 Huiru Zhang1,2,3,4†
Huiru Zhang1,2,3,4† Xianjun Yu
Xianjun Yu Weiding Wu
Weiding Wu- 1Department of Pancreatic Surgery, Shanghai Cancer Centre, Fudan University, Shanghai, China
- 2Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, China
- 3Shanghai Pancreatic Cancer Institute, Fudan University, Shanghai, China
- 4Pancreatic Cancer Institute, Fudan University, Shanghai, China
Pancreatic cancer has an exclusive inhibitory tumor microenvironment characterized by a dense mechanical barrier, profound infiltration of immunosuppressive cells, and a lack of penetration of effector T cells, which constitute an important cause for recurrence and metastasis, resistance to chemotherapy, and insensitivity to immunotherapy. Neoadjuvant therapy has been widely used in clinical practice due to its many benefits, including the ability to improve the R0 resection rate, eliminate tumor cell micrometastases, and identify highly malignant tumors that may not benefit from surgery. In this review, we summarize multiple aspects of the effect of neoadjuvant therapy on the immune microenvironment of pancreatic cancer, discuss possible mechanisms by which these changes occur, and generalize the theoretical basis of neoadjuvant chemoradiotherapy combined with immunotherapy, providing support for the development of more effective combination therapeutic strategies to induce potent immune responses to tumors.
1 Introduction
Pancreatic cancer, which is highly malignant and difficult to treat, is ranked fourth among cancer-related deaths in the United States and is anticipated to be the second highest contributor to cancer death by 2030 (1, 2). Surgery is the only potential cure for pancreatic cancer. However, due to the high primary irresectability rate and many postoperative complications, the 5-year survival rate is less than 30% in patients who undergo surgery (3). Adjuvant therapy increases survival benefits to some extent but may cause bone marrow suppression and immune deficiency (4). In addition, a proportion of patients fail to receive the designed adjuvant therapy because of postoperative complications, delayed surgical recovery, early disease recurrence, and other reasons (5, 6). Neoadjuvant therapy is considered to be a promising therapeutic strategy, which is mainly reflected in the following: a) it can transform the initial marginal or locally unresectable tumors into resectable tumors and increase the number of surgical candidates, b) it can improve the R0 resection rate and overall survival, c) it can eliminate circulating tumor cells and distant micrometastases as well as reduce postoperative recurrence, and d) it can identify patients with progressive disease unsuitable for surgical intervention (7–11). First-line neoadjuvant therapies include a combination chemotherapy regimens consisting of oxaliplatin, irinotecan, fluorouracil, and leucovorin (FOLFIRINOX) or gemcitabine combined with nab-paclitaxel (AG), and many clinical trials have reported their beneficial effects on pancreatic cancer (12–14).
Cancer development is the result of complex and dynamic interactions between tumor cells and the tumor microenvironment (TME). The TME is a highly complex system consisting of tumor cells, immune cells, stromal cells, and non-cellular components, including extracellular matrix and soluble products, such as chemokines and cytokines (15). Pancreatic cancer has an exclusive inhibitory TME characterized by a dense physical barrier and the profound infiltration of immunosuppressive cells (16). Malignant epithelial cells account for only approximately 20% of the tumor bulk, while the desmoplastic stroma formed by cancer-associated fibroblasts (CAFs), collagens, and hyaluronic acid accounts for approximately 80% of the tumor mass (17). In addition, a large number of myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), and regulatory T cells (Tregs), as well as limited effector T cells, may contribute to the immunosuppressive characteristics of the TME (18). These components constitute important causes of pancreatic cancer recurrence, metastasis, resistance to chemotherapy, and insensitivity to immunotherapy (16). Therefore, understanding how neoadjuvant therapy affects stromal components and the antitumor immune response will help to develop new combined immunotherapy strategies for treating this lethal cancer in the future.
This review concludes several aspects of neoadjuvant therapy-induced alterations in the pancreatic cancer microenvironment (Figure 1) and discusses the possible mechanisms of these changes, providing a basis for the development of more promising combination therapies with immunotherapy in a neoadjuvant setting. The methods for reference screening are summarized in Supplementary Figure 1.
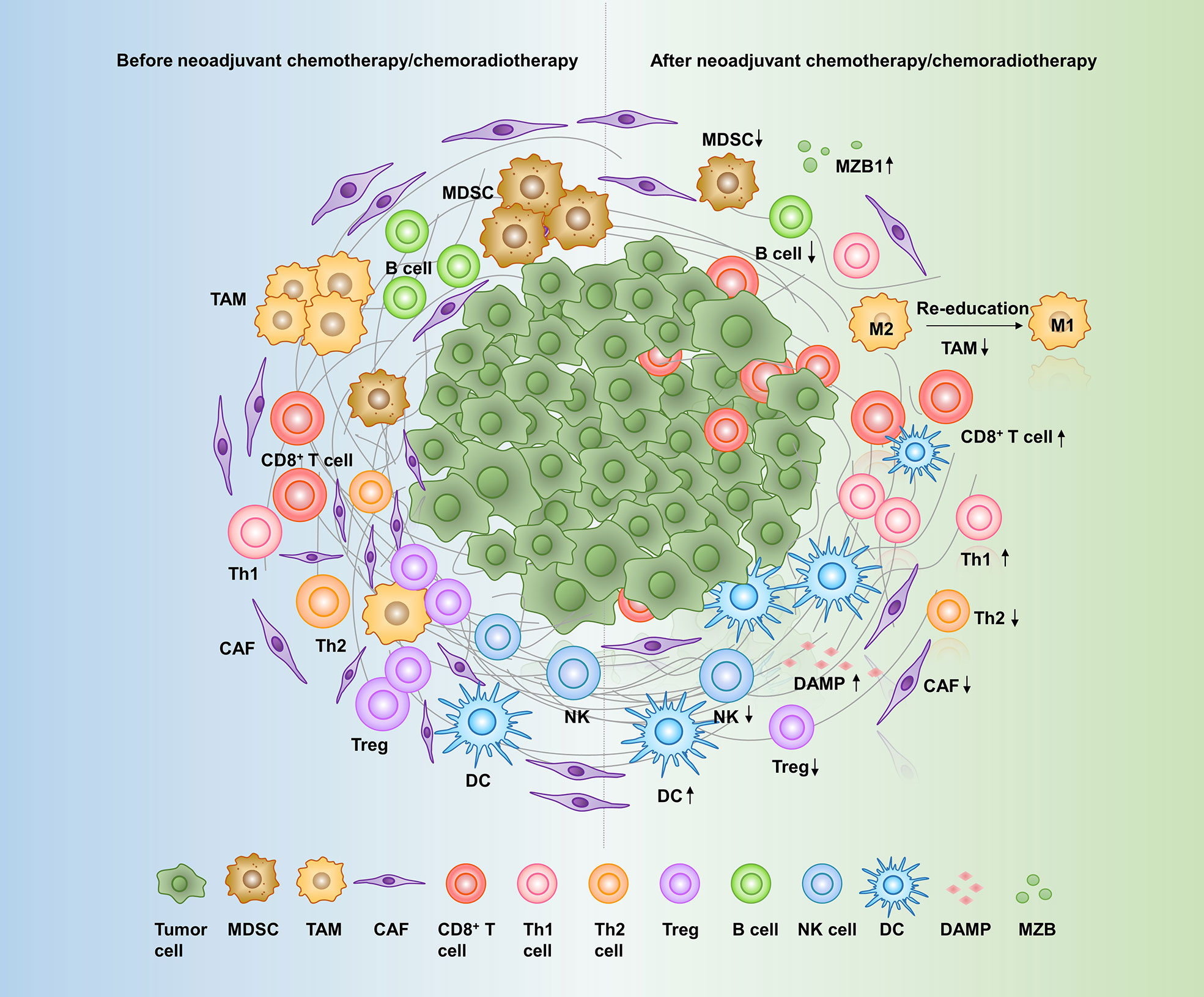
Figure 1 Effects of neoadjuvant therapy on the pancreatic cancer microenvironment. A variety of cells and molecules in the pancreatic cancer microenvironment are altered in response to neoadjuvant therapy: CD8+ T cells, Th1 cells, DCs, DAMPs, and MZB1 are increased, while CAFs, Th2 cells, Tregs, MDSCs, TAMs, NK cells, and B cells are decreased. MDSC, myeloid-derived suppressor cell; TAM, tumor-associated macrophage; CAF, cancer-associated fibroblast; Th cell, T helper cell; Treg, regulatory T cell; NK cell, natural killer cell; DC, dendritic cell; DAMPs, damage-associated molecular patterns; MZB1, Marginal zone B- and B1-cell-specific protein.
2 Effects of neoadjuvant therapy on the pancreatic tumor microenvironment
2.1 Cancer-associated fibroblasts and extracellular matrix
CAFs are the major cellular components of the pancreatic cancer stroma and can secrete cytokines, growth factors, and extracellular matrix (ECM) proteins that are involved in tumor growth and metastasis, ECM deposition, and immunosuppressive microenvironment formation (19). CAFs modify tumor immunity by polarizing the adaptive immune response toward a protumor phenotype and influencing innate immune cell recruitment and activation (20). Specifically, they favor the tumor-promoting function of CD4+ T helper 2 lymphocytes over the tumor-protective T helper 1 response (21) and directly reduce the activation of CD8+ cytotoxic T cells and natural killer (NK) cells (22). CAFs also promote M2 macrophage polarization and induce the differentiation of Tregs to form the protumor microenvironment (23, 24). Therefore, understanding the effects of neoadjuvant therapy on CAFs is important for understanding the immune microenvironment.
Many markers have been used to identify and characterize CAFs, including α-smooth muscle actin (αSMA), fibroblast activation protein, fibroblast-specific protein 1, and podoplanin (PDPN) (25). αSMA is considered to be a unique marker of activated CAFs compared with normal fibroblasts (26). A study analyzed CAFs and stromal activation in neoadjuvantly treated and resected primary tumors using αSMA. The results showed that neoadjuvant therapy (gemcitabine/gemcitabine + erlotinib/gemcitabine + oxaliplatin/FOLFIRINOX/other regimens (such as radiotherapy)) may induce stromal depletion and reduce the number of CAFs (27, 28). Interestingly, tumors treated with the AG regimen had less CAF and stromal contents than those treated with conventional preoperative chemotherapy (gemcitabine + S-1), and the main effects were due to nab-paclitaxel and not gemcitabine (28, 29).
PDPN is a small cell-surface mucin-like glycoprotein that plays an important role in fibroblasts, macrophages, T helper cells, and epithelial cells and is associated with poor prognosis in several cancers. It acts as an endogenous ligand for the platelet C-type lectin receptor CLEC-2, which can elicit powerful platelet aggregation (30, 31). Platelets are rich in several growth factors, such as transforming growth factor-β, vascular endothelial growth factor-A, and CD40 ligand, which cause epithelial–mesenchymal transition, immune paralysis, and drug resistance (32). The neoadjuvant AG regimen resulted in a significant reduction in PDPN+ CAF contents and a decrease in extravasated platelet activation through PDPN+ CAF depletion, thereby preventing aggressive tumor cells from spreading, invading, and developing resistance to drugs (33).
CAFs have previously been considered to have tumor-promoting functions. However, recent studies targeting the hedgehog pathway have suggested that αSMA+ CAFs may have tumor-restraining functions (34, 35), which may be due to more in-depth research on the heterogeneity level of CAFs. Elyada et al. (36) applied scRNA-seq to pancreatic cancer tissue and found three distinct CAF subtypes: myofibroblastic CAFs (myCAFs), inflammatory CAFs (iCAFs), and antigen-presenting CAFs (apCAFs). myCAFs have elevated expression of αSMA, are closely adjacent to tumor cells, and are associated with ECM deposition (35, 37, 38), immunosuppression (39, 40), and tumor restraint (34, 35). iCAFs lack increased αSMA expression and instead secrete interleukin-6 (IL-6) and are distant from tumor cells (41). They are involved in immunosuppression and tumor promotion (39, 41) and may have overlapping functions with senescent fibroblasts (37). apCAFs have high expression of MHC II family genes and may perform antigen-processing and antigen-presentation functions (42). Notably, most current studies on neoadjuvant chemotherapy/chemoradiotherapy and CAFs are considered to involve all CAFs rather than specific CAF subtypes, as αSMA is regarded as a universal marker of activated CAFs. Due to the complex functional characteristics of CAF subtypes in pancreatic cancer, we emphasize the need to determine the relationship between neoadjuvant chemoradiotherapy and specific CAF subtypes to better inform the use of matrix-targeted therapy following or in combination with neoadjuvant chemoradiotherapy.
CAFs produce various extracellular matrix components, including fibronectin, laminin, hyaluronan, and collagens, of which type I, IV, or V collagens promote the malignant features of pancreatic cancer (43–45). Collagen has been reported to increase tumor tissue stiffness, regulate tumor immunity, and promote metastasis (46). After neoadjuvant therapy (FOLFIRINOX/AG/gemcitabine/gemcitabine + S1/S1 + irinotecan + oxaliplatin/S1 + radiation(50 Gy)/gemcitabine + S1 + radiation (50 Gy)), the expression of type I, III, IV, and V collagens in pancreatic cancer tissue was decreased (47). The mechanism of this remodeling may be that these neoadjuvant regimens reduced the expression of ephrin-A5, the upstream regulatory gene of collagen synthesis in CAFs, which leads to significant alterations in the expression level of the collagen gene, thus inducing a reduction in collagen volume in the TME (47). The remodeling of CAFs and collagens by neoadjuvant chemotherapy/chemoradiotherapy is associated with tumor softening and tumor shrinkage, which may allow the surgical removal of pancreatic cancer, but this softening is often difficult to detect on radiologic imaging.
The effect of neoadjuvant chemoradiotherapy on the level of interstitial fibrosis is controversial. As mentioned above, some studies suggest that some neoadjuvant chemotherapy or chemoradiotherapy regimens can reduce interstitial fibrosis by reducing CAFs. However, several studies seemed to support the concept that neoadjuvant chemoradiotherapy increases pancreatic cancer stromal fibrosis (48, 49), which may be secondary to tissue damage caused by preoperative chemoradiotherapy and subsequent reconstruction and repair. Two cell-surface proteins, a disintegrin and metalloprotease 10 and ephrinB2, are considered drivers of fibrosis, ECM, and epithelial–mesenchymal transition after neoadjuvant stereotactic body radiation therapy (49). In addition, the urokinase plasminogen activator activates plasminogen to form plasminase, which is involved in tissue degradation and proteolysis (50). Neoadjuvant FOLFIRINOX reduced PLAU expression levels (51), which may reduce the degradation of ECM structural components, increase fibrosis, and reduce tumor invasion and metastasis. Matsuda et al. (52) evaluated the pathological tissues of surgically resected specimens from patients undergoing neoadjuvant AG therapy and found that encapsulating mature fibrosis after neoadjuvant AG chemotherapy was associated with improved prognosis in patients with pancreatic cancer. This mature fibrosis refers to fine collagen fibers stratified into multiple layers with few or no fibroblasts. It forms a tumor bed at the same site where cancer cells accumulate before neoadjuvant therapy. The presence of mature fibrosis in the peri-tumor area may suggest that neoadjuvant AG chemotherapy may maintain effects for a longer time, thereby improving prognosis.
2.2 Tumor-associated macrophages
TAMs, an important type of infiltrating immune cells in the TME, are primarily divided into the antitumor M1 phenotype (classically activated state) and the protumor M2 phenotype (alternatively activated state), reflecting the Th1–Th2 polarization of T cells. M1 macrophages can directly mediate cytotoxicity or kill tumor cells through antibody-dependent cell-mediated cytotoxicity; M2 macrophages can inhibit the T cell-mediated antitumor immune response, promote tumor angiogenesis, and lead to tumor progression and metastasis (53). High M2 macrophage infiltration is known to be an independent predictor of poor survival (54).
Neoadjuvant therapy (gemcitabine/gemcitabine + erlotinib/gemcitabine + oxaliplatin/FOLFIRINOX/FOLFIRINOX + radiotherapy/other regimens (such as radiotherapy)) has been reported to drive the depletion of MDSCs and M2 macrophages (27, 54). Gemcitabine, an important agent in neoadjuvant therapy regimens, is thought to lower M2 macrophage mediators, including growth factors, such as platelet growth factors A and B and placental growth factor, and matrix proteins, such as osteopontin and fibronectin (55). In addition, gemcitabine-treated macrophages modulated the polarization of TAMs into an antitumor phenotype by increasing the expression of several immunostimulatory cytokines, such as IL-12, IL-27, and IFN-γ, as well as proinflammatory mediators, including prostaglandin-endoperoxide synthase, Fas ligand, and TNF-α (55). M1 and M2 macrophages are highly plastic, and their classification is influenced by therapeutic intervention or changes in the TME. Neoadjuvant low-dose irradiation has been reported to program the differentiation of iNOS+ M1 macrophages by inducing endothelial cell activation and Th1 chemokine expression and by inhibiting the production of angiogenic factors, immunosuppressive factors, and tumor growth factors (56). Other studies have shown that during the early stage after irradiation, macrophages polarize into the M1 phenotype, but in later stages, resident and recruited macrophages tend to differentiate into the M2 phenotype following vascular disruption and tumor hypoxia (57). Therefore, the effects of neoadjuvant radiotherapy on TAMs remain controversial and need further study.
Matsuki et al. (58) found that the number of M2 macrophages was significantly lower in female patients than in male patients after neoadjuvant chemoradiotherapy (gemcitabine + S-1 + 30-Gy radiation), which may be related to increased sex-dependent interferon regulatory factor 5 (IRF-5) expression in female patients. IRF-5 is a transcription factor that has been shown to have tumor suppression functions and plays a central role in regulating TAM polarization (59, 60). IRF-5 is thought to promote M1 macrophage polarization by equipping the cells with an IL-12high IL-23high IL-10low cytokine profile and promoting Th1 and Th17 lymphocyte responses (61). Based on these findings, IRF-5 may be a key participant in the remodeling of the TME by neoadjuvant chemoradiotherapy, which warrants further study. In addition, the changes in immune cells after neoadjuvant chemoradiotherapy were related to their location in the TME. Okubo et al. (62) demonstrated that neoadjuvant chemoradiotherapy (S-1 + radiation) significantly reduced the M2 macrophage count in the nests of cancer cells, while little change was observed in the M2 macrophage count in the tumor stroma.
2.3 T cells
To evaluate the effect of neoadjuvant therapy on the immune microenvironment of pancreatic cancer, many researchers have examined resected specimens from patients who received neoadjuvant chemoradiotherapy and those who did not receive neoadjuvant therapy with immunohistochemical staining or flow cytometry. Homma et al. (63) found that the number of CD4+ and CD8+ lymphocytes in patients receiving neoadjuvant gemcitabine plus S-1 followed by radiation therapy was significantly higher than that in patients not receiving neoadjuvant therapy. Tsuchikawa et al. (64) found that neoadjuvant gemcitabine-based chemotherapy with or without chemoradiotherapy reduced the number of Foxp3+ Tregs but did not induce CD4+ or CD8+ lymphocyte infiltration. Shibuya and others suggested that the number of myeloid cells and Tregs in the pancreatic cancer microenvironment was significantly reduced after neoadjuvant chemotherapy (gemcitabine + docetaxel + capecitabine) followed by chemoradiotherapy. Although the number of CD8+ cells was also significantly decreased, the ratio of CD4+ and CD8+ cells to Tregs was significantly higher (65). Michelakos et al. (54) found that neoadjuvant FOLFIRINOX with or without chemoradiotherapy reduced human leukocyte antigen (HLA)-A defects, increased CD8+ cell density, and decreased Treg density. Mota Reyes et al. (27) found an effect of neoadjuvant therapy (gemcitabine/gemcitabine + erlotinib/gemcitabine + oxaliplatin/FOLFIRINOX/other regimens (such as radiotherapy)) on enriching antitumor immune cells in the TME, especially the selective deletion of Tregs and MDSCs, which is related to the increase in T-cell antitumor activity. Okubo and his colleagues showed that neoadjuvant S‐1 plus radiation therapy reduced the overall immune cell count, but these changes were heterogeneous in the cancer cell nests and cancer stroma. The number of CD4+ T cells, CD20+ B cells, and Foxp3+ T cells in the cancer stroma was significantly reduced in patients who received neoadjuvant S‐1 plus radiation therapy compared to those who did not, whereas the count of these cells in the cancer cell nests did not significantly differ between groups (62).
The effect of neoadjuvant chemoradiotherapy on T cells is complex and important. The depletion of Tregs and the increase in the CD8+ T/Treg ratio by the aforementioned neoadjuvant regimen would improve the survival benefit. Low Treg frequency, high CD8+ T-cell frequency, high CD4+ T-cell frequency, and high CD8+ T/Treg ratio can be used as predictors for pancreatic cancer patients treated with neoadjuvant chemotherapy (66, 67). Targeting the TME to increase CD4+ and CD8+ T-cell counts and the CD8+/FOXP3+ cell ratio could decrease tumor recurrence and improve clinical outcomes (68). In addition, the effects of neoadjuvant chemotherapy on CD4+ and CD8+ T cells differed among these studies, which may be related to drug types, treatment cycles, detection methods, and heterogeneity of the patient population. Notably, T-cell subtypes have different functional characteristics, and thus, more unique markers need to be tested to further determine the effect of neoadjuvant chemotherapy on different T-cell subsets. In summary, clinical responders to neoadjuvant therapy may present an immunologic landscape of increased antitumor T lymphocytes and decreased Tregs, the mechanisms of which are discussed below.
2.3.1 Neoadjuvant therapy activates T-cell effector molecules
Neoadjuvant FOLFIRINOX has been found to increase the expression of CD44 (a T-cell activation marker) and granzyme B (a protease related to the killing function of cytotoxic T cells), reflecting increased CD8+ T-cell activation (69). Patients who received the neoadjuvant AG and FOLFIRINOX regimens exhibited a strong tendency to show decreased expression of programmed death-ligand 1 (PD-L1) and CD47 on tumor cells, which restored the attack of T cells and macrophages on tumors, respectively (70). After neoadjuvant stereotactic body radiation and IL-12 combination therapy, intratumoral interferon-γ production increased, which initiated inhibitory cell reprogramming and subsequently increased CD8+ T-cell activation (71). Furthermore, gemcitabine enhanced the killing function of cytotoxic T lymphocytes by upregulating Fas expression on pancreatic cancer cells (72). Paclitaxel not only directly activated effector T cells by raising the production of the Th1 cytokines interferon-γ and IL-2 and by increasing the expression of the activator marker CD44 in CD4+ and CD8+ T cells but also indirectly reduced the inhibitory function of Tregs by upregulating CD95 (73, 74). Delitto et al. (75) found higher levels of IL-2 in fresh pancreatic cancer surgical specimens after neoadjuvant gemcitabine-based chemotherapy, which was associated with T-cell proliferation and activation.
2.3.2 Neoadjuvant therapy regulates T-cell infiltration
High interstitial pressure and vascular collapse of the pancreatic TME limit T-cell entry to the tumor sites (76). Abnormal vasculature reduces oxygen supply, resulting in hypoxia in the TME. Hypoxia promotes CAF recruitment, connective tissue hyperplasia and activation (77), and transforming growth factor-β expression in CAFs, resulting in the exclusion of CD8+ T cells from the tumor parenchyma to hinder antitumor immunity following immunotherapy (78, 79). Hypoxia also facilitates the recruitment of MDSCs and inhibits the activity of T lymphocytes (80). The reduction in CAFs and interstitial activation induced by the above neoadjuvant regimens may facilitate T-cell infiltration into the tumor sites. Neoadjuvant local low-dose γ irradiation normalizes the aberrant vasculature and induces iNOS expression in TAMs, thereby enhancing tumor-specific T-cell recruitment and T cell-mediated tumor rejection in pancreatic cancer (56). Chemotherapy has been reported to induce intratumoral chemokine expression, favoring T-cell infiltration (81). Because T-cell infiltration in pancreatic cancer is directly correlated with a distinct panel of four chemokines, CCL4, CCL5, CXCL9, and CXCL10 (82), we hypothesize that neoadjuvant therapy increases T-cell infiltration by influencing these key chemokines, which needs to be confirmed by more basic experiments.
2.3.3 Neoadjuvant therapy promotes antigen presentation
Dying cancer cells release damage-associated molecular patterns (DAMPs), such as calreticulin, heat shock protein (HSP), and high mobility group box protein 1, into the TME, which promote the recruitment and activation of antigen-presenting cells (APCs) by acting on different pattern recognition receptors (83, 84). Mature APCs can phagocytize antigenic material, migrate to lymph nodes, present antigens on MHC molecules to T cells, and trigger a T-cell immune response (85). Considered “eat me signals”, Hsp70 and calreticulin can interact with a variety of APC surface receptors, such as CD91 and CD40, to promote the cross-presentation of antigens on MHC class I molecules, thus triggering a tumor-specific CD8+ T-cell response (86–88). MHC class I-related chains A and B (MICA/B), as a component of DAMPs, enhance the cytolytic responses of NK cells, γδ T cells, and CD8+ αβ T cells by binding to natural killer group 2 member D (89, 90).
Neoadjuvant chemoradiotherapy (gemcitabine + S-1 followed by radiation therapy (30 Gy)) has been reported to induce the overexpression of DAMPs (such as MICA/B, calreticulin, and Hsp70), increase the number of CD8+ and CD4+ tumor-infiltrating T lymphocytes, and decrease the ratio of Treg/tumor-infiltrating T lymphocytes (90). This finding implies that this neoadjuvant chemoradiotherapy may stimulate T-cell immune responses by promoting antigen presentation through DAMPs (such as calreticulin and Hsp70) or directly enhance the cytolytic responses of CD8+ αβ T cells, NK cells, and γδ T cells by inducing the overexpression of MICA/B. In addition, radiation can also enhance antigen processing and calreticulin exposure, resulting in enhanced T-cell killing (91).
2.3.4 Neoadjuvant therapy depletes tumorigenic immune cells and induces neoantigen release
Neoadjuvant chemotherapy (such as FOLFIRINOX and some gemcitabine-based regimens) aims to increase the accumulation of tumor-specific T cells in the peritumoral niche by selectively depleting protumorigenic immune cells (27, 65). Treg ablation restores immunogenic tumor-associated CD11c+ dendritic cells (DCs) and activates CD8+ T cells (92). MDSC depletion may also be a mechanism of neoadjuvant chemotherapy-induced increase in T cells because MDSCs can control T-cell responses by inducible nitric oxide synthase and arginase 1 (93).
Mutation-associated neoantigens have been shown to be the target of powerful antitumor T-cell responses. Due to the presence of these neoantigens, patients can mount endogenous immune responses, and the antigen-specific T cells are expanded (94). Neoadjuvant chemoradiotherapy induces new mutations in rectal cancer, resulting in the generation of neoantigens and changing the immune function of patients (95). Radiotherapy is thought to increase the expression of genes encoding immunogenic mutated proteins that are presented by MHC-I and MHC-II molecules, thereby triggering CD8+ and CD4+ T-cell responses (96). Therefore, we predict that the T-cell response promoted by neoadjuvant chemoradiotherapy may be related to the expression of strong neoantigens in pancreatic cancer.
2.4 B cells and T follicular helper cells
B lymphocytes play a dual role in tumor immunity. As a promotive effect, B cells produce antibodies to participate in humoral immunity and activate T cells to participate in cellular immunity. Alternatively, IL-10 from B cells inhibits cytotoxic T-cell activation to play a critical role in immunosuppression (97). The number and function of CD19+CD20+ B cells decreased in response to neoadjuvant therapy (gemcitabine/gemcitabine + erlotinib/gemcitabine + oxaliplatin/FOLFIRINOX/S-1/other regimens (such as radiotherapy)) (27, 69, 98). Notably, MZB1 (marginal zone B- and B1-cell-specific protein) was detected in the tumor stroma after neoadjuvant chemoradiotherapy and was positively correlated with elevated accumulation of CD8+ T lymphocytes (99). MZB1 upregulated tumor-specific antibody secretion and decreased IL-10 expression by regulating the storage of calcium ions in the endoplasmic reticulum, which activated antitumor immunity (99). According to our previous study, T follicular helper cells (Tfhs) recruited CD8+ T cells and B cells by secreting CXCL13 and promoted the maturation of B cells into antibody-producing plasma cells by secreting IL-21, thereby promoting the formation of an immunostimulatory TME (100). The function of Tfhs was inhibited by the PD-L1/programmed cell death protein 1 (PD-1) signaling pathway in pancreatic cancer, which was reversed after neoadjuvant AG chemotherapy (100).
2.5 Other immune cells and tertiary lymphoid organs
DCs, considered the most effective APCs in tumor immunity, have exhibited marked expansion after neoadjuvant therapy (gemcitabine/gemcitabine + erlotinib/gemcitabine + oxaliplatin/FOLFIRINOX/other regimens (such as radiotherapy)) (27). As mentioned earlier, neoadjuvant chemoradiotherapy (gemcitabine + S-1 followed by radiation therapy (30 Gy)) triggered the release of immunostimulatory DAMPs, which activated DCs through toll-like receptor 4 (101). Gemcitabine-based neoadjuvant therapy was found to increase FMS-like tyrosine kinase 3 ligand (Flt3L) concentrations (75). Flt3L is a growth factor, and the main effect of the Flt3/Flt3L axis is to regulate DC generation, development, maturation, expansion, and homeostasis (102, 103). Flt3L might synergize successfully with other therapies that promote tumor antigen availability by increasing the number of DCs.
Intratumoral NK-cell infiltration constitutes an independent prognostic factor for pancreatic cancer (27). After neoadjuvant therapy (gemcitabine/gemcitabine + erlotinib/gemcitabine + oxaliplatin/FOLFIRINOX/S-1/other regimens (such as radiotherapy)), the number of CD56+ NK cells was significantly reduced (27), and their functions were notably decreased (98). The mechanism of the alterations in NK cells after neoadjuvant therapy is unclear. However, studies have shown that neoadjuvant chemoradiotherapy (gemcitabine + S-1 followed by radiation therapy (30 Gy)) induces the overexpression of MHC class I-related chain A/B (90), a membrane protein expressed in cancer cells that is induced by irradiation or chemotherapy, and interacts with natural killer group 2 member D to enhance the killing effect of NK cells on cancer cells (89, 104, 105).
Neoadjuvant FOLFIRINOX therapy prevents tumor-associated neutrophil (TAN) accumulation by affecting the CXCL5–CXCR2 axis and decreasing intratumoral IL-8 levels (51). CXCL5 is a chemokine for neutrophils, and CXCR2 is essential for the recruitment of TANs (106). Similarly, IL-8 can elicit massive neutrophil accumulation and activation (107). In the absence of neutrophils, activated and functional T cells infiltrate pancreatic tumors otherwise devoid of effector T cells (108). However, few reports have examined the effects of neoadjuvant therapy on eosinophils, basophils, or mast cells.
Tertiary lymphoid organs (TLOs) are accumulations of lymphoid cells that recognize antigens and produce antigen-specific antibodies in the immune system (109). The proportion of CD8+ T lymphocytes, PNAd+ high endothelial venules, CD163+ macrophages, and Ki‐67+ cells within the TLO was higher after neoadjuvant chemotherapy (gemcitabine/S-1/gemcitabine + S-1/FOLFIRINOX) with/without radiation, while the proportion of PD-1+ immunosuppressive lymphocytes within the TLO was lower (110). By inducing T-cell infiltration and the development of TLO in the TME, “nonimmunogenic” tumors can be transformed into “immunogenic” tumors (111).
3 More promising treatment strategies
In summary, neoadjuvant therapy alters the pancreatic cancer microenvironment by reducing CAFs and interstitial fibrosis; decreasing the infiltration of immunosuppressive cells, including M2 macrophages, MDSCs, TANs, and Tregs; and increasing cytotoxic T-cell recruitment. This change creates favorable conditions for immunotherapy. We list some clinical trials of neoadjuvant chemotherapy or chemoradiotherapy combined with immunotherapy (Table 1). We speculate that neoadjuvant chemoradiotherapy combined with immunotherapy is promising, and this strategy is discussed below (Figure 2). Notably, the efficacy of neoadjuvant combination strategies needs to be tested in more clinical trials.

Table 1 Clinical trials of neoadjuvant chemotherapy or chemoradiotherapy combined with immunotherapy.
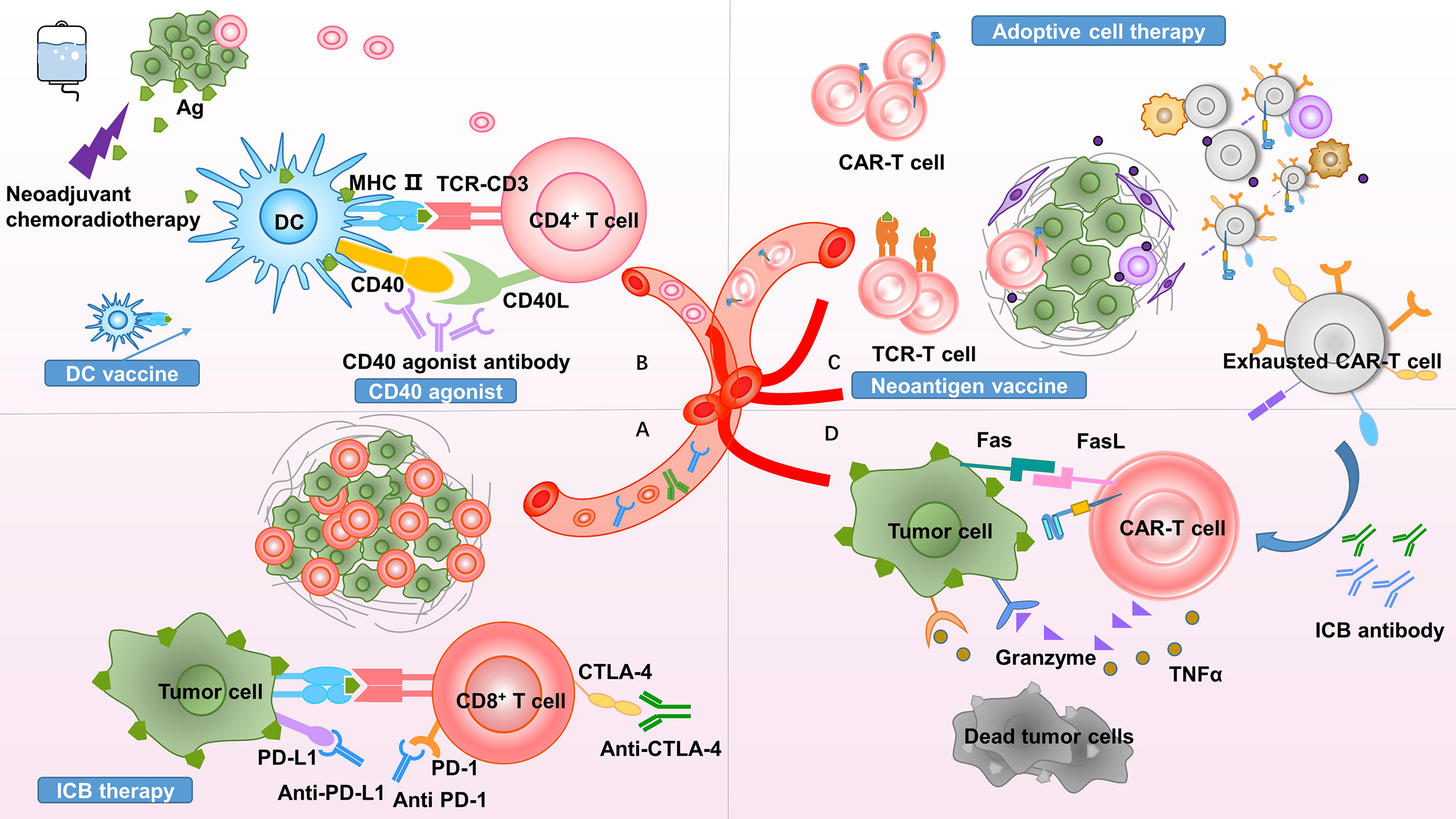
Figure 2 Theoretical basis of neoadjuvant combination therapy. Proposed rationales to clarify the promise of neoadjuvant combination therapy. (A) Neoadjuvant therapy increases immunotherapy targets by promoting effector T-cell infiltration and increasing ICB antibody delivery. (B) Due to the presence of the tumor burden serving as an antigen source, neoadjuvant therapy promotes antigen presentation and improves responses to CD40 agonists. (C) Neoadjuvant therapy increases the infiltration of CAR-T cells and TCR-T cells into the tumor site. The inhibitory TME exhausts T cells, upregulating inhibitory molecular receptors, such as PD-1, TIM-3, and LAG-3. (D) ICB reverses T-cell exhaustion and restores antitumor immunity. PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1; CTLA-4, cytotoxic T-lymphocyte-associated antigen 4; MHC, major histocompatibility complex; Ag, antigen; DC, dendritic cell; TCR, T-cell receptor; CAR, chimeric antigen receptor; TIM-3, T-cell immunoglobulin and mucin-domain containing-3; LAG-3, lymphocyte-activation gene 3; ICB, immune checkpoint blockade; TNF, tumor necrosis factor; TME, tumor microenvironment.
3.1 Immune checkpoint blockade therapy
Currently known immunotherapy targets focus on cytotoxic T cells, including PD-1, cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), T-cell immunoglobulin and mucin-domain containing-3 (TIM-3), lymphocyte-activation gene 3 (LAG-3), and T-cell immunoglobulin and ITIM domain (TIGIT). The premise of effective immunotherapy is efficient T-cell infiltration, and the level of T-cell infiltration at baseline is of great significance for the prediction and prognosis of immune checkpoint blockade (ICB) (112, 113). Although activated T cells are observed on the periphery of tumors, they will not respond to ICB if they cannot infiltrate the tumor itself (114). Neoadjuvant chemoradiotherapy may increase T-cell infiltration into the TME by increasing tumor antigen release and normalizing vasculature, providing targets for ICB. Chemotherapy combined with immunotherapy is predicted to reinforce and synergistically increase the antitumor effects of either therapy alone (115). A phase Ib study of pembrolizumab, an anti-PD-1 antibody, combined with chemotherapy enrolled 49 patients with advanced metastatic solid tumors, including 11 pancreatic cancer patients. The results showed that the clinical response rate was approximately 92%, and standard doses of pembrolizumab could be safe in combination with gemcitabine and nab-paclitaxel (116). Radiotherapy also provides targets for immunotherapy by regulating the expression of vascular endothelial cell adhesion molecules and chemokines to enhance lymphocyte infiltration (117). Many clinical trials concerning ICB therapy combined with neoadjuvant therapy in pancreatic cancer are recruiting or are about to begin recruiting patients (NCT02305186 and NCT05132504), and their results are to be expected. In addition, the antistromal effect of neoadjuvant chemoradiotherapy may increase ICB antibody delivery, enabling ICB-resistant patients to respond again.
3.2 CD40 agonist therapy
CD40 is a member of the tumor necrosis factor receptor superfamily expressed on the surface of various immune cells, including APCs, and in some tumor cells (118). CD40 agonists enable DCs to increase the levels of critical T-cell stimulatory cytokines, including IL-12, which potentiates T-cell activation (119). The addition of chemotherapy prior to administering agonist CD40 monoclonal antibody (mAb) has been shown to prime APCs by releasing tumor-associated antigens through cytotoxic cell death, improving responses to CD40 agonists (120). To test this hypothesis, a phase I trial combining an agonist CD40 mAb (CP-870,893) with gemcitabine was conducted on patients with advanced pancreatic cancer (121). The combination treatment was well tolerated and associated with objective tumor responses in 22 (19%) patients (121). In another phase 1b study, the CD40 agonistic mAb APX005M (sotigalimab) was combined with gemcitabine and nab-paclitaxel, with or without nivolumab, in patients with metastatic pancreatic ductal adenocarcinoma (122). Combination therapy achieved a 58% response rate among 24 patients (122).
Moreover, CD40 mAb synergizes with chemotherapy and radiotherapy to sensitize tumors that had previously resisted treatment with anti-CTLA-4 or anti-PD-1/PD-L1 mAb, further stimulating tumor regression and improving survival (123, 124). Byrne et al. (125) conducted a phase 1 trial and demonstrated that the neoadjuvant agonist CD40 mAb (selicrelumab) with or without chemotherapy can modulate the TME in pancreatic cancer, including reducing tumor stromal density, activating DCs, re-educating macrophages, and increasing T-cell infiltration. In addition, they found that the expression of LAG-3, PD-1, and TIGIT was significantly increased on CD4+ and CD8+ T cells in the TME, implying that combined ICB therapy may provide greater clinical synergistic benefit.
3.3 Adoptive cell therapy
Adoptive cell therapy is a type of immunotherapy for eliminating cancer by directly delivering in vitro activated and expanded tumor-specific or non-specific killer cells to patients. In addition to the well-known T cells (126), γδT cells (127), NK cells (128), NKT cells (129), and even macrophages (130) are engineered to express antigen-specific T-cell receptors or chimeric antigen receptors (CARs) (131). To date, CAR-T-cell therapy has achieved success mainly in hematological malignancies. Despite extensive research, CAR-T-cell therapy has not proven effective in solid tumors (132). The trafficking and infiltration of CAR-T cells into tumor cells limit the effectiveness of CAR-T-cell therapy (133). As mentioned above, the number of T cells in the TME of some patients increased after neoadjuvant chemotherapy, and these increased T cells may come from tissue-resident T cells or may be recruited from the bone marrow or periphery, which reflects that the TME after neoadjuvant therapy may be conducive to the existence of T cells. In addition, the reduction in CAFs after neoadjuvant therapy may break down the dense mechanical barrier of pancreatic cancer and promote T-cell infiltration into the tumor site, which may improve the function of CAR T cells. However, once CAR-T cells enter the tumor, sustained antigen exposure, suppressive immune cells, and suppressive cytokines in the TME cause the function of T cells to gradually degenerate (134). This condition, known as “exhaustion”, is characterized by attenuated effector cytotoxicity, reduced cytokine production, and increased expression of multiple inhibitory molecular receptors, such as PD-1, TIM-3, and LAG-3 (135). Exhausted CAR-T cells with impaired proliferation and persistence cannot efficiently kill malignant clones, usually leading to treatment failure (133). The reduction in MDSCs and Tregs after neoadjuvant therapy attenuates the immunosuppressive state of the TME, creating an improved TME to alleviate CAR-T exhaustion. In addition, targeting immune checkpoint pathways (such as PD-1 and CTLA-4) can relieve the exhaustion of CD8+ T cells and renew their priming (136, 137). Combining CAR-T-cell therapy with ICB or depleting other suppressive factors in the TME has shown very promising results for mitigating the phenomenon of T-cell exhaustion (132).
3.4 Dendritic cell therapy
DCs enhance the immune response by presenting antigens to T cells. Based on their powerful antigen presentation capability, DCs are designed as vaccines loaded with tumor antigens to promote antigen-specific antitumor T-cell immunity. Recent studies suggest that DC vaccines in combination with chemotherapy or ICB or CD40 agonists may be a promising therapeutic approach. Reducing tumors by neoadjuvant chemotherapy is considered to improve DC therapy because DC therapy is most effective in the case of low tumor burden (138). In addition, the depletion of suppressive immune cells, including Tregs and MDSCs, also facilitates the synergistic effect of neoadjuvant chemoradiotherapy combined with DC therapy. Promoting tumor-specific immunity with DC vaccines and then amplifying it with ICB is an attractive direction. Clinical evidence suggests that the combination of DC therapy and ICB is more effective than using DC therapy as monotherapy (139, 140). The blockade of PD-L1 on DCs promotes the activation of T cells by DCs, downregulates IL-10 secretion by Th2 cells, and upregulates IL-2 secretion by Th1 cells (141). Lau et al. (142) investigated the efficacy of a DC vaccine in combination with CD40 agonism in a mouse model of pancreatic cancer with poor immunogenicity. The results showed that although CD40 agonist monotherapy ameliorated intratumoral T-cell infiltration, these cells showed marked exhaustion. CD40 agonists combined with DC vaccines can improve T-cell exhaustion and reduce tumor progression.
3.5 Neoantigen vaccination
Neoantigens are somatic mutations expressed only by tumor cells (143). Because of their specific expression in tumors, neoantigens can only destroy tumor cells without causing excessive damage to important healthy tissues (144). Therefore, neoantigens are ideal targets for cancer immunotherapy. DCs loaded with neoantigens have been reported to trigger specific T-cell responses in melanoma patients (145). Another approach to target neoantigens is to transfer engineered T cells expressing the tumor-specific T-cell receptor into the host. In a recent study, a patient with progressive metastatic pancreatic cancer was treated with autologous T cells that had been genetically engineered to clonally express two allogeneic HLA-C*08:02-restricted T-cell receptors (TCRs) targeting mutant KRAS G12D expressed by the tumors. Visceral metastases regressed in this patient, and the response was ongoing at 6 months (146). Since most neoantigens are derived from patient-specific mutations, neoantigen T-cell receptor gene therapy is highly personalized (147). In conclusion, we believe that cancer immunotherapy targeting neoantigens is a promising treatment option, but methods to reduce the cost of this highly personalized treatment warrant more research.
4 Conclusions
The beneficial effects of neoadjuvant chemotherapy do not depend on the direct cytotoxicity of chemotherapeutic agents, as traditionally thought. Instead, these effects may be mediated by the restoration of antitumor responses via alterations in the phenotype of the immune microenvironment. These alterations are caused by remodeling antigen exposure and the distribution of immune cells in the TME. This review summarizes the changes and possible mechanisms of the pancreatic cancer immune microenvironment in response to neoadjuvant therapy. We found that many neoadjuvant regimens (such as AG or FOLFIRINOX) led to an immunological shift toward a more effective antitumor immune response in the TME, providing an impetus for the possibility of combining neoadjuvant chemotherapy or chemoradiotherapy with immunotherapy and highlighting the urgent need for combined neoadjuvant therapeutic strategies. Theoretically, the impact of neoadjuvant therapy on the immune microenvironment is heterogeneous in the population. Some patient groups will follow the direction of promoting inflammation and anticancer activity, while others will not. The selection of subsets of patients who can benefit from neoadjuvant therapy remains a problem to be solved. We suggest that the short time between cancer diagnostic biopsy and primary surgery can be used to screen for neoadjuvant therapies that induce a response-associated TME. Current studies have not been able to include the changes of all components in the TME, and we look forward to more discoveries in this area to develop more effective treatments to eradicate the disease in the future.
Author contributions
All authors contributed to the article and approved the submitted version.
Funding
This study was jointly supported by the National Natural Science Foundation of China (U21A20374, 82173091, and 81701630), Shanghai Municipal Science and Technology Major Project (21JC1401500), Scientific Innovation Project of Shanghai Education Committee (2019-01-07-00-07-E00057), Clinical Research Plan of Shanghai Hospital Development Center (SHDC2020CR1006A), Xuhui District Artificial Intelligence Medical Hospital Cooperation Project (2021-011), Shanghai Natural Science Foundation (22ZR1412900), Research Project of Shanghai Municipal Health Commission (20214Y0396, 20194Y0375), and Shanghai Pujiang Program (2021PJD014). The funding agencies had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.956984/full#supplementary-material
Supplementary Figure 1 | The flowchart of searching strategies.
References
1. Islami F, Ward EM, Sung H, Cronin KA, Tangka FKL, Sherman RL, et al. Annual report to the nation on the status of cancer, part 1: National cancer statistics. J Natl Cancer Inst (2021) 113(12):1648–99. doi: 10.1093/jnci/djab131
2. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the united states. Cancer Res (2014) 74(11):2913–21. doi: 10.1158/0008-5472.CAN-14-0155
3. Strobel O, Neoptolemos J, Jager D, Buchler MW. Optimizing the outcomes of pancreatic cancer surgery. Nat Rev Clin Oncol (2019) 16(1):11–26. doi: 10.1038/s41571-018-0112-1
4. Barreto JN, McCullough KB, Ice LL, Smith JA. Antineoplastic agents and the associated myelosuppressive effects: A review. J Pharm Pract (2014) 27(5):440–6. doi: 10.1177/0897190014546108
5. Mayo SC, Gilson MM, Herman JM, Cameron JL, Nathan H, Edil BH, et al. Management of patients with pancreatic adenocarcinoma: National trends in patient selection, operative management, and use of adjuvant therapy. J Am Coll Surg (2012) 214(1):33–45. doi: 10.1016/j.jamcollsurg.2011.09.022
6. Merkow RP, Bilimoria KY, Tomlinson JS, Paruch JL, Fleming JB, Talamonti MS, et al. Postoperative complications reduce adjuvant chemotherapy use in resectable pancreatic cancer. Ann Surg (2014) 260(2):372–7. doi: 10.1097/SLA.0000000000000378
7. Schorn S, Demir IE, Reyes CM, Saricaoglu C, Samm N, Schirren R, et al. The impact of neoadjuvant therapy on the histopathological features of pancreatic ductal adenocarcinoma - a systematic review and meta-analysis. Cancer Treat Rev (2017) 55:96–106. doi: 10.1016/j.ctrv.2017.03.003
8. Ishikawa O, Ohigashi H, Imaoka S, Sasaki Y, Iwanaga T, Matayoshi Y, et al. Is the long-term survival rate improved by preoperative irradiation prior to whipple’s procedure for adenocarcinoma of the pancreatic head? Arch Surg (1994) 129(10):1075–80. doi: 10.1001/archsurg.1994.01420340089017
9. Roland CL, Yang AD, Katz MH, Chatterjee D, Wang H, Lin H, et al. Neoadjuvant therapy is associated with a reduced lymph node ratio in patients with potentially resectable pancreatic cancer. Ann Surg Oncol (2015) 22(4):1168–75. doi: 10.1245/s10434-014-4192-6
10. Youngwirth LM, Nussbaum DP, Thomas S, Adam MA, Blazer DG 3rd, Roman SA, et al. Nationwide trends and outcomes associated with neoadjuvant therapy in pancreatic cancer: An analysis of 18 243 patients. J Surg Oncol (2017) 116(2):127–32. doi: 10.1002/jso.24630
11. Wysocka O, Kulbacka J, Saczko J. Adjuvant, neoadjuvant, and experimental regimens in overcoming pancreatic ductal adenocarcinoma. Prz Gastroenterol (2016) 11(3):155–62. doi: 10.5114/pg.2016.61438
12. Oba A, Ho F, Bao QR, Al-Musawi MH, Schulick RD, Del Chiaro M. Neoadjuvant treatment in pancreatic cancer. Front Oncol (2020) 10:245. doi: 10.3389/fonc.2020.00245
13. Jang JY, Han Y, Lee H, Kim SW, Kwon W, Lee KH, et al. Oncological benefits of neoadjuvant chemoradiation with gemcitabine versus upfront surgery in patients with borderline resectable pancreatic cancer: A prospective, randomized, open-label, multicenter phase 2/3 trial. Ann Surg (2018) 268(2):215–22. doi: 10.1097/SLA.0000000000002705
14. He J, Schulick RD, Del Chiaro M. Landmark series: Neoadjuvant treatment in borderline resectable pancreatic cancer. Ann Surg Oncol (2021) 28(3):1514–20. doi: 10.1245/s10434-020-09535-x
15. Giraldo NA, Sanchez-Salas R, Peske JD, Vano Y, Becht E, Petitprez F, et al. The clinical role of the tme in solid cancer. Br J Cancer (2019) 120(1):45–53. doi: 10.1038/s41416-018-0327-z
16. Ren B, Cui M, Yang G, Wang H, Feng M, You L, et al. Tumor microenvironment participates in metastasis of pancreatic cancer. Mol Cancer (2018) 17(1):108. doi: 10.1186/s12943-018-0858-1
17. Lin HJ, Lin J. Seed-in-Soil: Pancreatic cancer influenced by tumor microenvironment. Cancers (Basel) (2017) 9(7):93. doi: 10.3390/cancers9070093
18. Ho WJ, Jaffee EM, Zheng L. The tumour microenvironment in pancreatic cancer - clinical challenges and opportunities. Nat Rev Clin Oncol (2020) 17(9):527–40. doi: 10.1038/s41571-020-0363-5
19. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer (2016) 16(9):582–98. doi: 10.1038/nrc.2016.73
20. Piersma B, Hayward MK, Weaver VM. Fibrosis and cancer: A strained relationship. Biochim Biophys Acta Rev Cancer (2020) 1873(2):188356. doi: 10.1016/j.bbcan.2020.188356
21. De Monte L, Reni M, Tassi E, Clavenna D, Papa I, Recalde H, et al. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J Exp Med (2011) 208(3):469–78. doi: 10.1084/jem.20101876
22. Nazareth MR, Broderick L, Simpson-Abelson MR, Kelleher RJ Jr., Yokota SJ, Bankert RB. Characterization of human lung tumor-associated fibroblasts and their ability to modulate the activation of tumor-associated T cells. J Immunol (2007) 178(9):5552–62. doi: 10.4049/jimmunol.178.9.5552
23. Zhang A, Qian Y, Ye Z, Chen H, Xie H, Zhou L, et al. Cancer-associated fibroblasts promote M2 polarization of macrophages in pancreatic ductal adenocarcinoma. Cancer Med (2017) 6(2):463–70. doi: 10.1002/cam4.993
24. Kinoshita T, Ishii G, Hiraoka N, Hirayama S, Yamauchi C, Aokage K, et al. Forkhead box P3 regulatory T cells coexisting with cancer associated fibroblasts are correlated with a poor outcome in lung adenocarcinoma. Cancer Sci (2013) 104(4):409–15. doi: 10.1111/cas.12099
25. von Ahrens D, Bhagat TD, Nagrath D, Maitra A, Verma A. The role of stromal cancer-associated fibroblasts in pancreatic cancer. J Hematol Oncol (2017) 10(1):76. doi: 10.1186/s13045-017-0448-5
26. Kuzet SE, Gaggioli C. Fibroblast activation in cancer: When seed fertilizes soil. Cell Tissue Res (2016) 365(3):607–19. doi: 10.1007/s00441-016-2467-x
27. Mota Reyes C, Teller S, Muckenhuber A, Konukiewitz B, Safak O, Weichert W, et al. Neoadjuvant therapy remodels the pancreatic cancer microenvironment Via depletion of protumorigenic immune cells. Clin Cancer Res (2020) 26(1):220–31. doi: 10.1158/1078-0432.CCR-19-1864
28. Miyashita T, Tajima H, Makino I, Okazaki M, Yamaguchi T, Ohbatake Y, et al. Neoadjuvant chemotherapy with gemcitabine plus nab-paclitaxel reduces the number of cancer-associated fibroblasts through depletion of pancreatic stroma. Anticancer Res (2018) 38(1):337–43. doi: 10.21873/anticanres.12227
29. Alvarez R, Musteanu M, Garcia-Garcia E, Lopez-Casas PP, Megias D, Guerra C, et al. Stromal disrupting effects of nab-paclitaxel in pancreatic cancer. Br J Cancer (2013) 109(4):926–33. doi: 10.1038/bjc.2013.415
30. Pula B, Witkiewicz W, Dziegiel P, Podhorska-Okolow M. Significance of podoplanin expression in cancer-associated fibroblasts: A comprehensive review. Int J Oncol (2013) 42(6):1849–57. doi: 10.3892/ijo.2013.1887
31. Lowe KL, Navarro-Nunez L, Watson SP. Platelet clec-2 and podoplanin in cancer metastasis. Thromb Res (2012) 129 Suppl 1:S30–7. doi: 10.1016/S0049-3848(12)70013-0
32. Takagi S, Sato S, Oh-hara T, Takami M, Koike S, Mishima Y, et al. Platelets promote tumor growth and metastasis Via direct interaction between Aggrus/Podoplanin and clec-2. PloS One (2013) 8(8):e73609. doi: 10.1371/journal.pone.0073609
33. Miyashita T, Tajima H, Gabata R, Okazaki M, Shimbashi H, Ohbatake Y, et al. Impact of extravasated platelet activation and podoplanin-positive cancer-associated fibroblasts in pancreatic cancer stroma. Anticancer Res (2019) 39(10):5565–72. doi: 10.21873/anticanres.13750
34. Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell (2014) 25(6):719–34. doi: 10.1016/j.ccr.2014.04.005
35. Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell (2014) 25(6):735–47. doi: 10.1016/j.ccr.2014.04.021
36. Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discovery (2019) 9(8):1102–23. doi: 10.1158/2159-8290.CD-19-0094
37. Biffi G, Tuveson DA. Diversity and biology of cancer-associated fibroblasts. Physiol Rev (2021) 101(1):147–76. doi: 10.1152/physrev.00048.2019
38. Lee JJ, Perera RM, Wang H, Wu DC, Liu XS, Han S, et al. Stromal response to hedgehog signaling restrains pancreatic cancer progression. Proc Natl Acad Sci U.S.A. (2014) 111(30):E3091–100. doi: 10.1073/pnas.1411679111
39. Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, et al. Il1-induced Jak/Stat signaling is antagonized by tgfbeta to shape caf heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discovery (2019) 9(2):282–301. doi: 10.1158/2159-8290.CD-18-0710
40. Dominguez CX, Muller S, Keerthivasan S, Koeppen H, Hung J, Gierke S, et al. Single-cell rna sequencing reveals stromal evolution into Lrrc15(+) myofibroblasts as a determinant of patient response to cancer immunotherapy. Cancer Discovery (2020) 10(2):232–53. doi: 10.1158/2159-8290.CD-19-0644
41. Ohlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med (2017) 214(3):579–96. doi: 10.1084/jem.20162024
42. Geng X, Chen H, Zhao L, Hu J, Yang W, Li G, et al. Cancer-associated fibroblast (Caf) heterogeneity and targeting therapy of cafs in pancreatic cancer. Front Cell Dev Biol (2021) 9:655152. doi: 10.3389/fcell.2021.655152
43. Armstrong T, Packham G, Murphy LB, Bateman AC, Conti JA, Fine DR, et al. Type I collagen promotes the malignant phenotype of pancreatic ductal adenocarcinoma. Clin Cancer Res (2004) 10(21):7427–37. doi: 10.1158/1078-0432.CCR-03-0825
44. Berchtold S, Grunwald B, Kruger A, Reithmeier A, Hahl T, Cheng T, et al. Collagen type V promotes the malignant phenotype of pancreatic ductal adenocarcinoma. Cancer Lett (2015) 356(2 Pt B):721–32. doi: 10.1016/j.canlet.2014.10.020
45. Ohlund D, Franklin O, Lundberg E, Lundin C, Sund M. Type iv collagen stimulates pancreatic cancer cell proliferation, migration, and inhibits apoptosis through an autocrine loop. BMC Cancer (2013) 13:154. doi: 10.1186/1471-2407-13-154
46. Xu S, Xu H, Wang W, Li S, Li H, Li T, et al. The role of collagen in cancer: From bench to bedside. J Transl Med (2019) 17(1):309. doi: 10.1186/s12967-019-2058-1
47. Nakajima K, Ino Y, Naito C, Nara S, Shimasaki M, Ishimoto U, et al. Neoadjuvant therapy alters the collagen architecture of pancreatic cancer tissue Via ephrin-A5. Br J Cancer (2022) 126(4):628–39. doi: 10.1038/s41416-021-01639-9
48. Hartley C, Rowan D, Chen X, Gomez-Arellano L, West AM, Oshima K, et al. Increased sparc expression is associated with neoadjuvant therapy in resectable pancreatic ductal adenocarcinoma. Pract Lab Med (2020) 21:e00171. doi: 10.1016/j.plabm.2020.e00171
49. Mueller AC, Piper M, Goodspeed A, Bhuvane S, Williams JS, Bhatia S, et al. Induction of Adam10 by radiation therapy drives fibrosis, resistance, and epithelial-to-Mesenchyal transition in pancreatic cancer. Cancer Res (2021) 81(12):3255–69. doi: 10.1158/0008-5472.CAN-20-3892
50. Cantero D, Friess H, Deflorin J, Zimmermann A, Brundler MA, Riesle E, et al. Enhanced expression of urokinase plasminogen activator and its receptor in pancreatic carcinoma. Br J Cancer (1997) 75(3):388–95. doi: 10.1038/bjc.1997.63
51. Wu MY, Shen M, Xu MD, Yu ZY, Tao M. Folfirinox regulated tumor immune microenvironment to extend the survival of patients with resectable pancreatic ductal adenocarcinoma. Gland Surg (2020) 9(6):2125–35. doi: 10.21037/gs-20-828
52. Matsuda Y, Inoue Y, Hiratsuka M, Kawakatsu S, Arai T, Matsueda K, et al. Encapsulating fibrosis following neoadjuvant chemotherapy is correlated with outcomes in patients with pancreatic cancer. PloS One (2019) 14(9):e0222155. doi: 10.1371/journal.pone.0222155
53. Pan Y, Yu Y, Wang X, Zhang T. Tumor-associated macrophages in tumor immunity. Front Immunol (2020) 11:583084. doi: 10.3389/fimmu.2020.583084
54. Michelakos T, Cai L, Villani V, Sabbatino F, Kontos F, Fernandez-Del Castillo C, et al. Tumor microenvironment immune response in pancreatic ductal adenocarcinoma patients treated with neoadjuvant therapy. J Natl Cancer Inst (2021) 113(2):182–91. doi: 10.1093/jnci/djaa073
55. Di Caro G, Cortese N, Castino GF, Grizzi F, Gavazzi F, Ridolfi C, et al. Dual prognostic significance of tumour-associated macrophages in human pancreatic adenocarcinoma treated or untreated with chemotherapy. Gut (2016) 65(10):1710–20. doi: 10.1136/gutjnl-2015-309193
56. Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, et al. Low-dose irradiation programs macrophage differentiation to an Inos(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell (2013) 24(5):589–602. doi: 10.1016/j.ccr.2013.09.014
57. Wu Q, Allouch A, Martins I, Modjtahedi N, Deutsch E, Perfettini JL. Macrophage biology plays a central role during ionizing radiation-elicited tumor response. BioMed J (2017) 40(4):200–11. doi: 10.1016/j.bj.2017.06.003
58. Matsuki H, Hiroshima Y, Miyake K, Murakami T, Homma Y, Matsuyama R, et al. Reduction of gender-associated M2-like tumor-associated macrophages in the tumor microenvironment of patients with pancreatic cancer after neoadjuvant chemoradiotherapy. J Hepatob Pancreat Sci (2021) 28(2):174–82. doi: 10.1002/jhbp.883
59. Chistiakov DA, Myasoedova VA, Revin VV, Orekhov AN, Bobryshev YV. The impact of interferon-regulatory factors to macrophage differentiation and polarization into M1 and M2. Immunobiology (2018) 223(1):101–11. doi: 10.1016/j.imbio.2017.10.005
60. Yanai H, Chen HM, Inuzuka T, Kondo S, Mak TW, Takaoka A, et al. Role of ifn regulatory factor 5 transcription factor in antiviral immunity and tumor suppression. Proc Natl Acad Sci U.S.A. (2007) 104(9):3402–7. doi: 10.1073/pnas.0611559104
61. Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, Sahgal N, et al. Irf5 promotes inflammatory macrophage polarization and Th1-Th17 responses. Nat Immunol (2011) 12(3):231–8. doi: 10.1038/ni.1990
62. Okubo S, Suzuki T, Hioki M, Shimizu Y, Toyama H, Morinaga S, et al. The immunological impact of preoperative chemoradiotherapy on the tumor microenvironment of pancreatic cancer. Cancer Sci (2021) 112(7):2895–904. doi: 10.1111/cas.14914
63. Homma Y, Taniguchi K, Murakami T, Nakagawa K, Nakazawa M, Matsuyama R, et al. Immunological impact of neoadjuvant chemoradiotherapy in patients with borderline resectable pancreatic ductal adenocarcinoma. Ann Surg Oncol (2014) 21(2):670–6. doi: 10.1245/s10434-013-3390-y
64. Tsuchikawa T, Hirano S, Tanaka E, Matsumoto J, Kato K, Nakamura T, et al. Novel aspects of preoperative chemoradiation therapy improving anti-tumor immunity in pancreatic cancer. Cancer Sci (2013) 104(5):531–5. doi: 10.1111/cas.12119
65. Shibuya KC, Goel VK, Xiong W, Sham JG, Pollack SM, Leahy AM, et al. Pancreatic ductal adenocarcinoma contains an effector and regulatory immune cell infiltrate that is altered by multimodal neoadjuvant treatment. PloS One (2014) 9(5):e96565. doi: 10.1371/journal.pone.0096565
66. Nejati R, Goldstein JB, Halperin DM, Wang H, Hejazi N, Rashid A, et al. Prognostic significance of tumor-infiltrating lymphocytes in patients with pancreatic ductal adenocarcinoma treated with neoadjuvant chemotherapy. Pancreas (2017) 46(9):1180–7. doi: 10.1097/MPA.0000000000000914
67. Karakhanova S, Ryschich E, Mosl B, Harig S, Jager D, Schmidt J, et al. Prognostic and predictive value of immunological parameters for chemoradioimmunotherapy in patients with pancreatic adenocarcinoma. Br J Cancer (2015) 112(6):1027–36. doi: 10.1038/bjc.2015.72
68. Liu C, Cheng H, Luo G, Lu Y, Jin K, Guo M, et al. Circulating regulatory T cell subsets predict overall survival of patients with unresectable pancreatic cancer. Int J Oncol (2017) 51(2):686–94. doi: 10.3892/ijo.2017.4032
69. Farren MR, Sayegh L, Ware MB, Chen HR, Gong J, Liang Y, et al. Immunologic alterations in the pancreatic cancer microenvironment of patients treated with neoadjuvant chemotherapy and radiotherapy. JCI Insight (2020) 5(1):e130362. doi: 10.1172/jci.insight.130362
70. Papalampros A, Vailas M, Ntostoglou K, Chiloeches ML, Sakellariou S, Chouliari NV, et al. Unique spatial immune profiling in pancreatic ductal adenocarcinoma with enrichment of exhausted and senescent T cells and diffused Cd47-sirpalpha expression. Cancers (Basel) (2020) 12(7):1825. doi: 10.3390/cancers12071825
71. Mills BN, Connolly KA, Ye J, Murphy JD, Uccello TP, Han BJ, et al. Stereotactic body radiation and interleukin-12 combination therapy eradicates pancreatic tumors by repolarizing the immune microenvironment. Cell Rep (2019) 29(2):406–21 e5. doi: 10.1016/j.celrep.2019.08.095
72. Pei Q, Pan J, Ding X, Wang J, Zou X, Lv Y. Gemcitabine sensitizes pancreatic cancer cells to the ctls antitumor response induced by bcg-stimulated dendritic cells Via a fas-dependent pathway. Pancreatology (2015) 15(3):233–9. doi: 10.1016/j.pan.2015.04.001
73. Javeed A, Ashraf M, Riaz A, Ghafoor A, Afzal S, Mukhtar MM. Paclitaxel and immune system. Eur J Pharm Sci (2009) 38(4):283–90. doi: 10.1016/j.ejps.2009.08.009
74. Zhang L, Dermawan K, Jin M, Liu R, Zheng H, Xu L, et al. Differential impairment of regulatory T cells rather than effector T cells by paclitaxel-based chemotherapy. Clin Immunol (2008) 129(2):219–29. doi: 10.1016/j.clim.2008.07.013
75. Delitto D, Black BS, Sorenson HL, Knowlton AE, Thomas RM, Sarosi GA, et al. The inflammatory milieu within the pancreatic cancer microenvironment correlates with clinicopathologic parameters, chemoresistance and survival. BMC Cancer (2015) 15:783. doi: 10.1186/s12885-015-1820-x
76. Stylianopoulos T, Martin JD, Chauhan VP, Jain SR, Diop-Frimpong B, Bardeesy N, et al. Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc Natl Acad Sci U.S.A. (2012) 109(38):15101–8. doi: 10.1073/pnas.1213353109
77. Chen IX, Chauhan VP, Posada J, Ng MR, Wu MW, Adstamongkonkul P, et al. Blocking Cxcr4 alleviates desmoplasia, increases T-lymphocyte infiltration, and improves immunotherapy in metastatic breast cancer. Proc Natl Acad Sci U.S.A. (2019) 116(10):4558–66. doi: 10.1073/pnas.1815515116
78. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. Tgfbeta attenuates tumour response to pd-L1 blockade by contributing to exclusion of T cells. Nature (2018) 554(7693):544–8. doi: 10.1038/nature25501
79. Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. Tgfbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature (2018) 554(7693):538–43. doi: 10.1038/nature25492
80. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. Pd-L1 is a novel direct target of hif-1alpha, and its blockade under hypoxia enhanced mdsc-mediated T cell activation. J Exp Med (2014) 211(5):781–90. doi: 10.1084/jem.20131916
81. Hong M, Puaux AL, Huang C, Loumagne L, Tow C, Mackay C, et al. Chemotherapy induces intratumoral expression of chemokines in cutaneous melanoma, favoring T-cell infiltration and tumor control. Cancer Res (2011) 71(22):6997–7009. doi: 10.1158/0008-5472.CAN-11-1466
82. Romero JM, Grunwald B, Jang GH, Bavi PP, Jhaveri A, Masoomian M, et al. A four-chemokine signature is associated with a T-Cell-Inflamed phenotype in primary and metastatic pancreatic cancer. Clin Cancer Res (2020) 26(8):1997–2010. doi: 10.1158/1078-0432.CCR-19-2803
83. Fucikova J, Kepp O, Kasikova L, Petroni G, Yamazaki T, Liu P, et al. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis (2020) 11(11):1013. doi: 10.1038/s41419-020-03221-2
84. Zhou J, Wang G, Chen Y, Wang H, Hua Y, Cai Z. Immunogenic cell death in cancer therapy: Present and emerging inducers. J Cell Mol Med (2019) 23(8):4854–65. doi: 10.1111/jcmm.14356
85. Galluzzi L, Vitale I, Warren S, Adjemian S, Agostinis P, Martinez AB, et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J Immunother Cancer (2020) 8(1):e000337. doi: 10.1136/jitc-2019-000337
86. Garg AD, Krysko DV, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T, et al. A novel pathway combining calreticulin exposure and atp secretion in immunogenic cancer cell death. EMBO J (2012) 31(5):1062–79. doi: 10.1038/emboj.2011.497
87. Tesniere A, Panaretakis T, Kepp O, Apetoh L, Ghiringhelli F, Zitvogel L, et al. Molecular characteristics of immunogenic cancer cell death. Cell Death Differ (2008) 15(1):3–12. doi: 10.1038/sj.cdd.4402269
88. Garg AD, Nowis D, Golab J, Vandenabeele P, Krysko DV, Agostinis P. Immunogenic cell death, damps and anticancer therapeutics: An emerging amalgamation. Biochim Biophys Acta (2010) 1805(1):53–71. doi: 10.1016/j.bbcan.2009.08.003
89. Chen D, Gyllensten U. Mica polymorphism: Biology and importance in cancer. Carcinogenesis (2014) 35(12):2633–42. doi: 10.1093/carcin/bgu215
90. Murakami T, Homma Y, Matsuyama R, Mori R, Miyake K, Tanaka Y, et al. Neoadjuvant chemoradiotherapy of pancreatic cancer induces a favorable immunogenic tumor microenvironment associated with increased major histocompatibility complex class I-related chain a/B expression. J Surg Oncol (2017) 116(3):416–26. doi: 10.1002/jso.24681
91. Gameiro SR, Jammeh ML, Wattenberg MM, Tsang KY, Ferrone S, Hodge JW. Radiation-induced immunogenic modulation of tumor enhances antigen processing and calreticulin exposure, resulting in enhanced T-cell killing. Oncotarget (2014) 5(2):403–16. doi: 10.18632/oncotarget.1719
92. Jang JE, Hajdu CH, Liot C, Miller G, Dustin ML, Bar-Sagi D. Crosstalk between regulatory T cells and tumor-associated dendritic cells negates anti-tumor immunity in pancreatic cancer. Cell Rep (2017) 20(3):558–71. doi: 10.1016/j.celrep.2017.06.062
93. Bronte V, Serafini P, Mazzoni A, Segal DM, Zanovello P. L-arginine metabolism in myeloid cells controls T-lymphocyte functions. Trends Immunol (2003) 24(6):302–6. doi: 10.1016/s1471-4906(03)00132-7
94. Danilova L, Anagnostou V, Caushi JX, Sidhom JW, Guo H, Chan HY, et al. The mutation-associated neoantigen functional expansion of specific T cells (Manafest) assay: A sensitive platform for monitoring antitumor immunity. Cancer Immunol Res (2018) 6(8):888–99. doi: 10.1158/2326-6066.CIR-18-0129
95. Ji D, Yi H, Zhang D, Zhan T, Li Z, Li M, et al. Somatic mutations and immune alternation in rectal cancer following neoadjuvant chemoradiotherapy. Cancer Immunol Res (2018) 6(11):1401–16. doi: 10.1158/2326-6066.CIR-17-0630
96. Lhuillier C, Rudqvist NP, Yamazaki T, Zhang T, Charpentier M, Galluzzi L, et al. Radiotherapy-exposed Cd8+ and Cd4+ neoantigens enhance tumor control. J Clin Invest (2021) 131(5):e138740. doi: 10.1172/JCI138740
97. Inoue S, Leitner WW, Golding B, Scott D. Inhibitory effects of b cells on antitumor immunity. Cancer Res (2006) 66(15):7741–7. doi: 10.1158/0008-5472.CAN-05-3766
98. Hane Y, Tsuchikawa T, Nakamura T, Hatanaka KC, Saito T, Tanaka K, et al. Immunological gene signature associated with the tumor microenvironment of pancreatic cancer after neoadjuvant chemotherapy. Pancreas (2020) 49(9):1240–5. doi: 10.1097/MPA.0000000000001665
99. Miyake K, Mori R, Homma Y, Matsuyama R, Okayama A, Murakami T, et al. Mzb1 in borderline resectable pancreatic cancer resected after neoadjuvant chemoradiotherapy. J Surg Res (2017) 220:391–401. doi: 10.1016/j.jss.2017.07.003
100. Lin X, Ye L, Wang X, Liao Z, Dong J, Yang Y, et al. Follicular helper T cells remodel the immune microenvironment of pancreatic cancer via secreting Cxcl13 and il-21. Cancers (Basel) (2021) 13(15):3678. doi: 10.3390/cancers13153678
101. Fang H, Ang B, Xu X, Huang X, Wu Y, Sun Y, et al. Tlr4 is essential for dendritic cell activation and anti-tumor T-cell response enhancement by damps released from chemically stressed cancer cells. Cell Mol Immunol (2014) 11(2):150–9. doi: 10.1038/cmi.2013.59
102. Cueto FJ, Sancho D. The Flt3l/Flt3 axis in dendritic cell biology and cancer immunotherapy. Cancers (Basel) (2021) 13(7):1525. doi: 10.3390/cancers13071525
103. Waskow C, Liu K, Darrasse-Jeze G, Guermonprez P, Ginhoux F, Merad M, et al. The receptor tyrosine kinase Flt3 is required for dendritic cell development in peripheral lymphoid tissues. Nat Immunol (2008) 9(6):676–83. doi: 10.1038/ni.1615
104. Morisaki T, Onishi H, Koya N, Kiyota A, Tanaka H, Umebayashi M, et al. Combinatorial cytotoxicity of gemcitabine and cytokine-activated killer cells in hepatocellular carcinoma Via the Nkg2d-Mica/B system. Anticancer Res (2011) 31(7):2505–10.
105. Miyashita T, Miki K, Kamigaki T, Makino I, Nakagawara H, Tajima H, et al. Low-dose gemcitabine induces major histocompatibility complex class I-related chain a/B expression and enhances an antitumor innate immune response in pancreatic cancer. Clin Exp Med (2017) 17(1):19–31. doi: 10.1007/s10238-015-0394-x
106. Zhang R, Liu Q, Peng J, Wang M, Li T, Liu J, et al. Cxcl5 overexpression predicts a poor prognosis in pancreatic ductal adenocarcinoma and is correlated with immune cell infiltration. J Cancer (2020) 11(9):2371–81. doi: 10.7150/jca.40517
107. Baggiolini M, Clark-Lewis I. Interleukin-8, a chemotactic and inflammatory cytokine. FEBS Lett (1992) 307(1):97–101. doi: 10.1016/0014-5793(92)80909-z
108. Chao T, Furth EE, Vonderheide RH. Cxcr2-dependent accumulation of tumor-associated neutrophils regulates T-cell immunity in pancreatic ductal adenocarcinoma. Cancer Immunol Res (2016) 4(11):968–82. doi: 10.1158/2326-6066.CIR-16-0188
109. van de Pavert SA, Mebius RE. New insights into the development of lymphoid tissues. Nat Rev Immunol (2010) 10(9):664–74. doi: 10.1038/nri2832
110. Kuwabara S, Tsuchikawa T, Nakamura T, Hatanaka Y, Hatanaka KC, Sasaki K, et al. Prognostic relevance of tertiary lymphoid organs following neoadjuvant chemoradiotherapy in pancreatic ductal adenocarcinoma. Cancer Sci (2019) 110(6):1853–62. doi: 10.1111/cas.14023
111. Lutz ER, Wu AA, Bigelow E, Sharma R, Mo G, Soares K, et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol Res (2014) 2(7):616–31. doi: 10.1158/2326-6066.CIR-14-0027
112. Daud AI, Loo K, Pauli ML, Sanchez-Rodriguez R, Sandoval PM, Taravati K, et al. Tumor immune profiling predicts response to anti-Pd-1 therapy in human melanoma. J Clin Invest (2016) 126(9):3447–52. doi: 10.1172/JCI87324
113. Disis ML. Immunologic biomarkers as correlates of clinical response to cancer immunotherapy. Cancer Immunol Immunother (2011) 60(3):433–42. doi: 10.1007/s00262-010-0960-8
114. Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K, et al. Endothelin b receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med (2008) 14(1):28–36. doi: 10.1038/nm1699
115. Smyth MJ, Ngiow SF, Ribas A, Teng MW. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat Rev Clin Oncol (2016) 13(3):143–58. doi: 10.1038/nrclinonc.2015.209
116. Weiss GJ, Waypa J, Blaydorn L, Coats J, McGahey K, Sangal A, et al. A phase ib study of pembrolizumab plus chemotherapy in patients with advanced cancer (Pembroplus). Br J Cancer (2017) 117(1):33–40. doi: 10.1038/bjc.2017.145
117. Sharabi AB, Lim M, DeWeese TL, Drake CG. Radiation and checkpoint blockade immunotherapy: Radiosensitisation and potential mechanisms of synergy. Lancet Oncol (2015) 16(13):e498–509. doi: 10.1016/S1470-2045(15)00007-8
118. van Kooten C, Banchereau J. Cd40-Cd40 ligand. J Leukoc Biol (2000) 67(1):2–17. doi: 10.1002/jlb.67.1.2
119. Vonderheide RH. Cd40 agonist antibodies in cancer immunotherapy. Annu Rev Med (2020) 71:47–58. doi: 10.1146/annurev-med-062518-045435
120. Byrne KT, Vonderheide RH. Cd40 stimulation obviates innate sensors and drives T cell immunity in cancer. Cell Rep (2016) 15(12):2719–32. doi: 10.1016/j.celrep.2016.05.058
121. Beatty GL, Torigian DA, Chiorean EG, Saboury B, Brothers A, Alavi A, et al. A phase I study of an agonist Cd40 monoclonal antibody (Cp-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin Cancer Res (2013) 19(22):6286–95. doi: 10.1158/1078-0432.CCR-13-1320
122. O’Hara MH, O’Reilly EM, Varadhachary G, Wolff RA, Wainberg ZA, Ko AH, et al. Cd40 agonistic monoclonal antibody Apx005m (Sotigalimab) and chemotherapy, with or without nivolumab, for the treatment of metastatic pancreatic adenocarcinoma: An open-label, multicentre, phase 1b study. Lancet Oncol (2021) 22(1):118–31. doi: 10.1016/S1470-2045(20)30532-5
123. Winograd R, Byrne KT, Evans RA, Odorizzi PM, Meyer AR, Bajor DL, et al. Induction of T-cell immunity overcomes complete resistance to pd-1 and ctla-4 blockade and improves survival in pancreatic carcinoma. Cancer Immunol Res (2015) 3(4):399–411. doi: 10.1158/2326-6066.CIR-14-0215
124. Rech AJ, Dada H, Kotzin JJ, Henao-Mejia J, Minn AJ, Twyman-Saint Victor C, et al. Radiotherapy and Cd40 activation separately augment immunity to checkpoint blockade in cancer. Cancer Res (2018) 78(15):4282–91. doi: 10.1158/0008-5472.CAN-17-3821
125. Byrne KT, Betts CB, Mick R, Sivagnanam S, Bajor DL, Laheru DA, et al. Neoadjuvant selicrelumab, an agonist Cd40 antibody, induces changes in the tumor microenvironment in patients with resectable pancreatic cancer. Clin Cancer Res (2021) 27(16):4574–86. doi: 10.1158/1078-0432.CCR-21-1047
126. Stern LA, Jonsson VD, Priceman SJ. Car T cell therapy progress and challenges for solid tumors. Cancer Treat Res (2020) 180:297–326. doi: 10.1007/978-3-030-38862-1_11
127. Rozenbaum M, Meir A, Aharony Y, Itzhaki O, Schachter J, Bank I, et al. Gamma-delta car-T cells show car-directed and independent activity against leukemia. Front Immunol (2020) 11:1347. doi: 10.3389/fimmu.2020.01347
128. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of car-transduced natural killer cells in Cd19-positive lymphoid tumors. N Engl J Med (2020) 382(6):545–53. doi: 10.1056/NEJMoa1910607
129. Xu X, Huang W, Heczey A, Liu D, Guo L, Wood M, et al. Nkt cells coexpressing a Gd2-specific chimeric antigen receptor and Il15 show enhanced in vivo persistence and antitumor activity against neuroblastoma. Clin Cancer Res (2019) 25(23):7126–38. doi: 10.1158/1078-0432.CCR-19-0421
130. Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M, et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol (2020) 38(8):947–53. doi: 10.1038/s41587-020-0462-y
131. Xie G, Dong H, Liang Y, Ham JD, Rizwan R, Chen J. Car-nk cells: A promising cellular immunotherapy for cancer. EBioMedicine (2020) 59:102975. doi: 10.1016/j.ebiom.2020.102975
132. Martinez M, Moon EK. Car T cells for solid tumors: New strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front Immunol (2019) 10:128. doi: 10.3389/fimmu.2019.00128
133. Ye B, Stary CM, Li X, Gao Q, Kang C, Xiong X. Engineering chimeric antigen receptor-T cells for cancer treatment. Mol Cancer (2018) 17(1):32. doi: 10.1186/s12943-018-0814-0
134. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol (2015) 15(8):486–99. doi: 10.1038/nri3862
135. Zeng Z, Wei F, Ren X. Exhausted T cells and epigenetic status. Cancer Biol Med (2020) 17(4):923–36. doi: 10.20892/j.issn.2095-3941.2020.0338
136. Dolina JS, Van Braeckel-Budimir N, Thomas GD, Salek-Ardakani S. Cd8(+) T cell exhaustion in cancer. Front Immunol (2021) 12:715234. doi: 10.3389/fimmu.2021.715234
137. Farhood B, Najafi M, Mortezaee K. Cd8(+) cytotoxic T lymphocytes in cancer immunotherapy: A review. J Cell Physiol (2019) 234(6):8509–21. doi: 10.1002/jcp.27782
138. Garg AD, Coulie PG, Van den Eynde BJ, Agostinis P. Integrating next-generation dendritic cell vaccines into the current cancer immunotherapy> landscape. Trends Immunol (2017) 38(8):577–93. doi: 10.1016/j.it.2017.05.006
139. Sadeghzadeh M, Bornehdeli S, Mohahammadrezakhani H, Abolghasemi M, Poursaei E, Asadi M, et al. Dendritic cell therapy in cancer treatment; the state-of-the-Art. Life Sci (2020) 254:117580. doi: 10.1016/j.lfs.2020.117580
140. Zaidi N, Quezada SA, Kuroiwa JMY, Zhang L, Jaffee EM, Steinman RM, et al. Anti-Ctla-4 synergizes with dendritic cell-targeted vaccine to promote il-3-Dependent Cd4(+) effector T cell infiltration into murine pancreatic tumors. Ann N Y Acad Sci (2019) 1445(1):62–73. doi: 10.1111/nyas.14049
141. Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med (2003) 9(5):562–7. doi: 10.1038/nm863
142. Lau SP, van Montfoort N, Kinderman P, Lukkes M, Klaase L, van Nimwegen M, et al. Dendritic cell vaccination and Cd40-agonist combination therapy licenses T cell-dependent antitumor immunity in a pancreatic carcinoma murine model. J Immunother Cancer (2020) 8(2):e000772. doi: 10.1136/jitc-2020-000772
143. Lu YC, Robbins PF. Cancer immunotherapy targeting neoantigens. Semin Immunol (2016) 28(1):22–7. doi: 10.1016/j.smim.2015.11.002
144. Yamamoto TN, Kishton RJ, Restifo NP. Developing neoantigen-targeted T cell-based treatments for solid tumors. Nat Med (2019) 25(10):1488–99. doi: 10.1038/s41591-019-0596-y
145. Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, et al. Cancer immunotherapy. a dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science (2015) 348(6236):803–8. doi: 10.1126/science.aaa3828
146. Leidner R, Sanjuan Silva N, Huang H, Sprott D, Zheng C, Shih YP, et al. Neoantigen T-cell receptor gene therapy in pancreatic cancer. N Engl J Med (2022) 386(22):2112–9. doi: 10.1056/NEJMoa2119662
147. Kim SP, Vale NR, Zacharakis N, Krishna S, Yu Z, Gasmi B, et al. Adoptive cellular therapy with autologous tumor-infiltrating lymphocytes and T-cell receptor-engineered T cells targeting common P53 neoantigens in human solid tumors. Cancer Immunol Res (2022) 10(8):932–46. doi: 10.1158/2326-6066.CIR-22-0040
Keywords: pancreatic cancer, tumor environment (TME), immune, immunology, neoadjuvant therapy
Citation: Zhang H, Ye L, Yu X, Jin K and Wu W (2022) Neoadjuvant therapy alters the immune microenvironment in pancreatic cancer. Front. Immunol. 13:956984. doi: 10.3389/fimmu.2022.956984
Received: 30 May 2022; Accepted: 08 September 2022;
Published: 26 September 2022.
Edited by:
Rakesh K. Singh, University of Nebraska Medical Center, United StatesReviewed by:
Akhil Chawla, Northwestern Medicine, United StatesCasper van Eijck, Erasmus University Rotterdam, Netherlands
Copyright © 2022 Zhang, Ye, Yu, Jin and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weiding Wu, d3V3ZWlkaW5nQGZ1ZGFucGNpLm9yZw==; Kaizhou Jin, amlua2FpemhvdUBmdWRhbnBjaS5vcmc=; Xianjun Yu, eXV4aWFuanVuQGZ1ZGFucGNpLm9yZw==
†These authors have contributed equally to this work