
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 12 August 2022
Sec. Inflammation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.967193
This article is part of the Research TopicInsights in Inflammation: 2022View all 13 articles
 Lin Wang1,2
Lin Wang1,2 Chengqi He1,2*
Chengqi He1,2*Macrophages are the most abundant immune cells within the synovial joints, and also the main innate immune effector cells triggering the initial inflammatory responses in the pathological process of osteoarthritis (OA). The transition of synovial macrophages between pro-inflammatory and anti-inflammatory phenotypes can play a key role in building the intra-articular microenvironment. The pro-inflammatory cascade induced by TNF-α, IL-1β, and IL-6 is closely related to M1 macrophages, resulting in the production of pro-chondrolytic mediators. However, IL-10, IL1RA, CCL-18, IGF, and TGF are closely related to M2 macrophages, leading to the protection of cartilage and the promoted regeneration. The inhibition of NF-κB signaling pathway is central in OA treatment via controlling inflammatory responses in macrophages, while the nuclear factor erythroid 2-related factor 2 (Nrf2) signaling pathway appears not to attract widespread attention in the field. Nrf2 is a transcription factor encoding a large number of antioxidant enzymes. The activation of Nrf2 can have antioxidant and anti-inflammatory effects, which can also have complex crosstalk with NF-κB signaling pathway. The activation of Nrf2 can inhibit the M1 polarization and promote the M2 polarization through potential signaling transductions including TGF-β/SMAD, TLR/NF-κB, and JAK/STAT signaling pathways, with the regulation or cooperation of Notch, NLRP3, PI3K/Akt, and MAPK signaling. And the expression of heme oxygenase-1 (HO-1) and the negative regulation of Nrf2 for NF-κB can be the main mechanisms for promotion. Furthermore, the candidates of OA treatment by activating Nrf2 to promote M2 phenotype macrophages in OA are also reviewed in this work, such as itaconate and fumarate derivatives, curcumin, quercetin, melatonin, mesenchymal stem cells, and low-intensity pulsed ultrasound.
Osteoarthritis (OA) is a highly prevalent musculoskeletal disorder characterized by pain, deformity, and functional deficits. Globally, it is a major medical and socioeconomic burden (1). Histologically, OA is characterized by cartilage degeneration, synovial lining thickening, and subchondral sclerosis (2). In pathophysiology, a low-grade, chronic inflammation predominantly leads to synovial joint deterioration as a result of an innate immune response (3). In vivo imaging evidence in patients with OA indicates a crucial role for activated macrophages, which is linked to its severity. Moreover, the disruption of macrophage transition may contribute to chronic and irreversible inflammatory changes in OA-affected joints (4). The activation of macrophages with heterogeneous phenotypes, which can exert pro- and anti-inflammatory effects on articular tissues during OA, has been suggested as a potential therapeutic target (5).
The macrophages within synovial joints include resident and interstitial subsets. The interstitial population of recruited monocyte-derived macrophages could exert functions of joint inflammation. While, a tight-junction-mediated shield composing of the subset of epithelial-like CX3CR1+ tissue-resident macrophages could restrict the inflammatory response (6, 7). OA mainly results from innate immune response induced by macrophages that are marked by CD14 and F4/80 as general surface antigens (8, 9). Furthermore, the plastic heterogeneous phenotypes of macrophages include classically activated pro-inflammatory phenotypes (CD80, CD86, and CD11b as M1 surface markers) and alternatively activated anti-inflammatory phenotypes (CD163, and CD206 as M2 surface markers) (10–12). There are several subtypes of M2 macrophages, including M2a marked by CD206, M2b marked by CD86, M2c marked by CD163 (13, 14), and M2d marked by CD68 (15–17). Induced by IL-4 and IL-13, M2a plays a vital role in anti-inflammation response and wound-healing via up-regulating the expression of IL-1RA, IL-10, CCL-18, and TGF-β. M2b is the immune regulator between M1 and M2a, which serves in both pro-and anti-inflammation responses. Besides, IL-10, TGF-β, or glucocorticoids can induce M2c activation, which causes the secretion of IL-10, CCL-18, and TGF-β, and M2c also has a powerful phagocytosis function (18, 19). While, M2d, also called tumor-associated macrophages (TAMs), plays a vital role in potent immunosuppression, angiogenesis, wound healing, and cancer metastasis (15, 16). However, most experimental therapies or mechanisms for OA use the balance between M1 and M2 as a key point (5, 20–22). In this review, the polarization state of macrophages will be dichotomized without discussing specific subtypes of M2. In addition, synovia in OA patients has a substantial proportion of M1 macrophages, which is consistent with CD14 and CD163 expression levels (23). Macrophage phenotypes are influenced by intracellular redox metabolism, as evidenced by increasing studies, leading to metabolic reprogramming from glycolysis in M1 to oxidative phosphorylation (OxPhos) in M2 (5, 24–26).
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a transcription factor expressed in most tissues and cells at a low level in the cytoplasm under homeostatic conditions through binding to Kelch-like ECH-associated protein 1 (KEAP1). Nrf2 is released in response to stress signals sensed by KEAP1, translocates to the nucleus, accumulates, and binds to antioxidant response elements (AREs) of target gene promoters (27). Finally, heme oxygenase-1 (HO-1), and glutathione S-transferase (GST) are transcribed, and reactive oxygen species (ROS) removal systems are initiated to protect cells from oxidative stress-induced damages and maintain redox homeostasis (28). Besides, there is increasing evidence that quite different metabolic characteristics and inflammation phenotypes between M1 and M2 macrophages are highly dependent on Nrf2. The negative regulation of Nrf2-related signalings for other transcription factors, such as nuclear factor-κB (NF-κB), may shed light on the link between defense against oxidative stress and reducing inflammation through Nrf2 signaling (24, 29). For example, cyclooxygenase-2 (COX-2), and hypoxia-inducible factor-1α (HIF-1α), which are closely related to the M1 phenotype, could be suppressed via the activation of Nrf2 signaling (30). Moreover, the activation of Nrf2 in macrophage could increase the levels of cysteine and glutathione (GSH), by regulating the transporter between cysteine and glutamate, and the GSH-synthesizing enzyme (31, 32). GSH could suppress ROS as a major cellular antioxidant via activating HO-1. It has also been reported that the accumulation of GSH could induce an increase in inflammatory factors including NO, IL-1β, IL-4, IL-10, TNF-α, and PGE2, which are related to the M1 phenotype (33). However, in tubular injury resulting from oxidative stress and inflammatory response, the deletion of ROS could promote M2 polarization (34). Similarly, inflammatory response and oxidative stress could be reduced in the cardiac injury induced by LPS, during which process the Nrf2/HO-1 pathway is activated, and the GSH is accumulated (35). Furthermore, it has been reported that the activated Nrf2 could suppress IL-1β without NF-κB or GSH in alveolar macrophages (36). In summary, the relationship is complicated, between the macrophage polarization and metabolic adaptation of macrophages upon inflammatory response and oxidative stress, including OA. As potential mechanisms underlying the protective effects of Nrf2 activation in macrophages, the Nrf2/HO-1 signaling pathway, and the negative regulation of NF-κB signaling will be discussed later.
Furthermore, it has been shown that there is extensive crosstalk between transcriptional pathways involving Nrf2 and NF-κB during oxidative stress and inflammation (29, 32). Generally, Nrf2 signaling negatively regulates NF-κB signaling in oxidative stress and inflammatory response, especially NF-κB (P65) pathway (37). That is, through Nrf2 transcriptional activation, the redox status and metabolism of macrophages changes, resulting in an anti-inflammatory phenotype (38). Stimulation inducing inflammatory M1 can simultaneously initiate NF-κB-dependent transcriptional pathway inducing the secretion of inflammatory factors quickly, and initiate Nrf2-dependent transcriptional pathway at the same time to cytoprotective response slowly (39). In summary, the action of Nrf2 has a potential role in preventing M1 polarization, and then promoting chondral protection and inhibiting OA progress. For example, in OA, Ca2+ influx evoked by transient receptor potential vanilloid 1 (TRPV1) mediated inhibition of M1 macrophage polarization through the phosphorylation of calmodulin-dependent protein kinase II (CaMKII), while the specific inhibitor of Nrf2 counteracted the anti-inflammatory effect (40).
However, the role of Nrf2 in macrophage reprogramming for OA treatment is still unclear to a large extent. Therefore, this review synthesizes evidence of macrophage reprogramming induced Nrf2 inhibition or activation in the progression or treatment of OA.
The intra-articular microenvironment is characterized by an inflammatory infiltrate largely composed of synovial macrophages. Inflammatory macrophages are believed to be responsible for the presence of OA (23). Damage-associated molecular patterns (DAMPs) are molecules or fragments produced by initial harmful factors that can trigger innate immunity. The DAMPs could activate pattern-recognition receptors for macrophage activation, for example, the toll-like receptor (TLR) 4, and the ligands could be cartilage matrix fragments, or plasma proteins into the articular cavity in OA (41, 42). In contrast, the production of macrophage-derived pro-inflammatory cytokines, such as TNF-α and IL-1β, was greatly reduced by depleting CD14-positive synovial macrophages specifically from OA synovial cells (43).
M1 macrophages tend to secrete pro-inflammatory cytokines including TNF-α and L-1β (44). A series of events triggered by TNF-α and IL-1β can cause cartilage degeneration, where chondrocyte death and cartilage matrix degradation are accelerated while synthesis and regeneration are inhibited (45). Cartilage mainly consists of chondrocytes and extracellular matrix (ECM). Apoptosis of chondrocytes and degeneration of aggrecan (ACAN) and type II collagen (COL2) in ECM are prominent pathological changes in OA (46). It is believed that autocrine TNF-α and IL-1β trigger the pro-inflammatory events in chondrocytes and the catabolic cascades in fibroblast synoviocytes through NF-κB signaling, resulting in the production of IL-6, NO, and prostaglandin E2 (PGE2) (3, 47). The release of IL-6 from macrophages induced by IL-1β can stimulate STAT3 signaling in macrophages, enhancing inflammation responses (48). As a result of IL-6 stimulation, chondrocytes and synovial fibroblasts produce PGE2 and collagenase (49). The high level of NO can inhibit the synthesis of ECM and enhance the activity of matrix metalloproteases (MMPs) (45). By degrading collagen and digesting matrix proteins, MMP1, 3 and 13 can result in skeletal cartilage absorption. The metabolic product of activated COX is arachidonic acid, the substrate of PGE2 biosynthesis (50). PGE2 can also stimulate the release of IL-6 through activating NF-κB pathways (49).
The increase of expression of one of the specific receptor of TNF-α, called TNF receptor I (TNFRI or p55), has been found on OA chondrocytes and synovial fibroblasts (47). In fact, TNF-α plays a central role in the intra-inflammation cascades of OA. TNF-α can break down the cartilage by inhibiting the synthesis of proteoglycan and COL2, and also promoting the apoptosis of chondrocytes. The death domain (DD) in the TNF receptor superfamily is a cytosolic domain and a cysteine-rich extracellular domain. The extrinsic apoptosis pathway is governed by TNF-α, which binds to and interacts with DD, acting on downstream caspases, ultimately leading to apoptosis (45). Similarly, the expression of the specific receptors of IL-1β, called IL-1 receptor type I (IL-1RI), has been found to be increased in human chondrocytes and synovial fibroblasts affected by OA (47). Furthermore, NO can decrease the level of IL-1RA (the antagonist of IL-1R) leading to an increase in IL-1 production (51). IL-1β can also induce apoptosis of chondrocytes relying on endogenous NO in reverse (45). In synovial fibroblasts, IL-1β activates NF-κB (P65) and promotes transcription of IL-6 and PGE2 (52). In chondrocytes, IL-1β can up-regulate the expression of IL-6 through phosphorylating STAT1 and STAT3 (45).
MMPs are a superfamily of proteases that can remodel and degrade ECM in connective tissues. By stimulating the release of MMP1, MMP3, and MMP13 in chondrocytes, TNF-α and IL-1β can affect the synthesis of proteoglycans, connexins, and type II collagen (47). MMP1 and MMP13 are collagenases, while MMP3 is matrix lyases. Apart from that, ADAMTS-4 and ADAMTS-5 belong to the disintegrin and metalloproteinase with thrombospondin motifs family (ADAMTS), which destroys ECM independently from MMPs (53). In chondrocytes, TNF-α and IL-1β can induce an increase in the release of ADAMTS-4 and ADAMTS-5 (45, 54). In addition, direct evidence shows the M1 inflammatory secretion due to interferon-γand TNF-α inhibits chondrogenic differentiation and cartilage repair by up-regulating IL-1β, IL-6, NO, MMP13, and ADAMTS5, and down-regulating ACAN and COL2 (55). To sum up, DAMPs-induced activation of M1 macrophages mainly dependent on NF-κB signaling mediates intra-articular inflammation and cartilage degeneration in OA.
The failure in the appropriate proportion of M1 and M2 phenotypes can be the main cause of OA-related low-grade inflammation (56). After all, M2 macrophages are anti-inflammatory and help to repair cartilage in contrast to M1 macrophages. In particular, M2 macrophages can secrete anti-inflammatory factors including IL-10, IL-1RA, chemokine (CeC motif) ligand (CCL18), and pro-chondrogenic mediators including transforming growth factor β (TGF-β) and insulin-like growth factor (IGF) (57). M2 phenotype can be activated by IL-4, IL-10, IL-13, TGF-β, and CCL18, which lead to a positive feedback loop to resolute inflammation (58, 59). Besides, M2 can also regulate collagen turnover pathways in cartilage to promote collagen remodeling (60). The secretion of M2 type can promote cartilage repair by up-regulating COL2 and glycosaminoglycan, inhibiting MMP13, and inhibiting apoptosis of chondrocytes (57).
The previous evidence indicates that IL-10 can protect and repair cartilage, and contributes to the regenerative microenvironment. In patients with OA, IL-10 reduces the specific receptors for TNF-α, and the effects of TNF-α on the fibroblasts by down-regulating PEG2, COX-2, and PLA2 (61). By modulating mitochondrial apoptotic pathways, IL-10 can also inhibit chondrocyte apoptosis by reducing caspase activity and the Bax/Bcl-2 ratio (62). Besides, IL-10 can promote the repairing of chondrocytes and ECM. After IL-10 is administered to compressed articular cartilage in vitro, the cell death of chondrocytes, the release of glycosaminoglycans, NO, and molecules that promote ECM degradation and inhibit its syntheses, such as MMP3, MMP13, ADAMTS-4, and inducible nitric oxide synthase (iNOS) are significantly reduced. Moreover, the subsequent study confirmed that the ECM protective effects of IL-10 can be time-dependent (63). Moreover, the overexpression of IL-10 can antagonize the characteristics of cartilage catabolism (MMP3 and MMP13) and the down-regulation of COL2 gene expression induced by TNF-α (64). In addition, the conditioned medium of M1 macrophages decreased the expression of COL2 and ACAN genes in mesenchymal stem cells (MSCs), which are genes associated with chondrogenic differentiation, while M2 macrophages did not exhibit similar inhibition (65).
Furthermore, the increase in IL-10 and IL-1RA can be induced by IL-4, causing the M2 activation in macrophages. The increased IL-10 and IL-1RA, as an anti-inflammatory response, also contribute to responding to the transcription of TNF-α induced by IFN-γ in macrophages. However, in OA synovial fluid, the expression of both is inhibited (66). IL-1RA can be produced by chondrocytes, monocytes, and fibroblasts, belonging to the IL-1 family, and competes with IL-1β to combine with IL-1R type I and II, without triggering the IL-1β-relative downstream inflammatory responses. Although, the production of endogenous IL-1RA needs to be 10-1000 times that of IL-1β to effectively block the binding of IL-1β (67). Additionally, CCL18 is a T-cell chemokine subset associated with the Th2 adaptive responses to IL-4, IL-10, and IL-13. CCL18 has since been identified as a mediator secreted by M2 and an introducer of M2 type (68). Besides, CCL18 can also stimulate fibroblast proliferation and collagen production independent of TGF-β (69). The role of CCL18 in OA is not extremely beneficial, because it has been seen that the relative higher CCL18 levels in synovial fluid of knee OA patients with more serious pathological structural changes (70). Besides, CCL18 can induce the significant enhancement of MMP-3 in fibroblast-like synoviocytes (71).
IGF-1 is a small polypeptide (~7 kDa) belonging to the growth factor family, of which the structure is related to insulin by 50% sequence homology (72, 73). IGF-1 can inhibit chondrocyte apoptosis induced by IL- 1β in vitro and can reduce synovitis in OA models (74). Furthermore, IGF-1 inhibits the degradation of cartilage ECM by down-regulating MMP-1, MMP-3, IL-1, and TNF-α (75). Notably, IGF-1 can also play a key role in cartilage anabolism by promoting COL2 and ECM proteoglycan synthesis and decreasing MMP13 to protect the cartilage (76). Through NF-κB signaling, IGF-1 can inhibit the pro-catabolism effects of IL-1β on cartilage, inhibit the apoptosis of chondrocytes (marked by caspase-3), and suppress inflammation (77–79). Moreover, it has been observed that IGF-1 promotes chondrogenic differentiation of adipose-derived MSCs through the expression of COL2, ACAN, and SOX9 (80). IGF-1 can positively regulate chondrogenesis in bone marrow-derived MSCs, and the chondroinductive effects of IGF-1 are independent of TGF-β1 (81). Furthermore, the combination of TGF and IGF can promote chondrogenesis in fracture models in vivo (82).
TGF-β family of growth factors and cytokines plays a critical role in skeletogenesis, and can be divided into two major subfamilies, the TGF-β/Activin/Nodal family and the bone morphogenetic protein (BMP) family (83, 84). Signaling by TGF-β1, 2, and 3 is mediated by membrane-bound receptor complexes, which are activated by SMAD proteins intracellularly. Activated type I receptor (also known as ALK5) phosphorylates SMAD2 and SMAD3, which can regulate the expression of target genes related to cartilage anabolism, COL2 for example. However, when TGF-β signals through the ALK1 receptor, phosphorylated SMADs 1/5/8 can be targeted to up-regulate cartilage catabolism genes, like MMP13 (85). The role of TGF-β in OA seems to have two sides, and the administration of TGF-β is equally controversial because the choice between phosphorylation of SMAD2/3 or SMAD5/8 has an unknown mechanism (86).
The conversion of the phenotype of macrophages in OA could play a role in treatment because the secreted cytokines regulate inflammation and cartilage metabolism (Figure 1).
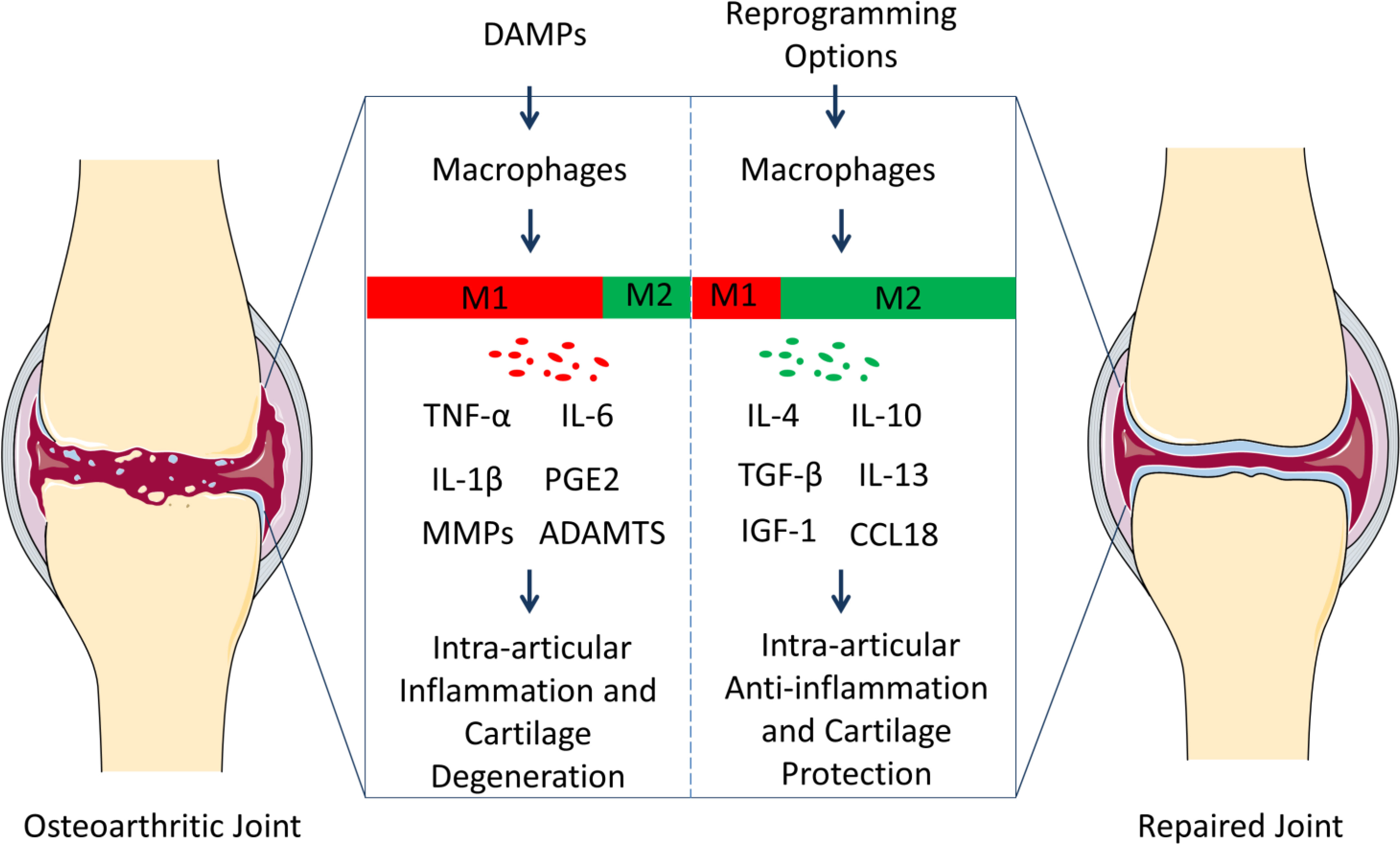
Figure 1 Polarization of macrophages in knee osteoarthritis (OA) pathology and repair. Resident macrophages in the synovium have two general phenotypes, M1 and M2. Macrophages tend to serve as M1 phenotype under the inflammation responses caused by damage-associated molecular patterns (DAMPs). M1 subgroups can contribute to OA progression through releasing inflammatory and degenerative molecules including TNF-α, IL-6, IL-1β, PGE2, MMPs, ADAMTS, which leads to synovitis, the death of chondrocytes, and the degradation of extracellular matrix (ECM). However, macrophages can be reprogrammed, and the M2 population can contribute to OA treatment via up-regulating anti-inflammatory and regenerative molecules (IL-4, IL-10, TGF-β, IL-13, IGF-1, and CCL18). And the microenvironment can be constructed, where the tissues in the osteoarthritic joints can repair. We suggest that reprograming macrophage phenotypes can be therapeutic targets for the prevention and treatment of OA.
The polarization of macrophages dynamically adapts to changes in the microenvironment, and macrophage reprogramming has a complex mechanism. The most studied several pathways relative to reprogramming of macrophages include TGF-β/SMAD, TLR/NF-κB, and JAK/STAT signaling pathways, with the regulation or cooperation of Notch, NLRP3, PI3K/Akt, and MAPK signaling (Figure 2).
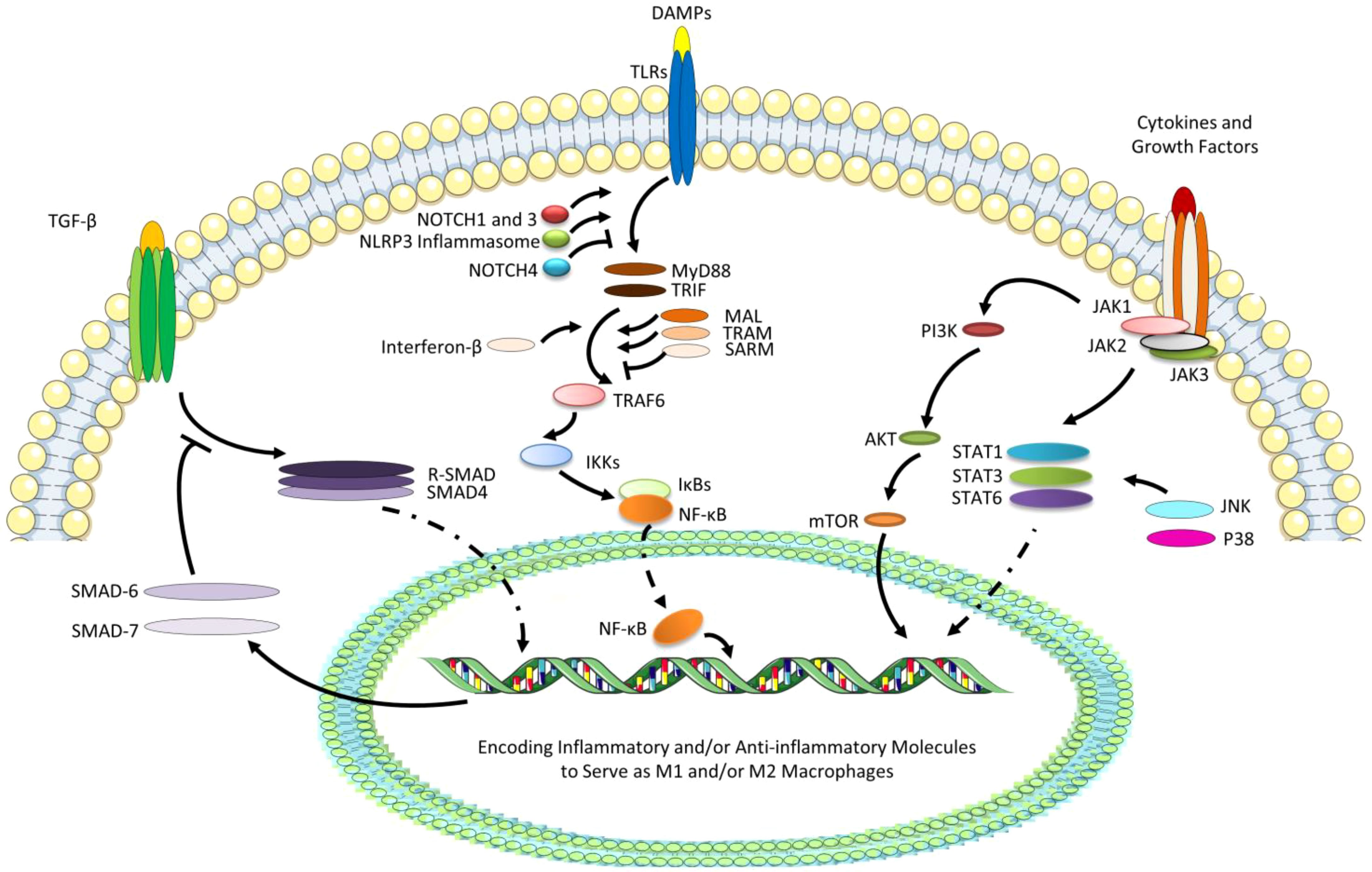
Figure 2 The most studied several pathways relative to reprogramming of macrophages. TGF-βcan interact with phosphorylated type II and type I receptors activating receptor-regulated SMADs (R-SMADs, a heterodimer of SMAD2 and SMAD3) to form the heteromeric trimer with SMAD4. The nuclear translocation of R-SMAD can promote the M2 polarization, while SMAD6 and SMAD7 as the negative regulators of TGF-β signaling transduction are also the target genes. Besides, due to the ligation of DAMPs with TLRs, especially TLR4, MyD88 and TRIF, TLR adaptors containing a Toll/IL-1 receptor (TIR) domain, can bind directly to TLRs and recruit MAL and TRAM, while SARM negatively can regulate the pathways. TIR-domain-containing adaptor-inducing interferon-β (dependent on TRIF) can be recruited and then activates TRAF6, and IκBs (especially IκBα) can degrade leading to the release of NF-κB (P50/65) and its translocation to the nucleus, which can promote the M1 polarization. TLR signaling cascade can also activate NOTCH and NLRP3 signaling, which can regulate pro-inflammatory responses via the regulated transcription of NF-κB. However, NOTCH4 can negatively regulate the TLR/NF-κB signaling. Finally, STAT/JAK signaling pathway start from tyrosine kinase-associated receptors binding various cytokines and growth factors, followed by the phosphorylation of these receptors and JAKs, and the initiation of the phosphorylation and activation of STATs. Generally, the activation of STAT6 signaling can promote M2 phenotype, while STAT1 and STAT3 can perform complicatedly under the effects of various cytokines or growth factors. JAKs can also activate PI3K/AKT signaling pathway, promoting M2 polarization, which can play a collaborative role in JAK/STAT6 signaling pathway. In addition, STATs can also be activated by JNK and P38, which belongs to extracellular signal-regulated kinases (ERKs), promoting M1 polarization.
Collagen, fibronectins, and fibrinoproteins can form complexes with TGF-β family proteins in the ECM (87). Under the effects of MMPs and/or serine proteases (for example, cathepsins), TGF-β proteins are released from the complex of ECM (86). TGF-β intracellular signaling is mediated by SMAD family members after interaction with two membrane receptors (sequentially phosphorylated type II and type I receptors) (88, 89). Receptor-regulated SMADs (R-SMADs) consisting of SMAD2 and SMAD3 are activated via the phosphorylated type I receptor. As a result of the heteromeric trimer with SMAD4, activated R-SMADs can translocate to the nucleus and trigger transcription of target genes (90), including more than 100 transcriptional/signaling regulators, immune modulators, and atherosclerosis-related genes. Dexamethasone-induced M2 polarization is enhanced by TGF-β/SMAD signaling, as type II receptors are elevated, which increases TGF-β response in macrophages. Several genes are associated with the M2 phenotype, including ID3, RGS1, ALOX5AP, TREM1, IL-17RB, JUNB, ELK3, RUNX3, ELL2, TLE3, BCOR, and FOS, and these genes encode functional molecules that are involved in immune responses, inhibiting apoptosis, and maintaining terminal differentiation (91).
In addition, the negative regulators of signal transduction (such as SMAD6 and SMAD7) are also the target genes of TGF-β, which regulates cell homeostasis (92). Of note, A number of E3 ubiquitin ligases, for example, the SMAD ubiquitin regulatory factors and the deubiquitinating enzymes, play a crucial role in recognition and degradation of R-SMADs, SMAD6, SMAD7, and TGF-β receptors. E3 ubiquitin ligases can induce proteasomal degradation via the catalysis of their substrates and self-ubiquitination (93, 94). In contrast, deubiquitinating enzymes can antagonize the ubiquitination of E3 ubiquitin ligases, for example USP15 (95). The TGF-β pathway has been studied in OA since it has the potential to regulate cartilage anabolism (86).
TLRs are prototype pattern-recognition receptors (PRRs) that recognize pathogen-associated molecular patterns (PAMPs) from microorganisms or DAMPs from damaged tissue (96). TLRs are highly expressed on immune cells, including monocytes, macrophages, and dendritic cells, and can also be up-regulated in response to IL-1 or TLR-4 stimulation in other cells. Through ligation of TLRs, endogenous molecules produced during OA, such as glycoprotein, fibronectin, and hyaluronan of ECM components, have been implicated in activating immune responses (97). Also, plasma proteins can be recognized as DAMPs, such as fibrinogen, which signals through TLR4 to induce inflammatory cytokines (42). There is strong evidence that synovial macrophages and chondrocytes express TLR2 and TLR4, while TLR4 senses more DAMPs than TLR2 in both OA and rheumatoid arthritis (RA). TLR4 plays a key role in the DAMP recognition and the signaling promotion, in the forms of homodimerization or heterodimerization (for example TLR4-TLR6 heterodimers), and the co-receptors, or accessory molecules (such as CD14 and CD36) (41).
There are five TLR adaptors containing a Toll/IL-1 receptor (TIR) domain, among which MyD88 and TRIF bind directly to TLRs and recruit MAL and TRAM, respectively, while SARM negatively regulates these pathways. When the MyD88-dependent TLR4 signaling pathway mediated by TRAM is activated, TIR-domain-containing adaptor-inducing interferon-β (dependent on TRIF) can be recruited and then activates a cascade of proteins including TRAF6, which finally induces the degradation of IκBs, and the release of NF-κB (P50/65) and its translocation to the nucleus (98). NF-κB is central to all macrophage TLR-medicated inflammation responses. IκBs inhibit NF-κB in the cytoplasm by forming complexes with NF-κB. Of note, the regulation of TLR/NF-κB signaling is mainly induced by the ubiquitination/deubiquitination of TRAF6 (99). Macrophages tend to show M1 polarization secreting TNF-α, IL-1β, IL-8, and COX-2. It has been seen that the block of TLR4/NF-κB signaling pathway can inhibit M1 polarization (100–102).
TLR signaling cascade can also activate NOTCH signaling, which regulates pro-inflammatory responses via the regulated transcription of NF-κB (103). The Notch gene family encodes evolutionarily highly conserved, single-pass, type I transmembrane heterodimers of 300 kDa called NOTCH receptors (NOTCH1-4 in mammals) which control macrophage activation and polarization via TLRs (104). It has been shown that macrophages up-regulate NOTCH1 upon the activation of TLRs, which implies that NOTCH1 mainly contributes to pro-inflammatory activation (105). In particular, signaling transduction enhances transcriptional activity by phosphorylating and degrading IkBα (106). Similarly, the activation of NOTCH3 by delta-like 4 (Dll4) in macrophages leads to enhanced responses to LPS or IL-1β via TLR4/NF-κB pathway (107). Furthermore, NOTCH1 and NOTCH3 cooperate to control the expression of NF-κB-dependent pro-inflammatory genes following TLR-4 activation, with NOTCH3 dominating for the first hours and NOTCH1 later (104). By contrast, the inhibition of NOTCH1 can induce a decrease in M1 polarization and an increase in M2 polarization (108). However, the activation of TLRs/NF-κB pathway in macrophages is negatively regulated by NOTCH4, depending on the phosphorylation of STAT3 and the weakness of STAT1, which activates STAT3/JAK2 signaling (109). Taken together, the regulation of Notch signaling has roles with two sides in TLR/NF-κB pathway in macrophages.
Furthermore, the NLRP3 inflammasome can be activated by DAMPs/TLRs/NF-κB pathway. NLRP3 consists of microparticles, ATP, cholesterol, and microbial toxins, acting as a key sensor of tissue damage and activating sterile inflammation (110). NLRP3 interacts with adapter apoptosis-associated speck-like protein (ASC), and pro-caspase-1 is recruited as an effector, resulting in the formation of NLRP3 inflammasome in the cytosol (111). Different from NOTCH, the role of NLRP3 signaling seems to be pro-inflammation as its effects on processing interleukin precursors (such as pro-IL-1β and pro-IL-18) into mature and secreted interleukin forms (112).
Although the TLR4/NF-κB pathway in chondrocytes has been emphasized as it induces cartilage catabolism (41, 113), the signaling pathway in macrophages has also been targeted. Specifically, chondroitin sulphate inhibits NF-κB and IL-1β secretion from macrophages through inhibition of TLR4 and DAMP interactions (114). On the contrary, lumican, a glycoprotein in adult articular cartilage, has been shown to be up-regulated in OA, and induce the inflammation related to macrophages via TLR4 pathway (115).
Signal transducer and activator of transcriptions (STATs) including STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B and STAT6 are latent in cytoplasm, and can be phosphorylated and activated via tyrosine phosphorylation and dimerization, finally leading to translocation to the nucleus, binding to promoter sequences and the activation of transcription. The transformation of STATs from latent to active is dependent on Janus kinases (JAKs), which belong to the family of tyrosine kinases (TYKs). JAK1, JAK2, JAK3, and TYK2 have been indicated as JAKs, which can act as tyrosine kinase and bind tyrosine kinase-associated receptors intracellularly. Tyrosine kinase-associated receptors can bind various cytokines and growth factors, followed by the phosphorylation of these receptors and JAKs, and the initiation of the phosphorylation and activation of STATs (116–118).
In macrophages, IL-13 and IL-4 can activate the M2 phenotype by activating JAK2/STAT3 signaling, where IL-4 phosphorylates STAT3 and STAT6, as well as up-regulating DNA binding activity of STAT3, and IL-13 initiates Tyk2 to cascade STAT1 and STAT6, and also to increase DNA binding activity of STAT1 (119). Moreover, STAT6 effects have been widely elucidated in M2 polarization induced by IL-4 and IL-13 (120, 121). IL-10 can activate STAT3 via JAK1 and Tyk2 (122). However, LPS and IL-6 can activate the M1 phenotype through JAK2/STAT3 signaling (123, 124). Besides, IFN-γ has an important role in phosphorylation and dimerization of STAT1, leading to M1 phenotype of macrophages (122, 125). Furthermore, STAT1 and STAT3 play antagonistic roles in pro- and anti-inflammation response in macrophages (126), whereas they can also cross-regulate each other in some immune responses, like the additional activation of STAT3 along with the pro-inflammation activation of STAT1 induced by IFN-γ (127).
Besides STATs, JAKs activate the phosphatidylinositol 3-kinase (PI3K) signaling pathway, for example, GM-CSF-induced activation of JAK2 (128). PI3Ks are lipid-signaling kinases that phosphorylate phosphoinositides to form PIP3, 4, and 5 (129, 130). After PI3K activation, 3-phosphoinositide-dependent kinase (PDK1) is recruited and activated. PDK1 phosphorylates and activates protein kinase B (AKT) (131). PI3K activation inhibits macrophage programming into M1, while AKT activation is a critical condition for M2 polarization (132). LPS induces the M1 phenotype in AKT1 ablation, while LPS induces the M2 phenotype in Akt2 ablation (133). The mechanistic target of rapamycin (mTOR) is an evolutionarily conserved PI3K family member, and contributes to the core of the downstream target signaling complexes of PI3K/AKT pathway, called mTORC1 and mTORC2 (130). In LPS-activated M1 polarization, activation of mTORC1 has been demonstrated. Furthermore, mTORC1-mediated feedback inhibition of mTORC2 activity in Akt signaling leads to inhibition of M2 polarization (134, 135). In addition, specific destruction of PI3K/AKT signaling pathway has little effect on JAK/STAT6 signaling, indicating a collaborative role for PI3K/AKT signaling pathway (136).
STATs can also be activated by serine threonine kinases other than JAKs, such as extracellular signal-regulated kinase (ERK) (137). ERK belongs to mitogen-activated protein kinase (MAPK) modules, which also include c-Jun N-terminal kinase (JNK) and P38 (also known as stress-activated protein kinases) (138), and mediate the protein kinase cascades (139). MAPK activation results in nuclear translocation of a number of transcription factors, including activator protein-1 (AP-1), activating transcription factor (ATF)-2, cAMP-responsive element binding protein (CBP), and members of the ETS family (140). In macrophages, the activation of TLR4 is induced by the phosphorylation cascade of JNK, P38, and ERK, leading to the transcription of NF-κB and AP-1, the increase in the expression of TNF-α and IL-6, forming the activation of M1 polarization (141–143). Among these, pro-inflammatory effects are mainly due to the signaling mediated by JNK and p38 (139). However, the activation of ERK1/2 can inhibit the nuclear translocation of p65 subunit of NF-κB, which results in anti-inflammation response (144, 145). Likewise, the activation of CBP/P300 can mediate the serine phosphorylation of STAT6 induced by IL-4, which can give rise to the M2 phenotype (146, 147).
Nrf2 belongs to the Cap’n’collar basic leucine zipper transcription factor family, within which the 605 amino acids act their roles as seven highly conserved functional NRF2-ECH homology (Neh) domains (148). It is the Neh2 domain in Nrf2 closest to the N-terminal that binds KEAP1 and is responsible for stabilizing the cytoplasm and degenerating it through ubiquitination. KEAP1 inhibits Nrf2, resulting in stable Nrf2 localization in the cytosol. A homodimer of KEAP1 can also be a stress sensor, recruiting and adapting the E3 ubiquitin ligase cullin-3 (CUL3). In turn, CUL3 can polyubiquitinate Neh2 lysine residues, finally resulting in degradation by ubiquitin proteasomes (149, 150).
Next, from the N-terminal to the C-terminal, there are Neh4, Neh5, Neh7, Neh6, Neh1, and Neh3, respectively. Neh4, Neh5, and Neh3 are transactivation domains that mediate the interaction of Nrf2 with other coactivators. Neh4 and Neh5 can bind CBP/P300 (151, 152), while Neh3 binds chromodomain-helicase-DNA binding 6 (CHD6), contributing to transcription. However, Neh7 and Neh6 are negative regulatory domains for Nrf2, which can bind a β-transducin repeat-containing protein (β-TrCP) and retinoic X receptor α (RXRα), separately (153, 154). The DNA binding domain of Neh1 is mediated by heterodimerization with transcription factors, such as small musculoaponeurotic fibrosarcoma (sMAF) (155).
In cells exposed to stress or electrophilic agents, the alignment of Nrf2 lysine residues is disrupted by the specific thiol residues, leading to the lysine residues being modified by electrophiles within the weak interaction with KEAP1, preventing ubiquitination, and ultimately releasing Nrf2 (156, 157). Nrf2 can translocate to the nucleus after dissociating from KEAP1. With the accumulation of Nrf2 in the nucleus, it forms a heterodimer with sMAF, which binds ARE, a trans-acting DNA enhancer motif. The Nrf2 binding ARE promotes genes encoding cytoprotective proteins, such as GSH-related enzymes, NAD(P)H dehydrogenase quinone 1 (NQO1), and HO-1, to prevent oxidative stress, electrophilic toxicity, and inflammation, and to maintain mitochondrial function and metabolism (158).
Furthermore, polarized macrophages can be used to target synovial inflammation caused by OA (5, 20). There is evidence that Nrf2 activation can inhibit M1 macrophage polarization in OA, which indicates that Nrf2 has a protective role in OA synovitis (40). To a large extent, however, the mechanism linking Nrf2-activation in macrophages remains unknown in the condition of OA. In addition to the existing macrophage polarization mechanism and the treatment strategy of OA, several possible pathways of Nrf2 activation controlling macrophage reprogramming will be discussed (Figure 3).
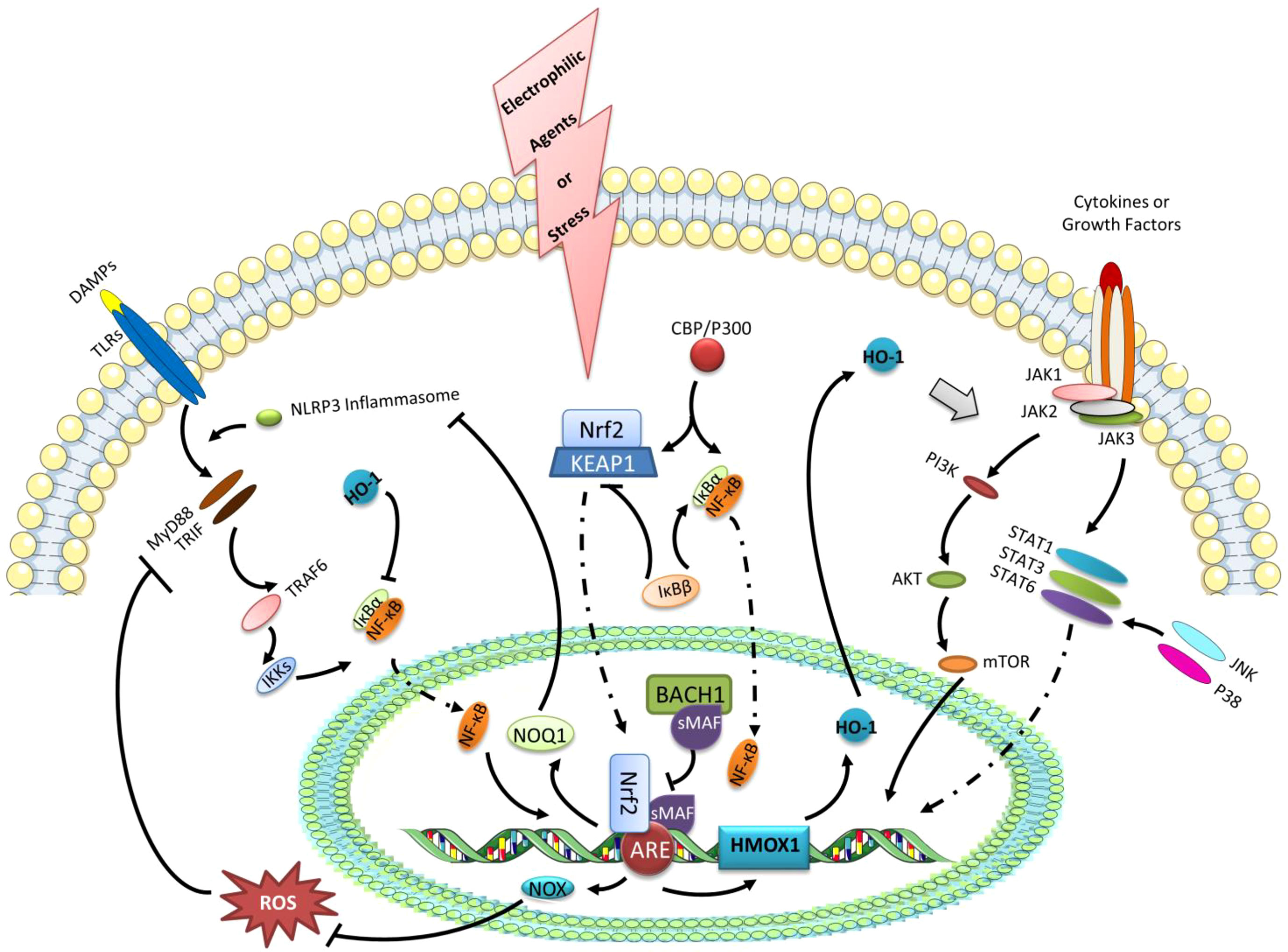
Figure 3 The expression of HO-1 and the negative regulation of NF-κB transcription as the possible pathways of Nrf2 activation controlling macrophage reprogramming. Under the stress or electrophilic agents, Nrf2 can translocate to the nucleus after dissociating from KEAP1. With the accumulation of Nrf2 in the nucleus, it forms a heterodimer with sMAF, which binds ARE, a trans-acting DNA enhancer motif. The Nrf2 binding ARE promotes genes encoding cytoprotective proteins, such as GSH-related enzymes, NAD(P)H dehydrogenase quinone 1 (NQO1), and HO-1. BACH1 is a negative regulator of the inducible HO-1 gene expression, that binds sMAF to inhibit HMOX1 transcription by Nrf2.HO-1 can regulate the polarization of macrophages via the STATs signaling pathway. Besides, HO-1 also has negative effects on the transcription of NF-κB. Furthermore, the up-regulated NOX1/2 and NOQ1 by Nrf2 activation can suppress NF-κB transcription via eliminating ROS and inhibiting NLRP3 inflammasome, respectively. In addition, IκBβ can stabilize KEAP1, while IκBβ can activate IκBα. Likewise, the competitive or antagonistic relationships also exist in the binding of IκBα and KEAP1 to the common activator CBP/P300.
Heme oxygenase (HO) is a microsomal enzyme that degrades heme to carbon monoxide (CO), iron, and biliverdin, which plays a protective role in intracellular detoxification during tissue injury. HO regulates a range of anti-inflammatory, antioxidant, and anti-apoptotic pathways through heme degradation, and HO-1 is responsible for most intracellular detoxification among all the HO members (159, 160). The transcription of HO-1 is regulated by the Nrf2/KEAP1/ARE pathway (161), transcription repressor BACH1, AP-1, and several protein kinase signalings (162). Furthermore, HO-1 expression plays a key role in M2 polarization in macrophages (163). For example, activating HO-1 after myocardial infarction can switch M1 macrophages into M2 macrophages (164).
BACH1 plays an important role in Nrf2/HO-1 transcriptional activity (165). BACH1 is a negative regulator of the inducible HO-1 gene expression, that binds sMAF to inhibit HMOX1 transcription by Nrf2, while losing its function in high concentrations of heme (166). BACH1 and Nrf2 competed with each other to regulate ARE-mediated gene expression (167). BACH1-deficient peritoneal macrophages express HO-1 and Arginase-1, Fizz-1, Ym1, and MRC1, which are M2 macrophage markers (168). Nrf2/KEAP1-BACH1 equilibrium has been identified in pulmonary emphysema patients, whereby high levels of BACH1 and KEAP1 result in reduced stress response, mediated by MAPKs, including JNK and ERK (169, 170). The evidence indicates that HO-1 expression mediated by Nrf2 can be regulated strictly by KACH1 in macrophages, and several signaling cascades can control the balance between both, for example MAPKs (171).
The promoter regions of HO-1 genes contain many AP-1 functional sites (163). AP-1 is made up of heterodimers of members of c-Fos and c-Jun (172). Classical AP-1 is located in the consensus sequence of ARE, and the region of ARE is also the main site for Nrf2 to interact with the HMOX1 promoter (173). A growing body of evidence suggests that inhibition of activated AP-1 helps induce HO-1 expression through Nrf2 (174, 175), which is induced by inhibiting c-Fos (176). It is not clear exactly what role AP-1 plays in Nrf-2 activity, but after all, AP-1’s interaction with c-Jun can activate the transcription of GSH-related enzymes and NQO1 (177).
Anti-inflammatory responses in monocytes/macrophages are mediated by an increase in HO-1. Activation of STAT3 and p38/PI3K signaling is necessary to induce HO-1 expression by LPS and IL-10 in rodents (178). LPS-induced activation of HO-1 in M1 phenotypes induces the production of IL-10, as well as the down-regulation of COX-2, iNOS, TNF-α, and IL-6 (179, 180). Inhibition of M1 phenotype with down-regulation of TNF-α induced by globular adiponectin is dependent on IL-10/STAT3/HO-1 pathway in Kupffer cells (181). However, the increase in IL-10 and the decrease in HO-1 have been observed in macrophages stimulated by LPS in humans (182). M1 polarization can be inhibited by HO-1, although this dichotomy suggests that HO-1 controls IL-10 expression in a complex way. Additionally, HO-1 is known to promote M2 phenotype via anti-inflammation response and cell protection. HO-1 expression induced by sphingosine-1-phosphate in the supernatant derived from apoptotic cells has been indicated to play a vital role in M2 polarization of macrophages, via the STAT1/STAT3 heterodimer (183). Full-length adiponectin induces an M2 phenotype via IL-4/STAT6/HO-1, with a decrease in macrophage sensitivity to stimulation by TLR4 ligands, and an increase in M2 markers (184).
Transcription factor NF-κB has a central role in inflammation, responsible for the promotion of pro-inflammatory mediators and cytokines (281). Nrf2 and NF-κB signaling regulate the redox homeostasis and inflammation responses, that is, the activation of Nrf2 has been identified to functionally couple the inhibition of NF-κB transcriptional activity (29, 185). For example, the up-regulated HO-1 is also a mediator induced by Nrf2 activation with negative regulatory effects on NF-κB (186). On the one hand, HO-1 can inhibit the degeneration of IκB-α leading to the stabilization of NF-κB in cytoplasm (187). On the other hand, CO induced by HO-1 can inhibit the TLR/NF-κB signaling via binding downstream transcription factor IRF3 and interfering in the downstream pathway (188). Besides, NF-κB family consists of two types, among which p65 has RelA, RelB, and c-Rel transactivation domains, while p50 and p52 do not have transactivation domains. And for transcription, P50 and P52 need to form heterodimerization with the Rel proteins (189). Notably, novel treatments for inflammatory diseases can inhibit NF-κB and activate Nrf2 to alleviate inflammation (190, 191).
The highly electronegative oxygen can accept electrons generated by normal oxidative metabolism within cells, and ROS are produced, which include superoxide anion (O2 -.), hydrogen peroxide (H2O2), hydroxyl radical, and singlet oxygen. ROS generated in cytoplasm and mitochondria act their key roles as signaling molecules to regulate physiological processes of macrophages. The ROS generated in cytoplasm and mitochondria serve as important signaling molecules to regulate macrophage physiological processes. NADPH oxidases (NOXs) transfer one electron from NADPH to oxygen to produce cytosolic ROS. Besides, the production of mitochondrial ROS is higher under the biochemical activities of ETC, monoamine oxidases, and P66shc (192). Macrophages could undergo pro-inflammatory cycles promoted by ROS. The targets of ROS include ROS/p38/NF-κB signaling (193), and ROS/p38/STAT1 axis (194), leading to M1 characteristics. And ROS can exacerbate inflammatory response related to NF-κB (p65) (195). However, the role of ROS has two sides. After all, it has been reported that ROS plays a critical role in M2 polarization, during which process the ROS produced by NOX1/NOX2 could contribute to monocyte-to-macrophage differentiation via activating of ERK and JNK (196).
Additionally, the activation of Nrf2 can regulate the antioxidant response by controlling the expression of detoxifying enzymes to buffer ROS. Thereby, the decreased ROS results in the inhibition of the activation of NF-κB mediated by oxidative stress (193, 197). Additionally, removing ROS also inhibits STAT1 phosphorylation and conversely activates STAT6 (198, 199). By reducing ROS levels within cells, Nrf2 inhibits oxidative stress-mediated activation, leading to an anti-inflammatory M2 phenotype. Additionally, given the NLRP3 inflammasome signaling as another NF-κB activator, Nrf2 can negatively regulate NLRP3 inflammasome activation (200, 201), especially in the regulative process of ROS. Similarly, Nrf2 can increase NQO1 production and lead to negative effects on NLRP3 inflammasome activation (200). In addition, Hippo-yes-associated protein signaling can also be viewed as a mechanism of Nrf2 activation alleviating inflammation related to NLRP3 inflammasome activation (202).
The mechanisms at a molecular level underlying Nrf2 inhibiting NF-κB are complex. On the one hand, IκB-α is the main inhibitor of NF-κB, which can be phosphorylated by IkB kinase (IKK) β resulting in the release of NF-κB in the pro-inflammation micro-environment. IKKβ can bind KEAP1 via the ETGE motif to mediate the ubiquitin and proteasome degradation. KEAP1 is the main inhibitor of Nrf2. When IKKβ is stabilized, KEAP1 can be inhibited, while IκB-α can be phosphorylated, which results in the inhibition of Nrf2 and the activation of NF-κB (203–205). On the other hand, the p65 subunit of NF-κB has been indicated as a partner of KEAP1, and the interaction between the both can inhibit Nrf2-ARE pathway (206). NF-κB (p65) also exerts negative effects on Nrf2 signal transduction by competing for binding with CBP/P300 between p65 and Nrf2. CBP/p300 is not only a co-activator of NF-κB and Nrf2, but also can negatively regulate the biological activity of ARE in the process of transcription under the action of p65 (207, 208). Conversely, the interaction between p65 and KEAP1, and the role of CBP have also been reported as the mechanisms by which Nrf2 negatively regulates NF-κB (209, 210). As a result, the Nrf2 activation can inhibit the production of pro-inflammatory cytokines induced by NF-κB (39, 211). The activated Nrf2 can decrease the release of IL-6 and IL-1β from macrophages by blocking pro-inflammatory cytokine transcription in macrophages (211). Furthermore, M2 polarization induced by the activation of Nrf2 has been reported to treat OA (212).
Fumarate is a mitochondrial metabolite acting as the terminal electron acceptor in the ETC of mammalians (213). In the Krebs cycle, succinate dehydrogenase (SDH) catalyzes the oxidation of succinate to fumarate (214). SDH-induced succinate oxidation can switch the production of ROS and the activation of M1 macrophages (215). And then, the isocitrate dehydrogenase in the Krebs cycle is blocked, inhibiting the canalization of cis-aconitate to isocitrate (216). The accumulated cis-aconitate binding to immune-responsive gene 1 can generate itaconate via decarboxylation (214, 217). Itaconate is an endogenous metabolite produced during the TCA cycle, which activates Nrf2 via the alkylation of KEAP1 to influence macrophage function (218).
4-octyl itaconate (OI) is an esterified itaconate derivative that can be converted into itaconate by a direct modification of intracellular cysteines (219, 220). Dimethyl fumarate (DMF) is an esterified fumarate derivative, which can be rapidly metabolized into monomethylfumarate (MMF) in vivo (220, 221). In macrophages, OI and DMF inhibit pro-IL-1β and NLRP3 signaling pathways activated by TLR4 binding DAMPs (222). However, different from OI, the promoting effect of DMF on KEAP1 alkylation and Nrf2 nuclear accumulation comes from its metabolite MMF (223).
OI and DMF have protective effects on the cartilage in OA. More specifically, OI-induced transcription of Nrf2 in chondrocytes results in the high expression of HO-1, NQO1, and GCLC, and the low secretion of IL-6, IL-10, MCP-1, and TNF-α, which can switch the prevention from cell death and apoptosis of chondrocytes via decreasing oxidative stress and inflammation responses, and put a brake on the progress of OA in vivo (224, 225). Similarly, dimethyl fumarate (DMF) can suppress the production of MMP-1, MMP-3, and MMP-13 and the destruction of COL2 induced by TNF-α in OA, which appears to work by inhibiting JAK/STAT3 signaling (226). Moreover, recent evidence shows that exogenous itaconate promotes polarization of M2 macrophages and reduces apoptosis in chondrocytes, a process in which Nrf2 activation-induced stimulator of interferon genes (STING) suppresses transcription of NF-κB (212).
Curcumin is a yellow-colored lipid-soluble polyphenol that is the main active ingredient extracted from the rhizome of Curcuma longa (also known as turmeric) (227). Curcumin has been shown to reduce inflammation and alleviate oxidative stress, where Nrf2 plays a vital role (228). By increasing Nrf2 transcription and HO-1 synthesis, curcumin protects cells from ROS-induced damage and reduces COX-2 production (229, 230). Of note, macrophage COX-2 is an inflammatory enzyme catalyzing the formation of prostaglandins and thromboxane, which is upregulated during pro-inflammatory conditions (231).
The effects of curcumin in OA can also be independent of the Nrf2/HO-1 axis in reducing inflammation, preventing ECM degradation, and promoting cartilage synthesis (232, 233). The effects on inflammation can be induced by suppressing the expression of pro-inflammatory mediators, such as TNF-α, IL-1β, IL-6, IL-17and TGF-β (233–236), which are mainly regulated by NF-κB signaling. Besides, the degeneration of IκBα and the expression of COX-2 in macrophages are reduced by curcumin, resulting in the reduced transcription of NF-κB lower responses to LPS and M1 polarization (236), implying the activation of Nrf2. Curcumin can upregulate the expression of COL2 and downregulate MMP1, MMP3, and MMP13 in ECM protection (235, 237). In addition, the higher level of mRNA related to cartilage anabolism, including COL2A1 and ACAN, can characterize the effect of curcumin on cartilage regeneration (232).
Similar to curcumin, quercetin is one of the most studied and abundant flavonoids found in tea, vegetables, and fruits (238). Quercetin has a high antioxidant activity due to its capacity to up-regulate the transcription of Nrf2, via promoting the degeneration of KEAP1 (239). Quercetin can induce the conversion of macrophages from M1 to M2, characterized by lower levels of iNOS-positive cells and inflammatory mediators, and higher levels of CD206-positive cells in diabetic wound healing (240). By scavenging ROS, quercetin-loaded ceria nanocomposite can also increase the M2/M1 ratio of macrophage polarization in periodontal inflammation models (241). Besides, quercetin has been suggested to suppress the expression of iNOS and COX-2, as well as the secretion of IL-6, TNF-α, and IL-1β of M1 macrophages with the decrease in ROS and chemokines related to M1 polarization, including CCL2 and CXCL10, while IL-10 of M2 macrophages has been found up-regulated, with the increase in HO-1 and NQO1 via AMPK and AKT signaling pathways (242). Notably, potential mechanisms underlying the effects of quercetin in the immunoregulation of macrophages can also involve the inhibitive effects on the activity of NLRP3 inflammasome via TLR2/Myd88/NF-κB and ROS/AMPK pathway (243).
In OA, the effects of quercetin on anti-inflammation response and cartilage protection have been addressed (244). Quercetin can act its anti-inflammatory role in suppressing NO, TNF-α, and IL-1β through inhibiting the NLRP3 signaling pathway, p38 activation, and endoplasmic reticulum stress (245–247). In addition, the activated SIRT1/AMPK signaling pathway can not only suppress the apoptosis of chondrocytes related to endoplasmic reticulum stress, but can also mediate the reversion of mitochondrial dysfunctions and the elimination of ROS in chondrocytes (246, 248). Along aside with anti-inflammation response, the inhibition of cartilage degeneration and the promotion of cartilage regeneration can be characterized by the down-regulation of MMP3, MMP9, MMP13, and ADAMTS-5, while the up-regulation of COL2 (247, 249). Of note, it has been implied that the effects of quercetin can be better loaded by Nano-materials (249, 250). Furthermore, the synovial level of TGF-β1 and TGF-β2 has been found up-regulated after the administration of quercetin due to the increase in M2 macrophages, which can also promote the production of IGF and build a microenvironment promoting chondrogenesis (248).
N-acetyl-5-methoxytryptamine, also known as Melatonin (Mel), is an indolamine with numerous functions in neural, endocrine, and immune physiological activities, playing a versatile role in the regulation of circadian rhythms, the defense against oxidative and inflammation, and the modulation of mitochondrial homeostasis. Mel is synthesized from tryptophan under 5-hydroxytryptamine in multiple extrapineal tissues (251, 252). In OA, Mel has been considered a novel treatment for OA due to its effects on the protection of chondrocytes from apoptosis, the promotion of anabolic metabolism, and the suppression of catabolic metabolism in cartilage, the restoration of redox balance, and the regulation of sirtuin signaling pathways (253).
The role of Mel in the immunoregulation of macrophages has not been verified, however, several studies support this idea. In macrophages, the administration of Mel can induce the activation of Nrf2 and the increased expression of HO-1, while the expression of iNOS and COX-2, and the production of TNF-α, IL-1β and IL-6 can be reduced through the inhibition of NF-κB transcription (254, 255). Besides, the reduced activation of STAT1 and the increased STAT3 can drive the transformation from M1 to M2 phenotype (256). Five potential mechanisms underlying shaping polarization of macrophages have been implied by the review, including through cellular pathways of JAK/STAT, cellular metabolism, miRNAs, mitochondrial dynamics, and mitophagy (257). Furthermore, the most recent study indicates that the interaction of Mel and MT1 receptors can activate PI3K/Akt and ERK signaling pathways in synovial fibroblasts leading to the up-regulation of microRNA-185a, which can reverse OA-induced pathology in animal models through reducing the secretion of TNF-α, IL-8, and vascular endothelial growth factors (258).
Mesenchymal stem cells (MSCs) are heterogeneous stromal cells commonly sourced from adipose tissue, bone marrow and umbilical cord blood. MSCs have the capacity of differentiating into adipocytes, chondrocytes, and osteoblasts. Except for the capacity of multidirectional differentiation, MSCs exert broad immunoregulatory abilities, which can induce the specific polarization (M1 to M2) of macrophages to promote the repair of damaged tissues via cell-to-cell contact and paracrine actions (259–262). The mechanisms and evidence of MSCs mediating the alterations of macrophage phenotypes have been reviewed. Furthermore, the Hippo pathway activating Yes-associated proteins was pointed out as a new mechanism of inhibiting NLRP3 signaling underlying MSCs regulating macrophages (263).
In OA, several studies have revealed the potential effects of MSCs on regulating intra-articular inflammation via M2 macrophage polarization. MSCs derived from bone marrow, which are labeled by iron oxide nanoparticles, can induce the increase in CD206-positive cells out of F4/80-positive macrophages, while can also induce the decrease in iNOS-positive cells out of F4/80-positive macrophages in animals experiencing OA due to destabilization of medical meniscus (264). Besides, the cell-free fat extract as a derivative of adipose-derived stem cells, which is rich in cytokines and nutrients, has been reported to be dose-dependently effective in relieving pain (tested by behavioral tests of rats), protecting cartilage, and increasing the ratio of M2 phenotype in the synovium (CD206-positive macrophages) in rat models with sodium iodoacetate-induced OA. The same study also revealed that the cell-free fat can decrease the ratio of CD86-positive cells, and reduce iNOS and COX-2 induced by LPS and IFN-γ in Raw 264.7. And then IL-6 and ADAMTs-5 were reduced, while the expression of SOX-9 was promoted in chondrocytes administrated by the cell-free fat. In addition, ROS can be regulated in these processes (265).
Notably, specifically pre-conditioned MSCs seem to show better regulation of macrophages. MSCs-derived extracellular vesicles (EVs) with antioxidative characteristics via over-expressing Nrf2 in adipose MSCs have effects on anti-inflammation and antioxidation. These EVs can induce increased levels of M2 macrophages, and decreased IL-6 and TNF-α (266). According to the International Society for Extracellular Vesicles, EVs are lipid bilayer membrane particles naturally released by the cells. EVs contain proteins, lipids, and nucleic acids, while without a functional nucleus (267). For MSCs, the production of EVs contributes to regulating the activation states of macrophages. The main active substances that play a regulatory role are miRNA and mitochondria transferred through EVs. miRNAs could target various transcription factors (for example, NF-κB) and adaptor proteins (for example, IL-1β) at the post-transcriptional level. Besides, the mitochondrial transfer could be related to the promotion of oxidative phosphorylation and the repair of oxidative stress function (268, 269). The acute lung injury has been ameliorated by MSC-derived small EVs (MSC-EVs) via activating Nrf2, during which process the increase in immune and redox mediators, including TLR4, Arg1, and HO-1 could be revealed (270). Besides, in a recent study, nanoparticles simulating EVs are effective for OA by promoting the polarization from M1 to M2. The structure of these EVs is oxidative stress-responsive bilirubin grafted polylysine biomaterial vesicles containing immunoglobulin IgG and berberine (271). However, there is still a knowledge gap in MSC-EVs regulating macrophage polarization via Nrf2 in OA treatment. Besides, the expression of Nrf2 in MSCs was promoted by hypoxic preconditioning, while the expression of NF-κB was reduced. And the intrarenal transplantation of these hypoxic preconditioned MSCs was more effective in the activation of HIF-1α/VEGF/Nrf2 signaling to reduce glomerular apoptosis, autophagy, and inflammation (272). However, the evidence of the Nrf2 activation in macrophage has been clearly pointed out in another study. Bone marrow-derived MSCs pre-conditioned by FNDC5, a transmembrane protein acting a crucial role in inflammation diseases, have been found to produce more exosomes. These pre-conditioned exosomes have shown the effects of promoting M2 macrophages and anti-inflammation response in myocardial infarction via the inhibition of NF-κB signaling pathway and the activation of Nrf2/HO-1 axis (273).
Low-intensity pulsed ultrasound (LIPUS) outputs in a pulse wave mode of ultrasound with a non-thermal effect, at an intensity lower than 3 W/cm2 (274). The use of LIPUS in OA treatment has been reported as an effective manner in protecting cartilage from degeneration via reducing the expression of MMP3, MMP13, and TGF-β1 (275). LIPUS has been considered an effective strategy for patients with OA, which was verified by a recent randomized clinical trial (276).
Besides, LIPUS can promote the cartilage differentiation of bone marrow-derived MSCs due to promoting the nuclear localization of SOX9 dependent on the phosphorylation of ERK1/2 (277). The combination of nanoparticles and LIPUS has been implied to have better effects on OA through inhibiting the degeneration of cartilage (278, 279). Of note, the effects of LIPUS on immunoregulation in macrophages have been reviewed as a potential mechanism of treating OA (20), however, there was little direct evidence. A most recent study has revealed that LIPUS can significantly suppress the secretion of IL-1β, IL-6, and TNF-α induced by LPS in macrophages derived from bone marrow, which can be attributed to increasing the level of intracellular itaconate and the expression of Nrf2 (280).
In this review, we reviewed the role of macrophage phenotypes in driving and relieving inflammation due to OA and summarized how NF-κB, Nrf2, and their crosstalk shape macrophage polarization. As Nrf2 signaling is believed to impact cellular metabolism, studying the effects of activators of Nrf2 on macrophage metabolism and phenotype related to inflammation could reveal how OA can be treated through reprogramming macrophage functions. Furthermore, studying the effect of Nrf2 activation on macrophages in the OA microenvironment may suggest a potential anti-inflammatory therapy target. Therefore, it is imperative to identify the relationship between Nrf2, macrophage function, and the progression of inflammatory diseases.
Conception and design of study, LW and CH. Drafting of article, LW. Revision of draft, CH. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (81972146 to CH), the Department of Science and Technology of Sichuan Province (2020YJ0210 and 2021YFS0004 to CH).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Neogi T. The epidemiology and impact of pain in osteoarthritis. Osteoarthritis Cartilage (2013) 21(9):1145–53. doi: 10.1016/j.joca.2013.03.018
2. Glyn-Jones S, Palmer AJ, Agricola R, Price AJ, Vincent TL, Weinans H, et al. Osteoarthritis. Lancet (2015) 386(9991):376–87. doi: 10.1016/s0140-6736(14)60802-3
3. Robinson WH, Lepus CM, Wang Q, Raghu H, Mao R, Lindstrom TM, et al. Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat Rev Rheumatol (2016) 12(10):580–92. doi: 10.1038/nrrheum.2016.136
4. Kraus VB, McDaniel G, Huebner JL, Stabler TV, Pieper CF, Shipes SW, et al. Direct in vivo evidence of activated macrophages in human osteoarthritis. Osteoarthritis Cartilage (2016) 24(9):1613–21. doi: 10.1016/j.joca.2016.04.010
5. Zhang H, Cai D, Bai X. Macrophages regulate the progression of osteoarthritis. Osteoarthritis Cartilage (2020) 28(5):555–61. doi: 10.1016/j.joca.2020.01.007
6. Culemann S, Grüneboom A, Nicolás-Ávila J, Weidner D, Lämmle KF, Rothe T, et al. Locally renewing resident synovial macrophages provide a protective barrier for the joint. Nature (2019) 572(7771):670–5. doi: 10.1038/s41586-019-1471-1
7. Haubruck P, Pinto MM, Moradi B, Little CB, Gentek R. Monocytes, macrophages, and their potential niches in synovial joints - therapeutic targets in post-traumatic osteoarthritis? Front Immunol (2021) 12:763702. doi: 10.3389/fimmu.2021.763702
8. Danks L, Sabokbar A, Gundle R, Athanasou NA. Synovial macrophage-osteoclast differentiation in inflammatory arthritis. Ann Rheum Dis (2002) 61(10):916–21. doi: 10.1136/ard.61.10.916
9. Zhang H, Lin C, Zeng C, Wang Z, Wang H, Lu J, et al. Synovial macrophage M1 polarisation exacerbates experimental osteoarthritis partially through r-spondin-2. Ann Rheum Dis (2018) 77(10):1524–34. doi: 10.1136/annrheumdis-2018-213450
10. Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol (2013) 229(2):176–85. doi: 10.1002/path.4133
11. Cutolo M, Campitiello R, Gotelli E, Soldano S. The role of M1/M2 macrophage polarization in rheumatoid arthritis synovitis. Front Immunol (2022) 13:867260. doi: 10.3389/fimmu.2022.867260
12. Skronska-Wasek W, Durlanik S, Garnett JP, Pflanz S. Polarized cytokine release from airway epithelium differentially influences macrophage phenotype. Mol Immunol (2021) 132:142–9. doi: 10.1016/j.molimm.2021.01.029
13. Wang LX, Zhang SX, Wu HJ, Rong XL, Guo J. M2b macrophage polarization and its roles in diseases. J Leukoc Biol (2019) 106(2):345–58. doi: 10.1002/jlb.3ru1018-378rr
14. Spiller KL, Anfang RR, Spiller KJ, Ng J, Nakazawa KR, Daulton JW, et al. The role of macrophage phenotype in vascularization of tissue engineering scaffolds. Biomaterials (2014) 35(15):4477–88. doi: 10.1016/j.biomaterials.2014.02.012
15. Ferrante CJ, Leibovich SJ. Regulation of macrophage polarization and wound healing. Adv Wound Care (New Rochelle) (2012) 1(1):10–6. doi: 10.1089/wound.2011.0307
16. Anders CB, Lawton TMW, Ammons MCB. Metabolic immunomodulation of macrophage functional plasticity in nonhealing wounds. Curr Opin Infect Dis (2019) 32(3):204–9. doi: 10.1097/qco.0000000000000550
17. Galdiero MR, Garlanda C, Jaillon S, Marone G, Mantovani A. Tumor associated macrophages and neutrophils in tumor progression. J Cell Physiol (2013) 228(7):1404–12. doi: 10.1002/jcp.24260
18. Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci (2008) 13:453–61. doi: 10.2741/2692
19. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol (2018) 233(9):6425–40. doi: 10.1002/jcp.26429
20. Sun Y, Zuo Z, Kuang Y. An emerging target in the battle against osteoarthritis: Macrophage polarization. Int J Mol Sci (2020) 21(22):8513. doi: 10.3390/ijms21228513
21. Yin J, Zeng H, Fan K, Xie H, Shao Y, Lu Y, et al. Pentraxin 3 regulated by miR-224-5p modulates macrophage reprogramming and exacerbates osteoarthritis associated synovitis by targeting CD32. Cell Death Dis (2022) 13(6):567. doi: 10.1038/s41419-022-04962-y
22. Dravid AA, MD K, Agarwal S, Agarwal R. Resolvin D1-loaded nanoliposomes promote M2 macrophage polarization and are effective in the treatment of osteoarthritis. Bioeng Transl Med (2022) 7(2):e10281. doi: 10.1002/btm2.10281
23. Wang D, Chai XQ, Hu SS, Pan F. Joint synovial macrophages as a potential target for intra-articular treatment of osteoarthritis-related pain. Osteoarthritis Cartilage (2022) 30(3):406–15. doi: 10.1016/j.joca.2021.11.014
24. Galván-Peña S, O'Neill LA. Metabolic reprograming in macrophage polarization. Front Immunol (2014) 5:420. doi: 10.3389/fimmu.2014.00420
25. Mills EL, O'Neill LA. Reprogramming mitochondrial metabolism in macrophages as an anti-inflammatory signal. Eur J Immunol (2016) 46(1):13–21. doi: 10.1002/eji.201445427
26. Ryan DG, O'Neill LAJ. Krebs Cycle reborn in macrophage immunometabolism. Annu Rev Immunol (2020) 38:289–313. doi: 10.1146/annurev-immunol-081619-104850
27. Marchev AS, Dimitrova PA, Burns AJ, Kostov RV, Dinkova-Kostova AT, Georgiev MI. Oxidative stress and chronic inflammation in osteoarthritis: can NRF2 counteract these partners in crime? Ann N Y Acad Sci (2017) 1401(1):114–35. doi: 10.1111/nyas.13407
28. Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci (2014) 39(4):199–218. doi: 10.1016/j.tibs.2014.02.002
29. Saha S, Buttari B, Panieri E, Profumo E, Saso L. An overview of Nrf2 signaling pathway and its role in inflammation. Molecules (2020) 25(22):5474. doi: 10.3390/molecules25225474
30. Kim SR, Seong KJ, Kim WJ, Jung JY. Epigallocatechin gallate protects against hypoxia-induced inflammation in microglia via NF-κB suppression and nrf-2/HO-1 activation. Int J Mol Sci (2022) 23(7):4004. doi: 10.3390/ijms23074004
31. Diotallevi M, Checconi P, Palamara AT, Celestino I, Coppo L, Holmgren A, et al. Glutathione fine-tunes the innate immune response toward antiviral pathways in a macrophage cell line independently of its antioxidant properties. Front Immunol (2017) 8:1239. doi: 10.3389/fimmu.2017.01239
32. Cuadrado A, Manda G, Hassan A, Alcaraz MJ, Barbas C, Daiber A, et al. Transcription factor NRF2 as a therapeutic target for chronic diseases: A systems medicine approach. Pharmacol Rev (2018) 70(2):348–83. doi: 10.1124/pr.117.014753
33. Kwon DH, Lee H, Park C, Hong SH, Hong SH, Kim GY, et al. Glutathione induced immune-stimulatory activity by promoting M1-like macrophages polarization via potential ROS scavenging capacity. Antioxidants (Basel) (2019) 8(9):413. doi: 10.3390/antiox8090413
34. Lu H, Sun X, Jia M, Sun F, Zhu J, Chen X, et al. Rosiglitazone suppresses renal crystal deposition by ameliorating tubular injury resulted from oxidative stress and inflammatory response via promoting the Nrf2/HO-1 pathway and shifting macrophage polarization. Oxid Med Cell Longev (2021) 2021:5527137. doi: 10.1155/2021/5527137
35. Li D, Wang M, Ye J, Zhang J, Xu Y, Wang Z, et al. Maresin 1 alleviates the inflammatory response, reduces oxidative stress and protects against cardiac injury in LPS-induced mice. Life Sci (2021) 277:119467. doi: 10.1016/j.lfs.2021.119467
36. Wall SB, Li R, Butler B, Burg AR, Tse HM, Larson-Casey JL, et al. Auranofin-mediated NRF2 induction attenuates interleukin 1 beta expression in alveolar macrophages. Antioxidants (Basel) (2021) 10(5):632. doi: 10.3390/antiox10050632
37. Rushworth SA, Shah S, MacEwan DJ. TNF mediates the sustained activation of Nrf2 in human monocytes. J Immunol (2011) 187(2):702–7. doi: 10.4049/jimmunol.1004117
38. Brüne B, Dehne N, Grossmann N, Jung M, Namgaladze D, Schmid T, et al. Redox control of inflammation in macrophages. Antioxid Redox Signal (2013) 19(6):595–637. doi: 10.1089/ars.2012.4785
39. Cuadrado A, Martín-Moldes Z, Ye J, Lastres-Becker I. Transcription factors NRF2 and NF-κB are coordinated effectors of the rho family, GTP-binding protein RAC1 during inflammation. J Biol Chem (2014) 289(22):15244–58. doi: 10.1074/jbc.M113.540633
40. Lv Z, Xu X, Sun Z, Yang YX, Guo H, Li J, et al. TRPV1 alleviates osteoarthritis by inhibiting M1 macrophage polarization via Ca(2+)/CaMKII/Nrf2 signaling pathway. Cell Death Dis (2021) 12(6):504. doi: 10.1038/s41419-021-03792-8
41. Gómez R, Villalvilla A, Largo R, Gualillo O, Herrero-Beaumont G. TLR4 signalling in osteoarthritis–finding targets for candidate DMOADs. Nat Rev Rheumatol (2015) 11(3):159–70. doi: 10.1038/nrrheum.2014.209
42. Sohn DH, Sokolove J, Sharpe O, Erhart JC, Chandra PE, Lahey LJ, et al. Plasma proteins present in osteoarthritic synovial fluid can stimulate cytokine production via toll-like receptor 4. Arthritis Res Ther (2012) 14(1):R7. doi: 10.1186/ar3555
43. Bondeson J, Wainwright SD, Lauder S, Amos N, Hughes CE. The role of synovial macrophages and macrophage-produced cytokines in driving aggrecanases, matrix metalloproteinases, and other destructive and inflammatory responses in osteoarthritis. Arthritis Res Ther (2006) 8(6):R187. doi: 10.1186/ar2099
44. Bondeson J, Blom AB, Wainwright S, Hughes C, Caterson B, van den Berg WB. The role of synovial macrophages and macrophage-produced mediators in driving inflammatory and destructive responses in osteoarthritis. Arthritis Rheum (2010) 62(3):647–57. doi: 10.1002/art.27290
45. Hwang HS, Kim HA. Chondrocyte apoptosis in the pathogenesis of osteoarthritis. Int J Mol Sci (2015) 16(11):26035–54. doi: 10.3390/ijms161125943
46. Pattoli MA, MacMaster JF, Gregor KR, Burke JR. Collagen and aggrecan degradation is blocked in interleukin-1-treated cartilage explants by an inhibitor of IkappaB kinase through suppression of metalloproteinase expression. J Pharmacol Exp Ther (2005) 315(1):382–8. doi: 10.1124/jpet.105.087569
47. Kapoor M, Martel-Pelletier J, Lajeunesse D, Pelletier JP, Fahmi H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol (2011) 7(1):33–42. doi: 10.1038/nrrheum.2010.196
48. Limagne E, Lançon A, Delmas D, Cherkaoui-Malki M, Latruffe N. Resveratrol interferes with IL1-β-Induced pro-inflammatory paracrine interaction between primary chondrocytes and macrophages. Nutrients (2016) 8(5):280. doi: 10.3390/nu8050280
49. Wang P, Zhu F, Konstantopoulos K. Prostaglandin E2 induces interleukin-6 expression in human chondrocytes via cAMP/protein kinase a- and phosphatidylinositol 3-kinase-dependent NF-kappaB activation. Am J Physiol Cell Physiol (2010) 298(6):C1445–56. doi: 10.1152/ajpcell.00508.2009
50. Smith WL, Garavito RM, DeWitt DL. Prostaglandin endoperoxide h synthases (cyclooxygenases)-1 and -2. J Biol Chem (1996) 271(52):33157–60. doi: 10.1074/jbc.271.52.33157
51. Pelletier JP, Mineau F, Ranger P, Tardif G, Martel-Pelletier J. The increased synthesis of inducible nitric oxide inhibits IL-1ra synthesis by human articular chondrocytes: possible role in osteoarthritic cartilage degradation. Osteoarthritis Cartilage (1996) 4(1):77–84. doi: 10.1016/s1063-4584(96)80009-4
52. Lee CH, Chiang CF, Kuo FC, Su SC, Huang CL, Liu JS, et al. High-Molecular-Weight hyaluronic acid inhibits IL-1β-Induced synovial inflammation and macrophage polarization through the GRP78-NF-κB signaling pathway. Int J Mol Sci (2021) 22(21):11917. doi: 10.3390/ijms222111917
53. Stanton H, Rogerson FM, East CJ, Golub SB, Lawlor KE, Meeker CT, et al. ADAMTS5 is the major aggrecanase in mouse cartilage in vivo and in vitro. Nature (2005) 434(7033):648–52. doi: 10.1038/nature03417
54. Wang T, He C. Pro-inflammatory cytokines: The link between obesity and osteoarthritis. Cytokine Growth Factor Rev (2018) 44:38–50. doi: 10.1016/j.cytogfr.2018.10.002
55. Utomo L, Bastiaansen-Jenniskens YM, Verhaar JA, van Osch GJ. Cartilage inflammation and degeneration is enhanced by pro-inflammatory (M1) macrophages in vitro, but not inhibited directly by anti-inflammatory (M2) macrophages. Osteoarthritis Cartilage (2016) 24(12):2162–70. doi: 10.1016/j.joca.2016.07.018
56. Xie J, Huang Z, Yu X, Zhou L, Pei F. Clinical implications of macrophage dysfunction in the development of osteoarthritis of the knee. Cytokine Growth Factor Rev (2019) 46:36–44. doi: 10.1016/j.cytogfr.2019.03.004
57. Dai M, Sui B, Xue Y, Liu X, Sun J. Cartilage repair in degenerative osteoarthritis mediated by squid type II collagen via immunomodulating activation of M2 macrophages, inhibiting apoptosis and hypertrophy of chondrocytes. Biomaterials (2018) 180:91–103. doi: 10.1016/j.biomaterials.2018.07.011
58. Mantovani A, Sica A, Locati M. New vistas on macrophage differentiation and activation. Eur J Immunol (2007) 37(1):14–6. doi: 10.1002/eji.200636910
59. Ruytinx P, Proost P, Van Damme J, Struyf S. Chemokine-induced macrophage polarization in inflammatory conditions. Front Immunol (2018) 9:1930(SEP). doi: 10.3389/fimmu.2018.01930
60. Madsen DH, Leonard D, Masedunskas A, Moyer A, Jürgensen HJ, Peters DE, et al. M2-like macrophages are responsible for collagen degradation through a mannose receptor-mediated pathway. J Cell Biol (2013) 202(6):951–66. doi: 10.1083/jcb.201301081
61. Alaaeddine N, Di Battista JA, Pelletier JP, Kiansa K, Cloutier JM, Martel-Pelletier J. Inhibition of tumor necrosis factor alpha-induced prostaglandin E2 production by the antiinflammatory cytokines interleukin-4, interleukin-10, and interleukin-13 in osteoarthritic synovial fibroblasts: distinct targeting in the signaling pathways. Arthritis Rheum (1999) 42(4):710–8. doi: 10.1002/1529-0131(199904)42:4<710::Aid-anr14>3.0.Co;2-4
62. John T, Müller RD, Oberholzer A, Zreiqat H, Kohl B, Ertel W, et al. Interleukin-10 modulates pro-apoptotic effects of TNF-alpha in human articular chondrocytes. Vitro Cytokine (2007) 40(3):226–34. doi: 10.1016/j.cyto.2007.10.002
63. Behrendt P, Preusse-Prange A, Klüter T, Haake M, Rolauffs B, Grodzinsky AJ, et al. IL-10 reduces apoptosis and extracellular matrix degradation after injurious compression of mature articular cartilage. Osteoarthritis Cartilage (2016) 24(11):1981–8. doi: 10.1016/j.joca.2016.06.016
64. Müller RD, John T, Kohl B, Oberholzer A, Gust T, Hostmann A, et al. IL-10 overexpression differentially affects cartilage matrix gene expression in response to TNF-α in human articular chondrocytes in vitro. Cytokine (2008) 44(3):377–85. doi: 10.1016/j.cyto.2008.10.012
65. Fahy N, de Vries-van Melle ML, Lehmann J, Wei W, Grotenhuis N, Farrell E, et al. Human osteoarthritic synovium impacts chondrogenic differentiation of mesenchymal stem cells via macrophage polarisation state. Osteoarthritis Cartilage (2014) 22(8):1167–75. doi: 10.1016/j.joca.2014.05.021
66. Lopa S, Leijs MJ, Moretti M, Lubberts E, van Osch GJ, Bastiaansen-Jenniskens YM. Arthritic and non-arthritic synovial fluids modulate IL10 and IL1RA gene expression in differentially activated primary human monocytes. Osteoarthritis Cartilage (2015) 23(11):1853–7. doi: 10.1016/j.joca.2015.06.003
67. Camargo Garbin L, Morris MJ. A comparative review of autologous conditioned serum and autologous protein solution for treatment of osteoarthritis in horses. Front Vet Sci (2021) 8:602978. doi: 10.3389/fvets.2021.602978
68. Schutyser E, Richmond A, Van Damme J. Involvement of CC chemokine ligand 18 (CCL18) in normal and pathological processes. J Leukoc Biol (2005) 78(1):14–26. doi: 10.1189/jlb.1204712
69. Cardoso AP, Pinto ML, Castro F, Costa ÂM, Marques-Magalhães Â, Canha-Borges A, et al. The immunosuppressive and pro-tumor functions of CCL18 at the tumor microenvironment. Cytokine Growth Factor Rev (2021) 60:107–19. doi: 10.1016/j.cytogfr.2021.03.005
70. Zhou Y, Chen J, Yang G. Serum and synovial fluid levels of CCL18 are correlated with radiographic grading of knee osteoarthritis. Med Sci Monit (2015) 21:840–4. doi: 10.12659/msm.892409
71. Takayasu A, Miyabe Y, Yokoyama W, Kaneko K, Fukuda S, Miyasaka N, et al. CCL18 activates fibroblast-like synoviocytes in patients with rheumatoid arthritis. J Rheumatol (2013) 40(6):1026–8. doi: 10.3899/jrheum.121412
72. Bendall SC, Stewart MH, Menendez P, George D, Vijayaragavan K, Werbowetski-Ogilvie T, et al. IGF and FGF cooperatively establish the regulatory stem cell niche of pluripotent human cells in vitro. Nature (2007) 448(7157):1015–21. doi: 10.1038/nature06027
73. Youssef A, Aboalola D, Han VK. The roles of insulin-like growth factors in mesenchymal stem cell niche. Stem Cells Int (2017) 2017:9453108. doi: 10.1155/2017/9453108
74. Fortier LA, Mohammed HO, Lust G, Nixon AJ. Insulin-like growth factor-I enhances cell-based repair of articular cartilage. J Bone Joint Surg Br (2002) 84(2):276–88. doi: 10.1302/0301-620x.84b2.11167
75. Cho H, Kim J, Kim S, Jung YC, Wang Y, Kang B-J, et al. Dual delivery of stem cells and insulin-like growth factor-1 in coacervate-embedded composite hydrogels for enhanced cartilage regeneration in osteochondral defects. J Controlled Release (2020) 327:284–95. doi: 10.1016/j.jconrel.2020.08.002
76. Zhang M, Zhou Q, Liang QQ, Li CG, Holz JD, Tang D, et al. IGF-1 regulation of type II collagen and MMP-13 expression in rat endplate chondrocytes via distinct signaling pathways. Osteoarthritis Cartilage (2009) 17(1):100–6. doi: 10.1016/j.joca.2008.05.007
77. Fosang AJ, Tyler JA, Hardingham TE. Effect of interleukin-1 and insulin like growth factor-1 on the release of proteoglycan components and hyaluronan from pig articular cartilage in explant culture. Matrix (1991) 11(1):17–24. doi: 10.1016/s0934-8832(11)80223-4
78. Montaseri A, Busch F, Mobasheri A, Buhrmann C, Aldinger C, Rad JS, et al. IGF-1 and PDGF-bb suppress IL-1β-induced cartilage degradation through down-regulation of NF-κB signaling: involvement of Src/PI-3K/AKT pathway. PloS One (2011) 6(12):e28663. doi: 10.1371/journal.pone.0028663
79. Oh CD, Chun JS. Signaling mechanisms leading to the regulation of differentiation and apoptosis of articular chondrocytes by insulin-like growth factor-1. J Biol Chem (2003) 278(38):36563–71. doi: 10.1074/jbc.M304857200
80. Zhou Q, Li B, Zhao J, Pan W, Xu J, Chen S. IGF-I induces adipose derived mesenchymal cell chondrogenic differentiation in vitro and enhances chondrogenesis in vivo. In Vitro Cell Dev Biol Anim (2016) 52(3):356–64. doi: 10.1007/s11626-015-9969-9
81. Longobardi L, O'Rear L, Aakula S, Johnstone B, Shimer K, Chytil A, et al. Effect of IGF-I in the chondrogenesis of bone marrow mesenchymal stem cells in the presence or absence of TGF-beta signaling. J Bone Miner Res (2006) 21(4):626–36. doi: 10.1359/jbmr.051213
82. Wildemann B, Schmidmaier G, Ordel S, Stange R, Haas NP, Raschke M. Cell proliferation and differentiation during fracture healing are influenced by locally applied IGF-I and TGF-beta1: comparison of two proliferation markers, PCNA and BrdU. J BioMed Mater Res B Appl Biomater (2003) 65(1):150–6. doi: 10.1002/jbm.b.10512
83. Hinck AP, Mueller TD, Springer TA. Structural biology and evolution of the TGF-β family. Cold Spring Harb Perspect Biol (2016) 8(12):a022103. doi: 10.1101/cshperspect.a022103
84. Kingsley DM. The TGF-beta superfamily: new members, new receptors, and new genetic tests of function in different organisms. Genes Dev (1994) 8(2):133–46. doi: 10.1101/gad.8.2.133
85. Blaney Davidson EN, Remst DF, Vitters EL, van Beuningen HM, Blom AB, Goumans MJ, et al. Increase in ALK1/ALK5 ratio as a cause for elevated MMP-13 expression in osteoarthritis in humans and mice. J Immunol (2009) 182(12):7937–45. doi: 10.4049/jimmunol.0803991
86. Cherifi C, Monteagudo S, Lories RJ. Promising targets for therapy of osteoarthritis: a review on the wnt and TGF-β signalling pathways. Ther Adv Musculoskelet Dis (2021) 13:1759720x211006959. doi: 10.1177/1759720x211006959
87. Derynck R, Budi EH. Specificity, versatility, and control of TGF-β family signaling. Sci Signal (2019) 12(570):eaav5183. doi: 10.1126/scisignal.aav5183
88. Moustakas A. Smad signalling network. J Cell Sci (2002) 115(17):3355–6. doi:10.1242/jcs.115.17.3355%J
89. Nakao A, Imamura T, Souchelnytskyi S, Kawabata M, Ishisaki A, Oeda E, et al. TGF-beta receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J (1997) 16(17):5353–62. doi:10.1093/emboj/16.17.5353
90. Brandes ME, Wakefield LM, Wahl SM. Modulation of monocyte type I transforming growth factor-beta receptors by inflammatory stimuli. J Biol Chem (1991) 266(29):19697–703.
91. Gratchev A, Kzhyshkowska J, Kannookadan S, Ochsenreiter M, Popova A, Yu X, et al. Activation of a TGF-beta-specific multistep gene expression program in mature macrophages requires glucocorticoid-mediated surface expression of TGF-beta receptor II. J Immunol (2008) 180(10):6553–65. doi:10.4049/jimmunol.180.10.6553
92. Itoh S, ten Dijke P. Negative regulation of TGF-beta receptor/Smad signal transduction. Curr Opin Cell Biol (2007) 19(2):176–84. doi:10.1016/j.ceb.2007.02.015
93. Imamura T, Oshima Y, Hikita A. Regulation of TGF-β family signalling by ubiquitination and deubiquitination. J Biochem (2013) 154(6):481–9. doi:10.1093/jb/mvt097%J
94. Inoue Y, Imamura T. Regulation of TGF-β family signaling by E3 ubiquitin ligases. Cancer Sci (2008) 99(11):2107–12. doi:10.1111/j.1349-7006.2008.00925.x
95. Eichhorn PJ, Rodón L, Gonzàlez-Juncà A, Dirac A, Gili M, Martínez-Sáez E, et al. USP15 stabilizes TGF-β receptor I and promotes oncogenesis through the activation of TGF-β signaling in glioblastoma. Nat Med (2012) 18(3):429–35. doi:10.1038/nm.2619
96. O'Neill LAJ, Golenbock D, Bowie AG. The history of toll-like receptors — redefining innate immunity. Nat Rev Immunol (2013) 13(6):453–60. doi: 10.1038/nri3446
97. Scanzello CR, Plaas A, Crow MK. Innate immune system activation in osteoarthritis: is osteoarthritis a chronic wound? Curr Opin Rheumatol (2008) 20(5):565–72. doi: 10.1097/BOR.0b013e32830aba34
98. Chen XX, Tang L, Fu YM, Wang Y, Han ZH, Meng JG. Paralemmin-3 contributes to lipopolysaccharide-induced inflammatory response and is involved in lipopolysaccharide-toll-like receptor-4 signaling in alveolar macrophages. Int J Mol Med (2017) 40(6):1921–31. doi: 10.3892/ijmm.2017.3161
99. Hamidzadeh K, Christensen SM, Dalby E, Chandrasekaran P, Mosser DM. Macrophages and the recovery from acute and chronic inflammation. Annu Rev Physiol (2017) 79:567–92. doi: 10.1146/annurev-physiol-022516-034348
100. Gong J, Li J, Dong H, Chen G, Qin X, Hu M, et al. Inhibitory effects of berberine on proinflammatory M1 macrophage polarization through interfering with the interaction between TLR4 and MyD88. BMC Complement Altern Med (2019) 19(1):314. doi: 10.1186/s12906-019-2710-6
101. Wang C, Ma C, Gong L, Guo Y, Fu K, Zhang Y, et al. Macrophage polarization and its role in liver disease. Front Immunol (2021) 12:803037. doi: 10.3389/fimmu.2021.803037
102. Xiang P, Chen T, Mou Y, Wu H, Xie P, Lu G, et al. NZ Suppresses TLR4/NF-κB signalings and NLRP3 inflammasome activation in LPS-induced RAW264.7 macrophages. Inflammation Res (2015) 64(10):799–808. doi: 10.1007/s00011-015-0863-4
103. Palaga T, Buranaruk C, Rengpipat S, Fauq AH, Golde TE, Kaufmann SHE, et al. Notch signaling is activated by TLR stimulation and regulates macrophage functions. Eur J Immunol (2008) 38(1):174–83. doi: 10.1002/eji.200636999
104. López-López S, Monsalve EM, Romero de Ávila MJ, González-Gómez J, Hernández de León N, Ruiz-Marcos F, et al. NOTCH3 signaling is essential for NF-κB activation in TLR-activated macrophages. Sci Rep (2020) 10(1):14839. doi: 10.1038/s41598-020-71810-4
105. Monsalve E, Pérez MA, Rubio A, Ruiz-Hidalgo MJ, Baladrón V, García-Ramírez JJ, et al. Notch-1 up-regulation and signaling following macrophage activation modulates gene expression patterns known to affect antigen-presenting capacity and cytotoxic activity. J Immunol (2006) 176(9):5362–73. doi: 10.4049/jimmunol.176.9.5362
106. Monsalve E, Ruiz-García A, Baladrón V, Ruiz-Hidalgo MJ, Sánchez-Solana B, Rivero S, et al. Notch1 upregulates LPS-induced macrophage activation by increasing NF-kappaB activity. Eur J Immunol (2009) 39(9):2556–70. doi: 10.1002/eji.200838722
107. Fung E, Tang SM, Canner JP, Morishige K, Arboleda-Velasquez JF, Cardoso AA, et al. Delta-like 4 induces notch signaling in macrophages: implications for inflammation. Circulation (2007) 115(23):2948–56. doi: 10.1161/circulationaha.106.675462
108. Singla RD, Wang J, Singla DK. Regulation of notch 1 signaling in THP-1 cells enhances M2 macrophage differentiation. Am J Physiol Heart Circ Physiol (2014) 307(11):H1634–42. doi: 10.1152/ajpheart.00896.2013
109. López-López S, Romero de Ávila MJ, Hernández de León NC, Ruiz-Marcos F, Baladrón V, Nueda ML, et al. NOTCH4 exhibits anti-inflammatory activity in activated macrophages by interfering with interferon-γ and TLR4 signaling. Front Immunol (2021) 12:734966. doi: 10.3389/fimmu.2021.734966
110. Zhong Z, Sanchez-Lopez E, Karin M. Autophagy, NLRP3 inflammasome and auto-inflammatory/immune diseases. Clin Exp Rheumatol (2016) 34(4 Suppl 98):12–6.
111. Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol (2009) 27:229–65. doi: 10.1146/annurev.immunol.021908.132715
112. Afonina IS, Zhong Z, Karin M, Beyaert R. Limiting inflammation–the negative regulation of NF-κB and the NLRP3 inflammasome. Nat Immunol (2017) 18(8):861–9. doi: 10.1038/ni.3772
113. Syx D, Tran PB, Miller RE, Malfait AM. Peripheral mechanisms contributing to osteoarthritis pain. Curr Rheumatol Rep (2018) 20(2):9. doi: 10.1007/s11926-018-0716-6
114. Stabler TV, Huang Z, Montell E, Vergés J, Kraus VB. Chondroitin sulphate inhibits NF-κB activity induced by interaction of pathogenic and damage associated molecules. Osteoarthritis Cartilage (2017) 25(1):166–74. doi: 10.1016/j.joca.2016.08.012
115. Barreto G, Senturk B, Colombo L, Brück O, Neidenbach P, Salzmann G, et al. Lumican is upregulated in osteoarthritis and contributes to TLR4-induced pro-inflammatory activation of cartilage degradation and macrophage polarization. Osteoarthritis Cartilage (2020) 28(1):92–101. doi: 10.1016/j.joca.2019.10.011
116. Banerjee S, Biehl A, Gadina M, Hasni S, Schwartz DM. JAK-STAT signaling as a target for inflammatory and autoimmune diseases: Current and future prospects. Drugs (2017) 77(5):521–46. doi: 10.1007/s40265-017-0701-9
117. O'Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med (2015) 66:311–28. doi: 10.1146/annurev-med-051113-024537
118. Xin P, Xu X, Deng C, Liu S, Wang Y, Zhou X, et al. The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int Immunopharmacol (2020) 80:106210. doi: 10.1016/j.intimp.2020.106210
119. Bhattacharjee A, Shukla M, Yakubenko VP, Mulya A, Kundu S, Cathcart MK. IL-4 and IL-13 employ discrete signaling pathways for target gene expression in alternatively activated monocytes/macrophages. Free Radic Biol Med (2013) 54:1–16. doi: 10.1016/j.freeradbiomed.2012.10.553
120. Chomarat P, Banchereau J. Interleukin-4 and interleukin-13: their similarities and discrepancies. Int Rev Immunol (1998) 17(1-4):1–52. doi: 10.3109/08830189809084486
121. Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol (2009) 27:451–83. doi: 10.1146/annurev.immunol.021908.132532
122. Hu X, Chakravarty SD, Ivashkiv LB. Regulation of interferon and toll-like receptor signaling during macrophage activation by opposing feedforward and feedback inhibition mechanisms. Immunol Rev (2008) 226:41–56. doi: 10.1111/j.1600-065X.2008.00707.x
123. Heinrich PC, Behrmann I, Müller-Newen G, Schaper F, Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J (1998) 334(2):297–314. doi: 10.1042/bj3340297
124. Okugawa S, Ota Y, Kitazawa T, Nakayama K, Yanagimoto S, Tsukada K, et al. Janus kinase 2 is involved in lipopolysaccharide-induced activation of macrophages. Am J Physiol Cell Physiol (2003) 285(2):C399–408. doi: 10.1152/ajpcell.00026.2003
125. Wang F, Zhang S, Jeon R, Vuckovic I, Jiang X, Lerman A, et al. Interferon gamma induces reversible metabolic reprogramming of M1 macrophages to sustain cell viability and pro-inflammatory activity. EBioMedicine (2018) 30:303–16. doi: 10.1016/j.ebiom.2018.02.009
126. Butturini E, Carcereri de Prati A, Mariotto S. Redox regulation of STAT1 and STAT3 signaling. Int J Mol Sci (2020) 21(19):7034. doi: 10.3390/ijms21197034
127. Kim HS, Kim DC, Kim H-M, Kwon H-J, Kwon SJ, Kang S-J, et al. STAT1 deficiency redirects IFN signalling toward suppression of TLR response through a feedback activation of STAT3. Sci Rep (2015) 5(1):13414. doi: 10.1038/srep13414
128. Petrina M, Martin J, Basta S. Granulocyte macrophage colony-stimulating factor has come of age: From a vaccine adjuvant to antiviral immunotherapy. Cytokine Growth Factor Rev (2021) 59:101–10. doi: 10.1016/j.cytogfr.2021.01.001
129. Cheung TT, McInnes IB. Future therapeutic targets in rheumatoid arthritis? Semin Immunopathol (2017) 39(4):487–500. doi: 10.1007/s00281-017-0623-3
130. Liu G, Yang H. Modulation of macrophage activation and programming in immunity. J Cell Physiol (2013) 228(3):502–12. doi: 10.1002/jcp.24157
131. Monick MM, Carter AB, Robeff PK, Flaherty DM, Peterson MW, Hunninghake GW. Lipopolysaccharide activates akt in human alveolar macrophages resulting in nuclear accumulation and transcriptional activity of beta-catenin. J Immunol (2001) 166(7):4713–20. doi: 10.4049/jimmunol.166.7.4713
132. Caescu CI, Guo X, Tesfa L, Bhagat TD, Verma A, Zheng D, et al. Colony stimulating factor-1 receptor signaling networks inhibit mouse macrophage inflammatory responses by induction of microRNA-21. Blood (2015) 125(8):e1–13. doi: 10.1182/blood-2014-10-608000
133. Arranz A, Doxaki C, Vergadi E, Martinez de la Torre Y, Vaporidi K, Lagoudaki ED, et al. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc Natl Acad Sci U.S.A. (2012) 109(24):9517–22. doi: 10.1073/pnas.1119038109
134. Byles V, Covarrubias AJ, Ben-Sahra I, Lamming DW, Sabatini DM, Manning BD, et al. The TSC-mTOR pathway regulates macrophage polarization. Nat Commun (2013) 4:2834. doi: 10.1038/ncomms3834
135. Festuccia WT. Regulation of adipocyte and macrophage functions by mTORC1 and 2 in metabolic diseases. Mol Nutr Food Res (2021) 65(1):e1900768. doi: 10.1002/mnfr.201900768
136. Liu R, Fan T, Geng W, Chen YH, Ruan Q, Zhang C. Negative immune regulator TIPE2 promotes M2 macrophage differentiation through the activation of PI3K-AKT signaling pathway. PloS One (2017) 12(1):e0170666. doi: 10.1371/journal.pone.0170666
137. Jain N, Zhang T, Fong SL, Lim CP, Cao X. Repression of Stat3 activity by activation of mitogen-activated protein kinase (MAPK). Oncogene (1998) 17(24):3157–67. doi: 10.1038/sj.onc.1202238
138. Morrison DK. MAP kinase pathways. Cold Spring Harb Perspect Biol (2012) 4(11):a011254. doi: 10.1101/cshperspect.a011254
139. Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev (2001) 22(2):153–83. doi: 10.1210/edrv.22.2.0428
140. Chan ED, Riches DWH. IFN-γ + LPS induction of iNOS is modulated by ERK, JNK/SAPK, and p38 mapk in a mouse macrophage cell line. Am J Physiol Cell Physiol (2001) 280(3):C441–C50. doi: 10.1152/ajpcell.2001.280.3.C441
141. Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol (2004) 4(7):499–511. doi: 10.1038/nri1391
142. Dong C, Davis RJ, Flavell RA. MAP kinases in the immune response. Annu Rev Immunol (2002) 20:55–72. doi: 10.1146/annurev.immunol.20.091301.131133
143. Sabio G, Davis RJ. TNF and MAP kinase signalling pathways. Semin Immunol (2014) 26(3):237–45. doi: 10.1016/j.smim.2014.02.009
144. Li ST, Dai Q, Zhang SX, Liu YJ, Yu QQ, Tan F, et al. Ulinastatin attenuates LPS-induced inflammation in mouse macrophage RAW264.7 cells by inhibiting the JNK/NF-κB signaling pathway and activating the PI3K/Akt/Nrf2 pathway. Acta Pharmacol Sin (2018) 39(8):1294–304. doi: 10.1038/aps.2017.143
145. Wang W, Guan WJ, Huang RQ, Xie YQ, Zheng JP, Zhu SX, et al. Carbocisteine attenuates TNF-α-induced inflammation in human alveolar epithelial cells in vitro through suppressing NF-κB and ERK1/2 MAPK signaling pathways. Acta Pharmacol Sin (2016) 37(5):629–36. doi: 10.1038/aps.2015.150
146. Gingras S, Simard J, Groner B, Pfitzner E. p300/CBP is required for transcriptional induction by interleukin-4 and interacts with Stat6. Nucleic Acids Res (1999) 27(13):2722–9. doi: 10.1093/nar/27.13.2722
147. Yu T, Gan S, Zhu Q, Dai D, Li N, Wang H, et al. Modulation of M2 macrophage polarization by the crosstalk between Stat6 and Trim24. Nat Commun (2019) 10(1):4353. doi: 10.1038/s41467-019-12384-2
148. Moi P, Chan K, Asunis I, Cao A, Kan YW. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci U.S.A. (1994) 91(21):9926–30. doi: 10.1073/pnas.91.21.9926
149. Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol (2004) 24(16):7130–9. doi: 10.1128/mcb.24.16.7130-7139.2004
150. Krajka-Kuźniak V, Paluszczak J, Baer-Dubowska W. The Nrf2-ARE signaling pathway: An update on its regulation and possible role in cancer prevention and treatment. Pharmacol Rep (2017) 69(3):393–402. doi: 10.1016/j.pharep.2016.12.011
151. Ganner A, Pfeiffer ZC, Wingendorf L, Kreis S, Klein M, Walz G, et al. The acetyltransferase p300 regulates NRF2 stability and localization. Biochem Biophys Res Commun (2020) 524(4):895–902. doi: 10.1016/j.bbrc.2020.02.006
152. Katoh Y, Itoh K, Yoshida E, Miyagishi M, Fukamizu A, Yamamoto M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells (2001) 6(10):857–68. doi: 10.1046/j.1365-2443.2001.00469.x
153. Rada P, Rojo AI, Evrard-Todeschi N, Innamorato NG, Cotte A, Jaworski T, et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/β-TrCP axis. Mol Cell Biol (2012) 32(17):3486–99. doi: 10.1128/mcb.00180-12
154. Wang H, Liu K, Geng M, Gao P, Wu X, Hai Y, et al. RXRα inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res (2013) 73(10):3097–108. doi: 10.1158/0008-5472.Can-12-3386
155. Igarashi K, Kataoka K, Itoh K, Hayashi N, Nishizawa M, Yamamoto M. Regulation of transcription by dimerization of erythroid factor NF-E2 p45 with small maf proteins. Nature (1994) 367(6463):568–72. doi: 10.1038/367568a0
156. Hayes JD, McMahon M, Chowdhry S, Dinkova-Kostova AT. Cancer chemoprevention mechanisms mediated through the Keap1-Nrf2 pathway. Antioxid Redox Signal (2010) 13(11):1713–48. doi: 10.1089/ars.2010.3221
157. Kansanen E, Jyrkkänen HK, Levonen AL. Activation of stress signaling pathways by electrophilic oxidized and nitrated lipids. Free Radic Biol Med (2012) 52(6):973–82. doi: 10.1016/j.freeradbiomed.2011.11.038
158. Nioi P, McMahon M, Itoh K, Yamamoto M, Hayes JD. Identification of a novel Nrf2-regulated antioxidant response element (ARE) in the mouse NAD(P)H:quinone oxidoreductase 1 gene: reassessment of the ARE consensus sequence. Biochem J (2003) 374(Pt 2):337–48. doi: 10.1042/bj20030754
159. Ryter SW. Heme oxygenase-1: An anti-inflammatory effector in cardiovascular, lung, and related metabolic disorders. Antioxidants (Basel) (2022) 11(3):555. doi: 10.3390/antiox11030555
160. Sanada Y, Tan SJO, Adachi N, Miyaki S. Pharmacological targeting of heme oxygenase-1 in osteoarthritis. Antioxidants (Basel) (2021) 10(3):419. doi: 10.3390/antiox10030419
161. Paine A, Eiz-Vesper B, Blasczyk R, Immenschuh S. Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem Pharmacol (2010) 80(12):1895–903. doi: 10.1016/j.bcp.2010.07.014
162. Bloom DA, Jaiswal AK. Phosphorylation of Nrf2 at Ser40 by protein kinase c in response to antioxidants leads to the release of Nrf2 from INrf2, but is not required for Nrf2 stabilization/accumulation in the nucleus and transcriptional activation of antioxidant response element-mediated NAD(P)H:quinone oxidoreductase-1 gene expression. J Biol Chem (2003) 278(45):44675–82. doi: 10.1074/jbc.M307633200
163. Naito Y, Takagi T, Higashimura Y. Heme oxygenase-1 and anti-inflammatory M2 macrophages. Arch Biochem Biophysics (2014) 564:83–8. doi: 10.1016/j.abb.2014.09.005
164. Ben-Mordechai T, Kain D, Holbova R, Landa N, Levin L-P, Elron-Gross I, et al. Targeting and modulating infarct macrophages with hemin formulated in designed lipid-based particles improves cardiac remodeling and function. J Controlled Release (2017) 257:21–31. doi: 10.1016/j.jconrel.2017.01.001
165. Sudan K, Vijayan V, Madyaningrana K, Gueler F, Igarashi K, Foresti R, et al. TLR4 activation alters labile heme levels to regulate BACH1 and heme oxygenase-1 expression in macrophages. Free Radic Biol Med (2019) 137:131–42. doi: 10.1016/j.freeradbiomed.2019.04.024
166. Zenke-Kawasaki Y, Dohi Y, Katoh Y, Ikura T, Ikura M, Asahara T, et al. Heme induces ubiquitination and degradation of the transcription factor Bach1. Mol Cell Biol (2007) 27(19):6962–71. doi: 10.1128/MCB.02415-06
167. Dhakshinamoorthy S, Jain AK, Bloom DA, Jaiswal AK. Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H:quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J Biol Chem (2005) 280(17):16891–900. doi: 10.1074/jbc.M500166200
168. Harusato A, Naito Y, Takagi T, Uchiyama K, Mizushima K, Hirai Y, et al. BTB and CNC homolog 1 (Bach1) deficiency ameliorates TNBS colitis in mice: Role of M2 macrophages and heme oxygenase-1. Inflammation Bowel Dis (2013) 19(4):740–53. doi: 10.1097/MIB.0b013e3182802968
169. Goven D, Boutten A, Leçon-Malas V, Boczkowski J, Bonay M. Prolonged cigarette smoke exposure decreases heme oxygenase-1 and alters Nrf2 and Bach1 expression in human macrophages: roles of the MAP kinases ERK(1/2) and JNK. FEBS Lett (2009) 583(21):3508–18. doi: 10.1016/j.febslet.2009.10.010
170. Goven D, Boutten A, Leçon-Malas V, Marchal-Sommé J, Amara N, Crestani B, et al. Altered Nrf2/Keap1-Bach1 equilibrium in pulmonary emphysema. Thorax (2008) 63(10):916–24. doi: 10.1136/thx.2007.091181
171. Park SY, Jin ML, Yi EH, Kim Y, Park G. Neochlorogenic acid inhibits against LPS-activated inflammatory responses through up-regulation of Nrf2/HO-1 and involving AMPK pathway. Environ Toxicol Pharmacol (2018) 62:1–10. doi: 10.1016/j.etap.2018.06.001
172. Hess J, Angel P, Schorpp-Kistner M. AP-1 subunits: quarrel and harmony among siblings. J Cell Sci (2004) 117(Pt 25):5965–73. doi: 10.1242/jcs.01589
173. Alam J, Stewart D, Touchard C, Boinapally S, Choi AM, Cook JL. Nrf2, a Cap'n'Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem (1999) 274(37):26071–8. doi: 10.1074/jbc.274.37.26071
174. Soo Kim H, Young Park S, Kyoung Kim E, Yeon Ryu E, Hun Kim Y, Park G, et al. Acanthopanax senticosus has a heme oxygenase-1 signaling-dependent effect on porphyromonas gingivalis lipopolysaccharide-stimulated macrophages. J Ethnopharmacol (2012) 142(3):819–28. doi: 10.1016/j.jep.2012.06.006
175. Yang XJ, Liu F, Feng N, Ding XS, Chen Y, Zhu SX, et al. Berberine attenuates cholesterol accumulation in macrophage foam cells by suppressing AP-1 activity and activation of the Nrf2/HO-1 pathway. J Cardiovasc Pharmacol (2020) 75(1):45–53. doi: 10.1097/fjc.0000000000000769
176. Alam MB, Chowdhury NS, Sohrab MH, Rana MS, Hasan CM, Lee SH. Cerevisterol alleviates inflammation via suppression of MAPK/NF-κB/AP-1 and activation of the Nrf2/HO-1 signaling cascade. Biomolecules (2020) 10(2):199. doi: 10.3390/biom10020199
177. Levy S, Jaiswal AK, Forman HJ. The role of c-jun phosphorylation in EpRE activation of phase II genes. Free Radical Biol Med (2009) 47(8):1172–9. doi: 10.1016/j.freeradbiomed.2009.07.036
178. Ricchetti GA, Williams LM, Foxwell BMJ. Heme oxygenase 1 expression induced by IL-10 requires STAT-3 and phosphoinositol-3 kinase and is inhibited by lipopolysaccharide. J Leukoc Biol (2004) 76(3):719–26. doi: 10.1189/jlb.0104046
179. Kapturczak MH, Wasserfall C, Brusko T, Campbell-Thompson M, Ellis TM, Atkinson MA, et al. Heme oxygenase-1 modulates early inflammatory responses: Evidence from the heme oxygenase-1-Deficient mouse. Am J Pathol (2004) 165(3):1045–53. doi: 10.1016/S0002-9440(10)63365-2
180. Otterbein LE, Bach FH, Alam J, Soares M, Lu HT, Wysk M, et al. Carbon monoxide has anti-inflammatory effects involving the mitogen- activated protein kinase pathway. Nat Med (2000) 6(4):422–8. doi: 10.1038/74680
181. Mandal P, Park PH, McMullen MR, Pratt BT, Nagy LE. The anti-inflammatory effects of adiponectin are mediated via a heme oxygenase-1-dependent pathway in rat kupffer cells. Hepatology (2010) 51(4):1420–9. doi: 10.1002/hep.23427
182. Miyazaki T, Kirino Y, Takeno M, Samukawa S, Hama M, Tanaka M, et al. Expression of heme oxygenase-1 in human leukemic cells and its regulation by transcriptional repressor Bach1. Cancer Sci (2010) 101(6):1409–16. doi: 10.1111/j.1349-7006.2010.01550.x
183. Weis N, Weigert A, Von Knethen A, Brüne B. Heme oxygenase-1 contributes to an alternative macrophage activation profile induced by apoptotic cell supernatants. Mol Biol Cell (2009) 20(5):1280–8. doi: 10.1091/mbc.E08-10-1005
184. Mandal P, Pratt BT, Barnes M, McMullen MR, Nagy LE. Molecular mechanism for adiponectin-dependent m2 macrophage polarization link between the metabolic and innate immune activity of full-length adiponectin. J Biol Chem (2011) 286(15):13460–9. doi: 10.1074/jbc.M110.204644
185. Ahmed SMU, Luo L, Namani A, Wang XJ, Tang X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim Biophys Acta (BBA) - Mol Basis Dis (2017) 1863(2):585–97. doi: 10.1016/j.bbadis.2016.11.005
186. Banning A, Brigelius-Flohé R. NF-kappaB, Nrf2, and HO-1 interplay in redox-regulated VCAM-1 expression. Antioxid Redox Signal (2005) 7(7-8):889–99. doi: 10.1089/ars.2005.7.889
187. Chen LG, Zhang YQ, Wu ZZ, Hsieh CW, Chu CS, Wung BS. Peanut arachidin-1 enhances Nrf2-mediated protective mechanisms against TNF-α-induced ICAM-1 expression and NF-κB activation in endothelial cells. Int J Mol Med (2018) 41(1):541–7. doi: 10.3892/ijmm.2017.3238
188. Koliaraki V, Kollias G. A new role for myeloid HO-1 in the innate to adaptive crosstalk and immune homeostasis. Adv Exp Med Biol (2011) 780:101–11. doi: 10.1007/978-1-4419-5632-3_9
189. Gilmore TD. The Rel/NF-kappaB signal transduction pathway: introduction. Oncogene (1999) 18(49):6842–4. doi: 10.1038/sj.onc.1203237
190. Kim SL, Choi HS, Ko YC, Yun BS, Lee DS. 5-hydroxymaltol derived from beetroot juice through lactobacillus fermentation suppresses inflammatory effect and oxidant stress via regulating NF-kB, MAPKs pathway and NRF2/HO-1 expression. Antioxidants (Basel) (2021) 10(8):1324. doi: 10.3390/antiox10081324
191. Ouyang J, Sun L, Pan J, Zeng Z, Zeng C, Zeng F, et al. A targeted nanosystem for detection of inflammatory diseases via Fluorescent/Optoacoustic imaging and therapy via modulating Nrf2/NF-κB pathways. Small (2021) 17(42):e2102598. doi: 10.1002/smll.202102598
192. Canton M, Sánchez-Rodríguez R, Spera I, Venegas FC, Favia M, Viola A, et al. Reactive oxygen species in macrophages: Sources and targets. Front Immunol (2021) 12:734229. doi: 10.3389/fimmu.2021.734229
193. Hwang J, Jin J, Jeon S, Moon SH, Park MY, Yum DY, et al. SOD1 suppresses pro-inflammatory immune responses by protecting against oxidative stress in colitis. Redox Biol (2020) 37:101760. doi: 10.1016/j.redox.2020.101760
194. Kim HS, Lee MS. Essential role of STAT1 in caspase-independent cell death of activated macrophages through the p38 mitogen-activated protein kinase/STAT1/reactive oxygen species pathway. Mol Cell Biol (2005) 25(15):6821–33. doi: 10.1128/mcb.25.15.6821-6833.2005
195. Wenzel P, Kossmann S, Münzel T, Daiber A. Redox regulation of cardiovascular inflammation - immunomodulatory function of mitochondrial and nox-derived reactive oxygen and nitrogen species. Free Radic Biol Med (2017) 109:48–60. doi: 10.1016/j.freeradbiomed.2017.01.027
196. Xu Q, Choksi S, Qu J, Jang J, Choe M, Banfi B, et al. NADPH oxidases are essential for macrophage differentiation. J Biol Chem (2016) 291(38):20030–41. doi: 10.1074/jbc.M116.731216
197. Soares MP, Seldon MP, Gregoire IP, Vassilevskaia T, Berberat PO, Yu J, et al. Heme oxygenase-1 modulates the expression of adhesion molecules associated with endothelial cell activation. J Immunol (2004) 172(6):3553–63. doi: 10.4049/jimmunol.172.6.3553
198. He C, Ryan AJ, Murthy S, Carter AB. Accelerated development of pulmonary fibrosis via Cu,Zn-superoxide dismutase-induced alternative activation of macrophages. J Biol Chem (2013) 288(28):20745–57. doi: 10.1074/jbc.M112.410720
199. Padgett LE, Burg AR, Lei W, Tse HM. Loss of NADPH oxidase-derived superoxide skews macrophage phenotypes to delay type 1 diabetes. Diabetes (2015) 64(3):937–46. doi: 10.2337/db14-0929
200. Liu X, Zhang X, Ding Y, Zhou W, Tao L, Lu P, et al. Nuclear factor E2-related factor-2 negatively regulates NLRP3 inflammasome activity by inhibiting reactive oxygen species-induced NLRP3 priming. Antioxid Redox Signal (2017) 26(1):28–43. doi: 10.1089/ars.2015.6615
201. Liu X, Zhou W, Zhang X, Lu P, Du Q, Tao L, et al. Dimethyl fumarate ameliorates dextran sulfate sodium-induced murine experimental colitis by activating Nrf2 and suppressing NLRP3 inflammasome activation. Biochem Pharmacol (2016) 112:37–49. doi: 10.1016/j.bcp.2016.05.002
202. Wang P, Ni M, Tian Y, Wang H, Qiu J, You W, et al. Myeloid Nrf2 deficiency aggravates non-alcoholic steatohepatitis progression by regulating YAP-mediated NLRP3 inflammasome signaling. iScience (2021) 24(5):102427. doi: 10.1016/j.isci.2021.102427
203. Ganesh Yerra V, Negi G, Sharma SS, Kumar A. Potential therapeutic effects of the simultaneous targeting of the Nrf2 and NF-κB pathways in diabetic neuropathy. Redox Biol (2013) 1(1):394–7. doi: 10.1016/j.redox.2013.07.005
204. Kim MJ, Jeon JH. Recent advances in understanding Nrf2 agonism and its potential clinical application to metabolic and inflammatory diseases. Int J Mol Sci (2022) 23(5):2846. doi: 10.3390/ijms23052846
205. Lee DF, Kuo HP, Liu M, Chou CK, Xia W, Du Y, et al. KEAP1 E3 ligase-mediated downregulation of NF-kappaB signaling by targeting IKKbeta. Mol Cell (2009) 36(1):131–40. doi: 10.1016/j.molcel.2009.07.025
206. Yu M, Li H, Liu Q, Liu F, Tang L, Li C, et al. Nuclear factor p65 interacts with Keap1 to repress the Nrf2-ARE pathway. Cell Signal (2011) 23(5):883–92. doi: 10.1016/j.cellsig.2011.01.014
207. Hwang YJ, Lee EW, Song J, Kim HR, Jun YC, Hwang KA. MafK positively regulates NF-κB activity by enhancing CBP-mediated p65 acetylation. Sci Rep (2013) 3:3242. doi: 10.1038/srep03242
208. Liu GH, Qu J, Shen X. NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim Biophys Acta (2008) 1783(5):713–27. doi: 10.1016/j.bbamcr.2008.01.002
209. Bao H, Qu Q, Zhang W, Wang X, Fang J, Xue J, et al. NRF2 exerts anti-inflammatory effects in LPS-induced gEECs by inhibiting the activation of the NF-κB. Mediators Inflammation (2021) 2021:9960721. doi: 10.1155/2021/9960721
210. Rius-Pérez S, Pérez S, Martí-Andrés P, Monsalve M, Sastre J. Nuclear factor kappa b signaling complexes in acute inflammation. Antioxid Redox Signal (2020) 33(3):145–65. doi: 10.1089/ars.2019.7975
211. Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H, et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun (2016) 7:11624. doi: 10.1038/ncomms11624
212. Ni L, Lin Z, Hu S, Shi Y, Jiang Z, Zhao J, et al. Itaconate attenuates osteoarthritis by inhibiting STING/NF-κB axis in chondrocytes and promoting M2 polarization in macrophages. Biochem Pharmacol (2022) 198:114935. doi: 10.1016/j.bcp.2022.114935
213. Spinelli JB, Rosen PC, Sprenger HG, Puszynska AM, Mann JL, Roessler JM, et al. Fumarate is a terminal electron acceptor in the mammalian electron transport chain. Science (2021) 374(6572):1227–37. doi: 10.1126/science.abi7495
214. Yin M, O'Neill LAJ. The role of the electron transport chain in immunity. FASEB J (2021) 35(12):e21974. doi: 10.1096/fj.202101161R
215. Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell (2016) 167(2):457–70.e13. doi: 10.1016/j.cell.2016.08.064
216. Mehta MM, Weinberg SE, Chandel NS. Mitochondrial control of immunity: beyond ATP. Nat Rev Immunol (2017) 17(10):608–20. doi: 10.1038/nri.2017.66
217. Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity (2015) 42(3):419–30. doi: 10.1016/j.immuni.2015.02.005
218. Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature (2018) 556(7699):113–7. doi: 10.1038/nature25986
219. Hooftman A, Angiari S, Hester S, Corcoran SE, Runtsch MC, Ling C, et al. The immunomodulatory metabolite itaconate modifies NLRP3 and inhibits inflammasome activation. Cell Metab (2020) 32(3):468–78.e7. doi: 10.1016/j.cmet.2020.07.016
220. Muri J, Wolleb H, Broz P, Carreira EM, Kopf M. Electrophilic Nrf2 activators and itaconate inhibit inflammation at low dose and promote IL-1β production and inflammatory apoptosis at high dose. Redox Biol (2020) 36:101647. doi: 10.1016/j.redox.2020.101647
221. Mrowietz U, Morrison PJ, Suhrkamp I, Kumanova M, Clement B. The pharmacokinetics of fumaric acid esters reveal their In vivo effects. Trends Pharmacol Sci (2018) 39(1):1–12. doi: 10.1016/j.tips.2017.11.002
222. Hoyle C, Green JP, Allan SM, Brough D, Lemarchand E. Itaconate and fumarate derivatives inhibit priming and activation of the canonical NLRP3 inflammasome in macrophages. Immunology (2022) 165(4):460–80. doi: 10.1111/imm.13454
223. Linker RA, Lee DH, Ryan S, van Dam AM, Conrad R, Bista P, et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain (2011) 134(Pt 3):678–92. doi: 10.1093/brain/awq386
224. Zhang P, Wang X, Peng Q, Jin Y, Shi G, Fan Z, et al. Four-octyl itaconate protects chondrocytes against H(2)O(2)-induced oxidative injury and attenuates osteoarthritis progression by activating Nrf2 signaling. Oxid Med Cell Longev (2022) 2022:2206167. doi: 10.1155/2022/2206167
225. Zhang Q, Bai X, Wang R, Zhao H, Wang L, Liu J, et al. 4-octyl itaconate inhibits lipopolysaccharide (LPS)-induced osteoarthritis via activating Nrf2 signalling pathway. J Cell Mol Med (2022) 26(5):1515–29. doi: 10.1111/jcmm.17185
226. Li Y, Tang J, Hu Y. Dimethyl fumarate protection against collagen II degradation. Biochem Biophys Res Commun (2014) 454(2):257–61. doi: 10.1016/j.bbrc.2014.10.005
227. Jayaprakasha GK, Jaganmohan Rao L, Sakariah KK. Antioxidant activities of curcumin, demethoxycurcumin and bisdemethoxycurcumin. Food Chem (2006) 98(4):720–4. doi: 10.1016/j.foodchem.2005.06.037
228. Shahcheraghi SH, Salemi F, Peirovi N, Ayatollahi J, Alam W, Khan H, et al. Nrf2 regulation by curcumin: Molecular aspects for therapeutic prospects. Molecules (2021) 27(1):167. doi: 10.3390/molecules27010167
229. Boyanapalli SS, Paredes-Gonzalez X, Fuentes F, Zhang C, Guo Y, Pung D, et al. Nrf2 knockout attenuates the anti-inflammatory effects of phenethyl isothiocyanate and curcumin. Chem Res Toxicol (2014) 27(12):2036–43. doi: 10.1021/tx500234h
230. Lin X, Bai D, Wei Z, Zhang Y, Huang Y, Deng H, et al. Curcumin attenuates oxidative stress in RAW264.7 cells by increasing the activity of antioxidant enzymes and activating the Nrf2-Keap1 pathway. PloS One (2019) 14(5):e0216711. doi: 10.1371/journal.pone.0216711
231. Seibert K, Zhang Y, Leahy K, Hauser S, Masferrer J, Perkins W, et al. Pharmacological and biochemical demonstration of the role of cyclooxygenase 2 in inflammation and pain. Proc Natl Acad Sci U.S.A. (1994) 91(25):12013–7. doi: 10.1073/pnas.91.25.12013
232. Jiang C, Luo P, Li X, Liu P, Li Y, Xu J. Nrf2/ARE is a key pathway for curcumin-mediated protection of TMJ chondrocytes from oxidative stress and inflammation. Cell Stress Chaperones (2020) 25(3):395–406. doi: 10.1007/s12192-020-01079-z
233. Yan D, He B, Guo J, Li S, Wang J. Involvement of TLR4 in the protective effect of intra-articular administration of curcumin on rat experimental osteoarthritis. Acta Cir Bras (2019) 34(6):e201900604. doi: 10.1590/s0102-865020190060000004
234. Kang C, Jung E, Hyeon H, Seon S, Lee D. Acid-activatable polymeric curcumin nanoparticles as therapeutic agents for osteoarthritis. Nanomedicine (2020) 23:102104. doi: 10.1016/j.nano.2019.102104
235. Park S, Lee LR, Seo JH, Kang S. Curcumin and tetrahydrocurcumin both prevent osteoarthritis symptoms and decrease the expressions of pro-inflammatory cytokines in estrogen-deficient rats. Genes Nutr (2016) 11:2. doi: 10.1186/s12263-016-0520-4
236. Wang Q, Ye C, Sun S, Li R, Shi X, Wang S, et al. Curcumin attenuates collagen-induced rat arthritis via anti-inflammatory and apoptotic effects. Int Immunopharmacol (2019) 72:292–300. doi: 10.1016/j.intimp.2019.04.027
237. Wang J, Wang X, Cao Y, Huang T, Song DX, Tao HR. Therapeutic potential of hyaluronic acid/chitosan nanoparticles for the delivery of curcuminoid in knee osteoarthritis and an in vitro evaluation in chondrocytes. Int J Mol Med (2018) 42(5):2604–14. doi: 10.3892/ijmm.2018.3817
238. D'Andrea G. Quercetin: A flavonol with multifaceted therapeutic applications? Fitoterapia (2015) 106:256–71. doi: 10.1016/j.fitote.2015.09.018
239. Yao P, Nussler A, Liu L, Hao L, Song F, Schirmeier A, et al. Quercetin protects human hepatocytes from ethanol-derived oxidative stress by inducing heme oxygenase-1 via the MAPK/Nrf2 pathways. J Hepatol (2007) 47(2):253–61. doi: 10.1016/j.jhep.2007.02.008
240. Fu J, Huang J, Lin M, Xie T, You T. Quercetin promotes diabetic wound healing via switching macrophages from M1 to M2 polarization. J Surg Res (2020) 246:213–23. doi: 10.1016/j.jss.2019.09.011
241. Wang Y, Li C, Wan Y, Qi M, Chen Q, Sun Y, et al. Quercetin-loaded ceria nanocomposite potentiate dual-directional immunoregulation via macrophage polarization against periodontal inflammation. Small (2021) 17(41):e2101505. doi: 10.1002/smll.202101505
242. Tsai CF, Chen GW, Chen YC, Shen CK, Lu DY, Yang LY, et al. Regulatory effects of quercetin on M1/M2 macrophage polarization and Oxidative/Antioxidative balance. Nutrients (2021) 14(1):67. doi: 10.3390/nu14010067
243. Luo X, Bao X, Weng X, Bai X, Feng Y, Huang J, et al. The protective effect of quercetin on macrophage pyroptosis via TLR2/Myd88/NF-κB and ROS/AMPK pathway. Life Sci (2022) 291:120064. doi: 10.1016/j.lfs.2021.120064
244. Sirše M. Effect of dietary polyphenols on osteoarthritis-molecular mechanisms. Life (Basel) (2022) 12(3):436. doi: 10.3390/life12030436
245. Li W, Wang Y, Tang Y, Lu H, Qi Y, Li G, et al. Quercetin alleviates osteoarthritis progression in rats by suppressing inflammation and apoptosis via inhibition of IRAK1/NLRP3 signaling. J Inflammation Res (2021) 14:3393–403. doi: 10.2147/jir.S311924
246. Qiu L, Luo Y, Chen X. Quercetin attenuates mitochondrial dysfunction and biogenesis via upregulated AMPK/SIRT1 signaling pathway in OA rats. BioMed Pharmacother (2018) 103:1585–91. doi: 10.1016/j.biopha.2018.05.003
247. Wang XP, Xie WP, Bi YF, Wang BA, Song HB, Wang SL, et al. Quercetin suppresses apoptosis of chondrocytes induced by IL-1β via inactivation of p38 MAPK signaling pathway. Exp Ther Med (2021) 21(5):468. doi: 10.3892/etm.2021.9899
248. Hu Y, Gui Z, Zhou Y, Xia L, Lin K, Xu Y. Quercetin alleviates rat osteoarthritis by inhibiting inflammation and apoptosis of chondrocytes, modulating synovial macrophages polarization to M2 macrophages. Free Radic Biol Med (2019) 145:146–60. doi: 10.1016/j.freeradbiomed.2019.09.024
249. Permatasari DA, Karliana D, Iskandarsyah I, Arsianti A, Bahtiar A. Quercetin prevent proteoglycan destruction by inhibits matrix metalloproteinase-9, matrix metalloproteinase-13, a disintegrin and metalloproteinase with thrombospondin motifs-5 expressions on osteoarthritis model rats. J Adv Pharm Technol Res (2019) 10(1):2–8. doi: 10.4103/japtr.JAPTR_331_18
250. Mok SW, Fu SC, Cheuk YC, Chu IM, Chan KM, Qin L, et al. Intra-articular delivery of quercetin using thermosensitive hydrogel attenuate cartilage degradation in an osteoarthritis rat model. Cartilage (2020) 11(4):490–9. doi: 10.1177/1947603518796550
251. Acuña-Castroviejo D, Escames G, Venegas C, Díaz-Casado ME, Lima-Cabello E, López LC, et al. Extrapineal melatonin: sources, regulation, and potential functions. Cell Mol Life Sci (2014) 71(16):2997–3025. doi: 10.1007/s00018-014-1579-2
252. Zhang HM, Zhang Y. Melatonin: a well-documented antioxidant with conditional pro-oxidant actions. J Pineal Res (2014) 57(2):131–46. doi: 10.1111/jpi.12162
253. Zhang Y, Liu T, Yang H, He F, Zhu X. Melatonin: A novel candidate for the treatment of osteoarthritis. Ageing Res Rev (2022) 78:101635. doi: 10.1016/j.arr.2022.101635
254. Mayo JC, Sainz RM, Tan DX, Hardeland R, Leon J, Rodriguez C, et al. Anti-inflammatory actions of melatonin and its metabolites, N1-acetyl-N2-formyl-5-methoxykynuramine (AFMK) and N1-acetyl-5-methoxykynuramine (AMK), in macrophages. J Neuroimmunol (2005) 165(1-2):139–49. doi: 10.1016/j.jneuroim.2005.05.002
255. Xia MZ, Liang YL, Wang H, Chen X, Huang YY, Zhang ZH, et al. Melatonin modulates TLR4-mediated inflammatory genes through MyD88- and TRIF-dependent signaling pathways in lipopolysaccharide-stimulated RAW264.7 cells. J Pineal Res (2012) 53(4):325–34. doi: 10.1111/j.1600-079X.2012.01002.x
256. Yi WJ, Kim TS. Melatonin protects mice against stress-induced inflammation through enhancement of M2 macrophage polarization. Int Immunopharmacol (2017) 48:146–58. doi: 10.1016/j.intimp.2017.05.006
257. Xia Y, Chen S, Zeng S, Zhao Y, Zhu C, Deng B, et al. Melatonin in macrophage biology: Current understanding and future perspectives. J Pineal Res (2019) 66(2):e12547. doi: 10.1111/jpi.12547
258. Liu SC, Tsai CH, Wang YH, Su CM, Wu HC, Fong YC, et al. Melatonin abolished proinflammatory factor expression and antagonized osteoarthritis progression in vivo. Cell Death Dis (2022) 13(3):215. doi: 10.1038/s41419-022-04656-5
259. Bernardo ME, Fibbe WE. Mesenchymal stromal cells: sensors and switchers of inflammation. Cell Stem Cell (2013) 13(4):392–402. doi: 10.1016/j.stem.2013.09.006
260. Dimarino AM, Caplan AI, Bonfield TL. Mesenchymal stem cells in tissue repair. Front Immunol (2013) 4:201. doi: 10.3389/fimmu.2013.00201
261. Hata N, Shinojima N, Gumin J, Yong R, Marini F, Andreeff M, et al. Platelet-derived growth factor BB mediates the tropism of human mesenchymal stem cells for malignant gliomas. Neurosurgery (2010) 66(1):144–56; discussion 56-7. doi: 10.1227/01.Neu.0000363149.58885.2e
262. Luque-Campos N, Bustamante-Barrientos FA, Pradenas C, García C, Araya MJ, Bohaud C, et al. The macrophage response is driven by mesenchymal stem cell-mediated metabolic reprogramming. Front Immunol (2021) 12:624746. doi: 10.3389/fimmu.2021.624746
263. Li C, Jin Y, Wei S, Sun Y, Jiang L, Zhu Q, et al. Hippo signaling controls NLR family pyrin domain containing 3 activation and governs immunoregulation of mesenchymal stem cells in mouse liver injury. Hepatology (2019) 70(5):1714–31. doi: 10.1002/hep.30700
264. Hamilton AM, Cheung WY, Gómez-Aristizábal A, Sharma A, Nakamura S, Chaboureau A, et al. Iron nanoparticle-labeled murine mesenchymal stromal cells in an osteoarthritic model persists and suggests anti-inflammatory mechanism of action. PloS One (2019) 14(12):e0214107. doi: 10.1371/journal.pone.0214107
265. Jia Z, Kang B, Cai Y, Chen C, Yu Z, Li W, et al. Cell-free fat extract attenuates osteoarthritis via chondrocytes regeneration and macrophages immunomodulation. Stem Cell Res Ther (2022) 13(1):133. doi: 10.1186/s13287-022-02813-3
266. Gao Y, Huang X, Lin H, Zhao M, Liu W, Li W, et al. Adipose mesenchymal stem cell-derived antioxidative extracellular vesicles exhibit anti-oxidative stress and immunomodulatory effects under PM(2.5) exposure. Toxicology (2021) 447:152627. doi: 10.1016/j.tox.2020.152627
267. Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the international society for extracellular vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles (2018) 7(1):1535750. doi: 10.1080/20013078.2018.1535750
268. Wang J, Xia J, Huang R, Hu Y, Fan J, Shu Q, et al. Mesenchymal stem cell-derived extracellular vesicles alter disease outcomes via endorsement of macrophage polarization. Stem Cell Res Ther (2020) 11(1):424. doi: 10.1186/s13287-020-01937-8
269. Arabpour M, Saghazadeh A, Rezaei N. Anti-inflammatory and M2 macrophage polarization-promoting effect of mesenchymal stem cell-derived exosomes. Int Immunopharmacol (2021) 97:107823. doi: 10.1016/j.intimp.2021.107823
270. Zhao R, Wang L, Wang T, Xian P, Wang H, Long Q. Inhalation of MSC-EVs is a noninvasive strategy for ameliorating acute lung injury. J Control Release (2022) 345:214–30. doi: 10.1016/j.jconrel.2022.03.025
271. Kou L, Huang H, Tang Y, Sun M, Li Y, Wu J, et al. Opsonized nanoparticles target and regulate macrophage polarization for osteoarthritis therapy: A trapping strategy. J Control Release (2022) 347:237–55. doi: 10.1016/j.jconrel.2022.04.037
272. Chang HH, Hsu SP, Chien CT. Intrarenal transplantation of hypoxic preconditioned mesenchymal stem cells improves glomerulonephritis through anti-oxidation, anti-ER stress, anti-inflammation, anti-apoptosis, and anti-autophagy. Antioxidants (Basel) (2019) 9(1):2. doi: 10.3390/antiox9010002
273. Ning H, Chen H, Deng J, Xiao C, Xu M, Shan L, et al. Exosomes secreted by FNDC5-BMMSCs protect myocardial infarction by anti-inflammation and macrophage polarization via NF-κB signaling pathway and Nrf2/HO-1 axis. Stem Cell Res Ther (2021) 12(1):519. doi: 10.1186/s13287-021-02591-4
274. Chiang CK, Loh JZ, Yang TH, Huang KT, Wu CT, Guan SS, et al. Prevention of acute kidney injury by low intensity pulsed ultrasound via anti-inflammation and anti-apoptosis. Sci Rep (2020) 10(1):14317. doi: 10.1038/s41598-020-71330-1
275. Gurkan I, Ranganathan A, Yang X, Horton WE Jr., Todman M, Huckle J, et al. Modification of osteoarthritis in the guinea pig with pulsed low-intensity ultrasound treatment. Osteoarthritis Cartilage (2010) 18(5):724–33. doi: 10.1016/j.joca.2010.01.006
276. Sawitzke AD, Jackson CG, Carlson K, Bizien MD, Leiner M, Reda DJ, et al. Effect of pulsed low-intensity ultrasonography on symptom relief and tibiofemoral articular cartilage thickness among veterans affairs enrollees with knee osteoarthritis: A randomized clinical trial. JAMA Netw Open (2022) 5(3):e220632. doi: 10.1001/jamanetworkopen.2022.0632
277. Sahu N, Budhiraja G, Subramanian A. Preconditioning of mesenchymal stromal cells with low-intensity ultrasound: influence on chondrogenesis and directed SOX9 signaling pathways. Stem Cell Res Ther (2020) 11(1):6. doi: 10.1186/s13287-019-1532-2
278. Chen CH, Kuo SM, Tien YC, Shen PC, Kuo YW, Huang HH. Steady augmentation of anti-osteoarthritic actions of rapamycin by liposome-encapsulation in collaboration with low-intensity pulsed ultrasound. Int J Nanomed (2020) 15:3771–90. doi: 10.2147/ijn.S252223
279. Zuo D, Tan B, Jia G, Wu D, Yu L, Jia L. A treatment combined prussian blue nanoparticles with low-intensity pulsed uinltrasound alleviates cartilage damage in knee osteoarthritis by initiating PI3K/Akt/mTOR pathway. Am J Transl Res (2021) 13(5):3987–4006.
280. Yamaguchi A, Maeshige N, Ma X, Uemura M, Noguchi H, Matsuda M, et al. Pulsed-ultrasound irradiation induces the production of itaconate and attenuates inflammatory responses in macrophages. J Inflammation Res (2022) 15:2387–95. doi: 10.2147/jir.S361609
Keywords: Nrf2, macrophage, osteoarthritis, intra-articular, anti-inflammation
Citation: Wang L and He C (2022) Nrf2-mediated anti-inflammatory polarization of macrophages as therapeutic targets for osteoarthritis. Front. Immunol. 13:967193. doi: 10.3389/fimmu.2022.967193
Received: 12 June 2022; Accepted: 27 July 2022;
Published: 12 August 2022.
Edited by:
Simone Mader, Ludwig Maximilian University of Munich, GermanyReviewed by:
João Alfredo Moraes, Federal University of Rio de Janeiro, BrazilCopyright © 2022 Wang and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chengqi He, aHhrZmhjcTIwMTVAMTI2LmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.