
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 15 August 2022
Sec. Viral Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.963923
This article is part of the Research Topic Antiviral Innate Immune Sensing, Regulation, and Viral Immune Evasion View all 48 articles
 Zhijie Jian1†
Zhijie Jian1† Rui Ma1†
Rui Ma1† Ling Zhu1,2
Ling Zhu1,2 Huidan Deng1Fengqin Li1,3
Huidan Deng1Fengqin Li1,3 Jun Zhao1,2Lishuang Deng1Siyuan Lai1Xiangang Sun1
Jun Zhao1,2Lishuang Deng1Siyuan Lai1Xiangang Sun1 Huaqiao Tang1
Huaqiao Tang1 Zhiwen Xu1,2*
Zhiwen Xu1,2*IFN is the most potent antiviral cytokine required for the innate and adaptive immune responses, and its expression can help the host defend against viral infection. Arteriviruses have evolved strategies to antagonize the host cell’s innate immune responses, interfering with IFN expression by interfering with RIG, blocking PRR, obstructing IRF-3/7, NF-κB, and degrading STAT1 signaling pathways, thereby assisting viral immune evasion. Arteriviruses infect immune cells and may result in persistence in infected hosts. In this article, we reviewed the strategies used by Arteriviruses to antagonize IFN production and thwart IFN-activated antiviral signaling, mainly including structural and nonstructural proteins of Arteriviruses encoding IFN antagonists directly or indirectly to disrupt innate immunity. This review will certainly provide a better insight into the pathogenesis of the arthritis virus and provide a theoretical basis for developing more efficient vaccines.
The mammalian immune system can effectively detect and fight against viral infections by inducing the production of type I interferon, which forms the first line of defense. The type I interferon response consists of two parts. The first part is triggered by viral stimulation when cells produce type I interferon and secrete IFN. In the second part of the response, both the IFN-producing cell and adjacent cells sense IFN, leading to the production of IFN-stimulated genes (ISG) (1).
Arteriviruses include porcine reproductive and respiratory syndrome virus (PRRSV), equine arteritis virus (EAV), lactate dehydrogenase-elevating virus (LDV), simian hemorrhagic fever virus (SHFV), and swing possum virus (SPV). They can persist in infected animals, PRRSV can persist for six months in pigs, EAV can persist for life in horses, LDV can usually persist in mice without pathological consequences for the host, and SHFV can show different symptoms in macaques and baboons, with macaques showing fatal hemorrhagic fever but baboons showing only persistent asymptomatic infection (2–5). EAV and PRRSV are considered important pathogens in veterinary studies among these arteriviruses. They can cause significant economic losses in the equine and swine industries, share similar molecular characteristics, and cause reproductive disorders in livestock (6). Therefore, effective Arterivirus control and prevention methods are urgently needed. This review summarizes research advances for the different pathways of anti-IFN responses to Arteriviruses (Figure 1). We want to provide creative insights to guide the development of innovative strategies to achieve Arteriviruses prevention and control.
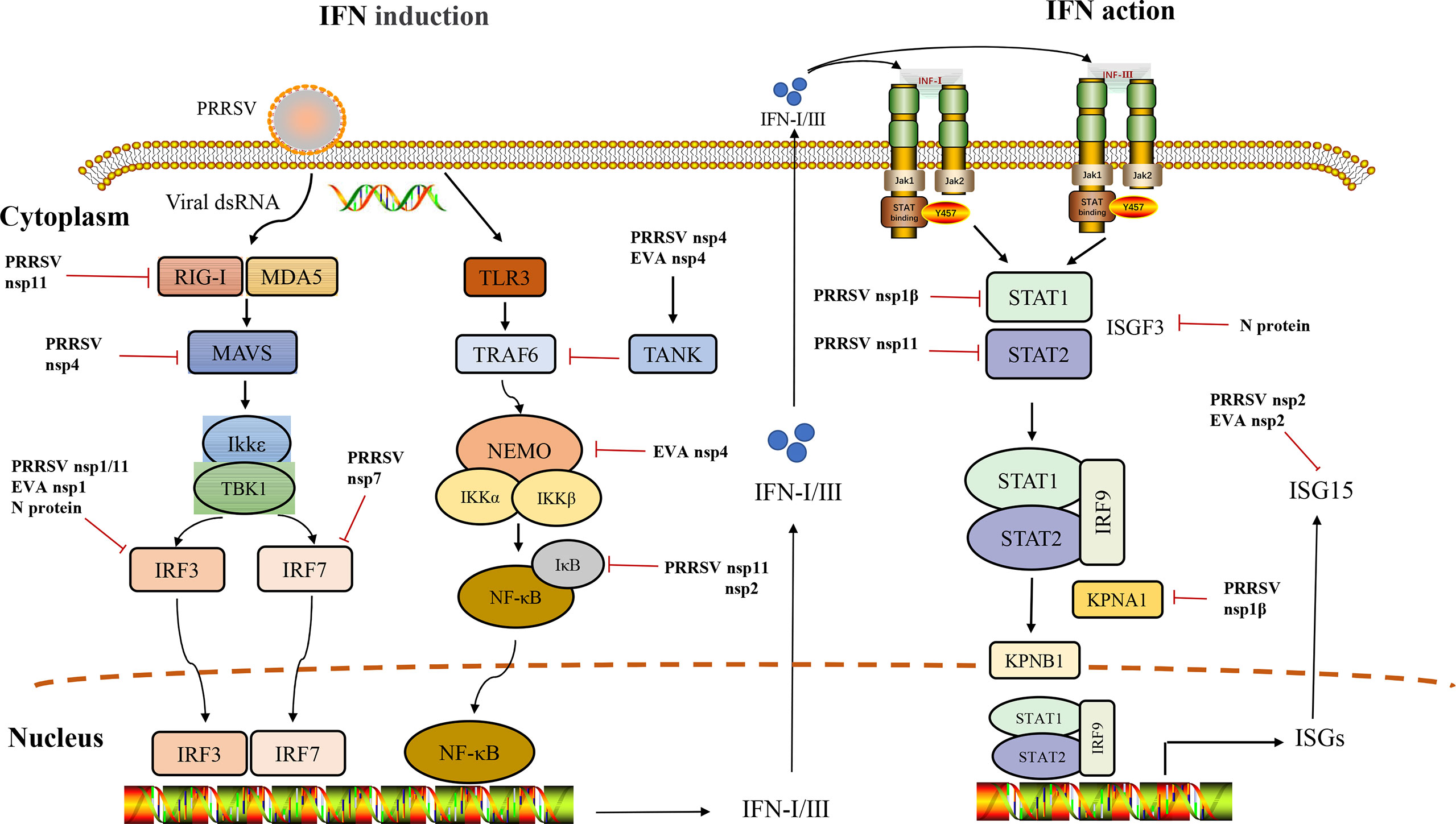
Figure 1 Interference of IFN induction and its downstream signaling pathway by Arteriviruses.
IFN is a soluble factor discovered in 1957 in viral infections and is named for its ability to interfere in viral replication (7). Interferons are classified into types I, II, and III IFNs (IFN-I, II, and III). In mammals, IFN-I is composed of 19 IFN proteins: 14 IFN-α subtypes (IFN-α1 to α14), IFN-ω, IFN-ε, IFN-τ, IFN-κ, and IFN-β and IFN-I signaling is mediated through the IFN-I receptor (IFNAR), which is a common cell surface receptor. IFN-II family is mainly produced by T lymphocytes and natural killer cells (NK cells), which are mediated by IFNGR (a receptor composed of IFNGR1 and IFNGR2). IFN-III comprises 4 subtypes, IFN-λ1, IFN-λ2, IFN-λ3, and IFN-λ4, and it is mediated by IFNLR (a receptor composed of IFNLR1 and IL10R2) (8–12). IFN-III is associated with IFN-I and IL-10, which have antiviral activity (10). IFN-λ is an epithelial cytokine that limits viral replication in epithelial cells and forms an additional protective layer at mucosal sites (13).
The activation of IFN-I response is divided into three phases: ①pattern recognition receptors (PRRs) on the cell membrane or cytoplasm PRRs recognize pathogen-associated molecular patterns (PAMPs); ②IFN triggers JAK-STAT via paracrine or autocrine signaling; ③expression of a large number of antiviral ISG genes, which puts the host into an antiviral state (14).
Most PRRs in the innate immune system of vertebrates can be classified into the following five types: Toll-like receptors (TLRs), retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), nucleotide oligomerization domain (NOD)-like receptors (NLRs), C-type lectin receptors (CLRs), and absent in melanoma-2 (AIM2)-like receptors (ALRs) (15). We will discuss two classes of viral sensing PRRs in this review. These include TLRs and RLRs, which are important for inducing the type I IFN response. TLRs primarily recognize viral RNA or DNA in the endosomes, and RLRs primarily recognize viral RNA in the cytoplasm. They play a key role in the induction of host IFN expression (16–18). Still, another is a set of structurally unrelated viral DNA sensors (Cyclic GMP-AMP synthase) and IFI16 located in the cytoplasm and/or nucleus, and it also plays a critical role in inducing the expression of host IFN (19). Interferon is normally secreted and binds to cell surface receptors in response to viral infection and activates a JAK/STAT-dependent signaling cascade that produces ISG and puts the cell in a state of resistance (20).
The prolonged infection caused by arteriviruses has a greater association with immune evasion, mainly through the suppression of interferon by various pathways to promote viral proliferation and long-term infection. Clarifying the antagonism between arteriviruses and interferon is important to understand the pathogenesis and find relevant targets as a basis for vaccine development. Therefore, this review will summarize the underlying immune evasion mechanisms by arteriviruses.
Arteriviruses use different mechanisms to suppress interferon responses to evade the host’s innate immune response. Early studies have shown that PRRSV infection in pigs leads to a weak induction of the natural immune response. Detection of interferon in alveolar lavage fluid reveals that interferon is maintained at very low levels, suggesting that PRRSV can interfere with IFN-I transcription directly at the level of IFN-β gene transcription in the early stages of infection (21–23). Before the challenge, IFN-pretreatment of pigs in vivo reduced PRRSV-induced symptoms. However, it appears that IFN therapy could not rescue PRRSV-infected swine from death, but it extended survival time (24). In vitro study, the inhibition of IFNs expression by PRRSV was similarly observed in MARC-145 cells infected with PRRSV and PAMs cells (23, 25).
Similarly, IFN-β production in Equine endothelial cells (EECS) was significantly inhibited after EAV infection, in contrast to SeV infection, which stimulated high levels of IFN-I expression, and EAV infection also significantly inhibited SeV-induced IFN-I production (26). All of the above studies suggest that Arteriviruses induce IFN inhibition, and the main mechanisms responsible for this phenomenon are reviewed next.
Different arteritis virus structural and nonstructural proteins exercise different functions in IFN inhibition (Table 1).

Table 1 Arteriviruses proteins inhibit IFN downstream signaling pathway.
ORF2, 2a, 3, 4, 5, 6, and 7 of PRRSV encode GP2, E, GP3, GP4, GP5, M protein, and N protein, respectively (41). Mechanistic studies of PRRSV antagonism to type I interferon have focused on IFN-α/β, two factors that play a major role in the fight against PRRSV infection. At least two structural proteins (M and N proteins) and four nonstructural proteins of PRRSV, nsp1, nsp2, nsp11, and nsp4, have been identified as exhibiting inhibitory effects on IFN-β promoter activation, with nsp1 showing the strongest inhibitory effect and self-cleavage of nsp1 during infection to produce NSP1α and NSP1β. NSP1β can inhibit IRF3 phosphorylation and NF-κB-dependent nuclear translocation (30, 31). Nsp4 is a 3C-like serine protease that antagonizes type I interferon production by cleaving mitochondria antiviral signaling protein (MAVS) and NF-κB essential regulators (NEMO) (32, 33, 42). PRRSV nsp7 inhibits IRF7 expression, downregulates IFN and downstream ISG expression, and promotes viral replication (38). Nsp11 can suppress the activation of IFN-β by cleaving the mRNA of MAVS (also known as IPS-1, Cardif, and VISA) via the endoribonuclease domain (43). PRRSV N protein is distributed in both cytoplasm and nucleus, suggesting that altered localization of N protein may affect its IFNs inhibitory activity (28). Some studies have demonstrated that N protein prevents IFN-β induction like that of nsp2 (44). IFN-γ plays an important role in the immune response against PRRSV. The duration of viremia and the degree of morbidity did not correlate well, but ELISA experiments showed that N, M protein, and nsp2 were indeed associated with PRRSV-specific induction of IFN-γ secretion by lymphocytes (27).
Among the EAV nsp, four nonstructural proteins, nsp1, nsp2, nsp4, and nsp11, have been identified as potential interferon antagonists. It was shown that the homolog of PRRSV nsp1α/β, EAV nsp1, has the strongest ability to inhibit type I IFN synthesis (26). EAV nsp2-encoded papain-like proteinase (PLP2) inhibits Ub- and ISG15-dependent innate immune responses (36). Similarly, EAV nsp4 can inhibit virus-induced IFN-β production by targeting NEMO for protein cleavage, and cleavage occurs at four sites, including E166, E171, Q205, and E349, consistent with PRRSV cleavage sites (34).
The RLRs group consisted of RIG-I, melanoma differentiation-associated gene 5 (MDA5), and Laboratory of Genetics and Physiology 2 (LGP2). RIG-I recognizes the triphosphate and diphosphate at the stem end of dsRNA, which is the hallmark of viral RNA of most RNA viruses (45). MDA5 perceives long dsRNA, which is believed to represent the intermediate replication product of many RNA viruses (46). LGP2, a protein structurally related to both RIG-I and MDA5, appears to be a cofactor for viral RNA sensing by a mechanism that is not completely understood and likely involves making viral RNA more accessible to RIG-I or MDA5 (47). RIG-I and MDA5 are important sensors for IFN-I production in the porcine innate immune system (48). RIG-1 and MDA-5 detect specific viral RNA PAMPs, while LGP2 negatively regulates RIG-I signaling and promotes RNA binding to MDA5 (49). RIG-I-like receptor-mediated type I IFN production plays an important role in the host’s defense against viral invasion (50). dsRNA is a specific secondary structure of viral RNA detected by RIG-I/MDA5 and induces IFN-α/β production through cascade activation of the RLR pathway (51). Viral dsRNA can trigger RIG-I, and the CARD domain of RIG-I interacts with the CARD domain of MAVS, and activation of MAVS recruits multiple downstream signaling components to the mitochondria, leading to activation of κ-B kinase inhibitor ϵ (Iκκϵ) and TANK-binding kinase 1 (TBK1), which in turn causes IRF3 phosphorylation. Phosphorylated IRF3 forms a dimer and translocates to the nucleus, activating transcription of the IFN-I gene (52, 53).
PRRSV infection inhibits IFN-β production mainly by interfering with MAVS activation in the RIG-I signaling pathway (54). The porcine reproductive and respiratory syndrome virus (PRRSV) 3C-like protease (3CLSP), by contrast, cleaves MAVS in a proteasome- and caspase-independent manner at Glu268 (E268/G269). Both cleavage products fail to activate the type I IFN response (55). Further studies showed that the highly pathogenic porcine reproductive and respiratory syndrome virus (HP-PRRSV) protein nsp4 cleaves MAVS and blocks RLR signaling, and causes specific downregulation of the MAVS, but nsp4 in the typical PRRSV strain CH-1a has no effect on MAVS, so this may be a strategy evolved by the virulent strain (32). Nsp11 reduces RIG-I mRNA dependent on its endoribonuclease activity. Nsp11 inhibits IRF3 and NF-κB activity when stimulated with dsRNA analogs and TNF-α, respectively, suggesting that this inhibition also depends on RLR (56).
Recent studies have shown that MDA5 senses the EAV genome to induce IFN expression (57).
Interferon regulatory factors (IRFs) are a family of transcription factors with 9 members identified so far. IRF4, 5, and 6 have no substantial role in IFN regulation and are also not described. IRF-1 and IRF-2 mRNA were expressed in multiple cell types, whereas IRF-8 expression was restricted to myeloid and lymphoid cell lines, and its mRNA was significantly upregulated in response to viral infection or IFN stimulation (58, 59). IRF-9 was originally identified as the DNA-binding subunit of ISGF3 and was proven essential for the antiviral response to IFN-α/β and IFN-γ (60, 61). IRF-3 and IRF-7 are closely related in their primary structure, and recent studies have identified an important and distinct role for these two factors in IFN-α/β gene induction in arteritis virus infection.
It has been suggested that IFN-λ expression is more flexible than IFN-α/β expression, which may allow IFN-III to be expressed in response to a wider range of stimuli than IFN-I, and would potentially make IFN-III expression less sensitive to microbial evasion strategies targeting the IRF pathway (62, 63). IRF3 is a target factor for various viruses and can impair natural immune signaling. Most viruses inhibit IRF3 phosphorylation and thus also IRF3 dimerization and translocation. In the absence of IRF-3 activation and IFN-β production, alternative pathways allow IFN-λ to be induced without IRF-3 activation. IRF-3 is a virus targeting factor and can impair innate immune signaling. Most viruses inhibit IRF3 phosphorylation, dimerization, and nuclear translocation. TBK1 and IKKϵ can induce IRF3 and IRF7 phosphorylation and be affected by K63-linked polyubiquitination (64, 65). The ubiquitin chain may serve as a platform for the assembly of the TBK1 signaling complex, so for TBK1, polyubiquitination of the K63 linkage appears to be important for TLR and RLR-induced IFN production (65, 66). Activated TBK1/IKKϵ phosphorylates IRF3 and/or IRF7 at specific serine residues in the cell membrane, which are subsequently transferred to the nucleus to recruit the coactivator CBP/p300 and form a complex to bind the IRF-3 response element of the IFN-β promoter (PRD I and III) (67–69). Interestingly, IRF7 was induced during IFN signaling at low levels in most cells, suggesting that IRF7 can strongly enhance IFN production (70).
Viral proteins target TBK1 to block IFNβ production by preventing TBK1 activation from MAVS or inhibiting IRF3 activation from TBK1. Once activated, MAVS signaling recruits multiple kinases, ubiquitin ligases, and adapters, leading to phosphorylation and activation of potential transcription factors involved in IFN promoter activation. These transcription factors, IRF factors, especially IRF3 and IRF7, are essential for IFN induction (71, 72). In addition, IRFs are also required for IFN induction during TLR activation. Therefore, it is unsurprising that virally encoded IFN antagonists can inhibit IRFs.
PRRSV nsp1 is the most potent interferon repressor protein among the nonstructural proteins. Studies have shown that the inhibition of type I IFN is due to PRRSV nsp1α/β blocking dsRNA-induced activation of IRF-3. In the presence of nsp1α/β, phosphorylation of IRF-3 and its nuclear translocation occurred normally, but the association of IRF3 with cAMP response element-binding protein(CBP) in the nucleus was inhibited, thereby blocking IRF-3 activation (73, 74). Nsp4 was reported to inhibit IRF-3-mediated activation of the IFN-β promoter, an inhibition derived from the hydrolytic activity of the nsp4 3C-like serine protease (75, 76). Recently, it has been shown that N proteins can inhibit poly(I:C)-mediated IRF-3 phosphorylation and nuclear translocation, thereby suppressing the expression of IFN-β (44). Therefore, IRF3 can be a direct viral target to block IFN production and a key target for vaccine development. IRF7 is another important regulator in the interferon signaling pathway. IRF7 can inhibit the early replication of PRRSV. While PRRSV nsp7 significantly down-regulates IRF7 expression, nsp4 and nsp5 do not down-regulate IRF7 expression. Instead, nsp11 upregulates IRF7 expression, which may result from complex virus-protein interactions (38).
Similarly, EVA nsp1α and NSP1β mediated the inactivation of MAVS, leading to inhibition of IRF-3 activity, which is similar to the role of PRRSV nsp1 (77). It was also found that EAV nsp1 blocked every signaling step upstream of IRF-3, suggesting that EAV nsp1 acts downstream of all these tested steps in this signaling pathway and, interestingly, does not have much effect on the nuclear accumulation of IRF-3, presumably having an effect on the IFN-β promoter in the nucleus (26).
Pathogen-associated molecular patterns in viral RNAs are recognized by various pattern recognition receptors, such as TLR3. TLR-3, -7, -8, and -9 are all capable of inducing type I IFN gene expression, and they exercise the function of detecting different forms of nucleic acids. They scan the extracellular and endosomal space to detect RNA and DNA, detect the viral genome from extracellularly lysis viral particles and initiate signaling cascades that lead to the secretion of IFN and other proinflammatory molecules, such as TLR3 recognition of dsRNA, initiating a TRIF-dependent signaling cascade (52, 78).
Suppressors of cytokine signaling (SOCS) are intracellular family proteins involved in the negative regulation of the immune response (79). Lung epithelial cells can induce IFN-β production and are the first to interact with pathogens, and plasmacytoid dendritic cells (PDCs) can rapidly establish a connection with TLR7 and induce IFN-I expression (80, 81). Recent studies have also shown that SOCS1 and SOCS3 strongly inhibit TLR7-mediated IFN-I production (82, 83) and that PRRSV N proteins can significantly activate SOCS1 promoter activity and induce SOCS1 expression at the protein level in Marc-145 cells, ultimately leading to IFN inhibition (84). Interestingly, TLR3-mediated IFN production after infection with Herpes simplex virus 1 (HSV-1) is cell type-dependent, with TLR3 limiting HSV-1 replication in mouse fibroblasts and CNS-resident cells (neurons, astrocytes), whereas no such protective mechanism is produced in mouse macrophages (85).
TLR3 interacts with TRIF by interacting with upstream adaptors. TRIF undergoes conformational changes and recruits the downstream TNF receptor-associated factor (TRAF)6 (86). The kinase receptor-interacting protein-1 (RIP-1) is part of the signaling pathways downstream of TLR3 and RIG-I. It can interact with TRIF to induce NF-κB activation (87). In its inactive state, the transcription factor NFκB is complexed with its inhibitor IκB (88). Upon stimulation, IκB is phosphorylated by the IκB kinase (IKK) complex, which is composed of two catalytic subunits, such as IKKα and IKKβ, and a regulatory subunit, such as NF-κB essential modulator (NEMO) (89). NF-κB regulates more than 100 genes that play key roles in inflammation, the innate immune response, and the initiation of adaptive immunity (90).
PRRSV nsp1 and nsp2 inhibit the NF-κB signaling pathway to antagonize IFN-β production (91, 92). Nsp1α inhibits the phosphorylation of IκBα, resulting in the nuclear localization of p65 being blocked, thereby aborting NF-κB function, which is associated with its C-terminal 14 amino acids (92). The nsp2 ovarian tumor protease (OUT) structural domain has deubiquitination activity, and IκB degradation is a necessary step for NF-κB activation, which can act on the IκB polyubiquitination process to prevent its degradation and ultimately inhibit NF-κB-mediated production of IFNs (91). PRRSV nsp4 cleaves TRAF family member-associated NFκB activator (TANK), which inhibits TRAF6-mediated NFκB activation (93). PRRSV nsp4 can also block NF-κB signaling targeting NEMO at a single locus E349 (33). Interestingly, the cleaved fragment of NEMO (1-349) still activates IFN and NF-κB promoter production, suggesting that nsp4 may fail to completely prevent NEMO-mediated IFN-β activation via cleavage at NEMO E349 (34). PRRSV nsp11 has also been reported to inhibit the NF-κB signaling pathway in response to deubiquitination activity (39).
EAV nsp1 inhibits IFN-β activation mainly through the NF-κB-dependent signaling pathway, which blocks each signaling step upstream of NF-κB activation, but nsp1 has little effect on NF-κB nuclear accumulation. It is speculated that EAV nsp1 may affect the IFN-β promoter in the nucleus (26). It has also been shown that EAV Nsp4 can cleave TANK to inhibit NF-κB expression (93).
Interferons are normally produced and secreted upon viral infection, and secreted IFN binds to the IFN receptor and activates Janus kinase 1 (JAK1) and tyrosine kinase 2 (TYK2) which phosphorylate signal transducers and activators of transcription proteins (STAT1 and STAT2) (94). Phosphorylated STAT1 and STAT2 form heterodimers that bind to IRF9 to form IFN-stimulated gene factor 3 (ISGF3). ISGF3 translocates to the nucleus and binds to the IFN-stimulated response element (ISRE), triggering the expression of hundreds of ISGs with antiviral functions and putting the cell in an antiviral state (20). Antiviral ISG plays a crucial role in eliminating viral infections (95). Many ISGs are signaling molecules or regulatory proteins in innate and adaptive immunity, and their induction of ISGs can further amplify and develop immune responses (including IFN responses) (96, 97).
PRRSV inhibits the IFN-activated JAK/STAT signal transduction and ISG expression in both MARC-145 and PAM cells (29, 98). Further research found that PRRSV nsp1β could block the nuclear translocation of STAT1 and significantly inhibit the expression of ISGs (35). IFN induces IFN-stimulated gene expression by activating phosphorylation of STAT1 and STAT2, which can form a heterotrimer with IRF9 (ISGF3) and translocate to the nucleus. Severe acute respiratory syndrome (SARS) and PRRSV both interfere with the host innate immune responses. Still, mechanisms that block nuclear translocation of ISGF3 are different, and SARS ORF6 can block nuclear translocation of STAT1 by sequestering KPNA2 alone (99). However, no interaction between nsp1β and any KPNAs was found in PRRSV-infected cells. PRRSV VR2385 can inhibit IFN-α signaling in MARC-145 and PAMs by interfering with ISGF3 nuclear translocation, but PRRSV modified live virus (MLV) infection of PAMs can directly activate IFN signaling, suggesting that there may be different effects of IFN induction between the two PRRSV strains, which may provide reference implications for PRRSV vaccine design (35). PRRSV nsp11 can induce STAT2 degradation directly via the ubiquitin-proteasome degradation pathway, in which amino acid residue K59 in nsp11 plays a key role but does not depend on endoribonuclease activity (40). Similarly, N proteins can inhibit interferon-induced elevation of STAT2 levels and ISGF3 nuclear translocation (29). PAM cells are affected by IFN-γ and microbial products such as lipopolysaccharide (LPS) and viral infection, and LPS-activated PAMs inhibit PRRSV replication, and genes in the JAK/STAT signaling pathway were found to be significantly upregulated, suggesting that it might play a key role in cellular activation (100).
Among the antiviral ISGs, the best-studied ones are ISG15, 2 ‘,5’-oligoadenylate synthetases (OASs), ribonuclease L (RNaseL), the dsRNA-activated protein kinase (PKR), p56 [ISG56, interferon-induced protein with tetratricopeptide repeats 1 (IFIT1)], and Mx1 (Myxovirus (influenza virus) resistance 1), and IFNs induce upregulation of transcriptional expression of several hundred interferon-stimulated genes (101, 102). ISG15 is a ubiquitin-like antiviral protein [59, 60]. ISG15 conjugation (ISGylation) to substrate proteins follows a process similar to ubiquitin conjugation (103, 104). Many viruses target STAT1 and STAT2 to inhibit the induction of ISG. ISG can inhibit nucleic acid nuclear input and RNA and protein synthesis or enhance viral degradation (102). ISG15 and ISGylation act in different cellular pathways, particularly in regulating antiviral innate immune responses. PRRSV nsp2 was previously identified as a potential antagonist of ISG15 production and ISGylation, overexpression of ISG15 inhibited PRRSV replication in cell culture, and the antiviral activity of interferon was reduced by inhibition of ISG15 binding (37). Interestingly, the pseudoknot region of the 3’ untranslated region (UTR) of the PRRSV genome can be recognized by RIG-I and TLR3 and strongly induces the expression of ISGs in PAMs, and importantly, similar structures predicted for other arterivirus members, including EAV, LDV, and SHFV, also show strong IFN-inducing activity (105).
The interferon-induced PKR plays an important role in antiviral response. PKR mediates translational control by phosphorylating the protein translation initiation factor eIF2α, inhibiting protein synthesis and viral replication (106). The addition inhibitor of PKR (2-AP) restored PRRSV replication in IFN-γ-treated cells (107). Research shows that PRRSV inhibited PKR activation during its early stage infection of PAMs (108).
Arteriviruses have evolved much to evade the host’s innate immune system to better survive in the host over the long term. The sustained low level of interferon expression is a fundamental reason for their ability to persist. Current studies have identified at least six viral proteins identified as IFN antagonists of PRRSV, further understanding of the immune regulation of viruses and strategies to evade the host immune system is necessary. The development of antiviral drugs can be facilitated by understanding the relationship between Arteriviruses and IFN antagonism to identify key immune evasion proteins. Also, understanding current antiviral strategies can enhance known antiviral pathways and further facilitate the development of safe and effective vaccine strains.
LZ, MR, and ZX conceived the scope of the review article writing, HD and FL assisted with language revisions. JZ, LD, SL, XS, and HT assisted with reviewing relevant literature. ZJ wrote the review with the help of other authors. All authors contributed to the article and approved the submitted version.
This article was supported by the Sichuan Province’s “14th Five-Year Plan” Sichuan Pig Major Science and Technology Project (No. 2021ZDZX0010) and the Key R&D Program in Rural Areas of Sichuan Provincial Department of Science and Technology (No. 2020YFN0147).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Nagarajan U. Induction and function of IFNβ during viral and bacterial infection. Crit Rev Immunol (2011) 31(6):459–74. doi: 10.1615/CritRevImmunol.v31.i6.20
2. Anderson GW, Rowland RR, Palmer GA, Even C, Plagemann PG. Lactate dehydrogenase-elevating virus replication persists in liver, spleen, lymph node, and testis tissues and results in accumulation of viral RNA in germinal centers, concomitant with polyclonal activation of b cells. J Virol (1995) 69(8):5177–85. doi: 10.1128/jvi.69.8.5177-5185.1995
3. Vatter HA, Brinton MA. Differential responses of disease-resistant and disease-susceptible primate macrophages and myeloid dendritic cells to simian hemorrhagic fever virus infection. J Virol (2014) 88(4):2095–106. doi: 10.1128/JVI.02633-13
4. Plagemann PG, Rowland RR, Even C, Faaberg KS. Lactate dehydrogenase-elevating virus: an ideal persistent virus? Springer Semin Immunopathol (1995) 17(2-3):167–86. doi: 10.1007/BF00196164
5. Gulyaeva A, Dunowska M, Hoogendoorn E, Giles J, Samborskiy D, Gorbalenya AE. Domain organization and evolution of the highly divergent 5' coding region of genomes of arteriviruses, including the novel possum nidovirus. J Virol (2017) 91(6):JVI.02096-16. doi: 10.1128/JVI.02096-16
6. Socha W, Rola J, Żmudziński JF. Variability of nonstructural proteins of equine arteritis virus during persistent infection of the stallion. Pol J Vet Sci (2015) 18(2):255–9. doi: 10.1515/pjvs-2015-0033
7. Isaacs A, Lindenmann J, Valentine RC. Virus interference. II. some properties of interferon. Proc R Soc Lond Ser B Biol Sci (1957) 147(927):268–73. doi: 10.1098/rspb.1957.0049
8. Sadler AJ, Williams BR. Interferon-inducible antiviral effectors. Nat Rev Immunol (2008) 8(7):559–68. doi: 10.1038/nri2314
9. Donnelly RP, Kotenko SV. Interferon-lambda: a new addition to an old family. J Interferon Cytokine Res (2010) 30(8):555–64. doi: 10.1089/jir.2010.0078
10. Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, et al. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol (2003) 4(1):69–77. doi: 10.1038/ni875
11. Swiecki M, Colonna M. Type I interferons: diversity of sources, production pathways and effects on immune responses. Curr Opin Virol (2011) 1(6):463–75. doi: 10.1016/j.coviro.2011.10.026
12. Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev (2004) 202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x
13. Zanoni I, Granucci F, Broggi A. Interferon (IFN)-λ takes the helm: Immunomodulatory roles of IFN-III. Front Immunol (2017) 8:1661. doi: 10.3389/fimmu.2017.01661
14. Takeuchi O, Akira S. Innate immunity to virus infection. Immunol Rev (2009) 227(1):75–86. doi: 10.1111/j.1600-065X.2008.00737.x
15. Li D, Wu M. Pattern recognition receptors in health and diseases. Signal Transduct Target Ther (2021) 6(1):291. doi: 10.1038/s41392-021-00687-0
16. Rathinam VAK, Fitzgerald KA. Cytosolic surveillance and antiviral immunity. Curr Opin Virol (2011) 1(6):455–62. doi: 10.1016/j.coviro.2011.11.004
17. Yoneyama M, Onomoto K, Jogi M, Akaboshi T, Fujita T. Viral RNA detection by RIG-i-like receptors. Curr Opin Immunol (2015) 32:48–53. doi: 10.1016/j.coi.2014.12.012
18. Kawai T, Akira S. Toll-like receptor and RIG-i-like receptor signaling. Ann N Y Acad Sci (2008) 1143:1–20. doi: 10.1196/annals.1443.020
19. Sparrer KM, Gack MU. Intracellular detection of viral nucleic acids. Curr Opin Microbiol (2015) 26:1–9. doi: 10.1016/j.mib.2015.03.001
20. Pervolaraki K, Stanifer ML, Münchau S, Renn LA, Albrecht D, Kurzhals S, et al. Type I and type III interferons display different dependency on mitogen-activated protein kinases to mount an antiviral state in the human gut. Front Immunol (2017) 8:459. doi: 10.3389/fimmu.2017.00459
21. Albina E, Carrat C, Charley B. Interferon-alpha response to swine arterivirus (PoAV), the porcine reproductive and respiratory syndrome virus. J Interferon Cytokine Res (1998) 18(7):485–90. doi: 10.1089/jir.1998.18.485
22. Van Reeth K, Labarque G, Nauwynck H, Pensaert M. Differential production of proinflammatory cytokines in the pig lung during different respiratory virus infections: correlations with pathogenicity. Res Vet Sci (1999) 67(1):47–52. doi: 10.1053/rvsc.1998.0277
23. Miller LC, Laegreid WW, Bono JL, Chitko-McKown CG, Fox JM. Interferon type I response in porcine reproductive and respiratory syndrome virus-infected MARC-145 cells. Arch Virol (2004) 149(12):2453–63. doi: 10.1007/s00705-004-0377-9
24. Dong S, Yin Y, Shen S, Guo Y, Gao M, Zhang W, et al. Inhibitory effects of recombinant porcine interferon-α on high- and low-virulence porcine reproductive and respiratory syndrome viruses. Res Vet Sci (2012) 93(2):1060–5. doi: 10.1016/j.rvsc.2011.12.006
25. Buddaert W, Van Reeth K, Pensaert M. In vivo and in vitro interferon (IFN) studies with the porcine reproductive and respiratory syndrome virus (PRRSV). Adv Exp Med Biol (1998) 440:461–7. doi: 10.1007/978-1-4615-5331-1_59
26. Go YY, Li Y, Chen Z, Han M, Yoo D, Fang Y, et al. Equine arteritis virus does not induce interferon production in equine endothelial cells: identification of nonstructural protein 1 as a main interferon antagonist. BioMed Res Int (2014) 2014:420658. doi: 10.1155/2014/420658
27. Molina RM, Cha SH, Chittick W, Lawson S, Murtaugh MP, Nelson EA, et al. Immune response against porcine reproductive and respiratory syndrome virus during acute and chronic infection. Vet Immunol Immunopathol (2008) 126(3-4):283–92. doi: 10.1016/j.vetimm.2008.08.002
28. Rowland RR, Kervin R, Kuckleburg C, Sperlich A, Benfield DA. The localization of porcine reproductive and respiratory syndrome virus nucleocapsid protein to the nucleolus of infected cells and identification of a potential nucleolar localization signal sequence. Virus Res (1999) 64(1):1–12. doi: 10.1016/S0168-1702(99)00048-9
29. Wang R, Nan Y, Yu Y, Zhang YJ. Porcine reproductive and respiratory syndrome virus Nsp1β inhibits interferon-activated JAK/STAT signal transduction by inducing karyopherin-α1 degradation. J Virol (2013) 87(9):5219–28. doi: 10.1128/JVI.02643-12
30. Beura LK, Sarkar SN, Kwon B, Subramaniam S, Jones C, Pattnaik AK, et al. Porcine reproductive and respiratory syndrome virus nonstructural protein 1beta modulates host innate immune response by antagonizing IRF3 activation. J Virol (2010) 84(3):1574–84. doi: 10.1128/JVI.01326-09
31. Ke H, Yoo D. The viral innate immune antagonism and an alternative vaccine design for PRRS virus. Vet Microbiol (2017) 209:75–89. doi: 10.1016/j.vetmic.2017.03.014
32. Huang C, Du Y, Yu Z, Zhang Q, Liu Y, Tang J, et al. Highly pathogenic porcine reproductive and respiratory syndrome virus Nsp4 cleaves VISA to impair antiviral responses mediated by RIG-i-like receptors. Sci Rep (2016) 6:28497. doi: 10.1038/srep28497
33. Huang C, Zhang Q, Guo XK, Yu ZB, Xu AT, Tang J, et al. Porcine reproductive and respiratory syndrome virus nonstructural protein 4 antagonizes beta interferon expression by targeting the NF-κB essential modulator. J Virol (2014) 88(18):10934–45. doi: 10.1128/JVI.01396-14
34. Chen J, Wang D, Sun Z, Gao L, Zhu X, Guo J, et al. Arterivirus nsp4 antagonizes interferon beta production by proteolytically cleaving NEMO at multiple sites. J Virol (2019) 93(12):1568–72. doi: 10.1128/JVI.00385-19
35. Patel D, Nan Y, Shen M, Ritthipichai K, Zhu X, Zhang YJ. Porcine reproductive and respiratory syndrome virus inhibits type I interferon signaling by blocking STAT1/STAT2 nuclear translocation. J Virol (2010) 84(21):11045–55. doi: 10.1128/JVI.00655-10
36. van Kasteren PB, Bailey-Elkin BA, James TW, Ninaber DK, Beugeling C, Khajehpour M, et al. Deubiquitinase function of arterivirus papain-like protease 2 suppresses the innate immune response in infected host cells. Proc Natl Acad Sci U S A (2013) 110(9):E838–47. doi: 10.1073/pnas.1218464110
37. Sun Z, Li Y, Ransburgh R, Snijder EJ, Fang Y. Nonstructural protein 2 of porcine reproductive and respiratory syndrome virus inhibits the antiviral function of interferon-stimulated gene 15. J Virol (2012) 86(7):3839–50. doi: 10.1128/JVI.06466-11
38. Liu K, Ma G, Liu X, Lu Y, Xi S, Ou A, et al. Porcine reproductive and respiratory syndrome virus counteracts type I interferon-induced early antiviral state by interfering IRF7 activity. Vet Microbiol (2019) 229:28–38. doi: 10.1016/j.vetmic.2018.12.015
39. Wang D, Fan J, Fang L, Luo R, Ouyang H, Ouyang C, et al. The nonstructural protein 11 of porcine reproductive and respiratory syndrome virus inhibits NF-κB signaling by means of its deubiquitinating activity. Mol Immunol (2015) 68(2 Pt A):e00385-19. doi: 10.1016/j.molimm.2015.08.011
40. Yang L, He J, Wang R, Zhang X, Lin S, Ma Z, et al. Nonstructural protein 11 of porcine reproductive and respiratory syndrome virus induces STAT2 degradation to inhibit interferon signaling. J Virol (2019) 93(22):997–1001. doi: 10.1128/JVI.01352-19
41. Meng XJ, Paul PS, Halbur PG, Lum MA. Characterization of a high-virulence US isolate of porcine reproductive and respiratory syndrome virus in a continuous cell line, ATCC CRL11171. J Vet Diagn Invest (1996) 8(3):374–81. doi: 10.1177/104063879600800317
42. Tian X, Lu G, Gao F, Peng H, Feng Y, Ma G, et al. Structure and cleavage specificity of the chymotrypsin-like serine protease (3CLSP/nsp4) of porcine reproductive and respiratory syndrome virus (PRRSV). J Mol Biol (2009) 392(4):977–93. doi: 10.1016/j.jmb.2009.07.062
43. Shi X, Wang L, Li X, Zhang G, Guo J, Zhao D, et al. Endoribonuclease activities of porcine reproductive and respiratory syndrome virus nsp11 was essential for nsp11 to inhibit IFN-β induction. Mol Immunol (2011) 48(12-13):1568–72. doi: 10.1016/j.molimm.2011.03.004
44. Sagong M, Lee C. Porcine reproductive and respiratory syndrome virus nucleocapsid protein modulates interferon-β production by inhibiting IRF3 activation in immortalized porcine alveolar macrophages. Arch Virol (2011) 156(12):2187–95. doi: 10.1007/s00705-011-1116-7
45. Pichlmair A, Schulz O, Tan CP, Näslund TI, Liljeström P, Weber F, et al. RIG-i-mediated antiviral responses to single-stranded RNA bearing 5'-phosphates. Science (2006) 314(5801):997–1001. doi: 10.1126/science.1132998
46. Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature (2006) 441(7089):101–5. doi: 10.1038/nature04734
47. Venkataraman T, Valdes M, Elsby R, Kakuta S, Caceres G, Saijo S, et al. Loss of DExD/H box RNA helicase LGP2 manifests disparate antiviral responses. J Immunol (2007) 178(10):6444–55. doi: 10.4049/jimmunol.178.10.6444
48. Dong XY, Liu WJ, Zhao MQ, Wang JY, Pei JJ, Luo YW, et al. Classical swine fever virus triggers RIG-I and MDA5-dependent signaling pathway to IRF-3 and NF-κB activation to promote secretion of interferon and inflammatory cytokines in porcine alveolar macrophages. Virol J (2013) 10:286. doi: 10.1186/1743-422X-10-286
49. Chan YK, Gack MU. Viral evasion of intracellular DNA and RNA sensing. Nat Rev Microbiol (2016) 14(6):360–73. doi: 10.1038/nrmicro.2016.45
50. Yoneyama M, Fujita T. RIG-I family RNA helicases: cytoplasmic sensor for antiviral innate immunity. Cytokine Growth Factor Rev (2007) 18(5-6):545–51. doi: 10.1016/j.cytogfr.2007.06.023
51. Jin HS, Suh HW, Kim SJ, Jo EK. Mitochondrial control of innate immunity and inflammation. Immune Netw (2017) 17(2):77–88. doi: 10.4110/in.2017.17.2.77
52. Fitzgerald KA, Rowe DC, Barnes BJ, Caffrey DR, Visintin A, Latz E, et al. LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J Exp Med (2003) 198(7):1043–55. doi: 10.1084/jem.20031023
53. Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science (2015) 347(6227):aaa2630. doi: 10.1126/science.aaa2630
54. Luo R, Xiao S, Jiang Y, Jin H, Wang D, Liu M, et al. Porcine reproductive and respiratory syndrome virus (PRRSV) suppresses interferon-beta production by interfering with the RIG-I signaling pathway. Mol Immunol (2008) 45(10):2839–46. doi: 10.1016/j.molimm.2008.01.028
55. Dong J, Xu S, Wang J, Luo R, Wang D, Xiao S, et al. Porcine reproductive and respiratory syndrome virus 3C protease cleaves the mitochondrial antiviral signalling complex to antagonize IFN-β expression. J Gen Virol (2015) 96(10):3049–58. doi: 10.1099/jgv.0.000257
56. Sun Y, Ke H, Han M, Chen N, Fang W, Yoo D. Nonstructural protein 11 of porcine reproductive and respiratory syndrome virus suppresses both MAVS and RIG-I expression as one of the mechanisms to antagonize type I interferon production. PloS One (2016) 11(12):e0168314. doi: 10.1371/journal.pone.0168314
57. van Kasteren PB, Beugeling C, Ninaber DK, Frias-Staheli N, van Boheemen S, García-Sastre A, et al. Arterivirus and nairovirus ovarian tumor domain-containing deubiquitinases target activated RIG-I to control innate immune signaling. J Virol (2012) 86(2):773–85. doi: 10.1128/JVI.06277-11
58. Miyamoto M, Fujita T, Kimura Y, Maruyama M, Harada H, Sudo Y, et al. Regulated expression of a gene encoding a nuclear factor, IRF-1, that specifically binds to IFN-beta gene regulatory elements. Cell (1988) 54(6):903–13. doi: 10.1016/S0092-8674(88)91307-4
59. Nelson N, Kanno Y, Hong C, Contursi C, Fujita T, Fowlkes BJ, et al. Expression of IFN regulatory factor family proteins in lymphocytes. induction of stat-1 and IFN consensus sequence binding protein expression by T cell activation. J Immunol (1996) 156(10):3711–20. doi: 10.1016/j.bcp.2013.01.007
60. Bluyssen AR, Durbin JE, Levy DE. ISGF3 gamma p48, a specificity switch for interferon activated transcription factors. Cytokine Growth Factor Rev (1996) 7(1):11–7. doi: 10.1016/1359-6101(96)00005-6
61. Taniguchi T, Tanaka N, Ogasawara K, Taki S, Sato M, Takaoka A. Transcription factor IRF-1 and its family members in the regulation of host defense. Cold Spring Harb Symp Quant Biol (1999) 64:465–72. doi: 10.1101/sqb.1999.64.465
62. Iversen MB, Paludan SR. Mechanisms of type III interferon expression. J Interferon Cytokine Res (2010) 30(8):573–8. doi: 10.1089/jir.2010.0063
63. Levy DE, Marié IJ, Durbin JE. Induction and function of type I and III interferon in response to viral infection. Curr Opin Virol (2011) 1(6):476–86. doi: 10.1016/j.coviro.2011.11.001
64. Verhelst K, Verstrepen L, Carpentier I, Beyaert R. IκB kinase ϵ (IKKϵ): a therapeutic target in inflammation and cancer. Biochem Pharmacol (2013) 85(7):873–80. doi: 10.1016/j.bcp.2013.01.007
65. Weil R, Laplantine E, Génin P. Regulation of TBK1 activity by optineurin contributes to cell cycle-dependent expression of the interferon pathway. Cytokine Growth Factor Rev (2016) 29:23–33. doi: 10.1016/j.cytogfr.2016.03.001
66. Zhao W. Negative regulation of TBK1-mediated antiviral immunity. FEBS Lett (2013) 587(6):542–8. doi: 10.1016/j.febslet.2013.01.052
67. Randall RE, Goodbourn S. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol (2008) 89(Pt1):1–47. doi: 10.1099/vir.0.83391-0
68. Yoneyama M, Suhara W, Fukuhara Y, Fukuda M, Nishida E, Fujita T. Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J (1998) 17(4):1087–95. doi: 10.1093/emboj/17.4.1087
69. Yang H, Ma G, Lin CH, Orr M, Wathelet MG. Mechanism for transcriptional synergy between interferon regulatory factor (IRF)-3 and IRF-7 in activation of the interferon-beta gene promoter. Eur J Biochem (2004) 271(18):3693–703. doi: 10.1111/j.1432-1033.2004.04310.x
70. Ikushima H, Negishi H, Taniguchi T. The IRF family transcription factors at the interface of innate and adaptive immune responses. Cold Spring Harb Symp Quant Biol (2013) 78:105–16. doi: 10.1101/sqb.2013.78.020321
71. Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature (2005) 434(7034):772–7. doi: 10.1038/nature03464
72. Sato M, Tanaka N, Hata N, Oda E, Taniguchi T. Involvement of the IRF family transcription factor IRF-3 in virus-induced activation of the IFN-beta gene. FEBS Lett (1998) 425(1):112–6. doi: 10.1016/S0014-5793(98)00210-5
73. Kim O, Sun Y, Lai FW, Song C, Yoo D. Modulation of type I interferon induction by porcine reproductive and respiratory syndrome virus and degradation of CREB-binding protein by nonstructural protein 1 in MARC-145 and HeLa cells. Virology (2010) 402(2):315–26. doi: 10.1016/j.virol.2010.03.039
74. Han M, Du Y, Song C, Yoo D. Degradation of CREB-binding protein and modulation of type I interferon induction by the zinc finger motif of the porcine reproductive and respiratory syndrome virus nsp1α subunit. Virus Res (2013) 172(1-2):54–65. doi: 10.1016/j.virusres.2012.12.012
75. Zhang J, Hu MM, Shu HB, Li S. Death-associated protein kinase 1 is an IRF3/7-interacting protein that is involved in the cellular antiviral immune response. Cell Mol Immunol (2014) 11(3):245–52. doi: 10.1038/cmi.2013.65
76. Chen Z, Li M, He Q, Du J, Zhou L, Ge X, et al. The amino acid at residue 155 in nonstructural protein 4 of porcine reproductive and respiratory syndrome virus contributes to its inhibitory effect for interferon-β transcription in vitro. Virus Res (2014) 189:226–34. doi: 10.1016/j.virusres.2014.05.027
77. Chen Z, Lawson S, Sun Z, Zhou X, Guan X, Christopher-Hennings J, et al. Identification of two auto-cleavage products of nonstructural protein 1 (nsp1) in porcine reproductive and respiratory syndrome virus infected cells: nsp1 function as interferon antagonist. Virology (2010) 398(1):87–97. doi: 10.1016/j.virol.2009.11.033
78. O'Neill LA, Golenbock D, Bowie AG. The history of toll-like receptors - redefining innate immunity. Nat Rev Immunol (2013) 13(6):453–60. doi: 10.1038/nri3446
79. Linossi EM, Calleja DJ, Nicholson SE. Understanding SOCS protein specificity. Growth Factors (Chur Switzerland) (2018) 36(3-4):104–17. doi: 10.1080/08977194.2018.1518324
80. Crotta S, Davidson S, Mahlakoiv T, Desmet CJ, Buckwalter MR, Albert ML, et al. Type I and type III interferons drive redundant amplification loops to induce a transcriptional signature in influenza-infected airway epithelia. PloS Pathog (2013) 9(11):e00099-20. doi: 10.1371/journal.ppat.1003773
81. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol (2010) 11(5):373–84. doi: 10.1038/ni.1863
82. Yu CF, Peng WM, Schlee M, Barchet W, Eis-Hübinger AM, Kolanus W, et al. SOCS1 and SOCS3 target IRF7 degradation to suppress TLR7-mediated type I IFN production of human plasmacytoid dendritic cells. J Immunol (2018) 200(12):4024–35. doi: 10.4049/jimmunol.1700510
83. Olière S, Hernandez E, Lézin A, Arguello M, Douville R, Nguyen TL, et al. HTLV-1 evades type I interferon antiviral signaling by inducing the suppressor of cytokine signaling 1 (SOCS1). PloS Pathog (2010) 6(11):e1001177. doi: 10.1016/s0969-2126(99)80002-1
84. Luo X, Chen XX, Qiao S, Li R, Xie S, Zhou X, et al. Porcine reproductive and respiratory syndrome virus enhances self-replication via AP-1-Dependent induction of SOCS1. J Immunol (2020) 204(2):394–407. doi: 10.4049/jimmunol.1900731
85. Zhu H, Zheng C. The race between host antiviral innate immunity and the immune evasion strategies of herpes simplex virus 1. Microbiol Mol Biol Rev (2020) 84(4):7832–46. doi: 10.1128/MMBR.00099-20
86. Hyun J, Kanagavelu S, Fukata M. A unique host defense pathway: TRIF mediates both antiviral and antibacterial immune responses. Microbes Infect (2013) 15(1):1–10. doi: 10.1016/j.micinf.2012.10.011
87. Meylan E, Burns K, Hofmann K, Blancheteau V, Martinon F, Kelliher M, et al. RIP1 is an essential mediator of toll-like receptor 3-induced NF-kappa b activation. Nat Immunol (2004) 5(5):503–7. doi: 10.1038/ni1061
88. Cramer P, Müller CW. A firm hand on NF-kappaB: structures of the IkappaBalpha-NFkappaB complex. Structure (London Engl 1993) (1999) 7(1):R1–6. doi: 10.1074/jbc.M115.660761
89. Yamamoto Y, Gaynor RB. IkappaB kinases: key regulators of the NF-kappaB pathway. Trends Biochem Sci (2004) 29(2):72–9. doi: 10.1016/j.tibs.2003.12.003
90. Santoro MG, Rossi A, Amici C. NF-kappaB and virus infection: who controls whom. EMBO J (2003) 22(11):651–62. doi: 10.1093/emboj/cdg267
91. Sun Z, Chen Z, Lawson SR, Fang Y. The cysteine protease domain of porcine reproductive and respiratory syndrome virus nonstructural protein 2 possesses deubiquitinating and interferon antagonism functions. J Virol (2010) 84(15):7832–46. doi: 10.1128/JVI.00217-10
92. Song C, Krell P, Yoo D. Nonstructural protein 1α subunit-based inhibition of NF-κB activation and suppression of interferon-β production by porcine reproductive and respiratory syndrome virus. Virology (2010) 407(2):268–80. doi: 10.1016/j.virol.2010.08.025
93. Huang L, Liu Q, Zhang L, Zhang Q, Hu L, Li C, et al. Encephalomyocarditis virus 3C protease relieves TRAF family member-associated NF-κB activator (TANK) inhibitory effect on TRAF6-mediated NF-κB signaling through cleavage of TANK. J Biol Chem (2015) 290(46):27618–32. doi: 10.1074/jbc.M115.660761
94. Levy DE. STATs: transcriptional control and biological impact. Nat Rev Mol Cell Biol (2002) 164:493–503. doi: 10.1038/nrm909
95. Schoggins JW, Rice CM. Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol (2011) 1(6):519–25. doi: 10.1016/j.coviro.2011.10.008
96. Yang E, Li MMH. All about the RNA: Interferon-stimulated genes that interfere with viral RNA processes. Front Immunol (2020) 11:605024. doi: 10.3389/fimmu.2020.605024
97. Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol (2014) 32:513–45. doi: 10.1146/annurev-immunol-032713-120231
98. Wang R, Nan Y, Yu Y, Yang Z, Zhang YJ. Variable interference with interferon signal transduction by different strains of porcine reproductive and respiratory syndrome virus. Vet Microbiol (2013) 166(3-4):e01352-19. doi: 10.1016/j.vetmic.2013.07.022
99. Frieman M, Yount B, Heise M, Kopecky-Bromberg SA, Palese P, Baric RS. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J Virol (2007) 81(18):9812–24. doi: 10.1128/JVI.01012-07
100. Liu Q, Zhang YL, Hu W, Hu SP, Zhang Z, Cai XH, et al. Transcriptome of porcine alveolar macrophages activated by interferon-gamma and lipopolysaccharide. Biochem Biophys Res Commun (2018) 503(4):2666–72. doi: 10.1016/j.bbrc.2018.08.021
101. Samuel CE. Antiviral actions of interferons. Clin Microbiol Rev (2001) 14(4):778–809, table of contents. doi: 10.1128/CMR.14.4.778-809.2001
102. Fensterl V, Sen GC. Interferons and viral infections. BioFactors (Oxford England) (2009) 35(1):14–20. doi: 10.1002/biof.6
103. Recht M, Borden EC, Knight E Jr. A human 15-kDa IFN-induced protein induces the secretion of IFN-gamma. J Immunol (1991) 147(8):2617–23. doi: 10.1073/pnas.0607038104
104. Lenschow DJ, Lai C, Frias-Staheli N, Giannakopoulos NV, Lutz A, Wolff T, et al. IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and sindbis viruses. Proc Natl Acad Sci U S A (2007) 104(4):1371–6. doi: 10.1073/pnas.0607038104
105. Xie S, Chen XX, Qiao S, Li R, Sun Y, Xia S, et al. Identification of the RNA pseudoknot within the 3' end of the porcine reproductive and respiratory syndrome virus genome as a pathogen-associated molecular pattern to activate antiviral signaling via RIG-I and toll-like receptor 3. J Virol (2018) 92(12):e00097-18. doi: 10.1128/JVI.00097-18
106. Clemens MJ, Elia A. The double-stranded RNA-dependent protein kinase PKR: structure and function. J Interferon Cytokine Res (1997) 17(9):503–24. doi: 10.1089/jir.1997.17.503
107. Rowland RR, Robinson B, Stefanick J, Kim TS, Guanghua L, Lawson SR, et al. Inhibition of porcine reproductive and respiratory syndrome virus by interferon-gamma and recovery of virus replication with 2-aminopurine. Arch Virol (2001) 146(3):539–55. doi: 10.1007/s007050170161
Keywords: arteriviruses, interferon (IFN), viral proteins, innate immunity, immune evasion
Citation: Jian Z, Ma R, Zhu L, Deng H, Li F, Zhao J, Deng L, Lai S, Sun X, Tang H and Xu Z (2022) Evasion of interferon-mediated immune response by arteriviruses. Front. Immunol. 13:963923. doi: 10.3389/fimmu.2022.963923
Received: 08 June 2022; Accepted: 13 July 2022;
Published: 15 August 2022.
Edited by:
Chenhe Su, Wistar Institute, United StatesReviewed by:
Zhonghan Li, Sichuan University, ChinaCopyright © 2022 Jian, Ma, Zhu, Deng, Li, Zhao, Deng, Lai, Sun, Tang and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhiwen Xu, YWJ0Y3h6d0AxMjYuY29t
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.