
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 11 August 2022
Sec. Autoimmune and Autoinflammatory Disorders
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.961197
 Jaeyong Jung1†
Jaeyong Jung1† Lucy Eunju Lee2,3†Hanna Kim3Ji Eun Kim3
Lucy Eunju Lee2,3†Hanna Kim3Ji Eun Kim3 Sung Hoon Jang3Jong Seong Roh4Beomgu Lee5William H. Robinson6,7
Sung Hoon Jang3Jong Seong Roh4Beomgu Lee5William H. Robinson6,7 Dong Hyun Sohn5*‡
Dong Hyun Sohn5*‡ Jae-Chul Pyun1*‡
Jae-Chul Pyun1*‡ Jason Jungsik Song3,8*‡
Jason Jungsik Song3,8*‡Although recent studies have demonstrated a proinflammatory effect of extracellular histones in sepsis via endothelial cytotoxicity, little is known about their contribution to autoimmune arthritis. Therefore, we investigated the role of extracellular histones in autoimmune arthritis and their cytotoxic effect on synoviocytes and macrophages. We measured histones in the synovial fluid of patients with rheumatoid arthritis (RA) and evaluated arthritis severity in a serum-transfer arthritis (STA) mouse model with intraperitoneal histone injection. Histone-induced cytotoxicity was measured using SYTOX green staining in the synoviocyte cell line MH7A and macrophages differentiated from the monocytic cell line THP-1, and the production of damage-associated molecular patterns (DAMPs) was measured by HMGB1 and ATP. Furthermore, we performed RNA-seq analysis of THP-1 cells stimulated with H2B-α1 peptide or with its citrullinated form. The levels of histones were elevated in RA synovial fluid, and histones aggravated arthritis in the STA model. Histones induced cytotoxicity and DAMP production in synoviocytes and macrophages. Chondroitin sulfate reduced histone-induced cytotoxicity, while lipopolysaccharides aggravated cytotoxicity. Moreover, the cytotoxicity decreased when the arginines in H2B-α1 were replaced with citrullines, which demonstrated its electrostatic nature. In transcriptome analysis, H2B-α1 changed the gene expression pattern of THP-1 cells involving chemokines, interleukin-1, -4, -10, -13, and toll-like receptor (TLR) signaling pathways. Extracellular histones were increased in RA synovial fluid and aggravated synovitis in STA. They induced lytic cell death through electrostatic interaction with synoviocytes and macrophages, leading to the secretion of DAMPs. These findings suggest that histones play a central role in autoimmune arthritis.
Histones are DNA-binding proteins that constitute nucleosomes, the basic subunits of chromatin. The four classes of core histones (H2A, H2B, H3, and H4) have similar structures, with a preserved histone fold domain and an unstructured amino acid tail (1). Positively charged histones and negatively charged phosphate groups of DNA combine to form a stable structure (2). In addition to their function in the nucleus, studies have highlighted that DNA–histone complexes are released into the extracellular space during cell death processes like necrosis and NETosis (3, 4).
NETosis is one of the lytic forms of cell death that releases neutrophil extracellular traps (NETs) that can capture and kill pathogens (5). The immune response against DNA, histones, and other proteins associated with NETs can be induced to form autoantibodies (6–8), and these intracellular components can induce inflammatory responses (9, 10). Histones, which comprise 70% of the protein components of NETs (9), cause cell injury and death in endothelial cells (11) and organ failure when released into the extracellular space (12). Proinflammatory properties of extracellular histone have been demonstrated in sepsis (4), chronic obstructive pulmonary disease (COPD) (13), systemic lupus erythematosus (14), disseminated intravascular coagulation (DIC) (15), and malignancies (16, 17), but the molecular mechanisms have not been well understood.
Rheumatoid arthritis (RA) is characterized by chronic synovial inflammation with the detection of autoantibodies that recognize citrullinated proteins (18). Recent studies have shown the role of citrullinated histones as autoantigens in RA (19). Activation of PAD4 gives rise to citrullinated histones, making NETosis a key driver of autoantigen production in RA (20, 21). However, the role of extracellular histones in RA remains largely unexplored. In this study, we investigated the influence of histones released from synovial cells during cell death processes on synovial inflammation.
Human synovial fluid samples were obtained under a protocol for discarded specimens approved by the Stanford University School of Medicine Institutional Review Boards that does not require consent. The samples were stored frozen at -80 °C until the time of analysis. Patients with RA that met the American College of Rheumatology criteria for the disease were included in the study (22). The sample donors were patients with knee osteoarthritis (OA) and over 45 years of age with radiographic Kellgren/Lawrence grade ≥ 3 changes (23). The levels of H2A, H2B, H3, and H4 were determined using the enzyme-linked immunosorbent assay (ELISA) kit for each histone (antibodies-online, Aachen, Germany) according to the manufacturer’s protocol.
Eight-week-old male C57BL/6 mice (Koatech, Pyeongtaek, Korea) were housed under controlled conditions at 23 ± 2°C and 50 ± 10% humidity on a 12 h light/dark cycle and had free access to a standard chow diet and water. STA was induced via intraperitoneal injection of 150 μL of K/BxN serum into C57BL/6 mice on day 0 and day 2. The mice were intraperitoneally injected with 200 μg of calf histone (Roche, Basel, Switzerland) or phosphate-buffered saline on day 1 and day 3. We used native histones purified by calf thymus. It is a mixture of histones H1, H2A, H2B, H3, and H4, and we used it as it is without any additional procedures. The histone treatment dose for intraperitoneal injection was determined based on the previously published method in a sepsis model, which demonstrated that the intravenously injected calf thymus histones were lethal at a concentration of 75-100 mg/kg and sublethal at 50 mg/kg in C57BL/6 mice (12). To investigate the effect of histones in arthritis, we injected 200 μg of calf thymus histones (equivalent to 10 mg/kg) intraperitoneally into each mouse based on our preliminary dose titration study (data not shown). The clinical arthritis score was determined as previously described (19): 0, no swelling or erythema; 1, mild swelling and erythema or digit inflammation; 2, moderate swelling and erythema confined to the region distal to the mid-paw; 3, pronounced swelling and erythema with extension to the ankle; and 4, severe swelling, erythema, and joint rigidity of the ankle, foot, and digits. The severity of arthritis in each limb was graded on a scale of 0–4 (a maximum possible score of 16 for each mouse) by a nonblinded examiner. Hind paw thickness was measured with a dial thickness gauge. The mouse joint tissues were fixed with 10% formalin, decalcified in 5% formic acid, and embedded in paraffin. Sections of the paraffin-embedded tissue were stained with hematoxylin and eosin (H&E), and scored by a blinded examiner for synovitis, pannus formation, and erosion of bone and/or cartilage based on a previously described scoring system (24). For each experimental condition, the joints of ten mice were evaluated and scored. The sample size was determined based on our pilot experiments. Mice were assigned to each group at random, and the order of treatments was randomized. All mice were included in the analysis. All animal experiments and protocols were approved by the Institutional Animal Care and Use Committee at Pusan National University (Miryang, Korea) and performed in accordance with the institutional and national guidelines for the care and use of laboratory animals.
Human synovial fibroblast cell line MH7A was cultured in RPMI-1640 (Hyclone, Logan, UT, USA) containing 25 mM HEPES and L-glutamine (Hyclone), supplemented with heat-inactivated 10% fetal bovine serum (FBS; Hyclone), 100 units/mL penicillin (Hyclone), and 100 μg/mL streptomycin (Hyclone). THP1-HMGB1-Lucia cells (THP-1 cells, InvivoGen, San Diego, CA, USA) were cultured in RPMI-1640, supplemented with 25 mM HEPES, L-glutamate, heat-activated 10% FBS, 100 units/mL penicillin, 100 μg/mL streptomycin, and 100 μg/mL Normocin (InvivoGen). For all assays, THP-1 cells were seeded at a density of 1 × 105 cells/well in 96-well plates and differentiated into adherent macrophages with 25 ng/mL phorbol 12-myristate 13-acetate (PMA, Sigma-Aldrich, MO, USA) overnight. Cells were treated with calf thymus histones (Abnova, Taipei, Taiwan) or recombinant histones, H2A, H2B, H3, and H4 (New England BioLabs, Cambridge, MA, USA) or histone peptides (Peptron, Daejeon, Korea, Table 1).

Table 1 Sequences of histone peptides.
The level of LDH released into the medium from cells was measured using an LDH assay kit (DoGenBio, Seoul, Korea). This kit measures the amount of a water-soluble tetrazolium salt (WST) to determine the cytotoxicity of cells. Cells were seeded onto 96-well plates at a density of 1 × 105 cells/well in serum-free media. After incubation with different concentrations of histones, 10 μL supernatant of each sample and 100 μL LDH Reaction Mix (WST substrate mix, LDH assay buffer) were incubated at 25°C in the dark for 30 min, and the optical density of soluble WST was then measured at 450 nm using FlexStation3 microplate reader (Molecular Devices, San Jose, CA, USA).
MH7A cells and THP-1 cells were placed at a density of 1 × 105 cells/well and 5 × 105 cells/well, respectively. The cells were treated with 5 μM Hoechst 33342 stain (ThermoFisher Scientific, Waltham, MA, USA) for 1 h, then treated with various concentrations of histones and/or lipopolysaccharide (LPS, Sigma-Aldrich) and stained with a plasma membrane-impermeable DNA-binding dye, SYTOX Green (Invitrogen), according to the manufacturer’s protocol, in a serum-free condition. Before histone addition, the cells were treated with or without 10 mM sodium perchlorate (Sigma-Aldrich) for 24 h. The live images of the cells were obtained under a fluorescence microscope (Eclipse Ti2, Nikon Instruments, Tokyo, Japan) or by an automatic cell imaging system (Operetta CLS™, PerkinElmer, Waltham, MA, USA). For images obtained from the Operetta cell imaging system, image analysis and cell counting were performed according to the manufacturer’s assay protocol using Harmony® software (Version 4.9, PerkinElmer).
The level of HMGB1::Lucia luciferase released from THP-1 cells was measured with a luminometer (FlexStation3) using luciferin. A sample from each well was mixed with QUANTI-Luc (InvivoGen) according to the manufacturer’s instructions, and the luciferase reading was measured.
ATPlite (PerkinElmer) is a luciferase-based assay used for the quantitative evaluation of cell proliferation and cytotoxicity. For this assay, cells were seeded onto a 96-well plate at a density of 5 × 105 cells/well. The cells were treated with different concentrations of histones for 1 h. Then, the ATPlite buffer and the lyophilized substrate solution were mixed. Finally, cells in each well were incubated with 50 μL of this mixture for 10 min, followed by a luminescence measurement.
?A3B2 tlsb .2pt?>To measure the cellular binding of FITC labeled poly-L-lysine (Sigma-Aldrich), THP-1 cells were seeded at a density of 2 × 105 cells/well and treated with 100 ng/mL LPS for 24 h. Then, 10 μg/mL poly-L-lysine-FITC was added to the cells and incubated for 30 min at 4°C. Cells were observed under a fluorescence microscope (Eclipse Ti2) to obtain fluorescence images. Quantification of fluorescence intensity of images was performed using ImageJ (https://imagej.nih.gov).
To detect cell membrane damage in flow cytometry, a DNA-staining fluorescent marker 7-AAD (Invitrogen) was used. MH7A and THP-1 cells were seeded at a density of 5 × 105 cells/well and treated with various concentrations of histones for 1 h. Subsequently, 1 μg/mL 7-AAD was added to the cells and incubated for 30 min. Cells were analyzed on BD LSRII SORP (BD Biosciences, San Jose, CA, USA) to detect 7-AAD.
IL-1β (eBioscience, San Diego, CA, USA), TNF-α (Invitrogen, San Diego, CA, USA), and CCL4 (R&D Systems, Minneapolis, MN, USA) levels were measured using ELISA kits as per the manufacturer’s instructions.
Total RNA was isolated from THP-1 cells treated with H2B-α1 peptide or cit-H2B-α1 peptide, using TRIzol reagent (Invitrogen). Samples of 3 μg of total RNA were prepared from each condition with three experimental replicates, and gene expression profiling was performed using the QuantSeq 3′ mRNA-Seq service (Ebiogen, Seoul, Korea). After quality checks using Bioanalyzer 2100 system (Agilent, Santa Clara, CA, USA), an oligo-dT primer set containing Illumina-compatible sequences at its 5′ end was hybridized to the prepared RNA, and reverse transcription was performed. High-throughput sequencing was performed as single-end sequencing using Illumina NextSeq 500 (San Diego, CA, USA). The resulting reads were aligned using Bowtie2 (http://bowtie-bio.sourceforge.net/bowtie2) with the human reference genome hg19 (25). The DEGs were determined based on the counts from alignments using coverage in Bedtools (https://bedtools.readthedocs.io). The read count data were processed based on the quantile normalization method using EdgeR (https://bioconductor.org). Gene enrichment analysis was performed by MATLAB (MathWorks, Natick, MA, USA) using the Reactome pathway database (https://reactome.org) and Gene Ontology Consortium database (http://geneontology.org).
Statistical analysis was performed using Prism 9 software (GraphPad, San Diego, CA, USA). Data are presented as mean ± standard error of the mean or median ± interquartile range. In addition, we performed an unpaired t-test or Mann–Whitney test to analyze the statistical differences between the two groups. One-way or two-way ANOVA with Tukey–Kramer multiple comparison tests were used for comparisons between multiple groups. Statistical significance level was set at P < 0.05.
The levels of histones H2A, H2B, H3, and H4 were higher in RA compared to those in OA synovial fluid samples, though the difference in H2B was not statistically significant (Figure 1A). We tested whether extracellular histones could aggravate inflammatory arthritis. When mice were induced for STA, co-injection of histone aggravated arthritis (Figure 1B). Histopathological analyses also showed that histones increased synovitis, pannus formation, and erosion of bone and cartilage (Figures 1C, D). These results showed that histones have a deleterious effect on arthritis.
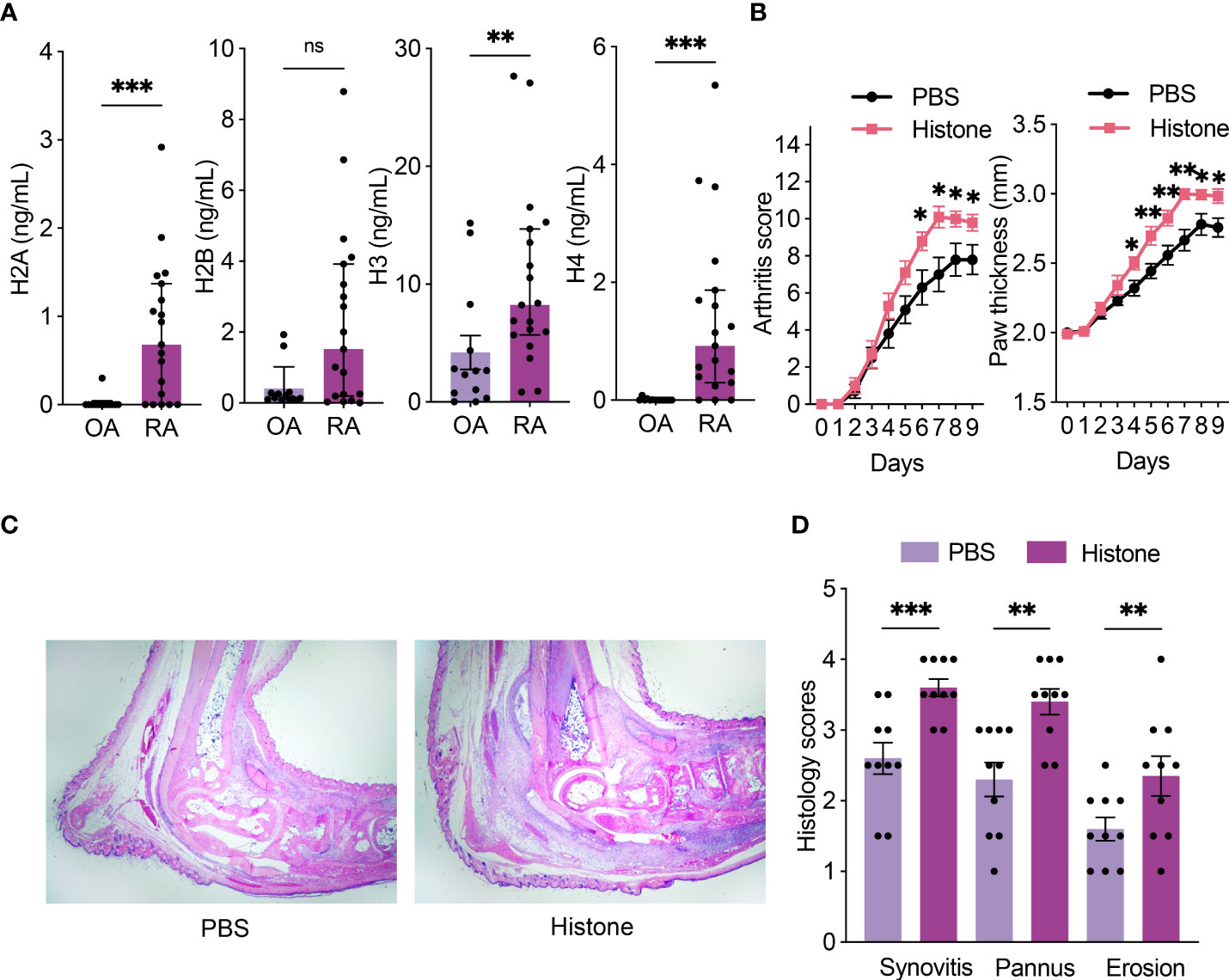
Figure 1 Histones are increased in RA synovial fluid and aggravate arthritis in STA mice. (A) Levels of histones (H2A, H2B, H3, and H4) in synovial fluid from osteoarthritis (OA; n = 13) and rheumatoid arthritis (RA; n = 19) patients. (B) Serum-transfer arthritis (STA) severity and paw thickness in mice (n = 10 per group) with intraperitoneal histone or phosphate-buffered saline (PBS) injection. K/BxN serum was given on days 0 and 2, and 200 μg of histone or PBS was given on days 1 and 3. (C) Representative images of hematoxylin and eosin (H&E) stained sections of ankle joints (×20 magnification). (D) Histologic scores of synovitis, pannus formation, and erosions of ankle joints. Data are shown as the median ± interquartile range (A) or the mean ± standard error of the mean (B and D). The P-values were calculated by Mann–Whitney tests (A) or unpaired t-tests (B, D). *P < 0.05, **P < 0.01, and ***P < 0.001. ns, not significant..
We investigated if the extracellular histones could induce cell injury and death in synoviocytes and macrophages. We observed that histones increased cytotoxicity in MH7A (synoviocytes) and PMA-differentiated THP-1 (macrophages) cells, as visualized by SYTOX Green dead cell staining (Figures 2A, B; Supplementary Movies 1A and B). Notably, histone-induced lytic cell death was more rapid and prominent in THP-1 cells than in MH7A cells (Supplementary Figures S1, S2).
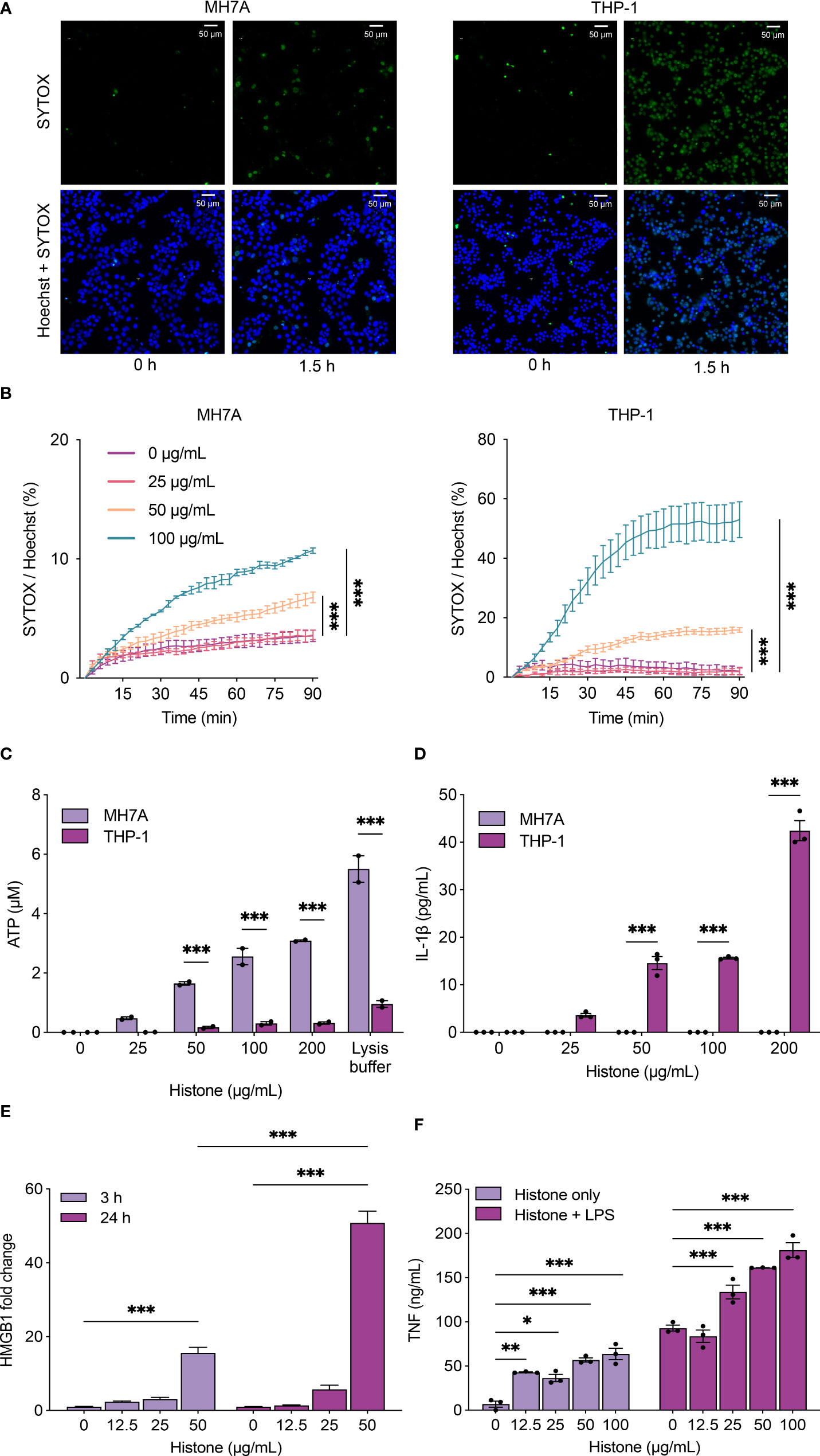
Figure 2 Histones induce cytotoxicity and the release of damage-associated molecular patterns (DAMPs). (A) Histone-induced cytotoxicity in MH7A and THP-1 cells detected by SYTOX Green. Representative fluorescence microscopy images of MH7A (left) and THP-1 cells (right) with 100 μg/mL of histones. See also Supplement Movies 1A, B (B) Time-course plot of dead cells detected by SYTOX green with the Operetta cell imaging system, MH7A (left) and THP-1 cells (right). (C) Levels of extracellular ATP from MH7A and THP-1 cells treated with histones for 1 h. (D) Levels of extracellular IL-1β from MH7A and THP-1 cells treated with histones for 1 h. (E) Levels of HMGB1 from THP-1 cells treated with histones for 3 h or 24 h. (F) Levels of TNF-α from THP-1 cells treated with histones and/or lipopolysaccharides (LPS; 10 ng/mL) for 24 h. Data are expressed as the mean ± standard error of the mean. ANOVA with Tukey–Kramer multiple comparison tests were conducted. *P < 0.05, **P < 0.01, and ***P < 0.001. The experiments were performed with technical replicates of three wells per condition.
Synovial macrophages are one of the most abundant inflammatory cell types in the synovium of RA patients, and activated macrophages produce various cytokines and chemokines to recruit immune cells (26, 27). To investigate the mechanism of histone-induced cytotoxicity and its consequence in inflamed joints, we determined the level of ATP, one of the molecules released in cell damage. Extracellular ATP, which can act as a DAMP, was increased by histones in both MH7A cells and THP-1 cells (Figure 2C). In contrast, IL-1β was induced by histones only in THP-1 cells, not in MH7A cells (Figure 2D). The level of HMGB1, a well-studied DAMP, was also increased by histones in THP-1 cells (Figure 2E). We also measured the level of TNF-α when THP-1 cells were treated by histones with or without LPS. The increased level of TNF-α under histone-treatment demonstrated that histones cause a pro-inflammatory effect that is additive with LPS (Figure 2F).
Histones exhibit cytotoxicity by binding to cells through their strong positive charge (28). Since anionic polysaccharides and peptides neutralize the cytotoxicity of histones (29), we tested if chondroitin sulfate, one of the charged molecules in synovial fluid, could attenuate histone-induced cytotoxicity. Histone-induced cytotoxicity and HMGB1 production were strongly inhibited by chondroitin sulfate (Figures 3A, B).
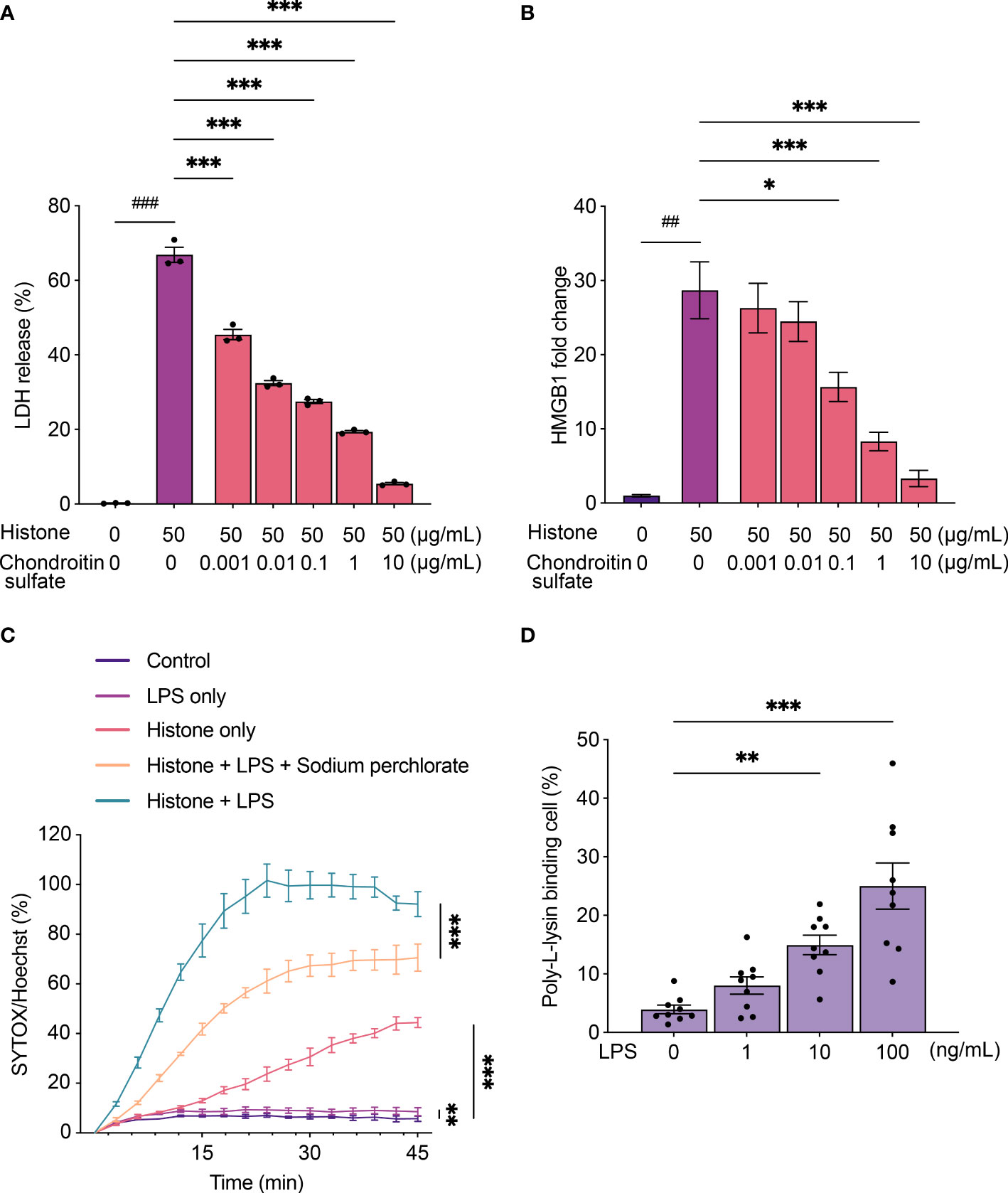
Figure 3 Chondroitin sulfate and lipopolysaccharides (LPS) regulate histone-induced cytotoxicity. (A) Lactate dehydrogenase (LDH) release assay using THP-1 cells to monitor histone-induced cytotoxicity for 24 h with chondroitin sulfate. (B) Levels of HMGB1 from THP-1 cells to monitor histone-induced cytotoxicity for 24 h with chondroitin sulfate. (C) Time-course plot of dead cells stained with SYTOX green evaluated by the Operetta cell imaging system after treatment with histone (100 μg/mL) and LPS (200 ng/mL) with sodium perchlorate (10 mM). (D) Poly-L-lysine binding to LPS-stimulated THP-1 cells. Data are expressed as the mean ± standard error of the mean. An unpaired t-test or a one-way ANOVA (A, B) or a two-way ANOVA (C, D) with Tukey–Kramer multiple comparison tests were conducted. *P < 0.05, **P < 0.01, and ***P < 0.001. ##P < 0.01, and ###P < 0.001. Different statistical tests were presented with different symbols.. The experiments were performed with technical replicates of three wells per condition.
We also analyzed the effect of histones on cells treated with LPS. SYTOX Green staining of THP-1 cells revealed that histone-induced cytotoxicity increased when cells were treated with LPS and histones than with histones alone (Figure 3C). To verify the electrostatic interaction during the inflammatory process of THP-1 cells, we evaluated the binding of positively charged poly-L-lysine to cells when they were stimulated by LPS. When treated with LPS, increased poly-L-lysine binding was observed (Figure 3D), and treatment with sodium perchlorate, a sulfation inhibitor, reduced the cytotoxicity (Figure 3C).
We investigated the classes of histones to reveal those that induce cytotoxicity. When THP-1 cells were treated with each histone, all histones induced cytotoxicity (Figure 4A), and all significantly increased HMGB1 production except for H4 (Figure 4B). As the recombinant histones were purified from E. coli, the levels of LPS in histones were measured and a significant amount of LPS was detected in H2B and H3 (Supplementary Figure S3). Since the LPS content of each histone was not proportional to the levels of LDH or HMGB1, LPS is not the major source of the cytotoxicity of the histones. However, the additive effect of the contaminated LPS in varying amounts should be considered. Referring to the previously studied histone structure (30, 31), we tested the cytotoxicity of α1 domain of each protein, H2A, H2B, H3, and H4 (Table 1). It has been suggested that histones bind to phospholipid-phosphodiester bonds using their DNA-binding α1 helices (32). When cells were treated with the α1 domain peptide of each histone, H2B-α1 peptide strongly induced cytotoxicity and HMGB1 production in THP-1 cells (Figures 4C, D). Since the binding of histones can be altered by modification of their positive charge, we replaced the positively charged arginines in H2B-α1 with citrullines (Table 1). Citrullination of H2B-α1 inhibited H2B-α1-induced cytotoxicity and HMGB1 production (Figures 4E, F).
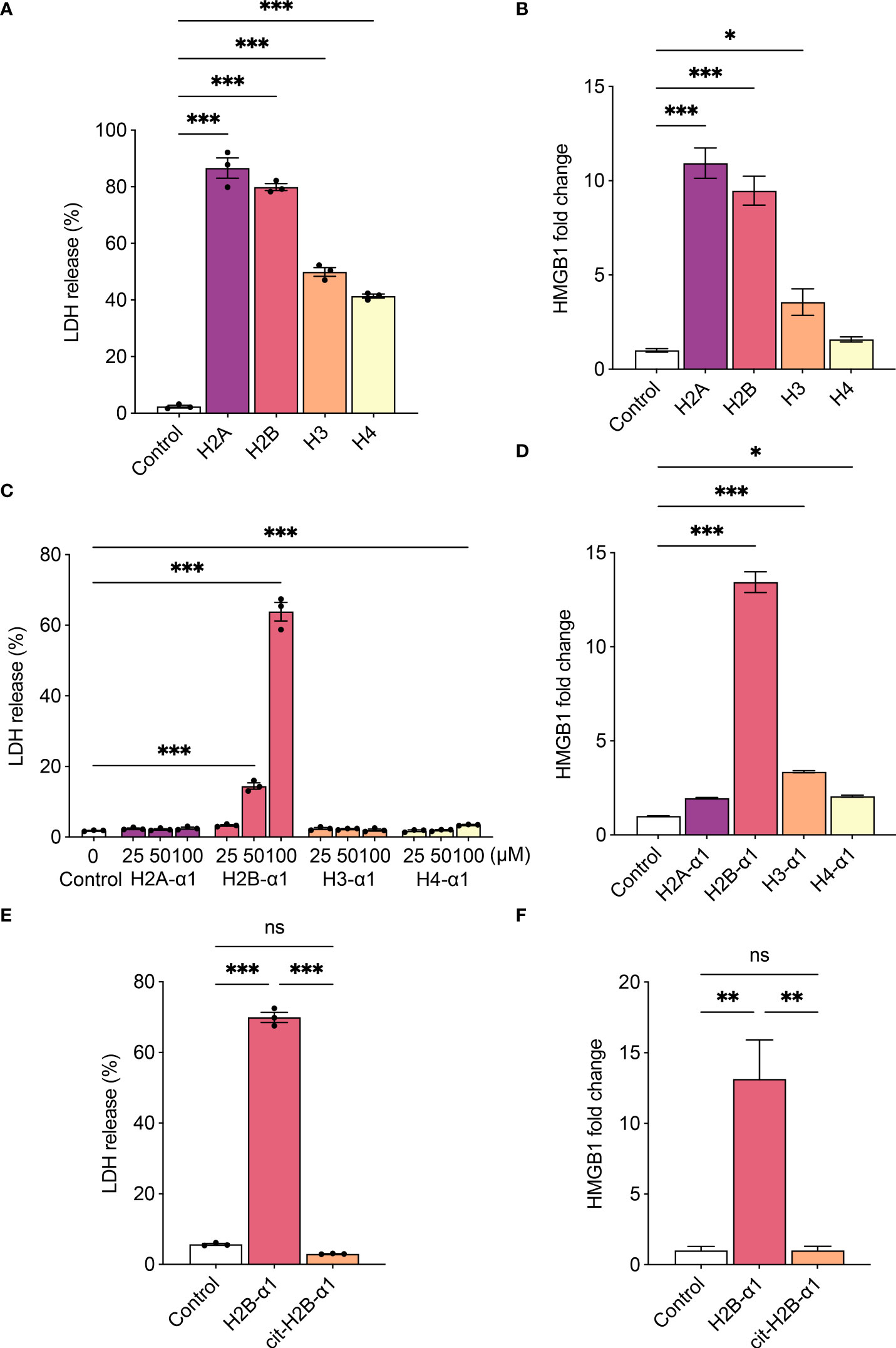
Figure 4 H2B-α1 peptide induces cytotoxicity in macrophage. (A), (B) Histone-induced cytotoxicity (3 h) with each class of recombinant histone proteins (4 μM). (C), (D) Histone-induced cytotoxicity (24 h) with each class of histone α1 peptides. (E), (F) Histone-induced cytotoxicity (24 h) with native or citrullinated (cit) H2B-α1 peptide (50 μM). Cytotoxicity levels were measured by lactate dehydrogenase (LDH) release assay (A, C, E), and HMGB1 release was measured by luciferase assay (B, D, F). Data are expressed as the mean ± standard error of the mean. ANOVA with Tukey–Kramer multiple comparison tests were conducted. *P < 0.05, **P < 0.01, and ***P < 0.001. ns, not significant.. The experiments were performed with technical replicates of three wells per condition. See table 1 for sequences of peptides.
To examine the cellular effects of histones, we analyzed the RNA-seq data of THP-1 cells treated with native or citrullinated H2B-α1 peptide. The clustergram representing the normalized expression of DEGs is shown (Figure 5A). Compared to THP-1 cells treated with H2B-α1 peptide, the transcriptome of the cells treated with citrullinated H2B-α1 peptide did not shift considerably from the expression pattern of the non-treated control cells. Applying principal component analysis (PCA) on 1420 DEGs, we verified two principal components, PC1 and PC2, explaining 76.9% and 14.5% of the variances, respectively (Figure 5B). In the two-dimensional PC space, representations of three replicates of each treatment group were exclusively distributed and completely separated. Transcriptome data was summarized as a Venn diagram (Figure 5C).
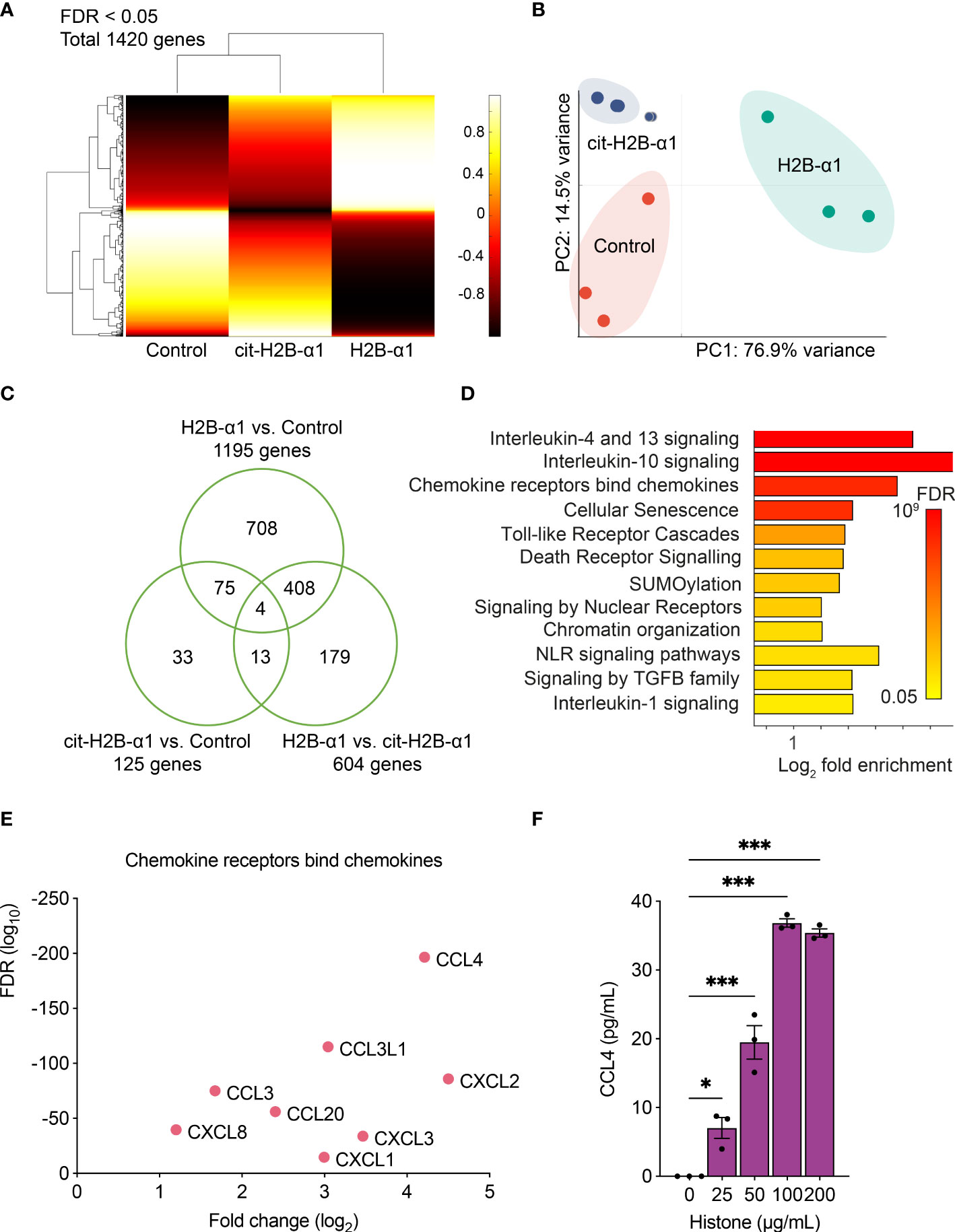
Figure 5 Transcriptome analysis of THP-1 cells with H2B-α1 peptide. (A) Expression clustering of RNA-seq data of THP-1 cells treated with H2B-α1 peptide or cit-H2B-α1 peptide for 3 h. (B) Principal component analysis of differentially expressed genes (DEGs). (C) Venn diagram of DEGs. (D) Significant gene ontology pathways from DEGs. (E) Top DEGs in chemokine pathway. (F) Levels of CCL4 with histone stimulation on THP-1 cells. Data in (F) are expressed as the mean ± standard error of the mean. ANOVA with Tukey–Kramer multiple comparison tests were conducted. *P < 0.05 and ***P < 0.001. The experiments were performed with technical replicates of three wells per condition.
Gene ontology and pathway analysis were performed to examine the molecular pathways related to the effect of native histones. The most enriched Reactome pathways are listed in Figure 5D, and for each pathway, the top DEGs are shown based on the false discovery rate (FDR) and expression fold change (Figure 5E and Supplementary Figures S4A–K) (33). H2B-α1 peptide stimulation changed the gene expression pattern of THP-1 cells involving chemokines, IL-1, -4, -10, -13 signaling, and TLR signaling pathways. Citrullination of positively charged arginines of cytotoxic H2B-α1 peptide decreased histone-induced proinflammatory gene expression. The histone-induced expression of CCL4, one of the DEGs with the largest fold change, was demonstrated in THP-1 cells (Figure 5F). This result showed that histone toxicity was related to inflammatory and apoptosis pathways and that arginine residues in the H2B-α1 peptide play an important role in histone toxicity.
Extracellular histones are major components of NETs, and their pro-inflammatory effect is known in various inflammatory processes (11, 12, 34, 35); however, the role of extracellular histones in synovial inflammation remains to be elucidated. In this study, we showed that there is an increase in the levels of histones in the synovial fluid of patients with RA compared to those in patients with OA. Previously, it has been demonstrated that the synovial fluid of patients with RA contains a large number of activated neutrophils and macrophages involved in synovial inflammation (26, 27, 36). In a sepsis model, histones released by NETosis trigger innate immune cells, and histones bound to endothelial cells cause endothelial injury (37). Based on the inflammatory nature of NETosis and its resulting extracellular histones, histones from neutrophils in RA synovial fluid would interact with other cells in the joint space, including macrophages and synoviocytes. To validate the pro-inflammatory effect of extracellular histones in arthritis, we demonstrated that intraperitoneal injection of histones aggravated arthritis severity in the STA mouse model.
To confirm the histone-induced cytotoxicity in cells in synovitis, we showed histone-induced cell death in synoviocytes and macrophages through direct treatment of the cells with histones. The results suggested that histones might contribute to synovial inflammation by inducing lytic cell death of synoviocytes and macrophages. Interestingly, time-course dead cell stain images indicated that THP-1 cells are more susceptible to histone-induced cytotoxicity than MH7A cells. Consistent with previous findings, our study demonstrated that histones induce the production of DAMPs, including ATP, HMGB1, and IL-1β, and increase the secretion of TNF-α (38–40). Extracellular ATP released from damaged cells is known to recruit macrophages and promote the secretion of IL-1β by dendritic cells, accelerating the inflammatory process (41). Histone-induced TNF-α production was further augmented by LPS. Therefore, it may have an augmented pro-inflammatory effect in patients with RA.
In addition, we verified that histone-induced cytotoxicity is regulated by electrostatic interaction. It is known that positively charged histones react with the negatively charged cell membrane, thereby causing cytotoxicity (42). Further, pro-inflammatory cytokines such as TNF-α are known to increase surface anionic molecules (43). In our study, LPS increased the binding of positively charged poly-L-lysine on cell surfaces. Furthermore, we found that anionic molecules such as chondroitin sulfate in the synovial fluid could inhibit histone-induced cytotoxicity. In addition, histone-induced cytotoxicity could be mitigated by adding sodium perchlorate, a sulfation inhibitor (44, 45). Considering that sulfation amplifies the anionic nature of proteins, these results suggested that electrostatic conditions of cell surface could regulate histone toxicity by inhibiting or enhancing the binding of histones. To further verify the electrostatic nature of the cytotoxicity of extracellular histones, we modified the cytotoxic α1 peptide of H2B by citrullinating its positively charged arginine residues. The citrullinated H2B-α1 peptide was less cytotoxic to THP-1 cells.
We found that all four classes of recombinant histone proteins induced cell death as the concentration increased, with H2A and H2B being the most cytotoxic. We also showed that H2B-α1 peptide was the most cytotoxic among histone α1 helix peptides from the four classes of histones. This is consistent with previous findings, which showed that H2B derived from frogs have bactericidal properties due to disruption of the bacterial membrane by the cationic nature of its N-terminal region (46, 47).
Using RNA-seq, we have shown that histones changed the expression of specific signaling pathways in THP-1 cells, including IL-1, 4, 10, 13 signaling, chemokines, TLR, and NOD-like receptor (NLR) signaling pathways. The chemokine signaling pathway had several DEGs with the highest degree of fold change, such as CCL4 and CXCL2. It is also known that expression of CCL3, CCL4, CXCL2, and CXCL8 is increased in RA synovial fluid, and that these chemokines are suggested to help neutrophils become resident within the joint in RA (48).
It has been known that NETosis is increased in RA synovium, which induces citrullination of histones by PADs (20, 21). However, there are citrullinated and uncitrullinated parts in extracellular histones derived from the synovial fluids of RA patients (9, 20). Although the hypercitrullinated milieu in RA might reduce the histone-mediated cellular injury (cytotoxicity, DAMP release, chemotaxis), which makes translating our observations into RA pathogenesis less interesting, there are several possibilities that uncitrullinated parts of extracellular histones are relevant to current understanding of RA pathogenesis. First, they might be involved in the breaking of immune tolerance on citrullinated histone during a very early stage of RA. Although citrullination occurs during physiological processes such as apoptosis and NETosis, autoantibody reactivity toward citrullinated antigens is an unusual pathologic process for which the molecular mechanism is unknown. Uncitrullinated parts of extracellular histones may play a role as an adjuvant. Second, they might be involved in local joint inflammation during the established stage of RA. Because there is no association between the ACPA titer and disease activity, the amount of citrullinated antigen is not associated with disease activity. Uncitrullinated parts of extracellular histones may play a role during an acute flare of arthritis. Third, they might be involved in seronegative RA. Seronegative RA patients displayed elevated IgG reactivity to native histone H2B compared to controls, but no citrulline-specific reactivity (49).
The limitations of this study include that the effects of post-translational modification of histones in RA were not fully investigated. Additionally, the influence of anionic proteins such as chondroitin sulfate in synovial fluid that binds to histones and mitigates their cytotoxic effects must be considered in future research. Another limitation is the lack of research into the association between extracellular histones and the formation of anti-citrullinated protein antibodies. Although we established the cytotoxicity and induction of DAMPs by extracellular histones, further research is needed to determine the net effects of extracellular histones on the development of autoimmunity in RA.
In conclusion, our study demonstrated that extracellular histones induce the production of DAMPs, such as ATP, HMGB1, and IL-1β, by causing lytic cell death of synoviocytes and macrophages. Our findings suggest a novel mechanism for the pathogenesis of RA in which extracellular histones play a proinflammatory role in synovitis. Furthermore, our results from charge-altering experiments imply that targeting the electrostatic effect of extracellular histones may be a possible therapeutic option for RA.
All data generated or analyzed during this study are included in this article and its supplementary information files. The RNA-seq data presented in the study is deposited in NCBI’s Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo) and Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra). The data is accessible through GEO accession number GSE205096 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE205096).
The studies involving human participants were reviewed and approved by Stanford University School of Medicine Institutional Review Boards. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements. The animal study was reviewed and approved by the Institutional Animal Care and Use Committee at Pusan National University (Miryang, Korea).
DS, J-CP, and JS designed the study, and JJ, HK, and JK conducted the experiments and analytical methods. JR, BL, and DS performed the animal experiments and joint histology. JJ, LL, HK, JK, JR, BL, WR, DS, J-CP, and JS discussed and interpreted the data. JJ, LL, HK, JK, JR, and BL made the figures and wrote the manuscript. DS, J-CP, and JS developed the hypothesis, secured funding, coordinated, and directed the project. All authors reviewed and edited the manuscript and had final approval of the submitted manuscript.
This study was supported by the Basic Science Research Program (grant number NRF-2021R1F1A1059528 to JS, NRF-2020R1A2B5B01002187 to J-CP, and NRF-2018R1A5A2023879 to DS) through the National Research Foundation of Korea, and funded by the Ministry of Education, Science, and Technology.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.961197/full#supplementary-material
1. Bednar J, Horowitz RA, Grigoryev SA, Carruthers LM, Hansen JC, Koster AJ, et al. Nucleosomes, linker DNA, and linker histone form a unique structural motif that directs the higher-order folding and compaction of chromatin. Proc Natl Acad Sci (1998) 95(24):14173–8. doi: 10.1073/pnas.95.24.14173
2. Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature (1997) 389(6648):251–60. doi: 10.1038/38444
3. Kumar SV, Kulkarni OP, Mulay SR, Darisipudi MN, Romoli S, Thomasova D, et al. Neutrophil extracellular trap-related extracellular histones cause vascular necrosis in severe gn. J Am Soc Nephrol (2015) 26(10):2399–413. doi: 10.1681/asn.2014070673
4. Chen R, Kang R, Fan XG, Tang D. Release and activity of histone in diseases. Cell Death Dis (2014) 5(8):e1370–e. doi: 10.1038/cddis.2014.337
5. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303(5663):1532–5. doi: 10.1126/science.1092385
6. Liu CL, Tangsombatvisit S, Rosenberg JM, Mandelbaum G, Gillespie EC, Gozani OP, et al. Specific post-translational histone modifications of neutrophil extracellular traps as immunogens and potential targets of lupus autoantibodies. Arthritis Res Ther (2012) 14(1):R25. doi: 10.1186/ar3707
7. Price JV, Tangsombatvisit S, Xu G, Yu J, Levy D, Baechler EC, et al. On silico peptide microarrays for high-resolution mapping of antibody epitopes and diverse protein-protein interactions. Nat Med (2012) 18(9):1434–40. doi: 10.1038/nm.2913
8. Hakkim A, Fürnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U.S.A. (2010) 107(21):9813–8. doi: 10.1073/pnas.0909927107
9. Pinegin B, Vorobjeva N, Pinegin V. Neutrophil extracellular traps and their role in the development of chronic inflammation and autoimmunity. Autoimmun Rev (2015) 14(7):633–40. doi: 10.1016/j.autrev.2015.03.002
10. Kessenbrock K, Krumbholz M, Schönermarck U, Back W, Gross WL, Werb Z, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med (2009) 15(6):623–5. doi: 10.1038/nm.1959
11. Abrams ST, Zhang N, Manson J, Liu T, Dart C, Baluwa F, et al. Circulating histones are mediators of trauma-associated lung injury. Am J Respir Crit Care Med (2013) 187(2):160–9. doi: 10.1164/rccm.201206-1037OC
12. Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, et al. Extracellular histones are major mediators of death in sepsis. Nat Med (2009) 15(11):1318–21. doi: 10.1038/nm.2053
13. Barrero CA, Perez-Leal O, Aksoy M, Moncada C, Ji R, Lopez Y, et al. Histone 3.3 participates in a self-sustaining cascade of apoptosis that contributes to the progression of chronic obstructive pulmonary disease. Am J Respir Crit Care Med (2013) 188(6):673–83. doi: 10.1164/rccm.201302-0342OC
14. Amoura Z, Piette JC, Chabre H, Cacoub P, Papo T, Wechsler B, et al. Circulating plasma levels of nucleosomes in patients with systemic lupus erythematosus: Correlation with serum antinucleosome antibody titers and absence of clear association with disease activity. Arthritis Rheum (1997) 40(12):2217–25. doi: 10.1002/art.1780401217
15. Nakahara M, Ito T, Kawahara K, Yamamoto M, Nagasato T, Shrestha B, et al. Recombinant thrombomodulin protects mice against histone-induced lethal thromboembolism. PloS One (2013) 8(9):e75961. doi: 10.1371/journal.pone.0075961
16. Holdenrieder S, Stieber P, Bodenmüller H, Busch M, Fertig G, Fürst H, et al. Nucleosomes in serum of patients with benign and malignant diseases. Int J Cancer (2001) 95(2):114–20. doi: 10.1002/1097-0215(20010320)95:2<114::aid-ijc1020>3.0.co;2-q
17. Trejo-Becerril C, Pérez-Cárdenas E, Treviño-Cuevas H, Taja-Chayeb L, García-López P, Segura-Pacheco B, et al. Circulating nucleosomes and response to chemotherapy: An in vitro, in vivo and clinical study on cervical cancer patients. Int J Cancer (2003) 104(6):663–8. doi: 10.1002/ijc.11003
18. Schellekens GA, Visser H, De Jong BAW, Van Den Hoogen FHJ, Hazes JMW, Breedveld FC, et al. The diagnostic properties of rheumatoid arthritis antibodies recognizing a cyclic citrullinated peptide. Arthritis Rheumatism (2000) 43(1):155–63. doi: 10.1002/1529-0131(200001)43:1<155::AID-ANR20>3.0.CO;2-3
19. Sohn DH, Rhodes C, Onuma K, Zhao X, Sharpe O, Gazitt T, et al. Local joint inflammation and histone citrullination in a murine model of the transition from preclinical autoimmunity to inflammatory arthritis. Arthritis Rheumatol (2015) 67(11):2877–87. doi: 10.1002/art.39283
20. Curran AM, Naik P, Giles JT, Darrah E. Pad enzymes in rheumatoid arthritis: Pathogenic effectors and autoimmune targets. Nat Rev Rheumatol (2020) 16(6):301–15. doi: 10.1038/s41584-020-0409-1
21. Aggarwal A, Srivastava R, Agrawal S. T Cell responses to citrullinated self-peptides in patients with rheumatoid arthritis. Rheumatol Int (2013) 33(9):2359–63. doi: 10.1007/s00296-013-2731-2
22. Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American rheumatism association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum (1988) 31(3):315–24. doi: 10.1002/art.1780310302
23. Kellgren JH, Lawrence JS. Radiological assessment of osteo-arthrosis. Ann Rheum Dis (1957) 16(4):494–502. doi: 10.1136/ard.16.4.494
24. Tang W, Lu Y, Tian QY, Zhang Y, Guo FJ, Liu GY, et al. The growth factor progranulin binds to tnf receptors and is therapeutic against inflammatory arthritis in mice. Science (2011) 332(6028):478–84. doi: 10.1126/science.1199214
25. Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods (2012) 9(4):357–9. doi: 10.1038/nmeth.1923
26. Haringman JJ, Gerlag DM, Zwinderman AH, Smeets TJ, Kraan MC, Baeten D, et al. Synovial tissue macrophages: A sensitive biomarker for response to treatment in patients with rheumatoid arthritis. Ann Rheum Dis (2005) 64(6):834–8. doi: 10.1136/ard.2004.029751
27. Kinne RW, Bräuer R, Stuhlmüller B, Palombo-Kinne E, Burmester G-R. Macrophages in rheumatoid arthritis. Arthritis Res Ther (2000) 2(3):189. doi: 10.1186/ar86
28. Marsman G, Zeerleder S, Luken BM. Extracellular histones Or nucleosomes: Differences in immunostimulation. Cell Death Dis (2016) 7(12):e2518–e. doi: 10.1038/cddis.2016.410
29. Meara CHO, Coupland LA, Kordbacheh F, Quah BJC, Chang C-W, Simon Davis DA, et al. Neutralizing the pathological effects of extracellular histones with small polyanions. Nat Commun (2020) 11(1):6408. doi: 10.1038/s41467-020-20231-y
30. Birkemo GA, Lüders T, Andersen Ø, Nes IF, Nissen-Meyer J. Hipposin, a histone-derived antimicrobial peptide in Atlantic halibut (Hippoglossus hippoglossus l.). Biochim Biophys Acta (2003) 1646(1-2):207–15. doi: 10.1016/s1570-9639(03)00018-9
31. Bustillo ME, Fischer AL, LaBouyer MA, Klaips JA, Webb AC, Elmore DE. Modular analysis of hipposin, a histone-derived antimicrobial peptide consisting of membrane translocating and membrane permeabilizing fragments. Biochim Biophys Acta (2014) 1838(9):2228–33. doi: 10.1016/j.bbamem.2014.04.010
32. Silk E, Zhao H, Weng H, Ma D. The role of extracellular histone in organ injury. Cell Death Dis (2017) 8(5):e2812–e. doi: 10.1038/cddis.2017.52
33. Gillespie M, Jassal B, Stephan R, Milacic M, Rothfels K, Senff-Ribeiro A, et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res (2022) 50(D1):D687–d92. doi: 10.1093/nar/gkab1028
34. Allam R, Scherbaum CR, Darisipudi MN, Mulay SR, Hägele H, Lichtnekert J, et al. Histones from dying renal cells aggravate kidney injury Via Tlr2 and Tlr4. J Am Soc Nephrol (2012) 23(8):1375–88. doi: 10.1681/asn.2011111077
35. Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: A predominant role of histones. PloS One (2012) 7(2):e32366. doi: 10.1371/journal.pone.0032366
36. Wittkowski H, Foell D, af Klint E, De Rycke L, De Keyser F, Frosch M, et al. Effects of intra-articular corticosteroids and anti-tnf therapy on neutrophil activation in rheumatoid arthritis. Ann Rheum Dis (2007) 66(8):1020–5. doi: 10.1136/ard.2006.061507
37. Li Y, Wan D, Luo X, Song T, Wang Y, Yu Q, et al. Circulating histones in sepsis: Potential outcome predictors and therapeutic targets. Front Immunol (2021) 12:650184. doi: 10.3389/fimmu.2021.650184
38. Yang R, Zou X, Tenhunen J, Tønnessen TI. Hmgb1 and extracellular histones significantly contribute to systemic inflammation and multiple organ failure in acute liver failure. Mediators Inflammation (2017) 2017:5928078. doi: 10.1155/2017/5928078
39. Allam R, Darisipudi MN, Tschopp J, Anders HJ. Histones trigger sterile inflammation by activating the Nlrp3 inflammasome. Eur J Immunol (2013) 43(12):3336–42. doi: 10.1002/eji.201243224
40. Yamaguchi A, Jitsuishi T, Hozumi T, Iwanami J, Kitajo K, Yamaguchi H, et al. Temporal expression profiling of damps-related genes revealed the biphasic post-ischemic inflammation in the experimental stroke model. Mol Brain (2020) 13(1):57. doi: 10.1186/s13041-020-00598-1
41. Junger WG. Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol (2011) 11(3):201–12. doi: 10.1038/nri2938
42. Gillrie MR, Lee K, Gowda DC, Davis SP, Monestier M, Cui L, et al. Plasmodium falciparum histones induce endothelial proinflammatory response and barrier dysfunction. Am J Pathol (2012) 180(3):1028–39. doi: 10.1016/j.ajpath.2011.11.037
43. Hunt JA, Flanagan BF, McLaughlin PJ, Strickland I, Williams DF. Effect of biomaterial surface charge on the inflammatory response: Evaluation of cellular infiltration and tnf alpha production. J BioMed Mater Res (1996) 31(1):139–44. doi: 10.1002/(sici)1097-4636(199605)31:1<139::Aid-jbm15>3.0.Co;2-i
44. Baeuerle PA, Huttner WB. Chlorate–a potent inhibitor of protein sulfation in intact cells. Biochem Biophys Res Commun (1986) 141(2):870–7. doi: 10.1016/s0006-291x(86)80253-4
45. Engelbrektson A, Hubbard CG, Tom LM, Boussina A, Jin YT, Wong H, et al. Inhibition of microbial sulfate reduction in a flow-through column system by (Per)Chlorate treatment. Front Microbiol (2014) 5:315. doi: 10.3389/fmicb.2014.00315
46. Kawasaki H, Isaacson T, Iwamuro S, Conlon JM. A protein with antimicrobial activity in the skin of schlegel's green tree frog rhacophorus schlegelii (Rhacophoridae) identified as histone H2b. Biochem Biophys Res Commun (2003) 312(4):1082–6. doi: 10.1016/j.bbrc.2003.11.052
47. Kawasaki H, Koyama T, Conlon JM, Yamakura F, Iwamuro S. Antimicrobial action of histone H2b in escherichia coli: Evidence for membrane translocation and DNA-binding of a histone H2b fragment after proteolytic cleavage by outer membrane proteinase T. Biochimie (2008) 90(11):1693–702. doi: 10.1016/j.biochi.2008.07.003
48. Wright HL, Lyon M, Chapman EA, Moots RJ, Edwards SW. Rheumatoid arthritis synovial fluid neutrophils drive inflammation through production of chemokines, reactive oxygen species, and neutrophil extracellular traps. Front Immunol (2020) 11:584116. doi: 10.3389/fimmu.2020.584116
49. Lloyd KA, Wigerblad G, Sahlström P, Garimella MG, Chemin K, Steen J, et al. Differential acpa binding to nuclear antigens reveals a pad-independent pathway and a distinct subset of acetylation cross-reactive autoantibodies in rheumatoid arthritis. Front Immunol (2018) 9:3033. doi: 10.3389/fimmu.2018.03033
Keywords: histone, cytotoxicity, DAMP, chemokine, synoviocyte, macrophage, inflammation, arthritis
Citation: Jung J, Lee LE, Kim H, Kim JE, Jang SH, Roh JS, Lee B, Robinson WH, Sohn DH, Pyun J-C and Song JJ (2022) Extracellular histones aggravate autoimmune arthritis by lytic cell death. Front. Immunol. 13:961197. doi: 10.3389/fimmu.2022.961197
Received: 04 June 2022; Accepted: 21 July 2022;
Published: 11 August 2022.
Edited by:
Marko Radic, University of Tennessee College of Medicine, United StatesReviewed by:
Paul Thompson, The Scripps Research Institute, United StatesCopyright © 2022 Jung, Lee, Kim, Kim, Jang, Roh, Lee, Robinson, Sohn, Pyun and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jason Jungsik Song, anNrc29uZ0B5dWhzLmFj; Dong Hyun Sohn, ZGhzb2huQHB1c2FuLmFjLmty; Jae-Chul Pyun, amNweXVuQHlvbnNlaS5hYy5rcg==
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.