 Yadong Fan1†
Yadong Fan1† Wenjin Dong2†
Wenjin Dong2† Ying Wang1†Shan Zhu3Rundong Chai1
Ying Wang1†Shan Zhu3Rundong Chai1 Zhe Xu1
Zhe Xu1 Xiaoyu Zhang4
Xiaoyu Zhang4 Yiqi Yan3
Yiqi Yan3 Long Yang1,5*
Long Yang1,5* Yuhong Bian1*
Yuhong Bian1*- 1School of Integrative Medicine, Tianjin University of Traditional Chinese Medicine, Tianjin, China
- 2Department of Science and Education, Tianjin Union Medical Center, Tianjin, China
- 3State Key Laboratory of Component Traditional Chinese Medicine, Tianjin University of Traditional Chinese Medicine, Tianjin, China
- 4The Reproductive Center, First Teaching Hospital of Tianjin University of Traditional Chinese Medicine, Tianjin, China
- 5Research Center for Infectious Diseases, Tianjin University of Traditional Chinese Medicine, Tianjin, China
Macrophages are involved in hepatocyte steatosis and necroinflammation and play an important role in the pathogenesis of non-alcoholic fatty liver disease (NAFLD). Impaired autophagy function (decreased autophagy or blocked autophagic flow) leads to cell damage and death and promotes NAFLD progression. The experimental and clinical research of glycyrrhetinic acid (GA) in the treatment of NAFLD has gradually attracted attention with clear pharmacological activities such as immune regulation, antiviral, antitumor, antioxidant, liver protection, and anti-inflammatory. However, the effects of GA on the STAT3-HIF-1α pathway and autophagy in macrophages are still unclear, and its mechanism of action in the treatment of NAFLD remains to be further elucidated. We constructed a NAFLD mouse model through a high-fat and high-sugar diet to investigate the therapeutic effects of GA. The results showed that GA reduced weight, improved the pathological changes and hepatic lipid deposition of liver, and abnormally elevated the levels of serum biochemical (AST, ALT, TG, T-CHO, LDL-C, and HDL-C) and inflammatory indexes (IL-1β, IL-4, IL-6, MCP-1, and TNF-α) in NAFLD mice. Further examination revealed that GA ameliorates excessive hepatic macrophage infiltration and hepatocyte apoptosis. The results of the cell experiments further elaborated that GA modulated the PA-induced macrophage STAT3-HIF-1α pathway and ameliorated impaired autophagic flux (blockade of autophagosome–lysosome fusion) and overactivation of inflammation. Excessive hepatocyte apoptosis caused by the uncontrolled release of inflammatory cytokines was also suppressed by GA.
Conclusion: This study demonstrated that GA could regulate the STAT3-HIF-1α pathway of macrophages, ameliorate the impaired autophagy flux, and reduce the excessive production of inflammatory cytokines to improve the excessive apoptosis of liver cells, thus playing a therapeutic role on NAFLD.
Introduction
With changes in dietary patterns and lifestyles, a global pandemic of chronic metabolic diseases has emerged. In this context, non-alcoholic fatty liver disease (NAFLD) is a metabolic stress-induced liver injury closely related to genetic susceptibility and insulin resistance, whose incidence is increasing year by year. According to statistics, the global incidence of NAFLD is about 25% (1), and the prevalence in China is about 29.2% (2), accompanied by an increasing trend of younger age (3). Metabolic syndromes (Mets), especially hypertension, type 2 diabetes, cardiovascular disease, and obesity, are closely related to the pathophysiological process of NAFLD (4, 5). The pathogenesis of NAFLD is complex, with theories such as “two-hit” and “multi-hit”. Lipid metabolism imbalance, immune system imbalance, inflammasome activation, mitochondrial dysfunction, endoplasmic reticulum stress, and intestinal microbial disorders can affect the pathogenesis and progression of NAFLD (6). The high concealment of NAFLD, multisystem involvement, and heavy medical expenses have caused serious social medical burden (7).
In NAFLD, free fatty acids (FFAs) are important mediators of lipotoxicity, which lead to cellular damage through different pathways including endoplasmic reticulum stress (8). This process is balanced by the upregulation of autophagy pathways that maintain cell survival and homeostasis. Impaired autophagy is a key factor in hepatocyte injury in NAFLD (9). Macrophages are involved in hepatocyte steatosis and necroinflammation and play an important role in the pathogenesis of NAFLD. Excessive infiltration of macrophages and overproduction of proinflammatory cytokines in the liver contribute to the pathogenesis of NAFLD (10). STAT3 has been well regarded as a key regulatory target of the development and progression in liver inflammation, injury, regeneration, activation of hepatic stellate cells, and liver cancer (11–15). As a key regulator of hypoxia, HIF-1α can broadly regulate the expression of hypoxia-inducible genes and the activation of various signaling pathways (16, 17). In the early stage of a high-fat diet and obesity, high levels of HIF-1α expression and hypoxia in white adipose tissue occur before the onset of insulin resistance and inflammation (18). The study confirms that the HIF gene in the liver is significantly elevated in NAFLD. The activation of HIF-1α in the liver and hepatocytes can promote lipid accumulation and liver damage to accelerate disease progression in a methionine–choline-deficient diet-induced NAFLD mouse (19). Clinical studies have also shown that HIF-1α is functionally activated in children with NAFLD accompanied by nocturnal hypoxia, which induces the expression of key genes of epithelial–mesenchymal transition and accelerates the progression of NAFLD (20). A recent study shows that HIF-1α-mediated autophagy injury in macrophages increases IL-1β production, which contributes to choline-deficient diet-induced NAFLD. STAT3 signaling is considered an upstream regulator of HIF-1α (21), and the activated STAT3 combines with the HIF-1α promoter and induces increasing HIF-1α expression. Silencing STAT3 will block HIF-1α expression even under hypoxic conditions (22). In the pathogenesis and progression of NAFLD, the STAT3-HIF-1α pathway plays a key role and deserves further exploration.
At present, there is no clinically approved specific drug for NAFLD. Glycyrrhizic acid (GL) and glycyrrhetinic acid (GA) are extracted from the roots of Glycyrrhiza uralensis Fisch. It has immunomodulatory, antiviral, antitumor, antioxidant, liver protection, and anti-inflammatory effects (23). GL can regulate hepatocyte lipid metabolism, glucose homeostasis, and insulin sensitivity in NAFLD mice (24–26). GL also restores bile acid homeostasis in NAFLD mice and inhibits liver inflammatory injury (27). In addition, GA restores the retinol metabolic balance in NAFLD mice to achieve therapeutic purposes (28). The early result shows that GA can prevent hepatic failure induced by lipopolysaccharide/D-galactosamine, reduce mortality and alanine aminotransferase (ALT)/aspartate aminotransferase (AST) elevation, and improve liver pathological injury. Its mechanism is related to the upregulation of macrophage interleukin-1 receptor-associated kinases M, deactivation of NF-κB, and inhibition of TNF-α production (29). However, the effect of GA on the STAT3-HIF-1α pathway and autophagy in macrophages is still unclear, and its mechanism of action in the treatment of NAFLD remains to be further elucidated. We aimed to investigate whether GA could ameliorate high-fat and high-sugar diet-induced NAFLD by modulating macrophage autophagic flux through the STAT3-HIF-1α pathway.
Materials and methods
Experimental animals
A total of 24 male C57BL/6 mice (SPF; age 8 weeks; weight, 19.2 ± 1.2 g) were purchased from Beijing Weitong Lihua Laboratory Animal Technology Co., Ltd., and housed in the Laboratory Animal Center of Tianjin University of Traditional Chinese Medicine (animal license number: SCXK (Beijing) 2019-0009). The animals were maintained under standard conditions of temperature (22 ± 2°C) and humidity (50 ± 5%) in a 12-h light/dark cycle. Animals were allowed free access to food and water throughout the experimental period. The operation and feeding procedures of the experimental animals abided by the relevant regulations of the Tianjin University of Traditional Chinese Medicine on the feeding and use of experimental animals.
Establishment of the NAFLD mouse model
After 1 week of adaptive feeding, mice were randomly divided into different experimental groups. Normal mice were fed with standard chow for 12 weeks. Mice in the NAFLD model group were fed a NAFLD/NASH high-fat-rich diet and drinking water containing fructose (with 55% of fructose and 45% sucrose by weight, 42 g/l of carbohydrates was mixed in drinking water, filter sterilized) for 12 weeks (30–32). On the basis of the model group, the experimental group was administered α-GA (Lot: 20170520, Tianjin Zhongyi Pharmaceutical Co., Ltd.) by intragastric administration (60 mg/kg) for 2 weeks. The intragastric administration volume was 0.01 ml/g, and the drug was dissolved by ultrasound with 0.5% sodium carboxymethylcellulose. The mice of the normal group and the model group were gavaged with co-solvent.
Cell treatments
Cells were purchased from the ATCC Cell Bank. The RAW264.7 cell culture condition was 10% fetal bovine serum (FBS) + 90% DMEM. The Kupffer cell culture condition was 10% FBS + 90% RPMI-1640. The AML-12 cell culture condition was 10% FBS + 90% DMEM/F12 + 10 µg/ml insulin + 5.5 µg/ml transferrin + 5 ng/ml selenium + 40 ng/ml dexamethasone. Cells were cultured in an incubator at 37°C with 5% CO2 concentration. Trypsin (0.25%) was used for digestion and passage. For experimental design, cells with 80%–90% cell fusion degree during the growth period in good condition were taken. Before the experiment, the CCK8 method was used to investigate the effects of palmitic acid (PA) (Lot: P0500, Sigma) coupled with bovine serum protein (without fatty acids) (Lot: A8850, Beijing Solaibao Technology Co., Ltd.) and α-GA on macrophage activity. In selected studies, cells were treated with the autophagy inhibitor 3-MA (5 mM, Lot: M833793, Macklin) for 30 min prior to PA and/or α-GA treatment, or bafilomycin (100 nM, Lot: A8510, Beijing Solaibao Technology Co., Ltd.) for the last 2 h of the PA and/or α-GA treatment. The mechanism study using the STAT3 small-molecule inhibitor (Stattic, 10 μm, Lot: S7024, Shanghai Selleck Biotechnology Co., Ltd.) and HIF-1α inhibitor Lificiguat (YC-1, 20 μM, Lot: S7958, Shanghai Selleck Biotechnology Co., Ltd.) complied with PA and/or α-GA treatment. CCK8 experiments were carried out in 96-well cell culture plates at a density of 5 × 10 (3) cells per well. The cells were seeded in a 12-well cell culture plate at a density of 1 × 10 (5) cells per well to design other experiments. After the cells were plated, the cells were cultured in an incubator for 24 h before intervention, and three replicate wells were set up in each group.
Hematoxylin–eosin and Oil Red O staining of the liver
During the experiment, the general state of the mice in each group was observed, and the body weight of the mice was recorded every week. After the last intragastric administration, the mice were fasted for 12 h and then weighed. After 15 weeks, the mice were anesthetized with 0.3% sodium pentobarbital solution, and the liver, thymus, and spleen of the mice were taken out. The organs were rinsed with normal saline and dried with filter paper to calculate the organ index. The same part of the liver of the mice in each group with a size of 2 cm (3) was excised. After fixation in 4% paraformaldehyde solution for 72 h, the tissues were cut into 5-μm slices and stained according to the standard hematoxylin–eosin (H&E) procedure. Another part of the liver tissue was prepared into frozen sections with a thickness of 10 μm. The prepared Oil Red O working solution (Oil Red O storage solution: distilled water = 3:2) was poured into the dye vat, the sections were submerged upright, stained for 30 min, and the excess dye was washed away with 60% isopropanol. The sections were rinsed three times with distilled water, differentiated with 75% ethanol, terminated with tap water, stained with hematoxylin for 90 s, and then mounted after returning to blue. All sections were photographed and examined microscopically.
Serum biochemical analysis
Before the mice were sacrificed, the blood of the mice was collected and serum samples were prepared. The biochemical indicators such as ALT, AST, high-density lipoprotein cholesterol (HDL-C), low-density lipoprotein cholesterol (LDL-C), total cholesterol (T-CHO), triglyceride (TG), creatinine (CRE), and blood urea nitrogen (BUN) were detected using an automatic biochemical analyzer (Microlab 300 from Rittal, the Netherlands). According to the instructions of the reagent manufacturer (Lot: 70-EK201B/3; 70-EK204/2; 70-EK2062/2; 70-EK206; 70-EK2822/2, Hangzhou Lianke Biotechnology Co., Ltd.), ELISA detection reagents of interleukin 1β (IL-1β), interleukin 4 (IL-4), interleukin 6 (IL-6), monocyte chemoattractant protein 1 (MCP-1), and tumor necrosis factor α (TNF-α) were used to detect their serum and cellular supernatant levels respectively, and absorbance at 450 nm was measured using a microplate spectrophotometer (Thermo Fisher Varioskan Flash). Before the experiment, the serum and supernatant in each group were thoroughly shaken and mixed.
Observation of macrophage infiltration in the liver
The paraffin sections were stained according to the immunohistochemical detection kit procedure (Lot: PK10006, Proteintech Group, Inc., China): deparaffinization to water, antigen heat retrieval (citric acid retrieval solution), endogenous peroxidase inactivation, 5% goat serum blocking, F4/80 antibody (1:200, Lot: 70076S, Cell Signaling Technology) overnight incubation, secondary antibody incubation for 1 h, DAB color development, hematoxylin counterstaining, dehydration and mounting, and microscopy.
Quantitative real-time PCR
Total RNA was extracted from mouse liver tissues or cells with TRIzol Reagent Kit (Lot: DP431, Tiangen Biochemical Technology (Beijing) Co., Ltd.), and the concentration and purity of RNA were examined using a spectrophotometer. cDNA was synthesized from 1 mg of total RNA using a reverse transcriptase kit (Lot: KR106, Tiangen Biochemical Technology (Beijing) Co., Ltd.). Primer sequences outlined in Table 1 were used to measure and quantify target mRNA levels by the quantitative real-time (RT)-PCR method. The relative mRNA expression levels of STAT3, HIF-1α, Beclin-1, BNIP3, TNF-α, MCP-1, IL-1β, and IL-6 genes were calculated by the 2-△△Ct method after standardization based on the β-actin transcription (Lot: FP206, Tiangen Biochemical Technology (Beijing) Co., Ltd.).

Table 1 Primer sequences to measure mRNA levels using quantitative RT-PCR.
Western blotting
Mouse liver tissue or experimental cells were dissolved in RIPA buffer for 30 min. The supernatant was collected after being centrifuged at 12,000 rpm and 4°C for 10 min. The total protein concentration was determined using the BCA method. The protein samples were separated by 10% SDS-PAGE and electrotransferred to polyvinylidene fluoride (PVDF) membranes. The PVDF membranes were blocked with 5% non-fat dry milk for 2 h at room temperature and then incubated with the primary antibodies (P-STAT3, STAT3, HIF-1α, p62, BNIP3, Beclin-1, LC3 A/B, and P-NF-κB p65 at 1:2,000 dilution, Lot: 9145S; 9139S; 36169S; 8025S; 3769S;3738S; 12741; 3033, Cell Signaling Technology or Bax, Bcl-2, cleaved caspase-3, and β-actin at 1:3,000 dilution, Lot: ab32503; ab59348; ab214430; ab8227, Abcam) overnight at 4°C. After incubation with the secondary antibody (1:10,000) for 1 h, the bands were visualized by an enhanced chemiluminescence system. Each group experiment was repeated three times. Quantitative analysis was performed with ImageJ software (National Institutes of Health, United States).
Fluorescence and flow cytometry
After the indicated treatments, cells were fixed with 4% paraformaldehyde and permeabilized in PBS containing 0.1% Triton X-100 (Sigma-Aldrich). Cells were incubated with rabbit anti-LC3 overnight at 4°C. The next day, a fluorescent secondary antibody (1:100) was added and incubated at 4°C for 1 h in the dark. Images were acquired using a fluorescence microscope. Six to eight different fields were randomly selected from each group, and the average LC3 region fluorescence intensity was analyzed and calculated by ImageJ software. After the culture, the cells in each group were collected and washed twice with PBS. According to the operation procedure of the PE-conjugated Annexin-V Apoptosis Detection Kit (Lot: 559763, Becton, Dickinson and Company), antibody incubation and FACSCalibur flow cytometer on-board detection (Becton, Dickinson and Company) were carried out.
Statistical analysis
GraphPad Prism 5 software was used to analyze the experimental data using a two-tailed Student’s t-test or one-way ANOVA test, and the measurement data were described as mean ± SEM. p < 0.05 indicated a statistically significant difference.
Results
α-GA improves the pathological changes of the liver and abnormal elevated levels of serum biochemical and inflammatory indexes in NAFLD mice
Compared with the control group, the body weight of mice in the NAFLD group increased significantly (p < 0.01), and α-GA could significantly inhibit the weight gain (p < 0.01). Compared with normal mice, a high-fat and high-sugar diet caused a slight increase in the liver index of NAFLD mice, and α-GA could alleviate this trend. However, there was no significant difference between the control group, NAFLD group, and α-GA group (p >0.05). The spleen index and thymus index of the mice in the NAFLD group were significantly higher than those in the control group (p < 0.01), indicating that significant immune enhancement and inflammatory response appeared in NAFLD mice. Compared with the NAFLD group, the spleen index in the α-GA group decreased, but there was no statistical significance (p>0.05). The intervention of α-GA could effectively reduce the abnormal increase of the thymus index in NAFLD mice (p < 0.01). This manifested that GA had certain immunomodulatory and anti-inflammatory effects (Figure 1).
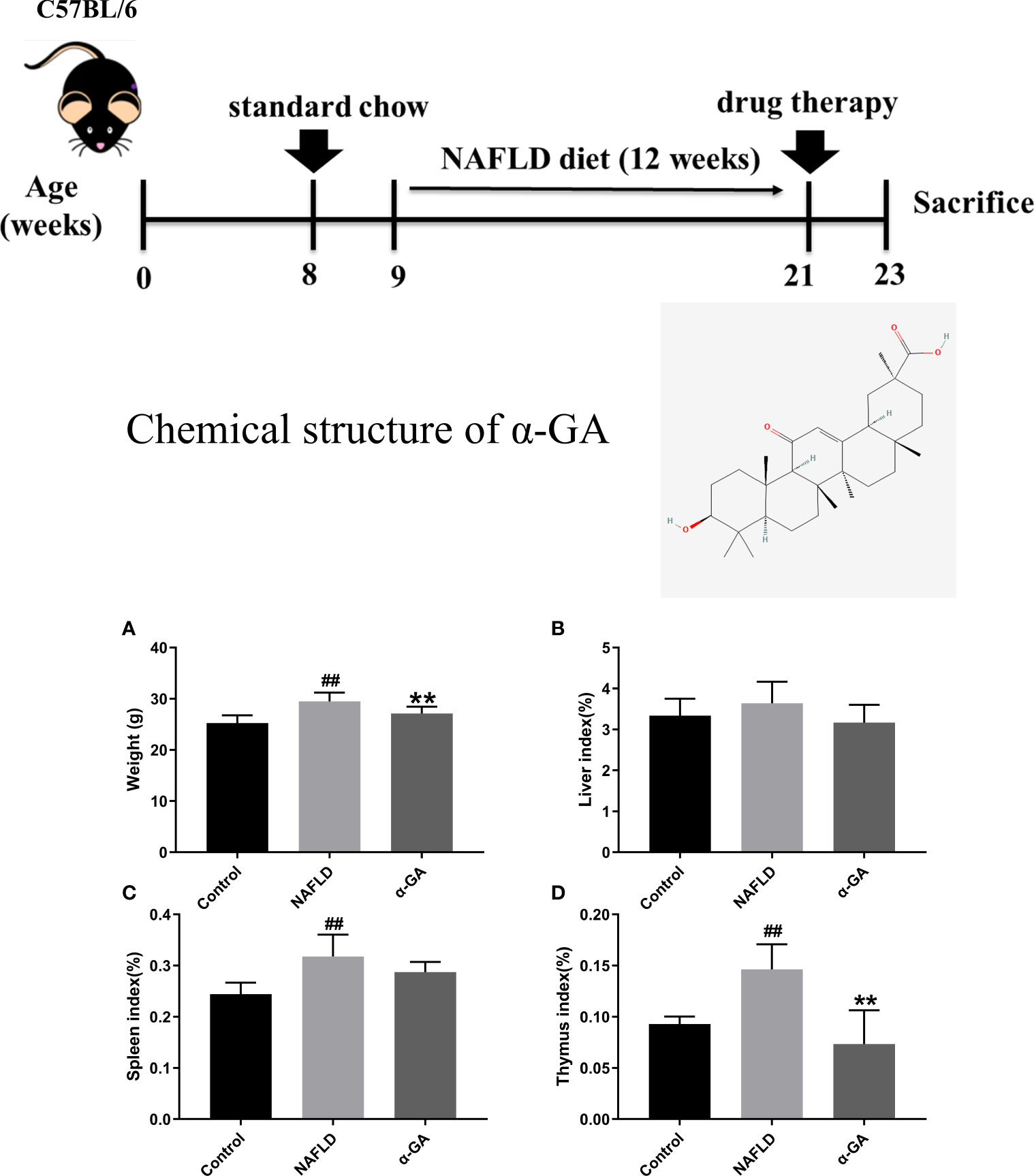
Figure 1 Weight and organ index of mice. (A) Body weight. (B) Liver index. (C) Spleen index. (D) Thymus index. mean ± SEM. #p < 0.05, ##p < 0.01, compared with the control group; *p < 0.05, **p < 0.01, compared with the NAFLD group. n = 6/group.
Hepatic tissue H&E and Oil Red O staining confirmed the significant fat deposition in the liver of NAFLD mice. After 2 weeks of intragastric administration of α-GA, hepatic lipid deposition was improved (Figure 2A). We found that serum AST, ALT, TG, T-CHO, and LDL-C levels were significantly increased (p < 0.01), and the HDL-C level was significantly decreased (p < 0.01) in the NAFLD group. After α-GA intervention, those abnormally elevated levels of serum biochemical indexes were significantly improved (p < 0.01) and had no significant effect on the renal function indexes of CRE and BUN in mice (p > 0.05). α-GA regulated liver lipid metabolism and improved liver cell damage (Figure 2B). Long-term liver fat deposition could lead to liver inflammation. Furthermore, it was found that the mice in the NAFLD group had a strong inflammatory response, with the levels of serum inflammatory indicators IL-1β, IL-4, IL-6, MCP-1, and TNF-α significantly increased (p < 0.01). α-GA demonstrated a good anti-inflammatory effect (p < 0.01) (Figure 2C).
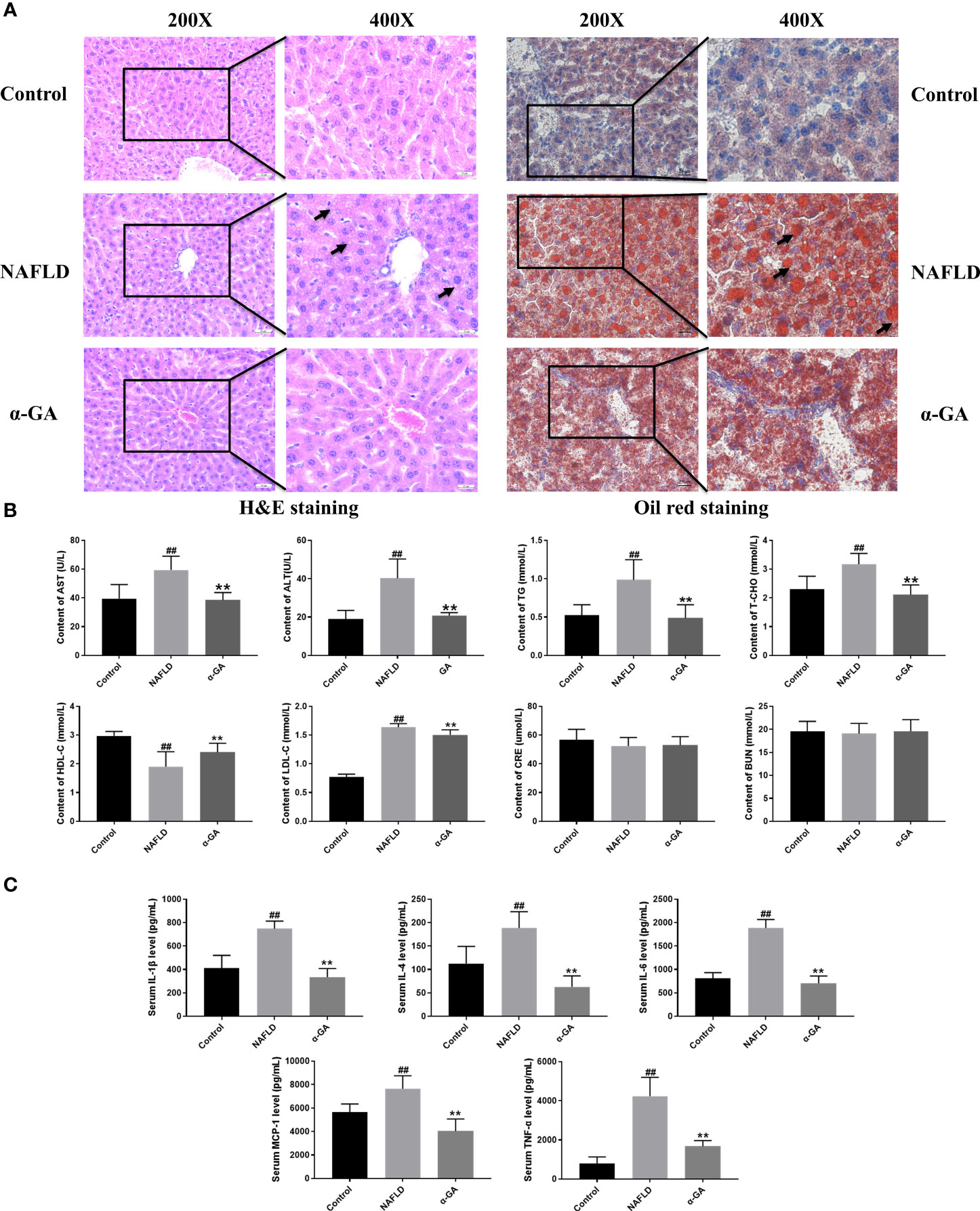
Figure 2 Effects of α-GA on pathological changes of liver and serum biochemical and inflammatory indexes in NAFLD mice. (A) H&E staining and Oil Red O staining. (B) Serum biochemical indexes. (C) Serum inflammatory indexes. mean ± SEM. #p < 0.05, ##p < 0.01, compared with the control group; *p < 0.05, **p < 0.01, compared with the NAFLD group. n = 6/group.
α-GA ameliorates excessive hepatic macrophage infiltration and apoptosis in NAFLD mice
Previous studies have shown that hepatic macrophages primarily expressing F4/80 are responsible for the aggressiveness of liver injury (33). Infiltration of macrophages into the liver is a hallmark and causes hepatic inflammatory injury (34). To investigate whether α-GA plays a potential role in the regulation of hepatic macrophages in NAFLD mice, we detected the expression of F4/80 in liver tissues. Compared with mice in the control group, the infiltration of macrophages in the liver tissues of the mice in the NAFLD group increased (p < 0.01), and the hepatic infiltration of macrophages was significantly decreased after α-GA treatment (p < 0.01). Given the important role of macrophages in NAFLD, it was suggested that the therapeutic effects of α-GA on NAFLD may be related to the regulation of mouse macrophages to exert anti-inflammatory immunity (Figure 3A). Previous results have also shown that GL can prevent neutrophils and macrophages from infiltrating in liver injury (35). At the same time, we also noticed that α-GA could indeed improve the apoptosis of liver tissue in NAFLD mice, accompanied by an increase in the level of Bcl-2 protein (p < 0.01) and a significant decrease in the level of Bax and cleaved-Caspase 3 protein (p < 0.01) (Figures 3B, C).
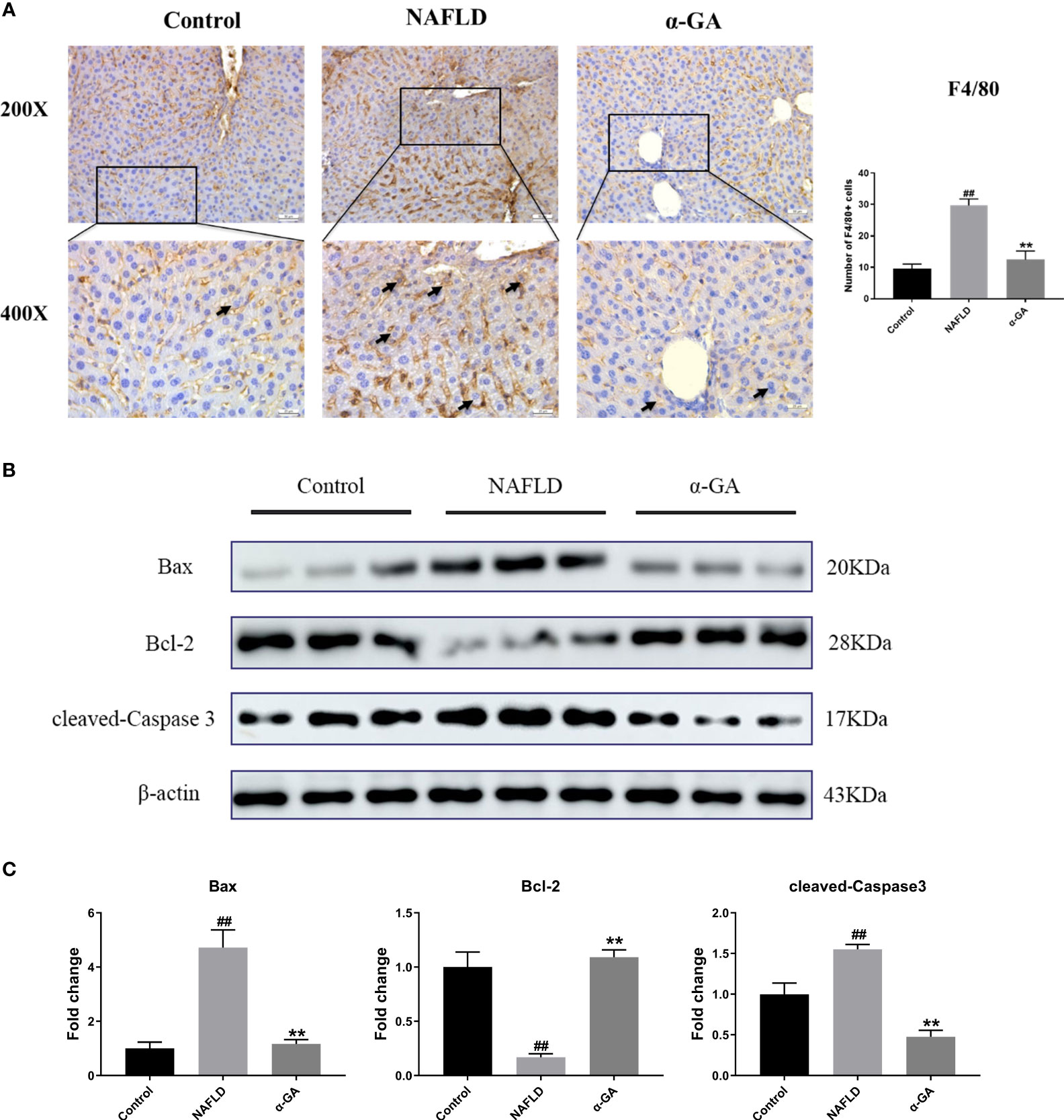
Figure 3 Effects of α-GA on hepatic macrophage infiltration and apoptosis in NAFLD mice. (A) Liver sections were subjected to F4/80 immunohistochemistry (black arrows indicate the F4/80-positive cells). The total number of F4/80-positive cells from five high-powered fields was counted per liver section by microscopy. At least three liver sections were included in each group. (B) Representative apoptosis-related protein expression bands (Bax, Bcl-2, and cleaved-Caspase 3). (C) Quantitative expressional analysis of WB bands. mean ± SEM. #p < 0.05, ##p < 0.01, compared with the control group; *p < 0.05, **p < 0.01, compared with the NAFLD group. n = 6/group.
α-GA modulates the hepatic STAT3-HIF-1α pathway and ameliorates impaired autophagic flux
As shown in Figure 4A, compared with the normal mice, the mRNA expression levels of STAT3, HIF-1α, Beclin-1, BNIP3, TNF-α, MCP-1, IL-1β, and IL-6 in the liver tissue of the NAFLD group mice were significantly increased (p < 0.01), and the expression levels of STAT3-HIF-1α pathway-related genes were significantly decreased after α-GA treatment (p < 0.01). The protein expression levels of STAT3, P-STAT3, HIF-1α, P-p65 (NF-κB activation marker), BNIP3 (the transcriptional target of HIF-1α), Beclin-1 (the autophagy-inducing protein), p62 (a marker of impaired autophagy flux), and LC3II (a marker of autophagy-inducible) were significantly increased (p < 0.05, p < 0.01). The increasing trend was significantly improved after α-GA intervention (p < 0.05, p < 0.01). The protein levels of P-STAT3 and HIF-1α in the liver tissue of NAFLD mice were significantly increased, and the expressions of autophagy markers BNIP3, Beclin-1, p62, and LC3II and the NF-κB activation marker P-p65 were consequently increased, indicating impaired hepatic autophagic flux and excessive inflammation. GA could regulate the STAT3-HIF-1α pathway to improve impaired autophagy flow and excessive inflammatory response in liver tissues of NAFLD mice (Figures 4B, C).
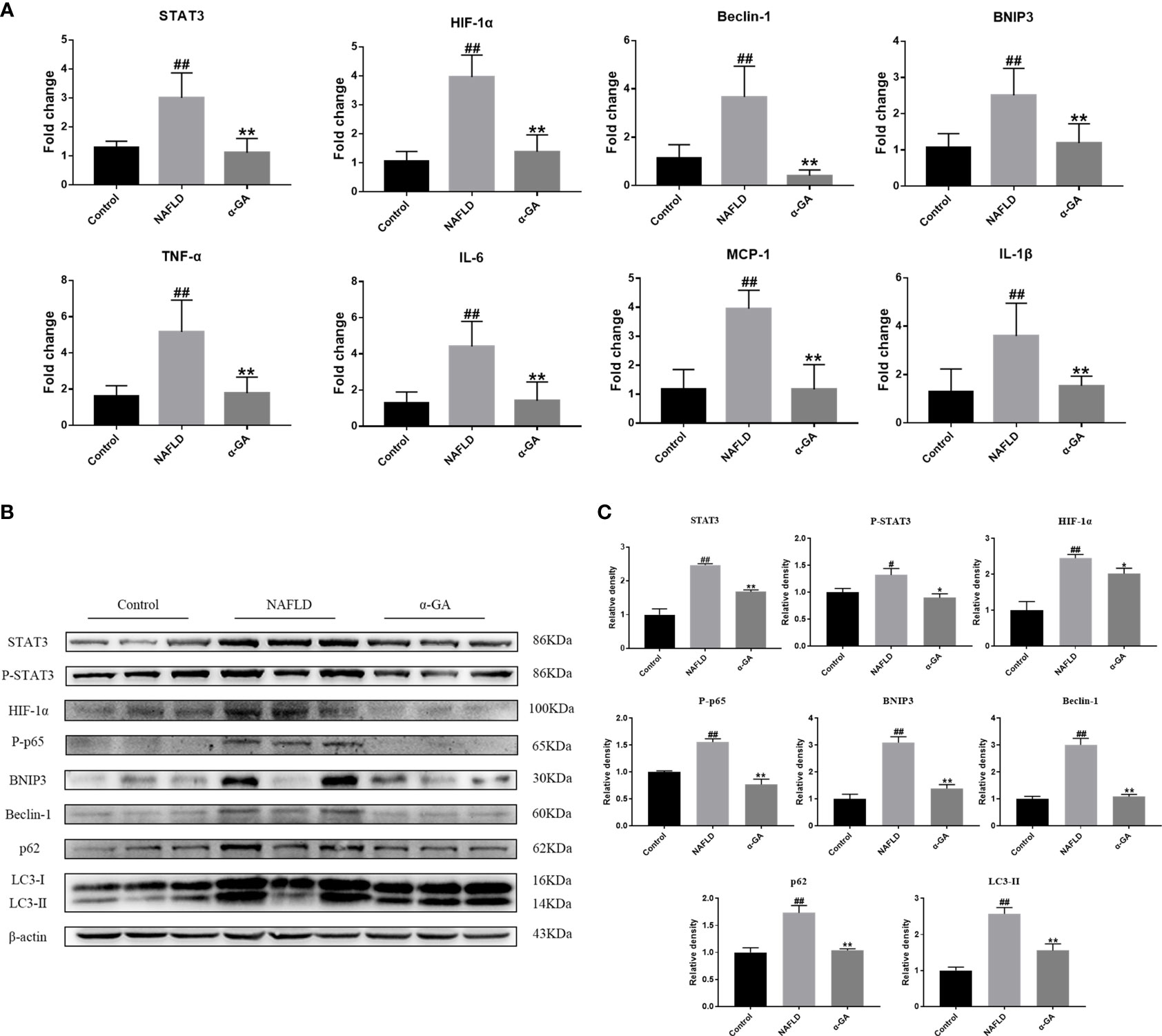
Figure 4 Effects of α-GA on the hepatic STAT3-HIF-1α pathway and autophagic flux. (A) The hepatic expression of STAT3-HIF-1α-related genes was measured by RT-PCR. (B) Representative apoptosis-related protein expression bands (STAT3, P-STAT3, HIF-1α, P-p65, BNIP3, Beclin-1, p62, and LC3II). (C) Quantitative expressional analysis of WB bands. Mean ± SEM. #p < 0.05, ##p < 0.01, compared with the control group; *p < 0.05, **p < 0.01, compared with the NAFLD group. n = 6/group.
α-GA modulates the PA-induced macrophage STAT3-HIF-1α pathway and ameliorates impaired autophagic flux
Hepatic macrophages play a key role in maintaining the homeostasis of the liver and the whole body through five major functions. These include removal of cellular debris and metabolic waste, maintenance of iron homeostasis, regulation of cholesterol homeostasis, modulation of antimicrobial defenses, and promotion of immune tolerance (36–39). To further elucidate the effect of α-GA on macrophages in the progression of NAFLD, first, the effects of PA and α-GA on the viability of RAW264.7 and Kupffer cells were investigated. The results showed that different concentration gradients of PA (100, 200, 300, 400 μM) had no significant effect on the viability (p > 0.05). Different concentrations of α-GA had no significant effect on the cell viability (p > 0.05), except that 20 μM had a certain inhibitory effect on RAW264.7 cell (p < 0.01) (Figure 5A). In order to further investigate the effect of FFAs on macrophages in vivo, the same dose (300 μM) of PA was used to induce macrophages in the subsequent experimental design. α-GA was set to low, medium, and high doses of 2.5, 5, and 10 μM, respectively.
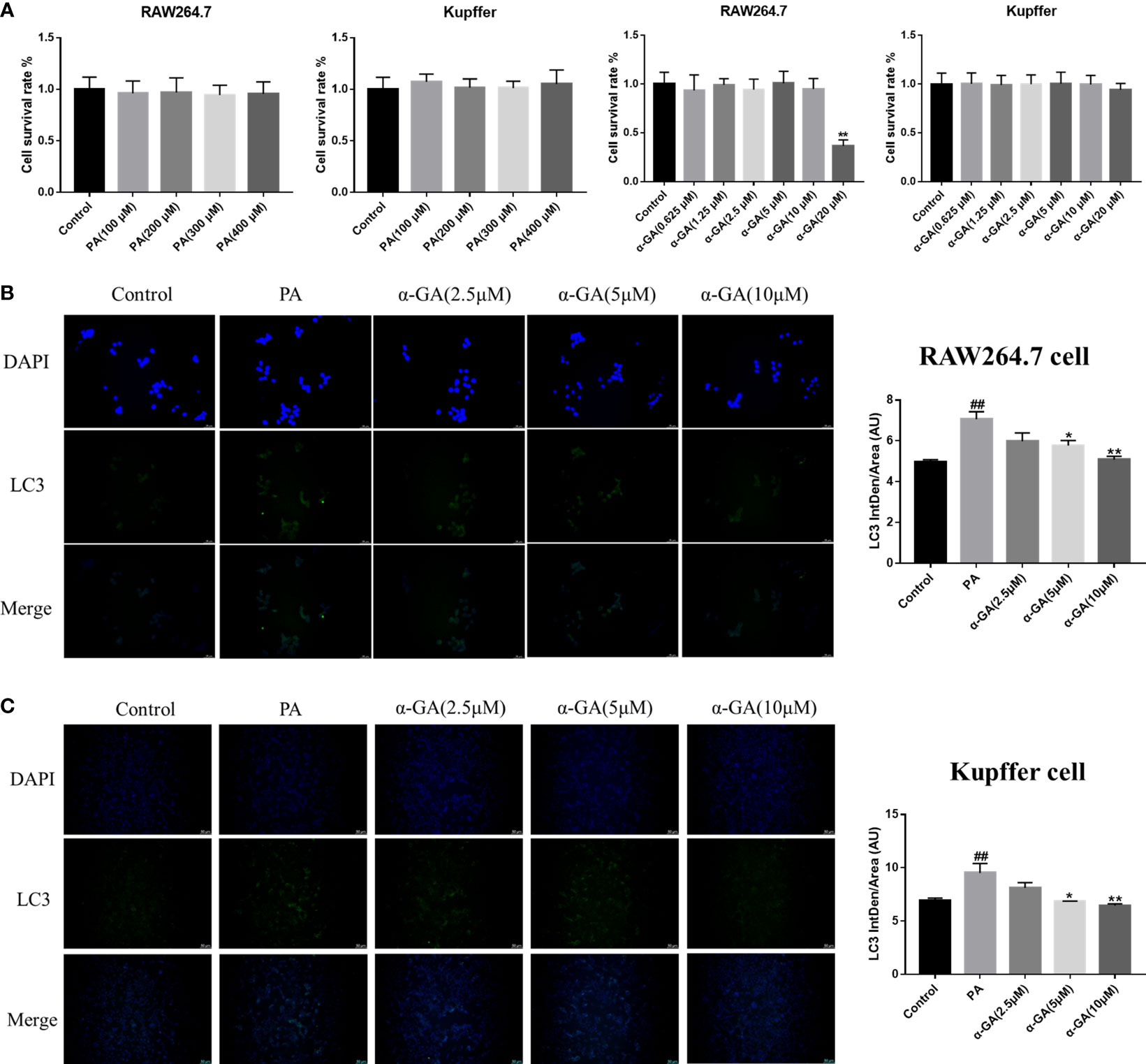
Figure 5 Effects of α-GA on the expression of PA-induced LC3 fluorescent protein in macrophages. (A) Effects of PA and α-GA on RAW264.7 and Kupffer cell viability measured by CCK8. (B) The detection of LC3 puncta in RAW264.7 cells was performed with anti-LC3 antibody and immunofluorescence staining (left). Quantification of LC3 fluorescence intensity (right). (C) The detection of LC3 puncta in Kupffer cells and immunofluorescence staining (left). Quantification of LC3 fluorescence intensity (right). Mean ± SEM. #p < 0.05, ##p < 0.01, compared with the control group; *p < 0.05, **p < 0.01, compared with the NAFLD group. n = 6/group.
After PA induction with/without α-GA intervention in RAW264.7 and Kupffer cells, the changes in the expression of autophagy marker LC3 fluorescent protein were observed. We found that compared with normal cells, the expression of LC3 fluorescent protein was significantly enhanced in PA-induced macrophages (p < 0.01), and α-GA could attenuate this enhanced trend in a concentration-dependent manner (Figures 5B, C).
To clarify whether the enhanced expression of LC3 fluorescent protein represents the activation of autophagy in macrophages, the study further explored the relationship between the STAT3-HIF-1α pathway, autophagy flux, and the pharmacological mechanism of α-GA in PA-induced macrophages. The levels of the STAT3-HIF-1α pathway and autophagy-related genes and proteins were significantly upregulated after PA induction and decreased in a concentration-dependent manner after α-GA intervention (Figures 6A–D). In addition, α-GA could improve the abnormal increase in the levels of inflammatory cytokines such as TNF-α, MCP-1, IL-1β, and IL-6 in the cell supernatant induced by PA (Figures 6E, F).
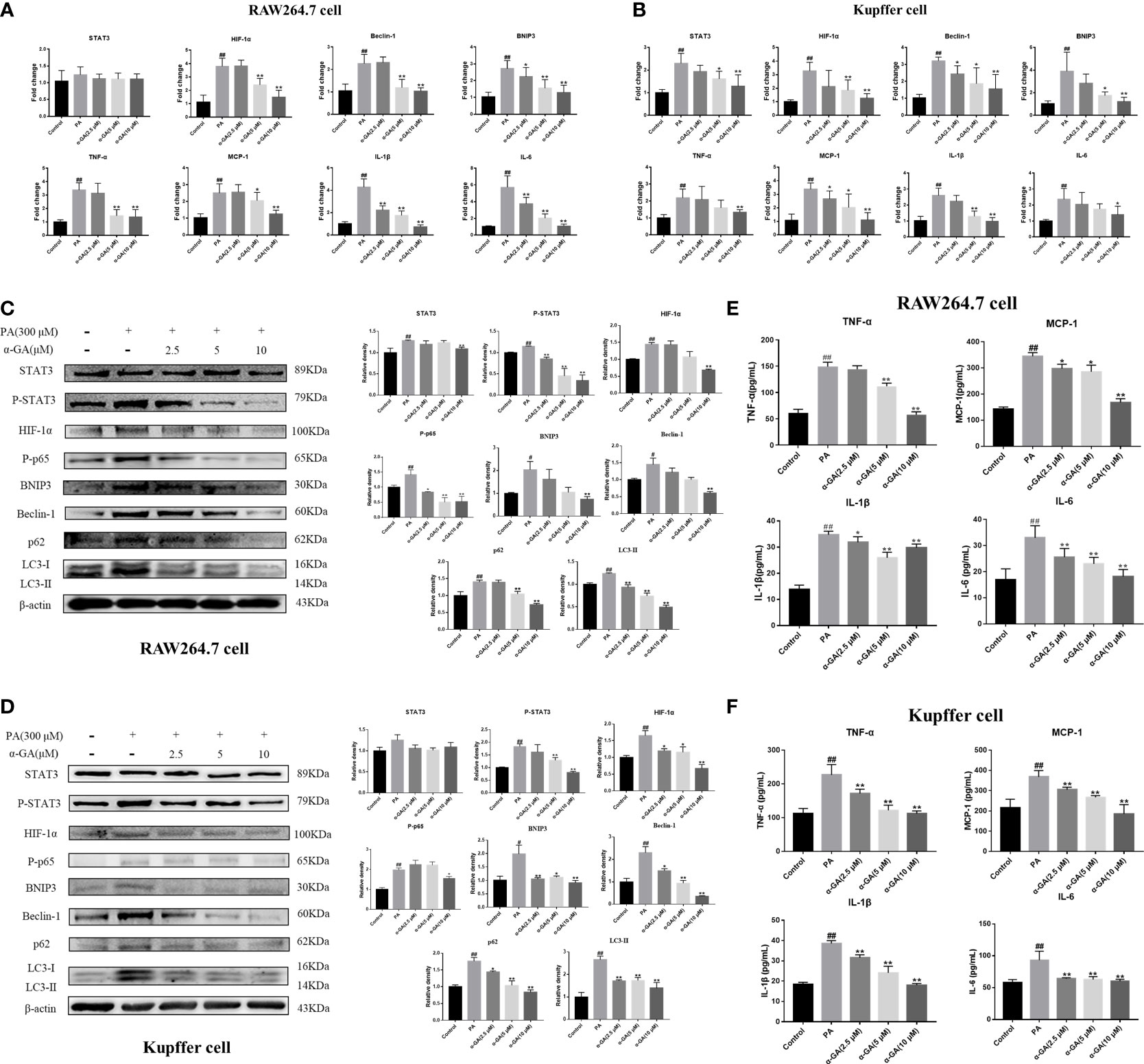
Figure 6 Effects of α-GA on the PA-induced macrophage STAT3-HIF-1α pathway and impaired autophagic flux. (A, B) Effects of α-GA on the STAT3-HIF-1α pathway and autophagy gene expression induced by PA in RAW264.7 and Kupffer cells (STAT3, HIF-1α, Beclin-1, BNIP3, TNF-α, MCP-1, IL-1β, and IL-6). (C, D) Effects of α-GA on the STAT3-HIF-1α pathway and autophagy protein expression induced by PA in RAW264.7 and Kupffer cells (STAT3, P-STAT3, HIF-1α, P-p65, BNIP3, Beclin-1, p62, and LC3II). Representative apoptosis-related protein expression bands (left). Quantitative expressional analysis of WB bands (right). (E, F) Effects of α-GA on the expression of inflammatory cytokines induced by PA in RAW264.7 and Kupffer cells (TNF-α, MCP-1, IL-1β, and IL-6). Mean ± SEM. #p < 0.05, ##p < 0.01, compared with the control group; *p < 0.05, **p < 0.01, compared with the NAFLD group. n = 6/group.
Furthermore, the inhibitors were used to investigate whether α-GA ameliorated the PA-induced impairment of autophagic flux in macrophages through the STAT3-HIF-1α pathway. PA could significantly increase the protein levels of P-STAT3, HIF-1α, p62, and LC3II in RAW264.7 cells (p < 0.05, p < 0.01), and α-GA treatment significantly decreased the expression of these proteins (p < 0.01). Compared with the PA group, the protein levels of P-STAT3, HIF-1α, p62, and LC3II were also significantly reduced after the intervention of the inhibitors Stattic and YC-1, indicating that the PA-induced impaired autophagic flux in macrophages was associated with enhanced STAT3 phosphorylation and HIF-1α hyperactivation. At the same time, there were no significant differences in the protein levels of P-STAT3, HIF-1α, p62, and LC3II between the inhibitor-only intervention group and α-GA with/without Stattic or YC-1 (p > 0.05). Similar findings were also found in the levels of inflammatory cytokines in the cell supernatant. These results suggested that α-GA had similar mechanisms of action to Stattic and YC-1. It could improve the PA-induced impairment of autophagic flux in macrophages and reduce the excessive production of inflammatory cytokines, possibly by inhibiting STAT3 phosphorylation and HIF-1α excessive activation (Figure 7).
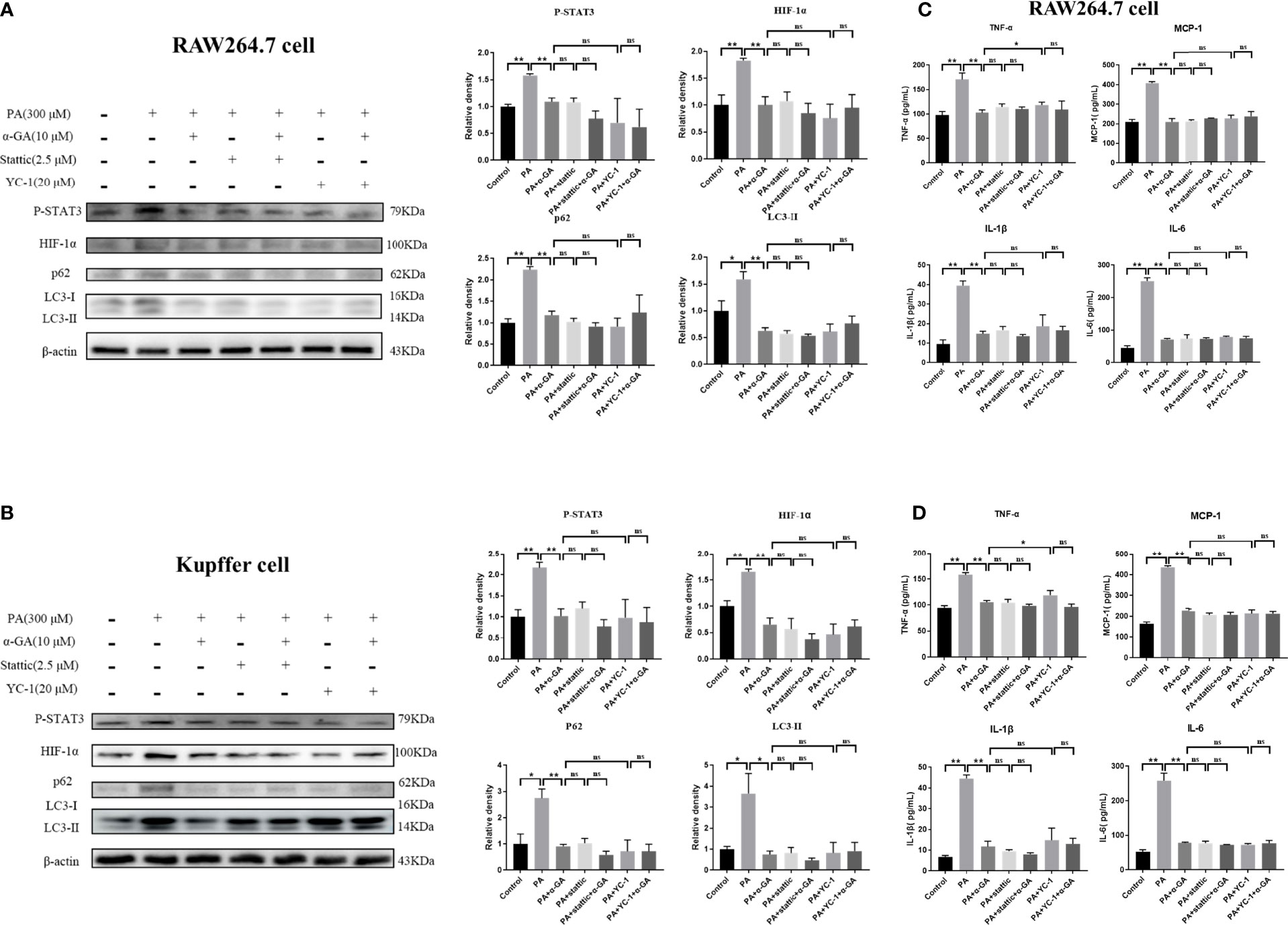
Figure 7 Effects of α-GA on PA-induced impaired autophagic flux in macrophages by regulating STAT3 and HIF-1α. (A, B) The protein expression of P-STAT3, HIF-1α, p62, and LC3II in RAW264.7 and Kupffer cells. Representative apoptosis-related protein expression bands (left). Quantitative expressional analysis of WB bands (right). (C, D) Effects of α-GA on the expression of inflammatory cytokines in RAW264.7 and Kupffer cells (TNF-α, MCP-1, IL-1β, and IL-6). Mean ± SEM. *p < 0.05, **p < 0.01, ns (no significance), p>0.05. n = 6/group.
α-GA ameliorates PA-induced blockade of autophagosome–lysosome fusion in macrophages and inhibits hepatocyte apoptosis
Autophagy of macrophages can significantly affect pathogen-recognition receptors (40). As ligands of Toll-like receptors, saturated fatty acids are associated with the pathogenesis of NAFLD. In order to further clarify the mechanism of PA on the impaired autophagic flux of macrophages and the regulatory effect of α-GA, bafilomycin A1 (interferes with the fusion of autophagosomes and lysosomes) and 3-MA (blocks autophagosome formation) were applied. In RAW264.7 cells, bafilomycin A1 or 3-MA combined with PA induction had no significant difference in p62 and LC3II protein levels compared with a single PA induction (p > 0.05), indicating that the PA-induced impairment of autophagic flux may be related to the blocking of autophagosomes formation and the fusion of autophagosomes and lysosomes. Compared with the bafilomycin A1 group, α-GA supplement could significantly reduce the p62 and LC3II protein levels (p < 0.05, p < 0.01). However, compared with the 3-MA group, α-GA supplement significantly decreased the LC3 II protein level (p < 0.01) but had no significant effect on the P62 protein level (p > 0.05), suggesting that α-GA regulated the above two autophagy processes but mainly improved the blockade of the autophagosome–lysosome fusion (Figure 8A).
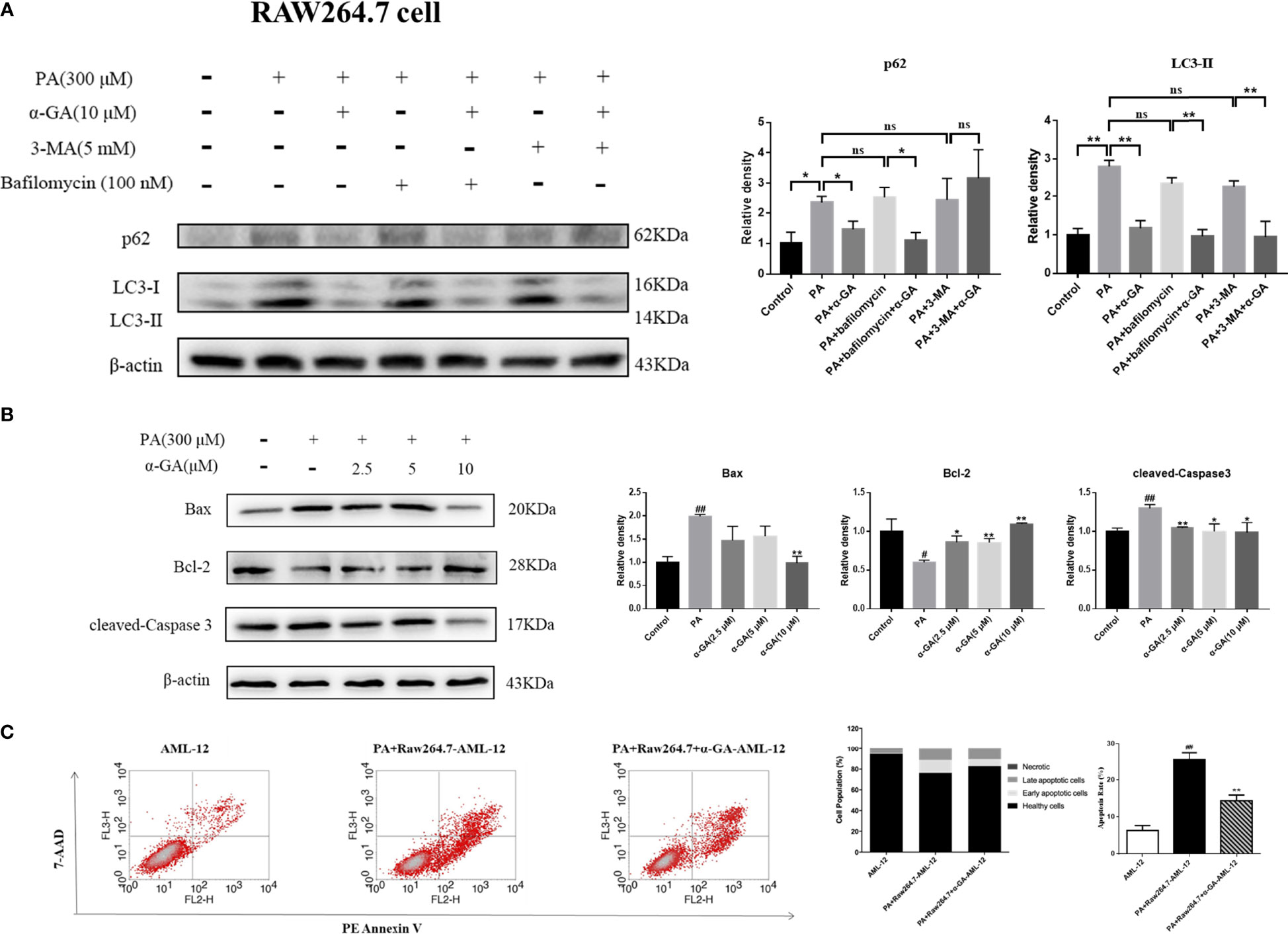
Figure 8 Effects of α-GA on hepatocyte apoptosis by regulating autophagic flux in RAW264.7 cells. (A) The protein expression of p62 and LC3II. The RAW264.7 cells were treated with bafilomycin A1 (interferes with the fusion of autophagosomes and lysosomes) and 3-MA (blocks autophagosome formation), with or without α-GA. Representative apoptosis-related protein expression bands (left). Quantitative expressional analysis of WB bands (right). *p < 0.05, **p < 0.01, ns (no significance), p>0.05. n = 6/group. (B) Effects of specifically treated RAW264.7 cell supernatants on the proteins associated with apoptosis in AML-12 cells (Bax, Bcl-2, and cleaved-Caspase 3). Representative apoptosis-related protein expression bands (left). Quantitative expressional analysis of WB bands (right). (C) The apoptosis rate of AML-12 cells was measured by flow cytometry. Mean ± SEM. #p < 0.05, ##p < 0.01, compared with the control group; *p < 0.05, **p < 0.01, compared with the NAFLD group. n = 6/group.
In Kupffer cells, there was no significant difference in p62 protein level between bafilomycin A1 or 3-MA combined with PA and PA alone (p > 0.05). Compared with PA alone, there was no significant difference in the LC3 II protein level in bafilomycin A1 combined with PA induction (p > 0.05), but there was an increase in 3-MA combined with PA induction (p < 0.05). These results indicated that the PA-induced impaired autophagy flux in Kupffer cells was mainly related to the blocking of the fusion of autophagosome and lysosome. Overall, α-GA could effectively ameliorate the PA-induced impairment of autophagic flux in macrophages, and its mechanism was mainly related to the promotion of interference with the fusion of autophagosomes and lysosomes (Figure 9A).
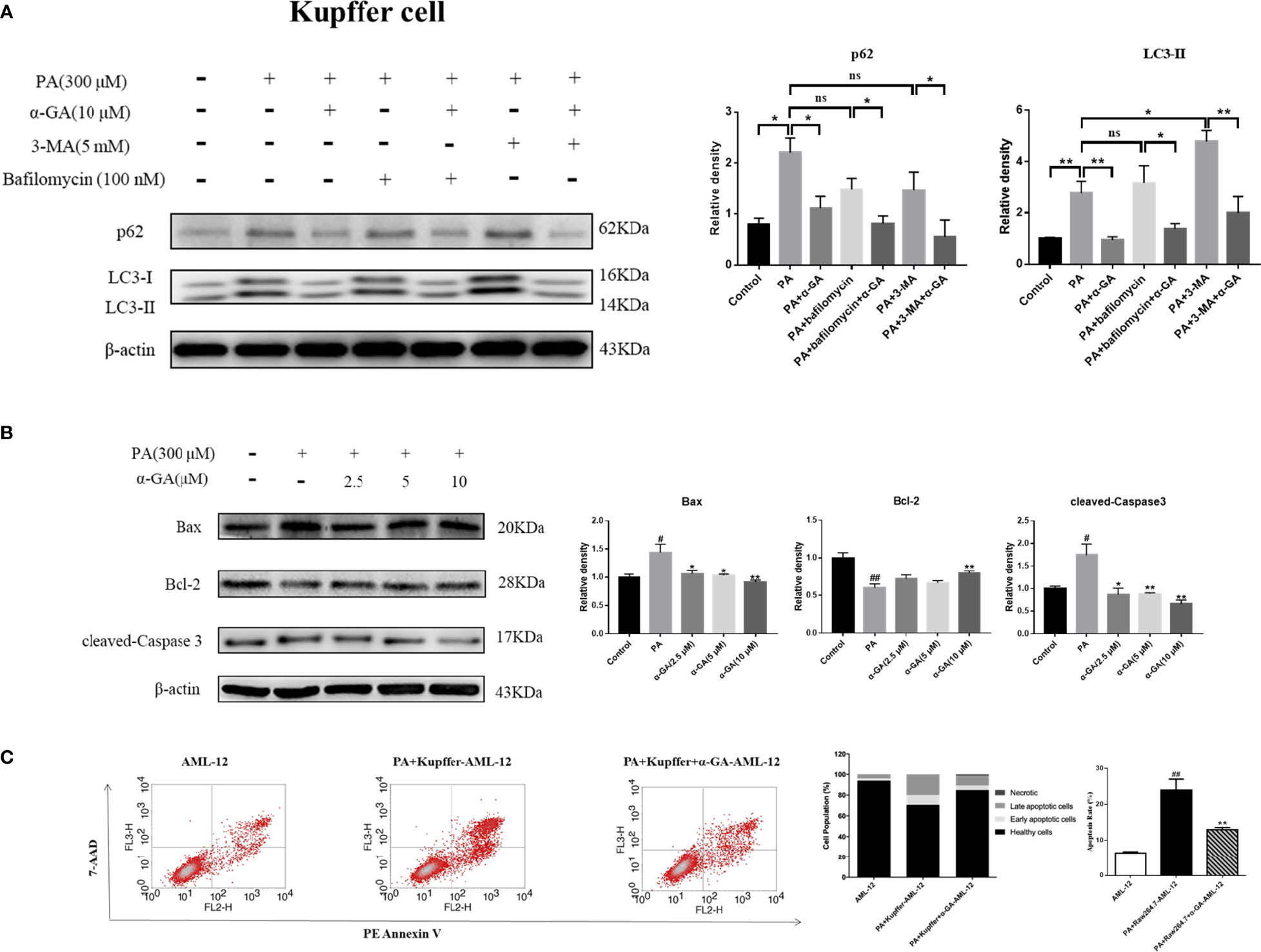
Figure 9 Effects of α-GA on hepatocyte apoptosis by regulating autophagic flux in Kupffer cells. (A) The protein expression of p62 and LC3II. The Kupffer cells were treated with bafilomycin A1 (interferes with the fusion of autophagosomes and lysosomes) and 3-MA (blocks autophagosome formation), with or without α-GA. Representative apoptosis-related protein expression bands (left). Quantitative expressional analysis of WB bands (right). *p < 0.05, **p < 0.01, ns (no significance), p>0.05. n = 6/group. (B) Effects of specific treated Kupffer cells supernatants on the proteins associated with apoptosis in AML-12 cells (Bax, Bcl-2, and cleaved-caspase 3). Representative apoptosis-related protein expression bands (left). Quantitative expressional analysis of WB bands (right). (C) The apoptosis rate of AML-12 cells was measured by flow cytometry. mean ± SEM. #p < 0.05, ##p < 0.01, compared with the control group; *p < 0.05, **p < 0.01, compared with the NAFLD group. n = 6/group.
What is the effect of α-GA on hepatocytes by improving PA-induced impairment to macrophage autophagy flux? After intervention for 24 h of normal medium, PA, and PA plus α-GA (2.5, 5, and 10 μM), the supernatant of macrophages was then extracted after 24 h of starvation treatment, which corresponded to the configuration of AML-12 hepatocyte medium. The supernatant of each group was mixed with normal AML-12 cell culture medium (1:1) and added to the cells for 24 h of intervention culture, and then the apoptosis was investigated. Macrophage supernatant induced by PA could lead to excessive apoptosis of hepatocytes; specifically, Bax and cleaved-caspase 3 protein levels were significantly increased and Bcl-2 protein levels were significantly decreased, which were significantly reversed after intervention with different doses of α-GA (Figure 8B, Figure 9B). Further, we analyzed the effect of two types of treated macrophage supernatants on hepatocyte apoptosis rate by flow cytometry. The specifically treated RAW264.7 cell and Kupffer cell supernatants caused 25.56% and 23.91% of hepatocyte apoptosis, respectively, while the supernatants treated with α-GA (10 μM) had significantly reduced apoptotic rates of 14.41% and 12.88% (Figure 8C, Figure 9C).
Discussion
Compared with other organs, the liver is rich in macrophages, and the sources include Kupffer and myeloid-derived monocytes/macrophages from the fetal yolk sac. Liver Kupffer cells and monocyte-derived macrophages integrate signals from the gut–liver axis, overnutrition, systemic low-grade inflammation, and steatosis, driving the progression of NAFLD and even liver fibrosis (10, 41). Clinically, NAFLD often has complications of obstructive sleep apnea, leading to periodic hypoxia. Obstructive sleep apnea and nocturnal hypoxia are important triggers for the progression of NAFLD (42). Intermittent hypoxia has been shown to cause tissue hypoxia and may contribute to abnormal hepatic lipid metabolism, mitochondrial dysfunction, oxidative stress, insulin resistance, inflammation, and hyperactivation of the sympathetic nervous system, contributing to the progression of NAFLD (43). HIF has been extensively studied in cancer, but there is evidence that this pathway has an important role in controlling metabolism and affecting NAFLD and Mets (44). Autophagy is an important regulatory mechanism to maintain cellular homeostasis and plays a key role in the occurrence and development of NAFLD. Impaired autophagy (decreased autophagy or blocked autophagic flux) leads to cell damage and death and promotes liver disease progression, a phenomenon well established in NAFLD livers, hepatocytes, endothelial cells, and hepatic stellate cells (45–48). Growing evidence from animal and clinical studies suggests that targeting autophagy of macrophages may be an effective therapeutic strategy for NAFLD and related Mets (49).
Research evidence shows that primary bone marrow-derived macrophages and peritoneal macrophage autophagic flux levels are significantly reduced in mice fed a high-fat diet, suggesting a general impairment of macrophage autophagy in obese mice. Excessive lipid accumulation reduces the level of autophagy, and impaired macrophage autophagy may promote the activation of innate immunity leading to obesity, mainly manifested in abnormal polarization of macrophages (increased M1 type and decreased M2 type). This suggests that autophagy has an important regulatory function in macrophage polarization and can downregulate inflammatory responses. Impaired macrophage autophagy may underlie the body’s inflammatory state, which in turn leads to the progression of liver inflammation and liver injury (50).
Our results suggested that GA could improve liver pathological changes and lipid metabolism abnormalities in NAFLD mice. GA reduced excessive infiltration of macrophages in the liver and excessive apoptosis of liver cells caused by a high-fat and high-sugar diet. Further investigation found that GA could improve the impaired autophagy of macrophages, which was related to the regulation of STA3-HIF-1α. (Figure 10). STAT3 is involved in the whole process of assembly and maturation from autophagosomes, and the activation of STAT3 transcriptional activity mainly depends on the phosphorylation of a single tyrosine residue Tyr705, which can be controlled by receptor tyrosine kinases such as MET, KDR, and EGFR, or non-receptor tyrosine kinases. It is directly catalyzed by tyrosine kinases such as JAKs, and phosphorylation at Ser727 of serine determines its maximal activation state. Nuclear STAT3 binds to specific DNA components to transcriptionally activate or repress target genes, such as BCL2, BECN1, PIK3C3, CTSB, CTSL, PIK3R1/p55α, PIK3R1/p50α, and MIR17HG and HIF-1α and BNIP3, depending on the cellular environment or stimulus, inhibit or stimulate autophagy. STAT3 monomers can also be transferred to mitochondria, interact with electron transport chain complexes I and II, inhibit the production of reactive oxygen species, reduce HIF-1α activity, and inhibit autophagy (51). The regulation of HIF-1α by STAT3 is divided into two aspects. On the one hand, STAT3 transcriptionally upregulates HIF-1α gene expression. On the other hand, STAT3 interacts with the C-terminal domain of HIF-1α and stabilizes the protein through von Hippel–Lindau-mediated ubiquitination. In NASH patients as well as mouse liver and cell models, the expression of STAT3 and p-STAT3 is increased, and autophagy is inhibited. Downregulation of STAT3 expression can activate autophagy and inhibit the inflammatory response of NASH (52, 53). In NASH, FFAs are important lipotoxic mediators that lead to cellular damage and induce liver damage through endoplasmic reticulum stress. Hypoxia enhances PA-induced activation of the pro-inflammatory state of human macrophages (54). Previous studies have also shown that in hepatic steatosis, HIF-1α and autophagy are involved in FFA-induced cellular stress in hepatocytes (55, 56). Recent results further confirmed that α-GA inhibits STAT3 Tyr705 phosphorylation by increasing protein tyrosine phosphatase 1 and 2 expressions and inhibits TGF-β-triggered hepatocellular carcinoma invasion and metastasis in vivo and in vitro (57). In addition, GA inhibits phosphorylation at Ser727 of STAT3 (58). This is consistent with previous reports that GA inhibits STAT3 phosphorylation in the treatment of colon cancer (59), psoriasis (60), and hepatocellular carcinoma (57). A recent study in China shows that GL reduces the expression level of HIF-1α, inhibits the release of inflammatory cytokines IL-6 and TNF-α, and plays protective roles in acute lung injury.
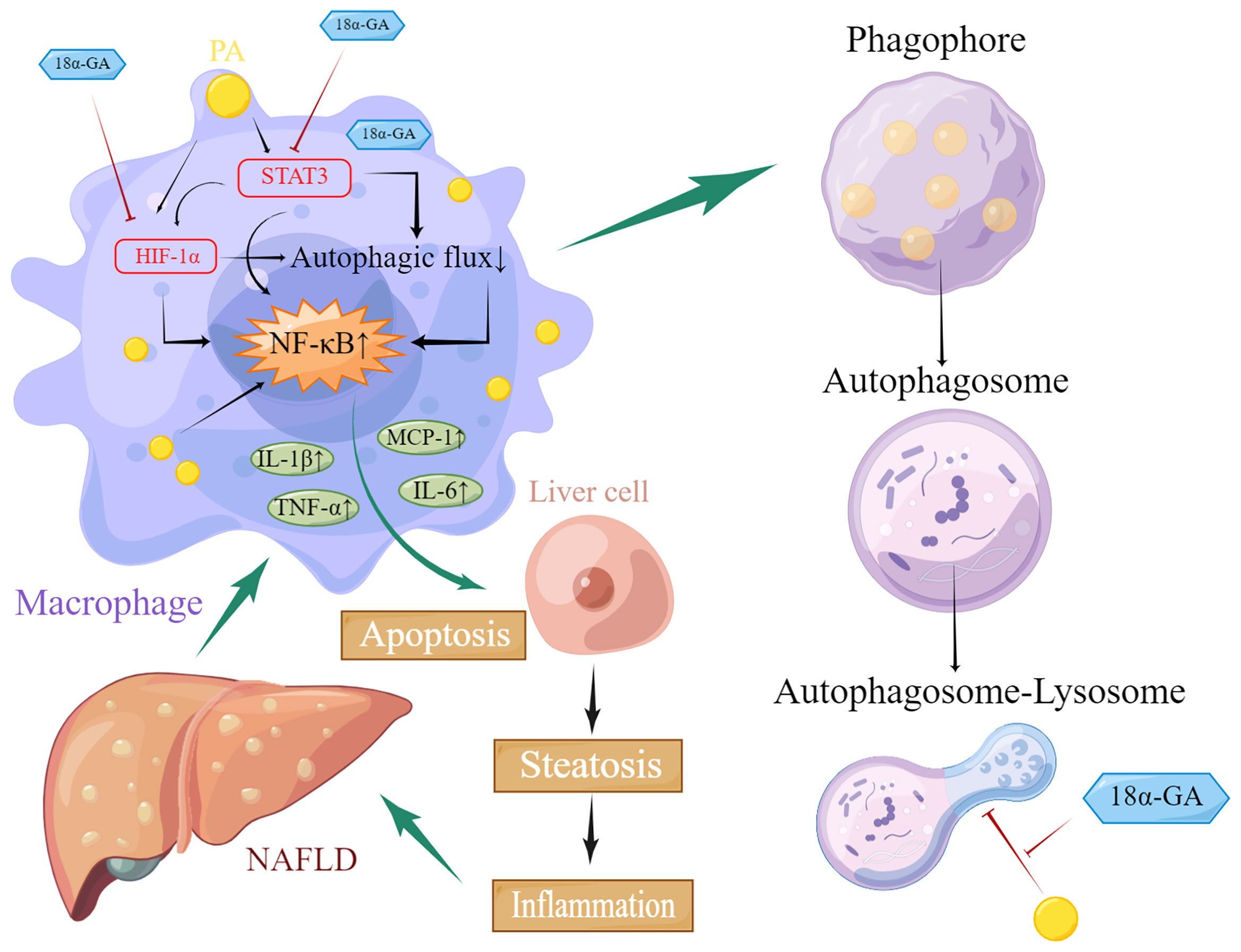
Figure 10 Schematic diagram of glycyrrhetinic acid (α-GA) regulating impaired macrophage autophagic flux in treatment of non-alcoholic fatty liver disease (NAFLD). Excess lipids (palmitic acid, PA) accumulate in macrophages, causing STAT3 phosphorylation and HIF-1α hyperactivation, impairing its autophagic flux, mainly hindering the fusion of autophagosomes and lysosomes. The subsequent enhancement of autoimmunity especially the overactivation of NF-κB led to the excessive release of inflammatory cytokines including TNF-α, MCP-1, IL-1β, and IL-6, which caused excessive apoptosis of hepatocytes and impaired functions of liver tissues through paracrine, thereby triggering NAFLD. α-GA can inhibit the impaired autophagic flux and excessive production of inflammatory cytokines in macrophages caused by NAFLD by regulating the overactivation of the STAT3-HIF-1α pathway in macrophages and improving the excessive apoptosis of hepatocytes, thereby exerting the therapeutic effect of NAFLD.
Due to their central position in the hepatic microenvironment, their long cytoplasmic protrusions, and the high density of pattern recognition receptors on their surface, hepatic macrophages act as initial sensors of liver injury (61, 62). The activation of hepatic macrophages and subsequent secretion of pro-inflammatory mediators (dependent on NLRP3 and NF-κB activation) lead to increased lipid accumulation and damage in hepatocytes, which are key events in NAFLD development and progression (63). During NAFLD/NASH, reactive oxygen species and damage-associated molecular patterns released from injured hepatocytes undergoing apoptosis or necrosis trigger macrophages secrete a variety of chemokines to recruit monocytes and other leukocytes, leading to insulin resistance and oversecretion of proinflammatory and chemokines such as TNF-α, IL-6, IL-1β, and MCP-1 (64). Hepatic macrophages are a major source of MCP-1, which will further recruit CCR2+ monocytes to diseased livers (65). Contributions of macrophage-derived cytokines (TNF-α, IFN-γ, and IL-1β), chemokines (MCP-1, IL-6), and reactive oxygen/nitrogen species lead to hepatocyte death and recruitment of additional immune cells that booster liver damage. Macrophages switch their functional characteristics from pro-inflammatory to anti-inflammatory, thereby promoting a resolution response to liver injury (66, 67). Autophagy is a lysosomal degradation pathway of cellular components and exhibits anti-inflammatory properties in macrophages. Studies have shown that macrophage autophagy can be used as a novel anti-inflammatory pathway to regulate NAFLD/NASH and liver fibrosis (68–71). GL/GA has been shown to play a key role in metabolic diseases, cancers, and respiratory diseases through the regulation of inflammatory signaling pathways such as NLRP3 and NF-κB (27, 72, 73). Our results also showed that GA regulated the STAT3-HIF-1α pathway to inhibit the phosphorylation of NF-κB. In the early stage of NAFLD, hepatic macrophages secrete MCP-1 to recruit monocyte-derived macrophages that promote inflammation and fibrosis. Meanwhile, impaired autophagy of hepatic macrophages leads to the release of pro-inflammatory cytokines and chemokines (TNF-α, IL-6, and IL-1β), leading to excessive apoptosis of liver cells and activation of hepatic stellate cells, which drives the progress of NAFLD/NASH (62).
Conclusion
In all, this study further demonstrated that α-GA could regulate the STAT3-HIF-1α pathway of macrophages, ameliorate the impaired autophagy flux, and reduce excessive production of inflammatory cytokines to improve the excessive apoptosis of liver cells, thus playing a therapeutic role in NAFLD.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
Ethics statement
The animal study was reviewed and approved by the Experimental animal ethics committee of the Institute of Tianjin University of Traditional Chinese Medicine. Mouse experimental Ethics: TCM-LAEC2021224.
Author contributions
YF, LY, and YB conceived and designed the experiments; YF, WD, YW, SZ, and XZ performed the experiments; YF, WD, YY, and RC analyzed the data; YF, YW, LY, and YB wrote the manuscript. All authors have read and approved the final manuscript.
Funding
This work was supported by the National Key R&D Program of China (2018YFC1706506), 2019 Graduate Innovation Fund of Tianjin University of Traditional Chinese Medicine School of Integrated Traditional Chinese and Western Medicine (ZXYCXLX201907), and 2020 Tianjin Postgraduate Research and Innovation Project (2020YJSB189 and YJSKC-20201003).
Acknowledgments
We are very grateful for the mice that were sacrificed in this experiment.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ye Q, Zou B, Yeo YH, Li J, Huang DQ, Wu Y, et al. Global prevalence, incidence, and outcomes of non-obese or lean non-alcoholic fatty liver disease: a systematic review and meta-analysis. Lancet Gastroenterol Hepatol (2020) 5(8):739–52. doi: 10.1016/S2468-1253(20)30077-7
2. Zhou F, Zhou J, Wang W, Zhang XJ, Ji YX, Zhang P, et al. Unexpected rapid increase in the burden of NAFLD in China from 2008 to 2018: A systematic review and meta-analysis. Hepatology. (2019) 70(4):1119–33. doi: 10.1002/hep.30702
3. Li J, Zou B, Yeo YH, Feng Y, Xie X, Lee DH, et al. Prevalence, incidence, and outcome of non-alcoholic fatty liver disease in Asia, 1999-2019: a systematic review and meta-analysis. Lancet Gastroenterol Hepatol (2019) 4(5):389–98. doi: 10.1016/S2468-1253(19)30039-1
4. Lonardo A, Nascimbeni F, Mantovani A, Targher G. Hypertension, diabetes, atherosclerosis and NASH: Cause or consequence? J Hepatol (2018) 68(2):335–52. doi: 10.1016/j.jhep.2017.09.021
5. Canfora EE, Meex RCR, Venema K, Blaak EE. Gut microbial metabolites in obesity, NAFLD and T2DM. Nat Rev Endocrinol (2019) 15(5):261–73. doi: 10.1038/s41574-019-0156-z
6. Loomba R, Friedman SL, Shulman GI. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell. (2021) 184(10):2537–64. doi: 10.1016/j.cell.2021.04.015
7. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med (2018) 24(7):908–22. doi: 10.1038/s41591-018-0104-9
8. Bernales S, McDonald KL, Walter P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol (2006) 4(12):e423. doi: 10.1371/journal.pbio.0040423
9. Czaja MJ. Function of autophagy in nonalcoholic fatty liver disease. Dig Dis Sci (2016) 61(5):1304–13. doi: 10.1007/s10620-015-4025-x
10. Tosello-Trampont AC, Landes SG, Nguyen V, Novobrantseva TI, Hahn YS. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-alpha production. J Biol Chem (2012) 287(48):40161–72. doi: 10.1074/jbc.M112.417014
11. He G, Karin M. NF-kappaB and STAT3 - key players in liver inflammation and cancer. Cell Res (2011) 21(1):159–68. doi: 10.1038/cr.2010.183
12. Dhar D, Antonucci L, Nakagawa H, Kim JY, Glitzner E, Caruso S, et al. Liver cancer initiation requires p53 inhibition by CD44-enhanced growth factor signaling. Cancer Cell (2018) 33(6):1061–77.e6. doi: 10.1016/j.ccell.2018.05.003
13. Lai CY, Yeh KY, Lin CY, Hsieh YW, Lai HH, Chen JR, et al. MicroRNA-21 plays multiple oncometabolic roles in the process of NAFLD-related hepatocellular carcinoma via PI3K/AKT, TGF-beta, and STAT3 signaling. Cancers (Basel) (2021) 13(5):940. doi: 10.3390/cancers13050940
14. Jung KH, Yoo W, Stevenson HL, Deshpande D, Shen H, Gagea M, et al. Multifunctional effects of a small-molecule STAT3 inhibitor on NASH and hepatocellular carcinoma in mice. Clin Cancer Res (2017) 23(18):5537–46. doi: 10.1158/1078-0432.CCR-16-2253
15. Banini BA, Kumar DP, Cazanave S, Seneshaw M, Mirshahi F, Santhekadur PK, et al. Identification of a metabolic, transcriptomic, and molecular signature of patatin-like phospholipase domain containing 3-mediated acceleration of steatohepatitis. Hepatology. (2021) 73(4):1290–306. doi: 10.1002/hep.31609
16. Peng PH, Chieh-Yu Lai J, Hsu KW, Wu KJ. Hypoxia-induced lncRNA RP11-390F4.3 promotes epithelial-mesenchymal transition (EMT) and metastasis through upregulating EMT regulators. Cancer Lett (2020) 483:35–45. doi: 10.1016/j.canlet.2020.04.014
17. Peng X, Gao H, Xu R, Wang H, Mei J, Liu C. The interplay between HIF-1alpha and noncoding RNAs in cancer. J Exp Clin Cancer Res (2020) 39(1):27. doi: 10.1186/s13046-020-1535-y
18. Lee YS, Kim JW, Osborne O, Oh DY, Sasik R, Schenk S, et al. Increased adipocyte O2 consumption triggers HIF-1alpha, causing inflammation and insulin resistance in obesity. Cell. (2014) 157(6):1339–52. doi: 10.1016/j.cell.2014.05.012
19. Csak T, Bala S, Lippai D, Satishchandran A, Catalano D, Kodys K, et al. microRNA-122 regulates hypoxia-inducible factor-1 and vimentin in hepatocytes and correlates with fibrosis in diet-induced steatohepatitis. Liver Int (2015) 35(2):532–41. doi: 10.1111/liv.12633
20. Sundaram SS, Swiderska-Syn M, Sokol RJ, Halbower AC, Capocelli KE, Pan Z, et al. Nocturnal hypoxia activation of the hedgehog signaling pathway affects pediatric nonalcoholic fatty liver disease severity. Hepatol Commun (2019) 3(7):883–93. doi: 10.1002/hep4.1354
21. Song S, Yuan P, Wu H, Chen J, Fu J, Li P, et al. Dendritic cells with an increased PD-L1 by TGF-beta induce T cell anergy for the cytotoxicity of hepatocellular carcinoma cells. Int Immunopharmacol (2014) 20(1):117–23. doi: 10.1016/j.intimp.2014.02.027
22. Niu G, Briggs J, Deng J, Ma Y, Lee H, Kortylewski M, et al. Signal transducer and activator of transcription 3 is required for hypoxia-inducible factor-1alpha RNA expression in both tumor cells and tumor-associated myeloid cells. Mol Cancer Res (2008) 6(7):1099–105. doi: 10.1158/1541-7786.MCR-07-2177
23. Chen K, Yang R, Shen FQ, Zhu HL. Advances in pharmacological activities and mechanisms of glycyrrhizic acid. Curr Med Chem (2020) 27(36):6219–43. doi: 10.2174/0929867325666191011115407
24. Sun X, Duan X, Wang C, Liu Z, Sun P, Huo X, et al. Protective effects of glycyrrhizic acid against non-alcoholic fatty liver disease in mice. Eur J Pharmacol (2017) 806:75–82. doi: 10.1016/j.ejphar.2017.04.021
25. Wang C, Duan X, Sun X, Liu Z, Sun P, Yang X, et al. Protective effects of glycyrrhizic acid from edible botanical glycyrrhiza glabra against non-alcoholic steatohepatitis in mice. Food Funct (2016) 7(9):3716–23. doi: 10.1039/c6fo00773b
26. Lu L, Hao K, Hong Y, Liu J, Zhu J, Jiang W, et al. Magnesium isoglycyrrhizinate reduces hepatic lipotoxicity through regulating metabolic abnormalities. Int J Mol Sci (2021) 22(11):5884. doi: 10.3390/ijms22115884
27. Yan T, Wang H, Cao L, Wang Q, Takahashi S, Yagai T, et al. Glycyrrhizin alleviates nonalcoholic steatohepatitis via modulating bile acids and meta-inflammation. Drug Metab Dispos (2018) 46(9):1310–9. doi: 10.1124/dmd.118.082008
28. Shi L, Guo S, Zhang S, Gao X, Liu A, Wang Q, et al. Glycyrrhetinic acid attenuates disturbed vitamin a metabolism in non-alcoholic fatty liver disease through AKR1B10. Eur J Pharmacol (2020) 883:173167. doi: 10.1016/j.ejphar.2020.173167
29. Yin XR, Gong X, Zhang L, Jiang R, Kuang G, Wang B, et al. Glycyrrhetinic acid attenuates lipopolysaccharide-induced fulminant hepatic failure in d-galactosamine-sensitized mice by up-regulating expression of interleukin-1 receptor-associated kinase-m. Toxicol Appl Pharmacol (2017) 320:8–16. doi: 10.1016/j.taap.2017.02.011
30. Kohli R, Kirby M, Xanthakos SA, Softic S, Feldstein AE, Saxena V, et al. High-fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology. (2010) 52(3):934–44. doi: 10.1002/hep.23797
31. Zhu C, Kim K, Wang X, Bartolome A, Salomao M, Dongiovanni P, et al. Hepatocyte notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Sci Transl Med (2018) 10(468):eaat0344. doi: 10.1126/scitranslmed.aat0344
32. Wang X, Zheng Z, Caviglia JM, Corey KE, Herfel TM, Cai B, et al. Hepatocyte TAZ/WWTR1 promotes inflammation and fibrosis in nonalcoholic steatohepatitis. Cell Metab (2016) 24(6):848–62. doi: 10.1016/j.cmet.2016.09.016
33. Shan Z, Ju C. Hepatic macrophages in liver injury. Front Immunol (2020) 11:322. doi: 10.3389/fimmu.2020.00322
34. Zhou HM, Zhou S, Shi Y, Wang Q, Wei S, Wang P, et al. TGR5/Cathepsin e signaling regulates macrophage innate immune activation in liver ischemia and reperfusion injury. Am J Transplant (2021) 21(4):1453–64. doi: 10.1111/ajt.16327
35. Yoshida T, Abe K, Ikeda T, Matsushita T, Wake K, Sato T, et al. Inhibitory effect of glycyrrhizin on lipopolysaccharide and d-galactosamine-induced mouse liver injury. Eur J Pharmacol (2007) 576(1-3):136–42. doi: 10.1016/j.ejphar.2007.08.012
36. Deppermann C, Kratofil RM, Peiseler M, David BA, Zindel J, Castanheira FVES, et al. Macrophage galactose lectin is critical for kupffer cells to clear aged platelets. J Exp Med (2020) 217(4):e20190723. doi: 10.1084/jem.20190723
37. Scott CL, Guilliams M. The role of kupffer cells in hepatic iron and lipid metabolism. J Hepatol (2018) 69(5):1197–9. doi: 10.1016/j.jhep.2018.02.013
38. Wang YN, van der Tuin S, Tjeerdema N, van Dam AD, Rensen SS, Hendrikx T, et al. Plasma cholesteryl ester transfer protein is predominantly derived from kupffer cells. Hepatology. (2015) 62(6):1710–22. doi: 10.1002/hep.27985
39. Heymann F, Peusquens J, Ludwig-Portugall I, Kohlhepp M, Ergen C, Niemietz P, et al. Liver inflammation abrogates immunological tolerance induced by kupffer cells. Hepatology. (2015) 62(1):279–91. doi: 10.1002/hep.27793
40. Saitoh T, Akira S. Regulation of innate immune responses by autophagy-related proteins. J Cell Biol (2010) 189(6):925–35. doi: 10.1083/jcb.201002021
41. Lefere S, Tacke F. Macrophages in obesity and non-alcoholic fatty liver disease: Crosstalk with metabolism. JHEP Rep (2019) 1(1):30–43. doi: 10.1016/j.jhepr.2019.02.004
42. Sundaram SS, Halbower A, Pan Z, Robbins K, Capocelli KE, Klawitter J, et al. Nocturnal hypoxia-induced oxidative stress promotes progression of pediatric non-alcoholic fatty liver disease. J Hepatol (2016) 65(3):560–9. doi: 10.1016/j.jhep.2016.04.010
43. Mesarwi OA, Loomba R, Malhotra A. Obstructive sleep apnea, hypoxia, and nonalcoholic fatty liver disease. Am J Respir Crit Care Med (2019) 199(7):830–41. doi: 10.1164/rccm.201806-1109TR
44. Gonzalez FJ, Xie C, Jiang C. The role of hypoxia-inducible factors in metabolic diseases. Nat Rev Endocrinol (2018) 15(1):21–32. doi: 10.1038/s41574-018-0096-z
45. Wu X, Poulsen KL, Sanz-Garcia C, Huang E, McMullen MR, Roychowdhury S, et al. MLKL-dependent signaling regulates autophagic flux in a murine model of non-alcohol-associated fatty liver and steatohepatitis. J Hepatol (2020) 73(3):616–27. doi: 10.1016/j.jhep.2020.03.023
46. Hammoutene A, Biquard L, Lasselin J, Kheloufi M, Tanguy M, Vion AC, et al. A defect in endothelial autophagy occurs in patients with non-alcoholic steatohepatitis and promotes inflammation and fibrosis. J Hepatol (2020) 72(3):528–38. doi: 10.1016/j.jhep.2019.10.028
47. Allaire M, Rautou PE, Codogno P, Lotersztajn S. Autophagy in liver diseases: Time for translation? J Hepatol (2019) 70(5):985–98. doi: 10.1016/j.jhep.2019.01.026
48. Galle-Treger L, Helou DG, Quach C, Howard E, Hurrell BP, Muench GRA, et al. Autophagy impairment in liver CD11c(+) cells promotes non-alcoholic fatty liver disease through production of IL-23. Nat Commun (2022) 13(1):1440. doi: 10.1038/s41467-022-29174-y
49. Weiskirchen R, Tacke F. Relevance of autophagy in parenchymal and non-parenchymal liver cells for health and disease. Cells. (2019) 8(1):16. doi: 10.3390/cells8010016
50. Liu K, Zhao E, Ilyas G, Lalazar G, Lin Y, Haseeb M, et al. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy. (2015) 11(2):271–84. doi: 10.1080/15548627.2015.1009787
51. You L, Wang Z, Li H, Shou J, Jing Z, Xie J, et al. The role of STAT3 in autophagy. Autophagy. (2015) 11(5):729–39. doi: 10.1080/15548627.2015.1017192
52. Li YL, Li XQ, Wang YD, Shen C, Zhao CY. Metformin alleviates inflammatory response in non-alcoholic steatohepatitis by restraining signal transducer and activator of transcription 3-mediated autophagy inhibition in vitro and in vivo. Biochem Biophys Res Commun (2019) 513(1):64–72. doi: 10.1016/j.bbrc.2019.03.077
53. Mohammed S, Nicklas EH, Thadathil N, Selvarani R, Royce GH, Kinter M, et al. Role of necroptosis in chronic hepatic inflammation and fibrosis in a mouse model of increased oxidative stress. Free Radic Biol Med (2021) 164:315–28. doi: 10.1016/j.freeradbiomed.2020.12.449
54. Snodgrass RG, Boss M, Zezina E, Weigert A, Dehne N, Fleming I, et al. Hypoxia potentiates palmitate-induced pro-inflammatory activation of primary human macrophages. J Biol Chem (2016) 291(1):413–24. doi: 10.1074/jbc.M115.686709
55. Cai N, Zhao X, Jing Y, Sun K, Jiao S, Chen X, et al. Autophagy protects against palmitate-induced apoptosis in hepatocytes. Cell Biosci (2014) 4:28. doi: 10.1186/2045-3701-4-28
56. Shen C, Dou X, Ma Y, Ma W, Li S, Song Z. Nicotinamide protects hepatocytes against palmitate-induced lipotoxicity via SIRT1-dependent autophagy induction. Nutr Res (2017) 40:40–7. doi: 10.1016/j.nutres.2017.03.005
57. Jie M, Zhang ZQ, Deng N, Liu QM, Wang C, Ge QY, et al. 18[Formula: see text]-glycyrrhetinic acid inhibits TGF-[Formula: see text]-induced epithelial-to-Mesenchymal transition and metastasis of hepatocellular carcinoma by targeting STAT3. Am J Chin Med (2022) 50(1):313–32. doi: 10.1142/S0192415X22500124
58. He SQ, Gao M, Fu YF, Zhang YN. Glycyrrhizic acid inhibits leukemia cell growth and migration via blocking AKT/mTOR/STAT3 signaling. Int J Clin Exp Pathol (2015) 8(5):5175–81.
59. Wang S, Shen Y, Qiu R, Chen Z, Chen Z, Chen W. 18 beta-glycyrrhetinic acid exhibits potent antitumor effects against colorectal cancer via inhibition of cell proliferation and migration. Int J Oncol (2017) 51(2):615–24. doi: 10.3892/ijo.2017.4059
60. Chen H, Liu H, Tang B, Chen Y, Han L, Yu J, et al. The protective effects of 18beta-glycyrrhetinic acid on imiquimod-induced psoriasis in mice via suppression of mTOR/STAT3 signaling. J Immunol Res (2020) 2020:1980456. doi: 10.1155/2020/1980456
61. Guillot A, Tacke F. Liver macrophages: Old dogmas and new insights. Hepatol Commun (2019) 3(6):730–43. doi: 10.1002/hep4.1356
62. Wen YK, Lambrecht J, Ju C, Tacke F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell Mol Immunol (2021) 18(1):45–56. doi: 10.1038/s41423-020-00558-8
63. Huang X, Yao Y, Hou X, Wei L, Rao Y, Su Y, et al. Macrophage SCAP contributes to metaflammation and lean NAFLD by activating STING-NF-kappaB signaling pathway. Cell Mol Gastroenterol Hepatol (2022) 14(1):1–26. doi: 10.1016/j.jcmgh.2022.03.006
64. Tacke F. Targeting hepatic macrophages to treat liver diseases. J Hepatol (2017) 66(6):1300–12. doi: 10.1016/j.jhep.2017.02.026
65. Heymann F, Tacke F. Immunology in the liver - from homeostasis to disease. Nat Rev Gastroenterol Hepatol (2016) 13(2):88–110. doi: 10.1038/nrgastro.2015.200
66. Sieweke MH, Allen JE. Beyond stem cells: Self-renewal of differentiated macrophages. Science. (2013) 342(6161):946. doi: 10.1126/science.1242974
67. Mossanen JC, Krenkel O, Ergen C, Govaere O, Liepelt A, Puengel T, et al. Chemokine (C-c motif) receptor 2-positive monocytes aggravate the early phase of acetaminophen-induced acute liver injury. Hepatology. (2016) 64(5):1667–82. doi: 10.1002/hep.28682
68. Lodder J, Denaes T, Chobert MN, Wan JH, El-Benna J, Pawlotsky JM, et al. Macrophage autophagy protects against liver fibrosis in mice. Autophagy. (2015) 11(8):1280–92. doi: 10.1080/15548627.2015.1058473
69. Liu WS, Baker RD, Bhatia T, Zhu LX, Baker SS. Pathogenesis of nonalcoholic steatohepatitis. Cell Mol Life Sci (2016) 73(10):1969–87. doi: 10.1007/s00018-016-2161-x
70. Cao Y, Mai WL, Li R, Deng SW, Li L, Zhou YX, et al. Macrophages evoke autophagy of hepatic stellate cells to promote liver fibrosis in NAFLD mice via the PGE2/EP4 pathway. Cell Mol Life Sci (2022) 79(6):303. doi: 10.1007/s00018-022-04319-w
71. Stahl EC, Haschak MJ, Popovic B, Brown BN. Macrophages in the aging liver and age-related liver disease. Front Immunol (2018) 9:2795. doi: 10.3389/fimmu.2018.02795
72. Yang Y, Huang L, Tian C, Qian B. Magnesium isoglycyrrhizinate inhibits airway inflammation in rats with chronic obstructive pulmonary disease. BMC Pulm Med (2021) 21(1):371. doi: 10.1186/s12890-021-01745-7
Keywords: glycyrrhetinic acid, nonalcoholic fatty liver disease, macrophage, autophagic flux, STAT3-HIF-1α pathway
Citation: Fan Y, Dong W, Wang Y, Zhu S, Chai R, Xu Z, Zhang X, Yan Y, Yang L and Bian Y (2022) Glycyrrhetinic acid regulates impaired macrophage autophagic flux in the treatment of non-alcoholic fatty liver disease. Front. Immunol. 13:959495. doi: 10.3389/fimmu.2022.959495
Received: 10 June 2022; Accepted: 01 July 2022;
Published: 28 July 2022.
Edited by:
Jia Xiao, Jinan University, ChinaReviewed by:
Hyo Youl Moon, Seoul National University, South KoreaZhe Shen, Zhejiang University, China
Copyright © 2022 Fan, Dong, Wang, Zhu, Chai, Xu, Zhang, Yan, Yang and Bian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Long Yang, bG9uZy55YW5nQHRqdXRjbS5lZHUuY24=; Yuhong bian, Ymlhbnl1aG9uZ18yMDEyQDE2My5jb20=
†These authors have contributed equally to this work