
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 05 September 2022
Sec. Viral Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.960544
This article is part of the Research TopicUpdates on Immunity to Influenza A Virus in Humans and AnimalsView all 5 articles
 Xinxin Li1,2Siya Liu1,2Kul Raj Rai1,2Wenzhuo Zhou1,2Song Wang1,2Xiaojuan Chi1,2Guijie Guo1,2Ji-Long Chen1,2*Shasha Liu1,2*
Xinxin Li1,2Siya Liu1,2Kul Raj Rai1,2Wenzhuo Zhou1,2Song Wang1,2Xiaojuan Chi1,2Guijie Guo1,2Ji-Long Chen1,2*Shasha Liu1,2*STAT2 is an important transcription factor activated by interferons (IFNs) upon viral infection and plays a key role in antiviral responses. Interestingly, here we found that phosphorylation of STAT2 could be induced by several viruses at early infection stage, including influenza A virus (IAV), and such initial activation of STAT2 was independent of type I IFNs and JAK kinases. Furthermore, it was observed that the early activation of STAT2 during viral infection was mainly regulated by the RIG-I/MAVS-dependent pathway. Disruption of STAT2 phosphorylation at Tyr690 restrained antiviral response, as silencing STAT2 or blocking STAT2 Y690 phosphorylation suppressed the expression of several interferon-stimulated genes (ISGs), thereby facilitating viral replication. In vitro experiments using overexpression system or kinase inhibitors showed that several kinases including MAPK12 and Syk were involved in regulation of the early phosphorylation of STAT2 triggered by IAV infection. Moreover, when MAPK12 kinase was inhibited, expression of several ISGs was clearly decreased in cells infected with IAV at the early infection stage. Accordingly, inhibition of MAPK12 accelerated the replication of influenza virus in host. These results provide a better understanding of how initial activation of STAT2 and the early antiviral responses are induced by the viral infection.
Influenza A virus (IAV) is widespread throughout the world and remains a major threat to human and animal health, causing significant annual mortality in severe cases (1, 2). Innate immunity is the front line of defense against viral infections. It uses evolutionarily highly conserved pattern recognition receptors (PRRs) to recognize specific components of bacteria or viruses to trigger immune responses (3). PRRs mainly include retinoic acid-induced gene type I receptors (RLRs), toll-like receptor (TLRs), nucleotide oligomeric domain (NOD)-like receptors (NLRs), C-type lectin receptors (CLRs), DNA-dependent activation of interferon regulatory factor 3 (DAI), and melanoma deficiency factor 2 (AIM2), etc (4–6). Activation of PRR-dependent pathway leads to the secretion of interferons (IFNs), inflammatory cytokines, and other mediators that recruit and activate effector cells, which contributes to pathogen elimination (7). In infected cells, panhandle structure of the IAV genome RNA can be detected by RIG-I (8). The genome of IAV can also be detected by other PRRs, such as TLR3 (9), TLR7 (10), and TLR8 (11). After binding to PRR, RIG-I can recruit mitochondrial antiviral signaling proteins (MAVS) by interacting with the CARD domain to initiate signaling pathways (12), thereby promoting the activation of multiple transcription factors, such as IRF3, IRF7, and NF-κB (13), which induce the production of cytokines and proinflammatory cytokines.
Signal transducer and activator of transcription 2 (STAT2) is a member of the STATs family, and like other members of the family, contains seven domains that are structurally and functionally conservative: N-terminal domain (NTD), coiled-helix domain (CCD), DNA-binding domain (DBD), crossover domain (LD), Src homologous 2 domain (SH2D), tyrosine phosphorylation site (pY), and transcriptional activation domain (TAD). STAT2, with a molecular weight of 113 kDa (14), has a variety of biological activities and is an important transcription factor implicated in the antiviral responses. Previous studies have shown that classical type I and type III IFNs bind to corresponding IFN receptors to activate Janus kinases (JAKs), including JAK1 and TYK2 (15, 16). Activated JAKs and STATs pathway mediates binding of the STAT1-STAT2 heterodimer with interferon regulator 9 (IRF9) to form the transcription factor complex ISGF3 (15, 17). ISGF3 binds to the IFN stimulating response element (ISRE) and controls the expression of numerous antiviral proteins (18), thereby establishing an antiviral state through a variety of mechanisms, including inhibition of viral transcription, translation and replication, and degradation of viral nucleic acids (19–21). In addition to its role in antiviral immunity, STAT2 has also been shown to be involved in the regulation of immune signaling. For example, STAT2 functions as a universal cytokine regulator by inhibiting STAT1 in multiple signaling pathways (22). Based on previous findings, Zika virus (ZIKV) NS5 protein induces the ubiquitination and degradation of STAT2, promotes STAT1 homodimer formation and enhances IFN-γ-induced ISG expression (23).
Although IFNs-activated JAK-STAT is a typical pathway for activation of innate immunity, many viruses have evolved strategies to evade the IFN signaling. For instance, paramyxovirus HPIV2 induces degradation of STAT2 protein (24). Similarly, V proteins from Hendra and Nipah viruses inactivate IFN signaling by directly inhibiting STAT1 and STAT2 functions (25, 26). The unique immune evasion mechanisms of these viruses suggest the importance of STAT2 for innate antiviral immunity. Previous reports have revealed that STAT2 is essential for TLR-induced inflammatory cytokines production in bone-marrow derived macrophage (BMDM) cell lines in a type I IFN receptor-independent manner, highlighting the prominent role of STAT2 independent of type I IFN (27). Moreover, there is evidence that some virus-induced ISGs are independent of IFN-mediated innate immune pathways (28, 29). Such as, the upregulation of ISG15 during HCMV infection occurred in an IFN-independent, IRF3-dependent manner (28, 30). These indicate that there exist IFN-independent mechanisms in response to viral infection. In addition, it is also thought that subcellular localization of MAVS determines the signaling activated during viral infection that controls ISG expression. For example, the expression of ISGs is mainly dependent on peroxisome-localized MAVS rather than mitochondria-localized MAVS during reovirus infection (31). Although extensive studies on STAT have been focused on the IFN-mediated JAKs over the past decades, a growing number of studies have suggested other pathways activating STAT. It is observed that cyclin-dependent kinase, as a positive regulator of IFN signaling, can directly affect STAT activation (32). SOCS1, an important suppressor of JAK-STAT signaling, is expressed earlier than cytokine secretion, implying that there may be a cytokine-independent pathway regulating STAT activity (33). Recently, it has also been shown that early activation of STAT1 during the viral infection is mediated by spleen tyrosine kinase (Syk) but not cytokine-activated JAK, providing new insights into the complex mechanisms underlying interaction between virus and host immune system (34).
In this study, we observed that virus-induced initial activation of STAT2 was independent of type I IFN signaling. Specifically, increased phosphorylation of STAT2 occurred at the early phase of infection with multiple viruses. Further experiments displayed that RIG-I/MAVS pathway regulated such activation of STAT2. In addition, we found that mitogen-activated protein kinase 12 (MAPK12) was involved in activating STAT2 and positively regulated the expression of some ISGs at the early phase of IAV infection. These results provide new evidence of how the early activation of STAT2-involved innate immunity is regulated during the viral infection.
A549 (ATCC CCL-185), 293T (CRL -11268), MDCK (ATCC CRL -2935), and PK15 cells used in this study were all from American Type Culture Collection (ATCC). Cells were grown in monolayer culture in Dulbecco Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (Gibco-BRL, Gaithersburg, MD, USA), 2 μg/mL trypsin, 100 U/mL penicillin and 100 g/mL streptomycin. Cells were cultured every two days, inoculated into porous plates one day before virus infection and further cultured at 37°C and 5% CO2.
In this study, the following antibodies were mainly used: anti-flag (Protein Tech, Wuhan, 66008-3-IG), anti-STAT2 (Cell Signaling Technology, Danvers, MA, USA, 72604), anti-pSTAT2 (Cell Signaling Technology, Danvers, MA, USA, 88410), and anti-β-actin (TransGen Biotech, Beijing, HC201). The antibodies against NP protein of IAV and GE of PRV were produced by our laboratory. IFN-β was purchased from Sino Biologicals (Beijing), SB203580, INCB-018424, and CP690550 were from Selleck (Houston, TX, USA).
IAV strains including A/WSN/33 (H1N1) (WSN), A/PR/8/34 (H1N1) (PR8), A/CA/04/09 (H1N1) (CA04), and H9N2 were cultured for 2 days in allantoic fluid of chicken embryo aged 9-10 days without specific pathogens. Pseudorabies virus (PRV) was propagated in MDCK cells and Muscovy Duck Reovirus (MDRV) was propagated in 293T cells, as previously described (35). For viral infection, cells were infected with virus at the indicated multiplicity of infection (MOI), mixed, 37°C adsorbed for 1 h, gently shaken every 15 min. Appropriate new maintenance solution was added to continue culture in 37°C and 5% CO2.
Lentiviral vector expressing specific shRNAs in pSIH-H1-GFP vector along with lentiviral packaging plasmids were transfected into 293T cells. The virus-containing supernatant was collected 48 h after transfection and infected A549 cells by centrifugation at 2200 rpm for 2 h, as previously described (36). The sequences used in the shRNAs targeting specific genes were shown in Table S1. A549 cell lines stably expressing STAT2-WT, STAT2-Y690F, or empty vector (EV) were generated by infecting the cells with retroviruses encoding these genes in pNL-CMV vector, as previously described (37).
Total RNA was isolated from cultured cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). According to the manufacturer’s method, reverse transcription of RNA (2 µg) into cDNA using a reverse transcription kit (Mei5bio, Beijing, MF949-01), followed by PCR using rTaq DNA polymerase and quantitative PCR (qPCR) using SYBR PremixEx TaqII (TaKaRa, Tokyo, Japan). GAPDH was chosen as a reference gene for internal standardization. The normalized ratio indicates that the ratio is automatically calculated by the Light Cycler system (Roche, Switzerland) using the ΔΔCT method. And the primers for RT-PCR and RT-qPCR were shown in Table S2.
Cell lysis was performed on collected cell samples using Beyotime (Shanghai, ST506). The protein concentration was quantified by the BCA protein assay kit (Beyotime, Shanghai, P0012). Finally, the protein samples were separated by SDS-PAGE and transferred to the NC membrane (Merck Millipore, HATF00010) for detection with an appropriate proportion of TBS diluted primary antibody.
For hemagglutinin (HA) assay, cell culture supernatant was collected at indicated time point after virus infection, adding 25 μL PBS, and then adding the 25 μL diluted virus sample to the first well, successively diluting it from left to right. Then added 25 μL 1% chicken red blood cells to all wells. At last, we incubated the samples for 20 minutes to read the results.
For plaque assay, MDCK cells were infected with supernatant of influenza virus culture and cultured at 37°C for 1 h. The cells were then washed with PBS to remove the virus inoculants and covered with a mixed medium containing low-melting-point agarose (Promega, Madison, WI, USA) and 1 µg/mL tosylamido phenylethyl chloromethyl ketone (TPCK)-treated trypsin (Sigma-Aldrich, St. Louis, MO, USA). Plaque was visible in 2 or 3 days at 37°C, and viral titers were counted and determined.
All data were represented by means and standard deviations (SD) from three independent experiments. GraphPad Prism 8 (GraphPad Software, Inc.) was used for all statistical evaluations. The student’s t-test was used to analyze the statistical difference between the two groups. P < 0.05 (shown by ∗), P < 0.01 (shown by ∗∗), P < 0.001 (shown by ∗∗∗), P < 0.0001 (shown by ∗∗∗∗). NS stands for not significant.
To investigate STAT2 Y690 phosphorylation (pSTAT2Y690) induced by viral infections, we performed a time course study in which levels of pSTAT2Y690 were examined in cells infected with various strains of IAV such as WSN, PR8, CA04, H9N2, another RNA virus, Muscovy duck reovirus (MDRV), and also DNA virus such as Pseudorabies virus (PRV). Increased pSTAT2Y690 was observed after WSN (Figure 1A), PR8 (Figure 1B), CA04 (Figure 1C), and H9N2 (Figure 1D) infection with respect to indicated hours of post infection (hpi). However, the STAT2 Y690 phosphorylation declined at the late hpi in the cells infected by various strains of IAV except infection with H9N2 (Figures 1A–D). Sustained activation of STAT2 even at 24 hpi following H9N2 infection may be related to the subtype or virulence of the strain. Of note, infection with several IAV strains tested (WSN, PR8, CA04, and H9N2) (Figures 1A–D) caused an increase in pSTAT2Y690 immediately at approximately 3 hpi. Next, we examined STAT2 phosphorylation induced by other RNA virus such as MDRV. It was found that early activation of STAT2 was also induced by infection of MDRV (Figure 1E). Additionally, elevated level of pSTAT2Y690 was observed in cells infected with a DNA virus PRV as early as 4 hpi (Figure 1F). Taken together, these data demonstrated that initial activation of STAT2 could be induced by infection of several types of viruses in various cell lines.
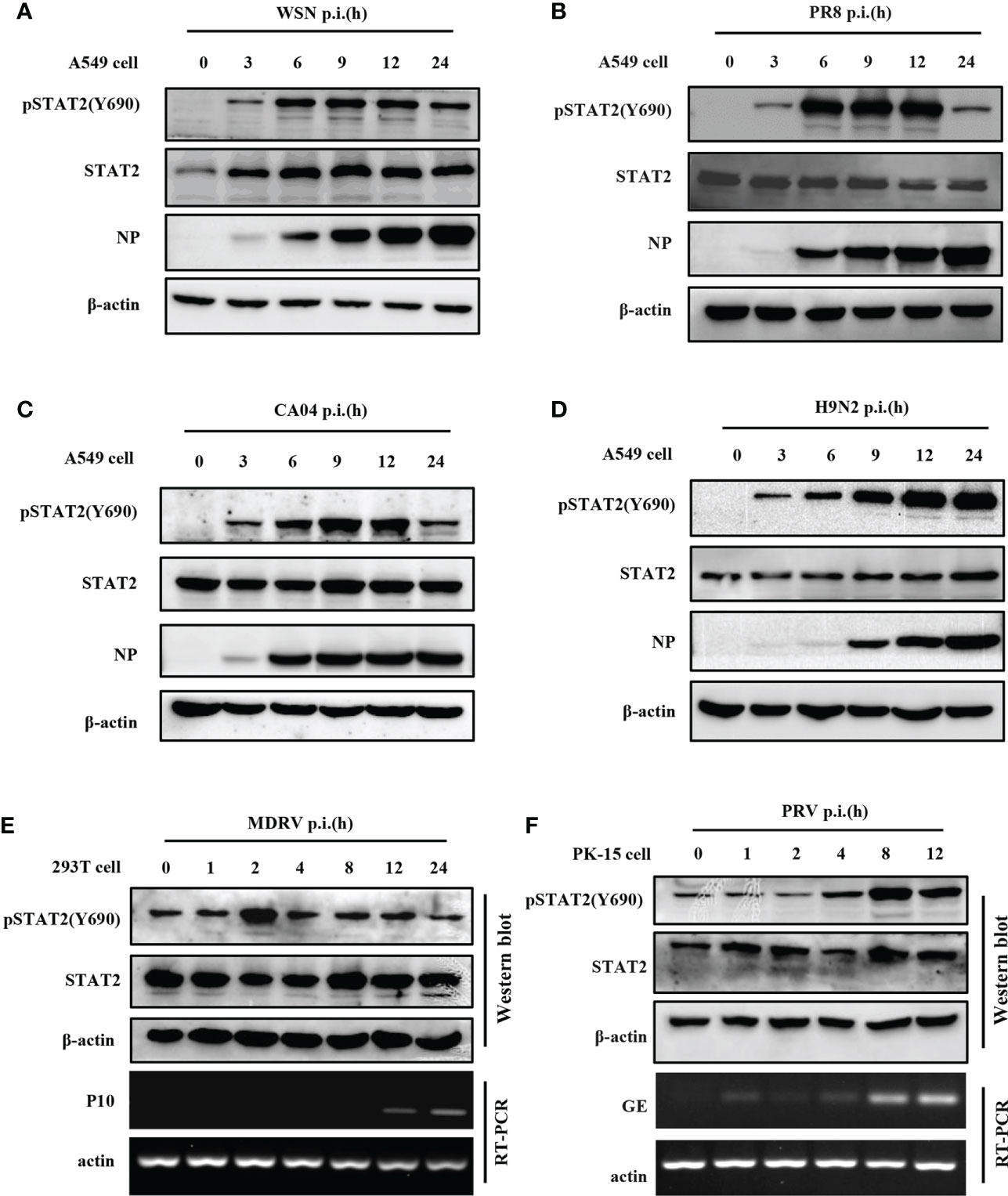
Figure 1 Phosphorylation of STAT2 can be induced by several viruses at early infection stage. (A–D) A549 cells were infected with IAV strains, including WSN (A), PR8 (B), CA04 (C), and H9N2 (D) at indicated time points. Cell lysates were collected to detect the corresponding protein by Western blotting. NP, IAV nucleoprotein. Diagram shows representative data from three independent experiments. (E, F) 293T cells were infected with MDRV (E) and PK15 cells infected with PRV (F) at corresponding time points. Corresponding proteins were detected by Western blotting, the corresponding mRNAs were detected by RT-PCR. P10, the non-structural protein of MDRV. GE, glycoprotein E of PRV.
It is well known that cytokines activate JAK/STAT signal transduction pathway (38). In particular, type I and type III IFNs are critical for activation of JAK/STAT signaling in innate antiviral immunity. Interestingly, previous report revealed that early activation of STAT1 is independent of cytokines (34). Therefore, we determined whether early phosphorylation of STAT2 Y690 upon viral infection was independent of these IFNs. A549 cells were infected by IAV at indicated time points and cell lysates were subjected to Western blotting for the examination of pSTAT2Y690 (Figure 2A). The results showed that phosphorylation of STAT2 Y690 appeared in cells at 3 hpi by WSN infection but was not caused by lysates of cells infected with the IAV (WCL) and cell culture supernatant (SN). Similarly, the STAT2 phosphorylation occurred earlier in A549 directly infected by other IAVs than that in A549 cells stimulated by virus-infected cell culture supernatant (Figures 2B–D). Furthermore, A549 cells were infected with WSN for different time points, and the phosphorylation of STAT1, STAT2 and IRF3 was examined. We observed that the phosphorylation of STAT2 and STAT1 occurred as early as 2 hpi and enhanced at 3 hpi. By contrast, the phosphorylation of IRF3 and expression of IFN-β was induced by the IAV infection at 5 hpi and later (Figures 2E, F). These results suggest that phosphorylation of STAT2 and STAT1 was likely induced before IRF3 activation and IFN production during IAV infection. Next, we employed cycloheximide (CHX) that can inhibit eukaryotic translation and eukaryotic protein synthesis. The data exhibited that increased phosphorylation of STAT2 still appeared at early infection stage (3 hpi) (Figure 2G). We further utilized type I IFN receptor knockout (IFNAR1 KO) cells and found that there was no difference in phosphorylation of STAT2 Y690 between IFNAR1 KO cells and WT A549 cells at 3 h post influenza virus infection, whereas IFN-β-induced pSTAT2Y690 was dramatically decreased (Figure 2H). Since STAT2 can also be activated by type III IFNs (16), we investigated whether type III IFNs are involved in virus-induced early phosphorylation of STAT2. For this, IFNLR1 siRNA was transfected into A549 cells for 24 h, followed by influenza virus infection, and protein and RNA samples were collected at the specified time. As compared with si-NC, the expression of IFNLR1 was significantly disrupted in si-IFNLR1 group (Figures 2I–J). However, Western blotting showed that silencing IFNLR1 had little effect on STAT2 activation at the early stage of influenza virus infection (Figure 2K). Together, these results suggest that type I and type III IFNs are likely not required for initial activation of STAT2 during the IAV infection.
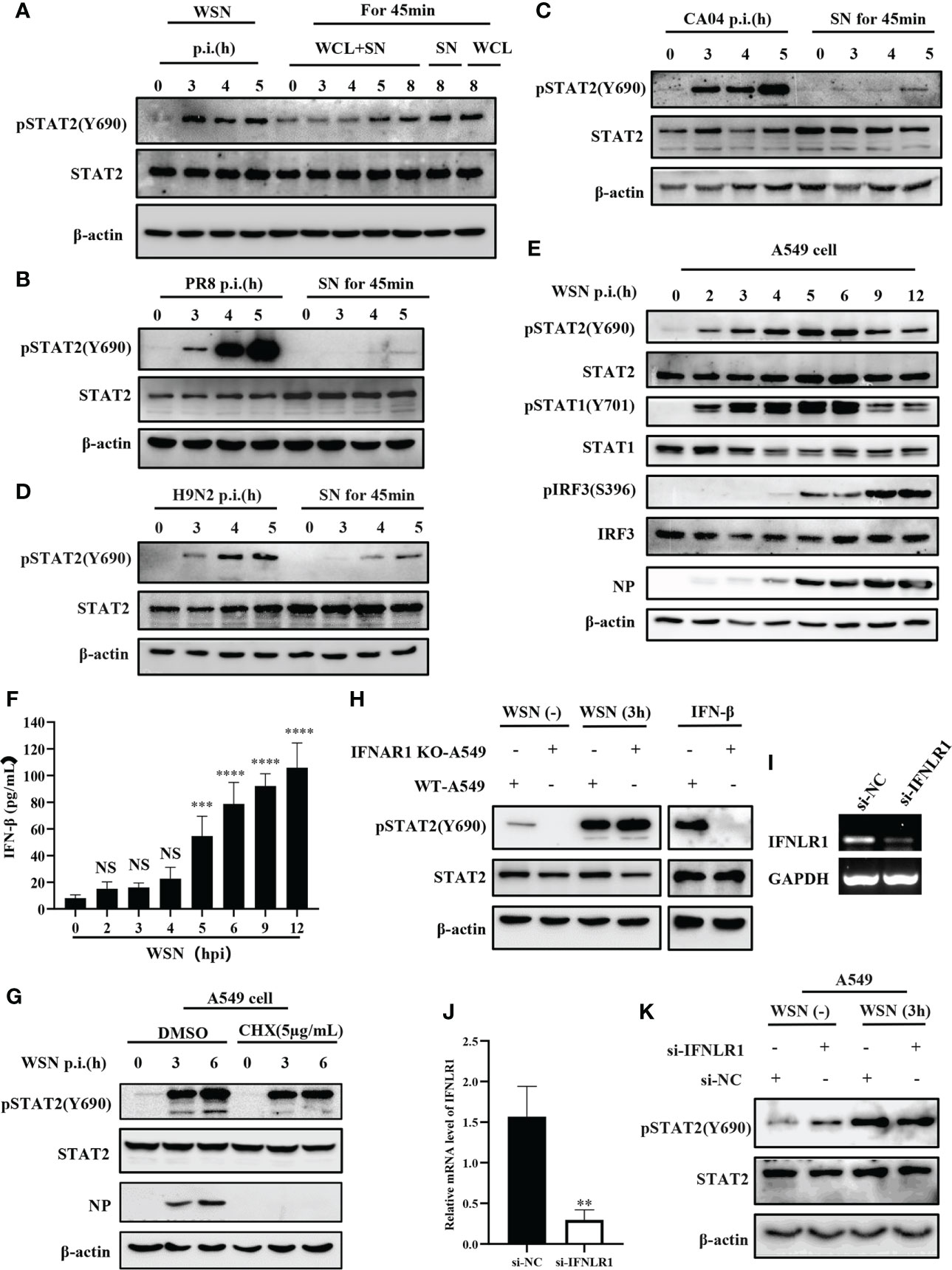
Figure 2 Initial activation of STAT2 is independent of type I and type III IFNs signaling at the early viral infection. (A) A549 cells were infected with WSN at the indicated time. Supernatant and whole cell lysate (WCL+SN) at the corresponding time point were collected to stimulate the naive cells for 45min, respectively. In the figure, SN and WCL were used as controls for the early phosphorylation of STAT2 Y690. The pSTAT2(Y690) was detected by Western blotting. (B–D) A549 cells were infected with different influenza strains PR8 (B), CA04 (C), and H9N2 (D). Corresponding cell supernatant (SN) cultures were collected to stimulate the naive cells for 45 min, respectively, followed by Western blotting with indicated antibodies. (E, F) A549 cells were infected with WSN for indicated time points. Western blotting analysis was performed to detect the phosphorylation of STAT1, STAT2, and IRF3 (E). ELISA was employed to examine the protein levels of IFN-β in the SN (F). (G) A549 cells were treated with DMSO or cycloheximide (CHX) (5µg/mL) for 20 min in advance, then infected with WSN for indicated time. pSTAT2 was detected by Western blotting. (H) IFNAR1 knockout (IFNAR1-KO-A549) or wild-type A549 (WT-A549) cells were infected with WSN for 3 h or treated with IFN-β for 45 min. pSTAT2 was analyzed by Western blotting. (I, J) Si-IFNLR1 or control si-NC was transfected into A549 cells for 24 (h) RT-PCR (I) and RT-qPCR (J) were used to detect the knockdown efficiency. (K) Si-IFNLR1 or si-NC -based A549 cells were infected with WSN for 3 h, and the level of pSTAT2(Y690) was detected by Western blotting. Data represent the mean ± SD from three independent experiments. **p < 0.01. The above results show the representative results of three independent experiments. ***p <0.001; ****p <0.0001; ns, no significance.
Next, we analyzed the effect of JAKs on the early phosphorylation of STAT2 Y690. First, we used the JAK inhibitor INCB018424 (INCB) to inhibit the activation of JAK1. We found that levels of the virus-induced pSTAT2Y690 were not significantly changed by JAK inhibitor treatment at the early stage of viral infection (Figure 3A). On contrast, phosphorylation of STAT2 Y690 decreased significantly in INCB treated cells followed by stimulation with IFN-β (Figures 3A–B). Similar results were observed when cells were treated with CP-690550 (CP), another inhibitor of JAK activation, followed by IAV infection or IFN-β treatment (Figures 3C–D). Furthermore, we used specific shRNAs to target JAK1 in A549 cells, and knockdown efficiency was determined by RT-qPCR (Figure 3E). Silencing JAK1 had little effect on pSTAT2Y690 at the early stage of viral infection (Figure 3F), but IFN-β-stimulated pSTAT2Y690 was significantly decreased upon disruption of JAK1 expression (Figure 3F). Previous studies have reported that the activation of STAT2 mainly depends on JAK1 and TYK2 (39). Therefore, we further explored whether TYK2 affected the early activation of STAT2. To this end, specific TYK2 shRNAs were employed to construct TYK2 knockdown A549 cells (Figure 3G). Similarly, the STAT2 phosphorylation was not significantly changed at the early stage of viral infection compared with the luciferase-targeted control (Figure 3H). As expected, the pSTAT2 was significantly reduced in IFN-β-stimulated sh-TYK2 A549 (Figure 3H). The data suggest that early phosphorylation of STAT2 may be independent of JAKs during the viral infection.
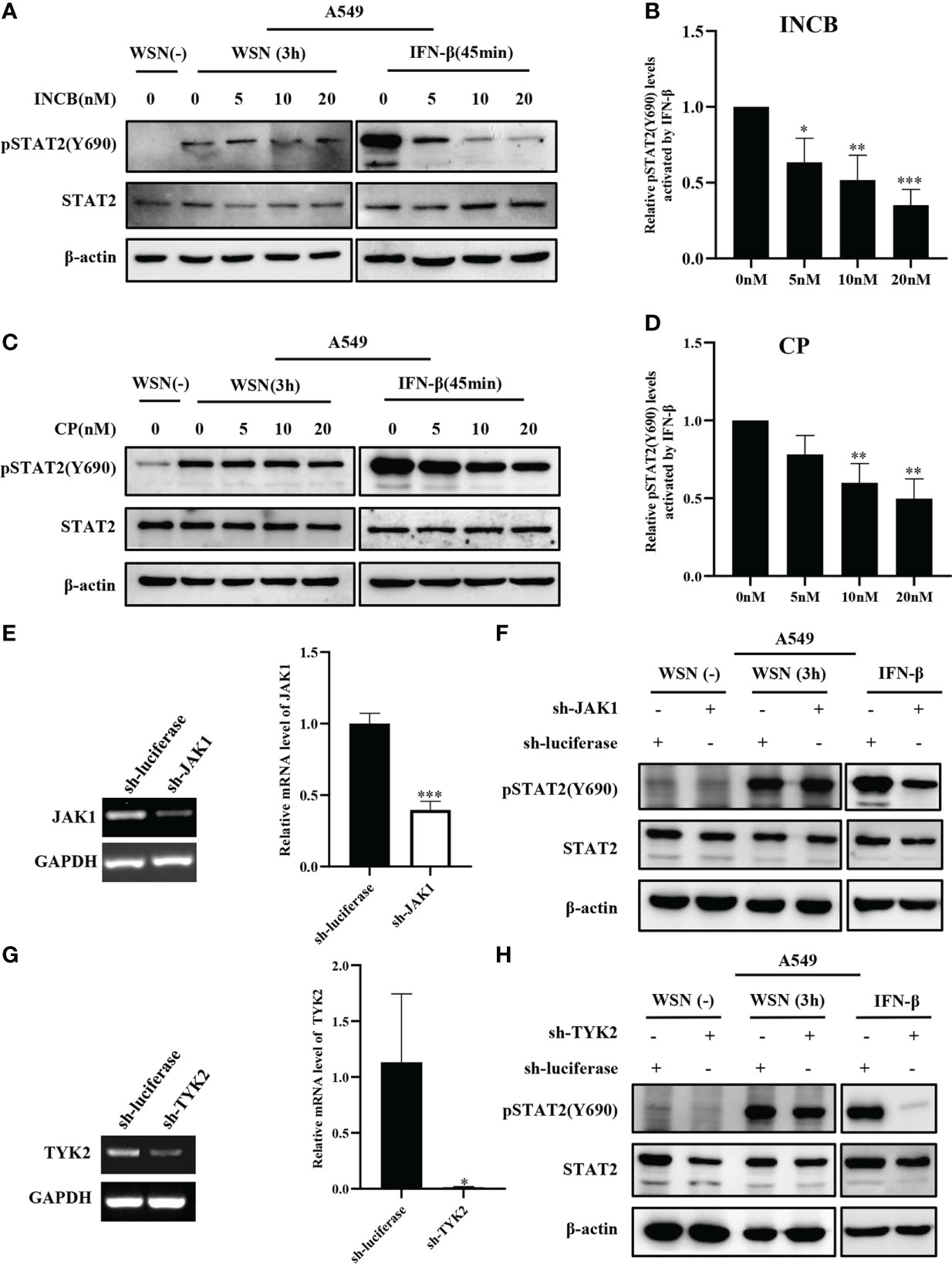
Figure 3 Inhibition and silence of JAKs have little effect on IAV-induced early phosphorylation of STAT2 at Y690. (A, C) A549 cells were pretreated with INCB018424 (INCB) (A) and CP-690550 (CP) (C) at different concentrations for 20 min, respectively, followed by WSN infection for 3 h or IFN-β (0.1 μg/mL) stimulation for 45 min. Western blotting analysis of pSTAT2 levels revealed representative blots from three independent experiments. (B, D) The level of pSTAT2 in (A), (C) was quantified by densitometry and normalized to STAT2 levels. The data are shown as the mean ± SD from three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001. (E, G) The interference efficiencies in JAK1- (E) and Tyk2- (G) ablated and control (luciferase) A549 cells were detected by RT-PCR (Left) and RT-qPCR (Right). Error bars represent the mean ± SD from three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001. (F, H) A549 cells as described in (E, G) were infected with WSN for 3 h or stimulated with IFN-β (0.1 μg/mL) for 45 min, respectively. The expression levels of specific proteins were detected by Western blotting. The data in the figures are representative images of three repetitions.
The tyrosine residue Tyr690 of STAT2 is directly adjacent to the domain SH2D and plays an important role in its interactions with other STAT proteins, especially STAT1 (40). In a typical IFN-I-induced signal transduction pathway, ISGF3 complex is formed by the interaction between STAT1 at Tyr701 and STAT2 at Tyr690 (17), which is crucial for antiviral immunity. Since STAT2 Y690 could be phosphorylated at the early stage of viral infection, we asked whether the phosphorylation of STAT2 had any effect on IAV replication. First, specific shRNAs were employed to target STAT2 in A549 cells (Figure 4A and Figures S1A–S1B). Supportively, STAT2 knockdown using sh-STAT2 significantly diminished IAV-induced activation of STAT2 at Tyr690 at 3 hpi (Figure 4A), and resulted in enhanced virus replication and higher viral titer as compared to the control (Figures 4B–C), suggesting that STAT2 is essential for establishment of antiviral response. To verify this observation, STAT2 Y690F, a dominant negative mutation of STAT2 (41) was generated (Figure 4D). We found that overexpression of STAT2 Y690F significantly enhanced the IAV replication as compared to that in cells expressing STAT2 WT or EV control (Figures 4E, F).
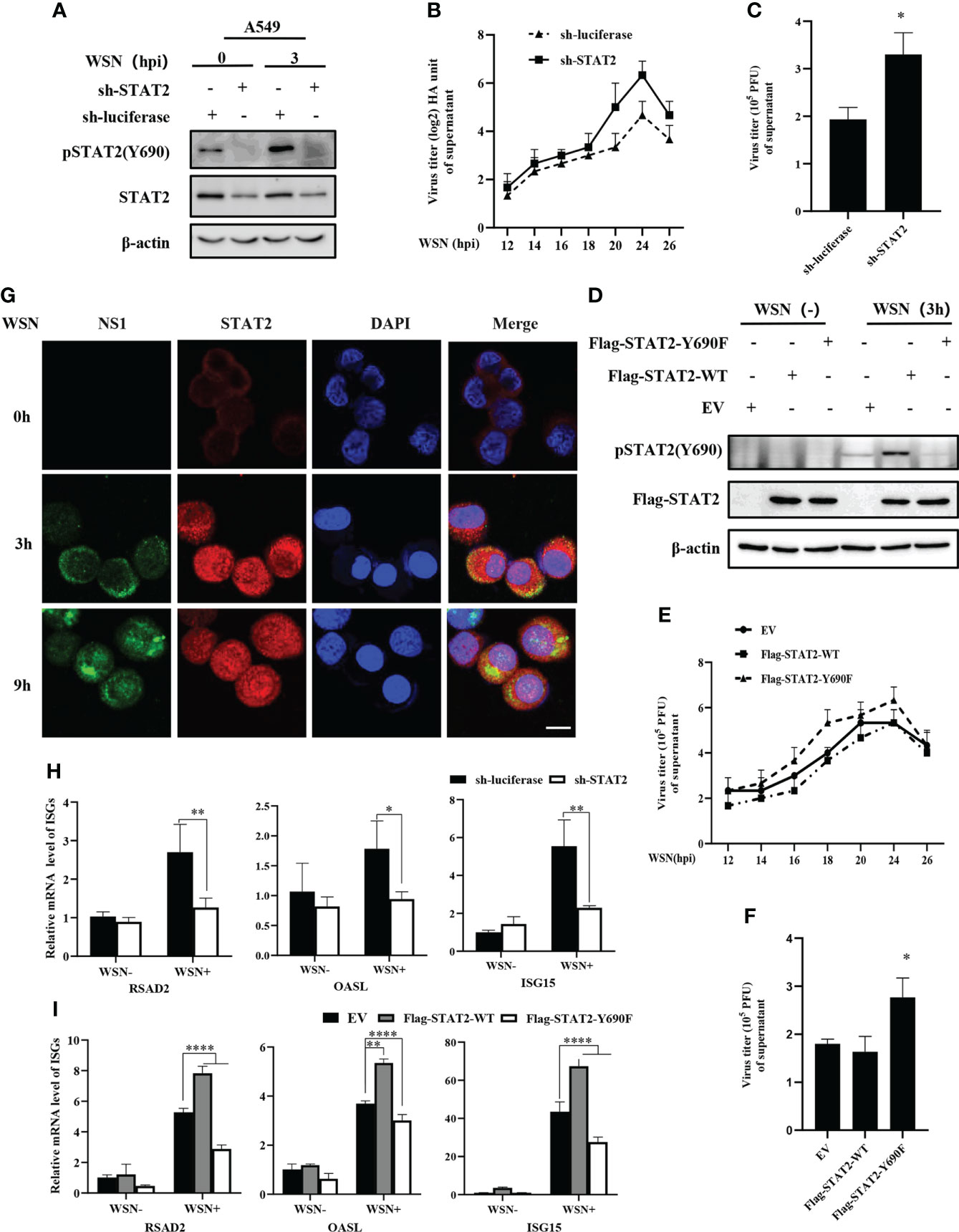
Figure 4 Disruption of STAT2 phosphorylation at Tyr690 promotes the IAV replication. (A) A549 cells stably expressing specific sh-RNA targeting STAT2 and control (luciferase) were infected with WSN virus for 3 (H) Cell lysates were analyzed by Western blotting with indicated antibodies. (B, C) STAT2- and luciferase- ablated A549 cells were infected with WSN (MOI = 1). Viral titers in supernatant of these cell cultures were examined by hemagglutinin (HA) assay at the indicated time points post infection (B). Sh-RNAs knockdown A549 cells or control cells were infected with WSN as described in (B). Viral titers in the supernatants of these cells were examined by plaque assay (24 h) (C). The mean ± SD of three independent experiments is given. (*P < 0.05) (D) A549 cells stably expressing STAT2-WT, STAT2-Y690F, and empty vector (EV) control were infected with or without WSN for 3 h and examined by Western blotting with the indicated antibodies. (E) A549 cells stably expressing STAT2-WT, STAT2-Y690F, and empty vector (EV) control were infected with WSN for indicated time point. Hemagglutinin (HA) assay were performed to examine the viral titers in the supernatant of cell culture. (F) The STAT2 overexpressing A549 cells or control cells as described in (D) were infected with WSN. Viral titers in the supernatant of these cells culture were examined by plaque assay (24 h). Data are represented as mean ± SD. (*P < 0.05.) (G) A549 cells were infected with or without WSN virus for 0 h, 3 h, and 9 (h) Immunofluorescence staining was performed to detect STAT2 (red) and NS1 (green). The nuclei were stained with DAPI (blue). Scale bar, 10 μm. (H) RT-qPCR was performed to detect the expression of RSAD2, OASL, and ISG15 in STAT2 knockdown A549 cells with IAV infection for 3 (h) (I) STAT2 overexpressing or control A549 cells as described in (D) were infected with WSN for 3 h, and the expression of RSAD2, OASL, and ISG15 was detected by RT-qPCR. Error bars represent the mean ± SD from three independent experiments. *p < 0.05; **p <0.01; ****p <0.0001.
Then, we examined the translocation of STAT2 into the nucleus after WSN infection. The accumulation of STAT2 and pSTAT2Y690 in the nucleus was observed as early as 3 hpi (Figure 4G, Figure S1C). To further address the relevance of early pSTAT2Y690 translocation into the nucleus with the activation of STAT2, we evaluated the expression of ISGs in STAT2 knockdown cells, and in the cells expressing WT or the dominant negative mutant of STAT2 (STAT2 Y690F) at the early time point post WSN infection. As shown in Figures 4H, I depletion of STAT2, or disruption of the STAT2 phosphorylation at Tyr690 led to a significant decrease in the expression of ISGs such as RSAD2, ISG15, and OASL at 3 hpi. These results suggest that STAT2 phosphorylation at Tyr690 accompanied by the initial activation of STAT2, contribute to the antiviral ISG response during early IAV infection. Overall, it was revealed that activation of STAT2 at Tyr690 played a critical role in host innate immune response to IAV infection.
The innate immune system triggers an antiviral response upon sensing various viral components, including double-stranded RNA by PRRs. Cytoplasmic helicase proteins RIG-I and MDA5 are main PPRs in sensing particular RNA viruses such as IAV (42). Although IAV genomic RNA panhandle structure has been shown to be recognized by RIG-I, TLR3, TLR7, and TLR8 (8, 43), it remains to be determined whether early phosphorylation of STAT2 is activated by the PRR-dependent signaling. To determine whether STAT2 Y690 can also be activated by dsRNA in host cells, we transfected A549 with different concentrations of poly (I:C), the dsRNA analogues for 6 h. Increased phosphorylation of STAT2 at Y690 was observed with respect to the increasing concentration of poly (I:C) (Figure 5A). Next, RIG-I, MDA5, or TLR3 knockdown A549 cell lines were generated, respectively (Figures S2A–S2D). The result showed that silencing RIG-I, but not MDA5 or TLR3, significantly reduced early pSTAT2Y690 levels (Figures 5B–E), suggesting that RIG-I was involved in IAV-indued early activation of STAT2. To study the specific pathway through which early activation of STAT2 occurred, we generated MAVS, IRF3 or IRF7 knockdown A549 cells followed by IAV infection (Figures S2E–G). The results showed that early STAT2 activation was significantly reduced in sh-MAVS cells (Figure 5F). However, there was no significant change of pSTAT2Y690 in sh-IRF3 and sh-IRF7 cell lines after the IAV infection (Figures 5G–H). In addition, we also found that the early phosphorylation of STAT2 was not affected when P65 of NF-κB was inhibited by BAY 11-7082 (Figure S2H). Therefore, the data suggest that the phosphorylation of STAT2 Y690 at the early stage of viral infection was mainly regulated through the RIG-I/MAVS signaling.
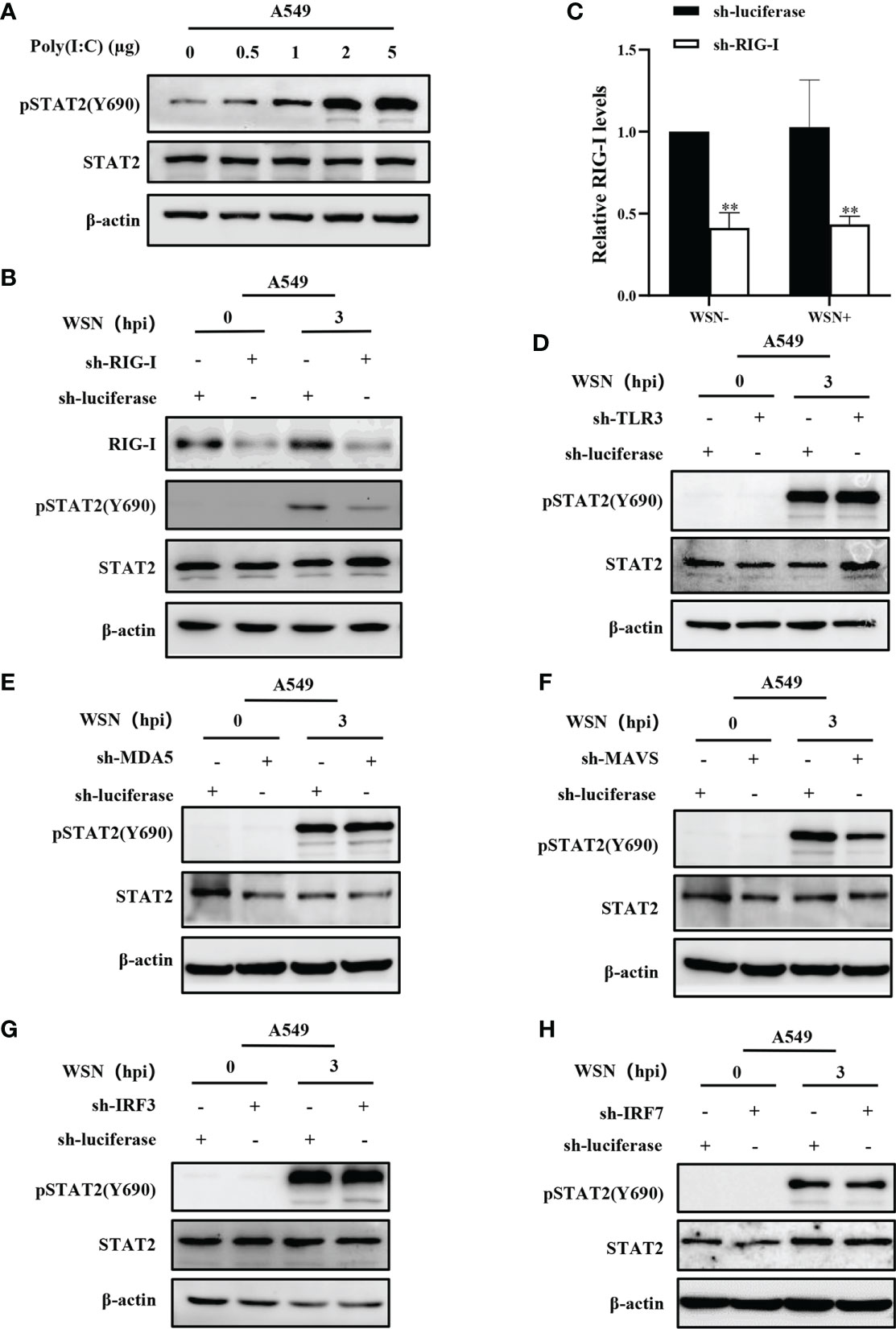
Figure 5 Early activation of STAT2 is mainly regulated by RIG-I/MAVS signaling. (A) A549 cells were transfected with different concentrations of Poly(I:C) for 6 h, and pSTAT2 were analyzed by Western blotting. (B ,C) Western blotting was performed to evaluate the indicated proteins in WSN-infected RIG-I or control (luciferase) knockdown A549 cells (B). The RIG-I levels were quantitated by densitometry as described in B (C). Error bars represent the mean ± SD from three independent experiments. **p < 0.01. (D–H) The sh-RNA based A549 cell lines stably expressing TLR3 (D), MDA5 (E), MAVS (F), IRF3 (G), IRF7 (H), and control (luciferase) were infected with WSN (MOI =1) for 3 h Cell lysates were collected and pSTAT2 levels were detected by Western blotting (D–H). The results shown in the figure are representative results of three independent experiments.
Since the virus-induced early activation of STAT2 is not regulated by JAKs, we identified which kinase(s) might regulate this process. Thus, we employed a kinase library and transfected the kinases into 293T cells to screen for kinases that could mediate the efficient phosphorylation of STAT2 Y690 (Data not shown). Then, RT-PCR was used to detect the kinases that could be expressed in A549 cells infected with or without WSN (Figure 6A). Syk, CDK9, and MAPK12 were preliminarily selected based on their relatively higher expression levels in A549 cells and ability to phosphorylate STAT2 in 293T cells (Figures 6A, B and Figure S3A). Furthermore, transfection of plasmids expressing these kinases in increasing concentrations caused an increased activation of STAT2 with respect to plasmid concentrations (Figures 6C–D and Figure S3B). To further investigate kinases involved in STAT2 activation at the early stage of viral infection, we constructed CDK9 or MAPK12 knockdown A549 cells using specific shRNAs, respectively (Figures S3C–S3E). The Syk knockout A549 cells were produced by our laboratory (44). The data showed that deleting Syk and silencing MAPK12 but not CDK9 caused a diminished early activation of STAT2 (Figures 6E–G), indicating that MAPK12 and Syk were key kinases for early activation of STAT2 induced by IAV infection.
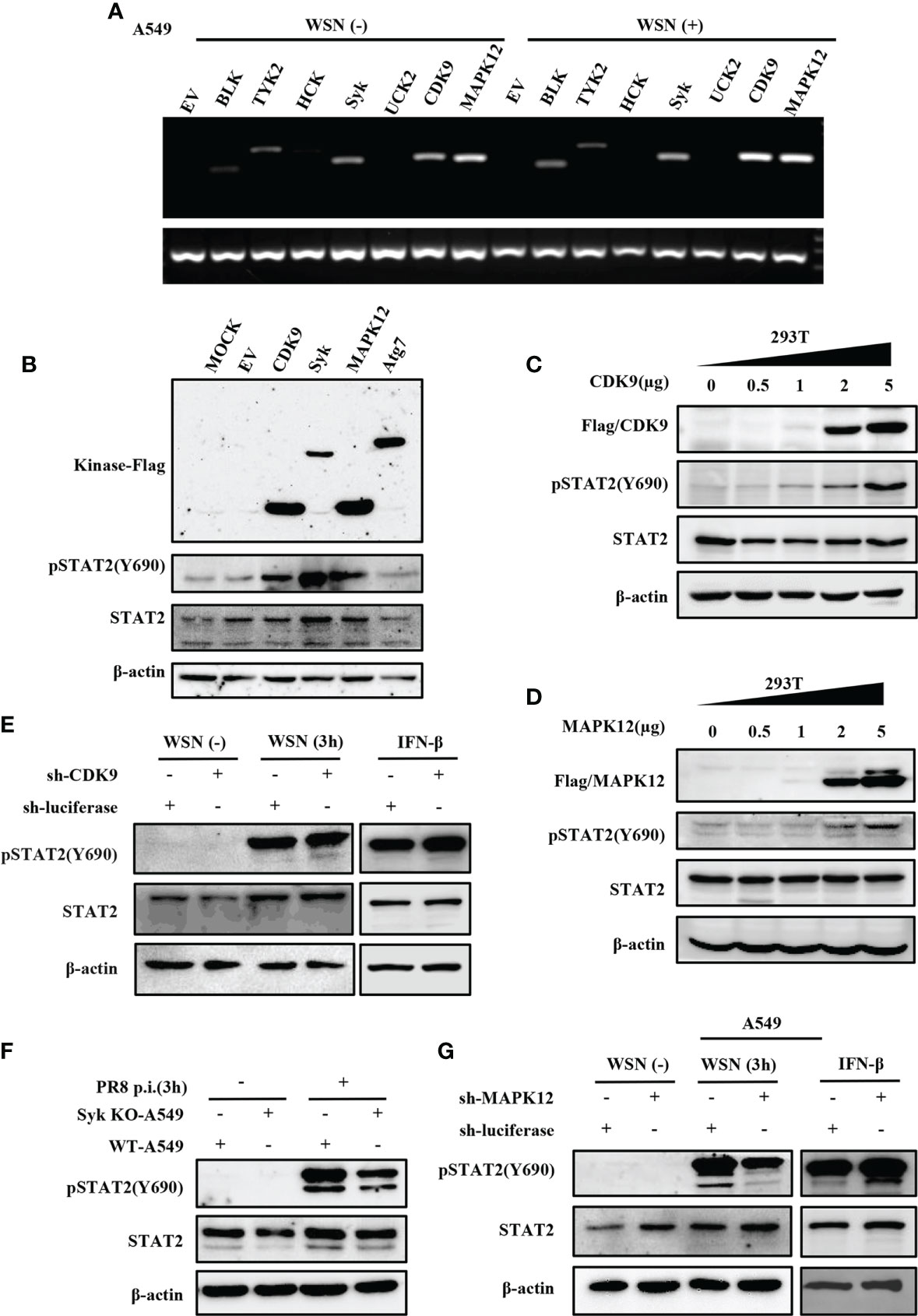
Figure 6 Several kinases are involved in early activation of STAT2 induced by IAV infection. (A) A549 cells were infected with or without WSN for 3 h, and the expression of various kinases was detected by RT-PCR. (B) 293T cell lines were transfected with different kinase plasmids for 24 h (MOCK as blank control, EV and Atg7 as negative control). Western blotting was performed to evaluate the pSTAT2 levels. (C, D) 293T cells were transfected with increasing amounts of MAPK12 (D) and CDK9 (C) -expressing plasmids as indicated or empty vector as control for 24 (h) Western blotting was performed with the indicated antibodies. Plotted are the representative data of three repeated experiments. (E, G) Sh-RNA-based A549 cells lines stably expressing CDK9 (E), MAPK12 (G), or control (luciferase) were infected with WSN with (MOI =1) for 3 h or stimulated by IFN-β (0.1 μg/mL) for 45 min, and the corresponding protein levels were detected by Western blotting. (F) Syk knockout A549 cells (Syk KO-A549) or control cells (WT-A549) were infected with PR8 (MOI =1) for 3 h and then protein samples were collected. The pSTAT2 was detected by Western blotting. The above data are representative results of three independent experiments.
MAPK12 is an important kinase involved in various signal transduction pathway. Considering the key role of MAPK12 in innate immune response, we further asked whether MAPK12 was involved in the regulation of antiviral genes during early viral infection. For this, we determined the expression of antiviral genes in cells treated with SB203580, an inhibitor of MAPK12 and control cells. It was found that inhibition of MAPK12 resulted in decreased expression of antiviral genes, such as RSAD2, OASL, and ISG15 (Figure 7A). The above results were further verified by RT-qPCR (Figures 7B–D). This indicated that MAPK12 positively regulates the expression of early antiviral genes. Next, we examined the early activation of STAT2 induced by IAV infection in A549 cells by treating with increasing concentration of SB203580. It was observed that the decreased phosphorylation level of STAT2 was correlated with the concentration of MAPK12 inhibitor (Figures 7E–F). Furthermore, we found that H9N2 induced pSTAT2Y690 was also decreased after MAPK12 inhibition (Figure 7G). It has been reported that RIG-I could activate p38 MAPK during SeV or Japanese encephalitis virus infection (45, 46). Our results showed that forced expression of MAVS led to a significant increase in the phosphorylation of STAT2 (Figure S4), implying that the RIG-I/MAVS signaling may mediate the activation of MAPK12 (p38γ), thereby enhancing early STAT2 phosphorylation. In addition, hemagglutinin assay showed that inhibition of MAPK12 remarkably promoted the replication of H9N2 virus (Figure 7H), suggesting that MAPK12 played an important role in host antiviral response. Taken together, MAPK12-mediated phosphorylation of STAT2 Y690 is critical for innate antiviral immunity at the early stage of influenza virus infection.
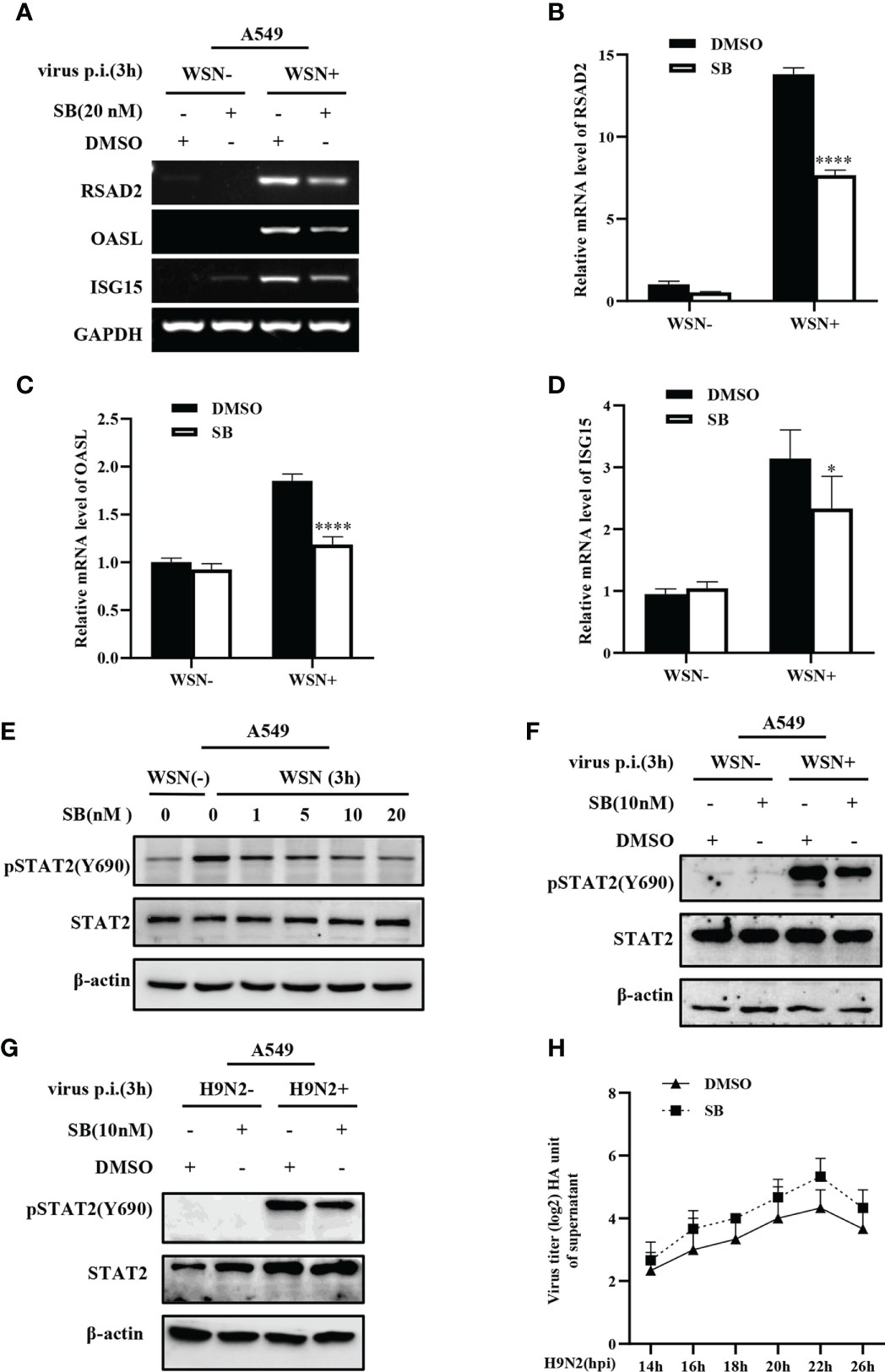
Figure 7 MAPK12 affects the expression of some ISGs during influenza virus infection. (A–D) SB203580 (SB) pretreated- A549 cells were infected with WSN for 3 h, and the expression of antiviral genes was detected by RT-PCR (A) and RT-qPCR (B-D). Error bars represent the mean ± SD from three independent experiments. *p < 0.05; ****p <0.0001. (E) A549 cells were pretreated with SB203580 (SB) at indicated concentrations for 20 min, then infected with WSN for 3 h, and pSTAT2 level was detected by Western blotting. Shown are representative data from three independent experiments. (F, G) SB203580 (SB) pretreated-A459 cells were infected with WSN (F) or H9N2 (G) for 3 h, and the level of pSTAT2 was detected by Western blotting. (H) SB203580 (SB) pretreated-A459 cells or control (DMSO) cell lines were infected with H9N2. Viral titers in supernatant of these cell culture were examined by hemagglutinin (HA) assay at indicated time point.
Our previous studies have shown that STAT1 can be activated through RIG-I/MAVS/Syk pathway at the early stage of viral infection, which is independent of cytokines and JAK signaling pathways (34). Similar to other members of the STAT family, STAT2 plays an important role in host defense against viral infections (17). In the JAK-STAT pathway mediated by type I IFN, phosphorylation of STAT1 and STAT2 leads to heterodimerization and interaction with IRF9, resulting in the formation of ISGF3 which then translocates into the nucleus and induces the transcription of ISGs (21, 47). STAT2 plays a key role in mediating antiviral immunity, as STAT2-/- mice exhibit increased susceptibility to viral infection and impaired response to type I IFN (48). Similar to STAT1-/- or IFNAR1-/- mice (49, 50), STAT2-/- mice display excessive inflammation, viral burden, and increased morbidity after infection with influenza virus (51). In addition, STAT2-/- hamsters have significantly elevated pulmonary viral titers after SARS-COV-2 infection (52). These observations indicate that disruption of STAT2 expression in vivo leads to a broad spectrum of viral susceptibility and higher mortality in animals. Besides, it has been shown that STAT2 positively regulates the induction of IL-6 in cooperation with the NF-κB pathway, and participates in the negative feedback of type I IFN signaling (53, 54). These results suggest that STAT2 plays a multitude of roles in regulating innate immunity and virus-host interactions.
Notably, it has been reported that IFN-like transcriptomes occur in the absence of IFN signaling in response to viral infection (55), which provides evidence for ISGs production in the absence of IFN during the early phase of viral infection. In this study, we observed that early phosphorylation of STAT2 at Y690 is activated by different IAV strain including WSN, PR8, CA04, and H9N2. Interestingly, initial activation of STAT2 was also detected upon infections with several other viruses such as dsRNA virus MDRV, and DNA virus PRV. Moreover, our results demonstrated that early activation of STAT2 was independent of JAKs, IFNAR1, and IFNLR1 following IAV infection, indicating that the initial activation of STAT2 is independent of the cytokine-mediated JAK signaling. This prompted us to probe signaling pathways that contribute to the early activation of STAT2. We found that depletion of RIG-I, but not TLR3 or MDA5, significantly dampened the initial activation of STAT2. As the primary mediator of RIG-I signaling (56), MAVS is required for the transduction of RIG-I signaling and consequent initiation of IFN responses (57, 58). Our results showed that silencing MAVS remarkably impaired the initial activation of STAT2, indicating that RIG-I/MAVS pathway mediates the early activation of STAT2 during IAV infection. NF-κB, IRF3, and IRF7, are the key transcription factors to induce the expression of type I and type III IFNs (59). By contrast, our results showed that silencing NF-κB, IRF3 or IRF7, had no significant effect on STAT2 phosphorylation at the early stage of influenza virus infection, supporting that cytokines are not required for the initial activation of STAT2 following IAV infection. Furthermore, IAV-induced phosphorylation of STAT2 Y690 plays an important role in antiviral response, as disruption of STAT2 expression, or forced expression of the dominant negative mutant of STAT2 (STAT2 Y690F) significantly decreased the expressions of several key ISGs such as RSAD2, ISG15, and OASL, thereby facilitating the replication of IAV.
In an attempt to identify potential kinase(s) that regulate the phosphorylation of STAT2 at Y690 at the early stage of viral infection, we found that Syk and MAPK12 may be involved in the early activation of STAT2 upon IAV infection, as depletion of Syk or MAPK12 significantly decreased STAT2 activation at the early stage of viral infection. MAPK12 (p38γ) is one of the p38 MAPKs which play an important role in the signaling cascades critical for innate immune responses (60–62). MAPK12-/- mice produced lower levels of cytokines such as IL-1β (63) and TNF-α upon LPS stimulation (62), thereby exhibiting lower symptom severity and joint damage compared with wild type mice. We then evaluated the functional involvement of MAPK12 in the host antiviral immunity in response to IAV infection. Our results showed that inhibition of MAPK12 dramatically impaired the expression of ISGs such as RSAD2, ISG15, and OASL, thereby promoting the replication of IAV. These results indicate that MAPK12 may be a kinase that mediates STAT2 activation at the early stage of influenza virus infection. Since MAPK12 is a serine/threonine kinase (64), it is possible that MAPK12 might modulate the activation of an unidentified tyrosine kinase that is responsible for the phosphorylation of STAT2 at Y690 during the early stage of viral infection. In addition, previous studies have shown that RIG-I and TLR3 can mediate the activation of p38 MAPKs during SeV or Japanese encephalitis virus infection (45, 46). Further studies are needed to determine the involvement of RIG-I/MAVS signaling in the activation of MAPK12 during IAV infection. In addition, precise mechanisms underlying the regulation of MAPK12 activation and phosphorylation of STAT2 by MAPK12 during viral infection deserve further investigation.
In summary, our results revealed an important role of STAT2 phosphorylation at the early stage of viral infection. Moreover, we demonstrated that the initial phosphorylation of STAT2 is dependent on RIG-I/MAVS/MAPK12 pathway. These findings proposed a novel mechanism by which STAT2 repressed viral infection, and provide new insights into interactions between viruses and host innate immunity.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
SSL, J-LC, and XL conceived and designed the experiment. XL, SYL, and WZ performed the experiments. KRR performed data analysis. SW and XC processed and typeset the figures. XL, SSL, J-LC, and GG contributed to the writing of the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by National Key Research and Development Program of China (2021YFD1800205) and National Natural Science Foundation of China (U1805231, 32030110 and 32102688).
We thank members of J-LC’s lab in College of Animal Sciences, Fujian Agriculture and Forestry University for technical assistance and useful discussions.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.960544/full#supplementary-material
1. Krammer F, Smith GJD, Fouchier RAM, Peiris M, Kedzierska K, Doherty PC, et al. Initial activation of STAT2 induced by IAV infection is critical for innate antiviral immunity. Influenza Nat Rev Dis Primers (2018) 4(1):3. doi: 10.1038/s41572-018-0002-y
2. Shin DL, Yang W, Peng JY, Sawatsky B, Von Messling V, Herrler G, et al. Avian influenza a virus infects swine airway epithelial cells without prior adaptation. Viruses (2020) 12(6):589. doi: 10.3390/v12060589
3. Park AIwasaki A. Type I and type III interferons - induction, signaling, evasion, and application to combat COVID-19. Cell Host Microbe (2020) 27(6):870–8. doi: 10.1016/j.chom.2020.05.008
4. Goubau D, Schlee M, Deddouche S, Pruijssers AJ, Zillinger T, Goldeck M, et al. Antiviral immunity via RIG-i-mediated recognition of RNA bearing 5’-diphosphates. Nature (2014) 514(7522):372–5. doi: 10.1038/nature13590
5. Acharya D, Liu GGack MU. Dysregulation of type I interferon responses in COVID-19. Nat Rev Immuno (2020) 20(7):397–8. doi: 10.1038/s41577-020-0346-x
6. Li G, Fan Y, Lai Y, Han T, Li Z, Zhou P, et al. Coronavirus infections and immune responses. J Med Virol (2020) 92(4):424–32. doi: 10.1002/jmv.25685
7. Iwasaki A, Foxman E FMolony RD. Early local immune defences in the respiratory tract. Nat Rev Immunol (2017) 17(1):7–20. doi: 10.1038/nri.2016.117
8. Liu G, Park HS, Pyo HM, Liu Q, Zhou Y. Influenza a virus panhandle structure is directly involved in RIG-I activation and interferon induction. J Virol (2015) 89(11):6067–79. doi: 10.1128/JVI.00232-15
9. Le Goffic R, Pothlichet J, Vitour D, Fujita T, Meurs E, Chignard M, et al. Cutting edge: Influenza a virus activates TLR3-dependent inflammatory and RIG-i-dependent antiviral responses in human lung epithelial cells. J Immunol (2007) 178(6):3368–72. doi: 10.4049/jimmunol.178.6.3368
10. Diebold SS, Kaisho T, Hemmi H, Akira SReis E Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science (2004) 303(5663):1529–31. doi: 10.1126/science.1093616
11. Lee N, Wong CK, Hui DS, Lee SK, Wong RY, Ngai KL, et al. Role of human toll-like receptors in naturally occurring influenza a infections. Influenza Other Respir Viruses (2013) 7(5):666–75. doi: 10.1111/irv.12109
12. Yu Q, Qu KModis Y. Cryo-EM structures of MDA5-dsRNA filaments at different stages of ATP hydrolysis. Mol Cell (2018) 72(6):999–1012.e6. doi: 10.1016/j.molcel.2018.10.012
13. Ludwig S, Hrincius E RBoergeling Y. The two sides of the same coin-influenza virus and intracellular signal transduction. Cold Spring Harb Perspect (2021) Med 11(1):a038513. doi: 10.1101/cshperspect.a038513
14. Stark GR, Darnell JE Jr. The JAK-STAT pathway at twenty. Immunity (2012) 36(4):503–14. doi: 10.1016/j.immuni.2012.03.013
15. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol (2014) 14(1):36–49. doi: 10.1038/nri3581
16. Lazear HM, Schoggins JW, Diamond MS. Shared and distinct functions of type I and type III interferons. Immunity (2019) 50(4):907–23. doi: 10.1016/j.immuni.2019.03.025
17. Lee CJ, An HJ, Cho ES, Kang HC, Lee JY, Lee HS, et al. Stat2 stability regulation: an intersection between immunity and carcinogenesis. Exp Mol Med (2020) 52(9):1526–36. doi: 10.1038/s12276-020-00506-6
18. Rengachari S, Groiss S, Devos JM, Caron E, Grandvaux NPanne D. Structural basis of STAT2 recognition by IRF9 reveals molecular insights into ISGF3 function. Proc Natl Acad Sci U.S.A. (2018) 115(4):e601–9. doi: 10.1073/pnas.1718426115
19. Macmicking JD. Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat Rev Immuno (2012) 12(5):367–82. doi: 10.1038/nri3210
20. Saka H AValdivia R. Emerging roles for lipid droplets in immunity and host-pathogen interactions. Annu Rev Cell Dev Biol (2012) 28:411–37. doi: 10.1146/annurev-cellbio-092910-153958
21. Blaszczyk K, Nowicka H, Kostyrko K, Antonczyk A, Wesoly JBluyssen HA. The unique role of STAT2 in constitutive and IFN-induced transcription and antiviral responses. Cytokine Growth Factor Rev (2016) 29:71–81. doi: 10.1016/j.cytogfr.2016.02.010
22. Ho J, Pelzel C, Begitt A, Mee M, Elsheikha HM, Scott DJ, et al. STAT2 is a pervasive cytokine regulator due to its inhibition of STAT1 in multiple signaling pathways. PloS Biol (2016) 14(10):e2000117. doi: 10.1371/journal.pbio.2000117
23. Chaudhary V, Yuen KS, Chan JF, Chan CP, Wang PH, Cai JP, et al. Selective activation of type II interferon signaling by zika virus NS5 protein. J Virol (2017) 91(14):e00163–17. doi: 10.1128/jvi.00163-17
24. Parisien JP, Lau JF, Rodriguez J, Ulane C MHorvath CM. Selective STAT protein degradation induced by paramyxoviruses requires both STAT1 and STAT2 but is independent of alpha/beta interferon signal transduction. J Virol (2002) 76(9):4190–8. doi: 10.1128/jvi.76.9.4190-4198.2002
25. Rodriguez JJ, Parisien J PHorvath CM. Nipah virus V protein evades alpha and gamma interferons by preventing STAT1 and STAT2 activation and nuclear accumulation. J Virol (2002) 76(22):11476–83. doi: 10.1128/jvi.76.22.11476-11483.2002
26. Rodriguez JJ, Wang L FHorvath CM. Hendra virus V protein inhibits interferon signaling by preventing STAT1 and STAT2 nuclear accumulation. J Virol (2003) 77(21):11842–5. doi: 10.1128/jvi.77.21.11842-11845.2003
27. Alazawi W, Heath H, Waters JA, Woodfin A, O’brien AJ, Scarzello AJ, et al. Stat2 loss leads to cytokine-independent, cell-mediated lethality in LPS-induced sepsis. Proc Natl Acad Sci U.S.A. (2013) 110(21):8656–61. doi: 10.1073/pnas.1221652110
28. Ashley CL, Abendroth A, Mcsharry B PSlobedman B. Interferon-independent upregulation of interferon-stimulated genes during human cytomegalovirus infection is dependent on IRF3 expression. Viruses (2019) 11(3):246. doi: 10.3390/v11030246
29. Ashley CL, Abendroth A, Mcsharry B PSlobedman B. Interferon-independent innate responses to cytomegalovirus. Front Immunol (2019) 10:2751. doi: 10.3389/fimmu.2019.02751
30. Stirnweiss A, Ksienzyk A, Klages K, Rand U, Grashoff M, Hauser H, et al. IFN regulatory factor-1 bypasses IFN-mediated antiviral effects through viperin gene induction. J Immunol (2010) 184(9):5179–85. doi: 10.4049/jimmunol.0902264
31. Dixit E, Boulant S, Zhang Y, Lee AS, Odendall C, Shum B, et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell (2010) 141(4):668–81. doi: 10.1016/j.cell.2010.04.018
32. Wu L, Qin Y, Xia S, Dai M, Han X, Wu Y, et al. Identification of cyclin-dependent kinase 1 as a novel regulator of type I interferon signaling in systemic lupus erythematosus. Arthritis Rheumatol (2016) 68(5):1222–32. doi: 10.1002/art.39543
33. Wei H, Wang S, Chen Q, Chen Y, Chi X, Zhang L, et al. Suppression of interferon lambda signaling by SOCS-1 results in their excessive production during influenza virus infection. PloS Pathog (2014) 10(1):e1003845. doi: 10.1371/journal.ppat.1003845
34. Liu S, Liao Y, Chen B, Chen Y, Yu Z, Wei H, et al. Critical role of syk-dependent STAT1 activation in innate antiviral immunity. Cell Rep (2021) 34(3):108627. doi: 10.1016/j.celrep.2020.108627
35. Li F, Chen Y, Zhang Z, Ouyang J, Wang Y, Yan R, et al. Robust expression of vault RNAs induced by influenza a virus plays a critical role in suppression of PKR-mediated innate immunity. Nucleic Acids Res (2015) 43(21):10321–37. doi: 10.1093/nar/gkv1078
36. Xiao M, Chen Y, Wang S, Liu S, Rai KR, Chen B, et al. Long noncoding RNA IFITM4P regulates host antiviral responses by acting as a competing endogenous RNA. J Virol (2021) 95(21):e0027721. doi: 10.1128/jvi.00277-21
37. Ma Y, Guo G, Li T, Wen F, Yang J, Chen B, et al. A novel imatinib-upregulated long noncoding RNA plays a critical role in inhibition of tumor growth induced by abl oncogenes. Mol Cancer (2022) 21(1):5. doi: 10.1186/s12943-021-01478-5
38. Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol (2014) 32:513–45. doi: 10.1146/annurev-immunol-032713-120231
39. Shemesh M, Lochte S, Piehler J, Schreiber G. IFNAR1 and IFNAR2 play distinct roles in initiating type I interferon-induced JAK-STAT signaling and activating STATs. Sci Signal (2021) 14(710):eabe4627. doi: 10.1126/scisignal.abe4627
40. Shuai K, Stark GR, Kerr IM, Darnell JE Jr. A single phosphotyrosine residue of Stat91 required for gene activation by interferon-gamma. Science (1993) 261(5129):1744–6. doi: 10.1126/science.7690989
41. Qureshi SA, Leung S, Kerr IM, Stark GR, Darnell JE Jr. Function of Stat2 protein in transcriptional activation by alpha interferon. Mol Cell Biol (1996) 16(1):288–93. doi: 10.1128/MCB.16.1.288
42. Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature (2006) 441(7089):101–5. doi: 10.1038/nature04734
43. Malik G, Zhou Y. Innate immune sensing of influenza a virus. Viruses (2020) 12(7):755. doi: 10.3390/v12070755
44. Li Y, Liu S, Chen Y, Chen B, Xiao M, Yang B, et al. Syk facilitates influenza a virus replication by restraining innate immunity at the late stage of viral infection. J Virol (2022) 96(7):e0020022. doi: 10.1128/jvi.00200-22
45. Jiang R, Ye J, Zhu B, Song Y, Chen H, Cao S. Roles of TLR3 and RIG-I in mediating the inflammatory response in mouse microglia following Japanese encephalitis virus infection. J Immunol Res 2014. (2014) p:787023. doi: 10.1155/2014/787023
46. Mikkelsen SS, Jensen SB, Chiliveru S, Melchjorsen J, Julkunen I, Gaestel M, et al. RIG-i-mediated activation of p38 MAPK is essential for viral induction of interferon and activation of dendritic cells: Dependence on TRAF2 and TAK1. J Biol Chem (2009) 284(16):10774–82. doi: 10.1074/jbc.M807272200
47. Schindler C, Levy DE, Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem (2007) 282(28):20059–63. doi: 10.1074/jbc.R700016200
48. Park C, Li S, Cha ESchindler C. Immune response in Stat2 knockout mice. Immunity (2000) 13(6):795–804. doi: 10.1016/s1074-7613(00)00077-7
49. Müller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, et al. Functional role of type I and type II interferons in antiviral defense. Science (1994) 264(5167):1918–21. doi: 10.1126/science.8009221
50. Meraz MA, White JM, Sheehan KC, Bach EA, Rodig SJ, Dighe AS, et al. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell (1996) 84(3):431–42. doi: 10.1016/s0092-8674(00)81288-x
51. Gopal R, Lee B, Mchugh KJ, Rich HE, Ramanan K, Mandalapu S, et al. STAT2 signaling regulates macrophage phenotype during influenza and bacterial super-infection. Front Immunol (2018) 9:2151. doi: 10.3389/fimmu.2018.02151
52. Boudewijns R, Thibaut HJ, Kaptein SJF, Li R, Vergote V, Seldeslachts L, et al. STAT2 signaling restricts viral dissemination but drives severe pneumonia in SARS-CoV-2 infected hamsters. Nat Commun (2020) 11(1):5838. doi: 10.1038/s41467-020-19684-y
53. Nan J, Wang Y, Yang J, Stark GR. IRF9 and unphosphorylated STAT2 cooperate with NF-κB to drive IL6 expression. Proc Natl Acad Sci U.S.A. (2018) 115(15):3906–11. doi: 10.1073/pnas.1714102115
54. Arimoto KI, Löchte S, Stoner SA, Burkart C, Zhang Y, Miyauchi S, et al. STAT2 is an essential adaptor in USP18-mediated suppression of type I interferon signaling. Nat Struct Mol Biol (2017) 24(3):279–89. doi: 10.1038/nsmb.3378
55. Schmid S, Mordstein M, Kochs G, García-Sastre A, Tenoever BR. Transcription factor redundancy ensures induction of the antiviral state. J Biol Chem (2010) 285(53):42013–22. doi: 10.1074/jbc.M110.165936
56. Thoresen D, Wang W, Galls D, Guo R, Xu L, Pyle AM. The molecular mechanism of RIG-I activation and signaling. Immunol Rev (2021) 304(1):154–68. doi: 10.1111/imr.13022
57. Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell (2005) 19(6):727–40. doi: 10.1016/j.molcel.2005.08.014
58. Ren Z, Ding T, Zuo Z, Xu Z, Deng J, Wei Z. Regulation of MAVS expression and signaling function in the antiviral innate immune response. Front Immunol (2020) 11:1030. doi: 10.3389/fimmu.2020.01030
59. Liu S, Chen J, Cai X, Wu J, Chen X, Wu YT, et al. MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. Elife (2013) 2:e00785. doi: 10.7554/eLife.00785
60. Ittner A, Block H, Reichel CA, Varjosalo M, Gehart H, Sumara G, et al. Regulation of PTEN activity by p38δ-PKD1 signaling in neutrophils confers inflammatory responses in the lung. J Exp Med (2012) 209(12):2229–46. doi: 10.1084/jem.20120677
61. González-Terán B, Cortés JR, Manieri E, Matesanz N, Verdugo, Rodríguez ME, et al. Eukaryotic elongation factor 2 controls TNF-α translation in LPS-induced hepatitis. J Clin Invest (2013) 123(1):164–78. doi: 10.1172/jci65124
62. Criado G, Risco A, Alsina-Beauchamp D, Pérez-Lorenzo MJ, Escós ACuenda A. Alternative p38 MAPKs are essential for collagen-induced arthritis. Arthritis Rheumatol (2014) 66(5):1208–17. doi: 10.1002/art.38327
63. Risco A, Del Fresno C, Mambol A, Alsina-Beauchamp D, Mackenzie KF, Yang HT, et al. p38γ and p38δ kinases regulate the toll-like receptor 4 (TLR4)-induced cytokine production by controlling ERK1/2 protein kinase pathway activation. Proc Natl Acad Sci U.S.A. (2012) 109(28):11200–5. doi: 10.1073/pnas.1207290109
Keywords: STAT2, influenza virus, kinase, MAPK12, innate immunity
Citation: Li X, Liu S, Rai KR, Zhou W, Wang S, Chi X, Guo G, Chen J-L and Liu S (2022) Initial activation of STAT2 induced by IAV infection is critical for innate antiviral immunity. Front. Immunol. 13:960544. doi: 10.3389/fimmu.2022.960544
Received: 03 June 2022; Accepted: 17 August 2022;
Published: 05 September 2022.
Edited by:
Aitor Nogales, Centro de Investigación en Sanidad Animal (CISA), SpainReviewed by:
Hiroyuki Oshiumi, Kumamoto University, JapanCopyright © 2022 Li, Liu, Rai, Zhou, Wang, Chi, Guo, Chen and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ji-Long Chen, chenjl@im.ac.cn; Shasha Liu, liushasha402@163.com
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.