 Tao Ming Sim
Tao Ming Sim Anselm Mak
Anselm Mak Sen Hee Tay
Sen Hee Tay
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 09 August 2022
Sec. Autoimmune and Autoinflammatory Disorders
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.957303
This article is part of the Research Topic The Role of Immune Cells in the Progression of Autoimmune Diseases Affecting the CNS View all 8 articles
Central nervous system (CNS) involvement of systemic lupus erythematosus (SLE), termed neuropsychiatric SLE (NPSLE), is a major and debilitating manifestation of the disease. While patients with SLE mostly complain of common neuropsychological symptoms such headache and mild mood disorders that may not even be technically attributed to SLE, many SLE patients present with life-threatening NPSLE syndromes such as cerebrovascular disease, seizures and psychosis that are equally challenging in terms of early diagnosis and therapy. While we are just beginning to unravel some mysteries behind the immunologic basis of NPSLE, advancements in the mechanistic understanding of the complex pathogenic processes of NPSLE have been emerging through recent murine and human studies. The pathogenic pathways implicated in NPSLE are multifarious and various immune effectors such as cell-mediated inflammation, autoantibodies and cytokines including type I interferons have been found to act in concert with the disruption of the blood-brain barrier (BBB) and other neurovascular interfaces. Beyond antimicrobial functions, neutrophils are emerging as decision-shapers during innate and adaptive immune responses. Activated neutrophils have been recognized to be involved in ischemic and infective processes in the CNS by releasing neutrophil extracellular traps (NETs), matrix metalloproteinase-9 and proinflammatory cytokines. In the context of NPSLE, these mechanisms contribute to BBB disruption, neuroinflammation and externalization of modified proteins on NETs that serve as autoantigens. Neutrophils that sediment within the peripheral blood mononuclear cell fraction after density centrifugation of blood are generally defined as low-density neutrophils (LDNs) or low-density granulocytes. LDNs are a proinflammatory subset of neutrophils that are increased with SLE disease activity and are primed to undergo NETosis and release cytokines such as interferon-α and tumor necrosis factor. This review discusses the immunopathogenesis of NPSLE with a focus on neutrophils as a core mediator of the disease and potential target for translational research in NPSLE.
Systemic lupus erythematosus (SLE) is a debilitating autoimmune disease characterized by a myriad of complex and heterogenous clinical manifestations, involving various organs such as the kidneys, skin and the central nervous system (CNS) (1). There is marked unpredictability in the disease course of SLE, often resulting in remissions and flares that lead to cumulative organ damage and mortality (2). The past 5 decades have seen increases in survival rates of SLE patients, though further improvements in survival have been hindered by organ damage to renal and neuropsychiatric systems (2).
Central nervous system involvement of SLE, termed neuropsychiatric SLE (NPSLE), is a major and debilitating manifestation of SLE which often masquerades as common and non-specific neuropsychological features such as headache and mild mood disorders. Such diverse, protean and often non-specific manifestations of NPSLE have made the clinical diagnosis and management of the condition challenging. Given this variability in disease phenotype, robust epidemiology studies are sorely lacking owing to the lack of consistency in inclusion criteria and case definition of neuropsychiatric (NP) events (3). While the true prevalence of NPSLE is unclear, CNS disease has been conservatively estimated to affect more than 20% of SLE patients (4). NPSLE is one of the most complex and challenging facets of SLE and can be mild or severe, focal or diffuse, acute or chronic, all posing a significant negative impact on quality of life (5) and has been found to be associated with an increase in mortality rate of as high as threefold as compared with SLE patients without NPSLE (6).
Research to elucidate the complex pathogenesis of NPSLE has seen recent advancements in mechanistic understanding and we are just beginning to unravel the mysteries behind the immunologic basis of NPSLE. The pathogenic pathways implicated in NPSLE are multifarious and various immune effectors such as cell-mediated inflammation, autoantibodies and cytokines including type I interferons (IFNs) have been found to act in concert with the disruption of the blood-brain barrier (BBB) and other neurovascular interfaces. Current management options for NPSLE are still inadequately optimized due to the lack of treatment targeting NPSLE-specific pathways. Nonetheless, advances in investigational techniques including lupus animal models and advances in neuroimaging have accelerated and augmented the study of NPSLE (7, 8) The aberrant adaptive immune response with loss of self-tolerance with activation of T and B cells leading to the production of pathogenic autoantibodies and systemic inflammation has long been advocated as one of the major mechanisms of the pathogenesis of SLE (9). The contribution of the innate immune system to SLE immunopathogenesis is yet less well understood.
Neutrophils are the most abundant leukocytes and are pivotal effector cells in the innate arm of the immune system, functioning as circulating phagocytes recruited to sites of infection to aid in the clearance of extracellular pathogens (10). Such effector functions of neutrophils are enabled by the presence of adhesion molecules such as selectins and integrins which enable interaction with the endothelium, allowing for chemotaxis, extravasation and migration into inflamed tissues. Neutrophils also display a range of pattern-recognition receptors and complement receptors which are critical in driving phagocytosis (11). Following phagocytosis, the fusion of the phagosome with neutrophil granules and the formation toxic reactive oxygen species enables the killing of engulfed pathogens. Neutrophils are also able to degrade virulence factors and kill bacteria through a unique cellular process involving the release of chromatin and granules as extracellular fibers called neutrophil extracellular traps (NETs) (12). While previously envisaged as a subset of non-specific leukocytes in the front line of immune defense, abnormalities of neutrophils have been found to be associated with the development of various autoimmune diseases such as SLE (13), rheumatoid arthritis and anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (14), making neutrophils increasingly recognized as more sophisticated cells of vital importance in the maintenance of immune homeostasis (15).
An overarching theme in the discussion of neuroimmune interactions are the unique properties of the brain vasculature and parenchyma. The CNS is an immune privileged site owing to the presence of the selective and restrictive BBB that limits exposure of the CNS to various toxins, pathogens, systemic immune mediators and cellular elements (16). Over the years, novel routes that enable the disruption of the BBB and consequent leukocyte infiltration to the CNS have been described (17–19). Disruption of the BBB also allows for other serum-borne immune effectors such as autoantibodies to cross into the CNS. Autoantibodies against a wide array of cellular antigens have been described in SLE and in particular, anti-neuronal antibodies have been found to be associated with NPSLE (20). Disruption of the BBB and increased penetrance of leukocytes and autoantibodies work in tandem to contribute to neuroinflammation in NPSLE.
This review discusses the current understanding of the pathogenic roles of neutrophils in SLE, proposing unifying mechanisms through which these intricate immune cells operate and contribute to the development of NPSLE. This review will also highlight the potential for translational research in NPSLE including therapies targeted at neutrophils and associated pathways.
In 1986, Hacbarth and Kajacsy-Balla were the first to discover low-density neutrophils (LDNs) or low-density granulocytes in the peripheral blood mononuclear cell (PBMC) preparations of adult SLE patients and found that the presence of this low buoyancy subset of neutrophils was correlated to SLE disease activity (21). They suggested that humoral factors in the plasma were responsible for in vitro activation of neutrophils, causing cellular degranulation and the formation of this unique phenotype of neutrophils in SLE patients. More recent studies of the nuclear morphology by transmission electron microscopy suggest that the low-density phenotype is due to LDNs being band or immature forms of neutrophils (22).
With the advent of transcriptome profiling, Bennett et al. performed microarray analysis of PBMCs isolated by density gradient in paediatric SLE patients and identified high expression of neutrophil-specific genes which was coined the ‘granulocyte signature’ (23). This ‘granulocyte signature’ was found to be associated with the increase in number of low-density neutrophils (LDNs) in the PBMC. These observations were echoed by another investigator who demonstrated that gene arrays performed on low-density cells isolated by Ficoll-Histopaque gradient centrifugation of bone marrow aspirates from SLE patients showed upregulation of granulopoiesis-related genes found in early stages of neutrophil development (24).
LDNs possess altered function as compared to normal neutrophils. LDNs exhibit enhanced capacity to synthesize cytokines including type I IFNs, decreased ability to phagocytize pathogens, and a marked propensity to spontaneously form NETs (25, 26). This altered physiology of LDNs is suggested to be responsible for autoimmunity and inflammation in SLE. NETs, which comprise chromatin fibers, histones and granular proteins, act as externalized autoantigens that activate the adaptive immune system and contribute to the pathogenesis of SLE (25). NETs synthesized by LDNs display increased externalization of immunostimulatory proteins such as interleukin-17 (IL-17), a vital cytokine associated with T cell activation, especially in chronic inflammation and autoimmunity (26, 27). The tendency of LDNs to spontaneously form NETs is thought to be due to an increase in the release of mitochondrial reactive oxygen species (28, 29). Oxidized mitochondrial DNA derived from LDN NETs is pro-inflammatory and is a potent stimulator of IFN genes, in particular it is able to induce type I IFN signaling in target cells (28). Mitochondrial DNA, unlike genomic DNA, contains hypomethylated CpG similar to bacterial DNA (30). Specifically, oxidized mitochondrial DNA in the form of NETs signals through the DNA sensor STING but via TLR9 in the form of extruded nucleoids from SLE neutrophils to mediate type I IFN activity (28, 30). Hence, the interferogenic capacity of oxidized mitochondrial DNA is clearly context dependent. In addition, NETs have also been found to cause endothelial damage and exposure of immunostimulatory molecules, further compounding the inflammatory reaction (26, 31).
LDNs have also been found to exhibit a distinct cytokine profile involving increased secretion of tumor necrosis factor alpha (TNF-α), IFN-γ and type I IFNs which are implicated in SLE pathogenesis (32). LDNs are also reported to express higher levels of TNF-α than high-density neutrophils (25). Dysregulation of type I IFN signature is well-characterized in SLE (33) and was previously attributed to increased type I IFN secretion by plasmacytoid dendritic cells (pDC) (34). Recent work have shown that neutrophils have the capacity to secrete type I IFN upon specific types of stimulation including granulocyte-colony stimulation factor and polyinosinic:polycytidylic acid (35, 36). In vitro, LDNs have been reported to synthesize type I IFNs in sufficient amounts to interfere with the differentiation of endothelial progenitor cells into mature endothelium (25, 31), a phenomenon observed in SLE and implicated in the development of early onset atherosclerosis (37). Interestingly, depletion of pDCs was not found to affect endothelial cell differentiation, suggesting that LDNs may contribute to endothelial disruption, mediated in part by a type I IFN effect (25).
Low-density neutrophils display aberrant properties from normal high-density neutrophils and result in exuberant NETosis and impaired clearance of NETs, providing a mechanism for stimulation of autoimmunity as the nuclear DNA, histones, and granule proteins released in the NETs serve as self-antigens (38).
Aberrant NETosis may explain the longstanding recognition of increased levels of circulating DNA in SLE patients (39). It has also been found that approximately 36% of SLE sera degrade NETs poorly, with inhibitors of DNase I detectable in some patients, while others displayed autoantibodies that coated NETs and mechanically protected against degradation (40). The significance of poor NET degradation is a potentially higher titer of anti-double-stranded DNA antibodies, and correspondingly more complement activation and end-organ damage (41). NETs have also been found to stimulate pDCs to release type I IFNs (42, 43), which lowers the threshold for autoreactivity of both antigen-presenting and antibody-producing cells (44). Further, SLE patients are shown to have autoantibodies to both the self-DNA and antimicrobial peptides in NETs, indicating that NETs serve as a source of novel intracellular autoantigens for subsequent B cell activation and production of autoantibodies (44). This phenomenon may be due to the fact that NETs display increased externalization of immunostimulatory proteins and autoantigens including double-stranded DNA, LL-37 and IL-17 (26).
The formation of NETs is a key mechanism in enabling neutrophils to control infection as NETs are a source of concentrated immune effectors including neutrophil elastase (NE), myeloperoxidase, cathepsin G and proteinase 3 which display microbicidal properties (45, 46). These released immune effectors in NETs are toxic to tissue and have been found to cause apoptosis in endothelial cells and contribute to glomerular injury in lupus nephritis (47, 48). Intrathecal NETosis may contribute to direct neuronal damage in NPSLE, which has been found to contribute to the pathogenesis of memory deficit in Alzheimer’s disease (49, 50). NE and cathepsin G found in NETs have the capacity to potently process and activate cytokines including IL-1α and IL-36 which promote inflammation (51) and may account for indirect damage to the CNS in NPSLE. There is also suggestion that NETs may be a source of vascular and organ damage in SLE as netting neutrophils have been shown to have capacity to cause endothelial damage and infiltrate organs such as skin and kidney (26). A murine study found that circulating neutrophils were able to be primed by antiphospholipid antibodies and promote thrombosis (52).
Matrix metalloproteinases (MMPs) are members of the metzincin group of proteases with a wide substrate spectrum which share the conserved zinc-binding motif in their catalytic active site (53). In biological systems, MMPs are zinc- and calcium-dependent endoproteinases that are crucial in degradation and remodelling of the extracellular matrix (ECM) and regulating extracellular tissue signaling networks (54, 55). MMPs are secreted as zymogens and are subsequently activated extracellularly by proteinase-facilitated or non-proteolytic mechanisms (56). MMP expression is closely regulated at the level of gene transcription, secretion, activation and inhibition by tissue inhibitors of metalloproteinases (TIMP) (56, 57).
Matrix metalloproteinase-9, also known as gelatinase B, is one of the most complex MMPs in the human proteasome and proteomic studies have identified possible substrates for MMP-9 that may reach a few hundred (58). MMP-9, which is found to be concentrated in the cerebellum, hippocampus and cerebral cortex of the brain, is responsible for degradation of ECM components including gelatine and type IV, V, XI and XVI collagens during tissue remodelling, which is postulated to influence the disruption of the BBB and neuroinflammation (58, 59). MMP-9 is secreted by a wide array of cells, including neutrophils, macrophages, lymphocytes and fibroblasts, with neutrophils being one of the richest sources of active MMP-9 (60, 61). This is attributed to neutrophils that contain various proteases such as serine proteases (elastase, proteinase 3 and cathepsin G) and urokinase plasminogen activator which promote MMP-9 activation (62).
The relationship between serum MMP-9 levels and SLE disease activity have been studied but results were inconsistent (63–66), possibly due to discrepancies in laboratory techniques employed or epigenetic polymorphisms (66). On the other hand, serum and CSF levels of MMP-9 have been found to be consistently elevated in SLE patients with NP involvement as compared to patients without (67). A study involving 123 patients with SLE who were evaluated clinically, with magnetic resonance imaging (MRI) and CSF analyses of MMP-9 found significant correlation between MMP-9 and intrathecal levels of tau and glial fibrillary acid protein, markers of neuronal degradation, suggesting ongoing neurodegeneration in the brains of NPSLE patients with high expression of MMP-9 (68). This study also found that intrathecal titers of IL-6 and IL-8 correlated with MMP-9 levels in the CSF of NPSLE patients. IL-6 and IL-8 are cytokines demonstrated to have elevated CSF levels in NPSLE patients (69, 70). These proinflammatory cytokines have been shown to stimulate the production and release of MMP-9 in other immune cells including macrophages (71–73). Taken together, a proinflammatory milieu conferred by proinflammatory cytokines such as IL-6 and IL-8 induces MMP-9 production, leading to the neurodegeneration and increased neuronal and astrocytic degradation productions observed in NPSLE (74, 75). IL-8 is also postulated to promote transmigration of activated inflammatory cells across the endothelium, amplifying the effects of ECM and BBB disruption by MMP-9 (68).
One study found that elevated levels of MMP-9 in patients with SLE were associated with MRI indices of cerebral infarcts and NP involvement, particularly cognitive dysfunction (64). Prior studies of giant cell arteritis elucidated that increased MMP-9 expression contributed to leukocyte migration into the vessel wall via damage to the internal elastic lamina of the vessels and inflammation (76). In the context of NPSLE, it is postulated that MMP-9 contributes to small-vessel vasculopathy via a similar mechanism, causing foci of cerebral ischaemia and infarcts derived from atherosclerosis in cerebral vessels, accounting for cognitive dysfunction (64). MMP-9 has also been shown to contribute directly to neuronal apoptosis and damage to brain parenchyma (77–80), through degradation of laminin which induces apoptosis and transient focal cerebral ischaemia (81).
Neutrophil gelatinase-associated lipocalin (NGAL) is a 25 kDa protein under the lipocalin superfamily. While NGAL was initially found in activated neutrophils, many other cell types including kidney tubular cells also express NGAL in response to noxious stimulants such as ischaemia and inflammation (82). NGAL is completely absent in normal brains but can be induced in the choroid plexus following infectious and inflammatory insults. NGAL potentiates the ability of MMP-9 to increase the permeability of the BBB, and NGAL levels correlate with microglia activation (83). In a prospective study of 60 children with SLE, increase in serum NGAL level was found to be associated with decline in psychomotor speed during an 18-month observation period (84). Nevertheless, the source of NGAL in this cohort was not ascertained. A study in a murine lupus model found that in the CNS, NGAL expression was increased more than 30-fold in lupus mice than normal mice and that NGAL promoted damage through mechanisms established to be linked to NPSLE such as disruption of the BBB and direct neurotoxicity (85). To establish the relevance of NGAL to human disease, the authors measured NGAL concentrations in the CSF of patients with NPSLE and healthy controls. It was found that CSF NGAL levels were significantly elevated in NPSLE, raising the potential for future clinical application of measuring NGAL levels in the CSF as a diagnostic biomarker for NPSLE.
In SLE, end-organ inflammation and damage mediated by immune-complex activation of complement cascade is well-described especially for lupus nephritis (86). However, the understanding of NPSLE is complicated by the presence of the BBB which serves as a physical and biochemical barrier of the CNS through its tight, adherens and gap junctions, specialized structures resulting from the unique interaction of cerebral endothelial cells, pericytes and astrocytic foot processes (87, 88). The CNS is an immune-privileged anatomic site due to the presence of this tightly regulated and highly restrictive BBB which serves as a functional and mechanical barrier, thereby preventing passive transfer of most immune cells, immune complexes, pathological antibodies and mediators including cytokines from the circulation to the CNS (16). BBB disruption is central to the altered homeostasis seen in a myriad of CNS disorders including neurodegenerative conditions such as Alzheimer’s disease and Parkinson’s disease and autoimmune conditions including multiple sclerosis and neuromyelitis optica (89, 90). Therefore, the question central to the study of SLE and its CNS manifestations is whether a breach of BBB integrity is implicated in NPSLE and if so, what the underlying mechanisms are.
Post-mortem autopsy studies of NPSLE brains have shown that histopathological lesions in NPSLE represent a continuum that ranges from non-specific lesions such as focal vasculopathy, to more specific lesions including C4d-C5b complement-associated microthrombi and diffuse vasculopathy (91). The presence of complement deposition is suggested to be a key factor in the interaction between circulating autoantibodies and the thrombo-ischaemic vascular lesions observed in NPSLE. Histopathology showing vasculitis with inflammatory infiltrates of predominantly mononuclear cells and depositions of immunoglobulin and complement, accompanied by fibrinoid necrosis have also been described in NPSLE (92).
The earliest reports of BBB impairment in NPSLE was by Winfield et al. in 1983 (93). Much of what is believed about the loss of BBB integrity in NPSLE is based on evidence from surrogate markers of leakage of serum proteins into the CNS such as Qalb (94, 95). However, lumbar puncture is required to obtain CSF which is an invasive procedure with associated risks, so its application has been relatively limited in the context of clinical diagnosis of NPSLE (96). BBB disruption can be inferred from regions of edema using computed tomography and MRI but these techniques lack spatial resolution to detect small leaks across the BBB (97). With the advent of advanced neuroimaging technique, various methods have been used to assess BBB permeability in SLE patients. They include non-gadolinium-based arterial spin labelled combined with diffusion-weighted imaging for paediatric patients and the gold standard, gadolinium-based dynamic contrast-enhanced MRI in adults (98–101).
Increased BBB permeability has been described in specific structures (e.g. right insular, Brodmann’s area 19,28,36 and 37) and the whole brain in SLE patients (101–103). Of note, increased BBB permeability in the right insular and Brodmann’s area 19 have been associated with anxiety/depression and worse psychomotor speed, respectively (102). In addition, extensive BBB leakage (affecting more than 9% of the brain volume) was associated with lower global cognitive scores and impairment in one or more cognitive tasks (101).
A seminal study involving ex vivo analyses of autopsied human brain after haemorrhagic conversion of an acute ischaemic stroke found a strong infiltration of infarcted brain by MMP-9-expressing neutrophils which were associated closely with regions with pronounced basal lamina collagen IV degradation and BBB breakdown (104). Depletion of neutrophils in a rat model of intracerebral hemorrhage displayed muted expression of MMP-9 and reduced neuroinflammation, BBB breakdown and axonal injury (61). A study of cerebrospinal fluid (CSF) and serum levels of active MMP-9 in patients with the multiple sclerosis found elevated levels of MMP-9 which correlated with disease activity and in particular, the MMP-9/TIMP ratio was found to be pivotal in modulating inflammatory disease activity (105). Taken together, this is suggestive that MMP-9-expressing neutrophils, not compensated by an adequate down-regulatory effect of TIMP, can facilitate BBB breakdown through disruption of the ECM and basal lamina, possibly potentiating invasion of the CNS by other immune cells and perpetuating neuroinflammation.
A two-hit mechanism for the development of NPSLE involving a breach in BBB and neuroinflammation caused by neurotoxic antibodies has been proposed by some authors (86). It is speculated that when BBB dysfunction occurs, the precise microenvironment of the CNS is perturbed, allowing pathologic autoantibodies and inflammatory cells to penetrate, resulting in diffuse NPSLE. Neutrophils are scarce in the CNS under normal conditions as the BBB restricts trafficking of these cells into brain parenchyma. Yet, the infiltration of the CNS by neutrophils in various CNS pathologies is a well-known phenomenon. Evaluation of MRL/lpr lupus mouse model found increased ICAM-1 and E-selectin on vasculature of the brain which preceded development of NPSLE (106). Neutrophils display ligands for these adhesion molecules such as LFA-1 which promote neutrophil arrest and adhesion prior to diapedesis (107). MMP-9-expressing neutrophils have been found to result in severe basal lamina type IV collagen degradation and blood extravasation, promoting BBB breakdown and enabling neuroinflammation (58, 59, 104). Pro-inflammatory cytokines released by netting neutrophils along with activation of the alternative complement pathway induce endothelial-to-mesenchymal transition which causes increased vascular leakage and permeability (47).These mechanisms may contribute to BBB disruption, with subsequent ingress of neutrophils and entry of neurotoxic mediators into the CNS. These effects are further augmented by the increased expression of adhesion molecules on the endothelium of the BBB in SLE which enhances adhesion and subsequent penetration of neutrophils into the CNS, with neutrophil products including MMP-9 and reactive oxygen species compounding BBB dysfunction and permeability to other inflammatory insults (Figure 1).
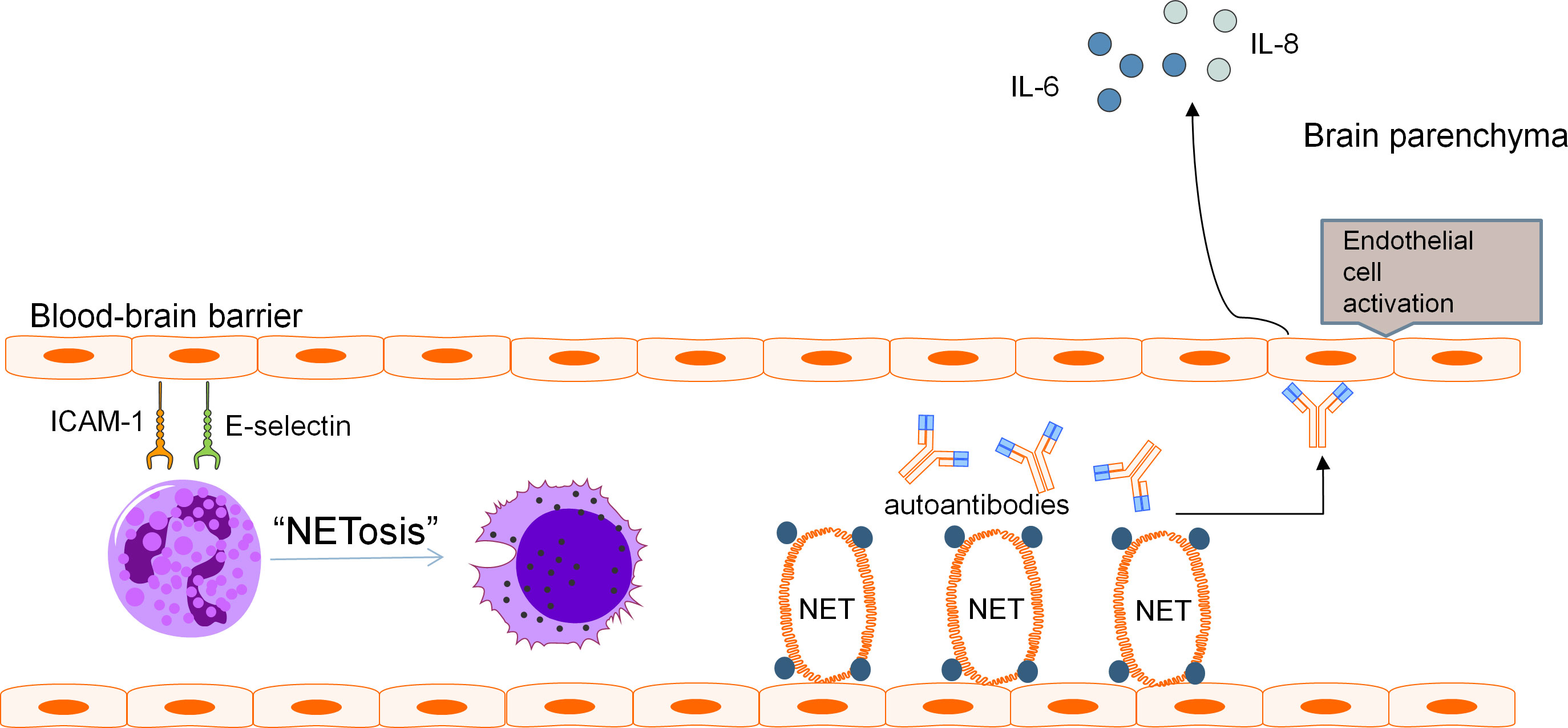
Figure 1 Pathogenic mechanism of BBB disruption. Blood-brain barrier disruption in NPSLE has been found to be related to various distinct but potentially complementary pathways involving neutrophils and their cellular products. Adhesion molecules ICAM-1 and E-selectin present on the vasculature of the brain promote neutrophil arrest and adhesion, leading to recruitment of pathogenic neutrophils to the BBB. These neutrophils produce NETs which cause direct endothelial damage and serve as a source of antigens for the development of autoantibodies including aPL and anti-NR2A/B antibodies. These autoantibodies can further aggravate endothelial damage and promote endothelial activation. The downstream effect is increased permeability of the BBB and secretion of pro-inflammatory cytokines including IL-6 and IL-8.
Complement is known to be strongly activated in SLE, resulting in hypocomplementemia being commonly seen in patients with active SLE. In fact, low total complement hemolytic activity and decreased levels of C3 and C4 as many as 90% of patients with diffuse proliferative glomerulonephritis (108). One major mechanism accounting for complement depletion in SLE is the formation of immune complexes which activates complement via the classical pathway (108). More recently, the complement system has been found to exhibit a dynamic interplay with neutrophils in the pathogenesis of SLE, the crosstalk facilitated by the presence of complement receptors on the surface of neutrophils including complement receptor 1 (CR1), C3a receptor (C3aR) and C5a receptor 1 (C5aR1) (109). In particular, complement activation has been shown to not only induce phagocytosis in neutrophils but also stimulate NETosis, an interaction possibly crucial in the development of NPSLE (110). Bacteria opsonized with C3b was found to be more potent NETosis inducers than non-opsonized bacteria, suggesting that C3b opsonization via the classical or alternative pathways facilitated NET formation (111). The results were corroborated by the finding that the addition of a CR1 antagonist prevented recognition of opsonized microbes by neutrophils, resulting in a decrease in NETosis (111). Similar results were documented from attempts to block C3 receptor via iC3b which resulted in marked inhibition of NETosis (112). As previously discussed, NETosis involves the extrusion of intracellular proteins which act as a source of autoantibody formation, and the crosstalk between complement and neutrophils may reinforce this pathogenic pathway in NPSLE.
The complement split product C5a which promotes chemotaxis of neutrophils through the C5a-C5aR1 axis has been found to participate in robust neuroinflammatory reactions. Indeed, factor B knockout MLR/lpr mice had decreased neutrophil infiltration into the brain compared to knockout MLR/lpr mice (113). The sensitivity of the CNS to effects of C5a is, at least in part, explained by the presence of C5aR1 receptors on the surface of numerous indigenous cell types including microglia, astrocytes and neurons which elicits downstream pro-inflammatory signaling pathways in these cells (114). C5a has been found to exert direct neurotoxic effects through these interactions with cells in the CNS, contributing to cellular apoptosis (115–117). C5a has also been found to contribute to BBB disruption in NPSLE, perturbing the delicate brain microenvironment (118). Signaling through the C5a-C5aR1 axis has been found to activate the NFκB pathway and mediate downstream effects on the expression proteins crucial for the maintenance of the BBB such as tight junction proteins, occludin and zona occludens-1 (ZO-1) (119, 120). The altered expression of these proteins was found to increase BBB permeability in a lupus murine model and an in vitro human BBB model (120). The NFκB pathway in endothelial cells also regulate the expression of adhesion molecules such as ICAM-1 and VCAM-1 which mediate the capture and adhesion of leukocytes to the endothelium (121). Increased expression of adhesion molecules on CNS endothelium magnifies the chemotactic properties of C5a on neutrophils and other immune cells, boosting the infiltration of these cells into the CNS through the disrupted BBB.
In a murine lupus model, exposure to pathogenic autoantibodies that bind both DNA and the N-methyl-D-aspartate (NMDA) receptor impaired spatial memory and cognition, inducing NPSLE (122). It was found from C1q knockout mice that dendritic complexity and spine density, but not acute neuronal loss, were better preserved in this model (122). Unlike wild-type mice, C1q-deficient mice behaved normally in an object-place memory task that tests spatial memory (122). In sum, complement proteins and neutrophils have an intricate and dynamic crosstalk which is enabled by the various complement receptors on neutrophils. The mechanisms of complement and neutrophils contributing to NPSLE are similar, and are likely to exist in tandem, amplifying pathogenic effects on the CNS in NPSLE.
Disruption of the BBB enables serum-borne effectors of inflammation to penetrate the CNS, which includes autoantibodies. A hallmark of SLE is the formation of autoantibodies. Some of which are found in the serum, CSF and neuronal tissues and are thought to be directly involved in disease pathways of NPSLE including antiphospholipid (aPL), anti-ribosomal P (anti-P) and anti-N-methyl-D-aspartate (NMDA) receptor subunit NR2A/B antibodies (123–125). Anti-endothelial cell antibodies have been described as having a crucial role in the recruitment of polymorphonuclear leukocytes to foci of inflammation by augmenting cellular adhesion to endothelium (126). Autoantibodies detected in the CSF are the result of either passive transfer of peripherally produced autoantibodies across the BBB or increased intrathecal production (127).
N-methyl-D-aspartate receptors are a class of L-glutamate receptors that play an important role in cognitive functions including memory and learning and are crucial for spatial memory (128). Antibodies against NMDA receptor NR2A/B subunits (anti-NR2A/B antibodies) are found in the sera of 30-40% of SLE patients and is a convincing candidate as a pathogenic factor in mediating cognitive dysfunction in SLE patients, fulfilling 4 out of 6 stringent pathogenicity criteria for autoantibodies (125, 129).
Affinity purified anti-NR2A/B antibodies have been found to cross-react with double-stranded DNA (dsDNA) (130). In vitro and in vivo experiments have found that anti-NR2A/B antibodies were capable of inducing apoptotic cell death in neuronal cultures and neuronal damage and loss of hippocampal neurons in a murine model (131, 132). Interestingly, functional NMDA receptors have also been found on the plasma membranes of rat neutrophils after activation (133). Although similar studies of NMDA receptor subunits on human neutrophils have not been reported, it would be a compelling hypothesis for NMDA receptor subsets elaborated on NETs to induce anti-NR2A/B antibodies that recognize both NR2A, NR2B and dsDNA.
In human studies, 57 SLE patients demonstrated an association between serum anti-NR2A/B antibodies and a reduction in short-term memory and depressed mood (134), while another cross-sectional study of 133 SLE patients revealed that serum anti-NR2A/B antibodies displayed impairments in sustained attention, planning abilities and executive function (135). A longitudinal study found an association between increasing titres of serum anti-NR2A/B antibodies with declining working memory function (84). CSF levels of anti-NR2A/B have also been shown to be associated with diffuse NPSLE, especially cognitive deficits (136) and correlate well with SLE disease severity (137). It is worth noting that some studies have not found a positive correlation between serum anti-NR2A/B antibodies and cognitive dysfunction (138). By contrast, CSF anti-NR2A/B antibodies have been consistently shown to be associated with diffuse and central NPSLE (136, 139). The discrepancies of results between CSF and serum anti-NR2A/B antibodies suggest that serum antibodies alone without other factors that disrupt the BBB and increase penetrance into the CNS may not be sufficient to induce neuropsychiatric damage and involvement of SLE (8).
Anti-phospholipid syndrome is characterized by arterial or venous thrombosis, adverse pregnancy outcomes and the positivity of one or more aPL antibodies, including anti-cardiolipin antibodies and anti-β2-glycoprotein 1 antibodies (140). Although this syndrome can occur in isolation, it is more prevalent amongst SLE patients and aPL antibodies are present in up to 30-40% of SLE patients (141). aPL antibodies are known to activate endothelial cells, monocytes and platelets, giving rise a prothrombotic milieu and have been linked with NPSLE (142), suggested by longitudinal studies which showed a correlation between declining cognitive function and persistently elevated aPL levels (143, 144). A study of 51 female SLE patients found a significant relation between persistent anti-cardiolipin antibody positivity with psychomotor speed reduction and decreased conceptual reasoning (143). Another study of 43 SLE patients assessing fronto-subcortical function over 1 decade discovered a significant association between worsening visuospatial functions with hyperlipidemia and positivity of lupus anticoagulant (145).
Among patients with SLE, aPL antibody positive patients are about twice as likely to develop NPSLE than patients who were negative (146) and aPL positivity has been long recognized as a strong risk factor for NPSLE development (147). While aPL antibodies accelerate atherosclerosis and cerebral ischaemia (148), aPL antibody positivity has also been linked with NPSLE syndromes that were not necessarily directly related to thrombotic events, such as cognitive dysfunction, chorea and seizures (149, 150). This suggests that aPL antibodies have a pathogenic role in NPSLE beyond their prothrombotic effects. Similar to anti-neuronal antibodies, there have been studies that showed binding of aPL antibodies to neurons and other CNS cells and induction of hyperactive behavior (151), thereby supporting a direct effect of these autoantibodies on the brain parenchyma. Nonetheless, the role of aPL antibodies in NPSLE has been largely ascribed to the sequelae of vascular occlusion and ischaemic events in regions of the brain including the frontal cortex, amygdala and hippocampus (152). Finally, local thrombotic events causing vascular injury is also hypothesized to insult the integrity of the BBB, enabling peripheral effectors including other neuropathic autoantibodies and immune cells to enter the CNS (Figure 2) (153). Notably, β2-glycoprotein 1 has been demonstrated on the surface of neutrophils and anti-β2-glycoprotein 1 IgG antibodies can induce NETosis in human neutrophils (154). This will allow this aPL antibody to partake in the neutrophil-related processes mentioned in the preceding sections to mediate NPSLE.
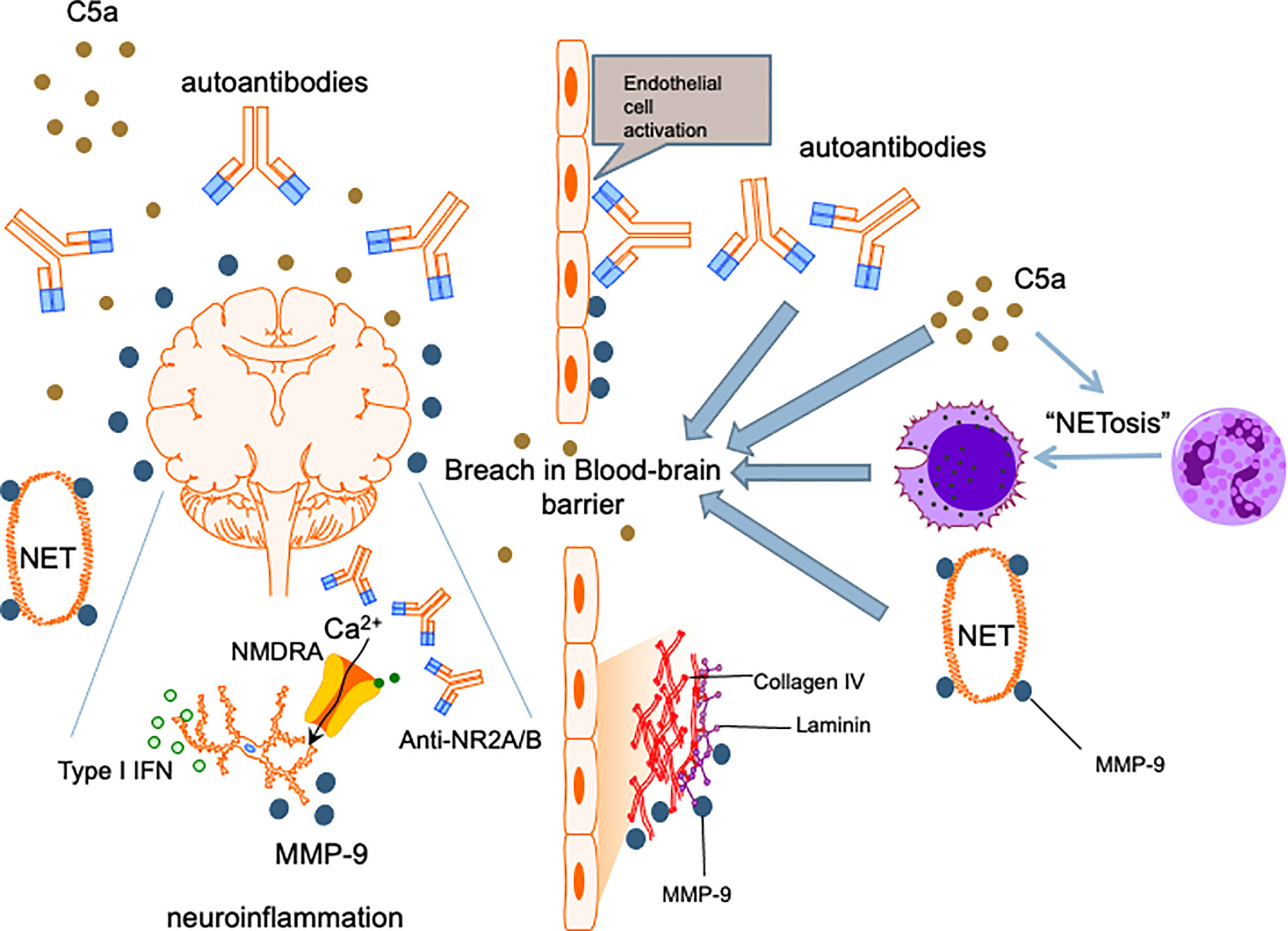
Figure 2 Proposed mechanism for NPSLE. Insults to the BBB allow penetration of the central nervous system by neutrophils and other inflammatory mediators. Cognitive deficits and other neuropsychiatric manifestations are caused by cytokines such as type I IFNs, anti-neuronal autoantibodies including anti-NR2A/B and matrix metalloproteinase-9 instigating neuronal damage and induction of apoptosis. C5a complement fragments are chemotactic and promote migration of immune cells including neutrophils into the CNS, an effect amplified by the ability of C5a to enhance NETosis and increase permeability of the BBB. C5a also exerts direct neurotoxic effects, contributing to further neuropsychiatric insults in NPSLE.
Several case reports and a case series described the involvement of neutrophils in the CNS of patients with SLE. For instance, a 14-year-old patient with SLE who presented with loss of consciousness revealed a vasculitic mass with histological features of perivasculitis associated with infiltration of leucocytes including neutrophils (155). CSF pleocytosis with neutrophil predominance was reported in a patient with cerebral lupus presented with acute episodes of haemorrhagic cerebral ischaemia (156). CSF neutrophil pleocytosis was also evident in a 35-year-old SLE patient who was diagnosed to have catastrophic transverse myelitis (157). While the proportion of neutrophils in the CSF of this patient continued to increase (from 76% of 683 white cells/μL to 84% of 1,585 white blood cells/μL) despite empirical antibiotic therapy, treatment with plasmapheresis, intravenous pulse methylprednisolone and cyclophosphamide resulted in a rapid reduction of white blood cell count (to 14 white blood cells/μL) and reversal of white blood cell proportion (from neutrophil predominance to 98% lymphocytes) in the CSF after 3 days, coupled with clinical and radiological improvement of myelitis (157). A 30-year-old patient with SLE who presented with mood and behavioral changes and transverse myelitis was noted to have multiple brain lesions including a large ring-enhancing right basal ganglion lesion on MRI of the brain. Biopsy of the right basal ganglion lesion revealed fibrinoid vascular necrosis and microthrombi with extensive neutrophilic vasculitis and cerebritis (158). Two distinct subtypes of myelitis were later described, comprising of grey and white matter myelitis (159). Patients with grey matter myelitis were more likely to have irreversible paraplegia, monophasic disease course, high SLE disease activity and neutrophilic pleocytosis on CSF analysis (159). In an observational study of 23 patients with SLE that aimed to observe the fluctuation of C3a and C5a in relation to SLE disease flares, in addition to the elevation of serum of C3a and C5a that was more prominent in patients with acute CNS dysfunction compared with patients with active SLE without CNS involvement, postmortem examination of 2 patients revealed neutrophil infiltration that occluded cerebral vessels (160). This observation implied that the release of circulating C3a and C5a may elicit vascular injury and activate neutrophil infiltration, contributing to cerebritis (160). In fact, the critical role of the C5a signaling pathway in mediating NPSLE via neutrophil infiltration was evident in an experimental study that demonstrated reduced cerebral neutrophil infiltration, neuronal apoptosis and expressions of p-JNK, pSTAT1 and p-ERK after treatment with C5a receptor antagonist in MRL/lpr lupus-prone mice (161). The potential role of targeting C5a receptor will be discussed further below.
Studies that specifically address the pathophysiological impact of neutrophils on clinical neuropsychiatric manifestations of SLE are scant. Clinical situations such as CNS infections and non-steroidal anti-inflammatory drugs (NSAIDs)-induced aseptic meningitis that cause CSF neutrophil pleocytosis may give rise to non-SLE related neuropsychiatric symptoms in patients with SLE (162–165). The use of glucocorticoids (GC) that often leads to peripheral neutrophilia, has been well described to induce a range of neuropsychiatric symptoms reminiscent of NPSLE including anxiety, sleep disturbance and psychosis (166). In patients with hypoalbuminaemia, the lack of circulating GC-binding protein facilitates access of unbound and free GC to the CNS, leading to neuropsychiatric syndromes in patients with SLE (167).
Circumferential evidence that implicates the mechanistic role of neutrophils in mediating NPSLE has been emerging since the 1980s. A study of sera from 53 patients with SLE found an increase in serum neutrophil aggregation, particularly in those with active disease (168). Intriguingly, a high level of neutrophil aggregation activity was prominent in patients with neuropsychiatric symptoms, suggesting the potential role of intravascular leukoaggregates in patient with NPSLE (168). More recently, NGAL (82–84) and NETs (169) in relation to NPSLE have been described although their roles in NPSLE require further elucidation and validation.
NETs have been detected in a number of CNS diseases including acute thrombotic strokes, particularly that NETs appear to promote coagulation and thrombosis (169). NETs were also observed to be involved in brain parenchymal oedema of traumatic brain injury in mice and gliomas (169). While a SLE patient with non-aPL antibody-mediated Libman-Sacks endocarditis who presented with seizures and psychosis was found to have active NETs formation in the mitral valve and peripheral circulation, no NETs remnant or heighted elastase activity was detected in the CSF (170). More observations and experiments are required to assess if NETs contribute a role to the pathogenesis of NPSLE in lupus patients.
Increasingly, there has been interest in targeting the neutrophil axis for therapeutic application in NPSLE. Inhibition of serine protease neutrophil elastase in a mouse model of SLE revealed reduced NETosis, lowered levels of some autoantibodies and conferred protection against arterial and venous thrombosis (171). Another murine study investigated the use of Cl-amidine, an inhibitor of peptidylarginine deiminases (PAD) which is an enzyme crucial in the formation of NETs (172). The study found that Cl-amidine led to robust improvements in vasculogenesis, endothelial function and thrombotic risk.
It is pertinent to remember that while NET formation functions as part of the innate immune system against pathogens, while excessive NETosis is implicated in the pathogenesis of SLE and other autoimmune and inflammatory disorders. Indeed, inhibition to or knock-out of PAD has been demonstrated to reduce the severity of autoimmune disease, degree of inflammation and production of autoantibodies through a mechanism dependent on inhibition of NET formation (173). Furthermore, PAD inhibition is also associated with a decrease in T-helper cell inflammatory responses mediated by the Th1 and Th17 subtypes (174–176). Given the intricate balance between maintaining immune homeostasis and autoimmunity, inhibition of NETosis may potentially impair protective host immune responses (177). PAD-deficient mice have been found to be more susceptible to bacterial infections than healthy mice (178). Another study reported that PAD-knock-out mice developed systemic inflammation and bacterial keratitis (179). Cyclosporine is an immunosuppressive agent which can inhibit NETosis through inhibition of calcium flux (180). It is well known that the use of immunosuppressive drugs such as cyclosporine is associated with infections (181), contributed in part by an inhibition of neutrophil-related pathways. Hence, inhibition of NET formation has to be undertaken with caution since they may predispose patients to potentially deleterious infective complications.
Neutrophils bear receptors such as C5a receptor that enable them to coordinate their effector functions with other elements of the innate immune system including the complements (182). Avacopan is an orally administered small-molecule C5a receptor antagonist which selectively blocks stimulatory effects of C5a, namely neutrophil chemoattraction and activation (183). A clinical trial on patients with ANCA-associated vasculitis found that avacopan was comparable to tapering doses of prednisone with respect to remission at week 26 but with fewer glucocorticoid-induced toxic effects (183). In the same trial, avacopan was also shown to confer renal protective effects as evidenced by improvements in renal function and albuminuria which are corroborated by effects seen in other human trials and murine studies (184–186). The potent renal protective pharmacological action of avacopan is attributed to the blockade of the C5a-C5a receptor axis which arrests chemoattraction and activation of neutrophils in glomeruli (184, 187). While clinical studies using avacopan in the treatment of SLE have not been performed, we speculate that this medication would be efficacious in neutrophil-mediated lupus manifestations in the CNS. Complement activation has also been implicated in antiphospholidpid syndrome (188). Eculizumab inhibits the cleavage of C5 to C5a and C5b, interrupting the complement mediated proinflammatory and prothrombotic environment (189). Indeed, eculizumab has been used in refractory catastrophic antiphospholipid syndrome (189). It has been proposed that blocking C5 activation and inhibiting C5aR1 signaling would limit the inflammatory damage mediated by neutrophils (190). Microfluidic studies have demonstrated that both avacopan and eculizumab inhibit both neutrophil chemotaxis and swarming functions (190). However, avacopan but not eculizumab preserves neutrophil phagocytosis of S. aureus bioparticles (190). Considering the known complication of increased susceptibility to encapsulated bacteria by eculizumab due to the inhibition of the distal complement components, avacopan appears to have better safety profile compared to eculizumab (183).
Neutrophils have been recognized as instigators of autoimmunity and effectors of tissue damage in SLE, therefore it is plausible that inhibition of neutrophil effector functions can be investigated as a potential therapeutic strategy for NPSLE. Breach of the BBB, neutrophil activation, transmigration and subsequent intrathecal and peripheral release of NETs are thought to act in concert with neuropathogenic autoantibodies including anti-NR2A/B antibodies to cause inflammation and neuronal cell death, leading to neuropsychiatric manifestations of SLE (191). LDNs represent a unique subset of neutrophils in SLE and their production of proinflammatory cytokines and other signaling molecules may have a role in orchestrating neurotoxic insults and other end-organ damage in SLE (192). Cellular products of LDNs such as MMP-9 and NGAL also play an instrumental role in disrupting the BBB and causing neuroinflammation in NPSLE. The diversity of clinical phenotypes and features of NPSLE is reflection that NP syndromes are a complex facet of SLE which involves distinct yet interwoven pathways and mechanisms. Given the central role that neutrophils play in the pathogenesis of NPSLE, therapeutic agents targeting the neutrophil axis warrant investigation for they could hold the key to the treatment and even prevention of NPSLE.
Conceptualization, TS and ST. Writing—original draft preparation, TS, AM, and ST. Writing—review and editing, TS, AM, and ST. Supervision, AM and ST. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Kaul A, Gordon C, Crow MK, Touma Z, Urowitz MB, van Vollenhoven R, et al. Systemic lupus erythematosus. Nat Rev Dis Primers (2016) 2:16039. doi: 10.1038/nrdp.2016.39
2. Mak A, Cheung MW-L, Chiew HJ, Liu Y, Ho R-m. Global trend of survival and damage of systemic lupus erythematosus: Meta-analysis and meta-regression of observational studies from the 1950s to 2000s. Semin Arthritis rheumatism WB Saunders (2012) pp:830–9. doi: 10.1016/j.semarthrit.2011.11.002
3. Govoni M, Hanly JG. The management of neuropsychiatric lupus in the 21st century: still so many unmet needs? Rheumatology (2020) 59:v52–62. doi: 10.1093/rheumatology/keaa404
4. Bertsias GK, Boumpas DT. Pathogenesis, diagnosis and management of neuropsychiatric SLE manifestations. Nat Rev Rheumatol (2010) 6:358–67. doi: 10.1038/nrrheum.2010.62
5. Hanly J, Urowitz M, Su L, Bae S, Gordon C, Wallace D, et al. Prospective analysis of neuropsychiatric events in an international disease inception cohort of patients with systemic lupus erythematosus. Ann rheumatic Dis (2010) 69:529–35. doi: 10.1136/ard.2008.106351
6. Zirkzee E, Huizinga T, Bollen E, Buchem MV, Middelkoop H, Wee N, et al. Mortality in neuropsychiatric systemic lupus erythematosus (NPSLE). Lupus (2014) 23:31–8. doi: 10.1177/0961203313512540
7. Pikman R, Kivity S, Levy Y, Arango M, Chapman J, Yonath H, et al. From animal model to human. Lupus (2017) 26:470–7. doi: 10.1177/0961203317694261
8. Seet D, Allameen NA, Tay SH, Cho J, Mak A. Cognitive dysfunction in systemic lupus erythematosus: Immunopathology, clinical manifestations, neuroimaging and management. Rheumatol Ther (2021) 8:651–79. doi: 10.1007/s40744-021-00312-0
9. Choi J, Kim ST, Craft J. The pathogenesis of systemic lupus erythematosus–an update. Curr Opin Immunol (2012) 24:651–7. doi: 10.1016/j.coi.2012.10.004
10. Mayadas TN, Cullere X, Lowell CA. The multifaceted functions of neutrophils. Annu Rev Pathology: Mech Dis (2014) 9:181–218. doi: 10.1146/annurev-pathol-020712-164023
11. Watts ER, Walmsley SR, Getting DAMP. (s) wets the whistle for neutrophil recruitment. Immunity (2018) 48:846–8. doi: 10.1016/j.immuni.2018.04.027
12. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303:1532–5. doi: 10.1126/science.1092385
13. Kaplan MJ. Neutrophils in the pathogenesis and manifestations of SLE. Nat Rev Rheumatol (2011) 7:691–9. doi: 10.1038/nrrheum.2011.132
14. Kaplan MJ. Role of neutrophils in systemic autoimmune diseases. Arthritis Res Ther (2013) 15:1–9. doi: 10.1186/ar4325
15. Wirestam L, Arve S, Linge P, Bengtsson AA. Neutrophils–important communicators in systemic lupus erythematosus and antiphospholipid syndrome. Front Immunol (2019) 10:2734. doi: 10.3389/fimmu.2019.02734
16. Hanly JG, Kozora E, Beyea SD, Birnbaum J. Nervous system disease in systemic lupus erythematosus: Current status and future directions. Arthritis Rheumatol (2019) 71:33–42. doi: 10.1002/art.40591
17. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature (2015) 523:337–41. doi: 10.1038/nature14432
18. Yang L, Kress BT, Weber HJ, Thiyagarajan M, Wang B, Deane R, et al. Evaluating glymphatic pathway function utilizing clinically relevant intrathecal infusion of CSF tracer. J Trans Med (2013) 11:1–9. doi: 10.1186/1479-5876-11-107
19. Engelhardt B, Vajkoczy P, Weller RO. The movers and shapers in immune privilege of the CNS. Nat Immunol (2017) 18:123–31. doi: 10.1038/ni.3666
20. How A, Dent PB, Liao SK, Denburg JA. Antineuronal antibodies in neuropsychiatric systemic lupus erythematosus. Arthritis Rheumatism: Off J Am Coll Rheumatol (1985) 28:789–95. doi: 10.1002/art.1780280710
21. Hacbarth E, Kajdacsy-Balla A. Low density neutrophils in patients with systemic lupus erythematosus, rheumatoid arthritis, and acute rheumatic fever. Arthritis Rheumatism (1986) 29:1334–42. doi: 10.1002/art.1780291105
22. Carmona-Rivera C, Kaplan MJ. Low-density granulocytes: a distinct class of neutrophils in systemic autoimmunity. Semin immunopathology Springer (2013) pp:455–63. doi: 10.1007/s00281-013-0375-7
23. Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med (2003) 197:711–23. doi: 10.1084/jem.20021553
24. Nakou M, Knowlton N, Frank MB, Bertsias G, Osban J, Sandel CE, et al. Gene expression in human lupus: bone marrow differentiates active from inactive patients and displays apoptosis and granulopoiesis signatures. Arthritis rheumatism (2008) 58:3541. doi: 10.1002/art.23961
25. Denny MF, Yalavarthi S, Zhao W, Thacker SG, Anderson M, Sandy AR, et al. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J Immunol (2010) 184:3284–97. doi: 10.4049/jimmunol.0902199
26. Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol (2011) 187:538–52. doi: 10.4049/jimmunol.1100450
27. Beringer A, Noack M, Miossec P. IL-17 in chronic inflammation: From discovery to targeting. Trends Mol Med (2016) 22:230–41. doi: 10.1016/j.molmed.2016.01.001
28. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med (2016) 22:146–53. doi: 10.1038/nm.4027
29. Blanco LP, Pedersen HL, Wang X, Lightfoot YL, Seto N, Carmona-Rivera C, et al. Improved mitochondrial metabolism and reduced inflammation following attenuation of murine lupus with coenzyme Q10 analog idebenone. Arthritis Rheumatol (2020) 72:454–64. doi: 10.1002/art.41128
30. Caielli S, Athale S, Domic B, Murat E, Chandra M, Banchereau R, et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med (2016) 213:697–713. doi: 10.1084/jem.20151876
31. Mak A, Chan JKY. Endothelial function and endothelial progenitor cells in systemic lupus erythematosus. Nat Rev Rheumatol (2022) 18:286–300. doi: 10.1038/s41584-022-00770-y
32. Rahman S, Sagar D, Hanna RN, Lightfoot YL, Mistry P, Smith CK, et al. Low-density granulocytes activate T cells and demonstrate a non-suppressive role in systemic lupus erythematosus. Ann rheumatic Dis (2019) 78:957–66. doi: 10.1136/annrheumdis-2018-214620
33. Sim TM, Ong SJ, Mak A, Tay SH. Type I interferons in systemic lupus erythematosus: A journey from bench to bedside. Int J Mol Sci (2022) 23:2505. doi: 10.3390/ijms23052505
34. Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci (2003) 100:2610–5. doi: 10.1073/pnas.0337679100
35. Shirafuji N, Matsuda S, Ogura H, Tani K, Kodo H, Ozawa K, et al. Granulocyte colony-stimulating factor stimulates human mature neutrophilic granulocytes to produce interferon-α. Blood (1990) 75:17–9. doi: 10.1182/blood.V75.1.17.17
36. Tamassia N, Le Moigne V, Rossato M, Donini M, McCartney S, Calzetti F, et al. Activation of an immunoregulatory and antiviral gene expression program in poly (I: C)-transfected human neutrophils. J Immunol (2008) 181:6563–73. doi: 10.4049/jimmunol.181.9.6563
37. Kaplan MJ, Salmon JE. How does IFN-α insult the vasculature? let me count the ways. Arthritis rheumatism (2011) 63:334. doi: 10.1002/art.30161
38. Salemme R, Peralta LN, Meka SH, Pushpanathan N, Alexander JJ. The role of NETosis in systemic lupus erythematosus. J Cell Immunol (2019) 1:33. doi: 10.33696/immunology.1.008
39. Tan E, Schur P, Carr R, Kunkel H. Deoxybonucleic acid (DNA) and antibodies to DNA in the serum of patients with systemic lupus erythematosus. J Clin Invest (1966) 45:1732–40. doi: 10.1172/JCI105479
40. Hakkim A, Fürnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci (2010) 107:9813–8. doi: 10.1073/pnas.0909927107
41. Leffler J, Martin M, Gullstrand B, Tydén H, Lood C, Truedsson L, et al. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J Immunol (2012) 188:3522–31. doi: 10.4049/jimmunol.1102404
42. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Trans Med (2011) 3:73ra20–0. doi: 10.1126/scitranslmed.3001201
43. Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med (2011) 3(73):73ra19. doi: 10.1126/scitranslmed.3001180
44. Banchereau J, Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity (2006) 25:383–92. doi: 10.1016/j.immuni.2006.08.010
45. Delgado-Rizo V, Martinez-Guzman MA, Iñiguez-Gutierrez L, García-Orozco A, Alvarado-Navarro A, Fafutis-Morris M. Neutrophil extracellular traps and its implications in inflammation: An overview. Front Immunol (2017) 8:81. doi: 10.3389/fimmu.2017.00081
46. Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, et al. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against candida albicans. PloS Pathog (2009) 5:e1000639. doi: 10.1371/journal.ppat.1000639
47. Pieterse E, Rother N, Garsen M, Hofstra JM, Satchell SC, Hoffmann M, et al. Neutrophil extracellular traps drive endothelial-to-mesenchymal transition. Arteriosclerosis thrombosis Vasc Biol (2017) 37:1371–9. doi: 10.1161/ATVBAHA.117.309002
48. Jansen MP, Emal D, Teske GJ, Dessing MC, Florquin S, Roelofs JJ. Release of extracellular DNA influences renal ischemia reperfusion injury by platelet activation and formation of neutrophil extracellular traps. Kidney Int (2017) 91:352–64. doi: 10.1016/j.kint.2016.08.006
49. Zenaro E, Pietronigro E, Bianca VD, Piacentino G, Marongiu L, Budui S, et al. Neutrophils promote alzheimer's disease–like pathology and cognitive decline via LFA-1 integrin. Nat Med (2015) 21:880–6. doi: 10.1038/nm.3913
50. Pietronigro EC, Della Bianca V, Zenaro E, Constantin G. NETosis in alzheimer's disease. Front Immunol (2017) 8:211. doi: 10.3389/fimmu.2017.00211
51. Clancy DM, Henry CM, Sullivan GP, Martin SJ. Neutrophil extracellular traps can serve as platforms for processing and activation of IL-1 family cytokines. FEBS J (2017) 284:1712–25. doi: 10.1111/febs.14075
52. Meng H, Yalavarthi S, Kanthi Y, Mazza LF, Elfline MA, Luke CE, et al. In vivo role of neutrophil extracellular traps in antiphospholipid antibody–mediated venous thrombosis. Arthritis Rheumatol (2017) 69:655–67. doi: 10.1002/art.39938
53. Gomis-Rüth FX. Catalytic domain architecture of metzincin metalloproteases. J Biol Chem (2009) 284:15353–7. doi: 10.1074/jbc.R800069200
54. Singh D, Srivastava SK, Chaudhuri TK, Upadhyay G. Multifaceted role of matrix metalloproteinases (MMPs). Front Mol Biosci (2015) 2:19. doi: 10.3389/fmolb.2015.00019
55. Löffek S, Schilling O, Franzke C-W. Biological role of matrix metalloproteinases: a critical balance. Eur Respir J (2011) 38:191–208. doi: 10.1183/09031936.00146510
56. Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res (2003) 92:827–39. doi: 10.1161/01.RES.0000070112.80711.3D
57. Liu J, Khalil RA. Matrix metalloproteinase inhibitors as investigational and therapeutic tools in unrestrained tissue remodeling and pathological disorders. Prog Mol Biol Trans Sci (2017) 148:355–420. doi: 10.1016/bs.pmbts.2017.04.003
58. Bronisz E, Kurkowska-Jastrzębska I. Matrix metalloproteinase 9 in epilepsy: The role of neuroinflammation in seizure development. Mediators Inflammation (2016) 2016:7369020. doi: 10.1155/2016/7369020
59. Liu Y, Liu H, Luo X, Deng J, Pan Y, Liang H. Overexpression of SMYD3 and matrix metalloproteinase-9 are associated with poor prognosis of patients with gastric cancer. Tumor Biol (2015) 36:4377–86. doi: 10.1007/s13277-015-3077-z
60. Yabluchanskiy A, Ma Y, Iyer RP, Hall ME, Lindsey ML. Matrix metalloproteinase-9: Many shades of function in cardiovascular disease. Physiology (2013) 28:391–403. doi: 10.1152/physiol.00029.2013
61. Moxon-Emre I, Schlichter LC. Neutrophil depletion reduces blood-brain barrier breakdown, axon injury, and inflammation after intracerebral hemorrhage. J Neuropathology Exp Neurol (2011) 70:218–35. doi: 10.1097/NEN.0b013e31820d94a5
62. Tschesche H, Zölzer V, Triebel S, Bartsch S. The human neutrophil lipocalin supports the allosteric activation of matrix metalloproteinases. Eur J Biochem (2001) 268:1918–28. doi: 10.1046/j.1432-1327.2001.02066.x
63. Robak E, Wierzbowska A, Chmiela M, Kulczycka L, Sysa-Jędrejowska A, Robak T. Circulating total and active metalloproteinase-9 and tissue inhibitor of metalloproteinases-1 in patients with systemic lupus erythomatosus. Mediators Inflammation (2006) 2006(1):17898. doi: 10.1155/MI/2006/17898
64. Ainiala H, Hietaharju A, Dastidar P, Loukkola J, Lehtimäki T, Peltola J, et al. Increased serum matrix metalloproteinase 9 levels in systemic lupus erythematosus patients with neuropsychiatric manifestations and brain magnetic resonance imaging abnormalities. Arthritis Rheumatism (2004) 50:858–65. doi: 10.1002/art.20045
65. Lesiak A, Narbutt J, Sysa-Jedrzejowska A, Lukamowicz J, McCauliffe D, Wózniacka A. Effect of chloroquine phosphate treatment on serum MMP-9 and TIMP-1 levels in patients with systemic lupus erythematosus. Lupus (2010) 19:683–8. doi: 10.1177/0961203309356455
66. Bahrehmand F, Vaisi-Raygani A, Kiani A, Rahimi Z, Tavilani H, Ardalan M, et al. Matrix metalloproteinase 9 polymorphisms and systemic lupus erythematosus: correlation with systemic inflammatory markers and oxidative stress. Lupus (2015) 24:597–605. doi: 10.1177/0961203314559085
67. Kwieciński J, Kłak M, Trysberg E, Blennow K, Tarkowski A, Jin T. Relationship between elevated cerebrospinal fluid levels of plasminogen activator inhibitor 1 and neuronal destruction in patients with neuropsychiatric systemic lupus erythematosus. Arthritis Rheumatism (2009) 60:2094–101. doi: 10.1002/art.24603
68. Trysberg E, Blennow K, Zachrisson O, Tarkowski A. Intrathecal levels of matrix metalloproteinases in systemic lupus erythematosus with central nervous system engagement. Arthritis Res Ther (2004) 6:1–6. doi: 10.1186/ar1228
69. Alcocer-Varela J, Aleman-Hoey D, Alarcon-Segovia D. Interleukin-1 and interleukin-6 activities are increased in the cerebrospinal fluid of patients with CNS lupus erythematosus and correlate with local late T-cell activation markers. Lupus (1992) 1:111–7. doi: 10.1177/096120339200100209
70. Trysberg E, Carlsten H, Tarkowski A. Intrathecal cytokines in systemic lupus erythematosus with central nervous system involvement. Lupus (2000) 9:498–503. doi: 10.1177/096120330000900704
71. Kothari P, Pestana R, Mesraoua R, Elchaki R, Khan KM, Dannenberg AJ, et al. IL-6-mediated induction of matrix metalloproteinase-9 is modulated by JAK-dependent IL-10 expression in macrophages. J Immunol (2014) 192:349–57. doi: 10.4049/jimmunol.1301906
72. Okada Y, Tsuchiya H, Shimizu H, Tomita K, Nakanishi I, Sato H, et al. Induction and stimulation of 92-kDa gelatinase/type IV collagenase production in osteosarcoma and fibrosarcoma cell lines by tumor necrosis factor α. Biochem Biophys Res Commun (1990) 171:610–7. doi: 10.1016/0006-291X(90)91190-4
73. Lotz M, Guerne P. Interleukin-6 induces the synthesis of tissue inhibitor of metalloproteinases-1/erythroid potentiating activity (TIMP-1/EPA). J Biol Chem (1991) 266:2017–20. doi: 10.1016/S0021-9258(18)52202-X
74. Trysberg E, Nylen K, Rosengren LE, Tarkowski A. Neuronal and astrocytic damage in systemic lupus erythematosus patients with central nervous system involvement. Arthritis Rheumatism (2003) 48:2881–7. doi: 10.1002/art.11279
75. Trysberg E, Höglund K, Svenungsson E, Blennow K, Tarkowski A. Decreased levels of soluble amyloid β-protein precursor and β-amyloid protein in cerebrospinal fluid of patients with systemic lupus erythematosus. Arthritis Res Ther (2004) 6:1–8. doi: 10.1186/ar1040
76. Nikkari ST, Höyhtyä M, Isola J, Nikkari T. Macrophages contain 92-kd gelatinase (MMP-9) at the site of degenerated internal elastic lamina in temporal arteritis. Am J Pathol (1996) 149:1427.
77. Rosenberg GA, Navratil M, Barone F, Feuerstein G. Proteolytic cascade enzymes increase in focal cerebral ischemia in rat. J Cereb Blood Flow Metab (1996) 16:360–6. doi: 10.1097/00004647-199605000-00002
78. Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA, et al. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J Neurosci (2001) 21:7724–32. doi: 10.1523/JNEUROSCI.21-19-07724.2001
79. Gu Z, Kaul M, Yan B, Kridel SJ, Cui J, Strongin A, et al. S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science (2002) 297:1186–90. doi: 10.1126/science.1073634
80. Horstmann S, Kalb P, Koziol J, Gardner H, Wagner S. Profiles of matrix metalloproteinases, their inhibitors, and laminin in stroke patients: influence of different therapies. Stroke (2003) 34:2165–70. doi: 10.1161/01.STR.0000088062.86084.F2
81. Gu Z, Cui J, Brown S, Fridman R, Mobashery S, Strongin AY, et al. A highly specific inhibitor of matrix metalloproteinase-9 rescues laminin from proteolysis and neurons from apoptosis in transient focal cerebral ischemia. J Neurosci (2005) 25:6401–8. doi: 10.1523/JNEUROSCI.1563-05.2005
82. Devarajan P. Neutrophil gelatinase-associated lipocalin (NGAL): a new marker of kidney disease. Scand J Clin Lab Invest Suppl (2008) 241:89–94. doi: 10.1080/00365510802150158
83. Jha MK, Suk K. Glia-based biomarkers and their functional role in the CNS. Expert Rev Proteomics (2013) 10:43–63. doi: 10.1586/epr.12.70
84. Brunner HI, Klein-Gitelman MS, Zelko F, Beebe DW, Foell D, Lee J, et al. Blood-based candidate biomarkers of the presence of neuropsychiatric systemic lupus erythematosus in children. Lupus Sci Med (2014) 1:e000038. doi: 10.1136/lupus-2014-000038
85. Mike EV, Makinde HM, Gulinello M, Vanarsa K, Herlitz L, Gadhvi G, et al. Lipocalin-2 is a pathogenic determinant and biomarker of neuropsychiatric lupus. J Autoimmun (2019) 96:59–73. doi: 10.1016/j.jaut.2018.08.005
86. Diamond B, Volpe BT. A model for lupus brain disease. Immunol Rev (2012) 248:56–67. doi: 10.1111/j.1600-065X.2012.01137.x
87. Ota Y, Capizzano AA, Moritani T, Naganawa S, Kurokawa R, Srinivasan A. Comprehensive review of wernicke encephalopathy: pathophysiology, clinical symptoms and imaging findings. Japanese J Radiol (2020) 38:809–20. doi: 10.1007/s11604-020-00989-3
88. Sim TM, Tarini D, Dheen ST, Bay BH, Srinivasan DK. Nanoparticle-based technology approaches to the management of neurological disorders. Int J Mol Sci (2020) 21:6070. doi: 10.3390/ijms21176070
89. Desai BS, Monahan AJ, Carvey PM, Hendey B. Blood–brain barrier pathology in alzheimer's and parkinson's disease: implications for drug therapy. Cell Transplant (2007) 16:285–99. doi: 10.3727/000000007783464731
90. Tomizawa Y, Yokoyama K, Saiki S, Takahashi T, Matsuoka J, Hattori N. Blood–brain barrier disruption is more severe in neuromyelitis optica than in multiple sclerosis and correlates with clinical disability. J Int Med Res (2012) 40:1483–91. doi: 10.1177/147323001204000427
91. Cohen D, Rijnink EC, Nabuurs RJ, Steup-Beekman GM, Versluis MJ, Emmer BJ, et al. Brain histopathology in patients with systemic lupus erythematosus: identification of lesions associated with clinical neuropsychiatric lupus syndromes and the role of complement. Rheumatology (2017) 56(1):77–86. doi: 10.1093/rheumatology/kew341
92. Rowshani A, Remans P, Rozemuller A, Tak P. Cerebral vasculitis as a primary manifestation of systemic lupus erythematosus. Ann rheumatic Dis (2005) 64:784–6. doi: 10.1136/ard.2004.026542
93. Winfield JB, Shaw M, Silverman LM, Eisenberg RA, Wilson HA III, Koffler D. Intrathecal IgG synthesis and blood-brain barrier impairment in patients with systemic lupus erythematosus and central nervous system dysfunction. Am J Med (1983) 74:837–44. doi: 10.1016/0002-9343(83)91075-6
94. Schwartz N, Stock AD, Putterman C. Neuropsychiatric lupus: new mechanistic insights and future treatment directions. Nat Rev Rheumatol (2019) 15:137–52. doi: 10.1038/s41584-018-0156-8
95. Wen J, Doerner J, Weidenheim K, Xia Y, Stock A, Michaelson JS, et al. TNF-like weak inducer of apoptosis promotes blood brain barrier disruption and increases neuronal cell death in MRL/lpr mice. J Autoimmun (2015) 60:40–50. doi: 10.1016/j.jaut.2015.03.005
96. Engelborghs S, Niemantsverdriet E, Struyfs H, Blennow K, Brouns R, Comabella M, et al. Consensus guidelines for lumbar puncture in patients with neurological diseases. Alzheimer's Dementia: Diagnosis Assess Dis Monit (2017) 8:111–26. doi: 10.1016/j.dadm.2017.04.007
97. Tomkins O, Kaufer D, Korn A, Shelef I, Golan H, Reichenthal E, et al. Frequent blood–brain barrier disruption in the human cerebral cortex. Cell Mol Neurobiol (2001) 21:675–91. doi: 10.1023/A:1015147920283
98. Ercan E, Ingo C, Tritanon O, Magro-Checa C, Smith A, Smith S, et al. A multimodal MRI approach to identify and characterize microstructural brain changes in neuropsychiatric systemic lupus erythematosus. NeuroImage Clin (2015) 8:337–44. doi: 10.1016/j.nicl.2015.05.002
99. Gulati G, Jones JT, Lee G, Altaye M, Beebe DW, Meyers-Eaton J, et al. Altered blood-brain barrier permeability in patients with systemic lupus erythematosus: A novel imaging approach. Arthritis Care Res (2017) 69:299–305. doi: 10.1002/acr.22923
100. Chi JM, Mackay M, Hoang A, Cheng K, Aranow C, Ivanidze J, et al. Alterations in blood-brain barrier permeability in patients with systemic lupus erythematosus. AJNR Am J neuroradiology (2019) 40:470–7. doi: 10.3174/ajnr.A5990
101. Kamintsky L, Beyea SD, Fisk JD, Hashmi JA, Omisade A, Calkin C, et al. Blood-brain barrier leakage in systemic lupus erythematosus is associated with gray matter loss and cognitive impairment. Ann rheumatic Dis (2020) 79:1580–7. doi: 10.1136/annrheumdis-2020-218004
102. Gulati G, Jones JT, Lee G, Altaye M, Beebe DW, Meyers-Eaton J, et al. Altered blood–brain barrier permeability in patients with systemic lupus erythematosus: a novel imaging approach. Arthritis Care Res (2017) 69:299–305. doi: 10.1002/acr.22923
103. Wang X, Ma L, Luo Y, Yang Y, Upreti B, Cheng Y, et al. Increasing of blood brain barrier permeability and the association with depression and anxiety in systemic lupus erythematosus patients. Front Med (2022) 9. doi: 10.3389/fmed.2022.852835
104. Rosell A, Cuadrado E, Ortega-Aznar A, Hernández-Guillamon M, Lo EH, Montaner J. MMP-9–positive neutrophil infiltration is associated to blood–brain barrier breakdown and basal lamina type iv collagen degradation during hemorrhagic transformation after human ischemic stroke. Stroke (2008) 39:1121–6. doi: 10.1161/STROKEAHA.107.500868
105. Fainardi E, Castellazzi M, Bellini T, Manfrinato M, Baldi E, Casetta I, et al. Cerebrospinal fluid and serum levels and intrathecal production of active matrix metalloproteinase-9 (MMP-9) as markers of disease activity in patients with multiple sclerosis. Multiple Sclerosis J (2006) 12:294–301. doi: 10.1191/135248506ms1274oa
106. Stielke S, Keilhoff G, Kirches E, Mertens PR, Neumann K-H, Tsokos GC, et al. Adhesion molecule expression precedes brain damages of lupus-prone mice and correlates with kidney pathology. J Neuroimmunology (2012) 252:24–32. doi: 10.1016/j.jneuroim.2012.07.011
107. Gorina R, Lyck R, Vestweber D, Engelhardt B. β2 integrin–mediated crawling on endothelial ICAM-1 and ICAM-2 is a prerequisite for transcellular neutrophil diapedesis across the inflamed blood–brain barrier. J Immunol (2014) 192:324–37. doi: 10.4049/jimmunol.1300858
108. Valentijn R, Van Overhagen H, Hazevoet H, Hermans J, Cats A, Daha M, et al. The value of complement and immune complex determinations in monitoring disease activity in patients with systemic lupus erythematosus. Arthritis Rheumatism (1985) 28:904–13. doi: 10.1002/art.1780280810
109. Vandendriessche S, Cambier S, Proost P, Marques PE. Complement receptors and their role in leukocyte recruitment and phagocytosis. Front Cell Dev Biol (2021) 9:624025. doi: 10.3389/fcell.2021.624025
110. de Bont CM, Boelens WC, Pruijn GJ. NETosis, complement, and coagulation: a triangular relationship. Cell Mol Immunol (2019) 16:19–27. doi: 10.1038/s41423-018-0024-0
111. Palmer L, Damgaard C, Holmstrup P, Nielsen C. Influence of complement on neutrophil extracellular trap release induced by bacteria. J periodontal Res (2016) 51:70–6. doi: 10.1111/jre.12284
112. Neeli I, Dwivedi N, Khan S, Radic M. Regulation of extracellular chromatin release from neutrophils. J innate Immun (2009) 1:194–201. doi: 10.1159/000206974
113. Alexander JJ, Jacob A, Vezina P, Sekine H, Gilkeson GS, Quigg RJ. Absence of functional alternative complement pathway alleviates lupus cerebritis. Eur J Immunol (2007) 37:1691–701. doi: 10.1002/eji.200636638
114. Woodruff TM, Ager RR, Tenner AJ, Noakes PG, Taylor SM. The role of the complement system and the activation fragment C5a in the central nervous system. Neuromolecular Med (2010) 12:179–92. doi: 10.1007/s12017-009-8085-y
115. Niculescu T, Weerth S, Niculescu F, Cudrici C, Rus V, Raine CS, et al. Effects of complement C5 on apoptosis in experimental autoimmune encephalomyelitis. J Immunol (2004) 172:5702–6. doi: 10.4049/jimmunol.172.9.5702
116. Fonseca MI, Ager RR, Chu S-H, Yazan O, Sanderson SD, LaFerla FM, et al. Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of alzheimer’s disease. J Immunol (2009) 183:1375–83. doi: 10.4049/jimmunol.0901005
117. Uwai M, Terui Y, Mishima Y, Tomizuka H, Ikeda M, Itoh T, et al. A new apoptotic pathway for the complement factor b-derived fragment bb. J Cell Physiol (2000) 185:280–92. doi: 10.1002/1097-4652(200011)185:2<280::AID-JCP13>3.0.CO;2-L
118. Jacob A, Hack B, Chiang E, Garcia JG, Quigg RJ, Alexander JJ. C5a alters blood-brain barrier integrity in experimental lupus. FASEB J (2010) 24:1682–8. doi: 10.1096/fj.09-138834
119. Jacob A, Hack B, Chen P, Quigg RJ, Alexander JJ. C5a/CD88 signaling alters blood–brain barrier integrity in lupus through nuclear factor-κB. J neurochemistry (2011) 119:1041–51. doi: 10.1111/j.1471-4159.2011.07490.x
120. Mahajan SD, Parikh NU, Woodruff TM, Jarvis JN, Lopez M, Hennon T, et al. C5a alters blood–brain barrier integrity in a human in vitro model of systemic lupus erythematosus. Immunology (2015) 146:130–43. doi: 10.1111/imm.12489
121. Arora V, Verma J, Dutta R, Marwah V, Kumar A, Das N. Reduced complement receptor 1 (CR1, CD35) transcription in systemic lupus erythematosus. Mol Immunol (2004) 41:449–56. doi: 10.1016/j.molimm.2004.03.004
122. Nestor J, Arinuma Y, Huerta TS, Kowal C, Nasiri E, Kello N, et al. Lupus antibodies induce behavioral changes mediated by microglia and blocked by ACE inhibitors. J Exp Med (2018) 215:2554–66. doi: 10.1084/jem.20180776
123. Abdel-Nasser AM, Ghaleb RM, Mahmoud JA, Khairy W, Mahmoud RM. Association of anti-ribosomal p protein antibodies with neuropsychiatric and other manifestations of systemic lupus erythematosus. Clin Rheumatol (2008) 27:1377–85. doi: 10.1007/s10067-008-0921-1
124. Afeltra A, Garzia P, Mitterhofer AP, Vadacca M, Galluzzo S, Del Porto F, et al. Neuropsychiatric lupus syndromes: relationship with antiphospholipid antibodies. Neurology (2003) 61:108–10. doi: 10.1212/01.WNL.0000058904.94330.A7
125. Gono T, Takarada T, Fukumori R, Kawaguchi Y, Kaneko H, Hanaoka M, et al. NR2-reactive antibody decreases cell viability through augmentation of Ca2+ influx in systemic lupus erythematosus. Arthritis Rheumatism (2011) 63:3952–9. doi: 10.1002/art.30616
126. Florey OJ, Johns M, Esho OO, Mason JC, Haskard DO. Antiendothelial cell antibodies mediate enhanced leukocyte adhesion to cytokine-activated endothelial cells through a novel mechanism requiring cooperation between FcγRIIa and CXCR1/2. Blood (2007) 109:3881–9. doi: 10.1182/blood-2006-08-044669
127. Hanly JG. Diagnosis and management of neuropsychiatric SLE. Nat Rev Rheumatol (2014) 10:338–47. doi: 10.1038/nrrheum.2014.15
128. Kumar A. NMDA receptor function during senescence: implication on cognitive performance. Front Neurosci (2015) 9:473. doi: 10.3389/fnins.2015.00473
129. Senécal J-L, Raymond Y. The pathogenesis of neuropsychiatric manifestations in systemic lupus erythematosus: a disease in search of autoantibodies, or autoantibodies in search of a disease? J Rheumatol (2004) 31:2093–8.
130. Yoshio T, Okamoto H, Hirohata S, Minota S. IgG anti–NR2 glutamate receptor autoantibodies from patients with systemic lupus erythematosus activate endothelial cells. Arthritis Rheumatism (2013) 65:457–63. doi: 10.1002/art.37745
131. DeGiorgio LA, Konstantinov KN, Lee SC, Hardin JA, Volpe BT, Diamond B. A subset of lupus anti-DNA antibodies cross-reacts with the NR2 glutamate receptor in systemic lupus erythematosus. Nat Med (2001) 7:1189–93. doi: 10.1038/nm1101-1189
132. Kowal C, DeGiorgio LA, Lee JY, Edgar MA, Huerta PT, Volpe BT, et al. Human lupus autoantibodies against NMDA receptors mediate cognitive impairment. Proc Natl Acad Sci (2006) 103:19854–9. doi: 10.1073/pnas.0608397104
133. Bryushkova EA, Vladychenskaya EA, Stepanova MS, Boldyrev AA. Effect of homocysteine on properties of neutrophils activated in vivo. Biochem (Mosc) (2011) 76:467–72. doi: 10.1134/S0006297911040109
134. Omdal R, Brokstad K, Waterloo K, Koldingsnes W, Jonsson R, Mellgren S. Neuropsychiatric disturbances in SLE are associated with antibodies against NMDA receptors. Eur J Neurol (2005) 12:392–8. doi: 10.1111/j.1468-1331.2004.00976.x
135. Massardo L, Bravo-Zehnder M, Calderon J, Flores P, Padilla O, Aguirre J, et al. Anti-N-methyl-D-aspartate receptor and anti-ribosomal-P autoantibodies contribute to cognitive dysfunction in systemic lupus erythematosus. Lupus (2015) 24:558–68. doi: 10.1177/0961203314555538
136. Arinuma Y, Yanagida T, Hirohata S. Association of cerebrospinal fluid anti–NR2 glutamate receptor antibodies with diffuse neuropsychiatric systemic lupus erythematosus. Arthritis Rheumatism (2008) 58:1130–5. doi: 10.1002/art.23399
137. Hirohata S, Arinuma Y, Yanagida T, Yoshio T. Blood-brain barrier damages and intrathecal synthesis of anti-N-methyl-D-aspartate receptor NR2 antibodies in diffuse psychiatric/neuropsychological syndromes in systemic lupus erythematosus. Arthritis Res Ther (2014) 16:1–7. doi: 10.1186/ar4518
138. Hanly JG, Robichaud J, Fisk JD. Anti-NR2 glutamate receptor antibodies and cognitive function in systemic lupus erythematosus. J Rheumatol (2006) 33:1553–8.
139. Yoshio T, Onda K, Nara H, Minota S. Association of IgG anti-NR2 glutamate receptor antibodies in cerebrospinal fluid with neuropsychiatric systemic lupus erythematosus. Arthritis Rheumatism (2006) 54:675–8. doi: 10.1002/art.21547
140. Limper M, Scirè CA, Talarico R, Amoura Z, Avcin T, Basile M, et al. Antiphospholipid syndrome: state of the art on clinical practice guidelines. RMD Open (2018) 4:e000785. doi: 10.1136/rmdopen-2018-000785
141. Ünlü O, Zuily S, Erkan D. The clinical significance of antiphospholipid antibodies in systemic lupus erythematosus. Eur J Rheumatol (2016) 3:75. doi: 10.5152/eurjrheum.2015.0085
142. Schreiber K, Sciascia S, De Groot PG, Devreese K, Jacobsen S, Ruiz-Irastorza G, et al. Antiphospholipid syndrome. Nat Rev Dis Primers (2018) 4:1–20. doi: 10.1038/nrdp.2017.103
143. Hanly JG, Hong C, Smith S, Fisk JD. A prospective analysis of cognitive function and anticardiolipin antibodies in systemic lupus erythematosus. Arthritis Rheumatism (1999) 42:728–34. doi: 10.1002/1529-0131(199904)42:4<728::AID-ANR16>3.0.CO;2-O
144. Menon S, Jameson-Shortall E, Newman S, Hall-Craggs M, Chinn R, Isenberg D. A longitudinal study of anticardiolipin antibody levels and cognitive functioning in systemic lupus erythematosus. Arthritis Rheumatism (1999) 42:735–41. doi: 10.1002/1529-0131(199904)42:4<735::AID-ANR17>3.0.CO;2-L
145. Ceccarelli F, Perricone C, Pirone C, Massaro L, Alessandri C, Mina C, et al. Cognitive dysfunction improves in systemic lupus erythematosus: results of a 10 years prospective study. PloS One (2018) 13:e0196103. doi: 10.1371/journal.pone.0196103
146. Ho RC, Thiaghu C, Ong H, Lu Y, Ho CS, Tam WW, et al. A meta-analysis of serum and cerebrospinal fluid autoantibodies in neuropsychiatric systemic lupus erythematosus. Autoimmun Rev (2016) 15:124–38. doi: 10.1016/j.autrev.2015.10.003
147. Mikdashi J, Handwerger B. Predictors of neuropsychiatric damage in systemic lupus erythematosus: data from the Maryland lupus cohort. Rheumatology (2004) 43:1555–60. doi: 10.1093/rheumatology/keh384
148. Narshi C, Giles I, Rahman A. The endothelium: an interface between autoimmunity and atherosclerosis in systemic lupus erythematosus? Lupus (2011) 20:5–13. doi: 10.1177/0961203310382429
149. Andrade R, Alarcon G, Gonzalez L, Fernandez M, Apte M, Vila L, et al. Seizures in patients with systemic lupus erythematosus: data from LUMINA, a multiethnic cohort (LUMINA LIV). Ann rheumatic Dis (2008) 67:829–34. doi: 10.1136/ard.2007.077594
150. Brey RL, Gharavi AE, Lockshin MD. Neurologic complications of antiphospholipid antibodies. Rheumatic Dis Clinics North America (1993) 19:833–50. doi: 10.1016/S0889-857X(21)00209-X
151. Katzav A, Ben-Ziv T, Blank M, Pick CG, Shoenfeld Y, Chapman J. Antibody-specific behavioral effects: intracerebroventricular injection of antiphospholipid antibodies induces hyperactive behavior while anti-ribosomal-P antibodies induces depression and smell deficits in mice. J Neuroimmunology (2014) 272:10–5. doi: 10.1016/j.jneuroim.2014.04.003
152. Borowoy AM, Pope JE, Silverman E, Fortin PR, Pineau C, Smith CD, et al. Neuropsychiatric lupus: the prevalence and autoantibody associations depend on the definition: results from the 1000 faces of lupus cohort. Semin Arthritis rheumatism Elsevier (2012) pp:179–85. doi: 10.1016/j.semarthrit.2012.03.011
153. Chi OZ, Hunter C, Liu X, Weiss HR. Effects of exogenous excitatory amino acid neurotransmitters on blood–brain barrier disruption in focal cerebral ischemia. Neurochemical Res (2009) 34:1249–54. doi: 10.1007/s11064-008-9902-7
154. Yalavarthi S, Gould TJ, Rao AN, Mazza LF, Morris AE, Núñez-Álvarez C, et al. Release of neutrophil extracellular traps by neutrophils stimulated with antiphospholipid antibodies: a newly identified mechanism of thrombosis in the antiphospholipid syndrome. Arthritis Rheumatol (Hoboken N.J.) (2015) 67:2990–3003. doi: 10.1002/art.39247
155. Huang C-H, Hung C-H, Chu Y-T, Hua Y-M. Tumor-like cerebral perivasculitis in a pediatric patient with systemic lupus erythematosus. Kaohsiung J Med Sci (2008) 24:218–22. doi: 10.1016/S1607-551X(08)70121-9
156. Abialmouna J, Shoemaker D, Pullicino P, Baer A. Marked cerebrospinal fluid pleocytosis in systemic lupus erythematosus related cerebral ischemia. J Rheumatol (1992) 19:626–9.
157. Heinlein A, Gertner E. Marked inflammation in catastrophic longitudinal myelitis associated with systemic lupus erythematosus. Lupus (2007) 16:823–6. doi: 10.1177/0961203307081846
158. Modjinou D, Gurin L, Chhabra A, Mikolaenko I, Lydon E, Smiles S. A case of systemic lupus erythematosus associated with longitudinal extensive transverse myelitis, cerebral neutrophilic vasculitis, and cerebritis. Bull Hosp Joint Dis (2014) 72(4):294–300.
159. Birnbaum J, Petri M, Thompson R, Izbudak I, Kerr D. Distinct subtypes of myelitis in systemic lupus erythematosus. Arthritis rheumatism (2009) 60:3378–87. doi: 10.1002/art.24937
160. Hopkins P, Michael Belmont H, Buyon J, Philips M, Weissmann G, Abramson SB. Increased levels of plasma anaphylatoxins in systemic lupus erythematosus predict flares of the disease and may elicit vascular injury in lupus cerebritis. Arthritis Rheumatism (1988) 31:632–41. doi: 10.1002/art.1780310508
161. Jacob A, Hack B, Bai T, Brorson JR, Quigg RJ, Alexander JJ. Inhibition of C5a receptor alleviates experimental CNS lupus. J neuroimmunology (2010) 221:46–52. doi: 10.1016/j.jneuroim.2010.02.011
162. Lammens M, Robberecht W, Waer M, Carton H, Dom R. Purulent meningitis due to aspergillosis in a patient with systemic lupus erythematosus. Clin Neurol Neurosurg (1992) 94:39–43. doi: 10.1016/0303-8467(92)90117-L
163. Thamtam VK, Uppin MS, Pyal A, Kaul S, Rani JY, Sundaram C. Fatal granulomatous amoebic encephalitis caused by acanthamoeba in a newly diagnosed patient with systemic lupus erythematosus. Neurol India (2016) 64:101. doi: 10.4103/0028-3886.173662
164. Iyer NR, McCune WJ, Wallace BI. West Nile Encephalitis mimicking neuropsychiatric lupus in a patient with systemic lupus erythematosus. BMJ Case Rep CP (2019) 12:e229537. doi: 10.1136/bcr-2019-229537
165. Moris G, Garcia-Monco JC. The challenge of drug-induced aseptic meningitis. Arch Internal Med (1999) 159:1185–94. doi: 10.1001/archinte.159.11.1185
166. Tay SH, Mak A. Diagnosing and attributing neuropsychiatric events to systemic lupus erythematosus: time to untie the Gordian knot? Rheumatology (2017) 56:i14–23. doi: 10.1093/rheumatology/kex018
167. Chau SY, Mok CC. Factors predictive of corticosteroid psychosis in patients with systemic lupus erythematosus. Neurology (2003) 61:104–7. doi: 10.1212/WNL.61.1.104
168. Abramson SB, Given WP, Edelson HS, Weissmann G. Neutrophil aggregation induced by sera from patients with active systemic lupus erythematosus. Arthritis Rheumatism (1983) 26:630–6. doi: 10.1002/art.1780260509
169. Guo Y, Zeng H, Gao C. The role of neutrophil extracellular traps in central nervous system diseases and prospects for clinical application. Oxid Med Cell Longevity (2021) 2021:9931742. doi: 10.1155/2021/9931742
170. Appelgren D, Dahle C, Knopf J, Bilyy R, Vovk V, Sundgren PC, et al. Active NET formation in libman–sacks endocarditis without antiphospholipid antibodies: a dramatic onset of systemic lupus erythematosus. Autoimmunity (2018) 51:310–8. doi: 10.1080/08916934.2018.1514496
171. Sule G, Mazza LF, Kazzaz NM, Yalavarthi S, Meng H, Knight JS. Inhibition of neutrophil elastase protects against glomerulonephritis and thrombosis in a mouse model of lupus. Arthritis Rheumatol (2017) 69(suppl 10).
172. Knight JS, Zhao W, Luo W, Subramanian V, O’Dell AA, Yalavarthi S, et al. Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J Clin Invest (2013) 123:2981–93. doi: 10.1172/JCI67390
173. Witalison EE, Thompson PR, Hofseth LJ. Protein arginine deiminases and associated citrullination: physiological functions and diseases associated with dysregulation. Curr Drug Targets (2015) 16:700–10. doi: 10.2174/1389450116666150202160954
174. Willis VC, Gizinski AM, Banda NK, Causey CP, Knuckley B, Cordova KN, et al. N-α-benzoyl-N5-(2-chloro-1-iminoethyl)-L-ornithine amide, a protein arginine deiminase inhibitor, reduces the severity of murine collagen-induced arthritis. J Immunol (2011) 186:4396–404. doi: 10.4049/jimmunol.1001620
175. Willis V, Banda N, Cordova K, Chandra P, Robinson W, Cooper D, et al. Protein arginine deiminase 4 inhibition is sufficient for the amelioration of collagen-induced arthritis. Clin Exp Immunol (2017) 188:263–74. doi: 10.1111/cei.12932
176. Kawalkowska J, Quirke A-M, Ghari F, Davis S, Subramanian V, Thompson PR, et al. Abrogation of collagen-induced arthritis by a peptidyl arginine deiminase inhibitor is associated with modulation of T cell-mediated immune responses. Sci Rep (2016) 6:1–12. doi: 10.1038/srep26430
177. Curran AM, Naik P, Giles JT, Darrah E. PAD enzymes in rheumatoid arthritis: pathogenic effectors and autoimmune targets. Nat Rev Rheumatol (2020) 16:301–15. doi: 10.1038/s41584-020-0409-1
178. Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med (2010) 207:1853–62. doi: 10.1084/jem.20100239
179. Thanabalasuriar A, Scott BNV, Peiseler M, Willson ME, Zeng Z, Warrener P, et al. Neutrophil extracellular traps confine pseudomonas aeruginosa ocular biofilms and restrict brain invasion. Cell Host Microbe (2019) 25:526–36. doi: 10.1016/j.chom.2019.02.007
180. Gupta AK, Giaglis S, Hasler P, Hahn S. Efficient neutrophil extracellular trap induction requires mobilization of both intracellular and extracellular calcium pools and is modulated by cyclosporine a. PloS One (2014) 9:e97088. doi: 10.1371/journal.pone.0097088
181. Keane J, Bresnihan B. Tuberculosis reactivation during immunosuppressive therapy in rheumatic diseases: diagnostic and therapeutic strategies. Curr Opin Rheumatol (2008) 20:443–9. doi: 10.1097/BOR.0b013e3283025ec2
182. Sengelov H. Complement receptors in neutrophils. Crit Rev Immunol (1995) 15(2):107–31. doi: 10.1615/CritRevImmunol.v15.i2.10
183. Jayne DR, Merkel PA, Schall TJ, Bekker P. Avacopan for the treatment of ANCA-associated vasculitis. New Engl J Med (2021) 384:599–609. doi: 10.1056/NEJMoa2023386
184. Xiao H, Dairaghi DJ, Powers JP, Ertl LS, Baumgart T, Wang Y, et al. C5a receptor (CD88) blockade protects against MPO-ANCA GN. J Am Soc Nephrol (2014) 25:225–31. doi: 10.1681/ASN.2013020143
185. Jayne DR, Bruchfeld AN, Harper L, Schaier M, Venning MC, Hamilton P, et al. Randomized trial of C5a receptor inhibitor avacopan in ANCA-associated vasculitis. J Am Soc Nephrol (2017) 28:2756–67. doi: 10.1681/ASN.2016111179
186. Merkel PA, Niles J, Jimenez R, Spiera RF, Rovin BH, Bomback A, et al. Adjunctive treatment with avacopan, an oral C5a receptor inhibitor, in patients with antineutrophil cytoplasmic antibody–associated vasculitis. ACR Open Rheumatol (2020) 2:662–71. doi: 10.1002/acr2.11185
187. Bekker P, Dairaghi D, Seitz L, Leleti M, Wang Y, Ertl L, et al. Characterization of pharmacologic and pharmacokinetic properties of CCX168, a potent and selective orally administered complement 5a receptor inhibitor, based on preclinical evaluation and randomized phase 1 clinical study. PLoS One (2016) 11:e0164646. doi: 10.1371/journal.pone.0164646
188. Davis WD, Brey RL. Antiphospholipid antibodies and complement activation in patients with cerebral ischemia. Clin Exp Rheumatol (1992) 10:455–60.
189. López-Benjume B, Rodríguez-Pintó I, Amigo MC, Erkan D, Shoenfeld Y, Cervera R, et al. Eculizumab use in catastrophic antiphospholipid syndrome (CAPS): Descriptive analysis from the "CAPS registry". Autoimmun Rev (2022) 21:103055. doi: 10.1016/j.autrev.2022.103055
190. Muldur S, Vadysirisack DD, Ragunathan S, Tang Y, Ricardo A, Sayegh CE, et al. Human neutrophils respond to complement activation and inhibition in microfluidic devices. Front Immunol (2021) 12:777932. doi: 10.3389/fimmu.2021.777932
191. Tay SH, Mak A. Anti-NR2A/B antibodies and other major molecular mechanisms in the pathogenesis of cognitive dysfunction in systemic lupus erythematosus. Int J Mol Sci (2015) 16:10281–300. doi: 10.3390/ijms160510281
Keywords: neutrophils, neuropsychiatric systemic lupus erythematosus, extracellular traps, bloodbrain barrier, systemic lupus erythematosus
Citation: Sim TM, Mak A and Tay SH (2022) Insights into the role of neutrophils in neuropsychiatric systemic lupus erythematosus: Current understanding and future directions. Front. Immunol. 13:957303. doi: 10.3389/fimmu.2022.957303
Received: 30 May 2022; Accepted: 19 July 2022;
Published: 09 August 2022.
Edited by:
Czeslawa Helena Kowal, Feinstein Institute for Medical Research, United StatesReviewed by:
Jessy J Alexander, University at Buffalo, United StatesCopyright © 2022 Sim, Mak and Tay. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sen Hee Tay, c2VuX2hlZV90YXlAbnVocy5lZHUuc2c=; Anselm Mak, bWRjYW1AbnVzLmVkdS5zZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.