferation-related gene MKI67. DCN induces apoptosis of A253 cells and differentiation of macrophages into the M1 phenotype, which is characterized by the expression of proinflammatory cytokines (102).
The salivary glands of pSS patients are characterized by chronic inflammation, and the lesions are mainly composed of T and B lymphocytes (26). In the initial stage of disease, lymphoid tissue initiator or inducer (LTi) cells, induced by precursors such as Epstein-Barr virus or cytomegalovirus, produce lymphotoxin, which promotes the expression of NF-κB signals in lymphoid tissue organizer cells by lymphotoxin-β receptors. This results in the enhanced expression of homeostatic chemokines and cytokines (CXCL13, CCL19, CCL21, RANKL, IL-17 and IL-22) (103, 104). At the same time, the virus induced the expression of interferon-γ and stimulated the expression of CXCL9 and CXCL10 in ductal epithelium. These cytokines and chemokines are involved in attracting T lymphocytes, B lymphocytes, and other immune cells to the site of inflammation and promote the formation and maintenance of organized lymphoid tissue (105–107). In addition, prolonged gland activation leads to the formation of FDC networks and the separation of T and B cells, and finally to the formation of ectopic lymphoid structures (ELS) with ectopic germinal centers as the core, T and B cell separation areas surrounding, and the formation of high endothelial venules (HEVs) as exchange channels with peripheral blood lymphocytes (103, 104, 108). The germinal center consists of a light zone and a dark zone. There are rapidly proliferating central blasts in the dark zone, and their Ig variable region genes can undergo somatic hypermutation, thereby protecting them from apoptosis. In the light zone, the central cells transformed from the central blast cells competitively bind to the antigen presented by FDC. The central B cells with high affinity BCR bound to the antigen were positively selected, and the cells that did not receive the antigen underwent apoptosis. Tfh cells regulate the apoptosis of positively selected central B cells or further development into memory B cells or plasma cells through Fas-FasL and CD40-CD40L (109). Tph further induces chemotaxis and conversion of memory B cells into plasma cells by producing IL-21 and CXCL13. Initiation and maintenance of ELS in pSS requires ectopic expression of lymphoid chemokines, including CXCL12, CXCL13, CCL19, and CCL21. These chemokines regulate lymphocyte trafficking and tissue localization by interacting with their unique receptors CXCR4 (for CXCL12), CXCR5(for CXCL13), and CCR7(for CCL19 and CCL21). CXCL12 is mainly produced by follicles and ducts, and its receptor is expressed in plasma cells (110). CXCL13 is mainly produced by stromal cells, memory CD4+ T cells, and non-immune cells (ductal epithelial and endothelial cells) in the FDC network. CCL21 is released from myofibroblast-like stromal cells and is closely associated with HEV formation (Figure 1).
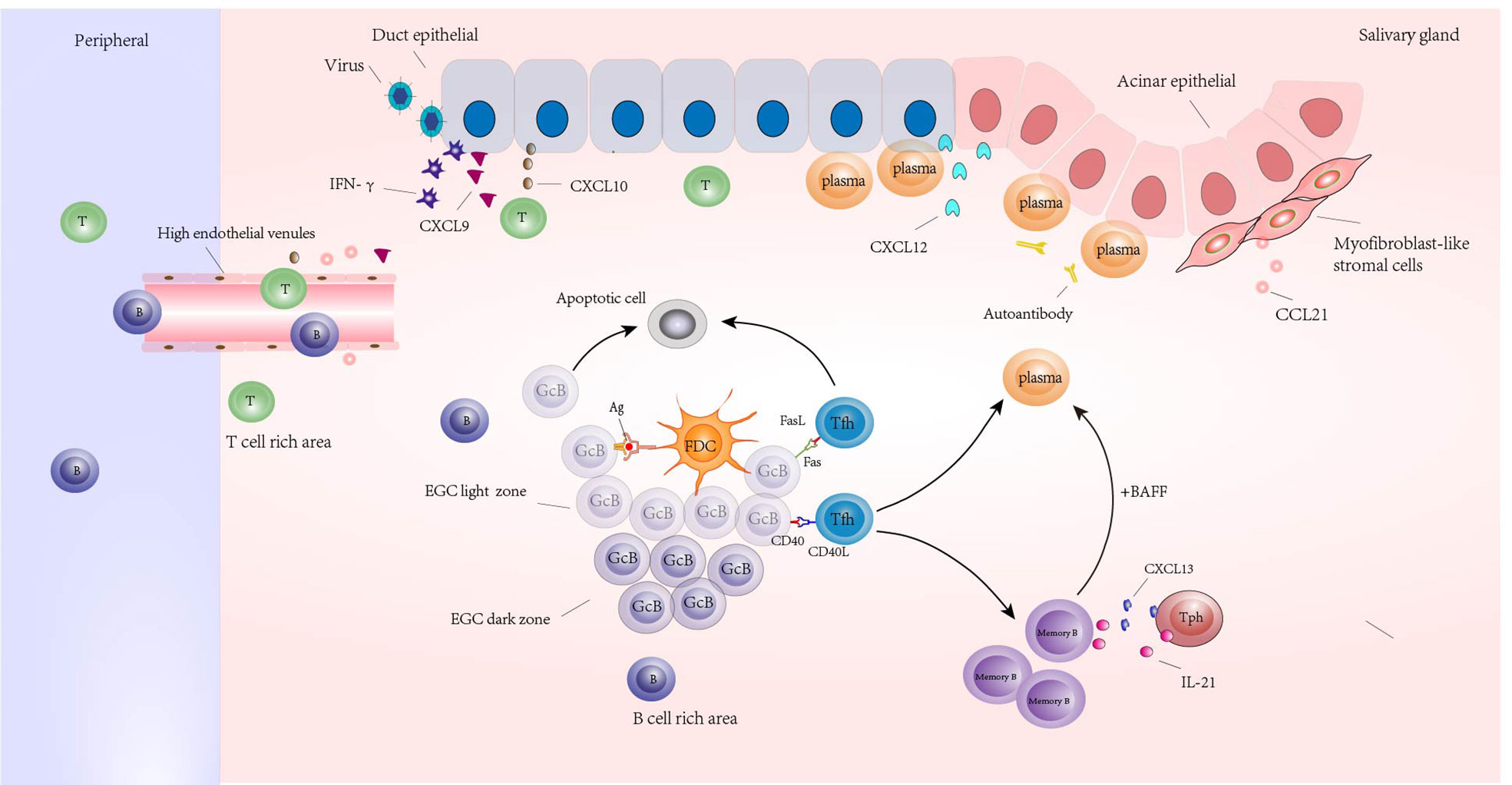
Figure 1 Ectopic lymphoid structures (ELSs) formed around the glands in the salivary glands of a patient with Sjogren’s syndrome. Its core is the ectopic germinal center (EGC), including the bright and dark regions; Germinal center B cells (GcB) were screened by follicular dendritic cells (FDC) in the bright region. The GcB cells that did not receive antigen presentation were apoptotic, and the GcB cells that received antigen presentation were transformed into memory B cells and plasma cells under the assistance of follicular helper T cells (Tfh) or were regulated by Tfh cells to undergo apoptosis. Under the regulation of IL-21 and CXCL13 produced by Tph, memory B cells were transformed into plasma cells under the combined action of BAFF and other factors. Plasma cells infiltrated around ducts and acinar epithelial cells expressing CXCL12 and produced autoantibodies. There are B cell rich areas and T cell rich areas around EGC. High endothelial venules (HEVs) appear in the periphery of lymphoid aggregates in T cell-rich areas, and HEV formation is influenced by CCL21 produced by myofibroblast-like stromal cells; Under the stimulation of virus and other inducements, tissues expressed IFN-γ and induced the release of CXCL9 and CXCL10; CXCL9 and CXCL10 promote the accumulation of peripheral T and B lymphocytes that enter the gland through HEV to the site of inflammation.
In the SG of pSS, upregulated genes are associated with lymphocyte chemotaxis, including IFN-induced chemokines such as CXCL10, and lymphocyte activation, such as TCR β-sites, which play a central role in T-cell activation. The MHC genes HLA-DR and HLA-DQ, which are related to antigen presentation, were also highly expressed in pSS. CXCL13 and CD3D genes were expressed in >90% of pSS patients and <10% of controls, confirming lymphocyte chemotaxis and activation in the SG of pSS patients. Lymphocyte-β (LTβ) is involved in ELS formation in inflammatory tissues and is one of the top 50 differentially expressed genes (DEGs) (111). Shimoyama et al., using single nucleotide polymorphism (SNP)-specific sequencing, found that the risk allele of human salivary gland GTF2I SNPs increased GTF2I expression and enhanced nuclear factor κB (NFκB) activation in human salivary gland cells via the NFκB P65 subunit (112). Inamo et al. used microarray technology to detect peripheral blood B cells of pSS patients and healthy controls and identified LINC00487 and SOX4 as key genes of B-cell disorder in pSS patients by the WGCNA algorithm (113). Many type I IFN genes associated with the response to viral infection were found in the first 200 genes with increased expression in pSS (114). These include IFNα-inducible protein 27, 9-27 (IFITM1), IFN stimulates gene 12 (ISG12), GBP2, and IFN regulator 8 (IRF8). Furthermore, the EBV-induced ligand (CCL19) and its receptor CCR7 genes were upregulated in pSS SGs. These two genes are involved in the activation of B and T cells.
In addition to infection, oxidative stress is an important cause of Sjögren’s syndrome, and reactive oxygen species (ROS) in oxidative stress mainly come from mitochondria. Mitochondria are organelles necessary to maintain homeostasis in cells, and their function is maintained by dynamic fine-tuning. Damaged mitochondria produce more ROS than healthy mitochondria. Changes in mitochondrial-endoplasmic reticulum contact sites (MERCs) can increase inflammatory signals and regulate stress responses and intracellular homeostasis, ultimately affecting cell fate (115). Damaged mitochondria produced more reactive oxygen species than healthy mitochondria, and the presence of mitochondria in pSS salivary gland cells resulted in severe ultrastructural changes (115). Recent scientific studies have shown that mitochondria-related differentially expressed genes (CD38, CMPK2, TBC1D9, and PYCR1) are closely related to the immune cell infiltration of salivary glands in pSS patients through real-time quantitative PCR (116).
Moreover, epigenetics, which includes DNA methylation, noncoding RNA and histone modifications, are involved in the regulation of inflammatory signals in pSS.
At present, there are many literature reports on DNA methylation and pSS (117). DNA methylation is catalyzed by DNA methyltransferase (DNMT) and refers to the presence of methyl radicals (methyl) in CpG dinucleotides from 5-methylcytosine (5-MC). A study on salivary gland methylation showed that the level of salivary gland epithelial cell methylation in pSS patients was lower than that in healthy individuals through DNA global methylation and real-time PCR detection (118). Coculture of human salivary gland cell lines established from irradiated tumor epithelial duct cells and Ramos human B-cell lines suggested that DNA demethylation is associated with lymphocyte infiltration, especially B-cell infiltration, and that the protein kinase C δ - extracellular signal-regulated kinase DNA methyltransferase 1 pathway may be involved in this phenomenon (118). However, DNA methylation is an epigenetic mechanism that includes the adjustment of gene expression and is heritable and reversible without modifying the DNA sequence. This provides another direction for the treatment of pSS.
MicroRNAs (miRNAs) are small noncoding RNAs (sRNAs) that alter gene expression by binding to target messenger RNAs (mRNAs) and inhibiting translation. A researcher (119) conducted sRNA analysis based on global next-generation sequencing (NGS) for pSS labial salivary glands (LSGs) and sicca control groups, and the results suggested that 30% of sRNA in pSS LSG was miRNA, and the miRNA with the most significant change was HSA-Mir-181D-5p compared with the control group. The mRNA level of TNF-α, a direct target of HSA-miR-181D-5p, was significantly increased and negatively correlated with the presence of HSA-miR-181D-5p. Downregulation of HSA-miR-181D-5p in the LSG of SS patients may promote the adenoinflammatory environment by deregulating its direct target TNF-α.
The N-terminal tail of histones protrudes from the nucleosome and is subject to various covalent posttranslational modifications, including acetylation, methylation, phosphorylation, ADP ribosylation, protein conjugation, β-N-acetylglucosamination, deimination/citrullination and ubiquitination/sumoylation. A variety of enzymes are involved in histone modification, such as histone deacetylases (HDACs), histone acetyl transferases (HATs) and histone methyl transferases (HMTs). It has been widely reported that HDACs are involved in immune reactions. HDAC4 negatively regulates the polarization of naive CD4+ T cells toward the Th17 phenotype (120), while HDAC6 induces IL-13 expression through AP-1, leading to the polarization of M2 macrophages (121). Sirtuin 2 (SIRT2), a member of the NAD+-dependent histone deacetylase family, promotes the deacetylation of p70S6K, activates the mTORC1/HIF-1α/RORγ T pathway, inhibits the production of IL-2 by CD4+ T cells, and promotes their differentiation into Th17 cells (122). Histone deacetylase inhibitors (HDACi) modulate the inhibitory T lymphocyte subsets of regulatory T cells (Tregs) and enhance FoxP3 acetylation, thereby protecting transcription factors from proteasome degradation. In vitro T-cell culture experiments in mice showed that HDACi reduced the proliferation of effector T cells and enhanced the inhibitory function of Tregs in coculture with effector T cells (123). HDAC6 inhibitors inhibit Th17-cell differentiation through the PKM2/STAT3 axis (124), while trastatin A (TSA), an HDAC inhibitor, inhibits dendritic cell maturation through downregulation of NF-κB (P65) (125). HDAC also plays an important role in B cells. Studies have shown that the development and survival of plasma cells depend on HDAC11, and the number of plasma cells in the peripheral blood of mice lacking HDAC11 is significantly reduced. In B cells lacking functional HDAC11, the differentiation of plasma cells in vitro is blocked, and HDAC11 is involved in the deacetylation of IRF4 at lysine 103 (126).
BAFF is a crucial cytokine of B cells, promoting B-cell maturation, proliferation, and survival. In pSS patients, salivary gland epithelial cells and innate immune cells can secrete BAFF (127, 128). Elevated LEVELS of BAFF were detected in salivary glands (127). Epithelial cells promote B-cell activation through BAFF, and BAFF may also promote epithelial cell survival through a member of the TNF receptor superfamily 13C (also known as the BAFF receptor) (129). BAFF is induced by type I and type II IFN (130). Therefore, TNFSF13B, encoding BAFF, can be considered the gene stimulated by IFN. IFN regulatory factors (IRFs) control the transcription of TNFSF13B: IRF1 and IRF2 are induction factors of TNFSF13B transcription, while IRF4 and IRF8 are inhibition factors (131). TNFSF13B transgenic mice overexpressing BAFF first developed a lupus-like phenotype and then acquired pSS characteristics, including reduced salivation. This finding supports the role of BAFF in promoting the pathogenesis of pSS.
The SG epithelium relies on a variety of signaling pathways to maintain homeostasis. One pathway is epithelial growth factor (EGF) signal transduction. For TLR signal transduction, EGF receptor (EGFR) activation has also been required for TLR3 signal transduction in the epithelial cell line and TLR-4-mediated downstream NFκB pathway activation, although in the cancer cell model system (132, 133). Conversely, TLR4 signaling also activates EGFR signaling, epithelial cell proliferation, and EGFR ligand expression (134).
The NFκB family is a group of transcription factors that activate a range of inflammatory downstream targets when they translocate to the nucleus. The NFκB pathway has been well demonstrated to be active in pSS SGECs. Phosphorylated IKKϵ (pIKKϵ), pIκBα, and pNFκB were highly expressed in the ductal epithelium of small SGs in pSS patients (135). In pSS SGECs, the expression of the NFκB inhibitor IκBα was significantly lower than that in healthy controls (135, 136), and IκBα inhibited NFκB activity by masking nuclear localization signals. Stimulation of TLR2 receptors in SGECs induces IL-2 production through the NFκB pathway in pSS SGECs (137, 138). In the human SG cell line, IL-6 upregulation is regulated by a set of pathways, including NFκB (139). Knockout of the natural NFκB inhibitor A20 in K14+ epithelial cells (thereby activating the constitutive pathway) is sufficient to trigger the initial stages of pSS, including reduced saliva production and lymphocyte invasion of SGs (54).
The transdifferentiation of epithelial cells into motile mesenchymal cells, a process known as epithelial-mesenchymal transformation (EMT), is essential for development, wound healing, and stem cell behavior and contributes pathologically to fibrosis and cancer progression (140). In primary epithelial tumors, the interaction between cells and the extracellular matrix is reshaped during EMT, resulting in the separation of epithelial cells from each other and the underlying basement membrane and the formation of migratory mesenchymal cells that migrate to different sites with blood flow (141). In pSS, the EMT process mainly involves fibrosis of epithelial cells. During fibrosis, EMT responds to the triggering of TGF-β1, and TGF-β family receptors mediate intracellular signaling cascades that activate SMAD family members through SMAD 2/3 phosphorylation (142). Sisto et al. exposed cultures of healthy salivary gland epithelial cells (SGECs) from healthy donors to TGF-β1 treatment. Semiquantitative RT−PCR, quantitative real-time PCR and Western blot analysis were performed to compare the related gene and protein levels of Smad2/3/4, Snail, e-cadherin, Vimentin and type I collagen (143). They observed higher expression of SMAD2, 3, and 4 and Snail in TGF-β 1-exposed SGECs than in untreated healthy SGECs at both the genetic and protein levels. Snail is the transcriptional repressor and promoter of EMT. Furthermore, compared with untreated SGEC, we found a significant decrease in the epithelial phenotypic marker e-cadherin and a significant increase in the mesenchymal phenotypic marker vimentin and type I collagen in the TGF-β 1-treated samples. This finding suggests that TGF-β1 induces EMT through the TGF-β1/SMAD/Snail signaling pathway, further confirming the existence of EMT in SGs. Concurrent use of the specific TGF-β1 inhibitor SB-431542 in healthy SGECs treated with TGF-β1 significantly reduced the fibrosis markers vimentin and type I collagen, while the epithelial marker e-cadherin returned to levels similar to those of untreated healthy SGECs. This further confirms that TGF-β1 plays an important role in EMT-dependent fibrosis. IL-17 and IL-22 play an important role in EMT. Through the study of salivary glands of pSS patients, researchers found that the expression of the epithelial marker E-cadherin was negatively correlated with the increase in tissue inflammation in pSS SG specimens, while the expression of mesenchymal vimentin and type I collagen was positively correlated. At the same time, they assessed the effect of IL-17 and IL-22 treatment on EMT-dependent SG fibrosis in primary human salivary gland epithelial cells (SGECs) isolated from healthy subjects. The results suggest that vimentin and type I collagen are upregulated after interleukin treatment, while e-cadherin expression is decreased, and the cooperation between IL-17 and IL-22 is required to induce EMT (144).
Recent results show that IFN-γ specifically inhibits the early steps of TGF-β-induced SMAD3 activation through the JAK/STAT pathway while inducing a rapid increase in SMAD7 expression (145, 146). SMAD7 binds to the TFG-β-receptor complex to inhibit TGF-β-mediated phosphorylation of SMAD3 and block TGF-β signaling (35), promoting SG precursor cell differentiation and saliva production (147). Pringle et al. recently demonstrated that SG progenitor cells respond to proinflammatory cytokines through proliferation and apparent cell death, likely through the JAK/STAT signaling pathway, suggesting that JAK/STAT signaling pathway inhibitors may interfere with SG epithelial homeostasis (35).
Microarray and real-time quantitative polymerase chain reaction (RT-qPCR) studies showed that IFN-stimulating genes were significantly upregulated in small salivary glands (MSG) in pSS patients compared with healthy controls (148, 149). Studies have shown that specific type I IFN-associated transcripts (IFIT-3) and type II IFN-associated transcripts (GFP-2) are expressed in MSGs, and IFIT-3 is mainly located in the duct epithelial cells of salivary glands. Gbp-2 is simultaneously located in ductal epithelial cells in lymphocyte aggregates and inflammatory cell infiltrates (150). Animal experiments demonstrated type I IFN dependence on SS development in female NOD mice and elevated pDC (the main producer of type I IFN) TYPE I IFN in their submandibular gland (SMG). After injection of pDCs consuming anti-BST2/CD317 antibodies into female NOD mice aged 4 to 7 weeks, the lack of pDCs hindered the development of SMG inflammation and secretion dysfunction and significantly reduced the number of TYPE I IFN mRNA, white blood cell count, and T and B lymphocytes in the SMG. The expression of IL-7, BAFF, TNF-α, IFN-γ, CXCL9, CXCL11, CD40, CD40 L, Lt-α, Lt-β and NOS2 decreased (92). A study confirmed that overexpression of both type I and type II interferon-induced genes (IFIG) was simultaneously observed in peripheral blood and MSG tissues of patients with pSS (13). Recent studies have suggested that type III IFN (also known as IFN-λ) may be involved in the pathogenesis of pSS. Epithelial IFN-λ2/IL-28a expression was increased in the MSGs of pSS patients compared with non-PSS controls (151). These results suggest the role of the IFN pathway in the pathogenesis of pSS.
BMP6 is a central cytokine that induces pSS-related secretion dysfunction. BMP6 can inhibit the water permeability of the salivary gland epithelial cell membrane by downregulating aquaporin 5 (AQP5), while local overexpression of BMP6 in the salivary gland or lacrimal gland can lead to loss of body fluid secretion in mice (152). HSP70 is an endogenous natural TLR4 ligand (TLR4 is an upstream regulator of BMP6) that stimulates BMP6 expression in pSS. The release of HSP70 from salivary epithelial cells may be triggered by overexpression of lysosome-associated membrane protein 3 (LAMP3). RT−PCR of small salivary gland RNA in pSS patients confirmed a positive correlation between BMP6 and LAMP3 expression. However, LAMP3 overexpression can induce BMP6 expression and a pSS phenotype in murine monocytes. The newly discovered LAMP3/HSP70/BMP6 axis provides an etiological model for SS gland dysfunction and autoimmunity (153).
Salivary gland inflammation in pSS is generally triggered by viral and bacterial infections in susceptible individuals, leading to initial tissue loss. Neutrophils and M2 macrophages clear the site of injury or infection when the decomposition mechanism works appropriately (154). However, when this mechanism is abnormal, dead cells are not cleared in time, leading to the formation of their antigens, increased levels of cytokines and chemokines, and lymphocyte infiltration (155). Specific pro-resolving mediators (SPMs, including liposomes, resolvins, marisins, and protectin) and their aspirin-triggered (AT) forms act as inflammatory mediators, promoting tissue regeneration by limiting uncontrolled inflammation while promoting its termination (156). Odusanwo et al. found that the RvD1 receptor ALX/FPR2 was present in fresh, isolated salivary gland cells and salivary-derived cell lines of 16-week-old C57BL/6 mice in animal experiments. RvD1 receptor activation eliminates tight junctions and cytoskeletal disruption caused by TNF-α by modulating the phosphatidylinositol 3-kinase (PI3K)/AkT signaling pathway, enhances the migration and polarity of salivary epithelial cells, and promotes inflammation regression and tissue repair in salivary epithelial cells (157). Parashar et al. showed that the gene expression of enzymes involved in SPM biosynthesis was changed in the submandibular glands of NOD/ShiLtJ female mice, in which 5-LOX and 12/15-LOX were downregulated and upregulated, respectively. Specific predecomposition mediator (SPM) lysosomal D1 (RvD1) promotes the breakdown of salivary gland inflammation, and mice lacking the RvD1 receptor ALX/FPR2 exhibit congenital and adaptive immune deficiencies in salivary glands. Female ALX/FPR2 KO mice showed increased autoantibody production and loss of salivary gland function with age. This suggests that underlying SPM maladjustment may lead to SS progression (158).
In the salivary glands of patients with Sjogren’s syndrome, upregulated TNF-α induces apoptosis of epithelial cells and disrupts barrier function controlled by tight junction proteins such as the Claudin superfamily, resulting in reduced salivary secretion and gland atrophy (159). TNF-induced apoptosis occurs through the binding of TNF type I receptors (TNFR1), which contain death domains that transmit apoptotic signals through caspase activation (160). In addition, TNFα significantly increased the levels of caspase 3, 8, 9 and cytochrome C, leading to a decrease in the level of Bcl-2 and induced apoptosis of SMG-C6 cells and human SMG tissues (161). Caspase 3 is considered the most important executor of apoptosis, and caspase 8 initiates the death receptor pathway of apoptosis. Caspase 9 is a key player in the mitochondrial pathway and is involved in various stimuli. Cytochrome C is released from damaged mitochondria and plays a key role in inducing apoptosis. miRNAs regulate the expression of target genes at the posttranscriptional level, and many miRNAs are involved in the regulation of apoptosis. To determine the role of these miRNAs in TNFα -induced acinar cell apoptosis, real-time PCR was used to measure their expression levels after cells were incubated with TNFα for a specified period of time. The results showed that TNFα could induce significantly increased levels of Mir-34a-5p, Mir-34a-3p, Mir-200b-5p and Mir-200b-3p simultaneously, while leT-7a-5P expression remained unchanged (161).
Interleukin-2 (IL-2) and high-affinity IL-2 receptor (IL-2R) are essential for the survival of regulatory T cells (Tregs), which are major players in immune tolerance and prevention of autoimmune diseases. Elevated IL-2R levels were found to be positively correlated with SS severity, as reflected by pathologically low salivary flow. Due to the impaired IL-2/IL-2R signaling ability in pSS patients, the immunosuppressive function of Tregs in SS patients was weakened, which may induce salivary gland infiltration of lymphocytes and induce and aggravate pSS (162).
Recent studies have found that interleukin-6 is significantly higher in pSS patients than in HCs patients and is associated with mononuclear cell infiltration in salivary gland tissues in these patients (163). Salivary gland epithelial cells are the primary cellular source of increased IL-6 secretion in these patients. In addition, IL-6 can induce the transformation of SGECs from morphological and phenotypic to mesenchymal phenotypes in a dose-dependent manner. Recent studies have shown that IL-6-treated SGECs have decreased e-cadherin expression and increased vimentin and type I collagen expression compared to control cells. The results confirmed that IL-6 dysregulation may lead to EMT-dependent fibrosis (164).
IL-7 is a 25 kDa soluble globular protein produced and secreted by nonhematopoietic cells such as stromal cells, epithelial cells and endothelial cells. IL-7R is widely expressed in T and B cells, and IL-7/IL7R signaling is critical for the development and maintenance of the entire lymphoid compartment. In vitro experiments have shown that IL-7 induces the production of Th1- and Th2-related cytokines, including IFNγ, monocytes induced by IFNγ (MIG), IFNγ-inducible 10-KD protein (IP-10) and IL-4 (165). Another in vitro cell study showed that IL-7 stimulation induced higher IFN-γ, IL-4, IL-17 and IL-21 production in CCR9+ Th cells and CXCR5+ Th cells (166).
Katsifis et al. showed that the expression of IL-17 protein in salivary glands increased gradually with increasing biopsy lesion score. Transforming growth factor β, IL-6, and IL-23 are essential promoters of Th17 differentiation and are abundant compared to the amounts in control tissues (167). Animal experiments showed that IL-17 inhibited acetylcholine-induced calcium migration and downregulated transient receptor type 1 expression in SG epithelial cells by promoting Nfkbiz mRNA stabilization. In addition, local IL-17 neutralization in SGs significantly reduced salivation and improved tissue inflammation in mice (168). These results suggest that IL-17 may lead to salivary gland dysfunction in Sjogren’s syndrome by inhibiting TRPC1-mediated calcium movement.
IL-21, a member of the recently discovered type I cytokine family, is mainly secreted by Tfh and Tph cells. IL-21R is expressed in B cells and activated CD4+ T cells. IL-21 costimulates B cells with BCR to promote their differentiation into plasma cells, which is also necessary for the formation of normal germinal centers (GCs). The addition of IL-21 to the coculture system blocked 90% of B cells from differentiating into plasma cells. Animal experiments showed that IL-21R knockout mice completely eliminated spontaneous accumulation of GC B cells and plasma cells in blood (169). In addition, IL-21R is required for Thef cell development. In general, IL-21R signaling is necessary for spontaneous accumulation of B and T-cell effector populations.
As a target organ most frequently involved in pSS patients, the impairment of salivary gland function can lead to an imbalance in oral microecology and severe discomfort for patients. An increasing number of treatments are available with further research on the pathogenesis of pSS. In the following sections, we will review the treatment of pSS from the perspective of the immune microenvironment.
As mentioned above, the salivary glands of pSS patients are chronically hypoxic, and blocking hypoxia development may be a potential treatment option. DMOG and FG-4497 hypoxic stabilizers have shown promising results in inflammatory bowel disease (IBD), reducing inflammation, reducing intestinal epithelial cell apoptosis, and enhancing intestinal barrier function (170–172). These may be potential drugs for improving salivary gland hypoxia. Moreover, drugs that inhibit PGE2, such as nonsteroidal drugs, may be equally effective in improving salivary gland function because of their role in hypoxia. In in vitro culture of human tubular HK-2 cells, cell death was mediated by COX-2-dependent PGE2 production, and the COX-2 inhibitor cefoxib prevented hypoxia-induced cell death (173). In another study of human retinal pigment epithelium, celecoxib also showed inhibition of HIF-1α under hypoxia (174)
Senescence makes salivary gland progenitor cells lose their ability to increase their value and differentiate, and the damaged salivary gland does not have enough regeneration potential to fully restore its function. Therefore, it is beneficial for pSS SGs to consume senescent cells and prevent the spread of senescence. Senolytics are a group of drugs that selectively eliminate senescent cells (175). In addition, pro-aging agents (such as navitoclax, dasatinib, and quercetin) work by inhibiting pro-survival pathways (such as Bcl-2 and Bcl-XL) to promote senescent cell death, thereby rejuvenating glandular cells and restoring glandular function (176). Selective removal of p16Ink4a-positive cells by ganciclovir or the antiaging drug ABT263 can eliminate senescent cells and improve the self-renewal ability of stem cells, thereby improving salivary gland function (177). Repressing cellular senescence contributes to the rescue of IR-induced hyposalivation by transient activation of Hh signaling, which is related to enhanced DNA repair and decreased oxidative stress in SMGs (178). Agonists of the Hh signaling pathway may be new targets for treating dryness. In addition, in an in vitro culture experiment of SGSCs after passage culture, the ROCK inhibitor Y-27632 inhibited the expression of senescence-related proteins and promoted cell proliferation (179). Another study showed that in C57BL/6 mice, loss of salivary function is closely related to cellular senescence, and radiation-induced loss of salivary gland function is dependent on IL-6, but IL-6 preconditioning can also prevent senescence and salivary gland hypofunction by enhancing DNA damage repair mechanisms (180). This suggests that IL-6 may play a dual role in Sjogren’s syndrome. A 6-month multicenter, double-blind, randomized placebo-controlled trial showed no improvement in systemic involvement and symptoms with toclizumab compared with placebo in patients with pSS (181). Currently, only toclizumab has been reported in the treatment of pSS with myelitis or refractory interstitial pneumonia (182, 183). However, the application of IL-6 in Sjogren’s syndrome remains to be explored.
As a novel small molecule immunomodulator, iguratimod was confirmed to inhibit B cells by reducing immunoglobulin production and various inflammatory cytokines, including IL-1, IL-6, IL-8, and TNF (184). Clinical studies have validated that iguratimod improved some dryness symptoms and disease activity in pSS patients, reducing BAFF and the percentage of plasma cells over 24 weeks. It can also inhibit PGE2 production by selectively inhibiting COX-2 and the NFκB pathway (185). In animal studies, iguratimod improved inflammatory infiltration of the submandibular gland in mice (186).
Rituximab (RTX) is a monoclonal antibody that targets CD20 on B cells. CD20 is involved in the regulation of B lymphocyte growth after activation. In their open study using RTX, Carubbi et al. (179, 187) found that RTX treatment reversed specific focal lymphocytic sialoadenitis into a nonspecific chronic sialoadenitis mode by depleting B cells, resulting in complete recovery of small salivary gland structure in patients with residual SG function. However, other studies suggested that RTX anti-CD20 treatment might not deplete B-cell infiltration of pSS MALT sites (188). Gong et al. demonstrated in a mouse model that the local production of BAFF is a key local factor in MALt-mediated anti-RTX-depleting B cells (189). B-cell depletion can be achieved only when anti-BAFF is combined with anti-CD20.
Belimumab inhibits soluble BAFF. A one-year open-label trial on belimumab showed that the reduction in B-cell activation biomarkers observed at week 28 continued to week 52, but there was no change in salivary flow, Schirmer test, or salivary biopsy lesion scores (190). Immunobiological evidence supports a sequential regimen of RTX prebelimumab administration designed to target microenvironment BAFF first to improve the success rate of subsequent rituximab depletion therapy in MALT pathological tissues (189). Ianalumab is a monoclonal antibody that consumes B cells and blocks the B-cell activator receptor. In a double-blind, placebo-controlled phase II single-center study, ianalumab (VAY736) resulted in rapid and sustained B-cell depletion and improved ESSDAI and ESSPRI scores, but the variability in salivation flow rate was high enough to make any comparison difficult (191)
In pSS mouse models, labial gland mesenchymal stem cell-derived Exos (LGMSC-EXOS) reduced inflammatory infiltration and restored salivary secretion in salivary glands (192). LGMSC EXO-derived microRNA-125B affects the plasma cells of pSS by directly binding to its target gene, PRDM1 (PR domain zinc finger protein 1, also known as BLIMP1), which may be developed as a target gene for the treatment of pSS.
Cyclosporine A inhibits the IL-2 activity of T cells by interfering with calcineurin required for IL-2 gene transcription (193, 194). Hydroxychloroquine (HCQ) reduces the production of type I IFN and blocks the activation of TLR7 and TLR9 receptors (195), thereby interfering with antigen processing and blocking T-cell activation (196). However, in randomized, double-blind controlled trials in patients with pSS, HCQ did not improve disease symptoms despite inhibiting type I IFN-induced gene expression (188). The effect of HCQ alone on improving glandular function remains controversial.
Abatacept (CTLA4-Ig) binds to the costimulatory molecule CD80/CD86 and blocks the binding of these molecules to CD28 on T cells (197). A recent 48-week trial of abatacept in patients with pSS showed significant improvement in clinical and dry eye symptoms but not in stimulated whole salivary flow (198). Studies have shown a reduction in GCs in lymphocytic lesions and SG lip biopsies after abatacept treatment (199, 200), but salivary and lacrimal gland function remained stable (201).
In a recent clinical trial, prezalumab (a nondepleting monoclonal antibody against ICOSL) had a significant biological effect on SG inflammation, with a significant reduction in the number of CD4+ICOS+Tfh-like cells compared with placebo, despite the failure of the primary endpoint. demonstrated the biological efficacy of targeting the ICOS/ICOS-L pathway in pSS (202).
Other researchers have mitigated pSS by blocking MHC class II IAg7 antigen presentation in NOD mice to prevent pathogenic T cells from recognizing their antigens. The results showed that tetraazatricyclo-dodecane (TATD) and 8-azaguanine (8-AZA) alleviated symptoms by improving saliva and lacrimal gland secretion, reducing autoantibody levels, and reducing the severity of lymphocyte infiltration in saliva and lacrimal glands (203).
Glucocorticoids are a widely used drug for chronic inflammatory autoimmune diseases. They bind to glucocorticoid receptors, resulting in increased transcription of anti-inflammatory genes, such as IL-10, and anti-inflammatory proteins that inhibit the expression of inflammatory genes. Studies have shown that glucocorticoid administration for 6 weeks improves saliva flow in patients but generally does not improve histological or functional parameters of SGs (204). However, a four-year long-term prospective study showed the opposite result: early pSS is characterized by a decline in salivary gland function, with or without steroid use, and a further decline in salivary gland function over time. Reduced salivary gland flow was not associated with corticosteroid use (205).
Leflunomide (LEF) inhibits pyrimidine biosynthesis and decreases naive and memory CD4+ T-cell and B-cell proliferation and NFκB activation (206, 207). In a phase II clinical trial involving 15 patients with early active PSS for 24 weeks, LEF treatment did not improve salivary flow (208). However, the combination of leflunomide and HCQ has been reported to increase salivary gland unstimulated significantly and stimulate total salivary production of pSS at certain time points (209, 210).
Drugs that treat pSS through the NF-κB signaling pathway, such as the novel synthetic DMARD drug iguratimod and the Syk signaling blocker GS-9876 (the Syk signaling pathway is upstream of IKK activation, and its blocking improves the release of NF-κB by its inhibitory complex), are currently in clinical trials. Their effect on the glands has yet to be tested.
In mouse models, Harim Tavares Dos Santos et al. found that hemolysin D1 (RvD1) and its aspirin-triggered AT-RVD1 effectively reduced inflammation and restored saliva flow before and after the onset of pSS. Resolvins are special proresolving mediators (SPMs) that can actively regulate inflammation. Furthermore, the expression of various SPM receptors (ALX/FPR2, BLT1, and CMKLR1) was found in human salivary glands, which may be a potential target for treating pSS patients (156).
CD40 is a transmembrane type I glycoprotein composed of 277 amino acids that belongs to the tumor necrosis factor (TNF) gene superfamily. The ligand CD40L/CD154 is a type II transmembrane protein and exists in a soluble (scd40L) or membrane-bound form. It is present on activated T cells, B cells, endothelial cells and epithelial cells (190). Compared with the control group, NOD mice treated with the CD40 DNA vaccine showed reduced lymphocyte infiltration and increased salivary secretion in salivary glands. At the same time, the expression levels of TNF-α and IL-6 in salivary glands decreased, the number of dendritic cells and plasma cells decreased, and the ANA level decreased (211). Iscalimab, an anti-CD40 antibody, has been shown to be safe and well tolerated at all doses in phase I clinical studies, with no clinically relevant changes in any of the safety parameters, including no evidence of thromboembolic events (212). However, its role in pSS patients remains to be further evaluated.
PSS patients have elevated levels of IL-7 and its receptor in salivary glands. Animal experiments showed that intraperitoneal injection of a blocking antibody against IL-7 receptor α chain (IL-7Rα) for 3 weeks in 10-week-old female NOD mice significantly improved characteristic SS pathology, including reduced salivary secretion and infiltration of leukocytes in the submandibular gland (SMG). Anti-IL-7r α treatment significantly reduced the amount of TNF-α in SMGs and increased the levels of Claudin-1 and aquaporin 5, two molecules essential for normal salivation (213). In phase I clinical trials of the monoclonal antibody GSK2618960 against interleukin-7 receptor α subunit (CD127), GSK2618960 was well tolerated and blocked IL-7 receptor signaling when fully targeted (214). This may be a new target for the future treatment of Sjogren’s syndrome.
Oral salivary gland reduction is one of the most common clinical manifestations of pSS, a disease that directly affects exocrine function. The onset of the disease is genetically susceptible in cells, viruses, and other environmental factors under stimulation through chronic hypoxia, cell senescence, local inflammation, and the production of autoantibodies and other pathways in salivary gland cells, so that their function is impaired. This paper systematically reviews the characteristics and regulatory pathways of the salivary gland microenvironment, hoping that more targeted treatments can be developed to restore gland function and improve dry mouth symptoms through an in-depth understanding of the local immune microenvironment.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
ZT wrote the manuscript. LW and XL participated in the modification. All authors contributed to the article and approved the submitted version.
This article was supported by the National Natural Science Foundation of China (No. 81871271 and No. U21A20365), the Fundamental Research Funds for the Central Universities (grant number WK 9110000148).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Qin B, Wang J, Yang Z, Yang M, Ma N, Huang F, et al. Epidemiology of primary sjögren's syndrome: A systematic review and meta-analysis. Ann Rheumatol Dis (2015) 74:1983–9. doi: 10.1136/annrheumdis-2014-205375
2. Bowman SJ, Ibrahim GH, Holmes G, Hamburger J, Ainsworth JR. Estimating the prevalence among Caucasian women of primary sjögren's syndrome in two general practices in Birmingham, U.K. scand. J Rheumatol (2004) 33:39–43. doi: 10.1080/03009740310004676
3. Bron AJ, Tomlinson A, Foulks GN, Pepose JS, Baudouin C, Geerling G, et al. Rethinking dry eye disease: A perspective on clinical implications. Ocul. Surf (2014) 12:1–31. doi: 10.1016/j.jtos.2014.02.002
4. Villa A, Abati S. Risk factors and symptoms associated with xerostomia: A cross-sectional study. Aust Dent J (2011) 56:290–5. doi: 10.1111/j.1834-7819.2011.01347.x
5. Thorne I, Sutcliffe N. Sjögren's syndrome. Br J Hosp. Med (Lond) (2017) 78(8):438–42. doi: 10.12968/hmed.2017.78.8.438
6. Ramos-Casals M, Brito-Zeron P, Solans R, Camps MT, Casanovas A, Sopeña B, et al. Systemic involvement in primary sjögren’s syndrome evaluated by the EULAR-SS disease activity index: Analysis of 921 Spanish patients (GEAS-SS registry). Rheumatol (Oxford) (2014) 53:321–31. doi: 10.1093/rheumatology/ket349
7. Flament T, Bigot A, Chaigne B, Henique H, Diot E, Marchand-Adam S. Pulmonary manifestations of sjögren’s syndrome. Eur Respir Rev (2016) 25:110–23. doi: 10.1183/16000617.0011-2016
8. Pavlakis PP, Alexopoulos H, Kosmidis ML, Kosmidis ML, Mamali I, Moutsopoulos HM, et al. Peripheral neuropathies in sjögren’s syndrome: A critical update on clinical features and pathogenetic mechanisms. J Autoimmun (2012) 39:27–33. doi: 10.1016/j.jaut.2012.01.003
9. Patel R, Shahane A. The epidemiology of sjögren’s syndrome. Clin Epidemiol (2014) 6:247–55. doi: 10.2147/CLEP.S47399
10. Renaudineau Y, Ballestar E. Epigenetics: DNA methylation signatures in sjogren syndrome. Nat Rev Rheumatol (2016) 12(10):565–6. doi: 10.1038/nrrheum.2016.144
11. Hernández-Molina G, Rodríguez-Pérez JM, Fernández-Torres J, Lima G, Pérez-Hernández N, López-Reyes A, et al. HIF1A (rs11549465) and AKNA (rs10817595) gene polymorphisms are associated with primary sjögren's syndrome. BioMed Res Int (2017) 2017:5845849. doi: 10.1155/2017/5845849
12. Rivière E, Pascaud J, Tchitchek N, Boudaoud S, Paoletti A, Ly B, et al. Salivary gland epithelial cells from patients with sjögren's syndrome induce b-lymphocyte survival and activation. Ann Rheum. Dis (2020) 79(11):1468–77. doi: 10.1136/annrheumdis-2019-216588
13. Nezos A, Gravani F, Tassidou A, Kapsogeorgou EK, Voulgarelis M, Koutsilieris M, et al. Type I and II interferon signatures in sjogren's syndrome pathogenesis: Contributions in distinct clinical phenotypes and sjogren's related lymphomagenesis. J Autoimmun (2015) 63:47–58. doi: 10.1016/j.jaut.2015.07.002
14. Atkinson JC, Wu AJ. Salivary gland dysfunction: Causes, symptoms, treatment. J Am Dent Assoc (1994) 125(4):409–16. doi: 10.14219/jada.archive.1994.0059
15. Haacke EA, Bootsma H, Spijkervet FKL, Visser A, Vissink A, Kluin PM, et al. Fcrl4+ b-cells in salivary glands of primary sjögren’s syndrome patients. J Autoimmun (2017) 81:90–8. doi: 10.1016/j.jaut.2017.03.012
16. Krüger B, Krick S, Dhillon N, Lerner SM, Ames S, Bromberg JS, et al. Donor toll-like receptor 4 contributes to ischemia and reperfusion injury following human kidney transplantation. Proc Natl Acad Sci USA (2009) 106:3390–5. doi: 10.1073/pnas.0810169106
17. Yamamoto K, Tomita N, Yoshimura S, Nakagami H, Taniyama Y, Yamasaki K, et al. Hypoxia-induced renal epithelial cell death through caspase-dependent pathway: Role of bcl-2, bcl-xL and bax in tubular injury. Int J Mol Med (2004) 14:633–40. doi: 10.3892/ijmm.14.4.633
18. Taylor CT, Lisco SJ, Awtrey CS, Colgan SP. Hypoxia inhibits cyclic nucleotide-stimulated epithelial ion transport: Role for nucleotide cyclases as oxygen sensors. J Pharmacol Exp Ther (1998) 284:568–75. doi: 10.1016/S1056-8719(98)00002-1
19. Xiaomin C, Shan J, Zihao Z, Xu X, Ying S, Du L, et al. Increased expression of interleukin-21-inducible genes in minor salivary glands are associated with primary sjögren's syndrome disease characteristics. Rheumatol (Oxford) (2021) 60(6):2979–89. doi: 10.1093/rheumatology/keaa695
20. Palazon A, Goldrath AW, Nizet V, Johnson RS. HIF transcription factors, inflammation, and immunity. Immunity (2014) 414:518–28. doi: 10.1016/j.immuni.2014.09.008
21. Dang EV, Barbi J, Yang H-Y, Jinasena D, Yu H, Zheng Y, et al. Control of TH17/Treg balance by hypoxia-inducible factor 1. Cell (2011) 146(5):772–84. doi: 10.1016/j.cell.2011.07.033
22. Sakai A, Sugawara Y, Kuroishi T, Sasano T, Sugawara S. Identification of IL-18 and Th17 cells in salivary glands of patients with sjögren's syndrome, and amplification of IL-17- mediated secretion of inflammatory cytokines from salivary gland cells by IL-18. J Immunol (2008) 1814:2898–906. doi: 10.4049/jimmunol.181.4.2898
23. Tomomasa K, Manabu S, Nao G. The HIF-1α pathway plays a critical role in salivary gland development in ex vivo organ cultures. FEBS Open Bio (2022) 12(2):460–9. doi: 10.1002/2211-5463.13351
24. Hernández-Molina G, Rodríguez-Pérez JM, Fernández-Torres J, Lima G, Pérez-Hernández N, López-Reyes A, et al. HIF1A (rs11549465) and AKNA (rs10817595) gene polymorphisms are associated with primary sjögren’s syndrome. BioMed Res Int (2017) 2017:1–8. doi: 10.1155/2017/5845849
25. Terrizzi AR, Conti MI, Martínez MP, Fernández-Solari J. The process of acclimation to chronic hypoxia leads to submandibular gland and periodontal alterations: An insight on the role of inflammatory mediators. Mediators Inflammation (2018) 2018:6794508. doi: 10.1155/2018/6794508
26. Christodoulou MI, Kapsogeorgou EK, Moutsopoulos HM. Characteristics of the minor salivary gland infiltrates in sjögren's syndrome. J Autoimmun (2010) 34:400–7. doi: 10.1016/j.jaut.2009.10.004
27. Bombardieri M, Barone F, Lucchesi D, Nayar S, van den Berg WB, Proctor G, et al. Inducible tertiary lymphoid structures, autoimmunity, and exocrine dysfunction in a novel model of salivary gland inflammation in C57BL/6 mice. J Immunol (2012) 189:3767–76. doi: 10.4049/jimmunol.1201216
28. Berra A, Sterin-Borda L, Bacman S, Borda E. Role of salivary IgA in the pathogenesis of sjögren syndrome. Clin Immunol (2002) 104(1):49–57. doi: 10.1006/clim.2002.5228
29. Shen L, Suresh L, Lindemann M, Xuan J, Kowal P, Malyavantham K, et al. Novel autoantibodies in sjögren's syndrome. Clin Immunol (2012) 145:251–5. doi: 10.1016/j.clim.2012.09.013
30. Maehara T, Moriyama M, Hayashida JN, Tanaka A, Shinozaki S, Kubo Y, et al. Selective localization of T helper subsets in labial salivary glands from primary sjögren's syndrome patients. Clin Exp Immunol (2012) 169(2):89–99. doi: 10.1111/j.1365-2249.2012.04606.x
31. Christodoulou MI, Kapsogeorgou EK, Moutsopoulos NM, Moutsopoulos HM. Foxp3+ T-regulatory cells in sjögren's syndrome: Correlation with the grade of the autoimmune lesion and certain adverse prognostic factors. Am J Pathol (2008) 173(5):1389–96. doi: 10.2353/ajpath.2008.080246
32. Streckfus C, Bigler L, Navazesh M, Al-Hashimi I. Cytokine concentrations in stimulated whole saliva among patients with primary sjögren's syndrome, secondary sjögren's syndrome, and patients with primary sjögren's syndrome receiving varying doses of interferon for symptomatic treatment of the condition: A preliminary study. Clin Oral Investig (2001) 5(2):133–5. doi: 10.1007/s007840100104
33. Kang EH, Lee YJ, Hyon JY, Yun PY, Song YW. Salivary cytokine profifiles in primary sjögren's syndrome differ from those in non-sjögren sicca in terms of TNF-alpha levels and Th-1/Th-2 ratios. Clin Exp Rheumatol (2011) 29(6):970–6.
34. Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat Med (2015) 21:1424–35. doi: 10.1038/nm.4000
35. Pringle S, Wang X, Verstappen GMPJPJ, Terpstra JH, Zhang CK, He A, et al. Salivary gland stem cells age prematurely in primary sjögren's syndrome. Arthritis Rheumatol (2018) 71:133–42. doi: 10.1002/art.40659
36. Wang X, Bootsma H, Terpstra J, Vissink A, van der Vegt B, Spijkervet FKL, et al. Progenitor cell niche senescence reflects pathology of the parotid salivary gland in primary sjögren's syndrome. Rheumatology (2020) 59:3003–13. doi: 10.1093/rheumatology/keaa012
37. Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol (2013) 15:978–90. doi: 10.1038/ncb2784
38. Kurosawa M, Shikama Y, Furukawa M, Arakaki R, Ishimaru N, Matsushita K. Chemokines up-regulated in epithelial cells control senescence-associated T cell accumulation in salivary glands of aged and sjögren's syndrome model mice. Int J Mol Sci (2021) 22(5):2302. doi: 10.3390/ijms22052302
39. Kong L, Ogawa N, Nakabayashi T, Liu GT, D'Souza E, McGuff HS, et al. Fas and fas ligand expression in the salivary glands of patients with primary sjögren's syndrome. Arthritis Rheumatol (1997) 40(1):87–97. doi: 10.1002/art.1780400113
40. Manganelli P, Quaini F, Andreoli AM, Lagrasta C, Pilato FP, Zuccarelli A, et al. Quantitative analysis of apoptosis and bcl-2 in sjögren's syndrome. J Rheumatol (1997) 24:1552–7.
41. Kong L, Ogawa N, McGUFF HS, Nakabayashi T, Sakata KM, Masago R, et al. Bcl-2 family expression in salivary glands from patients with primary sjögren's syndrome: Involvement of bax in salivary gland destruction. Clin Immunol Immunopathol (1998) 88:133–41. doi: 10.1006/clin.1998.4556
42. Katsiougiannis S, Tenta R, Skopouli FN. Endoplasmic reticulum stress causes autophagy and apoptosis leading to cellular redistribution of the autoantigens Ro/Sjögren's syndrome-related antigen a (SSA) and La/SSB in salivary gland epithelial cells. Clin Exp Immunol (2015) 181:244–52. doi: 10.1111/cei.12638
43. Kyriakidis NC, Kapsogeorgou EK, Gourzi VC, Konsta OD, Baltatzis GE, Tzioufas AG, et al. Toll-like receptor 3 stimulation promotes Ro52/TRIM21 synthesis and nuclear redistribution in salivary gland epithelial cells, partially via type I interferon pathway. Clin Exp Immunol (2014) 178:548–60. doi: 10.1111/cei.12432
44. Arellano-Garcia ME, Misuno K, Tran SD, Hu S. Interferon-γ induces immunoproteasomes and the presentation of MHC I-associated peptides on human salivary gland cells. PloS One (2014) 9:e10287. doi: 10.1371/journal.pone.0102878
45. Franco A, Valesini G, Barnaba V, Silvagni C, Tiberti A, Balsano F, et al. Class II MHC antigen expression on epithelial cells of salivary glands from patients with sjögren's syndrome. Clin Exp Rheumatol (1987) 5(3):199–203. doi: 10.1074/jbc.M111.277046
46. Kapsogeorgou EK, Manoussakis MN. Salivary gland epithelial cells (SGEC): Carriers of exquisite B7-2 (CD86) co-stimulatory molecules. J Autoimmun (2010) 35:188–91. doi: 10.1016/j.jaut.2010.06.006
47. Kawakami A, Nakashima K, Tamai M, Nakamura H, Iwanaga N, Fujikawa K, et al. Toll-like receptor in salivary glands from patients with sjögren's syndrome: Functional analysis by human salivary gland cell line. J Rheumatol (2007) 34:1019–26.
48. Dimitriou ID, Kapsogeorgou EK, Abu-Helu RF, Abu-Helu RF, Moutsopoulos HM, Manoussakis MN, et al. Establishment of a convenient system for the long-term culture and study of non-neoplastic human salivary gland epithelial cells. Eur J Oral Sci (2002) 110:21–30. doi: 10.1034/j.1600-0722.2002.00152.x
49. Ohlsson M, Szodoray P, Loro LL, Johannessen AC, Jonsson R. CD40, CD154, bax and bcl-2 expression in sjögren's syndrome salivary glands: A putative antiapoptotic role during its effector phases. Scand J Immunol (2002) 56:561–71. doi: 10.1046/j.1365-3083.2002.01168.x
50. Sakai A, Sugawara Y, Kuroishi T, Sasano T, Sugawara S. Identification of IL-18 and Th17 cells in salivary glands of patients with sjögren's syndrome, and amplification of IL-17-mediated secretion of inflammatory cytokines from salivary gland cells by IL-18. J Immunol (2008) 181:2898–906. doi: 10.4049/jimmunol.181.4.2898
51. Szyszko EA, Brokstad KA, Oijordsbakken G, Jonsson MV, Jonsson R, Skarstein K. Salivary glands of primary sjögren's syndrome patients express factors vital for plasma cell survival. Arthritis Res Ther (2011) 13(1):R2. doi: 10.1186/ar3220
52. Sfriso P, Oliviero F, Calabrese F, Miorin M, Facco M, Contri A, et al. Epithelial CXCR3-b regulates chemokines bioavailability in normal, but not in sjögren's syndrome, salivary glands. J Immunol (2006) 176:2581–9. doi: 10.4049/jimmunol.176.4.2581
53. Xanthou G, Polihronis M, Tzioufas AG, Paikos S, Sideras P, Moutsopoulos HM, et al. "Lymphoid" chemokine messenger RNA expression by epithelial cells in the chronic inflammatory lesion of the salivary glands of sjögren's syndrome patients: possible participation in lymphoid structure formation. Arthritis Rheumatol (2001) 44:408–18. doi: 10.1002/1529-0131(200102)44:2<408::AID-ANR60>3.0.CO;2-0
54. Wang X, Shaalan A, Liefers S, Coudenys J, Elewaut D, Proctor GB, et al. Dysregulation of NF-kB in glandular epithelial cells results in sjögren's-like features. PloS One (2018) 13:e0200212. doi: 10.1371/journal.pone.0200212
55. Spachidou MP, Bourazopoulou E, Maratheftis CI, Kapsogeorgou EK, Moutsopoulos HM, Tzioufas AG, et al. Expression of functional toll-like receptors by salivary gland epithelial cells: Increased mRNA expression in cells derived from patients with primary sjögren's syndrome. Clin Exp Immunol (2007) 147:497–503. doi: 10.1111/j.1365-2249.2006.03311.x
56. Rusakiewicz S, Nocturne G, Lazure T, Semeraro M, Flament C, Caillat-Zucman S, et al. NCR3/NKp30 contributes to pathogenesis in primary sjögren's syndrome. Sci Transl Med (2013) 5:195ra96. doi: 10.1126/scitranslmed.300572
57. Amft N, Curnow SJ, Scheel-Toellner D, Devadas A, Oates J, Crocker J, et al. Ectopic expression of the b cell-attracting chemokine BCA-1 (CXCL13) on endothelial cells and within lymphoid follicles contributes to the establishment of germinal center-like structures in sjögren's syndrome. Arthritis Rheumatol (2001) 44:2633–41. doi: 10.1002/1529-0131(200111)44:11<2633::AID-ART443>3.0.CO;2-9
58. Manoussakis MN, Spachidou MP, Maratheftis CI. Salivary epithelial cells from sjögren's syndrome patients are highly sensitive to anoikis induced by TLR-3 ligation. J Autoimmun (2010) 35:212–8. doi: 10.1016/j.jaut.2010.06.010
59. Fox RI, Kang HI, Ando D, Abrams J, Pisa E. Cytokine mRNA expression in salivary gland biopsies of sjögren's syndrome. J Immunol (1994) 152:5532–9.
60. Gong Y-Z, Nititham J, Taylor K, Miceli-Richard C, Sordet C, Wachsmann D, et al. Differentiation of follicular helper T cells by salivary gland epithelial cells in primary sjögren's syndrome. J Autoimmun (2014) 51:57–66. doi: 10.1016/j.jaut.2013.11.003
61. Ha Y-J, Choi YS, Kang EH, Chung JH, Cha S, Song YW, et al. Increased expression of interferon-λ in minor salivary glands of patients with primary sjögren's syndrome and its synergic effect with interferon-α on salivary gland epithelial cells. Clin Exp Rheumatol (2018) 36(Suppl. 112):S31–40.
62. Verstappen GM, Pringle S, Bootsma H, Kroese FGM. Epithelial–immune cell interplay in primary sjögren syndrome salivary gland pathogenesis. Nat Rev Rheumatol (2021) 17:333–48. doi: 10.1038/s41584-021-00605-2
63. Morva A, Kapsogeorgou EK, Konsta OD, Moutsopoulos HM, Tzioufas AG. Op0079 salivary gland epithelial cells (SGECS) promote the differentiation of b cells. Ann Rheum. Dis (2013) 72:A77–7. doi: 10.1136/annrheumdis-2013-eular.284
64. Barone F, Bombardieri M, Rosado MM, Morgan PR, Challacombe SJ, De Vita S, et al. CXCL13, CCL21, and CXCL12 expression in salivary glands of patients with sjögren's syndrome and MALT lymphoma: Association with reactive and malignant areas of lymphoid organization. J Immunol (2008) 180:5130–40. doi: 10.4049/jimmunol.180.7.5130
65. Dean S, Wang CS, Nam K, Maruyama CL, Trump BG, Baker OJ. Aspirin triggered resolvin D1 reduces inflammation and restores saliva secretion in a sjögren's syndrome mouse model. Rheumatol (Oxford) (2019) 58(7):1285–92. doi: 10.1093/rheumatology/kez072
66. Aggarwal S, Pittenger MF. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood (2005) 105(4):1815–22. doi: 10.1182/blood-2004-04-1559
67. McCoy SS, Giri J, Das R, Paul PK, Pennati A, Parker M, et al. Minor salivary gland mesenchymal stromal cells derived from patients with Sjö;gren's syndrome deploy intact immune plasticity. Cytotherapy (2021) 23(4):301–10. doi: 10.1016/j.jcyt.2020.09.008
68. Park CS, Choi YS. How do follicular dendritic cells interact intimately with b cells in the germinal centre? Immunology (2005) 114(1):2–10. doi: 10.1111/j.1365-2567.2004.02075.x
69. Christodoulou MI, Kapsogeorgou EK, Moutsopoulos HM. Characteristics of the minor salivary gland infifiltrates in sjögren's syndrome. J Autoimmun (2010) 34(4):400–7. doi: 10.1016/j.jaut.2009.10.004
70. Manoussakis MN, Boiu S, Korkolopoulou P, Kapsogeorgou EK, Kavantzas N, Ziakas P, et al. Rates of infifiltration by macrophages and dendritic cells and expression of interleukin-18 and interleukin-12 in the chronic inflflammatory lesions of sjögren's syndrome: Correlation with certain features of immune hyperactivity and factors associated with high risk of lymphoma development. Arthritis Rheum (2007) 56(12):3977–88. doi: 10.1002/art.23073
71. Verstappen GM, Ice JA, Bootsma H, Pringle S, Haacke EA, de Lange K, et al. Gene expression profifiling of epithelium-associated Fcrl4+ b cells in primary sjögren's syndrome reveals a pathogenic signature. J Autoimmun (2020) 109:102439. doi: 10.1016/j.jaut.2020.102439
72. Peck AB, Nguyen CQ, Ambrus J. Early covert appearance of marginal zone b cells in salivary glands of sjögren's syndrome-susceptible mice: Initiators of subsequent overt clinical disease. Int J Mol Sci (2021) 22(4):1919. doi: 10.3390/ijms22041919
73. Shen L, Gao C, Suresh L, Xian Z, Song N, Chaves LD, et al. Central role for marginal zone b cells in an animal model of sjögren's syndrome. Clin Immunol (2016) 168:30–6. doi: 10.1016/j.clim.2016.04.008
74. Daridon C, Pers J-O, Devauchelle V, Martins-Carvalho C, Hutin P, Pennec Y-L, et al. Identifification of transitional type II b cells in the salivary glands of patients with sjögren’s syndrome. Arthritis Rheum (2006) 54:2280–8. doi: 10.1002/art.21936
75. Hansen A, Odendahl M, Reiter K, Jacobi AM, Feist E, Scholze J, et al. Diminished peripheral blood memory b cells and accumulation of memory b cells in the salivary glands of patients with sjögren's syndrome. Arthritis Rheum (2002) 46:2160–71. doi: 10.1002/art.10445
76. Hansen A, Reiter K, Ziprian T, Jacobi A, Hoffmann A, Gosemann M, et al. Dysregulation of chemokine receptor expression and function by b cells of patients with primary sjögren's syndrome. Arthritis Rheum (2005) 52:2109–19. doi: 10.1002/art.21129
77. Hansen A, Lipsky PE, Dörner T. B cells in sjögren's syndrome: Indications for disturbed selection and differentiation in ectopic lymphoid tissue. Arthritis Res Ther (2007) 9:218. doi: 10.1186/ar2210
78. Skarstein K, Jensen JL, Galtung H, Jonsson R, Brokstad K, Aqrawi LA. Autoantigen-specific b cells and plasma cells are prominent in areas of fatty infiltration in salivary glands of patients with primary sjögren's syndrome. Autoimmunity (2019) 52(7-8):242–50. doi: 10.1080/08916934.2019.1684475
79. Cui L, Elzakra N, Xu S, Xiao GG, Yang Y, Hu S. Investigation of three potential autoantibodies in sjogren's syndrome and associated MALT lymphoma. Oncotarget (2017) 8(18):30039–49. doi: 10.18632/oncotarget.15613
80. Singh N, Cohen PL. The T cell in sjogren's syndrome: Force majeure, not spectateur. J Autoimmun (2012) 39(3):229–33. doi: 10.1016/j.jaut.2012.05.019
81. Verstappen GM, Corneth OBJ, Bootsma H, Kroese FGM. Th17 cells in primary sjögren's syndrome: Pathogenicity and plasticity. J Autoimmun (2018) 87:16–25. doi: 10.1016/j.jaut.2017.11.003
82. Blokland SLM, van Vliet-Moret FM, Hillen MR, Pandit A, Goldschmeding R, Kruize AA, et al. Epigenetically quantified immune cells in salivary glands of sjögren's syndrome patients: A novel tool that detects robust correlations of T follicular helper cells with immunopathology. Rheumatol (Oxford) (2020) 59:335–43. doi: 10.1093/rheumatology/kez268
83. Pontarini E, Murray-Brown WJ, Croia C, Lucchesi D, Conway J, Rivellese F, et al. Unique expansion of IL-21+ tfh and tph cells under control of ICOS identifies sjögren's syndrome with ectopic germinal centres and MALT lymphoma, Ann. Rheumatol Dis (2020) 79:1588–99. doi: 10.1136/annrheumdis-2020-217646
84. Crotty S. Follicular helper CD4 t cells (TFH), annu. Rev Immunol (2011) 29:621–63. doi: 10.1146/annurev-immunol-031210-101400
85. Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, et al. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity (2011) 34:932–46. doi: 10.1016/j.immuni.2011.03.023
86. Weinstein JS, Bertino SA, Hernandez SG, Poholek AC, Teplitzky TB, Nowyhed HN, et al. B cells in T follicular helper cell development and function: separable roles in delivery of ICOS ligand and antigen. J Immunol (2014) 192:3166–79. doi: 10.4049/jimmunol.1302617
87. Sage PT, Sharpe AH. T Follicular regulatory cells. Immunol Rev (2016) 271:246–59. doi: 10.1111/imr.12411
88. Rao DA, Gurish MF, Marshall JL, Slowikowski K, Fonseka CY, Liu Y, et al. Pathologically expanded peripheral T helper cell subset drives b cells in rheumatoid arthritis. Nature (2017) 542:110–4. doi: 10.1038/nature20810
89. Dupré A, Pascaud J, Rivière E, Paoletti A, Ly B, Mingueneau M, et al. Association between T follicular helper cells and T peripheral helper cells with b-cell biomarkers and disease activity in primary sjögren syndrome. RMD Open (2021) 7(1):e001442. doi: 10.1136/rmdopen-2020-001442
90. Pontarini E, Murray-Brown WJ, Croia C, Murray-Brown WJ, Croia C, Lucchesi D, Conway J, Rivellese F, et al. Unique expansion of IL-21+ tfh and tph cells under control of ICOS identifies sjögren's syndrome with ectopic germinal centres and MALT lymphoma. Ann Rheum. Dis (2020) 79(12):1588–99. doi: 10.1136/annrheumdis-2020-217646
91. Lowin B, Hahne M, Mattmann C, Tschopp J. Cytolytic T-cell cytotoxicity is mediated through perforin and fas lytic pathways. Nature (1994) 370(6491):650–2. doi: 10.1038/370650a0
92. Zhou J, Zhang X, Yu Q. Plasmacytoid dendritic cells promote the pathogenesis of sjögren's syndrome. Biochim Biophys Acta Mol Basis Dis (2022) 1868(2):166302. doi: 10.1016/j.bbadis.2021.166302
93. Ozaki Y, Ito T, Son Y, Amuro H, Shimamoto K, Sugimoto H, et al. Decrease of blood dendritic cells and increase of tissue-infiltrating dendritic cells are involved in the induction of sjögren's syndrome but not in the maintenance. Clin Exp Immunol (2010) 159(3):315–26. doi: 10.1111/j.1365-2249.2009.04071.x
94. Novkovic M, Onder L, Cupovic J, Abe J, Bomze D, Cremasco V, et al. Topological small-world organization of the fibroblastic reticular cell network determines lymph node functionality. PloS Biol (2016) 14(7):e1002515. doi: 10.1371/journal.pbio.1002515
95. Nayar S, Campos J, Smith CG, Iannizzotto V, Gardner DH, Mourcin F, et al. Immunofibroblasts are pivotal drivers of tertiary lymphoid structure formation and local pathology. Proc Natl Acad Sci U S A (2019) 116(27):13490–7. doi: 10.1073/pnas.1905301116
96. Korsunsky I, Wei K, Pohin M, Kim EY, Barone F, Major T, et al. Cross-tissue, single-cell stromal atlas identifies shared pathological fibroblast phenotypes in four chronic inflammatory diseases. Med (N Y) (2022) 3(7):481–518.e14. doi: 10.1016/j.medj.2022.05.002
97. Nguyen HN, Noss EH, Mizoguchi F, Huppertz C, Wei KS, Watts GFM, et al. Autocrine loop involving IL-6 family member LIF, LIF receptor, and STAT4 drives sustained fibroblast production of inflammatory mediators. Immunity (2017) 46(2):220–32. doi: 10.1016/j.immuni.2017.01.004
98. Patel S. Danger-associated molecular patterns (DAMPs): The derivatives and triggers of inflammation. Curr Allergy Asthma Rep (2018) 18(11):63. doi: 10.1007/s11882-018-0817-3
99. Castro I, Albornoz N, Aguilera S, Barrera MJ, González S, Núñez M, et al. Aberrant MUC1 accumulation in salivary glands of sjögren's syndrome patients is reversed by TUDCA in vitro. Rheumatol (Oxford) (2020) 59:742–53. doi: 10.1093/rheumatology/kez316
100. Schaefer L. Complexity of danger: The diverse nature of damage-associated molecular patterns. J Biol Chem (2014) 289(51):35237–45. doi: 10.1074/jbc.R114.619304
101. Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signalling. Mediators Inflflamm (2010) 2010:672395. doi: 10.1155/2010/672395
102. Gao R, Tang J, Dong Y, Ming B, Yu Z, Zhong J, et al. The aberrant levels of decorin induce damages of human salivary gland epithelial cells and polarization of macrophages. Mod. Rheumatol (2022), roac034. doi: 10.1093/mr/roac034
103. TD RANDALL, MEBIUS RE. The development and function of mucosal lymphoid tissues: A balancing act with micro-organisms. Mucosal Immunol (2014) 7:455–66. doi: 10.1038/mi.2014.11
104. Kivity S, Arango MT, Ehrenfeld M, Tehori O, Shoenfeld Y, Anaya JM, et al. Infection and autoimmunity in sjögren's syndrome: A clinical study and comprehensive review. J Autoimmun (2014) 51:17–22. doi: 10.1016/j.jaut.2014.02.008
105. Kroese FG, Abdulahad WH, Haacke E, Bos NA, Vissink A, Bootsma H. B-cell hyperactivity in primary sjögren's syndrome. Expert Rev Clin Immunol (2014) 10:483–99. doi: 10.1586/1744666X.2014.891439
106. Ogawa N, Ping L, Zhenjun L, Takada Y, Sugai S. Involvement of the interferon-gamma-induced T cell-attracting chemokines, interferon-gamma-inducible10-kd protein (CXCL10) and monokine induced by interferon-gamma (CXCL9), in the salivary gland lesions of patients with sjögren's syndrome. Arthritis Rheumatol (2002) 46:2730–41. doi: 10.1002/art.10577
107. Bombardieri M, Lewis M, Pitzalis C. Ectopic lymphoid neogenesis in rheumatic autoimmune diseases. Nat Rev Rheumatol (2017) 13:141–54. doi: 10.1038/nrrheum.2016.217
108. Gommerman JL, Browning JL. Lymphotoxin/light, lymphoid microenvironments and autoimmune disease. Nat Rev Immunol (2003) 3:642–55. doi: 10.1038/nri1151
109. van Eijk M, Defrance T, Hennino A, de Groot C. Death-receptor contribution to the germinal-center reaction. Trends Immunol (2001) 22(12):677–82. doi: 10.1016/s1471-4906(01)02086-5
110. Vanbervliet B, Bendriss-Vermare N, Massacrier C, Homey B, de Bouteiller O, Brière F, et al. The inducible CXCR3 ligands control plasmacytoid dendritic cell responsiveness to the constitutive chemokine stromal cell-derived factor 1 (SDF-1)/CXCL12. J Exp Med (2003) 198(5):823–30. doi: 10.1084/jem.20020437
111. Pontarini E, Coleby R, Bombardieri M. Cellular and molecular diversity in sjogren's syndrome salivary glands: Towards a better definition of disease subsets. Semin Immunol (2021) 101547. doi: 10.1016/j.smim.2021.101547
112. Shimoyama S, Nakagawa I, Jiang JJ, Matsumoto I, Chiorini JA, Hasegawa Y, et al. Sjögren's syndrome-associated SNPs increase GTF2I expression in salivary gland cells to enhance inflammation development. Int Immunol (2021) 33(8):423–34. doi: 10.1093/intimm/dxab025
113. Inamo J, Suzuki K, Takeshita M, Kassai Y, Takiguchi M, Kurisu R, et al. Identification of novel genes associated with dysregulation of b cells in patients with primary sjögren's syndrome. Arthritis Res Ther (2020) 22(1):153. doi: 10.1186/s13075-020-02248-2
114. Li H, Ice JA, Lessard CJ, Sivils KL. Interferons in sjögren's syndrome: genes, mechanisms, and effects. Front Immunol (2013) 4:290. doi: 10.3389/fimmu.2013.00290
115. Barrera MJ, Aguilera S, Castro I, Carvajal P, Jara D, Molina C, et al. Dysfunctional mitochondria as critical players in the inflammation of autoimmune diseases: Potential role in sjögren's syndrome. Autoimmun. Rev (2021) 20(8):102867. doi: 10.1016/j.autrev.2021.102867
116. Li N, Li Y, Hu J, Wu Y, Yang J, Fan H, et al. A link between mitochondrial dysfunction and the immune microenvironment of salivary glands in primary sjögren's syndrome. Front Immunol (2022) 13:845209. doi: 10.3389/fimmu.2022.845209
117. Guimarães JR, Coêlho MC, de Oliveira NFP. Contribution of DNA methylation to the pathogenesis of sjögren's syndrome: A review. Autoimmunity (2022) 55:215–22. doi: 10.1080/08916934.2022.2062593
118. Thabet Y, Le Dantec C, Ghedira I, Devauchelle V, Cornec D, Pers JO, et al. Epigenetic dysregulation in salivary glands from patients with primary sjögren's syndrome may be ascribed to infiltrating b cells. J Autoimmun (2013) 41:175–81. doi: 10.1016/j.jaut.2013.02.002
119. Castro I, Carvajal P, Jara D, Aguilera S, Heathcote B, Barrera MJ, et al. Small RNA expression profiling reveals hsa-miR-181d-5p downregulation associated with TNF-α overexpression in sjögren's syndrome patients. Front Immunol (2022) 13:870094. doi: 10.3389/fimmu.2022.870094
120. Dou B, Ma F, Jiang Z, Zhao L. Blood HDAC4 variation links with disease activity and response to tumor necrosis factor inhibitor and regulates CD4+ T cell differentiation in ankylosing spondylitis. Front Med (Lausanne) (2022) 9:875341. doi: 10.3389/fmed.2022.875341
121. Tseng CC, Huang SY, Tsai HP, Wu CW, Hsieh TH. HDAC6 is a prognostic biomarker that mediates IL-13 expression to regulate macrophage polarization through AP-1 in oral squamous cell carcinoma. Sci Rep (2022) 12(1):10513. doi: 10.1038/s41598-022-14052-w
122. Hisada R, Yoshida N, Umeda M, Burbano C, Bhargava R, Scherlinger M, et al. The deacetylase SIRT2 contributes to autoimmune disease pathogenesis by modulating IL-17A and IL-2 transcription. Cell Mol Immunol (2022) 19(6):738–50. doi: 10.1038/s41423-022-00874-1
123. Ratay ML, Balmert SC, Bassin EJ, Little SR. Controlled release of an HDAC inhibitor for reduction of inflammation in dry eye disease. Acta Biomater (2018) 71:261–70. doi: 10.1016/j.actbio.2018.03.002
124. Chang Q, Yin D, Li H, Du X, Wang Z, Liu Y, et al. HDAC6-specific inhibitor alleviates hashimoto's thyroiditis through inhibition of Th17 cell differentiation. Mol Immunol (2022) 149:39–47. doi: 10.1016/j.molimm.2022.05.004
125. Yu Y, Liu B, Chen S, Wang J, Chen F, Liu T, et al. Trichostatin a inhibits dendritic cell maturation through down-regulating NF-κ b (p65) pathway. Mol Biol Rep (2022) 49(4):2619–27. doi: 10.1007/s11033-021-07065-7
126. Mostofa A, Distler A, Meads MB, Sahakian E, Powers JJ, Achille A, et al. Plasma cell dependence on histone/protein deacetylase 11 reveals a therapeutic target in multiple myeloma. JCI Insight (2021) 6(24):e151713. doi: 10.1172/jci.insight.151713
127. Lavie F, Miceli-Richard C, Quillard J, Roux S, Leclerc P, Mariette X, et al. Expression of BAFF (BLyS) in T cells infiltrating labial salivary glands from patients with sjögren's syndrome. J Pathol (2004) 202:496–502. doi: 10.1002/path.1533
128. Daridon C, Devauchelle V, Hutin P, Le Berre R, Martins-Carvalho C, Bendaoud B, et al. Aberrant expression of BAFF by b lymphocytes infiltrating the salivary glands of patients with primary sjögren's syndrome. Arthritis Rheum (2007) 56:1134–44. doi: 10.1002/art.22458
129. Lahiri A, Varin MM, Le Pottier L, Pochard P, Bendaoud B, Youinou P, et al. Specific forms of BAFF favor BAFF receptor-mediated epithelial cell survival. J Autoimmun (2014) 51:30–7. doi: 10.1016/j.jaut.2014.02.004
130. Litinskiy MB, Nardelli B, Hilbert DM, He B, Schaffer A, Casali P, et al. DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat Immunol (2002) 3:822–9. doi: 10.1038/ni829
131. Sjöstrand M, Johansson A, Aqrawi L, Olsson T, Wahren-Herlenius M, Espinosa A, et al. The expression of BAFF is controlled by IRF transcription factors. J Immunol (2016) 196:91–6. doi: 10.4049/jimmunol.1501061
132. De S, Zhou H, DeSantis D, Croniger CM, Li X, Stark GR, et al. Erlotinib protects against LPS-induced endotoxicity because TLR4 needs EGFR to signal. Proc Natl Acad Sci U S A (2015) 112:9680–5. doi: 10.1073/pnas.1511794112
133. Yamashita M, Chattopadhyay S, Fensterl V, Saikia P, Wetzel JL, Sen GC, et al. Epidermal growth factor receptor is essential for toll-like receptor 3 signaling. Sci Signal (2012) 5:ra50–0. doi: 10.1126/scisignal.2002581
134. Hsu D, Fukata M, Hernandez YG, Sotolongo JP, Goo T, Maki J, et al. Toll-like receptor 4 differentially regulates epidermal growth factor-related growth factors in response to intestinal mucosal injury. Lab Invest (2010) 90:1295–305. doi: 10.1038/labinvest.2010.100
135. Sisto M, Lisi S, Lofrumento DD, Ingravallo G, De Lucro R, D'Amore M, et al. Salivary gland expression level of IκBα regulatory protein in sjögren's syndrome. J Mol Histol (2013) 44:447–54. doi: 10.1007/s10735-013-9487-6
136. Nakamura H, Kawakami A, Ida H, Koji T, Eguchi K. EGF activates PI3K-akt and NF-κB via distinct pathways in salivary epithelial cells in sjögren's syndrome. Rheumatol. Int (2007) 28:127–36. doi: 10.1007/s00296-007-0411-9
137. Kwok SK, Cho ML, Her YM, Oh HJ, Park MK, Lee SY, et al. TLR2 ligation induces the production of IL-23/IL-17 via IL-6, STAT3 and NF-kB pathway in patients with primary sjögren's syndrome. Arthritis Res Ther (2012) 14:R64. doi: 10.1186/ar3780
138. Sisto M, Lorusso L, Lisi S. TLR2 signals via NF-κB to drive IL-15 production in salivary gland epithelial cells derived from patients with primary sjögren's syndrome. Clin Exp Med (2017) 17:341–50. doi: 10.1007/s10238-016-0429-y
139. Wei L, Xiong H, Li W, Li B, Cheng Y. Upregulation of IL-6 expression in human salivary gland cell line by IL-17 via activation of p38 MAPK, ERK, PI3K/Akt, and NF-κB pathways. J Oral Pathol Med (2018) 47:847–55. doi: 10.1111/jop.12765
140. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol (2014) 15(3):178–96. doi: 10.1038/nrm3758
141. Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol (2019) 20(2):69–84. doi: 10.1038/s41580-018-0080-4
142. Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res (2009) 19:156–72. doi: 10.1038/cr.2009.5
143. Sisto M, Lorusso L, Ingravallo G, Tamma R, Ribatti D, Lisi S. The TGF-β1 signaling pathway as an attractive target in the fibrosis pathogenesis of sjögren's syndrome. Mediators Inflamm (2018) 2018:1965935. doi: 10.1155/2018/1965935
144. Sisto M, Lorusso L, Tamma R, Ingravallo G, Ribatti D, Lisi S. Interleukin-17 and -22 synergy linking inflammation and EMT-dependent fibrosis in sjögren's syndrome. Clin Exp Immunol (2019) 198(2):261–72. doi: 10.1111/cei.13337
145. Higashi K, Inagaki Y, Fujimori K, Nakao A, Kaneko H, Nakatsuka I. Interferon-gamma interferes with transforming growth factor-beta signaling through direct interaction of YB-1 with SMAD3. J Biol Chem (2003) 278:43470–9. doi: 10.1074/jbc.M302339200
146. Ulloa L, Doody J, Massagué J. Inhibition of transforming growth factor-β/SMAD signalling by the interferon-γ/STAT pathway. Nature (1999) 397:710–3. doi: 10.1038/17826
147. Moustakas A, Pardali K, Gaal A, Heldin CH. Mechanisms of TGF-βsignaling in regulation of cell growth and differentiation. Immunol Lett (2002) 82:85–91. doi: 10.1016/S0165-2478(02)00023-8
148. Gottenberg JE, Cagnard N, Lucchesi C, Letourneur F, Mistou S, Lazure T, et al. Activation of IFN pathways and plasmacytoid dendritic cell recruitment in target organs of primary sjögren's syndrome. Proc Natl Acad Sci USA (2006) 103:2770–5. doi: 10.1073/pnas.0510837103
149. Hjelmervik TOR, Petersen K, Jonassen I, Jonsson R, Bolstad AI. Gene expression profifiling of minor salivary glands clearly distinguishes primary sjögren's syndrome patients from healthy control subjects. Arthritis Rheumatol (2005) 52:1534–44. doi: 10.1002/art.21006
150. Hall JC, Baer AN, Shah AA, Criswell LA, Shiboski CH, Rosen A, et al. Molecular subsetting of interferon pathways in sjögren's syndrome. Arthritis Rheumatol (2015) 67:2437–46. doi: 10.1002/art.39204
151. Apostolou E, Kapsogeorgou EK, Konsta OD, Giotakis I, Saridaki MI, Andreakos E, et al. Expression of type III interferons (IFNλs) and their receptor in sjögren's syndrome. Clin Exp Immunol (2016) 186:304–12. doi: 10.1111/cei.12865
152. Yin H, Cabrera-Perez J, Lai Z, Michael D, Weller M, Swaim WD, et al. Association of bone morphogenetic protein 6 with exocrine gland dysfunction in patients with sjögren's syndrome and in mice. Arthritis Rheumatol (2013) 65(12):3228–38. doi: 10.1002/art.38123
153. Mo YQ, Nakamura H, Tanaka T, Odani T, Perez P, Ji Y, et al. Lysosomal exocytosis of HSP70 stimulates monocytic BMP6 expression in sjögren's syndrome. J Clin Invest (2022) 132(6):e152780. doi: 10.1172/JCI152780
154. Atri C, Guerfali FZ, Laouini D. Role of human macrophage polarization in inflammation during infectious diseases. Int J Mol Sci (2018) 19:1801. doi: 10.3390/ijms19061801
155. Saito I, Terauchi K, Shimuta M, Nishiimura S, Yoshino K, Takeuchi T, et al. Expression of cell adhesion molecules in the salivary and lacrimal glands of sjögren's syndrome. J Clin Lab Anal (1993) 7:180–7. doi: 10.1002/jcla.1860070309
156. Dos Santos HT, Nam K, Maslow F, Trump B, Baker OJ. Specialized pro-resolving receptors are expressed in salivary glands with sjögren's syndrome. Ann Diagn Pathol (2022) 56:151865. doi: 10.1016/j.anndiagpath.2021.151865
157. Odusanwo O, Chinthamani S, McCall A, Duffey ME, Baker OJ. Resolvin D1 prevents TNF-α- mediated disruption of salivary epithelial formation. Am J Physiol Cell Physiol (2012) 302:C1331– 45. doi: 10.1152/ajpcell.00207.2011
158. Parashar K, Schulte F, Hardt M, Baker OJ. Sex-mediated elevation of the specialized pro-resolving lipid mediator levels in a sjögren's syndrome mouse model. FASEB J (2020) 34(6):7733–44. doi: 10.1096/fj.201902196R
159. Barrera MJ, Bahamondes V, Sepúlveda D, Quest AF, Castro I, Cortés J, et al. Sjögren's syndrome and the epithelial target: A comprehensive review, J. Autoimmun (2013) 42:7–18. doi: 10.1016/j.jaut.2013.02.001
160. Mei M, Qu LH, Cong X, Zhang Y, Xiang RL, Yu GY, et al. CTRP3 promotes TNF-α-induced apoptosis and barrier dysfunction in salivary epithelial cells. Cell Signal (2021) 85:110042. doi: 10.1016/j.cellsig.2021.110042
161. Qu LH, Hong X, Zhang Y, Cong X, Xiang RL, Mei M, et al. C1q/tumor necrosis factor-related protein-6 attenuates TNF-α-induced apoptosis in salivary acinar cells via AMPK/SIRT1-modulated miR-34a-5p expression. J Cell Physiol (2021) 236(8):5785–800. doi: 10.1002/jcp.30262
162. Keindl M, Davies R, Bergum B, Brun JG, Hammenfors D, Jonsson R, et al. Impaired activation of STAT5 upon IL-2 stimulation in tregs and elevated sIL-2R in sjögren's syndrome. Arthritis Res Ther (2022) 24(1):101. doi: 10.1186/s13075-022-02769-y
163. Benchabane S, Boudjelida A, Toumi R, Belguendouz H, Youinou P, Touil-Boukoffa C. A case for IL-6, IL-17A, and nitric oxide in the pathophysiology of sjögren's syndrome. Int J Immunopathol Pharmacol (2016) 29(3):386–97. doi: 10.1177/0394632016651273
164. Sisto M, Tamma R, Ribatti D, Lisi S. IL-6 contributes to the TGF-β1-Mediated epithelial to mesenchymal transition in human salivary gland epithelial cells. Arch Immunol Ther Exp (Warsz) (2020) 68(5):27. doi: 10.1007/s00005-020-00591-5
165. Bikker A, van Woerkom JM, Kruize AA, Wenting-van Wijk M, de Jager W, Bijlsma JW, et al. Increased expression of interleukin-7 in labial salivary glands of patients with primary sjögren's syndrome correlates with increased inflammation. Arthritis Rheumatol (2010) 62(4):969–77. doi: 10.1002/art.27318
166. Blokland SLM, Hillen MR, Kruize AA, Meller S, Homey B, Smithson GM, et al. Increased CCL25 and T helper cells expressing CCR9 in the salivary glands of patients with primary sjögren's syndrome: Potential new axis in lymphoid neogenesis. Arthritis Rheumatol (2017) 69(10):2038–51. doi: 10.1002/art.40182
167. Katsifis GE, Rekka S, Moutsopoulos NM, Pillemer S, Wahl SM. Systemic and local interleukin-17 and linked cytokines associated with sjögren's syndrome immunopathogenesis. Am J Pathol (2009) 175(3):1167–77. doi: 10.2353/ajpath.2009.090319
168. Xiao F, Du W, Zhu X, Tang Y, Liu L, Huang E, et al. IL-17 drives salivary gland dysfunction via inhibiting TRPC1-mediated calcium movement in sjögren's syndrome. Clin Transl Immunol (2021) 10(4):e1277. doi: 10.1002/cti2.1277
169. Rankin AL, Guay H, Herber D, Bertino SA, Duzanski TA, Carrier Y, et al. IL-21 receptor is required for the systemic accumulation of activated b and T lymphocytes in MRL/MpJ-Fas(lpr/lpr)/J mice. J Immunol (2012) 188(4):1656–67. doi: 10.4049/jimmunol.1003871
170. Tambuwala MM, Cummins EP, Lenihan CR, Kiss J, Stauch M, Scholz CC, et al. Loss of prolyl hydroxylase-1 protects against colitis through reduced epithelial cell apoptosis and increased barrier function. Gastroenterology (2010) 139:2093–101. doi: 10.1053/j.gastro.2010.06.068
171. Cummins EP, Seeballuck F, Keely SJ, Mangan NE, Callanan JJ, Fallon PG, et al. The hydroxylase inhibitor dimethyloxalylglycine is protective in a murine model of colitis. Gastroenterology (2008) 134:156–165.e1. doi: 10.1053/j.gastro.2007.10.012
172. Robinson A, Keely S, Karhausen J, Gerich ME, Furuta GT, Colgan SP, et al. Mucosal protection by hypoxia-inducible factor prolyl hydroxylase inhibition. Gastroenterology (2008) 134:145–55. doi: 10.1053/j.gastro.2007.09.033
173. Liu NN, Zhao N, Cai N. The effect and mechanism of celecoxib in hypoxia-induced survivin up-regulation in HUVECs. Cell Physiol Biochem (2015) 37(3):991–1001. doi: 10.1159/000430225
174. Sun YZ, Cai N, Liu NN. Celecoxib down-regulates the hypoxia-induced expression of HIF-1α and VEGF through the PI3K/AKT pathway in retinal pigment epithelial cells. Cell Physiol Biochem (2017) 44(4):1640–50. doi: 10.1159/000485764
175. Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, et al. Senolytics improve physical function and increase lifespan in old age. Nat Med (2018) 24:1246–56. doi: 10.1038/s41591-018-0092-9
176. Fuhrmann-Stroissnigg H, Ling YY, Zhao J, McGowan SJ, Zhu Y, Brooks RW, et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat Commun (2017) 8:422. doi: 10.1038/s41467-017-00314-z
177. Peng X, Wu Y, Brouwer U, van Vliet T, Wang B, Demaria M, et al. Cellular senescence contributes to radiation-induced hyposalivation by affecting the stem/progenitor cell niche. Cell Death Dis (2020) 11(10):854. doi: 10.1038/s41419-020-03074-9
178. Hai B, Zhao Q, Deveau MA, Liu F. Delivery of sonic hedgehog gene repressed irradiation-induced cellular senescence in salivary glands by promoting DNA repair and reducing oxidative stress. Theranostics (2018) 8(4):1159–67. doi: 10.7150/thno.23373
179. Carubbi F, Cipriani P, Di Benedetto P, Ruscitti P, Alunno A, Gerli R, et al. Persistence of focal lymphocytic sialadenitis in patients with primary sjögren's syndrome treated with rituximab: A possible role for glandular BAFF. Clin Exp Rheumatol (2016) 34(6):1123–4.
180. Marmary Y, Adar R, Gaska S, Wygoda A, Maly A, Cohen J, et al. Radiation-induced loss of salivary gland function is driven by cellular senescence and prevented by IL6 modulation. Cancer Res (2016) 765:1170–80:2846. doi: 10.1158/0008-5472.CAN-15-1671
181. Felten R, Devauchelle-Pensec V, Seror R, Duffau P, Saadoun D, Hachulla E, et al. Interleukin 6 receptor inhibition in primary sjögren syndrome: A multicentre double-blind randomised placebo-controlled trial. Ann Rheum. Dis (2021) 80(3):329–38. doi: 10.1136/annrheumdis-2020-218467
182. Justet A, Ottaviani S, Dieudé P, Taillé C. Tocilizumab for refractory organising pneumonia associated with sjögren's disease. BMJ Case Rep (2015) 2015:bcr2014209076. doi: 10.1136/bcr-2014-209076
183. Ishikawa Y, Hattori K, Ishikawa J, Fujiwara M, Kita Y. Refractory sjögren's syndrome myelopathy successfully treated with subcutaneous tocilizumab: A case report. Med (Baltimore) (2019) 98(27):e16285. doi: 10.1097/MD.0000000000016285
184. Xu Y, Zhu Q, Song J, Liu H, Miao Y, Yang F, et al. Regulatory effect of iguratimod on the balance of Th subsets and inhibition of inflammatory cytokines in patients with rheumatoid arthritis. Mediators Inflamm (2015) 2015:356040. doi: 10.1155/2015/356040
185. Shao Q, Wang S, Jiang H, Liu L. Efficacy and safety of iguratimod on patients with primary sjögren's syndrome: A randomized, placebo-controlled clinical trial. Scand J Rheumatol (2021) 50(2):143–52. doi: 10.1080/03009742.2020.1809701
186. Qi X, Tian Yu, Sun C, Gao L, Guo H. Effects of iramod on submandibular gland inflammation and NF-κB signaling pathway in sjögren's syndrome mice. Chin J Immunol (2021) 37(08):907–11. doi: 10.3969/j.issn.1000-484X.2021.08.003
187. Carubbi F, Cipriani P, Marrelli A, Benedetto P, Ruscitti P, Berardicurti O, et al. Effificacy and safety of rituximab treatment in early primary sjögren's syndrome: A prospective, multi-center, follow-up study. Arthritis Res Ther (2013) 15(5):R172. doi: 10.1186/ar4359
188. Gandolfo S, De Vita S. Double anti-B cell and anti-BAFF targeting for the treatment of primary Sj�gren's syndrome. Clin Exp Rheumatol (2019) 118:199–208.
189. Gong Q, Ou Q, Ye S, Lee WP, Cornelius J, Diehl L, et al. Importance of cellular microenvironment and circulatory dynamics in b cell immunotherapy. J Immunol (2005) 174:817–26. doi: 10.4049/jimmunol.174.2.817
190. Toubi E, Shoenfeld Y. The role of CD40–CD154 interactions in autoimmunity and the benefit of disrupting this pathway. Autoimmunity (2004) 37(6-7):457–64. doi: 10.1080/08916930400002386
191. Dörner T, Posch MG, Li Y, Petricoul O, Cabanski M, Milojevic JM, et al. Treatment of primary sjögren's syndrome with ianalumab (VAY736) targeting b cells by BAFF receptor blockade coupled with enhanced, antibody-dependent cellular cytotoxicity. Ann Rheum. Dis (2019) 78(5):641–7. doi: 10.1136/annrheumdis-2018-214720
192. Xing Y, Li B, He J, Hua H. Labial gland mesenchymal stem cell derived exosomes-mediated miRNA-125b attenuates experimental sjögren's syndrome by targeting PRDM1 and suppressing plasma cells. Front Immunol (2022) 13:871096. doi: 10.3389/fimmu.2022.871096
193. Kronke M, WJ L, Depper JM. Cyclosporin a inhibits T-cell growth factor gene expression at the level of mRNA transcription. Proc Natl Acad Sci USA (1984) 81:5214–8. doi: 10.1073/pnas.81.16.5214
194. Power WJ, Mullaney P, Farrell M, Collum LM. Effect of topical cyclosporin a on conjunctival T cells in patients with secondary sjögren's syndrome. Cornea (1993) 12:507–11. doi: 10.1097/00003226-199311000-00008
195. De Vita S, Quartuccio L, Seror R, Salvin S, Ravaud P, Fabris M, et al. Efficacy and safety of belimumab given for 12 months in primary sjögren's syndrome: The BELISS open-label phase II study. Rheumatol (Oxford) (2015) 54:2249–56. doi: 10.1093/rheumatology/kev257
196. Goldman FD, Gilman AL, Hollenback C, Kato RM, Premack BA, Rawlings DJ, et al. Hydroxychloroquine inhibits calcium signals in T cells: A new mechanism to explain its immunomodulatory properties. Blood (2000) 95:3460–6. doi: 10.1182/blood.V95.11.3460
197. Tsuboi H, Matsumoto I, Hagiwara S, Hirota T, Takahashi H, Ebe H, et al. Efficacy and safety of abatacept for patients with sjögren's syndrome associated with rheumatoid arthritis: Rheumatoid arthritis with orencia trial toward sjögren's syndrome endocrinopathy (ROSE) trial–an open-label, one-year, prospective study–interim analysis of 32 patients for 24 weeks. Mod. Rheumatol (2015) 25:187–93. doi: 10.3109/14397595.2014.951144
198. de Wolff L, van Nimwegen JF, Mossel E, van Zuiden GS, Stel AJ, Majoor KI, et al. Long-term abatacept treatment for 48 weeks in patients with primary sjögren's syndrome: The open-label extension phase of the ASAP-III trial. Semin Arthritis Rheumatol (2022) 53:151955. doi: 10.1016/j.semarthrit.2022.151955
199. Haacke EA, van der Vegt B, Meiners PM, Vissink A, Spijkervet FK, Bootsma H, et al. Abatacept treatment of patients with primary sjögren's syndrome results in a decrease of germinal centres in salivary gland tissue, clin. Exp Rheumatol (2017) 35:317–20.
200. Verstappen GM, Meiners PM, Corneth OBJ, Visser A, Arends S, Abdulahad WH, et al. Attenuation of follicular helper T cell-dependent b cell hyperactivity by abatacept treatment in primary sjögren's syndrome, arthritis rheumatol. Arthritis Rheumatol (2017) 69:1850–61. doi: 10.1002/art.40165
201. Meiners PM, Vissink A, Kroese FGM, Spijkervet FK, Smitt-Kamminga NS, Abdulahad WH, et al. Abatacept treatment reduces disease activity in early primary sjögren's syndrome (open-label proof of concept ASAP study). Ann Rheum Dis (2014) 73:1393–6. doi: 10.1136/annrheumdis-2013-204653
202. Pontarini E, Verstappen GM, Grigoriadou S, Kroese FGM, Bootsma H, Bombardieri M, et al. Blocking T cell co-stimulation in primary Sjögren's syndrome: Rationale, clinical efficacy and modulation of peripheral and salivary gland biomarkers. Clin Exp Rheumatol (2020) 126:222–7.
203. Gupta S, Li D, Ostrov DA, Nguyen CQ. Blocking IAg7 class II major histocompatibility complex by drug-like small molecules alleviated sjögren's syndrome in NOD mice. Life Sci (2022) 288:120182. doi: 10.1016/j.lfs.2021.120182
204. Fox PC, Datiles M, Atkinson JC, Macynski AA, Scott J, Fletcher D, et al. Prednisone and piroxicam for treatment of primary sjögren's syndrome. Clin Exp Rheumatol (1993) 11:149–56.
205. Pijpe J, Kalk WWI, Bootsma H, Spijkervet FK, Kallenberg CG, Vissink A, et al. Progression of salivary gland dysfunction in patients with sjögren's syndrome. Ann Rheum Dis (2006) 66:107–12. doi: 10.1136/ard.2006.052647
206. Bruneau JM, Yea CM, Spinella-Jaegle SPurification of human dihydroorotate dehydrogenase and its inhibition by A77 1726, the active metabolite of leflunomide. Biochem J (1998) 336:299–303. doi: 10.1042/bj3360299
207. Manna SK, Aggarwal BB. Immunosuppressive leflunomide metabolite (A77 1726) blocks TNF-dependent nuclear factor-kappa b activation and gene expression. J Immunol (1999) 162:2095–102.
208. van Woerkom JM, Kruize AA, Geenen R, van Roon EN, Goldschmeding R, Verstappen SM, et al. Safety and efficacy of leflunomide in primary sjögren's syndrome: A phase II pilot study. Ann Rheum. Dis (2007) 66:1026–32.
209. Fox RI, Dixon R, Guarrasi V, Krubel S. Treatment of primary sjögren's syndrome with hydroxychloroquine: A retrospective, open-label study. Lupus (1996) 5:31–6. doi: 10.1177/0961203396005001081
210. Gottenberg JE, Ravaud P, Puéchal X, Le Guern V, Sibilia J, Goeb V, et al. Effects of hydroxychloroquine on symptomatic improvement in primary sjögren syndrome: The JOQUER randomized clinical trial. JAMA (2014) 312:249–58. doi: 10.1001/jama.2014.7682
211. Zhou YB, Yuan X, Wang QK, Zhang H, Wang GS, Li XP, et al. Injection of CD40 DNA vaccine ameliorates the autoimmune pathology of non-obese diabetic mice with sjögren's syndrome. Immunol Lett (2020) 226:62–70. doi: 10.1016/j.imlet.2020.07.005
212. Espié P, He Y, Koo P, Sickert D, Dupuy C, Chokoté E, et al. First-in-human clinical trial to assess pharmacokinetics, pharmacodynamics, safety, and tolerability of iscalimab, an anti-CD40 monoclonal antibody. Am J Transplant (2020) 20(2):463–73. doi: 10.1111/ajt.15661
213. Zhou J, Yu Q. Anti-IL-7 receptor-α treatment ameliorates newly established sjögren's-like exocrinopathy in non-obese diabetic mice. Biochim Biophys Acta Mol Basis Dis (2018) 1864(7):2438–47. doi: 10.1016/j.bbadis.2018.04.010
Keywords: immune microenvironment, salivary gland, Sjögren’s syndrome, senility, chronic inflammation, hypoxia
Citation: Tan Z, Wang L and Li X (2022) Composition and regulation of the immune microenvironment of salivary gland in Sjögren’s syndrome. Front. Immunol. 13:967304. doi: 10.3389/fimmu.2022.967304
Received: 12 June 2022; Accepted: 23 August 2022;
Published: 13 September 2022.
Edited by:
Jing Ni, Anhui Medical University, ChinaReviewed by:
Huaxun Wu, Anhui Medical University, ChinaCopyright © 2022 Tan, Wang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaomei Li, bGl4aWFvbWVpQHVzdGMuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.