 Qingzhou Meng
Qingzhou Meng Haixiang Sun
Haixiang Sun Jianghuai Liu
Jianghuai Liu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 26 August 2022
Sec. Primary Immunodeficiencies
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.960348
This article is part of the Research Topic Advances in Therapeutic Strategies of Inborn Errors of Immunity View all 9 articles
Rapid advances in high throughput sequencing have substantially expedited the identification and diagnosis of inborn errors of immunity (IEI). Correction of faulty genes in the hematopoietic stem cells can potentially provide cures for the majority of these monogenic immune disorders. Given the clinical efficacies of vector-based gene therapies already established for certain groups of IEI, the recently emerged genome editing technologies promise to bring safer and more versatile treatment options. Here, we review the latest development in genome editing technologies, focusing on the state-of-the-art tools with improved precision and safety profiles. We subsequently summarize the recent preclinical applications of genome editing tools in IEI models, and discuss the major challenges and future perspectives of such treatment modalities. Continued explorations of precise genome editing for IEI treatment shall move us closer toward curing these unfortunate rare diseases.
Recent developments in sequencing technologies have significantly accelerated the identification and determination of human monogenic immune defects. It has become evident that the collective prevalence of such genetic diseases is much higher than previously considered (at a scale of 1/1000 to 1/5000 births) (1). Traditionally known as “primary immunodeficiencies”, these defects are now commonly designated as inborn errors of immunity (IEI), a term better reflecting the presentation of a broad spectrum of immune-related phenotypes. Indeed, IEI patients can feature not only susceptibility to infections, but also autoimmunity/autoinflammation, allergy and malignancy (1–3). Additionally, even patients with the same mutations may manifest heterogeneous phenotypes. Active investigations of this expanding catalog of disorders have provided invaluable genetic and mechanistic insights on the development, activation and regulation of the human immune system, and its interactions with pathogens.
Such efforts have also brought advances to the treatment outlook for IEI. The Increased understandings to the molecular underpinning of various forms of IEIs have established the basis for some targeted therapeutic approaches, by which the defective components and pathways in immunity may be supplemented or pharmacologically targeted (4, 5). On the other hand, since a majority of IEI affecting the immune cells, hematopoietic stem cell transplantation (HSCT)-based therapies are considered to be curative for IEI (4, 6, 7). Nonetheless, allogeneic HSCT has long been limited by the need for finding the donors with optimal HLA genotypes, and by the potential risks of graft-versus-host-disease (GVHD) with severe consequence. Alternatively, the use of gene-corrected autologous hematopoietic stem cells (HSCs) removes such immunological barriers (7, 8). Indeed, viral vector-based gene therapies aimed at correcting the faulty genes in the autologous HSC compartment have made steady progress over the last 25 years (9). Particularly, the recent revolution in genome editing technologies has provided more advanced tools toward the promise of achieving precise and long-lasting management of IEIs (9, 10).
Genome editing refers to the technologies that enable programmed genetic modifications at specific locations in the genome (11). Fundamentally, these technologies depend on the ingenious design of different forms of programmable, sequence-specific nucleases (Figure 1). Targeted cleavage on the genome would drive DNA repair at the locus, which may subsequently lead to intended genetic changes (12). The first two major systems are the Zinc-finger nucleases (ZFN) and Transcription activator-like effector nucleases (TALEN) (13, 14). Therein, the Fokl nuclease is respectively fused to the assembled arrays of modular Zinc-fingers and Transcription activator-like motifs, which in turn mediate specific sequence recognition. Not long after the development of ZFN and TALEN, a third system originated from the bacterial defense mechanism exploded into the scene. This system is referred to as the Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-associated protein (CRISPR/Cas), corresponding to the nomenclature of the related genetic components in the prokaryotes (15). In general, the CRISPR/Cas platform is assembled with the Cas nuclease and a guide RNA component that contains a programmable sequence motif (a spacer of ~ 20-nt) complementary to the specific DNA target. Besides its potent targeting activity, this system also features unprecedented convenience and versatility in applications (16, 17). It is currently adopted as the most popular framework for constructing genome editing tools. Since its introduction in 2012, CRISPR/Cas technology has undergone extraordinary developments. As a result, these revolutionary tools have made strong impacts in all fields of life sciences.
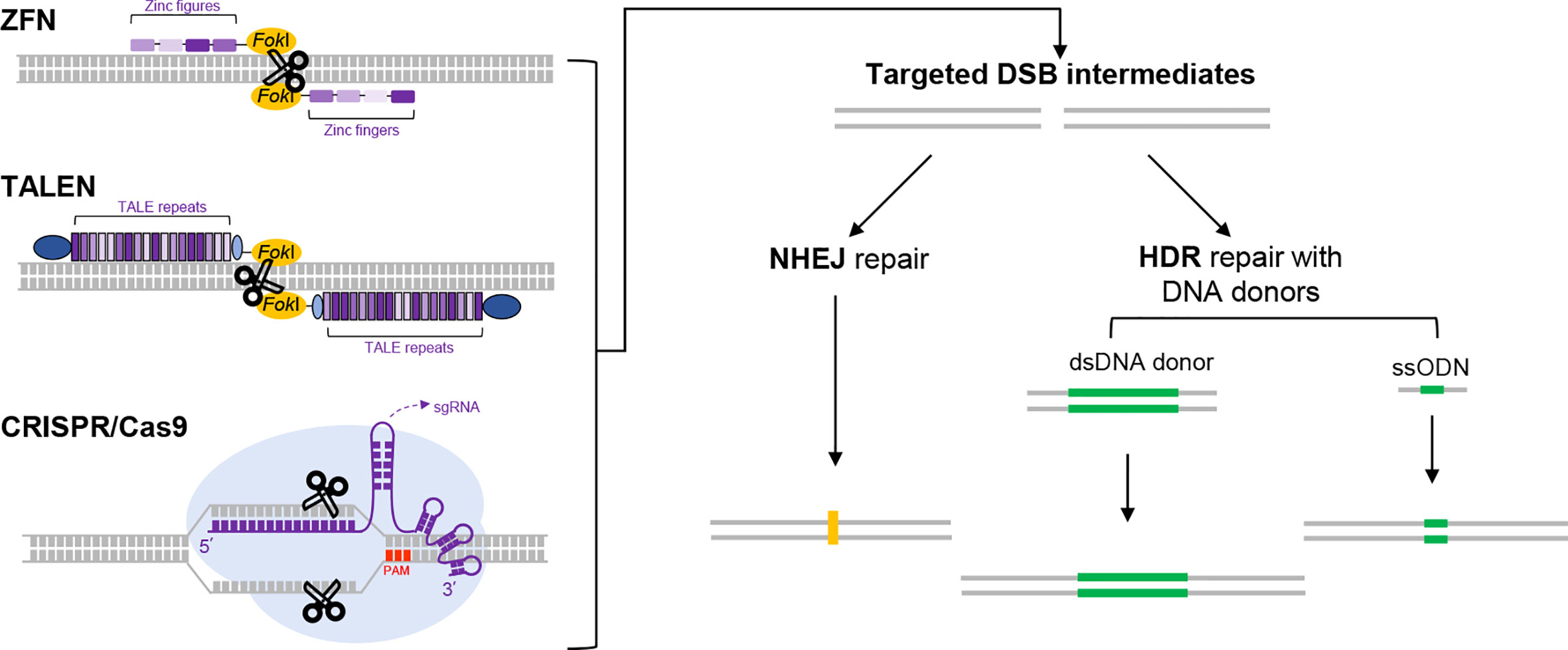
Figure 1 The use of programmable nucleases for genome editing. Three major programmable nucleases, i.e., ZFNs, TALENs and CRISPR/Cas9 are schematically illustrated on the left part. ZFNs and TALENs are respectively composed of sequence-specific DNA-binding protein modules linked to a nonspecific DNA cleavage domain (derived from FokI endonuclease). In ZFNs, an array of zinc figures (small blocks in different shades of purple), each specifically recognizing a 3-bp of DNA sequence, are assembled in tandem to program a specific DNA-binding event. In TALENs, variants of TALE repeats correspond to each of the four single bases. Therefore, programmed DNA binding by TALEN is enabled by an assembled array of TALE repeats (dense purple bands). Two other constant domains in TALEN are also depicted in dark and light blue, respectively. For DNA cleavage to occur, the FokI domain requires dimerization. As a result, only two adjacent targeting events by a pair of targeting ZFN or TALEN monomers in correct orientations would trigger double-strand break (DSB) at a specific locus. The mechanistic principle for CRISPR/Cas9 (a prototype for many CRISPR/Cas tools) is different from those for ZFNs and TALENs. It employs a Cas9 protein and an engineered guide RNA. The Cas9 protein features two nuclease domains, each responsible for cleaving one strand of DNA. The engineered guide RNA (in a form of single guide RNA, sgRNA [purple]) directs Cas9 to the DNA target by base-pairing, which results in generation of a targeted DSB. The target sequence for CRISPR/Cas9 also requires an immediately adjacent PAM sequence (shown in red). Regardless of the nuclease tools used, the DSBs generated would stimulate cellular DNA repair mechanisms (right part), in the form of the error-prone nonhomologous end joining (NHEJ) or homology-directed repair (HDR). The former pathway often leads to uncontrolled insertions or deletions of bases (indels, shown in yellow) around the break site, which tends to cause gene disruptions. The latter pathway engages precise repair directed by homologous donor DNA. In practice, when the cells are supplied with a donor DNA template (either as dsDNA or single strand oligodeoxynucleotides [ssODN] as indicated) containing the desired edits (green) and flanking homologous regions, the HDR pathway can lead to precise genome editing outcomes.
The powerful advances in genome editing technology have reshaped the landscape for future treatments of genetic diseases. For IEIs, in particular, considering the clinical feasibility of using genetically corrected autologous HSCs to normalize immunity, it is anticipated that the genome editing-based strategy shall evolve into promising treatment options (9, 10). A brief survey of the ClinVar database (18), with the keyword of “immunodeficiency”, shows that the majority of the associated pathogenic variants belong to the categories of single nucleotide variants (SNV), deletions and/or insertions (Figure 2). Indeed, many of the available genome editing tools have shown the capacity to make (or reverse) these types of genetic changes (11, 19). Here, we will introduce the latest developments in CRISPR/Cas tools for precise genome editing. We will provide an overview for the latest preclinical applications of genome editing for IEI treatments. Furthermore, the challenges and future prospects for translating genome editing technology to the bedside of IEI patients will be discussed. Due to the focused theme of the present work and space limitations, we sincerely apologize to those authors whose valuable contributions to the field are not included in this review.
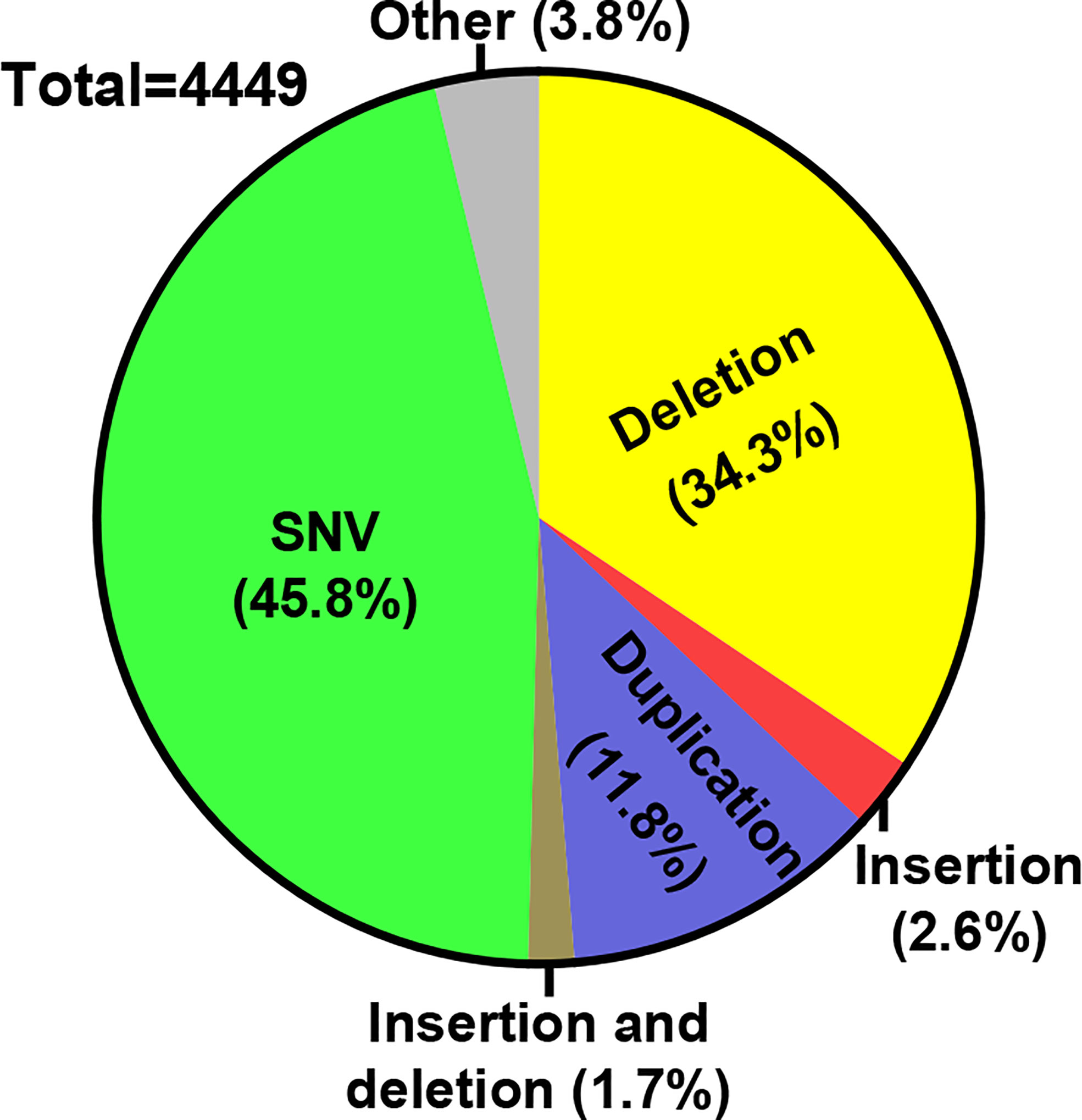
Figure 2 Distribution of IEI-associated variants. A survey of ClinVar database was conducted (June, 2022) for the distribution of the pathogenic variants associated with a keyword of “immunodeficiency”. The numbers of different variant types belonging to each of the indicated categories were filtered. Subsequently, the percentages for each category of variants within all different variants are marked on the pie graph.
The list of different forms of IEIs has greatly expanded over the last decade, thanks to the revolution in sequencing technologies. Based on the recent classification by International Union of Immunological Societies in 2019, IEI disorders encompass 430 distinct defects (1). According to their overall phenotypic features, these disorders can be divided into 10 broad categories: “combined immunodeficiencies”, “combined immunodeficiencies with syndromic features”, “predominantly antibody deficiencies”, “diseases of immune dysregulation”, “congenital defects of phagocytes”, “defects in intrinsic and innate immunity”, “autoinflammatory diseases”, “complement deficiencies”, “bone marrow failure”, and “phenocopies of IEI”. It is also clear from the accumulated knowledge regarding these genetic diseases, certain gene defects (or given alleles) may drive a spectrum of phenotypes, whereas similar phenotypes may also be attributed to defects in different genes (1, 3). Additionally, some of the defects would exhibit incomplete penetrance. The significantly improved understandings toward the genetic basis of IEI have not only opened up many options of targeted therapies, but also highlighted the potential of developing curative genetic therapies.
The majority of IEI defects primarily affect the immune cell compartments originated from HSCs (5, 7). Indeed, HSCT treatments have shown successes against the most severe forms of IEI that are refractory to other medical therapies, including severe combined immunodeficiencies (SCID), combined immunodeficiencies, chronic granulomatous diseases (CGD), Wiskott-Aldrich syndrome (WAS), and bone marrow failures (6, 7, 9). However, allogenic HSCT is limited by the difficulties of finding the optimal HLA-matched donors. When alternative donor strategies are used, the managements against conditioning morbidities, graft rejection and later GVHD present substantial challenges (7). On the other hand, the transplant of autologous, genetically engineered hematopoietic stem and progenitor cells (HSPCs) would avoid such immunological barriers. In principle, the defective HSPCs from the patients may be stably transduced ex vivo via a viral vector that produce the functional gene product, and subsequently are engrafted back to reconstitute (at least partially) the hematopoietic system. Over the past several decades, marked progresses have been made on the development of ex vivo manipulation of HSCs via viral gene vectors, and the subsequent autologous HSCT in clinical settings (8, 9, 20). Currently, the use of self-inactivating lentiviral vectors has shown good promise as a safe and effective platform for HSC gene therapy, as demonstrated in clinical trials for X-linked SCID, adenosine deaminase (ADA)-deficient SCID, Fanconi anemia (FA), CGD and WAS (21–25). Moreover, similar gene therapy approaches for other IEIs including RAG1-deficient SCID and Artemis -deficient SCID have also entered clinical stages (see NCT04797260 and NCT03538899), based on favorable outcomes from preclinical studies (26, 27). However, when implementing the current generation of HSC gene therapy tools, the permanent, semi-random integration of lentiviral vectors in the genome may still pose long-term risks. For genes whose expressions requires cell type- and/or activation status-dependent regulation (such as CD40L, mutated in X-linked hyper-IgM syndrome), the viral vector-dependent, non-physiological transgene expression can cause safety concerns (28), or conversely, may lack the required robustness upon activation (29). In addition, such “gene addition” approach is not suitable for correcting gain-of-function mutations, and may not be effective in rescuing certain dominant-negative defects (9). In this regard, precise mutation-targeted, “gene correction” methods are believed to represent the future tools-of-choice for gene-based therapy of IEIs.
Effective modification of the genomes in animal cells had been a challenging task until recently. The emergence of genome editing technologies in the late 2000s and early 2010s indeed marked the beginning of a new era (16, 17). Empowered by these ground-breaking technologies, making genetic changes in a cell has become quite accessible. The tremendous development of the genome editing toolbox over the last decade has also brought the hope that the ultimate cures for many genetic diseases, through somatic genome editing, are on the horizon.
The first generation of genome editing technologies are based on the engineered nucleases that can program a double stranded DNA break (DSB) at a site(s) of interest. The popular classes of programmable nucleases include ZFNs (30), TALENs (31, 32) and CRISPR/Cas9 (15, 33, 34), which emerged successively. Among these, the CRISPR-Cas9 has become the most widely used platform owing to its potent activity, unprecedented convenience and versatility. Originally identified as a bacterial adaptive defense system against phages, the CRISPR/Cas9 can specifically cleavage foreign DNA sequences based on the targeting information contained in a guide RNA moiety (formed with a tracrRNA and a crRNA). In the commonly used platform for genome editing, the Streptococcus pyogenes Cas9 protein (SpCas9) is directed by an engineered guide RNA (single guide RNA, sgRNA) to make DSB cut at the target DNA sequence (See Figure 1) (16, 17). Each sgRNA is simply customized by a stretch of a 20-nt spacer sequence that aligns with a target DNA sequence. One restraint for the target DNA sequence is its immediate adjacency to a downstream protospacer adjacent motif (PAM). For SpCas9, the classical PAM sequence is 5′-NGG-3′, which is found very frequently in a genome space. Other natural CRISPR/Cas systems (e.g., SaCas9 or Cas12a) operating in similar or slightly deviant manners have also been later harnessed for genome editing (35). Importantly, with their recognition of corresponding PAM motifs, these different tools further expand the targeting scope for CRISPR/Cas-based genome editing.
The nuclease-generated DSBs activate the cellular repair pathways (see Figure 1), which can result in non-homologous end-joining (NHEJ) (13, 33, 34, 36) or the template-dependent, homology-directed repair (HDR) (12, 33, 34, 37, 38). The first pathway operates actively in most cell types, which often drives formation of indels at the targeted sites. These indels are sometimes sufficient to cause loss-of-function in genes or regulatory elements (39–41). The use of paired sgRNAs can lead to more definitive knockouts (42). In the presence of a co-introduced homologous template, the cells may also choose the second repair option, which can lead to precise installation of designed sequence changes (33, 34, 38). Nevertheless, establishment of such HDR-dependent gene knock-in often requires screening or selection, owing to the relatively lower efficiency of HDR repair (43). Further technical advancements to enhance the preference of repair via HDR vs NHEJ represents an intensive area of research (19).
A major safety concern for genome editing is the off-target effects by the programmed nucleases. For instance, Cas9 may cause cleavage at certain off-target sites bearing high similarity to the sequence of sgRNA spacer (44). Studies have shown that minor mismatch, especially at the PAM-distal positions, can sometimes be tolerated (44, 45). In general, limiting the amount of the Cas9/sgRNA complex and/or preventing its prolonged presence can effectively reduce off-target cleavage (42). Other developments include the engineering of high-fidelity Cas9 (46–49), and the use of modified sgRNA architectures (50, 51) to lower the chances of Cas9 cleavage at imperfectly matched sites. Since the efficiency and accuracy for a given CRISPR/Cas9 platform tend to inversely correlate (52), to further seek strategies for reducing off-target cleavage while maintaining the on-target genome editing efficiencies remains an important goal.
Although Cas9 cleavage-dependent genome editing approaches have been widely applied in cells and model organisms, the generation of DSB intermediates poses concerns for applications that require greater levels of safety, such as therapeutic genome editing. Indeed, studies have reported that Cas9-generated DSBs may cause certain levels of large deletions, genomic rearrangements, and even potentially chromothripsis in different cell types including human HSCs (53, 54). The potential of off-target DSBs exacerbates such risks. Moreover, although the introduction of a repair template can lead to engagement of the HDR pathway, the co-existing NHEJ pathway often complicate the repair outcomes (11). To address these issues, a number of innovative, precise genome editing platforms that avoid the requirement of DSB formation have been established.
The emergence of base editors (BE) underscores the power of harnessing DNA base-modification activities for precise genetic manipulation. The common base editors can be divided into cytosine- or adenine-base editors (CBE and ABE), which respectively induces C-to-T and A-to-G base transitions in the target sequences (55–57). These base editors also mostly adopt the CRISPR/Cas9 framework for target binding, by the use of Cas9 form deficient in dsDNA cleavage activity (Figure 3). In principle, CBE is constructed by fusing the mutant Cas9 first with a cytidine deaminase enzyme, which recognizes the Cs within the mutant Cas9/sgRNA-exposed stretch of single-stranded DNA (ssDNA) as substrates (55). This subsequently leads to conversion of cytidine to uridine, the latter serving as a temporary surrogate for thymidine in base pairing properties. Furthermore, to inhibit the base excision repair for uracil removal, the CBE is also engineered to include an additional uracil glycosylase inhibitor domain. Thirdly, the deliberate use of a Cas9 nickase (nCas9 [D10A]), capable of nicking the unmodified single DNA strand, can direct the cellular repair pathways to “re-write” the sequence near the nick. This would eventually lead to a permanent change of C·G into a T·A base pair (Figure 3).
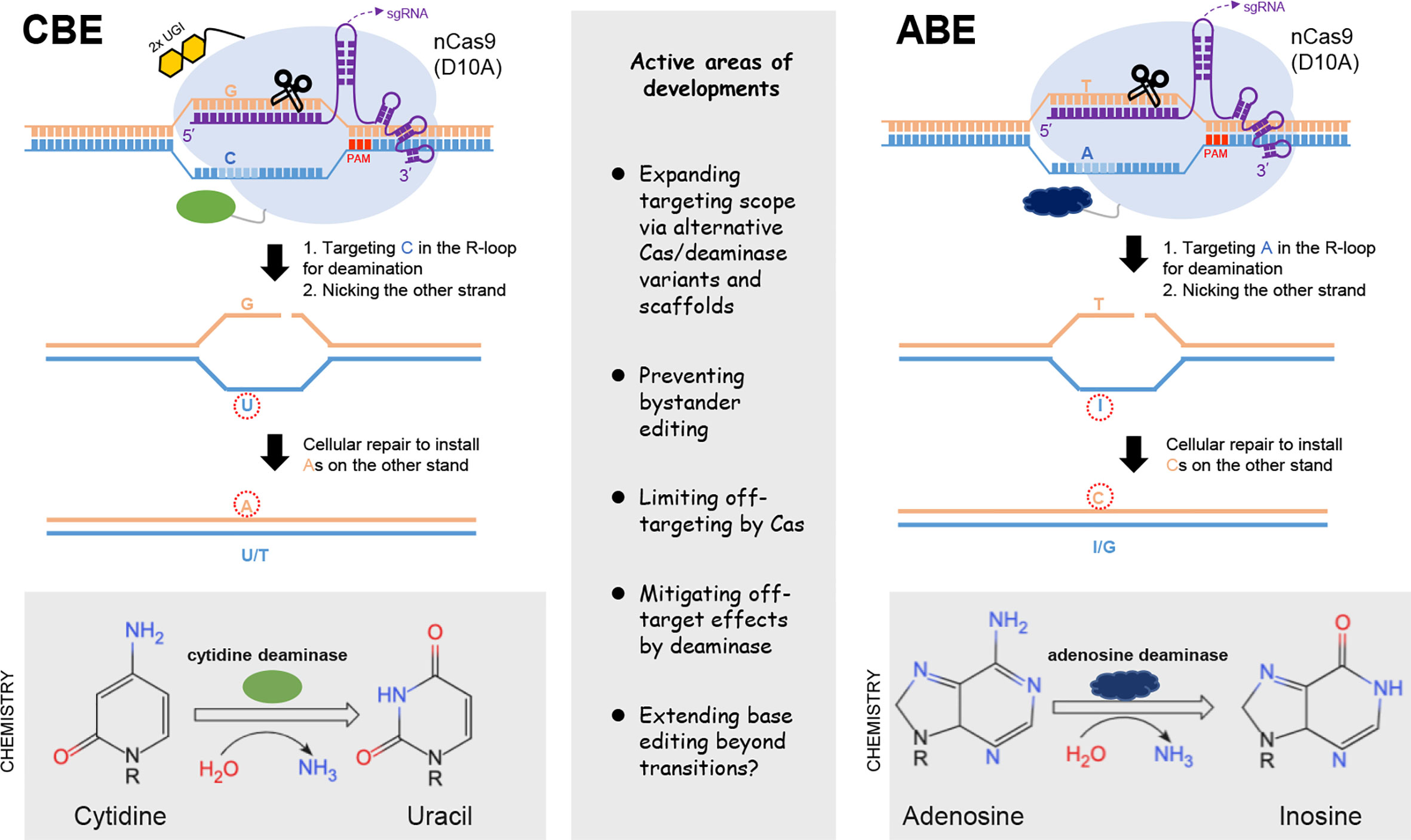
Figure 3 Base editing tools. The two major classes of base editors are illustrated on the left and right parts, respectively. CBE is constructed by fusing the Cas9 nickase (nCas9) with a cytidine deaminase enzyme, which targets the Cs within the exposed R-loop for conversion into U, the latter serving as a temporary surrogate for thymidine in base pairing properties. The uracil glycosylase inhibitor domains (2xUGI, in yellow) engineered to prevent base excision of the U are also shown. The activity window of the initially developed CBE often covers positions 4-8 (schematically indicated with light blue). The adoption of nCas9 in CBE is by design. Its activity would nick the unedited strand, thereby signaling the cellular repair to install the corresponding As on the complementary strand. The design of ABE follows similar principles, except for the employment of an evolved, DNA-targeting adenosine deaminase. Deamination of adenosine produces inosine, which serves as a temporary surrogate for guanosine. Note that no inhibitory domains against base excision of I are required for optimal ABE activities. Some key considerations for the current developments of base editors are summarized in the middle part.
The blueprint for the initial establishment of an ABE is similar as above, except for the employment of a specific deaminase activity to first convert an A to an inosine, the latter serving as a temporary surrogate for G. Since there is no natural enzyme that catalyze this reaction on a DNA strand, a directed evolution approach was applied toward an E. Coli tRNA adenosine deaminase (ecTadA). This led to the establishment of an engineered TadA that can catalyze adenosine deamination in the target sequence when fused to the Cas9 moiety (nCas9) (56). Interestingly, no requirement for an additional domain to inhibit base excision at inosine is required for activity enhancement of the ABE. Similar as above, the use of nCas9 in ABE would also shift the strand preference for the repair pathway to promote the eventual change of an A·T into a G·C pair (Figure 3). Collectively, the abilities the cytosine and adenine BE to mediate targeted base transitions have strong practical implications. Indeed, more than half of currently cataloged pathogenic SNP in the human genomes are base transitions in reference to the WT alleles (57). It is also important to note that, the initially described CBE and ABE tools already showed potent editing activities. Given their proper complementation to the DSB-dependent editing tools, the BEs platforms have undergone active development since their introductions.
The first nCas9-based CBE and ABE respectively showed activities against Cs and As situated in a particular window (approximately at position 4-8, when the corresponding PAM motif is counted as position 21-23) of the target sequence (57). For more flexibility with the target scope, many other base editors have been developed based on different Cas variants (19, 58–60). These BEs show different PAM requirements, and feature their respective editing windows. As more Cas domains with less restricted PAMs are becoming available (61), increasing proportions of C and A base positions (and by extension their complementary Gs and Ts) in the genome become targetable. It is estimated that, by the use of different Cas variants, the expanded targeting scope of the BEs are sufficient to cover most pathological base transitions cataloged in the ClinVar database (18, 19). Moreover, the choice of different natural/evolved deaminase domains (62–64) and other modifications on BE architectures (65, 66) can influence not only the BEs’ editing window features, but also their substrate context preferences (55, 67). Practically, while BEs with wider editing windows are more likely to cover a particular base target, they are linked with a higher risk of generating “bystander” mutations at unintended Cs or As within the targeting window. When the bystander base changes are detrimental and need to be avoided, BEs with narrower editing windows or requiring a more restricted sequence context may be employed (58, 68).
Off-target effects by BEs can be attributed to Cas9-dependent and Cas9-independent mechanisms. The base editors are recruited by the sgRNA to imperfectly aligned sites, potentially leading to deamination of Cs or As within the activity window. Since a subsequent nCas9-dependent strand nicking would significantly facilitate productive installation of base transitions at such unintended sites, the various strategies to mitigate Cas9’s off-target cleavage activities would also be effective in improving BE specificities (68–70). On the other hand, the Cas9-independent off-target base editing is believed to mainly result from the action of the deaminases on the transiently formed single-stranded DNA in the cells (71, 72). Such undesired effects can be substantially reduced by the adoption of certain mutant deaminase domains or by changes in BE architecture to install a stricter requirement of Cas9-mediated target binding for the deaminase actions (71, 72). In addition, the variously observed activities by BEs to edit cellular RNAs can also be mitigated by the use of further optimized deaminase domains (73, 74).
It needs to be noted that the current BE strategies are apparently not suited for making precise base transversions (a purine to a pyrimidine, or vice versa). Additionally, there is often a great need for installing other types of genetic modifications in research models and/or for translational applications. The prime editing (PE) platform are subsequently established to potentially fill such a gap.
The recently emerged PE technology enables installation of various types of point mutations and small insertions/deletions (75), which represents a significant breakthrough in the field (Figure 4A). The basic PE system is composed of a fusion protein of nCas9 (H840A) and a reverse transcriptase (RTase) domain, together with an engineered prime editing guide RNA (pegRNA). The pegRNA consists of a targeting sgRNA scaffold and an extended 3′ region. Particularly, this 3′ region features a primer-binding site (PBS), and an adjacent sequence containing the intended edits. The latter sequence would serve as a template for reverse transcription (RT) and is named RT template. Guided by the pegRNA, the nCas9 (H840A) makes a nick on the sgRNA spacer-displaced ssDNA. By design, the PBS segment in the 3′ portion of the pegRNA forms base pairs with the sequence upstream of the nick. The adjacent RT template can then readily direct reverse transcription by the nCas9-fused RTase, which employs the nick-created 3′-end as the “primer”. Next, due to homology, the newly reverse-transcribed ssDNA may displace the nick-downstream DNA from base-pairing with the unedited strand. Following removal of such 5′-flap structure by cellular nucleases, nick ligation concludes the editing on the first strand. Further DNA repair to resolve the one side-edited heteroduplex can lead to permanent installation of the edits (Figure 4A). In aggregate, this forms an applicable platform named PE2. A subsequent development of the system (i.e., PE3) is by inclusion of a second sgRNA for nicking the unedited strand, to bias the repair pathway toward productive incorporation of edits into this strand (75).
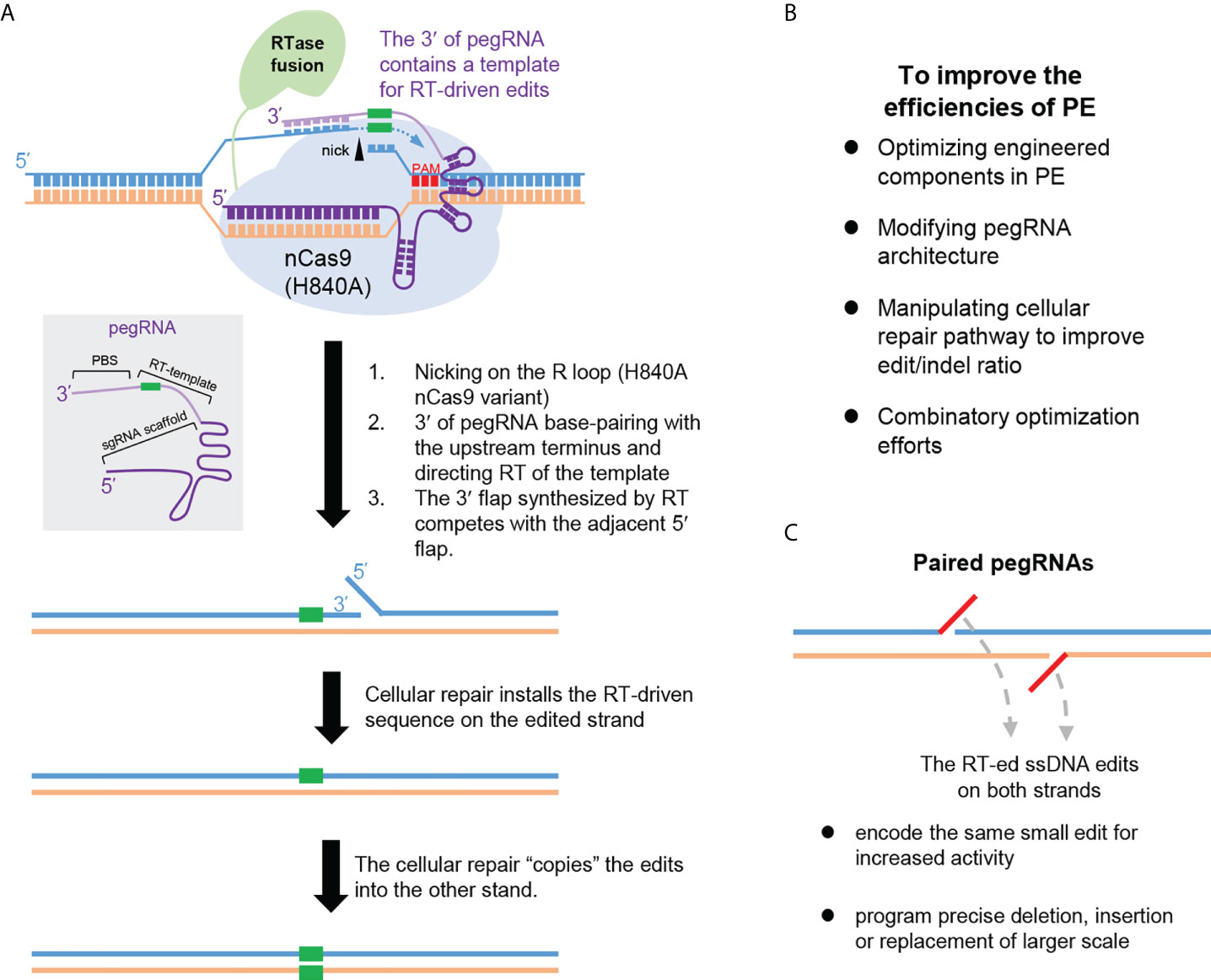
Figure 4 Prime editing. (A) The principle underlying the prime editor (PE) is illustrated. PE employs nCas9 (H840A) fused with a reverse transcriptase (RT) domain. PE is directed by pegRNA whose structure is schematically shown in a grey box. The pegRNA is composed of a targeting sgRNA scaffold and an extended 3′ region. This region can base pair with the nicked R-loop (via its terminal portion named primer binding sequence, PBS), and directs the ensuing reverse transcription (via its edits-containing segment named RT-template). The potential mechanistic stages underlying PE are depicted in order. (B) Major focuses for improving the PE efficiencies are summarized. (C) The advantages of PE with paired pegRNAs are summarized. Note that each pegRNA would enable reverse transcription of a segment of ssDNA (indicated in red). Programming the paired pegRNA sequences would enable increased PE activities, or empower precise editing of larger scales.
Due to the adoption of a templated, reverse transcription-based mechanism for editing, PE can support a broad scope of precise genetic modifications with high purity (avoiding bystander mutations) (75). Importantly, likely owing to the involvement of multiple hybridization events, the PE platform also feature very low genome-wide off-target effects (75–77). Nevertheless, applications of PE in various systems have revealed a general trend of its suboptimal and inconsistent efficiencies. This has stimulated an active line of investigations to enhance PE efficiencies (Figure 4B). Against various potential bottlenecks for PE actions, several optimization strategies have been undertaken. These include the strategies to improve the chromatin accessibility of the targets (78), to optimize the reverse transcriptase domain (79), to modify the pegRNA architectures for increased levels/activities (79–82), and to manipulate DNA repair mechanisms to facilitate productive incorporation of the edits (83, 84). Combination of more than one strategy may enable further enhancement effects (83).
Other major developments in PE include to harness the use of paired pegRNAs (Figure 4C) for either activity enhancement (85, 86), or for inducing precise sequence deletion or replacements (87–89). In the former case, dual pegRNAs encoding the same edits on opposing strands would be used, so that precise editing on both strands may be engaged (85, 86). A similar strategy can be adopted for the induction of precise deletions, via assembling sequences on either side of the deletion as respective RT templates in the paired pegRNAs (88). This would support much longer (in kb-scale) deletions in comparison to the technological limit allowed by the use of a single pegRNA. Furthermore, by the design of complementary new sequences in the RT templates of the paired pegRNAs, precise insertion of a new sequence (in the scale of hundred-bp) in place of the original DNA segment between the nicks can be also achieved (87, 89). For instance, Bxb1 recombinase-recognized attB or attP sites can be precisely installed in the genome. When applied in conjunction with the site-specific recombinase technology, this can drive precise insertion of large DNA fragments (in kb-scale) (89). One other useful PE-related tool is the WT Cas9-based variant of PE (PE-nuclease, PEn) (90–92). Compared to the original PE, the PEn generates a DSB intermediate that significantly promoted the rates of installing RT template-encoded edits. However, the parallelly induced imprecise edits from the error-prone NHEJ represent an apparent limitation that need to be further addressed. Collectively, the high versatility of precise genome editing brought by the PE technology holds great promise for future therapeutic applications.
The advances in genome editing technologies have suggested a promising direction for future treatments of IEIs. Similar to viral vector-based gene therapies, genome editing for correcting the mutant gene may be feasibly applied to the autologous HSPCs ex vivo, followed by transplantation of the treated cells back to the patients (8–10, 93). In this relatively new area of research, current investigations have focused on testing different genome-editing strategies in IEI-related preclinical models.
The mutations of the X-chromosome-located IL2RG gene are responsible for SCID-X1 (6). The resultant deficiencies in the common γ-chain-dependent cytokine signaling cause a complete block in the development of T and NK cells, in conjunction with functional defects in B cells. As substantial development had been made on SCID-X1 gene therapy, IL2RG genome editing in human HSPCs were carried out as proof-of-principle tests. A common correction strategy was adopted by various studies (94). Functional rescues of most pathogenic IL2RG mutations would be achieved by the targeted insertion of a WT IL2RG cDNA to the endogenous IL2RG locus via DSB-induced HDR. It was found that optimization of culture conditions to promote the ex vivo proliferation HSC was important, due to the low HDR activities in the normally slow-cycling HSCs (95). Initially, with the implementation of customized ZFNs (as mRNA) and a repair template in the form of an integrase-defective lentiviral vector, low but detectable levels of HDR-driven correction of IL2RG were achieved. Significant enhancements were later made by the use of an adenovirus-associated virus vector (i.e., AAV6)-based repair template (96, 97). By the use of CRISPR/Cas9 for DSB induction, together with the delivery of an AAV6 template, up to 45% of treated CD34+ HSPCs showed desired corrections (97). The in vivo multi-lineage repopulation by the gene-corrected cells was evidenced in the permissive, recipient mice. Collectively, these works have established an initial framework for the possibility of editing genetically deficient HSPCs for treatment of SCID-X1 and other IEIs.
The mutations affecting each of the 5 subunits of the NADPH oxidase system are responsible for CGD (6). Due to the role of NADPH oxidase in the generation of microbicidal reactive oxygen species in myeloid cells, patients are susceptible to recurrent severe infections, which reduces quality of life and cause mortality (98). The CGD is a challenging condition for gene therapy. A high rate of gene delivery to HSCs and an optimized level of engraftment are required, as the functionally restored myeloid cells do not show selective advantage in vivo (6, 10). Furthermore, the control of specific transgene expression in myeloid cells is desirable to limit ectopic transgene-induced toxicity to cells in other lineages (99).
The mutant CYBB gene (gp91 subunit) contributes to X-linked form of CGD (X-CGD). Previously, a lentiviral vector with a myeloid-specific chimeric promoter has been developed for gene rescue in X-CGD (99, 100). Recently, a clinical trial on such a X-CGD gene therapy platform has shown promising efficacy in 12 months of follow-up (21). Nevertheless, a gene correction strategy at the endogenous locus would more closely mimic physiological expression of the corrected genes, potentially enhancing the treatments’ efficacy/toxicity profiles. An earlier study using ZFN and an AAV6-based template for knocking-in CYBB gene to a genomic “safe harbor”, i.e., the AAVS1 locus, in human HSPCs paved the way for future developing gene-correction at the endogenous locus (101). In later investigations for correcting a point mutation in exon 7, CRISPR/Cas9 was used in conjunction with an ssDNA oligonucleotide repair template to achieve moderate levels of gene correction (102). By further promoting cellular HDR pathway via inhibition of 53BP1 activity, restoration of protein expression was achieved in high percentages (~ 65%) of myeloid cells derived either from in vitro edited HSPC, or from long-termed engrafted human BM cells (103). Such substantial correction rates by genome editing suggests its potential in future treatment of X-CGD. It needs to be noted that as CYBB is affected by a large number of pathogenic mutations (104), the feasibility for developing such in situ correction approaches into potential X-CGD treatment modalities would require future demonstration.
The autosomal forms of CGD include those caused by mutations affecting the p47 subunit of NADPH oxidase (p47-CGD) (105). Interestingly, one specific homozygous 2-nt deletion (ΔGT) in exon 2 the NCF1 genes (encoding p47) represents the predominant defect in p47-CGD, which indeed accounts for ~ 20% of all CGD cases. As two pseudogenes for NCF1, i.e., NCF1A and NCF1C harbor the same deletion, the patient-related ΔGT in NCF1 is likely to be caused by the pseudogene-related gene conversion events (106, 107). By adopting a system of ZFN coupled with an AAV6-based template, some in vitro myeloid progenies of edited HSPCs showed apparent rescue of p47 function. The work further provided evidence that functional p47 rescue may have arisen from the correction of the pseudogenes (108). Further tests for in vivo, long-term correction of p47 function are still warranted. Nevertheless, due to the relative prevalence of this specific ΔGT mutation in p47-CGD, further development of such an in situ correction strategy would have therapeutic implications.
Another group of intensively studied IEI is WAS. The X-linked WAS disease is caused by mutations in the WAS gene (encoding WASp), a regulator of actin polymerization with broad pattern of expression in hematopoietic cells (109). The deficiency of WASp function leads to dysfunctions in all mature hematopoietic cells, except the red blood cells. In accordance, WAS patients suffer from severe platelet defects and complex immunodeficiency (110). Clinical trials have been undertaken to assess lentiviral vector-based gene therapy for WAS (23, 111, 112). To ensure safety, a 1.6-kb endogenous WAS promoter was used in the self-inactivating lentiviral vector to control WAS expression. The results showed sustained clinical benefits for treated patients (23). Nevertheless, although the therapeutic vector largely corrected the dysregulated T lymphocyte compartment, the function of the platelet was only partially restored (23). One likely reason for such incomplete rescue is that the physiological WAS expression is not fully recapitulated under the control of the 1.6-kb promoter. Therefore, a recent study was carried out to insert a WT WAS cDNA to the endogenous WAS locus in human HSPCs via CRISPR/Cas9 and an AAV6-based template (113). Interestingly, this led to more efficient restoration of WASp expression, indicated by the positive percentage (~ 50%) and by per cell level (equivalent to WT level), as compared to the parallelly tested lentiviral transduction system (described above). Moreover, macrophages, platelets and T lymphocytes derived from the edited HSPCs in vitro showed restored functionality. The long-term (up to 26 weeks) multi-lineage repopulating ability (~ 37%) by the edited HSPCs were further established in transplanted mice (113). Overall, this high-performance gene-correction platform for WAS warrants further development, as HDR-mediated in situ cDNA insertion would enable functional restoration of WASp activity affected by many different mutations (> 300).
X-linked hyper-IgM syndrome (XHIM) is caused by mutations in the CD40LG gene encoding CD40 ligand (CD40L) (114). Activation of T cells lead to induced cell-surface expression of CD40L, which in turn acts via interaction with CD40 on the B cells to drive class-switch recombination at the immunoglobulin heavy-chain gene (115). Due to the lack of IgG/A/E antibodies, the XHIM patients are highly susceptible to infection and autoimmunity, and exhibit poor long-term prognosis (116). Lessons from earlier development of CD40L gene therapy models have underscored the unmet need for proper transcriptional control of CD40L, as ectopic expression of this transgene could cause abnormal lymphoproliferation (117). Recently, targeted insertion of a WT CD40LG cDNA to the endogenous locus in human HSPCs has been tested via the use of programmed nucleases and an AAV6-based template. The results showed reasonable rates of gene integration. Normal multilineage differentiation of the edited cells in vitro and in vivo was also demonstrated (118).
Another example of X-linked IEI is the IPEX (Immune dysregulation, polyendocrinopathy, enteropathy, X-linked) (119). This genetic autoimmune condition is caused by the mutations in the FOXP3 gene, which encodes an essential transcription factor for the maintenance and function the regulatory T cells (Treg). The effector T cells (Teff) also transiently up-regulate FOXP3 expression following TCR activation (120). Therefore, deficiency of FOXP3 profoundly impair the function of both Treg and Teff cells, driving severe autoimmune manifestations (121). The tight cell type- and activation status-dependent FOXP3 expression patterns has hampered development of viral vector-based HSPCs gene therapy for IPEX (122). Aimed at enabling physiological correction of FOXP3, a recent study tested the strategy of integrating FOXP3 cDNA to the endogenous locus, via genome editing with CRISPR/Cas9 and an AAV6 template. This led to functional restoration of Treg and Teff cells from IPEX patients. Additionally, FOXP3-edited HSPCs showed long-term engraftments and maintained multi-lineage differentiation potential in the recipient mice (123).
In another combined immunodeficiency disease, the X-linked MAGT1 deficiency with increased susceptibility to Epstein-Barr virus and N-linked glycosylation defect (XMEN), the mutated gene encodes an Mg2+ transporter and a non-catalytic subunit of an oligosaccharyltransferase (124). In addition to Mg2+ abnormalities, the deficient N-glycosylation of certain cellular proteins such as the NKG2D receptor on the CD8 and NK cells cause immune defects and the marked susceptibility to chronic EBV infections (125). Currently, effective therapeutic options for XMEN have not been developed. A recent work attempted to insert a WT MAGT1 cDNA to the endogenous locus in human HSPCs via CRISPR/Cas9 and an AAV6-based template (126). To enhance the efficiency/toxicity profile of precise editing in HSPC, combined inhibition of NHEJ pathway and p53-mediated damage response was applied. This led to efficient correction of MAGT1 in patient-derived HSPCs, and restoration of T and NK cell functions (126). The edited HSPCs also showed long-term engraftment to support multi-lineage development in transplanted mice. In addition, efficient direct correction of patient-derived T cells was demonstrated with the same targeting strategy, which suggested a potential, shorter-term T cell-centered treatment strategy.
FA represents the most common inherited bone marrow failure syndrome, and is caused by mutations in any of the > 20 genes in the FA DNA repair pathway (127). As the hematopoietic compartment is particularly sensitive to deficiency in this pathway, the FA patients show progressive exhaustion of bone marrow reserve early in life (128). Notably, mutations in FANCA gene account for more than 60% of total FA, which defines the subtype of FA-A (129). Lentiviral vector-based gene therapy for FA-A are under clinical investigations (25, 130). WT FANCA gene addition via genome editing has also been reported in patient-derived HSPCs (131). However, the commonly used DSB/HDR genome editing strategy is conceivably suboptimal in this particular context, due to the impaired HDR in cells with deficiencies of the FA pathway (132, 133). On the other hand, a recent study explored an alternative strategy of harnessing NHEJ to rescue the coding frame of the mutant FA gene, by programming a DSB near the original mutation (134). The relatively enhanced NHEJ activities (over HDR) in the FA HSC cells are also conducive to the implementation of this strategy. Importantly, the functionally corrected cells would exhibit selective advantages over the uncorrected or mis-corrected cells. Indeed, the authors showed that despite the imprecision in initial editing outcomes, cells with the therapeutic alleles at FANCA increased progressively in vitro and in vivo (134). Although the long-term safety and therapeutic effects in this model await to be investigated, these results suggest a convenient and economic approach for FA gene corrections, where a relatively low number of initially corrected HSPCs may become favorably selected to provide phenotypic rescue over time. Whether such a strategy may be suitable for other types of IEI conditions are exciting avenues for future research.
The remarkable developments in genome-editing technologies have opened up new research and therapeutic opportunities for IEIs. It is now generally convenient to establish precisely genome-edited animal or cell models carrying the patient-specific mutations (16). Such models provide invaluable tools for systematic exploration of mechanistic links between IEI genotypes and phenotypes, which shall subsequently suggest therapeutic strategies in a disease-specific manner. Importantly, as genome editing is moving into clinical applications for other blood disorders, i.e., β-thalassemia and sickle cell disease (135, 136), we have herein placed our main focus on the therapeutic aspect of genome editing in IEIs.
Despite the high burden for implementation, the vector-based gene therapies for some IEIs have currently reached clinical stages (9). Further exploration of genome-edited HSPC therapies should be driven by previous experiences from the conventional gene therapies. Based on current technical feasibility, ex vivo gene correction followed by in vivo transplantation shall remain the major treatment format (8).
In principle, various genome editing tools developed so far can be applied to drive targeted, somatic corrections of genetic defects underlying IEIs. Such a strategy may potentially exhibit advantages in safety, efficacy and application scope, compared to the current viral vector-based gene therapies (9, 10). Nevertheless, there are still many challenges that need to be carefully addressed, before these new technologies can be translated into valid IEI treatment options. It is important to note that a number of important considerations regarding HSPC collection, ex vivo HSPC culture, optimal patient conditioning regimens, and transplanted HSPC have been thoroughly discussed recently by some excellent gene therapy-related reviews (6, 9, 20). Therefore, only considerations that are specific to the genome editing approaches would be briefly covered below.
Despite a large list of available genome-editing tools (11), most of them are yet to become applicable for the clinics. For genome editing to develop into a common treatment option against the highly diverse IEI-causing mutations, its universal safety, adaptability, and effectiveness would need to be firmly established. Currently, the CRISPR/Cas9-based genome editing platform has undergone the most extensive and advanced developments (19). Many studies have explored the DSB/HDR mechanism for cDNA knock-in at the endogenous locus, potentially representing an “one-size-fits-all” type of correction strategy for a given gene mutated in IEI patients. One of the most efficient delivery routes for genome editing of HSPC is via electroporation of Cas9 machinery in the form of a ribonucleoprotein (RNP) complex (8, 93), where the sgRNAs may be chemically synthesized and modified for higher activities (137). To promote HDR knock-in while limiting the DNA template-triggered toxicity, the co-delivery of AAV6-based repair template is commonly adopted (95). However, it is often challenging to consistently achieve high rates of HDR in HSPCs, especially in the primitive HSC compartments, which would cause difficulties for long-term engraftment of corrected cells. Studies on the culture conditions or molecular manipulations to increase the proliferation/expansion of HSCs, and to promote their HDR activities continue to constitute an active area of research (93, 138). Additionally, clonal tracking investigations need to be carried out more extensively, so that protocols leading to optimized clonal repertoire of the gene-edited HSPCs can be established (139).
The off-target effects by Cas9 often raise concerns regarding unintended, detrimental modifications to the genomes. Considerable developments have been made on improving the fidelity of Cas9 (19). However, further investigations on the off-target events based on sensitive and unbiased detections in clinically relevant models, as well as on the long-term safety profiles of edited cells are still highly warranted (9, 11). Additionally, due to the possibility of DSB intermediates to induce large genomic abnormalities, careful testing and optimization for the current DSB/HDR strategy for gene correction in HSPCs still represent a priority issue (11). Furthermore, despite the fact that cDNAs placed under the endogenous promoters are under similar transcriptional control as the endogenous gene, this may not be sufficient to recapitulate the overall regulation of the gene, due to the likely omission of other regulatory elements such as the intronic elements (10). Genes with multiple functional splicing variants are also unlikely to be fully rescued by a single form of cDNA.
On the other hand, since most of the current preclinical investigations have focused on the Cas9-dependent knock-in strategies, they are further developed regarding feasibility and reliability. Given further improvements in the efficacy/toxicity profiles and reductions in practical costs associated with ex vivo editing, we anticipate that certain editing regimens may be translated to clinical stage in the near future.
In contrast to the Cas9-dependent genome-editing approach, the later developed base editors and prime editors provide opportunities for precise, in situ modification of the gene targets, without the requirement of DSB and introduction of a DNA template (19, 57). The fact that both BE and PE do not rely on HDR (inefficient in primitive HSCs) for installation of mutations also present important advantages for their potential application in HSPC therapy. Although yet to reach clinical stages for any indications, these newer tools already show strong potential for future therapeutic applications.
However, gene corrections by these approaches need to be designed on per mutation basis. This is currently a demanding task, considering that these editing platforms are still under extensive developments regarding their editing safety, on-target efficiency and purity profiles. The relative bulkiness of base editors and prime editors may impair their deliveries to the cells (11). Moreover, the relatively large sizes of pegRNAs and their activity-enhanced variants also become hurdles for PE applications that require chemically synthesized guide RNAs (81).
Most current base editors can only program base transitions, but not base transversions. Furthermore, it remains challenging to target a particular position without potentially engaging bystander editing. A possible solution against such editing impurities is by testing and selecting from base editors with reportedly different PAM restrictions, activity windows and context preferences (19). However, for such a strategy to become applicable, systematic and global characterizations and benchmarking of many different base editors would be required.
Compared to the base editors, the editing scope by the prime editors are apparently broader (75), which shall cover the majority of the pathogenic alleles in IEIs. However, their overall suboptimal and inconsistent activities still represent a major limiting factor. Besides further improving the architecture of PE tools, continued global-scale targeting experiments are required to further refine the rules governing the effectiveness by pegRNAs (140).
Despite the challenges, we anticipate that the BEs and PEs platforms shall bring some breakthroughs to the prospect of future IEI treatment. These in situ, precise genome editing platforms provide unprecedented opportunities to “seamlessly” correct a faulty gene, while currently exhibiting good safety features. Taking the recent history of genome editing as a reference, it is safe to predict that many of the current technical burdens for precise genome editing of HSPCs would be removed in the foreseeable future. Novel platforms shall also arise in a rapid pace. Coupled with continued experiences from the ongoing IEI gene therapy trials (9), the future outlook is certainly bright for precise genome editing to become a powerful treatment option for more patients with these devastating conditions.
QM, HS, and JL wrote and revised the manuscript. All authors contributed to the article and approved the submitted version.
This work is supported by grants from the National Key Research and Development Program of China (2021YFF1000704 and 2019YFA0802802 to JL).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human inborn errors of immunity: 2019 update on the classification from the international union of immunological societies expert committee. J Clin Immunol (2020) 40(1):24–64. doi: 10.1007/s10875-019-00737-x
2. Bousfiha A, Jeddane L, Picard C, Al-Herz W, Ailal F, Chatila T, et al. Human inborn errors of immunity: 2019 update of the iuis phenotypical classification. J Clin Immunol (2020) 40(1):66–81. doi: 10.1007/s10875-020-00758-x
3. Notarangelo LD, Bacchetta R, Casanova J-L, Su HC. Human inborn errors of immunity: An expanding universe. Sci Immunol (2020) 5(49):eabb1662. doi: 10.1126/sciimmunol.abb1662
4. Perez E. Future of therapy for inborn errors of immunity. Clin Rev Allergy Immunol (2022) 63(1):75-89. doi: 10.1007/s12016-021-08916-8
5. Bucciol G, Meyts I. Recent advances in primary immunodeficiency: From molecular diagnosis to treatment. F1000Research (2020) 9(194):1-15. doi: 10.12688/f1000research.21553.1
6. Fischer A, Hacein-Bey-Abina S. Gene therapy for severe combined immunodeficiencies and beyond. J Exp Med (2019) 217(2):e20190607. doi: 10.1084/jem.20190607
7. Castagnoli R, Delmonte OM, Calzoni E, Notarangelo LD. Hematopoietic stem cell transplantation in primary immunodeficiency diseases: Current status and future perspectives. Front Pediatr (2019) 7:295. doi: 10.3389/fped.2019.00295
8. Charlesworth CT, Hsu I, Wilkinson AC, Nakauchi H. Immunological barriers to haematopoietic stem cell gene therapy. Nat Rev Immunol (2022). doi: 10.1038/s41577-022-00698-0
9. Ferrari G, Thrasher AJ, Aiuti A. Gene therapy using haematopoietic stem and progenitor cells. Nat Rev Genet (2021) 22(4):216–34. doi: 10.1038/s41576-020-00298-5
10. Rai R, Thrasher AJ, Cavazza A. Gene editing for the treatment of primary immunodeficiency diseases. Hum Gene Ther (2021) 32(1-2):43–51. doi: 10.1089/hum.2020.185
11. Doudna JA. The promise and challenge of therapeutic genome editing. Nature (2020) 578(7794):229–36. doi: 10.1038/s41586-020-1978-5
12. Rouet P, Smih F, Jasin M. Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc Natl Acad Sci USA (1994) 91(13):6064–8. doi: 10.1073/pnas.91.13.6064
13. Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet (2010) 11(9):636–46. doi: 10.1038/nrg2842
14. Joung JK, Sander JD. Talens: A widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol (2013) 14(1):49–55. doi: 10.1038/nrm3486
15. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-Rna-Guided DNA endonuclease in adaptive bacterial immunity. Science (2012) 337(6096):816–21. doi: 10.1126/science.1225829
16. Hsu PD, Lander ES, Zhang F. Development and applications of crispr-Cas9 for genome engineering. Cell (2014) 157(6):1262–78. doi: 10.1016/j.cell.2014.05.010
17. Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with crispr-Cas9. Science (2014) 346(6213):1258096. doi: 10.1126/science.1258096
18. Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, et al. Clinvar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res (2016) 44(D1):D862–8. doi: 10.1093/nar/gkv1222
19. Anzalone AV, Koblan LW, Liu DR. Genome editing with crispr-cas nucleases, base editors, transposases and prime editors. Nat Biotechnol (2020) 38(7):824–44. doi: 10.1038/s41587-020-0561-9
20. Naldini L. Genetic engineering of hematopoiesis: Current stage of clinical translation and future perspectives. EMBO Mol Med (2019) 11(3):e9958. doi: 10.15252/emmm.201809958
21. Kohn DB, Booth C, Kang EM, Pai S-Y, Shaw KL, Santilli G, et al. Lentiviral gene therapy for X-linked chronic granulomatous disease. Nat Med (2020) 26(2):200–6. doi: 10.1038/s41591-019-0735-5
22. Kohn DB, Hershfield MS, Puck JM, Aiuti A, Blincoe A, Gaspar HB, et al. Consensus approach for the management of severe combined immune deficiency caused by adenosine deaminase deficiency. J Allergy Clin Immunol (2019) 143(3):852–63. doi: 10.1016/j.jaci.2018.08.024
23. Magnani A, Semeraro M, Adam F, Booth C, Dupré L, Morris EC, et al. Long-term safety and efficacy of lentiviral hematopoietic Stem/Progenitor cell gene therapy for wiskott–Aldrich syndrome. Nat Med (2022) 28(1):71–80. doi: 10.1038/s41591-021-01641-x
24. Ravin SSD, Wu X, Moir S, Kardava L, Anaya-O’Brien S, Kwatemaa N, et al. Lentiviral hematopoietic stem cell gene therapy for X-linked severe combined immunodeficiency. Sci Trans Med (2016) 8(335):335ra57–ra57. doi: 10.1126/scitranslmed.aad8856
25. Río P, Navarro S, Wang W, Sánchez-Domínguez R, Pujol RM, Segovia JC, et al. Successful engraftment of gene-corrected hematopoietic stem cells in non-conditioned patients with fanconi anemia. Nat Med (2019) 25(9):1396–401. doi: 10.1038/s41591-019-0550-z
26. Garcia-Perez L, van Eggermond M, van Roon L, Vloemans SA, Cordes M, Schambach A, et al. Successful preclinical development of gene therapy for recombinase-activating gene-1-Deficient scid. Mol Ther Methods Clin Dev (2020) 17:666–82. doi: 10.1016/j.omtm.2020.03.016
27. Punwani D, Kawahara M, Yu J, Sanford U, Roy S, Patel K, et al. Lentivirus mediated correction of Artemis-deficient severe combined immunodeficiency. Hum Gene Ther (2017) 28(1):112–24. doi: 10.1089/hum.2016.064
28. Sacco MG, Ungari M, Catò EM, Villa A, Strina D, Notarangelo LD, et al. Lymphoid abnormalities in Cd40 ligand transgenic mice suggest the need for tight regulation in gene therapy approaches to hyper immunoglobulin m (Igm) syndrome. Cancer Gene Ther (2000) 7(10):1299–306. doi: 10.1038/sj.cgt.7700232
29. Romero Z, Torres S, Cobo M, Muñoz P, Unciti JD, Martín F, et al. A tissue-specific, activation-inducible, lentiviral vector regulated by human Cd40l proximal promoter sequences. Gene Ther (2011) 18(4):364–71. doi: 10.1038/gt.2010.144
30. Urnov FD, Miller JC, Lee YL, Beausejour CM, Rock JM, Augustus S, et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature (2005) 435(7042):646–51. doi: 10.1038/nature03556
31. Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by tal effectors. Science (2009) 326(5959):1501. doi: 10.1126/science.1178817
32. Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, et al. Breaking the code of DNA binding specificity of tal-type iii effectors. Science (2009) 326(5959):1509–12. doi: 10.1126/science.1178811
33. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using Crispr/Cas systems. Science (2013) 339(6121):819–23. doi: 10.1126/science.1231143
34. Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. Rna-guided human genome engineering via Cas9. Science (2013) 339(6121):823–6. doi: 10.1126/science.1232033
35. Knott GJ, Doudna JA. Crispr-cas guides the future of genetic engineering. Science (2018) 361(6405):866–9. doi: 10.1126/science.aat5011
36. Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, et al. Targeting DNA double-strand breaks with tal effector nucleases. Genetics (2010) 186(2):757–61. doi: 10.1534/genetics.110.120717
37. Jasin M, Rothstein R. Repair of strand breaks by homologous recombination. Cold Spring Harb Perspect Biol (2013) 5(11):a012740. doi: 10.1101/cshperspect.a012740
38. Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. Rna-programmed genome editing in human cells. Elife (2013) 2:e00471. doi: 10.7554/eLife.00471
39. Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the crispr-Cas9 system. Science (2014) 343(6166):80–4. doi: 10.1126/science.1246981
40. Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, et al. Genome-scale crispr-Cas9 knockout screening in human cells. Science (2014) 343(6166):84–7. doi: 10.1126/science.1247005
41. Korkmaz G, Lopes R, Ugalde AP, Nevedomskaya E, Han R, Myacheva K, et al. Functional genetic screens for enhancer elements in the human genome using crispr-Cas9. Nat Biotechnol (2016) 34(2):192–8. doi: 10.1038/nbt.3450
42. Kim S, Kim D, Cho SW, Kim J, Kim JS. Highly efficient rna-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res (2014) 24(6):1012–9. doi: 10.1101/gr.171322.113
43. Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem (2010) 79:181–211. doi: 10.1146/annurev.biochem.052308.093131
44. Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, et al. High-frequency off-target mutagenesis induced by crispr-cas nucleases in human cells. Nat Biotechnol (2013) 31(9):822–6. doi: 10.1038/nbt.2623
45. Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, et al. DNA Targeting specificity of rna-guided Cas9 nucleases. Nat Biotechnol (2013) 31(9):827–32. doi: 10.1038/nbt.2647
46. Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science (2016) 351(6268):84–8. doi: 10.1126/science.aad5227
47. Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, et al. High-fidelity crispr-Cas9 nucleases with no detectable genome-wide off-target effects. Nature (2016) 529(7587):490–5. doi: 10.1038/nature16526
48. Chen JS, Dagdas YS, Kleinstiver BP, Welch MM, Sousa AA, Harrington LB, et al. Enhanced proofreading governs crispr-Cas9 targeting accuracy. Nature (2017) 550(7676):407–10. doi: 10.1038/nature24268
49. Hu JH, Miller SM, Geurts MH, Tang W, Chen L, Sun N, et al. Evolved Cas9 variants with broad Pam compatibility and high DNA specificity. Nature (2018) 556(7699):57–63. doi: 10.1038/nature26155
50. Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving crispr-cas nuclease specificity using truncated guide rnas. Nat Biotechnol (2014) 32(3):279–84. doi: 10.1038/nbt.2808
51. Kocak DD, Josephs EA, Bhandarkar V, Adkar SS, Kwon JB, Gersbach CA. Increasing the specificity of crispr systems with engineered rna secondary structures. Nat Biotechnol (2019) 37(6):657–66. doi: 10.1038/s41587-019-0095-1
52. Kim D, Luk K, Wolfe SA, Kim JS. Evaluating and enhancing target specificity of gene-editing nucleases and deaminases. Annu Rev Biochem (2019) 88:191–220. doi: 10.1146/annurev-biochem-013118-111730
53. Leibowitz ML, Papathanasiou S, Doerfler PA, Blaine LJ, Sun L, Yao Y, et al. Chromothripsis as an on-target consequence of crispr-Cas9 genome editing. Nat Genet (2021) 53(6):895–905. doi: 10.1038/s41588-021-00838-7
54. Kosicki M, Tomberg K, Bradley A. Repair of double-strand breaks induced by crispr-Cas9 leads to Large deletions and complex rearrangements. Nat Biotechnol (2018) 36(8):765–71. doi: 10.1038/nbt.4192
55. Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature (2016) 533(7603):420–4. doi: 10.1038/nature17946
56. Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, et al. Programmable base editing of a•T to G•C in genomic DNA without DNA cleavage. Nature (2017) 551(7681):464–71. doi: 10.1038/nature24644
57. Rees HA, Liu DR. Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet (2018) 19(12):770–88. doi: 10.1038/s41576-018-0059-1
58. Kim YB, Komor AC, Levy JM, Packer MS, Zhao KT, Liu DR. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat Biotechnol (2017) 35(4):371–6. doi: 10.1038/nbt.3803
59. Li X, Wang Y, Liu Y, Yang B, Wang X, Wei J, et al. Base editing with a Cpf1-cytidine deaminase fusion. Nat Biotechnol (2018) 36(4):324–7. doi: 10.1038/nbt.4102
60. Richter MF, Zhao KT, Eton E, Lapinaite A, Newby GA, Thuronyi BW, et al. Phage-assisted evolution of an adenine base Editor with improved cas domain compatibility and activity. Nat Biotechnol (2020) 38(7):883–91. doi: 10.1038/s41587-020-0453-z
61. Collias D, Beisel CL. Crispr technologies and the search for the Pam-free nuclease. Nat Commun (2021) 12(1):555. doi: 10.1038/s41467-020-20633-y
62. Ma Y, Zhang J, Yin W, Zhang Z, Song Y, Chang X. Targeted aid-mediated mutagenesis (Tam) enables efficient genomic diversification in mammalian cells. Nat Methods (2016) 13(12):1029–35. doi: 10.1038/nmeth.4027
63. Nishimasu H, Shi X, Ishiguro S, Gao L, Hirano S, Okazaki S, et al. Engineered crispr-Cas9 nuclease with expanded targeting space. Science (2018) 361(6408):1259–62. doi: 10.1126/science.aas9129
64. Wang X, Li J, Wang Y, Yang B, Wei J, Wu J, et al. Efficient base editing in methylated regions with a human Apobec3a-Cas9 fusion. Nat Biotechnol (2018) 36(10):946–9. doi: 10.1038/nbt.4198
65. Oakes BL, Fellmann C, Rishi H, Taylor KL, Ren SM, Nadler DC, et al. Crispr-Cas9 circular permutants as programmable scaffolds for genome modification. Cell (2019) 176(1-2):254–67. doi: 10.1016/j.cell.2018.11.052
66. Tan J, Zhang F, Karcher D, Bock R. Engineering of high-precision base editors for site-specific single nucleotide replacement. Nat Commun (2019) 10(1):439. doi: 10.1038/s41467-018-08034-8
67. Liu Z, Chen S, Shan H, Jia Y, Chen M, Song Y, et al. Precise base editing with cc context-specificity using engineered human Apobec3g-Ncas9 fusions. BMC Biol (2020) 18(1):111. doi: 10.1186/s12915-020-00849-6
68. Gehrke JM, Cervantes O, Clement MK, Wu Y, Zeng J, Bauer DE, et al. An Apobec3a-Cas9 base Editor with minimized bystander and off-target activities. Nat Biotechnol (2018) 36(10):977–82. doi: 10.1038/nbt.4199
69. Rees HA, Komor AC, Yeh WH, Caetano-Lopes J, Warman M, Edge ASB, et al. Improving the DNA specificity and applicability of base editing through protein engineering and protein delivery. Nat Commun (2017) 8:15790. doi: 10.1038/ncomms15790
70. Kim D, Kim DE, Lee G, Cho SI, Kim JS. Genome-wide target specificity of crispr rna-guided adenine base editors. Nat Biotechnol (2019) 37(4):430–5. doi: 10.1038/s41587-019-0050-1
71. Doman JL, Raguram A, Newby GA, Liu DR. Evaluation and minimization of Cas9-independent off-target DNA editing by cytosine base editors. Nat Biotechnol (2020) 38(5):620–8. doi: 10.1038/s41587-020-0414-6
72. Yu Y, Leete TC, Born DA, Young L, Barrera LA, Lee SJ, et al. Cytosine base editors with minimized unguided DNA and rna off-target events and high on-target activity. Nat Commun (2020) 11(1):2052. doi: 10.1038/s41467-020-15887-5
73. Gaudelli NM, Lam DK, Rees HA, Solá-Esteves NM, Barrera LA, Born DA, et al. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat Biotechnol (2020) 38(7):892–900. doi: 10.1038/s41587-020-0491-6
74. Rees HA, Wilson C, Doman JL, Liu DR. Analysis and minimization of cellular rna editing by DNA adenine base editors. Sci Adv (2019) 5(5):eaax5717. doi: 10.1126/sciadv.aax5717
75. Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, et al. Search-and-Replace genome editing without double-strand breaks or donor DNA. Nature (2019) 576(7785):149–57. doi: 10.1038/s41586-019-1711-4
76. Schene IF, Joore IP, Oka R, Mokry M, van Vugt AHM, van Boxtel R, et al. Prime editing for functional repair in patient-derived disease models. Nat Commun (2020) 11(1):5352. doi: 10.1038/s41467-020-19136-7
77. Kim DY, Moon SB, Ko JH, Kim YS, Kim D. Unbiased investigation of specificities of prime editing systems in human cells. Nucleic Acids Res (2020) 48(18):10576–89. doi: 10.1093/nar/gkaa764
78. Park SJ, Jeong TY, Shin SK, Yoon DE, Lim SY, Kim SP, et al. Targeted mutagenesis in mouse cells and embryos using an enhanced prime Editor. Genome Biol (2021) 22(1):170. doi: 10.1186/s13059-021-02389-w
79. Zong Y, Liu Y, Xue C, Li B, Li X, Wang Y, et al. An engineered prime Editor with enhanced editing efficiency in plants. Nat Biotechnol (2022). doi: 10.1038/s41587-022-01254-w
80. Liu Y, Yang G, Huang S, Li X, Wang X, Li G, et al. Enhancing prime editing by Csy4-mediated processing of pegrna. Cell Res (2021) 31(10):1134–6. doi: 10.1038/s41422-021-00520-x
81. Nelson JW, Randolph PB, Shen SP, Everette KA, Chen PJ, Anzalone AV, et al. Engineered pegrnas improve prime editing efficiency. Nat Biotechnol (2022) 40(3):402–10. doi: 10.1038/s41587-021-01039-7
82. Zhang G, Liu Y, Huang S, Qu S, Cheng D, Yao Y, et al. Enhancement of prime editing via xrrna motif-joined pegrna. Nat Commun (2022) 13(1):1856. doi: 10.1038/s41467-022-29507-x
83. Chen PJ, Hussmann JA, Yan J, Knipping F, Ravisankar P, Chen PF, et al. Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell (2021) 184(22):5635–52.e29. doi: 10.1016/j.cell.2021.09.018
84. Ferreira da Silva J, Oliveira GP, Arasa-Verge EA, Kagiou C, Moretton A, Timelthaler G, et al. Prime editing efficiency and fidelity are enhanced in the absence of mismatch repair. Nat Commun (2022) 13(1):760. doi: 10.1038/s41467-022-28442-1
85. Zhuang Y, Liu J, Wu H, Zhu Q, Yan Y, Meng H, et al. Increasing the efficiency and precision of prime editing with guide rna pairs. Nat Chem Biol (2022) 18(1):29–37. doi: 10.1038/s41589-021-00889-1
86. Lin Q, Jin S, Zong Y, Yu H, Zhu Z, Liu G, et al. High-efficiency prime editing with optimized, paired pegrnas in plants. Nat Biotechnol (2021) 39(8):923–7. doi: 10.1038/s41587-021-00868-w
87. Wang J, He Z, Wang G, Zhang R, Duan J, Gao P, et al. Efficient targeted insertion of Large DNA fragments without DNA donors. Nat Methods (2022) 19(3):331–40. doi: 10.1038/s41592-022-01399-1
88. Choi J, Chen W, Suiter CC, Lee C, Chardon FM, Yang W, et al. Precise genomic deletions using paired prime editing. Nat Biotechnol (2022) 40(2):218–26. doi: 10.1038/s41587-021-01025-z
89. Anzalone AV, Gao XD, Podracky CJ, Nelson AT, Koblan LW, Raguram A, et al. Programmable deletion, replacement, integration and inversion of Large DNA sequences with twin prime editing. Nat Biotechnol (2022) 40(5):731–40. doi: 10.1038/s41587-021-01133-w
90. Jiang T, Zhang XO, Weng Z, Xue W. Deletion and replacement of long genomic sequences using prime editing. Nat Biotechnol (2022) 40(2):227–34. doi: 10.1038/s41587-021-01026-y
91. Adikusuma F, Lushington C, Arudkumar J, Godahewa Gelshan I, Chey YCJ, Gierus L, et al. Optimized nickase- and nuclease-based prime editing in human and mouse cells. Nucleic Acids Res (2021) 49(18):10785–95. doi: 10.1093/nar/gkab792
92. Tao R, Wang Y, Hu Y, Jiao Y, Zhou L, Jiang L, et al. Wt-pe: Prime editing with nuclease wild-type Cas9 enables versatile Large-scale genome editing. Signal Transduct Target Ther (2022) 7(1):108. doi: 10.1038/s41392-022-00936-w
93. Haltalli MLR, Wilkinson AC, Rodriguez-Fraticelli A, Porteus M. Hematopoietic stem cell gene editing and expansion: State-of-the-Art technologies and recent applications. Exp Hematol (2022) 107:9–13. doi: 10.1016/j.exphem.2021.12.399
94. Lombardo A, Genovese P, Beausejour CM, Colleoni S, Lee YL, Kim KA, et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol (2007) 25(11):1298–306. doi: 10.1038/nbt1353
95. Genovese P, Schiroli G, Escobar G, Tomaso TD, Firrito C, Calabria A, et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature (2014) 510(7504):235–40. doi: 10.1038/nature13420
96. Schiroli G, Ferrari S, Conway A, Jacob A, Capo V, Albano L, et al. Preclinical modeling highlights the therapeutic potential of hematopoietic stem cell gene editing for correction of scid-X1. Sci Transl Med (2017) 9(411):eaan0820. doi: 10.1126/scitranslmed.aan0820
97. Pavel-Dinu M, Wiebking V, Dejene BT, Srifa W, Mantri S, Nicolas CE, et al. Gene correction for scid-X1 in long-term hematopoietic stem cells. Nat Commun (2019) 10(1):1634. doi: 10.1038/s41467-019-09614-y
98. Güngör T, Teira P, Slatter M, Stussi G, Stepensky P, Moshous D, et al. Reduced-intensity conditioning and hla-matched haemopoietic stem-cell transplantation in patients with chronic granulomatous disease: A prospective multicentre study. Lancet (2014) 383(9915):436–48. doi: 10.1016/s0140-6736(13)62069-3
99. Santilli G, Almarza E, Brendel C, Choi U, Beilin C, Blundell MP, et al. Biochemical correction of X-cgd by a novel chimeric promoter regulating high levels of transgene expression in myeloid cells. Mol Ther (2011) 19(1):122–32. doi: 10.1038/mt.2010.226
100. Brendel C, Rothe M, Santilli G, Charrier S, Stein S, Kunkel H, et al. Non-clinical efficacy and safety studies on G1xcgd, a lentiviral vector for ex vivo gene therapy of X-linked chronic granulomatous disease. Hum Gene Ther Clin Dev (2018) 29(2):69–79. doi: 10.1089/humc.2017.245
101. De Ravin SS, Reik A, Liu PQ, Li L, Wu X, Su L, et al. Targeted gene addition in human Cd34(+) hematopoietic cells for correction of X-linked chronic granulomatous disease. Nat Biotechnol (2016) 34(4):424–9. doi: 10.1038/nbt.3513
102. De Ravin SS, Li L, Wu X, Choi U, Allen C, Koontz S, et al. Crispr-Cas9 gene repair of hematopoietic stem cells from patients with X-linked chronic granulomatous disease. Sci Transl Med (2017) 9(372):eaah3480. doi: 10.1126/scitranslmed.aah3480
103. De Ravin SS, Brault J, Meis RJ, Liu S, Li L, Pavel-Dinu M, et al. Enhanced homology-directed repair for highly efficient gene editing in hematopoietic Stem/Progenitor cells. Blood (2021) 137(19):2598–608. doi: 10.1182/blood.2020008503
104. Roos D, Kuhns DB, Maddalena A, Roesler J, Lopez JA, Ariga T, et al. Hematologically important mutations: X-linked chronic granulomatous disease (Third update). Blood Cells Mol Dis (2010) 45(3):246–65. doi: 10.1016/j.bcmd.2010.07.012
105. Roos D. Chronic granulomatous disease. In: Knaus UG, Leto TL, editors. Nadph oxidases: Methods and protocols. New York, NY: Springer New York (2019). p. 531–42.
106. Görlach A, Lee PL, Roesler J, Hopkins PJ, Christensen B, Green ED, et al. A P47-phox pseudogene carries the most common mutation causing P47-phox- deficient chronic granulomatous disease. J Clin Invest (1997) 100(8):1907–18. doi: 10.1172/jci119721
107. Roesler J, Curnutte JT, Rae J, Barrett D, Patino P, Chanock SJ, et al. Recombination events between the P47-phox gene and its highly homologous pseudogenes are the main cause of autosomal recessive chronic granulomatous disease. Blood (2000) 95(6):2150–6. doi: 10.1182/blood.V95.6.2150
108. Merling RK, Kuhns DB, Sweeney CL, Wu X, Burkett S, Chu J, et al. Gene-edited pseudogene resurrection corrects P47(Phox)-deficient chronic granulomatous disease. Blood Adv (2017) 1(4):270–8. doi: 10.1182/bloodadvances.2016001214
109. Jin Y, Mazza C, Christie JR, Giliani S, Fiorini M, Mella P, et al. Mutations of the wiskott-Aldrich syndrome protein (Wasp): Hotspots, effect on transcription, and translation and Phenotype/Genotype correlation. Blood (2004) 104(13):4010–9. doi: 10.1182/blood-2003-05-1592
110. Worth AJ, Thrasher AJ. Current and emerging treatment options for wiskott-Aldrich syndrome. Expert Rev Clin Immunol (2015) 11(9):1015–32. doi: 10.1586/1744666x.2015.1062366
111. Aiuti A, Biasco L, Scaramuzza S, Ferrua F, Cicalese MP, Baricordi C, et al. Lentiviral hematopoietic stem cell gene therapy in patients with wiskott-Aldrich syndrome. Science (2013) 341(6148):1233151. doi: 10.1126/science.1233151
112. Hacein-Bey Abina S, Gaspar HB, Blondeau J, Caccavelli L, Charrier S, Buckland K, et al. Outcomes following gene therapy in patients with severe wiskott-Aldrich syndrome. JAMA (2015) 313(15):1550–63. doi: 10.1001/jama.2015.3253
113. Rai R, Romito M, Rivers E, Turchiano G, Blattner G, Vetharoy W, et al. Targeted gene correction of human hematopoietic stem cells for the treatment of wiskott - aldrich syndrome. Nat Commun (2020) 11(1):4034. doi: 10.1038/s41467-020-17626-2
114. Notarangelo LD, Hayward AR. X-Linked immunodeficiency with hyper-igm (Xhim). Clin Exp Immunol (2000) 120(3):399–405. doi: 10.1046/j.1365-2249.2000.01142.x
115. Seyama K, Nonoyama S, Gangsaas I, Hollenbaugh D, Pabst HF, Aruffo A, et al. Mutations of the Cd40 ligand gene and its effect on Cd40 ligand expression in patients with X-linked hyper igm syndrome. Blood (1998) 92(7):2421–34. doi: 10.1182/blood.V92.7.2421
116. Levy J, Espanol-Boren T, Thomas C, Fischer A, Tovo P, Bordigoni P, et al. Clinical spectrum of X-linked hyper-igm syndrome. J Pediatr (1997) 131(1 Pt 1):47–54. doi: 10.1016/s0022-3476(97)70123-9
117. Brown MP, Topham DJ, Sangster MY, Zhao J, Flynn KJ, Surman SL, et al. Thymic lymphoproliferative disease after successful correction of Cd40 ligand deficiency by gene transfer in mice. Nat Med (1998) 4(11):1253–60. doi: 10.1038/3233
118. Kuo CY, Long JD, Campo-Fernandez B, de Oliveira S, Cooper AR, Romero Z, et al. Site-specific gene editing of human hematopoietic stem cells for X-linked hyper-igm syndrome. Cell Rep (2018) 23(9):2606–16. doi: 10.1016/j.celrep.2018.04.103
119. d'Hennezel E, Ben-Shoshan M, Ochs HD, Torgerson TR, Russell LJ, Lejtenyi C, et al. Foxp3 forkhead domain mutation and regulatory T cells in the ipex syndrome. N Engl J Med (2009) 361(17):1710–3. doi: 10.1056/NEJMc0907093
120. McMurchy AN, Gillies J, Gizzi MC, Riba M, Garcia-Manteiga JM, Cittaro D, et al. A novel function for Foxp3 in humans: Intrinsic regulation of conventional T cells. Blood (2013) 121(8):1265–75. doi: 10.1182/blood-2012-05-431023
121. Bacchetta R, Barzaghi F, Roncarolo MG. From ipex syndrome to Foxp3 mutation: A lesson on immune dysregulation. Ann N Y Acad Sci (2018) 1417(1):5–22. doi: 10.1111/nyas.13011
122. Santoni de Sio FR, Passerini L, Valente MM, Russo F, Naldini L, Roncarolo MG, et al. Ectopic Foxp3 expression preserves primitive features of human hematopoietic stem cells while impairing functional T cell differentiation. Sci Rep (2017) 7(1):15820. doi: 10.1038/s41598-017-15689-8
123. Goodwin M, Lee E, Lakshmanan U, Shipp S, Froessl L, Barzaghi F, et al. Crispr-based gene editing enables Foxp3 gene repair in ipex patient cells. Sci Adv (2020) 6(19):eaaz0571. doi: 10.1126/sciadv.aaz0571
124. Ravell JC, Chauvin SD, He T, Lenardo M. An update on xmen disease. J Clin Immunol (2020) 40(5):671–81. doi: 10.1007/s10875-020-00790-x
125. Ravell J, Chaigne-Delalande B, Lenardo M. X-Linked immunodeficiency with magnesium defect, Epstein-Barr virus infection, and neoplasia disease: A combined immune deficiency with magnesium defect. Curr Opin Pediatr (2014) 26(6):713–9. doi: 10.1097/mop.0000000000000156
126. Brault J, Liu T, Bello E, Liu S, Sweeney CL, Meis RJ, et al. Crispr-targeted Magt1 insertion restores xmen patient hematopoietic stem cells and lymphocytes. Blood (2021) 138(26):2768–80. doi: 10.1182/blood.2021011192
127. Bagby G. Recent advances in understanding hematopoiesis in fanconi anemia. F1000Res (2018) 7:105. doi: 10.12688/f1000research.13213.1
128. Garaycoechea JI, Patel KJ. Why does the bone marrow fail in fanconi anemia? Blood (2014) 123(1):26–34. doi: 10.1182/blood-2013-09-427740
129. Antonio Casado J, Callén E, Jacome A, Río P, Castella M, Lobitz S, et al. A comprehensive strategy for the subtyping of patients with fanconi anaemia: Conclusions from the Spanish fanconi anemia research network. J Med Genet (2007) 44(4):241–9. doi: 10.1136/jmg.2006.044719
130. Kelly PF, Radtke S, von Kalle C, Balcik B, Bohn K, Mueller R, et al. Stem cell collection and gene transfer in fanconi anemia. Mol Ther (2007) 15(1):211–9. doi: 10.1038/sj.mt.6300033
131. Diez B, Genovese P, Roman-Rodriguez FJ, Alvarez L, Schiroli G, Ugalde L, et al. Therapeutic gene editing in Cd34(+) hematopoietic progenitors from fanconi anemia patients. EMBO Mol Med (2017) 9(11):1574–88. doi: 10.15252/emmm.201707540
132. Du W, Amarachintha S, Wilson AF, Pang Q. Hyper-active non-homologous end joining selects for synthetic lethality resistant and pathological fanconi anemia hematopoietic stem and progenitor cells. Sci Rep (2016) 6:22167. doi: 10.1038/srep22167
133. Pace P, Mosedale G, Hodskinson MR, Rosado IV, Sivasubramaniam M, Patel KJ. Ku70 corrupts DNA repair in the absence of the fanconi anemia pathway. Science (2010) 329(5988):219–23. doi: 10.1126/science.1192277
134. Román-Rodríguez FJ, Ugalde L, Álvarez L, Díez B, Ramírez MJ, Risueño C, et al. Nhej-mediated repair of crispr-Cas9-Induced DNA breaks efficiently corrects mutations in hspcs from patients with fanconi anemia. Cell Stem Cell (2019) 25(5):607–21.e7. doi: 10.1016/j.stem.2019.08.016
135. Frangoul H, Altshuler D, Cappellini MD, Chen YS, Domm J, Eustace BK, et al. Crispr-Cas9 gene editing for sickle cell disease and Β-thalassemia. N Engl J Med (2021) 384(3):252–60. doi: 10.1056/NEJMoa2031054
136. Wu Y, Zeng J, Roscoe BP, Liu P, Yao Q, Lazzarotto CR, et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat Med (2019) 25(5):776–83. doi: 10.1038/s41591-019-0401-y
137. Hendel A, Bak RO, Clark JT, Kennedy AB, Ryan DE, Roy S, et al. Chemically modified guide rnas enhance crispr-cas genome editing in human primary cells. Nat Biotechnol (2015) 33(9):985–9. doi: 10.1038/nbt.3290
138. Charlesworth CT, Camarena J, Cromer MK, Vaidyanathan S, Bak RO, Carte JM, et al. Priming human repopulating hematopoietic stem and progenitor cells for Cas9/Sgrna gene targeting. Mol Ther Nucleic Acids (2018) 12:89–104. doi: 10.1016/j.omtn.2018.04.017
139. Ferrari S, Jacob A, Beretta S, Unali G, Albano L, Vavassori V, et al. Efficient gene editing of human long-term hematopoietic stem cells validated by clonal tracking. Nat Biotechnol (2020) 38(11):1298–308. doi: 10.1038/s41587-020-0551-y
Keywords: inborn errors of immunity (IEI), hematopoietic stem cell (HSC), gene therapy, genome editing, base editing, prime editing
Citation: Meng Q, Sun H and Liu J (2022) Precise somatic genome editing for treatment of inborn errors of immunity. Front. Immunol. 13:960348. doi: 10.3389/fimmu.2022.960348
Received: 02 June 2022; Accepted: 09 August 2022;
Published: 26 August 2022.
Edited by:
Maurizio Miano, Giannina Gaslini Institute, (IRCCS), ItalyReviewed by:
Gesmar Rodrigues Silva Segundo, Federal University of Uberlandia, BrazilCopyright © 2022 Meng, Sun and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianghuai Liu, bGl1amlhbmdodWFpQG5qdS5lZHUuY24=; Haixiang Sun, c3RldmVuc3VuekAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.