 Jiahao Chen
Jiahao Chen Qinhui Liu
Qinhui Liu Jinhan He
Jinhan He Yanping Li
Yanping Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 15 August 2022
Sec. Inflammation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.958790
This article is part of the Research Topic New Insights of Immune Cells in Cardiovascular and Metabolic Disorders View all 36 articles
Diabetic nephropathy (DN) is a chronic, inflammatory disease affecting millions of diabetic patients worldwide. DN is associated with proteinuria and progressive slowing of glomerular filtration, which often leads to end-stage kidney diseases. Due to the complexity of this metabolic disorder and lack of clarity about its pathogenesis, it is often more difficult to diagnose and treat than other kidney diseases. Recent studies have highlighted that the immune system can inadvertently contribute to DN pathogenesis. Cells involved in innate and adaptive immune responses can target the kidney due to increased expression of immune-related localization factors. Immune cells then activate a pro-inflammatory response involving the release of autocrine and paracrine factors, which further amplify inflammation and damage the kidney. Consequently, strategies to treat DN by targeting the immune responses are currently under study. In light of the steady rise in DN incidence, this timely review summarizes the latest findings about the role of the immune system in the pathogenesis of DN and discusses promising preclinical and clinical therapies.
Diabetic nephropathy (DN) occurs in 20-50% of patients with diabetes and is the major risk for end-stage kidney disease (ESKD) (1). In 2019, 2.6 million new cases of DN were reported worldwide, and this incidence is predicted to increase in the future (2). Given the financial burden and lower quality of life associated with DN, understanding its molecular causes is of important for effective intervention and prevention.
DN is a clinical syndrome characterized by persistent albuminuria and a progressive decline in renal function, and it presents a typical pattern of glomerular disease (3). DN involves both changes in renal structure and function (4). Structurally, DN pathological features consist of glomerular mesangial expansion, basement membrane thickening, podocytes loss, nodular glomerulosclerosis and endothelial cells destruction (5). In the early stage of DN, there is tubular hypertrophy, but it is eventually processes to interstitial fibrosis with tubular atrophy. In the advanced stage, the injured kidney is infiltrated by immune cells (6). Functionally, DN shows increased albumin excretion and impaired glomerular filtration rate (7).
Historically, DN has not been considered an immune-mediated disease, but rather a disorder mediated by metabolic and hemodynamic factors (8). The progression of DN is highly unpredictable and it often occurs slowly over many years. In many countries, renal biopsy is rarely performed in patients with diabetes. It is only investigated when there is a significant increase in albuminuria or substantial decrease in renal function, which allow physicians to determine whether there is another kidney problem or comorbidities (1, 2, 9). Consequently, renal biopsies are usually performed in advanced stages of DN. This has severely hindered researchers to elucidate the role of immune system in progression of DN. Nevertheless, studies have been able to uncover a central role for immune-mediated inflammation in DN, involving both the innate and adaptive branches (7, 8, 10–12). Macrophages, as the predominant innate immune cells in DN, are commonly observed in the glomeruli and interstitium in experimental DN models and clinical trials at all stages of DN (13–15). The adaptive immune system mainly comprises T cells and B cells. The progression of DN correlates with activation of T cells in the blood and elevated numbers of CD4+ T cells in the kidney (11, 16, 17).
The immune pathogenic mechanism of DN is complex and involves the interaction of multiple pathways (Figure 1). In a diabetic mellitus, hyperglycemia and high lipid levels, including oxidative stress, reactive oxygen species (ROS), and oxidized lipids, damage kidney cells, leading to the release of damage-associated molecular patterns (DMAPs), and then trigger the pro-inflammatory signaling pathways (18). Besides, glycated proteins, such as advanced glycation end products (AGEs), can directly activate the complement system and trigger pro-inflammatory signaling (19). In response to continuous activation of innate immune injury, renal mesangial cells, endothelial cells and podocytes produce a variety of inflammatory mediators, including cytokines, chemokines, and adhesion molecules. These activate and recruit monocytes and macrophages, leading to further inflammatory cascade responses (7). The sustained chronic inflammation eventually drives the remodeling of renal structure and tubulointerstitial fibrosis (20–23).
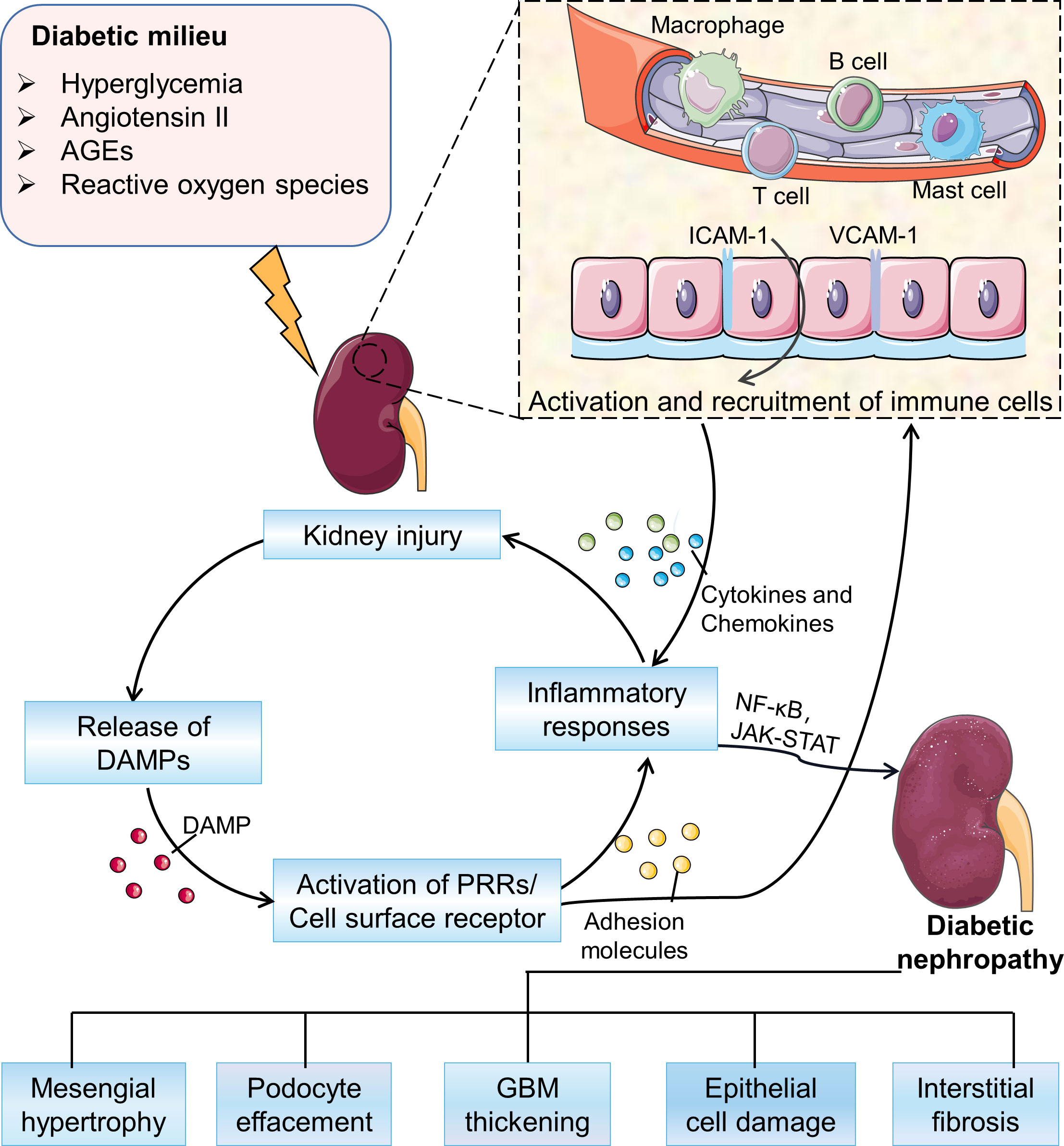
Figure 1 Overview of the pathogenesis of DN. In the diabetic milieu, hyperglycemia, advanced glycation end-products (AGEs), angiotensin II, and oxidative stress activate a variety of signaling cascades driving the recruitment and activation of immune cells to promote the development of inflammation and ultimately leading to a series of pathological changes in DN. AGEs, advanced glycation end products; DAMPs, damage associated molecular patterns; PRRs, pattern recognition receptors; GBM, glomerular basement membrane.
Various subsets of kidney cells in DN overexpress cell adhesion molecules, which are proteins on the cell surface to bind or attach immune cells to ECM. These cell adhesion molecules recruit immune cells to the kidney (24). The immune cells express transcription factors as well as secrete cytokines and chemokines that work together to induce a pro-inflammatory response to exacerbate disease pathology (4). These insights of the involvement of the immune system in DN may lead to more effective treatments than the current strategies of blood glucose control and inhibition of the renin-angiotensin system. In this review, we provide an overview of the contribution by the immune system to DN pathogenesis, and we explore current efforts to treat the disease by targeting immune-related factors.
Macrophages are the most important type of infiltrating immune cells in renal biopsies from experimental animal models and clinical patients with DN (25). The accumulation of F4/80- or CD68-positive macrophages detected by immunohistochemical staining or flow cytometry has been a characteristic feature of DN (26, 27). In mice with type 1 or 2 diabetes, macrophages accumulate in kidneys and become activated, which is associated with persistent hyperglycemia, deposition of glomerular immune complex, and increased production of chemokine, ultimately leading to renal injury and fibrosis (14, 15). Although detailed molecular mechanisms of macrophage migration and homing to the kidney have not been fully elucidated, cell adhesion molecules and chemokines/chemokine receptors are involved in this process. The vascular endothelium overexpresses cell adhesion molecules in its surface, such as intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1), which capture circulating macrophage precursors (28, 29). Mesangial cells, podocytes, and tubular epithelial cells are stimulated to secrete monocyte chemoattractant protein-1 (MCP-1) and osteopontin to facilitate migration of macrophages across the vascular endothelium and within the kidney (30–33). Renal parenchymal cells in diabetic mice also produce macrophage colony stimulating factor 1 (CSF-1), which promotes proliferation of kidney macrophages (Figure 2) (15, 25, 34).
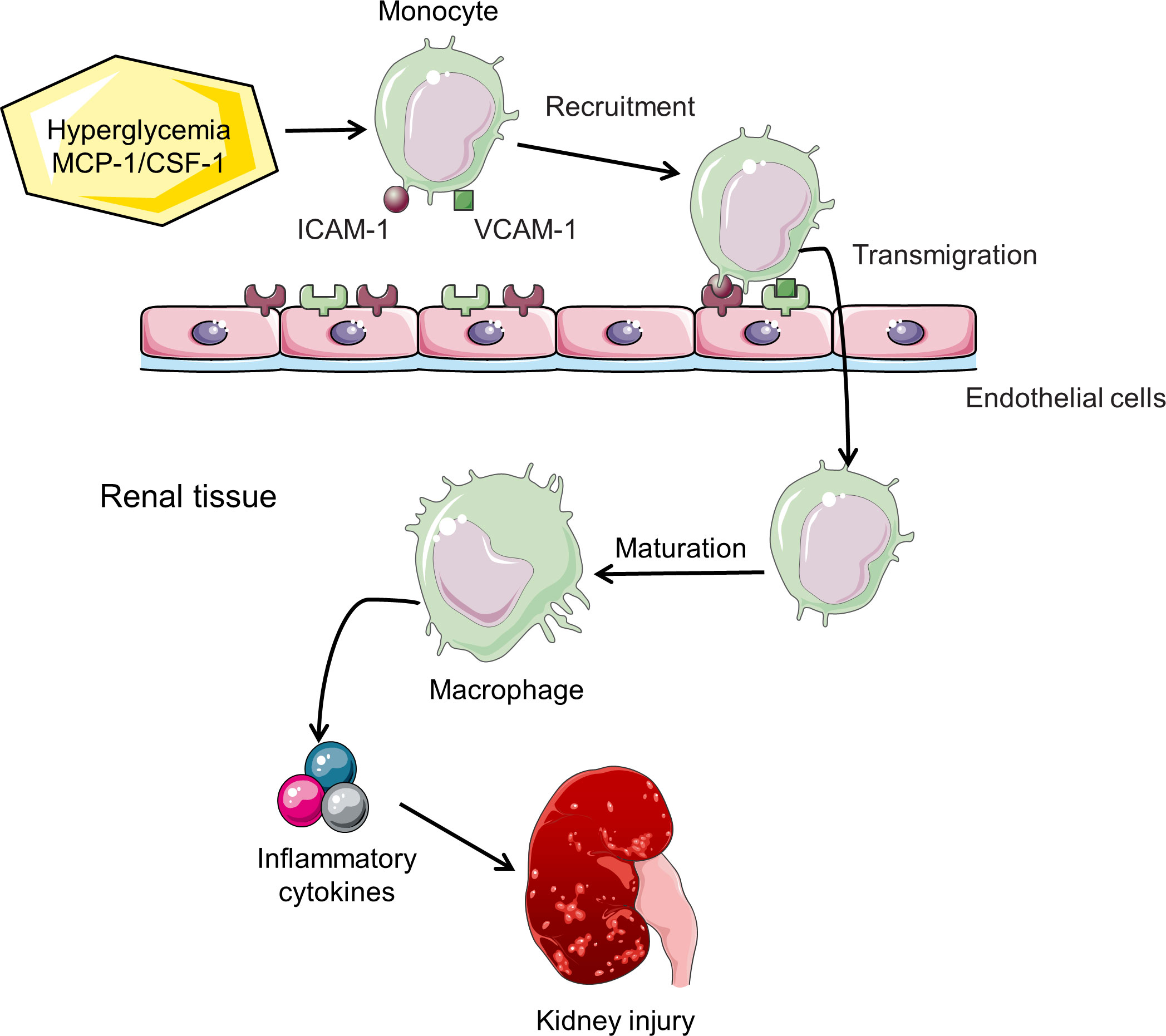
Figure 2 Macrophage recruitment and activation in DN. Hyperglycemia induces increased expression of cell adhesion molecules (ICAM-1/VCAM-1) and chemokines (MCP-1/CSF-1), thereby enhancing the recruitment of monocytes to the kidney. Chemokines also promote transendothelial migration. Monocytes mature into macrophages and subsequently release inflammatory cytokines, leading to the progression of DN.
Several factors promote the homing of macrophages to the kidney in the diabetic environment. Hyperglycemia and AGEs stimulate renal tubular cells expressing ICAM-1 and MCP-1 in the diabetic milieu, which promotes the recruitment of macrophages (32, 35). Once macrophages recruit to the diabetic kidney, local high glucose levels, AGEs and oxidized low-density lipoprotein (Ox-LDL) stimulate macrophages to release inflammatory cytokines (24). Other factors by which macrophages promote DN progression include production of ROS and proteases (24). These processes will aggravate tissue injury and ultimately lead to renal fibrosis.
Macrophages are plastic, pluripotent cells whose functions can change dramatically according to the microenvironment. Macrophages are classified as being “classically activated” (type M1) or “alternatively activated” (type M2) (36, 37). M1 macrophages perform immune surveillance function by secreting pro-inflammatory cytokines and chemokines and presenting antigen on their surface in order to stimulate other immune cells. M2 macrophages play an important role in immune regulation by secreting inhibitory cytokines and down-regulating immune response; they are inefficient at presenting antigens (38). Macrophages at sites of diabetic kidney injury are mainly of the M1 type (14, 15, 39).
Studies have shown that increased numbers of M1 macrophages are associated with severe DN lesions in mice lacking cyclooxygenase-2 (COX-2), an enzyme involved in metabolic processes preceding inflammation (40). The “triggering receptor expressed on myeloid cells”-1 (TREM-1) is an activating receptor of the immunoglobulin superfamily present on human myeloid cells. It can polarize macrophages toward the M2 type, thus reducing renal inflammation in vitro and in vivo (41). Mesenchymal stem cells (MSCs) also polarize macrophages towards the M2 phenotype and prevent renal injury in mouse models of DN. Interestingly, these effects are abolished in DN mouse models that have been treated with clodronate liposomes to deplete macrophages, suggesting that M2-type macrophages are necessary for renal protection. The ability of MSCs to polarize macrophages towards M2 appears to involve the activity of transcription factor EB (TFEB), which restores intracellular lysosomal function and autophagy activity, helping MSCs suppress the inflammatory response and alleviate renal injuries (42).
T cells, which recruit to the diabetic kidney accompanying by the recruitment of macrophages, also contributes to the progression of DN. Although several previous studies have shown that the number of CD4+ T cells in renal interstitium correlates with the albuminuria level in DN animal models (17, 43, 44), the mechanism by which T cells home to the kidney in diabetes is poorly understood. Adhesion molecules and chemokines are reported to be involved in T cell recruitment (24). Leukocyte function-associated antigen 1 (LFA-1), which is expressed on T cells, could combine with ICAM-1 expressed on renal endothelial cells, tubular epithelial cells, and mesangial cells to promote T cell migration to kidney (45). CD4+ T cells were increased in the glomeruli of db/db diabetic mice, but this increase was abolished in the kidneys of ICAM-1 knockout db/db mice (35), suggesting that the interaction of LFA-1 with ICAM-1 plays a significant role in the recruitment of T cells to kidney. Activated T cells will secrete inflammatory cytokines such as interferon gamma (IFN-γ) and TNF-α (17). These inflammatory cytokines directly damage the kidney through cytotoxic effects and indirectly promote the homing and activation of macrophages (16). In addition, AGEs can bind to the AGE receptor expressed on T cells, which in turn stimulates T cells to secrete IFN-γ, leading to kidney inflammation (46).
T cells can be divided into many subsets according to their function and specific markers. Flow cytometry, immunohistochemistry, and immunofluorescence staining techniques are generally used to distinguish different T cell subtypes (11, 39). It is well-known that CD4+ T cells can differentiate into T-helper (Th) 1 cells, Th2 cells, Th17 cells, and Treg cells, which mainly produce IFN-γ, interleukin (IL)-4, IL-17 and Foxp3, respectively (47, 48). As the many subsets of T cells indicates, their roles are varied when the adaptive immune response is activated in DN pathogenesis (49, 50). The Th1 cell response precedes and accompanies type 1 diabetes (51). Increased levels of ICAM-1, P-selectin, IFN-γ and migration inhibitory factor in the kidney of mice with diabetes mellitus are associated with the homing of effector Th1 cells to the glomerulus (16, 52). Similarly, T-helper 17 cells secret IL-17 to elicit a strong pro-inflammatory response (53). Neutralization of IL-17A blocks NF-kB activation and the subsequent upregulation of proinflammatory genes, which in turn inhibits infiltration of the kidney by inflammatory cells (54). In contrast, Th2 cells produce IL-4 to promote humoral immunity, inhibit Th1 activation, and inhibit inflammation and fibrosis, providing an overall immunosuppressive effect (55). Furthermore, transfer of CD4+-Foxp3+ Treg cells improves insulin resistance and ameliorates DN pathogenesis in mice by tipping the balance toward anti-inflammation and suppressing CD8+ T cells infiltration in the kidneys and adipose tissue (47, 56, 57). CD8+ T cells are predominantly cytotoxic and damage the kidney by direct cell-cell signaling via surface molecules and indirect signaling via cytokines (58).
There are limited literature about the role of B cells in the pathogenesis of DN. IgG+ B cells shown modestly increased in glomeruli of non-obese diabetic mice (59). After depletion B cells in these mice, the re-emerging B cells exhibit an immunosuppressive phenotype and inhibit the onset of diabetes (60). Studies have shown that CD20+ B cells were observed in the renal interstitium of patients with type 1 or 2 diabetes mellitus, suggesting the possibility of B cell participation in DN progression (61).
In the diabetic milieu, hyperglycemia and AGEs stimulate NF-κB signaling, which plays an important role in the development and function of B cells. It has been reported that the hyperglycemic environment might directly increase the number of both antibody- and cytokine-producing B cells, and contribute to the development of DN (59). Currently, the mechanism of B cells regulating DN is poorly understood. The role of B cells contributing to DN is most likely due to the antibodies produced by B cells. These antibodies can direct against antigens such as oxLDL and AGEs and lead to the formation of immune complexes, triggering inflammation and glomerulonephritis (59). Further studies are urgently needed to uncover the function and regulatory mechanism of B cells in DN pathogenesis.
Mast cells are multipotent bone marrow-derived cells rich in growth factors and inflammatory mediators (62). Regarding the production of tryptase and chymase, mast cells were divided into MCT subtype and MCTC subtype in humans. MCT subtype only produces tryptase, whereas MCTC subtype produces both tryptase and chymase (63). In the experimental animal model of DN, there is evidence that mast cells infiltrate the kidney (64). In patients with DN, the number of mast cells increased with the progression of DN (62). Increased mast cell numbers and degranulation levels were significantly associated with tubulointerstitial injury, suggesting the mast cells are involved in development of DN (65).
Mast cells can be activated in several ways, including the well-known classical pathway, IgE-FcϵR cross-linking, and alternative pathways, such as the complement pathway and toll-like receptors pathway (65). C3a complement, the most potent activator of mast cells, has been reported to increase in DN (19). Thus, research suggests that the increased complement activation in diabetic mellitus may contribute to the recruitment and activation of mast cells. Once mass cells infiltrate into the kidney, they contribute to the pathogenesis of DN by releasing TGF-β, chymase, tryptase, renin, histamine, and inflammatory cytokines (4, 64). Specifically, mast cells may aggravate tubular interstitial fibrosis by synthesizing and releasing TGF-β and reninto initiate and promote tubular inflammation through releasing TGF-β and TNF-α (64). Further studies are needed to confirm the possible involvement of mediators by which mast cells affect the complex pathogenesis of DN.
Cytokines are a group of low-molecular-weight peptides with pharmacological activities. They have characteristic functions in autocrine and paracrine signaling, and they are important effectors of the immune system (Table 1).
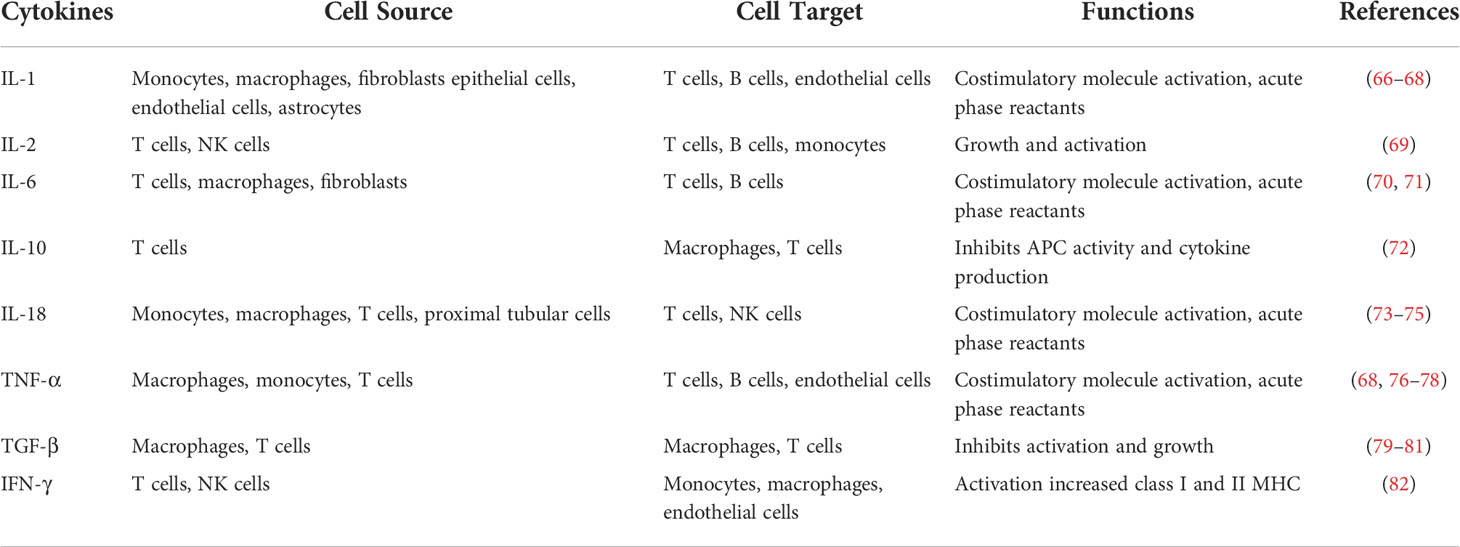
Table 1 Cytokines involved in DN pathogenesis.
IL-1 can be induced by almost all nucleated cell types, but it is mainly produced by activated macrophages and is a potent mediator of inflammation (67). In an experimental model of DN, renal IL-1 expression was found to be elevated, which was followed by expression of chemokines and adhesion molecules (66, 67). IL-1 helps drive mesangial cell proliferation and matrix synthesis, it increases vascular endothelial permeability, and it is linked to hemodynamic abnormalities within the glomerulus (83). It also upregulates ICAM-1 in certain subsets of kidney cells, such as mesangial cells, endothelial cells, and renal tubular epithelial cells (34).
Renal biopsies from DN patients show infiltration of the mesangium, stroma, and tubules by cells expressing IL-6 (67). In addition, a positive relationship was found between the severity of diabetic glomerular lesions (mesangial dilatation) and IL-6 mRNA levels in glomerular mesangial cells and podocytes, indicating that IL-6 may positively influence the dynamics of the ECM accumulation in the kidney (70). Interestingly, one study found that IL-6 regulates the differentiation of M1 macrophages into M2 macrophages through IL-4-STAT6 signaling. This finding identifies IL-6 signaling as an important determinant of macrophage activation, conferring on IL-6 an unexpected homeostatic role in limiting inflammation (71).
Among those cytokines involved in DN, IL-18 seems to be the most important one to DN pathogenesis. Elevated IL-18 levels in serum and urine have been reported in DN patients, and urinary excretion of β-2 microglobulin, a marker of tubular interstitial injury, positively correlates with serum levels of IL-18 (75, 76). Increased levels of IL-18 were found in the renal biopsies of diabetic patients in proximal tubules and epithelial cells. Serum IL-18 levels were also greater in DN patients than in healthy subjects. IL-18 is closely related to many pathogenic molecular mechanisms involved in DN. As a potent inflammatory cytokine, IL-18 promotes the production of other inflammatory cytokines, such as IL-1 and TNF-α (73). IL‐18 can also upregulate the expression of ICAM‐1, VCAM‐1, and IFN-γ in endothelial cells (74). IL-18-dependent apoptosis may play a critical role in apoptosis-induced injury in DN. Besides, IL-18 activation may lead to increased free radical production and oxidative damage (84, 85). Thus, IL-18-induced oxidative stress may be an additional mechanism by which IL-18 contributes to DN progression. Considering the vital l role of IL-18 in DN, it may become a novel therapeutic target for the prevention and therapy of DN.
TNF-α, a pleiotropic inflammatory cytokine, is mainly produced by monocytes, macrophages, and T cells (66). Renal cells such as mesangial cells, glomerular cells, endothelial cells, and renal tubular cells can also secrete TNF-α in response to hyperglycemia and AGEs (86–88). The role of TNF-α in DN is supported by the detection of increased levels of the cytokine in urine from diabetic patients, and by the correlations between those levels and clinical markers of DN and disease progression (76, 77). TNF-α participates in DN progression through multiple mechanisms. TNF-α is cytotoxic to kidney cells and can induce cell apoptosis and production of ROS, as well as alter hemodynamic balance between vasoconstriction and vasodilatation (68). TNF-α increases ROS production and vice versa, which amplifies the inflammatory response (78). In rats with streptozotocin-induced diabetes, elevated TNF-α increases oxidative stress, leading to urinary albumin excretion, a marker of kidney injury (83). Other studies have shown that TNF-α significantly promotes the development of renal hypertrophy and sodium retention, both of which are characteristic alterations during early DN (34, 77, 89).
TGF-β is a major regulator of ECM production and accumulation in the diabetic kidney (90). It forwards the two milestones of DN progression, which are renal cell hypertrophy and ECM accumulation (91). Many factors of diabetic mellitus stimulate TGF-β production in the kidney. Hyperglycemia, angiotensin II, mitogen-activated protein kinase, and PKC have been shown to regulate TGF-β expression (92–95). A few studies have proven that ROS in diabetic conditions can directly or indirectly promote the production of TGF-β. Once TGF-β is activated in kidneys, it induces the production of fibronectin and collagen types I, III, and IV (79); it restrains matrix metalloproteinases, such as plasminogen activator, collagenase, elastase, and stromelysin; and it activates proteases inhibitors, such as tissue inhibitors of metalloproteinases and plasminogen activator inhibitor 1, which blocks ECM degradation (80). TGF-β positively regulates its own expression while also stimulating the deposition of ECM, thus amplifying the fibrosis response (79–81). A high glucose environment induces TGF-β expression and activation, thus pushing podocytes into the apoptosis process, which impairs filtration barrier and renal function (96). Therefore, studies targeting TGF-β signaling disruption, such as knockout of the type 2 TGF-β receptor or the downstream signaling molecular Smad3, and administration of anti-TGF-β antibodies, suspend mesangial matrix expansion and deterioration of renal function in mice (97, 98).
ICAM-1 is an adhesion molecule (Table 2) expressed in endothelial, mesangial and epithelial cells and has been directly associated with kidney injury and DN progression in a rat model (99, 100). ICAM-1 can bind to integrins on the surface of leukocytes to promote their adhesion to endothelial cells and transmigration (68). ICAM-1 expression is upregulated in response to pro-inflammatory factors, especially TNF-α (105). Altered hemodynamic conditions resulting from TGF-β-induced ECM accumulation are also one of the factors contributing to ICAM-1 up-regulation. In addition, oxidative stress can also promote ICAM-1 expression (106). In renal mesangial and endothelial cells, AGEs induce the production of ROS, which activates NF-κB and promotes the release of pro-inflammatory cytokines and adhesion molecules (107). ICAM-1 plays a critical role in the leukocytes migration, especially T cells to the kidney (101). Deleting ICAM-1 in diabetic mice ameliorated symptoms of DN, such as glomerular hypertrophy, mesangial matrix expansion, and proteinuria (101).

Table 2 The type and function of adhesion molecules.
Similar to ICAM-1, VCAM-1 also involved in the leukocyte-endothelial adhesion that helps recruit leukocytes to the kidney during inflammation. In kidney interstitium of diabetic KKAy mice, VCAM-1 is upregulated on the endothelial cells of venules, and it is expressed in infiltrating cells (103). In DN patients, VCAM-1 is upregulated in kidney and as a soluble form in plasma (29). VACM-1 levels correlate with the number of infiltrating immune cells in kidney and are associated with severity and progression of albuminuria (22, 24, 104).
Previous in vivo and in vitro studies have shown that differential expression of chemokines and their receptors precisely orchestrate molecular mechanisms that lead to immune cell migration in DN progression. Among them, MCP-1, also known as CC chemokine ligand 2 (CCL2), has been proposed as marker of the degree of tubular injury and renal inflammation in DN (108). In mice model of diabetes-induced renal injury, MCP-1 levels progressively increase in the kidney. Furthermore, in vitro studies indicate that MCP-1 expression increases in the presence of high amounts of glucose (109), and animal models of type 1 and 2 diabetes show reduced renal damage after knockout of MCP-1 (4, 15, 31). In the clinic, urinary MCP-1 levels are obviously higher in patients with microalbuminuria or albuminuria diabetes than in patients with normoalbuminuria diabetes or in healthy controls. Moreover, urinary MCP-1 levels increase as DN progresses, and they are significantly associated with other risk factors for DN (110).
Several factors were associated with the expression of MCP-1, such as hyperglycemia, TGF-β, NF-κB, PKC, ROS, and AGEs (34). There is evidence that angiotensin II also promotes MCP-1 expression. Blocking renin-angiotensin system with angiotensin converting enzyme inhibitors or angiotensin II receptor blockers significantly down-regulated the MCP-1 level in kidney cells. MCP-1 promotes the transmigration of macrophages across endothelial cells to kidney, which is the main process in the homing of macrophages in DN (111, 112). It also promotes the migration of T cells and dendritic cells to the diabetic kidney (113, 114).
Previous studies have thoroughly reviewed transcription factors involved in DN, including NF- kB, Janus kinase-signal transducer and activator of transcription (JAK-STAT), upstream stimulatory factors 1 and 2, activator protein 1, cAMP-response-element-binding protein, nuclear factor of activated T cells, and stimulating protein 1 (115). In this review, we will briefly discuss the two most vital transcription factors, NF-κB and JAK/STAT, and their roles in DN.
NF-κB is believed to be a master switch in the control of inflammation and is involved in the transcription of numerous genes involved in the pathogenesis of DN (Figure 3) (116), such as those giving rise to angiotensinogen, cytokines, and adhesion molecules (117–119). In diabetic rat models, NF-κB activation upregulates the levels of pro-inflammatory cytokines TNF-α and IL-1β (116). Upregulation of NF-κB has been indicated in monocytes of peripheral blood from patients with diabetes, and the extent of upregulation correlates with DN severity (120). Activation of NF-κB and transcription of certain pro-inflammatory chemokines in tubular epithelial cells are markers of progressive DN. Albuminuria may be one of the major pro-inflammatory phenotypes resulting from NF-κB activation (121).
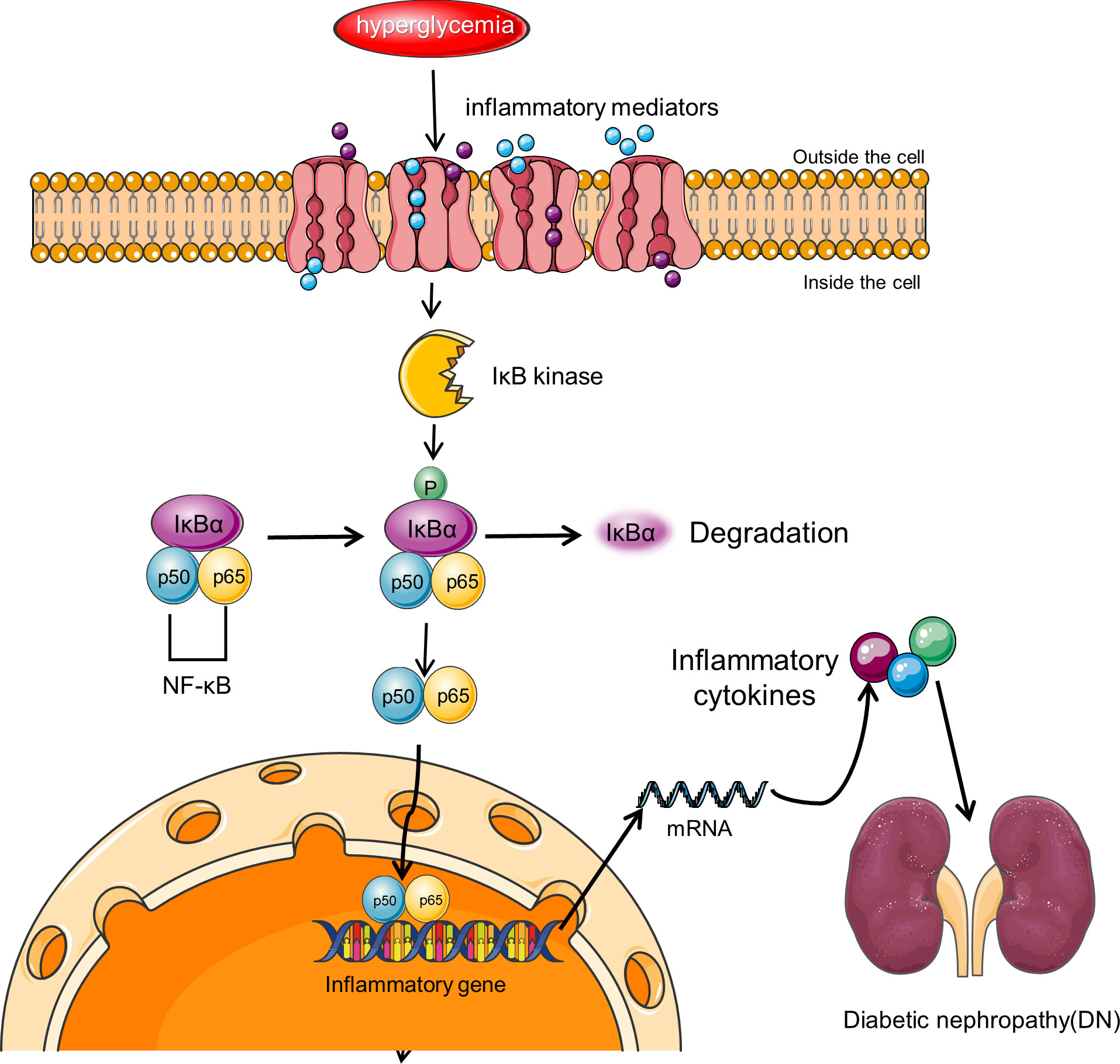
Figure 3 NF-κB signaling pathway in DN. NF-κB is a transcriptional regulator expressed in the cytoplasm of almost all cell types, and its activity is controlled by the IκB regulatory protein family. Activation of NF-κB involves the inhibitory protein IκB kinase being phosphorylated by specific IκB and subsequently degraded by proteolysis. Free NF-κB translocates to the nucleus, binds to promoter and enhancer sites, and activates transcription. NF-κB signaling pathway leads to increased transcription of target genes encoding inflammatory cytokines and other target genes associated with this complication, resulting in renal inflammation.
The JAK-STAT signaling pathway includes a family of intracellular signaling molecules that initiate activation of target genes encoding growth factors, hormones, and cytokines (Figure 4) (122). Studies have shown that high glucose can activate the JAK-STAT signaling in rat renal mesangial cells and in mice renal cortex at early stages of DN (123, 124). Genome-wide transcriptome analysis of DN patients showed upregulation of JAK1/2 and STAT1/3 (125). In diabetic mice, the JAK-STAT signaling is over-expressed, as is its downstream target gene encoding “suppressor of cytokine signaling (SOCS) 3”, and its upstream regulatory gene SIRT1 (126).
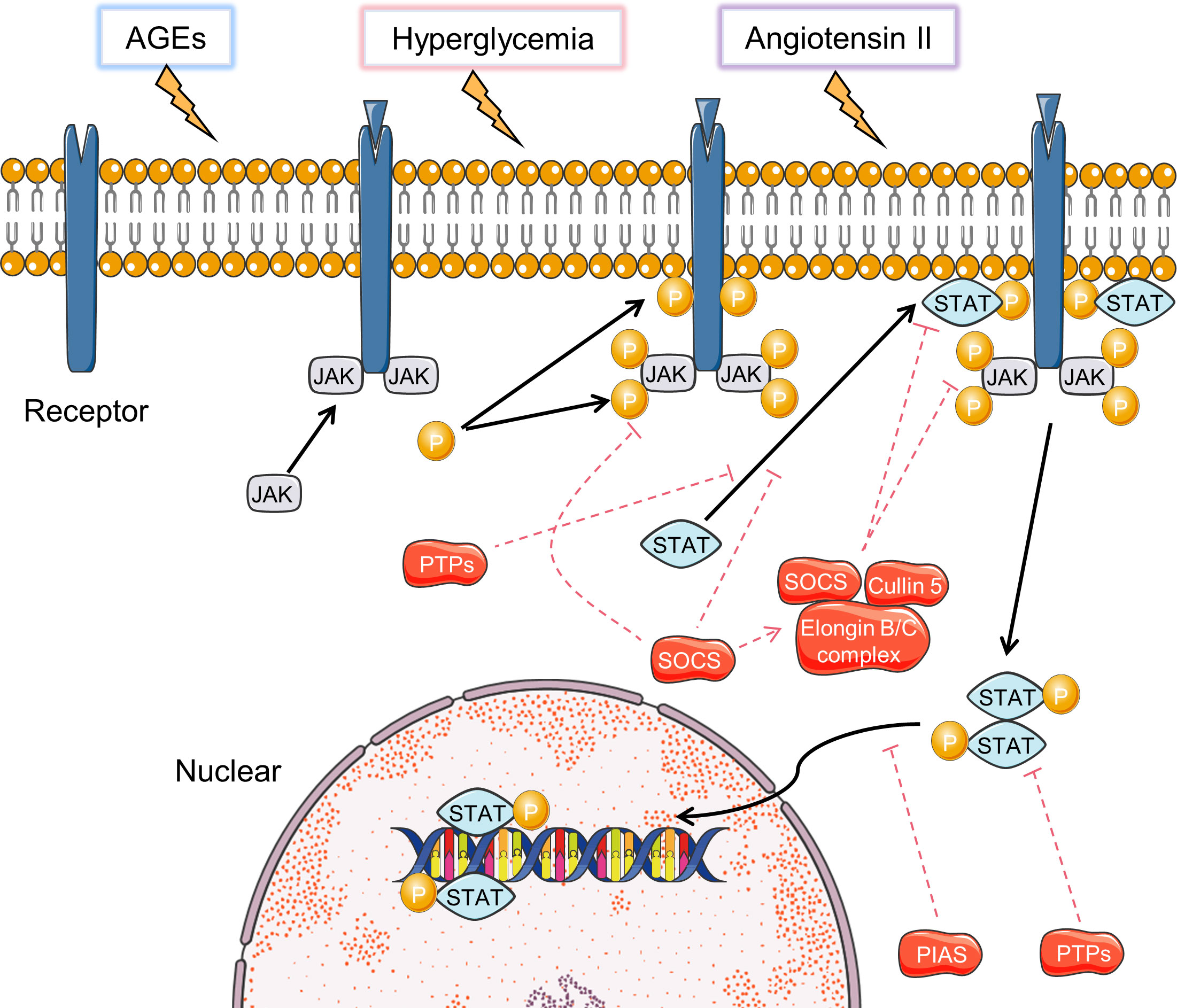
Figure 4 Activation and inhibition of JAK-STAT signaling pathways. Black arrows indicate the activation process and the red dotted arrows indicated inhibition process.
Hyperglycemia-induced JAK-STAT activation is a vital mechanism of renal injury in DN (127). Hyperglycemia can increase the production of angiotensin II, which in turn induces JAK2 through enhanced oxidative stress. ROS has been suggested as a mediator of hyperglycemia to regulate JAK protein activation (127). In diabetic environment, AEGs and MAPK activation can promote the acetylation and phosphorylation of STAT3 in mice and human diabetic kidneys, leading to enhanced STAT3 transcriptional activity (128–130). Transgenic mice with reduced STAT3 activation ability are protected from inflammation and injury in the diabetic kidney (131). Overexpression of SOCS-1 and SOCS-3, which are negative regulators of JAK-STAT signaling, reduce macrophage infiltrations, levels of pro-inflammatory cytokines, renal injury in rodents with DN (126). The current researches mainly focus on JAK1/2 and STAT3. Therefore, future studies on the role of other JAKs and STATs may aid in revealing novel regulatory mechanisms of DN.
The complement system is an essential part of the innate immune systems, which can enhance the ability of antibodies and phagocytes to clear microbes and damaged cells (8). The complement system also promotes inflammation (23). Growing evidence has shown that complement system is involved in the progression of DN (19). According to transcriptome and immunohistochemical analysis of renal biopsies, 50-60% of DN patients have glomerular deposition of complement component C3, and such deposition is associated with severity of glomerulosclerosis (125). The glomerular deposition of complement C3 is also a characteristic of DN animal models associated with type 1 or 2 diabetes, and such deposition has been linked to glomerular deposition of immunoglobulin G (IgG), which induces inflammation and damages the kidney tissue by producing chemokines (4, 132, 133).
The inflammasome assembles during DN immune responses in a way that drives the pathology of kidney diseases. NLRP3 is by far the best characterized inflammasome in the kidney (21, 134). The activation of NLRP3 inflammasome in immune cells generally requires two steps: priming and activation. The priming step is stimulated by the binding of pathogen-associated molecular patterns (PAMPs) and/or DAMPs to toll-like receptors and/or cytokine receptors. This step often involves the activation of NF-κB signaling and regulation of downstream genes that increase the expression of inflammasome-associated genes and substrates (10, 21). Following priming, the activation step involves NLRP3 oligomerization and the assembly of inflammasome components into a complex. By cleaving pro-caspase-1 into caspase-1, the resulting complex promotes the maturation and secretion of IL-1β and IL-18, further leading to the accumulation of mesangial cells, podocyte damage, and albuminuria (10, 134).
The expression of NLRP3 is elevated in the glomerulus of mouse DN models. Thus, NLRP3-knockout animal models are necessary to reveal the vital role of NLRP3 inflammasome in DN. Indeed, one study demonstrated that level of IL-1β in podocytes was significantly upregulated in STZ-induced diabetic mice, which was reversed in NLRP3 knockout mice (135). Deleting NLRP3 significantly prevented the accumulation of glomerular neutral lipid and cholesterol in diabetic mice (136).
These studies make clear that the immune system plays an essential role in the progression of DN. Below, we review promising therapeutic targets in DN as well as therapeutic agents already under development.
Among the inflammatory mediators associated with DN, TNF-α has perhaps been best studied for its therapeutic potential: several studies have examined how its inhibition can slow DN progression (137). Infliximab is a chimeric immunoglobulin G1κ murine/human monoclonal antibody developed as a therapeutic agent against rheumatoid arthritis and Crohn’s disease (138, 139). Infliximab reduced the expression of TNF-α and improved DN symptoms in diabetic mice (140). The TNF-α inhibitor SKF86002 markedly decreased glomerulus TNF-α level and improved kidney function in patients with DN (141). Pentoxifylline (PTX), originally created to treat intermittent claudication caused by peripheral vascular diseases (142–144), has shown potential for mitigating proteinuria and restoring glomerular filtration in the context of diabetic kidney disease. PTX inhibits TNF-α expression as well as the activity of other inflammatory mediators, such as IL-1, IL-6, IFN-γ, VCAM-1 and ICAM-1 (145–147). Future studies are needed to clarify whether PTX can improve renal outcomes in DN.
Direct inhibitors of TGF-β can efficiently block the progression of DN (148). But indirect inhibition has also shown benefit (149, 150). Melatonin, a hormone secreted by the pineal gland, may improve kidney inflammation and interstitial fibrosis in DN by inhibiting the TLR4 and TGF-β/Smad3 signaling pathways (150). Given that melatonin is also capable of reducing urinary excretion and protecting podocytes (151), it may prove a promising therapeutic in DN. Sitagliptin is a dipeptidyl peptidase-4 (DPP-4) inhibitor best known for its hypoglycemic properties (152). In diabetic mice, sitagliptin improved renal function by inhibiting the TGF-β/Smad signaling pathway (153). Dencichine is a non-protein amino acid, originally extracted from Panax notoginseng (154), that may treat DN by reducing hyperglycemia, restoring metabolic disorder, reducing ECM deposition, increasing the activity of enzymes that degrade the ECM, and down-regulating TGF-β/Smad signalling in DN glomeruli (155).
Breviscapine and triptolide act as MCP-1 receptor antagonists in animal models of DN, reducing downstream signaling pathways that induce ROS production and inflammation (156). Breviscapine, extracted from the Chinese herb Erigeron breviscapus, may indirectly mitigate DN by reducing albuminuria (156). In contrast, triptolide regulates the proportion of Th1/Th2 cells, reduces MCP-1 expression, and inhibits macrophage infiltration as well as expression of related inflammatory factors in the kidney (157–160). Other inhibitors, such as the CCR2 inhibitor CCX140-B and the MCP-1/CCL2 inhibitor NOX-E36, are currently in pre-clinical studies or clinical trials (161–164). In a murine model of DN, NOX-E36 significantly reduced glomerulosclerosis and improved glomerular filtration rate (163), while CCX140-B significantly reduced proteinuria in DN patients (161).
Inhibitors of NF-κB have been used to mitigate DN and inflammatory injury of the kidney, as well as improve kidney function (165). Thiazolidinediones, agonists of peroxisome proliferator-activated receptor (PPAR)-γ, are widely used as insulin sensitizer in diabetes therapy (166, 167). These ligands repress renal injury in an experimental rat DN model by inhibiting NF-κB activity (168). Cultured renal tubular epithelial cells pretreatment with15a, a derivative of salviadione, prevented high glucose induced NF-κB activation and expression of inflammatory cytokines (169). In mice with streptozotocin-induced diabetes, the antioxidant tocotrienol suppressed NF-κB activation, reduced TNF-α and TGF-β levels and reversed renal dysfunction (4, 170). Treating these animal model with BAY-110782, an inhibitor of IκB, or pyrrolidine dithiocarbamate, an inhibitor of NF-κB, reduced NF-κB activation, renal macrophage infiltration and production of the inflammatory cytokines MCP-1, TNF-α, IL-1β and IL-6 (171, 172).
Various drugs and compounds may show anti-inflammatory effects in DN by inhibiting JAK-STAT signaling (131). Paeoniflorin, a monoterpene glycoside extracted from the dried root of P.lactiflora Pall, downregulates the phosphorylation of JAK2 and STAT3 in diabetic kidney (173). Baricitinib, a selective inhibitor of JAK1 and JAK2, reduced albuminuria in patients with DN associated with type 2 diabetes in phase 2 randomized clinical trials (174, 175). Others inhibitors of various JAK proteins, such as ruxolitinib and tofacitinib, have already been approved for clinical use by the US Food and Drug Administration (175, 176).
To date, only a few studies have reported the efficacy of blocking complement system in DN (19, 23). The lectin-like domain of thrombomodulin constrained glucose-induced complement activation on podocytes an endothelial cells and ameliorated albuminuria and glomerular damage in mice (177). Treatment with receptors of the complement fragments C3a/C5a may ameliorate DN by partially blocking the endothelial-myofibroblast transition and fibrosis through inhibition of the Wnt/β-catenin signaling pathway (178). Similarly, in a diabetic rat model, administration of C3a receptor improved DN pathogenesis by inhibiting IκBα phosphorylation and TGF-β/Smad3 signaling, which reduced the cytokine release and ECM accumulation (179).
Abnormal regulation of the complement cascade leads to immune and non-immune types of kidney damage (19). This insight into the pathological mechanisms related to complement and regulators will aid the development of new therapies. Monoclonal humanized antibody eculizumab, that binds C5 and prevents assembly of the membrane attack complex (C5b-9), is already in clinical use (180). Complement-targeting therapy is expected to exert a more important role in the treatment of DN in the future.
MCC950, a small molecule inhibitor of NLRP3, can specifically and potently inhibits NLRP3 inflammasome activation (181). MCC950 is reported to improve podocyte injury in DN by inhibiting lipid accumulation, ROS production and p65 activation (135). CY-09 is another NLRP3-specific inhibitor, and it blocks oligomerization of the NLRP3 inflammasome (182). Furthermore, it downregulates blood glucose and insulin level, improves glucose tolerance and decreases hepatic steatosis in diabetic mice, suggesting that it may exert therapeutic effects against type 2 diabetes. In fact, CY-09 reduces the levels of IL-1β in the serum, liver and adipose tissue of diabetic mice, without affecting metabolic parameters in control mice (183). Oridonin is a the main ingredient of the traditional Chinese herb R.rubescens that significantly attenuates diabetes-induced renal injury by dampening inflammatory responses, based on studies in vitro and in vivo (183). Oridonin appears to prevent NF-κB from binding DNA and turning genes on (184). Tranilast is a cell membrane stabilizer that has been widely used in the treatment of inflammatory diseases because it inhibits the release of histamine and other chemical mediators (185). Tranilast prevents NLRP3 assembly by inhibiting interactions of NLRP3 with other NLRP3 molecules or with apoptosis-associated speck-like protein containing a C-terminal caspase activation and recruitment domain. Tranilast blocks the ability of a high fat diet to upregulate IL-1β in the serum, liver, or adipose tissues of diabetic mice. Tranilast also suppresses caspase-1 cleavage in diabetic mice, suggesting that the drug can inhibit metabolic stress-induced inflammasome activation (186).
SGLT2 inhibitors, which alleviate hyperglycemia by stimulating the excretion of glucose into urine, have been approved for the treatment of type 2 diabetes (187). Since persistent hyperglycemia is a central cause of DN progression, SGLT2 may also be effective against that renal complication (188). SGLT2 blocks glucose reabsorption at the proximal tubule, leading to glucosuria and lowering of blood glucose levels, which is independent of insulin (189). Treating diabetic animals with empagliflozin or ipragliflozin reduces their hyperglycemia and reduces levels of pro-inflammatory cytokines and chemokines, NF-κB and C-reactive protein in kidney or plasma (190–192). Dagagliazine mitigates hyperglycemia and diabetic tubulointerstitial injury by suppressing inflammatory markers and oxidative stress in the renal tissues of diabetic mice (193). Similarly, dapagliflozin blocks oxidative stress, inflammation and apoptosis induced by high glucose, and it promotes renal function and angiogenesis by upregulating vascular endothelial growth factor (194). Canagliflozin decreases plasma levels of IL-6, matrix metalloproteinase-7, TNF receptor 1, and fibronectin 1 in human, suggesting that it may mitigate inflammation, ECM deposition and fibrosis in DN (195).
MicroRNAs are important mediators of the post-transcriptional feedback control mechanism and participate in metabolism and inflammation regulation. Pioneering work with microRNAs has provided a new outlook on molecules and signaling pathways involved in DN pathogenesis (Table 3). MicroRNAs are non-coding RNAs that regulate gene expression through epigenetic mechanisms and may therefore allow design of drugs that could prevent DN before it appears (222–224). Both miR-192 and miR-21 have been implicated in renal fibrosis, albeit through different mechanisms (196, 225). Of note, miR-192 is involved in a negative feedback loop with TGF-β signaling (226). Thus, these miRNAs deserve further investigation as targets in the treatment of DN. Indeed, knockdown of miR-21 in the kidneys of diabetic db/db mice improved renal function and inhibited renal fibrosis and inflammation during DN associated with type 2 diabetes (197). Induction of renal protective miRNAs and silencing of injury-induced miRNAs in patients with DN have been shown to restore renal function (222). Currently, several miRNAs-based preparations have entered clinical trials, such as Miravirsen, an inhibitor of miR-122 for hepatitis C treatment (227), and MRX24, a liposome-based miR-34 mimic for the treatment of cancer (228). We believe that microRNAs-based preparations may also apply to the treatment of DN in the future.
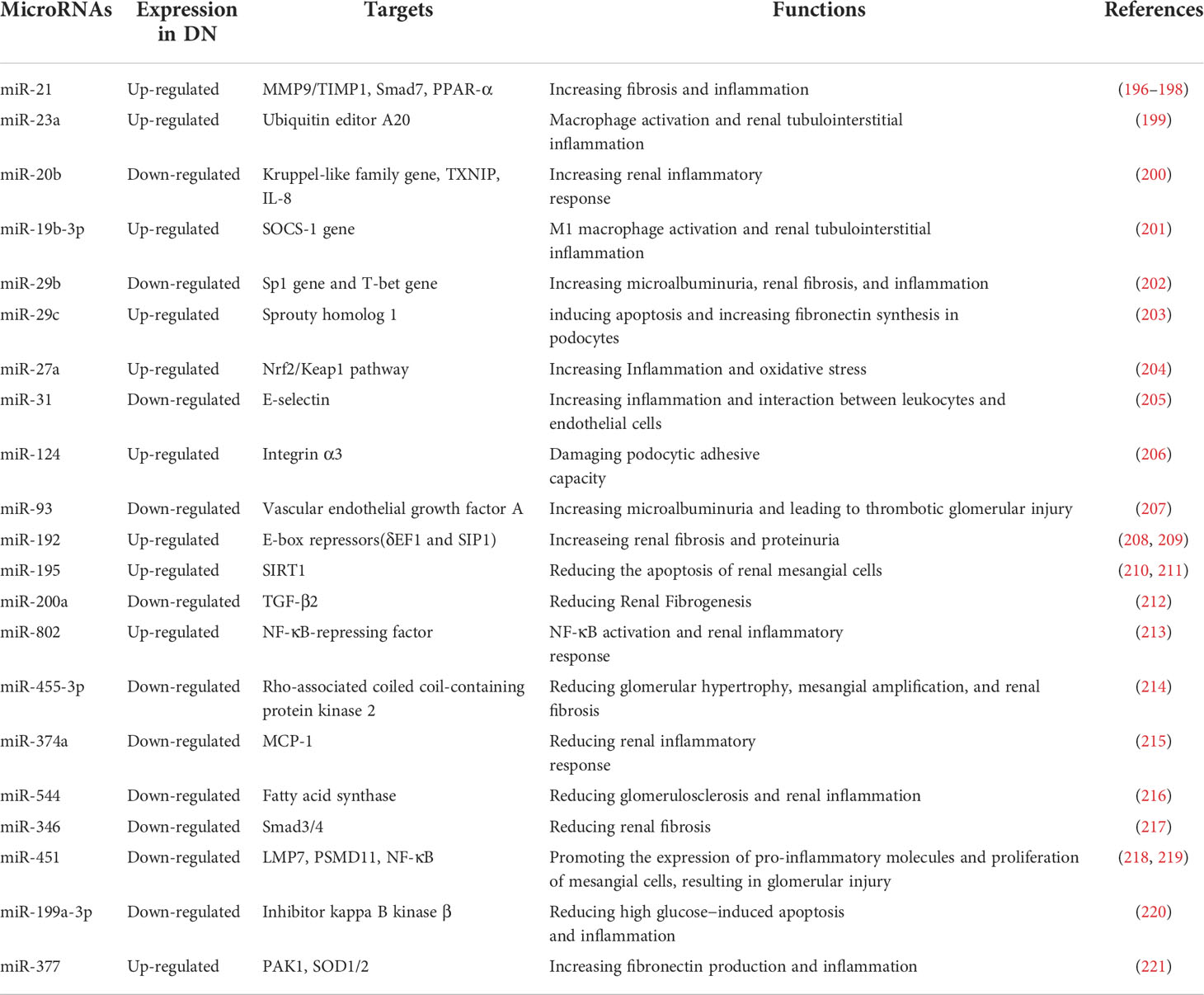
Table 3 The miRNAs involved in regulating the immune mechanism of DN.
Stem cells are a class of cells that have the ability to renew themselves indefinitely and differentiate into multiple cell lineages (229). Stem cells can be classified according to their differentiation capability: (1) pluripotent stem cells; (2) multipotent stem cells; (3) unipotent stem cells (230). Mesenchymal stem cells (MSCs) are one of the most widely studied pluripotent stem cells in DN (231). Among these stem cells, MSCs have several advantages to apply in DN therapy, such as easy harvesting, multi-lineage differentiation potential, strong immunosuppression, and no immune rejection (232). MSCs come from a wide range of sources, including bone marrow, adipose tissue, umbilical cord blood, peripheral blood, and amniotic fluid, among which bone marrow is the most abundant source (233–237). MSCs can differentiate into glomerular mesangial cells, tubular epithelial cells, endothelial cells, and podocytes (238–240). In STZ-induced rat DN model, MSCs injection can upregulate anti-inflammatory factors such as IL-10 and EGF, downregulate pro-inflammatory factors, and inhibit macrophage activation (241). In addition, administration of MSCs reduced pathological damage, collagen deposition, and fibrosis in the kidney (242). Although the safety and efficacy of MSCs therapy have been evaluated in clinical trials for kidney transplantation, liver fibrosis, and Crohn’s disease, the clinical trials of MSCs in DN are still ongoing (229, 243–245).
Recently, microvesicles secreted by MSCs, known as exosomes, have been widely studied in animal experiments and have demonstrated their roles in DN therapy (246–248). Exosomes containing functional proteins and RNA (microRNA and mRNA) can be detected in the MSCs medium supernatant, which contributes to cell-to-cell communication in paracrine manners (248). Therefore, many studies have focused on the role of exosomes as a key factor in the paracrine action of MSCs in DN (246, 247, 249, 250). Exosomes isolated from MSCs conditioned medium by ultrafiltration-combined purification method were administrated to STZ-induced DN rat model. The result showed significantly reduced mTOR pathway expression and fibrosis markers in renal tissue (249). Intravenously administration of MSC-conditioned medium to high-fat diet (HFD) and STZ-induced diabetic mice showed decreased proteinuria and proinflammatory cytokines expression, and significantly ameliorated tubulointerstitial fibrosis (247). Research in the coming years will focus on this secretion as a possible treatment option without significant side effects. Future studies are needed to clarify the molecular mechanism of mesenchymal-derived exosomes in improving DN.
Due to impaired glomerular filtration and tubular secretion function in DN, drugs can hardly reach the injured kidneys efficiently. Therefore, the treatment of kidney diseases requires high doses of the drug, which are usually associated with serious adverse effects. In recent years, the application of nanomedicines is gradually emerging in the treatment of renal diseases. Owing to the superior targetability and improved pharmacokinetic properties of nanomedicine, kidney-targeted nanomedicine carrying drug candidates can help to address the challenges associated with DN pharmacotherapy (251).
Numerous nanomedicine-based drug delivery systems have been developed to deliver therapeutic agents specifically to the kidney (252). For example, drug nanocomplexes containing low-molecular-weight chitosan bind the megalin-cubilin receptor in proximal tubules (251). Albumin nanoparticles with specific size target mesangial cells. Wu et al. reported that albumin-methylprednisolone nanoconjugates with a size of about 10 nm can specifically target the podocytes (253). These nanoconjugates avoid the side effects of glucocorticoids in patients with DN. Another investigation developed a nanoconjugate of baicalin and lysozyme with good renal targetability. This conjugate successfully ameliorated renal fibrosis and inflammation via NF-κB, TGF-β1/Smad3, and IGF-1/p38 MAPK signaling pathways. Manna et al. developed and studied the effect of pomegranate peel extract-stabilized gold nanoparticles (PPE-AuNPs) on the STZ-induced DN mice model (254). In DN mice, PPE-AuNPs significantly improved renal fibrosis and glomerular sclerosis. Specifically, it alleviated renal inflammation by modulating the MAPK/NF-kB/STAT3/cytokine axis.
As evidenced by the large number of nanoparticle formulations already on the market and many more in clinical trials, nanomedicines will surely take a large market share soon. Novel strategies to develop nanomedicine-based platforms with superior efficacy and safety for DN-targeted drug delivery hold great promising for the treatment of DN in the future.
The global burden of diabetes seems certain to increase dramatically in the future, coinciding with the rise in obesity. This implies a corresponding increase in the incidence of DN. Despite the efficacy of hypoglycemic drugs, they will be insufficient to halt disease onset and progression as the number of new cases. Therefore, new strategies and targets against DN are urgently needed. Emerging knowledge about immune responses and inflammation as bridges in the pathogenesis between abnormal metabolism and DN offers new promising for targeted therapies. Already under investigation are therapies focusing on the regulation of inflammatory pathways and, involving targets such as immune cells, pro-inflammatory cytokines, adhesion molecules, chemokines, JAK-STAT signaling, or NF-κB signaling. Additional promising targets may be the complement system, microRNAs and downstream targets of specific inflammatory signaling pathways. It is clear that the role of the immune response in DN pathogenesis is quite complex and multi-faceted, which highlights the need to explore combination therapies.
JC and YL wrote the manuscript. QL and JH contributed to the discussion and reviewed the manuscript. JH and YL obtained funding. JC, JH and YL are the guarantors of this work and as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
This study was supported by the National Natural Science Foundation of China (82025007, 82170874, 81930020, and 81870599).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Selby NM, Taal MW. An updated overview of diabetic nephropathy: Diagnosis, prognosis, treatment goals and latest guidelines. Diabetes Obes Metab (2020) 22 Suppl 1:3–15. doi: 10.1111/dom.14007
2. Deng Y, Li N, Wu Y, Wang M, Yang S, Zheng Y, et al. Global, regional, and national burden of diabetes-related chronic kidney disease from 1990 to 2019. Front Endocrinol (Lausanne) (2021) 12:672350. doi: 10.3389/fendo.2021.672350
3. Kalantar-Zadeh K, Jafar TH, Nitsch D, Neuen BL, Perkovic V. Chronic kidney disease. Lancet (2021) 398(10302):786–802. doi: 10.1016/S0140-6736(21)00519-5
4. Wada J, Makino H. Inflammation and the pathogenesis of diabetic nephropathy. Clin Sci (Lond) (2013) 124(3):139–52. doi: 10.1042/CS20120198
5. Barrera-Chimal J, Jaisser F. Pathophysiologic mechanisms in diabetic kidney disease: A focus on current and future therapeutic targets. Diabetes Obes Metab (2020) 22 Suppl 1:16–31. doi: 10.1111/dom.13969
6. Kikkawa R, Koya D, Haneda M. Progression of diabetic nephropathy. Am J Kidney Diseases (2003) 41(3):S19–21. doi: 10.1053/ajkd.2003.50077
7. Tang SCW, Yiu WH. Innate immunity in diabetic kidney disease. Nat Rev Nephrol (2020) 16(4):206–22. doi: 10.1038/s41581-019-0234-4
8. Tesch GH. Diabetic nephropathy - is this an immune disorder? Clin Sci (Lond) (2017) 131(16):2183–99. doi: 10.1042/CS20160636
9. Cockwell P, Fisher L-A. The global burden of chronic kidney disease. Lancet (2020) 395(10225):662–4. doi: 10.1016/S0140-6736(19)32977-0
10. Wan S, Wan S, Jiao X, Cao H, Gu Y, Yan L, et al. Advances in understanding the innate immune-associated diabetic kidney disease. FASEB J (2021) 35(2):e21367. doi: 10.1096/fj.202002334R
11. Kong L, Andrikopoulos S, MacIsaac RJ, Mackay LK, Nikolic-Paterson DJ, Torkamani N, et al. Role of the adaptive immune system in diabetic kidney disease. J Diabetes Investig (2022) 13(2):213–26. doi: 10.1111/jdi.13725
12. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell (2006) 124(4):783–801. doi: 10.1016/j.cell.2006.02.015
13. Guiteras R, Sola A, Flaquer M, Manonelles A, Hotter G, Cruzado JM. Exploring macrophage cell therapy on diabetic kidney disease. J Cell Mol Med (2019) 23(2):841–51. doi: 10.1111/jcmm.13983
14. Chow F, Ozols E, Nikolic-Paterson DJ, Atkins RC, Tesch GH. Macrophages in mouse type 2 diabetic nephropathy: Correlation with diabetic state and progressive renal injury. Kidney Int (2004) 65(1):116–28. doi: 10.1111/j.1523-1755.2004.00367.x
15. Chow FY, Nikolic-Paterson DJ, Atkins RC, Tesch GH. Macrophages in streptozotocin-induced diabetic nephropathy: potential role in renal fibrosis. Nephrol Dial Transplant (2004) 19(12):2987–96. doi: 10.1093/ndt/gfh441
16. Wu CC, Sytwu HK, Lu KC, Lin YF. Role of T cells in type 2 diabetic nephropathy. Exp Diabetes Res (2011) 2011:514738. doi: 10.1155/2011/514738
17. Moon JY, Jeong KH, Lee TW, Ihm CG, Lim SJ, Lee SH. Aberrant recruitment and activation of T cells in diabetic nephropathy. Am J Nephrol (2012) 35(2):164–74. doi: 10.1159/000334928
18. Kanwar YS, Sun L, Xie P, Liu FY, Chen S. A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annu Rev Pathol (2011) 6:395–423. doi: 10.1146/annurev.pathol.4.110807.092150
19. Flyvbjerg A. The role of the complement system in diabetic nephropathy. Nat Rev Nephrol (2017) 13(5):311–8. doi: 10.1038/nrneph.2017.31
20. Zheng Z, Zheng F. Immune cells and inflammation in diabetic nephropathy. J Diabetes Res (2016) 2016:1841690. doi: 10.1155/2016/1841690
21. Komada T, Muruve DA. The role of inflammasomes in kidney disease. Nat Rev Nephrol (2019) 15(8):501–20. doi: 10.1038/s41581-019-0158-z
22. Pichler R, Afkarian M, Dieter B, Tuttle K. Immunity and inflammation in diabetic kidney disease: translating mechanisms to biomarkers and treatment targets. Am J Physiol Renal Physiol (2017) 312(4):F716–F31. doi: 10.1152/ajprenal.00314.2016
23. Hickey F, Martin F. Role of the immune system in diabetic kidney disease. Curr Diabetes Rep (2018) 18(4):20. doi: 10.1007/s11892-018-0984-6
24. Hickey FB, Martin F. Diabetic kidney disease and immune modulation. Curr Opin Pharmacol (2013) 13(4):602–12. doi: 10.1016/j.coph.2013.05.002
25. Tesch GH. Macrophages and diabetic nephropathy. Semin Nephrol (2010) 30(3):290–301. doi: 10.1016/j.semnephrol.2010.03.007
26. Awad AS, You H, Gao T, Cooper TK, Nedospasov SA, Vacher J, et al. Macrophage-derived tumor necrosis factor-alpha mediates diabetic renal injury. Kidney Int (2015) 88(4):722–33. doi: 10.1038/ki.2015.162
27. Yu J, Dong R, Da J, Li J, Yu F, Zha Y. High-mobility group nucleosome-binding protein 1 mediates renal fibrosis correlating with macrophages accumulation and epithelial-to-Mesenchymal transition in diabetic nephropathy mice model. Kidney Blood Press Res (2019) 44(3):331–43. doi: 10.1159/000499877
28. Sugimoto H, Shikata K, Hirata K, Akiyama K, Matsuda M, Kushiro M, et al. Increased expression of intercellular adhesion molecule-1 (ICAM-1) in diabetic rat glomeruli: glomerular hyperfiltration is a potential mechanism of ICAM-1 upregulation. Diabetes (1997) 46(12):2075–81. doi: 10.2337/diab.46.12.2075
29. Seron D, Cameron JS, Haskard DO. Expression of VCAM-1 in the normal and diseased kidney. Nephrol Dial Transplant (1991) 6(12):917–22. doi: 10.1093/ndt/6.12.917
30. Yamaguchi H, Igarashi M, Hirata A, Tsuchiya H, Sugiyama K, Morita Y, et al. Progression of diabetic nephropathy enhances the plasma osteopontin level in type 2 diabetic patients. Endocr J (2004) 51(5):499–504. doi: 10.1507/endocrj.51.499
31. Wada T, Furuichi K, Sakai N, Iwata Y, Yoshimoto K, Shimizu M, et al. Up-regulation of monocyte chemoattractant protein-1 in tubulointerstitial lesions of human diabetic nephropathy. Kidney Int (2000) 58(4):1492–9. doi: 10.1046/j.1523-1755.2000.00311.x
32. Chow FY, Nikolic-Paterson DJ, Ma FY, Ozols E, Rollins BJ, Tesch GH. Monocyte chemoattractant protein-1-induced tissue inflammation is critical for the development of renal injury but not type 2 diabetes in obese db/db mice. Diabetologia (2007) 50(2):471–80. doi: 10.1007/s00125-006-0497-8
33. Hsieh TJ, Chen R, Zhang SL, Liu F, Brezniceanu ML, Whiteside CI, et al. Upregulation of osteopontin gene expression in diabetic rat proximal tubular cells revealed by microarray profiling. Kidney Int (2006) 69(6):1005–15. doi: 10.1038/sj.ki.5000206
34. Navarro-Gonzalez JF, Mora-Fernandez C, Muros de Fuentes M, Garcia-Perez J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat Rev Nephrol (2011) 7(6):327–40. doi: 10.1038/nrneph.2011.51
35. Chow FY, Nikolic-Paterson DJ, Ozols E, Atkins RC, Tesch GH. Intercellular adhesion molecule-1 deficiency is protective against nephropathy in type 2 diabetic db/db mice. J Am Soc Nephrol (2005) 16(6):1711–22. doi: 10.1681/ASN.2004070612
36. Gordon S. Alternative activation of macrophages. Nat Rev Immunol (2003) 3(1):23–35. doi: 10.1038/nri978
37. Mosser DM. The many faces of macrophage activation. J Leukocyte Biol (2003) 73(2):209–12. doi: 10.1189/jlb.0602325
38. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol (2004) 25(12):677–86. doi: 10.1016/j.it.2004.09.015
39. Cucak H, Nielsen Fink L, Hojgaard Pedersen M, Rosendahl A. Enalapril treatment increases T cell number and promotes polarization towards M1-like macrophages locally in diabetic nephropathy. Int Immunopharmacol (2015) 25(1):30–42. doi: 10.1016/j.intimp.2015.01.003
40. Wang X, Yao B, Wang Y, Fan X, Wang S, Niu A, et al. Macrophage cyclooxygenase-2 protects against development of diabetic nephropathy. Diabetes (2017) 66(2):494–504. doi: 10.2337/db16-0773
41. Zhang X, Yang Y, Zhao Y. Macrophage phenotype and its relationship with renal function in human diabetic nephropathy. PloS One (2019) 14(9):e0221991. doi: 10.1371/journal.pone.0221991
42. Yuan Y, Li L, Zhu L, Liu F, Tang X, Liao G, et al. Mesenchymal stem cells elicit macrophages into M2 phenotype via improving transcription factor EB-mediated autophagy to alleviate diabetic nephropathy. Stem Cells (2020) 38(5):639–52. doi: 10.1002/stem.3144
43. Herrera M, Soderberg M, Sabirsh A, Valastro B, Molne J, Santamaria B, et al. Inhibition of T-cell activation by the CTLA4-fc abatacept is sufficient to ameliorate proteinuric kidney disease. Am J Physiology-Renal Physiol (2017) 312(4):F748–F59. doi: 10.1152/ajprenal.00179.2016
44. Lim AKH, Ma FY, Nikolic-Paterson DJ, Kitching AR, Thomas MC, Tesch GH. Lymphocytes promote albuminuria, but not renal dysfunction or histological damage in a mouse model of diabetic renal injury. Diabetologia (2010) 53(8):1772–82. doi: 10.1007/s00125-010-1757-1
45. Galkina E, Ley K. Leukocyte recruitment and vascular injury in diabetic nephropathy. J Am Soc Nephrol (2006) 17(2):368–77. doi: 10.1681/ASN.2005080859
46. Imani F, Horii Y, Suthanthiran M, Skolnik EY, Makita Z, Sharma V, et al. ADVANCED GLYCOSYLATION ENDPRODUCT-SPECIFIC RECEPTORS ON HUMAN AND RAT T-LYMPHOCYTES MEDIATE SYNTHESIS OF INTERFERON-GAMMA - ROLE IN TISSUE REMODELING. J Exp Med (1993) 178(6):2165–72. doi: 10.1084/jem.178.6.2165
47. Eller K, Kirsch A, Wolf A, Sopper S, Tagwerker A, Stanzl U, et al. Potential role of regulatory T cells in reversing obesity-linked insulin resistance and diabetic nephropathy. Diabetes (2011) 60(11):2954–62. doi: 10.2337/db11-0358
48. Kim SM, Lee SH, Lee A, Kim DJ, Kim YG, Kim SY, et al. Targeting T helper 17 by mycophenolate mofetil attenuates diabetic nephropathy progression. Transl Res (2015) 166(4):375–83. doi: 10.1016/j.trsl.2015.04.013
49. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol (2010) 28:445–89. doi: 10.1146/annurev-immunol-030409-101212
50. Mosmann TR, Coffman RL. TH1 and TH2 cells: Different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol (1989) 7:145–73. doi: 10.1146/annurev.iy.07.040189.001045
51. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol (2011) 29:621–63. doi: 10.1146/annurev-immunol-031210-101400
52. Mosmann TR, Sad S. The expanding universe of T-cell subsets: Th1, Th2 and more. Immunol Today (1996) 17(3):138–46. doi: 10.1016/0167-5699(96)80606-2
53. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol (2009) 27:485–517. doi: 10.1146/annurev.immunol.021908.132710
54. Lavoz C, Matus YS, Orejudo M, Carpio JD, Droguett A, Egido J, et al. Interleukin-17A blockade reduces albuminuria and kidney injury in an accelerated model of diabetic nephropathy. Kidney Int (2019) 95(6):1418–32. doi: 10.1016/j.kint.2018.12.031
55. Zhu J. T Helper 2 (Th2) cell differentiation, type 2 innate lymphoid cell (ILC2) development and regulation of interleukin-4 (IL-4) and IL-13 production. Cytokine (2015) 75(1):14–24. doi: 10.1016/j.cyto.2015.05.010
56. Beissert S, Schwarz A, Schwarz T. Regulatory T cells. J Invest Dermatol (2006) 126(1):15–24. doi: 10.1038/sj.jid.5700004
57. Bilate AM, Lafaille JJ. Induced CD4+Foxp3+ regulatory T cells in immune tolerance. Annu Rev Immunol (2012) 30:733–58. doi: 10.1146/annurev-immunol-020711-075043
58. Zhang F, Wang C, Wen X, Chen Y, Mao R, Cui D, et al. Mesenchymal stem cells alleviate rat diabetic nephropathy by suppressing CD103(+) DCs-mediated CD8(+) T cell responses. J Cell Mol Med (2020) 24(10):5817–31. doi: 10.1111/jcmm.15250
59. Smith M, Simmons K, Cambier J. B cells in type 1 diabetes mellitus and diabetic kidney disease. Nat Rev Nephrol (2017) 13(11):712–20. doi: 10.1038/nrneph.2017.138
60. Fiorina P, Vergani A, Dada S, Jurewicz M, Wong M, Law K, et al. Targeting CD22 reprograms b-cells and reverses autoimmune diabetes. Diabetes (2008) 57(11):3013–24. doi: 10.2337/db08-0420
61. Kleffel S, Vergani A, Tezza S, Ben Nasr M, Niewczas M, Wong S, et al. Interleukin-10+ regulatory b cells arise within antigen-experienced CD40+ b cells to maintain tolerance to islet autoantigens. Diabetes (2015) 64(1):158–71. doi: 10.2337/db13-1639
62. Balakumar P, Reddy J, Singh M. Do resident renal mast cells play a role in the pathogenesis of diabetic nephropathy? Mol Cell Biochem (2009) 330(1-2):187–92. doi: 10.1007/s11010-009-0132-3
63. Ehara T, Shigematsu H. Mast cells in the kidney. Nephrology (2003) 8(3):130–8. doi: 10.1046/j.1440-1797.2003.00153.x
64. Jones S, Kelly D, Cox A, Zhang Y, Gow R, Gilbert R. Mast cell infiltration and chemokine expression in progressive renal disease. Kidney Int (2003) 64(3):906–13. doi: 10.1046/j.1523-1755.2003.00183.x
65. Holdsworth SR, Summers SA. Role of mast cells in progressive renal diseases. J Am Soc Nephrol (2008) 19(12):2254–61. doi: 10.1681/ASN.2008010015
66. Navarro-González JF, Mora-Fernández C. The role of inflammatory cytokines in diabetic nephropathy. J Am Soc Nephrol (2008) 19(3):433–42. doi: 10.1681/ASN.2007091048
67. Wu CC, Sytwu HK, Lin YF. Cytokines in diabetic nephropathy. Adv Clin Chem (2012) 56:55–74. doi: 10.1016/B978-0-12-394317-0.00014-5
68. Elmarakby AA, Sullivan JC. Relationship between oxidative stress and inflammatory cytokines in diabetic nephropathy. Cardiovasc Ther (2012) 30(1):49–59. doi: 10.1111/j.1755-5922.2010.00218.x
69. Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol (2014) 192(12):5451–8. doi: 10.4049/jimmunol.1490019
70. Suzuki D, Miyazaki M, Naka R, Koji T, Yagame M, Jinde K, et al. In situ hybridization of interleukin 6 in diabetic nephropathy. Diabetes (1995) 44(10):1233–8. doi: 10.2337/diab.44.10.1233
71. Mauer J, Chaurasia B, Goldau J, Vogt MC, Ruud J, Nguyen KD, et al. Signaling by IL-6 promotes alternative activation of macrophages to limit endotoxemia and obesity-associated resistance to insulin. Nat Immunol (2014) 15(5):423–30. doi: 10.1038/ni.2865
72. Sziksz E, Pap D, Lippai R, Beres NJ, Fekete A, Szabo AJ, et al. Fibrosis related inflammatory mediators: Role of the IL-10 cytokine family. Mediators Inflamm (2015) 2015:764641. doi: 10.1155/2015/764641
73. Dai SM, Matsuno H, Nakamura H, Nishioka K, Yudoh K. Interleukin-18 enhances monocyte tumor necrosis factor alpha and interleukin-1beta production induced by direct contact with T lymphocytes: implications in rheumatoid arthritis. Arthritis Rheumatol (2004) 50(2):432–43. doi: 10.1002/art.20064
74. Morel JC, Park CC, Woods JM, Koch AE. A novel role for interleukin-18 in adhesion molecule induction through NF kappa b and phosphatidylinositol (PI) 3-kinase-dependent signal transduction pathways. J Biol Chem (2001) 276(40):37069–75. doi: 10.1074/jbc.M103574200
75. Moriwaki Y, Yamamoto T, Shibutani Y, Aoki E, Tsutsumi Z, Takahashi S, et al. Elevated levels of interleukin-18 and tumor necrosis factor-alpha in serum of patients with type 2 diabetes mellitus: relationship with diabetic nephropathy. Metabolism (2003) 52(5):605–8. doi: 10.1053/meta.2003.50096
76. Navarro JF, Mora C, Muros M, Garcia J. Urinary tumour necrosis factor-alpha excretion independently correlates with clinical markers of glomerular and tubulointerstitial injury in type 2 diabetic patients. Nephrol Dial Transplant (2006) 21(12):3428–34. doi: 10.1093/ndt/gfl469
77. DiPetrillo K, Coutermarsh B, Gesek F. Urinary tumor necrosis factor contributes to sodium retention and renal hypertrophy during diabetes. Am J Physiol Renal Physiol (2003) 284(1):F113–21. doi: 10.1152/ajprenal.00026.2002
78. Rivero A, Mora C, Muros M, Garcia J, Herrera H, Navarro-Gonzalez JF. Pathogenic perspectives for the role of inflammation in diabetic nephropathy. Clin Sci (Lond) (2009) 116(6):479–92. doi: 10.1042/CS20080394
79. Tsuchida K, Cronin B, Sharma K. Novel aspects of transforming growth factor-beta in diabetic kidney disease. Nephron (2002) 92(1):7–21. doi: 10.1159/000064486
80. Chiarelli F, Gaspari S, Marcovecchio ML. Role of growth factors in diabetic kidney disease. Horm Metab Res (2009) 41(8):585–93. doi: 10.1055/s-0029-1220752
81. Lee H. Pathogenic role of TGF-β in the progression of podocyte diseases. Histol Histopathol (2011) 26(1):107–16. doi: 10.14670/HH-26.107
82. Tannenbaum CS, Hamilton TA. Immune-inflammatory mechanisms in IFNgamma-mediated anti-tumor activity. Semin Cancer Biol (2000) 10(2):113–23. doi: 10.1006/scbi.2000.0314
83. Navarro JF, Milena FJ, Mora C, Leon C, Garcia J. Renal pro-inflammatory cytokine gene expression in diabetic nephropathy: effect of angiotensin-converting enzyme inhibition and pentoxifylline administration. Am J Nephrol (2006) 26(6):562–70. doi: 10.1159/000098004
84. Roland L, Gagne A, Belanger MC, Boutet M, Julien P, Bilodeau JF. Plasma interleukin-18 (IL-18) levels are correlated with antioxidant vitamin coenzyme Q(10) in preeclampsia. Acta Obstet Gynecol Scand (2010) 89(3):360–6. doi: 10.3109/00016340903576020
85. Jung MK, Song HK, Kim KE, Hur DY, Kim T, Bang S, et al. IL-18 enhances the migration ability of murine melanoma cells through the generation of ROI and the MAPK pathway. Immunol Lett (2006) 107(2):125–30. doi: 10.1016/j.imlet.2006.08.004
86. Jevnikar AM, Brennan DC, Singer GG, Heng JE, Maslinski W, Wuthrich RP, et al. STIMULATED KIDNEY TUBULAR EPITHELIAL-CELLS EXPRESS MEMBRANE-ASSOCIATED AND SECRETED TNF-ALPHA. Kidney Int (1991) 40(2):203–11. doi: 10.1038/ki.1991.201
87. Sugimoto H, Shikata K, Wada J, Horiuchi S, Makino H. Advanced glycation end products-cytokine-nitric oxide sequence pathway in the development of diabetic nephropathy: aminoguanidine ameliorates the overexpression of tumour necrosis factor-alpha and inducible nitric oxide synthase in diabetic rat glomeruli. Diabetologia (1999) 42(7):878–86. doi: 10.1007/s001250051241
88. Dong X, Swaminathan S, Bachman LA, Croatt AJ, Nath KA, Griffin MD. Resident dendritic cells are the predominant TNF-secreting cell in early renal ischemia-reperfusion injury. Kidney Int (2007) 71(7):619–28. doi: 10.1038/sj.ki.5002132
89. Wong CK, Ho AWY, Tong PCY, Yeung CY, Kong APS, Lun SWM, et al. Aberrant activation profile of cytokines and mitogen-activated protein kinases in type 2 diabetic patients with nephropathy. Clin Exp Immunol (2007) 149(1):123–31. doi: 10.1111/j.1365-2249.2007.03389.x
90. Diamond-Stanic MK, You YH, Sharma K. Sugar, sex, and TGF-beta in diabetic nephropathy. Semin Nephrol (2012) 32(3):261–8. doi: 10.1016/j.semnephrol.2012.04.005
91. Chen S, Jim B, Ziyadeh F. Diabetic nephropathy and transforming growth factor-beta: transforming our view of glomerulosclerosis and fibrosis build-up. Semin Nephrol (2003) 23(6):532–43. doi: 10.1053/S0270-9295(03)00132-3
92. Rocco MV, Chen Y, Goldfarb S, Ziyadeh FN. ELEVATED GLUCOSE STIMULATES TGF-BETA GENE-EXPRESSION AND BIOACTIVITY IN PROXIMAL TUBULE. Kidney Int (1992) 41(1):107–14. doi: 10.1038/ki.1992.14
93. Chen S, Cohen MP, Lautenslager GT, Shearman CW, Ziyadeh FN. Glycated albumin stimulates TGF-beta 1 production and protein kinase c activity in glomerular endothelial cells. Kidney Int (2001) 59(2):673–81. doi: 10.1046/j.1523-1755.2001.059002673.x
94. Kumar Pasupulati A, Chitra PS, Reddy GB. Advanced glycation end products mediated cellular and molecular events in the pathology of diabetic nephropathy. Biomolecular Concepts (2016) 7(5-6):293–309. doi: 10.1515/bmc-2016-0021
95. Campbell KN, Raij L, Mundel P. Role of angiotensin II in the development of nephropathy and podocytopathy of diabetes. Curr Diabetes Rev (2011) 7(1):3–7. doi: 10.2174/157339911794273973
96. Ziyadeh F, Hoffman B, Han D, Iglesias-De La Cruz M, Hong S, Isono M, et al. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proc Natl Acad Sci United States America (2000) 97(14):8015–20. doi: 10.1073/pnas.120055097
97. Chen S, Iglesias-de la Cruz M, Jim B, Hong S, Isono M, Ziyadeh F. Reversibility of established diabetic glomerulopathy by anti-TGF-beta antibodies in db/db mice. Biochem Biophys Res Commun (2003) 300(1):16–22. doi: 10.1016/S0006-291X(02)02708-0
98. Wang A, Ziyadeh F, Lee E, Pyagay P, Sung S, Sheardown S, et al. Interference with TGF-beta signaling by Smad3-knockout in mice limits diabetic glomerulosclerosis without affecting albuminuria. Am J Physiol Renal Physiol (2007) 293(5):F1657–65. doi: 10.1152/ajprenal.00274.2007
99. Watson A, Gray S, Jiaze L, Soro-Paavonen A, Wong B, Cooper M, et al. Alagebrium reduces glomerular fibrogenesis and inflammation beyond preventing RAGE activation in diabetic apolipoprotein e knockout mice. Diabetes (2012) 61(8):2105–13. doi: 10.2337/db11-1546
100. Matsui H, Suzuki M, Tsukuda R, Iida K, Miyasaka M, Ikeda H. Expression of ICAM-1 on glomeruli is associated with progression of diabetic nephropathy in a genetically obese diabetic rat, wistar fatty. Diabetes Res Clin Pract (1996) 32(1-2):1–9. doi: 10.1016/0168-8227(96)01209-0
101. Okada S, Shikata K, Matsuda M, Ogawa D, Usui H, Kido Y, et al. Intercellular adhesion molecule-1-deficient mice are resistant against renal injury after induction of diabetes. Diabetes (2003) 52(10):2586–93. doi: 10.2337/diabetes.52.10.2586
102. Lyck R, Enzmann G. The physiological roles of ICAM-1 and ICAM-2 in neutrophil migration into tissues. Curr Opin Hematol (2015) 22(1):53–9. doi: 10.1097/MOH.0000000000000103
103. Ina K, Kitamura H, Okeda T, Nagai K, Liu Z, Matsuda M, et al. Vascular cell adhesion molecule-1 expression in the renal interstitium of diabetic KKAy mice. Diabetes Res Clin Practice (1999) 44(1):1–8. doi: 10.1016/S0168-8227(99)00011-X
104. Murakami H, Tamasawa N, Matsui J, Yamato K, JingZhi G, Suda T. Plasma levels of soluble vascular adhesion molecule-1 and cholesterol oxidation product in type 2 diabetic patients with nephropathy. J Atheroscl Thrombosis (2001) 8(1):21–4. doi: 10.5551/jat1994.8.21
105. Sumagin R, Sarelius IH. TNF-alpha activation of arterioles and venules alters distribution and levels of ICAM-1 and affects leukocyte-endothelial cell interactions. Am J Physiology-Heart Circulatory Physiol (2006) 291(5):H2116–H25. doi: 10.1152/ajpheart.00248.2006
106. Sucosky P, Balachandran K, Elhammali A, Jo H, Yoganathan AP. Altered shear stress stimulates upregulation of endothelial VCAM-1 and ICAM-1 in a BMP-4-and TGF-beta 1-dependent pathway. Arterioscler Thromb Vasc Biol (2009) 29(2):254–60. doi: 10.1161/ATVBAHA.108.176347
107. Heidland A, Sebekova K, Schinzel R. Advanced glycation end products and the progressive course of renal disease. Am J Kidney Dis Off J Natl Kidney Foundation (2001) 38:S100–6. doi: 10.1053/ajkd.2001.27414
108. Chang TT, Chen JW. The role of chemokines and chemokine receptors in diabetic nephropathy. Int J Mol Sci (2020) 21(9):3172. doi: 10.3390/ijms21093172
109. Park J, Ryu DR, Li JJ, Jung DS, Kwak SJ, Lee SH, et al. MCP-1/CCR2 system is involved in high glucose-induced fibronectin and type IV collagen expression in cultured mesangial cells. Am J Physiol Renal Physiol (2008) 295(3):F749–57. doi: 10.1152/ajprenal.00547.2007
110. Shoukry A, Bdeer Sel A, El-Sokkary RH. Urinary monocyte chemoattractant protein-1 and vitamin d-binding protein as biomarkers for early detection of diabetic nephropathy in type 2 diabetes mellitus. Mol Cell Biochem (2015) 408(1-2):25–35. doi: 10.1007/s11010-015-2479-y
111. Janiak P, Bidouard J, Cadrouvele C, Poirier B, Gouraud L, Grataloup Y, et al. Long-term blockade of angiotensin AT1 receptors increases survival of obese zucker rats. Eur J Pharmacol (2006) 534:271–9. doi: 10.1016/j.ejphar.2006.01.032
112. Amann B, Tinzmann R, Angelkort B. ACE inhibitors improve diabetic nephropathy through suppression of renal MCP-1. Diabetes Care (2003) 26(8):2421–5. doi: 10.2337/diacare.26.8.2421
113. Carr M, Roth S, Luther E, Rose S, Springer T. Monocyte chemoattractant protein 1 acts as a T-lymphocyte chemoattractant. Proc Natl Acad Sci United States America (1994) 91(9):3652–6. doi: 10.1073/pnas.91.9.3652
114. Xu L, Warren M, Rose W, Gong W, Wang J. Human recombinant monocyte chemotactic protein and other c-c chemokines bind and induce directional migration of dendritic cells in vitro. J Leukocyte Biol (1996) 60(3):365–71. doi: 10.1002/jlb.60.3.365
115. Sanchez AP, Sharma K. Transcription factors in the pathogenesis of diabetic nephropathy. Expert Rev Mol Med (2009) 11:e13. doi: 10.1017/S1462399409001057
116. Barnes P, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. New Engl J Med (1997) 336(15):1066–71. doi: 10.1056/NEJM199704103361506
117. Liu R, Zhong Y, Li X, Chen H, Jim B, Zhou M, et al. Role of transcription factor acetylation in diabetic kidney disease. Diabetes (2014) 63(7):2440–53. doi: 10.2337/db13-1810
118. Mezzano S, Aros C, Droguett A, Burgos ME, Ardiles L, Flores C, et al. NF-kappaB activation and overexpression of regulated genes in human diabetic nephropathy. Nephrol Dial Transplant (2004) 19(10):2505–12. doi: 10.1093/ndt/gfh207
119. Warren AM, Knudsen ST, Cooper ME. Diabetic nephropathy: an insight into molecular mechanisms and emerging therapies. Expert Opin Ther Targets (2019) 23(7):579–91. doi: 10.1080/14728222.2019.1624721
120. Soetikno V, Sari F, Veeraveedu P, Thandavarayan R, Harima M, Sukumaran V, et al. Curcumin ameliorates macrophage infiltration by inhibiting NF-κB activation and proinflammatory cytokines in streptozotocin induced-diabetic nephropathy. Nutr Metab (2011) 8(1):35. doi: 10.1186/1743-7075-8-35
121. Yi B, Hu X, Zhang H, Huang J, Liu J, Hu J, et al. Nuclear NF-kappaB p65 in peripheral blood mononuclear cells correlates with urinary MCP-1, RANTES and the severity of type 2 diabetic nephropathy. PloS One (2014) 9(6):e99633. doi: 10.1371/journal.pone.0099633
122. Xin P, Xu X, Deng C, Liu S, Wang Y, Zhou X, et al. The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int Immunopharmacol (2020) 80:106210. doi: 10.1016/j.intimp.2020.106210
123. Sun M, Bu W, Li Y, Zhu J, Zhao J, Zhang P, et al. Danzhi jiangtang capsule ameliorates kidney injury via inhibition of the JAK-STAT signaling pathway and increased antioxidant capacity in STZ-induced diabetic nephropathy rats. Biosci Trends (2019) 12(6):595–604. doi: 10.5582/bst.2018.01255
124. Zhang N, Zheng Q, Wang Y, Lin J, Wang H, Liu R, et al. Renoprotective effect of the recombinant anti-IL-6R fusion proteins by inhibiting JAK2/STAT3 signaling pathway in diabetic nephropathy. Front Pharmacol (2021) 12:681424. doi: 10.3389/fphar.2021.681424
125. Woroniecka K, Park A, Mohtat D, Thomas D, Pullman J, Susztak K. Transcriptome analysis of human diabetic kidney disease. Diabetes (2011) 60(9):2354–69. doi: 10.2337/db10-1181
126. Zitman-Gal T, Einbinder Y, Ohana M, Katzav A, Kartawy A, Benchetrit S. Effect of liraglutide on the janus kinase/signal transducer and transcription activator (JAK/STAT) pathway in diabetic kidney disease in db/db mice and in cultured endothelial cells. J Diabetes (2019) 11(8):656–64. doi: 10.1111/1753-0407.12891
127. Modesti A, Bertolozzi I, Gamberi T, Marchetta M, Lumachi C, Coppo M, et al. Hyperglycemia activates JAK2 signaling pathway in human failing myocytes via angiotensin II-mediated oxidative stress. Diabetes (2005) 54(2):394–401. doi: 10.2337/diabetes.54.2.394
128. Banes-Berceli A, Shaw S, Ma G, Brands M, Eaton D, Stern D, et al. Effect of simvastatin on high glucose- and angiotensin II-induced activation of the JAK/STAT pathway in mesangial cells. Am J Physiol Renal Physiol (2006) 291(1):F116–21. doi: 10.1152/ajprenal.00502.2005
129. Li R, Yang N, Zhang L, Huang Y, Zhang R, Wang F, et al. Inhibition of Jak/STAT signaling ameliorates mice experimental nephrotic syndrome. Am J Nephrol (2007) 27(6):580–9. doi: 10.1159/000108102
130. Amiri F, Shaw S, Wang X, Tang J, Waller J, Eaton D, et al. Angiotensin II activation of the JAK/STAT pathway in mesangial cells is altered by high glucose. Kidney Int (2002) 61(5):1605–16. doi: 10.1046/j.1523-1755.2002.00311.x
131. Marrero MB, Banes-Berceli AK, Stern DM, Eaton DC. Role of the JAK/STAT signaling pathway in diabetic nephropathy. Am J Physiol Renal Physiol (2006) 290(4):F762–8. doi: 10.1152/ajprenal.00181.2005
132. Wada T, Nangaku M. Novel roles of complement in renal diseases and their therapeutic consequences. Kidney Int (2013) 84(3):441–50. doi: 10.1038/ki.2013.134
133. Xiao X, Ma B, Dong B, Zhao P, Tai N, Chen L, et al. Cellular and humoral immune responses in the early stages of diabetic nephropathy in NOD mice. J Autoimmun (2009) 32(2):85–93. doi: 10.1016/j.jaut.2008.12.003
134. Franchi L, Munoz-Planillo R, Nunez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol (2012) 13(4):325–32. doi: 10.1038/ni.2231
135. Wu M, Yang Z, Zhang C, Shi Y, Han W, Song S, et al. Inhibition of NLRP3 inflammasome ameliorates podocyte damage by suppressing lipid accumulation in diabetic nephropathy. Metabolism (2021) 118:154748. doi: 10.1016/j.metabol.2021.154748
136. Shahzad K, Bock F, Dong W, Wang H, Kopf S, Kohli S, et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int (2015) 87(1):74–84. doi: 10.1038/ki.2014.271
137. Donate-Correa J, Luis-Rodriguez D, Martin-Nunez E, Tagua VG, Hernandez-Carballo C, Ferri C, et al. Inflammatory targets in diabetic nephropathy. J Clin Med (2020) 9(2):458. doi: 10.3390/jcm9020458
138. Maini R, Breedveld F, Kalden J, Smolen J, Davis D, Macfarlane J, et al. Therapeutic efficacy of multiple intravenous infusions of anti-tumor necrosis factor alpha monoclonal antibody combined with low-dose weekly methotrexate in rheumatoid arthritis. Arthritis Rheumatism (1998) 41(9):1552–63. doi: 10.1002/1529-0131(199809)41:9<1552::AID-ART5>3.0.CO;2-W
139. Baert F, D'Haens G, Peeters M, Hiele M, Schaible T, Shealy D, et al. Tumor necrosis factor alpha antibody (infliximab) therapy profoundly down-regulates the inflammation in crohn's ileocolitis. Gastroenterology (1999) 116(1):22–8. doi: 10.1016/S0016-5085(99)70224-6
140. Moriwaki Y, Inokuchi T, Yamamoto A, Ka T, Tsutsumi Z, Takahashi S, et al. Effect of TNF-alpha inhibition on urinary albumin excretion in experimental diabetic rats. Acta Diabetol (2007) 44(4):215–8. doi: 10.1007/s00592-007-0007-6
141. Prichett W, Hand A, Sheilds J, Dunnington D. Mechanism of action of bicyclic imidazoles defines a translational regulatory pathway for tumor necrosis factor alpha. J Inflammation (1995) 45(2):97–105.
142. De Sanctis M, Cesarone M, Belcaro G, Nicolaides A, Griffin M, Incandela L, et al. Treatment of intermittent claudication with pentoxifylline: A 12-month, randomized trial–walking distance and microcirculation. Angiology (2002) 53:S7–12.
143. Aviado D, Dettelbach H. Pharmacology of pentoxifylline, a hemorheologic agent for the treatment of intermittent claudication. Angiology (1984) 35(7):407–17. doi: 10.1177/000331978403500703
144. Aviado D, Porter J. Pentoxifylline: a new drug for the treatment of intermittent claudication. mechanism of action, pharmacokinetics, clinical efficacy and adverse effects. Pharmacotherapy (1984) 4(6):297–307. doi: 10.1002/j.1875-9114.1984.tb03380.x
145. Doherty G, Jensen J, Alexander H, Buresh C, Norton J. Pentoxifylline suppression of tumor necrosis factor gene transcription. Surgery (1991) 110(2):192–8.
146. Han J, Thompson P, Beutler B. Dexamethasone and pentoxifylline inhibit endotoxin-induced cachectin/tumor necrosis factor synthesis at separate points in the signaling pathway. J Exp Med (1990) 172(1):391–4. doi: 10.1084/jem.172.1.391
147. Donate-Correa J, Tagua VG, Ferri C, Martin-Nunez E, Hernandez-Carballo C, Urena-Torres P, et al. Pentoxifylline for renal protection in diabetic kidney disease. a model of old drugs for new horizons. J Clin Med (2019) 8(3):287. doi: 10.3390/jcm8030287
148. Gagliardini E, Benigni A. Role of anti-TGF-beta antibodies in the treatment of renal injury. Cytokine Growth Factor Rev (2006) 17(1-2):89–96. doi: 10.1016/j.cytogfr.2005.09.005
149. Winiarska K, Dzik J, Labudda M, Focht D, Sierakowski B, Owczarek A, et al. Melatonin nephroprotective action in zucker diabetic fatty rats involves its inhibitory effect on NADPH oxidase. J Pineal Res (2016) 60(1):109–17. doi: 10.1111/jpi.12296
150. Rashed LA, Elattar S, Eltablawy N, Ashour H, Mahmoud LM, El-Esawy Y. Mesenchymal stem cells pretreated with melatonin ameliorate kidney functions in a rat model of diabetic nephropathy. Biochem Cell Biol = Biochimie Biologie Cellulaire (2018) 96(5):564–71. doi: 10.1139/bcb-2017-0230
151. Wei J, Wang Y, Qi X, Fan Z, Wu Y. Melatonin ameliorates hyperglycaemia-induced renal inflammation by inhibiting the activation of TLR4 and TGF-β1/Smad3 signalling pathway. Am J Trans Res (2020) 12(5):1584–99.
152. Bae E. DPP-4 inhibitors in diabetic complications: role of DPP-4 beyond glucose control. Arch Pharmacal Res (2016) 39(8):1114–28. doi: 10.1007/s12272-016-0813-x
153. Li L, Lian X, Wang Z, Zheng J, Liu J, Chu Y, et al. The dipeptidyl peptidase-4 inhibitor sitagliptin ameliorates renal injury in type 1 diabetic mice via inhibiting the TGF-beta/Smad signal pathway. Pharmazie (2019) 74(4):239–42. doi: 10.1681/ph.2019.8918
154. Xiang C, Zhou R, Zhang Y, Zhang J, Yang H. [Research progress on saponins in panax notoginseng and their molecular mechanism of anti-cerebral ischemia]. Zhongguo Zhong yao Za Zhi = Zhongguo Zhongyao Zazhi = China J Chin Materia Medica (2020) 45(13):3045–54. doi: 10.19540/j.cnki.cjcmm.20200302.403
155. Jie L, Pengcheng Q, Qiaoyan H, Linlin B, Meng Z, Fang W, et al. Dencichine ameliorates kidney injury in induced type II diabetic nephropathy via the TGF-beta/Smad signalling pathway. Eur J Pharmacol (2017) 812:196–205. doi: 10.1016/j.ejphar.2017.06.024
156. Xu XX, Zhang W, Zhang P, Qi XM, Wu YG, Shen JJ. Superior renoprotective effects of the combination of breviscapine with enalapril and its mechanism in diabetic rats. Phytomedicine (2013) 20(10):820–7. doi: 10.1016/j.phymed.2013.03.027
157. Wei X, Gong J, Zhu J, Niu L, Zhu W, Li N, et al. Therapeutic effects of triptolide on interleukin-10 gene-deficient mice with colitis. Int Immunopharmacol (2008) 8:1808–12. doi: 10.1016/j.intimp.2008.08.019
158. Lin N, Liu C, Xiao C, Jia H, Imada K, Wu H, et al. Triptolide, a diterpenoid triepoxide, suppresses inflammation and cartilage destruction in collagen-induced arthritis mice. Biochem Pharmacol (2007) 73(1):136–46. doi: 10.1016/j.bcp.2006.08.027
159. Ma R, Liu L, Liu X, Wang Y, Jiang W, Xu L. Triptolide markedly attenuates albuminuria and podocyte injury in an animal model of diabetic nephropathy. Exp Ther Med (2013) 6(3):649–56. doi: 10.3892/etm.2013.1226
160. Jiang CB, Wei MG, Tu Y, Zhu H, Li CQ, Jing WM, et al. Triptolide attenuates podocyte injury by regulating expression of miRNA-344b-3p and miRNA-30b-3p in rats with adriamycin-induced nephropathy. Evid Based Complement Alternat Med (2015) 2015:107814. doi: 10.1155/2015/107814
161. de Zeeuw D, Bekker P, Henkel E, Hasslacher C, Gouni-Berthold I, Mehling H, et al. The effect of CCR2 inhibitor CCX140-b on residual albuminuria in patients with type 2 diabetes and nephropathy: a randomised trial. Lancet Diabetes Endocrinol (2015) 3(9):687–96. doi: 10.1016/S2213-8587(15)00261-2
162. Gale JD, Gilbert S, Blumenthal S, Elliott T, Pergola PE, Goteti K, et al. Effect of PF-04634817, an oral CCR2/5 chemokine receptor antagonist, on albuminuria in adults with overt diabetic nephropathy. Kidney Int Rep (2018) 3(6):1316–27. doi: 10.1016/j.ekir.2018.07.010
163. Menne J, Eulberg D, Beyer D, Baumann M, Saudek F, Valkusz Z, et al. C-c motif-ligand 2 inhibition with emapticap pegol (NOX-E36) in type 2 diabetic patients with albuminuria. Nephrol Dial Transplant (2017) 32(2):307–15. doi: 10.1093/ndt/gfv459
164. Ninichuk V, Clauss S, Kulkarni O, Schmid H, Segerer S, Radomska E, et al. Late onset of Ccl2 blockade with the spiegelmer mNOX-E36-3'PEG prevents glomerulosclerosis and improves glomerular filtration rate in db/db mice. Am J Pathol (2008) 172(3):628–37. doi: 10.2353/ajpath.2008.070601
165. Sharma D, Bhattacharya P, Kalia K, Tiwari V. Diabetic nephropathy: New insights into established therapeutic paradigms and novel molecular targets. Diabetes Res Clin Pract (2017) 128:91–108. doi: 10.1016/j.diabres.2017.04.010
166. Mirza A, Althagafi I, Shamshad H. Role of PPAR receptor in different diseases and their ligands: Physiological importance and clinical implications. Eur J Medicinal Chem (2019) 166:502–13. doi: 10.1016/j.ejmech.2019.01.067
167. Ohga S, Shikata K, Yozai K, Okada S, Ogawa D, Usui H, et al. Thiazolidinedione ameliorates renal injury in experimental diabetic rats through anti-inflammatory effects mediated by inhibition of NF-kappaB activation. Am J Physiol Renal Physiol (2007) 292(4):F1141–50. doi: 10.1152/ajprenal.00288.2005
168. Ding C, Chen H, Liang B, Jiao M, Liang G, Zhang A. Biomimetic synthesis of the natural product salviadione and its hybrids: discovery of tissue-specific anti-inflammatory agents for acute lung injury. Chem Sci (2019) 10(17):4667–72. doi: 10.1039/C9SC00086K
169. Li L, Ding C, Zou C, Xiong Z, Zhu W, Qian J, et al. A novel salviadione derivative, compound 15a, attenuates diabetes-induced renal injury by inhibiting NF-kappaB-mediated inflammatory responses. Toxicol Appl Pharmacol (2020) 409:115322. doi: 10.1016/j.taap.2020.115322
170. Ahn KS, Sethi G, Krishnan K, Aggarwal BB. Gamma-tocotrienol inhibits nuclear factor-kappaB signaling pathway through inhibition of receptor-interacting protein and TAK1 leading to suppression of antiapoptotic gene products and potentiation of apoptosis. J Biol Chem (2007) 282(1):809–20. doi: 10.1074/jbc.M610028200
171. Kolati SR, Kasala ER, Bodduluru LN, Mahareddy JR, Uppulapu SK, Gogoi R, et al. BAY 11-7082 ameliorates diabetic nephropathy by attenuating hyperglycemia-mediated oxidative stress and renal inflammation via NF-kappaB pathway. Environ Toxicol Pharmacol (2015) 39(2):690–9. doi: 10.1016/j.etap.2015.01.019
172. Lee FT, Cao Z, Long DM, Panagiotopoulos S, Jerums G, Cooper ME, et al. Interactions between angiotensin II and NF-kappaB-dependent pathways in modulating macrophage infiltration in experimental diabetic nephropathy. J Am Soc Nephrol (2004) 15(8):2139–51. doi: 10.1097/01.ASN.0000135055.61833.A8
173. Li X, Wang Y, Wang K, Wu Y. Renal protective effect of paeoniflorin by inhibition of JAK2/STAT3 signaling pathway in diabetic mice. Biosci Trends (2018) 12(2):168–76. doi: 10.5582/bst.2018.01009
174. Tuttle KR, Brosius FC 3rd, Adler SG, Kretzler M, Mehta RL, Tumlin JA, et al. JAK1/JAK2 inhibition by baricitinib in diabetic kidney disease: results from a phase 2 randomized controlled clinical trial. Nephrol Dial Transplant (2018) 33(11):1950–9. doi: 10.1093/ndt/gfx377
175. O'Shea J, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity (2012) 36(4):542–50. doi: 10.1016/j.immuni.2012.03.014
176. O'Shea J, Schwartz D, Villarino A, Gadina M, McInnes I, Laurence A. The JAK-STAT pathway: Impact on human disease and therapeutic intervention. Annu Rev Med (2015) 66:311–28. doi: 10.1146/annurev-med-051113-024537
177. Wang H, Vinnikov I, Shahzad K, Bock F, Ranjan S, Wolter J, et al. The lectin-like domain of thrombomodulin ameliorates diabetic glomerulopathy via complement inhibition. Thromb Haemost (2012) 108(6):1141–53. doi: 10.1160/TH12-07-0460
178. Li L, Chen L, Zang J, Tang X, Liu Y, Zhang J, et al. C3a and C5a receptor antagonists ameliorate endothelial-myofibroblast transition via the wnt/β-catenin signaling pathway in diabetic kidney disease. Metabolism (2015) 64(5):597–610. doi: 10.1016/j.metabol.2015.01.014
179. Li L, Yin Q, Tang X, Bai L, Zhang J, Gou S, et al. C3a receptor antagonist ameliorates inflammatory and fibrotic signals in type 2 diabetic nephropathy by suppressing the activation of TGF-beta/smad3 and IKBalpha pathway. PloS One (2014) 9(11):e113639. doi: 10.1371/journal.pone.0113639
180. Eliesen GAM, van Drongelen J, van den Broek PHH, Sarlea A, van der Heijden OWH, Langemeijer S, et al. Placental disposition of eculizumab, C5 and C5-eculizumab in two pregnancies of a woman with paroxysmal nocturnal haemoglobinuria. Br J Clin Pharmacol (2021) 87(4):2128–31. doi: 10.1111/bcp.14565
181. Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med (2015) 21(3):248–55. doi: 10.1038/nm.3806
182. Jiang H, He H, Chen Y, Huang W, Cheng J, Ye J, et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J Exp Med (2017) 214(11):3219–38. doi: 10.1084/jem.20171419
183. Vong CT, Tseng HHL, Yao P, Yu H, Wang S, Zhong Z, et al. Specific NLRP3 inflammasome inhibitors: promising therapeutic agents for inflammatory diseases. Drug Discov Today (2021) 26(6):1394–408. doi: 10.1016/j.drudis.2021.02.018
184. Li J, Bao L, Zha D, Zhang L, Gao P, Zhang J, et al. Oridonin protects against the inflammatory response in diabetic nephropathy by inhibiting the TLR4/p38-MAPK and TLR4/NF-κB signaling pathways. Int Immunopharmacol (2018) 55:9–19. doi: 10.1016/j.intimp.2017.11.040
185. Darakhshan S, Pour AB. Tranilast: a review of its therapeutic applications. Pharmacol Res (2015) 91:15–28. doi: 10.1016/j.phrs.2014.10.009
186. Huang Y, Jiang H, Chen Y, Wang X, Yang Y, Tao J, et al. Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol Med (2018) 10(4):e8689. doi: 10.15252/emmm.201708689
187. Chao E. SGLT-2 inhibitors: A new mechanism for glycemic control. Clin Diabetes Publ Am Diabetes Assoc (2014) 32(1):4–11. doi: 10.2337/diaclin.32.1.4
188. Tahara A, Takasu T. Prevention of progression of diabetic nephropathy by the SGLT2 inhibitor ipragliflozin in uninephrectomized type 2 diabetic mice. Eur J Pharmacol (2018) 830:68–75. doi: 10.1016/j.ejphar.2018.04.024
189. Thomas MC, Cherney DZI. The actions of SGLT2 inhibitors on metabolism, renal function and blood pressure. Diabetologia (2018) 61(10):2098–107. doi: 10.1007/s00125-018-4669-0
190. Vallon V, Gerasimova M, Rose MA, Masuda T, Satriano J, Mayoux E, et al. SGLT2 inhibitor empagliflozin reduces renal growth and albuminuria in proportion to hyperglycemia and prevents glomerular hyperfiltration in diabetic akita mice. Am J Physiol Renal Physiol (2014) 306(2):F194–204. doi: 10.1152/ajprenal.00520.2013
191. Gembardt F, Bartaun C, Jarzebska N, Mayoux E, Todorov VT, Hohenstein B, et al. The SGLT2 inhibitor empagliflozin ameliorates early features of diabetic nephropathy in BTBR ob/ob type 2 diabetic mice with and without hypertension. Am J Physiol Renal Physiol (2014) 307(3):F317–25. doi: 10.1152/ajprenal.00145.2014
192. Ashrafi Jigheh Z, Ghorbani Haghjo A, Argani H, Roshangar L, Rashtchizadeh N, Sanajou D, et al. Empagliflozin attenuates renal and urinary markers of tubular epithelial cell injury in streptozotocin-induced diabetic rats. Indian J Clin Biochem (2020) 35(1):109–14. doi: 10.1007/s12291-018-0790-6
193. Cai T, Ke Q, Fang Y, Wen P, Chen H, Yuan Q, et al. Sodium-glucose cotransporter 2 inhibition suppresses HIF-1α-mediated metabolic switch from lipid oxidation to glycolysis in kidney tubule cells of diabetic mice. Cell Death Dis (2020) 11(5):390. doi: 10.1038/s41419-020-2544-7
194. Elkazzaz SK, Khodeer DM, El Fayoumi HM, Moustafa YM. Role of sodium glucose cotransporter type 2 inhibitors dapagliflozin on diabetic nephropathy in rats; inflammation, angiogenesis and apoptosis. Life Sci (2021) 280:119018. doi: 10.1016/j.lfs.2021.119018
195. Heerspink HJL, Perco P, Mulder S, Leierer J, Hansen MK, Heinzel A, et al. Canagliflozin reduces inflammation and fibrosis biomarkers: a potential mechanism of action for beneficial effects of SGLT2 inhibitors in diabetic kidney disease. Diabetologia (2019) 62(7):1154–66. doi: 10.1007/s00125-019-4859-4
196. Zhong X, Chung AC, Chen HY, Dong Y, Meng XM, Li R, et al. miR-21 is a key therapeutic target for renal injury in a mouse model of type 2 diabetes. Diabetologia (2013) 56(3):663–74. doi: 10.1007/s00125-012-2804-x
197. Wang J, Gao Y, Ma M, Li M, Zou D, Yang J, et al. Effect of miR-21 on renal fibrosis by regulating MMP-9 and TIMP1 in kk-ay diabetic nephropathy mice. Cell Biochem Biophys (2013) 67(2):537–46. doi: 10.1007/s12013-013-9539-2
198. Chau BN, Xin C, Hartner J, Ren S, Castano AP, Linn G, et al. MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci Transl Med (2012) 4(121):121ra18. doi: 10.1126/scitranslmed.3003205
199. Li ZL, Lv LL, Tang TT, Wang B, Feng Y, Zhou LT, et al. HIF-1alpha inducing exosomal microRNA-23a expression mediates the cross-talk between tubular epithelial cells and macrophages in tubulointerstitial inflammation. Kidney Int (2019) 95(2):388–404. doi: 10.1016/j.kint.2018.09.013
200. Zitman-Gal T, Green J, Pasmanik-Chor M, Golan E, Bernheim J, Benchetrit S. Vitamin d manipulates miR-181c, miR-20b and miR-15a in human umbilical vein endothelial cells exposed to a diabetic-like environment. Cardiovasc Diabetol (2014) 13:8. doi: 10.1186/1475-2840-13-8
201. Lv LL, Feng Y, Wu M, Wang B, Li ZL, Zhong X, et al. Exosomal miRNA-19b-3p of tubular epithelial cells promotes M1 macrophage activation in kidney injury. Cell Death Differ (2020) 27(1):210–26. doi: 10.1038/s41418-019-0349-y
202. Chen HY, Zhong X, Huang XR, Meng XM, You Y, Chung AC, et al. MicroRNA-29b inhibits diabetic nephropathy in db/db mice. Mol Ther (2014) 22(4):842–53. doi: 10.1038/mt.2013.235
203. Long J, Wang Y, Wang W, Chang BH, Danesh FR. MicroRNA-29c is a signature microRNA under high glucose conditions that targets sprouty homolog 1, and its in vivo knockdown prevents progression of diabetic nephropathy. J Biol Chem (2011) 286(13):11837–48. doi: 10.1074/jbc.M110.194969
204. Song J, Zhang H, Sun Y, Guo R, Zhong D, Xu R, et al. Omentin-1 protects renal function of mice with type 2 diabetic nephropathy via regulating miR-27a-Nrf2/Keap1 axis. BioMed Pharmacother (2018) 107:440–6. doi: 10.1016/j.biopha.2018.08.002
205. Rovira-Llopis S, Escribano-Lopez I, Diaz-Morales N, Iannantuoni F, Lopez-Domenech S, Andújar I, et al. Downregulation of miR-31 in diabetic nephropathy and its relationship with inflammation. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol (2018) 50(3):1005–14. doi: 10.1159/000494485
206. Li D, Lu Z, Jia J, Zheng Z, Lin S. MiR-124 is related to podocytic adhesive capacity damage in STZ-induced uninephrectomized diabetic rats. Kidney Blood Press Res (2013) 37(4-5):422–31. doi: 10.1159/000355721
207. Long J, Wang Y, Wang W, Chang BH, Danesh FR. Identification of microRNA-93 as a novel regulator of vascular endothelial growth factor in hyperglycemic conditions. J Biol Chem (2010) 285(30):23457–65. doi: 10.1074/jbc.M110.136168
208. Kato M, Zhang J, Wang M, Lanting L, Yuan H, Rossi J, et al. MicroRNA-192 in diabetic kidney glomeruli and its function in TGF-beta-induced collagen expression via inhibition of e-box repressors. Proc Natl Acad Sci USA (2007) 104(9):3432–7. doi: 10.1073/pnas.0611192104
209. Putta S, Lanting L, Sun G, Lawson G, Kato M, Natarajan R. Inhibiting microRNA-192 ameliorates renal fibrosis in diabetic nephropathy. J Am Soc Nephrol (2012) 23(3):458–69. doi: 10.1681/ASN.2011050485
210. Chen YQ, Wang XX, Yao XM, Zhang DL, Yang XF, Tian SF, et al. Abated microRNA-195 expression protected mesangial cells from apoptosis in early diabetic renal injury in mice. J Nephrol (2012) 25(4):566–76. doi: 10.5301/jn.5000034
211. Shan L, Zhang H, Han Y, Kuang R. Expression and mechanism of microRNA 195 in diabetic retinopathy. Endocr J (2021) 69(5):529–37. doi: 10.1507/endocrj.EJ21-0231
212. Wang B, Koh P, Winbanks C, Coughlan M, McClelland A, Watson A, et al. miR-200a prevents renal fibrogenesis through repression of TGF-β2 expression. Diabetes (2011) 60(1):280–7. doi: 10.2337/db10-0892
213. Sun D, Chen J, Wu W, Tang J, Luo L, Zhang K, et al. MiR-802 causes nephropathy by suppressing NF-κB-repressing factor in obese mice and human. J Cell Mol Med (2019) 23(4):2863–71. doi: 10.1111/jcmm.14193
214. Wu J, Liu J, Ding Y, Zhu M, Lu K, Zhou J, et al. MiR-455-3p suppresses renal fibrosis through repression of ROCK2 expression in diabetic nephropathy. Biochem Biophys Res Commun (2018) 503(2):977–83. doi: 10.1016/j.bbrc.2018.06.105
215. Yang Z, Guo Z, Dong J, Sheng S, Wang Y, Yu L, et al. miR-374a regulates inflammatory response in diabetic nephropathy by targeting MCP-1 expression. Front Pharmacol (2018) 9:900. doi: 10.3389/fphar.2018.00900
216. Sun T, Liu Y, Liu L, Ma F. MicroRNA-544 attenuates diabetic renal injury via suppressing glomerulosclerosis and inflammation by targeting FASN. Gene (2020) 723:143986. doi: 10.1016/j.gene.2019.143986
217. Zhang Y, Xiao HQ, Wang Y, Yang ZS, Dai LJ, Xu YC. Differential expression and therapeutic efficacy of microRNA-346 in diabetic nephropathy mice. Exp Ther Med (2015) 10(1):106–12. doi: 10.3892/etm.2015.2468
218. Sun Y, Peng R, Peng H, Liu H, Wen L, Wu T, et al. miR-451 suppresses the NF-kappaB-mediated proinflammatory molecules expression through inhibiting LMP7 in diabetic nephropathy. Mol Cell Endocrinol (2016) 433:75–86. doi: 10.1016/j.mce.2016.06.004
219. Wei H, Li J, Li Y, Song J. MicroRNA-451 inhibits inflammation and proliferation of glomerular mesangial cells through down-regulating PSMD11 and NF-kappaB p65. Biosci Rep (2019) 39(10):BSR20191455. doi: 10.1042/BSR20191455
220. Zhang R, Qin L, Shi J. MicroRNA−199a−3p suppresses high glucose−induced apoptosis and inflammation by regulating the IKKβ/NF−κB signaling pathway in renal tubular epithelial cells. Int J Mol Med (2020) 46(6):2161–71. doi: 10.3892/ijmm.2020.4751
221. Wang Q, Wang Y, Minto AW, Wang J, Shi Q, Li X, et al. MicroRNA-377 is up-regulated and can lead to increased fibronectin production in diabetic nephropathy. FASEB J (2008) 22(12):4126–35. doi: 10.1096/fj.08-112326
222. Kato M, Natarajan R. MicroRNAs in diabetic nephropathy: functions, biomarkers, and therapeutic targets. Ann N Y Acad Sci (2015) 1353:72–88. doi: 10.1111/nyas.12758
223. Zhou H, Ni WJ, Meng XM, Tang LQ. MicroRNAs as regulators of immune and inflammatory responses: Potential therapeutic targets in diabetic nephropathy. Front Cell Dev Biol (2020) 8:618536. doi: 10.3389/fcell.2020.618536
224. Wu H, Kong L, Zhou S, Cui W, Xu F, Luo M, et al. The role of MicroRNAs in diabetic nephropathy. J Diabetes Res (2014) 2014:1–12. doi: 10.1155/2014/871439
225. Kato M, Dang V, Wang M, Park JT, Deshpande S, Kadam S, et al. TGF-beta induces acetylation of chromatin and of ets-1 to alleviate repression of miR-192 in diabetic nephropathy. Sci Signal (2013) 6(278):ra43. doi: 10.1038/s41419-020-2544-7
226. Tang J, Yao D, Yan H, Chen X, Wang L, Zhan H. The role of MicroRNAs in the pathogenesis of diabetic nephropathy. Int J Endocrinol (2019) 2019:8719060. doi: 10.1155/2019/8719060
227. Janssen HLA, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, et al. Treatment of HCV infection by targeting MicroRNA. New Engl J Med (2013) 368(18):1685–94. doi: 10.1056/NEJMoa1209026
228. Ling H, Fabbri M, Calin GA. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat Rev Drug Discov (2013) 12(11):847–65. doi: 10.1038/nrd4140
229. Pan B, Fan G. Stem cell-based treatment of kidney diseases. Exp Biol Med (Maywood NJ) (2020) 245(10):902–10. doi: 10.1177/1535370220915901
230. Shi Y, Wang Y, Li Q, Liu K, Hou J, Shao C, et al. Immunoregulatory mechanisms of mesenchymal stem and stromal cells in inflammatory diseases. Nat Rev Nephrol (2018) 14(8):493–507. doi: 10.1038/s41581-018-0023-5
231. Liu D, Zheng W, Pan S, Liu Z. Concise review: current trends on applications of stem cells in diabetic nephropathy. Cell Death Dis (2020) 11(11):1000. doi: 10.1038/s41419-020-03206-1
232. Singer NG, Caplan AI. Mesenchymal stem cells: Mechanisms of inflammation. In: Abbas AK, Galli SJ, Howley PM, editors. Annu Rev Pathol (2011) 6:457–78:226. Annual Review of Pathology-Mechanisms of Disease.
233. Bunnell B. Adipose tissue-derived mesenchymal stem cells. Cells (2021) 10(12):3433. doi: 10.3390/cells10123433
234. Romanov Y, Svintsitskaya V, Smirnov V. Searching for alternative sources of postnatal human mesenchymal stem cells: candidate MSC-like cells from umbilical cord. Stem Cells (Dayton Ohio) (2003) 21(1):105–10. doi: 10.1634/stemcells.21-1-105
235. Chong P, Selvaratnam L, Abbas A, Kamarul T. Human peripheral blood derived mesenchymal stem cells demonstrate similar characteristics and chondrogenic differentiation potential to bone marrow derived mesenchymal stem cells. J Orthopaedic Res Off Publ Orthopaedic Res Society (2012) 30(4):634–42. doi: 10.1002/jor.21556
236. Loukogeorgakis S, De Coppi P. Stem cells from amniotic fluid–potential for regenerative medicine. Best Pract Res Clin Obstetrics Gynaecol (2016) 31:45–57. doi: 10.1016/j.bpobgyn.2015.08.009
237. Liu Y, Tang S. Recent progress in stem cell therapy for diabetic nephropathy. Kidney Dis (Basel Switzerland) (2016) 2(1):20–7. doi: 10.1159/000441913
238. Imasawa T, Utsunomiya Y, Kawamura T, Zhong Y, Nagasawa R, Okabe M, et al. The potential of bone marrow-derived cells to differentiate to glomerular mesangial cells. J Am Soc Nephrol JASN (2001) 12(7):1401–9. doi: 10.1681/ASN.V1271401
239. Poulsom R, Forbes S, Hodivala-Dilke K, Ryan E, Wyles S, Navaratnarasah S, et al. Bone marrow contributes to renal parenchymal turnover and regeneration. J Pathol (2001) 195(2):229–35. doi: 10.1002/path.976
240. Tögel F, Westenfelder C. Mesenchymal stem cells: a new therapeutic tool for AKI. Nat Rev Nephrol (2010) 6(3):179–83. doi: 10.1038/nrneph.2009.229
241. Li Y, Liu J, Liao G, Zhang J, Chen Y, Li L, et al. Early intervention with mesenchymal stem cells prevents nephropathy in diabetic rats by ameliorating the inflammatory microenvironment. Int J Mol Med (2018) 41(5):2629–39. doi: 10.3892/ijmm.2018.3501
242. Lin L, Lin H, Wang D, Bao Z, Cai H, Zhang X. Bone marrow mesenchymal stem cells ameliorated kidney fibrosis by attenuating TLR4/NF-kappaB in diabetic rats. Life Sci (2020) 262:118385. doi: 10.1016/j.lfs.2020.118385
243. Molendijk I, Bonsing B, Roelofs H, Peeters K, Wasser M, Dijkstra G, et al. Allogeneic bone marrow-derived mesenchymal stromal cells promote healing of refractory perianal fistulas in patients with crohn's disease. Gastroenterology (2015) 149(4):918–27.e6. doi: 10.1053/j.gastro.2015.06.014
244. Tan J, Wu W, Xu X, Liao L, Zheng F, Messinger S, et al. Induction therapy with autologous mesenchymal stem cells in living-related kidney transplants: a randomized controlled trial. JAMA (2012) 307(11):1169–77. doi: 10.1001/jama.2012.316
245. Lin B, Chen J, Qiu W, Wang K, Xie D, Chen X, et al. Allogeneic bone marrow-derived mesenchymal stromal cells for hepatitis b virus-related acute-on-chronic liver failure: A randomized controlled trial. Hepatol (Baltimore Md) (2017) 66(1):209–19. doi: 10.1002/hep.29189
246. Zhang Y, Xu J, Liu S, Lim M, Zhao S, Cui K, et al. Embryonic stem cell-derived extracellular vesicles enhance the therapeutic effect of mesenchymal stem cells. Theranostics (2019) 9(23):6976–90. doi: 10.7150/thno.35305
247. Nagaishi K, Mizue Y, Chikenji T, Otani M, Nakano M, Konari N, et al. Mesenchymal stem cell therapy ameliorates diabetic nephropathy via the paracrine effect of renal trophic factors including exosomes. Sci Rep (2016) 6:34842. doi: 10.1038/srep34842
248. Yu B, Zhang X, Li X. Exosomes derived from mesenchymal stem cells. Int J Mol Sci (2014) 15(3):4142–57. doi: 10.3390/ijms15034142
249. Ebrahim N, Ahmed I, Hussien N, Dessouky A, Farid A, Elshazly A, et al. Mesenchymal stem cell-derived exosomes ameliorated diabetic nephropathy by autophagy induction through the mTOR signaling pathway. Cells (2018) 7(12). doi: 10.3390/cells7120226
250. Cai X, Zou F, Xuan R, Lai XY. Exosomes from mesenchymal stem cells expressing microribonucleic acid-125b inhibit the progression of diabetic nephropathy via the tumour necrosis factor receptor-associated factor 6/Akt axis. Endocrine J (2021) 68(7):817–28. doi: 10.1507/endocrj.EJ20-0619
251. Lin B, Ma Y, Wang J. Nano-technological approaches for targeting kidney diseases with focus on diabetic nephropathy: Recent progress, and future perspectives. Front Bioeng Biotechnol (2022) 10:870049. doi: 10.3389/fbioe.2022.870049
252. Pelaz B, Alexiou C, Alvarez-Puebla R, Alves F, Andrews A, Ashraf S, et al. Diverse applications of nanomedicine. ACS Nano (2017) 11(3):2313–81. doi: 10.1021/acsnano.6b06040
253. Wu L, Chen MY, Mao HJ, Wang NN, Zhang B, Zhao XF, et al. Albumin-based nanoparticles as methylprednisolone carriers for targeted delivery towards the neonatal fc receptor in glomerular podocytes. Int J Mol Med (2017) 39(4):851–60. doi: 10.3892/ijmm.2017.2902
Keywords: diabetic nephropathy, immune responses, therapeutic target, inflammation, pathogenesis
Citation: Chen J, Liu Q, He J and Li Y (2022) Immune responses in diabetic nephropathy: Pathogenic mechanisms and therapeutic target. Front. Immunol. 13:958790. doi: 10.3389/fimmu.2022.958790
Received: 01 June 2022; Accepted: 28 July 2022;
Published: 15 August 2022.
Edited by:
Yingmei Feng, Capital Medical University, ChinaReviewed by:
Nehal Mohsen Elsherbiny, Mansoura University, EgyptCopyright © 2022 Chen, Liu, He and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinhan He, amluaGFuaGVAc2N1LmVkdS5jbg==; Yanping Li, bGl5YW5waW5nXzUxMkAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.