 Sagar Paudel
Sagar Paudel- 1Center for Lung Biology and Disease, Louisiana State University (LSU) School of Veterinary Medicine, Baton Rouge, LA, United States
- 2Department of Pathobiological Sciences, Louisiana State University (LSU) School of Veterinary Medicine, Baton Rouge, LA, United States
- 3Section of Pulmonary and Critical Care, Department of Medicine, LSU Health Sciences Center, New Orleans, LA, United States
During acute infectious and inflammatory conditions, a large number of neutrophils are in high demand as they are consumed in peripheral organs. The hematopoietic system rapidly responds to the demand by turning from steady state to emergency granulopoiesis to expedite neutrophil generation in the bone marrow (BM). How the hematopoietic system integrates pathogenic and inflammatory stress signals into the molecular cues of emergency granulopoiesis has been the subject of investigations. Recent studies in the field have highlighted emerging concepts, including the direct sensing of pathogens by BM resident or sentinel hematopoietic stem and progenitor cells (HSPCs), the crosstalk of HSPCs, endothelial cells, and stromal cells to convert signals to granulopoiesis, and the identification of novel inflammatory molecules, such as C/EBP-β, ROS, IL-27, IFN-γ, CXCL1 with direct effects on HSPCs. In this review, we will provide a detailed account of emerging concepts while reassessing well-established cellular and molecular players of emergency granulopoiesis. While providing our views on the discrepant results and theories, we will postulate an updated model of granulopoiesis in the context of health and disease.
Introduction
Ever since Metchnikoff first observed phagocytosis of bacteria in white blood cells in 1882 (1), we have been fascinated by neutrophils as they have taken the spotlight of the innate immune system. Neutrophils play a central role in host defense against bacterial, viral, and fungal infections. Patients with neutropenia and clinical disorders (severe congenital neutropenia, leukocyte adhesive deficiency, and chronic granulomatous diseases) suffer from severe infections, which highlights the important role of neutrophils in host immunity. On the flip side, uncontrolled neutrophil extravasation may lead to excessive collateral inflammatory damage found in conditions such as acute respiratory distress syndrome.
Neutrophils are short-lived and therefore the human body generates about 0.5-1 x 1011 neutrophils per day in the bone marrow (BM) to maintain the circulating pools during steady-state conditions (2). Neutrophils develop from the dormant but self-renewing hematopoietic stem cells (HSCs), which reside in a specialized BM niche and represent the topmost cells in the hierarchy of the hematopoietic system (3, 4). Long-term HSCs first differentiate to short-term HSCs (ST-HSCs), which then give rise to multi-potent but non-renewing differentiated cells, multi-potent progenitors (MPPs). Further downstream, MPPs can differentiate into oligopotent common myeloid progenitors (CMPs) and common lymphoid progenitors (CLPs). Then, CMPs advance as either granulocyte monocyte progenitors (GMPs) giving rise to neutrophils and macrophages or megakaryocyte/erythrocyte progenitor (MEP) giving rise to platelets and erythrocytes. HSC, its niche and lineage specifications in health and disease has been the recent subject of excellent reviews published elsewhere (5–7). A comprehensive review on neutrophil ontogeny and biology with specific emphasis on the differences between neonatal and adult neutrophil populations has been recently offered by Lawrence et al. (8).
During hematopoietic stress such as systemic infections or physical insults, neutrophils are used up in larger quantities and the demand for neutrophils in the peripheral blood increases by several folds higher than steady state. The hematopoietic system rapidly senses this neutrophil demand and integrates these pathogen signals to activate dormant HSCs to proliferate and differentiate into neutrophil progenitors (Figure 1). This distinct program of accelerated de novo production of neutrophils from amplification of neutrophil progenitors is known as ‘emergency granulopoiesis’, which is usually a protective hematopoietic immune response to fatal infection. This process of differentiation and then amplification of multipotent HSCs into mature, segmented neutrophils is highly sophisticated and governed by a complex network of molecular and cellular players (Figure 1). Clinical manifestations of patients with heightened emergency granulopoiesis are elevated levels of acute phase proteins, leukocytosis, neutrophilia, and appearance of immature myeloid precursor cells (left shift).
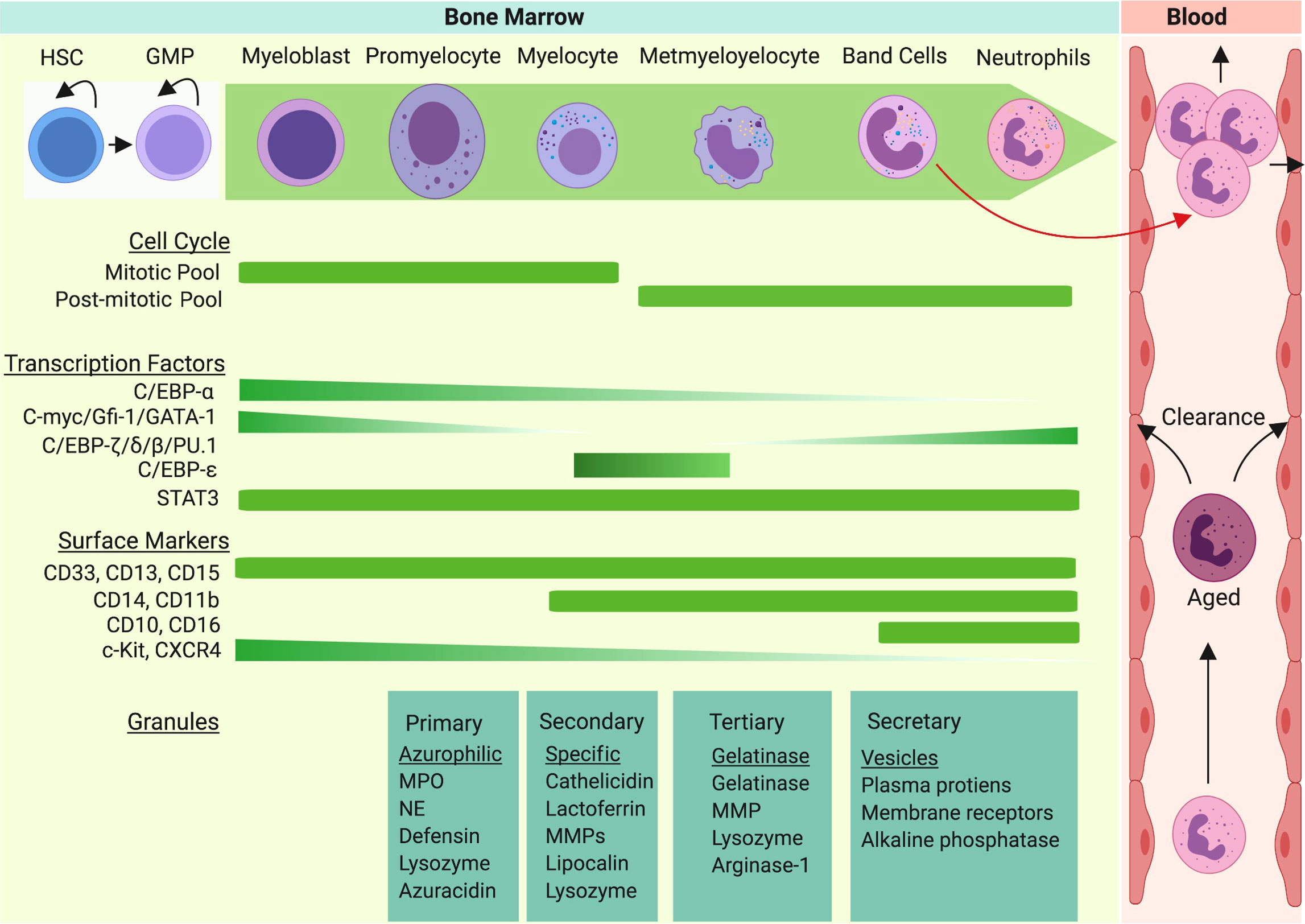
Figure 1 Neutrophil granulopoiesis is a hierarchical program of the hematopoietic system. Granulopoiesis, the process of neutrophil generation and maturation, is characterized by sequential development of distinct granules, surface proteins, and nuclear segmentation. Homeostatic granulopoiesis starts with generation of myeloblasts and promyelocytes (rich in primary azurophilic granules) which depends on expression of GATA-1, C/EBP-α, Gfi-1, and c-Myc genes. The development of secondary specific granules appears in myelocytes (last proliferative stage). C-Myc and C/EBP-ϵ contribute to formation of secondary granules. The development of metamyelocytes with gelatinase granules marks terminal differentiation of neutrophil granulopoiesis. As expression of C/EBP-β, C/EBP-δ, and C/EBP-ζ are upregulated in terminal neutrophil stages, gelatinase granules and secretary vesicles appear and nuclear segmentation intensifies. During emergency granulopoiesis, C/EBP-β takes over the process and an excess amount of neutrophils are generated and released to the bloodstream along with immature neutrophils (band cells).
In this review, we provide a detailed examination of the cellular and molecular players involved in activation of emergency granulopoiesis, the molecular messengers that integrate pathogen signals into HSC activation, proliferation and differentiation into neutrophils, and regulatory molecular and environmental mechanisms that control emergency granulopoiesis in the context of infectious and inflammatory conditions.
Granulopoiesis: Steady state vs emergency
As neutrophils are mitotically inactive and relatively short lived, their counts in circulation during physiological conditions are constantly replenished through a process known as ‘steady-state granulopoiesis’. Localized mild-grade bacterial infections do not invoke strong hematopoietic pressure, as the host rapidly resolves infections, limiting their systemic dissemination. Consequently, HSCs do not sense the demand and BM granulopoiesis remains unaltered from the steady state. However, overwhelming systemic infections put much stronger stress to hematopoietic systems, as large quantities of neutrophils are lost in an attempt to limit dissemination. To meet urgent needs of depleting neutrophils, the BM hematopoietic system rapidly switches steady-state granulopoiesis to a well-coordinated process of neutrophil generation in a large scale that involves activation and proliferation of myeloid progenitors known as ‘emergency granulopoiesis’. Studies suggest BM niche adjust to allow space for the heightened kinetics and magnitude of emergency granulopoiesis at the expense of erythropoiesis and lymphopoiesis, as inflammatory signals are shown to reduce the expression of lymphoid expansion and retention signals in BM (9–11). Besides systemic infections, iatrogenic conditions such as the irradiation or chemotherapy-induced myeloablation results in profound neutropenic conditions (12, 13) and can trigger emergency granulopoiesis to replenish neutrophil homeostasis. Despite their differences in stimuli, many cellular and molecular events of granulopoiesis are shared. However, accumulating evidence shows that infection-induced emergency granulopoiesis may have a major evolutionary impact on the response of the hematopoietic system to stress. This notion of evolution of the hematopoietic system in response to stress became clearer as HSCs were shown to circulate, sense, and respond to pathogen recognition (14–16). With the discovery of CCAAT/enhancer-binding protein (C/EBP) in the regulation of granulopoiesis (9, 17, 18), we now know that emergency granulopoiesis is differentially regulated at the transcriptional level from steady state.
Pathogen sensing system in emergency granulopoiesis
The hematopoietic system usually responds to infection by enhancing cellular output, but how local/systemic infections are sensed and subsequently messaged to the BM is not very clear. Given that the granulopoietic BM niche is spatially disconnected from the site of pathogen entry, it was long believed that activation of emergency granulopoiesis is an indirect process and relies on sensing of pathogen by germ-line encoded pathogen recognition receptors (PRRs) by immune and resident cells. Toll-like receptors (TLRs)-expressing cells are first to detect pathogens or their products (Figure 2). These cells do not generate neutrophils by themselves but secrete hematopoietic cytokines (such as G-CSF, IL-6) which indirectly influence differentiation and proliferation of neutrophil precursors (19). However, recent reports show that HSPCs can express pattern recognition receptors (11, 20, 21) as well as multiple traffic molecules, and egress and re-enter the BM niche (14, 15), thereby suggesting immediate and direct activation of emergency granulopoiesis by these precursor cells (Figure 2). In the following section, we will dive deeper into understanding how the hematopoietic system has evolved to sense pathogens and to rapidly message the bone marrow niche.
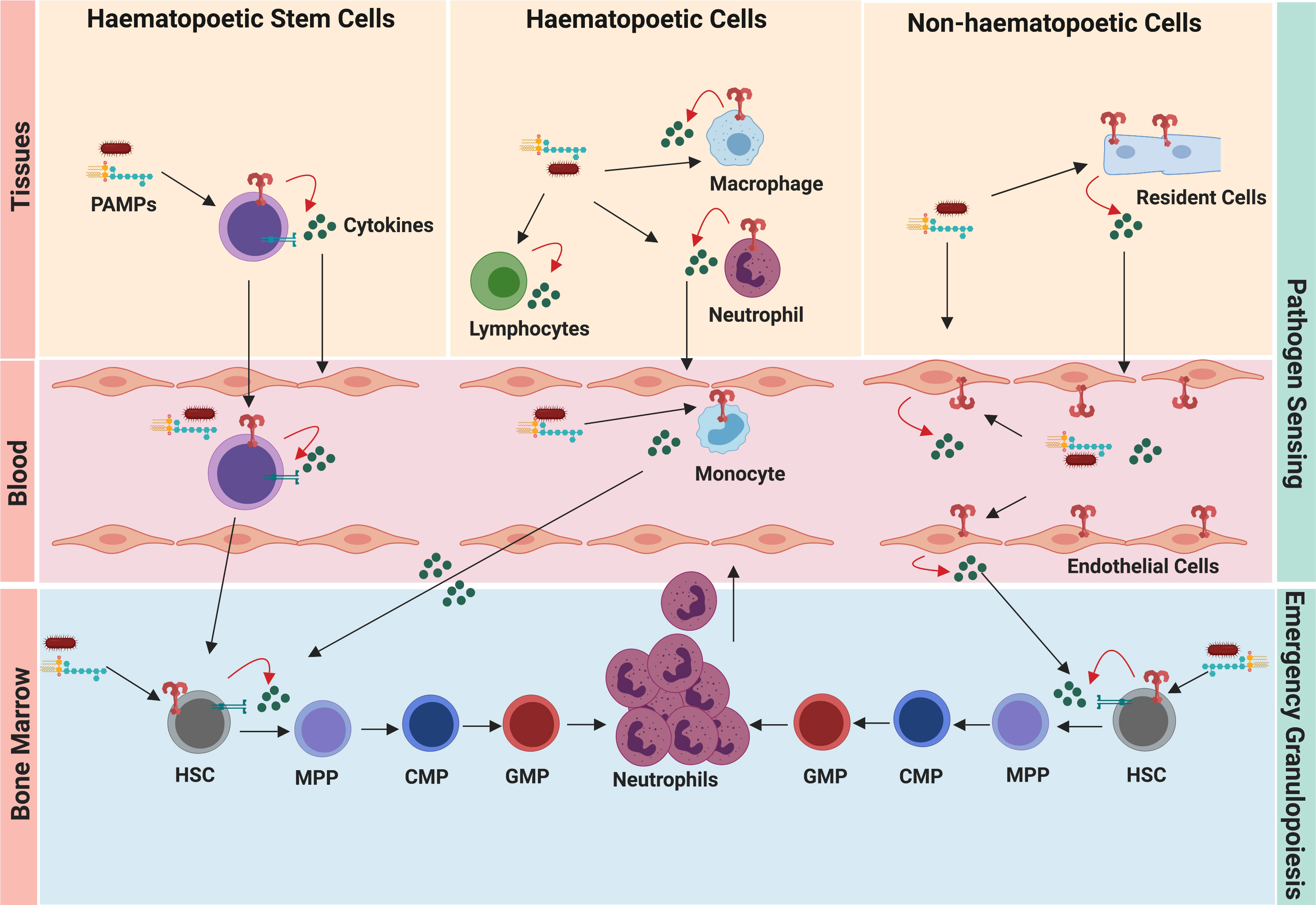
Figure 2 Pathogen sensing system of emergency granulopoiesis. During infection and inflammation, the hematopoietic cells, resident cells, and circulating HSCs sense disseminating pathogens and danger signals. Pathogen-induced granulopoiesis heavily depends upon TLR-signaling, which translates pathogen signals into molecular cues of granulopoiesis. TLR engagement on these cells results in the secretion of several granulopoietic growth factors (such as G-CSF, IL-6, CXCL1), which initiate signaling cascades in HSCs and neutrophil progenitors, thereby activating emergency granulopoiesis. Expression of TLRs by HSCs and endothelial cells facilitates more direct ways of initiating granulopoiesis compared to indirect activation of immune cells at the site of infection/inflammation.
HSPCs
Immature HSCs and early progenitors express PRRs and initiate granulopoiesis directly as they can actively participate in recognition of pathogen-derived products (20–22). Nagai et al. were first to demonstrate that HSCs (Lin-c-Kit+ subset) express TLR2, TLR4 and their co-receptors (MD-2, CD14) and GMPs (Lin-c-Kit+Sca-1-CD34+FcγR+subset) express TLR2 (20). Furthermore, ex vivo stimulation of HSCs with TLR2 agonist (Pam3CSK4) or with TLR4 agonist (LPS) drove MyD88-dependent but cytokine-independent differentiation to produce myeloid lineage cells (20). Immediately, Massberg et al. revealed that lymph borne HSPCs originate in the BM and possess extensive multi-lineage potential even at peripheral organs when encountering pathogens (15). Specifically, these HSPCs are shown to proliferate to myeloid cells within the kidney when implanted under the kidney capsule in response to TLR agonist (15). Human CD34+ HSPCs also express functional TLRs and whose ligation with agonists induces the differentiation into myeloid cells without addition of cytokine, in some cases, at the expense of lymphopoiesis (23–25). Since then, numerous studies have shown TLR ligation as an important step in HSC differentiation to myeloid cells (11, 26–28). The functional consequence of TLR engagement was further elucidated when an elegant study demonstrated that LPS-stimulated HSPCs secrete IL-6 and that HSPCs derived from IL-6-/- mice failed to differentiate to myeloid cells under neutropenic conditions achieved with the treatment of either chemotherapy (5-fluorouracil) or irradiation (29). Besides TLR2 and TLR4, a recent study demonstrated that TLR7 signaling drove type I IFN-dependent myeloid differentiation of CMP in vitro and peripheral expansion of monocytes in vivo (30). A recent study highlighted that the in vivo response of HSC to pathogens is much more complex and redundant, as MyD88/Triff-/- mice or C3H/HeJ (loss of function mutation for TLR4) mice had normal expansion of HSPCs during S. aureus infection (31). Collectively, these studies clearly demonstrate that the hematopoietic system has evolved to detect and respond to pathogens directly through TLRs and these encounters are all geared to HSC differentiation, amplification of progenitors, and generation of progeny cells at the BM niche or at the site of infection in some cases.
Hematopoietic and non-hematopoietic cells
A long prevailing hypothesis is that emergency granulopoiesis is indirectly activated by hematopoietic and resident cells via inflammatory mediators. Tissue resident macrophages and circulating monocytes are equipped with PRRs, sense pathogens at the site of entry, and release massive amounts of granulopoietic cytokines (such as G-CSF, GM-CSF, IL-6) during local or systemic infection. With the discovery of IL-17A-producing innate lymphoid cells and regulatory properties of neutrophils (32), we speculate that monocytes and macrophages are not the sole players. Although there is lack of careful in vivo experimentation proving direct roles of these cells in emergency granulopoiesis, the abundance of data suggests their role is largely mediated by cytokine and growth factors which can activate HSC differentiation, regulate cell survival, and generate lineage committed myeloid cells. Experiments with antibody-mediated depletion have broadened our knowledge of these cells but careful interpretation of data should be made, as the observation could be a positive feedback granulopoiesis. For instance, anti-Ly6G depleted neutropenic mice or LysMCre/wt Mcl-1f/f mice (lacking mature neutrophils) during steady state elicited elevated levels of G-CSF resulting in enhanced HPSC proliferation and GMP differentiation (33). Soon after, Bugl et al. (34) not only confirmed the enhanced G-CSF and HSC expansion in neutropenic mice but also showed G-CSF-mediated feedback granulopoiesis was dependent on BM neutrophil mass. In contrast to popular belief, using clodronate liposome or reciprocal TLR4-/- chimera, Boettcher et al. were first to reveal that selective TLR4 expression within the hematopoietic compartment failed to induce emergency granulopoiesis and neutrophilia during systemic LPS injection (35). TLR4 signaling in non-hematopoietic but not in the hematopoietic compartment was an absolute requirement for LPS-induced, G-CSF-mediated granulopoiesis (35). In another elegant work, Boettcher et al., using endothelial cell (EC) specific MyD88-/- mice in bone marrow chimeras, demonstrated that EC-intrinsic MyD88 signaling dominantly contributed to G-CSF production and induced GMP expansion and neutrophil generation after in vivo LPS or Escherichia coli challenge (36). As the highest G-CSF expression was noted in BM EC after LPS challenge, it is convincing that vasculature lining ECs could be an important layer of local signal to the BM niche to initiate granulopoiesis.
NOD-like receptors
Until recently, the role of intracellular NLRs in hematopoiesis was elusive. Burberry et al. showed that NOD1 and NOD2 cooperate with TLR4 receptors to lower CXCL12 in BM and induce G-CSF, thereby mobilizing HSC to give rise to neutrophils and monocytes during E. coli infection (37). Furthermore, NLRP3 inflammasome regulates HSC mobilization to peripheral blood through IL-1β and IL-18 production following administration of G-CSF and AMD3100 (38). However, genetic ablation of caspase-1/11 in neonatal mice resulted in the enhanced emergency myelopoiesis possibly through expansion of BM and splenic HSC and higher G-CSF/M-CSF levels, implicating an NLR/caspase/IL-1β axis in granulopoiesis (39). Mice deficient in NLRC4 and NLRP6 have enhanced neutrophil recruitment and elevated levels of hematopoietic cytokines during pulmonary and systemic infection (40–42), but whether these mice also present granulopoietic defects in BM was not explored. More directed studies are needed to identify if these intracellular sensors are actually a class of negative regulator of TLR-mediated granulopoiesis.
Molecular translation of pathogen signals into granulopoiesis
The hematopoietic system must translate pathogen sensing into the process of emergency granulopoiesis (Figure 3). Although little emerging evidence suggests HSPCs can respond to direct pathogen sensing, a vast number of studies unequivocally demonstrate that the coordinated action of myeloid growth factors, cytokines, chemokines, and transcription factors mediate emergency granulopoiesis. Recent evidence reveals these mediators can instruct lineage choice for uncommitted HSPCs rather than just enhancing proliferation and survival of the precursor. In this section, we dissect how these myeloid growth factors mediate HSPC differentiation, lineage choices, proliferation, and survival of myeloid progenitors during emergency granulopoiesis.
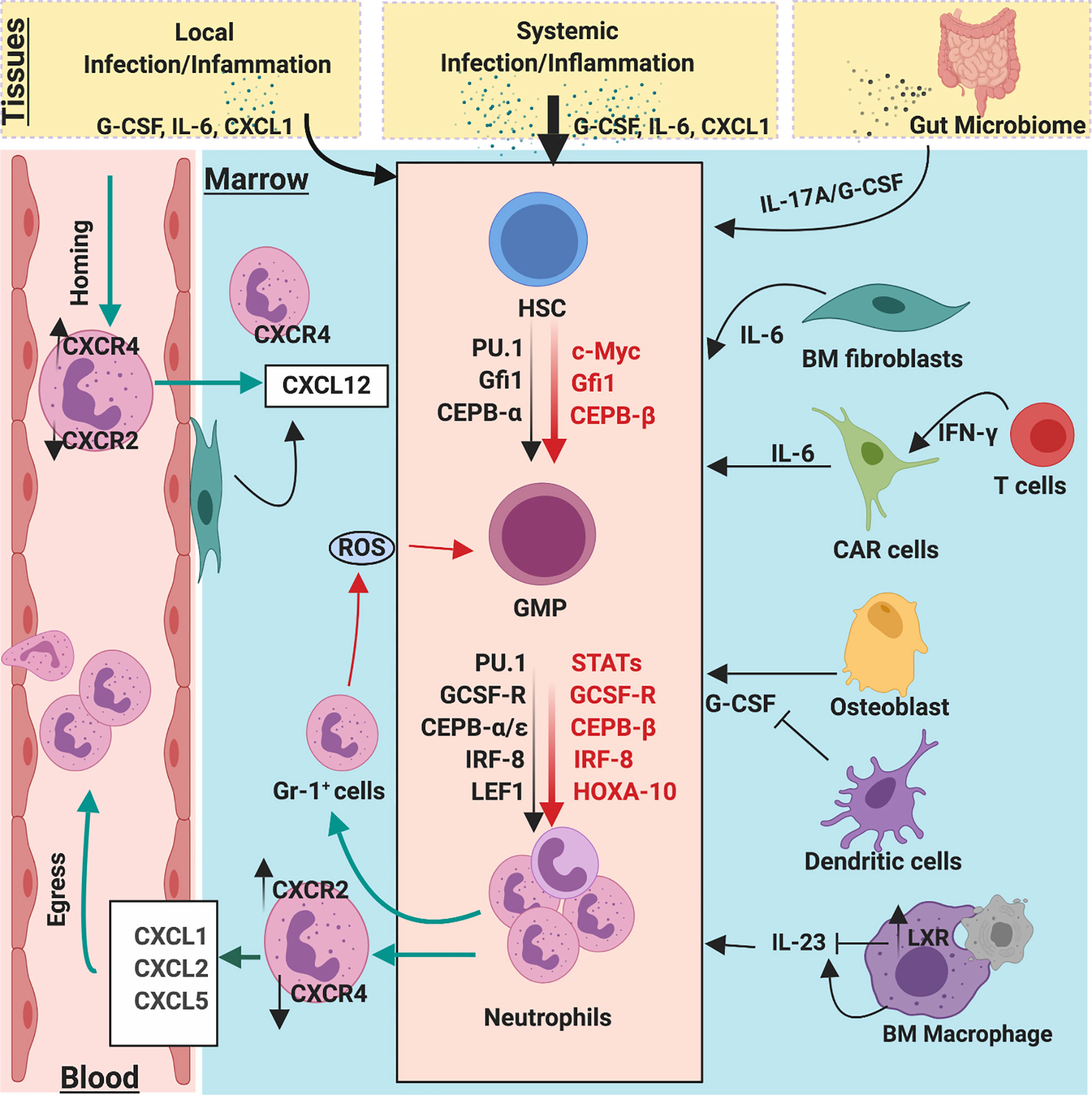
Figure 3 Molecular and cellular regulators of emergency and homeostatic granulopoiesis. Granulopoiesis is a tightly regulated process by several molecular and cellular players. During the steady state, C/EBPα is a primary transcription factor that regulates proliferation of neutrophil progenitors by limiting the expression of cell cycle gene such as c-Myc, CDK2, and CDK4. However, during emergency granulopoiesis, C/EBPβ directly replaces C/EBPα at the MYC promoter and expedites c-Myc expression to enhance the generation of neutrophils. Robust C/EBPβ activity relies on strong activation of JAK/STAT pathways by G-CSF, which is massively released during infection/inflammation. During steady state, clearance of apoptotic neutrophils by macrophages activates LXR expression, which downregulates IL-23 secretion, thereby limiting the activity of G-CSF/IL-17A. Bone marrow stromal fibroblast and CXCL12 abundance reticular (CAR) cells also secrete IL-6 which regulate granulopoiesis. Additionally, ROS release from BM GR-1+ cells also regulate proliferation and differentiation of GMPs into neutrophil lineages. Gut microbiome provides PAMPs to activate and regulate steady-state and emergency granulopoiesis through the IL-23/G-CSF/IL-17A axis.
Colony stimulating factors
Myeloid growth factors such as G-CSF (and its receptor G-CSFR), GM-CSF, M-CSF are the best-studied colony stimulating factors in context of granulopoiesis. Hematopoietic cells, including HSPCs, are known to express receptors for these cytokines (11). In vivo daily administration of G-CSF or GM-CSF with stem cell factor resulted into neutrophilia and a synergistic GMP expansion in bone marrow (43). GM-CSF stimulates the expansion of myeloid lineage cells both in vivo and in vitro (44, 45). Daily in vivo administration of M-CSF expanded the number of blood monocytes and resident macrophages in spleen and liver (46). Mice deficient in G-CSF (Gcsf-/-) and G-CSFR (Gcsfr-/-) displayed a diminished level of peripheral neutrophils and a corresponding decrease in neutrophil progenitors in the BM niche during steady state (47, 48). Exogenous administration of G-CSF into uninfected Gcsf-/- mice augmented the level of circulating neutrophils (47). However, the role of GM-CSF in baseline appeared redundant as mice deficient in GM-CSF (Gmcsf-/-) showed no major defect in steady-state granulopoiesis and maintained normal pools of myeloid cells and their precursors in blood, marrow, and spleen (49). However, the usage of these mice in infection-induced granulopoiesis has yielded some discrepant results. Earlier reports demonstrated that both Gcsf-/- and Gmcsf-/- mice had impaired emergency granulopoiesis and host defense against Listeria monocytogenes infection (47, 50). In contrast, emergency granulopoiesis in Gcsf-/- mice were not dampened but indistinguishable from control mice when challenged with Candida albicans (51). These Gcsf-/- mice have a strikingly high level of IL-6 but mice deficient in both G-CSF and IL-6 also developed neutrophilia and BM expansion of GMPs (51). Furthermore, mice deficient in GM-CSF (Gmcsf-/-) have reduced circulating pools of hematopoietic cells but surprisingly displayed subtle changes in BM precursors during Mycobacterium avium infection (52). Additionally, mice deficient in all three colony stimulating factors (G-CSF, GM-CSF, M-CSF) have reduced circulating myeloid cells during the resting state, but they were surprisingly able to mount emergency granulopoiesis during thioglycolate-induced peritonitis (53). Accumulating evidence shows intriguing functional consequences of these cytokines on cellular targets. Mechanistically, G-CSF acts as a proliferative signal for HSCs but impairs renewal ability, which is dependent on TLR signaling and host microbiome (54). Additionally, G-CSF disrupts the HSC niche resulting in their mobilization (55), suggesting a G-CSF-induced defect in repopulation could be in fact increased HSCs mobilization from the BM niche. Using long-term bio-imaging observations at the single-cell level, Rieger et al. (56) demonstrated that bipotent GMPs responded with generation of exclusively mature neutrophilic granulocytic or monocytic cells in response to G-CSF and GM-CSF. More insights on the instructive action of cytokines came into light as Mossadegh-Keller et al. (57) utilized time-lapse imaging and single cell gene expression analysis to reveal that M-CSF directly stimulates HSCs to activate transcription factor PU.1, which promotes differentiation into the myeloid cell lineage. Additionally, Jack et al. (58) have demonstrated that M-CSF targets ERK to elevate c-Fos and phospho-C/EBPalpha whereas G-CSF influences SHP2 activity in marrow progenitors to impact the lineage specification. Epstein-Barr virus infected humanized NOG mice demonstrate increased granulopoiesis along with elevated levels of GM-CSF (59). Finally, Bacillus Calmette–Guérin (BCG) vaccination-induced rapid protection from neonatal sepsis is mediated by G-CSF-induced emergency granulopoiesis (60).
CXC and CC-chemokines
Although chemokines and their receptors have long been described on their ability to recruit leukocytes, recent reports reveal that chemokine systems are increasing involved in the differentiation, proliferation, and mobilization of hematopoietic progenitors (61, 62). Copiously produced by the various cells of BM niche (63), CXCL12 (or SDF1) and its receptor CXCR4 axis is a representative homeostatic chemokine system primarily regulating HSC retention (64). Mice deficient in CXCL12 or CXCR4 displayed a virtual absence of myelopoiesis in BM, reduced number of myeloid cells in fetal liver, and impaired B-lymphopoiesis (65, 66). Recently, CXCL12 has been shown to regulate mitochondrial respiration in early HSCs, which are critical for maintaining their undifferentiated state (67). CCL3, a CC-chemokine that has long been implicated for hematologic malignancies (68), has been shown to enhance myelopoietic activity for mature progenitors or suppress myelopoietic activity for more immature progenitors (69). Using CCR1-deficient mice, CCL3 signaling through CCR1 receptor was shown to play a dominant role for the proliferation but not suppression of lineage-committed myeloid progenitors (70). In contrast, mice lacking CCL3 (Ccl3-/-) have profound increases in HSPCs and immature myeloid progenitors but reduced numbers of committed myeloid progenitors without altering BM microenvironmental populations (71). In contract to CXCL12/CXCR4 and CCL3/CCR1, CCR2 and its ligands (CCL2/12/13) have long been known to have myelosuppressive effects (72). Using RFP-CCR2 reporter mice, the profound expression of CCR2 was observed in neutrophils (73). Additionally, mice lacking CCR2 displayed increased sequestration of all subsets of myeloid cells in BM and their corresponding reduced numbers in blood and spleen (73). Recently, another CC-chemokine, CCL5/RANTES, has been demonstrated to influence the BM niche cell subtypes and lineage skewing phenotype (74). Indeed, mice lacking CCL5 (Ccl5-/-) displayed reduced levels of myeloid-biased HSCs and myeloid progenitors but concomitant increases in lymphoid-biased HSCs and T cells progeny (74). However, the retroviral expression of CCL5 in BM progenitors or a brief ex vivo stimulation of HSCs with CCL5 resulted in T cell deficiency but the expansion of myeloid progenitors (74).
Among CXC-chemokines, CXCL1 and CXCL2 are also known to suppress the ex vivo proliferation of myeloid progenitors by activation of CXCR2 (75, 76). The myelosuppressive activity of CXCR2 signaling was confirmed as mice lacking CXCR2 (Cxcr2−/−) have enhanced expansion of myeloid progenitors in BM and neutrophilia in peripheral blood (76–78). Although Cxcr2−/− stem/progenitor cells showed initial expansion in vivo, these cells had exhausted self-renewal capacity in a serial transplantation assay (78). However, Cxcr2−/− mice displayed comparable numbers and cytokine responsiveness of myeloid progenitors in BM, but abnormal retention of mature neutrophils (79). In another elegant study, Mei, J. et al. (80) showed that Cxcr2−/− or Cxcl5−/− mice had IL-17A/G-CSF and commensal bacteria dependent-neutrophil hyperplasia in BM and mild neutrophilia in peripheral blood during steady state. Using chimeric mice, the authors delineate that the loss of CXCR2 in the hematopoietic compartment contributed to enhanced granulopoiesis while enterocytes and BM resident cells supplied CXCL5 under basal conditions (80). Recently, we examined the role of CXC chemokine in context of emergency granulopoiesis. Contrastingly, we showed that mice deficient in CXCL1 (Cxcl1-/-) displayed reduced blood neutrophilia and recruitment to lungs and a corresponding defect in generation of neutrophils in BM (81). Using multi-parametric flow cytometry, Cxcl1-/- mice showed reduced numbers of hematopoietic stem cells and granulocyte-monocyte progenitors, which can be corrected by exogenous administration of other CXCR2 ligands such as CXCL2, CXCL5 (81).
Interleukins
IL-6 is the most studied interleukin in the context of hematopoiesis. Although mice deficient in IL-6 (Il-6-/-) have no apparent deficit in neutrophil number during the basal state (82), they displayed reduced neutrophilia and neutrophil response during Listeria and Candida infections (83, 84). Since enhanced granulopoiesis in Gcsf-/- and Gmcsf-/- mice were associated with elevated IL-6 levels (51), the usage of mice with triple deficiency of IL-6, G-CSF, GM-CSF implicated IL-6 as a rescue cytokine that can take over granulopoiesis in the absence of classical granulopoietic cytokines, G-CSF and GM-CSF (85). Since these triple knock-out mice died shortly after birth, an in vitro granulopoiesis using embryonic fibroblasts revealed an additive effect for IL-6 (85). The neutrophil promoting activity of medium conditioned by Gcsf-/-/Gmcsf-/-/Il-6-/- fibroblasts was reduced by 40% compared to Gcsf-/-/Gmcsf-/- fibroblasts (85). Zhao et al. (29) and Schurch et al. (86) demonstrated that HSPCs and MSC produced IL-6 in response to inflammatory signals that stimulate HSC mobilization and myeloid differentiation in neutropenia or to promote clearance of the infection. More recently, it has been shown that TLR4 signaling-derived IL-6 promotes emergency granulopoiesis by phosphorylating C/EBP-α/β (87).
The role of IL-3 in emergency granulopoiesis is discrepant (88, 89). Innate response activator (IRA) B cells are the main producers of IL-3 during polymicrobial sepsis, and it is essential for emergency hematopoiesis, but dampens host survival benefit (89). In contrast, the specific depletion of IRA B cells in sepsis was detrimental as it leads to loss of GM-CSF signaling, increase in bacterial burden, and elicits cytokine storm (90). Unlike IL-6 and IL-3, the involvement of the IL-1 family and their receptor IL-1R in hematopoiesis have not been explored extensively until recently. Mice deficient in IL-1R (Il-1r-/-) have reduced proliferation of HSCs, MPPs, and GMP during alum-induced inflammatory granulopoiesis (91). Indeed, Pietras et al. (92) demonstrated that acute exposure of IL-1 enhanced the HSC proliferation and differentiation along the myeloid lineages through activation of transcription factor PU.1, but its chronic exposure impaired HSC functions and self-renewal capacity. IL-27, one of the IL-6/IL-12 family cytokines, has been recently shown to specifically promote the expansion and differentiation of LSK cells, particularly MPPs, into myeloid lineages in synergy with stem cell factor (93). Similarly, IL-27 transgenic mice displayed enhanced myelopoiesis and impaired development of B cell lineages (94). These results suggest that IL-27 is one of the limited cytokines that play a role in HSC regulation.
Interferons
Type I IFNs (IFN-α/β) and Type II IFNs (IFN-γ) are pro-inflammatory cytokines that regulate early hematopoiesis. Early reports using a model of virus-induced transient pancytopenia revealed that mice lacking α/β (IFN-α/βR-/-) or γ (IFN-γR-/-) receptors have a different degree of BM cellularity (95). Indeed, during virus-induced pancytopenia, BM cells (specifically pluripotent and committed progenitors) were substantially lost in WT and IFN-γR-/-, but not in IFN-α/βR-/- mice (95). Later, two elegant reports confirmed that IFN-α stimulates the exit and proliferation of HSCs in two different models (96, 97). Essers et al. (96) reported that the in vivo treatment of IFN-α enhanced the HSC proliferation in mice and HSCs deficient in IFN-α/βR were unresponsive to the treatment. Similarly, Sato et al. (97) discovered mice deficient in interferon response factor 2 (Irf2-/-), a transcriptional suppressor of IFN-α signaling, have an enhanced proportion of proliferating HSCs and Irf2-/- HSCs have accelerated cell division and failed to generate hematopoietic cells. Unlike Type I IFNs, the role of IFN-γ appears to be controversial in HSC proliferation. IFN-γ is sufficient to induce HSCs proliferation in vivo and HSCs from Ifnγr-/- mice have a reduced rate of proliferation (94). In stark contrast, IFN-γ has been shown to impair HSC proliferation and restoration during viral infection through inducing SOCS1 expression, thereby suppressing STAT5 phosphorylation (98). Moreover, the anti-proliferative activity of IFN-γ was shown to be dependent on integrin β3 signaling as it promotes serine phosphorylation of STAT1 in HSCs (99). Additionally, de Bruin et al. demonstrated that IFN-γ determined the fate of GMPs to monocytes over neutrophils during viral infection as IFN-γ enhanced the expression of PU.1 and IRF8 but dampened STAT3 phosphorylation (100).
Reactive oxygen species
ROS content of HSCs has been known to regulate their self-renewal, migration, and development (101–105). While ROS-low-HSCs are long-term repopulating and low cell cycling, ROS-high-HSCs are short-term repopulating, cell cycling, and differentiating (102, 103). Recently, the cell extrinsic effect of ROS on hematopoiesis has been explored in thioglycolate and infection-elicited granulopoiesis (106, 107). BM myeloid (GR-1+) cells produced NADPH oxidase-dependent ROS, which regulates the expansion of GMPs via a PTEN oxidation-dependent manner during E. coli-induced emergency granulopoiesis (107). Later, another study by the same group demonstrated that ROS-producing BM GR-1+ cells are also critical for neutrophil homeostasis in sterile inflammation-induced granulopoiesis (106). However, the contribution of pathogen sensing into ROS production by the BM GR-1+ niche and its cell specific proliferation of GMPs, but not MEP and CEP as shown by these studies needs further investigation.
Regulation of emergency granulopoiesis
At the molecular level, several transcription factors are involved in cell fate decision, differentiation, and proliferation.
CCAAT/Enhancer-binding proteins (C/EBPs)
Several studies have shown that transcription factors CCAAT/enhancer-binding proteins (C/EBPs) -α, -β, and -ϵ have major regulatory roles in neutrophil granulopoiesis (8, 108, 109). C/EBP-α, the prototypical basic-region leucine zipper (bZIP) transcription factor, is mostly expressed in myeloid cells within the hematopoietic system and known to regulate steady state granulopoiesis (17, 18). Mice deficient in C/EBP-α (C/EBP-α-/-) displayed impaired steady-state granulopoiesis, as they selectively lacked mature granulocytes but retain monocytes (18). Furthermore, these mice were unresponsive to exogenous IL-6 and G-CSF since expression of IL-6 and G-CSF receptors were compromised with loss of C/EBP-α (18, 110). C/EBP-α-/- fetal liver progenitors displayed high plating capacity for HSPCs but a striking block in differentiation of MPP into bipotent GMPs both in vivo and in vitro (111). Mechanistically, C/EBP-α negatively regulates the expression of c-Myc, a cell proliferative factor, through an E2F binding site, thereby altering myeloblasts differentiation to CMPs (112). Moreover, C/EBP-α directly causes proliferation arrest by inhibiting cyclin-dependent kinase 2 (Cdk2) and Cdk4 (113). C/EBP-α also decides cell fate as it directs a myeloid gene expression program through interacting factors involved in the expression of Il-6r, Csf3r, Gfi-1, Irf-8, and Klf5 (110, 114–116). For instance, the expression of C/EBPα in HSCs or in vivo activation of C/EBPα in mice resulted in increases in myeloid progenitors and granulocytes but concomitant decreases in erythroid and/or non-myeloid progenitors (117, 118). Further, the conditional silencing of C/EBPα in HSPCs led to the loss of expression of myeloid genes but aberrant expression of T cell-related genes such as Cd7 and Lck (119). C/EBP-ϵ regulates terminal differentiation of neutrophil development (120). Although C/EBP-ϵ-/- mice have no defect in generation of all blood cell lineages, they failed to develop functional neutrophils (121).
Conversely, C/EBP-β is upregulated in hematopoietic sub-populations during infectious or inflammatory states and orchestrates emergency granulopoiesis (9, 33, 122, 123). C/EBP-β can be induced in C/EBP-α-/- cells in vitro with IL-3, GM-CSF stimulation, or transduction with G-CSF to generate neutrophils, suggesting the existence of C/EBP-α-independent pathways for granulocyte generation (110, 124). Hirai et al. (9) were first to show conclusively that a large number of mature granulocytes can be generated from C/EBP-α-/- progenitors following cytokine (G-CSF, GM-CSF, and IL-3) treatment. Following C. albicans or cytokine administration, the sustained upregulation of C/EBP-β, but not C/EBPs-α, -δ, and –ϵ, was observed in granulocyte progenitors, and C/EBP-β-/- progenitors exhibit impaired proliferation and differentiation during cytokine- or fungal–induced emergency granulopoiesis (9, 123). Unlike C/EBPα, C/EBP-β-mediated emergency granulopoiesis may be dependent on prolonged expression of c-Myc in progenitor cells (9). How pathogen sensing during systemic infection leads to switch from C/EBP-α to C/EBP-β to promote emergency granulopoiesis is unknown.
STAT proteins
The exact mechanism of the switch from C/EBP-α-dependent steady state to C/EBP-β-mediated emergency granulopoiesis during the infectious state is poorly understood. However, the heightened level of granulocytic cytokines (G-CSF, GM-CSF, IL-6) and their interactions with receptors and cellular targets have revealed several other transcription factors that may act as a bridge between pathogen sensing and molecular translation during emergency granulopoiesis. For examples, the engagement of G-CSF signaling induces JAK-STAT pathways that mostly involve activation of STAT3 but also to a lesser extent STAT1 and STAT5. Gcsfr-/- mice displayed impaired activation of STAT3 and STAT5 and a defect in G-CSF-induced granulocytic differentiation in vivo and in vitro (125). Furthermore, STAT3 directly regulates expression of C/EBP-β in Gr-1+ granulocytes in L. monocytogenes-induced granulopoiesis, and STAT3 favors increase in occupancy of C/EBP-β over C/EBP-α to the c-Myc promoter during G-CSF-induced granulopoiesis (126). However, mice harboring STAT3 deletion in hematopoietic progenitors exhibited enhanced output of granulocyte precursors and functional neutrophils in G-CSF-induced granulopoiesis, providing direct evidence of a STAT3-dependent negative feedback loop (127). Mechanistically, deficiency of STAT3 resulted in impaired induction of suppressor of cytokine signaling (SOCS3) which otherwise would act as negative regulator of JAK catalytic activity during G-CSF-induced granulopoiesis (127). Similarly, Socs3-/- neutrophilic granulocytes displayed enhanced STAT3 activity in vitro (128) and Socs3-/- mice develop neutrophilia when injected with G-CSF (128, 129). The GM-CSF receptor pathways also participate in emergency granulopoiesis, and it is mediated through STAT5A and STAT5B (50, 130). Stat5a/b-/- mice exhibit defects in GMP generation and neutrophil survival during GM-CSF-induced granulopoiesis or 5FU-mediated myelosuppression (130).
Other myeloid transcription factors
Besides C/EBPs and STATs, Ets transcription factors PU.1, Runt-related transcription factor 1 (Runx1), zinc finger transcription factor (Gfi-1), lymphoid enhancer-binding factor 1 (LEF1), Interferon regulatory factor 8 (IRF8), and homeobox gene-A10 (HOXA10) are other important transcription factors regulating granulopoiesis. PU.1 is critical for development of all hematopoietic lineages (131) and the expression level of PU.1 determines lineage choice as low concentration of PU.1 allows C/EBP-α to favor granulocytes generation over monocytes from myeloblasts during steady-state granulopoiesis (132). One study reported that Runx1-/- mice exhibit a mild myeloproliferative phenotype with enhanced neutrophilia and expanded myeloid progenitors (133), while another study demonstrated that deletion or dominant inhibition of Runx1 reduces C/EBP-α and favors monopoiesis over granulopoiesis (134). Gfi1 antagonizes PU.1 (135) and is essential for steady-state and emergency granulopoiesis, and consequently Gfi1-/- mice are highly susceptible to bacterial infection (136). LEF1 directly controls C/EBP-α expression and mediates generation and differentiation of neutrophils (137). IRF8 influences the expression of Fanconi DNA repair proteins (i.e., FNCC and FNCF) which are critical for genomic stability during S phase (138, 139). Irf8-/- mice displayed decreased FAS sensitivity in myeloid progenitors and sustained granulocyte generation during alum-induced emergency granulopoiesis (140). Mice deficient in Fanconi-C were neutropenic and exhibited impaired HSC survival and function in alum-induced granulopoiesis (138). Like SOCS3 and IRF8, HOXA10 is a negative regulator of emergency granulopoiesis as Hoxa10-/- mice develop uncontrolled neutrophilia associated with heightened activity of Triad1, an anti-proliferative E3 ubiquitin ligase (141, 142). More recently, secretory leukocyte protease inhibitor (SLPI), a natural repressor of neutrophil elastase, has been shown to regulate the expression of several transcription factors in G-CSF-triggered emergency granulopoiesis and it is severely reduced in congenital neutropenic patients (143). Transducing SLPI-shRNA into human BM CD34+ progenitors and leukemic NB4 cells, Klimenkova et al. demonstrated that SLPI regulates G-CSF-induced granulopoiesis by regulating NFκB levels, ERK1/2 phosphorylation and activation of LEF-1, and c-Myc-dependent signaling pathways (143). Recently, Danek et al. demonstrate the β-Catenin-TCF/LEF directly regulates G-CSF receptor, thereby consequently controlling steady and emergency granulopoiesis (144).
Microbiota
As crosstalk between host microbiome and immunity is well recognized, a large body of evidence suggests that gut microbiota can directly regulate neutrophil homeostasis and contribute to priming host immune responses. Antibiotic-treated and germ-free mice exhibit reduced numbers of granulocyte progenitors in BM and spleen, and a decline in blood neutrophils (80, 145–149). Mice replenished and enriched with gut microbiota exhibit restored granulopoiesis (145, 150, 151). Microbiota-derived products such as peptidoglycan or lipoprotein can make their way into the BM niche, thereby allowing HSPCs and myeloid precursors to detect them and initiate hematopoiesis directly (148, 151–153). Another possibility is that HSPCs can exit the BM, patrol peripheral organs or even reside there for few days, and encounter gut microbiota-derived pathogens/their products before re-entering the BM (14, 15). On other hand, gut microbial products such as LPS can activate TLR4/MyD88 pathways to induce IL-17A and G-CSF, thereby indirectly activating granulopoiesis (145). Granulopoiesis in germ-free (GF) mice is also dependent on TLR signaling; as Myd88-/-/Ticam-/- GF mice failed to restore microbiota-driven granulopoiesis when transferred with microbiota or serum from colonized mice (151). The malnourished host has impaired hematopoiesis, and supplementation of immunobiotics such as Lactobacillus rhamnosus CRL1505 in the host restored the granulopoiesis and improved immune responses (154). Thus, it is tempting to speculate that microbiota may have educated the hematopoietic system to respond to peripheral demands along a continuum, and manipulation of microbiota or immunobiotics could improve host immunity in neutropenic patients.
Macrophages
The macrophage’s inability to clear apoptotic neutrophils regulates steady-state granulopoiesis (155–157). After phagocytosis of aged neutrophils, macrophages secrete less IL-23 thereby curbing down IL-17/G-CSF axis and steady-state granulopoiesis (155). Mice harboring a conditional deletion of the anti-apoptotic gene cellular FLICE-like inhibitory protein (c-FLIP) in myeloid cells (c-FLIPf/fLysM-Cre) lacks macrophages specifically the in marginal zone and BM stroma (156). c-FLIPf/fLysM-Cre mice exhibited delayed clearance of circulating neutrophils and developed severe neutrophilia in a G-CSF-dependent but not IL-17-dependent manner (156). Mechanistically, the engulfment of senescent neutrophils activates liver x receptors (LXR) in macrophages which directly suppress the IL-23/IL-17/G-CSF granulopoietic cascade (157). Recently, mast cells have been shown to enhance macrophage phagocytosis, and mice lacking mast cells (KitW‐sh) exhibited elevated IL-17/G-SCF activity, aberrant intramedullary myelopoiesis, and peripheral blood neutrophilia (158).
Non-hematopoietic BM stromal cells
With recent advancements in reporter mouse models, BM chimeras, imaging, and cell sorting techniques, the presence of the non-hematopoietic cellular compartment has been mapped out and appreciated as a critical regulator of granulopoiesis and immunity (159). BM endothelial cells express TLR4 and MyD88 at the basal state and respond with heightened G-CSF expression upon LPS stimulation (36). TLR4 signaling confined to the non-hematopoietic compartment only is sufficient to promote LPS-induced granulopoiesis (35). In particular, VCAM+CD146low BM stromal fibroblasts produce IL-6 which contributes to expansion of GMPs at the expense of erythropoiesis during Toxoplasma gondii infection (160). Similarly, BM CXCL12-abundant reticular (CAR) cells secrete IL-6 in response to IFN-γ by CD8 T cells during viral infection, and IL-6 in turn favors HSPCs differentiation toward myeloid lineages (86).
Special notes on neutrophil circadian rhythms
Circadian control of the immune system is an emerging concept of the regulation of immune homeostasis (161) (Figure 4). A recent study has shown that, like HSCs (162) and monocytes (163), circulating neutrophils also follow daily circadian fluctuations, as they age in peripheral blood and re-enter the BM for clearance (164). Mechanistically, daily aged circulating neutrophils are characterized by expression of low levels of L-selectin (CD62L) but high levels of CXCR4, guiding their way back to the BM within 12 hours (164). Using an organism-wide circadian screening approach, an elegant study has identified circadian expression of trafficking molecules (ICAM-1, VCAM-1, CD11a, CD49d, CXCR4) for neutrophils and other leukocytes, regulating the rhythmic emigration of leukocytes from blood (165). More specifically, chronopharmacological targeting of these migratory molecules has revealed that CXCR4, L-selectin, and ICAM-1 govern rhythmic neutrophil migration to the bone marrow, spleen, and liver respectively (165). Following an acute LPS exposure, the blood counts of neutrophils and other leukocytes exhibited a dramatic drop but their time-of-day differences between day and night were preserved, extending the notion of neutrophil rhythmic oscillation to the inflammatory state (165). Although circadian control of neutrophil oscillation is still in its infancy, we can speculate that it is a critical mechanism of immune sentinels to maintain neutrophil homeostasis and determine the strength of the immune response during infection and inflammation.
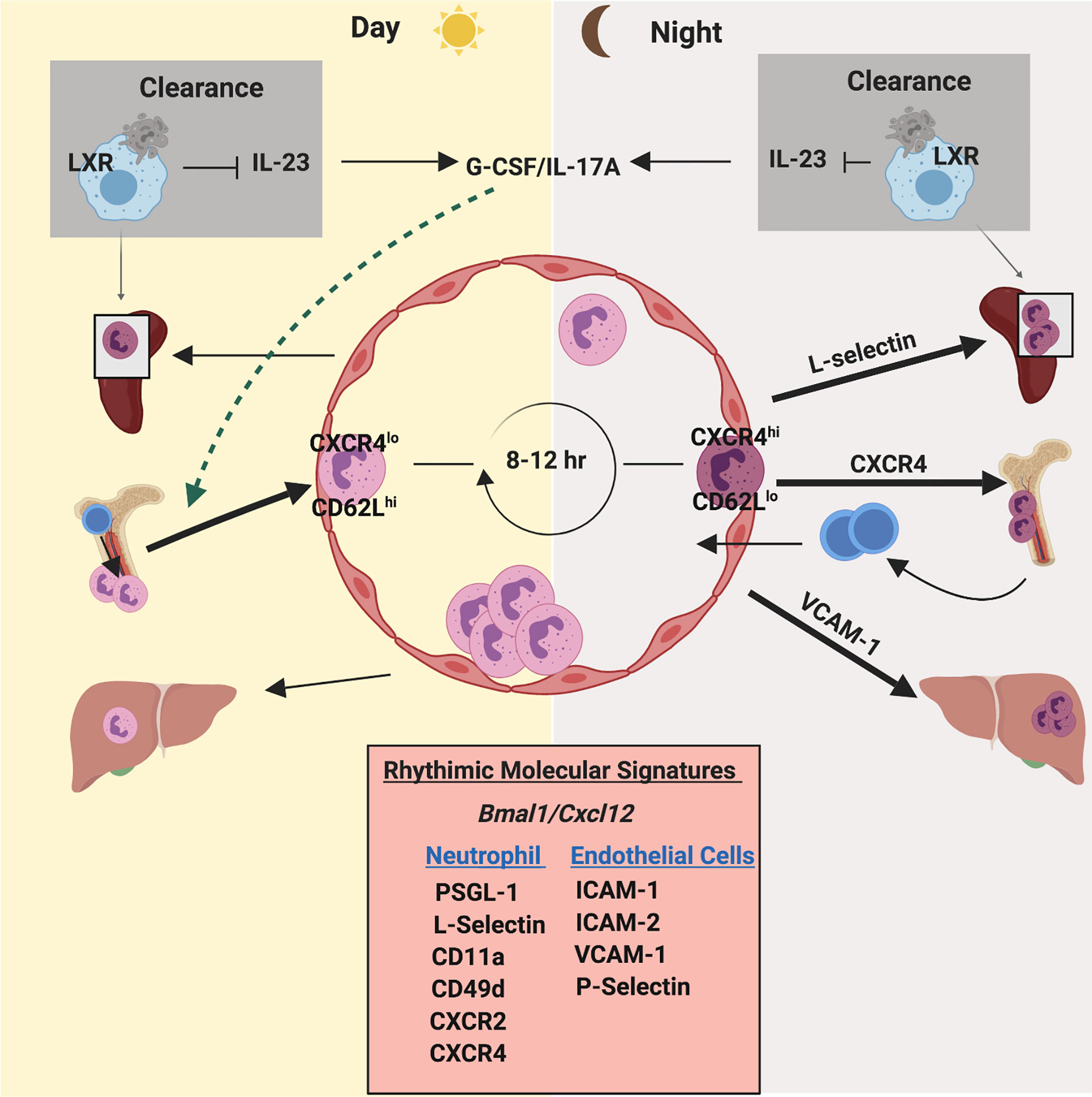
Figure 4 Circadian rhythm regulates neutrophil homeostasis and function. Neutrophils exhibit an oscillation in numbers in blood and tissues, which coincides with daily circadian rhythms. Neutrophil numbers in blood peak after few hours of initiation of light and then drop during the light phase. Under the influence of clock genes (such as Bmal1) and granulopoietic cytokines (mainly G-CSF), CXCL12/CXCR4 regulates daily release and retention of neutrophils. Furthermore, a circadian fluctuation of the expression of CD62L and CXCR4 on the surface of neutrophils dictates their counts in blood during steady state. CD62LhighCXCR4low neutrophils enter circulation during the light phase. After 8-12 hours, aged neutrophils (CD62LlowCXCR4high) enter the bone marrow for clearance coinciding with daily circadian rhythms. Additionally, oscillatory expression of pro-inflammatory and adhesive molecules on the endothelium and on neutrophils exhibit a robust circadian rhythmicity during neutrophil release and egress.
Trained immunity
Recently, several epidemiological studies have shown that vaccination or prior exposure to a disease provide non-specific heterologous protection to non-related diseases through epigenetic imprinting of immunological memory of innate cells, a phenomenon termed as “Trained immunity’ or “Innate immune memory” (166). Trained immunity is orchestrated by epigenetic imprinting, which involves histone modifications, as well as, transcriptional and functional reprogramming of innate and non-immune cells following vaccination and infection (167). Kalafati et al. have shown that beta-glucan-induced trained immunity is associated with transcriptomic and epigenetic imprinting of granulopoiesis, which has an anti-tumor effect (168).
Human vs mouse granulopoiesis
Our current knowledge of granulopoiesis largely depends on data from murine models. However, we recognize there are significant differences in the maturation, migration, heterogeneity, and function of human and mouse neutrophils. Neutrophils accounts for up to ∼70% of all peripheral leukocytes in humans and ∼10–20% in mice (169). Furthermore, human and mouse neutrophils differ in neutrophil molecules (selectins, FcαRI, serine proteases) (170–172), expression of cytokines (IL-10, IL-17) (173), activation pathways of ROS (174), and cytotoxic granular content (defensins, bactericidal enzymes) (175) that can collectively alter the overall biology and immunology of these cells between species. On the other hand, a recent study utilizing single-cell transcriptomics to investigate human and mouse lung cancers revealed conservation of neutrophil subsets between species (176, 177).
Concluding remarks and future directions
In summary, it is very clear now that neutrophil homeostasis during the infectious state is a complex and tightly regulated process. Neutrophil homeostasis involves the sensing of pathogens or other stimuli by hematopoietic and non-hematopoietic cells (TLR/MyD88 pathways), the release of growth factors (G-CSFs, IL-6, CXCs) to activate several transcription machineries, and the ultimate instruction to activate dormant HSC and generate neutrophils in the BM (C/EBP-β, STATs), all gearing to mount a protective hematopoietic immune response. C/EBP-β mediates emergency granulopoiesis and C/EBP-α regulates steady-state granulopoiesis.
Accumulating data from several preclinical models have deepened our knowledge regarding mechanisms of neutrophil homeostasis. In a few instances, however, they have generated conflicting data and authors have sometimes postulated different and contradictory mechanisms of neutrophil homeostasis. These discrepancies can be explained by differences in experimental model (infection or cytokine-induced), types and inoculum of pathogens (bacterial vs. fungal), route of infections, timing of analysis, endpoints to measure granulopoiesis (BM vs. peripheral blood output, host survival vs. recruitment), and even composition of microbiota. We suggest that these discrepancies can be resolved by the development of appropriate experimental models to test clearly defined endpoints to study emergency granulopoiesis.
It is now obvious that the hematopoietic system has emerged to detect invading pathogens in a very fast and versatile way. A diverse spectrum of microbial messaging to the bone marrow is in place. For instance, direct pathogen sensing by HSC and myeloid progenitors allows the hematopoietic system to rapidly respond with early granulopoiesis to control imminent infection. How this pathway of direct activation differs from indirect activation of HSPCs in terms of conversion of pathogen signal to molecular cues during granulopoiesis is largely unknown. Does waking up HSC with TLR agonists or PAMPs have clinical use? Furthermore, future studies should be directed to investigate the relative contributions and differences of direct versus indirect HSPC activation. With the discovery of ECs regulating the emergency granulopoietic response, it is possible that vasculature linings ECs are a critical layer of the hematopoietic system that convert systemic spreading pathogen signals to molecular cues. Additionally, various bone marrow stromal cells, macrophages, and aged neutrophils appear to control granulopoiesis. Is there any crosstalk between hematopoietic and resident cells during granulopoiesis? What will be their contributions? Do they initiate distinct signaling mechanisms? An emerging challenge to hematologists and immunologists would be to solve how these diverse pathogen signals, picked up by different cells, are converted by HSC to launch a mechanism of emergency granulopoiesis.
Growth factors, cytokines, and chemokines activate HSCs, instruct lineage choice, and regulate the generation of neutrophils, thereby making them excellent molecular targets for therapeutics. G-CSF and GM-CSF are already in use for clinical applications to regenerate myeloid cells. However, decades of preclinical studies have demonstrated that these signaling pathways are extremely redundant. For instance, IL-6 can surrogate the function of all three colony-stimulating factors during emergency granulopoiesis. Experimental models with gain and loss of function of these specific pathways are warranted to understand their specific role. Thus, understanding the common cellular targets of these molecular messengers would be of great interest for therapeutic efficiency.
In addition, we feel that there is limited knowledge on key cellular, molecular, and environmental players that regulate emergency granulopoiesis. Several outstanding questions remain unanswered. What are the mechanisms in place to prevent an uncontrolled granulopoietic response? Could dysregulation of the granulopoietic response or direct HSC activation contribute to BM failure and development of hematologic diseases? How do we translate our findings from preclinical studies into human infectious and hematologic diseases? A systems biology and single-cell approach may be needed to understand how hematopoietic systems integrate molecular signals into a single and unique granulopoietic response. The evolving role of the host microbiome in shaping and priming the hematopoietic response is another promising area to explore.
A better understanding of sensors, mediators, and regulators of emergency granulopoiesis may inspire novel therapeutic approaches for neonatal, pediatric, and adult infectious diseases. Signaling pathways used during emergency granulopoiesis could be examined and exploited for development of therapeutic targets for hematologic as well as malignant diseases.
Author contributions
SP wrote the draft along with LG and LJ. DJ and SJ edited the draft. All authors contributed to the article and approved the submitted version.
Acknowledgments
We would like to acknowledge members of the Center for Lung Biology and Diseases, including Tirumalai Rangasamy, Joseph DeCorte, John T. Le and Dinesh Bhattarai, for helpful discussions. This work is supported by R01AI157353 (SJ), R01AI140500 (SJ) and P20GM130555 (SJ) from the National Institute of Health.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Mechnikov II. Immunity in infective diseases. by il'ia il'ich mechnikov, 1905. Rev Infect Dis (1988) 10:223–7. doi: 10.1093/clinids/10.1.223
2. Dancey JT, Deubelbeiss KA, Harker LA, Finch CA. Neutrophil kinetics in man. J Clin Invest (1976) 58:705–15. doi: 10.1172/JCI108517
3. Trumpp A, Essers M, Wilson A. Awakening dormant haematopoietic stem cells. Nat Rev Immunol (2010) 10:201–9. doi: 10.1038/nri2726
4. Seita J, Weissman IL. Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdiscip Rev Syst Biol Med (2010) 2:640–53. doi: 10.1002/wsbm.86
5. Pouzolles M, Oburoglu L, Taylor N, Zimmermann VS. Hematopoietic stem cell lineage specification. Curr Opin Hematol (2016) 23:311–7. doi: 10.1097/MOH.0000000000000260
6. Eaves CJ. Hematopoietic stem cells: concepts, definitions, and the new reality. Blood (2015) 125:2605–13. doi: 10.1182/blood-2014-12-570200
7. Hoggatt J, Kfoury Y, Scadden DT. Hematopoietic stem cell niche in health and disease. Annu Rev Pathol (2016) 11:555–81. doi: 10.1146/annurev-pathol-012615-044414
8. Lawrence SM, Corriden R, Nizet V. The ontogeny of a neutrophil: Mechanisms of granulopoiesis and homeostasis. Microbiol Mol Biol Rev (2018) 82. doi: 10.1128/MMBR.00057-17
9. Hirai H, Zhang P, Dayaram T, Hetherington CJ, Mizuno S, Imanishi J, et al. C/EBPbeta is required for 'emergency' granulopoiesis. Nat Immunol (2006) 7:732–9. doi: 10.1038/ni1354
10. Ueda Y, Kondo M, Kelsoe G. Inflammation and the reciprocal production of granulocytes and lymphocytes in bone marrow. J Exp Med (2005) 201:1771–80. doi: 10.1084/jem.20041419
11. Takizawa H, Boettcher S, Manz MG. Demand-adapted regulation of early hematopoiesis in infection and inflammation. Blood (2012) 119:2991–3002. doi: 10.1182/blood-2011-12-380113
12. Kuderer NM, Dale DC, Crawford J, Cosler LE, Lyman GH. Mortality, morbidity, and cost associated with febrile neutropenia in adult cancer patients. Cancer (2006) 106:2258–66. doi: 10.1002/cncr.21847
13. Hasan S, Naqvi AR, Rizvi A. Transcriptional regulation of emergency granulopoiesis in leukemia. Front Immunol (2018) 9:481. doi: 10.3389/fimmu.2018.00481
14. Wright DE, Wagers AJ, Gulati AP, Johnson FL, Weissman IL. Physiological migration of hematopoietic stem and progenitor cells. Science (2001) 294:1933–6. doi: 10.1126/science.1064081
15. Massberg S, Schaerli P, Knezevic-Maramica I, Kollnberger M, Tubo N, Moseman EA, et al. Immunosurveillance by hematopoietic progenitor cells trafficking through blood, lymph, and peripheral tissues. Cell (2007) 131:994–1008. doi: 10.1016/j.cell.2007.09.047
16. Boettcher S, Manz MG. Regulation of inflammation- and infection-driven hematopoiesis. Trends Immunol (2017) 38:345–57. doi: 10.1016/j.it.2017.01.004
17. Zhang P, Iwasaki-Arai J, Iwasaki H, Fenyus ML, Dayaram T, Owens BM, et al. Enhancement of hematopoietic stem cell repopulating capacity and self-renewal in the absence of the transcription factor C/EBP alpha. Immunity (2004) 21:853–63. doi: 10.1016/j.immuni.2004.11.006
18. Zhang DE, Zhang P, Wang ND, Hetherington CJ, Darlington GJ, Tenen DG. Absence of granulocyte colony-stimulating factor signaling and neutrophil development in CCAAT enhancer binding protein alpha-deficient mice. Proc Natl Acad Sci U.S.A. (1997) 94:569–74. doi: 10.1073/pnas.94.2.569
20. Nagai Y, Garrett KP, Ohta S, Bahrun U, Kouro T, Akira S, et al. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity (2006) 24:801–12. doi: 10.1016/j.immuni.2006.04.008
21. Schmid MA, Takizawa H, Baumjohann DR, Saito Y, Manz MG. Bone marrow dendritic cell progenitors sense pathogens via toll-like receptors and subsequently migrate to inflamed lymph nodes. Blood (2011) 118:4829–40. doi: 10.1182/blood-2011-03-344960
22. Zhang NN, Shen SH, Jiang LJ, Zhang W, Zhang HX, Sun YP, et al. RIG-I plays a critical role in negatively regulating granulocytic proliferation. Proc Natl Acad Sci U.S.A. (2008) 105:10553–8. doi: 10.1073/pnas.0804895105
23. De Luca K, Frances-Duvert V, Asensio MJ, Ihsani R, Debien E, Taillardet M, et al. The TLR1/2 agonist PAM(3)CSK(4) instructs commitment of human hematopoietic stem cells to a myeloid cell fate. Leukemia (2009) 23:2063–74. doi: 10.1038/leu.2009.155
24. Sioud M, Floisand Y, Forfang L, Lund-Johansen F. Signaling through toll-like receptor 7/8 induces the differentiation of human bone marrow CD34+ progenitor cells along the myeloid lineage. J Mol Biol (2006) 364:945–54. doi: 10.1016/j.jmb.2006.09.054
25. Chicha L, Jarrossay D, Manz MG. Clonal type I interferon-producing and dendritic cell precursors are contained in both human lymphoid and myeloid progenitor populations. J Exp Med (2004) 200:1519–24. doi: 10.1084/jem.20040809
26. Esplin BL, Shimazu T, Welner RS, Garrett KP, Nie L, Zhang Q, et al. Chronic exposure to a TLR ligand injures hematopoietic stem cells. J Immunol (2011) 186:5367–75. doi: 10.4049/jimmunol.1003438
27. Zhao Y, Ling F, Wang HC, Sun XH. Chronic TLR signaling impairs the long-term repopulating potential of hematopoietic stem cells of wild type but not Id1 deficient mice. PloS One (2013) 8:e55552. doi: 10.1371/journal.pone.0055552
28. Megias J, Yanez A, Moriano S, O'Connor JE, Gozalbo D, Gil ML. Direct toll-like receptor-mediated stimulation of hematopoietic stem and progenitor cells occurs in vivo and promotes differentiation toward macrophages. Stem Cells (2012) 30:1486–95. doi: 10.1002/stem.1110
29. Zhao JL, Ma C, O'Connell RM, Mehta A, DiLoreto R, Heath JR, et al. Conversion of danger signals into cytokine signals by hematopoietic stem and progenitor cells for regulation of stress-induced hematopoiesis. Cell Stem Cell (2014) 14:445–59. doi: 10.1016/j.stem.2014.01.007
30. Buechler MB, Akilesh HM, Hamerman JA. Cutting edge: Direct sensing of TLR7 ligands and type I IFN by the common myeloid progenitor promotes mTOR/PI3K-dependent emergency myelopoiesis. J Immunol (2016) 197:2577–82. doi: 10.4049/jimmunol.1600813
31. Scumpia PO, Kelly-Scumpia KM, Delano MJ, Weinstein JS, Cuenca AG, Al-Quran S, et al. Cutting edge: bacterial infection induces hematopoietic stem and progenitor cell expansion in the absence of TLR signaling. J Immunol (2010) 184:2247–51. doi: 10.4049/jimmunol.0903652
32. Cossio I, Lucas D, Hidalgo A. Neutrophils as regulators of the hematopoietic niche. Blood (2019) 133:2140–8. doi: 10.1182/blood-2018-10-844571
33. Cain DW, Snowden PB, Sempowski GD, Kelsoe G. Inflammation triggers emergency granulopoiesis through a density-dependent feedback mechanism. PloS One (2011) 6:e19957. doi: 10.1371/journal.pone.0019957
34. Bugl S, Wirths S, Radsak MP, Schild H, Stein P, Andre MC, et al. Steady-state neutrophil homeostasis is dependent on TLR4/TRIF signaling. Blood (2013) 121:723–33. doi: 10.1182/blood-2012-05-429589
35. Boettcher S, Ziegler P, Schmid MA, Takizawa H, van Rooijen N, Kopf M, et al. Cutting edge: LPS-induced emergency myelopoiesis depends on TLR4-expressing nonhematopoietic cells. J Immunol (2012) 188:5824–8. doi: 10.4049/jimmunol.1103253
36. Boettcher S, Gerosa RC, Radpour R, Bauer J, Ampenberger F, Heikenwalder M, et al. Endothelial cells translate pathogen signals into G-CSF-driven emergency granulopoiesis. Blood (2014) 124:1393–403. doi: 10.1182/blood-2014-04-570762
37. Burberry A, Zeng MY, Ding L, Wicks I, Inohara N, Morrison SJ, et al. Infection mobilizes hematopoietic stem cells through cooperative NOD-like receptor and toll-like receptor signaling. Cell Host Microbe (2014) 15:779–91. doi: 10.1016/j.chom.2014.05.004
38. Lenkiewicz AM, Adamiak M, Thapa A, Bujko K, Pedziwiatr D, Abdel-Latif AK, et al. The Nlrp3 inflammasome orchestrates mobilization of bone marrow-residing stem cells into peripheral blood. Stem Cell Rev (2019) 15:391–403. doi: 10.1007/s12015-019-09890-7
39. Gentile LF, Cuenca AL, Cuenca AG, Nacionales DC, Ungaro R, Efron PA, et al. Improved emergency myelopoiesis and survival in neonatal sepsis by caspase-1/11 ablation. Immunology (2015) 145:300–11. doi: 10.1111/imm.12450
40. Paudel S, Ghimire L, Jin L, Baral P, Cai S, Jeyaseelan S. NLRC4 suppresses IL-17A-mediated neutrophil-dependent host defense through upregulation of IL-18 and induction of necroptosis during gram-positive pneumonia. Mucosal Immunol (2019) 12:247–57. doi: 10.1038/s41385-018-0088-2
41. Anand PK, Malireddi RK, Lukens JR, Vogel P, Bertin J, Lamkanfi M, et al. NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature (2012) 488:389–93. doi: 10.1038/nature11250
42. Ghimire L, Paudel S, Jin L, Baral P, Cai S, Jeyaseelan S. NLRP6 negatively regulates pulmonary host defense in gram-positive bacterial infection through modulating neutrophil recruitment and function. PloS Pathog (2018) 14:e1007308. doi: 10.1371/journal.ppat.1007308
43. Ulich TR, del Castillo J, McNiece IK, Yi ES, Alzona CP, Yin SM, et al. Stem cell factor in combination with granulocyte colony-stimulating factor (CSF) or granulocyte-macrophage CSF synergistically increases granulopoiesis in vivo. Blood (1991) 78:1954–62. doi: 10.1182/blood.V78.8.1954.1954
44. Gasson JC. Molecular physiology of granulocyte-macrophage colony-stimulating factor. Blood (1991) 77:1131–45. doi: 10.1182/blood.V77.6.1131.1131
45. Metcalf D, Begley CG, Williamson DJ, Nice EC, De Lamarter J, Mermod JJ, et al. Hemopoietic responses in mice injected with purified recombinant murine GM-CSF. Exp Hematol (1987) 15:1–9.
46. Hume DA, Pavli P, Donahue RE, Fidler IJ. The effect of human recombinant macrophage colony-stimulating factor (CSF-1) on the murine mononuclear phagocyte system in vivo. J Immunol (1988) 141:3405–9.
47. Lieschke GJ, Grail D, Hodgson G, Metcalf D, Stanley E, Cheers C, et al. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood (1994) 84:1737–46. doi: 10.1182/blood.V84.6.1737.1737
48. Liu F, Wu HY, Wesselschmidt R, Kornaga T, Link DC. Impaired production and increased apoptosis of neutrophils in granulocyte colony-stimulating factor receptor-deficient mice. Immunity (1996) 5:491–501. doi: 10.1016/S1074-7613(00)80504-X
49. Stanley E, Lieschke GJ, Grail D, Metcalf D, Hodgson G, Gall JA, et al. Granulocyte/macrophage colony-stimulating factor-deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proc Natl Acad Sci U.S.A. (1994) 91:5592–6. doi: 10.1073/pnas.91.12.5592
50. Zhan Y, Lieschke GJ, Grail D, Dunn AR, Cheers C. Essential roles for granulocyte-macrophage colony-stimulating factor (GM-CSF) and G-CSF in the sustained hematopoietic response of listeria monocytogenes-infected mice. Blood (1998) 91:863–9. doi: 10.1182/blood.V91.3.863
51. Basu S, Hodgson G, Zhang HH, Katz M, Quilici C, Dunn AR. "Emergency" granulopoiesis in G-CSF-deficient mice in response to candida albicans infection. Blood (2000) 95:3725–33. doi: 10.1182/blood.V95.12.3725
52. Zhan Y, Cheers C. Haemopoiesis in mice genetically lacking granulocyte-macrophage colony stimulating factor during chronic infection with mycobacterium avium. Immunol Cell Biol (2000) 78:118–23. doi: 10.1046/j.1440-1711.2000.00891.x
53. Hibbs ML, Quilici C, Kountouri N, Seymour JF, Armes JE, Burgess AW, et al. Mice lacking three myeloid colony-stimulating factors (G-CSF, GM-CSF, and m-CSF) still produce macrophages and granulocytes and mount an inflammatory response in a sterile model of peritonitis. J Immunol (2007) 178:6435–43. doi: 10.4049/jimmunol.178.10.6435
54. Schuettpelz LG, Borgerding JN, Christopher MJ, Gopalan PK, Romine MP, Herman AC, et al. G-CSF regulates hematopoietic stem cell activity, in part, through activation of toll-like receptor signaling. Leukemia (2014) 28:1851–60. doi: 10.1038/leu.2014.68
55. Semerad CL, Christopher MJ, Liu F, Short B, Simmons PJ, Winkler I, et al. G-CSF potently inhibits osteoblast activity and CXCL12 mRNA expression in the bone marrow. Blood (2005) 106:3020–7. doi: 10.1182/blood-2004-01-0272
56. Rieger MA, Hoppe PS, Smejkal BM, Eitelhuber AC, Schroeder T. Hematopoietic cytokines can instruct lineage choice. Science (2009) 325:217–8. doi: 10.1126/science.1171461
57. Mossadegh-Keller N, Sarrazin S, Kandalla PK, Espinosa L, Stanley ER, Nutt SL, et al. M-CSF instructs myeloid lineage fate in single haematopoietic stem cells. Nature (2013) 497:239–43. doi: 10.1038/nature12026
58. Jack GD, Zhang L, Friedman AD. M-CSF elevates c-fos and phospho-C/EBPalpha(S21) via ERK whereas G-CSF stimulates SHP2 phosphorylation in marrow progenitors to contribute to myeloid lineage specification. Blood (2009) 114:2172–80. doi: 10.1182/blood-2008-11-191536
59. Katahira Y, Higuchi H, Matsushita H, Yahata T, Yamamoto Y, Koike R, et al. Increased granulopoiesis in the bone marrow following Epstein-Barr virus infection. Sci Rep (2019) 9:13445. doi: 10.1038/s41598-019-49937-w
60. Brook B, Harbeson DJ, Shannon CP, Cai B, He D, Ben-Othman R, et al. BCG Vaccination-induced emergency granulopoiesis provides rapid protection from neonatal sepsis. Sci Transl Med (2020) 12. doi: 10.1126/scitranslmed.aax4517
61. Bonavita O, Mollica Poeta V, Massara M, Mantovani A, Bonecchi R. Regulation of hematopoiesis by the chemokine system. Cytokine (2018) 109:76–80. doi: 10.1016/j.cyto.2018.01.021
62. Broxmeyer HE. Chemokines in hematopoiesis. Curr Opin Hematol (2008) 15:49–58. doi: 10.1097/MOH.0b013e3282f29012
63. Mendelson A, Frenette PS. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat Med (2014) 20:833–46. doi: 10.1038/nm.3647
64. Karpova D, Bonig H. Concise review: CXCR4/CXCL12 signaling in immature hematopoiesis–lessons from pharmacological and genetic models. Stem Cells (2015) 33:2391–9. doi: 10.1002/stem.2054
65. Ma Q, Jones D, Borghesani PR, Segal RA, Nagasawa T, Kishimoto T, et al. Impaired b-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc Natl Acad Sci U.S.A. (1998) 95:9448–53. doi: 10.1073/pnas.95.16.9448
66. Nagasawa T, Hirota S, Tachibana K, Takakura N, Nishikawa S, Kitamura Y, et al. Defects of b-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature (1996) 382:635–8. doi: 10.1038/382635a0
67. Messina-Graham S, Broxmeyer H. SDF-1/CXCL12 modulates mitochondrial respiration of immature blood cells in a bi-phasic manner. Blood Cells Mol Dis (2016) 58:13–8. doi: 10.1016/j.bcmd.2016.01.008
68. Baba T, Naka K, Morishita S, Komatsu N, Hirao A, Mukaida N. MIP-1alpha/CCL3-mediated maintenance of leukemia-initiating cells in the initiation process of chronic myeloid leukemia. J Exp Med (2013) 210:2661–73. doi: 10.1084/jem.20130112
69. Broxmeyer HE, Sherry B, Lu L, Cooper S, Oh KO, Tekamp-Olson P, et al. Enhancing and suppressing effects of recombinant murine macrophage inflammatory proteins on colony formation in vitro by bone marrow myeloid progenitor cells. Blood (1990) 76:1110–6. doi: 10.1182/blood.V76.6.1110.1110
70. Broxmeyer HE, Cooper S, Hangoc G, Gao JL, Murphy PM. Dominant myelopoietic effector functions mediated by chemokine receptor CCR1. J Exp Med (1999) 189:1987–92. doi: 10.1084/jem.189.12.1987
71. Staversky RJ, Byun DK, Georger MA, Zaffuto BJ, Goodman A, Becker MW, et al. The chemokine CCL3 regulates myeloid differentiation and hematopoietic stem cell numbers. Sci Rep (2018) 8:14691. doi: 10.1038/s41598-018-32978-y
72. Youn BS, Mantel C, Broxmeyer HE. Chemokines, chemokine receptors and hematopoiesis. Immunol Rev (2000) 177:150–74. doi: 10.1034/j.1600-065X.2000.17701.x
73. Fujimura N, Xu B, Dalman J, Deng H, Aoyama K, Dalman RL. CCR2 inhibition sequesters multiple subsets of leukocytes in the bone marrow. Sci Rep (2015) 5:11664. doi: 10.1038/srep11664
74. Ergen AV, Boles NC, Goodell MA. Rantes/Ccl5 influences hematopoietic stem cell subtypes and causes myeloid skewing. Blood (2012) 119:2500–9. doi: 10.1182/blood-2011-11-391730
75. Sanchez X, Suetomi K, Cousins-Hodges B, Horton JK, Navarro J. CXC chemokines suppress proliferation of myeloid progenitor cells by activation of the CXC chemokine receptor 2. J Immunol (1998) 160:906–10.
76. Broxmeyer HE, Cooper S, Cacalano G, Hague NL, Bailish E, Moore MW. Involvement of interleukin (IL) 8 receptor in negative regulation of myeloid progenitor cells in vivo: evidence from mice lacking the murine IL-8 receptor homologue. J Exp Med (1996) 184:1825–32. doi: 10.1084/jem.184.5.1825
77. Cacalano G, Lee J, Kikly K, Ryan AM, Pitts-Meek S, Hultgren B, et al. Neutrophil and b cell expansion in mice that lack the murine IL-8 receptor homolog. Science (1994) 265:682–4. doi: 10.1126/science.8036519
78. Sinclair A, Park L, Shah M, Drotar M, Calaminus S, Hopcroft LE, et al. CXCR2 and CXCL4 regulate survival and self-renewal of hematopoietic stem/progenitor cells. Blood (2016) 128:371–83. doi: 10.1182/blood-2015-08-661785
79. Eash KJ, Greenbaum AM, Gopalan PK, Link DC. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J Clin Invest (2010) 120:2423–31. doi: 10.1172/JCI41649
80. Mei J, Liu Y, Dai N, Hoffmann C, Hudock KM, Zhang P, et al. Cxcr2 and Cxcl5 regulate the IL-17/G-CSF axis and neutrophil homeostasis in mice. J Clin Invest (2012) 122:974–86. doi: 10.1172/JCI60588
81. Paudel S, Baral P, Ghimire L, Bergeron S, Jin L, DeCorte JA, et al. CXCL1 regulates neutrophil homeostasis in pneumonia-derived sepsis caused by streptococcus pneumoniae serotype 3. Blood (2019) 133:1335–45. doi: 10.1182/blood-2018-10-878082
82. Kopf M, Baumann H, Freer G, Freudenberg M, Lamers M, Kishimoto T, et al. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature (1994) 368:339–42. doi: 10.1038/368339a0
83. Dalrymple SA, Lucian LA, Slattery R, McNeil T, Aud DM, Fuchino S, et al. Interleukin-6-deficient mice are highly susceptible to listeria monocytogenes infection: correlation with inefficient neutrophilia. Infect Immun (1995) 63:2262–8. doi: 10.1128/iai.63.6.2262-2268.1995
84. Romani L, Mencacci A, Cenci E, Spaccapelo R, Toniatti C, Puccetti P, et al. Impaired neutrophil response and CD4+ T helper cell 1 development in interleukin 6-deficient mice infected with candida albicans. J Exp Med (1996) 183:1345–55. doi: 10.1084/jem.183.4.1345
85. Walker F, Zhang HH, Matthews V, Weinstock J, Nice EC, Ernst M, et al. IL6/sIL6R complex contributes to emergency granulopoietic responses in G-CSF- and GM-CSF-deficient mice. Blood (2008) 111:3978–85. doi: 10.1182/blood-2007-10-119636
86. Schurch CM, Riether C, Ochsenbein AF. Cytotoxic CD8+ T cells stimulate hematopoietic progenitors by promoting cytokine release from bone marrow mesenchymal stromal cells. Cell Stem Cell (2014) 14:460–72. doi: 10.1016/j.stem.2014.01.002
87. Sasaki Y, Guo YM, Goto T, Ubukawa K, Asanuma K, Kobayashi I, et al. IL-6 generated from human hematopoietic stem and progenitor cells through TLR4 signaling promotes emergency granulopoiesis by regulating transcription factor expression. J Immunol (2021) 207:1078–86. doi: 10.4049/jimmunol.2100168
88. Yang YC, Ciarletta AB, Temple PA, Chung MP, Kovacic S, Witek-Giannotti JS, et al. Human IL-3 (multi-CSF): identification by expression cloning of a novel hematopoietic growth factor related to murine IL-3. Cell (1986) 47:3–10. doi: 10.1016/0092-8674(86)90360-0
89. Weber GF, Chousterman BG, He S, Fenn AM, Nairz M, Anzai A, et al. Interleukin-3 amplifies acute inflammation and is a potential therapeutic target in sepsis. Science (2015) 347:1260–5. doi: 10.1126/science.aaa4268
90. Rauch PJ, Chudnovskiy A, Robbins CS, Weber GF, Etzrodt M, Hilgendorf I, et al. Innate response activator b cells protect against microbial sepsis. Science (2012) 335:597–601. doi: 10.1126/science.1215173
91. Ueda Y, Cain DW, Kuraoka M, Kondo M, Kelsoe G. IL-1R type I-dependent hemopoietic stem cell proliferation is necessary for inflammatory granulopoiesis and reactive neutrophilia. J Immunol (2009) 182:6477–84. doi: 10.4049/jimmunol.0803961
92. Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat Cell Biol (2016) 18:607–18. doi: 10.1038/ncb3346
93. Furusawa J, Mizoguchi I, Chiba Y, Hisada M, Kobayashi F, Yoshida H, et al. Promotion of expansion and differentiation of hematopoietic stem cells by interleukin-27 into myeloid progenitors to control infection in emergency myelopoiesis. PloS Pathog (2016) 12:e1005507. doi: 10.1371/journal.ppat.1005507
94. Seita J, Asakawa M, Ooehara J, Takayanagi S, Morita Y, Watanabe N, et al. Interleukin-27 directly induces differentiation in hematopoietic stem cells. Blood (2008) 111:1903–12. doi: 10.1182/blood-2007-06-093328
95. Binder D, Fehr J, Hengartner H, Zinkernagel RM. Virus-induced transient bone marrow aplasia: major role of interferon-alpha/beta during acute infection with the noncytopathic lymphocytic choriomeningitis virus. J Exp Med (1997) 185:517–30. doi: 10.1084/jem.185.3.517
96. Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature (2009) 458:904–8. doi: 10.1038/nature07815
97. Sato T, Onai N, Yoshihara H, Arai F, Suda T, Ohteki T. Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon-dependent exhaustion. Nat Med (2009) 15:696–700. doi: 10.1038/nm.1973
98. de Bruin AM, Demirel O, Hooibrink B, Brandts CH, Nolte MA. Interferon-gamma impairs proliferation of hematopoietic stem cells in mice. Blood (2013) 121:3578–85. doi: 10.1182/blood-2012-05-432906
99. Umemoto T, Matsuzaki Y, Shiratsuchi Y, Hashimoto M, Yoshimoto T, Nakamura-Ishizu A, et al. Integrin alphavbeta3 enhances the suppressive effect of interferon-gamma on hematopoietic stem cells. EMBO J (2017) 36:2390–403. doi: 10.15252/embj.201796771
100. de Bruin AM, Libregts SF, Valkhof M, Boon L, Touw IP, Nolte MA. IFNgamma induces monopoiesis and inhibits neutrophil development during inflammation. Blood (2012) 119:1543–54. doi: 10.1182/blood-2011-07-367706
101. Ludin A, Gur-Cohen S, Golan K, Kaufmann KB, Itkin T, Medaglia C, et al. Reactive oxygen species regulate hematopoietic stem cell self-renewal, migration and development, as well as their bone marrow microenvironment. Antioxid Redox Signal (2014) 21:1605–19. doi: 10.1089/ars.2014.5941
102. Jang YY, Sharkis SJ. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood (2007) 110:3056–63. doi: 10.1182/blood-2007-05-087759
103. Ito K, Hirao A, Arai F, Matsuoka S, Takubo K, Hamaguchi I, et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature (2004) 431:997–1002. doi: 10.1038/nature02989
104. Ito K, Hirao A, Arai F, Takubo K, Matsuoka S, Miyamoto K, et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med (2006) 12:446–51. doi: 10.1038/nm1388
105. Juntilla MM, Patil VD, Calamito M, Joshi RP, Birnbaum MJ, Koretzky GA. AKT1 and AKT2 maintain hematopoietic stem cell function by regulating reactive oxygen species. Blood (2010) 115:4030–8. doi: 10.1182/blood-2009-09-241000
106. Zhu H, Kwak HJ, Liu P, Bajrami B, Xu Y, Park SY, et al. Reactive oxygen species-producing myeloid cells act as a bone marrow niche for sterile inflammation-induced reactive granulopoiesis. J Immunol (2017) 198:2854–64. doi: 10.4049/jimmunol.1602006
107. Kwak HJ, Liu P, Bajrami B, Xu Y, Park SY, Nombela-Arrieta C, et al. Myeloid cell-derived reactive oxygen species externally regulate the proliferation of myeloid progenitors in emergency granulopoiesis. Immunity (2015) 42:159–71. doi: 10.1016/j.immuni.2014.12.017
108. Ward AC, Loeb DM, Soede-Bobok AA, Touw IP, Friedman AD. Regulation of granulopoiesis by transcription factors and cytokine signals. Leukemia (2000) 14:973–90. doi: 10.1038/sj.leu.2401808
109. Cirilli M, Bereshchenko O, Ermakova O, Nerlov C. Insights into specificity, redundancy and new cellular functions of C/EBPa and C/EBPb transcription factors through interactome network analysis. Biochim Biophys Acta Gen Subj (2017) 1861:467–76. doi: 10.1016/j.bbagen.2016.10.002
110. Zhang P, Iwama A, Datta MW, Darlington GJ, Link DC, Tenen DG. Upregulation of interleukin 6 and granulocyte colony-stimulating factor receptors by transcription factor CCAAT enhancer binding protein alpha (C/EBP alpha) is critical for granulopoiesis. J Exp Med (1998) 188:1173–84. doi: 10.1084/jem.188.6.1173
111. Heath V, Suh HC, Holman M, Renn K, Gooya JM, Parkin S, et al. C/EBPalpha deficiency results in hyperproliferation of hematopoietic progenitor cells and disrupts macrophage development in vitro and in vivo. Blood (2004) 104:1639–47. doi: 10.1182/blood-2003-11-3963
112. Johansen LM, Iwama A, Lodie TA, Sasaki K, Felsher DW, Golub TR, et al. C-myc is a critical target for c/EBPalpha in granulopoiesis. Mol Cell Biol (2001) 21:3789–806. doi: 10.1128/MCB.21.11.3789-3806.2001
113. Wang H, Iakova P, Wilde M, Welm A, Goode T, Roesler WJ, et al. C/EBPalpha arrests cell proliferation through direct inhibition of Cdk2 and Cdk4. Mol Cell (2001) 8:817–28. doi: 10.1016/S1097-2765(01)00366-5
114. Friedman AD. Transcriptional control of granulocyte and monocyte development. Oncogene (2007) 26:6816–28. doi: 10.1038/sj.onc.1210764
115. Robinson C, Shore RC, Kirkham J. Tuft protein: its relationship with the keratins and the developing enamel matrix. Calcif Tissue Int (1989) 44:393–8. doi: 10.1007/BF02555967
116. Federzoni EA, Humbert M, Torbett BE, Behre G, Fey MF, Tschan MP. CEBPA-dependent HK3 and KLF5 expression in primary AML and during AML differentiation. Sci Rep (2014) 4:4261. doi: 10.1038/srep04261
117. Suh HC, Gooya J, Renn K, Friedman AD, Johnson PF, Keller JR. C/EBPalpha determines hematopoietic cell fate in multipotential progenitor cells by inhibiting erythroid differentiation and inducing myeloid differentiation. Blood (2006) 107:4308–16. doi: 10.1182/blood-2005-06-2216
118. Fukuchi Y, Shibata F, Ito M, Goto-Koshino Y, Sotomaru Y, Ito M, et al. Comprehensive analysis of myeloid lineage conversion using mice expressing an inducible form of C/EBP alpha. EMBO J (2006) 25:3398–410. doi: 10.1038/sj.emboj.7601199
119. Wouters BJ, Jorda MA, Keeshan K, Louwers I, Erpelinck-Verschueren CA, Tielemans D, et al. Distinct gene expression profiles of acute myeloid/T-lymphoid leukemia with silenced CEBPA and mutations in NOTCH1. Blood (2007) 110:3706–14. doi: 10.1182/blood-2007-02-073486
120. Lekstrom-Himes JA. The role of C/EBP(epsilon) in the terminal stages of granulocyte differentiation. Stem Cells (2001) 19:125–33. doi: 10.1634/stemcells.19-2-125
121. Yamanaka R, Barlow C, Lekstrom-Himes J, Castilla LH, Liu PP, Eckhaus M, et al. Impaired granulopoiesis, myelodysplasia, and early lethality in CCAAT/enhancer binding protein epsilon-deficient mice. Proc Natl Acad Sci U.S.A. (1997) 94:13187–92. doi: 10.1073/pnas.94.24.13187
122. Hirai H, Yokota A, Tamura A, Sato A, Maekawa T. Non-steady-state hematopoiesis regulated by the C/EBPbeta transcription factor. Cancer Sci (2015) 106:797–802. doi: 10.1111/cas.12690
123. Satake S, Hirai H, Hayashi Y, Shime N, Tamura A, Yao H, et al. C/EBPbeta is involved in the amplification of early granulocyte precursors during candidemia-induced "emergency" granulopoiesis. J Immunol (2012) 189:4546–55. doi: 10.4049/jimmunol.1103007
124. Zhang P, Nelson E, Radomska HS, Iwasaki-Arai J, Akashi K, Friedman AD, et al. Induction of granulocytic differentiation by 2 pathways. Blood (2002) 99:4406–12. doi: 10.1182/blood.V99.12.4406
125. McLemore ML, Grewal S, Liu F, Archambault A, Poursine-Laurent J, Haug J, et al. STAT-3 activation is required for normal G-CSF-dependent proliferation and granulocytic differentiation. Immunity (2001) 14:193–204. doi: 10.1016/S1074-7613(01)00101-7
126. Zhang H, Nguyen-Jackson H, Panopoulos AD, Li HS, Murray PJ, Watowich SS. STAT3 controls myeloid progenitor growth during emergency granulopoiesis. Blood (2010) 116:2462–71. doi: 10.1182/blood-2009-12-259630
127. Lee CK, Raz R, Gimeno R, Gertner R, Wistinghausen B, Takeshita K, et al. STAT3 is a negative regulator of granulopoiesis but is not required for G-CSF-dependent differentiation. Immunity (2002) 17:63–72. doi: 10.1016/S1074-7613(02)00336-9
128. Croker BA, Metcalf D, Robb L, Wei W, Mifsud S, DiRago L, et al. SOCS3 is a critical physiological negative regulator of G-CSF signaling and emergency granulopoiesis. Immunity (2004) 20:153–65. doi: 10.1016/S1074-7613(04)00022-6
129. Kimura A, Kinjyo I, Matsumura Y, Mori H, Mashima R, Harada M, et al. SOCS3 is a physiological negative regulator for granulopoiesis and granulocyte colony-stimulating factor receptor signaling. J Biol Chem (2004) 279:6905–10. doi: 10.1074/jbc.C300496200
130. Kimura A, Rieger MA, Simone JM, Chen W, Wickre MC, Zhu BM, et al. The transcription factors STAT5A/B regulate GM-CSF-mediated granulopoiesis. Blood (2009) 114:4721–8. doi: 10.1182/blood-2009-04-216390
131. Scott EW, Simon MC, Anastasi J, Singh H. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science (1994) 265:1573–7. doi: 10.1126/science.8079170
132. Bjerregaard MD, Jurlander J, Klausen P, Borregaard N, Cowland JB. The in vivo profile of transcription factors during neutrophil differentiation in human bone marrow. Blood (2003) 101:4322–32. doi: 10.1182/blood-2002-03-0835
133. Growney JD, Shigematsu H, Li Z, Lee BH, Adelsperger J, Rowan R, et al. Loss of Runx1 perturbs adult hematopoiesis and is associated with a myeloproliferative phenotype. Blood (2005) 106:494–504. doi: 10.1182/blood-2004-08-3280
134. Guo H, Ma O, Speck NA, Friedman AD. Runx1 deletion or dominant inhibition reduces cebpa transcription via conserved promoter and distal enhancer sites to favor monopoiesis over granulopoiesis. Blood (2012) 119:4408–18. doi: 10.1182/blood-2011-12-397091
135. Dahl R, Iyer SR, Owens KS, Cuylear DD, Simon MC. The transcriptional repressor GFI-1 antagonizes PU.1 activity through protein-protein interaction. J Biol Chem (2007) 282:6473–83. doi: 10.1074/jbc.M607613200
136. Hock H, Hamblen MJ, Rooke HM, Traver D, Bronson RT, Cameron S, et al. Intrinsic requirement for zinc finger transcription factor gfi-1 in neutrophil differentiation. Immunity (2003) 18:109–20. doi: 10.1016/S1074-7613(02)00501-0
137. Skokowa J, Cario G, Uenalan M, Schambach A, Germeshausen M, Battmer K, et al. LEF-1 is crucial for neutrophil granulocytopoiesis and its expression is severely reduced in congenital neutropenia. Nat Med (2006) 12:1191–7. doi: 10.1038/nm1474
138. Hu L, Huang W, Hjort E, Eklund EA. Increased fanconi c expression contributes to the emergency granulopoiesis response. J Clin Invest (2013) 123:3952–66. doi: 10.1172/JCI69032
139. Saberwal G, Horvath E, Hu L, Zhu C, Hjort E, Eklund EA. The interferon consensus sequence binding protein (ICSBP/IRF8) activates transcription of the FANCF gene during myeloid differentiation. J Biol Chem (2009) 284:33242–54. doi: 10.1074/jbc.M109.010231
140. Hu L, Huang W, Hjort EE, Bei L, Platanias LC, Eklund EA. The interferon consensus sequence binding protein (Icsbp/Irf8) is required for termination of emergency granulopoiesis. J Biol Chem (2016) 291:4107–20. doi: 10.1074/jbc.M115.681361
141. Wang H, Bei L, Shah CA, Hu L, Eklund EA. HoxA10 terminates emergency granulopoiesis by increasing expression of Triad1. J Immunol (2015) 194:5375–87. doi: 10.4049/jimmunol.1401909
142. Wang H, Bei L, Shah CA, Horvath E, Eklund EA. HoxA10 influences protein ubiquitination by activating transcription of ARIH2, the gene encoding Triad1. J Biol Chem (2011) 286:16832–45. doi: 10.1074/jbc.M110.213975
143. Klimenkova O, Ellerbeck W, Klimiankou M, Unalan M, Kandabarau S, Gigina A, et al. A lack of secretory leukocyte protease inhibitor (SLPI) causes defects in granulocytic differentiation. Blood (2014) 123:1239–49. doi: 10.1182/blood-2013-06-508887
144. Danek P, Kardosova M, Janeckova L, Karkoulia E, Vanickova K, Fabisik M, et al. Beta-Catenin-TCF/LEF signaling promotes steady-state and emergency granulopoiesis via G-CSF receptor upregulation. Blood (2020) 136:2574–87. doi: 10.1182/blood.2019004664
145. Deshmukh HS, Liu Y, Menkiti OR, Mei J, Dai N, O'Leary CE, et al. The microbiota regulates neutrophil homeostasis and host resistance to escherichia coli K1 sepsis in neonatal mice. Nat Med (2014) 20:524–30. doi: 10.1038/nm.3542
146. Goris H, de Boer F, van der Waaij D. Myelopoiesis in experimentally contaminated specific-pathogen-free and germfree mice during oral administration of polymyxin. Infect Immun (1985) 50:437–41. doi: 10.1128/iai.50.2.437-441.1985
147. Tada T, Yamamura S, Kuwano Y, Abo T. Level of myelopoiesis in the bone marrow is influenced by intestinal flora. Cell Immunol (1996) 173:155–61. doi: 10.1006/cimm.1996.0261
148. Clarke TB, Davis KM, Lysenko ES, Zhou AY, Yu Y, Weiser JN. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat Med (2010) 16:228–31. doi: 10.1038/nm.2087
149. Mittrucker HW, Seidel D, Bland PW, Zarzycka A, Kaufmann SH, Visekruna A, et al. Lack of microbiota reduces innate responses and enhances adaptive immunity against listeria monocytogenes infection. Eur J Immunol (2014) 44:1710–5. doi: 10.1002/eji.201343927
150. Damlund DS, Metzdorff SB, Hasselby JP, Wiese M, Lundsager M, Nielsen DS, et al. Postnatal hematopoiesis and gut microbiota in NOD mice deviate from C57BL/6 mice. J Diabetes Res (2016) 2016:6321980. doi: 10.1155/2016/6321980
151. Balmer ML, Schurch CM, Saito Y, Geuking MB, Li H, Cuenca M, et al. Microbiota-derived compounds drive steady-state granulopoiesis via MyD88/TICAM signaling. J Immunol (2014) 193:5273–83. doi: 10.4049/jimmunol.1400762
152. Zeng MY, Cisalpino D, Varadarajan S, Hellman J, Warren HS, Cascalho M, et al. Gut microbiota-induced immunoglobulin G controls systemic infection by symbiotic bacteria and pathogens. Immunity (2016) 44:647–58. doi: 10.1016/j.immuni.2016.02.006
153. Salva S, Alvarez S. The role of microbiota and immunobiotics in granulopoiesis of immunocompromised hosts. Front Immunol (2017) 8:507. doi: 10.3389/fimmu.2017.00507
154. Herrera M, Salva S, Villena J, Barbieri N, Marranzino G, Alvarez S. Dietary supplementation with lactobacilli improves emergency granulopoiesis in protein-malnourished mice and enhances respiratory innate immune response. PloS One (2014) 9:e90227. doi: 10.1371/journal.pone.0090227
155. Stark MA, Huo Y, Burcin TL, Morris MA, Olson TS, Ley K. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity (2005) 22:285–94. doi: 10.1016/j.immuni.2005.01.011
156. Gordy C, Pua H, Sempowski GD, He YW. Regulation of steady-state neutrophil homeostasis by macrophages. Blood (2011) 117:618–29. doi: 10.1182/blood-2010-01-265959
157. Hong C, Kidani Y, A-Gonzalez. N, Phung T, Ito A, Rong X, et al. Coordinate regulation of neutrophil homeostasis by liver X receptors in mice. J Clin Invest (2012) 122:337–47. doi: 10.1172/JCI58393
158. Jachetti E, D'Inca F, Danelli L, Magris R, Dal Secco C, Vit F, et al. Frontline science: Mast cells regulate neutrophil homeostasis by influencing macrophage clearance activity. J Leukoc Biol (2019) 105:633–44. doi: 10.1002/JLB.4HI1018-390R
159. Nombela-Arrieta C, Isringhausen S. The role of the bone marrow stromal compartment in the hematopoietic response to microbial infections. Front Immunol (2016) 7:689. doi: 10.3389/fimmu.2016.00689
160. Chou DB, Sworder B, Bouladoux N, Roy CN, Uchida AM, Grigg M, et al. Stromal-derived IL-6 alters the balance of myeloerythroid progenitors during toxoplasma gondii infection. J Leukoc Biol (2012) 92:123–31. doi: 10.1189/jlb.1011527
161. Scheiermann C, Kunisaki Y, Frenette PS. Circadian control of the immune system. Nat Rev Immunol (2013) 13:190–8. doi: 10.1038/nri3386
162. Mendez-Ferrer S, Lucas D, Battista M, Frenette PS. Haematopoietic stem cell release is regulated by circadian oscillations. Nature (2008) 452:442–7. doi: 10.1038/nature06685
163. Nguyen KD, Fentress SJ, Qiu Y, Yun K, Cox JS, Chawla A. Circadian gene Bmal1 regulates diurnal oscillations of Ly6C(hi) inflammatory monocytes. Science (2013) 341:1483–8. doi: 10.1126/science.1240636
164. Casanova-Acebes M, Pitaval C, Weiss LA, Nombela-Arrieta C, Chevre R, A-Gonzalez. N, et al. Rhythmic modulation of the hematopoietic niche through neutrophil clearance. Cell (2013) 153:1025–35. doi: 10.1016/j.cell.2013.04.040
165. He W, Holtkamp S, Hergenhan SM, Kraus K, de Juan A, Weber J, et al. Circadian expression of migratory factors establishes lineage-specific signatures that guide the homing of leukocyte subsets to tissues. Immunity (2018) 49:1175–1190 e7. doi: 10.1016/j.immuni.2018.10.007
166. Benn CS, Netea MG, Selin LK, Aaby P. A small jab - a big effect: nonspecific immunomodulation by vaccines. Trends Immunol (2013) 34:431–9. doi: 10.1016/j.it.2013.04.004
167. Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, et al. Trained immunity: A program of innate immune memory in health and disease. Science (2016) 352:aaf1098. doi: 10.1126/science.aaf1098
168. Kalafati L, Kourtzelis I, Schulte-Schrepping J, Li X, Hatzioannou A, Grinenko T, et al. Innate immune training of granulopoiesis promotes anti-tumor activity. Cell (2020) 183:771–785 e12. doi: 10.1016/j.cell.2020.09.058
169. Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol (2004) 172:2731–8. doi: 10.4049/jimmunol.172.5.2731
170. Zollner O, Lenter MC, Blanks JE, Borges E, Steegmaier M, Zerwes HG, et al. L-selectin from human, but not from mouse neutrophils binds directly to e-selectin. J Cell Biol (1997) 136:707–16. doi: 10.1083/jcb.136.3.707
171. Hajjar E, Broemstrup T, Kantari C, Witko-Sarsat V, Reuter N. Structures of human proteinase 3 and neutrophil elastase–so similar yet so different. FEBS J (2010) 277:2238–54. doi: 10.1111/j.1742-4658.2010.07659.x
172. Aleyd E, Heineke MH, van Egmond M. The era of the immunoglobulin a fc receptor FcalphaRI; its function and potential as target in disease. Immunol Rev (2015) 268:123–38. doi: 10.1111/imr.12337
173. Tecchio C, Micheletti A, Cassatella MA. Neutrophil-derived cytokines: facts beyond expression. Front Immunol (2014) 5:508. doi: 10.3389/fimmu.2014.00508
174. Bagaitkar J, Matute JD, Austin A, Arias AA, Dinauer MC. Activation of neutrophil respiratory burst by fungal particles requires phosphatidylinositol 3-phosphate binding to p40(phox) in humans but not in mice. Blood (2012) 120:3385–7. doi: 10.1182/blood-2012-07-445619
175. Ganz T. Defensins: antimicrobial peptides of innate immunity. Nat Rev Immunol (2003) 3:710–20. doi: 10.1038/nri1180
176. Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, et al. Single-cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity (2019) 50:1317–1334 e10. doi: 10.1016/j.immuni.2019.03.009
Keywords: granulopoiesis, infection, transcription factors, cytokines, chemokines
Citation: Paudel S, Ghimire L, Jin L, Jeansonne D and Jeyaseelan S (2022) Regulation of emergency granulopoiesis during infection. Front. Immunol. 13:961601. doi: 10.3389/fimmu.2022.961601
Received: 04 June 2022; Accepted: 15 August 2022;
Published: 05 September 2022.
Edited by:
Rudolf Lucas, Augusta University, United StatesReviewed by:
Alan D. Friedman, Johns Hopkins University, United StatesRavi Misra, University of Rochester, United States
Copyright © 2022 Paudel, Ghimire, Jin, Jeansonne and Jeyaseelan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Samithamby Jeyaseelan, jey@lsu.edu